
Catalytic hydroesterification of lignin: a versatile and efficient entry into fully biobased tunable materials†
Ruqaya
Buhaibeh
a,
Tiphaine
Richard
ab,
Régis M.
Gauvin
 ac,
Mathieu
Sauthier
a and
Clément
Dumont
*ab
ac,
Mathieu
Sauthier
a and
Clément
Dumont
*ab
aUniv. Lille, CNRS, Centrale Lille, ENSCL, Univ. Artois, UMR 8181-UCCS-Unité de Catalyse et Chimie du Solide, F-59000 Lille, France. E-mail: clement.dumont@icam.fr
bICAM site de Lille, 6 rue Auber, 59016 Lille Cedex, France
cChimie ParisTech, PSL University, CNRS, Institut de Recherche de Chimie Paris, 75005 Paris, France
First published on 20th January 2023
Abstract
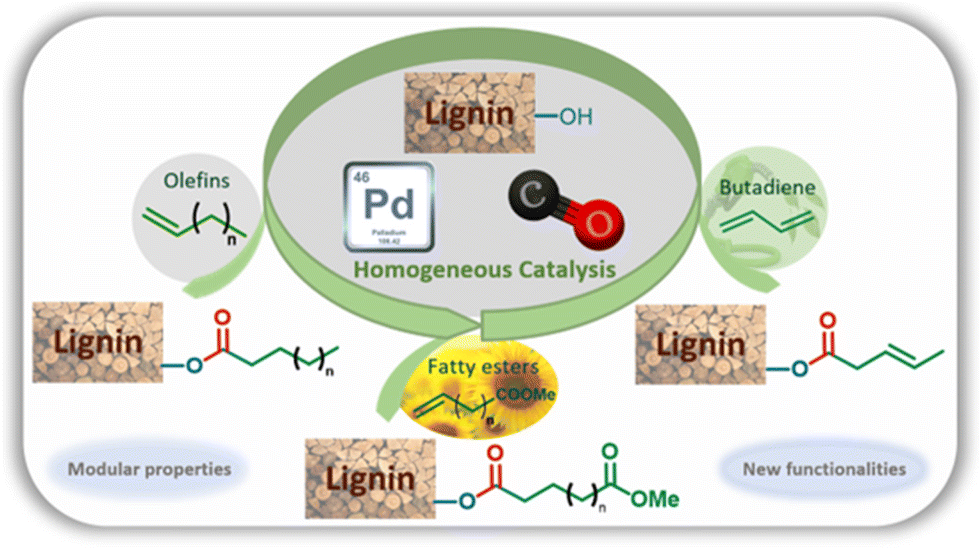
Hydroxyl groups of several industrial grade lignins are esterified by palladium-catalyzed hydroesterification reactions with CO and olefins, fatty esters or 1,3-butadiene. The reaction is efficient and salt-free and allows the introduction of aliphatic chains to lignin through the ester linkage. Technical and modified lignins are characterized by NMR, IR, and SEC and their thermal properties are evaluated by DSC. The esterified lignins show modified glass transition temperatures that depend on the substitution degree of the hydroxyl groups and on the nature of the aliphatic chains that have been introduced. The implementation of this catalysis also provides the possibility of inserting new reactive functionalities such as esters or unsaturations from abundant and/or biobased reactants.
Introduction
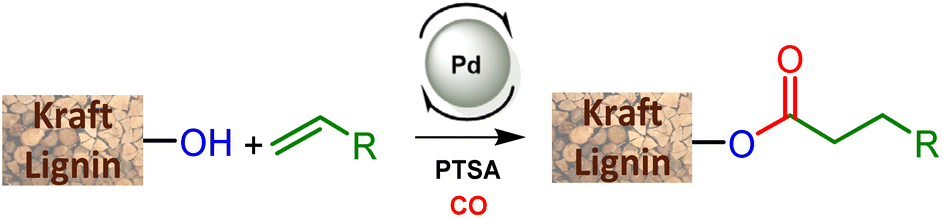
Non-renewable fossil resources continue to play a major role in the chemical industry, materials manufacturing, and energy production. Their consumption releases increasingly more CO2 into the atmosphere, playing a negative role in climate change and depleting the planet's resources. For a more sustainable economy, the use of renewable carbon for the production of materials and chemicals is particularly attractive as it allows recycling and storage of CO2. Lignocellulosic biomass is one of the major alternative sources to oil, is not an edible resource and is available in large quantities. Lignin represents about 20% of lignocellulosic biomass and 100 million tons of lignin are currently industrially produced as a side product of the paper industry and within second-generation bioethanol production plants. Lignin is a bio-based polymer with high aromatic content and is clearly very attractive for the plastic industry. Still, the compound is essentially used as a low-value fuel and is currently 98% burned to generate electricity and heat.1 The valorization of lignins for the production of materials is indeed largely impeded by their weak mechanical properties and poor compatibility with synthetic polymers. To overcome these limitations, some research fields involve the functionalization of phenolic and aliphatic hydroxyl groups of lignin to improve its properties and thus offer access to new valorization routes.2 Among the chemical transformations of lignin, esterification reactions are particularly used in the development of new material solutions. The esterification of hydroxyl groups present in lignins has been for example used to improve the compatibility of lignin in blends,3 to use lignin as a macromonomer,4 or to graft polymerization initiators.5 Other fields of application also use esters such as adhesives,6 lubricants,7 anti-UV films8 or energy production.9 However, if aliphatic alcohols can be readily involved in esterification reactions with carboxylic acids, the phenol functions of lignins show a much lower reactivity.10 Thus, these transformations currently involve “activated carboxylic acids” through the use of the Steglich reaction11 (with DCC/DMAP), acid halides12 or anhydrides.13 Nevertheless, these reactions have the disadvantage of using harmful reagents generating salts or deleterious co-products in stoichiometric amounts. Consequently, this significantly increases the environmental impact of these transformations.As an environmentally friendly pathway, ester functionalization methodologies using homogeneous catalysis such as the carbonylative telomerization reaction14 have shown high efficiency for efficient lignin functionalization (Scheme 1).
 | ||
| Scheme 1 Overall scheme for Pd-catalyzed hydroesterification on lignin. | ||
The present article proposes to open a new lignin functionalization pathway via the implementation of ester synthesis through the hydroesterification reaction of olefins. This catalytic transformation affords esters that are thus readily obtained from the combination of carbon monoxide, an alkene and a nucleophilic alcohol. This reaction is atom-economical and avoids the formation of stoichiometric amounts of salts.15 Different kinds of catalysts have been studied for this reaction, including base metals such as cobalt16 and nickel17 as developed by W. Reppe in the 1960s. However, these catalysts require drastic conditions of temperature and pressure, which significantly counterbalance the interest in their use. On the other hand, palladium catalysis proceeds under softer reaction conditions, is more tolerant to functionalities and thus appears to be highly suitable to convert a functionalized derivative such as lignin without prior purification.18 Interestingly, the practicality of this industrially relevant reaction opens the way to efficient and atom-economical syntheses of esterified lignins from olefins, either as bulk products from a large scale petroleum-based process, or as bio-based terpenes and unsaturated fatty acids or esters.
Results and discussion
This study aiming at the synthesis of esterified lignins from the hydroesterification reaction was initiated with guaiacol and benzyl alcohol as substrates to check the efficiency of the reaction towards both aliphatic and phenolic hydroxyl groups. Such functionalities are found in lignin (Scheme 2) and the use of such simple molecules is highly suitable to collect spectroscopic references for the assessment of lignin grafting by 1H NMR (see below). For this purpose, a simple and industrially relevant olefin, 1-hexene, was used as the co-reagent. Standard reaction conditions classically used for the hydroesterification of olefins with simple aliphatic alcohols were initially applied for the transformation of the substrates.18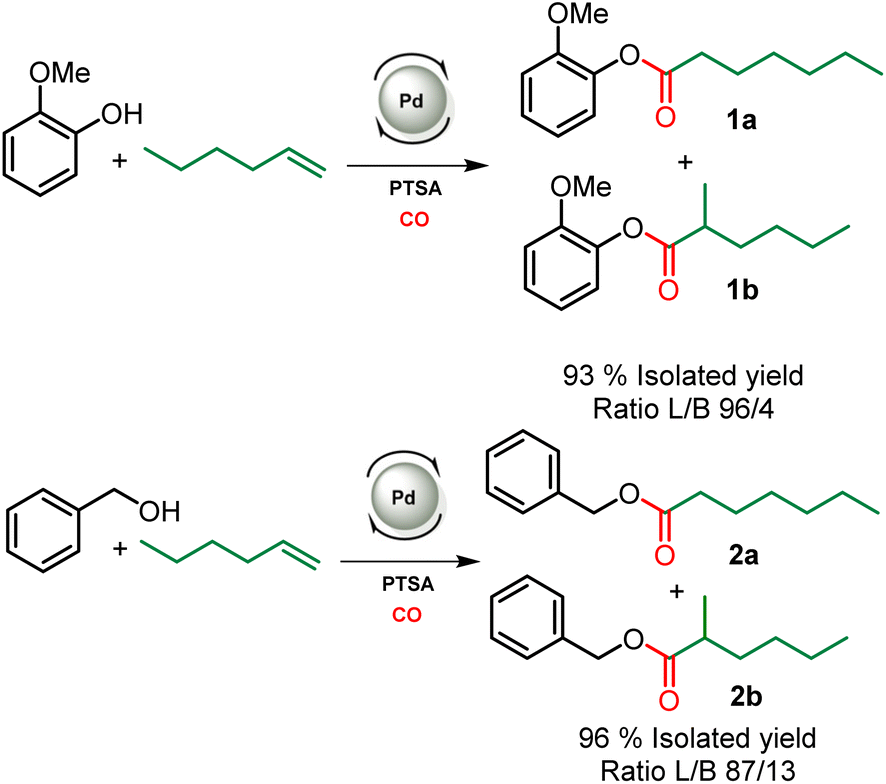 | ||
| Scheme 2 Hydroesterification of 1-hexene with guaiacol (top) and benzyl alcohol (bottom). Reaction conditions: [Pd]: Pd(OAc)2/PPh3 – 120 °C, 22 h, 30 bar CO, THF. L/B ratio determined in 1H NMR, see Fig. SI1.† | ||
The catalyst is prepared in situ by combining 1 mol% Pd(OAc)2 with 0.25 equivalents with respect to alcohols of both PPh3 and para-toluenesulfonic acid (PTSA). With these reaction conditions, a very high conversion (99%) of phenol was obtained and the ester was isolated with 93% yield. 1H NMR of the isolated product evidences the presence of two isomers with linear (L) 1a and branched (B) 1b structures with a measured L/B ratio of 96/4. The yield of ester 2 derived from benzyl alcohol was 96% and a slightly higher proportion of the branched isomer was obtained (L/B = 87/13). These molecules validate the possibility of carrying out the hydroesterification reaction of olefins with hydroxyl groups in THF which is known as a suitable solvent for lignin. Interestingly, both types of alcohols present within the lignin structure are amenable to efficient conversion into esters. The same reaction parameters were then used for the hydroesterification of Kraft lignins with 1-hexene. This industrially available material was used as received for all the catalytic tests. As the polymeric structure of the industrial grade Kraft lignin is very different from that of the pure simple compounds used, a first optimization study was initiated in order to maximize the conversion of the alcohols (Table 1).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Hexene (eq./OH) | PTSA (eq./OH) | Temp (°C) | Solvent | time (h) | Conv.a OHtotal | %Weightb |
| Conditions: 0.5 g of Kraft lignin, 10 mL of solvent, Pd(OAc)2 1 mol% in respect to OH group, PPh3 0.25 eq. per OH group, 30 bar of CO.a Conversion of total OH groups on the recovered part of lignin – comparison of the amount of chains grafted on the lignin with the quantity of available hydroxyl groups, determined by 1H NMR with pentafluorobenzaldehyde as internal standard.b Weight of lignin recovered in proportion to the amount of starting lignin. | |||||||
| 1 | 8 | 0.25 | 90 | THF | 22h | 28% | 32% |
| 2 | 8 | 0.25 | 100 | THF | 22h | 51% | 33% |
| 3 | 8 | 0.25 | 120 | THF | 22h | 61% | 52% |
| 4 | 8 | 0.15 | 120 | THF | 22h | 42% | 24% |
| 5 | 8 | 0.08 | 120 | THF | 22h | 28% | 42% |
| 6 | 8 | 0 | 120 | THF | 22h | 0% | 54% |
| 7 | 8 | 0.25 | 120 | THF | 2 h | 14% | 21% |
| 8 | 8 | 0.25 | 120 | THF | 4 h | 42% | 33% |
| 9 | 8 | 0.25 | 120 | THF | 6 h | 61% | 26% |
| 10 | 3 | 0.25 | 120 | THF | 22h | 39% | 56% |
| 11 | 1.5 | 0.25 | 120 | THF | 22h | 22% | 72% |
| 12 | 8 | 0.25 | 120 | DMSO | 22h | 0% | 98% |
| 13 | 8 | 0.25 | 120 | Dioxane | 22h | 70% | 56% |
| 14 | 3 | 0.25 | 120 | Dioxane | 22h | 72% | 105% |
| 15 | 1.5 | 0.25 | 120 | Dioxane | 22h | 67% | 113% |
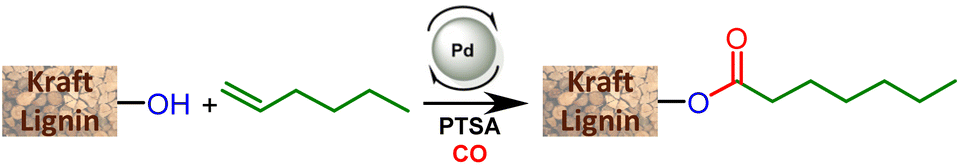
The impact of the reaction temperature on the conversion of the OH groups in lignin was first studied. At 90 °C, 28% of the total alcohols were converted (entry 1). Increasing the reaction temperature improves the substitution degree from 51% (100 °C) to 61% (120 °C) (entries 2 and 3). Although the reaction is rather efficient in terms of substitution degrees, it is noteworthy that large amounts of precipitated lignin were observed at the end of the catalytic runs. For example, at 120 °C, half of the initial lignin is lost as insoluble material. This precipitate proved to be also insoluble in commonly used organic solvents or water. The formation of this material during the reaction may be due to well reported condensation reactions in an acidic environment.19 This insoluble fraction is not drastically influenced by the reaction temperature, as 32% to 52% of the material is obtained between 90 °C and 120 °C. A reduced amount of acid leads to a decreased substitution degree that ranges from 61% with 0.25 equivalents of PTSA/OH to 28% with 0.08 (entries 3 to 5). As expected in the case of the hydroesterification reaction, a high acid concentration is clearly needed to promote the formation of catalytically relevant palladium hydrides and, hence, reach higher substitution degrees. The amount of PTSA does not drastically impact the formation of the insoluble lignin and rather similar amounts of functionalized lignin are obtained in most cases (for example, compare entries 3 and 5). A control test at 120 °C with lignin without PTSA in THF also generated insoluble compounds thus indicating that the observed insolubility does not come from the acid used at this temperature (entry 6).
A kinetic study performed with the reaction conditions of entry 3 indicates that 6 hours of reaction is necessary to reach 61% of substitution degree (entries 7–9 and 3) without improving the mass of recoverable lignin. Reducing the amount of the olefin decreases the conversion of hydroxyl groups from 61% to 39% and 22% with respectively 8 eq., 3 eq. and 1.5 eq. of 1-hexene. On the other hand, the decrease of olefin equivalents seems to play a role in the formation of the insoluble lignin fraction, as the %weight of functionalized lignin increased from 52% to 72% by reducing the amount of olefin from 8 eq. to 1.5 eq. (entries 3, 10 and 11). Lignin is notorious for poorly tolerating lipophilic chains and tending to precipitate in the presence of alkanes. It can thus be hypothesized that the starting lignin precipitated upon the addition of 1-hexene in the reactor and further cured within the precipitate under the reaction conditions used. Other solvents were thus explored in order to find a better solubilizing medium for all the reagents. The reaction was first carried out in DMSO but no conversion was observed due to the fact that 1-hexene is poorly soluble in this solvent (biphasic liquid/liquid system formed), thus limiting the contact between all the reagents and catalyst. It should be noted that with DMSO, no lignin was insoluble at the end of the reaction (entry 12). The use of dioxane allowed an excellent conversion of 70% and a slight improvement in the mass of lignin recovered (compare entries 3 and 13). With this solvent, a further decrease of the number of equivalents of olefin allowed a higher recovery of functionalized lignin (entries 14 and 15).
No insoluble lignin was observed after the reaction and the recovered masses of 105% and 113% are in agreement with the increase of mass due to the grafting of the aliphatic ester chains on the lignin.
Other parameters such as the pressure of CO, the catalytic charge in Pd and the amount of solvent were studied without a significant improvement of the substitution degrees nor the lignin insolubility (see ESI Table SI 1†).
Detailed characterization by 1H NMR of the product obtained in entry 15 of Table 1 evidences the presence of alkyl chains with the characteristic signals at 0.7–1.1 ppm corresponding to the terminal CH3, 1.1–1.6 ppm for the CH2 groups of the aliphatic chain and at 1.6–1.95 ppm for the CH2 in the β position of the ester (Fig. 1). These spectroscopic data correlate well with the data recorded for hydroesterified guaiacol and benzyl alcohol (Fig. SI 1 and SI 3†).
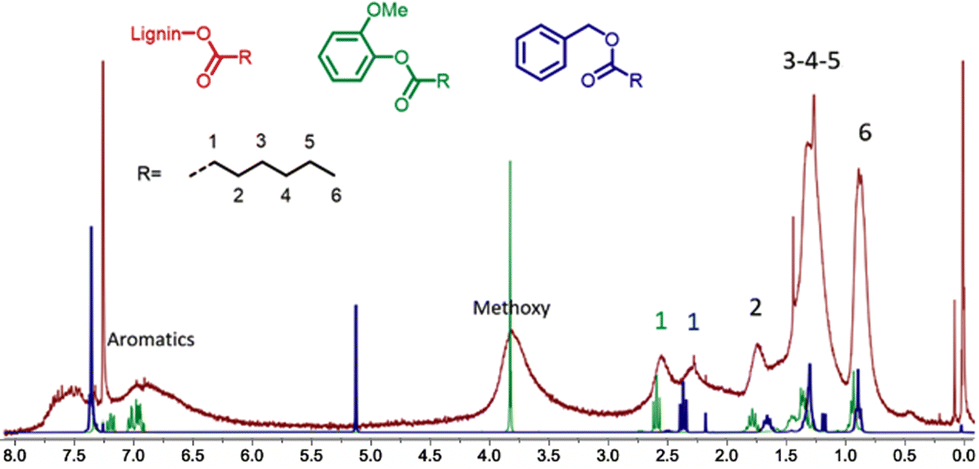 | ||
| Fig. 1 Comparison of the 1H NMR spectra of the hydroesterified lignin (red spectrum) with those of esters of guaïacol (green) and benzyl alcohol (blue). | ||
Most particularly, the presence of the peak at 2.15–2.85 ppm corresponding to the CH2 in the α position to the ester indicates the presence of esters grafted onto the phenolic groups of the lignin. The α-CH2 of esters from aliphatic alcohols are less deshielded and give signals at 2.10–2.45 ppm as confirmed by the 1H NMR data recorded for the hydroesterified benzyl alcohol. All the 1H NMR spectra of the compounds obtained during the optimization of the reaction have this same pattern. Only the intensity of the alkyl peaks with respect to the methoxy and aromatic groups of lignin differs according to the degree of substitution.
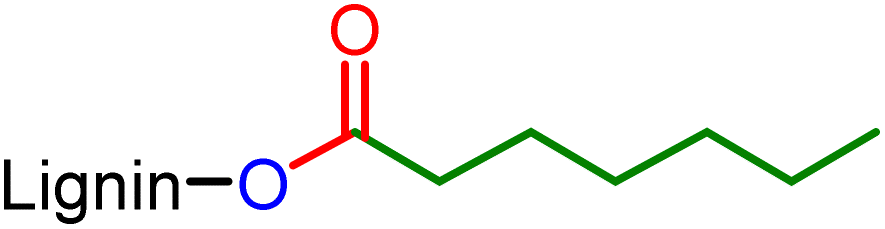
The comparative study of the infrared spectra of native and modified lignin highlights a band at 1728 cm−1 characteristic of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O elongations corresponding to the new ester functions grafted on lignin (Fig. 2). The decrease in intensity of the broad band at 3360 cm−1 (O–H stretching) evidences the conversion of the hydroxyl groups. Also note the decrease after the reaction of the bands at 1511 cm−1 and 1270 cm−1 corresponding respectively to the O–H deformations of phenolic alcohols and aliphatic alcohols. This suggests that the reaction occurs on both phenols and aliphatic alcohols. New intense characteristic bands also appear at 2928, 1121 and 679 cm−1 corresponding to the C–H and C–C elongations of the aliphatic chains grafted on lignin. The esterification of the hydroxyl groups is also confirmed by the 31P NMR analysis performed after the derivatization of the reaction product with 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane.20 The comparison with the analytical data recorded for the technical lignins (see ESI Table SI 2†) shows a conversion of 75% of the total OH of Kraft lignin, including 91% of phenolic alcohol and 68% of aliphatic alcohols (Table 2 entry 1). Up to 30% of the carboxylic acids are also substituted, possibly as a side reaction of internal esterification with aliphatic OH. Other complementary analyses are available in the ESI such as 2D HSQC NMR (Fig. SI 41†).
O elongations corresponding to the new ester functions grafted on lignin (Fig. 2). The decrease in intensity of the broad band at 3360 cm−1 (O–H stretching) evidences the conversion of the hydroxyl groups. Also note the decrease after the reaction of the bands at 1511 cm−1 and 1270 cm−1 corresponding respectively to the O–H deformations of phenolic alcohols and aliphatic alcohols. This suggests that the reaction occurs on both phenols and aliphatic alcohols. New intense characteristic bands also appear at 2928, 1121 and 679 cm−1 corresponding to the C–H and C–C elongations of the aliphatic chains grafted on lignin. The esterification of the hydroxyl groups is also confirmed by the 31P NMR analysis performed after the derivatization of the reaction product with 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane.20 The comparison with the analytical data recorded for the technical lignins (see ESI Table SI 2†) shows a conversion of 75% of the total OH of Kraft lignin, including 91% of phenolic alcohol and 68% of aliphatic alcohols (Table 2 entry 1). Up to 30% of the carboxylic acids are also substituted, possibly as a side reaction of internal esterification with aliphatic OH. Other complementary analyses are available in the ESI such as 2D HSQC NMR (Fig. SI 41†).
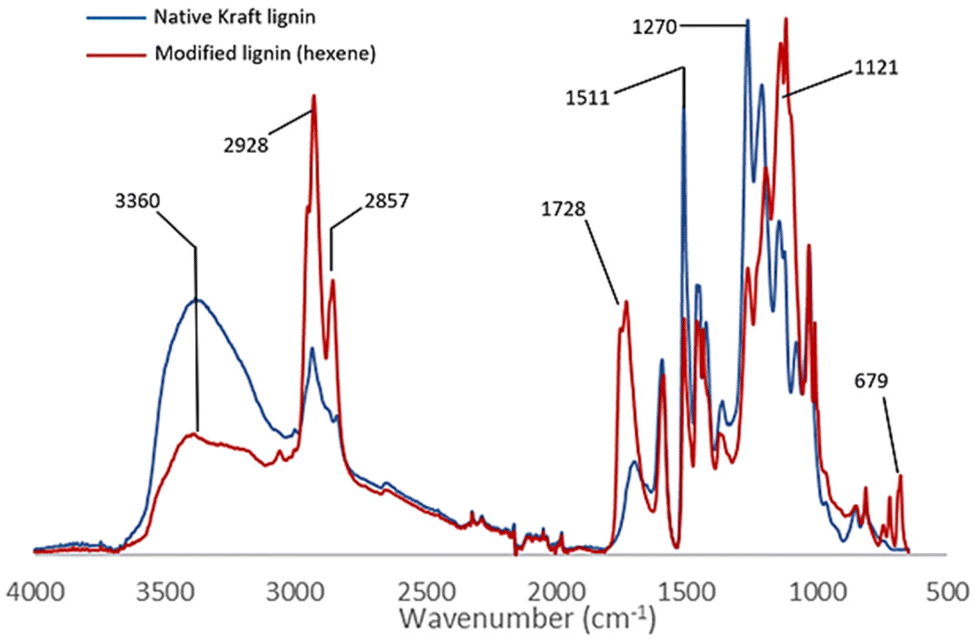 | ||
| Fig. 2 Comparison of IR spectra of technical (blue) and modified kraft lignin (red). | ||
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Lignin | Conversiona [%] | L/Bb | ICP (wt% Pd) | |||
| OHtot | OHal | OHph | COOH | ||||
| Conditions: 0.5 g of lignin, 10 mL of dioxane, Pd(OAc)2 1 mol% in respect to OH groups, 25 eq./Pd of PPh3 and PTSA, 3 eq./OH 1-hexene, 30 bar of CO.a Conversion of total, aliphatic, and phenolic OH groups, respectively, and of acid groups, determined by 31P NMR with a correction factor considering modified lignin content. See correction factor calculation after Table SI2.†b Determined by 13C NMR, see SI15.† | |||||||
| 1 | Kraft | 75 | 91 | 68 | 30 | 92/8 | 0.152 |
| 2 | Organosolv | 84 | 84 | 82 | 73 | 97/3 | 0.228 |
| 3 | Soda | 85 | 100 | 80 | 65 | 93/7 | 0.190 |
The optimized protocol with 1-hexene was applied to other commercially available lignins. Substitution degrees of 84% and 85% were respectively obtained with Organosolv and Soda lignins (Table 2 entries 2 and 3). This demonstrates the broad applicability of this methodology to other types of lignins. The linear to branched ratio of esters (L/B in Table 2) was quantified using quantitative 13C NMR and showed similar results to that of the simple molecules with less than 10% of branched isomers. The palladium content in the lignins was measured by ICP in order to evaluate the amount of metal that leached into the final material. The ICP titrations indicate that only 25% of the catalyst remains in the lignin. Thus, most of the catalyst was extracted from the lignin thanks to the purification steps. A major line of future development of our process is the isolation and recovery of the spent metal catalyst.
From Kraft lignin, other olefins were grafted to probe the impact of the chain size on the thermal properties of the new materials (Table 3). The reaction with 1-octene leads to conversions of 87% of total lignin alcohols (100% of aliphatics and 81% of phenolics – entry 2). 1-Decene gives slightly lower results with 70% conversion while 1-dodecene gives the best results with 91% alcohol functionalization (entries 3 and 4).
|
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Olefin | Conversiona [%] | DSC | SEC | |||||
| OHtot | OHal | OHph | COOH | T g (°C) | M n | M w | Đ | ||
| Conditions: 0.5 g of Kraft lignin, 10 mL of dioxane, Pd(OAc)2 1 mol% in respect to OH groups, 25 eq./Pd of PPh3 and PTSA, 3 eq./OH olefin, 30 bar of CO.a Conversion of total, aliphatic, and phenolic OH groups, respectively, and of acid groups, determined by 31P NMR with correction factor (see SI). | |||||||||
| 1 |
|
75 | 91 | 68 | 30 | 102 | 2600 | 4400 | 1.7 |
| 2 |
|
87 | 100 | 81 | 44 | 80 | 2900 | 5100 | 1.7 |
| 3 |
|
70 | 82 | 64 | 40 | 110 | 2500 | 4000 | 1.6 |
| 4 |
|
91 | 90 | 91 | 58 | 72 | 3100 | 6700 | 2.2 |
Regarding thermal properties, in Kraft lignins functionalized with esters carrying a 6-carbon chain issuing from 1-hexene, the glass transition temperature (Tg) of the compounds drops from 140 °C (technical Kraft lignin) to 102 °C (entry 1). With 8 and 12 carbon chains, Tg further decreased to 80 °C and 72 °C respectively (entries 2 and 4). For the 10-carbon chain from 1-decene, the glass transition temperature of the sample is the highest of the series (110 °C) and this can be explained by a lower degree of substitution (entry 3).
SEC measurements were run on the isolated materials to determine their number average molecular weight Mn and dispersity Đ (see ESI Table SI 4†). SEC analysis of used technical Kraft lignin after acetylation revealed a number average molecular weight Mn of 1800 g mol−1 with a dispersity Đ of 3.1. After hydroesterification with 1-hexene, a higher molecular weight (Mn = 2600 g mol−1) corresponding to the large amount of grafted chains on the macromolecule is observed. For similar conversion rates, the more carbons in the grafted chain, the higher the molecular weight, as expected. SEC analysis indicated Mn values of 2900 g mol−1 with the material obtained from the hydroesterification of 1-octene and of 3100 g mol−1 with 1-dodecene (Table 3 entries 2 and 4).
This new synthesis procedure opens up opportunities for bio-sourced fatty acid grafting through the transformation of the unsaturation while the carboxylic acid remains pending. The most abundant fatty acid, oleic acid, has been reacted under the optimized reaction conditions. With this internal olefin, 26% of the hydroxyl groups were converted (see Table 4 entry 1). A deeper analytical study showed that only the aliphatic hydroxyl groups are substituted. In addition, 1H NMR indicates the presence of untouched unsaturations (double bonds) within the product. Thus, the hydroesterification reaction has not occurred, and the grafting was essentially obtained from a classical esterification of aliphatic alcohols with oleic acid in the presence of PTSA. The reaction with methyl oleate results in a slightly improved alcohol conversion that reaches 29%. With this starting material, 12% of the hydroxyl groups have been esterified thanks to the carbonylation reaction (entry 2).
| Entry | Olefin | Conversiona [%] | Carbonylation ratiob | |||
|---|---|---|---|---|---|---|
| OHtot | OHal | OHph | COOH | |||
| Conditions: 0.5 g of Kraft lignin, 10 mL of dioxane, Pd(OAc)2 1 mol% in respect to the OH groups, 25eq./Pd of PPh3 and PTSA, 3 eq./OH of olefin, 30 bar of CO.a Conversion of total, aliphatic, and phenolic OH groups, respectively, and of acid groups, determined by 31P NMR with correction factor (see ESI†).b Comparison between direct esterification and carbonylation by hydroesterification – determined by 1H NMR considering the remaining unsaturations on the lignin (5.2–5.5 ppm). | ||||||
| 1 |

|
26 | 85 | 0 | 60 | 1% |
| 2 |

|
29 | 92 | 3 | 87 | 12% |
| 3 |
|
36 | 68 | 22 | 31 | 100% |
| 4 |

|
90 | 89 | 90 | 8 | 100% |
| 5 |

|
48 | 75 | 36 | 50 | 100% |
As internal olefins proved to be less reactive, we turned our attention towards 10-undecenoic acid, derived from renewable ricinoleic acid via the thermolytic pathway. Indeed, this terminal olefin exhibits a higher reactivity than oleic acid in carbonylative esterification. A conversion of 36% of the total alcohols was obtained, including 22% of the phenols, exclusively via hydroesterification reactions (entry 3). The highest conversions were obtained with the ester derivative methyl 10-undecenoate with 90% conversion of hydroxyl groups including 90% of phenolic groups (entry 4). As a further example, estragole as a terminal olefin bearing an aromatic group leads to 48% overall conversion of hydroxyl groups, including 36% substitution of phenol groups (entry 5). Remarkably, in the present study, this enhanced reactivity pattern of terminal olefins favours the grafting of the hydroxyl sites through the hydroesterification reaction rather than classical esterification or transesterification processes.
The 1H NMR analysis of lignin modified with 10-undecenoate shows the presence of methyl ester groups with a strong, sharp peak at 3.6 ppm (Fig. 3, label 6). The assignment is further evidenced by the sharper linewidth of this signal compared to others from the material, as expected by the higher chain mobility from the grafted ester chain ends. The protons of the carbon atom in the alpha position relative to the carbonyl groups are also observed in the range 2.2–2.7 ppm as two signals (Fig. 3, labels 1 and 5).
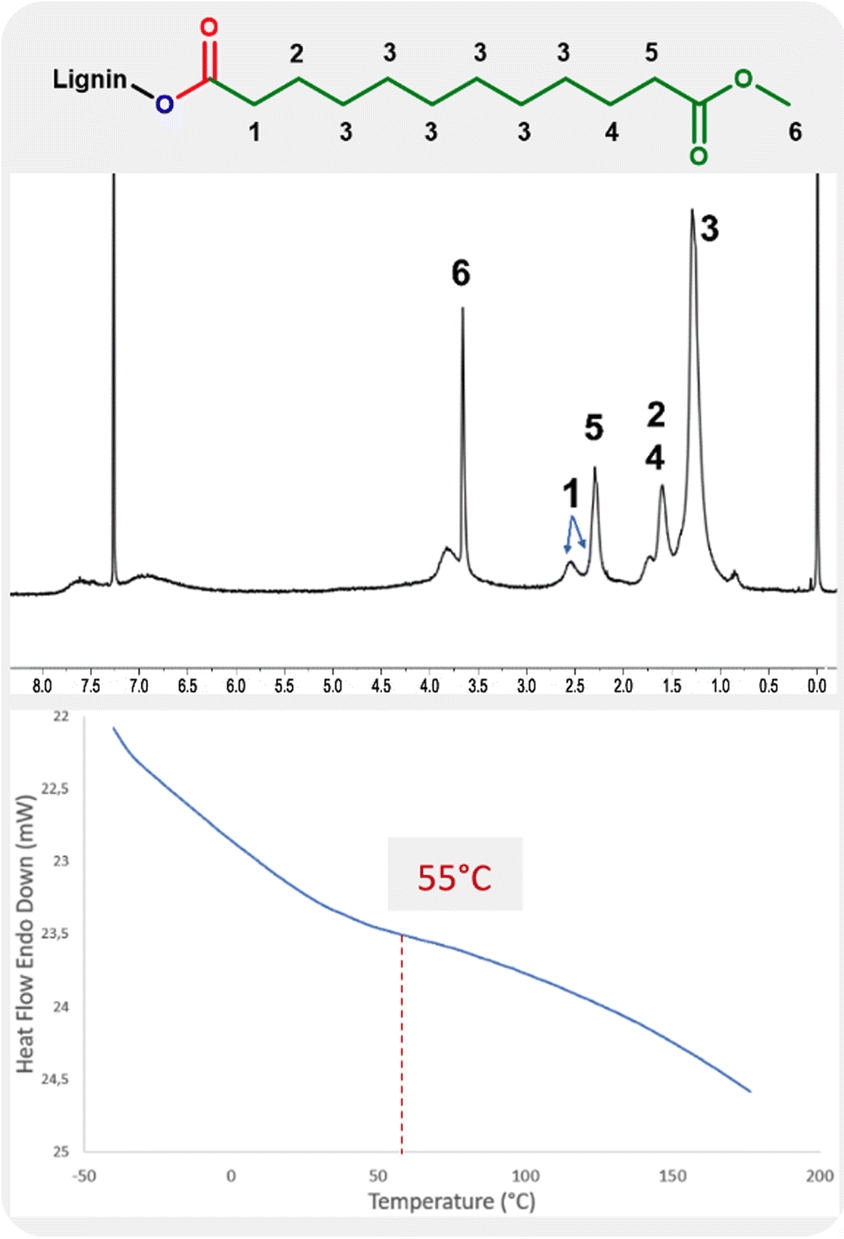 | ||
| Fig. 3 1H NMR and DSC analysis of the product of hydroesterification of methyl 10-undecenoate with Kraft lignin (Table 4, entry 4). | ||
The influence from the addition of this long aliphatic tail into the lignin is also evidenced by DSC with a glass transition temperature of 55 °C.
The practicality of our approach to lignin material functionalization with methyl 10-undecenoate was probed further through a scale-up of the process. It proved to be amenable to a 100 gram scale: the resulting fully biosourced lignin-vegetable oil-based resin could then be readily shaped on hot-plates (Fig. 4). This functionalized lignin is particularly interesting due to the improved compatibility provided by the long fatty chains, combined with the availability of the reactive ester functions which can be used in the formulation of blends or as a polyester macromonomer.
 | ||
| Fig. 4 Lignin shaping on hot plates after hydroesterification of methyl 10-undecenoate – upscaling of the experiment from Table 4 entry 4. | ||
Grafting of other polymerizable functionalities is also attractive for the valorization of lignin as a material. More specifically, unsaturations have aroused recent interest.21 Indeed, these alkenes, in addition to being directly polymerizable by metathesis, thiol–ene or radical polymerization, can also be used as a precursor for epoxy resins.22 In this regard, the hydroesterification reaction can be a clean solution for grafting unsaturations onto lignin using butadiene as a reagent. Butadiene has several advantages. As a gas under ambient conditions, the removal of excess butadiene is straightforward and the reagent is simply released at the reactor opening, easing up the purification procedures. Furthermore, although butadiene is nowadays mostly petroleum based, it can also be obtained from bioethanol.23 The methoxycarbonylation of 1,3-butadiene described by M. Beller24 shows the feasibility of using this gas as a reagent with aliphatic alcohols. As part of our study, this reaction was first performed with guaiacol as a phenol model to ensure the reactivity of phenols as nucleophile. In contrast to the hydroesterification reaction of non-conjugated olefins (vide supra), the hydroesterification of butadiene requires a different catalytic system and softer reaction conditions. A combination of Pd(OAc)2 with dppb (1,4-bis(diphenylphosphino)butane) is used to generate the catalyst, and benzoic acid is employed as the co-catalyst. The reaction with guaiacol was evaluated with 1 mol% of Pd(OAc)2, 6 equivalents per Pd of dppb at 120 °C for 24 h under 25 bar of carbon monoxide. At the end of the reaction, 90% conversion of the starting guaiacol was observed, with the formation of two isomers in a 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio. Their purification can be performed by silica gel column chromatography, although similar properties hinder the efficiency of the separation. The isomers differ in the position of the double bond on the aliphatic chain, mostly as the trans isomers from 1H and 13C NMR (see the ESI†). The β–δ isomer was isolated with 52% yield and the α–β isomer with 11% yield (Scheme 3). It should be noted that no additional isomers were observed during the study, the low isolated yields are due to the arduous separation of these two products as analytically pure compounds. In addition, the same procedure was carried out with benzylic alcohol as substrate. As in the case of gaiacol, a mixture of two isomers was obtained in 94% yield with ratio of 60% β–δ isomer and 40% α–β isomer (see ESI Fig. SI 9†).
:4 ratio. Their purification can be performed by silica gel column chromatography, although similar properties hinder the efficiency of the separation. The isomers differ in the position of the double bond on the aliphatic chain, mostly as the trans isomers from 1H and 13C NMR (see the ESI†). The β–δ isomer was isolated with 52% yield and the α–β isomer with 11% yield (Scheme 3). It should be noted that no additional isomers were observed during the study, the low isolated yields are due to the arduous separation of these two products as analytically pure compounds. In addition, the same procedure was carried out with benzylic alcohol as substrate. As in the case of gaiacol, a mixture of two isomers was obtained in 94% yield with ratio of 60% β–δ isomer and 40% α–β isomer (see ESI Fig. SI 9†).
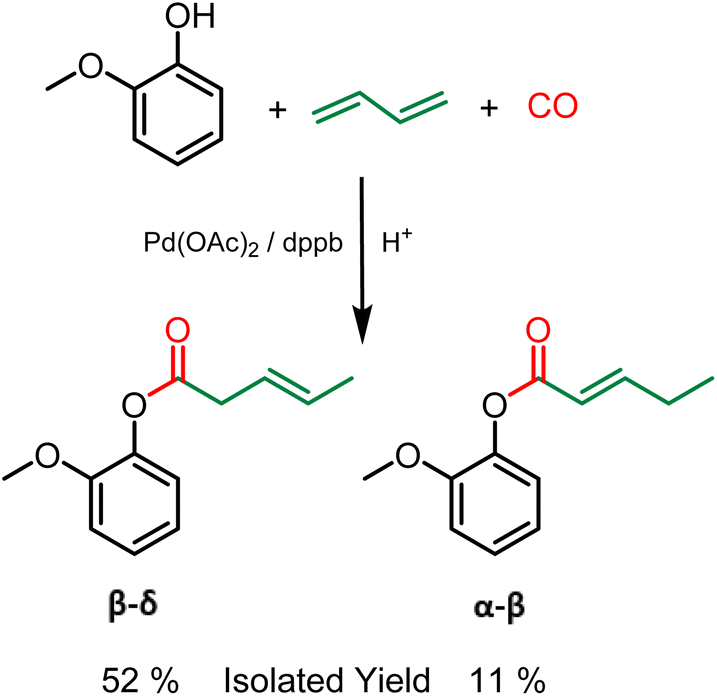 | ||
| Scheme 3 Hydroesterification of butadiene with guaiacol – Reaction conditions: Guaiacol 9 mmol, benzoic acid 0.3 eq., butadiene 7 eq., Pd(OAc)2 1 mol%, dppb 6 eq./Pd, 120 °C, 24 h, 25 bar CO, THF. | ||
The implementation of this procedure on Kraft lignin leads to a conversion of 43% of all types of alcohols, including 40% of phenols and 47% of aliphatic alcohols (Table 5 entry 1). As in the case of the simple molecules, two isomers are observed as shown by 1H NMR analysis of the product (see below for details). The integration of proton signals evidences a selectivity of 63% for β–δ and 37% for α–β isomers. Interestingly, increasing the reaction time does not improve the conversion, but does increase the ratio of α–β isomer (entry 2). The catalyst thus appears to initially functionalize the alcohols with the unsaturation in the β–δ position as the kinetic product of the reaction, and then subsequently isomerizes the double bond to the α–β position, thus forming the thermodynamic product. This assumption is confirmed as a reduced catalyst loading of 0.5% leads to a lower selectivity toward the isomer α–β which accounts for 14% of the mixture (entry 3). In order to maximize the functionalization of lignin alcohols, a higher catalytic loading up to 1.6 mol% of Pd was also investigated and allowed an increase of the total conversion to 67%. Under these conditions, 52% of the phenols and almost all of the aliphatic alcohols were converted (entry 4). Finally, as in the case of olefin hydroesterification described above, the use of dioxane significantly increases the efficiency of the reaction. With this solvent, the reaction reaches 95% conversion of the alcohols (93% of phenols – entry 5). Other optimization parameters such as the reaction time or temperature have also been studied and showed a limited impact on the conversion of lignin's hydroxyl groups (see ESI Table SI 3†).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Pd(OAc)2 (eq./OH) | time (h) | Conversiona [%] | Isomers | |||
| OHtot | OHal | OHph | Ratio β–δb | Ratio α–βb | |||
| Conditions: 0.5 g of Kraft lignin, 6 eq./Pd of dppb, 7 eq./OH of butadiene, 10 mL of degassed THF, 50 bar CO and 30 eq./Pd of benzoic acid, 110 °C.a Conversion of total OH groups: comparison of the amount of chains grafted onto the lignin with the quantity of available hydroxyl groups; determined by 1H NMR with pentafluorobenzaldehyde as internal standard.b Ratio calculated by 1H NMR from the integrals of signals between 2.17–2.4 and 2.7–3.45 ppm.c Reaction performed with dioxane instead of THF. | |||||||
| 1 | 1% | 15 | 43 | 47 | 40 | 63% | 37% |
| 2 | 1% | 48 | 46 | 51 | 43 | 47% | 53% |
| 3 | 0.5% | 15 | 13 | 28 | 6 | 86% | 14% |
| 4 | 1.6% | 15 | 67 | 96 | 52 | 36% | 64% |
| 5c | 1% | 15 | 95 | 99 | 93 | 95% | 5% |
Detailed analysis of the 1H NMR spectrum recorded for the material obtained from entry 5 of Table 5 shows extensive grafting of chains containing unsaturation into the lignin. The spectrum fits very well with the spectra recorded for the products obtained from simple compounds guaiacol and benzyl alcohol (Fig. 5A). The weak characteristic signals centered at 1.15 ppm, 2.25 ppm and 6.02 ppm (see labels e, g and h) show the presence of the α–β isomer as a minor component compared to the strong peak at 1.6 ppm (label d) attributed to the β–δ isomer. The presence of two signals at 2.75–3.25 ppm and 3.25–3.5 ppm corresponds to the α-CH2 of the ester resulting from respectively the aliphatic and phenolic groups (labels a and i). Note also the presence of low molecular weight polybutadiene in the product due to the polymerization of butadiene under this reaction temperature. The integration of the 1H NMR signals allowed a quantification of the amount of polymer, which is estimated between 5 to 10 wt%. The conversion of the two types of lignin alcohols is confirmed by the 31P NMR analysis of the product after phosphorylation (Fig. 5B). The 31P NMR spectrum recorded for the functionalized lignin shows very weak signals in the 146–150 ppm and 138–145 ppm ranges thus indicating that most of the aliphatic and phenolic hydroxyl groups have been converted. Carboxylic acids are also converted after reaction (signals between 134 and 136 ppm). This may be associated with the internal esterification reactions as seen above, the reaction being performed under acidic conditions. The SEC analysis reveals a molecular weight of 5100 g mol−1 and a polydispersity index of 2.3. The Mn value after modification with butadiene is significantly higher compared to those obtained above with other olefins most probably due to the presence of polybutadiene, whose shoulder is clearly observed in the SEC analysis at short retention times (see ESI Fig. SI 40†).
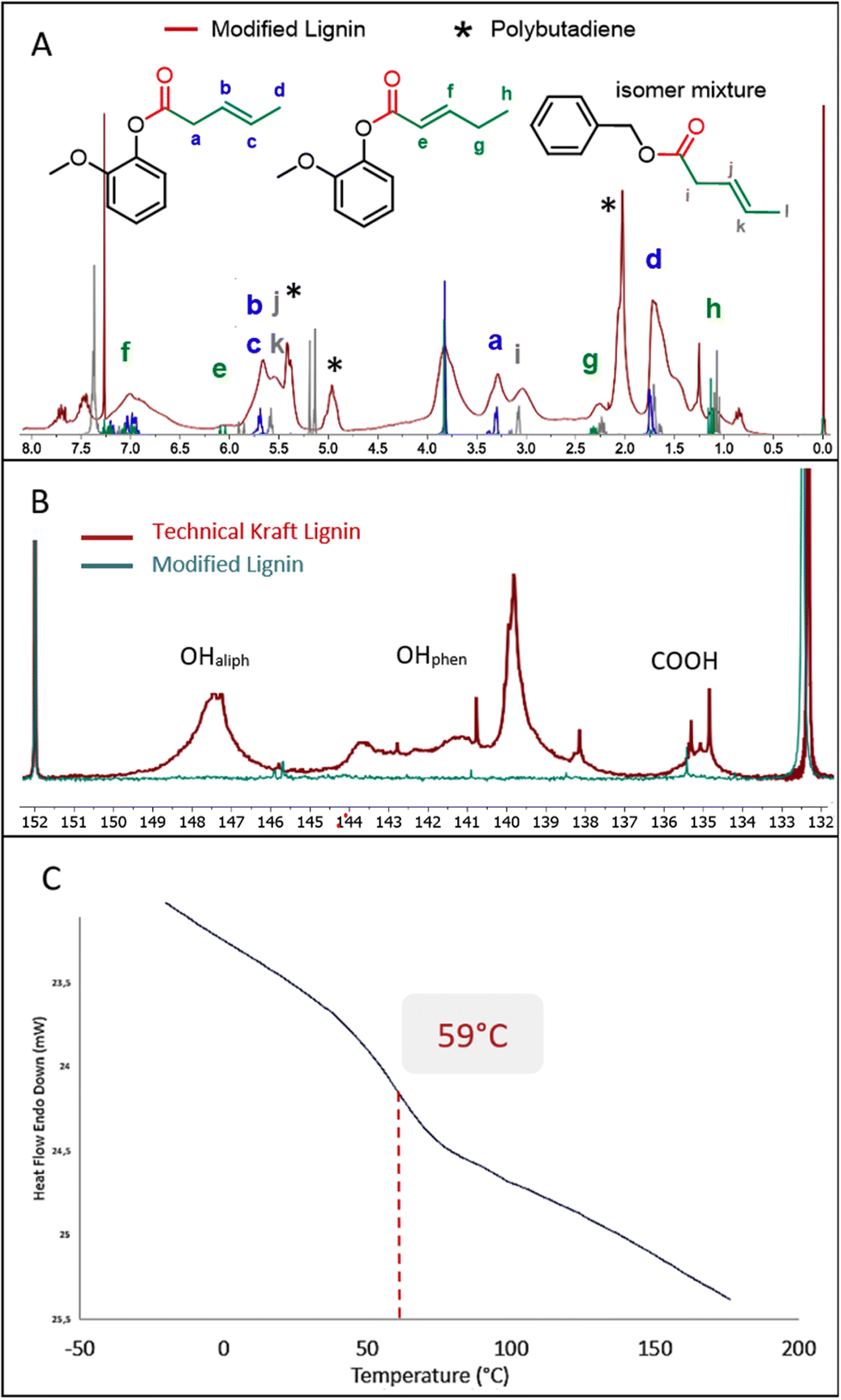 | ||
| Fig. 5 1H NMR (A), 31P NMR with intensities calibrated on the internal standard (B) and DSC (C) analysis of the product of hydroesterification of butadiene with Kraft lignin (Table 5, entry 5). | ||
Interestingly, the DSC analysis of this product gives a glass transition temperature of 59 °C, similar to that observed with methyl 10-undecenoate (Fig. 5C). Although the grafted chains are shorter, the presence of unsaturation and a slightly higher conversion seem to significantly impact the glass transition temperature. This modified lignin thus presents new interesting properties from a thermal point of view, with visibly improved compatibility with solvents. This material is for example soluble in toluene. Furthermore, the C–C unsaturations grafted via our catalytic functionalization protocol allow the modified lignin to be further chemically modified, for instance as a macromonomer for innovative green polymer materials.
Conclusions
A new method of lignin functionalization through the palladium-catalyzed hydroesterification reaction has been developed. The reaction readily converts hydroxyl groups of lignin into esters from a reaction with CO and an olefin without generating salts in stoichiometric amounts. Importantly, the method is effective for all types of alcohol functions, whether aliphatic or phenolic. The characterization of the esterified lignins was carried out using a combination of spectroscopic methods (NMR and IR). Thanks to a comparative study using simple molecules which provides a robust tool for the assessment of selectivity towards all alcohol types, lignin grafting with ester functions was fully determined. Under suitable reaction conditions and with widely available catalysts, up to about 90% of the hydroxyl groups in Kraft lignin have been readily converted. The process also proved to be suitable with other types of lignins such as Organosolv and Soda. Interestingly, the chain length of the grafted esters can be tuned by varying the nature of the starting olefin. DSC and SEC analyses demonstrated the modulation of the physicochemical properties of lignin depending on the length of the grafted chain. An ICP study also indicates that only a limited amount of palladium is contained in the final product thanks to its removal during the purification phase. Reagents such as butadiene or bio-based fatty esters have allowed the introduction of new reactive functions within lignin while significantly lowering glass transition temperatures. Furthermore, a scale-up to the 100 g scale was successfully carried out, resulting in a moldable bio-sourced material. Thanks to the versatility of this approach, new lignin-based shapable resins can thus be produced and these can be functionalized with cross-linkable functions such as double bonds or esters. Thus, our versatile catalytic process offers an efficient and selective entry towards new biosourced thermosetting polyester materials based on lignin with highly tunable properties.Materials and methods
General considerations
Kraft lignin was provided by UPM Biochemicals, Organosolv by ChemicalPoint and Soda by GreenValue Enterprises LLC. Lignins were dried at reduced pressure at 60 °C for 48 h before use. Chemicals were purchased from Aldrich, Alfa Aesar, Acros and Strem. Butadiene was purchased from Linde Gas France. Tetrahydrofuran was purified using a MBraun SPS-800 solvent purification system. DSC measurements were carried out using a PerkinElmer DSC 4000 differential scanning calorimeter. The measurements were made with a heating rate of 10 K min−1 in the temperature range from 243 K to 453 K, under nitrogen flow (20 ml min−1).NMR spectra were recorded in CDCl3 (99.8% atom D), purchased from Aldrich. 1H NMR spectra were obtained at either 300 and 400 MHz, 13C were recorded at 75 MHz with 1H decoupling and 101 MHz with both 1H and 31P decouplings, and 31P spectra were acquired at 121 and 162 MHz with 1H decoupling. Chemical shifts (1H and 13C) were referenced in parts of million relative to TMS (δ = 0.00 ppm) and were referenced to the residual solvent peak (1H, CDCl3 7.26 ppm). Diffuse reflectance infrared spectra were collected with a Harrick cell on a Nicolet Avatar spectrometer fitted with a MCT detector. Typically, 64 scans were accumulated for each spectrum (resolution of 4 cm−1).
The number-average (Mn), the weight-average (Mw) molecular weights and the dispersity (Đ) of the product were determined by size exclusion chromatography (SEC) in tetrahydrofuran at 40 °C at a flow rate of 1 mL min−1. Sample concentration was of 0.50 w%. Mn, Mw, and Đ were determined from the refractive index (RI) signal using a calibration curve based on polystyrene (PS) standards from Polymer Standards Service, on a Waters apparatus equipped with Waters Styragel columns HR2, HR3, HR5 and HR5E.
Nickel (231604 nm) and palladium (340458 nm) concentrations were determined using an Agilent's 5110 Vertical Dual View ICP-OES equipped with a OneNeb nebulizer, a quartz double pass spray chamber and a quartz torch (Agilent). Quantifications were carried out by an external calibration approach.
Titration of the hydroxyl groups in the technical lignins and modified lignins
31P NMR experiments were carried out following a previously reported method:25 lignin (30 mg) was dissolved in a CDCl3/pyridine mixture (1:1.6 v/v, 0.5 mL). The phosphitylation reagent, 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (150 μL), and the internal standard N-hydroxy-6-norbornene-2,3-dicarboximide (100 μL of 0.1 M solution in 1:1.6 v/v CDCl3/pyridine mixture) were added successively. Chromium(III) acetylacetonate (100 μL of 0.014 M solution) in the same CDCl3/pyridine mixture was added to the solution in order to homogenize and accelerate phosphorus relaxation. Spectra were recorded with a 15 s relaxation time and an average number of 512 scans. Chemical shifts are relative to the signal of the phospholane hydrolysis product at 132.2 ppm. The integrated value of the internal standard was used for calculation of the absolute amount of each functional group. Comparison between the amount of technical lignin functional group and the amount of hydroesterified lignin functional groups gave the substitution ratio of hydroxyl groups. This method includes a correction factor for integrations of modified lignins considering the reduced lignin content (see ESI†).
General procedure for olefins/fatty esters hydroesterification on lignin
An autoclave was charged with lignin (500 mg), palladium acetate (6 mg, 0.027 mmol), triphenylphosphine (175 mg, 0.67 mmol), and paratoluenesulfonic acid (115 mg, 0.67 mmol). After performing three vacuum/N2 cycles, degassed dioxane (10 mL) and degassed olefin (9.15 mmol) were added into the autoclave. The reactor was pressurized under 30 bar of carbon monoxide and then heated to 120 °C for 22 h under magnetic stirring with a cross magnet. After cooling, the excess gas was evacuated, the solvent was evaporated, and the crude was solubilized in 3 mL THF and precipitated with petroleum ether (30 mL). The product was washed 3 times with petroleum ether (10 mL) the resulting brown powder was dried at 60 °C under reduced pressure for 24 h.General procedure for butadiene hydroesterification on kraft lignin
Kraft lignin (500 mg), palladium acetate (6 mg, 0.026 mmol), 1,4-bis(diphenylphosphino)butane (70 mg, 0.16 mmol), and benzoic acid (100 mg, 0.82 mmol) were introduced into an autoclave. After performing three vacuum/N2 cycles, distilled and degassed THF (10 mL) is added to the reaction medium and the autoclave is cooled to −40 °C. Butadiene (2 mL, 1.2 g, 22 mmol) is condensed in a Schlenk tube kept cold by an acetone/dry ice mixture and then introduced into the reactor by cannula. The reactor is pressurized under 50 bar of carbon monoxide and then heated at 120 °C for 22 h under magnetic stirring with a cross-shaped magnet. After the reaction, the reactor is cooled to room temperature and the excess gas is evacuated. The solvent is extracted with a rotary evaporator. The crude is solubilized in 3 mL THF and the modified lignin is precipitated in 30 mL petroleum ether. After settling for one hour, the lignin is recovered and dried under vacuum at 80 °C overnight.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Ministère de l'Enseignement Supérieur, de la Recherche et de l'Innovation, the CNRS, the Hauts-de-France Region and Icam for their financial support. We acknowledge C. Delabre for HR-MS analysis. ICP analyses were performed in the “Spectrométrie par torche à plasma” platform of the Research Federation Michel-Eugène Chevreul hosted by the LASIRE laboratory. We would also like to acknowledge COST Action CA17128 (LignoCOST) for promoting exchange and dissemination of knowledge and expertise in the field of lignin valorization.References
- (a) D. S. Bajwa, G. Pourhashem, A. H. Ullah and S. G. Bajwa, Ind. Crops Prod., 2019, 139, 111526 CrossRef CAS; (b) S. Laurichesse and L. Avérous, Prog. Polym. Sci., 2014, 39, 1266–1290 CrossRef CAS.
- A. E. Kazzaz, Z. H. Feizi and P. Fatehi, Green Chem., 2019, 21, 5714–5752 RSC.
- O. Gordobil, I. Egüés, R. Llano-Ponte and J. Labidi, Polym. Degrad. Stab., 2014, 108, 330–338 CrossRef CAS.
- K. Huang, S. Ma, S. Wang, Q. Li, Z. Wu, J. Liu, R. Liu and J. Zhu, Green Chem., 2019, 21, 4964–4970 RSC.
- (a) Y. Xu, L. Yuan, Z. Wang, P. A. Wilbon, C. Wang, F. Chu and C. Tang, Green Chem., 2016, 18, 4974–4981 RSC; (b) C. Wang and R. A. Venditti, ACS Sustainable Chem. Eng., 2015, 3, 1839–1845 CrossRef CAS.
- R. Paul, B. John and S. K. Sahoo, Biomacromolecules, 2022, 23, 816–828 CrossRef CAS PubMed.
- M. A. Jedrzejczyk, S. Van den Bosch, J. Van Aelst, K. Van Aelst, P. D. Kouris, M. Moalin, G. R. M. M. Haenen, M. D. Boot, E. J. M. Hensen, B. Lagrain, B. F. Sels and K. V. Bernaerts, ACS Sustainable Chem. Eng., 2021, 9, 12548–12559 CrossRef CAS.
- J. Huang, Q. Guo, R. Zhu, Y. Liu, F. Xu and X. Zhang, Int. J. Biol. Macromol., 2021, 189, 635–640 CrossRef CAS PubMed.
- D. Wang, S. H. Lee, J. Kim and C. B. Park, ChemSusChem, 2020, 13, 2807–2827 CrossRef CAS PubMed.
- Difference of reactivity between aliphatic alcohol and phenol in esterification reaction through carbodiimide coupling: R. Shelkov, M. Nahmany and A. Melman, Org. Biomol. Chem., 2004, 2, 397–401 RSC.
- T. Saito, R. H. Brown, M. A. Hunt, D. L. Pickel, J. M. Pickel, J. M. Messman, F. S. Baker, M. Keller and A. K. Naskar, Green Chem., 2012, 14, 3295–3303 RSC.
- (a) S. Laurichesse, C. Huillet and L. Avérous, Green Chem., 2014, 16, 3958–3970 RSC; (b) P. Buono, A. Duval, L. Averous and Y. Habibi, ChemSusChem, 2017, 10, 984–992 CrossRef CAS PubMed.
- C. Scarica, R. Suriano, M. Levi, S. Turri and G. Griffini, ACS Sustainable Chem. Eng., 2018, 6, 3392–3401 CrossRef CAS.
- C. Dumont, F. Belva, R. M. Gauvin and M. Sauthier, ChemSusChem, 2018, 11, 3917–3922 CrossRef CAS PubMed.
- (a) J. F. Knifton, J. Org. Chem., 1976, 41, 2885–2890 CrossRef CAS; (b) J. F. Knifton, J. Am. Oil Chem. Soc., 1978, 55(5), 496–499 CrossRef CAS; (c) Shell (Drent, E.), EP 0271144, 1988; (d) Shell (Drent, E.), EP 0186228, 1986.
- W. Reppe, N. V. Kutepow, K. R. Rueppurr and H. Bille, U.S. Patent, 3014962, 1961, 18 Search PubMed.
- W. Reppe, N. V. Kutepow and W. Kölsch, DE 927090C. 1955.
- (a) G. Kiss, Chem. Rev., 2001, 101, 3435–3456 CrossRef CAS PubMed; (b) I. del Río, C. Claver and P. W. N. M. van Leeuwen, Eur. J. Inorg. Chem., 2001, 2001, 2719–2738 CrossRef.
- K. Shimada, S. Hosoya and T. Ikeda, J. Wood Chem. Technol., 1997, 17, 57–72 CrossRef CAS.
- (a) A. Granata and D. S. Argyropoulos, J. Agric. Food Chem., 1995, 43, 1538–1544 CrossRef CAS; (b) G. Gellerstedt, Ind. Crops Prod., 2015, 77, 845–854 CrossRef CAS.
- (a) L. C. Over and M. A. R. Meier, Green Chem., 2015, 18, 197–207 RSC; (b) A. Duval and L. Avérous, ChemSusChem, 2017, 10, 1813–1822 CrossRef CAS PubMed.
- M. Jawerth, M. Lawoko, S. Lundmark, C. Perez-Berumen and M. Johansson, RSC Adv., 2016, 6, 96281–96288 RSC.
- G. Pomalaza, P. A. Ponton, M. Capron and F. Dumeignil, Catal. Sci. Technol., 2020, 10, 4860–4911 RSC.
- M. Beller, A. Krotz and W. Baumann, Adv. Synth. Catal., 2002, 344, 517–524 CrossRef CAS.
- (a) A. Granata and D. S. Argyropoulos, J. Agric. Food Chem., 1995, 43, 1538–1544 CrossRef CAS; (b) G. Gellerstedt, Ind. Crops Prod., 2015, 77, 845–854 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for compounds’ catalytic reactivity, optimization of lignin catalytic modification, and additional characterization data (NMR, SEC, DSC). See DOI: https://doi.org/10.1039/d2gc03708d |
| This journal is © The Royal Society of Chemistry 2023 |