
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Analytical methodologies for oxidized organic compounds in the atmosphere†
Aleksi
Tiusanen
a,
Jose
Ruiz-Jimenez
 ab,
Kari
Hartonen
ab and
Susanne K.
Wiedmer
*a
ab,
Kari
Hartonen
ab and
Susanne K.
Wiedmer
*a
aDepartment of Chemistry, P.O. Box 55, FI-00014 University of Helsinki, Finland. E-mail: susanne.wiedmer@helsinki.fi
bInstitute for Atmospheric and Earth System Research, Chemistry, Faculty of Science, P.O. Box 55, FI-00014 University of Helsinki, Finland
First published on 26th July 2023
Abstract
Oxidized compounds in the atmosphere can occur as emitted primary compounds or as secondary products when volatile emitted precursors react with various oxidants. Due to the presence of polar functional groups, their vapor pressures decrease, and they condense onto small particles. Thereby, they have an effect on climate change by the formation of clouds and scattering solar radiation. The particles and oxidized compounds themselves can cause serious health problems when inhaled. Therefore, it is of utmost importance to study oxidized compounds in the atmosphere. Much ongoing research is focused on the discovery of new oxidized substances and on the evaluation of their sources and factors influencing their formation. Monitoring biogenic and anthropogenic primary oxidized compounds or secondary oxidized products in chamber experiments or field campaigns is common. New discoveries have been reported, including various oxidized compounds and a new group of compounds called highly oxidized organic molecules (HOMs). Analytics of HOMs are mainly focused on chromatography and high-resolution mass spectrometry employing chemical ionization for identifying and quantifying compounds at low concentrations. Oxidized compounds can also be monitored by spectrophotometric methods in which the determinations of total amounts are based on functional groups. This review highlights recent findings on oxidized organic compounds in the atmosphere and analytical methodologies used for their detection and quantification. The discussion includes gas and liquid chromatographic methods, sampling, extraction, concentration, and derivatization procedures involved, as well as mass spectrometric and spectrophotometric methods.
Environmental significanceOxidized compounds in the atmosphere can occur as emitted primary compounds or as secondary products when volatile emitted precursors react with various oxidants. These are currently under intensive investigation due to environmental and health aspects. The discovery of new oxidized species and subsequent evaluation of their sources and factors influencing their formation is an area of research that has recently received considerable attention. Monitoring of biogenic and anthropogenic primary oxidized compounds or secondary oxidized products in chamber experiments or field campaigns is common. A new group of compounds called highly oxidized organic molecules (HOMs) has recently been discovered. This review highlights the latest findings on oxidized organic compounds in the atmosphere and analytical methodologies used to detect and quantify them. |
1. Introduction
The atmosphere contains solid particles and liquid droplets that mix in the air we breathe and form atmospheric aerosols. Particulate matter (PM) consists of liquid and solid particles, and these can contain inorganic or organic constituents (compounds). The organic part can be divided into primary organic aerosols and secondary organic aerosols (SOAs).1–3 Primary organic aerosols contain organic compounds emitted directly from original sources and they are not formed in atmospheric processes.4 In contrast, SOAs are formed from organic matter in the atmosphere, usually through oxidation involving ozone, anthropogenic nitrogen oxides (NOx), and various radicals.1,5 Hydroxyl radicals (OH) are important oxidants during the daytime as they are formed under the influence of light.6 The reactions with precursors yield various reactive peroxide radicals which act as intermediates in the formation of oxidized compounds according to several proposed reaction mechanisms.7–11 Among the oxidation reactions occurring in the dark, the most essential are nitrate radicals (NO3), which are formed in the reaction between ozone and nitrogen dioxide.12 In addition to oxidants, atmospheric sulfuric acid increases SOA production by acting as an acidic catalyst enhancing reactions and by producing additional organosulfate compounds.13 Gaseous volatile organic compounds (VOCs) are typical emitted precursors that react with the above oxidants to form secondary products.Organic aerosols (OA) have an impact on climate change because of their capability to absorb and scatter solar radiation, and they affect cloud nucleation by aerosol–cloud interactions.14 The vapor pressures of the oxidized compounds are decreased compared to their precursors, allowing them to either participate in nucleation reactions that form new particles or to condense on the surfaces of existing particles. The presence of polar organic compounds can increase the hygroscopicity of particles and improve their ability to act as condensation nuclei.15,16 The formation also has negative health effects by increasing PM2.5 and causing e.g. asthma symptoms and lung cancer when inhaled.17 Alpha-pinene SOA components have recently been demonstrated to potentially cause oxidative stress to lung cells.18 The oxidized forms of polycyclic aromatic hydrocarbon compounds (PAH) are considered even more toxic and bioavailable compared to their original form.19,20
Modern mass spectrometric (MS) methods are the most important analytical methodologies in today's atmospheric studies. There are recent reviews discussing this topic as the technology is constantly developing and providing new information, for example, see ref. 21–23. Here, we discuss oxidized compounds commonly analyzed in the atmosphere or compounds generated in laboratory smog chamber experiments to assess emission sources and factors influencing their formation and occurrence. Several compounds have been monitored actively from the atmosphere for at least two decades,13 but the recently discovered new compounds are also discussed. All the most common analytical methods from sampling to the detection and identification of compounds are presented, assessing their advantages and disadvantages.
2. Oxidized compounds in the atmosphere
Oxidized compounds can be used as markers or tracers of their sources and precursors. The term tracer has a specific definition including being ideally a precursor and source specific, stable, and produced with high yield to be analyzed.24,25 We found only a few publications preferring and justifying the use of the term marker as the criterion for tracers are not fulfilled, especially regarding compound stability and source specificity.13,24 The impact of OA on climate and health is considerable, and the analysis of oxidized marker compounds is important as they provide information about the factors influencing the formation of OA, such as formation mechanisms and the origin of precursors.5,26 The concentrations of the markers vary locally and seasonally, which can be explained by possible different emission rates of precursors and meteorological conditions including e.g., temperature, relative humidity, intensity of solar radiation, and concentration of oxidants.27 In the eastern parts of the Mediterranean, the proportion of SOAs in PM has been between 65 and 78%, and sometimes more than 80%, due to increased concentrations of precursors and ozone in the summer.28,29 In the urban area of Hong Kong, the secondary formation has produced 51.4% of the total organic carbon (TOC) in the atmosphere.12 The secondary formation contributed to PM2.5 only 2.8% in the rural area of Nanjing (China), where emissions of primary compounds had greater contribution including sources such as biomass burning (77.4%), fungal spores (12.2%), plant debris (5.5%), and soil resuspension (2.1%).30 In addition to local sources, long-distance transportation has been assessed to have a significant effect on the prevalence of compounds in the studied areas.3,30,31Here, we present specific oxidation products of the most common biogenic and anthropogenic VOC compounds, as well as other compounds emitted as primary products or less specific products oxidized from various precursors. We are aware that our selected examples provide only a brief overview as there are many publications presenting a more diverse range of different markers. In addition, we introduce highly oxidized organic molecules (HOMs), a recently discovered group of oxidized compounds. We use the term marker to align and simplify the discussion.
2.1. Isoprene markers
Isoprene (2-methyl-1,3-butadiene) is the most common biogenic VOC (BVOC) emitted especially by deciduous trees.32 Marine phytoplankton has also been found to have a possible effect on increasing isoprene emission in the spring when photosynthesis intensifies.3 Isoprene reacts with OH, first forming a peroxy radical derivative, from which the resulting reaction pathways and products depend especially on atmospheric acidity and concentrations of NOx and sulfate.13,33 Ozonolysis also contributes to isoprene oxidation in which typical products are formed, albeit less compared to the OH-initiated reactions.34At low NOx concentrations, a more favored intermediate product is isoprene-epoxydiol (IEPOX), which has an epoxy ring that can react under acidic conditions with nucleophiles (Fig. S1†).10 At higher NOx concentrations, the peroxy radical derivative forms methacrolein that is oxidized further to methacrolein acid epoxide (MAE), which also reacts with nucleophiles under acidic conditions (Fig. S2†).11 Isoprene marker concentrations have generally been found to be the lowest in winter and the highest in the summer when isoprene emission and OH concentrations are at their peak values.12,27,35
The most commonly monitored IEPOX derivatives have been 2-methylerythritol and 2-methylthreitol (known as 2-methyltetrols, 2-ME),5,26,27,34 which are formed at low NOx concentrations by the addition of water with acidic catalysis. Recently,9,36 oxidation products of 2-MEs have been detected and identified, namely 2-methyltartaric acids, 2-methylerythronic acid, 2-methylthreonic acid, and their 3-methyl isomers. These compounds have been oxidized several times, based on which (and other corresponding higher generation oxidation products in general) SOA aging could be expressed.37 Other analyzed IEPOX derivatives, abundant at low NOx concentration, are 2-methyl-2,3,4-trihydroxy-1-butene and cis and trans-2-methyl-1,3,4-trihydroxy-1-butene (so-called C5-alkenetriols), which are formed in the same reaction path with 2-MEs.10,13,27,33 Aerosols containing sulfuric acid yield organosulfate derivatives of 2-ME (2-MEOS), which have been monitored frequently.33,38,39 These derivatives are sensitive and degraded into C5-alkenetriols and 2-ME form upon acid catalysis.13,38 As SOA aging indication, 2-MEOSs can react further by intermolecular rearrangement into cyclic compounds, producing tetrahydrofuran or lactone derivatives.13,40
2-Methylglyceric acid (2-MGA) is also a commonly monitored isoprene marker, the concentration of which typically increases at higher NOx concentrations.10,24,26 Higher relative amounts of 2-MGA to 2-MEs are due to traffic or by wood burning, which is common in the winter season.5,27 It has also been suggested that 2-MGA occurs in winter due to lower temperatures, as the reaction pathway forming 2-MGA is possibly more favored at lower temperatures.12 A reaction path for the formation of 2-MEs at increased NOx concentration has also been presented,41 as 2-MEs have been observed to be abundant in urban areas, despite increased NOx concentrations.12
2.2. Terpene markers
Terpenes are significant BVOCs emitted from vegetation, especially from coniferous trees.27 Alpha and beta-pinene as well as delta-3-carene and limonene are the most common monoterpenes that react with atmospheric oxidants such as ozone, OH, and NO3.42 SOAs derived from terpenes have been determined to be generally more abundant in Europe than in Asia or America, where isoprene had greater contribution to SOAs.24 In Alaska, isoprene has been found to be a more significant source of SOA than monoterpenes, despite the vegetation being mostly boreal coniferous forest.27 Unlike isoprene, no clear seasonal variation has been observed for monoterpene markers generally.24,35 The highest concentrations of markers have usually been observed in the spring,3,27 but increased concentrations have also been observed in the fall due to long-distance transport.3 In addition to monoterpenes, a common sesquiterpene beta-caryophyllene's oxidization marker beta-caryophyllinic acid (Fig. S3†) has been monitored, however, to a lesser extent because beta-caryophyllene is reactive and less abundant in the atmosphere due to its lower volatility.43 Beta-caryophyllinic acid concentrations have typically been highest in the winter, probably due to the release of beta-caryophyllene from the burning of biomass and wood.3,24,27The most common markers of monoterpenes (Fig. S3†) are the first-generation SOA markers cis-pinonic acid and pinic acid, and their second-generation oxidation products such as 3-hydroxyglutaric acid (3-HGA), 3-methylbutane-1,2,3-tricarboxylic acid (3-MBTCA), 3-hydroxy-4,4-dimethylglutaric acid, 3-acetylpentanedioic acid, and 3-isopropylpentanedioic acid.5,12,26,27,30,44 Malic acid has been proposed to be a higher-generation oxidation product of terpenes based on a high correlation with terpene markers in field studies and in chamber experiments.12,45 However, it is not a very specific marker as it can be formed from other precursors from anthropogenic and biogenic sources.45
The first-generation products have been found to form under atmospheric conditions with OH or ozone and at lower NOx concentrations, while an increase in NOx concentrations has yielded more higher-generation products.34,44 In smog chamber experiments by Müller et al.,44cis-pinonic acid was demonstrated to react with OH radicals, as pinic acid was not observed to produce 3-MTBCA. Occurring in the gas phase, cis-pinonic acid was observed to have higher volatility, and its fraction increased at higher chamber temperatures leading to higher 3-MBTCA concentrations. The monoterpene SOA has been found to be less aged mostly in the spring and winter time, as the relative amount of 3-MBTCA compared to the first-generation products has been smaller.27,35 Correspondingly, the concentrations of 3-MBTCA or 3-HGA have been at their highest level in the summer, indicating SOA aging.27,35 The higher ratio of 3-MBTCA to 3-HGA has been applied to assess alpha-pinene being the dominant monoterpene rather than beta-pinene and limonene.26,27 Typical markers to monitor d-limonene SOAs include limonic acid, limonoic acid, and 3-carboxyheptanedioic acid.46–48
2.3. Anthropogenic markers
Ubiquitous anthropogenic VOC and SVOC in the atmosphere include aliphatic hydrocarbons, BTEX compounds (benzene, toluene, ethylbenzene, and xylenes), other alkyl benzenes, and PAHs originating from anthropogenic sources, e.g., vehicle exhaust and biomass burning.49,50 Aromatic compounds and unsaturated hydrocarbons containing a double bond react with ozone and OH.19,34,49–51 Increased concentrations of anthropogenic markers (Fig. S4†) have generally been observed in the winter when biomass burning and other anthropogenic activities are more common.24,31 Increased anthropogenic NOx concentrations have also yielded oxidized nitro compounds which have been monitored and discovered recently.24,35,512,3-Dihydroxy-4-oxopentanoic acid is a commonly analyzed marker, which is formed when benzene and other alkylbenzenes are oxidized photochemically or with ozone, regardless of NOx concentrations.5,24,35,52–54 In general, this marker has been more abundant in urban areas, but increased concentrations have been observed in rural areas possibly due to biomass burning.26 Another frequently analyzed oxidation product of alkylated benzenes has been benzoic acid.24,31,34,55 Phthalic acid and 4-methylphthalic acid have been considered to be oxidation products of naphthalene and its methylated derivatives.5,35,45,54,56 Sato et al.51 recently discovered the formation of 3,5-dinitrosalicylic acid from toluene and 4-nitrophthalic acid from naphthalene. These new products are proposed to be more specific markers.
Anthropogenic aromatic and aliphatic hydrocarbons are typical precursors of dicarboxylic acids, which are very common in the atmosphere.50,57 Cyclic alkenes have been reported to form mostly dicarboxylic acids in ozonolysis, and to a lesser extent hydroxy and oxo acids as well as methyl and ethyl esters.58 The ratio between adipic acid and azelaic acid, which are formed from anthropogenic cyclohexene and biogenic oleic acid, has been applied to determine whether the acid sources are biogenic or anthropogenic.5
In addition to secondary formation, anthropogenic activities have a significant effect on primary emissions. There are numerous studies showing positive correlations between increased anthropogenic sources and marker concentrations. Levoglucosan is a very commonly monitored marker which concentrations have been higher in urban areas and during colder seasons due to the increased biomass burning.27,30,34,35 Terephthalic acid has been monitored as an oxidized marker emitted from burning plastics.35 Cresols and other phenolic compounds are emitted from biomass burning, and they can react with NOx to form methylnitrocatechols and nitroguaiacols, which have been observed at increased concentrations in the winter.24,54 The above-mentioned markers are common in combustion reactions; therefore, such natural processes can also be sources.
2.4. Other oxidized markers
Organic acids may not be precursor-specific since the same acids have been observed to form from different precursors or they have been primarily emitted from anthropogenic and biogenic sources.24,34,59 Many smaller dicarboxylic acids are formed in the oxidation of homologous larger dicarboxylic acids, which is why oxalic acid (C2) is generally the most abundant dicarboxylic acid followed by malonic acid (C3) and succinic acid (C4).60,61 Bikkina et al.62 demonstrated in their recent chamber experiments that the ozonolysis of isoprene yielded a great amount of oxalic acid and smaller amounts of other acids such as malonic acid, succinic acid, and glyoxylic acid. Sato et al.45 recently discovered meso-tartaric acid to be a potential marker for toluene since meso diastereomer occurs rarely in nature. In addition, DL-isomers of tartaric acid, malic acid, succinic acid, fumaric acid, and maleic acid were produced. Saturated and multifunctional dicarboxylic acids have been observed at increased concentrations in the summer time possibly due to their formation mainly by photochemical oxidation.31 On the other hand, increased concentrations of dicarboxylic acids in urban areas have been observed in the autumn and winter because of increased anthropogenic activities and pollution.12The atmosphere has been monitored for carbohydrates which are typically primary oxidized compounds from various sources e.g., biomass burning, plant debris, and fungal spores.30,63 Like levoglucosan, markers such as mannosan and galactosan have been typical markers for biomass burning.35 Increased concentrations of plant and fungal spore-originated markers arabitol, mannitol, and glucose have been observed in PM2.5 due to soil particles released into the air.35 Carbonyl compounds are common in the water-soluble fraction of PM, where they can act as intermediates of oxidation reactions and have harmful effects on health.64 Glyoxal and methylglyoxal are small dicarbonyl compounds frequently analyzed in the atmosphere,65–68 in which they occur as primarily emitted or being formed in oxidation reactions of several different precursors.49,66,69 These compounds can enhance SOA formation by oligomerization or by oxidation reactions yielding organic acids and generating more ozone and OH, increasing the oxidation capacity of the aerosol.65,69,70 Short-chain aldehydes and carbonyls have been analyzed as a result of traffic emissions.71 They have possibly been formed from HOMs decomposing in the particle phase.72
2.5. Highly oxidized organic molecules
Oxidation reactions in the atmosphere yield products that are rich in oxygen relative to the amount of carbon in the molecule. These compounds are called HOM compounds but the exact definition is still pending as this group of compounds is a relatively new discovery.73 Ehn et al.74 presented the term HOM for the first time referring to highly oxidized multifunctional compounds found in the oxidation reaction of alpha-pinene. The compounds were observed to form clusters with nitrate ions, and the oxygen–carbon ratio (O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C) of molecules was determined by high resolution mass spectrometry (HR-MS) to be between 0.7 and 1.3. The analyzed compounds were naturally charged and assumed to contain carbonyl groups and geminal diols, which is why the multifunctionality term was included in the name. The term HOM has also been referred to as highly oxidized organic compounds hereafter, where functional groups themselves are not exactly included in the definition.73 Due to the high degree of oxidation, the compounds have a lowered vapor pressure which is why the group has also been denoted as extremely low-volatility organic compounds, which further emphasizes their importance in the formation of SOAs.72,75
:C) of molecules was determined by high resolution mass spectrometry (HR-MS) to be between 0.7 and 1.3. The analyzed compounds were naturally charged and assumed to contain carbonyl groups and geminal diols, which is why the multifunctionality term was included in the name. The term HOM has also been referred to as highly oxidized organic compounds hereafter, where functional groups themselves are not exactly included in the definition.73 Due to the high degree of oxidation, the compounds have a lowered vapor pressure which is why the group has also been denoted as extremely low-volatility organic compounds, which further emphasizes their importance in the formation of SOAs.72,75
Bianchi et al.73 recently suggested three criteria to define HOMs and to distinguish them from other oxidized species. As the first criterion, HOMs should be formed via an autoxidation reaction mechanism involving peroxy radicals. They preferably possess at least one peroxide group, but this requirement has been relaxed in more recent publications.9,76,77 The second criterion states that HOM formation occurs only under atmospherically relevant conditions. These two criteria exclude oxygen-rich carbohydrates and multifunctional organic acids, which may be primary biogenic compounds formed in the atmosphere along with other reaction pathways or their formation occurs in combustion reactions.44,78 Hydroperoxide groups are thought to be a reason for a high O:C ratio in HOMs,75 and this is proposed as the third criterion for defining HOM compounds. A ratio exceeding 0.7 is considered to be the limit.72 However, Bianchi et al.73 proposed as their third criterion that the HOM molecule should contain six or more oxygen atoms, but in some cases only five. The higher absolute amount of oxygen is justified by the fact that the molecule would be more likely formed by the reaction pathway suggested for HOMs.
The formation of HOMs has been found to be involved in the reaction of peroxy radicals (autoxidation).7,75 This reaction and its mechanism presented in Fig. S5† would explain hydroperoxide and carbonyl functionalities to occur in HOMs and in SOA constituents in general. In the reaction, the OH and molecular oxygen form a peroxy radical, which further undergoes a proton transfer (H-shift) and reacts with oxygen. A new oxidized group is added to the original molecule, which in turn further increases the rate of autoxidation. HOMs have been found to form especially from VOC precursors with a cyclic structure and double bonds, such as monoterpenes and anthropogenic aromatic hydrocarbons.76 Wang et al.77 demonstrated in a recent study that even saturated hydrocarbons are able to form highly oxidized species by autoxidation under atmospheric and combustion engine's conditions. Also, the aldehydes employed as surrogates representing the first-generation oxidation products of alkanes were found to produce high yields of HOMs. This was a logical observation, since the initiation of autoxidation requires a polar functional group in the reacting molecule, as shown in Fig. S5.† Interestingly, the formation was also observed at higher NOx concentrations, where the autoxidation reaction is usually terminated.
The exact structures of HOM compounds are not known so far, but structures have been proposed by applying quantum mechanical computational methods which are not covered in this review.76,77 HOMs are, based on experimental evidence, assumed to contain other functional groups, such as hydroxyl and carbonyl groups, in addition to peroxide groups.72,73,76 Nitrogen-containing compounds are likely to contain organic nitrate functionalities.75,79,80 In sulfate-containing aerosols, HOMs might act as precursors producing highly oxidized organosulfate compounds condensed in the particle phase.72 HOMs occur as monomers or form dimers with their peroxide groups.76 The current criteria previously presented were determined by researchers specializing in HOM research, but due to the novelty of the compound group and the lack of experimental evidence, the criteria on the basis of which the classification has been made must be clearly defined for the time being.73 As an example, Jaoui et al.9 have recently presented that 2-methyltartaric acids formed from isoprene could be classified as HOMs. It was stated that they are formed by a mechanism similar to autoxidation under normal atmospheric conditions, and the amount of oxygen in them fulfilled the presented criteria. Wang et al.77 did not classify the highly oxidized species formed from saturated hydrocarbons as HOMs if they were produced at higher temperatures and/or possessed less than six oxygen atoms.
3. Analytical methods for oxidized compounds in the atmosphere
The most common analytical procedures and instrumental techniques to determine oxidized compounds in aerosol samples are presented in this section. Methods to analyze specific markers by chromatographic methods are introduced first. These offline analytical methods focus on particle phase compounds, in which sampling and sample preparation steps prior to analysis are also introduced. Capillary electromigration techniques are not covered in this review, because they have only been employed for the separation of some organic acids.31,61,81 Other techniques presented include HR-MS techniques to online detect gas phase HOMs and spectrophotometric applications to determine the total amount of oxidized compounds based on their functional groups.3.1. Analysis of markers by chromatographic methods
The analysis of aerosol samples is challenging due to their complexity, which is why gas chromatography (GC) methods have often been applied to the analysis of aerosol samples.82 One crucial requirement is the volatility of the analytes, which does not apply to highly oxidized compounds with reduced vapor pressure.83 Thereby, the compounds cannot be analyzed in their native form, but need to be derivatized to increase volatility. Some typical analyses performed with GC techniques are presented in Table 1.| Analytes | Extraction | Derivatization | GC technique | Concentrations measured (ng m−3) | Ref. |
|---|---|---|---|---|---|
| Organic acids | 3 × 10 min water by UAE. Filtration and pH adjusted with potassium hydroxide to 8.5–9.0 followed by evaporation to almost dryness with rotary vapor | 14% Boron trifluoride in butanol (100 °C, 1 h). Water and ACN were added and derivatives were extracted to n-hexane. Silylation of hydroxy groups with BSTFA (80 °C, 30 min) followed by drying with nitrogen flow and dilution into n-hexane | GC-EI-MS, GC-EI-TOF MS, and GC-FID. DB-5MS columns (30 m × 0.25 mm × 0.25 μm) | Hydroxycarboxylic acids: 0.1–27.3 | 86 |
| Oxaloacetic acids: <0.005–2 | |||||
| Dicarboxylic acids: 5.36–423 | |||||
| Quantitated by FID | |||||
| Carbonyl compounds | Quartz filters were extracted with 1:1 (v/v) ACN-DCM and concentration with rotary evaporator prior to derivatization steps |
Filters impregnated with BHA (gas phase) and quartz filters (particles) were immersed in ACN containing 4% BHA (room temperature, 24 h). Solvent exchange to ethyl acetate with rotary evaporator. The concentration with N2 was performed followed by silylation with BSTFA (60 °C, 1 h) | GC-EI-MS DB5-column (30 m × 0.32 mm × 0.5 μm) | Glycolaldehyde: 0–826 | 66 and 90 |
| Hydroxyacetone: 0–579 | |||||
| Glyoxal: 46–1200 | |||||
| Methylglyoxal: 88–2690 | |||||
| Nonanal: 0–500 | |||||
| Decanal: 0–230 | |||||
| Aged isoprene SOA markers | Tetracosane-d50 and cis-ketopinic acid were spiked on quartz-fiber filters prior to extraction. Sonication in 5 mL of MeOH for 1 h. Evaporation to dryness with nitrogen flow | Silylation in pyridine with BSTFA and 1% TMCS (70 °C, 1 h) | GC-ion trap MS EI and methane-CI RTx-5MS column (60 × 0.25 mm × 0.25 μm) | Methyltartaric acid: 1.52–6.27 | 36 |
| 2-Methylthreonic acid: 0.12–0.69 | |||||
| 3-Methylthreonic acid: 0.04–0.22 | |||||
| 2-Methylglyceric acid: 1.182–4.4 | |||||
| Terpene SOA and isoprene SOA markers | Quartz filter aliquots were extracted with 3 × 7 mL of DCM/MeOH (2:1 v/v) with sonification. Extracts were filtered and evaporated dryness with a rotary evaporator and nitrogen flow |
Silylation in pyridine with BSTFA and 1% TMSC (70 °C, 3 h). Diluted in n-hexane and tridecane was added as ISTD prior to GC analysis | GC-EI-MS DB-5MS column (30 m × 0.25 mm × 0.25 μm) | Monoterpene markers: 0.1–25.3 | 27 |
| Isoprene markers: 0.01–46.6 | |||||
| Beta-caryophyllinic acid: 0.1–4.0 | |||||
| Organic acids, terpene SOA markers, isoprene SOA markers, levoglucosan |
Cis-ketopinic acid was spiked on quartz filters prior to extraction. Filters were extracted with 3 × 30 ml of DCM/MeOH (1:1 v/v) by UAE for 3 × 20 minutes. Extracts were combined, filtered, and evaporated to dryness with a rotary evaporator and nitrogen flow |
Step 1: carbonyls were derivatized by adding a heated solution of MHA in ACN (70 °C, 1 h) | GC-EI-MS HP-5MS column | Monocarboxylic acids: 0.82–130 | 34 |
| Dicarboxylic acids: 0.30–88.2 | |||||
| Step 2: methylation of carboxylic groups by adding TMSD and MeOH (in an ultrasonic bath, 20 min) | Aromatic acids: 0.78–52.0 | ||||
| Hydroxycarboxylic acids 0.39–58.7 | |||||
| Step 3: silylation of hydroxyl groups with BSTFA, 1% TMCS, and pyridine addition (70 °C, 60 min) | Monoterpene markers 0.32–31.2 | ||||
| Isoprene markers 0.53–57.5 | |||||
| Levoglucosan 0.65–21.1 |
Liquid chromatographic (LC) techniques are also common analytical techniques for the determination of SOA markers (Table 2). Compared to GC methods, higher molar mass compounds like humic substances, and larger dimers can be analyzed by LC methods.84 Another advantage is that the decomposition of compounds and the formation of artifacts due to the high temperatures employed in GC can also be prevented. Cui et al.38 demonstrated artifact formation as a result of thermal decomposition of 2-MEOS resulting in up to 188% higher concentrations of 2-MEs by GC than by LC. In addition, C5-alkene triols and 3-methyltetrahydrofuran-3,4-diols were detected in the pure standard of sulfate derivatives when analyzed by GC-MS. These were not observed upon analysis by LC-MS. It is important to consider the thermal stability of compounds as ion sources are using high temperatures to ionize analytes. For example, organic acids can decarboxylate at high temperatures, which might complicate tentative identification or decrease the sensitivity in the targeted analysis. An additional advantage of LC techniques is that by choosing a column and a detector suitable for the analyzed compounds, laborious derivatization steps can be avoided.2,30,85
| Analytes | Extraction and possible derivatization | LC technique and columns | Concentrations measured (ng m3) | Ref. |
|---|---|---|---|---|
| Glyoxal and methyl glyoxal | 30 min UAE with 1:1 (v/v) water and MeOH. μ-SPE device with DNPH-coated C18 adsorbent was set to extracts and vortexed for 3 minutes and then dried. 150 μL of ACN was eluted through the device by centrifuging to perform on-sorbent derivatization and extraction |
LC-DAD (detection at 450 nm) C18-column (150 × 4.6 mm, 5 μm) | Glyoxal: 0.38 | 67 |
| Methylglyoxal: 0.41 | ||||
| 2-MEs and 2-MEOS | UAE with MeOH twice, cooling in between. Filtration and evaporation with nitrogen flow. Extract residues reconstituted to 150 μL ACN/water (95:5 v/v) and diluted by a factor of 20 or 30 |
LC-ESI(−)-Q-TOF MS BEH amide HILIC column (100 × 2.1 mm, 1.7 μm) | 2-MEs: 0.14–0.86 | 38 |
| 2-MEOS: 0.40–2.33 | ||||
| Organic acids and organosulfates | Extraction with 0.25 mL (13 mm quartz filters) or 3 mL (47 mm polycarbonate filters) of water with shaking for 2 h. 2.9 mL of 3 mL extract was diluted with 3.1 mL of water. All samples were filtered (0.2 μm) prior to analysis | IC-ESI(−)-Orbitrap MS AS19-4 μm hydroxide-selective anion-exchange column | Monoacids: 0.8–23.1 | 2 |
| Diacids: 0.1–98.2 | ||||
| Citric acid: 1.8 | ||||
| Terpene-derived acids: 0.2–3.0 | ||||
| Organosulfates: 0.3–17.8 | ||||
| Carbohydrates and monoterpene SOA markers | UAE (in an ice bath) of filters with 3 × 3 mL of MeOH for 15 min. Extracts were filtered (0.22 μm) and evaporated with N2. Reconstitution to 500 μL of 0.01% acetic acid:ACN solution (20:60 v/v to analyze carbohydrates and 80:20 v/v for organic acids) |
LC-ESI(−)-MS/MS | Levoglucosan: 32.27–201.85 | 30 |
| Monoterpene SOA markers: Thermo Hypersil C18 (100 × 2.1 mm, 3 μm) | Sugars: 0.47–2.63 | |||
| Carbohydrates: Prevail column (150 × 4.6 mm, 3 μm) | Sugar alcohols: 0.25–3.22 | |||
| Organic acids: 0.03–2.22 | ||||
| Glyoxal and methyl glyoxal (snow samples) | Spike of 4-fluorobenzaldehyde (ISTD) and BHA solution (derivatization agent) to melted samples. pH adjustment to 3.5 and PDMS coated stir bar was placed for 20 h and 1000 rpm. Oxime derivatives were desorbed by stirring (500 rpm) in 1.5 mL of ACN for 3 h | LC-ESI(+)-MS/MS (ion trap analyzer) | Glyoxal: 0.085–16.3 ng mL−1 | 68 |
| Pursuit XRs C8 column (150 × 2.0 mm, 3 μm) | Methylglyoxal: 0.126–3.6 ng mL−1 | |||
| C1–C10 aldehydes and cyclohexanone | UAE of filters with 2 × 10 mL water followed by dilution to 25 mL. 10 mL of extract were derivatized by adding acidic DNPH solution followed by mixing for 5 min | IT-SPME-cLC-μESI(−)-MS | Formaldehyde (C1): 20–30 | 64 |
| 2 mL of derivatized extract was applied through the IT-SPME column followed by 34 μL injection of water to clean unretained impurities | Zorbax C18-column (150 × 0.5 mm, 5 μm) | Acetaldehyde (C2): 10 | ||
| IT-SPME: TRB-35 GC-column (43 cm × 0.32 mm × 3 μm) integrated to injector | C3–C4 and C6–C8 aldehydes: 1 |
The filter material is typically pure quartz, often baked at a high temperature (400–500 °C) for several hours prior to sampling to remove impurities.30,38,86 There are alternate options such as polycarbonate filters by which Kwiezinski et al.2 obtained better recoveries for small and polar organic acids by water extraction, compared to quartz filters. Filters could be pre-weighed to determine gravimetrically the amount of collected particles after the sampling.34 In this case, it is also possible to determine the amount of TOC and elemental carbon, allowing for the estimation of the markers' proportions in the OA.27 Gas phase collectors typically contain an adsorbent like Amberlite XAD-2 (ref. 87 and 89) or Amberlite XAD-4.36,50
Polar compounds are water-soluble and condense into particles, allowing them to precipitate from the atmosphere into e.g., water or snow. Because of this, oxidized compounds of the atmosphere have been analyzed not only from aerosol samples but also from rainwater, snow samples, and ice core samples.68,90–92 There has been particular interest in snow samples to examine ice nucleation properties and the occurrence of markers in ice.68,91 Despite potential changes in compounds trapped in ice due to cosmic radiation,93 such compounds have been analyzed to provide information about the history of climate change. For example, analysis of ice core samples taken from Alaska has revealed information about the occurrences of isoprene and terpene markers, as well as climate change-related climate oscillations from the 1660s.94 For this, a sample was taken from a depth of 180 m, as from other upper layers depicted different eras.
:1 v/v) was employed for dissolving non-polar (e.g. C8–C10 aldehydes) and polar compounds (e.g. citric acid and tartaric acid). When choosing the extraction solvent, its suitability for the following analysis must also be considered. For example, anhydrides are converted into acids by water extraction, which will increase their concentration.82 Bateman et al.96 demonstrated artifact formation as carbonyl species and organic acids were converted into hemiacetals, acetals, and methyl esters when MeOH was employed in the extraction.
Ultrasound-assisted extractions (UAE) have been very common,34,38,64,86 but mechanical extraction with an orbital shaker or by vortexing has also been employed providing good and quantitative recoveries.2,66 Albinet et al.97 compared MeOH extractions using UAE and vortexing for the assessment of reference concentrations for 12 SOA markers in an urban dust standard reference material (SRM 1649b, NIST). Similar concentrations were obtained with both extraction techniques, and the reference values determined in the study made it possible to evaluate the efficiency of the extractions in a later study.24 Mutzel et al.98 observed the formation of peroxides when samples were extracted with water and employing UAE due to the possible formed reactive radicals. Compared to mechanically extracted samples, reduced concentrations of terpene-derived organic acids were observed in the UAE extracts, which was either caused by reactions with peroxides formed or by the decomposition of acids under ultrasound treatment. Similar formations of reactive radicals can occur not only in water but also in organic solvents and their aqueous mixtures.99
Microextraction techniques have been useful for concentrating the analytes and avoiding the aforementioned methods and their drawbacks. Naing and Lee67 employed a vortex-assisted micro solid phase extraction (VA-μSPE) technique to determine glyoxal and methylglyoxal. The μSPE device was prepared by enclosing the C18 adsorbent in a polypropylene envelope, after which the device was immersed in a 2,4-dinitrophenylhydrazine (DNPH) solution. The device was placed in the aqueous extract from which the compounds were on-sorbent derivatized and extracted simultaneously under stirring. The compounds were detected in the extracts at concentrations of 0.14–0.15 μg L−1 using LC with diode array detection (DAD).
Müller-Tautges et al.68 employed stir bar sorptive extraction (SBSE) to analyze compounds deposited from the atmosphere into snow. The compounds were derivatized to phenyl oximes in melted samples from which derivatives were enriched on a polydimethylsiloxane (PDMS) coated stirring rod. The compounds were desorbed into the solvent prior to LC-MS, providing limits of detection (LOD) of 0.21–0.24 μg L−1 in snow samples.
Prieto-Blanco et al.64 applied in-tube solid phase microextraction (IT-SPME) after the aqueous extraction of aldehydes, and conversion of the aldehydes to DNPH derivatives. In this column switching technique, the extract was applied to a GC column integrated into the injector of an LC. Analytes were concentrated and unretained impurities were washed away, after which the eluent was directed into the capillary to desorb the analytes for subsequent LC analysis. This method using capillary LC-MS demonstrated excellent sensitivity as LODs varied between 0.9 and 2.8 ng L−1. However, the required configuration was rather complex and difficult to implement.
3.1.4.1. Carbonyl compounds. A versatile range of different derivatization reagents for carbonyl compounds are available for analyses employing both GC and LC methods. In GC analyses, multistep derivatization applying silylation is usually initiated first by converting the carbonyl groups.34,86,87,100,101 This avoids the direct reaction of the carbonyl group and the silylation reagent, and the possible enolization of the carbonyl groups, which all could form artifacts that complicate and interfere with the analysis.102,103Fig. 1 demonstrates possible artifact formation between an aldehyde with α-hydrogen and a common silylation reagent N-methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA); the added trimethyl silane group (TMS) could lead to misidentification as hydroxyl or carboxylic species. Oxaloacetic acid is shown to represent keto–enol-tautomerization which has been observed to occur in the atmosphere when analyzed in field samples.86 The enol form produces a conjugated system spanning the whole molecule, which is why this form occurs. An unknown aldehyde in Fig. 1 can also produce such an unpredicted (in this case, unlike oxaloacetic acid) enol form and artifact with MSTFA.103
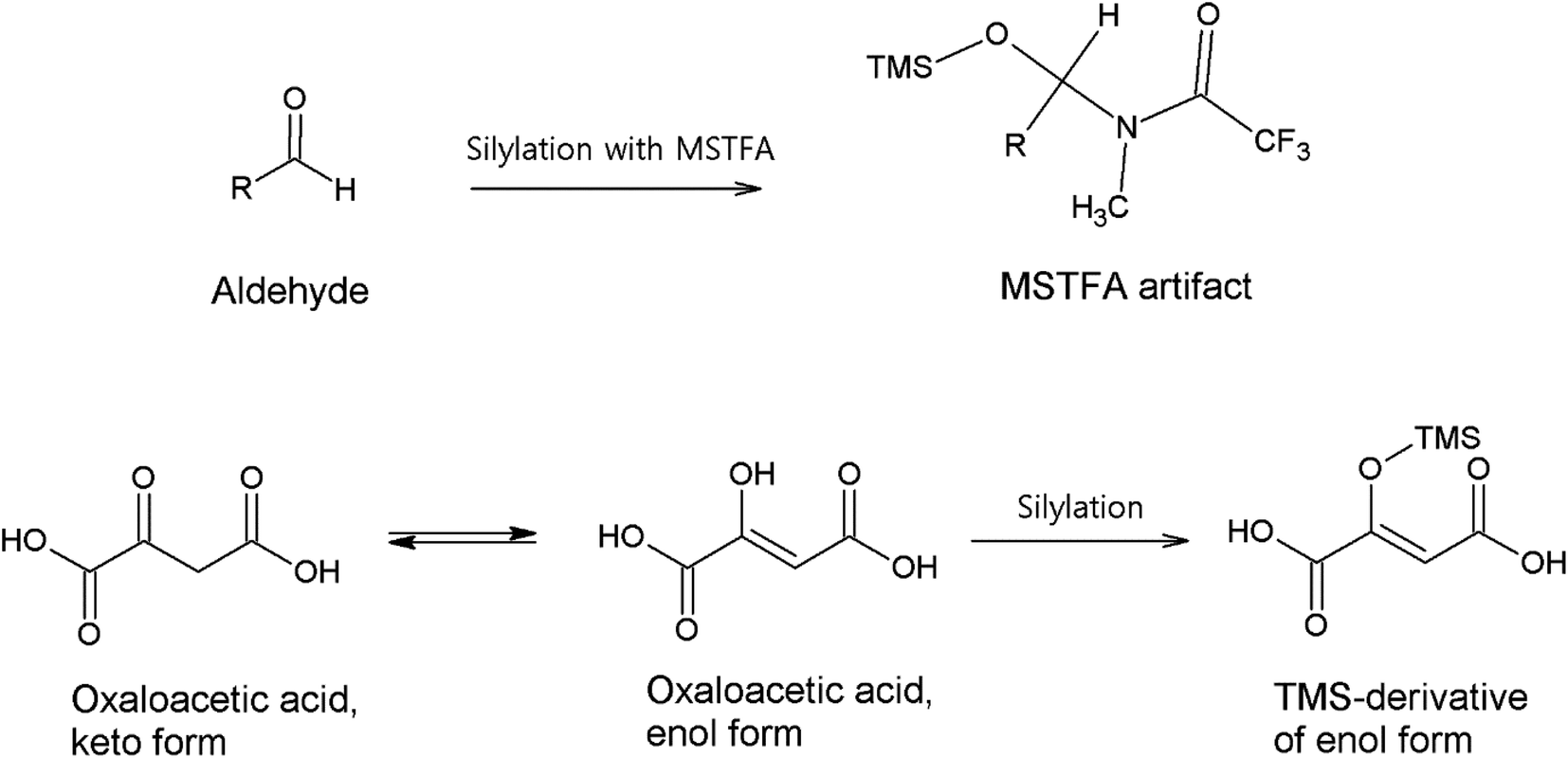 | ||
| Fig. 1 Possible artifact formation between an aldehyde and MSTFA, as well as keto–enol tautomerization of oxaloacetic acid. MSTFA artifact reproduced from ref. 103 with permission from Elsevier, copyright 1999. | ||
Ketones and aldehydes can be specifically converted to oximes, where the oxygen atoms of the group have a hydrophobic substituent depending on the reagent used. Possible reagents are O-methylhydroxylamine (MHA),34,87,89,95,101O-benzylhydroxylamine (BHA),66,68,90O-tert-butylhydroxylamine,104,105 and O-(2,3,4,5,6-pentafluorobenzyl)hydroxylamine (PFBHA).82,106 Oxime derivatives have been mainly applied for GC analyses, but they can also be employed in LC.68 Oxime derivatization can be performed in an organic solvent (MeOH, ACN, DCM, and their mixtures) or in an aqueous matrix.68,82,95,101,104 On-filter derivatization has been applied on filters impregnated with BHA for sampling of gaseous carbonyl compounds in the atmosphere.66 Reactions are slow and a minimum of 20 hours has been required for the reactions to proceed at room temperature.66,68,82 The reactions are even slower with ketones, and to achieve quantitative reaction, heating the reaction mixture at 60 °C (or higher) for two hours using MHA as a reagent has been suggested.95 Quantitative reaction has been achieved by heating at 70 °C for one hour,101 but recoveries have been about 70% or less for carbonyl compounds in field samples.87 Bulky substituent containing oxime derivatives provides more specific fragmentation in mass spectra but the drawback is that they are capable of forming only mono derivatives for alpha, beta-diketones due to the steric hindrance.82,104 Compounds containing more carbonyl groups could be better analyzed by GC-MS applying alkyl oximes, providing smaller derivative molecular masses compared to PFBHA.104
Asymmetric carbonyl compounds yield E and Z isomers of oximes which often appear in chromatograms as multiple peaks possessing similar electron impact (EI) mass spectra.104 An asymmetric ketone containing one carbonyl group has been observed to produce two peaks, of which the first eluting is clearly smaller than the latter, while the case is the opposite considering aldehydes.95 The exact number of carbonyl groups cannot necessarily be deduced from the number of peaks, as two to four different peaks have been observed for dicarbonyls.105 Accurate determination would require complete separation of all isomers. Possible molecular ions appear best in the EI mass spectrum with aromatic analytes or when employing an aromatic oxime reagent, otherwise the spectral identification has been based on ion fragments and ions corresponding to the alkoxy and alkyl groups of the oxime group.90,95,101,104,105 The possible involvement of the nitrogen atom of the oxime group in the analogous McLafferty rearrangement has been proposed to explain other specific peaks in the mass spectrum.95,101 Loss of an NO group [M-30]+ from the molecule derivatized with PFBHA has been observed.82
In addition to oxime derivatives, acetals and ketals have also been formed from carbonyls for GC analyses. The conversion has been executed in n-butanol solution containing 14% of boron trifluoride by heating at 100 °C for 1 hour.62,86,100 This strategy has the advantage that organic acids are simultaneously converted into butyl esters (discussed later). Higher incubation temperature is crucial as no dibutoxy derivatives have been observed to form at 65 °C.102 Dried solvents are recommended to avoid the formation of hemiacetals which produce an extra hydroxy moiety and misidentification of the compound.101 Derivatives produce fragmentations in the EI spectrum corresponding to the cleavage of butene and the butoxy group.100
DNPH mentioned earlier is a common derivatization reagent specific for carbonyls. The hydrazone derivative formed by DNPH is generally separated with LC and detected by UV at 360 nm or by MS with electrospray ionization (ESI) at negative polarity.64,67,83 Analysis with GC is possible, but it would require optimization of temperatures to prevent thermal degradation and the isolation of derivatives from the reaction mixture containing the acid and unreacted reagent with e.g., cation exchange SPE.71,107 Under strongly basic conditions, the hydrazone derivative forms a quinoid structure with strong absorption at 480 nm.83,108 Conditions have been assessed to be established only by the addition of a strong base (pH over 12), which is why this has not been applicable in LC techniques restricted by the durability of the column materials.83 Aimanant and Ziemann108 applied this higher pH in a spectrophotometric method (discussed later) to determine the total concentration of carbonyl compounds obtained in a chamber experiment after ozonolysis of alpha-pinene. The reaction with DNPH could be carried out like for oxime derivatization in an aqueous matrix in which the reaction could be completed in just a few minutes at room temperature.64,67 Compared to the oxime reaction, it is very fast, which is why DNPH derivatization should be considered for LC analyses. Girard's reagent T (trimethyl amino acetohydrazide chloride) is also a possible alternative to convert carbonyls into hydrazone derivatives. By introducing this functional group into the molecule, detection is possible either by UV absorption or by MS employing positive polarity.46,109
3.1.4.2. Carboxylic acids. Alkylation of carboxylic acids (i.e. esterification) is a common derivatization method for GC analysis. Without using highly toxic reagents such as diazomethane,95 esterification is often done by acid catalysis of samples dissolved in an alcohol. The most common acid is boron trifluoride, with MeOH or butanol as alkylating alcohols.34,86,87,100,101 Boron trichloride is a very similar catalyst that can be applied as an alternative.102 Silylation (discussed later) has also been applied to derivatize carboxyl groups, but esterification has been preferred in compound identification to distinguish between carboxyl and hydroxyl groups.86,87,95,100,101 For the targeted analysis, organic acids have been silylated reducing the sample pretreatment steps.9,27,36 These derivatives produce larger peaks compared to methyl esters, which enhances the sensitivity of the method.95 In addition to improving the sensitivity, silylation has been shown to be more reproducible than esterification.110 This derivatization strategy may not be recommended for small acids, such as oxalic acid, as sample loss may occur in the analysis when the resulting derivatives are highly volatile.45
Butanol has been more popular in acid-catalyzed esterification reactions because butyl esters are less volatile. The methyl esters of smaller acids, such as glycolic acid and pyruvic acid, evaporate easily after esterification and in the following extractions.102 Oxalic acid converted to dibutyl esters have been determined in yields of 75% or better62,86 and they separate better from the solvent peak.101 Quantitative esterification in butanol containing 14% of boron trifluoride has typically been achieved by heating at 100 °C for 30–60 minutes.62,86,100,111 After the reaction, the mixture is neutralized by adding a solvent mixture containing water, the butyl esters are extracted into n-hexane, followed by drying. Extracts are analyzed by GC or silylation is performed first to derivatize the remaining free hydroxyl groups. Esterification alone has been found to be inadequate for acids with two or more hydroxyl groups due to their high polarity.101,102
As an alternative method, trimethylsilyl diazomethane (TMSD) has been applied to methylate acids in the presence of MeOH.34,87,89,95,101 The reagent acts as a silylating agent if the amount of MeOH used in the reaction is too low.95 The reaction has been carried out after the addition of the reagents in an ultrasonic bath, where a 20-minute hold was sufficient. The advantage of using TMSD is that it does not require neutralization, thereby eliminating the need for extraction and possible evaporation of methyl esters for the next derivatization step, clearly reducing the risk of sample loss.
Ion fragmentation of the esters' alkoxy groups ([M-31]+ and [M-73]+) and corresponding alcohols from the molecular ion have been typically observed in EI-spectra.95,100,102 The ions formed in these processes possibly fragment further with the loss of carbon monoxide [−28]. Possible cleavage of butene [−56] by McLafferty rearrangement has been evaluated with butyl esters.100 Another typical cleavage with esters is alpha cleavage ([M-59]+ and [M-101]+) and beta cleavage ([M-73]+ and [M-115]+). Gowda et al.100 identified 3-hydroxyglutaric acid and 3-hydroxyadipic acid by beta cleavage, which distinguished acids from their 2-hydroxy isomers. The 74 m/z ion characteristic of long-chain methyl esters of mono- and dicarboxylic acids is formed by the McLafferty rearrangement.95 With methyl esters, in addition to the protonated molecular ion, the formation of a corresponding dimer has been observed using chemical ionization (CI) with methane.102
3.1.4.3. Hydroxyl compounds. Silylation is a derivatization method in which the active hydrogen attached to an electronegative atom is typically replaced with a TMS group. As mentioned in the section discussing the derivatization of carboxylic acids, hydroxyl groups have been derivatized by silylation when oxidized species have been analyzed in aerosol samples by GC methods. 4-(Dimethylamino)benzoyl chloride112 and 4-nitrobenzoyl chloride (4-NBC)83 have been proposed as alternatives to derivatize hydroxyl moieties in SOAs for LC separations. However, according to our knowledge, these have not (yet) been employed in real field studies.
Various silylation reagents are available and among these, N,O-bis(trimethylsilyl) trifluoroacetamide (BSTFA) is a powerful reagent that reacts with many types of hydroxyl groups.113 BSTFA has been the most common reagent for silylation with 1% of trimethylchlorosilane (TMCS) added as a catalyst.5,27,36,66,86,87,101 It is important to note that BSTFA is not capable of converting sterically hindered tertiary hydroxyl groups in, e.g., citric acid and tertiary alcohols.95 Another employed reagent is MSTFA,24,113 which is similar to BSTFA in its silylation properties.95 The use of N,O-bis(trimethylsilyl)acetamide has been avoided due to its tendency to easily produce undesired artifacts.95,103
Silylation requires careful optimization of several factors including the properties and amount of the reagents and solvents, temperatures, and reaction times.95,103 The derivatives react with water and protic polar solvents, which is why drying and solvent exchange is always performed prior to derivatization. The derivatization involves evaporation of the sample extracts to dryness, followed by the addition of a silylation reagent and pyridine as catalyst.9,34,82 The reaction has been accelerated by heating at 70–80 °C for 30–60 minutes.27,36,82,86 Silylation is possible to perform in the gas phase, which could occur during the injection into a hot GC injector.103
In EI spectra, the silylated compound generally has a high abundance of ion m/z 73, corresponding to the cleaved TMS group.90,101 Ion m/z 75 is also observed presenting the cleaved trimethyl siloxane group (TMSO) ion lacking a methyl group. If the molecular ion is not present, the molecular weight could possibly be assessed from the largest ion presenting the derivatized molecule that has lost a methyl from the TMS group [M-15]+.100 In addition, the corresponding molecular ion [M-89]+ can be observed resulting from the cleavage of the entire TMSO group.82 The ion m/z 147 provides an indication of two silylated groups, which has been observed for C2–C6 dicarboxylic acids and multifunctional acids such as tartaric acid.82,95,100,111 The ion is assumed to be formed when the TMS groups are in close proximity to the molecule.95 A fragment ion m/z 103 is a possible indication of a primary alcohol due to the loss of TMSO and a methylene group.90,95 If carboxylic acids were silylated, a fragment presenting alpha cleavage [M-117]+ can distinguish alcohols from acids.36
In LC methods, it is possible to employ a broad range of columns according to the analytes' properties. In reversed-phase liquid chromatography (RPLC), the most common column material is octadecyl (C18) or octyl silane for the separation of especially terpene-derived acids, their dimers, and DNPH-derived carbonyl compounds.30,67,68,114 A column containing phenyl groups has been applied for the analysis of aromatic compounds, such as nitroaromatic compounds.51 Hydrophilic interaction liquid chromatography (HILIC) columns have proven useful for more polar compounds such as saccharides, organosulfates, and organic acids including terpene derivatives, dicarboxylic acids and smaller multifunctional acids.39,63,85 Isomers of 2-MEs and 2-MEOS have been separated particularly well with HILIC, whereas separation was not possible with RPLC.38 The smallest and highly polar organic acids, on the other hand, have been analyzed by ion chromatography (IC). Kwiezinski et al.2 employed an IC-MS method employing an anion exchange column to determine oxalic acid, glycolic acid, malic acid, and citric acid with LODs of 2.5–10 μg L−1 and more hydrophobic acids with five and ten times lower detection limits. Carbohydrates, 2-MEs and other sugar alcohols have been analyzed by high performance anion exchange chromatography (HPAEC).115,116
In GC, EI is the most common ionization technique that provides information about the structure of compounds through characteristic ion patterns.27,36,86,100,101 The advantage of the softer CI is that the molecular ions are better visible in the spectra which helps to identify the compounds better.36,102 The most common ionization technique for LC has been ESI, which has been applied mostly in negative mode to detect non-derivatized sugars, organic acids, and organosulfates separated with RPLC, HILIC, and IC.2,30,38,64 Positive mode ESI is applicable for detection of carbohydrates.30 Compared to other LC works, the IC-ESI-MS configuration employed by Kwiezinski et al.2 required a separate suppressor and post-column addition of iso-propanol to avoid non-volatile salt stacking in the ion source and to increase the sensitivity, as the eluent was an aqueous solution of potassium hydroxide. With CI employing ammonia and isobutane, it has been possible to detect DNPH derivatives of carbonyls, hydroxyl group compounds derived with 4-nitrobenzonyl chloride, and organic acids derived with 2-nitrophenylhydrazine hydrochloride using positive polarity.83
A simple quadrupole mass analyzer has been commonly employed in low resolution mass spectrometers.27,64,66,86 An ion trap has also been employed as another low resolution mass analyzer.9,36,68 For accurate identifications of compounds, HR-MS is required to determine the elemental composition or to provide more selective detection of analytes. For those purposes, the most commonly employed HR-MS analyzer has been Orbitrap2,36 or time of flight (TOF) which has been employed as alone or in combination with a quadrupole (Q-TOF).38,86,87,100,101 This tandem mass spectrometry (MS/MS) has also been established by coupling three quadrupoles.30 MS/MS has also been referred for a single ion-trap analyzer that collides isolated ions with, e.g., helium to provide collisional induced dissociation and multiple reaction monitoring.68 In addition to quantitative analysis, MS/MS has been employed to identify compounds by applying neutral loss mode. For example, loss of water [M-18] and decarboxylation [M-44] have been monitored to identify carboxylic acids tentatively.117,118 With the same principle, other compounds with certain functional groups have been identified, such as organic hydroperoxides and peroxy acids, which could be identified by their characteristic loss of hydrogen peroxide [M-34].119
Detectors other than MS have been employed but to a lesser extent. Flame ionization detection (FID) in combination with GC has been employed for quantification of compounds when the qualitative information has been acquired by MS.86,100 FID is sensitive and has provided LODs of 5 pg m−3 for multifunctional organic acids using a high sample volume exceeding 7000 m3.86 A pulsed amperometric detector has been employed in HPAEC to detect carbohydrates and 2-MEs at low LOD of 0.05 ng m−3.115,116 Ultraviolet-visible (UV-VIS) has been employed for more selective detection of DNPH-derivatized carbonyl compounds in LC.67 The non-derivatized carbonyl or carboxyl group also acts as a chromophore, in which case UV detection can be applied also for compounds in their native form.61 However, UV detection is not so sensitive compared to MS detection, and, e.g., Prieto-Blanco et al.64 obtained 30–100 times lower detection limits for DNPH-derived aldehydes compared to those obtained by IT-SPME-cLC-diode array detection (LODs ranging from 30 to 198 ng L−1).120
| Analytes | Applied surrogate | Ref. |
|---|---|---|
| Methyltheronic acids | Citramalic acid | 36 |
| 2-Methyltartaric acids + 3-hydroxyglutaric acid | Tartaric acid | |
| 2-Methylglyceric acid | Pentanedioic acid | |
| 3-Carboxyheptanedioic acid | d-Arabitol | |
| 2-MEs | d-Threitol | |
| Aliphatic monocarboxylic acids | Dodecanoic acid | 87 and 101 |
| Aliphatic dicarboxylic acids | Glutaric acid, adipic acid, azelaic acid | |
| Ketocarboxylic acids | Cis-pinonic acid | |
| Aromatic carboxylic acids | p-Toluic acid | |
| Carbonyls | Nopinone | |
| Alcohols and phenols | Hexadecanol and p-hydroxybenzoic acid | |
| Carbohydrates and glycolics (polyols) | Levoglucosan and meso-erythritol | |
| Hydroxycarboxylic acids | Malic acid | |
| 2-MGA, 2-MEs, C5-alkene triols | meso-Erythritol | 27 |
| 3-MBTCA, beta-caryophyllinic acid | Pinic acid |
3.2. Ion mobility mass spectrometry
Ion mobility spectrometry (IMS) is an alternative separation technique integrated with MS for the analyses of atmospheric compounds. The separation efficiency with IMS is not that great as compared to chromatography, but the selectivity of the ionization technique used can be of great help for complex samples. Generally, analytes are ionized and separated by the electric field and opposing drift gas flow based on their ion mobility, which is highly dependent on the structure of the ion (mass, charge, and collision cross section) and its interactions with the added drift gas. In recent publications, the method has especially been applied to the separation of isomeric compounds. The methodology is applicable to online analyses of gaseous compounds and aerosol samples extracted into suitable solvents. Analysis of aerosol extracts without chromatographic separation can be challenging as other ions contained in the matrix may cause ion suppression and interfere with the separation by forming additional clusters.122–124The early application of IMS-MS in atmospheric research was reported by Krechmer et al.122 for the online analysis of a nitrate-rich gas phase with CI-IMS-TOF. The field measurements provided detection of BVOC oxidation products, agreeing well with previous results obtained with nitrate CI-MS methods (which will be discussed later in the section regarding HOM detection). The evaluation was focused on monoterpene-derived HOMs, of which monomeric species were efficiently detected. However, larger dimers were not detected, most probably due to poor ion transmission and sample losses due to interactions with the walls of the instrument. To analyze 2-MEOS isomers, aerosol phase samples were extracted and analyzed by ESI. Isomers of 2-MEOS were separated providing information on isomer occurrence, which depended on the isomeric form of the IEPOX intermediate reacting with sulfuric acid. The separation efficiency was observed to decrease, probably due to the extracted sulfuric acid which formed clusters with the analytes in IMS.
West et al.123 employed ESI-IMS-Q-TOF to separate alpha-pinene and limonene SOA oxidation products possessing similar mass spectra. Alpha-pinene products possessing cyclobutane rings were successfully separated from isomeric noncyclic limonene products. The collision cross sections in the employed nitrogen gas were demonstrated to be dependent on the ionization mode, showing the formation of positive sodium adducts being favorable for monomeric products and negative deprotonation for dimeric and trimeric products. Iinuma et al.125 characterized alpha-pinene-derived dimer esters by LC-ESI-IMS-TOF/TOF employing collision-induced dissociation to identify their acid components. Isomeric cis-norpinic acid and terpenylic acid did not separate in IMS using nitrogen as the buffer gas; however, the separation was successful switching into more polar carbon dioxide. Zhang et al.126 studied alkyl nitrates using ESI, with a focus on the optimal addition of anions to the extracts for adduct formation. Chloride and nitrate were found to form clusters most efficiently. For identification, a specific nitrate fragment was determined by collision-induced dissociation analysis, regardless of the adduct used. The correlation of the calculated collision cross section and the mass-charge ratio was also applied to the identification, as the ratio was found to behave differently compared to organic acids or organosulfate compounds.
3.3. Mass spectrometric detection of highly oxidized organic molecules
Oxidized species have been analyzed by MS without separation prior to detection. The analysis in this case is typically for compounds present in the gas phase, but also extracts of samples taken on filters can be analyzed by MS alone. No separate sampling is required for the gas phase compounds, so the measurements have been done online in real time. Emphasis has been on the examination of HOM formation in chamber experiments or their occurrence in field samples. Selected studies focusing on HOM compounds and other oxidized species are presented in Table 4.| Detected compounds | Information about study | Technique | Ref. |
|---|---|---|---|
| a Wang et al.77 do not classify compounds as HOMs if possessing less than six oxygen atoms and/or formed under conditions representing a combustion engine. b Monomers and dimers are included. | |||
| C8–10H12–18O4–9 (monomers) | Alpha-pinene-derived HOMs generated in chamber experiments were analyzed in both gas (online) and particle (offline) phase | Online: CIMS with iodide ionization | 133 |
| C16–20H24–36O8–14 (dimers) | Offline: ESI-IMS-TOF with sodium adducts [M + Na]+ | ||
| C8–10H12–16O6–11 (gas and particle phase) | Alpha-pinene ozonolysis chamber experiments and field measurements in rural areas surrounded by forest and agricultural pasture dominated by alpha-pinene emissions. The study involved the detection of HOMs in both phases including discoveries of highly oxidized organosulfate compounds and carbonyl functionalities in HOMs after DNPH derivatization | CI-APi-TOF with NO3− ionization | 72 |
| C7–10H8–14O7–11S (particle phase) | Particle phase samples were extracted and analyzed directly or with DNPH derivatization by LC-(−)ESI-TOF or UHPLC-(−)IMS-QTOF technique | ||
| C3–7H5–9O6–7N0–1 (lighter HOMs) | Field measurements in boreal forest area where monoterpenes were dominating BVOCs. HOMs formation and occurrence in daytime and night-time with different oxidants presents was assessed | CI-APi-TOF with NO3− ionization | 79 |
| C9–10H14–16O7–13N0–2 (monomers) | |||
| C16–20H28–32O9–19N0–2 (dimers) | |||
| C5–20H6–30O4–18 (monoterpenes)b | Field measurements in forest area providing possible markers for isoprene and monoterpene | CI-APi-TOF with NO3− ionization | 80 |
| C3–20H5–31N1–2O4–14 (monoterpenes)b | |||
| C4–15H6–20O5–22 (isoprene)b | |||
| C4–15H6–11N1–3O5–11 (isoprene)b | |||
| C6–20H11–38O4–14b | Oxidized carbonyl species, peroxy radicals and NO2 species produced from alkanes, alcohols, and carbonyls in chamber experiments representing conditions in the atmosphere and combustion engine | CI-APi-TOF with NO3− and ethylamine ionization | 77 |
| C10H21O1–4NO2 | |||
| C10H17O1–8NO2 | |||
| C6H11O2–5NO2 | |||
For the time being, HOM analytics mainly rely on MS methods where they are analyzed from the gas phase since the reactivity of their supposed peroxide groups makes them unstable and causes analytical problems.73,76 Peroxide compounds are challenging as they may degrade in SOAs within a few hours.127 Analytics are also limited because the exact structures of HOM compounds are still unknown.76 Mutzel et al.72 have shown by LC-MS, employing DNPH derivatization, that HOMs found in the gas phase occurred also in the particle phase, and they contained carbonyl groups. Zhao et al.128 applied iodometry (discussed later) to LC-ESI-MS, in which samples were qualitatively analyzed for peroxide functionalities containing species before and after iodometry. The spectra obtained were compared, in which the peaks of certain compounds had disappeared after treatment, indicating that the compound had a peroxide group. Peroxide compounds have also been successfully analyzed from the ozonolysis-generated alpha-pinene SOAs using LC, where the separated peroxides dimerized p-hydroxyphenylacetic acid in post-column derivatization followed by detection with a fluorescence detector.129,130 Despite successful peroxide compound detection with LC techniques, the use of chromatographic methods for HOM analyses is challenging, because HOMs are most likely too polar to be all separated and their original structures could change during the separation process by reacting with column materials.76
There are differences in the configurations of ion sources when employing different reagent gases, as presented in Fig. 2. The ion source of the CI-APi-TOF instrumentation is typically the so-called Eisele type ion source, which is practically a flow tube model source where, in addition to the sample, pure air and reagent gas are supplied to the source.73,134 Reactive gas species are formed from the reagent gas with the aid of a corona needle or a radiation source, such as an alpha-emitting Am-241 or an X-ray source.135–137 Voltages are applied to direct ionized analytes towards the mass analyzer system. In the design of ion source, it has been important to pay particular attention to sample losses of oxidized compounds due to the adsorption on the walls and inlets as compounds have lowered vapor pressures. To eliminate this, high sample flows are applied in the ion source and the structure of the ion source is designed to practically have no walls.75 The configuration employing iodine or acetate, in turn, has an ion-molecule reaction chamber (IMR) as an ion source, in which case instruments have been called CI-TOFMS or TOF-CIMS instead of CI-APi-TOF.138,139 This source type also has some radioactive material to generate the reagent gas, but unlike the Eisele type ion source, a reduced pressure is applied in the ionization chamber and the supply of reagent gas to the chamber is orthogonal to the sample flow.
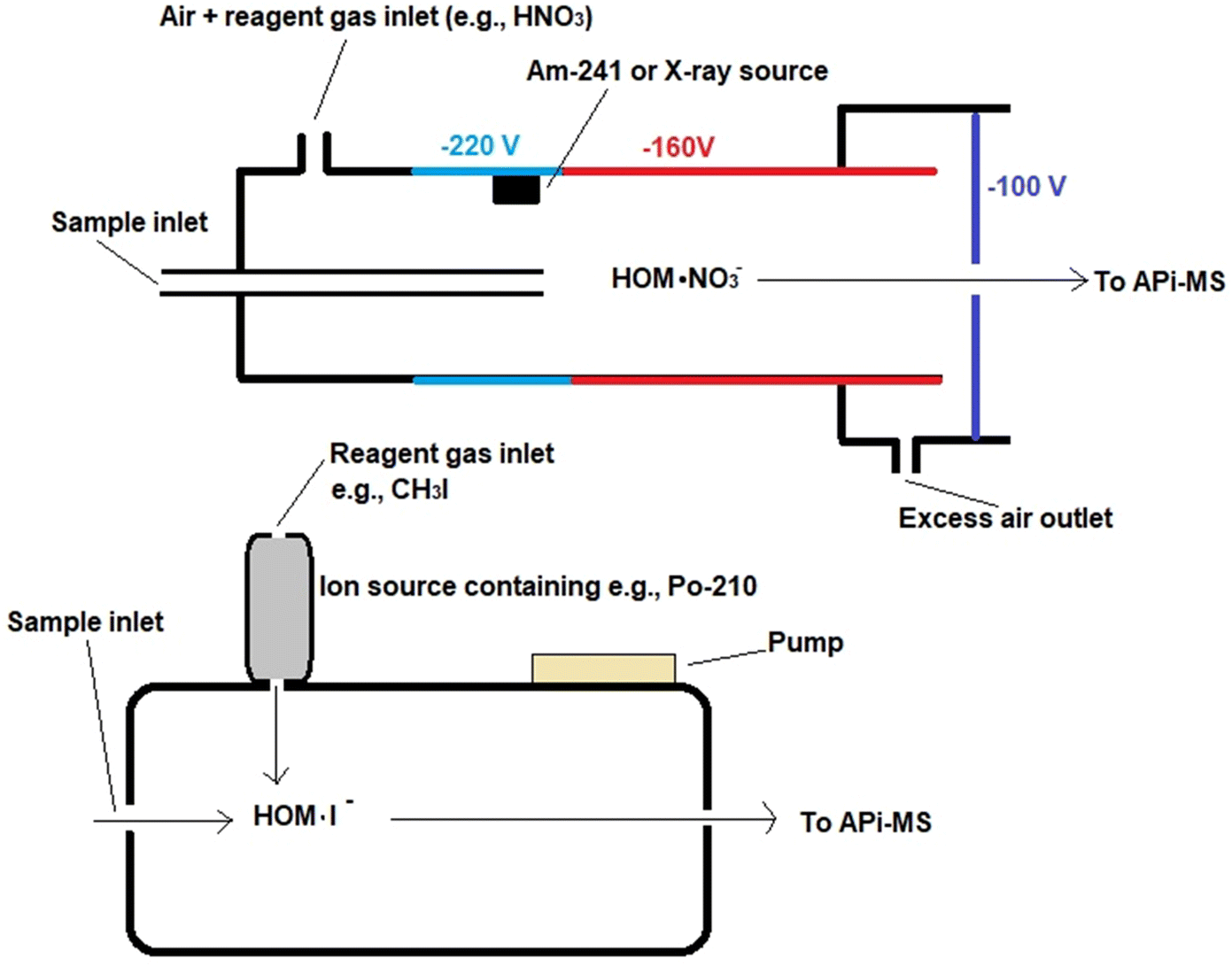 | ||
| Fig. 2 Schematic structure of an Eisele-type ion source, reproduced from ref. 137 with permission from National Academy of Sciences, copyright 2014. An IMR ion source, reproduced from ref. 139 with permission from Copernicus Publications, copyright 2011. | ||
Proton transfer reaction mass spectrometry (PTR-MS), which can be classified as a CI technique, is optimal only for volatile compounds, as found by Riva et al.,138 who compared five different CI techniques to detect about a thousand compounds formed in the ozonolysis of alpha-pinene. The comparison also included the so-called Vocus instrument, which has a discharge reagent-ion source and a focusing ion-molecule reactor compared to the traditional PTR-MS configuration.140 The Vocus instrument has faster response times which enables faster analyses, decreasing the loss of SVOC on the walls and inlets. This was verified as the Vocus instrument was able to detect SVOC with an oxygen content ranging from three to five with good agreement with the iodine-CI technique included in the comparison. Li et al.141 found that the Vocus instrument is still poorly suited for the detection of peroxide compounds, most likely due to the fragmentation of peroxides in the PTR process.
In APi-TOF, the ionized species entering the APi interface are directed by quadrupoles and ionic lens systems before entering the mass analyzer. TOF is clearly the most commonly used mass analyzer for studies of HOMs.72,79,80,133 As this analyzer can provide high mass resolution, exact elemental compositions of HOMs can be assessed and compounds distinguished from each other without any separation technique. Riva et al.142 compared CI-Orbitrap with CI-APi-TOF employing acetate and n-propylamine as reagent gases in both systems to detect peroxide radicals. Both mass analyzers gave results with good agreement regardless of the reagent gas, but the responses in the Orbitrap were observed to have poor linearity at the lowest concentrations.
| HOM + (HNO3)X × NO−3 → HOM × NO−3 + (HNO3)X | (1) |
The iodide adducts have also been studied, as iodide has been found to ionize easily oxidized and acidic compounds.144 The IMR source typically utilizes methyl iodide and a Po-210 alpha radiation source to form a reactive gas cluster I(H2O)− with water.138,144 One of the benefits using iodide adducts is the dehydroxylation of hydroperoxides observed in the mass spectra, which could be used as a qualitative tool for identifying the compounds.145 The iodide CI methodology is optimal for less oxidized semi-volatile organic compounds (SVOC), possessing oxygen atoms from three to five138 or having the O:C ratio of about 0.6.145
In addition to inorganic anions, adducts have also been formed with organic anions, such as acetate, pyruvate, and lactate.146 The Eisele-type ion source employing corresponding acids as dopant gases have been employed, but the IMR ion source has also been applied to acetate.139 Of the above anions, acetate has been the most commonly used in the studies of oxidized species. For example, Berndt et al.147 compared nitrate and acetate ionization to study HOM compounds formed in the ozonolysis of cycloalkenes. Acetate was found to be more favorable for the analysis of compounds containing one hydroperoxide group as systematically lower concentrations were observed using nitrate. The study also demonstrated acetate's ability to form detectable ions by deprotonating analytes and forming adducts with them. Because of those two possible ionization mechanisms, acetate is not favorable for qualitative analysis as the interpretation of the mass spectrum becomes more complex.148,149
The above-mentioned adducts have been detected with negative polarity, but oxidized compounds have also been analyzed with positive polarity. Methyl-, ethyl-, and n-propyl-amine as well as hydrazine have been employed to detect molecules containing peroxide radicals that form clusters with aminium and hydrazinium cations.150 Riva et al.138 compared different CI techniques and found that butylamine gave poorly correlated results to other CIMS techniques, which is thought to be due to reagent gas depletion, leading to non-linear responses. In addition to organic cationic adducts, the sodium adduct has been found to be particularly well suited for ionizing polyolic HOMs and those containing organic peroxides and ester groups.133
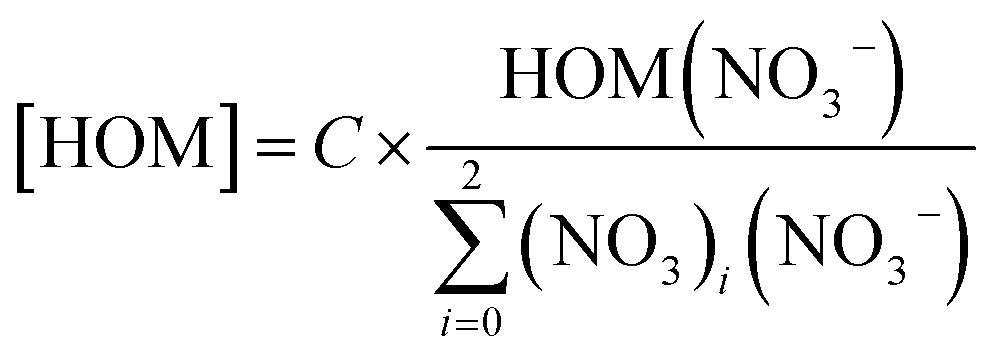 | (2) |
3.4. Spectrophotometric applications
Spectrophotometry has been introduced as an analytical method to study SOA constituents. Compared to chromatographic and MS methods, spectrophotometric methods are far less popular, mainly due to the lower sensitivity.108 Methods applied for SOA research provide only total concentrations of functional groups, not individual marker compounds. Selective functional group reactions have been applicable to LC methods, providing possible new alternatives for the derivatization reactions introduced above.83 In addition to these methods, Fourier-transform infrared spectroscopy (FTIR) has also provided similar qualitative information about functional group compositions.151–155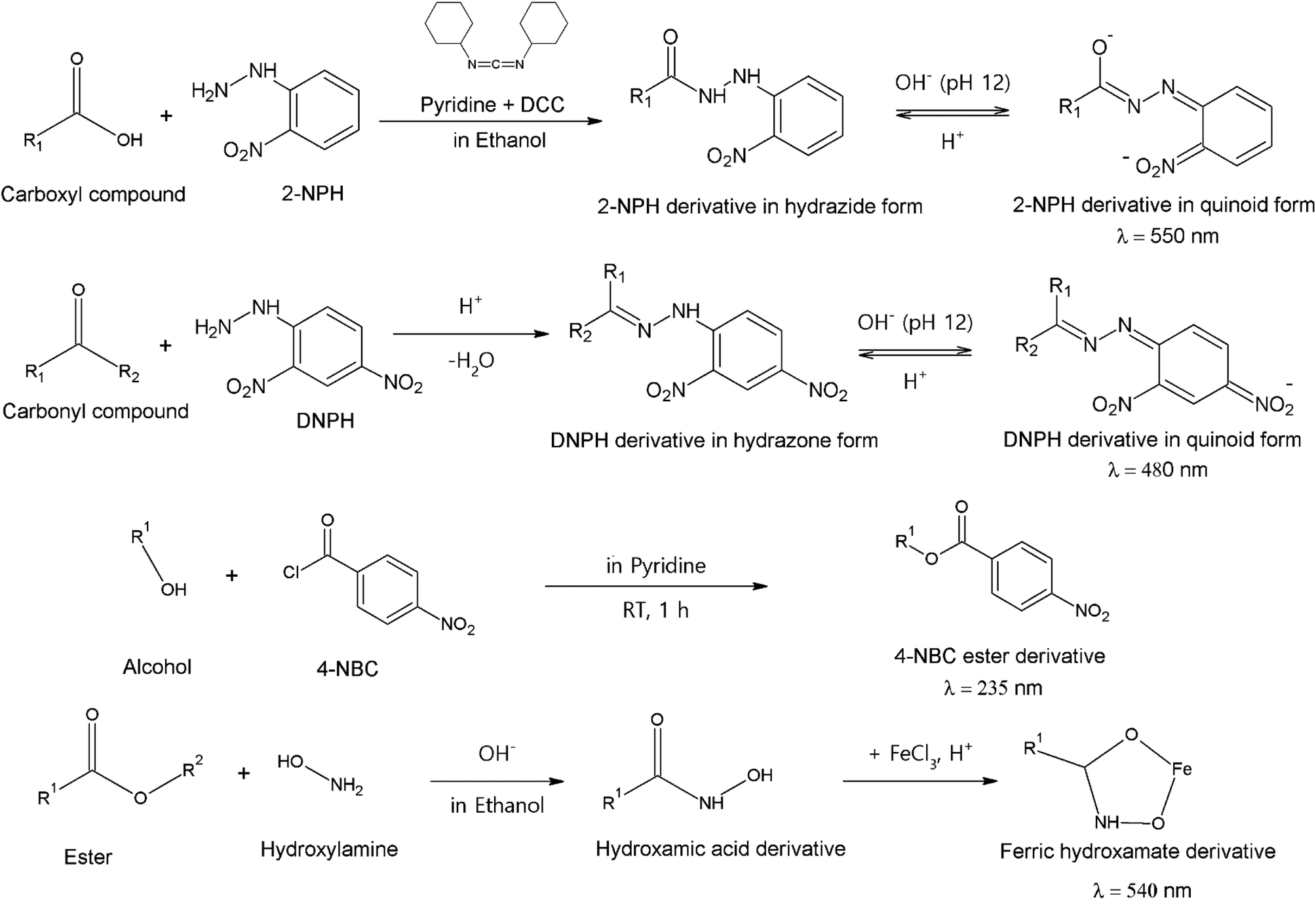 | ||
| Fig. 3 Functional group-specific reactions and measurement wavelengths (λ) applied in spectrophotometric application. Reproduced from ref. 108 with permission from Taylor & Francis Group, copyright 2013. | ||
This spectrophotometric method has also been applied in later studies analyzing functional group compositions obtained in chamber experiments.156,157 Recently, Claflin et al.152 have also applied this for real field samples. In that study, organonitrates were determined spectrophotometrically by measuring samples directly at 210 nm. The study also involved a comparison with the FTIR method in which functional groups were specifically detected from dried filters. The results provided justifications to determine the total concentrations of functional groups, as their abundance could indicate the origin of formed SOAs including precursors and oxidants.
Chamber test samples demonstrated the appearance of nitrates when SOAs were generated in the presence of NOx or NO3. Peroxide and methylene fractions increased as carboxylic, carbonyl, and ester species decreased when alpha-pinene oxidation involved OH instead of ozone. In reactions involving OH and NOx, the ratio of methylene to nitrate of 1 indicated isoprene of being a precursor and the ratio of 2 suggested that monoterpene is the precursor. Based on observations from chamber experiments, monoterpenes and possibly isoprene were suggested precursors in field samples producing SOAs in the presence of OH and ozone at low NOx concentration conditions. FTIR and spectrophotometry were found to provide comparable results when analyzing organonitrates, hydroxyl groups, and non-acid carbonyl groups. FTIR provided simpler and faster analyses, but spectral interferences and overlaps were the drawbacks encountered.
Iodometry has been applied to peroxide using long path absorption photometry (LOPAP), which is a spectrophotometric technique employing iodometry introduced by Mertes et al.159 The instrumentation is closed and pure nitrogen (5.0) is fed into the reaction chambers to eliminate the interference of oxygen. The pump inside dispenses solvents (ethanol and 0.1 M HCl in this application) into the reaction chamber, where the filter sample and solid potassium iodide have been added. After the reaction phase, the pump dispenses the sample through the filter into the long path detection chamber, called the liquid core waveguide (LCW). This is a thin capillary made of Teflon material with a refractive index lower than that of water and ethanol, which enables the light produced by the deuterium lamp to propagate in the capillary with total reflection. The absorbance is measured with a separate spectrophotometer integrated into the LOPAP instrument.
Mertes et al.159 used a 50 cm long LCW achieving a better sensitivity according to the Lambert–Beer law. The method was assessed to be suitable for peroxides poorly soluble in water and ethanol, as the absorbances produced by benzoyl peroxide and lauroyl peroxide were assessed to be practically the same as the absorbances produced by water soluble hydrogen peroxide and peracetic acid. Analyzed smog chamber samples revealed the mass fraction of peroxides being 34% in fresh alpha-pinene ozonolysis SOAs and decreasing to 12% after remaining stable for 6 hours. Mass fractions decreased when NOx concentration was increased, but a similar peroxide concentration time trend was observed. A molecular weight of 300 g mol−1 was applied for calculations based on an estimated mixture of peroxyhemiacetal and minor hydroperoxide species forming in monoterpene SOAs generated with ozone.160 Krapf et al.127 have utilized this application for the determination of HOM yields from laboratory-produced alpha-pinene SOA. Assuming that HOM contains on average two hydroperoxide groups, the HOM content contributing to alpha-pinene SOA was estimated to be 5%. Also, the same above-mentioned peroxide concentration time trend was observed demonstrating high instability with half-lives shorter than an hour.
4. Conclusion and prospects
New oxidized species are constantly being discovered in the atmosphere. One of the challenges in analyzing SOA markers is the diversity and large number of compounds, which complicates the analysis. Analytical standards are not available and must be synthesized to determine concentrations more accurately for compounds and confirm their identity. In addition to using surrogates, the concentrations have been dependent on the analytical method used and there have been variations at sampling points and times. Finding new and more specific compounds would be important, especially for the assessment of aged SOAs. In the absence of specific markers, monitoring of precursor VOCs is recommended for the characterization of SOAs. Chromatographic methods have been used to determine oxidized markers at a concentration level as low as pg m−3. In GC methods, it has been possible to determine a more diverse range of compounds with a single analysis. Due to the higher separation efficiency, it has been possible to better separate the isomers of the compounds compared to LC methods and to numerically detect more compounds. The disadvantage is the need for derivatization of oxidized compounds, which makes sample processing more laborious and can create interfering artifacts without proper optimization. LC methods, on the other hand, have generally been used for the analysis of compounds directly from extracts or with fewer sample processing steps. MS, as an independent analyzer or integrated as a detector after chromatographic separation, is essential in modern research and provides good sensitivity and qualitative information of the compounds. Spectrophotometric methods are simple and can be used with cheaper equipment. Their sensitivity is lower and they do not provide information on individual compounds, which is why they have not been mainstream techniques in SOA research. Iodometric determination by spectrophotometry of peroxides from laboratory-generated SOAs has been more commonly used. HOM compounds are a relatively new discovery that has not yet been the subject of much research. There are no harmonized and unambiguous definitions, so for the time being exact criteria and definitions for HOMs should be presented in the research articles. HOMs are challenging to analyze by chromatographic techniques, and authentic standards for quantitative analysis are not yet available, so the analysis of HOMs is so far qualitative and semiquantitative using HR-MS methods and chemical ionization. The hydroperoxide and other peroxide groups in HOMs are highly reactive, which complicates the analysis of the compounds due to the poor stability of the peroxide compounds and changes in SOA samples as the peroxides react with the matrix. Currently, analysis of HOM compounds is focused on CI-APi-TOF or corresponding methods, where different reagent gases with specificity to different functionalities could potentially provide new information on the structure of HOM compounds.Author contributions
Aleksi Tiusanen: writing – original draft, conceptualization, investigation, visualization; Jose Ruiz-Jimenez: writing – review & editing, conceptualization, supervision; Kari Hartonen: writing – review & editing, conceptualization, supervision; Susanne Wiedmer: writing – review & editing, conceptualization, supervision.Conflicts of interest
There are no conflicts of interest to declare.References
- M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. M. Donahue, C. George, A. H. Goldstein, J. F. Hamilton, H. Herrmann, T. Hoffmann, Y. Iinuma, M. Jang, M. E. Jenkin, J. L. Jimenez, A. Kiendler-Scharr, W. Maenhaut, G. McFiggans, T. F. Mentel, A. Monod, A. S. H. Prévôt, J. H. Seinfeld, J. D. Surratt, R. Szmigielski and J. Wildt, The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 2009, 9, 5155–5236 CrossRef CAS.
- C. Kwiezinski, C. Weller, D. van Pinxteren, M. Brüggemann, S. Mertes, F. Stratmann and H. Herrmann, Determination of highly polar compounds in atmospheric aerosol particles at ultra-trace levels using ion chromatography Orbitrap mass spectrometry, J. Sep. Sci., 2021, 44, 2343–2357 CrossRef CAS PubMed.
- C. Zhu, K. Kawamura and P. Fu, Seasonal variations of biogenic secondary organic aerosol tracers in Cape Hedo, Okinawa, Atmos. Environ., 2016, 130, 113–119 CrossRef CAS.
- B. Nozière, M. Kalberer, M. Claeys, J. Allan, B. D'Anna, S. Decesari, E. Finessi, M. Glasius, I. Grgić, J. F. Hamilton, T. Hoffmann, Y. Iinuma, M. Jaoui, A. Kahnt, C. J. Kampf, I. Kourtchev, W. Maenhaut, N. Marsden, S. Saarikoski, J. Schnelle-Kreis, J. D. Surratt, S. Szidat, R. Szmigielski and A. Wisthaler, The Molecular Identification of Organic Compounds in the Atmosphere: State of the Art and Challenges, Chem. Rev., 2015, 115, 3919–3983 CrossRef PubMed.
- M. Lewandowski, I. R. Piletic, T. E. Kleindienst, J. H. Offenberg, M. R. Beaver, M. Jaoui, K. S. Docherty and E. O. Edney, Secondary organic aerosol characterisation at field sites across the United States during the spring–summer period, Int. J. Environ. Anal. Chem., 2013, 93, 1084–1103 CrossRef CAS.
- R. Atkinson and J. Arey, Atmospheric Degradation of Volatile Organic Compounds, Chem. Rev., 2003, 103, 4605–4638 CrossRef CAS PubMed.
- J. D. Crounse, L. B. Nielsen, S. Jørgensen, H. G. Kjaergaard and P. O. Wennberg, Autoxidation of Organic Compounds in the Atmosphere, J. Phys. Chem. Lett., 2013, 4, 3513–3520 CrossRef CAS.
- S. Wang, R. Wu, T. Berndt, M. Ehn and L. Wang, Formation of Highly Oxidized Radicals and Multifunctional Products from the Atmospheric Oxidation of Alkylbenzenes, Environ. Sci. Technol., 2017, 51, 8442–8449 CrossRef CAS PubMed.
- M. Jaoui, I. R. Piletic, R. Szmigielski, K. J. Rudzinski, M. Lewandowski, T. P. Riedel and T. E. Kleindienst, Rapid production of highly oxidized molecules in isoprene aerosol via peroxy and alkoxy radical isomerization pathways in low and high NOx environments: Combined laboratory, computational and field studies, Sci. Total Environ., 2021, 775, 145592 CrossRef CAS PubMed.
- J. D. Surratt, A. W. H. Chan, N. C. Eddingsaas, M. Chan, C. L. Loza, A. J. Kwan, S. P. Hersey, R. C. Flagan, P. O. Wennberg and J. H. Seinfeld, Reactive intermediates revealed in secondary organic aerosol formation from isoprene, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6640–6645 CrossRef CAS PubMed.
- Y.-H. Lin, H. Zhang, H. O. T. Pye, Z. Zhang, W. J. Marth, S. Park, M. Arashiro, T. Cui, S. H. Budisulistiorini, K. G. Sexton, W. Vizuete, Y. Xie, D. J. Luecken, I. R. Piletic, E. O. Edney, L. J. Bartolotti, A. Gold and J. D. Surratt, Epoxide as a precursor to secondary organic aerosol formation from isoprene photooxidation in the presence of nitrogen oxides, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6718–6723 CrossRef CAS PubMed.
- Y. Cheng, Y. Ma and D. Hu, Tracer-based source apportioning of atmospheric organic carbon and the influence of anthropogenic emissions on secondary organic aerosol formation in Hong Kong, Atmos. Chem. Phys., 2021, 21, 10589–10608 CrossRef CAS.
- M. Claeys and W. Maenhaut, Secondary Organic Aerosol Formation from Isoprene: Selected Research, Historic Account and State of the Art, Atmosphere, 2021, 12, 728 CrossRef CAS.
- S. Bikkina, K. Kawamura and M. Sarin, Secondary Organic Aerosol Formation over Coastal Ocean: Inferences from Atmospheric Water-Soluble Low Molecular Weight Organic Compounds, Environ. Sci. Technol., 2017, 51, 4347–4357 CrossRef CAS PubMed.
- V.-M. Kerminen, Relative roles of secondary sulfate and organics in atmospheric cloud condensation nuclei production, J. Geophys. Res.: Atmos., 2001, 106, 17321–17333 CrossRef CAS.
- P. Saxena and L. M. Hildemann, Organics alter hygroscopic behavior of atmospheric particles, J. Geophys. Res.: Atmos., 1995, 100, 18755–18770 CrossRef.
- Y.-F. Xing, Y.-H. Xu, M.-H. Shi and Y.-X. Lian, The impact of PM2.5 on the human respiratory system, J. Thorac. Dis., 2016, 8, E69–E74 Search PubMed.
- F. Khan, K. Kwapiszewska, Y. Zhang, Y. Chen, A. T. Lambe, A. Kołodziejczyk, N. Jalal, K. Rudzinski, A. Martínez-Romero, R. C. Fry, J. D. Surratt and R. Szmigielski, Toxicological Responses of α-Pinene-Derived Secondary Organic Aerosol and Its Molecular Tracers in Human Lung Cell Lines, Chem. Res. Toxicol., 2021, 34, 817–832 Search PubMed.
- H. L. Runberg and B. J. Majestic, Hydroxyl radical (OH) formation during the photooxidation of anthracene and its oxidized derivatives, Atmos. Environ., 2022, 286, 119214 CrossRef CAS.
- J. N. Pitts, D. M. Lokensgard, P. S. Ripley, K. A. Van Cauwenberghe, L. Van Vaeck, S. D. Shaffer, A. J. Thill and W. L. Belser, ‘Atmospheric’ Epoxidation of Benzo[a]pyrene by Ozone: Formation of the Metabolite Benzo[a]pyrene-4,5-Oxide, Science, 1980, 210, 1347–1349 CrossRef CAS PubMed.
- D. J. Foreman and S. A. McLuckey, Recent Developments in Gas-Phase Ion/Ion Reactions for Analytical Mass Spectrometry, Anal. Chem., 2020, 92, 252–266 CrossRef CAS PubMed.
- G.-L. Hou and X.-B. Wang, Molecular Specificity and Proton Transfer Mechanisms in Aerosol Prenucleation Clusters Relevant to New Particle Formation, Acc. Chem. Res., 2020, 53, 2816–2827 CrossRef CAS PubMed.
- Y. Gao, K. Lu and Y. Zhang, Review of technologies and their applications for the speciated detection of RO2 radicals, J. Environ. Sci., 2023, 123, 487–499 CrossRef PubMed.
- G. M. Lanzafame, D. Srivastava, O. Favez, B. A. M. Bandowe, P. Shahpoury, G. Lammel, N. Bonnaire, L. Y. Alleman, F. Couvidat, B. Bessagnet and A. Albinet, One-year measurements of secondary organic aerosol (SOA) markers in the Paris region (France): Concentrations, gas/particle partitioning and SOA source apportionment, Sci. Total Environ., 2021, 757, 143921 CrossRef CAS PubMed.
- P. A. Sheppard, Atmospheric tracers and the study of the general circulation of the atmosphere, Rep. Prog. Phys., 1963, 26, 213 CrossRef CAS.
- X. Ding, Q.-F. He, R.-Q. Shen, Q.-Q. Yu and X.-M. Wang, Spatial distributions of secondary organic aerosols from isoprene, monoterpenes, β-caryophyllene, and aromatics over China during summer: secondary organic aerosols over China, J. Geophys. Res.: Atmos., 2014, 119, 11877–11891 CrossRef CAS.
- Md. M. Haque, K. Kawamura and Y. Kim, Seasonal variations of biogenic secondary organic aerosol tracers in ambient aerosols from Alaska, Atmos. Environ., 2016, 130, 95–104 CrossRef CAS.
- E. Kostenidou, K. Florou, C. Kaltsonoudis, M. Tsiflikiotou, S. Vratolis, K. Eleftheriadis and S. N. Pandis, Sources and chemical characterization of organic aerosol during the summer in the eastern Mediterranean, Atmos. Chem. Phys., 2015, 15, 11355–11371 CrossRef CAS.
- I. Stavroulas, A. Bougiatioti, G. Grivas, D. Paraskevopoulou, M. Tsagkaraki, P. Zarmpas, E. Liakakou, E. Gerasopoulos and N. Mihalopoulos, Sources and processes that control the submicron organic aerosol composition in an urban Mediterranean environment (Athens): a high temporal-resolution chemical composition measurement study, Atmos. Chem. Phys., 2019, 19, 901–919 CrossRef CAS.
- M. Cao, M. Chen, P. Ge, Y. Cui and W. Li, Seasonal variation, source contribution, and impact factors of biogenic organic aerosols in PM2.5 in Nanjing, China, Sci. Total Environ., 2022, 843, 156875 CrossRef CAS PubMed.
- M. Teich, D. van Pinxteren and H. Herrmann, A one year study of functionalised medium-chain carboxylic acids in atmospheric particles at a rural site in Germany revealing seasonal trends and possible sources, J. Atmos. Chem., 2019, 76, 115–132 CrossRef CAS.
- A. Guenther, T. Karl, P. Harley, C. Wiedinmyer, P. I. Palmer and C. Geron, Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature), Atmos. Chem. Phys., 2006, 6, 3181–3210 CrossRef CAS.
- L. D. Yee, G. Isaacman-VanWertz, R. A. Wernis, N. M. Kreisberg, M. Glasius, M. Riva, J. D. Surratt, S. S. de Sá, S. T. Martin, M. L. Alexander, B. B. Palm, W. Hu, P. Campuzano-Jost, D. A. Day, J. L. Jimenez, Y. Liu, P. K. Misztal, P. Artaxo, J. Viegas, A. Manzi, R. A. F. de Souza, E. S. Edgerton, K. Baumann and A. H. Goldstein, Natural and Anthropogenically Influenced Isoprene Oxidation in Southeastern United States and Central Amazon, Environ. Sci. Technol., 2020, 54, 5980–5991 CrossRef CAS PubMed.
- P. G. Kanellopoulos, E. Chrysochou, K. Koukoulakis, E. Vasileiadou, C. Kizas, C. Savvides and E. Bakeas, Secondary organic aerosol tracers and related polar organic compounds between urban and rural areas in the Eastern Mediterranean region: source apportionment and the influence of atmospheric oxidants, Environ. Sci.: Processes Impacts, 2020, 22, 2212–2229 CAS.
- F. Ikemori, R. Nishimura, S. Saito, M. Akiyama, S. Yamamoto, A. Iijima and S. Sugata, Organic Molecular Tracers in PM2.5 at Urban Sites during Spring and Summer in Japan: Impact of Secondary Organic Aerosols on Water-Soluble Organic Carbon, Atmosphere, 2021, 12, 579 CrossRef CAS.
- M. Jaoui, R. Szmigielski, K. Nestorowicz, A. Kolodziejczyk, K. Sarang, K. J. Rudzinski, A. Konopka, E. Bulska, M. Lewandowski and T. E. Kleindienst, Organic Hydroxy Acids as Highly Oxygenated Molecular (HOM) Tracers for Aged Isoprene Aerosol, Environ. Sci. Technol., 2019, 53, 14516–14527 CrossRef CAS PubMed.
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. H. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, E. J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Evolution of Organic Aerosols in the Atmosphere, Science, 2009, 326, 1525–1529 CrossRef CAS PubMed.
- T. Cui, Z. Zeng, E. O. dos Santos, Z. Zhang, Y. Chen, Y. Zhang, C. A. Rose, S. H. Budisulistiorini, L. B. Collins, W. M. Bodnar, R. A. F. de Souza, S. T. Martin, C. M. D. Machado, B. J. Turpin, A. Gold, A. P. Ault and J. D. Surratt, Development of a hydrophilic interaction liquid chromatography (HILIC) method for the chemical characterization of water-soluble isoprene epoxydiol (IEPOX)-derived secondary organic aerosol, Environ. Sci.: Processes Impacts, 2018, 20, 1524–1536 RSC.
- A. P. S. Hettiyadura, I. M. Al-Naiema, D. D. Hughes, T. Fang and E. A. Stone, Organosulfates in Atlanta, Georgia: anthropogenic influences on biogenic secondary organic aerosol formation, Atmos. Chem. Phys., 2019, 19, 3191–3206 CrossRef CAS.
- P. Wach, G. Spólnik, J. D. Surratt, K. Blaziak, K. J. Rudzinski, Y.-H. Lin, W. Maenhaut, W. Danikiewicz, M. Claeys and R. Szmigielski, Structural Characterization of Lactone-Containing MW 212 Organosulfates Originating from Isoprene Oxidation in Ambient Fine Aerosol, Environ. Sci. Technol., 2020, 54, 1415–1424 CrossRef CAS PubMed.
- K. Sato, Detection of nitrooxypolyols in secondary organic aerosol formed from the photooxidation of conjugated dienes under high-NO conditions, Atmos. Environ., 2008, 42, 6851–6861 CrossRef CAS.
- J. Zhang, X. He, X. Ding, J. Z. Yu and Q. Ying, Modeling Secondary Organic Aerosol Tracers and Tracer-to-SOA Ratios for Monoterpenes and Sesquiterpenes Using a Chemical Transport Model, Environ. Sci. Technol., 2022, 56, 804–813 CrossRef CAS PubMed.
- T. R. Duhl, D. Helmig and A. Guenther, Sesquiterpene emissions from vegetation: a review, Biogeosciences, 2008, 5, 761–777 CrossRef CAS.
- L. Müller, M.-C. Reinnig, K. H. Naumann, H. Saathoff, T. F. Mentel, N. M. Donahue and T. Hoffmann, Formation of 3-methyl-1,2,3-butanetricarboxylic acid via gas phase oxidation of pinonic acid – a mass spectrometric study of SOA aging, Atmos. Chem. Phys., 2012, 12, 1483–1496 CrossRef.
- K. Sato, F. Ikemori, S. Ramasamy, A. Fushimi, K. Kumagai, A. Iijima and Y. Morino, Four- and Five-Carbon Dicarboxylic Acids Present in Secondary Organic Aerosol Produced from Anthropogenic and Biogenic Volatile Organic Compounds, Atmosphere, 2021, 12, 1703 CrossRef CAS.
- J. Laskin, P. A. Eckert, P. J. Roach, B. S. Heath, S. A. Nizkorodov and A. Laskin, Chemical Analysis of Complex Organic Mixtures Using Reactive Nanospray Desorption Electrospray Ionization Mass Spectrometry, Anal. Chem., 2012, 84, 7179–7187 CrossRef CAS PubMed.
- M. Jaoui, E. Corse, T. E. Kleindienst, J. H. Offenberg, M. Lewandowski and E. O. Edney, Analysis of Secondary Organic Aerosol Compounds from the Photooxidation of d-Limonene in the Presence of NOX and their Detection in Ambient PM2.5, Environ. Sci. Technol., 2006, 40, 3819–3828 CrossRef CAS PubMed.
- M. Vestenius, H. Hellén, J. Levula, P. Kuronen, K. J. Helminen, T. Nieminen, M. Kulmala and H. Hakola, Acidic reaction products of monoterpenes and sesquiterpenes in atmospheric fine particles in a boreal forest, Atmos. Chem. Phys., 2014, 14, 7883–7893 CrossRef.
- S. Wang, M. J. Newland, W. Deng, A. R. Rickard, J. F. Hamilton, A. Muñoz, M. Ródenas, M. M. Vázquez, L. Wang and X. Wang, Aromatic Photo-oxidation, A New Source of Atmospheric Acidity, Environ. Sci. Technol., 2020, 54, 7798–7806 CrossRef CAS PubMed.
- K. Kristensen, M. Bilde, P. P. Aalto, T. Petäjä and M. Glasius, Denuder/filter sampling of organic acids and organosulfates at urban and boreal forest sites: Gas/particle distribution and possible sampling artifacts, Atmos. Environ., 2016, 130, 36–53 CrossRef CAS.
- K. Sato, F. Ikemori, S. Ramasamy, A. Iijima, K. Kumagai, A. Fushimi, Y. Fujitani, S. Chatani, K. Tanabe, A. Takami, H. Tago, Y. Saito, S. Saito, J. Hoshi and Y. Morino, Formation of secondary organic aerosol tracers from anthropogenic and biogenic volatile organic compounds under varied NO and oxidant conditions, Atmos. Environ.: X, 2022, 14, 100169 CAS.
- T. E. Kleindienst, M. Jaoui, M. Lewandowski, J. H. Offenberg, C. W. Lewis, P. V. Bhave and E. O. Edney, Estimates of the contributions of biogenic and anthropogenic hydrocarbons to secondary organic aerosol at a southeastern US location, Atmos. Environ., 2007, 41, 8288–8300 CrossRef CAS.
- R. Wang, Y. Huang and G. Cao, Heterogeneous oxidation of isoprene SOA and toluene SOA tracers by ozone, Chemosphere, 2020, 249, 126258 CrossRef CAS PubMed.
- I. M. Al-Naiema, J. H. Offenberg, C. J. Madler, M. Lewandowski, J. Kettler, T. Fang and E. A. Stone, Secondary Organic Aerosols from Aromatic Hydrocarbons and their Contribution to Fine Particulate Matter in Atlanta, Georgia, Atmos. Environ., 2020, 223, 117227 CrossRef CAS PubMed.
- I. Suh, R. Zhang, L. T. Molina and M. J. Molina, Oxidation Mechanism of Aromatic Peroxy and Bicyclic Radicals from OH–Toluene Reactions, J. Am. Chem. Soc., 2003, 125, 12655–12665 CrossRef CAS PubMed.
- M. Jang and S. R. McDow, Products of Benz[a]anthracene Photodegradation in the Presence of Known Organic Constituents of Atmospheric Aerosols, Environ. Sci. Technol., 1997, 31, 1046–1053 CrossRef CAS.
- D. van Pinxteren, C. Neusüβ and H. Herrmann, On the abundance and source contributions of dicarboxylic acids in size-resolved aerosol particles at continental sites in central Europe, Atmos. Chem. Phys., 2014, 14, 3913–3928 CrossRef.
- S. Gao, M. Keywood, N. L. Ng, J. Surratt, V. Varutbangkul, R. Bahreini, R. C. Flagan and J. H. Seinfeld, Low-Molecular-Weight and Oligomeric Components in Secondary Organic Aerosol from the Ozonolysis of Cycloalkenes and α-Pinene, J. Phys. Chem. A, 2004, 108, 10147–10164 CrossRef CAS.
- H. Guo, J. Zhou, L. Wang, Y. Zhou, J. Yuan and R. Zhao, Seasonal Variations and Sources of Carboxylic Acids in PM2.5 in Wuhan, China, Aerosol Air Qual. Res., 2015, 15, 517–528 CrossRef CAS.
- K. Kawamura and S. Bikkina, A review of dicarboxylic acids and related compounds in atmospheric aerosols: Molecular distributions, sources and transformation, Atmos. Res., 2016, 170, 140–160 CrossRef CAS.
- H. Adler and H. Sirén, Study on Dicarboxylic Acids in Aerosol Samples with Capillary Electrophoresis, J. Anal. Methods Chem., 2014, 2014, 498168 Search PubMed.
- S. Bikkina, K. Kawamura, Y. Sakamoto and J. Hirokawa, Low molecular weight dicarboxylic acids, oxocarboxylic acids and α-dicarbonyls as ozonolysis products of isoprene: Implication for the gaseous-phase formation of secondary organic aerosols, Sci. Total Environ., 2021, 769, 144472 CrossRef CAS PubMed.
- J. Ruiz-Jimenez, M. Okuljar, O.-M. Sietiö, G. Demaria, T. Liangsupree, E. Zagatti, J. Aalto, K. Hartonen, J. Heinonsalo, J. Bäck, T. Petäjä and M.-L. Riekkola, Determination of free amino acids, saccharides, and selected microbes in biogenic atmospheric aerosols – seasonal variations, particle size distribution, chemical and microbial relations, Atmos. Chem. Phys., 2021, 21, 8775–8790 CrossRef CAS.
- M. C. Prieto-Blanco, Y. Moliner-Martínez, P. López-Mahía and P. Campíns-Falcó, Determination of carbonyl compounds in particulate matter PM2.5 by in-tube solid-phase microextraction coupled to capillary liquid chromatography/mass spectrometry, Talanta, 2013, 115, 876–880 CrossRef CAS PubMed.
- Q. Li, D. Gong, H. Wang, Y. Wang, S. Han, G. Wu, S. Deng, P. Yu, W. Wang and B. Wang, Rapid increase in atmospheric glyoxal and methylglyoxal concentrations in Lhasa, Tibetan Plateau: Potential sources and implications, Sci. Total Environ., 2022, 824, 153782 CrossRef CAS PubMed.
- K. Kawamura, K. Okuzawa, S. G. Aggarwal, H. Irie, Y. Kanaya and Z. Wang, Determination of gaseous and particulate carbonyls (glycolaldehyde, hydroxyacetone, glyoxal, methylglyoxal, nonanal and decanal) in the atmosphere at Mt. Tai, Atmos. Chem. Phys., 2013, 13, 5369–5380 CrossRef.
- N. N. Naing and H. K. Lee, Determination of glyoxal and methylglyoxal in atmospheric fine particulate matter by vortex-assisted micro-solid phase extraction and liquid chromatography-diode array detection, J. Chromatogr. A, 2018, 1573, 42–47 CrossRef CAS PubMed.
- C. Müller-Tautges, A. Eichler, M. Schwikowski and T. Hoffmann, A new sensitive method for the quantification of glyoxal and methylglyoxal in snow and ice by stir bar sorptive extraction and liquid desorption-HPLC-ESI-MS, Anal. Bioanal. Chem., 2014, 406, 2525–2532 CrossRef PubMed.
- T.-M. Fu, D. J. Jacob, F. Wittrock, J. P. Burrows, M. Vrekoussis and D. K. Henze, Global budgets of atmospheric glyoxal and methylglyoxal, and implications for formation of secondary organic aerosols, J. Geophys. Res.: Atmos., 2008, 113(D15), 1–17 CrossRef.
- R. Volkamer, F. San Martini, L. T. Molina, D. Salcedo, J. L. Jimenez and M. J. Molina, A missing sink for gas-phase glyoxal in Mexico City: Formation of secondary organic aerosol, Geophys. Res. Lett., 2007, 34(19), 1–5 CrossRef.
- S. Dugheri, N. Mucci, G. Cappelli, A. Bonari, G. Garzaro, G. Marrubini, G. Bartolucci, M. Campagna and G. Arcangeli, Monitoring of Air-Dispersed Formaldehyde and Carbonyl Compounds as Vapors and Adsorbed on Particulate Matter by Denuder-Filter Sampling and Gas Chromatographic Analysis, Int. J. Environ. Res. Public Health, 2019, 16, 1969 CrossRef CAS PubMed.
- A. Mutzel, L. Poulain, T. Berndt, Y. Iinuma, M. Rodigast, O. Böge, S. Richters, G. Spindler, M. Sipilä, T. Jokinen, M. Kulmala and H. Herrmann, Highly Oxidized Multifunctional Organic Compounds Observed in Tropospheric Particles: A Field and Laboratory Study, Environ. Sci. Technol., 2015, 49, 7754–7761 CrossRef CAS PubMed.
- F. Bianchi, T. Kurtén, M. Riva, C. Mohr, M. P. Rissanen, P. Roldin, T. Berndt, J. D. Crounse, P. O. Wennberg, T. F. Mentel, J. Wildt, H. Junninen, T. Jokinen, M. Kulmala, D. R. Worsnop, J. A. Thornton, N. Donahue, H. G. Kjaergaard and M. Ehn, Highly Oxygenated Organic Molecules (HOM) from Gas-Phase Autoxidation Involving Peroxy Radicals: A Key Contributor to Atmospheric Aerosol, Chem. Rev., 2019, 119, 3472–3509 CrossRef CAS PubMed.
- M. Ehn, E. Kleist, H. Junninen, T. Petäjä, G. Lönn, S. Schobesberger, M. Dal Maso, A. Trimborn, M. Kulmala, D. R. Worsnop, A. Wahner, J. Wildt and Th. F. Mentel, Gas phase formation of extremely oxidized pinene reaction products in chamber and ambient air, Atmos. Chem. Phys., 2012, 12, 5113–5127 CrossRef CAS.
- M. Ehn, J. A. Thornton, E. Kleist, M. Sipilä, H. Junninen, I. Pullinen, M. Springer, F. Rubach, R. Tillmann, B. Lee, F. Lopez-Hilfiker, S. Andres, I.-H. Acir, M. Rissanen, T. Jokinen, S. Schobesberger, J. Kangasluoma, J. Kontkanen, T. Nieminen, T. Kurtén, L. B. Nielsen, S. Jørgensen, H. G. Kjaergaard, M. Canagaratna, M. D. Maso, T. Berndt, T. Petäjä, A. Wahner, V.-M. Kerminen, M. Kulmala, D. R. Worsnop, J. Wildt and T. F. Mentel, A large source of low-volatility secondary organic aerosol, Nature, 2014, 506, 476–479 CrossRef CAS PubMed.
- M. Rissanen, Anthropogenic Volatile Organic Compound (AVOC) Autoxidation as a Source of Highly Oxygenated Organic Molecules (HOM), J. Phys. Chem. A, 2021, 125, 9027–9039 CrossRef CAS PubMed.
- Z. Wang, M. Ehn, M. P. Rissanen, O. Garmash, L. Quéléver, L. Xing, M. Monge-Palacios, P. Rantala, N. M. Donahue, T. Berndt and S. M. Sarathy, Efficient alkane oxidation under combustion engine and atmospheric conditions, Commun. Chem., 2021, 4, 18 CrossRef CAS PubMed.
- M. Claeys, I. Kourtchev, V. Pashynska, G. Vas, R. Vermeylen, W. Wang, J. Cafmeyer, X. Chi, P. Artaxo, M. O. Andreae and W. Maenhaut, Polar organic marker compounds in atmospheric aerosols during the LBA-SMOCC 2002 biomass burning experiment in Rondônia, Brazil: sources and source processes, time series, diel variations and size distributions, Atmos. Chem. Phys., 2010, 10, 9319–9331 CrossRef CAS.
- C. Yan, W. Nie, M. Äijälä, M. P. Rissanen, M. R. Canagaratna, P. Massoli, H. Junninen, T. Jokinen, N. Sarnela, S. A. K. Häme, S. Schobesberger, F. Canonaco, L. Yao, A. S. H. Prévôt, T. Petäjä, M. Kulmala, M. Sipilä, D. R. Worsnop and M. Ehn, Source characterization of highly oxidized multifunctional compounds in a boreal forest environment using positive matrix factorization, Atmos. Chem. Phys., 2016, 16, 12715–12731 CrossRef CAS.
- P. Massoli, H. Stark, M. R. Canagaratna, J. E. Krechmer, L. Xu, N. L. Ng, R. L. Mauldin, C. Yan, J. Kimmel, P. K. Misztal, J. L. Jimenez, J. T. Jayne and D. R. Worsnop, Ambient Measurements of Highly Oxidized Gas-Phase Molecules during the Southern Oxidant and Aerosol Study (SOAS) 2013, ACS Earth Space Chem., 2018, 2, 653–672 CrossRef CAS.
- D. van Pinxteren, M. Teich and H. Herrmann, Hollow fibre liquid-phase microextraction of functionalised carboxylic acids from atmospheric particles combined with capillary electrophoresis/mass spectrometric analysis, J. Chromatogr. A, 2012, 1267, 178–188 CrossRef CAS PubMed.
- O. Pindado Jiménez, R. M. Pérez Pastor, M. G. Vivanco and M. Santiago Aladro, A chromatographic method to analyze products from photo-oxidation of anthropogenic and biogenic mixtures of volatile organic compounds in smog chambers, Talanta, 2013, 106, 20–28 CrossRef PubMed.
- A. P. Ranney and P. J. Ziemann, Identification and quantification of oxidized organic aerosol compounds using derivatization, liquid chromatography, and chemical ionization mass spectrometry, Aerosol Sci. Technol., 2017, 51, 342–353 CrossRef CAS.
- J. Klyta and M. Czaplicka, Determination of secondary organic aerosol in particulate matter – Short review, Microchem. J., 2020, 157, 104997 CrossRef CAS.
- Z. Kitanovski, I. Grgić and M. Veber, Characterization of carboxylic acids in atmospheric aerosols using hydrophilic interaction liquid chromatography tandem mass spectrometry, J. Chromatogr. A, 2011, 1218, 4417–4425 CrossRef CAS PubMed.
- D. Gowda and K. Kawamura, Seasonal variations of low molecular weight hydroxy-dicarboxylic acids and oxaloacetic acid in remote marine aerosols from Chichijima Island in the western North Pacific (December 2010–November 2011), Atmos. Res., 2018, 204, 128–135 CrossRef CAS.
- R. M. Flores and P. V. Doskey, Vapor- and aerosol-phase atmospheric organic matter in urban air of the Midwest USA, Atmos. Environ., 2021, 264, 118705 CrossRef CAS.
- P. Anttila, T. Hyötyläinen, A. Heikkilä, M. Jussila, J. Finell, M. Kulmala and M.-L. Riekkola, Determination of organic acids in aerosol particles from a coniferous forest by liquid chromatography–mass spectrometry, J. Sep. Sci., 2005, 28, 337–346 CrossRef CAS PubMed.
- R. M. Flores and P. V. Doskey, Using multidimensional gas chromatography to group secondary organic aerosol species by functionality, Atmos. Environ., 2014, 96, 310–321 CrossRef CAS.
- S. Matsunaga and K. Kawamura, Determination of α- and β-Hydroxycarbonyls and Dicarbonyls in Snow and Rain Samples by GC/FID and GC/MS Employing Benzyl Hydroxyl Oxime Derivatization, Anal. Chem., 2000, 72, 4742–4746 CrossRef CAS PubMed.
- R. Winterhalter, M. Kippenberger, J. Williams, E. Fries, K. Sieg and G. K. Moortgat, Concentrations of higher dicarboxylic acids C5–C13 in fresh snow samples collected at the High Alpine Research Station Jungfraujoch during CLACE 5 and 6, Atmos. Chem. Phys., 2009, 9, 2097–2112 CrossRef CAS.
- A. Beschnitt, M. Schwikowski and T. Hoffmann, Towards comprehensive non-target screening using heart-cut two-dimensional liquid chromatography for the analysis of organic atmospheric tracers in ice cores, J. Chromatogr. A, 2022, 1661, 462706 CrossRef CAS PubMed.
- A. J. Colussi and M. R. Hoffmann, In situ photolysis of deep ice core contaminants by Çerenkov radiation of cosmic origin, Geophys. Res. Lett., 2003, 30(4), 44 CrossRef.
- A. Pokhrel, K. Kawamura, K. Ono, O. Seki, P. Fu, S. Matoba and T. Shiraiwa, Ice core records of monoterpene- and isoprene-SOA tracers from Aurora Peak in Alaska since 1660s: Implication for climate change variability in the North Pacific Rim, Atmos. Environ., 2016, 130, 105–112 CrossRef CAS.
- K. Kowalewski and T. Gierczak, Multistep derivatization method for the determination of multifunctional oxidation products from the reaction of α-pinene with ozone, J. Chromatogr. A, 2011, 1218, 7264–7274 CrossRef CAS PubMed.
- A. P. Bateman, M. L. Walser, Y. Desyaterik, J. Laskin, A. Laskin and S. A. Nizkorodov, The Effect of Solvent on the Analysis of Secondary Organic Aerosol Using Electrospray Ionization Mass Spectrometry, Environ. Sci. Technol., 2008, 42, 7341–7346 CrossRef CAS PubMed.
- A. Albinet, G. M. Lanzafame, D. Srivastava, N. Bonnaire, F. Nalin and S. A. Wise, Analysis and determination of secondary organic aerosol (SOA) tracers (markers) in particulate matter standard reference material (SRM 1649b, urban dust), Anal. Bioanal. Chem., 2019, 411, 5975–5983 CrossRef CAS PubMed.
- A. Mutzel, M. Rodigast, Y. Iinuma, O. Böge and H. Herrmann, An improved method for the quantification of SOA bound peroxides, Atmos. Environ., 2013, 67, 365–369 CrossRef CAS.
- B. Miljevic, F. Hedayat, S. Stevanovic, K. E. Fairfull-Smith, S. E. Bottle and Z. D. Ristovski, To Sonicate or Not to Sonicate PM Filters: Reactive Oxygen Species Generation Upon Ultrasonic Irradiation, Aerosol Sci. Technol., 2014, 48, 1276–1284 CrossRef CAS.
- D. Gowda, K. Kawamura and E. Tachibana, Identification of hydroxy- and keto-dicarboxylic acids in remote marine aerosols using gas chromatography/quadruple and time-of-flight mass spectrometry, Rapid Commun. Mass Spectrom., 2016, 30, 992–1000 CrossRef CAS PubMed.
- R. M. Flores and P. V. Doskey, Evaluation of multistep derivatization methods for identification and quantification of oxygenated species in organic aerosol, J. Chromatogr. A, 2015, 1418, 1–11 CrossRef CAS PubMed.
- M. Jaoui, T. E. Kleindienst, M. Lewandowski and E. O. Edney, Identification and Quantification of Aerosol Polar Oxygenated Compounds Bearing Carboxylic or Hydroxyl Groups. 1. Method Development, Anal. Chem., 2004, 76, 4765–4778 CrossRef CAS PubMed.
- J. L. Little, Artifacts in trimethylsilyl derivatization reactions and ways to avoid them, J. Chromatogr. A, 1999, 844, 1–22 CrossRef CAS PubMed.
- J. R. Wells and J. E. Ham, A new agent for derivatizing carbonyl species used to investigate limonene ozonolysis, Atmos. Environ., 2014, 99, 519–526 CrossRef CAS PubMed.
- J. E. Ham, S. R. Jackson, J. C. Harrison and J. R. Wells, Gas-phase reaction products and yields of terpinolene with ozone and nitric oxide using a new derivatization agent, Atmos. Environ., 2015, 122, 513–520 CrossRef CAS PubMed.
- M. Rodigast, A. Mutzel, Y. Iinuma, S. Haferkorn and H. Herrmann, Characterisation and Optimisation of a Method for the Detection and Quantification of Atmospherically Relevant Carbonyl Compounds in Aqueous Medium, Aerosols/Laboratory Measurement/Validation and Intercomparisons, 2015 Search PubMed.
- J.-Z. Dong and S. C. Moldoveanu, Gas chromatography–mass spectrometry of carbonyl compounds in cigarette mainstream smoke after derivatization with 2,4-dinitrophenylhydrazine, J. Chromatogr. A, 2004, 1027, 25–35 CrossRef CAS PubMed.
- S. Aimanant and P. J. Ziemann, Development of Spectrophotometric Methods for the Analysis of Functional Groups in Oxidized Organic Aerosol, Aerosol Sci. Technol., 2013, 47, 581–591 CrossRef CAS.
- R. E. J. Mitchel and H. C. Birnboim, The use of Girard-T reagent in a rapid and sensitive method for measuring glyoxal and certain other α-dicarbonyl compounds, Anal. Biochem., 1977, 81, 47–56 CrossRef CAS PubMed.
- M. C. Pietrogrande, D. Bacco and M. Mercuriali, GC–MS analysis of low-molecular-weight dicarboxylic acids in atmospheric aerosol: comparison between silylation and esterification derivatization procedures, Anal. Bioanal. Chem., 2010, 396, 877–885 CrossRef CAS PubMed.
- J. Yu, R. C. Flagan and J. H. Seinfeld, Identification of Products Containing −COOH, −OH, and −CO in Atmospheric Oxidation of Hydrocarbons, Environ. Sci. Technol., 1998, 32, 2357–2370 CrossRef CAS.
- A. J. Kalafut-Pettibone and W. S. McGivern, Analytical Methodology for Determination of Organic Aerosol Functional Group Distributions, Anal. Chem., 2013, 85, 3553–3560 CrossRef CAS PubMed.
- G. Isaacman, N. M. Kreisberg, L. D. Yee, D. R. Worton, A. W. H. Chan, J. A. Moss, S. V. Hering and A. H. Goldstein, Online derivatization for hourly measurements of gas- and particle-phase semi-volatile oxygenated organic compounds by thermal desorption aerosol gas chromatography (SV-TAG), Atmos. Meas. Tech., 2014, 7, 4417–4429 CrossRef.
- C. M. Kenseth, N. J. Hafeman, Y. Huang, N. F. Dalleska, B. M. Stoltz and J. H. Seinfeld, Synthesis of Carboxylic Acid and Dimer Ester Surrogates to Constrain the Abundance and Distribution of Molecular Products in α-Pinene and β-Pinene Secondary Organic Aerosol, Environ. Sci. Technol., 2020, 54, 12829–12839 CrossRef CAS PubMed.
- Z.-S. Zhang, G. Engling, C.-Y. Chan, Y.-H. Yang, M. Lin, S. Shi, J. He, Y.-D. Li and X.-M. Wang, Determination of isoprene-derived secondary organic aerosol tracers (2-methyltetrols) by HPAEC-PAD: Results from size-resolved aerosols in a tropical rainforest, Atmos. Environ., 2013, 70, 468–476 CrossRef CAS.
- L. Liang, G. Engling, Z. Du, Y. Cheng, F. Duan, X. Liu and K. He, Seasonal variations and source estimation of saccharides in atmospheric particulate matter in Beijing, China, Chemosphere, 2016, 150, 365–377 CrossRef CAS PubMed.
- M. Glasius, M. Duane and B. R. Larsen, Determination of polar terpene oxidation products in aerosols by liquid chromatography–ion trap mass spectrometry, J. Chromatogr. A, 1999, 833, 121–135 CrossRef CAS.
- A. L. Vogel, M. Äijälä, A. L. Corrigan, H. Junninen, M. Ehn, T. Petäjä, D. R. Worsnop, M. Kulmala, L. M. Russell, J. Williams and T. Hoffmann, In situ submicron organic aerosol characterization at a boreal forest research station during HUMPPA-COPEC 2010 using soft and hard ionization mass spectrometry, Atmos. Chem. Phys., 2013, 13, 10933–10950 CrossRef.
- S. Zhou, J. C. Rivera-Rios, F. N. Keutsch and J. P. D. Abbatt, Identification of organic hydroperoxides and peroxy acids using atmospheric pressure chemical ionization–tandem mass spectrometry (APCI-MS/MS): application to secondary organic aerosol, Atmos. Meas. Tech., 2018, 11, 3081–3089 CrossRef CAS.
- M. C. Prieto-Blanco, P. López-Mahía and P. Campíns-Falcó, On-line analysis of carbonyl compounds with derivatization in aqueous extracts of atmospheric particulate PM10 by in-tube solid-phase microextraction coupled to capillary liquid chromatography, J. Chromatogr. A, 2011, 1218, 4834–4839 CrossRef CAS PubMed.
- S. Castillo, I. Mattila, J. Miettinen, M. Orešič and T. Hyötyläinen, Data Analysis Tool for Comprehensive Two-Dimensional Gas Chromatography/Time-of-Flight Mass Spectrometry, Anal. Chem., 2011, 83, 3058–3067 CrossRef CAS PubMed.
- J. E. Krechmer, M. Groessl, X. Zhang, H. Junninen, P. Massoli, A. T. Lambe, J. R. Kimmel, M. J. Cubison, S. Graf, Y.-H. Lin, S. H. Budisulistiorini, H. Zhang, J. D. Surratt, R. Knochenmuss, J. T. Jayne, D. R. Worsnop, J.-L. Jimenez and M. R. Canagaratna, Ion mobility spectrometry–mass spectrometry (IMS–MS) for on- and offline analysis of atmospheric gas and aerosol species, Atmos. Meas. Tech., 2016, 9, 3245–3262 CrossRef CAS.
- C. P. West, D. Mesa Sanchez, A. C. Morales, Y.-J. Hsu, J. Ryan, A. Darmody, L. V. Slipchenko, J. Laskin and A. Laskin, Molecular and Structural Characterization of Isomeric Compounds in Atmospheric Organic Aerosol Using Ion Mobility-Mass Spectrometry, J. Phys. Chem. A, 2023, 127, 1656–1674 CrossRef CAS PubMed.
- I. Kourtchev, P. Szeto, I. O'Connor, O. A. M. Popoola, W. Maenhaut, J. Wenger and M. Kalberer, Comparison of Heated Electrospray Ionization and Nanoelectrospray Ionization Sources Coupled to Ultra-High-Resolution Mass Spectrometry for Analysis of Highly Complex Atmospheric Aerosol Samples, Anal. Chem., 2020, 92, 8396–8403 CrossRef CAS PubMed.
- Y. Iinuma, S. Ramasamy, K. Sato, A. Kołodziejczyk and R. Szmigielski, Structural Characterisation of Dimeric Esters in α-Pinene Secondary Organic Aerosol Using N2 and CO2 Ion Mobility Mass Spectrometry, Atmosphere, 2020, 12, 17 CrossRef.
- X. Zhang, H. Zhang, W. Xu, X. Wu, G. S. Tyndall, J. J. Orlando, J. T. Jayne, D. R. Worsnop and M. R. Canagaratna, Molecular characterization of alkyl nitrates in atmospheric aerosols by ion mobility mass spectrometry, Atmos. Meas. Tech., 2019, 12, 5535–5545 CrossRef CAS.
- M. Krapf, I. El Haddad, E. A. Bruns, U. Molteni, K. R. Daellenbach, A. S. H. Prévôt, U. Baltensperger and J. Dommen, Labile Peroxides in Secondary Organic Aerosol, Chem, 2016, 1, 603–616 CAS.
- R. Zhao, C. M. Kenseth, Y. Huang, N. F. Dalleska and J. H. Seinfeld, Iodometry-Assisted Liquid Chromatography Electrospray Ionization Mass Spectrometry for Analysis of Organic Peroxides: An Application to Atmospheric Secondary Organic Aerosol, Environ. Sci. Technol., 2018, 52, 2108–2117 CrossRef CAS PubMed.
- H. Li, Z. Chen, L. Huang and D. Huang, Organic peroxides' gas-particle partitioning and rapid heterogeneous decomposition on secondary organic aerosol, Atmos. Chem. Phys., 2016, 16, 1837–1848 CrossRef CAS.
- Y. Gong, Z. Chen and H. Li, The oxidation regime and SOA composition in limonene ozonolysis: roles of different double bonds, radicals, and water, Atmos. Chem. Phys., 2018, 18, 15105–15123 CrossRef CAS.
- H. Junninen, M. Ehn, T. Petäjä, L. Luosujärvi, T. Kotiaho, R. Kostiainen, U. Rohner, M. Gonin, K. Fuhrer, M. Kulmala and D. R. Worsnop, A high-resolution mass spectrometer to measure atmospheric ion composition, Atmos. Meas. Tech., 2010, 3, 1039–1053 CrossRef CAS.
- M. Ehn, H. Junninen, T. Petäjä, T. Kurtén, V.-M. Kerminen, S. Schobesberger, H. E. Manninen, I. K. Ortega, H. Vehkamäki, M. Kulmala and D. R. Worsnop, Composition and temporal behavior of ambient ions in the boreal forest, Atmos. Chem. Phys., 2010, 10, 8513–8530 CrossRef CAS.
- X. Zhang, A. T. Lambe, M. A. Upshur, W. A. Brooks, A. Gray Bé, R. J. Thomson, F. M. Geiger, J. D. Surratt, Z. Zhang, A. Gold, S. Graf, M. J. Cubison, M. Groessl, J. T. Jayne, D. R. Worsnop and M. R. Canagaratna, Highly Oxygenated Multifunctional Compounds in α-Pinene Secondary Organic Aerosol, Environ. Sci. Technol., 2017, 51, 5932–5940 CrossRef CAS PubMed.
- F. L. Eisele and D. J. Tanner, Measurement of the gas phase concentration of H2SO4 and methane sulfonic acid and estimates of H2SO4 production and loss in the atmosphere, J. Geophys. Res.: Atmos., 1993, 98, 9001–9010 CrossRef CAS.
- T. Jokinen, M. Sipilä, H. Junninen, M. Ehn, G. Lönn, J. Hakala, T. Petäjä, R. L. Mauldin, M. Kulmala and D. R. Worsnop, Atmospheric sulphuric acid and neutral cluster measurements using CI-APi-TOF, Atmos. Chem. Phys., 2012, 12, 4117–4125 CrossRef CAS.
- O. Garmash, M. P. Rissanen, I. Pullinen, S. Schmitt, O. Kausiala, R. Tillmann, D. Zhao, C. Percival, T. J. Bannan, M. Priestley, Å. M. Hallquist, E. Kleist, A. Kiendler-Scharr, M. Hallquist, T. Berndt, G. McFiggans, J. Wildt, T. F. Mentel and M. Ehn, Multi-generation OH oxidation as a source for highly oxygenated organic molecules from aromatics, Atmos. Chem. Phys., 2020, 20, 515–537 CrossRef CAS.
- A. Kürten, T. Jokinen, M. Simon, M. Sipilä, N. Sarnela, H. Junninen, A. Adamov, J. Almeida, A. Amorim, F. Bianchi, M. Breitenlechner, J. Dommen, N. M. Donahue, J. Duplissy, S. Ehrhart, R. C. Flagan, A. Franchin, J. Hakala, A. Hansel, M. Heinritzi, M. Hutterli, J. Kangasluoma, J. Kirkby, A. Laaksonen, K. Lehtipalo, M. Leiminger, V. Makhmutov, S. Mathot, A. Onnela, T. Petäjä, A. P. Praplan, F. Riccobono, M. P. Rissanen, L. Rondo, S. Schobesberger, J. H. Seinfeld, G. Steiner, A. Tomé, J. Tröstl, P. M. Winkler, C. Williamson, D. Wimmer, P. Ye, U. Baltensperger, K. S. Carslaw, M. Kulmala, D. R. Worsnop and J. Curtius, Neutral molecular cluster formation of sulfuric acid–dimethylamine observed in real time under atmospheric conditions, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15019–15024 CrossRef PubMed.
- M. Riva, P. Rantala, J. E. Krechmer, O. Peräkylä, Y. Zhang, L. Heikkinen, O. Garmash, C. Yan, M. Kulmala, D. Worsnop and M. Ehn, Evaluating the performance of five different chemical ionization techniques for detecting gaseous oxygenated organic species, Atmos. Meas. Tech., 2019, 12, 2403–2421 CrossRef CAS.
- T. H. Bertram, J. R. Kimmel, T. A. Crisp, O. S. Ryder, R. L. N. Yatavelli, J. A. Thornton, M. J. Cubison, M. Gonin and D. R. Worsnop, A field-deployable, chemical ionization time-of-flight mass spectrometer, Atmos. Meas. Tech., 2011, 4, 1471–1479 CrossRef CAS.
- J. Krechmer, F. Lopez-Hilfiker, A. Koss, M. Hutterli, C. Stoermer, B. Deming, J. Kimmel, C. Warneke, R. Holzinger, J. Jayne, D. Worsnop, K. Fuhrer, M. Gonin and J. de Gouw, Evaluation of a New Reagent-Ion Source and Focusing Ion–Molecule Reactor for Use in Proton-Transfer-Reaction Mass Spectrometry, Anal. Chem., 2018, 90, 12011–12018 CrossRef CAS PubMed.
- H. Li, T. G. Almeida, Y. Luo, J. Zhao, B. B. Palm, C. D. Daub, W. Huang, C. Mohr, J. E. Krechmer, T. Kurtén and M. Ehn, Fragmentation inside proton-transfer-reaction-based mass spectrometers limits the detection of ROOR and ROOH peroxides, Atmos. Meas. Tech., 2022, 15, 1811–1827 CrossRef CAS.
- M. Riva, M. Brüggemann, D. Li, S. Perrier, C. George, H. Herrmann and T. Berndt, Capability of CI-Orbitrap for Gas-Phase Analysis in Atmospheric Chemistry: A Comparison with the CI-APi-TOF Technique, Anal. Chem., 2020, 92, 8142–8150 CrossRef CAS PubMed.
- N. Hyttinen, O. Kupiainen-Määttä, M. P. Rissanen, M. Muuronen, M. Ehn and T. Kurtén, Modeling the Charging of Highly Oxidized Cyclohexene Ozonolysis Products Using Nitrate-Based Chemical Ionization, J. Phys. Chem. A, 2015, 119, 6339–6345 CrossRef CAS PubMed.
- B. H. Lee, F. D. Lopez-Hilfiker, C. Mohr, T. Kurtén, D. R. Worsnop and J. A. Thornton, An Iodide-Adduct High-Resolution Time-of-Flight Chemical-Ionization Mass Spectrometer: Application to Atmospheric Inorganic and Organic Compounds, Environ. Sci. Technol., 2014, 48, 6309–6317 CrossRef CAS PubMed.
- S. Iyer, X. He, N. Hyttinen, T. Kurtén and M. P. Rissanen, Computational and Experimental Investigation of the Detection of HO 2 Radical and the Products of Its Reaction with Cyclohexene Ozonolysis Derived RO 2 Radicals by an Iodide-Based Chemical Ionization Mass Spectrometer, J. Phys. Chem. A, 2017, 121, 6778–6789 CrossRef CAS PubMed.
- T. Berndt, S. Richters, T. Jokinen, N. Hyttinen, T. Kurtén, R. V. Otkjær, H. G. Kjaergaard, F. Stratmann, H. Herrmann, M. Sipilä, M. Kulmala and M. Ehn, Hydroxyl radical-induced formation of highly oxidized organic compounds, Nat. Commun., 2016, 7, 13677 CrossRef CAS PubMed.
- T. Berndt, S. Richters, R. Kaethner, J. Voigtländer, F. Stratmann, M. Sipilä, M. Kulmala and H. Herrmann, Gas-Phase Ozonolysis of Cycloalkenes: Formation of Highly Oxidized RO 2 Radicals and Their Reactions with NO, NO 2 , SO 2 , and Other RO 2 Radicals, J. Phys. Chem. A, 2015, 119, 10336–10348 CrossRef CAS PubMed.
- P. Brophy and D. K. Farmer, Clustering, methodology, and mechanistic insights into acetate chemicalionization using high-resolution time-of-flight mass spectrometry, Atmos. Meas. Tech., 2016, 9, 3969–3986 CrossRef CAS.
- N. Hyttinen, M. P. Rissanen and T. Kurtén, Computational Comparison of Acetate and Nitrate Chemical Ionization of Highly Oxidized Cyclohexene Ozonolysis Intermediates and Products, J. Phys. Chem. A, 2017, 121, 2172–2179 CrossRef CAS PubMed.
- T. Berndt, N. Hyttinen, H. Herrmann and A. Hansel, First oxidation products from the reaction of hydroxyl radicals with isoprene for pristine environmental conditions, Commun. Chem., 2019, 2, 21 CrossRef.
- S. Liu, L. Ahlm, D. A. Day, L. M. Russell, Y. Zhao, D. R. Gentner, R. J. Weber, A. H. Goldstein, M. Jaoui, J. H. Offenberg, T. E. Kleindienst, C. Rubitschun, J. D. Surratt, R. J. Sheesley and S. Scheller, Secondary organic aerosol formation from fossil fuel sources contribute majority of summertime organic mass at Bakersfield, J. Geophys. Res.: Atmos., 2012, 117(D24), 1–21 CrossRef.
- M. S. Claflin, J. Liu, L. M. Russell and P. J. Ziemann, Comparison of methods of functional group analysis using results from laboratory and field aerosol measurements, Aerosol Sci. Technol., 2021, 55, 1042–1058 CrossRef CAS.
- M. P. DeVault, A. C. Ziola and P. J. Ziemann, Products and Mechanisms of Secondary Organic Aerosol Formation from the NO 3 Radical-Initiated Oxidation of Cyclic and Acyclic Monoterpenes, ACS Earth Space Chem., 2022, 6, 2076–2092 CrossRef CAS.
- M. P. DeVault, A. C. Ziola and P. J. Ziemann, Chemistry of Secondary Organic Aerosol Formation from Reactions of Monoterpenes with OH Radicals in the Presence of NOx, J. Phys. Chem. A, 2022, 126, 7719–7736 CrossRef CAS PubMed.
- S. L. Lewis, L. M. Russell, J. A. McKinsey and W. J. Harris, Small contributions of dust to PM2.5 and PM10 concentrations measured downwind of Oceano Dunes, Atmos. Environ., 2023, 294, 119515 CrossRef CAS.
- A. P. Ranney and P. J. Ziemann, Microscale spectrophotometric methods for quantification of functional groups in oxidized organic aerosol, Aerosol Sci. Technol., 2016, 50, 881–892 CrossRef CAS.
- M. S. Claflin, J. E. Krechmer, W. Hu, J. L. Jimenez and P. J. Ziemann, Functional Group Composition of Secondary Organic Aerosol Formed from Ozonolysis of α-Pinene Under High VOC and Autoxidation Conditions, ACS Earth Space Chem., 2018, 2, 1196–1210 CrossRef CAS.
- T. Gautam, S. Wu, J. Ma and R. Zhao, Potential Matrix Effects in Iodometry Determination of Peroxides Induced by Olefins, J. Phys. Chem. A, 2022, 126, 2632–2644 CrossRef CAS PubMed.
- P. Mertes, L. Pfaffenberger, J. Dommen, M. Kalberer and U. Baltensperger, Development of a sensitive long path absorption photometer to quantify peroxides in aerosol particles (Peroxide-LOPAP), Atmos. Meas. Tech., 2012, 5, 2339–2348 CrossRef CAS.
- K. S. Docherty, W. Wu, Y. B. Lim and P. J. Ziemann, Contributions of Organic Peroxides to Secondary Aerosol Formed from Reactions of Monoterpenes with O 3, Environ. Sci. Technol., 2005, 39, 4049–4059 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3em00163f |
| This journal is © The Royal Society of Chemistry 2023 |