
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering extracellular vesicles derived from macrophages for tumor therapy: a review
Ying
Yan†
b,
He
Zhang†
a,
Shiqi
Wei
acd,
Weimin
Xie
b,
Ying
Chen
acd and
Huaming
Yang
 *abcd
*abcd
aEngineering Research Center of Nano-Geomaterials of Ministry of Education, China University of Geosciences, Wuhan 430074, P. R. China. E-mail: hm.yang@cug.edu.cn; hmyang@csu.edu.cn; Fax: +86-27-68788733; Tel: +86-27-68788701
bHunan Key Laboratory of Mineral Materials and Application, School of Minerals Processing and Bioengineering, Central South University, Changsha 410083, P. R. China
cFaculty of Materials Science and Chemistry, China University of Geosciences, Wuhan 430074, P. R. China
dKey Laboratory of Functional Geomaterials in China Nonmetallic Minerals Industry, China University of Geosciences, Wuhan 430074, P. R. China
First published on 2nd February 2023
Abstract
In medicine, surgical operation, radiotherapy, chemotherapy and other treatment methods have achieved tumor treatment, but the treatment effect of these methods isn’t particularly ideal, and the tumor cannot be completely cured. Compared with traditional treatments, immunotherapy using nanoparticles can achieve lasting efficacy. Macrophages are important immune cells in living organisms, they play a key role in innate immunity or adaptive immunity. Tumor-associated macrophages (TAMs) are the key link between tumor immunosuppression and tumor progression, which play a vital role in the process of tumor occurrence, development, invasion and metastasis. Although many studies have shown that changing macrophage phenotype plays a role in tumor treatment, these treatments also cause greater immune rejection. Macrophage-derived extracellular vesicles (EVs) are small vesicles with nanoscale bilayer membrane structure secreted by macrophages in an organism. They can carry bioactive substances, such as proteins, lipids and nucleic acids containing parental cell information and mediate the information transmission among cells. However, EVs also have some disadvantages, such as low yield, insufficient targeting and too many ineffective components. Engineering EVs can be particularly useful in ameliorating these problems. Therefore, engineering EVs derived from macrophages can be used in tumor therapy. This paper briefly reviews the application of macrophage-derived EVs in tumor therapy.
1. Introduction
Every country expends a great deal of human, physical and financial resources on cancer therapy. Although achievements have been obtained, these achievements pale in comparison with investment. Cancer is one of the most common and high mortality diseases in the world due to the proliferation of various cancer triggers, such as widespread smoking and alcohol consumption, increasing water pollution and air pollution, and the current limited level of cancer treatment.1 For example, traditional cancer treatments including surgery, radiotherapy and chemotherapy, have associated side effects due to their lack of targeting to cancer cells, which may have a negative impact on the life quality.2,3 Surprisingly, nanoparticles mediated drug delivery platforms show great potential in significantly improving the delivery of tumor therapeutic drugs due to their small size and long circulation time. Indeed, these platforms have improved the therapeutic effect of some chemotherapeutic drugs, but their clinical applications are limited by the poor performance of nanoparticles, mainly including premature drug release, lack of specific tumor targeting, poor penetration, and so on.4 Therefore, there is imperative to develop more effective and more tolerant anticancer treatments. More and more new treatment strategies such as immunotherapy using nanoparticles have been established to obtain high survival rates and low adverse reactions in cancer patients.The occurrence and development of tumor are a complex process involving multiple factors, including excessive proliferation, invasion and migration of tumor cells, epithelial–mesenchymal transition and angiogenesis. Blood vessels provide nutrition and oxygen to tumor cells, which is a crucial link in the process of malignant progression of tumor.5 As the growth of tumor, tumor cells, peripheral immune cells, stromal cells, endothelial cells, other cellular components and noncellular components such as cytokines and chemokines all combine to form the tumor microenvironment (TME),6 that is, the place where neovascularization occurs. Macrophages in TME, also called TAMs, the largest number of cells except for tumor cells, play a significant role in tumor angiogenesis. Therefore, the effects of TAMs on tumor cells and tumor angiogenesis have been extensively studied and targeting macrophages in TME is an important anti-tumor strategy.
EVs are the collective name for structures surrounded by phospholipid bilayers that are naturally secreted by cells, which can be regarded as membrane lacking functionality. It is a collective term for many subtypes, including exosomes, microvesicles, granules, apoptotic bodies and oncosomes.7 Exosomes are membranous vesicles with a diameter of 30–150 nm that are released into the extracellular space by the fusion of multivesicular body (MVBs) formed by cell membrane invagination, Fig. 1 is a schematic diagram of the process of exosome generation and release.8 Microvesicles (MVs) have different sizes, ranging from 100 to 1000 nm in diameter. They sprout and shed from the cell membrane.9 These vesicles of varying sizes contain proteins, lipids, messenger RNA (mRNA), microRNA (miRNA), long non-coding RNA (lncRNA) and mitochondria.10,11 For specific EVs, it is difficult to determine their specific biogenic pathway, so the term EV recommended by the International Association of EVs is used in this review.12
 | ||
| Fig. 1 Exosome biogenesis and/or release. The plasma membrane invaginates and buds to form early endosomes, the early endosomes are internally acidified to form late endosomes, the late endosomes invaginate again to form luminal MVBs, and finally they fuse with the plasma membrane to release exosomes, which involve a variety of proteins in this process, or the luminal MVBs are degraded by lysosomes. Fig. 1 was reproduced from ref. 8 with permission from Springer Nature, Copyright 2017. | ||
EVs are released by various cell types including immune cells (macrophages, B lymphocytes, T lymphocytes and dendritic cells) and structural cells (endothelial cells, alveolar macrophages, type I and type II alveolar epithelial cells, fibroblasts and smooth muscle cells).13,14 Due to the diversity of sources, EVs can be detected in a variety of body fluids, including blood, urine, saliva, breast milk, amniotic fluid, pleural effusion and ascites, semen, cerebrospinal fluid and others.9,15 EVs transmit complex biological information to recipient cells through their bioactive substances to regulate their behaviors, which is a key medium for intercellular communication.16 In this way, EVs are involved in the pathogenesis and development of various diseases, and the number, type and carrier substances of EVs released by cells in the state of disease are different, which indicates that EVs have the potential as a new biomarker for diagnosis and prognosis.17 On the one hand, the engineering EVs can realize the multiple functions of vesicles, on the other hand, it can improve the treatment efficiency of diseases. This paper focuses on the important role of macrophages in tumorigenesis, the function of macrophage EVs in tumor, and current research on engineering macrophage-derived EVs in tumor therapy.
2. Outline of engineering EVs
2.1 EV subtypes and functions
EVs are a collection of heterogeneous membrane-bound vesicles with different sizes, carriers, membrane composition and bioactivity. EVs can also be classified by size: 30–150 nm, 100–1000 nm, and 1000–5000 nm which are referred to as exosomes, MVs, and apoptotic bodies, respectively. Because size, density, contents, membrane orientation and surface molecules of vesicle subtypes overlap, the existing separation and analysis methods are difficult to distinguish effectively.7–9 At present, there is no uniform standard nomenclature, so exosomes, MVs and extranuclear granules are collectively referred to as EVs in this paper. EV carriers are functional, including genetic materials, proteins, lipids and soluble mediators. MicroRNAs (miRNAs) are frequently studied, they are a kind of noncoding RNA with a length of about 22 nucleotides and can regulate post-transcriptional genes that bind to target mRNA, thereby affecting physiological processes and diseases.18 EVs can act as carriers to protect free miRNAs from degradation in body fluids. In addition to the encapsulated carriers, EV membrane proteins and lipids also play a role through autocrine, paracrine and endocrine signals.19 EV surface molecules can serve as specific binding sites after receptor and ligand binding target EVs. On the plasma membrane of receptor cells, for example, EVs activate downstream signal transduction, endocytosis or fusion with the plasma membrane. The role of EVs is of great importance to many health and disease states, such as metabolism, ischemia, inflammation, pain, and malignancy.2.2 The advantages and disadvantages of engineering EVs
The advantages of EVs are mainly reflected in their inanimate but unique bioactivity, which reflects their cellular origin and can be potentially applied to the treatment of various diseases. EVs are smaller, simpler, more stable and relatively easy to modify, manufacture and store than parental cells. Because of these properties, EVs with membranes provide protection against enzyme-induced and non-enzyme-induced degradation of their molecular contents and may be less carcinogenic and immunogenicity than directly transplanted cells.20 However, the disadvantages of EVs treatment are also apparent. EVs are relatively rapidly internalized by cells or other tissues at and/or around the delivery sites, often entering non-specific cell types and systemic circulation.21,22 Therefore, the main challenge in developing vesicle-based therapies is to determine whether EVs are tailored to specific cell types within tissues. Furthermore, it is important to evaluate the safety of vesicle-based therapies in clinical settings and study their biodistribution, toxicity, thrombosis and half-life. Along these lines, preclinical testing of the efficacy on EVs in large animal models (as opposed to rodent models) should be encouraged, and careful consideration should be given to clinical conversion. In addition, vesicle dosing and administration regimens must be carefully determined, as well as the optimal method for developing effective vesicle delivery. Before bringing vesicles into the clinic, it is also important to ensure that the mass supply of vesicles is feasible at recommended safety levels to meet clinical requirements. It is essential to find reliable sources for the production of large quantities of vesicles to increase overall EV production with clinical-grade purity (e.g., through genetic modification of production cells or EV generation in response to environmental conditions).2.3 Methods of engineering EVs
Given the above-mentioned shortcomings of natural EVs, the academic community has developed a variety of engineering EV transformation strategies such as EV membrane surface engineering to improve the targeting and stability of EVs, and the content loading transformation of drugs loaded into EVs. The EVs transformed by these strategies are collectively referred to as engineering EVs.23 The rational engineering transformation of natural EVs is a necessary condition for its clinical application. This paper introduces the common engineering transformation strategies for EVs. So far, there is no clear definition of engineering EVs, which can generally be understood as artificially modified EVs to meet the needs of various scientific research. The engineering transformation of EVs mainly includes surface modification and content loading. The main purpose of surface modification is to make EVs have specific targeting or reduce the probability of being cleared by the liver, while content loading allows EVs to be loaded with a variety of nucleic acids, proteins, small molecule compounds and other drugs. The surface modification of EVs is the molecular surface engineering of the phospholipid membrane of EVs. The principal methods include parental cell modification, non-covalent binding and covalent binding.24The principle of parental cell transformation strategy is to genetically modify the parent cells with the ability of EV secretion so that the target protein can be expressed on the surface of cell membrane, and the target proteins can be displayed on the surface of EV membrane. Usually, the target protein is a fusion protein consisting of a functional protein and a transmembrane structure. In theory, all transmembrane proteins on the cell membrane can serve as the transmembrane structure of the fusion protein. There are two basic types of transmembrane proteins: α-helix proteins exist in the inner membrane of bacterial cells or the plasma membrane of eukaryotic cells. β-Barrel proteins are only found on the outer membrane of Gram-negative bacteria, the cell wall of Gram-positive bacteria, and the outer membrane of mitochondria.25 When the functional protein of the fusion protein is a targeting protein or targeting peptide, the targeting of exosomes can be improved. Cheng et al. expressed the fusion proteins of anti-CD3 (CD3 antibody), anti-epidermal growth factor receptor (anti-EGFR) and PDGFR on the exosome membrane by transfecting Expi293F cells, so that the constructed engineering exosomes had the affinity of breast cancer cells and T cells at the same time. Therefore, they could specifically enrich T cells in the vicinity breast cancer cells to improve the targeting and killing ability of T cells to breast cancer cells.26 Non-covalent binding refers to the combination of specific substances with EV membrane through non-covalent bonds, including classical interaction and hydrophobic interaction. Non-covalent binding requires relatively mild reaction conditions and weak binding strength. Zhu et al. combined exosomes from human embryonic stem cells with cyclo (Arg–Gly–ASP–D-Tyr–Lys) peptide C (RGDyK) through post-insertion method. Nanocarriers modified with RGDyK could more effectively deliver paclitaxel (PTX) to cancer cells.27 Compared to non-covalent bonds, covalent bonds allow for the modification of exosomal membranes by introducing bioconjugation and “click chemistry” reactions without fear of their bioactivity. The reaction of “click chemistry” are fast and efficient with high conjugation site selectivity, good compatibility and high specificity. Therefore, the advancement of “click chemistry” technology is of great significance for the rapid and large-scale production of engineering exosomes.28 The comparison of the three surface modification methods is shown in Table 1.
| Surface modification method | Functional molecular binding site | Bonding strength | Advantages |
|---|---|---|---|
| Parental cell transformation | Cell membrane | Strong | The constructed cell line can express continuously and stably |
| Non-covalent binding | Cell membrane and EV membrane | Weak | The reaction conditions are mild |
| Covalent binding | EV membrane | Strong | High efficiency and high bonding strength |
There are two main approaches to load drugs into the exocrine bodies, passive diffusion and active encapsulation.29 Passive diffusion is widely used for loading of lipid-soluble small molecules. By incubating EVs or parental cells directly with the drug, the drug can diffuse from the EV outside to the EV inside along the concentration gradient to realize the drug loading. The loading efficiency is dependent on the hydrophobicity of the drug molecule. The greatest advantage of passive diffusion is that it does not cause any damage to cells, but the main disadvantage of passive diffusion is the low loading efficiency.
The method reported by Pascucci is that the PTX solution was co-cultured with SR4987 mesenchymal, PTX was loaded into cells using passive diffusion method, and after re-culturing the cells, the medium containing exosomes was collected and finally the engineering PTX-loaded exosomes were isolated. Compared to exosomes from cells in the untreated group, exosomes from cells in the treated groups showed significantly enhanced anti-proliferative viability against cystic fibrosis pancreatic cancer cells in vitro.30
Hydrophilic compounds and substances with large molecular weight are difficult to be loaded by passive diffusion. At present, the load of such substances is mostly achieved by active packaging.29 The methods of active packaging mainly include: ultrasonic method, extrusion method and electroporation method. The ultrasound method is to co-incubate EVs with drug molecules, and then deform the EV membrane through the mechanical shear force of ultrasound and ultrasonic probes, so that drugs enter the EVs to realize the loading of drug molecules. Kim et al. loaded the potent anticancer drug PTX into macrophage-derived exosomes by sonication, and the engineering EVs showed high anti-cancer efficacy in a mouse model of lung metastasis.31 The advantage of ultrasonic method is that both hydrophilic and hydrophobic drugs have high loading efficiency, but the disadvantage is that the drugs may adhere to the membrane surface.
The extrusion method is to mix EVs from donor cells with drugs and then load the mixture into a syringe-based lipid extruder with a 100–400 nm porous membrane at a controlled temperature (Fig. 2a). During the extrusion process, the EV membranes rupture and mix vigorously with the drug. It's unclear whether the harsh mechanical forces used in this method alter membrane properties such as zeta potential and membrane protein structure. Fuhrmann et al. used the extrusion method to load porphyrins into exosomes derived from MDA-MB231 breast cancer cells, which altered the zeta potential of the original exosomes and caused cytotoxicity, whereas porphyrins loaded into exosomes using other methods showed no obvious cytotoxicity.32 Zhang et al. used a chemically-induced membrane blebbing and extrusion combined method to induce triple-negative breast cancer (TNBC) cell apoptosis, which secrete a large number of apoptotic body analogue (ABA) vesicles for therapeutic drug delivery, which suggests the great potential of ABAs for targeted drug delivery therapy, in particular efficient TNBC treatment.33 But, the application of extrusion method is relatively few, and its effect on cytotoxicity needs to be further studied.
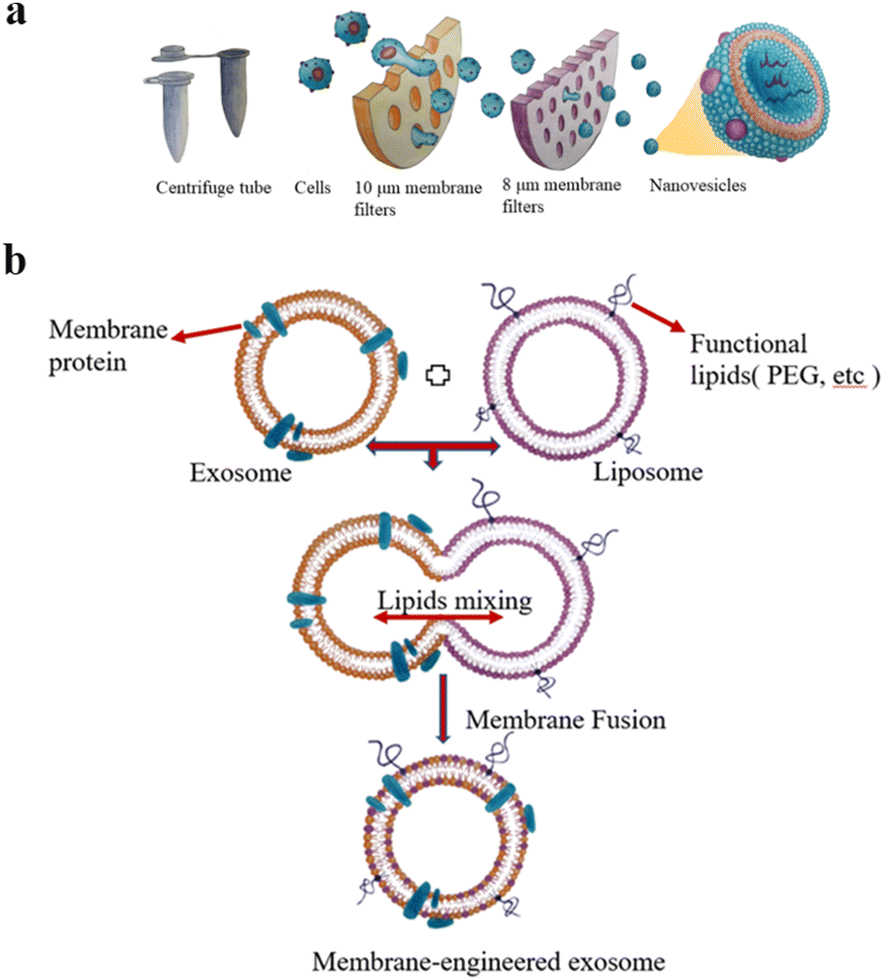 | ||
| Fig. 2 (a) Extrusion method: first, the cells are filtered through a series of polycarbonate membrane filters with gradually decreasing pore sizes, and then subjected to density gradient ultracentrifugation or size exclusion column chromatography. Finally, vesicles with the size of 100–200 nm are obtained. (b) The preparation of engineered-hybrid-exosomes by fusing the membrane proteins of exosomes and functional lipids of liposomes. Fig. 2 was reproduced from ref. 3 with permission from Elsevier, Copyright 2021. | ||
The principle of electroporation is to apply an electric field to the EVs suspended in a conductive solution, and the current interferes with the phospholipid bilayer of the EVs, forming temporary pores in the membrane so that drugs can diffuse into the EVs through the pores. Liang et al. expressed the fusion protein of human epidermal growth factor receptor 2 (Her2) and recombinant lysosomal associated membrane protein 2 (LAMP2) on the exosome membrane to effectively target colon cancer cells.34 The results showed that Her2-LAMP2 fusion protein expressed on the exosome surface, promoted targeted cellular uptake by HCT-116 cells through EGFR receptor-mediated endocytosis. The advantage of electroporation is that it does not introduce any chemical reagents and is very efficient in loading hydrophilic drugs such as DNA and RNA. The disadvantage is that it may lead to the fusion of the introduced drug with the cellular components. In addition, as shown in Fig. 2b, EV membrane proteins are combined with the lipids of multifunctional liposomes to achieve their fusion. This engineered-hybrid-vesicle is endowed with a variety of functions. The comparison of content loading methods is shown in Table 2.
| Content loading methods | Passive diffusion | Ultrasonic method | Extrusion method | Electroporation method |
|---|---|---|---|---|
| Advantages | The experimental operation is simple | The loading efficiency is high | High yield for large-scale application | No other substances are introduced, and the loading efficiency of hydrophilic drugs is high |
| Disadvantages | Only liposoluble drugs can be loaded and the loading efficiency is low | The drug may be loaded on the membrane surface | It may be toxic to cells | It may cause the drug to bind to the cell's components |
3. Macrophage introduction
3.1 The key findings of the relationship between macrophages and tumor cells, and macrophage types
As shown in Fig. 3, the relationship between macrophages and tumor cells is gradually discovered. In the 19th century, researchers found that tumor often occurred at sites of chronic inflammation, so they first suggested that inflammation was associated with cancer; for more than a century, epidemiological studies have shown that chronic inflammation can promote the occurrence of different types of cancer, such as colon cancer, prostate cancer, liver cancer, etc.; in the late 1970s, researchers found that tumor growth was induced by TAMs, the main component of infiltrating leukocytes in tumor; in 1992, based on the “macrophage homeostasis hypothesis”, Mantovani proposed that TAMs have dual effects of killing tumor and promoting tumor growth.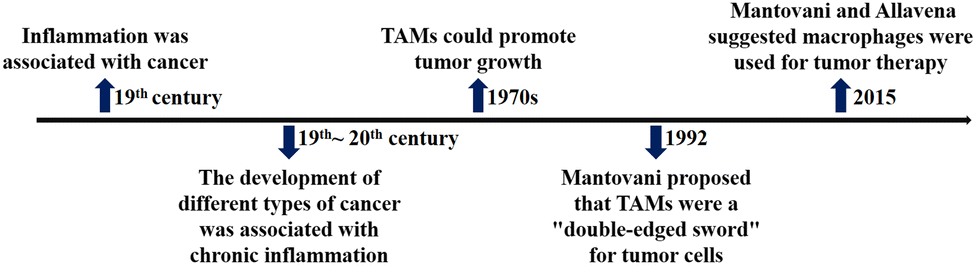 | ||
| Fig. 3 The timeline of key findings of relationship between macrophages and tumor cells. | ||
Tissue-resident macrophages originate from the prenatal stage of the yolk sac and fetal liver, whereas under inflammatory and homeostatic conditions in certain tissues, macrophages are derived from circulating Ly6C+CCR2+ monocytes such as colonic mucosal macrophages. In normal tissue, macrophages play a significant role in the body. In order to maintain tissue homeostasis, macrophages perform multiple nutrient functions including removing apoptotic debris, regulating blood vessels, promoting the formation of extracellular matrix (ECM), tissue generation and tissue remodelling. In tumors, breast, head, neck, thyroid, liver, pancreatic cells, kidney, bladder, ovary and uterus are densely infiltrated by macrophages, which is closely linked to the poor prognosis of cervical cancer, glioma, melanoma and non-Hodgkin's tumor. In most solid tumors, macrophages play a dominant role in the myeloid cell population. By studying the relationship between malignant tumor growth and macrophages in human and mice, it's consequently clear that Italian scientists Mantovani and Allavena proposed that macrophages are the ultimate direction for cancer therapy.35
Macrophages in the TME, called TAMs, are the most abundant inflammatory cell population in the tumor stroma, accounting for 30–50% of the total number of inflammatory cells. Their biological characteristics are closely related to the occurrence and development of tumors. As illustrated in Fig. 4, macrophages have two activated forms: (1) M1 type, classically activated macrophages. (2) M2 type, alternative activated macrophages.36,37 M2-Type macrophages can be divided into three types: (1) M2a type, which stimulates the activation of T helper 2 (Th2) cells, participates in type II inflammatory reaction and allergic reaction, and wraps and kills parasites. (2) M2b type, mainly mediates Th2 cell activation and immune regulation. (3) M2c type, mainly mediates immune regulation and participates in matrix deposition and tissue repair.38 As is known to all, M1-type macrophage activation requires dual signals: human interferon-γ (INF-γ) and exogenous tumor necrosis factor (TNF-α) or inducers of endogenous TNF [bacteria or its products liposaccharide (LPS)]; activation of M2-type macrophages don’t require dual signalling but require appropriate inducers, such as interleukin-4 (IL-4), IL-10, IL-13, glucocorticoid, vitamin D3, transforming growth factor-β (TGF-β), etc.39
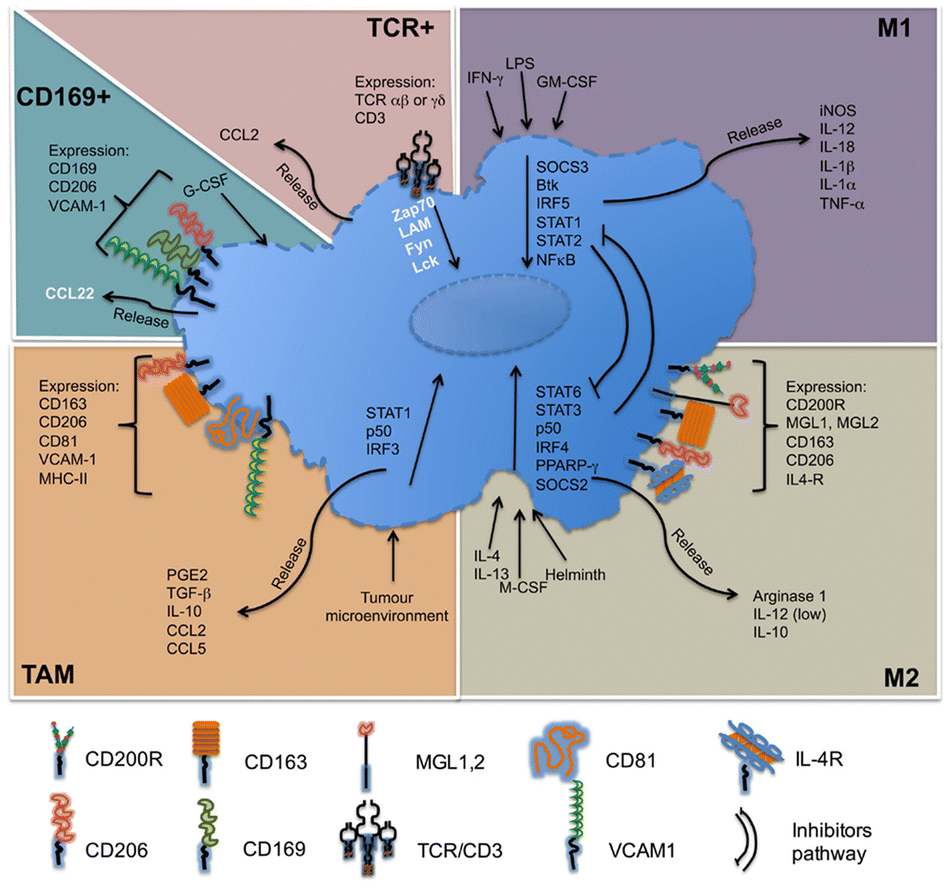 | ||
| Fig. 4 Molecules related to the classification of macrophages. M1-Type macrophages, also known as classical activated macrophages, are generally activated by interferon-γ and LPS activation; M2-type macrophages are called selective activated macrophages, which are activated by Th2 cytokines, such as IL-4, IL-13 and immune complexes; TAMs are mainly differentiated from monocytes. Chemokines such as CSF1 and CCL2 secreted by tumor cells can recruit monocytes from peripheral blood to TME, and the microenvironment responds and secretes cytokines, such as TGF-β and IL-10. CD169+ macrophages are involved in immune tolerance and erythropoiesis. TCR+ macrophages are a new subset of macrophages that release the chemokine CCL2 and play a role in inflammatory and infectious diseases. Fig. 4 was reproduced from ref. 37 with permission from Frontiers, Copyright 2015. | ||
3.2 Relationship between TAMs and tumors
The main functions of M1-type macrophages include the secretion of toxic intermediates, multiple inflammatory factors (such as IL-1, IL-6, IL-12, IL-23, TNF, etc.), chemokines (such as CCL1, CCL2, CCL3, CCL4, CCL5, etc.) and activation of Th1 cells, killing phagocytic microorganisms and tumor cells. And they participate in Th1 cell immune response by expressing a large number of major histocompatibility complex II and B7 molecules as important antigen-presenting cells, that is, tumor suppression. M2-Type macrophages can promote tumor growth, angiogenesis, invasion and metastasis, that is, tumor progression. Fig. 5a shows that there are two sources of TAMs in the TME: (1) embryonic stem cells or monocyte-derived tissue-resident tumor-associated macrophages (trTAMs) that change phenotype and/or activation status in the process of cancer, collectively referred to as trTAMs.40,41 (2) Monocytes undergo a distinct differentiation process, which is called tissue-induced tumor-associated macrophages (tiTAMs) due to tumor differentiation.42 Among them, trTAMs mainly take place in the early stage of tumor, while tiTAMs appear mostly in the later stages of tumor. It is important to emphasize that in the PyMT model, tiTAMs cannot exhibit M2 phenotype. TAM differentiation depends on the notch/recombination signalling of binding proteins in the immunoglobulin κJ (Rbpj) signalling pathway, and excision of Rbpj can lead to reducing TAMs and tumor growth.42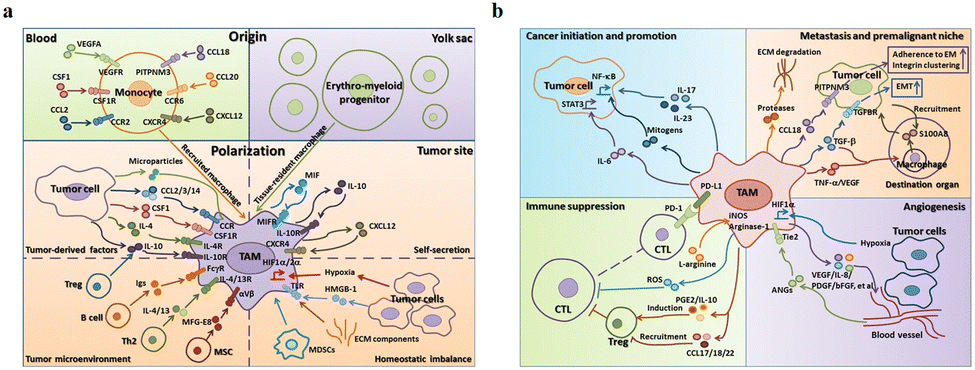 | ||
| Fig. 5 (a) Two sources of TAM in the tumor microenvironment are peripheral blood monocytes and yolk sac. These factors (MIF, IL-10 and CXCL12) secreted by TAMs result in tissue stress (hypoxia, tumor-derived HMGB-1 and ECM components). (b) The effects of TAMs on tumor progression. (1) TAMs can produce cytokines, such as IL-6, IL-17 and IL-23. (2) TAMs inhibit the proliferation of CTL cells through L-arginine-dependent iNOS or arginase metabolism, resulting in the production of reactive oxygen species. (3) TAMs promote tumor cell invasion in ectopic tissues through protease-dependent ECM remodelling. (4) Hypoxia environment induces HIF-1α expression in TAMs, which further regulates the transcription of angiogenesis-related genes. Fig. 5 was reproduced from ref. 41 with permission from Springer Nature, Copyright 2017. | ||
As shown in Fig. 5b, TAMs tend to be M2 phenotype and play an important role in tumor escape, mainly as follows: (1) secretion of tumor growth and survival factors, such as epidermal growth factor (EGF), IL-6 and CXCL-8. (2) TAMs participate in matrix remodelling. They in tumor stroma can produce a variety of matrix-degrading enzymes to promote matrix membrane dissolution, stroma digestion and remodelling, providing conditions for tumor invasion. (3) TAMs directly or indirectly involve in angiogenesis. They play an important role in angiogenesis, including the production of pro-angiogenic factors [such as vascular endothelial growth factor (VEGF), TGF-β, platelet-derived growth factor (PDGF) and many chemokines]. (4) TAMs participate in lymph node generation and lymph node metastasis. VEGF-C and VEGF-D secreted by TAMs are closely linked to tumor lymphatic vessel formation. The up-regulation of VEGF-C expression by TAMs can increase the density of lymphatic microvessels in breast cancer tissues and regenerate lymphatic epithelial cells, thus promoting lymphatic formation.40,43 VEGF-C expression induced by TAMs could promote lymph node metastasis of Lewis lung cancer.44 Wu et al. found that blocking the nuclei factor-κB (NF-κB) pathway could significantly reduce the expression of VEGF-C, suggesting that NF-κB pathway is one of the main pathways that mediate the up-regulation of TAMs to produce VEGF-C.45 (5) TAMs involve in immunosuppression. They produce a variety of immunosuppressive factors and chemokines to inhibit specific immune responses. For example, TAMs can produce TGF-β and IL-10, which inhibit the immunity of TME.46
3.3 TAMs and tumor immunotherapy
Immunotherapy is the application of immunological principles and methods to improve the immunogenicity of tumor cells and the sensitivity to effector cell killing, as well as to stimulate and enhance the body's anti-tumor immune response. Immunotherapy is largely dependent on the immune system and its underlying activity, early preclinical and clinical data show promising outcomes. At present, tumor macrophage immunotherapy includes TAM phenotype and anti-tumor therapy. The anti-tumor therapy methods are M2-type to M1-type transformation, directly killing TAMs in the TME, improving the ability of body's immune cell and monoclonal antibody (mAb) targeted therapy.(1) TAM phenotype and anti-tumor therapy, macrophages are highly plastic cells that adopt different activation phenotypes in response to changing microenvironments. Different phenotypes of TAMs in TME directly affect tumor development, such as T cell immunoglobulin and mucin-do-main-containing molecule-3 (Tim-3). And they can induce the differentiation of myeloid-derived suppressor cells (MDSCs) into TAMs and promote the viability and proliferation of leukemia stem cells, thereby promoting the progression of leukemia.47
(2) Anti-tumor therapy methods for M2-type to M1-type transformation, Mantovani and Sica et al. have pointed out in several review articles that M1, M2, and TAMs usually coexist in the TME; TAMs may tend to activate the M2 phenotype, showing the characteristics associated with M2-type macrophages; under certain external factors, the activation phenotype of TAMs can be dynamically switched. Therefore, converting M2-type TAMs in the TME to M1-type TAMs is also an ideal therapeutic approach.48,49 Previous studies have shown that the use of CpG DNA and IL-10 receptor antibodies can achieve the transformation of M2-type TAMs to M1-type.50
(3) Direct killing of TAMs in the TME, which are tended to be M2 phenotype and thus promote tumor development. Therefore, directly killing TAMs in TME also plays an important role in tumor inhibition, such as clodronate liposomes and zoledronic acid.51 In mouse models, CD204 is a cell surface molecule that is highly expressed in TAMs. In experiments, CD204-positive macrophages have an impact on the progression of canine breast tumors, indicating the potential of CD204 as a prognostic factor.52
(4) Improving the immunity of body's immune cells, this therapy can be directed by CD8+ T cells, the main cytotoxic immune cells, or indirectly by targeting macrophages. In treatment process of pancreatic cancer, clodronate liposomes could foster CD8+ T cell infiltration to alter macrophages and suppress tumor growth.53
(5) MAb targeted-therapy, a promising cancer treatment option for TAMs, is engage in antibody-dependent cytotoxicity/cell phagocytosis through the binding of their cell surface expression receptors to the antibody Fc fragment.54 In mice, FcγRI, FcγRIIa and FcγRIIIa are activated receptors, while FcγRIIb is an inhibitory receptor.55 Monoclonal antibody therapy and anti-tumor therapy are ineffective in the absence of one or more activated Fcγ receptors. It was observed in mice that the killing of tumor cells by antibodies was more dependent on FcγRIIb.56–58
Although corresponding research progress has been made in macrophage treatment, these method faces many difficulties: it is impossible to, for example, monitor or maintain the bioactivity of macrophages for a long time, which affects macrophage therapeutic effect to a certain extent. Primary macrophages are more inclined to be used in cell therapy, which are cumbersome to prepare and poor in reproducibility. The safety of macrophage therapy also needs to be considered, such as enhanced cytotoxicity may cause excess cytokine storm and excessive inflammatory environment. The TEM can polarize macrophages into the M2 tissue repair phenotype, further increasing the malignancy of the tumor.59 Blocking CSF1/CSF1R can enhance tumor sensitivity to other immunotherapies, such as PD-L1 blocking antibodies.60 But CSF1/CSF1R blockade leads to depletion of tissue-resident macrophages by increasing other signalling or increasing the bioactivity of Tregs in TME, which leads to serious consequences for tissues requiring CSF1R signalling for maintenance.61,62 CCL2/CCR2 blockade inhibits monocyte recruitment, but established TAMs can still promote tumor progression.63 After TAMs are recruited again at the tumor site, the failure of CCL2 blocking therapy is declared.64 Therefore, to circumvent the problems of cell therapy, EV therapeutic approach will be one of the most promising ways in the future.
3.4 Relationship between macrophage-derived EVs and tumors
The function of exosomes in intercellular communication, particularly during tumor development, has been extensively studied. Exosomes play a variety of roles in the microenvironment, such as remodelling the ECM, mediating intercellular signals and molecular transmission among cells.65 As shown in Fig. 6a, relevant studies have shown that exosomes play a “double-edged sword” role in the cancer development.66 Because exosomes are secreted vesicles by cells and are easily absorbed by cells, they have potential applications in cancer immunotherapy.67 A growing number of studies suggests that exosomes can alter the status of recipient cells through the transport of bioactive substances. Therefore, exosomes can be used as potential biomarkers for the diagnosis and prognosis of diseases.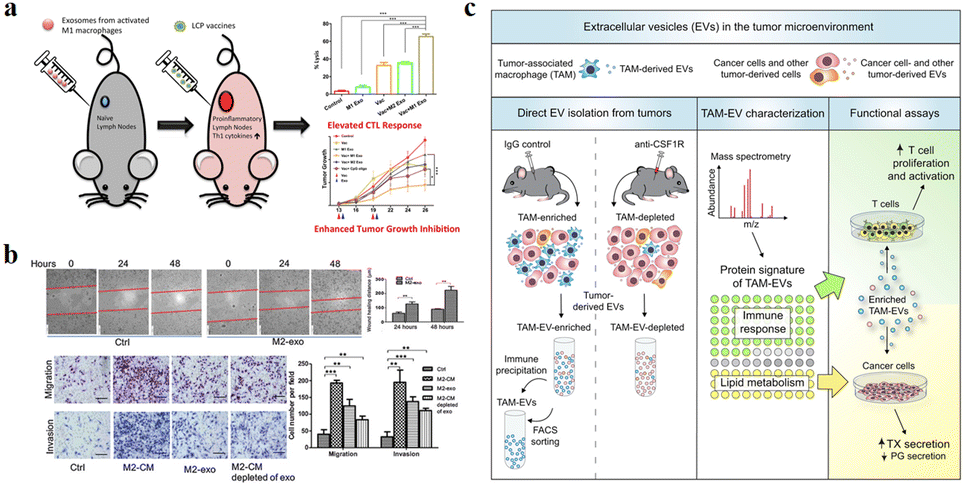 | ||
| Fig. 6 (a) Schematic illustration of exosomes derived from M1 macrophages enhancing cancer vaccines by creating pro-inflammatory microenvironment in lymph nodes. (b) Exosomes derived from M2 macrophages could regulate the migration and invasion of colon cancer cells. (c) Schematic illustration of TAM-EVs promoting T cell proliferation/activity in vitro, promoting and reducing TX and PG secretion from cancer cells, respectively. (a) was reproduced from ref. 66 with permission from Elsevier, Copyright 2017. (b) was reproduced from ref. 78 with permission from American Association for Cancer Research, Copyright 2019. (c) was reproduced from ref. 82 with permission from Elsevier, Copyright 2019. | ||
Circulating monocytes have a natural tendency to tumor and inflammatory tissues. The surface of these cells highly express ICAM-1 protein and interact with adhesion molecules on the leukocyte surface, thus exhibiting high affinity for pathological environments.68,69 Macrophages are tumor-targeted.70,71 They can migrate to the tumor site in a short time and secrete specific chemokines. The tumor has high permeability to chemokines, so macrophages can directionally aggregate and infiltrate into TEM. In addition, tumor tissues can also secrete high levels of chemokines, such as CCL2 and CCL5, to ensure that macrophages are recruited to tumor sites.72 M1 macrophages overexpress the surface chemokine receptor CCR2, while tumor tissue produce monocyte chemoattractant protein-1 (MCP-1 and CCL2), which is a member of the C–C chemokine family and can recruit monocytes/macrophages into TEM through the CCL2–CCR2 pathway.73 The EVs derived from macrophages are favored for the following reasons: (1) EVs can play a variety of functions similar to those of parental cells, and the EVs secreted by macrophages are also tumor-targeted, so they can be used as a tumor specific drug delivery system. Guo et al. co-incubated the M1 macrophage-derived EVs with Chinese hamster ovary cells and tumor cells (SK-OV3 cells), which could increase the cell uptake rate of tumor cells.74 (2) There are a lot of immune factors on the surface of EVs derived from macrophages, such as CD47, which sends a signal of “don’t eat me” to tumor cells, which helps to escape immune surveillance.75,76 (3) EVs are easy to circulate, have the ability to cross the biological barrier and reduce the risk of cell therapy, such as cytokine release synthesis, thus reducing the side effects on healthy tissues.77
Due to the high heterogeneity of macrophages, exosomes derived from different types of macrophages have different functions. Fig. 6a and b show that exosomes derived from M1-type macrophages generally have pro-inflammatory and anti-tumor functions, while those from M2-type macrophages play anti-inflammatory and tumor-promoting roles.66,78 Immune cell exosomes can inhibit tumor cell growth and migration by inhibiting the interaction between tumor cells and ECM. The exosomes of human mononuclear macrophages contain high levels of a disintegrin and metalloproteinase-15 (ADAM15), and their disintegrin domain contains Arg-Gly-Asp (RGD) sequences that bind integrin αvβ3. Integrin αvβ3 regulates the adhesion and migration of tumor cells in the ECM. Exosomes containing ADAM15 directly inhibit cell adhesion, growth and migration induced by cancer procoagulant and fibronectin, as well as tumor growth in vivo, without stimulating other immune cells.79,80 Soluble disintegrated proteins with the RGD motif have been demonstrated to inhibit tumor progression by inhibiting integrin-mediated ECM interactions.81 Proteomic and lipid analysis showed that exosomes of tumor-associated macrophages (TAMexos) carry regulatory factors of inflammation and lipid metabolism, which affect the characteristics of the tumor immune microenvironment. TAMexos can promote the proliferation and activation of T cells. Compared with TAMs, TAMexos have a molecular profile related to Th1/M1 polarization characteristics, enhancing inflammation and immune response, while having a better patient prognosis. As illustration in Fig. 6c, TAMexos also contain bioactive lipids and biosynthetic enzymes that may alter pro-inflammatory signals in cancer cells.82 Exosomes produced by LPS-stimulated mouse macrophages contain elevated inflammatory cytokines and miRNA to induce NF-κB activation in naive cells.83 A single injection of exosome alleviates hyperalgesia in a mouse model of inflammatory pain. Exosomes containing miR21-3p, miR146a and miR146b have been known to play a role in the prevention of innate immune overactivation by inhibiting TLR signalling involving NF-κB and other mRNAs.84,85
However, miR21 in the exosomes of TAMs (M2-type macrophages) can be directly transferred to gastric cancer cells, and down-regulation of PTEN endows gastric cancer cis-resistance, leading to more active proliferation-related PI3K-Akt signalling.86 This result can be supported by the role of miR21 in several cancers.87 In addition, macrophage-derived exosomes significantly reduced the sensitivity of pancreatic ductal adenocarcinoma (PDAC) cells to gemcitabine.88 However, natural exosomes have disadvantages such as difficult flux extraction, insufficient targeting and too many ineffective components, leading to poor therapeutic effect. Similarly, these problems are also applicable to macrophage-derived exosomes. Therefore, to solve the above problems, we should use the engineering exosomes to improve therapeutic effect.
4. Application of engineering macrophage-derived EVs in tumor therapy
4.1 Engineering EVs as drug carriers
The high heterogeneity of macrophages determines the different phenotypes of macrophages in different cell microenvironments. The EVs are derived from parental cells, so they are highly heterogeneous. For example, the polarization of macrophages into M1 or M2 type changes the function of exosomes, which can be regarded as simple engineering exosomes.62,78 Compared with other synthetic vectors, their biocompatibility is beyond doubt, and related proteins on the surface of exosomes are conducive to phagocytosis. Therefore, exosomes as drug carriers have attracted more and more attention and research. Kim et al. found that exosome-loaded PTX secreted by engineered macrophages could significantly enhance the anti-tumor effect of lung metastases in mice.31 As shown in Fig. 7, Wang et al. found that M1-exosomes (M1-Exos) (concentration of 40 μg mL−1) had few side effects on tumor cells alone in vitro, while PTX-M1-Exos could better inhibit the growth of tumor cells.89In vivo experiments, the M1-Exos group alone also showed a tendency to inhibit tumor growth. As a vector, M1-Exos cannot kill tumor cells but can play an indirect anti-tumor effect by activating immune cells in vivo.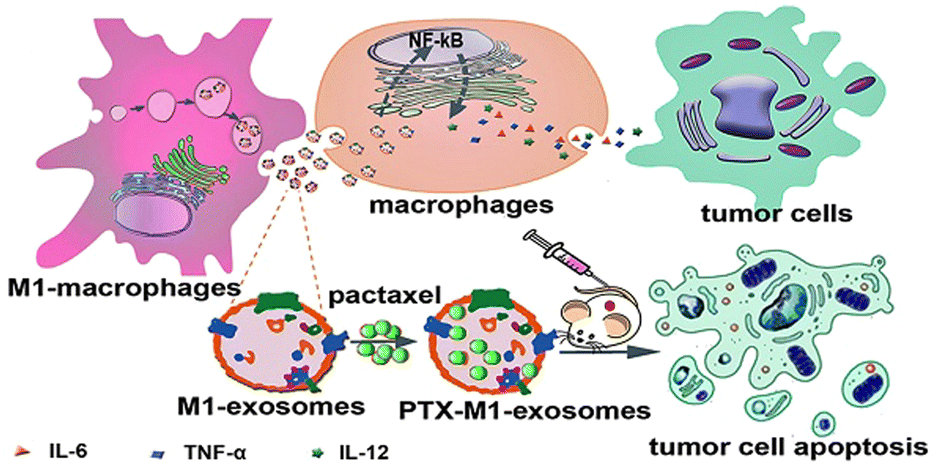 | ||
| Fig. 7 M1-Exos were used as the carrier of the anticancer drug, paclitaxel. M1-Exos could promote the production of pro-inflammatory factors in macrophages, and PTX-M1-Exos showed better antitumor effect than M1-type macrophages in mice. Fig. 7 was reproduced from ref. 89 with permission from Ivyspring International, Copyright 2019. | ||
EVs are also of great significance in the treatment of brain diseases. Sun et al. encapsulated the hydrophobic drug curcumin into exosomes to improve its solubility and bioavailability, and delivered it to the brain through a nasal drug delivery system to treat brain inflammation.90 Microglia are macrophages in the central nervous system that can be absorbed by gliomas and penetrate tumors. Du et al. used microglial cells (BV2 cells) as vectors to deliver PTX for the treatment of gliomas.91 A schematic diagram of the drug delivery system is illustrated in Fig. 8, in order to avoid the toxic effect of PTX on microglia, liposomes were used to isolate drugs from BV2 cells for the first time. Dipalmitoyl phosphatidylserine (DPPS) was doped into liposomes as a “eat me” signal to enhance microglia phagocytosis of liposomes. This study shows that engineered microglia can cross the blood-brain barrier, independently migrate to glioma and transfer cargoes to glioma cells. Notably, EVs and tunnel nanotubes are considered to provide a unique model of cargo transport between microglia and glioma cells. In vivo, engineered drug-delivery microglia has a high ability to target the brain, penetrate gliomas and inhibit tumor progression, supporting the idea that the use of engineered microglia is a potential strategy for treating gliomas.
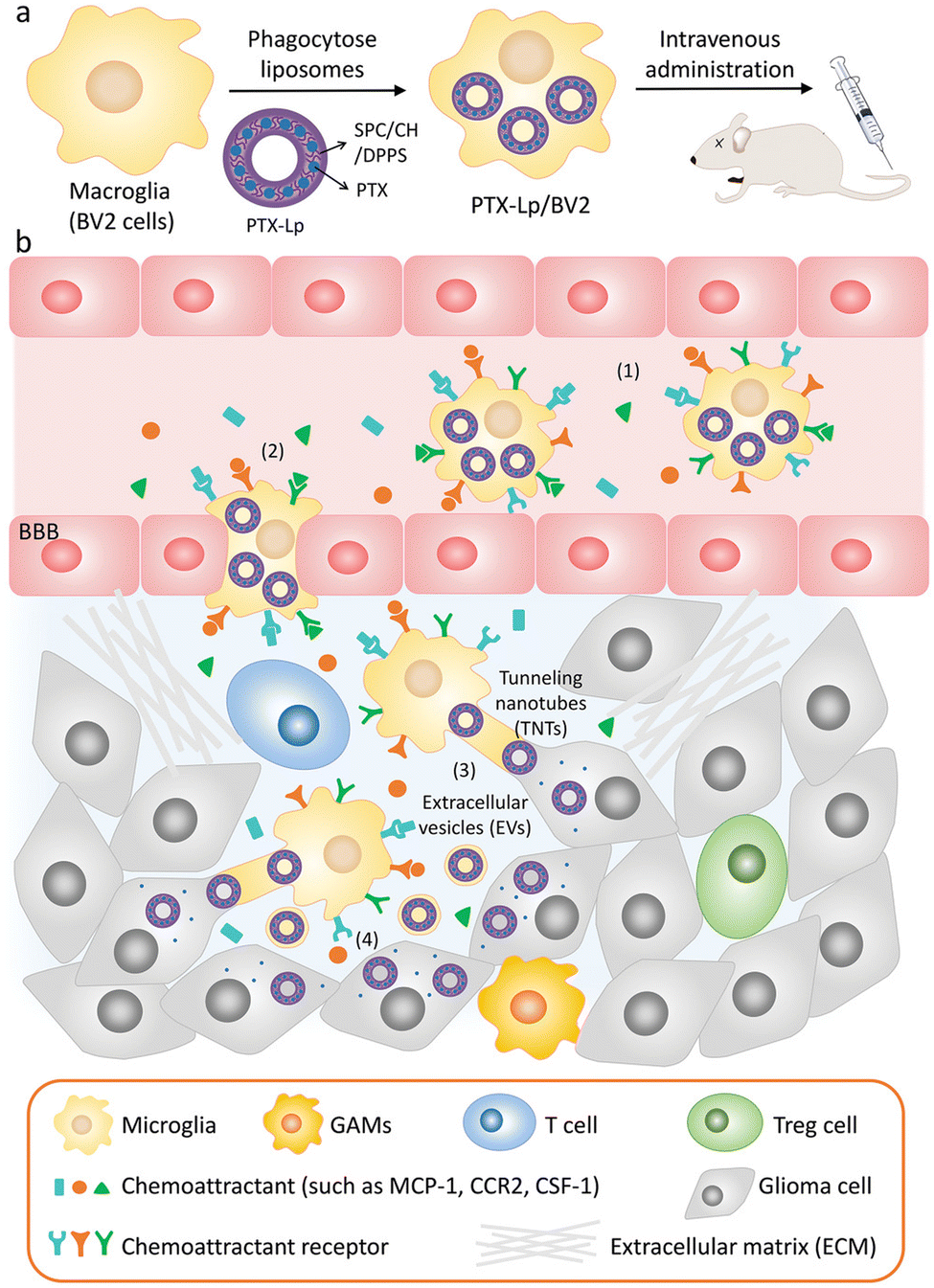 | ||
| Fig. 8 (a) Schematic illustration of the PTX-Lp/BV2 preparation. (b) Schematic diagram of PTX Lp/BV2 transport process after intravenous injection: (1) motility of PTX Lp/BV2 was guided by chemoattractant diffusion in the glioma microenvironment, (2) metastasis of PTX-Lp/BV2 cells to gliomas across the blood-brain barrier, (3) drugs were transported from BV2 cells to glioma cells through EV and tunnel nanotubes, (4) PTX produced anti-tumor effect at the site of tumor cells. Fig. 8 was reproduced from ref. 91 with permission from Wiley-VCH, Copyright 2021. | ||
The synergistic effects of immunotherapy, photodynamic therapy (PDT) and chemotherapy can produce powerful anticancer effects. As illustrated in Fig. 9, Ding et al. induced M1-macrophage-derived EVs (M1 EVs) to be simultaneously loaded with bis[2,4,5-trichloro-6-(pentyloxycarbonyl)phenyl] oxalate (CPPO), chlorin e6 (Ce6) and doxorubicin (Dox-EMCH).92 After drug administration, M1 EVs were mainly taken up by tumor cells due to their targeting, and M1 EVs repolarized M2-type to M1-type macrophages, producing not only immunotherapeutic effects but also H2O2. The reaction between H2O2 and CPPO generated chemical energy, which activated Ce6 and generated singlet oxygen (1O2) for chemiluminescence and PDT for imaging. Meanwhile, 1O2-induced membrane rupture resulted in Dox-EMCH release, which was activated and penetrated into the deep hypoxic region of the tumor.
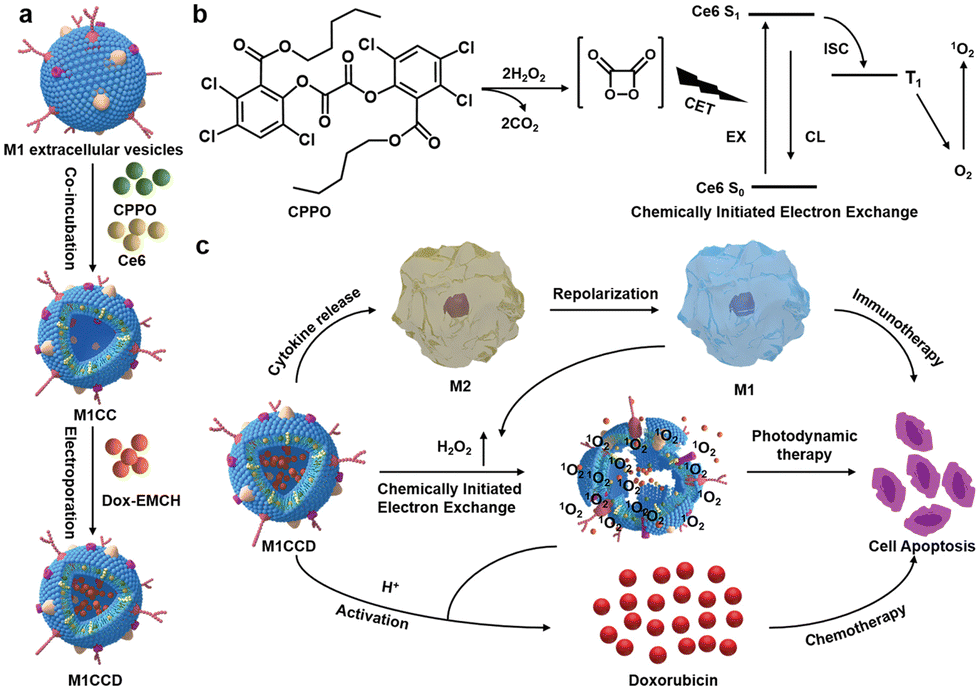 | ||
| Fig. 9 (a) Schematic diagram of the construction process of M1CCD. (b) The mechanism of chemiluminescence and chemical excitation of singlet oxygen. (c) Schematic diagram of the synergistic anti-tumor mechanism of M1CCD. Fig. 9 was reproduced from ref. 92 with permission from Wiley-VCH, Copyright 2021. | ||
In order to further monitor the transfer and release of EVs as smart nanocarriers. Su et al. prepared a pH-triggered EVs with fluorescence conversion by loading amphoteric ion fluorescent carbon dots (CDs) into EVs secreted by macrophages.93 The idea of the experiment is illustrated in Fig. 10, the amphoteric ion CDs contained in vesicles could tightly bind to the chemotherapy drug DOX through electrostatic interaction during blood circulation, thus avoiding premature drug unloading. The nanocarriers have a long blood circulation half-life of 15.12 ± 1.57 h and a high tumor volume of 9.88% ID g−1. At the same time, the fluorescence of CDs was “off” due to the fluorescence internal filter effect (IFE) between DOX and CDs. The low pH of the lysosome caused the charge reversal of the CDs when the nanosensors entered the tumor cells. DOX could be rapidly released by electrostatic repulsion and the fluorescence of CDs was turned “on” after drug released, improving drug delivery and tracking drug release in real-time.
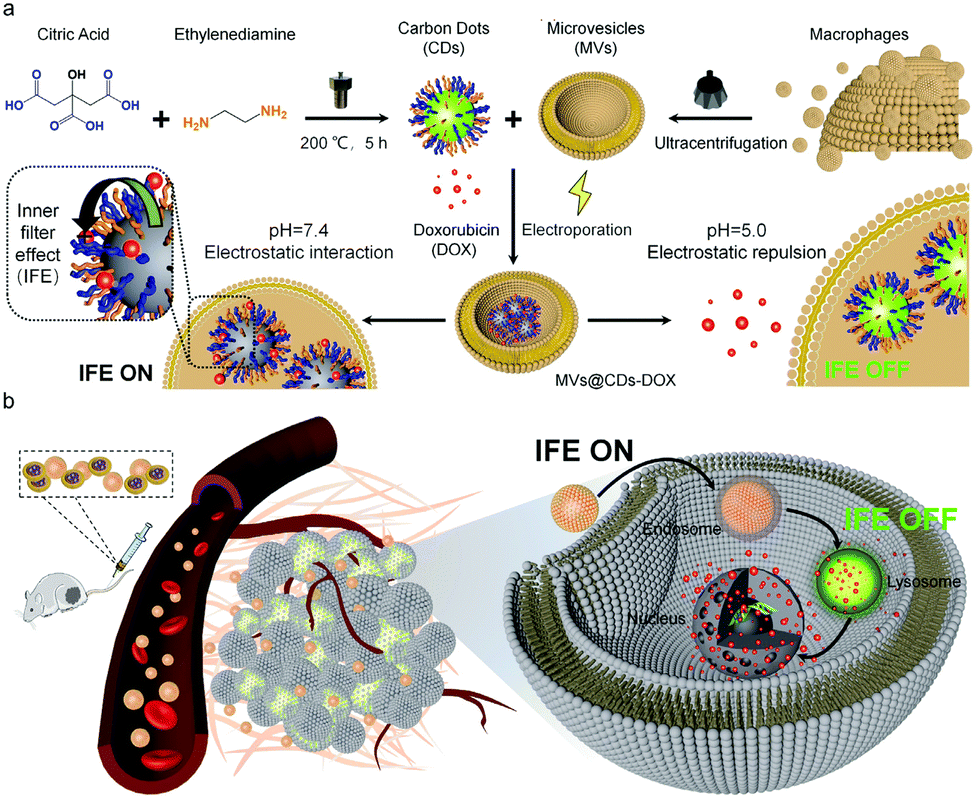 | ||
| Fig. 10 (a) Schematic illustration of the construction process of pH-triggered fluorescence-switchable EVs MVs@CDs-DOX and the interaction between CDs and DOX at different pH values. (b) Schematic diagram of drug delivery and release from MVs@CDs in tumor cells. Fig. 10 was reproduced from ref. 93 with permission from Royal Society of Chemistry, Copyright 2021. | ||
Modification of parental cells to change the function of exosomes is also commonly used in tumor therapy. As shown in Fig. 11, Wei et al. developed mannose-modified macrophage-derived microparticles (Man-MPs) loaded with metformin (Met@Man-MPs) that could effectively target M2-like TAMs and repolarify into M1 phenotype.94 Met@Man-MPs-reset TAMs reshaped the tumor immune microenvironment by increasing the recruitment of CD8+ T cells to tumor tissues and reducing the immunosuppressive infiltration of bone marrow-derived suppressor cells and regulatory T cells. More importantly, the collagen-degrading ability of Man-MPs contributed to the infiltration of CD8+ T cells into the tumor, enhancing tumor accumulation and infiltration of anti-PD-1 antibodies. These unique properties of Met@Man-MPs contribute to the strengthening of anti-PD-1 antibody therapy, improving anti-cancer efficacy and long-term memory immunity after combination therapy. In addition, extracellular vesicle-based nanomedicine carriers can be used for the treatment of melanoma. Jiang et al. combined exosomes derived from macrophages with TNF-related apoptosis-inducing ligands for anti-melanoma treatment.95 Trail-Exo enhanced the targeting of exosomes by specifically binding DR5 and induces apoptosis of tumor cells. Trail-Exo could be used as a suitable nanocarrier to load TPL and be effectively internalized into A375 cells through endocytosis. More importantly, TPL could be better delivered to tumor cells through Trail-Exo, and Trail-Exo/TPL can enhance cytotoxicity, inhibit tumor invasion and migration, and promote tumor apoptosis by activating both internal and external apoptosis pathways, thus improving the synergistic anti-melanoma effect.
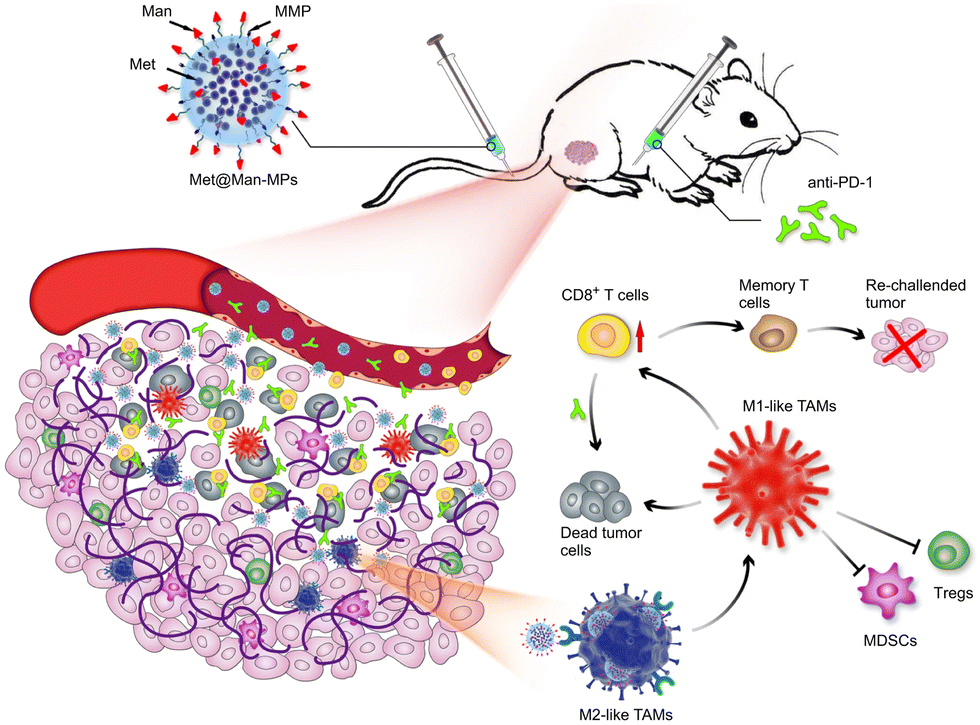 | ||
| Fig. 11 Schematic diagram of the mechanism of Met@Man-MPs for anti-PD-1 therapy. Met@Man-MPs with MMP activity effectively targeted M2 TAMs, transformed macrophages into M1-type, and at the same time it could degrade tumor collagen, helped CD8+ T cells infiltrate into tumors and enhanced anti-PD-1 antibody tumor accumulation and infiltration, while Met@Man-MPs synergistically inhibited tumor growth with anti-PD-1 antibody, enabling the organism to develop long-term memory immunity. Fig. 11 was reproduced from ref. 94 with permission from Springer Nature, Copyright 2021. | ||
4.2 Engineering EVs to improve their targeting
Targeting EVs can greatly improve the efficacy of anti-tumor therapy. Li et al. developed a macrophage-derived exosome-encapsulated poly(lactic-glycolic acid) nanoplatforms for targeted chemotherapy of TNBC.96 A peptide was modified on the surface of exosomes to target mesenchymal–epithelial transition factor overexpressed by TNBC cells, thereby significantly improving the cellular uptake efficiency and anti-tumor efficacy of Dox. In vivo studies showed that the nanocarriers exhibited significant tumor targeting, enhanced tumor growth inhibition and induced strong tumor apoptosis. Nucleic acids are likewise often loaded into EVs to target tumor therapy. Fan et al. designed functionalized DNA as a hinge to anchor quantum dots (QDs) on the surface of exosomes, enabling a modest and biocompatible labelling strategy.97 QD/DNA-labeled exosomes could be rapidly phagocytosed by tumor cells, indicating that exosomal QD/DNA could be used as specific tumor markers. Furthermore, M1-type macrophage artificial vesicles (M1 mv) were constructed by a pneumatic liposome extruder. The results showed that a single M1 mv could kill tumor cells and achieve ideal biological therapy.In order to enhance the anti-tumor efficacy and specificity of drug release of M1 mv, a targeted trigger drug delivery system is constructed to realize tumor vision therapy in response to specific miRNA. Liu et al. combined vesicular stomatitis virus glycoprotein (VSV-G), a pH-responsive viral fusion protein, and anti-PD-L1 siRNA (siPD-L1) into M1 EVs through electroporation method.98 The scheme of the experiment is shown in Fig. 12, this virus-mimicking nucleic acid-engineered EVs (siRNA@V-M1 EVs) were able to target tumor tissue after injection in CT26 tumor-bearing mice because of the natural tumor-homing properties of M1 EVs. Fusion of VSV-G to cells facilitated the direct release of siPD-L1 into the cytoplasm, triggering gene silencing and resulting in efficient blockade of the PD-L1/PD-1 interaction, which in turn increased the CD8+ T cell population. Meanwhile, M1 EVs and IFN-γ secreted by CD8+ T cells promote the repolarization of M2 TAMs into M1-type macrophages. Blockade of the PD-L1/PD-1 pathway, re-establishment of T-cell recognition, and repolarization of M1-type macrophages through multifunctional EVs can achieve satisfactory anti-tumor effects in tumor model, showing as a potential for new anti-tumor approaches. As shown in Fig. 13, engineering EVs generally aim to increase yield, targeting and increasing active ingredients, and the combination of the three enhances the anti-tumor therapeutic effect. In conclusion, engineering EVs can realize the synergistic effect of immunotherapy, PDT, and chemotherapy, which can produce powerful anticancer efficacy and provide new ideas for tumor therapy.
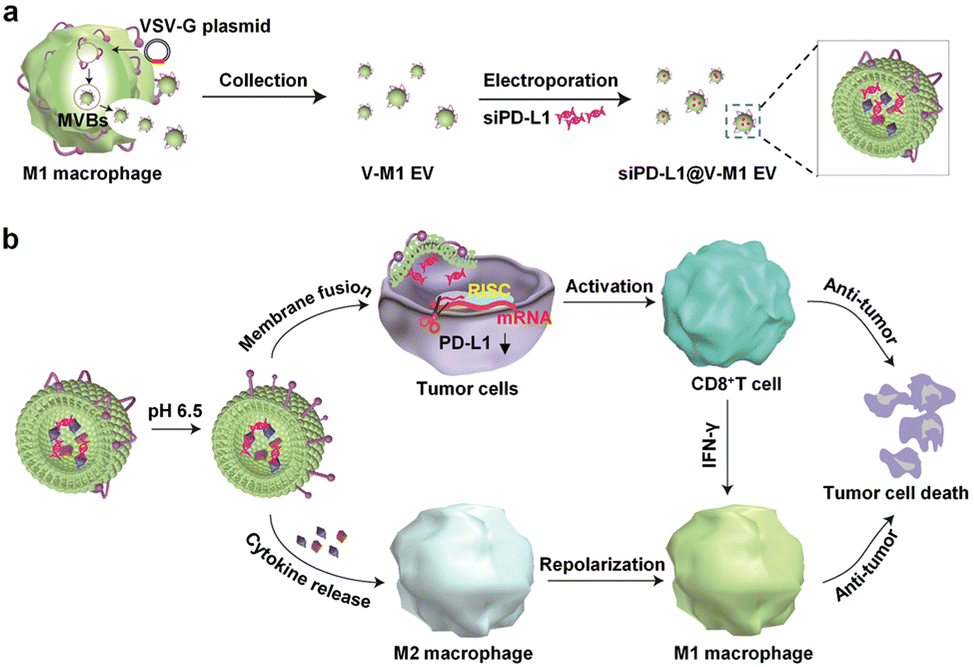 | ||
| Fig. 12 (a) Schematic illustration of the preparation process of siPD-L1@V-M1 EVs. VSV-G was loaded into EVs by engineering M1-type macrophages, and siPD-L1 was loaded into V-M1 EVs by electroporation. (b) Schematic illustration of antitumor activity of siPD-L1@V-M1 EVs. siPD-L1@V-M1 EVs released siPD-1 into tumor cells through membrane fusion, reducing PD-L1 expression, promoting CD8+ T cell proliferation and releasing IFN-γ, thus improving anti-tumor effect. Cytokines and IFN-γ in siPD-L1@V-M1 EVs facilitated the transformation of M2-type macrophages to M1-type, thereby strengthening the anti-tumor effect. Fig. 12 was reproduced from ref. 95 with permission from American Chemical Society, Copyright 2020. | ||
 | ||
| Fig. 13 Schematic diagram of drug loading and targeting improvement of engineered macrophage-derived EVs. | ||
5. Conclusions
To sum up, despite the low yield, low transmembrane efficiency and insufficient targeting of macrophage-derived EVs, they have unique advantages, such as good biocompatibility, easy modification and suitable size. At present, engineering macrophage-derived EVs have been used in laboratory research, and existing experimental results have confirmed that they can be used for anti-tumor therapy, providing a new research direction for clinical tumor treatment. Engineering EVs with surface modification and content loading modification show great potential in many fields, and progress has been made in disease diagnosis, drug delivery, vivo imaging, etc., but no engineering EVs have officially entered the clinic applications so far. Currently, the major challenge of engineering macrophage-derived EVs is that their yield cannot meet the needs of large-scale therapeutics. Although some attempts have been made to produce EVs on a large scale by cell extrusion, there are still some problems during EV production, such as cell blockage. In addition, there are still some problems in the clinical application of engineering EVs such as storage and design, which can be solved by studying the pharmacokinetics of engineered EVs in humans. Therefore, we should focus on bringing engineering EVs out of the laboratory and in combination with the clinic in the future.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the National Key R&D Program of China (2022YFC2904804), the CUG Scholar Scientific Research Funds at China University of Geosciences (Wuhan) (2019152), the Fundamental Research Funds for the Central Universities at China University of Geosciences (Wuhan) and the National Science Fund for Distinguished Young Scholars (51225403).References
- A. Jemal, R. Siegel, E. Ward, Y. Hao, J. Xu and M. J. Thun, Ca-Cancer J. Clin., 2009, 59, 225–249 CrossRef PubMed.
- J. O. Martinez, M. Evangelopoulos, R. Bhavane, S. Acciardo, F. Salvatore, X. Liu, M. Ferrari and E. Tasciotti, Curr. Drug Targets, 2015, 16, 1582–1590 CrossRef CAS PubMed.
- H. Liu, S. Deng, L. Han, Y. Ren, J. Gu, L. He, T. Liu and Z. Yuan, Colloids Surf., B, 2021, 209, 112163 CrossRef PubMed.
- X. Xu, P. E. Saw, W. Tao, Y. Li, X. Ji, S. Bhasin, Y. Liu, D. Ayyash, J. Rasmussen, M. Huo, J. Shi and O. C. Farokhzad, Adv. Mater., 2017, 29, 1700141 CrossRef PubMed.
- R. Paduch, Cell. Oncol., 2016, 39, 397–410 CrossRef CAS PubMed.
- R. Zhang, Q. Liu, T. Li, Q. Liao and Y. Zhao, Cancer Cell Int., 2019, 19, 300 CrossRef PubMed.
- J. Lotvall, A. F. Hill, F. Hochberg, E. I. Buzas, D. Di Vizio, C. Gardiner, Y. S. Gho, I. V. Kurochkin, S. Mathivanan, P. Quesenberry, S. Sahoo, H. Tahara, M. H. Wauben, K. W. Witwer and C. Thery, J. Extracell. Vesicles, 2014, 3, 26913 CrossRef PubMed.
- N. P. Hessvik and A. Llorente, Cell. Mol. Life Sci., 2018, 75, 193–208 CrossRef CAS PubMed.
- M. Colombo, G. Raposo and C. Thery, Annu. Rev. Cell Dev. Biol., 2014, 30, 255–289 CrossRef CAS PubMed.
- Y. Fujita, T. Kadota, J. Araya, T. Ochiya and K. Kuwano, Am. J. Respir. Cell Mol., 2018, 58, 560–565 CrossRef CAS PubMed.
- C. C. Horgan, A. Nagelkerke, T. E. Whittaker, V. Nele, L. Massi, U. Kauscher, J. Penders, M. S. Bergholt, S. R. Hood and M. M. Stevens, J. Mater. Chem. B, 2020, 8, 4447–4459 RSC.
- C. Thery, K. W. Witwer, E. Aikawa, M. Jose Alcaraz, J. D. Anderson, R. Andriantsitohaina, A. Antoniou, T. Arab, F. Archer and G. K. Atkin-Smith, et al. , J. Extracell. Vesicles, 2018, 7, 1535750 CrossRef PubMed.
- K. P. Hough, D. Chanda, S. R. Duncan, V. J. Thannickal and J. S. Deshane, Allergy, 2017, 72, 534–544 CrossRef CAS PubMed.
- S. P. Nana-Sinkam, M. Acunzo M, C. M. Croce and K. Wang, Am. J. Respir. Crit. Care Med., 2017, 196, 1510–1518 CrossRef CAS PubMed.
- A. Emelyanov, T. Shtam, R. Kamyshinsky, L. Garaeva, N. Verlov, I. Miliukhina, A. Kudrevatykh, G. Gavrilov, Y. Zabrodskaya, S. Pchelina and A. Konevega, PLoS One, 2020, 15, e0227949 CrossRef CAS PubMed.
- B. Sastre, J. A. Canas, J. M. Rodrigo-Munoz and V. del Pozo, Front. Immunol., 2017, 8, 826 CrossRef PubMed.
- J. Cheng, N. Zhu, Y. Zhang, Y. Yu, K. Kang, Q. Yi and Y. Wu, J. Mater. Chem. B, 2022, 10, 4059–4069 RSC.
- A. Montecalvo, A. T. Larregina, W. J. Shufesky, D. B. Stolz, M. L. G. Sullivan, J. M. Karlsson, C. J. Baty, G. A. Gibson, G. Erdos, Z. Wang, J. Milosevic, O. A. Tkacheva, S. J. Divito, R. Jordan, J. Lyons-Weiler, S. C. Watkins and A. E. Morelli, Blood, 2012, 119, 756–766 CrossRef CAS PubMed.
- V. Claudia, G. Martina and G. Paola, J. Lipid Res., 2018, 59, 1325–1340 CrossRef PubMed.
- I. K. Herrmann, J. A. W. Matthew and F. Gregor, Nat. Nanotechnol., 2021, 16, 746–759 CrossRef PubMed.
- R. C. de Abreu, H. Fernandes, P. A. D. Martins, S. Sahoo, C. Emanueli and L. Ferreira, Nat. Rev. Cardiol., 2020, 17, 685–697 CrossRef PubMed.
- Z. Zhang, B. Buller and M. Chopp, Nat. Rev. Neurol., 2019, 15, 193–203 CrossRef PubMed.
- M. Nawaz, Stem Cell Invest., 2017, 4, 83 CrossRef PubMed.
- Q. Zhu, M. Heon, Z. Zhao and M. He, Lab Chip, 2018, 18, 1690–1703 RSC.
- K. D. Tsirigos, S. Govindarajant, C. Bassot, A. Vastermark, J. Lamb, N. Shu and A. Elofsson, Curr. Opin. Struct. Biol., 2018, 50, 9–17 CrossRef CAS PubMed.
- Q. Cheng, X. J. Shi, M. L. Han, G. Smbatyan, H. J. Lenz and Y. Zhang, J. Am. Chem. Soc., 2018, 140, 16413–16417 CrossRef CAS PubMed.
- Q. Zhu, X. Ling, Y. Yang, J. Zhang, Q. Li, X. Niu, G. Hu, B. Chen, H. Li, Y. Wang and Z. Deng, Adv. Sci., 2019, 6, 1801899 CrossRef PubMed.
- T. Tian, H. Zhang, C. He, S. Fan, Y. Zhu, C. Qi, N. Huang, Z. Xiao, Z. Lu, B. A. Tannous and J. Gao, Biomaterials, 2018, 150, 137–149 CrossRef CAS PubMed.
- X. Luan, K. Sansanaphongpricha, I. Myers, H. Chen, H. Yuan and D. Sun, Acta Pharmacol. Sin., 2017, 38, 754–763 CrossRef CAS PubMed.
- L. Pascucci, V. Cocce, A. Bonomi, D. Ami, P. Ceccarelli, E. Ciusani, L. Vigano, A. Locatelli, F. Sisto, S. M. Doglia, E. Parati, M. E. Bernardo, M. Muraca, G. Alessandri, G. Bondiolotti and A. Pessina, J. Controlled Release, 2014, 192, 262–270 CrossRef CAS PubMed.
- M. S. Kim, M. J. Haney, Y. Zhao, D. Yuan, I. Deygen, N. L. Klyachko, A. V. Kabanov and E. V. Batrakova, Nanomed. Nanotechnol. Biol. and Med., 2018, 14, 195–204 CrossRef CAS PubMed.
- G. Fuhrmann, A. Serio, M. Mazo, R. Nair and M. M. Stevens, J. Controlled Release, 2015, 205, 35–44 CrossRef CAS PubMed.
- K. Zhang, H. Fu, C. Xing, Y. Luo, F. Cheng, Q. Fu, Y. Huang and L. Qiu, J. Mater. Chem. B, 2021, 9, 8472–8479 RSC.
- G. Liang, Y. Zhu, D. J. Ali, T. Tian, H. Xu, K. Si, B. Sun, B. Chen and Z. Xiao, J. Nanobiotechnol., 2020, 18, 10 CrossRef CAS PubMed.
- A. Mantovani and P. Allavena, J. Exp. Med., 2015, 212, 435–445 CrossRef CAS PubMed.
- J. P. Edwards, X. Zhang, K. A. Frauwirth and D. M. Mosser, J. Leukocyte Biol., 2006, 80, 1298–1307 CrossRef CAS PubMed.
- L. Chavez-Galan, M. L. Olleros, D. Vesin and I. Garcia, Front. Immunol., 2015, 6, 263 Search PubMed.
- A. Sica and A. Mantovani, J. Clin. Invest., 2012, 122, 787–795 CrossRef CAS PubMed.
- S. Gordon and F. O. Martinez, Immunity, 2010, 32, 593–604 CrossRef CAS PubMed.
- R. A. Franklin and M. O. Li, Trends Cancer, 2016, 2, 20–34 CrossRef PubMed.
- L. Yang and Y. Zhang., J. Hematol. Oncol., 2017, 10, 58 CrossRef PubMed.
- R. A. Franklin and M. O. Li, Oncoimmunology, 2014, 3, e955346 CrossRef PubMed.
- M. Ding, X. Fu, H. Tan, R. Wang, Z. Chen and S. Ding, Mol. Med. Rep., 2012, 6, 1023–1029 CrossRef CAS PubMed.
- B. Zhang, G. Yao, Y. Zhang, J. Gao, B. Yang, Z. Rao and J. Gao, Clinics, 2011, 66, 1879–1886 CrossRef PubMed.
- H. Wu, J. Xu, Y. He, J. Peng, H. Zhang, C. Chen, W. Li and S. Cai, J. Surg. Oncol., 2012, 106, 462–468 CrossRef CAS PubMed.
- N. L. Costa, M. C. Valadares, P. P. C. Souza, E. F. Mendonca, J. C. Oliveira, T. A. Silva and A. C. Batista, Oral Oncol., 2013, 49, 216–223 CrossRef CAS PubMed.
- L. Gao, S. Yu and X. Zhang, Cell Biochem. Biophys., 2014, 70, 273–277 CrossRef CAS PubMed.
- A. Mantovani, P. Allavena and A. Sica, Eur. J. Cancer, 2004, 40, 1660–1667 CrossRef CAS PubMed.
- A. Sica, T. Schioppa, A. Mantovani and P. Allavena, Eur. J. Cancer, 2006, 42, 717–727 CrossRef CAS PubMed.
- C. Guiducci, A. P. Vicari, S. Sangaletti, G. Trinchieri and M. P. Colombo, Cancer Res., 2005, 65, 3437–3446 CrossRef CAS PubMed.
- P. Tsagozis, F. Eriksson and P. Pisa, Cancer Immunol. Immun., 2008, 57, 1451–1459 CrossRef CAS PubMed.
- B. J. Seung, H. Y. Lim, J. I. Shin, H. W. Kim, S. H. Cho, S. H. Kim and J. H. Sur, Vet. Pathol., 2018, 55, 417–424 CrossRef CAS PubMed.
- X. Yang, J. Lin, G. Wang and D. Xu, Cancers, 2022, 14, 1474 CrossRef CAS PubMed.
- L. M. Weiner, J. C. Murray and C. W. Shuptrine, Cell, 2012, 148, 1081–1084 CrossRef CAS PubMed.
- F. Nimmerjahn, S. Gordan and A. Lux, Trends Immunol., 2015, 36, 325–336 CrossRef CAS PubMed.
- R. A. Clynes, T. L. Towers, L. G. Presta and J. V. Ravetch, Nat. Med., 2000, 6, 443–446 CrossRef CAS PubMed.
- V. Minard-Colin, Y. Xiu, J. C. Poe, M. Horikawa, C. M. Magro, Y. Hamaguchi, K. M. Haas and T. F. Tedder, Blood, 2008, 112, 1205–1213 CrossRef CAS PubMed.
- M. A. Otten, G. J. van der Bij, S. J. Verbeek, F. Nimmerjahn, J. V. Ravetch, R. H. J. Beelen, J. G. J. V. de Winkel and M. van Egmond, J. Immunol., 2008, 181, 6829–6836 CrossRef CAS PubMed.
- S. Lee, S. Kivimae, A. Dolor and F. C. Szoka, J. Controlled Release, 2016, 240, 527–540 CrossRef CAS PubMed.
- Y. Zhu, J. Yang, D. Xu, X. Gao, Z. Zhang, J. L. Hsu, C. Li, S. O. Lim, Y. Sheng, Y. Zhang, J. Li, Q. Luo, Y. Zheng, Y. Zhao, L. Lu, H. Jia, M. Hung, Q. Dong and L. Qin, Gut, 2019, 68, 1653–1666 CrossRef CAS PubMed.
- D. F. Quail and J. A. Joyce, Clin. Cancer Res., 2017, 23, 876–884 CrossRef CAS PubMed.
- D. Gyori, E. L. Lim, F. M. Grant, D. Spensberger, R. Roychoudhuri, S. J. Shuttleworth, K. Okkenhaug, L. R. Stephens and P. T. Hawkins, Jci. Insight, 2018, 3, e120631 CrossRef PubMed.
- Y. Zhu, J. M. Herndon, D. K. Sojka, K. W. Kim, B. L. Knolhoff, C. Zuo, D. R. Cullinan, J. Luo, A. R. Bearden, K. J. Lavine, W. M. Yokoyama, W. G. Hawkins, R. C. Fields, G. J. Randolph and D. G. DeNardo, Immunity, 2017, 47, 323–338 CrossRef CAS PubMed.
- L. Bonapace, M. M. Coissieux, J. Wyckoff, K. D. Mertz, Z. Varga, T. Junt and M. Bentires-Alj, Nature, 2014, 215, 130–133 CrossRef PubMed.
- X. Yu, Q. Zhang, X. Zhang, Q. Han, H. Li, Y. Mao, X. Wang, H. Guo, D. M. Irwin, G. Niu and H. Tan, J. Cancer, 2019, 10, 2892–2906 CrossRef CAS PubMed.
- L. Cheng, Y. Wang and L. Huang, Mol. Ther., 2017, 25, 1665–1675 CrossRef CAS PubMed.
- A. Lukic, C. J. E. Wahlund, C. Gomez, D. Brodin, B. Samuelsson, C. E. Wheelock, S. Gabrielsson and O. Radmark, Cancer Lett., 2019, 444, 1–8 CrossRef CAS PubMed.
- C. Belli, D. Trapani, G. Viale, P. D’Amico, B. A. Duso, P. Della Vigna, F. Orsi and G. Curigliano, Cancer Treat. Rev., 2018, 65, 22–32 CrossRef CAS PubMed.
- R. Molinaro, J. O. Martinez, A. Zinger, A. De Vita, G. Storci, N. Arrighetti, E. De Rosa, K. A. Hartman, N. Basu, N. Taghipour, C. Corbo and E. Tasciotti, Biomater. Sci., 2020, 8, 333–341 RSC.
- M. R. Jadus, M. C. Irwin, M. R. Irwin, R. D. Horansky, S. Sekhon, K. A. Pepper, D. B. Kohn and H. T. Wepsic, Blood, 1996, 87, 5232–5241 CrossRef CAS PubMed.
- C. E. Lewis and J. W. Ponard, Cancer Res., 2006, 66, 605–612 CrossRef CAS PubMed.
- S. Li, S. Feng, L. Ding, Y. Liu, Q. Zhu, Z. Qian and Y. Gu, Int. J. Nanomed., 2016, 11, 4107–4124 CrossRef CAS PubMed.
- L. Guo, Y. Zhang, Z. Yang, H. Peng, R. Wei, C. Wang and M. Feng, ACS Nano, 2019, 13, 1078–1096 CAS.
- L. Guo, Y. Zhang, R. Wei, X. Zhang, C. Wang and M. Feng, Theranostics, 2020, 10, 6581–6598 CrossRef CAS PubMed.
- S. Kaur, S. P. Singh, A. G. Elkahloun, W. Wu, M. S. Abu-Asab and D. D. Roberts, Matrix Biol., 2014, 37, 49–59 CrossRef CAS PubMed.
- S. Kamerkar, V. S. LeBleu, H. Sugimoto, S. Yang, C. F. Ruivo, S. A. Melo, J. J. Lee and R. Kalluri, Nature, 2017, 546, 498–503 CrossRef CAS PubMed.
- Y. Wang, M. Zhao, S. Liu, J. Guo, Y. Lu, J. Cheng and J. Liu, Cell Death Dis., 2020, 11, 924 CrossRef CAS PubMed.
- J. Lan, L. Sun, F. Xu, L. Liu, F. Hu, D. Song, Z. Hou, W. Wu, X. Luo, J. Wang, X. Yuan, J. Hu and G. Wang, Cancer Res., 2019, 79, 146–158 CrossRef CAS PubMed.
- T. G. Wolfsberg, P. Primakoff, D. G. Myles and J. M. White, J. Cell Biol., 1995, 131, 275–278 CrossRef CAS PubMed.
- H. D. Lee, B. H. Koo, Y. H. Kim, O. H. Jeon and D. S. Kim, FASEB J., 2012, 26, 3084–3095 CrossRef CAS PubMed.
- V. Trochon-Joseph, D. Martel-Renoir, L. M. Mir, A. Thomaidis, P. Opolon, E. Connault, H. Li, C. Grenet, F. Fauvel-Lafeve, J. Soria, C. Legrand, C. Soria, M. Perricaudet and H. Lu, Cancer Res., 2004, 64, 2062–2069 CrossRef CAS PubMed.
- C. Cianciaruso, T. Beltraminelli, F. Duval, S. Nassiri, R. Hamelin, A. Mozes, H. Gallart-Ayala, G. C. Torres, B. Torchia, C. H. Ries, J. Ivanisevic and M. Palma, Cell Rep., 2019, 27, 3062–3080 CrossRef CAS PubMed.
- M. K. McDonald, Y. Tian, R. A. Qureshi, M. Gormley, A. Ertel, R. Gao, E. A. Lopez, G. M. Alexander, A. Sacan, P. Fortina and S. K. Ajit, Pain, 2014, 155, 1527–1539 CrossRef CAS PubMed.
- S. D. Hsu, F. M. Lin, W. Wu, C. Liang, W. Huang, W. Chan, W. T. Tsai, G. Chen, C. J. Lee, C. M. Chiu, C. H. Chien, M. C. Wu, C. Huang, A. P. Tsou and H. D. Huang, Nucleic Acids Res., 2011, 39, D163–D169 CrossRef CAS PubMed.
- K. D. Taganov, M. P. Boldin, K. Chang and D. Baltimore., Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12481–12486 CrossRef CAS PubMed.
- P. Zheng, L. Chen, X. Yuan, Q. Luo, Y. Liu, G. Xie, Y. Ma and L. Shen, J. Exp. Clin. Canc. Res., 2017, 36, 53 CrossRef PubMed.
- S. Yang, C. Huang, X. Li, M. Yu, Y. He and J. Li, Toxicology, 2013, 306, 162–168 CrossRef CAS PubMed.
- Z. Yin, T. Ma, B. Huang, H. Lin, Y. Zhou, J. Yan, Y. Zou and S. Chen, J. Exp. Clin. Canc. Res., 2019, 38, 310 CrossRef PubMed.
- P. Wang, H. Wang, Q. Huang, C. Peng, L. Yao, H. Chen, Z. Qiu, Y. Wu, L. Wang and W. Chen, Theranostics, 2019, 9, 1714–1727 CrossRef CAS PubMed.
- D. Sun, X. Zhuang, X. Xiang, Y. Liu, S. Zhang, C. Liu, S. Barnes, W. Grizzle, D. Miller and H. Zhang, Mol. Ther., 2010, 18, 1606–1614 CrossRef CAS PubMed.
- Y. Du, Z. Yang, Q. Sun, M. Lin, R. Wang, Y. Peng, X. Chen and X. Qi, Adv. Healthcare Mater., 2021, 10, 2002200 CrossRef CAS PubMed.
- J. Ding, G. Lu, W. Nie, L. Huang, Y. Zhang, W. Fan, G. Wu, H. Liu and H. Xie, Adv. Mater., 2021, 33, 2005562 CrossRef CAS PubMed.
- R. Su, X. Xiong, Y. Li, X. Wei, S. Zheng, J. Zhao and S. Zhou, Biomater. Sci., 2021, 9, 5812–5823 RSC.
- Z. Wei, X. Zhang, T. Yong, N. Bie, G. Zhan, X. Li, Q. Liang, J. Li, J. Yu, G. Huang, Y. Yan, Z. Zhang, B. Zhang, L. Gan, B. Huang and X. Yang, Nat. Commun., 2021, 12, 440 CrossRef CAS PubMed.
- L. Jiang, Y. Gu, Y. Du, X. Tang, X. Wu and J. Liu, ACS Appl. Mater. Interfaces, 2021, 13, 42411–42428 CrossRef CAS PubMed.
- S. Li, Y. Wu, F. Ding, J. Yang, J. Li, X. Gao, C. Zhang and J. Feng, Nanoscale, 2020, 12, 10854–10862 RSC.
- Z. Fan, K. Xiao, J. Lin, Y. Liao and X. Huang, Small, 2019, 15, 1903761 CrossRef CAS PubMed.
- H. Liu, L. Huang, M. Mao, J. Ding, G. Wu, W. Fan, T. Yang, M. Zhang, Y. Huang and H. Xie, Adv. Funct. Mater., 2020, 30, 2006515 CrossRef CAS.
Footnote |
| † These authors have contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |