
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox-active ligands – a viable route to reactive main group metal compounds
Glen G.
Briand

Department of Chemistry and Biochemistry, Mount Allison University, Sackville, New Brunswick E4L 1G8, Canada. E-mail: gbriand@mta.ca
First published on 21st November 2023
Abstract
Anionic redox-active ligands such as o-amidophenolates, catecholates, dithiolenes, 1,2-benzendithiolates, 2-amidobenzenethiolates, reduced α-diimines, ferrocenyl and porphyrinates are capable of reversible oxidation and thus have the ability to act as sources of electrons for metal centres. These and other non-innocent ligands have been employed in coordination complexes of base transition metals to influence their redox chemistry and afford compounds with useful catalytic, optical, magnetic and conducting properties. Despite the focus in contemporary main group chemistry on designing reactive compounds with potential catalytic activity, comparatively few studies exploring the chemistry of main group metal complexes incorporating redox-active ligands have been reported. This article highlights relevant chemical reactivity and electrochemical studies that probe the oxidation/reduction of main group metal compounds possessing redox-active ligands and comments on the prospects for this relatively untapped avenue of research.
Introduction
A current focus of main group chemistry is the mimicry of the catalytic behavior of organometallic complexes of precious transition metals for organic transformations important to the fine chemicals and pharmaceuticals industries.1–4 Specifically, these metals are particularly suited for catalyzing coupling reactions as they often favour two-electron redox changes thus allowing for “oxidative addition” of substrates, rearrangement of ligands and subsequent “reductive elimination” of products. Heavy main group metals such as indium and bismuth are desirable alternatives to these due to their lower toxicity compared to these precious transition metals and other heavy main metals (e.g. Tl, Sn, Pb, Sb), lower cost and high Lewis acidity due to their large atomic radii. However, these metals are predominantly stable in one oxidation state and do not typically undergo reversible oxidation reactions. Indium is most stable in the +3 oxidation state, though mononuclear indium(I) species which feature a reactive valence lone pair of electrons may be isolated. Chemical oxidation of these compounds to indium(III) has been demonstrated, but the corresponding reduction reactions have not. Bismuth is also predominantly stable in the +3 state but possesses a lone pair of valence electrons that is relatively non-reactive due to the inert pair effect. Bismuth(V) compounds may be isolated with the appropriate choice of ligand and, more recently, Cornella et al. have employed a ligand framework that permits chemical oxidation of bismuth(III) to bismuth(V) species, followed by subsequent reductive elimination of products. This demonstrates the use of an organobismuth compound capable of a III/V redox couple in a catalytic cycle.5–7An alternative approach to designing heavy main group metal compounds capable of redox chemistry is through use of non-innocent “redox-active” ligands.8 Anionic ligands such as o-amidophenolates (AP), catecholates (Cat), dithiolenes (mnt), 1,2-benzenedithiolates (tdt), 2-amidothiophenolates (abt), reduced α-diimides (DAB, BIAN) and ferrocenyl ligands (Fc) are capable of reversible one-electron oxidation, while porphyrins (Por) are capable of two-electron processes (Fig. 1). Therefore, complexation with these ligands allows for potentially reversible oxidation of organometallic complexes possessing metal centres with inaccessible or no valence electrons. These ligands have found use in the synthesis of base transition metal complexes with useful catalytic properties by suppressing favourable one-electron redox processes and facilitating two-electron processes.9–15 They have also been employed in preparing complexes with a variety of interesting optical, magnetic and conducting properties and applications in new generations of optoelectronic devices.16
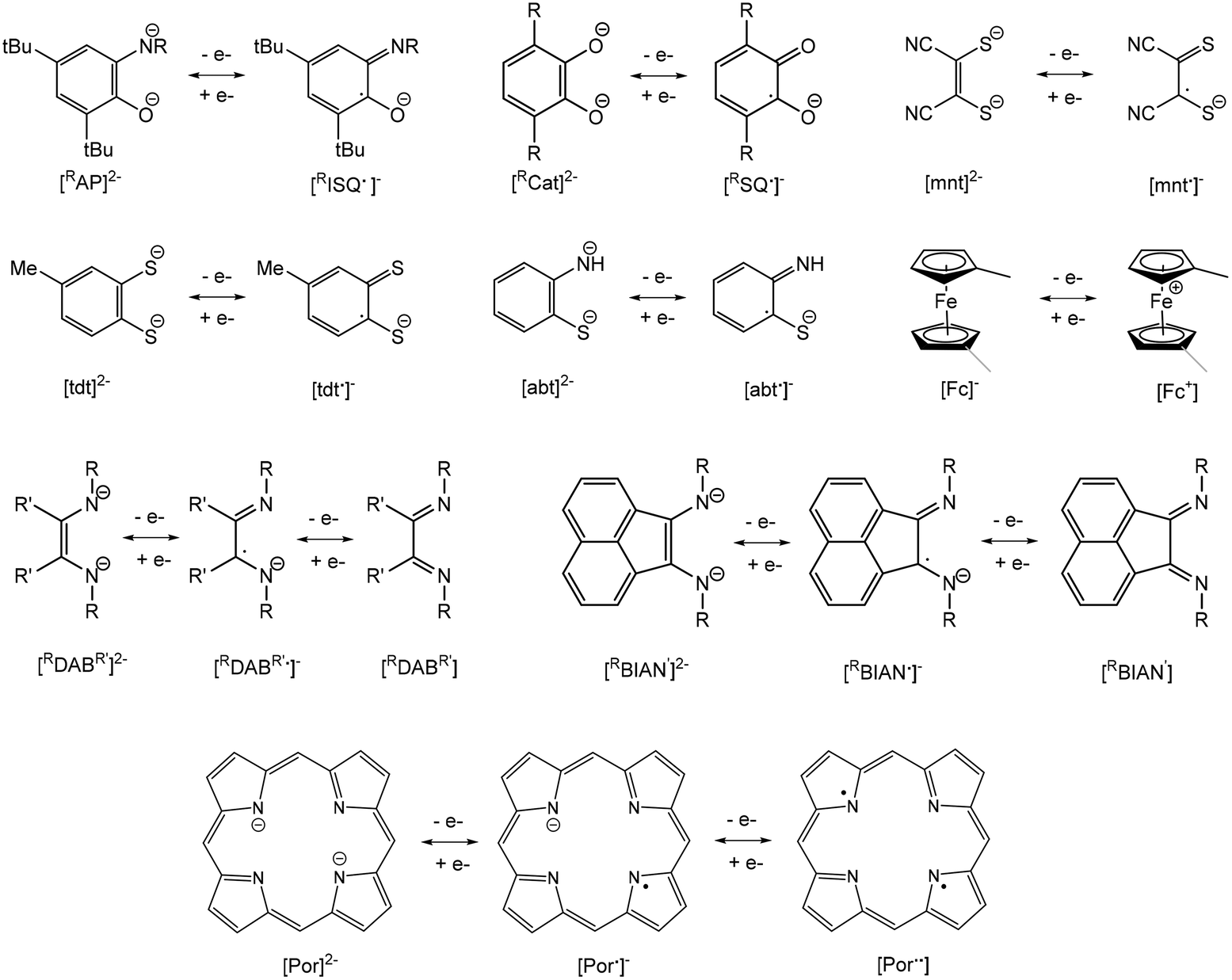 | ||
| Fig. 1 Redox couples of redox-active ligands capable of acting as electron reservoirs: o-amidophenolate (AP)/o-iminosemiquinone (ISQ), catecholate (Cat)/o-semiquinone (SQ), maleoniriledithiolate (mnt), 3,4-toluenedithiolate (tdt), 2-amidobenzenethiolate (abt), ferrocenyl (Fc), 1,4-diaza-1,3-butabiene (DAB), 1,2-bis(imino)acenaphthene (BIAN) and porphyrinato (Por). | ||
Chemical reactivity and electrochemistry
Heavy main group metal complexes of these redox-active ligands have been known for more than 50 years.17,18 Of the comparatively few reports of these complexes versus those of base transition metals, the majority are limited to synthetic or structural studies. As an illustration of the current state of the field, the following highlights relevant chemical reactivity or electrochemical studies that report oxidation/reduction of main group metal compounds.o-Amidophenolates (AP)
Structural drawings of select main group metal o-amidophenolate complexes are found in Fig. 2. Group 13 complexes of o-amidophenolate have been studied extensively by Piskunov and have been recently reviewed.19 The reaction of the heteroleptic complexes (DippAP)GaX (X = Me, I) (Dipp = 2,6-diisopropylphenyl) with single-electron oxidants (I2, O2, HgCl2, HgBr2, TMUDS = tetramethylthiuram disulfide) results in oxidation of the [DippAP]2− ligand and formation of a monoradical species, as confirmed by EPR studies, which spontaneously symmetrize to a paramagnetic neutral biradical species (DippISQ˙)2GaX/Y.20 Attempted synthesis of (DippAP)InI via metathesis reaction resulted in oxidation of the (DippAP)2− ligand and formation of the diradical species (DippISQ˙)2InI.21 However, the reduced ligand complex (DippAP)InI(tmeda) (1) (tmeda = N,N,N′,N′-tetramethylethylenediamine) could be isolated in the presence of the coordinating tmeda ligand.22 Reaction of 1 with oxidizing agents I2 or HgCl2 resulted in single oxidation of the (DippAP)2− ligand and isolation of (DippISQ˙)InX2(tmeda) (X = I or Cl, respectively).23 The stability of these complexes versus the corresponding gallium complexes is attributed to the coordination of the tmeda ligand to the metal centre. Alternatively, reaction of 1 with O2 or TMTUDS (tetramethylthiuramedisulfide) led to disproportionation and formation of the bis-ligand species (DippISQ˙)2InX (X = I or S2CNMe2, respectively), the latter of which proceeds through the expected (DippISQ˙)InI(S2CNMe2)(tmeda) intermediate as determined by EPR spectroscopy. Similar reactivity was observed for [(DippAP)InEt]2. Oxidation of the homoleptic monoanion of [(DME)3Na]+[(DippAP)2In]− (2) (DME = dimethoxyethane) with Hg2X2 (X = Cl, Br) gives the corresponding 2-electron oxidized products (DippISQ˙)2InX.24 Attempted activation of RI (R = Me, Et) with 2 in thf proceeds slowly at room temperature and more quickly at reflux temperatures to give diamagnetic 3.25 The reaction results in the formal addition of the alkyl cation group to the (DippAP)2− ligand to give (DippRAP)− and addition of the iodide anion to the indium centre. Activation of allyl halides (AllX, X = Cl, Br, I) with [(thf)2Na(DippAP)2Ga] yields analogous products to 3, with the reaction of AllI completed in a few hours and that with AllCl requiring more than two weeks.26 Attempts to isolate (DmaISQ˙)3Ga (H2DmaAp = 2-(3,5-dimethoxyanilino)-4,6-di-tert-butylphenol) from the reaction of GaCl3 and H2DmaAp in the presence of Et3N resulted in the formation of 4, which features C–H bond activation of the (DmaAP2)− ligand and the formation of new C–N bonds, presumably as a result of the absence of ortho-substituents in the N-[3,5-(MeO)2C6H3] group.27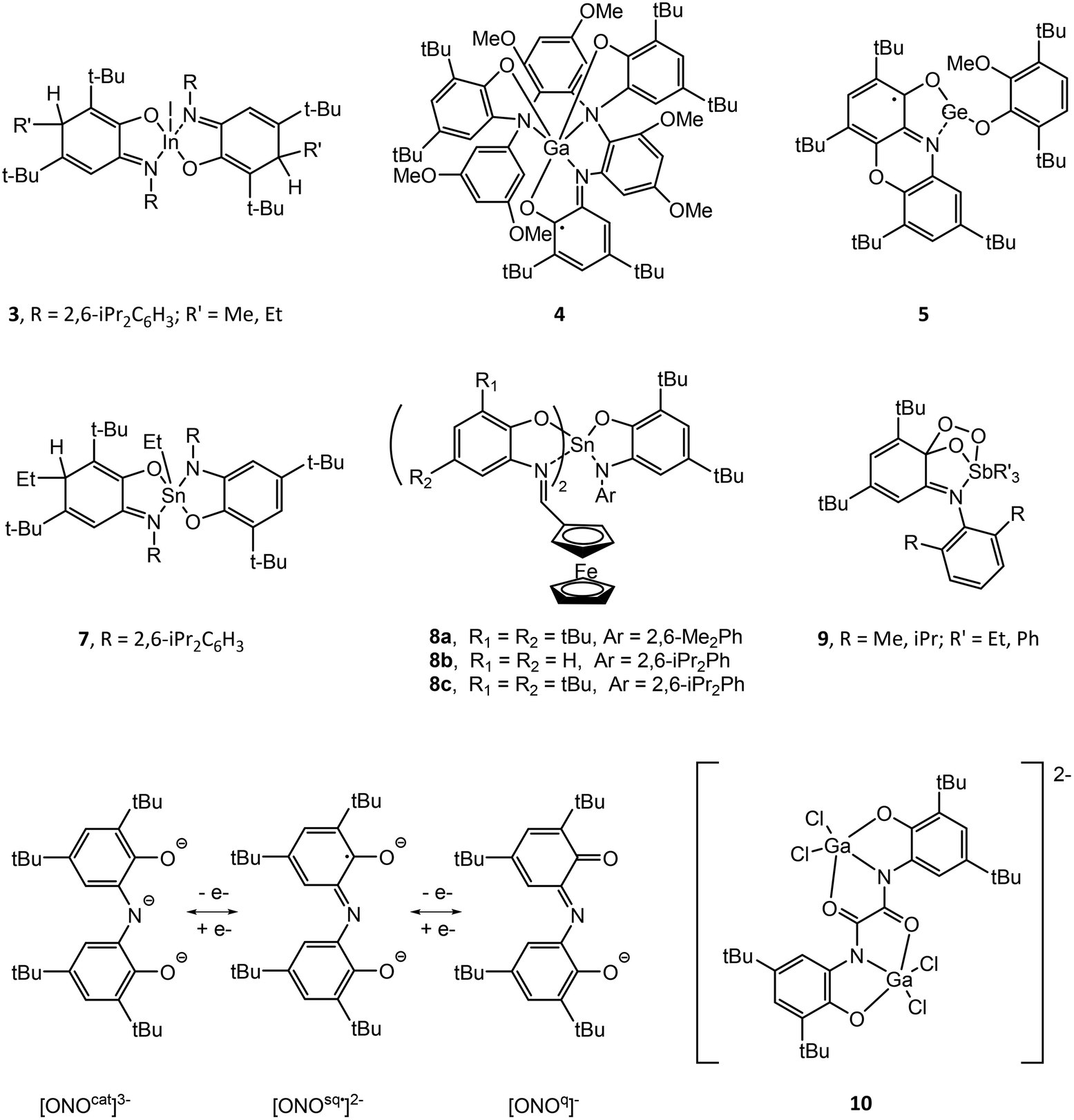 | ||
| Fig. 2 Structural drawings of select main group metal o-amidophenolate (AP) compounds and ligands. | ||
(PhenoxAP)GeII (PhenoxAP = 2,4,6,8-tetra-tert-butylphenoxazin-1-one) reacts with the one-electron oxidant 3,6-di-tert-butyl-2-methoxyphenoxyl radical (RO˙) to afford (PhenoxISQ˙)GeII(OR) (5), in which the (PhenoxAP)2− ligand versus the GeII centre is oxidized.28 Cyclic voltammetry (CV) studies of (RAP)GeII (R = Ad (6a), Ph (6b), tBu (6c)) show a single irreversible oxidation wave for 6a and 6c and a single quasi-reversible wave for 6b. Chemical oxidation of 6a–c with the 3,6-di-tert-butyl-2-ethoxyphenoxy radical allowed for detection of the 6a˙+ radical only by EPR spectroscopy.29 Oxidation of (RAP)SnII (R = Ad, tBu) with TMTDS results in oxidation of SnII and formation of (RAP)SnIV(S2CNMe2)2.30
The homoleptic compound (tBuAP)2SnIV has been shown to display redox isomerism with the tin(II) electromeric form (tBuISQ˙)2SnII in non-polar solvents, as determined by magnetochemistry, EPR, UV-visible spectroscopy and Mössbauer spectroscopy.31 The interconversion can be quenched through addition of a strong donor solvent such as pyridine, resulting in the octahedral compound (tBuAP)2SnIV(py)2. Reaction of the six-coordinate complex (DippAP)(DippISQ˙)2SnIV with air oxygen produces [(DippAP)(DippISQ˙)Sn]2O and (DippISQ˙)2Sn(OH)2 containing μ-oxo- and hydroxo- ligands, respectively.32 Oxidation of [(DippAP)PbII]2 with Hg2Br2 affords the radical species [(DippISQ˙)PbIIBr] as detected by EPR spectroscopy.33
Reaction of (DippAP)SnIVPh2 with dioxygen yields the ligand oxidized species (DippISQ˙)2SnIVPh2 and Ph2SnO, while oxidation with sulfur yields (DippAP)(DippISQ˙)SnPh and (Ph3Sn)S.34 Alternatively, reaction of [(DippAP)SnIVEt2(thf)] with dioxygen or sulfur yields (DippISQ˙)2SnIVEt2, which spontaneously undergoes alkyl elimination to give (DippAP)(DippISQ˙)SnIVEt, (DippAP)(DippEtAp)SnIVEt (7) and Et2SnE (E = O, S). Reaction of (DippAP)SnIVtBu2 with allylhalides AllX (X = Cl, Br, I) results in alkyl transfer to the ring carbon and a C–C bond formation similar to that observed for 7. Alternatively, reaction of (DippAP)SnII with AllX results in oxidation of the tin centre and formation of (DippAP)Sn(All)X followed by disproportionation to (DippAP)2Sn and a similar alkyl transfer to the ring carbon atoms.35 (DippAP)SnII reacts with TMTD (tetramethylthiuram disulfide) to give oxidation at tin and isolation of (DippAP)SnIV(S2CNMe2)2, while reaction with ethoxy-3,6-di-tert-butylphenoxy radical (OR˙) results in formation of (DippISQ˙)SnII(OR), as determined by EPR spectroscopy at low temperature.36
A series of tin(IV) complexes (DippAP)SnIV(IPFc)2 (IPFc = 2-(ferrocenylmethyleneamino)phenolate) (8a–c) containing both o-amidophenolate and ferrocenyl ligands have been studied by CV.37 Generally, these complexes show four quasi-reversible oxidations assigned to oxidation of (DippAP)2− to (DippISQ˙)− to neutral o-iminoquinone at lower potentials followed by the separate oxidation of each (IPFc) to (IPFc+) at higher potentials. Chemical oxidation of 8b and 8c with silver triflate and characterization of the resulting products by EPR supported the assignment of the first oxidation and the formation of [(DippISQ˙)SnIV(IP-Fc)2]+ (8b˙+ and 8c˙+).
The antimony compounds (RAP)SbVR3 (R = Dmp, Dipp; R′ = Et, Ph), which are prepared from the oxidative addition of neutral o-iminobenzoquinone and R′3SbIII, have been shown to reversibly bond dioxygen.38–42 The compounds are air stable but bind molecular oxygen in solution to yield (RAP)(O2)SbR′3 (9), which features a trioxastibolane ring as determined by X-ray crystallography. Moderate heating of solutions of 9 lead to elimination of O2 and isolation of (RAP)SbR′3. The proposed mechanism involves oxidation of the (RAP)2− ligand and formation of a [(RISQ˙)SbVR′3]+[O2˙]− ion pair as an initial step.
3,5-di-tert-Butyl-1,2-quinone-1-(2-hydroxy-3,5-di-tert-butylphenyl)imine (ONO) ligands can exist in three anionic oxidation states (Fig. 2).43 The complexes (ONOq)Al(AcacPh2)Cl (AcacPh2− = diphenylacetylacetonate) and (ONOq)Al(QuinO)Cl (QuinO− = 8-oxyquinoline) were reduced with KC8 to undergo a single-electron reduction in the presence of pyridine to give [(ONOsq˙)Al(AcacPh2)(py)]− and [(ONOsq˙)Al(QuinO)(py)]− monoradicals.44 Alternatively, oxidation of (HONOcat)AlCl(OEt2) with tetrachloro-1,2-quinone (o-O2C6Cl4), 9,10-phenanthrenequinone (o-O2C14H8) or pyrene-4,5-dione (o-O2C16H8) in the presence of pyridine results in expulsion of HCl and the formation of (ONOsq˙)Al(o-O2C6Cl4˙)(py), (ONOsq˙)Al(o-O2C14H8˙)(py) and (ONOsq˙)Al(o-O2C16H8˙)(py), respectively, which possess two redox-active ligands. The assignment of the radical semiquinone redox state of the ligands was determined using X-ray crystallography, UV-vis-NIR spectroscopy and magnetic measurements. The [N(nBu)4]+ salt of the binuclear dianionic gallium complex [(bbpo)(GaCl2)2]2− (10) (H4bbpo = 1,2-bis(3,5-di-tert-butyl-2-hydroxyphenyl)oxamine) has been studied by CV and exhibits two ligand-centred reversible one-electron oxidation waves.45 Magnetic measurements show two uncoupled (o-iminobenzosemiquinonate)gallium(III) units at room temperature.
Catecholates (Cat)
Structural drawings of select main group metal catecholate complexes are found in Fig. 3. The reaction of [(tBuCat)InI(thf)]2 (11) and [(tBuCat)InMe]3 (12) and with oxidizing agents was studied by EPR spectroscopy.46 Reaction of 11 with one equivalent of I2 in thf gave (tBuSQ˙)InI2(thf) (13), which features the monooxidized (tBuSQ˙)− ligand. Dissolution of 13 in diethyl ether lead to spontaneously disproportionation to (tBuSQ˙)2InI and InI3. Alternatively, dissolution of 13 in non-coordinating toluene or hexane solvent resulted in formation of the homoleptic triradical (tBuSQ˙)3In (14). This compound undergoes ligand exchange with tmeda to eliminate neutral o-benzoquinone and form (tBuCat)(tBuSQ˙)In(tmeda) via intramolecular electron transfer between (tBuSQ˙)− ligands. Reaction of [(tBuCat)InMe]3 (12) with 1.5 equivalents of I2 gave (tBuSQ˙)InMeI, which spontaneously disproportionates to (tBuSQ˙)3In (14), MeInI2 and Me2InI. This is presumably due to the weak coordination of thf in (tBuSQ˙)InMeI versus (tBuSQ˙)InI2(thf) (13). Similar reactivity is observed for 11 with TMTUDS to give the radical species (tBuSQ˙)InI(TMTUDS), which is stable in thf but further reacts in non-coordinating solvent to give (tBuSQ˙)3In (14). An analogous result is observed for the corresponding reaction of 12 and TMTUDS, though the product (tBuSQ˙)InMe(TMTUDS) slowly disproportionates in thf to (tBuSQ˙)2InMe. The products of these reactions depend on the reaction conditions, notably the solvent. | ||
| Fig. 3 Structural drawings of select main group metal catecholate (Cat) compounds and reaction schemes. | ||
As with (DippAP)SnII, (tBuCat)SnII also reacts with TMTD to give oxidation at SnII and isolation of (tBuCat)SnIV(S2CNMe2)2, while reaction with ethoxy-3,6-di-tert-butylphenoxy radical (OR˙) results in oxidation of the catecholate ligand and formation of (tBuSQ˙)SnII(OR), as determined by EPR spectroscopy at low temperature.36
Analogous to (RAP)SbVR′3 (R = Dmp, Dipp; R′ = Et, Ph), a series of antimony catecholate compounds (RCat)SbVR′3 (R′ = Et, Ph) (15) is prepared from the oxidative addition of neutral o-benzoquinone and R′3SbIII and reversibly bonds dioxygen to give (RCat)(O2)SbR′3 (16).38,42,47–49 In addition to computational studies on the effect of the electronic and steric properties of the catecholate or o-amidophenolate ligand on reactivity, it has been demonstrated experimentally that the redox potential of the ligand is crucial in allowing or forbidding the binding process. For example, (4,5-(MeO)2-tBuCat)SbPh3 (15b) (4,5-(MeO)2-tBuCat = 4,5-dimethoxy-3,6-di-tert-butyl-catecholato) and (4-MeO-tBuCat)SbPh3 (15c) (4-MeO-tBuCat = 4-methoxy-3,6-di-tert-butyl-catecholato) undergo reversible addition of dioxygen while (tBuCat)SbPh3 (15a) does not. This is a result of the electron donor ability of the methoxy group, which allows the catechol ligand to be more easily oxidized. (4,5-Cat)SbR′3 (4,5-Cat = 4,5-(1,1,4,4-tetramethyl-butane-1,4-diyl)-catecholato; R′ = Et, Ph) and [Ph4Sb]+[(4,5-Cat)2SbPh2]− have been studied by CV. (4,5-Cat)SbPh3 undergoes one reversible one-electron ligand oxidation to give [(4,5-SQ˙)SbPh3]+ and a second irreversible one-electron ligand oxidation as a result of dissociation of neutral o-benzoquinone. Alternatively, (4,5-Cat)SbEt3 undergoes two irreversible one-electron oxidations, while [(4,5-Cat)2SbPh2]− undergoes two reversible one-electron ligand oxidations to give [(4,5-Cat)(4,5-SQ˙)SbPh2] and [(4,5-SQ˙)2SbPh2]+.48
Similar to the o-amidophenolate (AP)SnIV(IP-Fc)2 compounds 8a–c, a series of tin(IV) complexes (Cat)SnIV(IPFc)2 (17a–f) containing both catecholate and ferrocenyl ligands have been studied by CV.37 The first two oxidations of 17a–f are similar, with oxidation of the catecholate ligand to give [(SQ˙)SnIV(IP-Fc)2]+, followed by oxidation of the IPFc ligand to give [(SQ˙)SnIV(IPFc)(IPFc+)2]2+. Compounds 17a, 17c and 17d then undergo an intramolecular electron transfer to give [(Q)SnIV(IPFc)2]2+ (Q = o-benzoquinone) followed by spontaneous decoordination of the neutral o-benzoquinone ligand to give [SnIV(IPFc)2]2+. For compound 17b and 17e, a third oxidation wave is assigned to further oxidation of the (SQ˙)− ligand, which is followed by decoordination of the neutral o-benzoquinone ligand to give [SnIV(IPFc)(IPFc+)]3+. Chemical oxidation of 17a and 17d with silver triflate and characterization of the resulting products by EPR supported the assignment of the first oxidation and the formation of [(SQ˙)Sn(IPFc)2]+.
α-Diimines – 1,4-diaza-1,3-butabienes (DAB) and 1,2-bis(imino)acenaphthenes (BIAN)
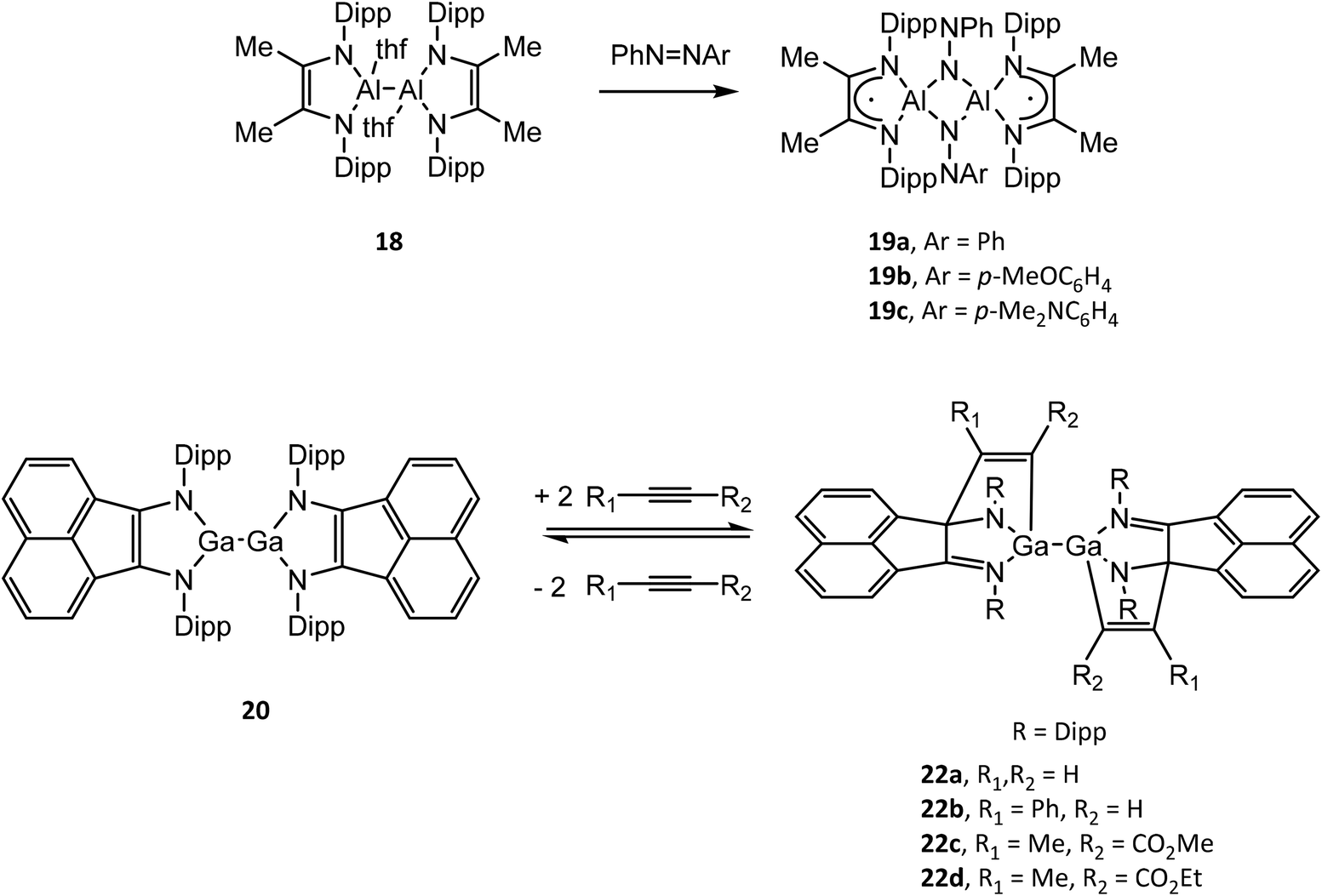
Structural drawings of select main group metal complexes with reduced α-diimine ligands are found in Fig. 4. The reactivity of (RDABR′)MII–MII(RDABR′) and (RBIAN)MII–MII(RBIAN) (M = Al, Ga) complexes has been studied and exhibit insertion reactions into the M–M bond to afford MIII species. For example (DippDABMe)AlII–AlII(DippDABMe) (18) reacts with symmetric and asymmetric azo compounds to form (DippDABMe˙)AlIII(μ2-NPh)(μ2-NAr)AlIII(DippDABMe˙) (19a–c) (Ar = Ph, p-MeOC6H4, p-Me2NC6H4) via oxidation of 18 and reduction of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond.50 Further, compound 19a–c could be reduced with Na give to the dianionic ligand complex [(DippDABMe)AlIII(μ2-NPh)(μ2-NAr)AlIII(DippDABMe)]2−. (DippBIAN)GaII–GaII(DippBIAN) (20) reacts with oxidizing agents (SCH2Ph)2, Me2N(S)CS–SC(S)NMe2 and I2 to afford (DippBIAN˙)GaIII(SCH2Ph)2, (DippBIAN)GaIII(S2CNMe2) and (DippBIAN˙)GaIII–GaIII(DippBIAN˙) (21), respectively.51,52 Compound 21 reversibly dissociates in pyridine solution to give (DippBIAN)GaIIII(py). The reversible addition of alkynes to 20, which involves direct bonding of the DippBIAN ligand with the substrate (22a–d), was exploited to facilitate the catalytic hydroamination and hydroarylation of phenylacetylene with anilines.53,54
N bond.50 Further, compound 19a–c could be reduced with Na give to the dianionic ligand complex [(DippDABMe)AlIII(μ2-NPh)(μ2-NAr)AlIII(DippDABMe)]2−. (DippBIAN)GaII–GaII(DippBIAN) (20) reacts with oxidizing agents (SCH2Ph)2, Me2N(S)CS–SC(S)NMe2 and I2 to afford (DippBIAN˙)GaIII(SCH2Ph)2, (DippBIAN)GaIII(S2CNMe2) and (DippBIAN˙)GaIII–GaIII(DippBIAN˙) (21), respectively.51,52 Compound 21 reversibly dissociates in pyridine solution to give (DippBIAN)GaIIII(py). The reversible addition of alkynes to 20, which involves direct bonding of the DippBIAN ligand with the substrate (22a–d), was exploited to facilitate the catalytic hydroamination and hydroarylation of phenylacetylene with anilines.53,54
 | ||
| Fig. 4 Structural drawings of select reduced α-diimine (DAB, BIAN) ligand main group metal compounds and reaction schemes. | ||
Reaction of MesDABMe (Mes = 2,4,6-trimethylphenyl) with InIOTf lead to formation of the adduct (MesDABMe)InI(OTf), while a similar reaction with less-electron-rich MesDABH resulted in formation of the reduced ligand diindane species [(MesDABH˙)InII(OTf)]2 (23).55 Compound 23 displays C–C coupling of the radical [MesDABH˙]− ligands in the solid-state. Similarly, the 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 reaction of DippDABH and InICl resulted in formation of the paramagnetic diindane [(DippDABH˙)InIICl]2 (24).56 Solution EPR data confirms the presence of a ligand based radical in both 23 and 24. (DippDABH˙)InIIICl2(thf) is formed in low yield from a disproportionation reaction of (DippDABH)Li2 and InICl, which also affords indium metal and neutral DippDABH as byproducts.57 Reactions of RDABH (R = (S) methylbenzyl, (R) 1′-chlorobutan-2′-yl, (S,S) 1′-chloro-1′-phenyl-propan-2′-yl or di-tert-butyl) with MCl3 (M = Al, In) in acetonitrile afford (RDABH)MCl3 adducts. The (RDABH)InCl3 adducts slowly decompose to 1,3-dialkyl-imidazolium and alkylammonium tetrachloroindate in thf solution.58 CV scans of the adduct (pBPBIAN)InCl3 (pBP = p-bromophenyl) shows two quasi-reversible reduction waves assigned to reduction of the InIII centre to InI, with subsequent reduction waves assigned to reduction of the pBPBIAN ligand.59 The adducts (RBIAN)InCl3 (R = 2,6-diisopropylphenyl (25), 2,6-dimethylphenyl and p-nitrophenyl) showed four irreversible reduction waves, likely due to the loss of Cl− ligands during reduction of the complex.60 Data suggested that mild reducing agents with potentials between −1.0 and −2.0 V can be used to reduce these complexes. Reduction of 25 with magnesium anthracene resulted in formation of reduced (DippBIAN)H2 and indium metal, while reduction of (pMP-BIAN)2InCl2[InCl4] (pMP = p-methoxyphenyl) with Cp2Co also resulted in formation of reduced (pMP-BIAN)H2 ligand.
:2 reaction of DippDABH and InICl resulted in formation of the paramagnetic diindane [(DippDABH˙)InIICl]2 (24).56 Solution EPR data confirms the presence of a ligand based radical in both 23 and 24. (DippDABH˙)InIIICl2(thf) is formed in low yield from a disproportionation reaction of (DippDABH)Li2 and InICl, which also affords indium metal and neutral DippDABH as byproducts.57 Reactions of RDABH (R = (S) methylbenzyl, (R) 1′-chlorobutan-2′-yl, (S,S) 1′-chloro-1′-phenyl-propan-2′-yl or di-tert-butyl) with MCl3 (M = Al, In) in acetonitrile afford (RDABH)MCl3 adducts. The (RDABH)InCl3 adducts slowly decompose to 1,3-dialkyl-imidazolium and alkylammonium tetrachloroindate in thf solution.58 CV scans of the adduct (pBPBIAN)InCl3 (pBP = p-bromophenyl) shows two quasi-reversible reduction waves assigned to reduction of the InIII centre to InI, with subsequent reduction waves assigned to reduction of the pBPBIAN ligand.59 The adducts (RBIAN)InCl3 (R = 2,6-diisopropylphenyl (25), 2,6-dimethylphenyl and p-nitrophenyl) showed four irreversible reduction waves, likely due to the loss of Cl− ligands during reduction of the complex.60 Data suggested that mild reducing agents with potentials between −1.0 and −2.0 V can be used to reduce these complexes. Reduction of 25 with magnesium anthracene resulted in formation of reduced (DippBIAN)H2 and indium metal, while reduction of (pMP-BIAN)2InCl2[InCl4] (pMP = p-methoxyphenyl) with Cp2Co also resulted in formation of reduced (pMP-BIAN)H2 ligand.
SnCl2 may be reacted with DippBIAN to afford the adduct (DippBIAN)SnIICl2, while the monoanionic ligand complex (DippBIAN˙)SnIICl may be prepared from DippBIAN and a five-fold excess of SnCl2 and KC8.61 Reaction of DippBIAN with one equivalents of SnCl2 and two equivalents of KC8 in the presence of one equivalent of [CpFe(CO)2]2 gives the dianionic ligand complex (DippBIAN)SnII.
It has been shown that the reaction of PnICl (Pn = P, As), generated in situ from PnCl3 and SnCl2 as reductant, with DippBIAN results in formation of N-heterocyclicphosphinium and -arsinium cations [(DippBIAN)PnIII][SnCl5·thf].62 The PnIII oxidation state was confirmed by 31P NMR and bond distances and angles elucidated from X-ray crystal structures. Similarly, reaction of PIBr, generated in situ from PBr3 and cyclohexene, with (RDABH) (R = Mes, Dipp) or (RBIAN) (R = Mes, Dipp) affords (RDABH)PBr or (RBIAN)PBr products, respectively.63 Alternatively, the redox reaction of PI3 and RDABR′ gives [(RDABR′)P][I3] (R = tBu, Dipp, Mes or cHex, R′ = H; R = p-tolyl, R′ = Me).64 While it has been shown that in situ generated “clamshell” ligand (PyBIAN) (Fig. 5a) adducts (PyBIAN)PnIIICl3 (Pn = P, As) may be reduced with cobaltocene to the corresponding N-heterocyclic chloropnictogens (PyBIAN)PnICl, similar reactions with Pn = Sb and Bi species afforded black material, PyBIAN and [Cp2Co]Cl.65
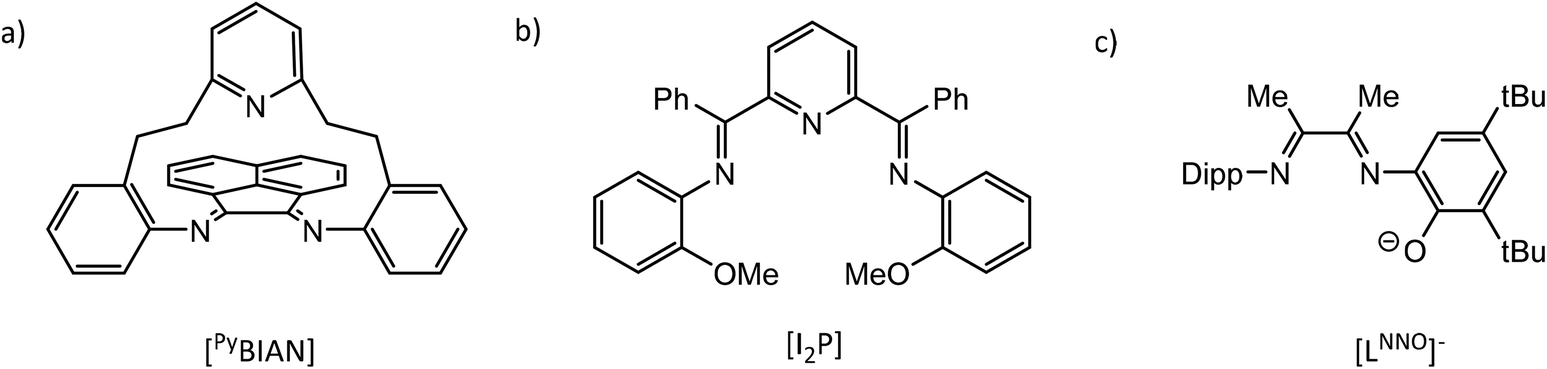 | ||
| Fig. 5 Structural drawings of α-diimine ligands: (a) [PyBIAN], (b) [I2P] and (c) [LNNO]−. | ||
The reactivity and electrochemistry of iminopyridine complexes of Al and Ga have been studied by Berben et al.66–69 and have been reviewed elsewhere.70 It has been shown that a series of octahedral group 13 complexes of 2,6-bis{1-[(2-methoxyphenyl)imino]-benzyl}pyridine (I2P, Fig. 5b) may be prepared that possess multiple ligand charge states: [(I2P0)(I2P−)M]2+, [(I2P−)2M]+, (I2P−)(I2P2−)M, [(I2P2−)2M]−, and [(I2P2−)(I2P3−)M]2−, and [(I2P3−)2Al]3− (M = Al, Ga, In).71 This is achieved through chemical oxidation or reduction of the various complexes with Fc(PF6) or Na, respectively. Further, cyclic voltammograms of (I2P−)(I2P2−)M display reversible redox events that span ligand-based charge states from [(I2P0)(I2P−)M]2+ up to [(I2P2−)2M]−. CV scans of (LNNO)TlMe2 (LNNO = 2,4-di-tert-butyl-6-(3-(2,6-di-iso-propylphenylimino)butan-2-ylidene)amino phenolato; Fig. 5c) showed an irreversible reduction wave which was assigned to reduction of the ligand and accompanied by with strong adsorption of the product to the electrode surface.72
Dithiolenes (mnt), 1,2-benzenedithiolates (tdt) and 2-amidobenzenethiolates (abt)
Early polarographic studies of [In(mnt)3]3−, [In(mnt)2]− and [In(tdt)2]− (mnt = maleonitriledithiolate, tdt = 3,4-toluenedithiolate) complexes showed that these can be reversibly reduced in the steps InIII → InII → InI → In0, though electrochemical or chemical oxidation of these species was not studied. A series of poorly characterized antimony and bismuth-tdt and -mnt complexes have been studied similarly, with tdt complexes showing a single reversible reduction wave and mnt complexes showing two reversible reduction waves.73 Cyclic voltammetry (CV) investigation of [NMe4]2[(bdt)2TlI2] (H2bdt = 1,2-benzenedithiol) showed an irreversible reduction wave due to reduction of Tl+ to Tl0 with deposition of metal on the electrode surface.74 Further, solutions of the compound undergo colour changes upon exposure to air. CV scans of [NEt4][(bdt)2TlIII] show a single irreversible oxidation peak. More recently, it has been shown via CV that indium compounds (NCN)In(tdt) [NCN = 2,6-bis(dimethylaminomethyl)phenyl] and dimeric [MeIn(tdt)(py)]2 undergo irreversible one-electron and two-electron electrochemical oxidations, respectively.75 Chemical oxidation of both compounds with diiodine did not give one-electron oxidation radical products (NCN)In(tdt˙)I and [MeIn(tdt˙)(py)]2, as observed for indium o-amidophenolate (1) and catecholate (11, 12) complexes. Instead, a spontaneous two-electron oxidation of the tdt ligand and formation of [tdt]n disulfide oligomers and RInI2 (R = NCN, Me) was observed, regardless of reaction stoichiometry. CV studies of the corresponding 2-amidobenzenethiolate (abt) analogues showed two very weak oxidation peaks for (NCN)In(abt) and only background current for [MeIn(abt)(py)]. Attempts to chemically oxidize both compounds with diiodine showed no reactivity. Reaction of [NBu4]2[PbII(mnt)2] with organohalides (RX, where R = 1° and 2° alkyl, X = Br, I) afforded [PbII(mnt)X]− and R2mnt, while no reaction was observed when R = 3° alkyl, aryl or X = Cl.76 Alkylation was found to proceed faster than for Na2(mnt).Ferrocenyl compounds (Fc)
Structural drawings of select main group metal ferrocenyl complexes are found in Fig. 6. A small number of ferrocenyl group 13 metal compounds have been synthesized and studied for their electrochemical properties via CV. The triel bridged [1.1]ferrocenophane complexes (RMFc)2 (Fc = ferrocenyl; R = (Me2NCH2)C6H4 (25); M = Al, Ga and In) exhibit a fully reversible two-electron redox process for M = Al and a conventional step-wise redox chemistry for M = Ga, In, indicating weak interaction between the iron centres.77,78 A similar two-electron process was observed for [Me(py)GaFc]2,78 while changing the ancillary ligand to R = C(SiMe3)2SiMe2NMe2 for the indium analogue results in a single irreversible oxidation wave.79 The splitting between the oxidation potentials in bis(ferrocenyl) compounds Fc2GaR (R = 5-Me3Si-2-(Me2NCH2)C6H3 and (2-C5H4N)Me2SiCH2) was found to be smaller than that in the [1.1]ferroceneophane complex (RGaFc)2 (R = 5-Me3Si-2-(Me2NCH2)C6H3), which is accredited to differences in electrostatic interactions between the two Fe centres in each class of complex and more effective solvation of the Fe-containing moieties in Fc2GaR.80 A trinuclear gallium-bridged ferrocenophane with a carousel structure [Fc3Ga2(py)2] (26) shows three reversible oxidation waves with half wave potentials lower than that of ferrocene, i.e. it is more easily oxidized than ferrocene.81,82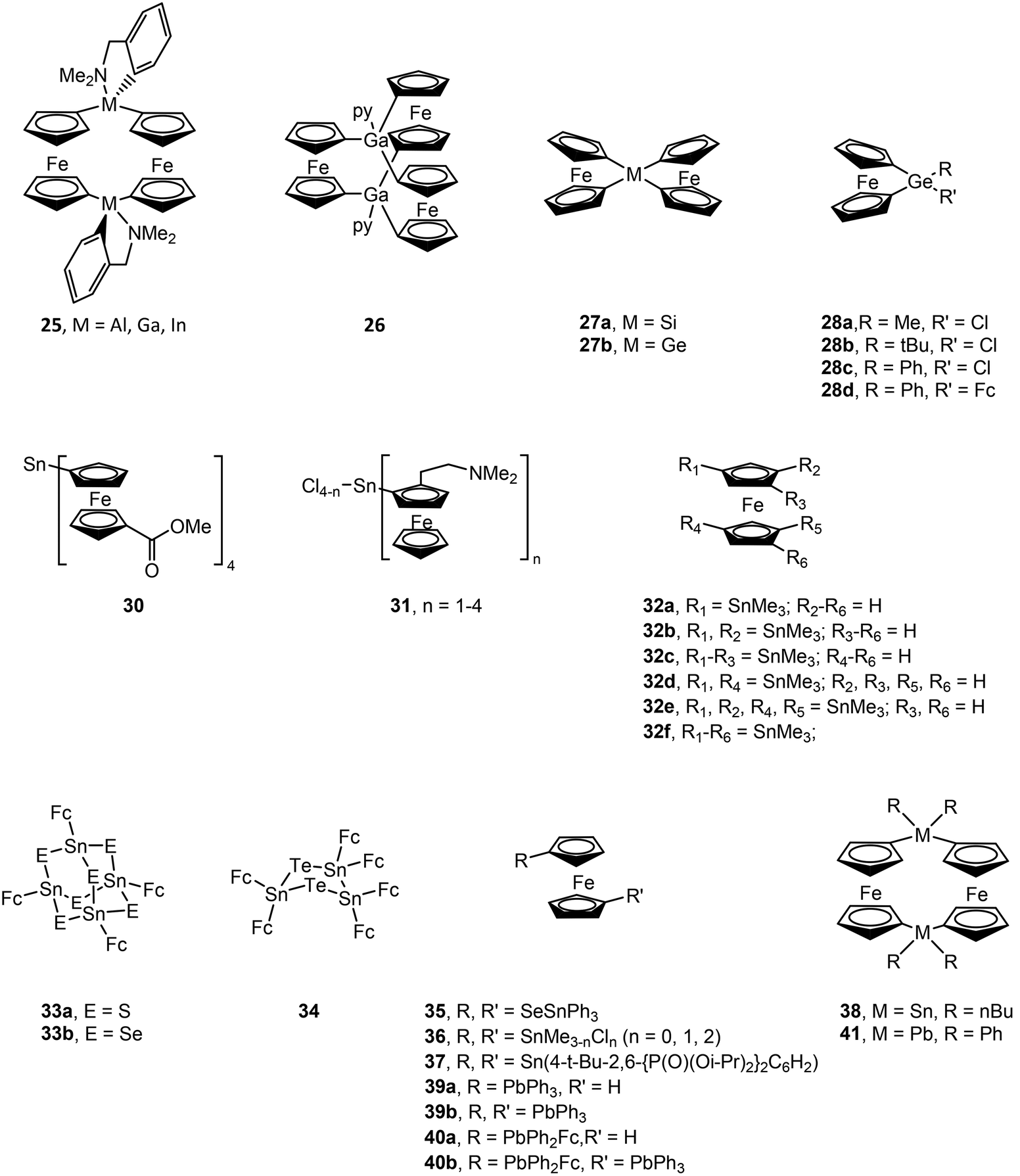 | ||
| Fig. 6 Structural drawings of select main group metal ferrocenyl (Fc) compounds. | ||
CV of the spirocyclic [1]sila- and germaferrocenophanes (Fc)2MIV (M = Si (27a), Ge (27b)) shows two one-electron oxidations, indicating Fe–Fe interactions.83 The first oxidation is reversible in both compounds, while the second is irreversible for 27a and reversible for 27b at fast scan rates. [1]Germylferrocenophane compounds 28a–28c show a reversible one-electron oxidation process with oxidation potentials significantly higher than that of ferrocene due to the electron withdrawing nature of the chloro group.84 Compound 28d exhibits two reversible one electron oxidation processes with the first redox process assigned to the pendant ferrocene group and the second to the ferrocenophane moiety. The bulky bis(ferrocenyl)germane compound (Fc*)2GeII (Fc* = 2,5-bis(3,5-di-tert-butylphenyl)-1-ferrocenyl) (29) was shown by CV to undergo two separate two-electron oxidations.85 Computations studies support that the first wave involves oxidation of GeII to GeIV, followed by facile intramolecular transfer from the Fe atoms in (Fc*)2Ge2+ to afford (Fc*+)2Ge: as a triplet dication. The stability of (Fc*+)2Ge: is afforded by the steric bulk of the Fc* ligands. Compound 29 exhibits characteristic reactivity of a germylene with oxidation of GeII to GeIV.
CV of (FcCO2Me)4SnIV (30) shows two sequential one-electron processes and one two-electron process.86 This compound could also be chemically oxidized in a step-wise manner to 30+, 302+ and 303+ using AgSbF6. CV and DPV (differential pulse voltammetry) data for a series of (FcCH2NMe2)nSnIVCl4−n (n = 1, 2, 3 or 4) (31) complexes shows one, two or three single electron oxidation steps for n = 1, 2 and 3, respectively. One two-electron oxidation step followed by a two one-electron oxidation steps is observed for n = 4. (FcCH2NMe2)2SnII shows two one-electron oxidation steps corresponding to oxidation of the ferrocenyl ligands versus the SnII centre.87 CV and square wave voltammetry (SWV) for a series of multiply-SnMe3 substituted ferrocenes (32a–f) show a reversible one-electron oxidation wave. The oxidation potential shifts to more negative values for each introduced stannyl group due their strong σ-donor and weak π-acceptor properties.88 CV and DPV studies of the adamantly-shaped [(FcSn)4E6] (E = S (33a), Se (33b)) and cyclic [(FcSn)3Te2] (34) complexes show a single four- or six-electron reversible oxidation peak, indicating four or six equivalent and independent redox sites, respectively.89 However, the broad oxidation peak indicates weak interaction among Fe centres, particularly for 34, as a result of short Fe–Fe distances. The oxidation potential decreases upon replacing E = S (33a) with E = Se (33b) due to decreased electron withdrawal from the Sn centres and an increase in electron density in the ferrocenyl ligands. Alternatively, the bridged 1,1′-ferrocenyl compound Fc(Sn(SePh)3)2 (35) decomposes to other products upon electrochemical oxidation, most likely due to Sn–Se bond cleavage.90 CV of 1,1′-bis(stannyl)ferrocenes Fc(SnIVMe3−nCln)2 (n = 0, 1, 2) (36) and Fc(SnIIR)2 (R = 4-t-Bu-2,6-{P(O)(Oi-Pr)2}2C6H2) (37) show reversible one-electron oxidations,91,92 as does 1,1′-bis(diphenylstibino)ferrocene.93 Polarographic measurements of the [1.1]stannylferrocenophane ((n-Bu)2SnFc)2 (38) shows two reversible one-electron oxidation waves.94 Oxidation potentials have also been obtained by CV for ferrocenyl lead compounds Ph3PbFc (39a), (Ph3Pb)2Fc (39b), Ph2PbFc2 (40a) and (Ph3Pb)Fc(Ph2Pb)Fc (40b) and lead [1.1]ferrocenophane (Ph2PbFc)2 (41).95 All compounds show separate one-electron oxidation wave for each Fc ligand. The first oxidation is at slightly higher potentials versus ferrocene for all compounds except for 40a where the first oxidation is at a slightly lower potential.
The 1,2-bis(ferrocenyl)dibismuthene Fc*MMFc* (Fc* = 2,5-(3,5-dimethylphenyl)-1-ferrocenyl; M = Sb, Bi) features two bulky Fc* ligands bridged by a MM π-spacer.96 CV shows three reversible one-electron oxidation couples, corresponding to oxidation of each of the Fc* ligands followed by the MM double bond. This suggests that the trication is stabilized by the ferrocenyl groups. CV scans of the 1,2-bis(ferrocenyl)digermene (E)-Trip(Fc)GeGe(Fc)Trip (Trip = 2,4,6-triisopropylphenyl) show a two-step reversible redox couple, suggesting coupling of the two ferrocenyl moieties through the GeGe π bond.97 Although ferrocene may be chemically oxidized to give the ferrocenium cation,98 no studies involving chemical oxidation of ferrocenyl indium or bismuth compounds have been reported.
Porphyrinates (Por)
Structural drawings of substituted porphyrinato ligands 5,10,15,20-tetraphenylporphyrinato (TPP), 5,10,15,20-tetra(pentafluorophenyl)porphyrinato (TPFP), 5,10,15,20-tetraferrocenylporphyrinato (TFcP), tetrakis[3,5-bis(trifluoromethyl)phenyl]porphyrinato (TArFP), 5,10,15,20-tetra-p-tolylporphyrinato (TTP), 5,10,15,20-tetramesitylporphyrinato (TMesP), 2,3,7,8,12,13,17,18-octaethylporphyrinato (OEP) and 2,3,12,13-tetrabromo-5,10,15,20-tetraphenylporphyrinato (Br4TPP) are found in Fig. 7. CV data has been reported for series of main group metal porphyrin complexes (TPP)AlR and (OEP)AlR (R = Me, nBu, Ph, C6F4H)99 and show one irreversible one-electron oxidation wave followed by two reversible one-electron oxidation waves. The first corresponds to the formation of [(TPP/OEP)AlR]˙+ followed by loss of R˙ to give [(TPP/OEP)Al]+. The second and third oxidation peaks correspond to oxidation of the porphyrin ligand. (TPP)MMe and (OEP)MMe (M = Ga, In) show similar redox behavior. However, cleavage of the M–Me does not occur for the M = Tl complexes and only the two reversible oxidation waves are observed. Al(TPP)Cl and Al(Por)Cl, react with Na/Hg in THF to yield the stable radicals [Al(TPP)(THF)2]˙ and [Al(Por)(THF)2]˙, respectively.100 Both are monomeric in solution with the unpaired electron delocalized throughout the ring system.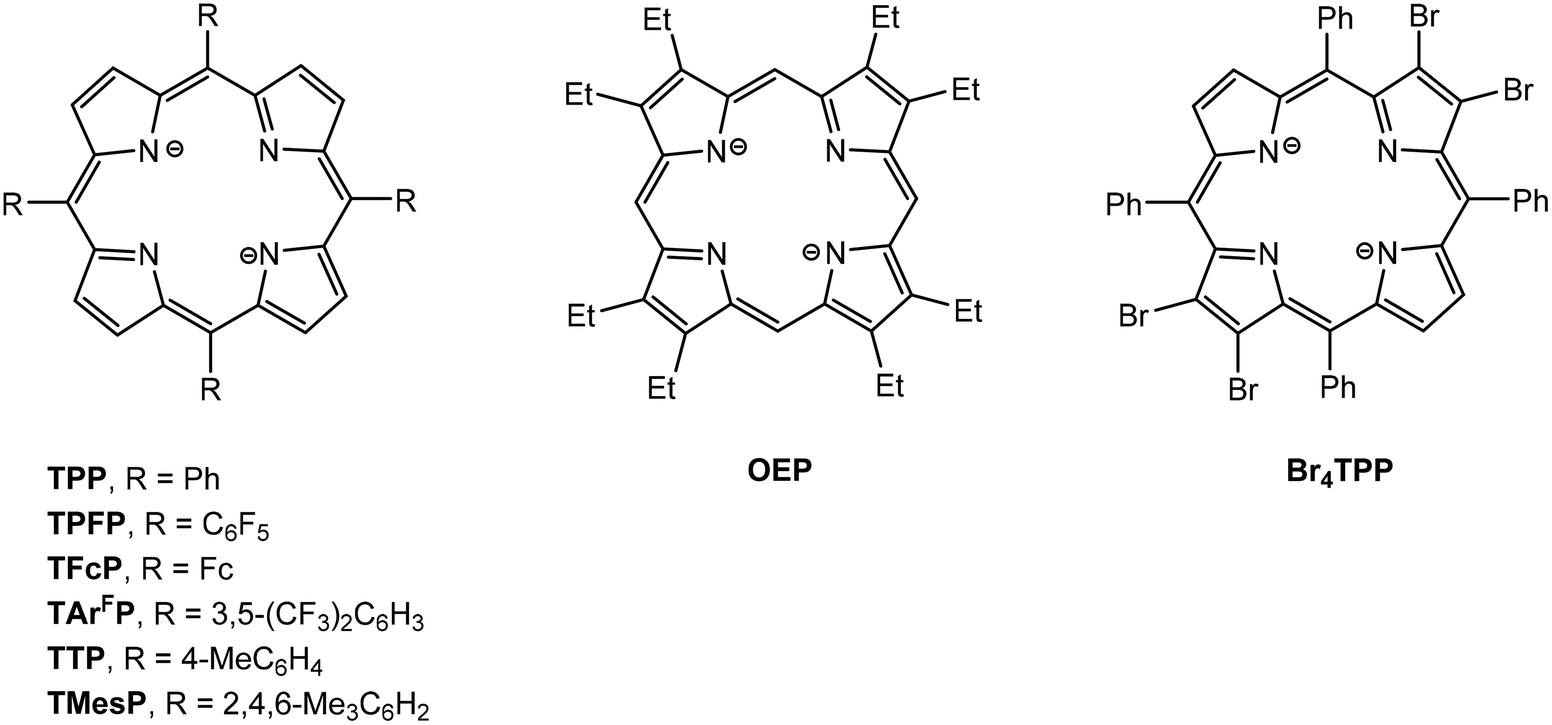 | ||
| Fig. 7 Structural drawings of porphyrinato (Por) ligands. | ||
(TPFP)GaCl (42) was shown to be a highly active and stable post-transition metal-based electrocatalyst for the hydrogen evolution reaction (HER).101 Electrochemical and spectroscopic studies in acidic media show that both the first and second reduction events of 42 are ligand-centered. The two-electron reduced form then reacts with a proton to give a GaIII–H species, which subsequently undergoes protonolysis to produce H2. Photolysis of (TPP)GaIII((CH2)4CHCH2) (43) in benzene solution results in isomerization to form (TPP)GaIII(CH2CH(CH2)4) as a result of homolytic cleavage of the Ga–C bond followed by cyclization of the 5-hexenyl radical.102 When the photolysis of (TPP)GaIII(C2H5) is carried out in the presence of dioxygen, the alky radical is trapped to form a alkylperoxy radical which recombines with the gallium radical to produce the peroxide (TPP)GaIII(OOC2H5). The formation of (TPP)GaIII(OO(CH2)4CHCH2) during the photolysis of 43 in the presence of dioxygen suggests that reaction with dioxygen occurs faster that cyclization.
CV, DPV, and SWV studies of (TFcP)InX (X = Cl, OH, Fc) show reversible oxidation of all four ferrocene substituents, and an irreversible porphyrin-centred oxidation at high positive potentials.103 The X = Fc compound shows a fifth reversible oxidation peak at lower potentials that was assigned to the axial In-ferrocenyl ligand using characteristic IVTC bands in the UV-vis-NIR spectra. These compounds were also chemically oxidized to the corresponding monocations using silver(I) triflate or dichloro-5,6-dicyanobenzoquinone (DDQ). Again, UV-vis-NIR spectroscopy was used to confirm oxidation of the axial In-ferrocenyl ligand.
The aromatic complex (TPP)GeII (44) is converted to antiaromatic (TPP)GeIV(py)2 (45) in pyridine solution.104 The reaction is reversed in benzene solution where pyridine dissociates from 45 to reform 44. This represents an unusual example of an oxidation-state change in a metal induced by coordination of a dative ligand. The complex (TArFP)GeII, which possesses a more electron withdrawing ligand, behaves similarly but favours the antiaromatic (TArFP)GeIV(py)2 product. Experimental observations and DFT studies indicate that formation of (TPP)MIV(py)2 from (TPP)MII in pyridine is most favourable for M = Si, borderline for M = Ge and unfavourable for M = Sn.
CV and DPV experiments on (TPP)SnIV(Fc)2 complexes reveal three reversible oxidation processes and two reversible reduction processes.105 The first two closely spaced oxidations were assigned to single-electron ferrocene-centered processes, while the third oxidation and both reduction couples are porphyrin core-centered single-electron processes. Both ferrocene substituents are oxidized at lower potentials compared to ferrocene. Chemical oxidation revealed two key processes associated with consecutive oxidation of the axial ferrocene substituents. The CV and SWV voltammograms of (TPP)SnIV(FcCOO)2 exhibit a single two-electron oxidative wave corresponding to simultaneous oxidation of the ferrocenecarboxylato ligands. Two reversible TPP ligand reduction waves were also observed.106 CV studies of the complexes of (Br4TPP)SnX (X = Cl, OH, OPh, p-(NO2)C6H4O) show two reversible reduction peaks, which are shifted to lower potentials versus non-brominated analogues due to the electron withdrawing effect of the β-bromine atoms.107
Tin porphyrin complexes were found to promote the oxidative cleavage of vicinal diols (Fig. 8a).108 Treatment of (TTP)SnIV(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh)2 (46) or (TTP)SnIV(NHtolyl)2 with pinacol and 2,3-diphenylbutane-2,3-diol afforded diolato complexes (TTP)SnIV[OC(Me)2C(Me)2O] (47a) and (TTP)SnIV[OC(Ph)(Me)C(Ph)(Me)O] (47b), respectively. Both complexes spontaneously undergo C–C cleavage reactions to give (TTP)SnII (48) and ketones OCMe2 and OCMe(Ph), respectively. In the presence of moist air, 48 is oxidized by dioxygen to (TTP)SnIV(OH)2 (49), which reacts with vicinal diols to reform (TTP)SnIV[OC(R)2C(R)2O] (47), thus completing the catalytic cycle. The oxidation state of the metal or TTP ligand in 48 is not confirmed in the study. In a subsequent study, the reaction of 49 with β-diketones acetylacetone, dibenzoylmethane, 1-phenylbutane-1,3-dione or 4,4-dimethyl-1-phenylpentane-1,3-dione afforded dicarboxylato tin(IV) porphyrin complexes (TPP)SnIV(O2CR)2 (R = Me (50a), Ph (50b), t-Bu (50c)) and ketonic compounds due to the C–C bond cleavage of β-diketones (Fig. 8b).109 The proposed mechanism involves nucleophilic attack of the OH ligands of 49, and a four-centred intermediate involving C–C and O–H bond cleavage and C–O and C–H bond formation. The enhanced nucleophilicity of hydroxo-ligands capable of the C–C bond cleavage of β-diketones is attributed to the electron-donating influence of porphyrin ligand to stabilize the high-valent SnIV center and increase the basicity of the hydroxo-ligands.
CPh)2 (46) or (TTP)SnIV(NHtolyl)2 with pinacol and 2,3-diphenylbutane-2,3-diol afforded diolato complexes (TTP)SnIV[OC(Me)2C(Me)2O] (47a) and (TTP)SnIV[OC(Ph)(Me)C(Ph)(Me)O] (47b), respectively. Both complexes spontaneously undergo C–C cleavage reactions to give (TTP)SnII (48) and ketones OCMe2 and OCMe(Ph), respectively. In the presence of moist air, 48 is oxidized by dioxygen to (TTP)SnIV(OH)2 (49), which reacts with vicinal diols to reform (TTP)SnIV[OC(R)2C(R)2O] (47), thus completing the catalytic cycle. The oxidation state of the metal or TTP ligand in 48 is not confirmed in the study. In a subsequent study, the reaction of 49 with β-diketones acetylacetone, dibenzoylmethane, 1-phenylbutane-1,3-dione or 4,4-dimethyl-1-phenylpentane-1,3-dione afforded dicarboxylato tin(IV) porphyrin complexes (TPP)SnIV(O2CR)2 (R = Me (50a), Ph (50b), t-Bu (50c)) and ketonic compounds due to the C–C bond cleavage of β-diketones (Fig. 8b).109 The proposed mechanism involves nucleophilic attack of the OH ligands of 49, and a four-centred intermediate involving C–C and O–H bond cleavage and C–O and C–H bond formation. The enhanced nucleophilicity of hydroxo-ligands capable of the C–C bond cleavage of β-diketones is attributed to the electron-donating influence of porphyrin ligand to stabilize the high-valent SnIV center and increase the basicity of the hydroxo-ligands.
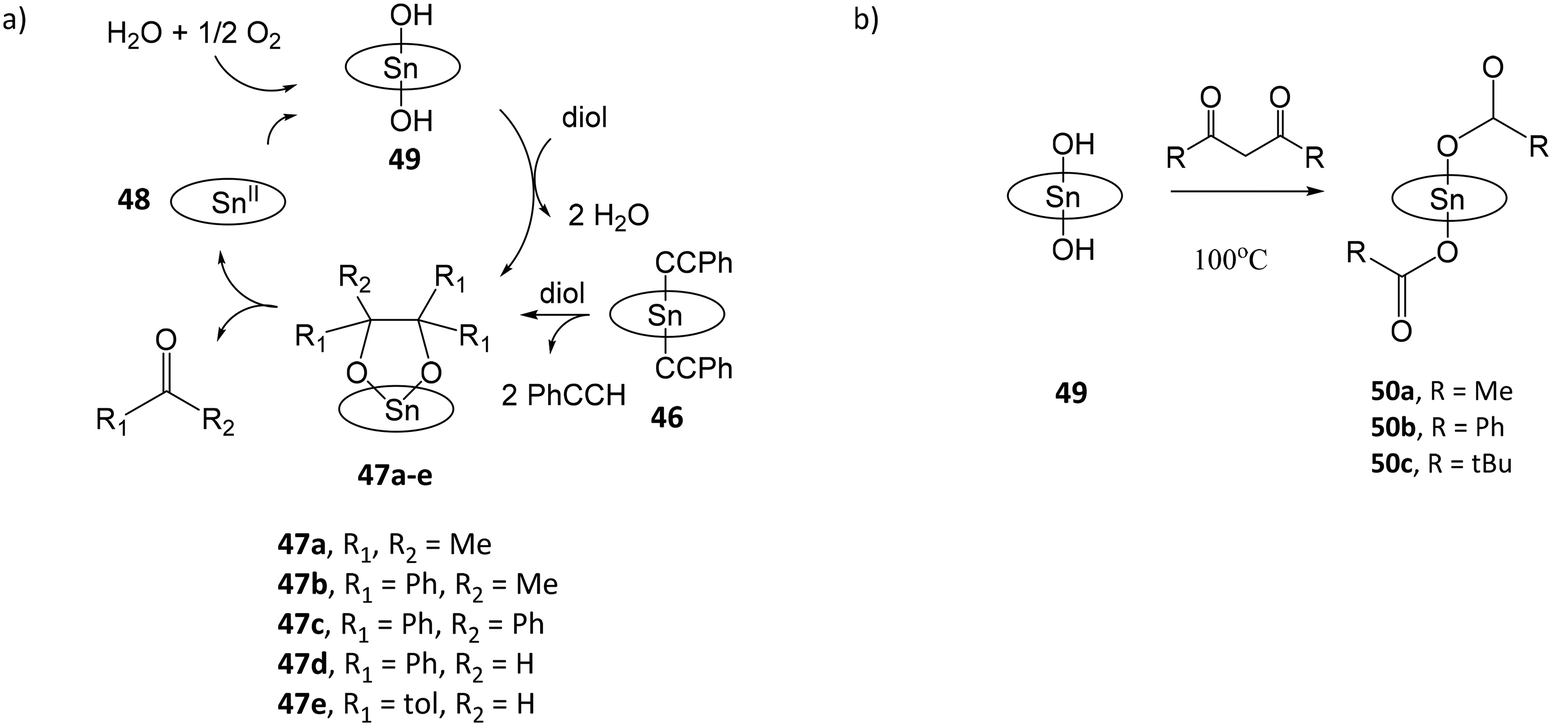 | ||
| Fig. 8 (a) Catalytic cycle for the oxidative cleavage of vicinal diols by tin porphyrin complexes; (b) reaction of (TTP)SnIV(OH)2 (49) with β-diketones to form dicarboxylato tin(IV) porphyrin complexes (50a–c). | ||
CV studies of (L)BiX (L = TPP, TTP, TMesP or OEP; X = SO3CF3, NO3) shows two reversible oxidation waves for each compound.110 UV-visible spectra of oxidized species confirm that only the porphyrin ligand is oxidized and there that is no evidence for oxidation of Bi(III) to Bi(V) in the complex. Oxidation potentials were found to vary little as a function of the counterion or solvent.
Conclusions and outlook
The reactivity and electrochemical studies presented herein highlight the potentially rich and diverse chemistry of main group metal compounds of incorporating redox-active ligands. For example, the oxidation of metal o-amidophenolate (RAP)2− and catecholate (RCat)2− compounds to produce stable o-iminosemiquinone (RISQ˙)− and o-semiquinone (RSQ˙)− compounds demonstrates the ability of these ligands to facilitate these reactions by acting as sources of electrons. Further, this reactivity can be tuned through choice of ligand substituents, which affects the redox potential of the ligand. The reversible addition of dioxygen to antimony(V) o-amidophenolates and catecholates and the reversible addition of alkynes to (RBIAN)2− supported digallanes provide examples of the corresponding reductive elimination reaction required for catalytic applications. The latter was further exploited to facilitate the catalytic hydroamination of phenylacetylenes with aniline. In both cases, bonding of the substrate was to both the metal center and the ligand, highlighting the potential for non-innocent participation of the redox-active ligand in chemical transformations. The non-innocent ligand behavior of aluminum bis(imino)pyridine compounds has been exploited by Berben et al. to mediate proton transfer for dehydrogenation catalysis. Alternatively, the catalytic oxidative C–C bond cleavage of visceral diols by a tin(IV) porphyrin complex does not involve direct participation by the porphyrin ligand, which primarily serves to promote oxidative addition and reductive elimination reactions at the tin center. Finally, the potential for reversible intramolecular electron transfer has also been shown in the redox isomerism observed in homoleptic bis(o-amidophenolato)tin(IV)/bis(o-iminosemiquinolato)tin(II) compounds and during electrochemical oxidation of mixed ligand catecholato/ferrocenyl tin(IV) compounds and ferrocenyl germanium(II) compounds.Despite these observations, information on the redox properties and reactivity of redox-active ligand compounds of main group metals, particularly those of indium and bismuth, remains relatively sparse. For example, the facile oxidation and spontaneous disproportionation reactions observed for indium o-amidophenolate and catecholate complexes demonstrate a high degree of reactivity. However, the chemical and electrochemical reversibility of these reactions has not yet been studied. Further, reactivity with a scope of substrates, including reactions with organohalides, small molecules and symmetric and asymmetric unsaturated species, has not been demonstrated. The spontaneous two-electron chemical oxidation of indium 3,4-toluenedithiolate species to yield oligomeric disulfides versus the controlled one-electron chemical oxidation of indium o-amidophenolates and catecholates shows the potential for varying reactivity with the choice of redox-active ligand. The ligand centered electrochemically reversible oxidation of ferrocenyl- and porphyrinato-indium and bismuth species suggest possible reversible chemical reactivity, while tunability of redox potentials is possible though modification of ligand substituents. Further, the synthesis of mixed redox active ligand systems leaves potential for expanding the scope of reactivity for these species.
The studies discussed herein confirm the ability of main group metal compounds of redox-active ligands to undergo “oxidative addition” type reactions to afford isolable species. However, it remains that only a very small number of these chemical reactions have been shown to be reversible. Therefore, the current challenge in the field is the identification of new reversible chemical reactions that also feature corresponding “reductive elimination” processes. This will ultimately require systematic studies involving informed selection of main group metal, redox-active ligand, substituents and substrate. In the quest to establish the potential utility of main group metal compounds in catalytic redox processes, the chemical reactivity and electrochemical properties of compounds containing redox-active ligands is an intriguing avenue of inquiry and one that should be fully explored.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Natural Sciences and Engineering Research Council of Canada (RGPIN-2019-05965), the New Brunswick Innovation Foundation, the Canadian Foundation for Innovation (Project No. 9211) and Mount Allison University are thanked for financial support.References
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- C. Weetman and S. Inoue, ChemCatChem, 2018, 10, 4213–4228 CrossRef CAS.
- M.-A. Légaré, C. Pranckevicius and H. Braunschweig, Chem. Rev., 2019, 119, 8231–8261 CrossRef PubMed.
- T. Chu and G. I. Nikonov, Chem. Rev., 2018, 118, 3608–3680 CrossRef CAS PubMed.
- O. Planas, F. Wang, M. Leutzsch and J. Cornella, Science, 2020, 367, 313–317 CrossRef CAS PubMed.
- O. Planas, V. Peciukenas and J. Cornella, J. Am. Chem. Soc., 2020, 142, 11382–11387 CrossRef CAS PubMed.
- H. W. Moon and J. Cornella, ACS Catal., 2022, 12, 1382–1393 CrossRef CAS PubMed.
- W. Kaim, Eur. J. Inorg. Chem., 2012, 2012, 343–348 CrossRef CAS.
- O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440–1459 RSC.
- T. Fogeron, T. K. Todorova, J.-P. Porcher, M. Gomez-Mingot, L.-M. Chamoreau, C. Mellot-Draznieks, Y. Li and M. Fontecave, ACS Catal., 2018, 8, 2030–2038 CrossRef CAS.
- V. K. K. Praneeth, M. R. Ringenberg and T. R. Ward, Angew. Chem., Int. Ed., 2012, 51, 10228–10234 CrossRef CAS PubMed.
- V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS.
- P. J. Chirik and K. Wieghardt, Science, 2010, 327, 794–795 CrossRef CAS PubMed.
- D. L. J. Broere, R. Plessius and J. I. van der Vlugt, Chem. Soc. Rev., 2015, 44, 6886–6915 RSC.
- J. I. van der Vlugt, Chem. – Eur. J., 2019, 25, 2651–2662 CrossRef CAS PubMed.
- B. Garreau-de Bonneval, K. I. Moineau-Chane Ching, F. Alary, T.-T. Bui and L. Valade, Coord. Chem. Rev., 2010, 254, 1457–1467 CrossRef CAS.
- R. O. Fields, J. H. Waters and T. J. Bergendahl, Inorg. Chem., 1971, 10, 2808–2810 CrossRef CAS.
- D. G. Tuck and M. K. Yang, J. Chem. Soc. A, 1971, 214–219 RSC.
- I. V. Ershova and A. V. Piskunov, Russ. J. Coord. Chem., 2020, 46, 154–177 CrossRef CAS.
- A. V. Piskunov, I. V. Ershova and G. K. Fukin, Russ. Chem. Bull., 2014, 63, 916–922 CrossRef CAS.
- A. V. Piskunov, I. N. Mescheryakova, A. S. Bogomyakov, G. V. Romanenko, V. K. Cherkasov and G. A. Abakumov, Inorg. Chem. Commun., 2009, 12, 1067–1070 CrossRef CAS.
- A. V. Piskunov, I. N. Mescheryakova, G. K. Fukin, V. K. Cherkasov and G. A. Abakumov, New J. Chem., 2010, 34, 1746–1750 RSC.
- A. V. Piskunov, I. N. Meshcheryakova, I. V. Ershova, A. S. Bogomyakov, A. V. Cherkasov and G. K. Fukin, RSC Adv., 2014, 4, 42494–42505 RSC.
- I. V. Ershova, A. S. Bogomyakov, G. K. Fukin and A. V. Piskunov, Eur. J. Inorg. Chem., 2019, 2019, 938–948 CrossRef CAS.
- A. V. Piskunov, I. N. Meshcheryakova, G. K. Fukin, A. S. Shavyrin, V. K. Cherkasov and G. A. Abakumov, Dalton Trans., 2013, 42, 10533–10539 RSC.
- A. V. Piskunov, I. V. Ershova, G. K. Fukin and A. S. Shavyrin, Inorg. Chem. Commun., 2013, 38, 127–130 CrossRef CAS.
- P. Chaudhuri, E. Bill, R. Wagner, U. Pieper, B. Biswas and T. Weyhermüller, Inorg. Chem., 2008, 47, 5549–5551 CrossRef CAS PubMed.
- K. V. Arsenyeva, A. V. Klimashevskaya, M. A. Zherebtsov, M. G. Chegerev, A. V. Cherkasov, I. A. Yakushev and A. V. Piskunov, Russ. J. Coord. Chem., 2022, 48, 464–477 CrossRef CAS.
- K. V. Arsenyeva, K. I. Pashanova, O. Y. Trofimova, I. V. Ershova, M. G. Chegerev, A. A. Starikova, A. V. Cherkasov, M. A. Syroeshkin, A. Y. Kozmenkova and A. V. Piskunov, New J. Chem., 2021, 45, 11758–11767 RSC.
- A. V. Piskunov, K. V. Tsys, M. G. Chegerev and A. V. Cherkasov, Russ. J. Coord. Chem., 2019, 45, 626–636 CrossRef CAS.
- M. G. Chegerev, A. V. Piskunov, A. A. Starikova, S. P. Kubrin, G. K. Fukin, V. K. Cherkasov and G. A. Abakumov, Eur. J. Inorg. Chem., 2018, 2018, 1087–1092 CrossRef CAS.
- A. V. Piskunov, I. N. Mescheryakova, G. K. Fukin, A. S. Bogomyakov, G. V. Romanenko, V. K. Cherkasov and G. A. Abakumov, Heteroat. Chem., 2009, 20, 332–340 CrossRef CAS.
- K. V. Tsys, M. G. Chegerev, G. K. Fukin and A. V. Piskunov, Mendeleev Commun., 2018, 28, 527–529 CrossRef CAS.
- A. V. Piskunov, I. N. Mescheryakova, G. K. Fukin, E. V. Baranov, M. Hummert, A. S. Shavyrin, V. K. Cherkasov and G. A. Abakumov, Chem. – Eur. J., 2008, 14, 10085–10093 CrossRef CAS PubMed.
- A. V. Piskunov, M. G. Chegerev and G. K. Fukin, J. Organomet. Chem., 2016, 803, 51–57 CrossRef CAS.
- M. G. Chegerev, A. V. Piskunov, A. V. Maleeva, G. K. Fukin and G. A. Abakumov, Eur. J. Inorg. Chem., 2016, 2016, 3813–3821 CrossRef CAS.
- S. V. Baryshnikova, A. I. Poddel'sky, E. V. Bellan, I. V. Smolyaninov, A. V. Cherkasov, G. K. Fukin, N. T. Berberova, V. K. Cherkasov and G. A. Abakumov, Inorg. Chem., 2020, 59, 6774–6784 CrossRef CAS PubMed.
- G. K. Fukin, E. V. Baranov, A. I. Poddel'sky, V. K. Cherkasov and G. A. Abakumov, ChemPhysChem, 2012, 13, 3773–3776 CrossRef CAS PubMed.
- G. A. Abakumov, A. I. Poddel'sky, E. V. Grunova, V. K. Cherkasov, G. K. Fukin, Y. A. Kurskii and L. G. Abakumova, Angew. Chem., Int. Ed., 2005, 44, 2767–2771 CrossRef CAS PubMed.
- A. I. Poddel'sky, Y. A. Kurskii, A. V. Piskunov, N. V. Somov, V. K. Cherkasov and G. A. Abakumov, Appl. Organomet. Chem., 2011, 25, 180–189 CrossRef.
- A. I. Poddel'sky, N. N. Vavilina, N. V. Somov, V. K. Cherkasov and G. A. Abakumov, J. Organomet. Chem., 2009, 694, 3462–3469 CrossRef.
- V. K. Cherkasov, G. A. Abakumov, E. V. Grunova, A. I. Poddel'sky, G. K. Fukin, E. V. Baranov, Y. V. Kurskii and L. G. Abakumova, Chem. – Eur. J., 2006, 12, 3916–3927 CrossRef CAS PubMed.
- P. Chaudhuri, M. Hess, K. Hildenbrand, E. Bill, T. Weyhermüller and K. Wieghardt, Inorg. Chem., 1999, 38, 2781–2790 CrossRef CAS PubMed.
- G. Szigethy and A. F. Heyduk, Dalton Trans., 2012, 41, 8144–8152 RSC.
- U. Beckmann, E. Bill, T. Weyhermüller and K. Wieghardt, Eur. J. Inorg. Chem., 2003, 2003, 1768–1777 CrossRef.
- A. V. Piskunov, A. V. Maleeva, I. N. Meshcheryakova and G. K. Fukin, Russ. J. Coord. Chem., 2013, 39, 245–256 CrossRef CAS.
- A. I. Poddel'sky, I. V. Smolyaninov, Y. A. Kurskii, G. K. Fukin, N. T. Berberova, V. K. Cherkasov and G. A. Abakumov, J. Organomet. Chem., 2010, 695, 1215–1224 CrossRef.
- A. I. Poddel'sky, I. V. Smolyaninov, N. V. Somov, N. T. Berberova, V. K. Cherkasov and G. A. Abakumov, J. Organomet. Chem., 2010, 695, 530–536 CrossRef.
- V. K. Cherkasov, E. V. Grunova, A. I. Poddel'sky, G. K. Fukin, Y. A. Kurskii, L. G. Abakumova and G. A. Abakumov, J. Organomet. Chem., 2005, 690, 1273–1281 CrossRef CAS.
- Y. Zhao, Y. Liu, L. Yang, J.-G. Yu, S. Li, B. Wu and X.-J. Yang, Chem. – Eur. J., 2012, 18, 6022–6030 CrossRef CAS PubMed.
- I. L. Fedushkin, A. A. Skatova, V. A. Dodonov, V. A. Chudakova, N. L. Bazyakina, A. V. Piskunov, S. V. Demeshko and G. K. Fukin, Inorg. Chem., 2014, 53, 5159–5170 CrossRef CAS PubMed.
- I. L. Fedushkin, A. S. Nikipelov, A. A. Skatova, O. V. Maslova, A. N. Lukoyanov, G. K. Fukin and A. V. Cherkasov, Eur. J. Inorg. Chem., 2009, 2009, 3742–3749 CrossRef.
- I. L. Fedushkin, A. S. Nikipelov and K. A. Lyssenko, J. Am. Chem. Soc., 2010, 132, 7874–7875 CrossRef CAS PubMed.
- I. L. Fedushkin, A. S. Nikipelov, A. G. Morozov, A. A. Skatova, A. V. Cherkasov and G. A. Abakumov, Chem. – Eur. J., 2012, 18, 255–266 CrossRef CAS PubMed.
- C. J. Allan, B. F. T. Cooper, H. J. Cowley, J. M. Rawson and C. L. B. Macdonald, Chem. – Eur. J., 2013, 19, 14470–14483 CrossRef CAS PubMed.
- R. J. Baker, R. D. Farley, C. Jones, M. Kloth and D. M. Murphy, Chem. Commun., 2002, 1196–1197 RSC.
- R. J. Baker, R. D. Farley, C. Jones, D. P. Mills, M. Kloth and D. M. Murphy, Chem. – Eur. J., 2005, 11, 2972–2982 CrossRef CAS PubMed.
- H. Rojas-Sáenz, G. V. Suárez-Moreno, I. Ramos-García, A. M. Duarte-Hernández, E. Mijangos, A. Peña-Hueso, R. Contreras and A. Flores-Parra, New J. Chem., 2013, 38, 391–405 RSC.
- J. Wang, R. Ganguly, L. Yongxin, J. Díaz, H. S. Soo and F. García, Dalton Trans., 2016, 45, 7941–7946 RSC.
- J. Wang, R. Ganguly, L. Yongxin, J. Díaz, H. S. Soo and F. García, Inorg. Chem., 2017, 56, 7811–7820 CrossRef CAS PubMed.
- V. A. Dodonov, O. A. Kushnerova, D. A. Razborov, E. V. Baranov, E. A. Ulivanova, A. N. Lukoyanov and I. L. Fedushkin, Russ. Chem. Bull., 2022, 71, 322–329 CrossRef CAS.
- G. Reeske, C. R. Hoberg, N. J. Hill and A. H. Cowley, J. Am. Chem. Soc., 2006, 128, 2800–2801 CrossRef CAS PubMed.
- J. W. Dube, G. J. Farrar, E. L. Norton, K. L. S. Szekely, B. F. T. Cooper and C. L. B. Macdonald, Organometallics, 2009, 28, 4377–4384 CrossRef CAS.
- G. Reeske and A. H. Cowley, Inorg. Chem., 2007, 46, 1426–1430 CrossRef CAS PubMed.
- A. L. Brazeau, N. D. Jones and P. J. Ragogna, Dalton Trans., 2012, 41, 7890–7896 RSC.
- T. W. Myers and L. A. Berben, Inorg. Chem., 2012, 51, 1480–1488 CrossRef CAS PubMed.
- T. M. Bass, C. R. Carr, T. J. Sherbow, J. C. Fettinger and L. A. Berben, Inorg. Chem., 2020, 59, 13517–13523 CrossRef CAS PubMed.
- T. J. Sherbow, L. W. T. Parsons, N. A. Phan, J. C. Fettinger and L. A. Berben, Inorg. Chem., 2020, 59, 17614–17619 CrossRef CAS PubMed.
- A. Arnold, T. J. Sherbow, A. M. Bohanon, R. I. Sayler, R. D. Britt, A. M. Smith, J. C. Fettinger and L. A. Berben, Chem. Sci., 2021, 12, 675–682 RSC.
- L. A. Berben, Chem. – Eur. J., 2015, 21, 2734–2742 CrossRef CAS PubMed.
- A. Arnold, T. J. Sherbow, R. I. Sayler, R. D. Britt, E. J. Thompson, M. T. Muñoz, J. C. Fettinger and L. A. Berben, J. Am. Chem. Soc., 2019, 141, 15792–15803 CrossRef CAS PubMed.
- E. N. Egorova, N. O. Druzhkov, A. S. Shavyrin, A. V. Cherkasov, G. A. Abakumov and A. Y. Fedorov, RSC Adv., 2015, 5, 19362–19367 RSC.
- G. Hunter, J. Chem. Soc., Dalton Trans., 1972, 1496–1498 RSC.
- B. E. Bosch, M. Eisenhawer, B. Kersting, K. Kirschbaum, B. Krebs and D. M. Giolando, Inorg. Chem., 1996, 35, 6599–6605 CrossRef CAS PubMed.
- G. G. Briand, T. George, G. A. MacNeil, J. D. Masuda, B. J. MacLean, M. W. R. Mosher, G. M. Sandala, P. Srinivasan, A. H. Stockli, R. L. Vanderkloet and C. J. Walsby, Eur. J. Inorg. Chem., 2023, 26, e202200542 CrossRef CAS.
- C. W. Allen, D. E. Lutes, E. J. Durhan and E. S. Bretschneider, Inorg. Chim. Acta, 1977, 21, 277–279 CrossRef CAS.
- J. A. Schachner, G. A. Orlowski, J. W. Quail, H.-B. Kraatz and J. Müller, Inorg. Chem., 2006, 45, 454–459 CrossRef CAS PubMed.
- A. Althoff, P. Jutzi, N. Lenze, B. Neumann, A. Stammler and H.-G. Stammler, Organometallics, 2003, 22, 2766–2774 CrossRef CAS.
- J. A. Schachner, C. L. Lund, I. J. Burgess, J. W. Quail, G. Schatte and J. Müller, Organometallics, 2008, 27, 4703–4710 CrossRef CAS.
- B. Bagh, N. C. Breit, K. Harms, G. Schatte, I. J. Burgess, H. Braunschweig and J. Müller, Inorg. Chem., 2012, 51, 11155–11167 CrossRef CAS PubMed.
- P. Jutzi, N. Lenze, B. Neumann and H.-G. Stammler, Angew. Chem., Int. Ed., 2001, 40, 1423–1427 CrossRef PubMed.
- A. Althoff, D. Eisner, P. Jutzi, N. Lenze, B. Neumann, W. W. Schoeller and H.-G. Stammler, Chem. – Eur. J., 2006, 12, 5471–5480 CrossRef CAS PubMed.
- M. J. MacLachlan, A. J. Lough, W. E. Geiger and I. Manners, Organometallics, 1998, 17, 1873–1883 CrossRef CAS.
- M. Castruita, F. Cervantes-Lee, J. S. Mahmoud, Y. Zhang and K. H. Pannell, J. Organomet. Chem., 2001, 637–639, 664–668 CrossRef CAS.
- Y. Suzuki, T. Sasamori, J.-D. Guo and N. Tokitoh, Chem. – Eur. J., 2018, 24, 364–368 CrossRef CAS PubMed.
- D. Siebler, C. Förster, T. Gasi and K. Heinze, Chem. Commun., 2010, 46, 4490–4492 RSC.
- K. Jacob, N. Seidel, F. Voigt, A. Fischer, C. Pietzsch, J. Holecek, A. Lycka, M. Fontani, E. Grigiotti and P. Zanello, J. Prakt. Chem., 2000, 342, 574–584 CrossRef CAS.
- N. Lenze, B. Neumann, A. Salmon, A. Stammler, H.-G. Stammler and P. Jutzi, J. Organomet. Chem., 2001, 619, 74–87 CrossRef CAS.
- Z. You, J. Bergunde, B. Gerke, R. Pöttgen and S. Dehnen, Inorg. Chem., 2014, 53, 12512–12518 CrossRef CAS PubMed.
- H. P. Nayek, G. Hilt and S. Dehnen, Eur. J. Inorg. Chem., 2009, 2009, 4205–4208 CrossRef.
- M. Herberhold, W. Milius, U. Steffl, K. Vitzithum, B. Wrackmeyer, R. H. Herber, M. Fontani and P. Zanello, Eur. J. Inorg. Chem., 1999, 1999, 145–151 CrossRef.
- M. Henn, M. Schürmann, B. Mahieu, P. Zanello, A. Cinquantini and K. Jurkschat, J. Organomet. Chem., 2006, 691, 1560–1572 CrossRef CAS.
- J. Schulz, J. Antala, I. Císařová and P. Štěpnička, Dalton Trans., 2023, 52, 1198–1211 RSC.
- T.-Y. Dong, M.-Y. Hwang, Y. Wen and W.-S. Hwang, J. Organomet. Chem., 1990, 391, 377–385 CrossRef CAS.
- G. Utri, K.-E. Schwarzhans and G. M. Allmaier, Z. Naturforsch., B: J. Chem. Sci., 1990, 45, 755–762 CrossRef CAS.
- M. Sakagami, T. Sasamori, H. Sakai, Y. Furukawa and N. Tokitoh, Chem. – Asian J., 2013, 8, 690–693 CrossRef CAS PubMed.
- T. Sasamori, H. Miyamoto, H. Sakai, Y. Furukawa and N. Tokitoh, Organometallics, 2012, 31, 3904–3910 CrossRef CAS.
- H. B. Gray, D. N. Hendrickson and Y. S. Sohn, Inorg. Chem., 1971, 10, 1559–1563 CrossRef CAS.
- R. Guilard, A. Zrineh, A. Tabard, A. Endo, B. C. Han, C. Lecomte, M. Souhassou, A. Habbou, M. Ferhat and K. M. Kadish, Inorg. Chem., 1990, 29, 4476–4482 CrossRef CAS.
- J. A. Cissell, T. P. Vaid and A. L. Rheingold, Inorg. Chem., 2006, 45, 2367–2369 CrossRef CAS PubMed.
- N. Wang, H. Lei, Z. Zhang, J. Li, W. Zhang and R. Cao, Chem. Sci., 2019, 10, 2308–2314 RSC.
- A. L. Balch, R. L. Hart and S. Parkin, Inorg. Chim. Acta, 1993, 205, 137–143 CrossRef CAS.
- S. J. Dammer, P. V. Solntsev, J. R. Sabin and V. N. Nemykin, Inorg. Chem., 2013, 52, 9496–9510 CrossRef CAS PubMed.
- J. A. Cissell, T. P. Vaid and G. P. A. Yap, J. Am. Chem. Soc., 2007, 129, 7841–7847 CrossRef CAS PubMed.
- P. V. Solntsev, J. R. Sabin, S. J. Dammer, N. N. Gerasimchuk and V. N. Nemykin, Chem. Commun., 2010, 46, 6581–6583 RSC.
- H. J. Kim, W. S. Jeon, J. H. Lim, C. S. Hong and H.-J. Kim, Polyhedron, 2007, 26, 2517–2522 CrossRef CAS.
- M. Ravikumar, A. Kathiravan, A. Neels and E. M. Mothi, Eur. J. Inorg. Chem., 2018, 2018, 3868–3877 CrossRef CAS.
- G. Du, A. Ellern and L. K. Woo, Inorg. Chem., 2004, 43, 2379–2386 CrossRef CAS PubMed.
- A. P. Singh, B. B. Park and H.-J. Kim, Tetrahedron Lett., 2012, 53, 6456–6459 CrossRef CAS.
- L. Michaudet, D. Fasseur, R. Guilard, Z. Ou, K. M. Kadish, S. Dahaoui and C. Lecomte, J. Porphyrins Phthalocyanines, 2000, 4, 261–270 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |