
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIridium complexes of an ortho-trifluoromethylphenyl substituted PONOP pincer ligand†
Ethan W.
Poole
a,
Itxaso
Bustos
 ab,
Thomas M.
Hood
a,
Jennifer E.
Smart
a and
Adrian B.
Chaplin
*a
ab,
Thomas M.
Hood
a,
Jennifer E.
Smart
a and
Adrian B.
Chaplin
*a
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry CV4 7AL, UK. E-mail: a.b.chaplin@warwick.ac.uk
bFacultad de Química de San Sebastián, Universidad del País Vasco (UPV/EHU), Apdo. 1072, 20080 San Sebastián, Spain
First published on 21st December 2022
Abstract
The synthesis and iridium coordination chemistry of a new pyridine-based phosphinito pincer ligand 2,6-(ArF2PO)2C5H3N (PONOP-ArF; ArF = 2-(CF3)C6H4) are described, where the P-donors have ortho-trifluoromethylphenyl substituents. The iridium(III) 2,2′-biphenyl (biph) derivative [Ir(PONOP-ArF)(biph)Cl] was obtained by reaction with [Ir(biph)(COD)Cl]2 (COD = 1,5-cyclooctadiene) and subsequent halide ion abstraction enabled isolation of [Ir(PONOP-ArF)(biph)]+ which features an Ir ← F–C bonding interaction in the solid state. Hydrogenolysis of the biphenyl ligand and formation of [Ir(PONOP-ArF)(H)2]+ was achieved by prolonged reaction of [Ir(PONOP-ArF)(biph)]+ with dihydrogen. This transformation paved the way for isolation and crystallographic characterisation of low valent iridium derivatives through treatment of the dihydride with tert-butylethylene (TBE). The iridium(I) π-complex [Ir(PONOP-ArF)(TBE)]+ is thermally stable but substitution of TBE can be achieved by reaction with carbon monoxide. The solid-state structure of the mono-carbonyl product [Ir(PONOP-ArF)(CO)]+ is notable for an intermolecular anagostic interaction between the metal centre and a pentane molecule which co-crystallises within a cleft defined by two aryl phosphine substituents.
Introduction
The transformation of organofluorine compounds, through selective functionalisation of the constituent and thermodynamically robust C–F bonds, is a formidable challenge relevant to contemporary organic synthesis and repurposing hydrofluorocarbon (HFC) refrigerants into synthons for pharmaceuticals, agrochemicals and materials.1 Insertion of a transition metal into a C–F bond is an attractive method, but the known organometallic chemistry of these reactions is almost exclusively limited to the activation of C(sp2)–F bonds. Recent advances in methodologies for enacting the cleavage of aliphatic C–F bonds have largely been associated with the application of main group reagents – with prominent examples involving oxidative addition to Al(I) complexes, across Mg(I)–Mg(I) dimers, or through the action of silylium Lewis acids – and are conspicuous for the under-representation of organotransition metal chemistry.2 Of the limited number of transition-metal-based examples, the C(sp3)–F bond activation reactions of iridium(I) pincer complex A – reported by Krogh-Jespersen, Goldman and co-workers in 2011 – are outstanding but notable for a mechanism that does not involve concerted C–F bond oxidative addition.3 Whilst thermodynamically favourable, calculations indicate this metal-based reactivity is associated with a prohibitively large kinetic barrier and instead a sequence involving concerted C–H bond oxidative addition and α-fluoride elimination (rate determining) was established; as illustrated in Fig. 1 for the activation of fluoromethane affording iridium(III) pincer complex B.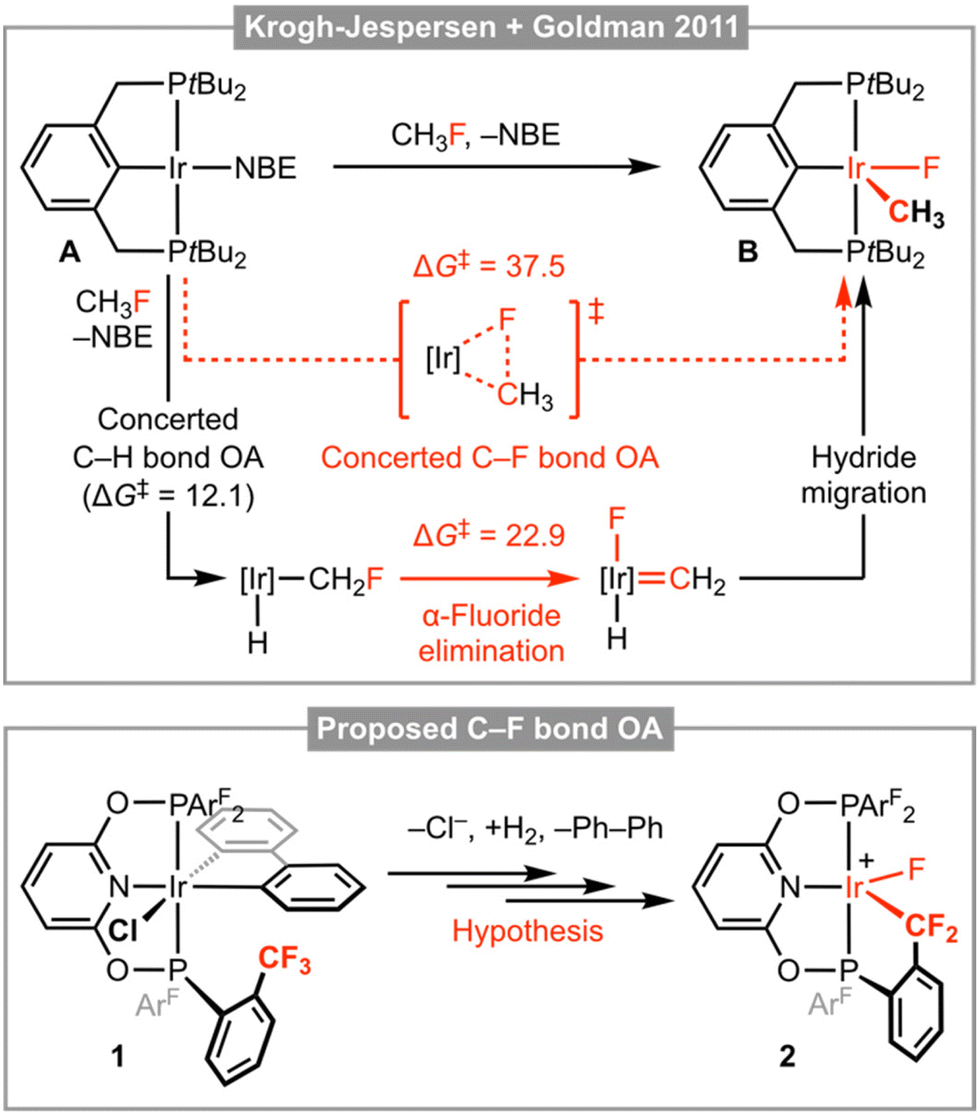 | ||
| Fig. 1 Activation of fluoromethane by an Ir(PCP) pincer complex and proposed intramolecular C(sp3)–F bond activation manifold. Calculated activation barriers in kcal mol−1 relative to A; NBE = norbornene. | ||
Motivated by the aforementioned chemistry established by Krogh-Jespersen, Goldman and co-workers and as part of our work with low coordinate group 9 pincer complexes,4–6 we speculated that the bespoke trifluoromethyl-functionalised PONOP pincer ligand 2,6-(ArF2PO)2C5H3N (PONOP-ArF; ArF = 2-(CF3)C6H4) could pre-organise interaction between a C(sp3)–F bond and reactive iridium(I) derivative, and thereby promote subsequent concerted oxidative addition by reducing the associated kinetic barrier. We herein present work evaluating this hypothesis, using 1 as a well-defined iridium(III) derivative of PONOP-ArF and synthon for the reactive three coordinate iridium(I) complex {Ir(PONOP-ArF)}+ by successive halide ion abstraction and hydrogenolysis of the constituent 2,2′-biphenyl ligand (biph; Fig. 1). Whilst iridium(III) oxidative addition product 2 was ultimately not obtained, during the course of this work iridium complexes featuring a coordinated trifluoromethyl appendage, tert-butylethylene (TBE), and an intermolecular anagostic interaction with pentane have been crystallographically characterised.
Results and discussion
The new phosphinito pincer ligand PONOP-ArF was prepared by adaptation of literature procedures for alkyl-substituted variants.7 Specifically, PONOP-ArF was obtained in 59% isolated yield following reaction between the known chlorophosphine PArF2Cl and doubly deprotonated 2,6-dihydroxypyridine in THF at elevated temperature (Fig. 2).8 Curiously, PONOP-ArF appears to be the first aryl-substituted pincer of the formulation 2,6-(R2PO)2C5H3N to be reported. Indeed, our attempts to prepare 2,6-(Ph2PO)2C5H3N under the same conditions resulted only in generation of O=PPh2Cl. The structure of PONOP-ArF was confirmed using a combination of multinuclear NMR spectroscopy and high-resolution ESI-MS, with a septet 31P resonance at δ 96.4 and corresponding doublet 19F resonance at δ −57.0 (4JPF = 64 Hz) the most remarkable spectroscopic features.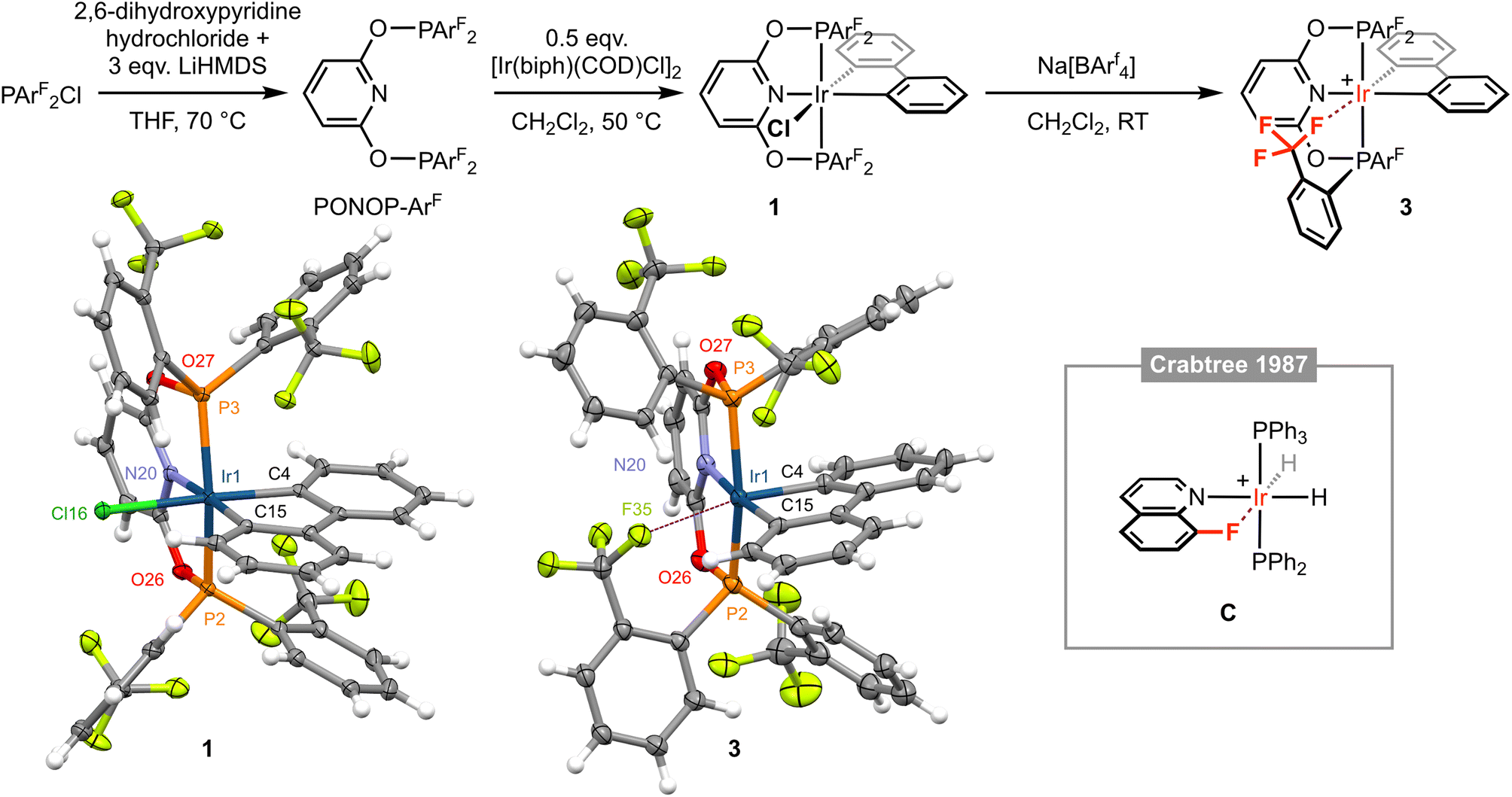 | ||
| Fig. 2 Synthesis and characterisation of iridium(III) complexes 1 and 3; [BArf4]− counter ions omitted for clarity. Solid-state structures of 1 and 3 drawn with thermal ellipsoids at 30% probability and without minor disordered components (1 × ArF substituent in 3) and solvent molecules. Selected metal-based bond lengths (Å) and angles (°): 1, Ir1–P2, 2.2800(8); Ir1–P3, 2.3050(8); Ir1–N20, 2.099(3); Ir1–C4, 2.066(3); Ir1–C15, 2.052(3); Ir1–Cl16, 2.4774(8); P2–Ir1–P3, 158.03(3); N20–Ir1–C15, 176.54(11); C4–Ir1–Cl16, 176.51(10); 3, Ir1–P2, 2.2795(9); Ir1–P3, 2.3075(8); Ir1–N20, 2.109(3); Ir1–C4, 2.026(3); Ir1–C15, 2.056(3); Ir1⋯F35, 2.543(2); P2–Ir1–P3, 160.17(3); N20–Ir–C15, 174.40(11); C4–Ir1⋯F35, 168.37(10); C, Ir⋯F, 2.514(8).12 | ||
Exploiting an iridium(III) precursor first described by Crabtree and informed by previous work in our laboratories involving {M(biph)Cl} (M = Rh, Ir) synthons,4,5,9–111 was synthesised through reaction of [Ir(biph)(COD)Cl]2 (COD = 1,5-cyclooctadiene) with PONOP-ArF in CH2Cl2 at 50 °C and isolated in 75% yield. In solution, 1 is characterised by time-averaged Cs symmetry and a significant downfield shift of the 31P resonance of the pincer ligand (δ 96.4 to 111.4), which is further notable for the loss of any significant coupling to 19F. Inequivalent 19F resonances are observed at very similar field to the free ligand: δ −57.1 and −57.3 (cf. δ −57.0), with the former significantly line broadened relative to the latter, presumably reflecting slower restricted rotation of the P–ArF bonds straddling the biphenyl ligand. The coordinated 13C resonances of the biphenyl ligand are located at δ 143.2 (m) and 140.2 (t, 2JPC = 9 Hz) with coupling to two equivalent 31P nuclei resolved for the latter. The structure of 1 was corroborated in the solid state by single crystal X-ray diffraction, exhibiting a pseudo-octahedral coordination geometry about the metal centre and unsymmetrically orientated ArF substituents (Fig. 2). The Ir1–C4 (2.066(3) Å) and Ir1–C15 (2.052(3) Å) distances are not statistically different, but the trend suggests in this system the chloride ligand exerts a greater trans influence than the pyridine-based donor.
Following our proposed retrosynthesis, 1 was treated with the halide ion abstracting agent Na[BArf4] (Arf = 3,5-(CF3)2C6H3), using the weakly coordinating solvent dichloromethane to accentuate the electrophilicity of the alkali metal and favour formation of a low-coordinate complex. The resulting derivative is formulated as [Ir(PONOP-ArF)(biph)][BArf4] 3 and was isolated from solution in 69% yield following filtration and crystallisation from CH2Cl2–hexane (Fig. 2). Crystallographic analysis of 3 indicates a dative bonding interaction between iridium and one of the triflouromethane groups is adopted in the solid state, as gauged by an Ir1⋯F35 contact of 2.543(2) Å, conferring a formally pseudo-octahedral metal coordination geometry (C4–Ir1–F35 = 168.37(10)°). This outcome is significant given the extremely limited number of well-defined transition metal examples of featuring M ← F–C bonding interactions. Indeed, a search of the CSD (v. 5.43) identified only iridium(III) 8-fluoroquinoline complex C with an Ir⋯F contact <2.8 Å (Fig. 2, Ir⋯F = 2.514(8) Å).12 Work by Togni and co-workers with PPh2(5,6,7,8-tetrafluoronaphthalenyl) is also notable, but the two iridium complexes studied in the solid state feature considerably more remote Ir⋯F contacts of ca. 3.0 Å.13 Moreover, preceding work in our group has involved characterisation of a series of rhodium(III) biph complexes of PPh2ArF with Rh⋯F contacts ranging from 2.363(2) to 2.459(2) Å.10 Consistent with the weakly interacting nature of the trifluoromethane group, the Ir1–C4 bond (2.026(3) Å) is significantly contracted relative to the Ir1–C15 (2.056(3) Å) bond distance. Indeed, isolated 3 adopts time-averaged C2v symmetry in CD2Cl2 solution at 298 K, indicating that rapid pseudorotation of the biph ligand and that Ir ← F–C bonding is not persistent on the NMR time scale.4,10 At this temperature, the pincer complex is characterised by single broad 31P and 19F resonances at δ 123.8 and δ −58.8, respectively (cf. δ 111.4 and δ −57.1/−57.3 for 1).
Placing a solution of 3 in CD2Cl2 under an atmosphere of CO (1 atm) resulted in immediate and quantitative formation of the carbonyl derivative [Ir(PONOP-ArF)(biph)(CO)][BArf4] 4 (Fig. 3), which adopts a static Cs symmetric structure characterised by a broad upfield-shifted 31P resonance at δ 103.3 (cf. δ 123.8 for 3) and two 19F signals at δ −57.4 and −57.5; the latter is line broadened relative to the former, presumably reflecting slower restricted rotation of the P–ArF bonds straddling the biphenyl ligand (cf.1). Coordination of CO is corroborated by a sharp triplet resonance at δ 172.6 (2JPC = 8 Hz) in the 13C{1H} NMR spectrum, IR spectroscopy with ν(CO) = 2062 cm−1, ESI-MS, and structural elucidation by single crystal X-ray diffraction (Fig. 3). The solid-state structure of 4 is rather routine, but the metrics associated with coordination of biph (Ir1–C4 = 2.092(3), Ir1–C15 = 2.072(3) Å) enable compilation of the following trend in ligand trans influence for this iridium(III) system by comparison to the structures of 1 and 3: CF3 < py < Cl− ≤ CO.
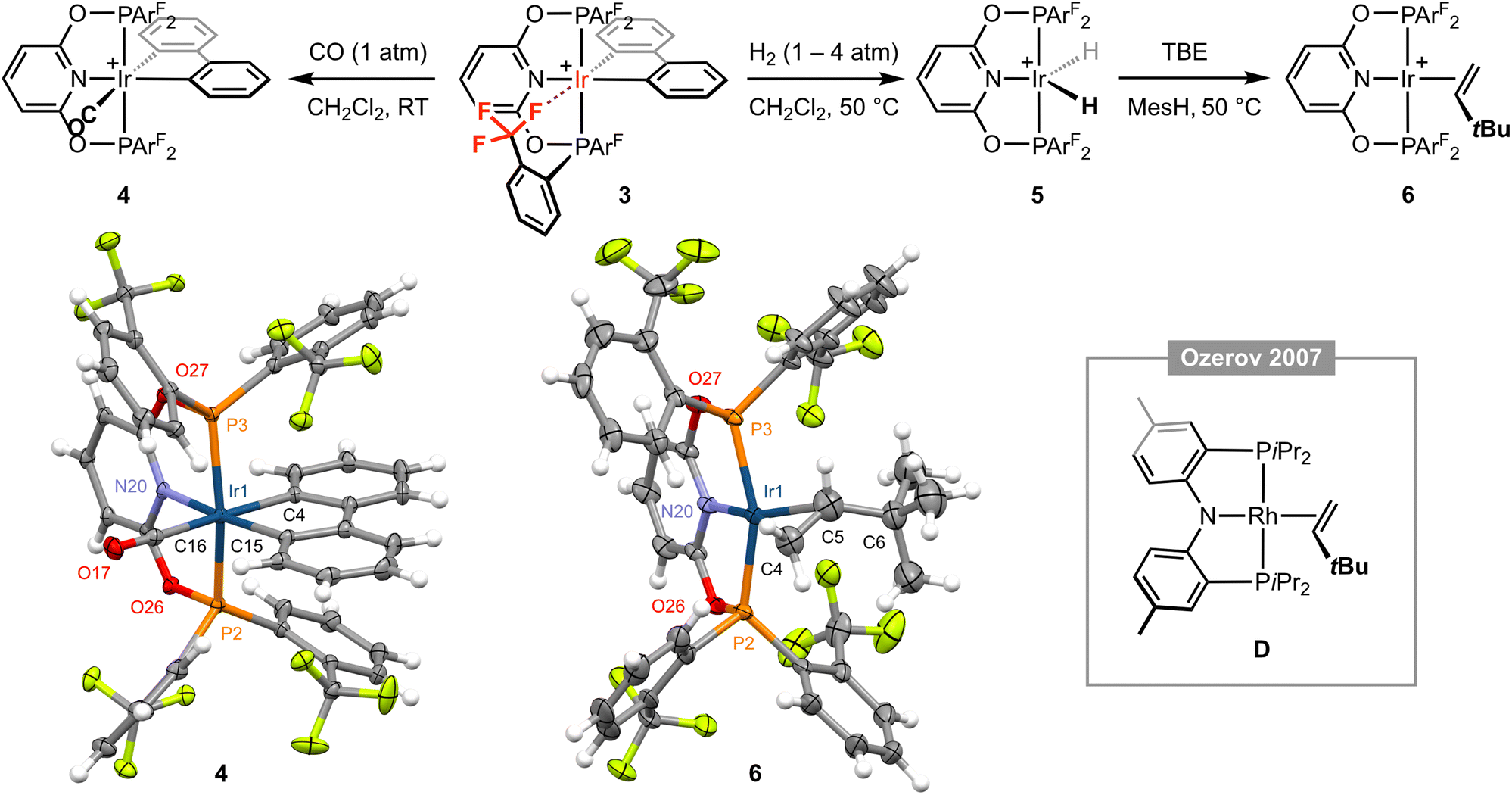 | ||
Fig. 3 Reactivity of complex 3; MesH = mesitylene and [BArf4]− counter ions omitted for clarity. Solid-state structures of 4 and 6 drawn with thermal ellipsoids at 30% probability and without solvent molecules. Selected metal-based bond lengths (Å) and angles (°): 4, Ir1–P2, 2.3300(7); Ir1–P3, 2.3243(7); Ir1–N20, 2.102(2); Ir1–C4, 2.092(3); Ir1–C15, 2.072(3); Ir1–C16, 1.958(3); C16–O17, 1.123(4); P2–Ir1–P3, 159.85(2); N20–Ir1–C15, 171.25(10); C4–Ir1–Cl16, 177.09(11); 6, Ir1–P2, 2.2657(11); Ir1–P3, 2.2704(12); Ir1–N20, 2.058(4); Ir1–Cnt(C4,C5), 2.110(12); C4–C5, 1.304(12), P2–Ir1–P3, 156.46(5); N20–Ir1–Cnt(C4,C5), 175.3(4); C4–C5–C6, 130.6(10); D, Rh–Cnt(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 2.051(3); CC, 1.360(10); N–Rh–Cnt(CC), 174.27(14); CC–C, 127.7(5).15 Cnt = centroid. C), 2.051(3); CC, 1.360(10); N–Rh–Cnt(CC), 174.27(14); CC–C, 127.7(5).15 Cnt = centroid. | ||
Moving on with our proposed retrosynthesis, 3 was heated in CH2Cl2 under an atmosphere of dihydrogen at 50 °C resulting in hydrogenolysis of the biph ligand and generation of the iridium(III) dihydride [Ir(PONOP-ArF)(H)2][BArf4] 5 within 8 days (1 atm H2)/3 days (4 atm H2), which is characterised by time-averaged C2v symmetry (δ31P 140.4; δ19F −57.0; δ1H −18.41 (br), T1 = 883 ± 17 ms (298 K, 600 MHz)) and was isolated in 84% yield (Fig. 3). Targeting in situ generation of the putative three coordinate iridium(I) complex {Ir(PONOP-ArF)}+, 5 was thereafter treated with 10 equivalents of the bulky sacrificial hydrogen acceptor TBE in the inert hydrocarbon solvent mesitylene.14 Heating at 50 °C resulted in quantitative formation of iridium(I) π-complex [Ir(PONOP-ArF)(TBE)][BArf4] 6 within 16 hours, which was isolated and fully characterised (Fig. 3). In d8-toluene solution, coordination of TBE is corroborated by downfield alkene 1H resonances at δ 4.55, 3.33 and 3.16, and conferral of asymmetry in the metal coordination sphere, viz. δ31P 151.9, 143.0 (2JPP = 380 Hz) and δ19F −56.6, −57.4, −58.6, −58.9. Crystals suitable for analysis by X-ray diffraction were obtained by recrystallisation from mesitylene–pentane and enable, to the best of our knowledge, structural elucidation of an iridium–TBE complex in the solid state for the first time (CSD v. 5.43). A neutral rhodium PNP pincer complex D reported by Ozerov and co-workers is the closest well-defined structural precedent (Fig. 3), but we are reluctant to make a detailed comparison of the metrics in 6 as the TBE ligand is not located with very high precision due to the quality of data collected (Rint = 0.0883).15
In attempt to induce formation of 2 by dissociation of TBE and C(sp3)–F bond cyclometallation, isolated 6 was heated in mesitylene at elevated temperature (120 °C, Fig. 4). Analysis of the reaction mixture by NMR spectroscopy after 24 hours, however, indicated that 2 was stable under these conditions with only ca. 10% decomposition of the anion apparent from the 19F{1H} NMR spectrum.16 Reactions involving C–H bond cyclometallation of tert-butyl and neo-pentyl substituted PNP pincer ligands have previously been reported for iridium and demonstrate that formation of iridacyclobutane and – most pertinent to this study – iridacyclopentane rings are possible.17
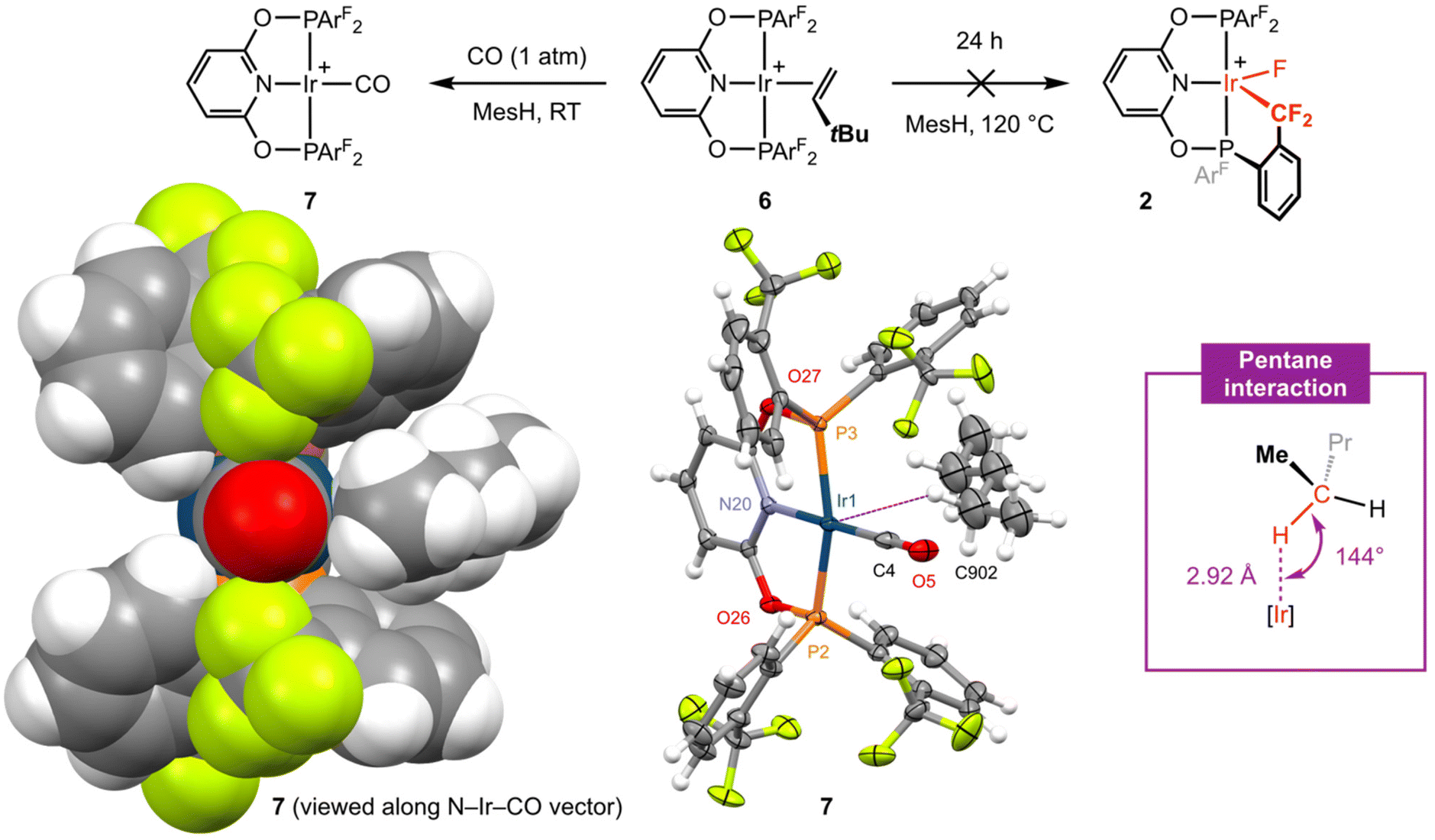 | ||
| Fig. 4 Reactivity of complex 6; MesH = mesitylene and [BArf4]− counter ions omitted for clarity. Solid-state structures of 7 drawn in space fill (left) and with thermal ellipsoids at 30% probability (middle); CH2Cl2 solvent molecule not shown. Selected metal-based bond lengths (Å) and angles (°): 7, Ir1–P2, 2.2720(7); Ir1–P3, 2.2828(7); Ir1–N20, 2.050(2); Ir1–C4, 1.858(3); C4–O5, 1.137(4); P2–Ir1–P3, 161.38(3); N20–Ir1–C4, 177.01(13); Ir1⋯C902, 3.759(11). | ||
Placing a solution of 6 in mesitylene under an atmosphere of CO (1 atm) resulted in immediate conversion into square planar iridium(I) carbonyl complex [Ir(PONOP-ArF)(CO)][BArf4] 7 (Fig. 4) at room temperature and shows that TBE can be readily substituted if the incoming ligand is a sufficiently strong donor (i.e. not a CF3 appendage). Complex 7 was isolated in 74% yield and the structure verified by a combination of NMR and IR spectroscopy. Sharp 31P and 19F singlet resonances δ 151.1 and δ −56.3 are consistent with C2v symmetry and coordination of CO is marked out by a sharp triplet resonance at δ 179.0 (2JPC = 9 Hz) in the 13C{1H} NMR spectrum (CD2Cl2). Analysis by IR spectroscopy identified a single carbonyl stretching band centred at ν(CO) = 2040 cm−1 and confirmed formation of a mono-carbonyl derivative. In line with the electron-withdrawing character of the phosphine substituents in 7, this band is significantly blue-shifted relative to alkyl substituted PONOP homologues such as [Ir(2,6-(tBu2PO)2C5H3N)(CO)][BArf4] (ν(CO) = 2010 cm−1).18 The structure of 7 is further substantiated in the solid state using single crystals grown from CH2Cl2−pentane (Fig. 4). The metal coordination geometry and associated metrics are in harmony with the spectroscopic data and otherwise unremarkable. Wider inspection of the unit cell, however, revealed an interesting intermolecular interaction between the iridium centre and a molecule of pentane which co-crystallises within a cleft defined by two aryl phosphine substituents (Fig. 4, left).19 Supported by the lattice, this anagostic interaction is characterised by an ![[I with combining low line]](https://www.rsc.org/images/entities/char_0049_0332.gif)
![[r with combining low line]](https://www.rsc.org/images/entities/char_0072_0332.gif) ⋯
⋯![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) –C contact of 2.92 Å and Ir⋯H–C angle of 144°.20,21 Whilst it is not uncommon for M⋯H–C interactions of this nature to be observed in solid-state structures of square planar platinum group complexes, a search of the CSD (v. 5.43) identified only 2 palladium(II) examples with intermolecular M⋯H contacts <3.0 Å with an alkane.22
–C contact of 2.92 Å and Ir⋯H–C angle of 144°.20,21 Whilst it is not uncommon for M⋯H–C interactions of this nature to be observed in solid-state structures of square planar platinum group complexes, a search of the CSD (v. 5.43) identified only 2 palladium(II) examples with intermolecular M⋯H contacts <3.0 Å with an alkane.22
Conclusions
The synthesis of a new pyridine-based phosphinito pincer ligand 2,6-(ArF2PO)2C5H3N (PONOP-ArF; ArF = 2-(CF3)C6H4) has been described, where the P-donors have ortho-trifluoromethylphenyl substituents. The iridium coordination chemistry of this ligand has been explored, starting from the iridium(III) 2,2′-biphenyl (biph) complex [Ir(PONOP-ArF)(biph)Cl] that was readily obtained by reaction with [Ir(biph)(COD)Cl]2 (COD = 1,5-cyclooctadiene). Subsequent halide ion abstraction enabled isolation of the cationic derivative [Ir(PONOP-ArF)(biph)]+ which is fluxional in solution but exhibits a distinct Ir ← F–C bonding interaction in the solid state as evidenced by an Ir⋯F contact of 2.543(2) Å. Hydrogenolysis of the biphenyl ligand and formation of [Ir(PONOP-ArF)(H)2]+ was achieved by prolonged reaction of [Ir(PONOP-ArF)(biph)]+ with dihydrogen at 50 °C and ultimately permitted access to low valent iridium derivatives through subsequent treatment of the dihydride with tert-butylethylene (TBE). The iridium(I) π-complex [Ir(PONOP-ArF)(TBE)]+ was isolated, crystallographically characterised, and is thermally stable in mesitylene solution. Substitution of TBE was achieved by reaction with carbon monoxide, with the solid-state structure of the mono-carbonyl product [Ir(PONOP-ArF)(CO)]+ notable for an intermolecular anagostic interaction between the metal centre and a pentane molecule which co-crystallises within a cleft defined by two aryl phosphine substituents (⋯–C = 2.92 Å).
Experimental
General methods
All manipulations were performed under an atmosphere of argon using Schlenk and glove box techniques unless otherwise stated. Glassware was oven dried at 150 °C overnight and flame-dried under vacuum prior to use. Molecular sieves were activated by heating at 300 °C in vacuo overnight. CD2Cl2 and TBE were freeze–pump–thaw degassed and dried over activated 3 Å molecular sieves. Mesitylene and d8-toluene were dried over sodium, distilled, freeze–pump–thaw degassed, and stored over activated 3 Å molecular sieves and a potassium mirror, respectively. Anhydrous CH2Cl2, THF, diethyl ether, hexane, and pentane were purchased from Acros Organics or Sigma-Aldrich, freeze–pump–thaw degassed and stored over activated 3 Å molecular sieves. PArF2Cl,8 [Ir(biph)(COD)Cl]2,9 and Na[BArf4]23 were synthesized according to published procedures, or minor variations thereof. All other reagents are commercial products and were used as received. NMR spectra were recorded on Bruker spectrometers under argon at 298 K unless otherwise stated. Chemical shifts are quoted in ppm and coupling constants in Hz. Virtual triplets (vt) are reported as the separation between the first and third lines.24 NMR spectra in non-deuterated solvents were recorded using an internal capillary of C6D6. High resolution (HR) ESI-MS were recorded on Bruker Maxis Plus instrument. IR spectra were recorded on a Bruker Alpha Platinum ATR FT-IR spectrometer.Preparation of PONOP-ArF
A suspension of 2,6-dihydroxypyridine hydrochloride (0.101 g, 0.684 mmol) and LiHMDS (0.343 g, 2.05 mmol) was refluxed in THF (20 mL) under vigorous stirring for 16 h at 70 °C. A solution of PArF2Cl (0.500 g, 1.40 mmol) in THF (15 mL) was added at ambient temperature and the resulting yellow solution heated at reflux for 16 h. Volatiles were removed in vacuo and the product extracted into toluene (20 mL). The solution was concentrated to dryness and residue washed with hexane (2 × 15 mL). Recrystallisation from diethyl ether afforded the pure product as a white solid. Yield: 0.301 g (0.401 mmol, 59%).1 H NMR (CD2Cl2, 500 MHz): δ 7.67 (dd, 3JHH = 7.4, 3JPH = 3.5, 4H, 3-ArF), 7.63 (t, 3JHH = 7.9, 1H, 4-py), 7.49 (t, 3JHH = 7.6, 4H, 4-ArF), 7.44 (t, 3JHH = 7.6, 4H, 5-ArF), 7.36 (dd, 3JHH = 7.3, 2JPH = 2.9, 4H, 6-ArF), 6.60 (d, 3JHH = 7.9, 2H, 3-py).
13 C{ 1 H} NMR (CD2Cl2, 126 MHz): δ 161.2 (d, 2JPC = 10, 2-py), 142.5 (s, 4-py), 137.9 (d, 1JPC = 40, 1-ArF), 133.1 (d, 2JPC = 3, 6-ArF), 132.8 (qd, 2JFC = 32, 2JPC = 25, 2-ArF), 132.0 (s, 5-ArF), 130.3 (s, 4-ArF), 126.7 (br, 3-ArF), 124.8 (q, 1JFC = 275, ArF{CF3}), 106.5 (d, 3JPC = 2, 3-py).
19 F{ 1 H} NMR (CD2Cl2, 376 MHz): δ −57.0 (d, 4JPF = 64).
31 P{ 1 H} NMR (CD2Cl2, 162 MHz): δ 96.4 (sept, 4JPF = 64).
HR ESI-MS (positive ion, 4 kV): 774.0588 ([M+Na]+, calcd 774.0592) m/z.
Attempted preparation of PONOP-Ph
A suspension of 2,6-dihydroxypyridine hydrochloride (0.50 g, 3.50 mmol) and LiHMDS (1.75 g, 10.0 mmol) was refluxed in THF (50 mL) under vigorous stirring for 16 h at 70 °C. A solution of chlorodiphenylphosphine (1.38 mL, 7.50 mmol) in THF (10 mL) was added at ambient temperature and the resulting yellow solution refluxed for 16 h. Volatiles were removed in vacuo and the product extracted into toluene (30 mL). The solution was concentrated to dryness and the residue washed with hexane (2 × 20 mL). Yield: 1.28 g. Analysis by NMR spectroscopy indicated formation of OPPh2Cl.
31 P{ 1 H} NMR (CD2Cl2, 162 MHz): δ 42.6 (s).
Preparation of [Ir(PONOP-ArF)(biph)Cl] 1
A suspension of [Ir(biph)(COD)Cl]2 (65.0 mg, 66.5 μmol) and PONOP-ArF (100 mg, 0.133 mmol) in CH2Cl2 (20 mL) was heated at 50 °C for 48 h. Volatiles were removed in vacuo and the crude product washed with hexane (2 × 15 mL) and dried. Colourless crystals of the product were obtained by recrystallisation from CH2Cl2–hexane. Yield: 113.0 mg (99.8 μmol, 75%).1 H NMR (CD2Cl2, 500 MHz): δ 9.06 (br, 2H, ArF), 8.60 (d, 3JHH = 7.5, 1H, 6-biph), 8.04 (t, 3JHH = 8.2, 1H, 4-py), 7.61–7.77 (m, 6H, ArF), 7.18 (d, 3JHH = 8.2, 2H, 3-py), 7.12–7.28 (m, 10H, 3,5-biph + ArF), 7.04 (t, 3JHH = 7.4, 1H, 4-biph), 6.68 (d, 3JHH = 7.4, 1H, 3′-biph), 6.15 (t, 3JHH = 7.4, 1H, 4′-biph), 5.86 (d, 3JHH = 7.5, 1H, 6′-biph), 5.76 (t, 3JHH = 7.5, 1H, 5′-biph).
13 C{ 1 H} NMR (CD2Cl2, 126 MHz): δ 159.8 (vt, JPC = 5, 2-py), 154.3 (s, 2-biph), 153.5 (s, 2′-biph), 143.9 (s, 4-py), 143.1–143.3 (m, 1-biph), 139.8 (t, 2JPC = 9, 1′-biph), 135.2 (s, 6′-biph), 134.6 (s, 6-biph), 133.2 (s, ArF), 132.9 (br, ArF), 131.4 (q, 2JFC = 33, 2/2′-ArF), 130.9 (s, ArF), 130.3–130.6 (m, ArF), 129.6 (q, 2JFC = 33, 2/2′-ArF), 127.6–127.8 (m, ArF), 127.1–127.3 (m, ArF), 126.1 (s, 5-biph), 125.3 (s, 5′-biph), 123.8 (q, 1JFC = 275, ArF{CF3}), 123.1 (s, 4-biph), 122.9 (q, 1JFC = 275, ArF{CF3}), 122.2 (s, 4′-biph), 120.3 (s, 3-biph), 119.6 (s, 3′-biph), 105.1 (vt, JPC = 5 3-py). Not all ArF signals unambiguously located.
19 F{ 1 H} NMR (CD2Cl2, 282 MHz): δ −57.1 (br, 6F), −57.3 (s, 6F).
31 P{ 1 H} NMR (CD2Cl2, 121 MHz): δ 111.4 (s).
HR ESI-MS (positive ion, 4 kV): 1096.0945 ([M–Cl]+, calcd 1096.0952) m/z.
Preparation of [Ir(PONOP-ArF)(biph)][BArf4] 3
A suspension of 1 (107.2 mg, 94.8 μmol) and Na[BArf4] (156.9 mg, 177.0 μmol) in CH2Cl2 was stirred at ambient temperature for 40 h. The solution was filtered and volatiles were removed in vacuo. Orange crystals of the product were obtained by recrystallisation from CH2Cl2–hexane. Yield: 127.3 mg (65.0 μmol, 69%).1 H NMR (CD2Cl2, 500 MHz, selected data): δ 8.27 (t, 3JHH = 8.3, 1H, 4-py), 7.40 (d, 3JHH = 8.3, 2H, 3-py). The spectrum is characterised by broad overlapping aromatic signals consistent with a fluxional time-averaged C2v symmetric structure on the timescale of the experiment.
13 C{ 1 H} NMR (CD2Cl2, 126 MHz, selected data): δ 160.0 (s, 2-py), 147.3 (s, 4-py), 131.2 (q, 2JFC = 33, 2-ArF), 123.3 (q, 1JFC = 275, ArF{CF3}), 106.8 (vt, JPC = 5, 3-py).
19 F{ 1 H} NMR (CD2Cl2, 376 MHz): δ −58.8 (br, 12F, ArF), −62.9 (s, 24F, Arf).
31 P{ 1 H} NMR (CD2Cl2, 162 MHz): δ 123.8 (s).
HR ESI-MS (positive ion, 4 kV): 1096.0963 ([M]+, calcd 1096.0952) m/z.
NMR scale reaction of 3 with CO
A solution of 3 (19.6 mg, 10.0 μmol) in CD2Cl2 (0.5 mL) within a J. Young valve NMR tube was freeze–pump–thaw degassed and placed under an atmosphere of CO (1 atm) at ambient temperature. An immediate colour change from dark orange to pale yellow was observed and analysis by NMR spectroscopy indicated quantitative formation of 4.Preparation of [Ir(PONOP-ArF)(biph)(CO)][BArf4] 4
A solution of 3 (48.1 mg, 24.5 μmol) in CH2Cl2 (1 mL) was freeze–pump–thaw degassed and placed under an atmosphere of CO (1 atm) at ambient temperature. An immediate colour change from dark orange to pale yellow was observed. The solution was freeze–pump–thaw degassed, placed under an atmosphere of argon, and layered with hexane to afford the product as colourless crystals upon diffusion. Yield: 45.9 mg (23.1 μmol, 94%).1 H NMR (CD2Cl2, 500 MHz): δ 8.33 (t, 3JHH = 8.3, 1H, 4-py), 7.91 (d, 3JHH = 7.2, 2H, 3-ArF), 7.65–7.80 (m, 15H, 2-Arf + 4,5,6-ArF + 6-biph), 7.61 (d, 3JHH = 7.6, 1H, 3-biph), 7.55 (br, 4H, 4-Arf), 7.48–7.54 (m, 2H, 3′-ArF), 7.44 (d, 3JHH = 8.3, 2H, 3-py), 7.40 (t, 3JHH = 7.8, 2H, 4′-ArF), 7.31 (t, 3JHH = 7.5, 1H, 4-biph), 7.18 (t, 3JHH = 7.4, 1H, 5-biph), 7.11 (d, 3JHH = 7.7, 1H, 3′-biph), 7.05 (t, 3JHH = 7.8, 2H, 5′-ArF), 6.92 (dvt, 3JHH = 8.0, JPH = 16, 2H, 6′-ArF), 6.60 (t, 3JHH = 7.5, 1H, 4′-biph), 6.32 (t, 3JHH = 7.4, 1H, 5′-biph), 5.45 (d, 3JHH = 7.5, 1H, 6′-biph).
13 C{ 1 H} NMR (CD2Cl2, 126 MHz): δ 172.6 (t, 2JPC = 7, CO), 162.2 (q, 1JCB = 50, 1-Arf), 159.1 (s, 2-py), 153.5 (s, 2′-biph), 151.9 (s, 2-biph), 148.2 (s, 4-py), 147.0 (t, 2JPC = 10, 1′-biph), 136.8 (s, 6-biph), 135.2 (s, 2-Arf), 134.7 (s, 6′-ArF), 133.4 (obscured t, 2JPC = 7, 1-biph), 133.3 (s, 4′-ArF), 132.5 (vt, JPC = 15, 5-ArF), 131.0 (vt, JPfC = 14, 5′-ArF), 130.7 (s, 6′-biph), 129.9 (s, 5-biph), 129.3 (qq, 2JFC = 32, 3JCB = 3, 3-Arf), 129.2–129.4 (m, 3-ArF), 128.3–128.4 (m, 3′-ArF), 128.0 (s, 5′-biph), 126.7 (s, 4′-biph), 126.4 (s, 4-biph), 125.0 (q, 1JFC = 273, Arf{CF3}), 125 (obscured, 1′-ArF), 123.5 (q, 1JFC = 274, ArF{CF3}), 123.4 (s, 3-biph), 123.3 (s, 3′-biph), 122.9 (q, 1JFC = 274, ArF{CF3}), 117.9 (sept, 3JFC = 4, 4-Arf), 107.2 (vt, JPC = 6, 3-py). Not all ArF signals unambiguously located.
19 F{ 1 H} NMR (CD2Cl2, 376 MHz): δ −57.4 (s, 6F, ArF), −57.5 (br, 6F, ArF), −62.9 (s, 24F, Arf).
31 P{ 1 H} NMR (CD2Cl2, 162 MHz): δ 103.3 (br s).
HR ESI-MS (positive ion, 4 kV): 1124.0902 ([M]+, calcd 1124.0901) m/z.
IR (ATR): ν(CO) 2062 cm−1.
NMR scale reaction of 3 with H2
A solution of 3 (19.6 mg, 10.0 μmol) in CD2Cl2 (0.5 mL) within a J. Young valve NMR tube was freeze–pump–thaw degassed and placed under an atmosphere of dihydrogen (1 atm). Analysis by NMR spectroscopy indicated quantitative formation of 5 with concomitant formation of biphenyl within 8 days at 50 °C.Preparation of [Ir(PONOP-ArF)(H)2][BArf4] 5
A solution of 3 (174 mg, 89.0 μmol) in CH2Cl2 (5 mL) was freeze–pump–thaw degassed and placed under an atmosphere of dihydrogen (4 atm). The solution was heated at 50 °C for 72 h, during which time a colour change from orange to yellow was observed. Volatiles were removed in vacuo and the pale-yellow product washed with pentane (2 × 15 mL) and dried. Yield: 136 mg (75.0 μmol, 84%).1 H NMR (CD2Cl2, 500 MHz): δ 8.22 (dvt, 3JHH = 8.0, JPH = 16, 4H, 6-ArF), 8.01 (t, 3JHH = 8.2, 1H, 4-py), 7.88 (d, 3JHH = 7.1, 4H, 3-ArF), 7.81 (t, 3JHH = 7.4, 4H, 5-ArF), 7.77 (t, 3JHH = 7.4, 4H, 4-ArF), 7.71–7.74 (m, 8H, 2-Arf), 7.55 (br, 4H, 4-Arf), 7.19 (d, 3JHH = 8.2, 2H, 3-py), −18.41 (br, 2H, Ir–H).
13 C{ 1 H} NMR (CD2Cl2, 126 MHz): δ 162.1 (q, 1JCB = 50, 1-Arf), 160.1 (br, 2-py), 145.7 (s, 4-py), 137.5 (br, 6-ArF), 135.2 (s, 2-Arf), 134.4 (s, 4-ArF), 132.5 (vt, JPC = 13, 5-ArF), 131.0 (q, 2JFC = 33, 2-ArF), 130.6 (vt, JPC = 58, 1-ArF), 129.3 (qq, 2JFC = 32, 3JCB = 3, 3-Arf), 129.1 (br, 3-ArF), 125.0 (q, 1JFC = 273, Arf{CF3}), 123.8 (q, 1JFC = 275, ArF{CF3}), 117.9 (sept, 3JFC = 4, 4-Arf), 105.8 (br, 3-py).
19 F{ 1 H} NMR (CD2Cl2, 376 MHz): δ −57.0 (s, 12F, ArF), −62.9 (s, 24F, Arf).
31 P{ 1 H} NMR (CD2Cl2, 162 MHz): δ 140.4 (s).
HR ESI-MS (positive ion, 4 kV): 946.0487 ([M]+, calcd 946.0481) m/z.
NMR scale reaction of 5 with TBE
To a solution of 5 (10.5 mg, 5.8 μmol) in mesitylene (0.5 mL) within a J. Young valve NMR tube was added TBE (7.5 μL, 58 μmol) at ambient temperature. The solution was heated at 50 °C for 16 h, during which time a colour change from yellow to orange was observed. Analysis by NMR spectroscopy indicated quantitative formation of 6.Preparation of [Ir(PONOP-ArF)(TBE)][BArf4] 6
To a solution of 5 (58.0 mg, 32.1 μmol) in mesitylene (3 mL) was added TBE (41.3 μL, 320 μmol) at ambient temperature. The solution was heated at 50 °C for 16 h, during which time a colour change from yellow to dark orange was observed. The dark orange product was precipitated with pentane, isolated by filtration, and dried in vacuo. Crystals suitable for X-ray crystallography were obtained through recrystallisation from mesitylene–pentane. Yield: 59.8 mg (31.6 μmol, 98%).
1
H NMR (d8-toluene, 500 MHz, selected data): δ 6.73 (t, 3JHH = 8.2, 1H, 4-py), 6.12–6.32 (m, 2H, 3/5-py), 4.48–4.62 (m, 1H, CHCH2), 3.38–3.28 (m, 1H, CHCHH), 3.16 (d, 3JHH = 8.1, 1H, CHCHH), −0.04 (s, 9H, tBu). The spectrum is characterised by very broad ArF signals consistent with a C1 symmetric structure on the timescale of the experiment.
13
C{
1
H} NMR (d8-toluene, 126 MHz, selected data): δ 161.5 (br, 2/6-py), 145.7 (s, 4-py), 104.2–104.8 (m, 3/5-py), 82.8 (br, ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) H=CH2, confirmed by HMQC), 44.4 (br, CHH2), 35.5 (s, tBu{C}), 28.0 (s, tBu{CH3}).
H=CH2, confirmed by HMQC), 44.4 (br, CHH2), 35.5 (s, tBu{C}), 28.0 (s, tBu{CH3}).
19 F{ 1 H} NMR (d8-toluene, 376 MHz): δ −56.6 (br, 3F, ArF), −57.4 (br, 3F, ArF), −58.6 (br, 3F, ArF), −58.9 (br, 3F, ArF), −62.9 (s, 24F, Arf).
31 P{ 1 H} NMR (d8-toluene, 162 MHz): δ 151.9 (d, 2JPP = 380, 1P), 143.0 (d, 2JPP = 380, 1P).
HR ESI-MS (positive ion, 4 kV): 1028.1251 ([M]+, 1028.1264 calcd) m/z.
NMR scale thermolysis of 6
A solution of 6 (18.9 mg, 10.0 μmol) in mesitylene (0.5 mL) within a J. Young valve NMR tube was heated at 120 °C for 24 h. No new phosphorous containing products were apparent by analysis by 31P NMR spectroscopy. A new peak was observed at δ −61.2 in the 19F{1H} NMR spectrum and ascribed to partial decomposition of the [BArf4]− anion (ca. 10% by integration relative to the signal for the intact anion).NMR scale reaction of 6 with CO; preparation of [Ir(PONOP-ArF)(CO)][BArf4] 7
A solution of 6 (18.9 mg, 10.0 μmol) in mesitylene (0.5 mL) within a J. Young valve NMR tube was freeze–pump–thaw degassed and placed under an atmosphere of CO (1 atm) at ambient temperature. An immediate colour change from dark orange to bright yellow was observed and analysis by NMR spectroscopy within 5 min indicated complete conversion of 6 into 7. Volatiles were removed under reduced pressure affording a yellow oil. Yellow crystals suitable for X-ray crystallography were obtained after recrystallisation from CH2Cl2–pentane. Yield: 13.6 mg (7.4 μmol, 74%).1 H NMR (CD2Cl2, 500 MHz): δ 8.13 (t, 3JHH = 8.3, 1H, 4-py), 8.02 (dvt, 3JHH = 8.2, JPH = 16, 4H, 6-ArF), 7.95 (d, 3JHH = 7.5, 4H, 3-ArF), 7.84 (t, 3JHH = 7.7, 4H, 4-ArF), 7.76 (t, 3JHH = 7.9, 4H, 5-ArF), 7.70–7.75 (m, 8H, 2-Arf), 7.54 (br, 4H, 4-Arf), 7.18 (d, 3JHH = 8.3, 2H, 3-py).
13 C{ 1 H} NMR (CD2Cl2, 126 MHz): δ 179.0 (t, 2JPC = 9, CO), 162.3 (vt, JPC = 9, 2-py), 162.2 (q, 1JCB = 50, 1-Arf), 148.7 (s, 4-py), 135.9 (vt, JPC = 20, 6-ArF), 135.2 (br, 2-Arf), 134.8 (s, 4-ArF), 133.1 (vt, JPC = 13, 5-ArF), 131.5 (qvt, 2JFC = 33, JPC = 7, 2-ArF), 129.6 (vt, JPC = 60. 1-ArF), 129.6 (obscured, 3-ArF), 129.3 (qq, 2JFC = 31, 3JCB = 3, 3-Arf), 125.0 (q, 1JFC = 272, Arf{CF3}), 123.8 (q, 1JFC = 275, ArF{CF3}), 117.9 (sept, 3JFC = 4, 4-Arf), 105.8 (vt, JPC = 7, 3-py).
19 F{ 1 H} NMR (CD2Cl2, 376 MHz): δ −56.3 (s, 12F, ArF), −62.9 (s, 24F, Arf).
31 P{ 1 H} NMR (CD2Cl2, 162 MHz): δ 151.1 (s).
HR ESI-MS (positive ion, 4 kV): 974.0448 ([M]+, 974.0431 calcd) m/z.
IR (ATR): ν(CO) 2040 cm−1.
Crystallography
Data were collected on a Rigaku Oxford Diffraction SuperNova AtlasS2 CCD diffractometer using graphite monochromated CuKα radiation and an Oxford Cryosystems N-HeliX cryostat (150 K). Data were collected and reduced using CrysAlisPro.25 The structures were solved using SHELXT and refined using SHELXL, through the Olex2 interface.26,27 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were placed in calculated positions and refined using the riding model. Full details for all structures reported are documented in the CIF format and have been deposited under CCDC 2217137–2217141.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We thank the Spanish Ministry of Universities and the European Union (Margarita Salas grant funded by the European Union-NextGenerationEU, I. B.), the European Research Council (ERC; grant agreement 637313, T. M. H. and A. B. C.), and Royal Society (RGF\EA\180128, J. E. S.; UF100592 and UF150675; A. B. C.) for financial support. High-resolution mass-spectrometry data were collected using instruments purchased through support from Advantage West Midlands and the European Regional Development Fund. Crystallographic data were collected using an instrument that received funding from the ERC under the European Union's Horizon 2020 research and innovation programme (grant agreement No 637313).References
- (a) D. J. Sheldon and M. R. Crimmin, Chem. Soc. Rev., 2022, 51, 4977–4995 RSC; (b) E. Pietrasiak and E. Lee, Chem. Commun., 2022, 58, 2799–2813 RSC; (c) S. D. Pike, M. R. Crimmin and A. B. Chaplin, Chem. Commun., 2017, 53, 3615–3633 RSC; (d) O. Eisenstein, J. Milani and R. N. Perutz, Chem. Rev., 2017, 117, 8710–8753 CrossRef CAS PubMed; (e) T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931–972 CrossRef CAS; (f) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119–2183 CrossRef CAS.
- (a) W. Chen, C. Bakewell and M. Crimmin, Synthesis, 2017, 810–821 CAS; (b) G. Coates, B. J. Ward, C. Bakewell, A. J. P. White and M. R. Crimmin, Chem. – Eur. J., 2018, 24, 16282–16286 CrossRef CAS; (c) M. R. Crimmin, M. J. Butler and A. J. P. White, Chem. Commun., 2015, 51, 15994–15996 RSC; (d) T. Chu, Y. Boyko, I. Korobkov and G. I. Nikonov, Organometallics, 2015, 34, 5363–5365 CrossRef CAS; (e) C. Douvris and O. V. Ozerov, Science, 2008, 321, 1188–1190 CrossRef CAS PubMed.
- J. Choi, D. Y. Wang, S. Kundu, Y. Choliy, T. J. Emge, K. Krogh-Jespersen and A. S. Goldman, Science, 2011, 332, 1545–1548 CrossRef CAS.
- (a) M. R. Gyton, A. E. Kynman, B. Leforestier, A. Gallo, J. R. Lewandowski and A. B. Chaplin, Dalton Trans., 2020, 49, 5791–5793 RSC; (b) T. M. Hood, B. Leforestier, M. R. Gyton and A. B. Chaplin, Inorg. Chem., 2019, 58, 7593–7601 CrossRef CAS.
- (a) T. M. Hood and A. B. Chaplin, Dalton Trans., 2021, 50, 2472–2482 RSC; (b) T. M. Hood, M. R. Gyton and A. B. Chaplin, Dalton Trans., 2020, 49, 2077–2086 RSC; (c) M. R. Gyton, B. Leforestier and A. B. Chaplin, Organometallics, 2018, 37, 3963–3971 CrossRef CAS.
- (a) A. Longcake, M. R. Lees, M. S. Senn and A. B. Chaplin, Organometallics, 2022, 41, 3557–3567 CrossRef CAS PubMed; (b) T. M. Hood and A. B. Chaplin, Dalton Trans., 2020, 49, 16649–16652 RSC; (c) M. R. Gyton, B. Leforestier and A. B. Chaplin, Angew. Chem., Int. Ed., 2019, 58, 15295–15298 CrossRef CAS; (d) M. R. Gyton, T. M. Hood and A. B. Chaplin, Dalton Trans., 2019, 48, 2877–2880 RSC.
- (a) H. Salem, L. J. W. Shimon, Y. Diskin-Posner, G. Leitus, Y. Ben-David and D. Milstein, Organometallics, 2009, 28, 4791–4806 CrossRef CAS; (b) W. H. Bernskoetter, S. K. Hanson, S. K. Buzak, Z. Davis, P. S. White, R. Swartz, K. I. Goldberg and M. Brookhart, J. Am. Chem. Soc., 2009, 131, 8603–8613 CrossRef CAS PubMed.
- G. Feng, M. P. Conley and R. F. Jordan, Organometallics, 2014, 33, 4486–4496 CrossRef CAS.
- Z. Lu, C.-H. Jun, S. R. de Gala, M. P. Sigalas, O. Eisenstein and R. H. Crabtree, Organometallics, 1995, 14, 1168–1175 CrossRef CAS.
- J. Emerson-King, I. Prokes and A. B. Chaplin, Chem. – Eur. J., 2019, 25, 6317–6319 CrossRef CAS PubMed.
- R. C. Knighton, J. Emerson-King, J. P. Rourke, C. A. Ohlin and A. B. Chaplin, Chem. – Eur. J., 2018, 24, 4927–4938 CrossRef CAS PubMed.
- R. J. Kulawiec, E. M. Holt, M. Lavin and R. H. Crabtree, Inorg. Chem., 1987, 26, 2559–2561 CrossRef CAS.
- K. Stanek, B. Czarniecki, R. Aardoom, H. Rüegger and A. Togni, Organometallics, 2010, 29, 2540–2546 CrossRef CAS.
- A. Kumar, T. M. Bhatti and A. S. Goldman, Chem. Rev., 2017, 117, 12357–12384 CrossRef CAS PubMed.
- S. Gatard, C. Guo, B. M. Foxman and O. V. Ozerov, Organometallics, 2007, 26, 6066–6075 CrossRef CAS.
- For precedents see: (a) J. A. Garduño, D. S. Glueck, R. E. Hernandez, J. S. Figueroa and A. L. Rheingold, Organometallics, 2022, 41, 1475–1479 CrossRef; (b) S. G. Weber, D. Zahner, F. Rominger and B. F. Straub, Chem. Commun., 2012, 48, 11325–11327 RSC; (c) H. Salem, L. J. W. Shimon, G. Leitus, L. Weiner and D. Milstein, Organometallics, 2008, 27, 2293–2299 CrossRef CAS; (d) W. V. Konze, B. L. Scott and G. J. Kubas, Chem. Commun., 1999, 1807–1808 RSC.
- (a) K. S. Lokare, R. J. Nielsen, M. Yousufuddin, W. A. Goddard III and R. A. Periana, Dalton Trans., 2011, 40, 9094–9097 RSC; (b) T. Yano, Y. Moroe, M. Yamashita and K. Nozaki, Chem. Lett., 2008, 37, 1300–1301 CrossRef CAS.
- J. Campos, S. Kundu, D. R. Pahls, M. Brookhart, E. Carmona and T. R. Cundari, J. Am. Chem. Soc., 2013, 135, 1217–1220 CrossRef CAS.
- There is a small amount of residual solvent disorder around the pentane molecule.
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908–6914 CrossRef CAS.
- The frontier orbitals of the metal complex do not permit 3-center–2-electron bonding with the C–H bond and 7·pentane should not be confused with a σ-alkane complex such as those reported by Weller and co-workers, e.g. S. D. Pike, A. L. Thompson, A. G. Algarra, D. C. Apperley, S. A. Macgregor and A. S. Weller, Science, 2012, 337, 1648–1651 CrossRef CAS PubMed.
- (a) G. Nawn, K. M. Waldie, S. R. Oakley, B. D. Peters, D. Mandel, B. O. Patrick, R. McDonald and R. G. Hicks, Inorg. Chem., 2011, 50, 9826–9837 CrossRef CAS PubMed; (b) E. K. Cope-Eatough, F. S. Mair, R. G. Pritchard, J. E. Warren and R. J. Woods, Polyhedron, 2003, 22, 1447–1454 CrossRef CAS.
- (a) A. J. Martínez-Martínez and A. S. Weller, Dalton Trans., 2019, 48, 3551–3554 RSC; (b) W. E. Buschman, J. S. Miller, K. Bowman-James and C. N. Miller, Inorg. Synth., 2002, 33, 83–91 Search PubMed.
- P. S. Pregosin, NMR in Organometallic Chemistry, Wiley-VCH, 2012, pp. 251–254 Search PubMed.
- CrysAlisPro (Rigaku Oxford Diffraction, Yarnton, UK) Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, C71, 3–8 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR, IR and ESI-MS spectra of isolated compounds. CCDC 2217137–2217141. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2dt03608h |
| This journal is © The Royal Society of Chemistry 2023 |