
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of imidazolium-based pentacoordinated organofluorosilicate and germanate salts†
Jan
Gnidovec
 ab,
Evelin
Gruden
a,
Melita
Tramšek
a,
Jernej
Iskra
c,
Jaroslav
Kvíčala
d and
Gašper
Tavčar
*ab
ab,
Evelin
Gruden
a,
Melita
Tramšek
a,
Jernej
Iskra
c,
Jaroslav
Kvíčala
d and
Gašper
Tavčar
*ab
aDepartment of Inorganic Chemistry and Technology, Jožef Stefan Institute, Jamova 39, 1000 Ljubljana, Slovenia. E-mail: gasper.tavcar@ijs.si
bJožef Stefan International Postgraduate School, Jamova 39, 1000 Ljubljana, Slovenia
cDepartment of Chemistry and Biochemistry, University of Ljubljana, Faculty of Chemistry and Chemical Technology, Večna pot 113, 1000 Ljubljana, Slovenia
dDepartment of Organic Chemistry, University of Chemistry and Technology, Prague, Technická 5, 166 28 Prague 6, Czech Republic
First published on 29th March 2023
Abstract
The syntheses of a new triphenyldifluorogermanate and various pentacoordinated organofluorosilicates are presented. The fluorogermane and fluorosilane compounds were obtained from the corresponding chlorosilanes and chlorogermane by halogen substitution with KF. Subsequent reaction with the imidazolium-based fluoride reagent [IPrH][F] (1,3-bis(2,6-diisopropylphenyl)imidazolium fluoride) led to the formation of [IPrH][Ph3SiF2] (1), [IPrH][Ph2SiF3] (3), [IPrH][Et2SiF3] (4), [IPrH][PhSiF4] (5), [IPrH][EtSiF4] (6) and [IPrH][Ph3GeF2] (7). All the products obtained were characterised by NMR, Raman spectroscopy and X-ray diffraction. The results were supported by DFT calculations of the structurally optimised compounds.
Introduction
Pentacoordinated organofluorosilicates ([R5−nSiFn]−; n = 1–4) are silicon-based compounds containing a combination of organic and fluoride substituents. They initially attracted great interest due to their excellent properties as fluoride ion donors.1,2 Their advantage is their improved solubility compared to readily available alkali metal fluorides and their increased stability compared to the widely used tetraalkylammonium fluorides.3 Notable examples are the [Me3SiF2]− and [Ph3SiF2]− anions which are an attractive choice for nucleophilic fluorination.2,4 Since the 1980s, when they were first structurally characterised, there have been numerous reports of unique pentacoordinated organofluorosilicate structures (Fig. 1).2,5–12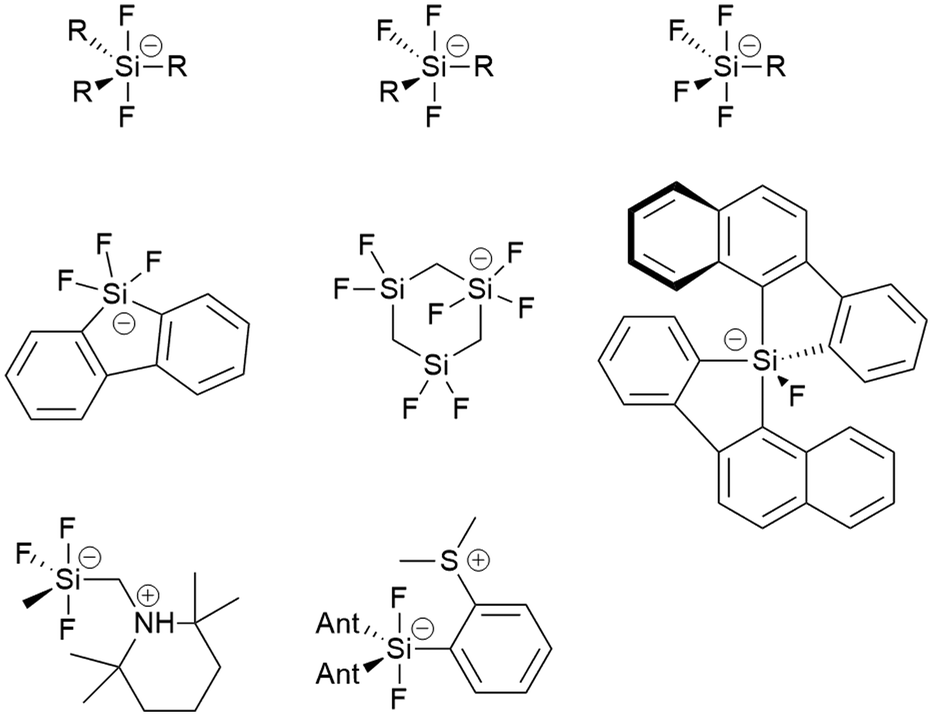 | ||
| Fig. 1 Examples of previously structurally characterised organofluorosilicates. | ||
Organofluorosilicate structures adopt a trigonal-bipyramidal configuration with the fluorine generally in the axial position.2 Only in one case has fluorine been observed in equatorial position, when the sterically demanding nature of a bulky bidentate ligand forced the fluorine atom into the equatorial plane.10 These pentacoordinated compounds contain 1 to 4 fluorine atoms, as shown in Fig. 1. The remaining positions are occupied by various organic substituents. Most of them are simple alkyl or aryl groups, although it has been shown that silicon can form heterocyclic structures with bidentate ligands.8–10,13 In certain cases, organofluorosilicates can even form the backbone of a coordination polymer.14 Due to the Lewis acidic properties of silanes, polydentate silicon-based ligand acceptors have been developed, leading to various hypervalent polysilicon species.9,15 In this way, the properties of fluorosilanes can be tuned to increase their efficiency in complexing fluoride anion, making them a useful fluoride sensor.12 Zwitterionic pentacoordinated organofluorosilicates are an example of neutral organofluorosilicates in which the silicon is negatively charged while an adjacent group contains a positive charge.11
Organofluorogermanates, on the other hand, are extremely rare. To our knowledge, there are only a few reports of structurally characterised organofluorogermanates [C23H17O][(CF3CF2)3GeF2] and [(CH3)4N][(CF3)3GeF2].16,17 Interestingly, all of them contain strongly electron-withdrawing groups. Like their silicon relatives, fluorogermanates prefer trigonal-bipyramidal configuration with fluorine atoms in the axial positions.
In 2016 the imidazolium-based fluorination reagent [IPrH][F] was developed in our research group.18 It has the ability to act as a free fluoride reagent and its reactivity has already been used for the synthesis of discrete [GeF5]−, [SiF5]−, [VOF4]−, [NbF6]−, [TaF6]−, [Me3AlF]− and [(n-Bu)3AlF]− anions.19–22 The preparation and structural characterisation of these anions was also possible due to the stabilising ability of the [IPrH]+ cation. The steric bulkiness of the cation and its interactions with the anions allow the stabilisation of discrete anions, that would otherwise form polynuclear species, by hindering their interactions with neighbouring anions. A good example of this is the stabilisation of discrete [GeF5]− anions in the structure of [IPrH][GeF5], which tends to form octahedrally coordinated [GeF6]2− units in the presence of sterically less demanding counterions.19 So far, the reactivity of [IPrH][F] has been tested on inorganic compounds known for their strong Lewis acidity20,22 as well as on SiF4 and GeF4.19
In this work, we were interested in reactivity of [IPrH][F] with even weaker Lewis acidic species. The formation and stability of organofluorosilicates depends strongly on the organic substituents attached to the silicon centre. Strong electron-withdrawing groups help in the formation of pentacoordinated anionic species. It is therefore not surprising that there is a distinct lack of organofluorosilicates with electron-donating groups. To this end, we have attempted to synthesise organofluorosilicates with electron donating ethyl groups. Due to the extremely small number of organofluorogermanate compounds, we also decided to test the reactivity towards an organofluorogermane.
Results and discussion
In this work we prepared 5 organofluorosilicate salts [IPrH][R4−nSiFn+1] (n = 1–3, R = Ph, Et) (1–6) and 1 phenylfluorogermanate [IPrH][Ph3GeF2] (7) starting from the corresponding organochlorosilanes or chlorogermane using KF and the imidazolium-based fluoride reagent [IPrH][F]. The products were prepared according to the synthetic procedures shown in Schemes 1 and 2.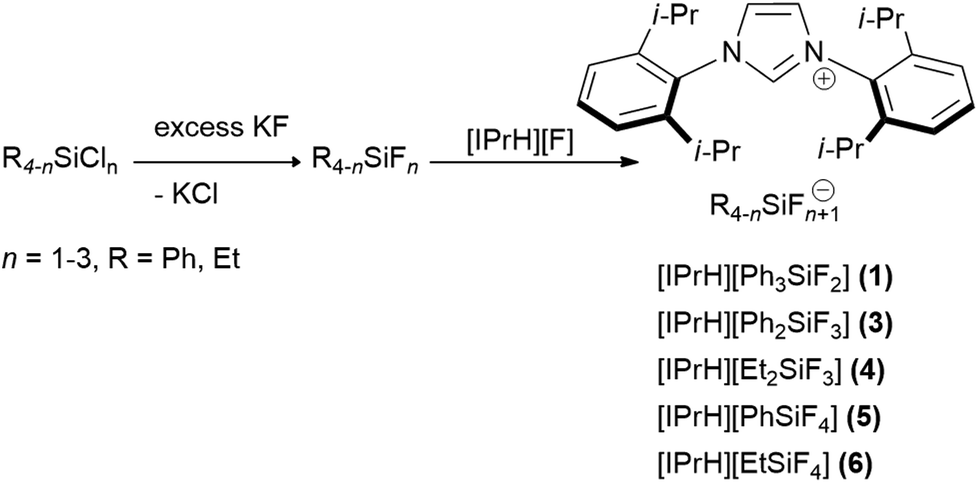 | ||
| Scheme 1 The general synthetic procedure for preparation of organofluorosilicates [IPrH][R4−nSiFn+1] (n = 1–3, R = Ph, Et) (1–6). | ||
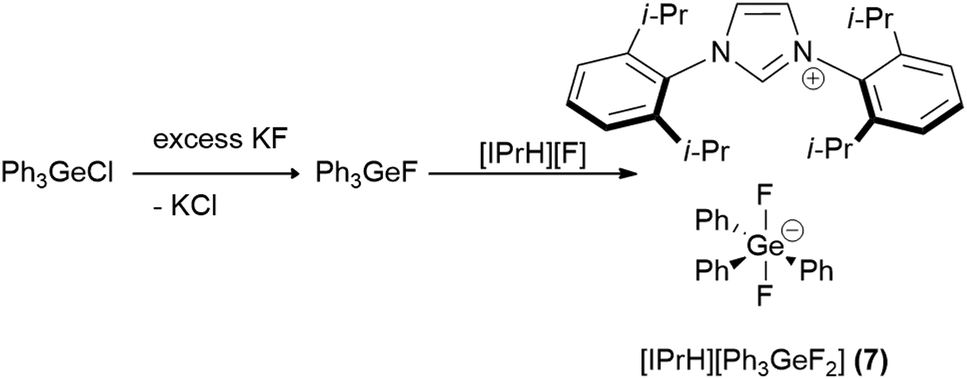 | ||
| Scheme 2 The synthetic procedure for preparation of [IPrH][Ph3GeF2] (7). | ||
In the first step, we converted organochlorosilanes [R4−nSiCln] (n = 1–3, R = Ph, Et) and triphenylchlorogermane [Ph3GeCl] to the corresponding organofluorosilanes [R4−nSiFn] (n = 1–3, R = Ph, Et) and triphenylfluorogermane [Ph3GeF] by using excess amounts of at least 5 equivalents of KF per chlorine atom of the starting compound. Excess amounts were used to ensure complete exchange of the halides. After completion of the reaction, the reaction mixture was filtered through PTFE filters to remove KCl and the remaining KF. In the second step, the filtrate was added to the equimolar solution of [IPrH][F] and stirred overnight. The crystals of the products were isolated by slowly evaporating the solvent in a closed system under static vacuum, the driving force being a temperature difference between 2 parts of the vessel. The crystals of compounds [IPrH][Ph3SiF2] (1), [IPrH][Ph2SiF3] (3), [IPrH][Et2SiF3] (4), [IPrH][PhSiF4] (5), [IPrH][EtSiF4] (6) and [IPrH][Ph3GeF2] (7) were obtained in satisfactory quality and were used for characterisation and further analysis. The ethylfluorosilicates proved to be significantly less stable than their phenyl counterparts. Under reduced dynamic pressure, the ethylfluorosilicates decomposed back into the reactants and the volatile fluorosilane was removed from the reaction mixture. As a result, only [IPrH][F] remained in the vessel, which was confirmed by NMR spectroscopy and X-ray diffraction.
In the next sections, the structural properties of triorganodifluorosilicates, diorganotrifluorosilicates and organotetrafluorosilicates are going to be presented simultaneously for the ethyl and phenyl counterparts. The properties of the triphenyldifluorogermanate are presented separately.
Triorganodifluorosilicates [IPrH][R3SiF2] (R = Ph, Et)
[IPrH][Ph3SiF2] (1) was prepared in quantitative yield according to the general procedure given above. Many attempts were made to synthesise [IPrH][Et3SiF2] (2), but all were unsuccessful. Unfortunately, we were not even able to observe the formation of 2 in solution. By following the progress of the reaction between [Et3SiF] and [IPrH][F] in MeCN in an NMR tube, we found that the silane [Et3SiF] did not react and only the reactants were visible in the reaction mixture. Our results indicate that 2 cannot be prepared under these conditions (Fig. S4 and S5 in the ESI†). Compound 1 crystallises in the monoclinic space group P21/c and its asymmetric unit is shown in Fig. 2. It contains an imidazolium cation [IPrH]+ and a triphenyldifluorosilicate anion [Ph3SiF2]−, which adopts a trigonal bipyramidal geometry with the fluorine atoms occupying the axial positions. The two Si–F bond distances in the anion are very similar. The Si(1)–F(1) has a bond distance of 1.743(1) Å, while Si(1)–F(2) has a distance of 1.746(1) Å. This difference is quite small compared to other crystal structures of this anion where this difference in Si–F bond length is more pronounced (1.734(1) and 1.721(1) Å; 1.753(2) and 1.718(2) Å; 1.732(1) and 1.726(1) Å).5,23,24 This could be due to the packing or the similarly strong hydrogen bonds that both fluorine atoms form with imidazolium cations. The [Ph3SiF2]− anion interacts with two adjacent cations to form 2 hydrogen bonds, each with one fluorine atom. The first hydrogen bond forms between F(1) and the hydrogen atom at the C2 position C(22)–H(22)⋯F(1) with an H⋯F distance of 2.0841(9) Å, while the second hydrogen bond forms between F(2) and the hydrogen atom on the backbone of the symmetrically generated unit C(24)–H(24)⋯F(2) with an H⋯F distance of 2.0383(9) Å. All significant interactions between cations and anions of the structurally characterised compounds are described with bond lengths and bond angles and are listed in Table S4 in the ESI.† Significant interactions were defined according to the calculations for [IPrH][GeF5] and [IPrH][SiF5].19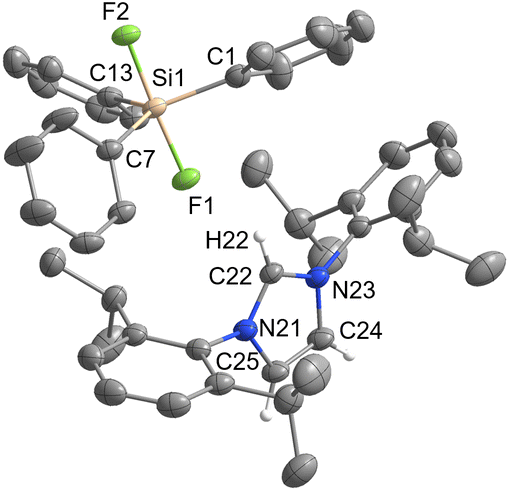 | ||
| Fig. 2 The crystal structure of [IPrH][Ph3SiF2] (1). The ellipsoids are drawn at 50% probability. All hydrogen atoms are omitted except for those on the imidazolium ring. Selected bond lengths (Å): Si(1)–F(1) 1.743(1), Si(1)–F(2) 1.746(1), Si(1)–C(1) 1.898(2), Si(1)–C(7) 1.906(2) and Si(1)–C(13) 1.907(2). | ||
Diorganotrifluorosilicates [IPrH][R2SiF3] (R = Ph, Et)
[IPrH][Ph2SiF3] (3) and [IPrH][Et2SiF3] (4) were prepared in quantitative yield according to the general procedure listed above. Compound 3 crystallises in the monoclinic space group P21/c, while 4 crystallises in the form of a solvate with MeCN (4·MeCN) in the monoclinic space group P21/n. The crystal structures of 3 and 4 are shown in Fig. 3 and 4, respectively. In the crystal structure of 3, the asymmetric unit is composed of two ion pairs. For clarity, only one is shown in Fig. 3.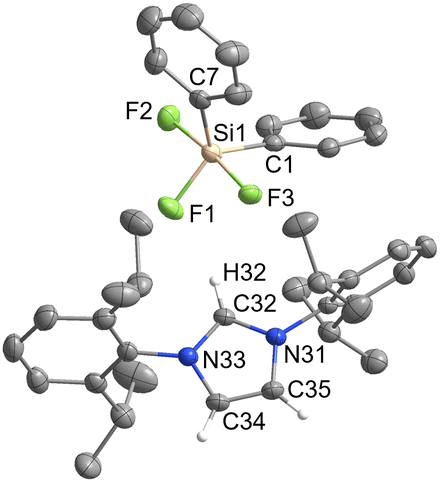 | ||
| Fig. 3 The crystal structure of [IPrH][Ph2SiF3] (3). The asymmetric unit contains two ion pairs of 3. For clarity only one ion pair is shown. The ellipsoids are drawn at 50% probability. All hydrogen atoms are omitted except for those on the imidazolium ring. Selected bond lengths (Å): Si(1)–F(1) 1.645(1), Si(1)–F(2) 1.691(1), Si(1)–F(3) 1.711(1), Si(1)–C(1) 1.897(2), Si(1)–C(7) 1.901(2). | ||
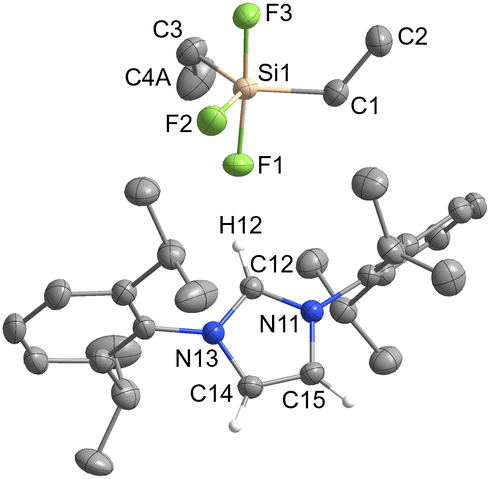 | ||
| Fig. 4 The crystal structure of [IPrH][Et2SiF3] (4). The ellipsoids are drawn at 50% probability. All hydrogen atoms are omitted except for those on the imidazolium ring. Selected bond lengths (Å): Si(1)–F(1) 1.733(1), Si(1)–F(2) 1.648(1), Si(1)–F(3) 1.729(1), Si(1)–C(1) 1.875(2), Si(1)–C(3) 1.885(2). | ||
The asymmetric unit of 4·MeCN contains one molecule of MeCN. For clarity only the structure of 4 is shown in Fig. 4. The complete asymmetric units of 3 and 4·MeCN are shown in Fig. S23 and S24 in the ESI.† The geometry of both structurally characterised [Et2SiF3]− and [Ph2SiF3]− anions is trigonal bipyramidal, with 2 fluorine atoms occupying the axial positions and the remaining fluorine atom located in the equatorial plane. In both structures, the equatorially bonded fluorine atoms have shorter Si–F bonds compared to the axially positioned ones. On average, the Si–F bond lengths in [Et2SiF3]− (1.703 Å) are longer than in [Ph2SiF3]− (1.682 Å). In both trifluorosilicates, the cation and the anion interact with each other and form 3 interactions. In compound 3, two hydrogen bonds are formed between the hydrogen atom at C2 position and the two nearest fluorine atoms (one axial and one equatorial). The hydrogen bond lengths of the fluorine atoms in the axial position (anion 1: F(3)⋯H(32) 2.233(1) Å and anion 2: F(6)⋯H(62) 2.226(1) Å) are similar to those in the equatorial plane (anion 1: F(1)⋯H(32) 2.298(1) Å and anion 2: F(4)⋯H(62) 2.343(1) Å). Furthermore, in compound 3, the fluorine atom in equatorial position interacts with hydrogen atoms on one of the adjacent phenyl rings (anion 1: F(1)⋯H(69) 2.368(1) Å and anion 2: F(4)⋯H(52) 2.424(1) Å).
In 4, 3 hydrogen bonds are formed, one between the hydrogen atom at the C2 position and the nearest fluorine atom in the axial position (F(1)⋯H(12) 2.058(1) Å), while the other two are formed between the remaining two fluorine atoms and the hydrogen atoms on the backbone of the symmetrically generated imidazolium ring (F(2)⋯H(15)I 2.307(1) Å and F(3)⋯H(14) 2.507(1) Å).
Organotetrafluorosilicates [IPrH][RSiF4] (R = Ph, Et)
[IPrH][PhSiF4] (5) and [IPrH][EtSiF4] (6) were prepared according to the general procedure listed above. To ensure quantitative conversion of chlorosilane to fluorosilane, the reaction time for halide exchange was doubled compared to other silanes. Compound 5 crystallises in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , while 6 crystallises in the monoclinic space group I2a. The crystal structures of 5 and 6 are shown in Fig. 5 and 6, respectively. The asymmetric units of the two compounds consist of two ion pairs. For clarity, only one of each is presented. The complete asymmetric units of 5 and 6 are shown in Fig. S25 and S27 in the ESI.† Both crystal structures feature a discrete [IPrH]+ cation and a discrete [PhSiF4]− or [EtSiF4]− anion, which have a trigonal bipyramidal geometry with 2 fluorine atoms in axial and 2 fluorine atoms in equatorial positions. In addition, the positions of the fluorine atoms in 5 are disordered. Two preferred orientations were modelled and refined, with domain A occupancy of 70% (anion 1) and 56% (anion 2) and domain B occupancy of 30% (anion 1) and 44% (anion 2). The disorder of the fluorine atoms is the result of two possible positions of the fluorine atoms in the anion due to the rotation of the fluorine atoms around the C(1A)–Si(1A) axis. The atoms in domain B are rotated by 90° compared to those in domain A. Due to high disorder in 5, the bond lengths and angles are not discussed further. However, they are given in Table S3 in the ESI.†
, while 6 crystallises in the monoclinic space group I2a. The crystal structures of 5 and 6 are shown in Fig. 5 and 6, respectively. The asymmetric units of the two compounds consist of two ion pairs. For clarity, only one of each is presented. The complete asymmetric units of 5 and 6 are shown in Fig. S25 and S27 in the ESI.† Both crystal structures feature a discrete [IPrH]+ cation and a discrete [PhSiF4]− or [EtSiF4]− anion, which have a trigonal bipyramidal geometry with 2 fluorine atoms in axial and 2 fluorine atoms in equatorial positions. In addition, the positions of the fluorine atoms in 5 are disordered. Two preferred orientations were modelled and refined, with domain A occupancy of 70% (anion 1) and 56% (anion 2) and domain B occupancy of 30% (anion 1) and 44% (anion 2). The disorder of the fluorine atoms is the result of two possible positions of the fluorine atoms in the anion due to the rotation of the fluorine atoms around the C(1A)–Si(1A) axis. The atoms in domain B are rotated by 90° compared to those in domain A. Due to high disorder in 5, the bond lengths and angles are not discussed further. However, they are given in Table S3 in the ESI.†
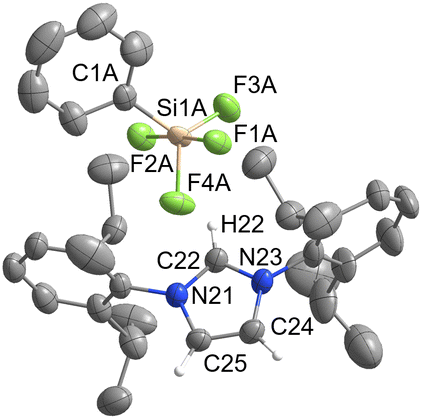 | ||
| Fig. 5 The crystal structure of [IPrH][PhSiF4] (5). The asymmetric unit contains two ion pairs of 5. For clarity only domain A of ion pair 1 is shown. The ellipsoids are drawn at 50% probability. All hydrogen atoms are omitted except for those on the imidazolium ring. Selected bond lengths (Å): Si(1A)–F(1A) 1.654(2), Si(1A)–F(2A) 1.718(2), Si(1A)–F(3A) 1.584(3), Si(1A)–F(4A) 1.660(2), Si(1A)–C(1A) 1.901(5). | ||
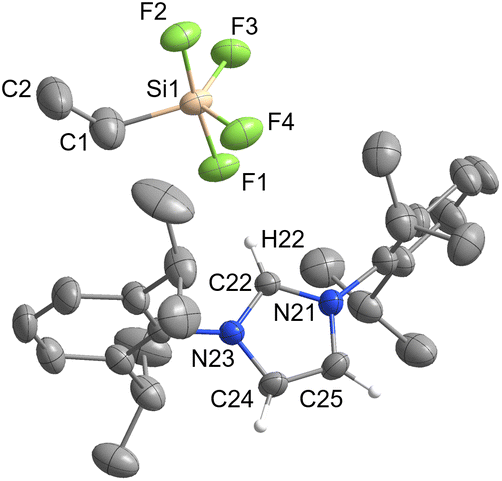 | ||
| Fig. 6 The crystal structure of [IPrH][EtSiF4] (6). The asymmetric unit contains two ion pairs of 6. For clarity only ion pair 1 is shown. The ellipsoids are drawn at 50% probability. All hydrogen atoms are omitted except for those on the imidazolium ring. Selected bond lengths (Å): Si(1)–F(1) 1.701(1), Si(1)–F(2) 1.673(1), Si(1)–F(3) 1.618(1), Si(1)–F(4) 1.614(1), Si(1)–C(1) 1.862(3). | ||
Compound 6 has longer Si–F bonds at axial positions (anion 1: Si(1)–F(1) 1.701(1) Å and Si(1)–F(2) 1.672(1) Å, anion 2: Si(2)–F(5) 1.698(1) Å, Si(2)–F(6) 1.670(1) Å) than in equatorial position (anion 1: Si(1)–F(3) 1.618(1) Å and Si(1)–F(4) 1.614(1), anion 2: Si(2)–F(7) 1.621(1) Å and Si(2)–F(8) 1.617(1) Å). Similar behaviour was observed in the structures of 3 and 4 and is consistent with the literature data for [MeSiF4]−.25 Moreover, the angles between the two axially positioned fluorine atoms (anion 1: F(1)–Si(1)–F(2) 174.87(7)°, anion 2: F(5)–Si(2)–F(6) 174.56(7)°) are smaller than the ideal angle of 180°. This distortion can be attributed to the influence of the organic substituent on the remaining fluorine atoms.
In 5 the [PhSiF4]− interacts with 3 imidazolium cations and forms several interactions. Due to high disorder, the interactions are not discussed in detail. All relevant interactions are provided in Table S4 in the ESI.† In the structure we have noticed the interactions between hydrogen atom at C2 position of the imidazolium ring and the closest axial and equatorial fluorine atoms. The other two fluorine atoms interact with the hydrogen atoms on the backbone of the imidazolium ring of the symmetrically generated unit.
In compound 6, the [EtSiF4]− anion interacts with 3 imidazolium cations and forms 5 relevant interactions. In each anion of the asymmetric unit, 2 fluorine atoms (one in axial and one in equatorial position) are oriented towards the hydrogen atom at C2 position of the cation and form 2 interactions (anion 1: F(1)⋯H(22) 2.050(1) Å and F(4)⋯H(22) 2.379(1) Å, anion 2: F(5)⋯H(52) 2.105(1) Å and F(8)⋯H(52) 2.317(1) Å). The other 2 fluorine atoms interact with hydrogen atoms on the backbone of the neighbouring imidazolium ring (anion 1: F(2)⋯H(55) 2.162(1) Å and F(3)⋯H(54) 2.280(1) Å, anion 2: F(6)⋯H(24) 2.200(1) Å and F(7)⋯H(25) 2.229(1) Å). There is also an interaction between the fluorine atom in the axial position and the hydrogen atoms of the neighbouring phenyl rings of the symmetrically generated unit (anion 1: F(2)⋯H(72) 2.460(1) Å, anion 2: F(6)⋯H(28) 2.478(1) Å).
Triphenyldifluorogermanate [IPrH][Ph3GeF2] (7)
Pentacoordinated organofluorogermanates are an interesting rarity, as very few structurally characterised compounds of this type exist.16,17 After initial success in the synthesis of various organofluorosilicates with [IPrH][F], we wanted to test the reactivity of analogous germanium compounds. In this way, [IPrH][Ph3GeF2] (7) was prepared in quantitative yield under the same reaction conditions used for the synthesis of organofluorosilicates. The crystal structure of 7 is shown in Fig. 7. Interestingly, the same crystallisation procedure produced 3 different types of crystals. The crystal structure of 7 shown in Fig. 7 crystallises without any solvent molecules in the triclinic space group P. The asymmetric unit consists of two ion pairs, but for clarity only one is shown in Fig. 7. In addition, we have structurally characterised 2 other structures of 7 in the form of solvates with MeCN 7a and 7b, each containing one molecule of MeCN. 7a crystallises in the triclinic space group P, while the 7b crystallises in the orthorhombic space group P212121. Due to the similarity of the anions with 7, the structural features of 7a and 7b are not discussed further. The complete asymmetric units of 7, 7a and 7b are shown in Fig. S28–S30 in the ESI.† The [Ph3GeF2]− anion in 7 retains the trigonal bipyramidal geometry typical of organofluorogermanates and organofluorosilicates with both fluorine atoms in axial position. Although it has the same geometry as the [Ph3SiF2]− anion, 1 and 7 are not isostructural and crystallise in different space groups of P and P21/c, respectively. The average Ge–F bond distance of the [Ph3GeF2]− anion of 1.920 Å is longer compared to other structurally characterised organofluorogermanate anions of [(C2F5)3GeF2]− and [(CF3)3GeF2]−, which have average Ge–F bond lengths of 1.834 Å and 1.810 Å, respectively.16,17 The Ge–F bond distances of the reported anions are probably shorter due to the strong electron-withdrawing groups attached to the germanium atom. Similarly to 1, [Ph3GeF2]− also interacts with 2 adjacent cations. One hydrogen bond is formed between the first fluorine atom and the hydrogen atom at the C2 position of the symmetrically generated unit (anion 1: F(1)⋯H(72) 1.982(2) Å, anion 2: F(4)⋯H(42) 1.921(2) Å), while the other is formed between the second fluorine atom and the hydrogen atom on the imidazolium backbone (anion 1: F(2)⋯H(45) 2.078(2) Å, anion 2: F(3A)⋯H(75) 1.93(1) Å and F(3B)⋯H(75) 2.07(2) Å). An additional interaction is formed between the fluorine atom F(2) and the hydrogen atom on the neighboring phenyl ring (F(2)⋯H(79) 2.373(2) Å).
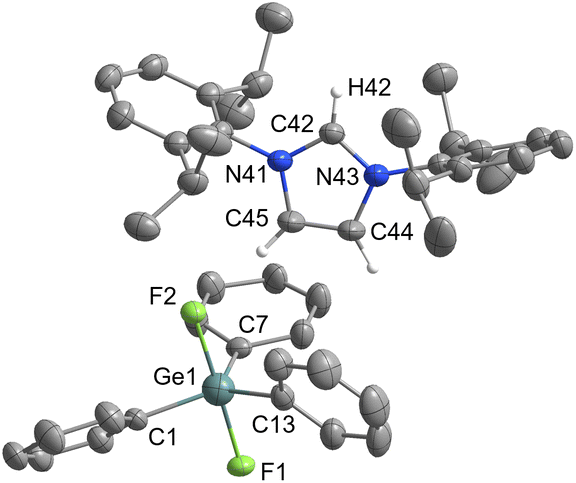 | ||
| Fig. 7 The crystal structure of [IPrH][Ph3GeF2] (7). The asymmetric unit contains two ion pairs of 7. For clarity only ion pair 1 is shown. The ellipsoids are drawn at 50% probability. All hydrogen atoms are omitted except for those on the imidazolium ring. Selected bond lengths (Å): Ge(1)–F(1) 1.930(1), Ge(1)–F(2) 1.923(1), Ge(1)–C(1) 1.946(3), Ge(1)–C(7) 1.949(3), Ge(1)–C(13) 1.963(3). | ||
NMR spectroscopy
1H, 13C and 19F spectra of all synthesised organofluorosilicate and organofluorogermanate salts are given in the ESI.† All spectra are consistent with the X-ray crystal structures of the compounds, except for 5. We attempted several times to record NMR spectra of 5 in the reaction mixture, but in each case, only HF2− and F− anions were observed. The same result was obtained regardless of the temperature that was used for the measurement.In the 1H NMR spectra, characteristic signals for the [IPrH]+ cation and typical signals for the ethyl and phenyl substituents are observed for all compounds. However, sometimes the signals of the substituents overlapped with the signals of the [IPrH]+ cation.
The 19F NMR spectra were initially measured at room temperature. But since some of the signals were not visible, the measurements were repeated at lower temperature. The appropriate signals were visible at −40 °C. The measured 19F NMR peaks of the characterised compounds are listed in Table 1 and are compared with literature data for structurally related compounds, although they were not measured in the same deuterated solvent. Since ethylfluorosilicates have not yet been characterised, the 19F values were compared with the related methylfluorosilicates. The 19F NMR chemical shifts of synthesised phenylfluorosilicates (1: −97.03 ppm and 3: −96.55 ppm and −134.21 ppm) are consistent with those previously reported ([Ph3SiF2]−: −96.6 ppm, [Ph2SiF3]−: −98.0 ppm and −134.0 ppm).4,26 The spectra of the trifluorosilicate salts 3 and 4 feature two signals, which are in ratio 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The signals that are more downfield belong to the fluorine atoms in axial positions, while the fluorine atoms in equatorial positions have signals shifted upfield. 19F–19F coupling between the axial and equatorial fluorine atoms was not observed. This is most likely due to the large peak widths of the signals, which measure about 100 Hz. Similar behaviour of this system was described previously.26 In 4, the signal for the fluorine atoms in the axial positions is significantly moved upfield, compared to its methyl equivalent.25 All other ethylfluorosilicates have similar values to their methyl-substituted relatives. In addition, the signal of 7 (−143.73 ppm) agrees with the values reported for [(CH3)3N][(CF3)3GeF2] (−137.6 ppm).16 Surprisingly, the two compounds have only slightly different signals, although the anions contain quite different substituents.
:1. The signals that are more downfield belong to the fluorine atoms in axial positions, while the fluorine atoms in equatorial positions have signals shifted upfield. 19F–19F coupling between the axial and equatorial fluorine atoms was not observed. This is most likely due to the large peak widths of the signals, which measure about 100 Hz. Similar behaviour of this system was described previously.26 In 4, the signal for the fluorine atoms in the axial positions is significantly moved upfield, compared to its methyl equivalent.25 All other ethylfluorosilicates have similar values to their methyl-substituted relatives. In addition, the signal of 7 (−143.73 ppm) agrees with the values reported for [(CH3)3N][(CF3)3GeF2] (−137.6 ppm).16 Surprisingly, the two compounds have only slightly different signals, although the anions contain quite different substituents.
| Compound | δ (19F) Si/Ge–F | δ (19F) Si/Ge–F in literature |
|---|---|---|
| a Axially positioned fluorine atom. | ||
| [IPrH][Ph3SiF2] (1) | −97.03 (298 K) | −96.6 ([(C4H9)4N][Ph3SiF2], CDCl3)4 |
| [IPrH][Ph2SiF3] (3) | −96.55 (233 K)a | −98.0 ([(C2H5)4N][Ph2SiF3], CD2Cl2)a26 |
| −134.21 (233 K) | −134.0 ([(C2H5)4N][Ph2SiF3], CD2Cl2)26 | |
| [IPrH][Et2SiF3] (4) | −85.99 (233 K)a | −66.3 ([PNP][Me2SiF3], CH3CH2CN, 193 K)a25 |
| −136.17 (233 K) | −132.9 ([PNP][Me2SiF3], CH3CH2CN, 193 K)25 | |
| [IPrH][PhSiF4] (5) | NA | −116.8 ([(CH3)4N][PhSiF4], DMSO-d8)26 |
| [IPrH][EtSiF4] (6) | −115.75 (233 K) | −110.5 ([(C4H9)4N][MeSiF4], CD2Cl2, 213 K)26 |
| [IPrH][Ph3GeF2] (7) | −143.73 (233 K) | −137.6 ([(CH3)3N][(CF3)3GeF2], MeCN-d3)16 |
Raman spectroscopy
Raman spectra were recorded for the compounds [IPrH][Ph3SiF2] (1), [IPrH][Ph2SiF3] (3), [IPrH][Et2SiF3] (4), [IPrH][EtSiF4] (6) and [IPrH][Ph3GeF2] (7). For the compounds with ethyl groups (4 and 6), the quality of the spectra is insufficient to determine peaks related to the vibrations of the anions. The reason for this is the significant fluorescence of these 2 samples. In the other compounds we could distinguish 2 peaks not related to the [IPrH]+ cation. In 7, we attributed the Ge–F vibration at 662 cm−1 to the symmetric stretching of GeF2 unit. The position of the Ge–F vibration agrees well with the published data for the structurally characterised [GeF5]− anions.19The phenylfluorosilicate compounds of 1 and 3 have similar signals at 667 and 663 cm−1, respectively. We attributed these vibrations to the symmetric stretching of Si–F bonds in SiF2 units. The signal at 999 cm−1 is present in all compounds with phenyl groups and we suspect that it may be related to Si–C and Ge–C vibrations. A similar signal has been observed in organofluoroaluminates for the Al–C vibration.22
Molecular calculations
The structures of [IPrH][R4−nSiFn+1] (n = 1–4, R = Ph, Et) and [IPrH][R4−nGeFn+1] (n = 1–4, R = Ph, Et) were used as a starting point for the DFT calculations of optimised structures, which were then compared with those obtained experimentally.Fig. 8 shows the optimised structures of [IPrH][Ph4−nSiFn+1] (n = 1–4), while Table 2 lists the experimental and calculated Si–C and Si–F bond distances of [IPrH][Ph4−nSiFn+1] (n = 1–4). The data for the ethylfluorosilicate and germanate counterparts can be found in Fig. S31, S32 and Tables S5, S6 in the ESI.†Table 2 shows that the calculated Si–C distances agree well with the experimental results, although they are slightly longer (1: calc. 1.92 Å, exp. 1.907(2) Å). On the other hand, the calculated Si–F values are significantly longer than the experimental values (1: calc. 1.85 Å, exp. 1.746(1) Å). The difference can be explained by the fact that the calculations were performed in the gas phase, where there is no effect of crystal packing. The [IPrH]+ cations tend to form strong hydrogen bonds and other electrostatic interactions with the silicate and germanate anions.19 Therefore, in the gas phase calculations, where no additional neighbouring cations and anions are present, the two ions arrange differently than in the crystal structure. We have also observed similar behaviour between the calculated and the structurally determined bond distances for organofluoroaluminate anions.22 Interestingly, according to DFT calculations the formation of interactions between the [IPrH]+ cation and the silicate or germanate anions result in the elongation of the Si–F or Ge–F bonds in axial position that are involved in the interactions.
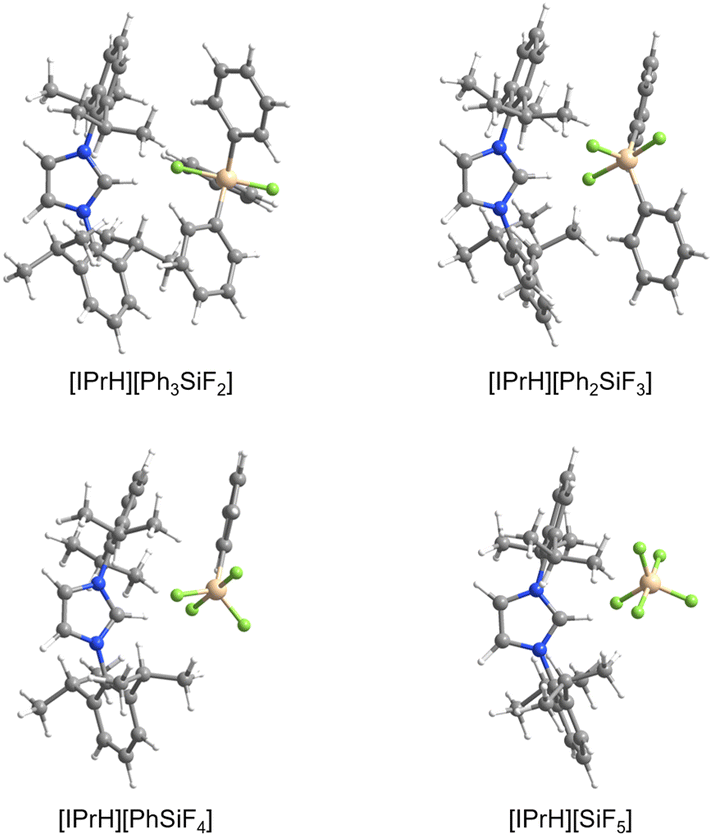 | ||
| Fig. 8 Optimised structures of [IPrH][Ph4−nSiFn+1] (n = 1–4). | ||
| Compound | d(Si–C)/Å | d(Si–F)/Å | ||||
|---|---|---|---|---|---|---|
| Experimental | Calculated | Experimental | Calculated | |||
| a Axial position of fluorine atoms. b Si–F distances, where fluorine atoms form the strongest interactions with the hydrogen atom at C2 position. c Si–C and Si–F distances for domain A, where the anions are disordered. d Values reported in ref. 19. | ||||||
| [IPrH][Ph3SiF2] (1) | 1.907(2) | 1.92 | 1.746(1)a | 1.85a,b | ||
| 1.906(2) | 1.92 | 1.743(1)a,b | 1.71a | |||
| 1.898(2) | 1.91 | |||||
| [IPrH][Ph2SiF3] (3) | 1.901(2) | 1.898(2) | 1.91 | 1.711(1)a,b | 1.710(1)a,b | 1.81a,b |
| 1.897(2) | 1.897(2) | 1.90 | 1.691(1)a | 1.689(1)a | 1.69a | |
| 1.645(1)b | 1.646(1)b | 1.68 | ||||
| [IPrH][PhSiF4] (5) | 1.901(5)c | 1.852(7)c | 1.90 | 1.718(2)b,c | 1.751(3)b,c | 1.77a,b |
| 1.660(2)a,c | 1.656(3)a,c | 1.68a | ||||
| 1.654(4)a,b,c | 1.625(4)a,b,c | 1.67 | ||||
| 1.584(3)c | 1.574(5)c | 1.63 | ||||
| [IPrH][SiF5]d | 1.62(1)a,b,c | 1.74a,b | ||||
| 1.644(4)a,c | 1.66a | |||||
| 1.59(1)b,c | 1.66 | |||||
| 1.594(4)c | 1.64 | |||||
| 1.585(4)c | 1.62 | |||||
In some cases, the calculations agree with experimentally observed results. For example, Table 2 shows that the Si–F bonds involved in interactions with hydrogen atoms at C2 position are particularly long (3: calc. 1.81 Å, exp. 1.711(1) Å). On the other hand, weaker interactions do not affect the Si–F or Ge–F bond distances as much as observed in the experimental values of [IPrH][SiF5].
The estimated energy released during the formation of organofluorosilicate and germanate salts was calculated according to the chemical reactions:
| [R4−nSiFn] + [IPrH][F] → [IPrH][R4−nSiFn+1] |
| [R4−nGeFn] + [IPrH][F] → [IPrH][R4−nGeFn+1]. |
The calculated energies of the reactions and the electronic energies of the optimised compounds are given in Table S7 in the ESI.† The calculated energies of the reactions are more exothermic with each additional fluorine atom in the compounds of the [IPrH][R4−nSiFn+1] and [IPrH][R4−nGeFn+1] series, where R = Ph, Et. It seems that the addition of fluorine atoms in the structures increases the stability of silicate and germanate anions, and thus favours the formation of more fluorinated products.
Conclusions
In this work we present a comprehensive study on the reactivity of organofluorosilanes and an organofluorogermane with the imidazolium-based naked fluoride reagent [IPrH][F]. It possesses a sterically demanding cation that has been shown to be able to stabilise discrete anions. For this reason, [IPrH][F] seemed to be a good choice for the preparation of related organofluorosilicates and germanates.Starting from commercially available organochlorosilanes, triphenylchlorogermane and potassium fluoride, a series of organofluorosilanes and Ph3GeF were prepared, which were further reacted with [IPrH][F] to form a triphenyldifluorogermanate [IPrH][Ph3GeF2] (7) and organofluorosilicates [IPrH][R4−nSiFn+1] (n = 1–3, R = Ph, Et) (1–6), with the exception of [IPrH][Et3SiF2] (2). The reactions described are simple and selective without the need for purification of the reaction mixtures. Where possible, the newly prepared compounds were characterised by X-ray structural analysis, NMR and Raman spectroscopy. Here we present the synthesis of ethyl-substituted organofluorosilicates (4, 6), which were previously unknown. Despite their weaker Lewis acidity, which is the result of its electron donating substituents, they were able to form stable organofluorosilicate salts. Unfortunately, compound 2 did not form, even in solution. An interesting discovery was also compound 7, which is an extremely rare example of an organofluorogermanate salt and the first structurally characterised one with aromatic substituents. In this work we have shown that [IPrH][F] is a suitable reagent for the formation of new organofluorogermanate and organofluorosilicate species. The experimentally obtained results are in agreement with the DFT calculations. The results show that in the [IPrH][R4−nSiFn] (n = 1–3, R = Ph, Et) series, the stability of the products increases with increasing number of fluorine atoms. Calculations for the germanium series have yielded similar results.
Experimental
General methods
All manipulations were performed under an argon atmosphere using Schlenk and glovebox techniques. To avoid possible side reactions of fluoride with glassware, homemade FEP (tetrafluoroethylene–hexafluoropropylene) reaction vessels with PTFE (polytetrafluoroethylene) valves were used. Before use, the FEP reaction vessels were dried under vacuum conditions at room temperature. MeCN was purified using Vigor solvent purification system. MeCN-d3 was dried over activated 3 Å molecular sieves. Molecular sieves were activated by heating at 150 °C in vacuo overnight. [IPrH][F] was synthesised according to the published procedure.18 KF was dried overnight in vacuo at 150 °C and stored in a glovebox before use. All other commercially available reagents were used as received.NMR spectroscopy
NMR samples were prepared in a glovebox under an inert atmosphere. To prevent the compounds from coming into contact with glass, the samples were measured using homemade FEP inlays for NMR tubes. The spectra were recorded at the Slovenian NMR Centre (National Institute of Chemistry) using a Bruker AVANCE NEO 600 or 400 MHz NMR spectrometer. Spectra were recorded at 298 or 233 K. The chemical shifts of 1H and 13C were referenced to the residual signals of the deuterated solvent and are given relative to tetramethylsilane (TMS). The 19F references were calculated according to IUPAC guidelines and are given relative to CFCl3.27Raman spectroscopy
Samples were loaded into 0.3 mm quartz capillaries in a glovebox. Raman spectra were recorded using a Horiba Jobin Yvon Labram-HR spectrometer coupled with an Olympus BXFM-ILHS microscope at room temperature. Samples were excited with the 633 nm emission line of a He–Ne laser.Crystal structure determination
Crystal data were collected on a Gemini A diffractometer equipped with an Atlas CCD detector using graphite monochromated Cu Kα radiation. All crystal data were collected at 150 K unless otherwise stated. Data were processed using the CrysAlisPro software package.28 An analytical absorption correction was applied to all data sets.29 Structures were solved using the SHELXT program.30 Structure refinement was performed using the SHELXL software30 implemented in the Olex2 program package.31 Figures were prepared using Diamond.32Molecular calculations
Molecular calculations of the crystal structures were performed in gas-phase with the Gaussian 16 program33 using the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional34 and the D3 empirical dispersion correction of Grimme35 with Becke–Johnson damping.36 Electrons were described with all-electron basis sets. We used the triple-ζ basis set with polarization functions, in particular Def2-TZVP.37,38Mass spectroscopy
10 μL of sample were injected directly through the electrospray ion source into the high-resolution ToF mass spectrometer at a mass resolution of 10000. Mass spectra were recorded in the negative ion mode in the mass range m/z 50 to m/z 1000.
Preparation of [IPrH][Ph3SiF2] (1)
A suspension of KF (0.291 g, 2.50 mmol) and Ph3SiCl (0.147 g, 0.50 mmol) was stirred in MeCN (5 mL) for 24 h at room temperature. Suspension was filtered and added to a solution of [IPrH][F] (0.204 g, 0.50 mmol) in MeCN (5 mL). Reaction mixture was stirred for another 24 h at room temperature. Volatiles were removed by slow evaporation under static vacuum conditions. Product was isolated in the form of yellowish crystals.1 H NMR (MeCN-d3, 600 MHz): δ 8.98 (s, 1H, NCHN), 7.97 (d, J = 7.3, 4H, Ph), 7.84 (s, 1H, NCHCHN), 7.65 (t, J = 7.8, 2H, p-CH), 7.47 (d, J = 7.8, 4H, m-CH), 7.32 (t, J = 7.0, 1H, Ph), 7.22–7.10 (m, 8H, Ph), 2.41 (m, J = 6.8, 4H, CHCH3), 1.27 (d, J = 6.8, 12H, CHCH3), 1.19 (d, J = 6.8, 12H, CHCH3).
13 C NMR (MeCN-d3, 151 MHz): δ 146.3, 137.8, 133.1, 127.3, 127.2, 125.7, 29.8, 24.4, 23.8.
19 F NMR (MeCN-d3, 565 MHz): δ −97.03 (t, JSi–F = 124.6).
Raman: ν(Si–F) 667 cm−1, ν(Si–C) 1000 cm−1.
HRMS (ESI − ): m/z = 297.0908 ([Ph3SiF2]− calculated for C18H15F228Si: 297.0911).
Attempted preparation of [IPrH][Et3SiF2] (2)
A suspension of KF (0.145 g, 1.25 mmol) and Et3SiCl (0.038 g, 0.25 mmol) was stirred in MeCN-d3 (3 mL) for 24 h at room temperature. Suspension was filtered and added to a solution of [IPrH][F] (0.102 g, 0.25 mmol) in MeCN-d3 (3 mL). Reaction mixture was stirred for another 24 h at room temperature. Aliquot of the reaction mixture was transferred to an NMR tube. 19F NMR of the reaction mixture showed the silane [Et3SiF] component unreacted.Preparation of [IPrH][Ph2SiF3] (3)
A suspension of KF (0.581 g, 5.00 mmol) and Ph2SiCl2 (0.127 g, 0.50 mmol) was stirred in MeCN (5 mL) for 24 h at room temperature. Suspension was filtered and added to a solution of [IPrH][F] (0.204 g, 0.50 mmol) in MeCN (5 mL). Reaction mixture was stirred for another 24 h at room temperature. Volatiles were removed by slow evaporation under static vacuum conditions. Product was isolated in the form of yellowish crystals.1 H NMR (MeCN-d3, 600 MHz) δ: 9.15 (s, 1H, NCHN), 7.85 (m, 2H, NCHCHN), 7.78 (m, 4H, Ph), 7.66 (t, J = 7.8, 2H, p-CH), 7.47 (d, J = 7.8, 4H, m-CH), 7.16 (m, 6H, Ph), 2.42 (m, 4H, CHCH3), 1.27 (d, J = 6.8 Hz, 12H, CHCH3), 1.19 (d, J = 6.9 Hz, 12H, CHCH3).
13 C NMR (MeCN-d3, 151 MHz,) δ: 146.3, 139.1, 137.8, 135.0, 133.1, 130.9, 128.9, 128.2, 127.4, 127.0, 125.7, 29.9, 24.5, 23.9.
19 F NMR (MeCN-d3, 565 MHz, 233 K) δ: −96.55 (s, axial F), −134.21 (s, equatorial F).
Raman: ν(Si–F) 663 cm−1, ν(Si–C) 999 cm−1.
HRMS (ESI − ): m/z = 239.0511 ([Ph2SiF3]− calculated for C12H10F328Si: 239.0504).
Preparation of [IPrH][Et2SiF3] (4)
A suspension of KF (0.581 g, 5.00 mmol) and Et2SiCl2 (0.079 g, 0.50 mmol) was stirred in MeCN (5 mL) for 24 h at room temperature. Suspension was filtered and added to a solution of [IPrH][F] (0.204 g, 0.50 mmol) in MeCN (5 mL). Reaction mixture was stirred for another 24 h at room temperature. Volatiles were removed by slow evaporation under static vacuum conditions. Product was isolated in the form of yellowish crystals.1 H NMR (MeCN-d3, 600 MHz) δ: 9.23 (s, 1H, NCHN), 7.90 (s, 2H, NCHCHN), 7.68 (m, 2H, p-CH), 7.51 (m, 4H, m-CH), 2.45 (m, 4H, CHCH3), 1.30 (d, J = 5.5, 12H, CHCH3), 1.23 (d, J = 5.5, 12H, CHCH3), 0.86 (m, 6H, CH3), 0.38 (m, 4H, CH2).
13 C NMR (MeCN-d3, 151 MHz,) δ: 146.3, 139.3, 133.1, 130.8, 127.0, 125.6, 29.8, 24.4, 23.8, 9.8, 7.8, 6.9.
19 F NMR (MeCN-d3, 565 MHz, 233 K) δ: −85.99 (s, axial F), −136.17 (s, equatorial F).
HRMS (ESI − ): m/z = 143.0500 ([Et2SiF3]− calculated for C4H10F328Si: 143.0504).
Preparation of [IPrH][PhSiF4] (5)
A suspension of KF (0.872 g, 7.50 mmol) and PhSiCl3 (0.106 g, 0.50 mmol) was stirred in MeCN (5 mL) for 24 h at room temperature. Suspension was filtered and added to a solution of [IPrH][F] (0.204 g, 0.50 mmol) in MeCN (5 mL). Reaction mixture was stirred for another 48 h at room temperature. Volatiles were removed by slow evaporation under static vacuum conditions. Product was isolated in the form of yellowish crystals. Only trace amounts of crystals were isolated, enabling single crystal X-ray diffraction but not enough for NMR, Raman or HRMS spectroscopy.Preparation of [IPrH][EtSiF4] (6)
A suspension of KF (0.872 g, 7.50 mmol) and EtSiCl3 (0.082 g, 0.50 mmol) was stirred in MeCN (5 mL) for 24 h at room temperature. Suspension was filtered and added to a solution of [IPrH][F] (0.204 g, 0.50 mmol) in MeCN (5 mL). Reaction mixture was stirred for another 48 h at room temperature. Volatiles were removed by slow evaporation under static vacuum conditions. Product was isolated in the form of yellowish crystals.1 H NMR (MeCN-d3, 600 MHz): δ 10.15 (s, 1H, NCHN), 7.61 (s, 3H), 7.44 (s, 5H), 2.48 (s, 4H), 1.25 (s, 12H), 1.18 (s, 12H), 0.85 (m, 3H, CH3), 0.44 (s, 2H, CH2).
13 C NMR (MeCN-d3, 151 MHz): δ 146.4, 132.7, 125.4, 29.7, 24.5, 23.7, 9.8.
19 F NMR (MeCN-d3, 565 MHz, 233 K): δ −115.75 (t, JSi–F = 111.6).
HRMS (ESI − ): m/z = 133.0089 ([EtSiF4]− calculated for C2H5F428Si: 133.0097).
Preparation of [IPrH][Ph3GeF2] (7)
A suspension of KF (0.291 g, 2.50 mmol) and Ph3GeCl (0.170 g, 0.50 mmol) was stirred in MeCN (5 mL) for 24 h at room temperature. Suspension was filtered and added to a solution of [IPrH][F] (0.204 g, 0.50 mmol) in MeCN (5 mL). Reaction mixture was stirred for another 24 h at room temperature. Volatiles were removed by slow evaporation under static vacuum conditions. Product was isolated in the form of yellowish crystals.1 H NMR (MeCN-d3, 600 MHz): δ 10.29 (s, 1H, NCHN), 7.86 (s, 2H, NCHCHN), 7.67 (d, J = 6.5, 4H, m-CH), 7.62 (t, J = 7.4, 2H, p-CH), 7.33–7.55 (m, 13H, Ph), 2.34 (m, 4H, CHCH3), 1.23 (d, J = 6.1 Hz, 12H, CHCH3), 1.13 (d, J = 6.2 Hz, 12H, CHCH3).
13 C NMR (MeCN-d3, 151 MHz): δ 145.8, 139.9, 138.8, 135.0, 132.6, 130.5, 130.4, 128.8, 126.2, 125.1, 29.4, 24.5, 23.1.
19 F NMR (MeCN-d3, 565 MHz, 233 K): δ −143.73 (s).
Raman: ν(Ge–F) 662 cm−1, ν(Ge–C) 999 cm−1.
HRMS (ESI − ): m/z = 343.0364 ([Ph3GeF2]− calculated for C18H15F274Ge: 343.0354).
Conflicts of interest
Authors declare that there are no conflicts of interest.Acknowledgements
The authors gratefully acknowledge the Slovenian Research Agency (ARRS) for financial support of the Research Programmes P1-0045 (Inorganic Chemistry and Technology), PR-09774 (Young Researcher Programme), and Project N1-0185 (advanced reagents for (asymmetric) nucleophilic fluorination). The authors also acknowledge the Czech Science Foundation for financial support of the Project No. 21-29531K (advanced reagents for (asymmetric) nucleophilic fluorination). The authors thank the Center for Mass Spectrometry (Jožef Stefan Institute) and Slovenian NMR Centre (National Institute of Chemistry) for their resources and support. Authors also thank Asst. Prof. Evgeny Goreshnik for help with the crystal structure determination and analysis.Notes and references
- W. J. Middleton, Org. Synth., 1986, 64, 221 CrossRef CAS.
- D. A. Dixon, W. B. Farnham, W. Heilemann, R. Mews and M. Noltemeyer, Heteroat. Chem., 1993, 4, 287–295 CrossRef CAS.
- K. O. Christe, W. W. Wilson, R. D. Wilson, R. Ban and J. A. Feng, J. Am. Chem. Soc., 1990, 112, 7619–7625 CrossRef CAS.
- A. S. Pilcher, H. L. Ammon and P. DeShong, J. Am. Chem. Soc., 1995, 117, 5166–5167 CrossRef CAS.
- S. Yamaguchi, S. Akiyama and K. Tamao, Organometallics, 1999, 18, 2851–2854 CrossRef CAS.
- D. Schomburg and R. Krebs, Inorg. Chem., 1984, 23, 1378–1381 CrossRef CAS.
- D. Schomburg, J. Organomet. Chem., 1981, 221, 137–141 CrossRef CAS.
- R. O. Day, C. Sreelatha, J. A. Deiters, S. E. Johnson, J. M. Holmes, L. Howe and R. R. Holmes, Organometallics, 1991, 10, 1758–1766 CrossRef CAS.
- D. Brondani, F. H. Carré, R. J. P. Corriu, J. J. E. Moreau and M. W. C. Man, Angew. Chem., Int. Ed. Engl., 1996, 35, 324–326 CrossRef CAS.
- L. J. P. Van Der Boon, L. Van Gelderen, T. R. De Groot, M. Lutz, J. C. Slootweg, A. W. Ehlers and K. Lammertsma, Inorg. Chem., 2018, 57, 12697–12708 CrossRef CAS PubMed.
- D. Kost, I. Kalikhman, S. Krivonos, R. Bertermann, C. Burschka, R. E. Neugebauer, M. Pülm, R. Willeke and R. Tacke, Organometallics, 2000, 19, 1083–1095 CrossRef CAS.
- Y. Kim, M. Kim and F. P. Gabbai, Org. Lett., 2010, 12, 600–602 CrossRef CAS PubMed.
- M. Kira, E. Kwon, C. Kabuto and K. Sakamoto, Chem. Lett., 1999, 1183–1184 CrossRef CAS.
- R. Pietschnig and K. Merz, Chem. Commun., 2001, 1, 1210–1211 RSC.
- K. Tamao, T. Hayashi and Y. Ito, J. Organomet. Chem., 1996, 506, 85–91 CrossRef CAS.
- D. J. Brauer, J. Wilke and R. Eujen, J. Organomet. Chem., 1986, 316, 261–269 CrossRef CAS.
- S. Pelzer, B. Neumann, H. G. Stammler, N. Ignat'ev and B. Hoge, Chem. – Eur. J., 2016, 22, 16460–16466 CrossRef CAS PubMed.
- B. Alič and G. Tavčar, J. Fluor. Chem., 2016, 192, 141–146 CrossRef.
- B. Alič, M. Tramšek, A. Kokalj and G. Tavčar, Inorg. Chem., 2017, 56, 10070–10077 CrossRef PubMed.
- Ž. Zupanek, M. Tramšek, A. Kokalj and G. Tavčar, Inorg. Chem., 2018, 57, 13866–13879 CrossRef PubMed.
- Ž. Zupanek, M. Tramšek, A. Kokalj and G. Tavčar, J. Fluor. Chem., 2019, 227, 109373 CrossRef.
- E. Gruden, M. Tramšek and G. Tavčar, Organometallics, 2022, 41, 41–51 CrossRef CAS.
- P. D. Prince, M. J. Bearpark, G. S. McGrady and J. W. Steed, Dalton Trans., 2007, 271–282 Search PubMed.
- A. F. Hill, J. S. Ward and Y. Xiong, Organometallics, 2015, 34, 5057–5064 CrossRef CAS.
- C. Bolli, J. Gellhaar, C. Jenne, M. Keßler, H. Scherer, H. Seeger and R. Uzun, Dalton Trans., 2014, 43, 4326–4334 RSC.
- F. Klanberg and E. L. Muetterties, Inorg. Chem., 1968, 7, 155–160 CrossRef CAS.
- R. K. Harris, E. D. Becker, S. M. De Cabral Menezes, P. Granger, R. E. Hoffman and K. W. Zilm, Magn. Reson. Chem., 2008, 46, 582–598 CrossRef CAS PubMed.
- CrysAlisPro, Version 1.171.37.33 (release 27-03-2014 CrysAlis171.NET), Agilent Technologies, (compiled Mar 27 2014, 17:12:48) Search PubMed.
- R. C. Clark and J. S. Reid, Acta Crystallogr., Sect. A: Found. Crystallogr., 1995, 51, 887–897 CrossRef.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- Diamond – Crystal and Molecular Structure Visualization, https://www.crystalimpact.com/diamond/, accessed 13 January 2023 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 (Version B01), Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, Raman and HRMS spectra of isolated compounds, crystal structure data and computational results. CCDC 2240916–2240923. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt00421j |
| This journal is © The Royal Society of Chemistry 2023 |