
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolybdate ionic liquids as halide-free catalysts for CO2 fixation into epoxides†
Nicola
Bragato
 ,
Alvise
Perosa
,
Maurizio
Selva
,
Giulia
Fiorani
* and
Roberto
Calmanti
*
,
Alvise
Perosa
,
Maurizio
Selva
,
Giulia
Fiorani
* and
Roberto
Calmanti
*
Dipartimento di Scienze Molecolari e Nanosistemi, Università Ca’ Foscari Venezia, Via Torino 155, 30172 Venezia, VE, Italy. E-mail: roberto.calmanti@unive.it; giulia.fiorani@unive.it
First published on 18th May 2023
Abstract
Herein we describe the syntheses of a series of molybdate and polyoxomolybdate ionic liquids (ILs), their full spectroscopic characterisation (FT-IR, 1H-, 13C-, 31P-, and 95Mo-NMR and ICP-MS), a comparison of their properties, and their applications as bifunctional catalysts for CO2 insertion into epoxides. The synthetic procedures to obtain ILs rely on anion exchange and acid–base reactions, including an innovative route for the synthesis of molybdate ionic liquids (Mo-ILs) using a halide-free organic IL precursor. The use of Mo-ILs as catalysts for CO2 fixation was investigated using 1,2-epoxyhexane as a model substrate. In the presence of 2.5 mol% of tetrabutylammonium molybdate, hexane carbonate was obtained in up to 86% yield at T = 120 °C, p0(CO2) = 30 bar in t = 9 h, under solventless conditions and without any added halide co-catalysts. The substrate scope was broadened to other 12 terminal and internal epoxides; moreover, the reaction was scaled up to 2 g of the substrate and catalyst recyclability was demonstrated up to 5 recycles.
Introduction
In the last few decades, carbon dioxide (CO2) anthropogenic emissions have gained alarming attention worldwide due to their steadily increasing concentration in the atmosphere (ca. 35 Gt per year), linked to global warming and climate change effects. CO2-based chemistry has drawn considerable attention in the recent past due to its use as an abundant, low-cost and renewable C1 carbon source. In fact, due to the limited availability of fossil fuels, it is expected that CO2 will become one of the main carbon sources in the future.1 Although CO2 chemical fixation is not efficient in terms of volume and storage timescale, its utilization as a renewable and non-toxic C1 feedstock represents a relevant synthetic strategy for the production of renewable-based added-value fine chemicals and energy vectors including ureas,2 carbamates,3 organic carbonates,4 polycarbonates,5 carboxylic acids,6 heterocycles,7 methanol,8etc.The challenge associated with CO2 chemical reactivity falls within its high thermodynamic and kinetic stabilities. Overcoming both barriers would require an adequate supply of energy and/or the presence of suitable high-energy co-reactants: in both cases, it would likely lead to CO2 emissions higher than its fixation into organic compounds.9 In contrast, overcoming the kinetic barrier requires only the use of an appropriate catalytic system.10 Common non-reductive routes to activate CO2 involve the use of Lewis bases such as organic superbases, amine-containing species,11,12 N-heterocyclic carbenes,13 or organometallic activation via the formation of transition metal–CO2 complexes.14 Activated CO2 can then react with a variety of organic and inorganic co-reactants, including unsaturated compounds (e.g. alkenes or alkynes), three- and four-membered strained heterocycles (e.g. oxiranes, oxetanes and aziridines) or organometallic species, to yield more thermodynamically-stable products.15 In this respect, the last decade has witnessed a steady development of bifunctional catalysts capable of simultaneously activating both CO2 and the chosen co-reactant(s).16
A remarkable case is represented by the design of catalytic systems active towards CO2 insertion into epoxides for the synthesis of cyclic organic carbonates (COCs). As depicted in Fig. 1, the accepted mechanism for the reaction requires two catalytically active moieties: an electrophilic one, typically a Lewis or Brønsted acid (A+), which activates the epoxide (I, Fig. 1), and a nucleophile (Nu−), which plays a dual role by initially promoting epoxide ring opening and the formation of alkoxide (II, Fig. 1) and hemicarbonate (III, Fig. 1) intermediates, while acting as a good leaving group in the final ring-closing step. Usually A+ is an onium cation, a metal-based Lewis acid (e.g. Fe, Cr, Co, Al, and Sn),17 or a species with H-bonding ability (e.g. –OH, –COOH, and –NH groups),18 while Nu− is a halide salt (typically bromide or iodide salts with organic countercations).16
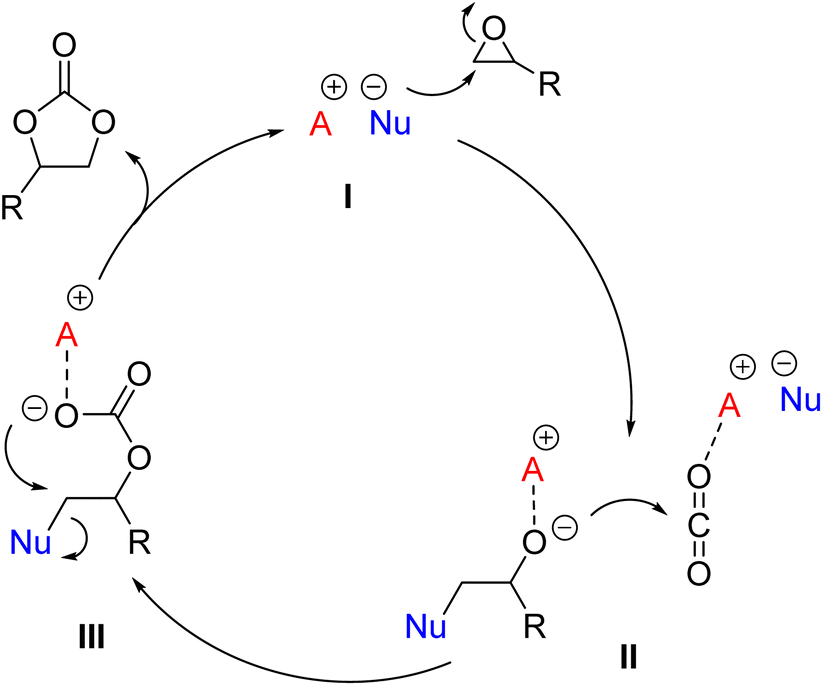 | ||
| Fig. 1 General mechanism for CO2 insertion into an epoxide. | ||
Comprehensive reviews on the subject have further confirmed that the use of bifunctional metal- and organo-catalysts for the reaction generally requires a co-catalyst or a halide salt to induce epoxide ring-opening.16,19 More recently, the development of halide-free CO2 insertion protocols has become an active and stimulating research theme in the field of CO2 valorisation.20–27
The term polyoxometalates (POMs) refers to a class of compounds based on early transition metal (groups 5 and 6) oxide building blocks with a general formula of (MOx)n, where M = Mo, W, V, Nb, Ta etc.28,29
In the past decades, POM-promoted CO2 capture and utilization has been widely explored and a plethora of POM-based compounds have been reported as efficient catalysts or promoters for CO2 fixation processes.30,31 Interestingly, a limited number of molybdenum-based active catalytic systems have been reported for CO2 fixation into epoxides. In 2011, Hu and co-workers developed a one-pot two-step epoxidation/CO2 insertion protocol for the synthesis of COCs starting from olefins, tert-butyl hydroperoxide as the oxidant and CO2 as the C1 source in the presence of a catalytic system composed of MoO2(acac)2 and tetrabutylammonium bromide, [N4444][Br].32 The authors demonstrated the conversion of terminal and internal olefins into the corresponding COCs, stating that the presence of MoO2(acac)2 was fundamental in the first epoxidation step while its presence practically did not affect the insertion step which was promoted by the halide salt co-catalyst. In 2016, Werner and co-workers developed a MoO3/tetrabutylphosphonium bromide catalytic system active towards the synthesis of bio-derived COCs starting from CO2 and epoxidized fatty acid esters.33 In 2018, Cheng and co-workers synthesized a novel binuclear molybdenum alkoxide catalyst, (Mo2(OtBu)6), which was employed in combination with [N4444][Br] for COC synthesis under mild conditions (rt and p0(CO2) = 0.1 MPa).34 More recently, Dai et al. optimised the preparation of three N-heterocyclic carbene–nitrogen molybdenum complexes, active for the preparation of 1,2-butylene carbonate from 2-ethyloxirane when used in combination with tetrabutylammonium iodide, [N4444][I] (cat: 0.5 mol%, [N4444][I] 2 mol%, T = 80 °C, t = 20 h, p0(CO2) = 5 bar).35 From these studies, it has been suggested that molybdenum species are mainly exploited as Lewis acid co-catalysts for the activation of the epoxide in combination with a tetraalkylammonium and/or phosphonium halide salt acting as a nucleophilic ring-opening promoter.
In this work, we report the preparation and characterization of different molybdate and polyoxomolybdate ionic liquids, which were evaluated as bifunctional catalysts for halide-free CO2 activation to synthesize COCs. Here we demonstrated the effective interaction between CO2 and Mo-ILs that allowed us to avoid the use of any halide co-catalysts in the CO2 insertion reaction. The focal point of this work is the sustainable synthesis of molybdate IL catalysts (Mo-ILs) starting from methylcarbonate ILs and their successful use for CO2 insertion with different epoxides under solventless conditions avoiding the presence of halide salt co-catalysts.
Results and discussion
Synthesis and characterization of Mo-ILs
A series of molybdenum salts (compounds 1–8, shown in Fig. 2) were synthesized in order to explore the correlation between their catalytic activity and the type of organic cation (ammonium, phosphonium, imidazolium and diazobicycloundecenium cations, characterised by distinctive lipophilic properties) as well as the structure of molybdenum-based anions, which represent the active site of the catalysts.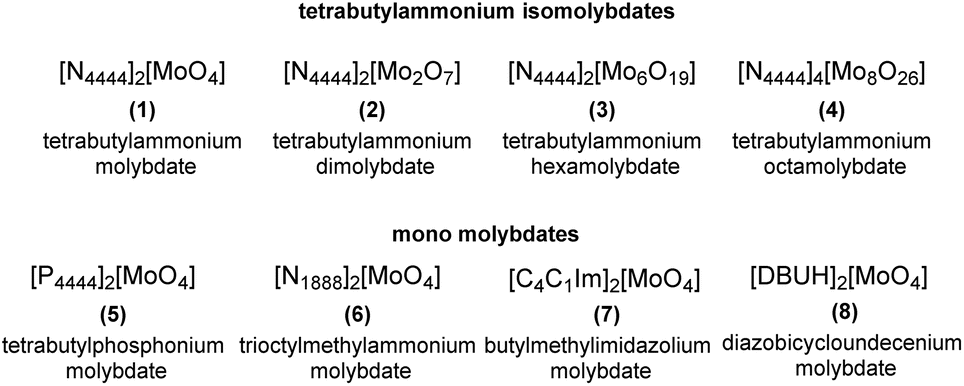 | ||
| Fig. 2 Mo-ILs used in this work. | ||
Compounds 1–8 were obtained through three different methodologies depicted in Fig. 3. Tetrabutylammonium isopolyoxomolybdates 2, 3 and 4 were synthesized according to a reported procedure based on the reaction between sodium molybdate dihydrate (Na2MoO4·2H2O) and [N4444][Br] in an acidic environment (Fig. 3a).36 ILs 1, 5 and 8 were prepared by acid–base neutralization between tetraalkylammonium, phosphonium or 1,8-diazabicyclo(5.4.0) undec-7-enenium hydroxide, [Q][OH] and molybdic acid (H2MoO4) (Fig. 3b).37 Catalysts 6 and 7 were synthesized upon metathesis of methylcarbonate ILs with H2MoO4, extending the applicability of a sustainable methodology previously developed by our group for the preparation of tungstate-based ILs to molybdenum-based ILs.38 As depicted in Fig. 3c, the latter methodology allowed the preparation of different Mo-ILs via a simple, halide-free and clean protocol avoiding work-up steps. The formation of methylcarbonic acid (i.e. the half ester of carbonic acid), which spontaneously decomposed to methanol and CO2 above −36 °C, is the driving force of the reaction.39
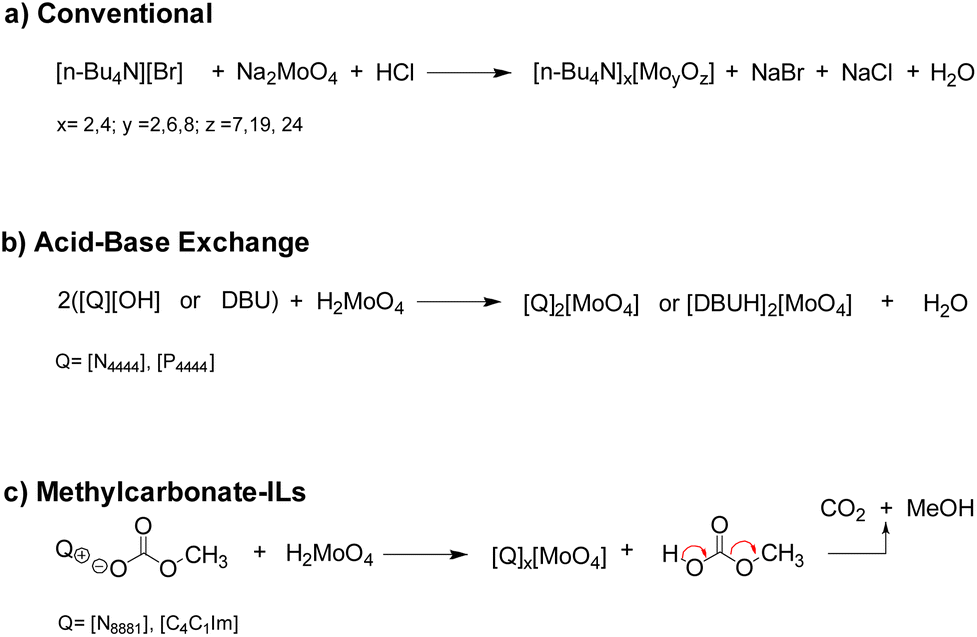 | ||
| Fig. 3 Synthetic routes for the preparation of molybdate ILs. | ||
The structures of Mo-ILs were confirmed by FT-IR, 1H-, 13C-, 31P- and 95Mo-NMR spectroscopy (see the ESI† for further details). In particular, the presence of molybdate species (for compounds 1 and 5–8) was confirmed by the large absorption band associated with the Mo–O stretching vibration observed between 831 and 826 cm−1 (see Fig. S10, S26, S31, S35 and S39†), while the dimolybdate one is characterized by a strong and broad absorption band at 883 cm−1 (see Fig. S14†).
Tetrabutylammonium isopolyoxomolybdates 3 and 4 showed a different kind of absorption due to a more complex polyoxoanion structure, more precisely, catalyst 3 is characterized by two strong absorption bands at 799 and 954 cm−1 while catalyst 4 is characterized by three characteristic absorption bands at 661, 807 and 922 cm−1 (see ESI Fig. S18 for compound 3 and S22 for compound 4†), in agreement with previously published results.3695Mo-NMR spectroscopy provides further confirmation of different oxomolybdenum species; in fact, the monomolybdate anions (catalysts 1, 5, 6, 7 and 8) present chemical shifts ranging from 5 to 13 ppm (see Fig. 4 and Fig. S9, S30, S25, S34 and S38†).40–42 These differences in the chemical shifts are ascribed to the interaction of the monomolybdate species with the cations, which influence the electronic environment. In some cases (Fig. S9 and S30†) the contemporary presence of dimolybdate species (−5 ppm) caused by the degradation process driven by the presence of water traces is evident.40
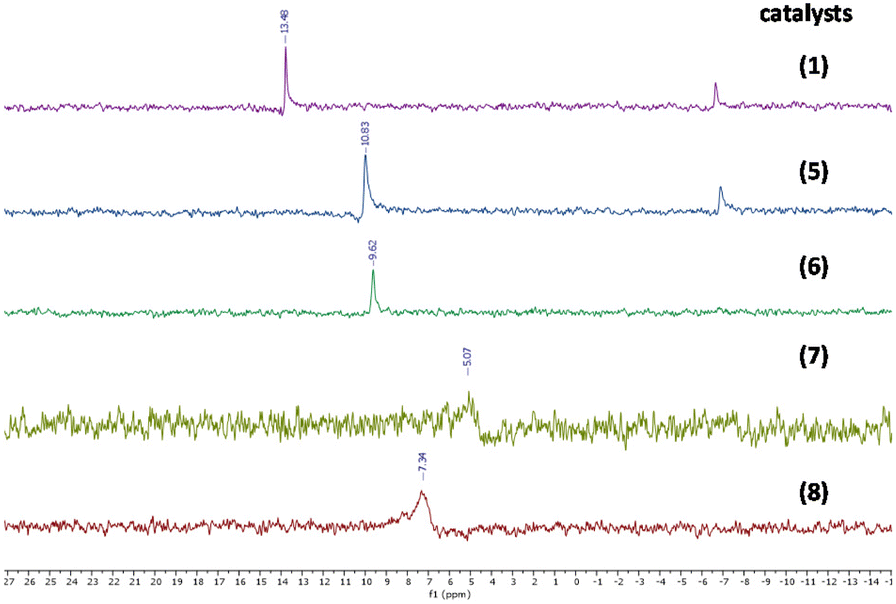 | ||
| Fig. 4 95Mo-NMR spectra of [MoO4]− ILs (see the ESI for the complete spectra, Fig. S9, S30, S25, S34 and S38†). | ||
95Mo-NMR spectra of tetrabutylammonium isopolyoxomolybdates showed single peaks between −5 ppm and 125 ppm as already reported in previous scientific reports,43 and confirmed the assumptions made through FT-IR spectroscopy (Fig. 5).
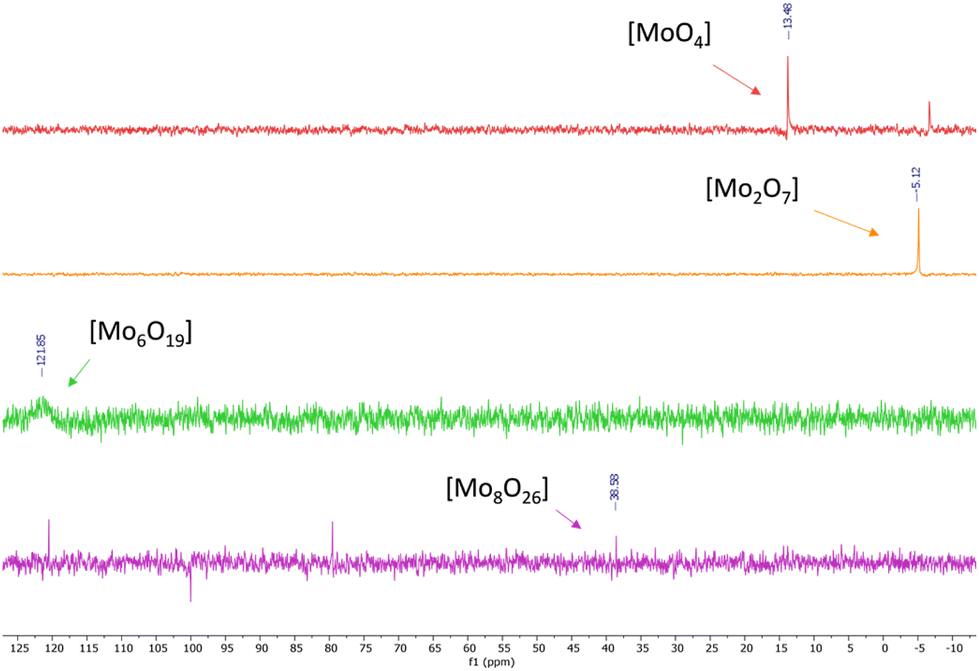 | ||
| Fig. 5 95Mo-NMR spectra of tetrabutylammonium isopolyoxomolybdate (see ESI Fig. S9, S13, S17 and S21†). | ||
ICP characterization further confirmed the structures and molecular weights of compounds 1, 2, 3 and 4 (see ESI Table S3†). It should be noted that compound 1 has a lower correspondence due to its peculiar hygroscopicity, as already observed.
1H-, 13C-, and 31P-NMR spectroscopy techniques were exploited to confirm the structure of organic cations. In the case of Mo-ILs 6 and 7, the disappearance of the peaks of the methyl carbonate anion of the precursors in the 1H-NMR spectra allowed us to verify the quantitative formation of the desired ILs obtained through path c, Fig. 3. Finally, 31P-NMR spectra confirmed the presence of a phosphonium cation in the case of catalyst 5 (see ESI Fig. S27†).
Mo-IL-catalyzed insertion of CO2 into epoxides
Initially, the role of different molybdate anions was investigated by performing the CO2 insertion reaction with 1,2-epoxyhexane (EH) as a model substrate to yield hexane carbonate (HC) under solventless conditions. Table 1 reports the results obtained by using 2.5 mol% of Mo with respect to the substrate, regardless of the Mo salt used.|
|
||||
|---|---|---|---|---|
| Entry | Catalysts | Conversionb (%) | Selectivityb (%) | Yieldb (%) |
| a Reaction conditions: 1,2-epoxyhexane (4 mmol), catalyst (2.5 mol%), p0(CO2) = 30 bar, T = 100 °C, t = 18 h, solventless reaction. b Conversion, selectivity and yield calculated by 1H NMR. | ||||
| 1 | [N4444]2[MoO4] | 93 | 68 | 63 |
| 2 | [N4444]2[Mo2O7] | 79 | 64 | 51 |
| 3 | [N4444]2[Mo6O19] | 0 | — | — |
| 4 | [N4444]2[Mo8O26] | 0 | — | — |
| 5 | [NH4]2[MoO4] | 0 | — | — |
| 6 | [Na]2[MoO4] | 0 | — | — |
[N4444]2[MoO4] (entry 1, Table 1) gave almost quantitative conversion and discrete selectivity towards HC (68%) while [N4444]2[Mo2O7] (entry 2, Table 1) showed poorer performance in terms of conversion of the substrate and selectivity towards HC. Tetrabutylammonium iso-polyoxomolybdates 3 and 4 (hexa- and octamolybdate) (entries 3 and 4, Table 1) did not exhibit significant conversion of the substrate: the molybdate atoms were likely poorly accessible and thus unable to interact with CO2 and promote the reaction, indicative of the importance of MoO4 species.
We suppose that the greater catalytic activity of ILs based on [MoO4]2− and [Mo2O7]2− can be ascribed to two main reasons: (i) interaction between CO2 and monomeric/dimeric Mo species demonstrated by 13C and 95Mo NMR spectra (see Fig. 9); this interaction was not observed with iso-polyoxometalates 3 and 4, in particular when recording 95Mo NMR spectra under a CO2 atmosphere (see Fig. S17 and S21†). (ii) Greater solubility of Mo-ILs 1 and 2 in the reaction mixture since they are soluble in neat epoxide while iso-polyoxomolybdates 3 and 4 are not soluble.
Ammonium and sodium molybdates were also tested (entries 5 and 6, Table 1), displaying negligible conversion, suggesting that the organic cation is fundamental to promoting the reaction. The inactivity of ammonium and sodium molybdates is probably due to their insolubility in the substrate and the non-interaction of these salts with CO2 (see Fig. S2†).
Catalyst 1 was chosen as a model catalyst to optimize the reaction conditions for the CO2 insertion reaction into 1,2-epoxyhexane. The influence of temperature (T = 50–160 °C) on the reaction of EH in the presence of 30 bar of CO2 and 2.5 mol% of catalyst for 18 h is reported in Fig. 6. Conducting the reaction at T ≥ 120 °C was crucial to obtaining a high conversion of EH, while lower temperatures did not allow adequate CO2 activation towards the formation of HC. However, temperatures >120 °C caused a decrease in selectivity due to the formation of polyethers as secondary products as observable from the typical chemical shift (3.90–3.30 ppm) in the 1H NMR spectra. The main by-product at T ≤ 120 °C was 1,2-hexanediol (according to 1H-NMR, 13C-NMR and GC-MS analysis, see ESI Fig. S42–S44†) but 1-(2-hydroxyhexoxy)hexan-2-ol was also observed as a smaller second by-product (see ESI Fig. S45–S47†). The formation of these compounds was ascribed to the presence of water and the ability of the molybdate anion to promote the hydration reaction of the epoxide.
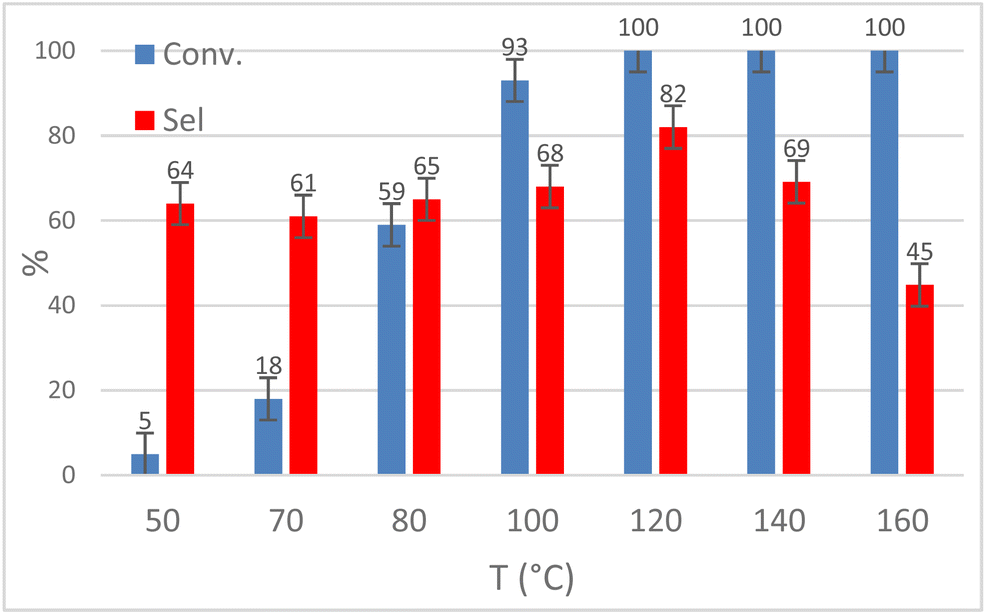 | ||
| Fig. 6 Effect of the temperature on CO2 insertion into 1,2-epoxyhexane (EH) to obtain hexane carbonate (HC). Reaction conditions: EH (4 mmol), 1 (2.5 mol%), CO2 (30 bar), t = 18 h, T = 50–160 °C, solventless reaction. Conversion, selectivity and yield were calculated by 1H-NMR. | ||
Crystallization of water is always observed in molybdate-based ionic liquids since they are very hygroscopic (see ESI Fig. S10†).
Having identified the optimal temperature (T = 120 °C), the kinetics of the reaction was monitored for t = 18 h and the product distribution is shown in Fig. 7. The conversion of EH was quantitative after 9 h and there was a slight degradation or side-reaction of the COC formed, as highlighted by the increase of by-products (i.e. others, grey line) after 9 h.
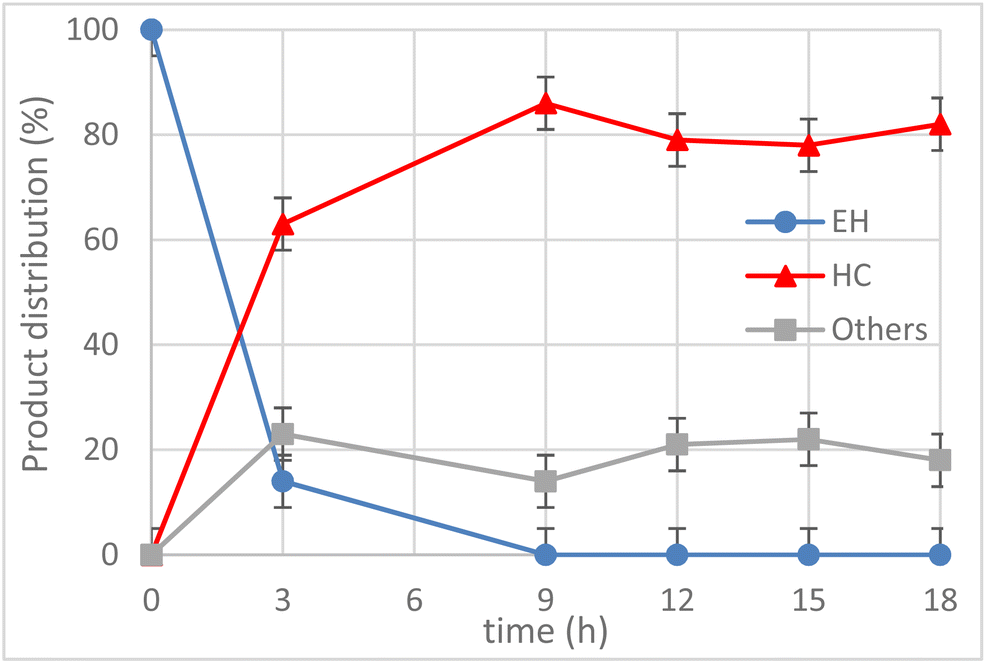 | ||
Fig. 7 Effect of the reaction time on CO2 insertion into 1,2-epoxyhexane (EH) to obtain hexane carbonate (HC). Reaction conditions: EH (4 mmol), 1 (2.5 mol%), CO2 (30 bar), T = 120 °C, t = 3–18 h, solventless conditions. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Determined by GC-FID. Determined by GC-FID. | ||
The influence of CO2 pressure (1–50 bar) was explored by performing the reaction at T = 120 °C for t = 9 h (Fig. 8). Although EH conversion was always quantitative, 30 bar of CO2 were required to achieve the best selectivity (86%) while poorer results were obtained with p(CO2) = 1–15 bar. Increasing the CO2 pressure to 50 bar led to a lower selectivity towards HC. This behavior could be explained by observing the endogenous pressure value in the autoclave, 65 bar, typical of quasi-critical conditions and likely causing a lower interaction between CO2 and the epoxide, hence promoting the formation of polyethers.
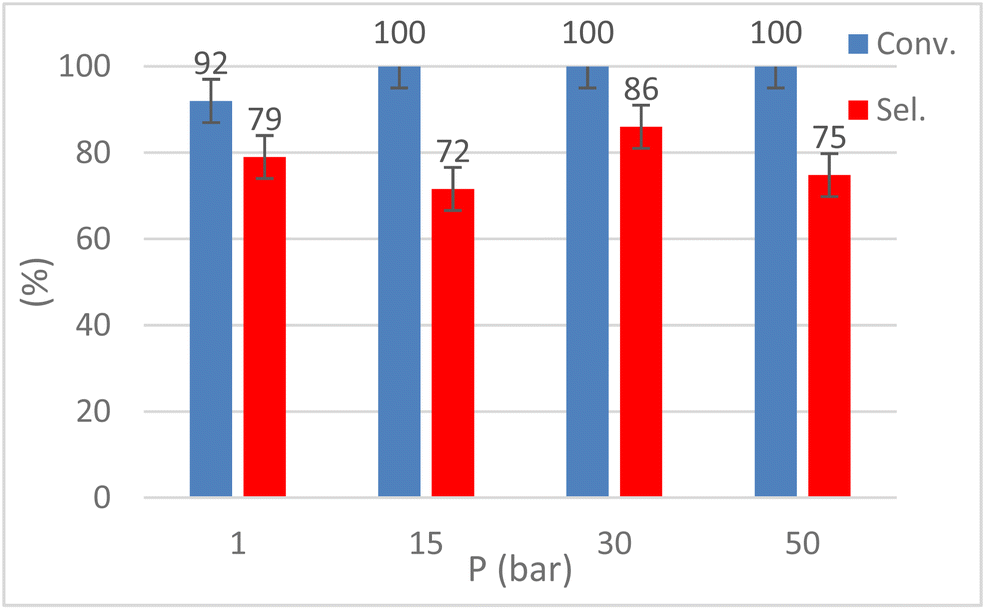 | ||
| Fig. 8 Pressure screening for CO2 insertion into 1,2-epoxyhexane (EH) to obtain hexane carbonate (HC). Reaction conditions: EH (4 mmol), 1 (2.5 mol%), T = 120 °C, t = 9 h, p = 1–50 bar (CO2), solventless conditions. Conversion and selectivity were calculated by 1H-NMR. | ||
Once it was proved that the mono-molybdate species is the most active for CO2 fixation into the epoxide, the effect of the organic onium cation was investigated. The catalytic activity of four different Mo-ILs 5, 6, 7, and 8 was tested in a model CO2 fixation reaction under the optimized operative conditions (T = 120 °C, p0(CO2) = 30 bar CO2, t = 9 h). The results are summarized in Table 2.
| Entry | Catalysts | Conversionb (%) | Selectivityb (%) | Yieldb (%) |
|---|---|---|---|---|
| a Reaction conditions: 1,2-epoxyhexane (4 mmol), p0(CO2) = 30 bar, T = 120 °C, t = 9 h, solventless conditions. b Conversion, selectivity and yield calculated by 1H-NMR. c Isolated yield. | ||||
| 1 | [N4444]2[MoO4] | >99 | 86 | 86 (83)c |
| 2 | [P4444]2[MoO4] | >99 | 79 | 79 |
| 3 | [N8881]2[MoO4] | 96 | 75 | 72 |
| 4 | [C4C1Im]2[MoO4] | 88 | 75 | 67 |
| 5 | [DBUH]2[MoO4] | 68 | 34 | 23 |
The reaction conversion showed a fluctuating trend consistent with that of the cation: the use of [N4444]2[MoO4] and [P4444]2[MoO4] allowed for achieving comparable conversion of EH while [N8881]2[MoO4] and [C4C1Im]2[MoO4] showed slightly poorer performance. It is worth noting that the conversion of EH and selectivity towards HC collapsed with DBUH2[MoO4] (68% and 34%, respectively, entry 5, Table 2). Probably the lipophilic character of catalysts 1 and 5–7 is crucial to ensure a good performance, as already remarked in Table 1 for [NH4]2[MoO4] and [Na]2[MoO4] (entries 5 and 6, Table 1). Additional control tests were performed with the catalyst precursors (e.g. [N4444]OH, [P4444]OH, [N1888][CH3OCOO] and [C1C4im][CH3OCOO]), further proving the improved performance of our Mo-ILs (see Table S1†); catalyst loading tests (see Table S2†) confirmed 2.5 mol% as the optimal loading for our Mo-IL 1. Hence, we decided to continue our investigation by using 1 as the catalyst since it allowed us to reach an excellent selectivity for the COC product (entry 1, Table 2), also leading to the maximum yield of HC. With the optimized conditions in hand, the scope of the coupling reaction was broadened by testing CO2 fixation into various linear and cyclic aliphatic epoxides with [N4444]2[MoO4] (Table 3).
|
|
||||
|---|---|---|---|---|
| Entry | Substrate | Conversionb (%) | Selectivityb (%) | Yieldb (%) |
|
a Reaction conditions: substrate (4 mmol), p0(CO2) = 30 bar, T = 120 °C, t = 9 h, solventless.
b Conversion, selectivity and yield calculated by 1H NMR.
c Isolated yield.
d
cis/trans ratio = 72:28.
e
cis/trans ratio = 16:84.
|
||||
| 1 |
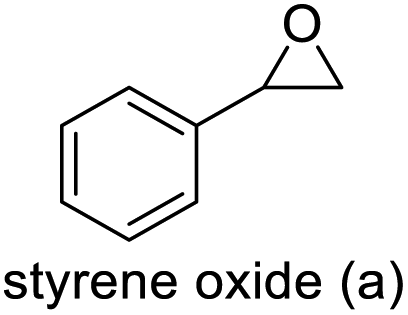
|
96 | 91 | 87 (76)c |
| 2 |
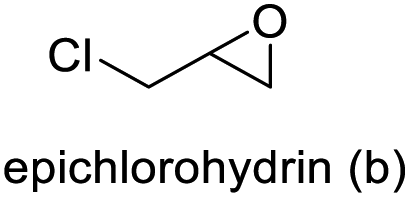
|
>99 | >99 | 99 (88) |
| 3 |
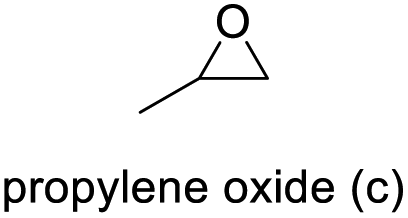
|
78 | 91 | 71 |
| 4 |
|
88 | 89 | 78 |
| 5 |
|
92 | 90 | 83 (78)c |
| 6 |
|
85 (61)c | 85 (61)c | 85 (61)c |
| 7 |
|
99 | 99 | 99 (75)c |
| 8 |
|
98 | 86 | 85 (61)c |
| 9 |
|
>99 | 63 | 63 |
| 10 |
|
>99 | 45 | 45 |
| 11 |
|
64 | 63d | 40 |
| 12 |
|
24 | 37e | 9 |
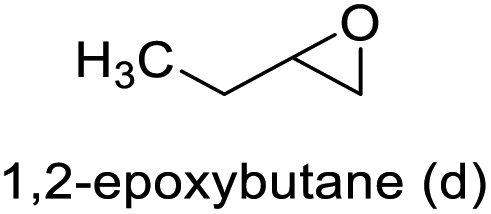
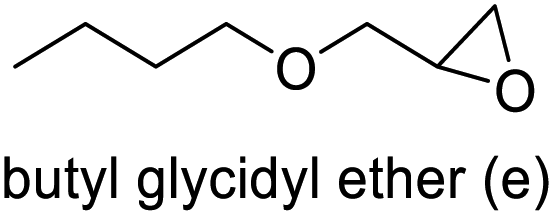
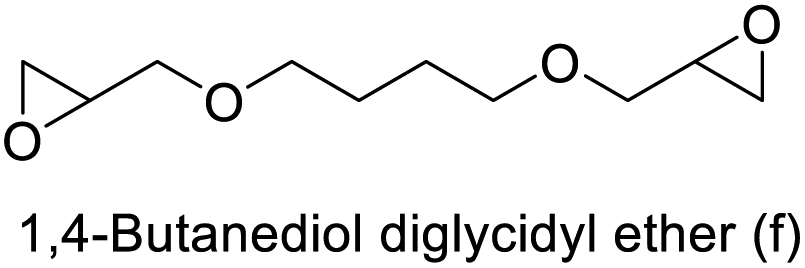
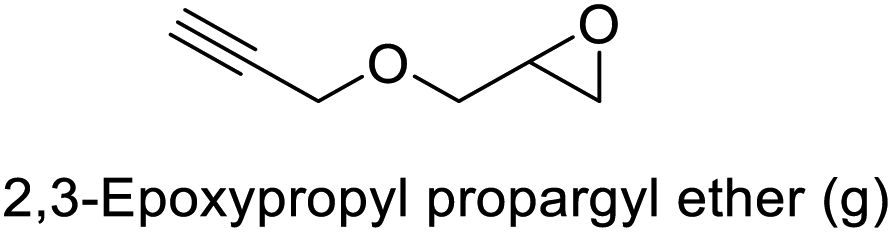
The results demonstrated its catalytic activity for CO2 fixation into terminal epoxides a–h (entries 1–8, Table 3): these compounds were converted into the corresponding COC products with conversion and selectivity similar to or higher than the one reported for 1,2-epoxyhexane. In particular, we have observed an excellent catalytic performance with industrially relevant epoxides such as epichlorohydrin (b) and propylene oxide (c). 1,2-Epoxybutane (d) shows a slightly higher conversion than c probably as a consequence of its lower volatility that enables a better interaction with Mo-ILs and its adduct with CO2. Terminal epoxides i and j were quantitatively converted but the selectivity towards the corresponding COCs decreased due to the formation of polyethers. The different behavior of i compared to b is probably due to the better leaving group ability of bromide compared to chloride, which allows the formation of polyethers. Nonetheless, Mo-ILs demonstrated significant tolerance to different functional groups such as in the cases of a, b, e, f, g and h, also demonstrating higher conversions compared to c, probably due to the greater occurrence of the latter in the gaseous phase under the reaction conditions investigated. Internal cyclic epoxides, such as k and l, were less reactive, presumably because of the steric factors. In particular for cyclohexene oxide (k, entry 11, Table 3), the selectivity towards the carbonates decreased in favour of polyethers and polycarbonates detected by 1H NMR analysis through the characteristic broad signals at 3.40 and 4.40 ppm in CDCl3, respectively (see Fig. S62†).44,45 The formation of cyclohexanone was not detected by 1H NMR analysis, hence excluding the occurrence of side products obtained via the Meinwald rearrangement.46 The presence of both cis and trans isomers (see Fig. S62†) is due to the possible ring closure of the five-membered ring and can occur by both SN1 and SN2 mechanisms, but with an evident preference for the formation of the cis product. This behaviour was already reported in the literature and by us in previous papers regarding the use of transition metal-based catalysts for CO2 fixation into internal epoxides.33,47 The presence of Mo-ILs evidently promotes an SN2 pathway and the formation of cis carbonate, while the formation of trans carbonate is favored by an SN1 pathway and the possible depolymerization of the polycarbonate by-product. In the case of limonene oxide (entry 12, Table 3), the high degree of substitution of the oxirane prevents its coordination to the active molybdenum species, leading to a low yield (8%) and selectivity towards the corresponding COC.
Molybdate anions are hence able to activate CO2 and catalyse its insertion into epoxides without the addition of a halide to promote ring-opening.
We then conducted further NMR experiments to better understand the interaction of molybdate with CO2 and delve into the reaction mechanism (see Fig. 9).
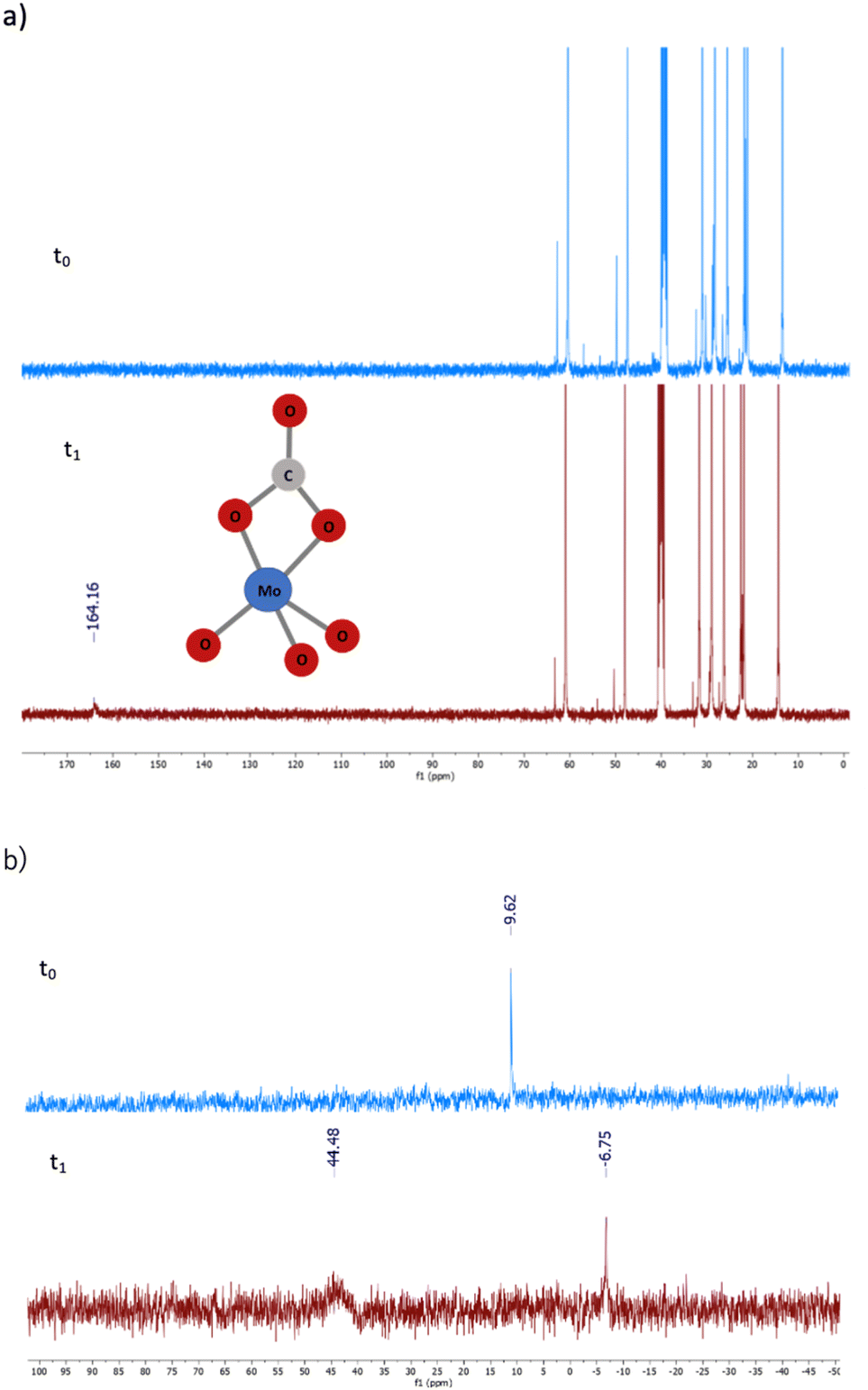 | ||
| Fig. 9 13C-NMR (a) and 95Mo-NMR (b) analyses of [N8881]2[MoO4] before (blue) and after (red) treatment with CO2. Operative conditions: [N8881]2[MoO4] 0.4 M in DMSO-d6, T = 25 °C, p0(CO2) = 5 bar. | ||
Upon addition of 5 bar of CO2 to a 0.4 M DMSO-d6 solution of catalysts 1 or 6 at room temperature, in a pressure-resistant NMR tube, the formation of a new species became quickly evident. As reported in Fig. 9a, the 13C NMR spectra showed the appearance of a peak at 164.16 ppm typical of a carbonyl group. 95Mo NMR analysis (Fig. 9b) highlighted the substantial disappearance of the initial molybdate species (9.62 ppm) and the formation of a new broad peak at 44.48 ppm distinctive of a molybdenum–CO2 adduct. These observations are in agreement with previous reports on the reversible binding of [MoO4]2− with CO2 through 95Mo NMR and single crystal X-ray diffraction analyses, demonstrating the formation of [MoO3(CO3)]2− carbonate complexes (95Mo-NMR = +46.7 ppm and 13C-NMR = 165.7 ppm).40
In our case, 95Mo-NMR analysis also clearly indicates the presence of [Mo2O7]2−, identified by a signal at −6.75 ppm.40
These observations led us to propose the mechanism described in Fig. 10 for CO2 insertion into epoxides catalyzed by Mo-ILs.
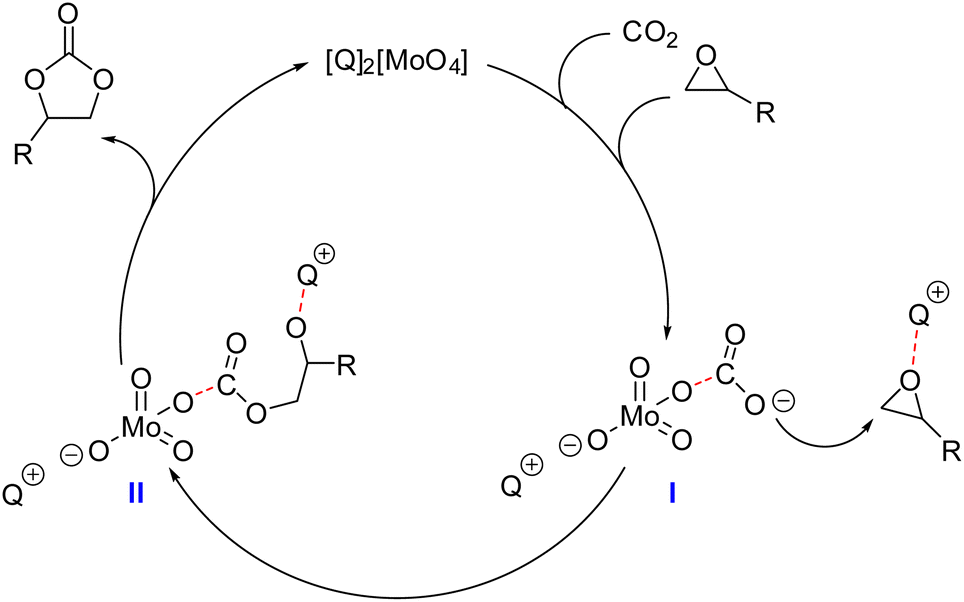 | ||
| Fig. 10 Mechanistic hypothesis for CO2 fixation into epoxides catalyzed by Mo-ILs. | ||
The molybdate–CO2 carbonate complex I formed in situ could itself act directly as the nucleophile and attack the cation-activated epoxide, yielding the hemicarbonate intermediate II, which can ring-close with the formation of a COC product and the release of the molybdate IL. This mechanism was coherent with the fact that CO2 insertion did not take place at T < 50 °C but only at T > 70 °C, where complex I was able to release activated CO2, as already observed for [MoO3(CO3)]2−.40 While most of the metal-based catalytic systems active towards CO2 fixation into epoxides require the presence of a halide (Cl−, Br−, I−) and/or a nucleophile co-catalyst to achieve any conversion,32–35 the present molybdate anions were active in the absence of a halide source or other external nucleophiles (Fig. 1). The mechanism proposed is supported by the evidence of CO2–molybdate complex formation described above, as well as by a comparison with other mechanisms reported for the synthesis of polycarbonates mediated by metal complexes containing an alkoxide or aryloxide group (−OR) that are able to undergo CO2 insertion into epoxides without requiring an external nucleophile.48
Finally, we evaluated the recyclability of our catalytic system and tested the scaling-up of our protocol. The results obtained from the first five recycles are reported in Fig. 11 while more details are reported in Fig. S63–S66 (see the ESI†).
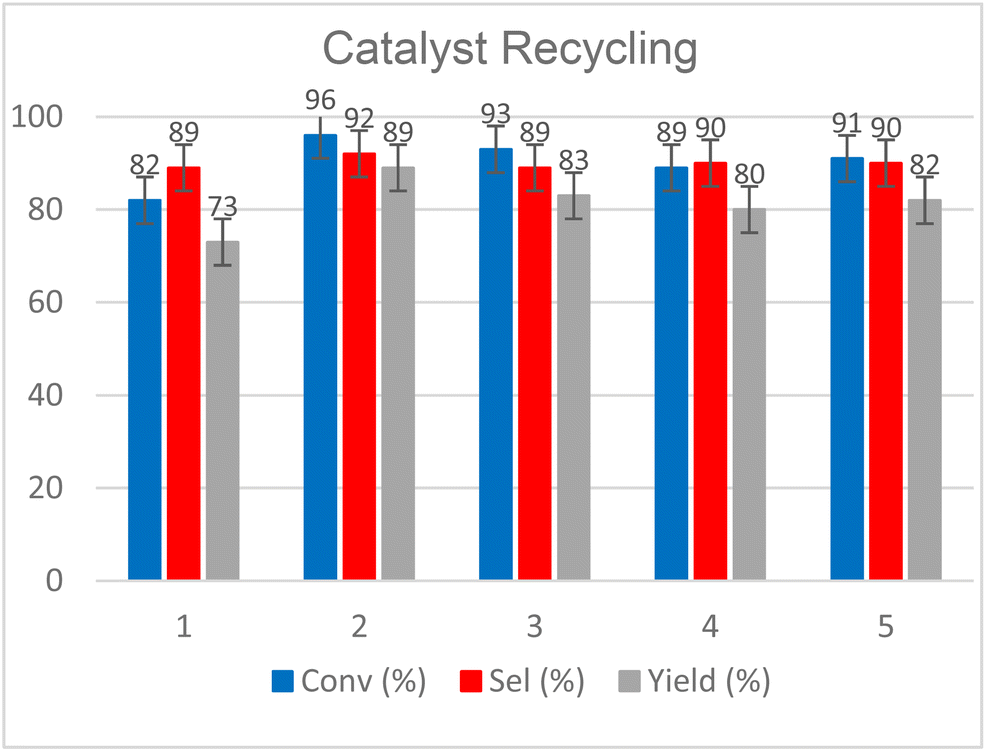 | ||
| Fig. 11 Catalyst recycling tests for CO2 insertion into 1,2-epoxyhexane (EH) to obtain hexane carbonate (HC). Reaction conditions: EH (20 mmol), 1 (2.5 mol%), T = 120 °C, t = 9 h, p0(CO2) = 30 bar, solventless conditions. Conversion, selectivity and yield were calculated by 1H-NMR. | ||
Reactions were performed starting from 2 g of 1,2-epoxyhexane (20 mmol) in the presence of 30 bar of CO2 and were conducted in a glass liner inserted into an autoclave at T = 120 °C for t = 9 h. At the end of each cycle, the products were separated through short path distillation apparatus (see Fig. S63,† left) while the catalyst was recovered and reused without further processing. The product mixture was recovered as a clear transparent liquid by distilling the vapors between T = 65 and 75 °C at p = 0.9 mbar (see Fig. S63,† right).
The distilled fraction was characterised by 1H NMR analysis, observing that it was mainly composed of the COC product, with traces of diol and dimerized ether 1-(2-hydroxyhexoxy)hexan-2-ol as the main by-products (see Fig. S64† for further details). Since our original objective was to demonstrate catalyst recyclability, we did not optimize the fractional distillation of each recycle run, nevertheless this system appeared suitable for both separation and recycling of the homogeneous catalyst. From Fig. 11, we can see how 1 could be used and recovered effectively for 5 consecutive cycles without any loss of activity. Except for step 1, in which a conversion and a lower selectivity were achieved due to the loss of the product inside the distillation system, the other steps maintained a conversion between 96 and 89% and a selectivity between 92 and 89%.
Our catalytic system remains efficient when compared to other halide-free (and solvent-free) protocols. Table 4 summarizes and compares our results with some recently reported systems for CO2 fixation into epichlorohydrin as an industrial key model epoxide.49 Mo-ILs showed a catalytic activity comparable to other homogeneous metal catalysts (entries 2–4, Table 4), heterogeneous metal catalysts, such as ZIFs, MOFs, and amorphous polymers (entries 5–9, Table 4), organic ILs (entries 10–13, Table 4), homogeneous organocatalysts (entries 14–17, Table 4), and heterogeneous organocatalysts (entries 18 and 19, Table 4). Interestingly, most catalysts are active under similar operative conditions (e.g. T = 60–150 °C, t = 3–24 h, p0(CO2) = 10–30 bar) and yields are usually >90%. Compared to other homogeneous catalysts (see entries 2–5 and 10–17, Table 4), Mo-ILs showed enhanced recyclability and were re-used up to 5 times without significant loss of catalytic activity. All the reported studies are relevant for the use of (renewable) CO2 but several other factors should be considered with a view to achieving sustainable organic transformations, such as synthesis routes, safety of the reactants, complexity, greenness and atom economy for the synthesis of catalysts, recyclability, the presence of co-catalysts, the presence of solvent, etc.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| No. | Catalyst | T (°C) | t (h) | CO2 (bar) | Yield (%) | Number of recycles | Ref. |
| a Metal homogeneous catalysts. b Metal heterogeneous catalysts. c Ionic liquids. d Homogeneous organocatalysts. e Heterogeneous organocatalysts. | |||||||
| 1a,c | [N4444][MoO4] – our work | 120 | 9 | 30 | 99 | 5 | |
| 2a,c | (DBUH)3NbO5 | 130 | 5 | 30 | 90 | 8 | 27 |
| 3a | [(C4H9)4N]3H7GeW9Mn3(H2O)3O37 | 150 | 3 | 30 | 93 | No | 51 |
| 4a | Cr(0) complex | 100 | 4 | 10 | 99 | No | 52 |
| 5b | HE-ZIF-PM | 100 | 8 | 10 | 97 | No | 53 |
| 6b | ZIF-67 | 120 | 6 | 10 | 95 | 3 | 54 |
| 7b | Zn/Co-ZIF-H2Ar-1000 | 140 | 8 | 8 | 93 | 11 | 55 |
| 8b | (Me2NH2)2[Zn8(Ad)4(DABA)6O]7 (MOF) | 100 | 24 | 10 | 99 | 5 | 56 |
| 9b | Aluminium-based polymeric (Al-PDC) | 100 | 20 | 1 | 93 | 11 | 57 |
| 10c | [P4444][bzim] | 100 | 3.5 | 10 | 88 | no | 58 |
| 11c | Pyridinium saccharinate (PPy·Sac) | 60 | 24 | 1 | 90 | no | 59 |
| 12c | Butyl methyl imidazolium acetate | 80 | 2 | 10 | 78 | no | 60 |
| 13c | [APBIm][Glu] | 105 | 3 | 5 | 98 | 5 | 61 |
| 14d | Salophen ligand | 120 | 24 | 10 | 76 | no | 62 |
| 15d | Acen-H catalyst | 110 | 4 | 10 | 99 | no | 63 |
| 16d | Carbodicarbene | 100 | 12 | 20 | 92 | no | 64 |
| 17d | 2-Aminopyridine | 60 | 16 | 10 | 90 | no | 65 |
| 18e | Chitosan biochar | 100 | 12 | 20 | 90 | no | 66 |
| 19e | N-doped shrimp shell carbon | 150 | 4 | 30 | 93 | 6 | 67 |
Mo-ILs are excellent catalysts since they are synthesized through a green, halide-free route starting from simple and cheap precursors compared to other catalysts reported in Table 4. They are easily recyclable and reusable. This is the first example in the literature of Mo-based catalysts effective for CO2 activation through a halide-free pathway. The downside of Mo-ILs is the presence of a metal since many recent studies focused on organocatalysts (cf. entries 10–19, Table 4) which may have a greener connotation. However, although the catalytic systems surveyed in Table 4 are useful for CO2 fixation, none are conceived to act in different consecutive catalytic transformations in a divergent approach.47,50 Here we established that Mo-ILs are active for both CO2 fixation into epoxides and the epoxidation of olefins.
Considering the well-known oxidation ability of molybdenum-based catalysts,68 we performed some preliminary tests to evaluate the epoxidation activity of our Mo-ILs, with a view to the direct conversion of an olefin in the corresponding COC. Epoxidation of 1-decene was chosen as the model reaction and performed in the presence of hydrogen peroxide as an environmentally benign oxidant, [N8881]2[MoO4] as a catalyst and H3PO4 as a co-catalyst. The best operative conditions are summarised in Scheme 1.
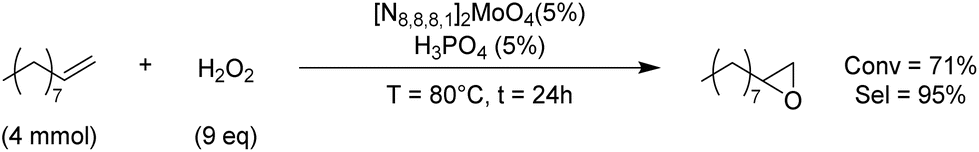 | ||
| Scheme 1 Epoxidation of a model substrate in the presence of hydrogen peroxide as an oxidant and a Mo-IL as a catalyst. Reaction conditions: 1-decene (4 mmol), H2O2 (30% w/wH2O, 9 eq.), [N8881]2[MoO4] (5 mol%), H3PO4 (5 mol%), T = 80 °C, t = 24 h. Mesitylene was added as an internal standard. Conversion and selectivity were calculated by GC analysis. | ||
Thus with these preliminary tests, we achieved 71% conversion of 1-decene and excellent 95% selectivity towards the formation of 1-decene epoxide. This result indicated the possibility of a one-pot olefin epoxidation–CO2 fixation by a halide-free route.
Conclusions
A set of novel molybdate ILs (Mo-ILs) was synthesized through a novel halide-free synthetic route and fully characterized with different spectroscopic techniques. These new compounds proved to be viable catalysts for CO2 fixation into epoxides; in particular, catalyst 1 ([N4,4,4,4]2[MoO4]) promoted the conversion of 1,2-epoxyhexane to the corresponding cyclic carbonate in 86% yield with p0(CO2) = 30 bar at T = 120 °C. To explain these results, a reaction mechanism based on the formation of a molybdate–carbonate intermediate as the active nucleophile was proposed in light of an evident interaction between CO2 and Mo established through 95Mo-NMR analysis. Although the investigated Mo-ILs were less efficient than other metal-based catalysts coupled with nucleophile sources, Mo-ILs exhibit a surprisingly higher catalytic activity with respect to other metals when used alone. To the best of our knowledge, this paper describes the first example in which simple Mo-ILs are successfully used for the cycloaddition of CO2 to epoxides without the use of any halide co-catalysts. Finally, our Mo-ILs were also active for the epoxidation of olefins with aqueous H2O2, with high selectivity towards the corresponding epoxide and high conversion of the olefin. The tandem olefin epoxidation–CO2 fixation processes are currently under investigation in our laboratory.Experimental section
Materials and methods
All chemicals were purchased from Sigma Aldrich (now Merck) and used as received. Trioctylmethylammonium methylcarbonate ([N1888][CH3OCOO]) and 1-butyl-3-methylimidazolium methylcarbonate ([C4C1im][CH3OCOO]) were synthesized following a procedure previously reported by our group.35 Qualitative and quantitative analyses and characterization of reaction mixtures and pure products were performed with: (a) a GC-MS system consisting of an Agilent 6890N GC equipped with an HP-5 capillary column (30 length, 0.32 mm internal diameter, and 0.25 mm film thickness) coupled with an Agilent Technologies 5975 mass detector operating at 70 eV, (b) a Bruker Ascend 400 (AV400) NMR spectrometer operating at 400 MHz for 1H nuclei, 100 MHz for 13C nuclei, 75 Hz for 31P nuclei and 26 MHz for 95Mo nuclei, and (c) a PerkinElmer Spectrum One FT-IR spectrometer operating at wavenumbers ranging from 400 to 4000 cm−1. GC-MS analyses were performed in EtOAc or Et2O, while 1H NMR and 13C NMR samples were prepared using deuterated solvents (CDCl3, MeOH-d4 or DMSO-d6, deuteration grade >99.9% D in all cases). In the 1H NMR spectra, the deuterated solvent residual signal was selected as the spectrum reference. ICP-MS analysis was conducted with a Nexion 350XX coupled with a seaFAST autosampler optimized in KED mode (4.4 ml of He) using Pt 195 as an internal standard. Further details about materials and methods are reported in the Experimental section of the ESI.†General synthesis of [N4444]2[MoO4], [P4444]2[MoO4] and [DBUH]2[MoO4]
These ILs were synthesized by adapting a reported procedure.37 In a 50 mL round bottom flask, 1.85 mmol of H2MoO4 were slowly added in portions to a solution of [N4,4,4,4][OH] (3.7 mmol, 2 equiv.) in 10 mL of H2O. The solution was then stirred for 3 h at room temperature. During this time the initial white suspension slowly dissolved, obtaining a clear, homogeneous solution accompanied by a pH variation from 13 to 9. Water was quantitatively removed from the mixture by evaporation in vacuo at T = 100 °C, yielding the corresponding [nBu4N]2[MoO4] as a white powder in a quantitative yield. A similar procedure was followed to synthesize [nBu4P]2[MoO4] and [DBUH]2[MoO4] starting from 3.7 mmol of [nBu4P][OH] or 1,8-diazabicyclo[5.4.0]undec-7-ene, respectively. The IL products were characterized by FT-IR, 1H-NMR, 13C-NMR, 31P-NMR (if suitable) and 95Mo-NMR spectroscopy.Synthesis of trioctylmethylammonium molybdate, [N1888]2[MoO4], for the corresponding methylcarbonate IL precursor
H2MoO4 (0.51 g, 3.16 mmol) was slowly added to an aqueous solution of [N8881][CH3OCOO] (2.80 g, 6.32 mmol, 2.0 equiv.). The solution was stirred for t = 3 h at T = 50 °C, and it was observed that the initially opalescent solution turned yellow. The solution was cooled to rt, and the product, [N8881]2[MoO4], was extracted with ethyl acetate. 2.49 g (2.78 mmol) of the IL was obtained (88% yield). The product was characterized by FT-IR, 1H-NMR, 13C-NMR and 95Mo-NMR spectroscopy.Synthesis of 1-butyl-3-methylimidazolium molybdate ([C4C1im]2[MoO4]) using the methylcarbonate IL precursor
[C4C1im]2[MoO4] was prepared following the same procedure as [N8881]2[MoO4] starting from 1-butyl-3-methylimidazolium methylcarbonate [C4C1Im]2[CH3OCOO] as reported in a previous paper.50 The ionic liquid product was characterized by IR, 1H-NMR, 13C-NMR and 95Mo-NMR spectroscopy.Typical procedure for CO2 insertion reactions
The selected epoxide (4 mmol, 1 equiv.), molybdate catalyst (2.5 mol%) and mesitylene as an internal standard (20 mol%) were charged in a 5 mL glass reactor equipped with a stirring bar and a pierced glass cap. The glass reactor was placed inside a 10 mL stainless steel autoclave. The autoclave was sealed, degassed via two vacuum/CO2 cycles and pressurized with CO2 (1–50 bar). The autoclave was then electrically heated at the desired temperature (50–160 °C) and the reaction mixture was magnetically stirred for the desired time (3–18 h). At the end of each run, the autoclave was rapidly cooled in an ice bath and vented. The final mixture was analysed by 1H-NMR (CDCl3) to calculate the conversion, yield and selectivity.Scaled-up procedure and recycling of the catalyst
2 g of 1,2-epoxyhexane (20 mmol, 1 equiv.) and catalyst 1 (0.05 mmol, 0.025 equiv.) were charged in a 5 mL glass reactor equipped with a stirring bar and a pierced glass cap. The glass reactor was placed inside a 100 mL stainless steel autoclave. The autoclave was sealed, degassed via two vacuum/CO2 cycles and pressurized with 30 bar of CO2. The autoclave was then heated at T = 120 °C under stirring for t = 9 h. At the end of each run, the autoclave was rapidly cooled in an ice bath and vented. The glass reactor was removed from the autoclave and equipped with short distillation apparatus to separate the catalyst from the reaction mixture. 0.8 mmol of mesitylene as an external standard were added to the distilled fraction which was then analyzed by 1H-NMR (CDCl3) to calculate the conversion, selectivity and yield. The glass liner containing the catalyst was recharged with 2 g (20 mmol) of 1,2-epoxyhexane for another catalytic run. The procedure was repeated 4 times (5 runs).Procedure for 13C-NMR and 95Mo-NMR tests under CO2 overpressure
A 0.4 M DMSO-d6 solution of [N4444]2[MoO4] or [N8881]2[MoO4] was introduced into a 3 mL glass inlet of an autoclave reactor. A glass reactor equipped with a pierced glass stopper and a stirring bar was introduced into a 10 mL stainless steel autoclave and charged with CO2 [p0(CO2) = 0.5 MPa]. The system was kept at room temperature for t = 4 h before transferring the mixture into a pressure-resistant tube which was then filled with CO2 [p0(CO2) = 0.5 MPa]. The 13C- and 95Mo-NMR spectra were recorded for 5120 scans.Procedure for the epoxidation of 1-decene
4 mmol of 1-decene, (N8,8,8,1)2(MoO4) (5 mol% molsubstrate−1), mesitylene as an internal standard (20 mol molsubstrate−1 and H3PO4 as an acid co-catalyst (5 mol% molsubstrate−1) were charged into a 10 ml glass reactor equipped with a magnetic stir bar and a condenser. The mixture was stirred for 1 minute and hydrogen peroxide (30% w/wH2O, 9 equiv.) was added through a syringe pump (0.15 mL min−1). The reactor was heated at the desired temperature (T = 80 °C) and the mixture was stirred at 800 rpm for 24 h. At the end, an aliquot of the reaction mixture was sampled and diluted with ethyl acetate, dried over Na2SO4, and then eluted through a Pasteur pipette filled with a plug of 2–3 cm of silica gel to remove the ionic catalyst from the reaction mixture. The aliquot was analysed by GC-MS to calculate the conversion and selectivity.Author contributions
The authors confirm contribution to the paper as follows: study conception and design: RC, GF and AP; data collection: NB; analysis and interpretation of results: NB, RC and GF; draft manuscript preparation: NB, RC, GF, AP and MS. All authors reviewed the results and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Prof. Marco Bortoluzzi for lending us some reagents. Dr Claudio Santo is gratefully acknowledged for his help in setting up the 95Mo-NMR experiments.References
- M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 CrossRef CAS PubMed.
- F. Barzagli, F. Mani and M. Peruzzini, J. CO2 Util., 2016, 13, 81–89 CrossRef CAS.
- Z.-Z. Yang, L.-N. He, J. Gao, A.-H. Liu and B. Yu, Energy Environ. Sci., 2012, 5, 6602–6639 RSC.
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353–1370 CrossRef.
- M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141–163 RSC.
- Y. Tsuji and T. Fujihara, Chem. Commun., 2012, 48, 9956–9964 RSC.
- K. Didehban, E. Vessally, M. Salary, L. Edjlali and M. Babazadeh, J. CO2 Util., 2018, 23, 42–50 CrossRef CAS.
- W.-H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed.
- N. Mac Dowell, P. S. Fennell, N. Shah and G. C. Maitland, Nat. Clim. Change, 2017, 7, 243–249 CrossRef CAS.
- Q.-W. Song, Z.-H. Zhou and L.-N. He, Green Chem., 2017, 19, 3707–3728 RSC.
- K. Takaishi, T. Okuyama, S. Kadosaki, M. Uchiyama and T. Ema, Org. Lett., 2019, 21, 1397–1401 CrossRef CAS PubMed.
- E. R. Pérez, R. H. Santos, M. T. Gambardella, L. G. de Macedo, U. P. Rodrigues-Filho, J.-C. Launay and D. W. Franco, J. Org. Chem., 2004, 69, 8005–8011 CrossRef PubMed.
- Y. Kayaki, M. Yamamoto and T. Ikariya, Angew. Chem., 2009, 121, 4258–4261 CrossRef.
- D. Yu, S. P. Teong and Y. Zhang, Coord. Chem. Rev., 2015, 293, 279–291 CrossRef.
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 RSC.
- J. W. Comerford, I. D. Ingram, M. North and X. Wu, Green Chem., 2015, 17, 1966–1987 RSC.
- F. Della Monica, M. Leone, A. Buonerba, A. Grassi, S. Milione and C. Capacchione, Mol. Catal., 2018, 460, 46–52 CrossRef CAS.
- M. Liu, X. Wang, Y. Jiang, J. Sun and M. Arai, Catal. Rev., 2019, 61(2), 214–269 CrossRef CAS.
- V. D'Elia, J. D. Pelletier and J. M. Basset, ChemCatChem, 2015, 7, 1906–1917 CrossRef.
- R. E. Barker, L. Guo, C. J. Mota, M. North, L. P. Ozorio, W. Pointer, S. Walberton and X. Wu, J. Org. Chem., 2022, 87, 16410–16423 CrossRef CAS PubMed.
- J. Poolwong, V. Aomchad, S. Del Gobbo, A. W. Kleij and V. D'Elia, ChemSusChem, 2022, 15, e202200765 CrossRef CAS PubMed.
- Z. Yue, T. Hu, H. Su, W. Zhao, Y. Li, H. Zhao, Y. Liu, Y. Liu, H. Zhang and L. Jiang, Fuel, 2022, 326, 125007 CrossRef CAS.
- L. Guo, K. J. Lamb and M. North, Green Chem., 2021, 23, 77–118 RSC.
- N. Zanda, A. Sobolewska, E. Alza, A. W. Kleij and M. A. Pericàs, ACS Sustainable Chem. Eng., 2021, 9, 4391–4397 CrossRef CAS.
- F. Zhang, Y. Wang, X. Zhang, X. Zhang, H. Liu and B. Han, Green Chem. Eng., 2020, 1, 82–93 CrossRef.
- A. W. Kleij, Curr. Opin. Green Sustain. Chem., 2020, 24, 72–81 CrossRef.
- A. Chen, C. Chen, Y. Xiu, X. Liu, J. Chen, L. Guo, R. Zhang and Z. Hou, Green Chem., 2015, 17, 1842–1852 RSC.
- S. Omwoma, C. T. Gore, Y. Ji, C. Hu and Y.-F. Song, Coord. Chem. Rev., 2015, 286, 17–29 CrossRef CAS.
- J. J. Borrás-Almenar, E. Coronado, A. Müller and M. Pope, Polyoxometalate molecular science, Springer Science & Business Media, 2003 Search PubMed.
- B. Yu, B. Zou and C.-W. Hu, J. CO2 Util., 2018, 26, 314–322 CrossRef CAS.
- M.-Y. Wang, R. Ma and L.-N. He, Sci. China: Chem., 2016, 59, 507–516 CrossRef CAS.
- F. Chen, T. Dong, T. Xu, X. Li and C. Hu, Green Chem., 2011, 13, 2518–2524 RSC.
- N. Tenhumberg, H. Büttner, B. Schäffner, D. Kruse, M. Blumenstein and T. Werner, Green Chem., 2016, 18, 3775–3788 RSC.
- J.-H. Chen, C.-H. Deng, S. Fang, J.-G. Ma and P. Cheng, Green Chem., 2018, 20, 989–996 RSC.
- F. Chen, S. Tao, N. Liu and B. Dai, Polyhedron, 2021, 196, 114990 CrossRef CAS.
- A. P. Ginsberg, Inorganic Syntheses, John Wiley & Sons, 1991, vol. 27 Search PubMed.
- Z. Song, W. Huang, Y. Zhou, Z.-Q. Tian, Z.-M. Li and D.-J. Tao, Green Chem., 2020, 22, 103–109 RSC.
- R. Calmanti, M. Selva and A. Perosa, Mol. Catal., 2020, 486, 110854 CrossRef CAS.
- M. Fabris, V. Lucchini, M. Noè, A. Perosa and M. Selva, Chem. – Eur. J., 2009, 15, 12273–12282 CrossRef CAS PubMed.
- I. Knopf, T. Ono, M. Temprado, D. Tofan and C. C. Cummins, Chem. Sci., 2014, 5, 1772–1776 RSC.
- H. Sunaba, K. Kamata and N. Mizuno, ChemCatChem, 2014, 6, 2333–2338 CrossRef CAS.
- S. Bank, S. Liu, S. N. Shaikh, X. Sun, J. Zubieta and P. D. Ellis, Inorg. Chem., 1988, 27, 3535–3543 CrossRef CAS.
- S. Gheller, M. Sidney, A. Masters, R. Brownlee, M. O'Conner and A. Wedd, Aust. J. Chem., 1984, 37, 1825–1832 CrossRef CAS.
- M. Taherimehr and P. P. Pescarmona, J. Appl. Polym. Sci., 2014, 131(21), 41141 CrossRef.
- C. Koning, J. Wildeson, R. Parton, B. Plum, P. Steeman and D. Darensbourg, Polymer, 2001, 42, 3995–4004 CrossRef CAS.
- T. Guo, Y. Gao, Z. Li, J. Liu and K. Guo, Synlett, 2019, 329–332 CAS.
- R. Calmanti, N. Sargentoni, M. Selva and A. Perosa, Catalysts, 2021, 11, 1477 CrossRef CAS.
- P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2, 2169–2187 RSC.
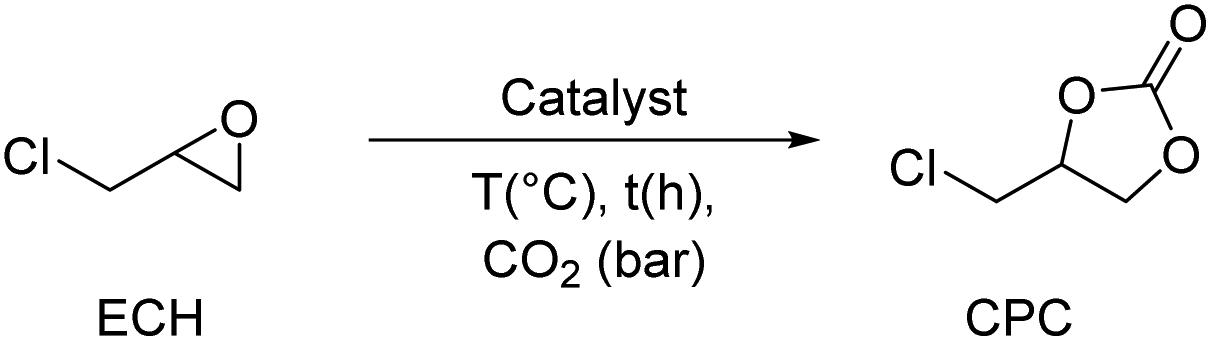
- ECH was chosen as a model substrate for comparison since many studies did not explore the use of EH.
- R. Calmanti, M. Selva and A. Perosa, Green Chem., 2021, 23, 7609–7619 RSC.
- F. W. Chen, T. Dong, X. F. Li, T. G. Xu and C. W. Hu, Chin. Chem. Lett., 2011, 22, 871–874 CrossRef CAS.
- Y. Kim, S. Ryu, W. Cho, M. Kim, M. H. Park and Y. Kim, Inorg. Chem., 2019, 58, 5922–5931 CrossRef CAS PubMed.
- W. Xu, H. Chen, K. Jie, Z. Yang, T. Li and S. Dai, Angew. Chem., Int. Ed., 2019, 58, 5018–5022 CrossRef CAS PubMed.
- R. R. Kuruppathparambil, T. Jose, R. Babu, G.-Y. Hwang, A. C. Kathalikkattil, D.-W. Kim and D.-W. Park, Appl. Catal., B, 2016, 182, 562–569 CrossRef CAS.
- S. Chaemchuen, X. Xiao, M. Ghadamyari, B. Mousavi, N. Klomkliang, Y. Yuan and F. Verpoort, J. Catal., 2019, 370, 38–45 CrossRef CAS.
- H. He, Q. Q. Zhu, J. N. Zhao, H. Sun, J. Chen, C. P. Li and M. Du, Chem. – Eur. J., 2019, 25, 11474–11480 CrossRef CAS PubMed.
- A. Mitra, T. Biswas, S. Ghosh, G. Tudu, K. S. Paliwal, S. Ghosh and V. Mahalingam, Sustainable Energy Fuels, 2022, 6, 420–429 RSC.
- P. Goodrich, H. N. Gunaratne, J. Jacquemin, L. Jin, Y. Lei and K. R. Seddon, ACS Sustainable Chem. Eng., 2017, 5, 5635–5641 CrossRef CAS.
- H. Tong, Y. Qu, Z. Li, J. He, X. Zou, Y. Zhou, T. Duan, B. Liu, J. Sun and K. Guo, Chem. Eng. J., 2022, 444, 135478 CrossRef CAS.
- L. Guglielmero, A. Mezzetta, C. S. Pomelli, C. Chiappe and L. Guazzelli, J. CO2 Util., 2019, 34, 437–445 CrossRef CAS.
- S. Yue, P. Wang, X. Hao and S. Zang, J. CO2 Util., 2017, 21, 238–246 CrossRef CAS.
- X. Wu, C. Chen, Z. Guo, M. North and A. C. Whitwood, ACS Catal., 2019, 9, 1895–1906 CrossRef CAS.
- Z. Yue, M. Pudukudy, S. Chen, Y. Liu, W. Zhao, J. Wang, S. Shan and Q. Jia, Appl. Catal., A, 2020, 601, 117646 CrossRef CAS.
- A. H. Liu, Y. L. Dang, H. Zhou, J. J. Zhang and X. B. Lu, ChemCatChem, 2018, 10, 2686–2692 CrossRef CAS.
- R. Azzouz, V. C. Moreno, C. Herasme-Grullon, V. Levacher, L. Estel, A. Ledoux, S. Derrouiche, F. Marsais and L. Bischoff, Synlett, 2020, 183–188 CAS.
- B.-H. Cheng, L.-J. Deng, J. Jiang and H. Jiang, Chem. Eng. J., 2022, 442, 136265 CrossRef CAS.
- D. Polidoro, A. Perosa, E. Rodríguez-Castellón, P. Canton, L. Castoldi, D. Rodríguez-Padrón and M. Selva, ACS Sustainable Chem. Eng., 2022, 10, 13835–13848 CrossRef CAS PubMed.
- Y. Shen, P. Jiang, P. T. Wai, Q. Gu and W. Zhang, Catalysts, 2019, 9, 31 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterization and reaction results. See DOI: https://doi.org/10.1039/d2gc04475g |
| This journal is © The Royal Society of Chemistry 2023 |