
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly stable amorphous silica-alumina catalysts for continuous bio-derived mesitylene production under solvent-free conditions†
Phillip
Reif
 a,
Navneet Kumar
Gupta
*ab and
Marcus
Rose
*a
a,
Navneet Kumar
Gupta
*ab and
Marcus
Rose
*a
aTechnical University of Darmstadt, Department of Chemistry, Alarich-Weiss-Straße 8, 64287 Darmstadt, Germany. E-mail: marcus.rose@tu-darmstadt.de
bCentre for Sustainable Technologies, Indian Institute of Science, Gulmohar Marg, Mathikere, 560012 Bengaluru, India. E-mail: nkgupta@iisc.ac.in
First published on 30th January 2023
Abstract
Aromatization of alkyl methyl ketones obtained from biorefinery streams is a viable and attractive catalytic pathway to renewable aromatics, precursors for various important monomers and chemicals. To achieve high catalytic activity and stability under continuous conditions, mesoporous amorphous silica-alumina (ASA) catalysts are studied for the acid-catalyzed self-condensation of biomass-derived acetone to mesitylene in solvent-free conditions using a fixed-bed reactor. The catalytic efficiency of ASA catalysts depends on their structure and intrinsic acidity. In comparison to pure alumina, ASA Siralox 30 exhibits a 2.2 times higher catalytic activity for acetone conversion and 3.8 times higher mesitylene yield, demonstrating the importance of Brønsted acid sites (BAS) generated in ASA catalysts. The detailed kinetic studies and catalyst characterization indicate that mesitylene formation is favored over BAS and that the formation rate is enhanced with the relative strength of BAS. We demonstrate here that Siralox 30 (total product selectivity = 66%, W/F = 12.5 gcat h mol−1) is an adequate and highly active catalyst for the continuous mesitylene synthesis with remarkable long-term operational stability (>50 hours-on-stream).
1. Introduction
The transition from fossil to renewable resources presents one of today's greatest challenges. Their steady depletion and contribution to global warming are driving factors for implementing a more sustainable circular economy which includes using biomass for chemicals and fuels production.1 Especially for household products that frequently contain polymers, there is a strong consumer demand for green alternatives due to environmental concerns and awareness.2 Many of these polymers possess aromatic monomers, e.g., p-xylene for polyethylene terephthalate (PET), styrene for polystyrene (PS), toluene diisocyanate for polyurethanes (PUR), derived from benzene, toluene, and xylene (BTX) of which more than 122 Mt are produced annually, also for use in solvent and fuel applications.3 As of today, aromatics are obtained by catalytic reforming of naphtha, a crude oil fraction.4 Thus, using biomass as raw material feedstock for their production, ideally conceived as a drop-in solution, opens the door to a large variety of renewably sourced products without requiring modification of the existing infrastructure downstream.Several routes to biomass-derived aromatics have been reported. Among those, one important pathway is the selective Diels–Alder cycloaddition of furanic compounds, especially furan derivatives, with dienophiles such as bio-ethylene.5 High costs associated with the production of furan and its derivatives from biomass are currently the biggest drawback.6 Another fundamental route is the depolymerization of lignin by (catalytic) fast pyrolysis which yields a “bio-oil”, a complex mixture of oxygenated aromatic compounds.7 These require extensive purification, separation, and deoxygenation to obtain alkyl aromatics suitable for downstream processing. A more selective but less explored pathway is the self-condensation of alkyl ketones. The C–C coupling reaction of alkanones via robust acid/base-catalyzed condensation is an efficient way to achieve deoxygenated aromatics from existing biorefinery streams, e.g., acetone from ABE-fermentation, in a single-step reaction without further need for metal-catalyzed dehydrogenation or upgrading via deoxygenation with hydrogen.8–11 Furthermore, gas-phase fermentation by autotrophic acetogens offers a highly promising carbon-negative route to acetone.12 Therefore, the efficient utilization of acetone via self-condensation for the formation of the aromatic product mesitylene (1,3,5-trimethylbenzene) is the focus of this study.
Previously, the self-condensation of acetone is mainly studied in the gas phase under atmospheric pressure, often at high reaction temperatures above 400 °C using various solid acid catalysts such as zeolites, titania, zirconia, niobia, mesoporous aluminosilicates, and tantalum phosphate.13–17 Despite the high catalytic activity, the formation of polycondensates on the strongly acidic catalysts leads to fast catalyst deactivation due to carbonaceous deposits which remains an inherent challenge.18 Faba et al., showed an increase in productivity for the gas-phase conversion of acetone over a mixed catalyst bed of TiO2 and Al-MCM-41 at 250 °C but lacked the proof of long-term stability.17
Recently, we showed that zeolite HY is stable for the mesitylene formation in the liquid phase over several hours at 190 °C.19 The larger pores of zeolite Y compared to other microporous zeolites proved to be beneficial for the catalyst stability.20 Additionally, the liquid phase conditions allowed continuous removal of products from the catalyst bed, thus reducing carbonaceous deposits.21 While zeolite HY was also applicable for the aromatization of larger alkyl methyl ketones, such as 2-butanone and 2-pentanone, its overall activity was still lacking regarding a potential process development for future integration into a biorefinery.19 In the course of the catalyst development, combining high activity with long-term operational stability remains an ongoing problem that we sought to solve by using large pore amorphous aluminosilicates as acid catalysts.
Herein, we report that ASA afforded remarkable activity and selectivity for mesitylene from acetone under continuous conditions not only in liquid but also in supercritical phase. The ASA catalysts were beneficial due to their combination of larger mesopores and moderate overall acid site density of medium strength with a lower number of strong Brønsted acid sites.22–25 The optimization of reaction conditions and long-term catalyst stability are examined for maximum mesitylene space–time-yield on ASA. Assessment of the acidity-activity-relationship for this material class is performed based on the degree of silica-doping.
2. Experimental
2.1 Materials
Commercial amorphous silica-alumina (denoted as ASA) were supplied by Sasol Germany GmbH and used after calcination in air at 550 °C for 6 h (2 K min−1, 100 NmL min−1). HY-5 catalyst was obtained by exchanging NaY three times with an aqueous solution of NH4NO3 (1 M, 60 °C, 1 h) and subsequent calcination in air (2 K min−1, 550 °C, 6 h, 100 NmL min−1). γ-Al2O3 (99.9%) was obtained from Alfa Aesar and Evonik Aeroperl 300/30 was used as pure SiO2.Acetone (99%) was obtained from Sigma-Aldrich and used without further purification. For the GC standard solution, 1,4-dioxane (99.5%, Roth) was diluted with 1-butanol (99%, Grüssing).
For GC-calibration, acetone (99.9%, Sigma Aldrich), mesityl oxide (97%, Sigma Aldrich), mesitylene (99%, Acros Organics), isophorone (97%, Sigma Aldrich), 2-butanone (99%, Sigma Aldrich), and 1,3,5-triethylbenzene (97%, Sigma Aldrich) were used.
2.2 Catalytic studies in fixed-bed flow reactor
Catalytic studies were performed in a previously described fixed-bed reactor in upward flow configuration (Fig. S1†).19 Briefly, 3 g of catalyst were placed in the isothermal zone in the center of the stainless-steel reactor (ID = 1.6 cm, length = 20 cm). The catalyst powders were pressed (8 ton, 5 min), crushed, and sieved to 100–200 micron particles to avoid mass transfer limitations (Fig. S2†). Neat acetone was fed into the reactor with an HPLC pump, and the reaction was performed at 200–300 °C and 75 bar to maintain liquid/supercritical conditions. The outlet of the reaction feed was continuously mixed with a standard solution of 1,4-dioxane (1.15 mol L−1) in 1-butanol downstream. The product solution was analyzed via online-gas chromatography (Shimadzu GC-2030, MEGA-5 column, 40–250 °C, 10 K min−1, H2) equipped with an FID (Fig. S3†). Acetone, mesityl oxide, mesitylene, isophorone, 2-butanone, and 1,3,5-triethylbenzene were calibrated with pure compounds while the other identified products were estimated via the concept of the effective carbon number combined with GC-MS.19,262.3 Catalyst characterization
Powder X-ray diffraction (XRD) measurements were performed on a Bruker D2-phaser using Cu-K-α-radiation (λ = 1.5406 Å) with a radiation tube voltage of 30 kV and current of 40 mA. Diffraction patterns were measured in 10–80° 2Θ at 0.02° intervals and 1 s step time.N2-physisorption was measured on a Quantachrome QuadraSorb at 77 K after evacuating the samples (approx. 30–100 mg) at 300 °C over night. The specific surface area (SBET) was obtained with the Brunauer–Emmett–Teller method. The micropore area was determined by the t-plot method.
Thermogravimetric analysis (TGA) of the spent catalysts was performed on a NETZSCH STA 449 FE Jupiter by heating (5 K min−1, 40–1000 °C) the samples (50 mg) in synthetic air (100 NmL min−1).
Temperature-programmed desorption of NH3 (NH3-TPD) was used to measure the total amount of acid sites on the silica-alumina catalysts. For this, samples (100 mg) were dried in N2 flow (100 NmL min−1) at 600 °C (10 K min−1, 3 h) and subsequently loaded with NH3 (2 vol% NH3 in N2, 20 mL min−1) at 140 °C. When the physisorbed NH3 was desorbed, the samples were heated to 600 °C (10 K min−1) in N2 flow (100 NmL min−1). The amount of chemisorbed NH3 was detected by FT-IR.
3. Results and discussion
3.1 Catalyst stability of zeolite HY compared to amorphous silica-alumina
In our previous work, zeolite HY was identified as a suitable solid acid catalyst for the efficient and stable liquid phase conversion of biomass-derived ketones to aromatics due to its larger pore size and high amount of acid sites.19 Catalytic activity of HY increased with temperature and at 190 °C steady-state conditions were obtained for the continuous conversion of acetone to mesitylene. However, catalyst productivity was relatively low with 3% mesitylene yield at a weight hourly space velocity (WHSV) of 7.8 h−1. When the reaction temperature was raised to 200 °C, the initial productivity significantly increased to about twofold but the activity steadily decreased with time-on-stream due to catalyst deactivation (Fig. 1). In contrast, ASA Siralox 30 is found to be very stable when converting acetone to mesitylene under similar reaction conditions (200 °C, WHSV = 7.8 h−1). While its activity at 200 °C is lower compared to zeolite HY, it shows a three-times increase in mesitylene yield to 4% for 220 °C. Even at this elevated temperature, the catalyst activity remains very stable for more than 7 hours-on-stream and shows no deactivation. To understand the difference in stability of HY-5 and Siralox 30, relevant properties of the solid acid catalysts are compared in Table 1. The surface area of the mostly microporous zeolite HY-5 is 778 m2 g−1 and four-times higher than the one of Siralox 30 which is completely mesoporous (Fig. S4†). The number of surface acid sites is higher on HY-5 with 0.53 mmol g−1 compared to 0.30 mmol g−1 on Siralox 30. This results in a significantly higher activity of HY-5 at reaction temperatures below 200 °C since mesitylene activity correlates with the number of acid sites.19 Moreover, the strength of the acid sites on HY-5 is also greater than on Siralox 30 as evidenced by the higher temperature of the maximum NH3 desorption (Fig. S5†). Thermogravimetric analysis of the spent catalysts in air determines a total mass loss of 20 wt% for HY-5 of which more than 17 wt% correspond to strongly bound, bulky carbonaceous deposits (Fig. S6†). Siralox 30, on the other hand, shows a minimal weight loss of 3.3 wt% from 200–600 °C which could be due to weakly bound deposits. Thus, the stronger and greater number of acid sites on HY-5 are not only more active, but also lead to increased carbon deposition.27 Those block the catalyst pores, and thus decrease the accessibility of active sites, as highlighted by the strongly diminished microporous surface area of spent HY-5 (Table 1). The observed slight increase in the mesoporous surface area of HY is due to carbon deposition as confirmed by TGA. In comparison, the surface area of spent Siralox 30 is almost completely retained due to its mesoporous structure and after calcination of the spent catalyst the number of surface acid sites is equivalent to the fresh sample.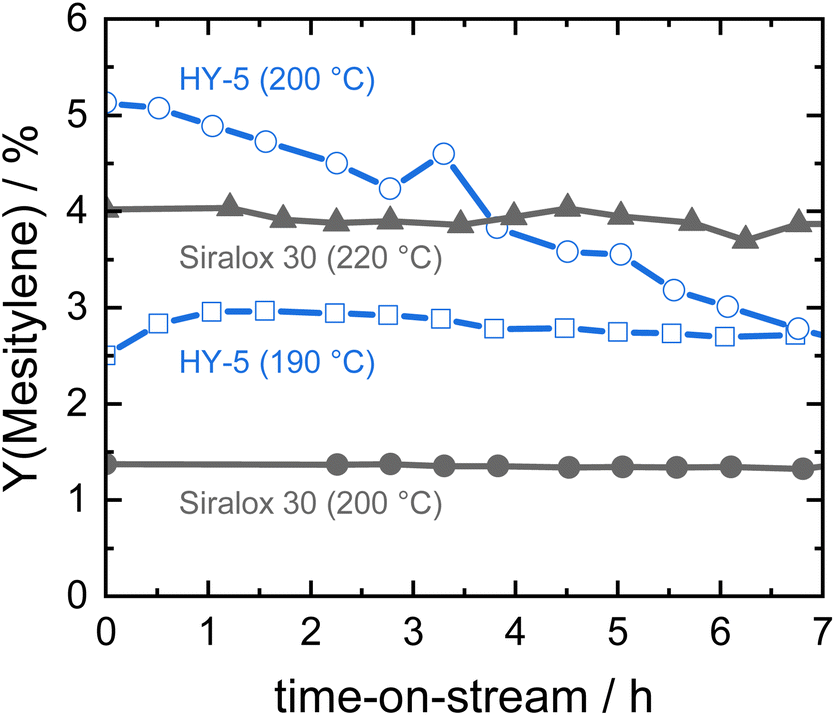 | ||
| Fig. 1 Catalyst stability of silica-alumina HY-5 and Siralox 30 based on mesitylene yield at different reaction temperatures (7.8 h−1, 40 bar). | ||
| Catalyst | Textural properties | Crystallinity | Total amount of acid sites/mmol NH3 g−1 | Mass loss/% | ||||
|---|---|---|---|---|---|---|---|---|
| BET surface area/m2 g−1 | Microporous area/m2 g−1 | Mesoporous area/m2 g−1 | Total pore volume/cm3 g−1 | Average pore diameter/nm | ||||
| HY-5 | 778 | 709 | 69 | 0.34 | 1.73 | Crystalline | 0.53 | |
| Siralox 30 | 199 | 0 | 199 | 0.59 | 12.3 | Amorphous | 0.30 | |
| HY-5 spent | 230 | 121 | 109 | 0.16 | 2.74 | Crystalline | 20 | |
| Siralox 30 spent | 190 | 0 | 190 | 0.58 | 11.6 | Amorphous | 3.3 | |
Hence, at higher reaction temperatures (more than 190 °C), mass transport out of the zeolite HY pores is slow compared to the formation of bulkier molecules favored by its stronger acid sites, resulting in quick deactivation of the catalyst. However, Siralox 30 remains stable even at higher temperatures due to its better mass transport capabilities and lower number of stronger acid sites.25 In this way, it is possible to greatly improve the activity of the catalyst with temperature without compromising stability. Therefore, the ASA Siralox 30 is a highly interesting solid acid catalyst for the liquid-phase aromatization of acetone and requires a deeper study of optimal reaction conditions for maximum activity and efficiency in the continuous flow process.
3.2 Process optimization for the efficient acetone aromatization using ASA
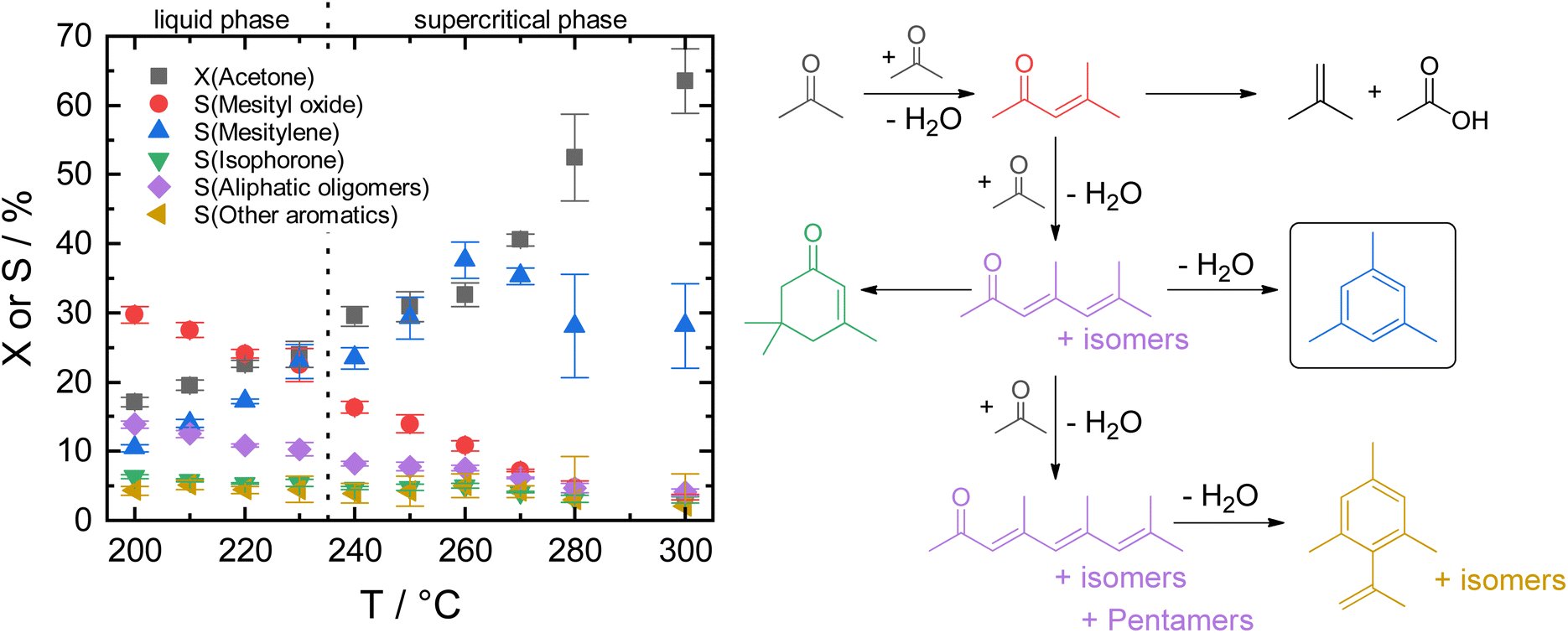 | ||
| Fig. 2 Left: acetone conversion and product selectivity for Siralox 30 at different reaction temperatures (WHSV = 7.8 h−1, 75 bar) in liquid and supercritical acetone. Error bars give the standard deviation of the GC measurements at steady-state conditions. Right: a simplified network of the acid-catalyzed self-condensation of acetone. | ||
By varying the reaction temperature, a strong influence on the mesitylene activity is found with a maximum selectivity at 260 °C for Siralox 30. The following experiments were therefore carried out at 260 °C.
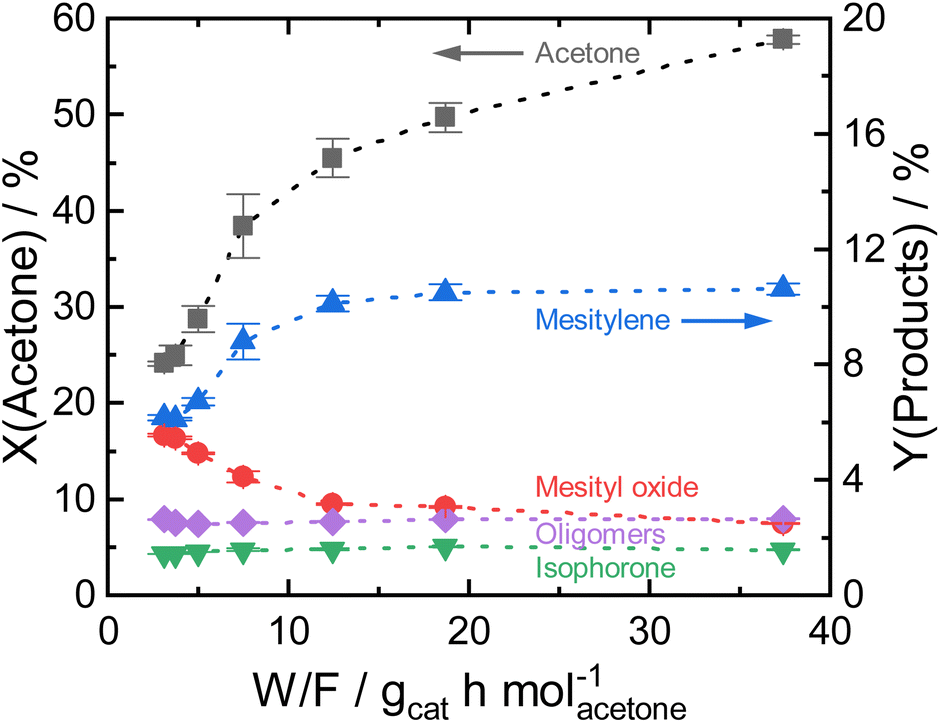 | ||
| Fig. 3 Acetone conversion and product yields as a function of contact time for Siralox 30 at 260 °C. The dotted lines are only for visual guidance. | ||
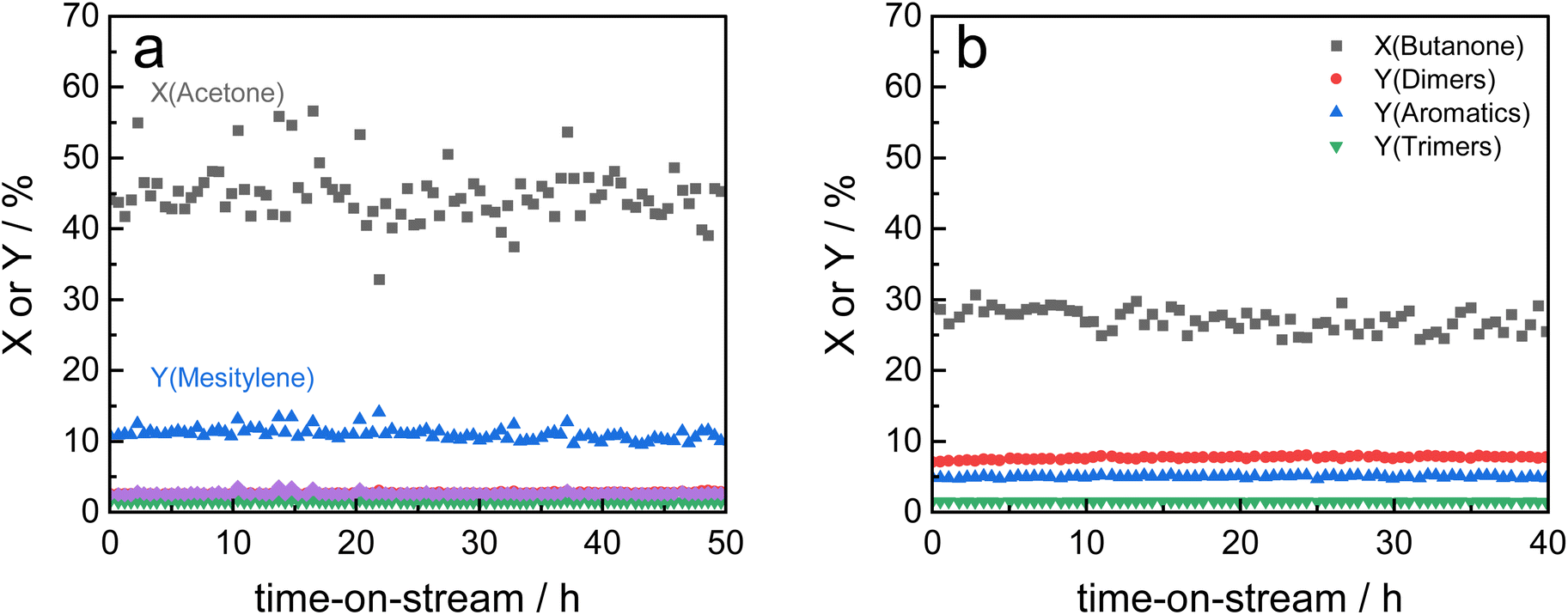 | ||
| Fig. 4 Stability test of Siralox 30 under optimized reaction conditions (260 °C, 12.5 gcat h mol−1, WHSV = 4.7 h−1) in the conversion of acetone (a) and 2-butanone (b). Other products depicted in 4a: ● Y(Mesityl oxide), ▼ Y(Isophorone), ◆ Y(Oligomers). | ||
Based on these findings, Siralox 30 was also tested for the conversion of 2-butanone under the optimized reaction conditions. The aromatic self-condensation product of 2-butanone is triethylbenzene that could be used as a potential precursor to styrene-type monomers. While Siralox 30 is stable for the conversion of this larger alkyl methyl ketone for more than 40 hours-on-stream, the catalytic activity is reduced compared to the conversion of acetone (Fig. 4b). For 2-butanone, the dimers present the main product with 8% yield whereas the yield of the aromatic triethylbenzenes is 5%. The lower catalyst activity is owed to the lower reactivity of 2-butanone and the steric hindrance due to its longer alkyl chain. The latter is also responsible that in comparison to acetone, a higher variety of isomers can be formed.19 The total carbon balance for the conversion of 2-butanone is therefore 80%, whereas the total product selectivity for dimers, aromatics and trimers is 48%. As a side product propionic acid was detected in low amounts.
Nevertheless, the mesoporous structure of Siralox 30 with its larger pores is advantageous for the condensation as it is suitable for larger products without deactivation.
3.3 Influence of silica-content on catalytic activity of ASA
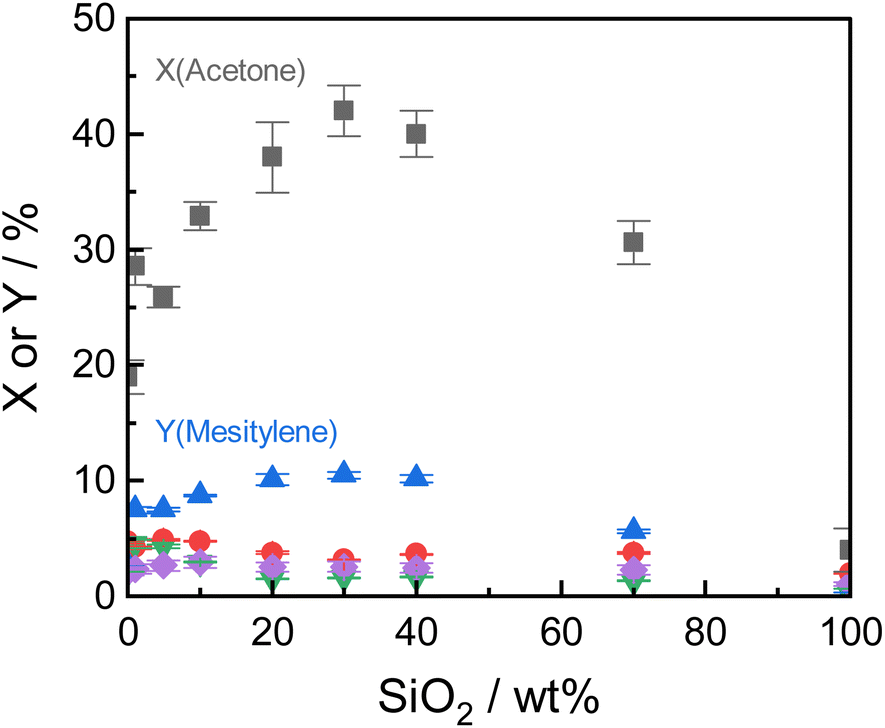 | ||
| Fig. 5 Steady-state acetone conversion and product yields (▲ Y(Mesitylene), ● Y(Mesityl oxide), ▼ Y(Isophorone), ◆ Y(Oligomers)) for varying silica-content at optimized reaction conditions (260 °C, 12.5 gcat h mol−1, 10 h-on-stream). | ||
Under the optimized reaction conditions, all tested Siralox materials were stable at steady-state conditions for more than 10 hours-on-stream (Fig. S9†). The silica-content therefore does not play a significant role for the stability of the catalysts under reaction conditions. The TGA measurements of the spent ASA from the flow reactor show that the mass loss is about 7 wt% for silica-loadings of 10–40 wt% (Fig. S10†) after reaction at 260 °C. For very high and low silica-loadings, the mass loss is lower than 7 wt% due to decreased catalyst activity. For flow conditions, ∼0.5 wt% less carbonaceous deposits are found compared to batch reactions which shows the efficiency of the continuous process with constant product removal from the catalyst bed.
The structural stability of the Siralox materials was assessed by XRD of the spent catalysts. The diffractograms (Fig. S11†) show that for the majority of materials, no significant change is visible. For Siralox 20 and 40, a small reflex at 49.2° can be observed, hinting to minor formation of the γ-AlO(OH) boehmite likely due to water formed in the aldol condensation. Surprisingly, Siralox 30 does not show the formation of boehmite which supports its suitability for scale-up in a continuous flow process.
ASA exhibit a predominant amount of various Lewis acid sites (LAS) but also possess BAS in lower amounts (Fig. S13†).22,25 The latter are created by doping the alumina with silica.31 With increasing silica content, the number of LAS decreases while the formation of BAS increases up to a maximum at 40 wt% silica.23,31 For silica-rich alumina of 90 wt% and above, no LAS are detected and the acidity is almost completely controlled by BAS. This is explained by the enrichment of the surface with silica which exceeds what would be expected from the bulk material composition. The majority of the material's surface at silica-loadings higher than 40 wt% is covered by silica and only contains small zones of the mixed aluminosilicate.31 In fact, Daniell et al., assessed the strength of the BAS and correlated it to the shift of the surface hydroxyl group Δv(OH) at ∼3748 cm−1 on adsorption of CO on the Siralox materials by FTIR-spectroscopy.31 The aluminosilicate surface shows a lower number of BAS which are strongly enhanced in strength by the addition of silica. Based on their measurement, we found that the formation of mesitylene correlates well (R2 = 0.78) with the measured Δv(OH) (Fig. 6). Accordingly, stronger BAS promote mesitylene formation under reaction conditions. For silica loadings higher than 60 wt%, the total number of acid sites and the strength of BAS decrease which results in the lower activities observed for these materials.23,31 Therefore, materials with silica contents of 30–40 wt% are most beneficial for the formation of mesitylene.
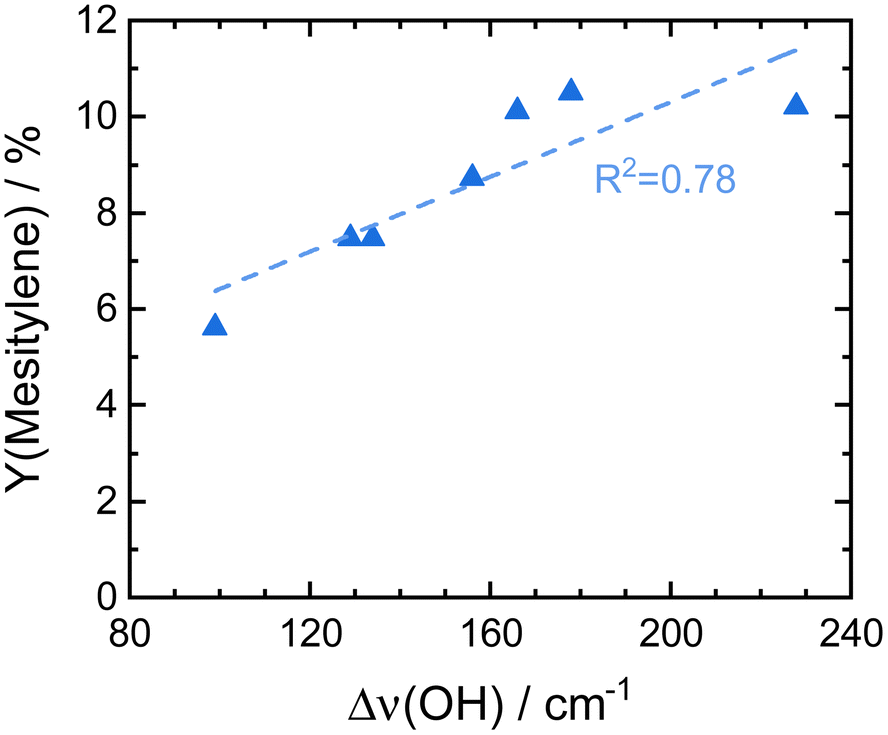 | ||
| Fig. 6 Comparison of the here reported mesitylene yields for Siralox materials based on literature data for the strength of their BAS marked by the shift of vibrational frequencies for surface hydroxyl groups Δν(OH) upon adsorption of CO on Siralox as determined by Daniell et al.31 | ||
The dimer mesityl oxide is preferably formed on ASA with low silica content which could stem from two effects: (1) mesityl oxide benefits from a higher number of LAS as observed by Panov and Fripiat34 and (2) a lack of BAS at low silica loadings which are required to promote the consecutive aromatization with another acetone molecule. This concludes to both LAS and BAS being beneficial for the formation of mesityl oxide and dimers, while BAS promote the consecutive formation of mesitylene.
3.4 Conceptual process design for the aromatization of acetone
The remarkable stability of Siralox 30 combined with the high total product selectivity of 66% renders it suitable for a potential scale-up and further assessment of the industrial application in a continuous flow process. Separation of products can be readily achieved via distillation based on the differences in boiling points. A benefit of the solvent-free process is the more energy-efficient separation, as no additional solvent needs to be vaporized and separated. Furthermore, recovered acetone and mesityl oxide can be recycled and added to the reactor feed to increase total efficiency (Fig. 7). Aromatics of the BTX-fraction can be obtained from mesitylene via industrially established transalkyation, thus offering a completely bio-based route for their production.35 Generation of added value is also possible from the side products by their transformation to various important intermediates used in the polymer industry, e.g., isophorone diisocyanate from isophorone for the polyurethane production.36 | ||
| Fig. 7 Conceptual process for a continuous conversion of acetone obtained via ABE-fermentation of biomass-derived sugars to aromatics. Condensation products can be readily separated via distillation and recovered acetone and mesityl oxide can be recycled for increased process efficiency. Also depicted is the potential end-use of the separated condensation products. Depiction of biomass conversion and separation processes is simplified. | ||
4. Conclusion
In conclusion, the continuous production of biomass-derived mesitylene from acetone can be achieved over ASA Siralox 30 under solvent-free continuous conditions in liquid as well as supercritical phase. At optimum conditions, Siralox 30 showed high stability without signs of deactivation for more than 50 hours-on-stream. Contributing to the stability is the mesoporous nature of Siralox 30 which facilitates mass transport and prevents deactivation by pore blocking from bulkier aromatic/aliphatic molecules as evidenced by the low amounts of carbonaceous deposits on the spent catalyst. Additionally, the larger pores allow the stable aromatization of 2-butanone to triethylbenzene with adequate yield, however activity is slightly decreased compared to the acetone-to-mesitylene conversion. Variation in the degree of doping with silica influences the strength, amount, and nature of acid sites. Thus, it was found that the strength of BAS facilitates the mesitylene yield with an optimum silica-loading of 30–40 wt%.Commercial availability of the low-cost catalyst, its remarkable stability, and a solvent-free continuous process present an excellent basis for a potential scale-up with the ASA Siralox 30. The fully integrated added value chains provide the opportunity for replacing fossil with biomass feedstock for a broad product spectrum with various applications.
Author contributions
PR: conceptualization, investigation, formal analysis, visualization, writing – original draft; NKG: conceptualization, funding acquisition, writing – review & editing; MR: supervision, funding acquisition, project administration, writing – review & editing.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
PR and MR thank the German Federal Ministry of Education and Research (BMBF) for funding (Grant No. 031B0680). NKG gratefully acknowledges the Alexander-von-Humboldt foundation for financial support. We thank Martin Lucas for continuous technical support, Sasol Germany GmbH for providing the Siralox materials, and Kiyotaka Nakajima for his valuable input.References
- J. Sherwood, The significance of biomass in a circular economy, Bioresour. Technol., 2020, 300, 122755, DOI:10.1016/j.biortech.2020.122755.
- Y. C. Yang, Consumer Behavior towards Green Products, J. Econ. Bus. Manag., 2017, 5(4), 160–167, DOI:10.18178/joebm.2017.5.4.505.
- Mordor Intelligence., Benzene-Toluene-Xylene (BTX) Market - Growth, Trends, COVID-19 Impact, and Forecasts (2022–2027). https://www.mordorintelligence.com/industry-reports/benzene-toluene-xylene-btx-market (accessed 2022-08-02).
- M. R. Rahimpour, M. Jafari and D. Iranshahi, Progress in catalytic naphtha reforming process: A review, Appl. Energy, 2013, 109, 79–93, DOI:10.1016/j.apenergy.2013.03.080.
- Y.-T. Cheng and G. W. Huber, Production of targeted aromatics by using Diels–Alder classes of reactions with furans and olefins over ZSM-5., Green Chem., 2012, 14(11), 3114, 10.1039/c2gc35767d.
- A. Maneffa, P. Priecel and J. A. Lopez-Sanchez, Biomass-Derived Renewable Aromatics: Selective Routes and Outlook for p-Xylene Commercialisation, ChemSusChem, 2016, 9(19), 2736–2748, DOI:10.1002/cssc.201600605.
- T. R. Carlson, G. A. Tompsett, W. C. Conner and G. W. Huber, Aromatic Production from Catalytic Fast Pyrolysis of Biomass-Derived Feedstocks, Top. Catal., 2009, 52(3), 241–252, DOI:10.1007/s11244-008-9160-6.
- S. Herrmann and E. Iglesia, Selective conversion of acetone to isobutene and acetic acid on aluminosilicates: Kinetic coupling between acid-catalyzed and radical-mediated pathways, J. Catal., 2018, 360, 66–80, DOI:10.1016/j.jcat.2018.01.032.
- E. R. Sacia, M. Balakrishnan, M. H. Deaner, K. A. Goulas, F. D. Toste and A. T. Bell, Highly Selective Condensation of Biomass-Derived Methyl Ketones as a Source of Aviation Fuel, ChemSusChem, 2015, 8(10), 1726–1736, DOI:10.1002/cssc.201500002.
- P. Reif, H. Rosenthal and M. Rose, Biomass–Derived Aromatics by Solid Acid–Catalyzed Aldol Condensation of Alkyl Methyl Ketones, Adv. Sustain. Syst., 2020, 4(10), 1900150, DOI:10.1002/adsu.201900150.
- T. J. Benson, P. R. Daggolu, R. A. Hernandez, S. Liu and M. G. White, Catalytic Deoxygenation Chemistry, Adv. Catal., 2013, 56, 187–353, DOI:10.1016/B978-0-12-420173-6.00003-6.
- F. E. Liew, R. Nogle, T. Abdalla, B. J. Rasor, C. Canter, R. O. Jensen, L. Wang, J. Strutz, P. Chirania, S. Tissera, A. P. de Mueller, Z. Ruan, A. Gao, L. Tran, N. L. Engle, J. C. Bromley, J. Daniell, R. Conrado, T. J. Tschaplinski, R. J. Giannone, R. L. Hettich, A. S. Karim, S. D. Simpson, S. D. Brown, C. Leang, M. C. Jewett and M. Köpke, Carbon-negative production of acetone and isopropanol by gas fermentation at industrial pilot scale, Nat. Biotechnol., 2022, 40(3), 335–344, DOI:10.1038/s41587-021-01195-w.
- M. Paulis, M. Martín, D. B. Soria, A. Díaz, J. A. Odriozola and M. Montes, Preparation and characterization of niobium oxide for the catalytic aldol condensation of acetone, Appl. Catal., A, 1999, 180(1), 411–420, DOI:10.1016/S0926-860X(98)00379-2.
- Z. Wu, J. Zhang, Z. Su, S. Lu, J. Huang, Y. Liang, T. Tan and F.-S. Xiao, Selective conversion of acetone to mesitylene over tantalum phosphate catalysts, Chem. Commun., 2022, 58(17), 2862–2865, 10.1039/d2cc00016d.
- S. K. Bej and L. T. Thompson, Acetone condensation over molybdenum nitride and carbide catalysts, Appl. Catal., A, 2004, 264(2), 141–150, DOI:10.1016/j.apcata.2003.12.051.
- S. Herrmann and E. Iglesia, Elementary steps in acetone condensation reactions catalyzed by aluminosilicates with diverse void structures, J. Catal., 2017, 346, 134–153, DOI:10.1016/j.jcat.2016.12.011.
- L. Faba, J. Gancedo, J. Quesada, E. Diaz and S. Ordóñez, One-Pot Conversion of Acetone into Mesitylene over Combinations of Acid and Basic Catalysts, ACS Catal., 2021, 11(18), 11650–11662, DOI:10.1021/acscatal.1c03095.
- L. Kubelkovå, J. Čjka, J. Nováková, V. Boszácek, I. Jirka and P. Jíaru, Acetone Conversion and Deactivation of Zeolites, in Studies in Surface Science and Catalysis, ed. P. A. Jacobs and R. A. van Santen, Elsevier, 1989, pp. 1203–1212. DOI:10.1016/S0167-2991(08)62006-6.
- P. Reif, N. K. Gupta and M. Rose, Liquid phase aromatization of bio-based ketones over a stable solid acid catalyst under batch and continuous flow conditions, Catal. Commun., 2022, 163, 106402, DOI:10.1016/j.catcom.2022.106402.
- J. Quesada, L. Faba, E. Díaz and S. Ordóñez, Effect of catalyst morphology and hydrogen co-feeding on the acid-catalysed transformation of acetone into mesitylene, Catal. Sci. Technol., 2020, 10(5), 1356–1367, 10.1039/C9CY02288K.
- H. Takaya, N. Todo, T. Hosoya, T. Minegishi, M. Yoneoka and H. Oshio, Cleaning Effects of the Reactant in the Liquid-phase Isomerization of m-Xylene over a Silica-Alumina Catalyst under Pressure, Bull. Chem. Soc. Jpn., 1971, 44(9), 2296–2301, DOI:10.1246/bcsj.44.2296.
- G. Crépeau, V. Montouillout, A. Vimont, L. Mariey, T. Cseri and F. Maugé, Nature, structure and strength of the acidic sites of amorphous silica alumina: an IR and NMR study, J. Phys. Chem. B, 2006, 110(31), 15172–15185, DOI:10.1021/jp062252d.
- S. Nassreddine, S. Casu, J. L. Zotin, C. Geantet and L. Piccolo, Thiotolerant Ir/SiO2−Al2O3 bifunctional catalysts: effect of support acidity on tetralin hydroconversion, Catal. Sci. Technol., 2011, 1(3), 408–412, 10.1039/c1cy00002k.
- Z. Wang, Y. Jiang, O. Lafon, J. Trébosc, K. D. Kim, C. Stampfl, A. Baiker, J.-P. Amoureux and J. Huang, Brønsted acid sites based on penta-coordinated aluminum species, Nat. Commun., 2016, 7, 13820, DOI:10.1038/ncomms13820.
- M. K. Mardkhe, K. Keyvanloo, C. H. Bartholomew, W. C. Hecker, T. M. Alam and B. F. Woodfield, Acid site properties of thermally stable, silica-doped alumina as a function of silica/alumina ratio and calcination temperature, Appl. Catal., A, 2014, 482, 16–23, DOI:10.1016/j.apcata.2014.05.011.
- J. T. Scanlon and D. E. Willis, Calculation of Flame Ionization Detector Relative Response Factors Using the Effective Carbon Number Concept, J. Chromatogr. Sci., 1985, 23(8), 333–340, DOI:10.1093/chromsci/23.8.333.
- M. Guisnet, L. Costa and F. R. Ribeiro, Prevention of zeolite deactivation by coking, J. Mol. Catal. A: Chem., 2009, 305(2), 69–83, DOI:10.1016/j.molcata.2008.11.012.
- D. Ambrose, C. Sprake and R. Townsend, Thermodynamic properties of organic oxygen compounds XXXIII. The vapour pressure of acetone, J. Chem. Thermodyn., 1974, 6(7), 693–700, DOI:10.1016/0021-9614(74)90119-0.
- A. Meyer, K. Noweck, A. Reichenauer and J. Schimanski, Process for the preparation of a catalyst carrier based on aluminosilicates. US5045519A, 1991.
- A. Gervasini, G. Bellussi, J. Fenyvesi and A. Auroux, Microcalorimetric and Catalytic Studies of the Acidic Character of Modified Metal Oxide Surfaces. 1. Doping Ions on Alumina, Magnesia, and Silica, J. Phys. Chem., 1995, 99(14), 5117–5125, DOI:10.1021/j100014a036.
- W. Daniell, U. Schubert, R. Glöckler, A. Meyer, K. Noweck and H. Knözinger, Enhanced surface acidity in mixed alumina–silicas: a low-temperature FTIR study, Appl. Catal., A, 2000, 196(2), 247–260, DOI:10.1016/S0926-860X(99)00474-3.
- E. J. Hensen, D. G. Poduval, V. Degirmenci, D. J. M. Ligthart, W. Chen, F. Maugé, M. S. Rigutto and J. R. van Veen, Acidity Characterization of Amorphous Silica–Alumina, J. Phys. Chem. C, 2012, 116(40), 21416–21429, DOI:10.1021/jp309182f.
- F. Lónyi and J. Valyon, On the interpretation of the NH3-TPD patterns of H-ZSM-5 and H-mordenite, Microporous Mesoporous Mater., 2001, 47(3), 293–301, DOI:10.1016/S1387-1811(01)00389-4.
- A. G. Panov and J. J. Fripiat, Acetone Condensation Reaction on Acid Catalysts, J. Catal., 1998, 178(1), 188–197, DOI:10.1006/jcat.1998.2142.
- Y. Li, H. Wang, M. Dong, J. Li, Z. Qin, J. Wang and W. Fan, Effect of zeolite pore structure on the diffusion and catalytic behaviors in the transalkylation of toluene with 1,2,4-trimethylbenzene, RSC Adv., 2015, 5(81), 66301–66310, 10.1039/C5RA09236A.
- H. Sardon, L. Irusta and M. J. Fernández-Berridi, Synthesis of isophorone diisocyanate (IPDI) based waterborne polyurethanes: Comparison between zirconium and tin catalysts in the polymerization process, Prog. Org. Coat., 2009, 66(3), 291–295, DOI:10.1016/j.porgcoat.2009.08.005.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc04116b |
| This journal is © The Royal Society of Chemistry 2023 |