
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGreen metrics in mechanochemistry
Nicolas
Fantozzi
a,
Jean-Noël
Volle
a,
Andrea
Porcheddu
 b,
David
Virieux
a,
Felipe
García
*cd and
Evelina
Colacino
*a
b,
David
Virieux
a,
Felipe
García
*cd and
Evelina
Colacino
*a
aICGM, Univ Montpellier, CNRS, ENSCM, 34293 Montpellier, France. E-mail: evelina.colacino@umontpellier.fr
bDipartimento di Scienze Chimiche e Geologiche, Università degli Studi di Cagliari, Cittadella Universitaria, 09042, Monserrato (CA), Italy
cDepartamento de Química Orgánica e Inorgánica, Facultad de Química, Universidad de Oviedo, Julián Claveria 8, Oviedo, 33006, Asturias, Spain. E-mail: garciafelipe@uniovi.es
dSchool of Chemistry, Monash University, Clayton, Victoria 3800, Australia. E-mail: Felipe.Garcia@monash.edu
First published on 11th September 2023
Abstract
The development of new green methodologies and their broader adoption for promoting sustainable development in chemistry laboratories and industry play a significant role in society, due to the economic importance of chemistry and its widespread presence in everyday life. Therefore, a sustainable approach to chemistry contributes to the well-being of the worldwide population and complies with the United Nations Sustainable Development Goals (UN SDGs) and the European Green Deal. The review highlights how batch and continuous mechanochemical methods are an eco-friendly approach for organic synthesis, with a lower environmental footprint in most cases, compared to solution-based procedures. The assessment is objectively based on the use of green metrics (e.g., atom and real atom economy, E-factor, process mass intensity, material parameter recovery, Eco-scale, stoichiometric factor, etc.) and indicators (e.g. DOZN tool and life cycle assessment, LCA, studies) applied to organic transformations such as synthesis of the amide bond, carbamates, heterocycles, active pharmaceutical ingredients (APIs), porphyrins, porous organic polymers (POPs), metal- or acid-catalysed processes, multicomponent and condensation reactions, rearrangements, etc. The generalized absence of bulk solvents, the precise control over the stoichiometry (i.e., using agents in a stoichiometrically rather than in excess), and the more selective reactions enabling simplified work-up procedures are the distinctive factors, marking the superiority of mechanochemical processes over solution-based chemistry.
 Nicolas Fantozzi | Nicolas FANTOZZI obtained his MSc in organic chemistry at Université Grenoble-Alpes. He completed his PhD in 2019 at Bordeaux University (France) on fluorescent supramolecular probes for neurotransmitters detection. Then, he moved to Université de Montpellier (France) as a postdoc position where he worked on the synthesis of active pharmaceutical ingredients by mechanochemistry. His research focuses on green and sustainable chemistry. |
 Jean-Noël Volle | Jean-Noël VOLLE obtained a Master of Science (DEA) at the University Lyon 1 in 1996, and he completed his PhD at the University of Lausanne in 2001 under the supervision of Prof. Manfred Schlosser. In 2002, he spent height months in the group of Martinus Gijs at Ecole Polytechnique Fédérale de Lausanne (EPFL), thus moved at National Graduate School of Chemistry of Montpellier (ENSCM) in 2003 as postdoctoral research fellow. Thereafter, he occupied a temporary teacher-researcher (ATER) at ENSCM for an eleven-month period in 2004–2005, and in late 2005, he became “Maître de Conférences” in the same academic institution. Currently, his researches are developed in the group of Prof. David VIRIEUX, located in the Institute Charles Gerhardt of Montpellier (ICGM) in the Molecular Chemistry and materials department. |
 Andrea Porcheddu | Andrea Porcheddu received a Chemistry degree with first-class honors from the University of Sassari (Italy) in 1995. He completed his PhD in 1999 under the supervision of Prof. Taddei and accomplished post-doctoral studies in 2000 at Louis Pasteur University (Strasbourg, Fr). In 2001, he moved back to Sassari University, where he was appointed Assistant Professor. In January 2015, he joined the Chemistry Department of the University of Cagliari (Italy), where he currently has a permanent position as a Full Professor. His expertise includes preparing compounds with complex molecular architecture and finding more environmentally friendly alternatives for synthesis using advanced technologies, Borrowing Hydrogen and Transfer Hydrogenation strategies, microwaves, blue-LEDs, IR-Irradiation. His main scientific interest currently covers mechanochemistry, focusing on value-added compounds for the industry and Active Pharmaceutical Ingredients. Prof. Porcheddu is the author or co-author of more than 125 scientific publications in refereed international journals (H index, Scopus = 39). |
 David Virieux | David Virieux completed his PhD in 1998 at the University of Montpellier, supervised by Prof. Henri-Jean Cristau. His doctoral research centered on polyphosphoryl compounds for selectively extracting actinides. He went to the USA at Rhône-Poulenc Ag Co to contribute to the development of small molecules potentially effective as insecticides. In late 1999, he rejoined Prof. Cristau's team at the ENSCM (Montpellier) and was later promoted to a full professorship in 2007. His research, conducted at the Institute Charles Gerhardt of Montpellier, revolves around specific facets of organophosphorus chemistry. These encompass a spectrum of applications within life sciences, including agrochemical and medicinal chemistry, and the development of new synthetic methodologies. Notably, his research places emphasis on organocatalysis, multicomponent reactions, and enantioselective synthesis. In 2014, he played a key role in founding Phost’In Therapeutics, an innovative player in the field of Glyco-Immuno-Oncology, currently advancing a drug candidate through phase 1 clinical trials. |
 Felipe García | Felipe García is originally from the coastal town of Gijón (Spain) and gained both his BSc and MSc degrees at Oviedo University. In 2001, he moved to the University of Cambridge to carry out his graduate studies as a Cambridge European Trust and Newton Trust Scholar under the supervision of Prof. D. S. Wright. He then gained a Junior Research Fellowship at Wolfson College (2005) and was appointed College Lecturer in Inorganic Chemistry at Newnham and Trinity Colleges (2006). In March 2011, he moved to Nanyang Technological University as an Assistant Professor. In 2022, he returned to the University of Oviedo as a Margarita Salas Senior researcher (funded by FiCYT). In 2024, he will transition to Monash University (Australia) to advance his research career. Felipe has published over 100 papers on Main Group Chemistry and maintains a strong interest in the synthesis of novel compounds for industrial and biological applications. |
 Evelina Colacino | Evelina Colacino received her double PhD (with European Label) in 2002 at the University of Montpellier II (France) and at the University of Calabria (Italy). She was Research Fellow at the Catholic University of Louvain (Belgium, 2003), Scientist at Sigma-Tau Pharmaceuticals (Italy, 2004) and Post-Doctoral Fellow at the University of Montpellier II (France, until 2007). Associate Professor (since 2013) of Organic and Green Chemistry, she develops eco-friendly methodologies by mechanochemistry, focusing on the preparation of Active Pharmaceutical Ingredients (medicinal mechanochemistry). She promotes sustainability in Higher Education by integrating the fundamentals and the practice of green chemistry and mechanochemistry in organic chemistry courses and teaching laboratories. Member of the Advisory Board of the Green Chemistry Commitment and the International Mechanochemical Association, she chairs the European Programmes COST Action CA18112 ‘Mechanochemistry for Sustainable Industry (2019–2023) and Horizon Project IMPACTIVE (Innovative Mechanochemical Processes to synthesise green ACTIVE pharmaceutical ingredients, 2022–2026). |
Introduction
Chemistry plays a significant role in society because of its economic importance and widespread presence in everyday life. Unfortunately, since it is everywhere, it is often overlooked. Products or processes taken for granted, such as drinking water, pasteurization, or medicines, were borne from advances in the Chemistry field – which is ultimately the science of change.Over the years, chemistry has evolved towards increasing complexity and diversity, from molecules to materials, from appealing structures to incredibly complex industrial processes.
However, chemistry is not only science but also industry, many chemical processes have an industrial edge, and consequently, it has an extraordinary impact on economic and social life. Therefore, it is unsurprising that “chemistry” is often summoned to deal with industrial and societal issues. Several costs and availability of raw materials, energy, safety in the use of products, community protection, and the battle against pollution, inter alia.
In the last century, the intensification of human tasks has involved chemistry with some disastrous results such as damage to the protective ozone layer, global warming, air pollution, and the limitless exploitation of natural resources. To address them, various measures have been taken over the past 50 years to reduce the adverse effects the production of chemicals can have on the environment.
The recognition of the need to reduce the adverse effects of the chemical industry on the environment to safeguard future generations has been the driving force behind the development of green chemistry. It is not a separate branch of chemistry but an aspect that permeates every process design stage.
Green chemistry is “the science that promotes the discovery, design, and use of chemicals and processes to reduce or remove the use and production of hazardous substances,” which can ultimately be summarized in one word: sustainability. The concept of sustainability is strongly connected to circular economy (i.e., an economic system based on reusing materials in subsequent productive cycles, reducing waste to a minimum). Another important aspect is the reduction of energy consumption. Have we gone far enough? Is it still possible to push chemistry toward an eco-friendlier future?
Applying the 12 principles of green chemistry – formulated by Anastas and Warner in In 19981 – to the industrial sector may seem challenging since many parameters can be considered. For instance, comparing different processes to the same product or evaluating them during development is not straightforward unless a shared metric is used. This difficulty, has been the driving force for the rapid emergence of green metrics over the last few years.
Green metrics
Green chemistry metrics are a collection of indicators used to describe several aspects relating to the principles of green chemistry for a given chemical process.These metrics allow measuring changes in a chemical process's performance by quantifying its overall efficiency or environmental impact. Notably, and directly related to mechanochemistry, central to this review, most efforts to minimize the environmental footprint of a chemical process have highlighted the need for using safer, less toxic, and more benign solvents or eliminating solvents. Also, reducing the number and quantities of reagents and auxiliaries is an effective way to minimize environmental impact.
However, the final evaluation heavily depends on a series of pressing questions: how do we define “greenness”? What are the appropriate indicators to measure the effectiveness of a chemical transformation while minimizing environmental impact? How to reduce waste production and/or energy consumption in a chemical process?2
These metrics generally encourage the development of new methodologies and facilitate the broader adoption of green chemistry technologies for promoting sustainable development in laboratories and industry. Table 1 reports the green chemistry metrics used to assess the greenness of mechanochemical processes.
| Metric | Abbreviation | Formula | Optimal value | Ref. |
|---|---|---|---|---|
| Atom economy | AE |
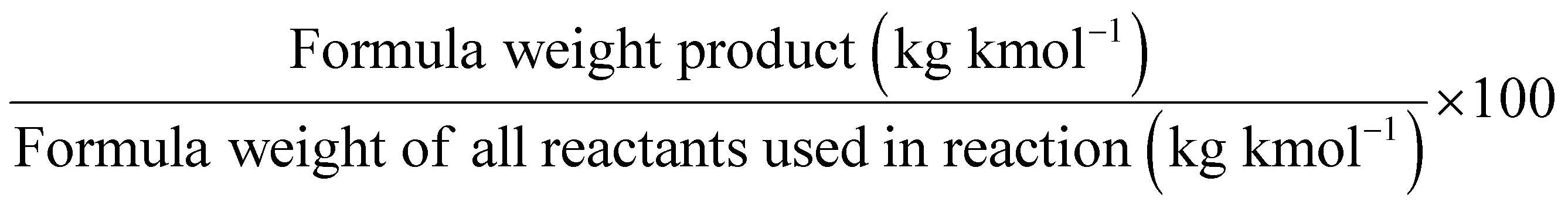
|
100% | 3 |
| FW: formula weight in g mol−1 | ||||
| Real atom economy | RAE |
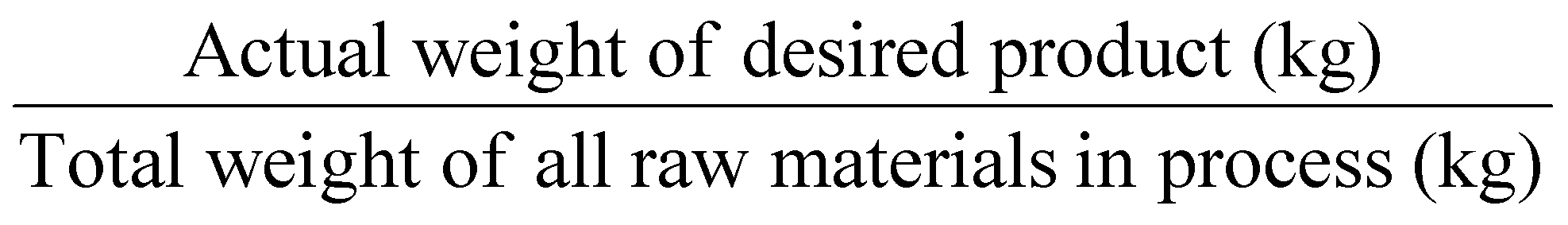
|
1 | 4 |
| Environmental factor | E-Factor |
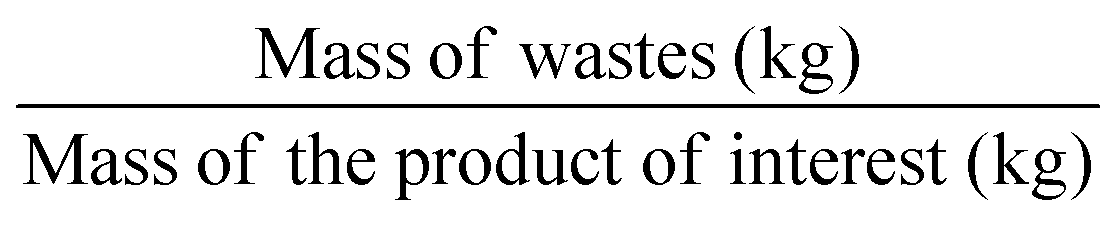
|
0 | 5 |
| Process mass intensity | PMI |
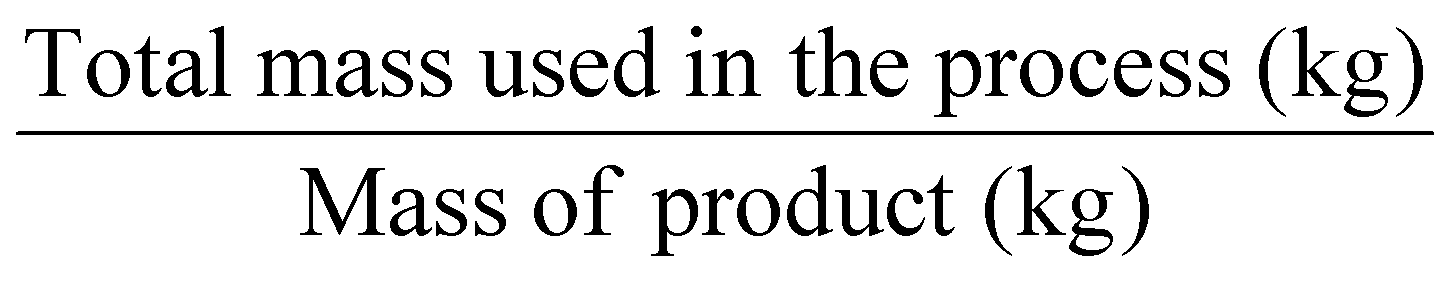
|
1 | 6 |
| Reaction mass efficiency | RME |
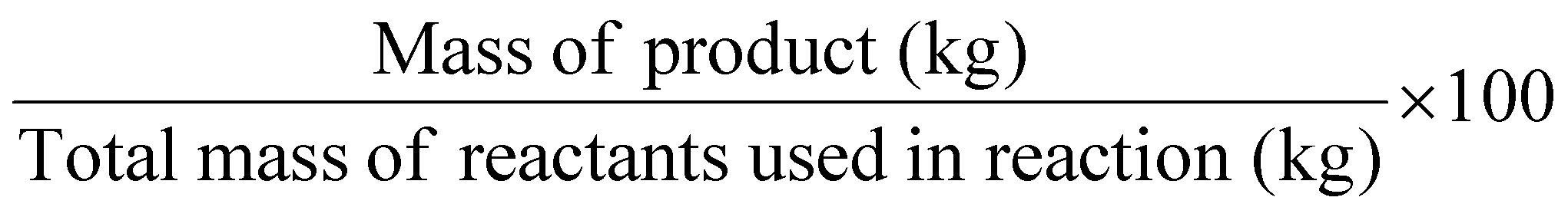
|
100% | 7 |
| Material recovery parameter | MRP |
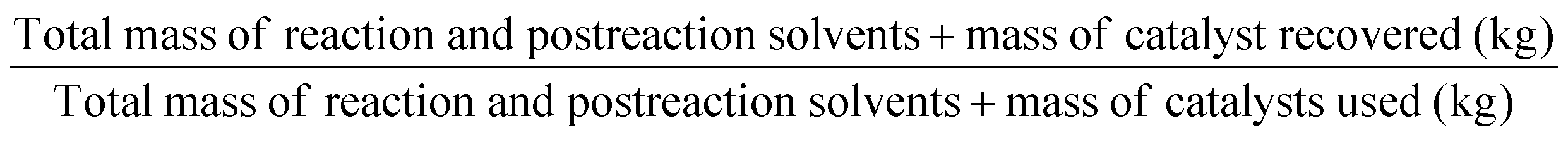
|
1 | 8 and 9 |
| 0 < MRP < 1 | ||||
| Stoichiometric factor | SF |
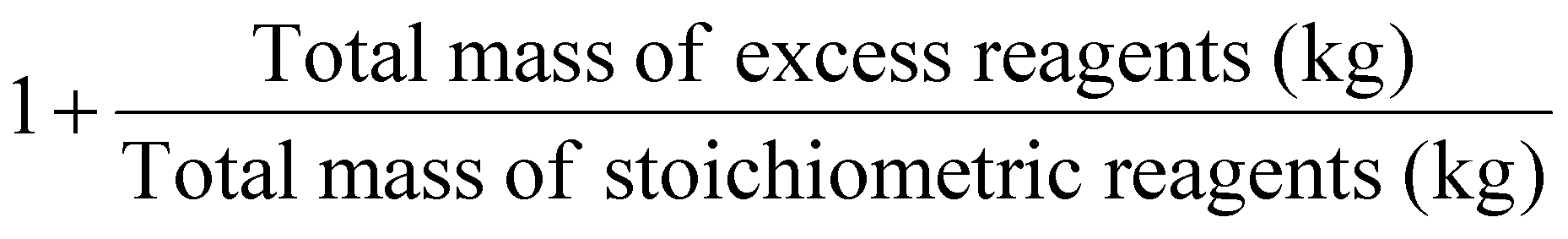
|
1 | 10 |
| Mass intensity | MI |

|
1 | 7 |
| Mass productivity | MP |
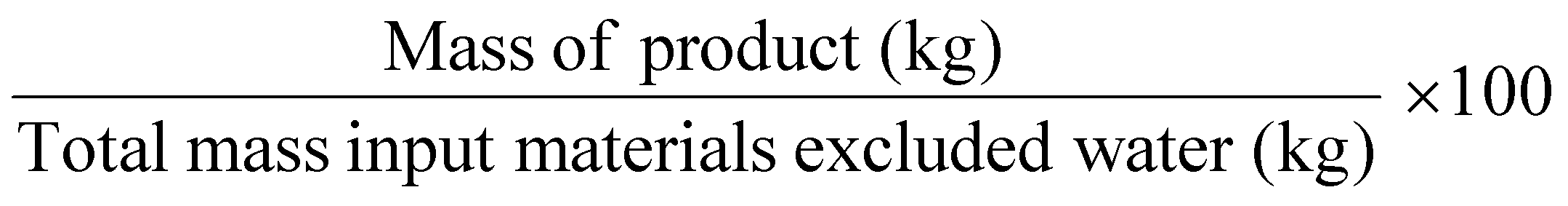
|
100% | 11 |
| Molar efficiency | Mol. E |
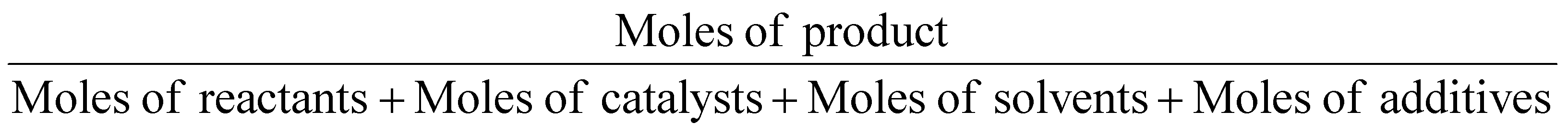
|
1 | 12 |
One of the most commonly used metrics is atom economy (AE) (Table 1, entry 1), also named atom efficiency, and identified as principle no. 2 among the 12 principles of green chemistry.1 Introduced by Barry Trost in 1991, AE is directly related to the search for synthetic efficiency, where the maximum number of atoms present in the reactants should be incorporated into the reaction products.3 On a scale between 0 and 100, the higher the value. The better is the AE of the process.
AE is a metric that can be calculated ‘a priori’ (i.e., before performing experiments). Therefore, when several methods are available to access the same target product, the calculation of the AE for each synthesis will drive the appropriate choice towards selecting the process displaying the highest AE value.
Related to AE and to take into account the reaction's yield and stoichiometry, Real Atom Economy (RAE) – Table 1, entry 2 – can also be calculated:4 to RAE should be close to 100%.
In contrast to AE, the environmental factor (E-factor) – Table 1, entry 3 – is an ‘a posteriori’ metric that can only be calculated once the experiment has been conducted. This parameter focuses on the waste(s) generated during a reaction,5 and it takes into account reagents, solvent losses throughout the synthesis, and work-up and purification steps, as well as all the additives used during the process (e.g., drying agents, silica gel, etc.) with respect to the formed product mass (which takes into account also the yield). While water is generally excluded from this calculation, energy losses should be usually included, which might not be trivial to be measured and calculated. Also, in its basic definition, fuel use has to be included. On a scale between 0 and 100, the E-factor has to be as close as possible to 0 to account for an environmentally-friendly process.
Another mass-based environmental process waste metric is process mass intensity (PMI), Table 1, entry 4, defined as the total mass in kg of raw materials (reagents, solvents, etc.) used (input of materials) to produce 1 kg of the product (output of the synthesis).6 The PMI is a metric used at the forefront of a process, and it can also be readily calculated from the E-factor (PMI = E-factor + 1). Therefore, the ideal value of PMI corresponds to 1. Therefore, when comparing two processes, the one with the lowest PMI will be the greenest.
The reaction mass efficiency (RME) – Table 1, entry 5 – is the percentage mass of the target product expressed in kg with respect to the mass of all reactants. Both AE and chemical yield are considered in the calculation.7 The higher the RME score, the more environmentally friendly the process will be.
As part of RME, the material recovery parameter (MRP), which considers solvent from reaction and extraction, indicates the possibility of reusing solvents. It should be between 0 and 1, the best value being 1.10 Stoichiometric factor (SF) allows to consider the excess reagents used throughout a process.7 The SF value of 1 corresponds to stoichiometric reactions (i.e., carried out with no excess reagents), whereas an SF > 1 indicates stoichiometric excess used.
Related to chemical production efficiency, Constable and Curzons developed the mass intensity (MI) and mass productivity (MP) parameters.7,11 Mass intensity and mass productivity are related to PMI and RME, respectively, excluding water. Therefore, for these indicators, the MI should be close to 1, and MP should be close to 100% efficiency.
In the same way, molar efficiency should be close to 100% and can be calculated following this equation.12
In 2006, Van Aken introduced the Eco-scale,13 a qualitative metric evaluating the quality of an organic transformation, taking into account yield, cost of the reaction components, safety, and conditions of reaction (temperature, duration), and it includes work-up and purification to give a score between 0 and 100 where 100 is the best score.
In addition to the 12 principles for greener chemistry and the parameters mentioned above, generic evaluation approaches such as life cycle assessment (LCA)14,15 or more chemically specific tools like DOZN 2.016 can be used for a quantitative evaluation of the environmental impact of chemical processes.
The DOZN 2.0 tool is a free web-based software able to quantitatively assess the greenness of a process or a product against the 12 principles of green chemistry – which are only qualitative. Therefore, each of the 12 green chemistry principles is scored by the DOZN 2.0 tool, taking into account data input from the reaction and process conditions and extracting data for reactants and chemicals from the globally harmonized system (GHS) and safety data sheet (SDS) information. Then, the 12 principles of green chemistry are divided into three subgroups: improved resource use (group 1), increased energy efficiency (group 2) and reduced human and environmental hazards (group 3). The software delivers an ‘aggregate score’ averaging and normalizing the scores obtained for each subgroup, ranging from 0 to 100. Generally speaking, an aggregate score below 1 indicates a green process. However, the closer to zero is the aggregate score, the greener the process will be. The DOZN 2.0 tool is a harmonized approach to greenness assessment, allowing to compare the greenness of any product or process by using the values obtained for their respective aggregate scores. Both these methods (LCA and DOZN) are more complex, considering additional parameters, such as global warming and ecotoxicity, for which advanced software is also required for the complete assessment.
Mechanochemical methods
Mechanochemical processes have been acknowledged by the International Union of Pure and Applied Chemistry (IUPAC) among the “top ten emerging technologies in chemistry,”17 responding to the growing need for sustainable reaction conditions and clean processes.The reactions are carried out by grinding reagents with ball-mill devices such as vibrating (VBM), planetary (PBM), SPEX mills, Tumbler ball-mill or single-screw device (SSD) using mechanical forces to enable chemical reactivity (Fig. 1).18
 | ||
| Fig. 1 Milling devices: (a) vibrational ball-mill, (b) planetary ball-mill, (c) SPEX 8000 shaker mill, (d) twin-screw Extrusion, (e) tumbler ball-mill, (f) single-screw device. Adapted with permission of the American Chemical Society from ref. 19–21. | ||
The use of mortar and pestle is a traditional and widely used tool for manual grinding and milling processes (e.g., chemistry, pharmacy, culinary arts, etc.). Mechanochemical reactions conducted with a mortar and pestle rely on the mechanical energy generated by manual grinding. The solid reagents are placed in the mortar, and the pestle is used to apply pressure and friction, causing the particles to collide and react. This grinding process facilitates solid-state reactions without additional solvents or heating.
The mortar and pestle technique offers several advantages. First, it allows precise control over the grinding process, enabling researchers to tailor the particle size and distribution. Using a mortar and pestle can also be advantageous in small-scale reactions or when specific reaction conditions are required.
However, it's important to note that the manual nature of the process can be time-consuming and labour-intensive, making it less suitable for large-scale or high-throughput applications. In such cases, automated ball mills are often preferred for their efficiency and ability to handle larger quantities.
Ball mills are versatile tools that have been used for wide range of milling applications. There are several types of mills that have been commonly used to perform mechanochemical reactions. Vibratory mills typically contain a jar that is shaken from side to side at high frequency causing the grinding media and reagents to impact and collide (Fig. 1a). This type of mill has been commonly used for the synthesis of organic and inorganic compounds and the mechanical alloying of materials.
The planetary ball mill action consists of one or more grinding jars that rotate around their axis while also rotating around a central axis (Fig. 1b). This dual rotation creates effective mixing and grinding of the reactants. In contrast to vibratory mils, which can only perform reactions at the multigram scale, these devices can work on scales of hundreds of grams.
The SPEX ball mill (Fig. 1c) operates similarly to the vibratory mill, with the vial and the grinding balls being shaken or agitated in a figure-of-eight motion. The mechanical action enables rapid and efficient grinding of various materials, including metals, ceramics, minerals, and polymers. This tool has been commonly used for mechanical alloying (blending elemental powders to form alloys), sample pulverization, and material synthesis.
Overall, ball mills are a versatile and powerful tool for mechanical milling and grinding, providing researchers with an efficient means to process and manipulate materials for various scientific and industrial purposes.
Twin-screw extruders are widely used in various industries, including polymer processing, food processing, pharmaceuticals, and chemical manufacturing (Fig. 1d and e). They are versatile tools that offer precise control over the mixing, compounding, and extrusion of materials. Twin-screw extruders consist of two intermeshing screws housed in a barrel. The screws rotate in the same direction but have opposite helical orientations. This design enables the screws to generate a strong forward conveying action while creating intense shear and mixing forces, which promotes intense mixing and thorough dispersion of additives (if used). In addition, the material can be forced through a die at the end of the barrel to shape and form the output materials.
Twin-screw extruders offer numerous advantages, including precise control over processing parameters, high productivity, efficient mixing and compounding, and the ability to process a wide range of materials. As a result, they are widely used in manufacturing plastic products, food processing (such as extruded snacks), pharmaceutical formulations, and various chemical processes where precise control over mixing, compounding, and extrusion is essential.
Finally, a drill press with a counter-clockwise rotating drill bit was recently used to demonstrate that excellent mechanochemistry can be achieved with a tight budget (Fig. 1f).
Controlling mechanochemical reactivity
There are several variables that have to be fine-tuned during any mechanochemical process (Fig. 2). In terms of batch processes, milling media (i.e., jars and balls), can be found in a variety of materials (stainless-steel, tungsten carbide, zirconia, Teflon, etc.). Each material has a specific density and hardness, which is a straightforward way to control the energy input – which ultimately influences chemical reactivity. Chemical resistance also differs between materials and will affect the potential leaching of metal ions (e.g., Fe, Co, etc.) or metal contamination resulting from prolonged milling. Additional ways to control the reaction is by changing the frequency or rotation speed of the mills, filling degree, number and size of balls, jar volume and geometry, etc.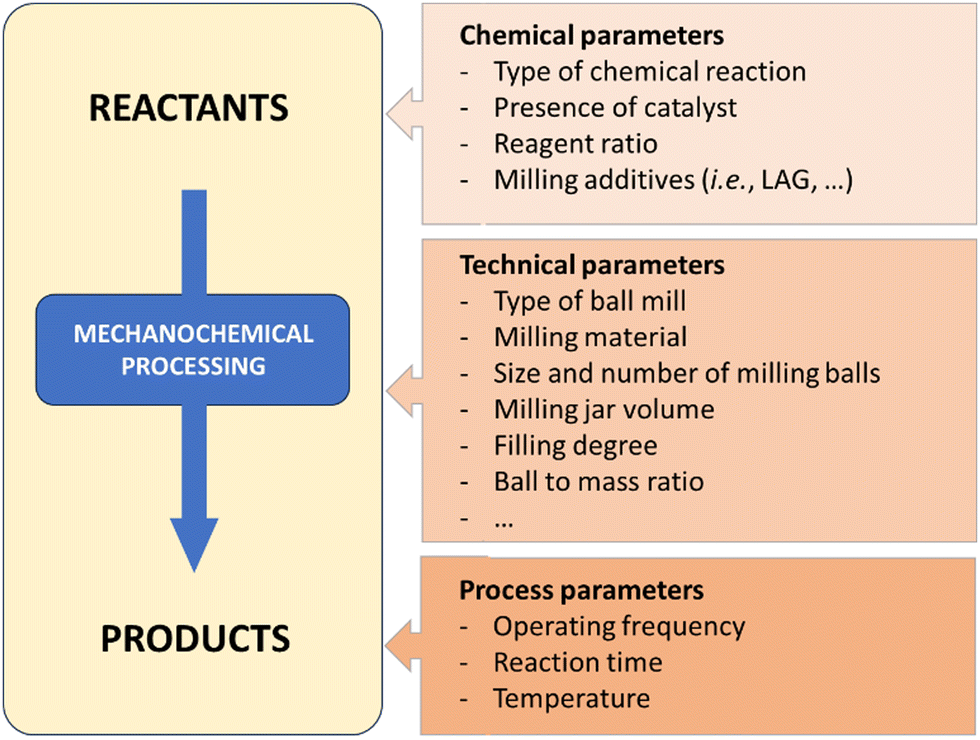 | ||
| Fig. 2 Selected parameters to be taken into consideration during a ball-mill mechanochemical process.22 | ||
Any chemical reactor requires ideal operating temperature to accommodate the nature (i.e., endothermic or exothermic) of the transformation taking place. Similar to standard batch solution-based reactors, batch mechanochemical vessel have an unfavourable surface area-to-volume ratio making heat removal challenging as the size of the reactor and/or the exothermicity of the reaction increases, which restricts the application of batch or semi-batch mechanochemical reactors to either laboratory-scale or transformations that are not overly exothermic and/or particularly heat sensitive. However, this limitation has been partially circumvented by developing custom-made temperature-controlled reactors and commercial equipment with temperature control for batch and continuous processes. This addresses and/or mitigates the challenges mentioned above with traditional mechanochemical techniques.23
Related to temperature effects, and despite most mechanochemical reactions reported in the literature relying on mechanical energy to initiate or promote chemical transformation without needing external heating at room temperature. Some mechanochemical reactions require the input of thermal energy for optimal results. Heating can provide to initiate the reaction with mechanochemical activation is not enough to surpass the activation energy barrier and proceed at a reasonable rate. In addition, applying heat can improve reaction kinetics, resulting in better yields and faster reaction rates.24–32
Moreover, the combination of heating and mechanical forces can cause solid-state transformations (e.g., phase transitions, structural changes, etc.), which can aid in achieving specific transformations or activating desired reaction pathways. Also, precise control of the reaction temperature implies better manipulation of the reaction conditions, which can result in optimised reaction rates, selectivities, and product yields.33
Mechanochemical synthesis can also be controlled by incorporating liquid additives into mechanochemical reactions (i.e., liquid assisted grinding, LAG). They act as lubricants, enhancing mixing and reaction kinetics. In this regard, it is important to note that the amount of liquids used is a very small quantity relative to the amounts used in their solutions-based counterparts, hence its addition does not constitute as bulk solvent use. LAG reactions are characterised by an empiric parameter called η (eta) (expressed in μL per mg of solid reactants) is used to describe, and normalise, the amount of liquid additive added. The formal definition allows to distinguish quantitatively among neat grinding, LAG conditions, slurry and reactions in ‘solution’. In particular, for η = 0 μL mg−1 the reaction occurs by neat grinding. For η values comprised between 0 and 1 μL mg−1, the reaction occurs under LAG, if η values are between 1 and 6 μL mg−1 the reaction is considered a slurry, while for η is above 6 μL mg−1, no mechanochemical effect can be evoked and the reaction is considered to occur in solution.34
In contrast to LAG, often, to optimise mechanochemical reactions, it is necessary to ensure that all reagents form a solid mixture, since the presence of liquid reagents can negatively affect the rheology of the mixture resulting in a paste-like consistency that hinders the energy transfer during the grinding process – thus rendering the process ineffective.35 In addition, extensive research has demonstrated that the mixture's rheology can significantly impact the kinetics of the chemical reaction. Factors such as particle size, surface area, and the presence of additives (liquid or solid) affect the texture of the mixture, consequently affecting the reaction kinetics (i.e., different reaction rates and pathways).36
Understanding and controlling the reaction mixture's texture is crucial for achieving the desired reaction rates and outcomes in mechanochemistry. Therefore, to improve the rheology and enhance mechanical action during impacts, it is common practice to include solid additives, such as silica and sodium sulfate, in the mixture. They are typically affordable solid green components that do not actively participate in the chemical reaction.
Advantages of mechanochemistry
In addition to the solvent-free, or virtually solvent-free, nature of mechanochemical reactions, which intrinsically reduces the process's environmental footprint. Mechanochemistry has shown other advantages – from higher yields and selectivity to access new compounds and “impossible molecules” – that have made it increasingly popular among synthetic chemists (Fig. 3).37 Moreover, the possibility of employing traditionally poorly insoluble reagents provides mechanochemical reactions an edge over conventional solution-based methods in a wide range of technologically relevant fields (i.e., perovskite synthesis, polyaromatics, etc.).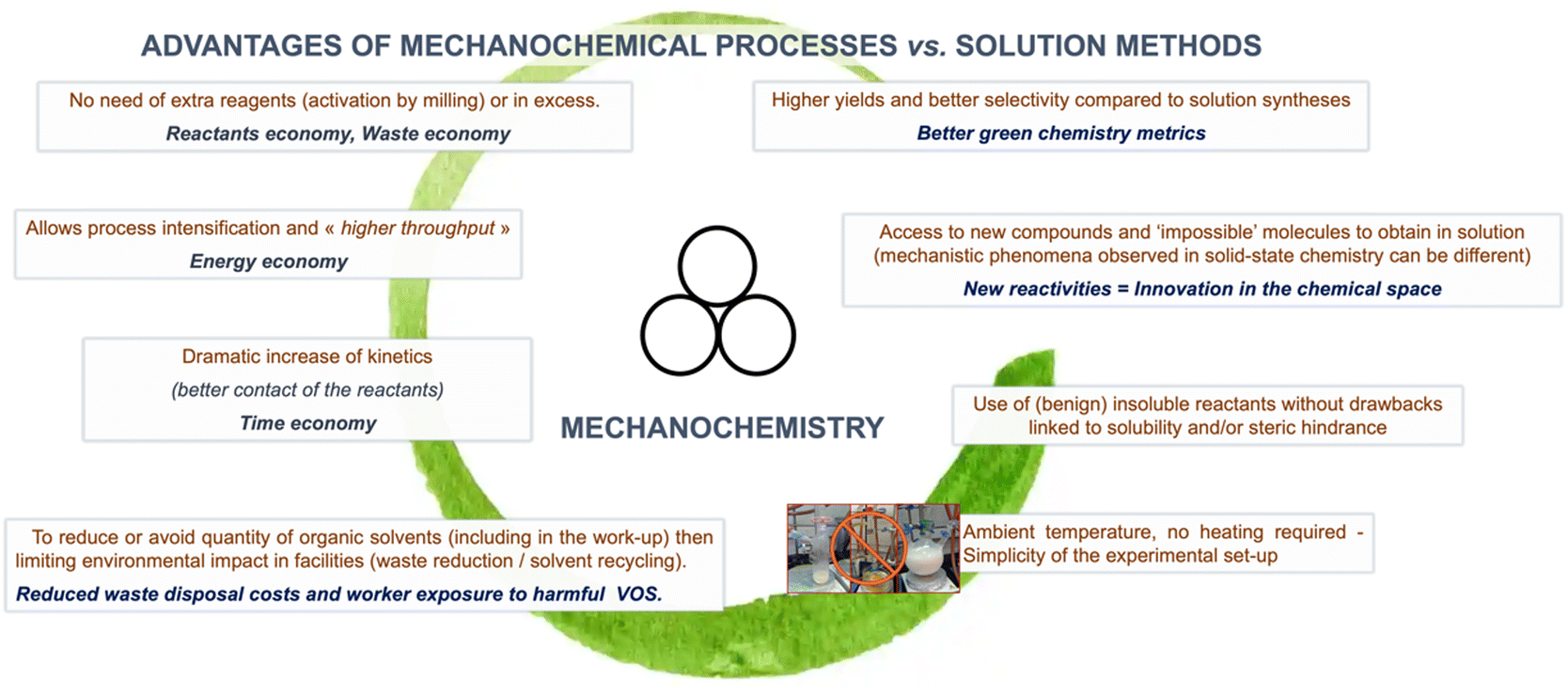 | ||
| Fig. 3 Advantages of mechanochemistry reported in the literature. Hanusa's formalism to represent reactions activated by mechanochemistry was used.38 | ||
Another aspect that is particularly relevant to this review, in addition to the elimination of bulk solvents, is that in contrast to traditional solution-based chemistry, where reactions often require excess reagents to achieve full conversion. Mechanochemical reactions can often be performed with a equimolar amount of reagents. This can be attributed to the efficient mixing of reagents caused by the milling action, which enhances reagent reactivity, allowing for complete utilization of the starting materials. This helps minimise the need for excess reagents, reducing waste generation, simplifying product purification, and enhancing atom economy.
Mechanochemistry and green chemistry
While the ball-milling devices mentioned above are limited to batch syntheses, twin-screw extrusion (TSE) is used as a larger-scale, continuous-flow mechanochemical method.39,40 However, even if mechanochemistry complies with several green chemistry principles, the quantitative assessment of the greenness of mechanochemical reactions and processes in comparison with solution based-approaches or other similar mechanochemical syntheses is possible only by a systematic calculation of green metrics.41–43Even though it is well accepted that mechanochemistry fits the ‘green toolbox’, green metrics calculations are not yet systematically undertaken for both batch and in continuous processes.
This review wishes to highlight this aspect, moving away from subjective assessments of the environmental footprint of mechanochemical reactions. Therefore, the reviewed articles provide a quantitative assessment and compare the environmental footprint of mechanochemical reactions in a quantitative way, which allows direct comparison with other synthetic methods. Even though several mass-based environmental process waste metrics exist, this review mentions only those applied in mechanochemical synthesis.
We would like to highlight at this stage that despite many mechanochemical reports claiming to be environmentally better than their solution-based counterparts, they could not be included since they dot provide the experimental details required for the calculation of green metrics. Moreover, since we only focus on the reports comprising green metric calculations many relevant examples from a wide range of areas (organic synthesis,44–48 catalysis,49,50 APIs,51 organometallic complexes,38,52 main group compounds,53 metal organic frameworks,54 co-crystals,55etc.) fall outside of scope of this review.
Green metrics used in mechanochemical reactions
Synthesis of amide bond: access to amides, peptides, and carbamates
Amide is one of the most common functional groups (FG) encountered in nature as it plays a critical role in the structure and properties of the “molecules of life,” such as peptides and proteins. This moiety is the most frequently encountered FG in bioactive molecules developed for pharmaceutical and agrochemical applications. A survey published in 2006 highlighted that throughout the synthesis of 128 drug candidates, the occurrence of N-acylation reactions to produce amide bonds was found to be 66% (i.e., 84/124).56 In 1999, an analysis of the comprehensive medicinal chemistry (CMC) database based on drug-like compounds underlined that the carboxamide functional group represented up to 27% of the bioactive molecules referenced.57 Amidation (N-acylation) represents a critical reaction in medicinal chemistry.58 It was consequently selected in the top green chemistry research priorities by the American Chemical Society Green Chemistry Pharmaceutical Roundtable (ACS GCIPR) in 2007 and 2018.59–61Classically, amide bond formation generally requires the activation of the carboxylic acid group by coupling reagents.62 Nevertheless, numerous non-classical methods were also developed.63 Most of these methods are solution-based, have low atom economy and present safety issues due to solvents and toxic reagents. This section reports the environmental benefits of amide-bond formation provided by mechanochemical processes.
In peptide chemistry, temporary protection of the α-amino function is generally required to enhance selectivity. tert-Butyloxycarbonyl (Boc), benzyl-oxycarbonyl (Cbz), 9-fluorenylmethyloxycarbonyl (Fmoc) are typical protecting groups; however, their use often requires the use of large quantities of toxic solvents (DMF, DCM, etc.). To solve this issue, Colacino et al. developed a solvent-free one-pot, two-step procedure for amino acid N-protection using a PBM (Scheme 1).64 In this approach, equimolar amounts of amino acids 1 are first transformed into the corresponding metal carboxylate 2 (M = Na, K) by milling with potassium carbonate (1 equiv.) in the presence of sodium chloride as a milling additive to act as a grinding agent and avoid paste-like mixture. The preparation of protected amino acids with Boc and Cbz groups was conducted in the PBM employing stainless steel or tungsten carbide (only for the Fmoc group) jars (12 mL), containing 24 balls (5 mm ∅) of the same material as the jar, and the mixture was ground at 500 rpm for 2 h. Thereafter, grafting of the protecting groups was accomplished by applying continuous (500 rpm) or cycled milling (300 rpm, three cycles of 1 h, 10 min intervals between each cycle), inversed rotation (reversal of regular rotation direction) of the deprotonated amino acids (1 equiv.) with the desired protecting reagent (1 equiv.) and NaCl for 2 to 3 h. NaCl was used as grinding auxiliary to keep the mixture as a floating powder, avoiding the formation of a sticky paste after the first milling step, making otherwise useless any milling effect.
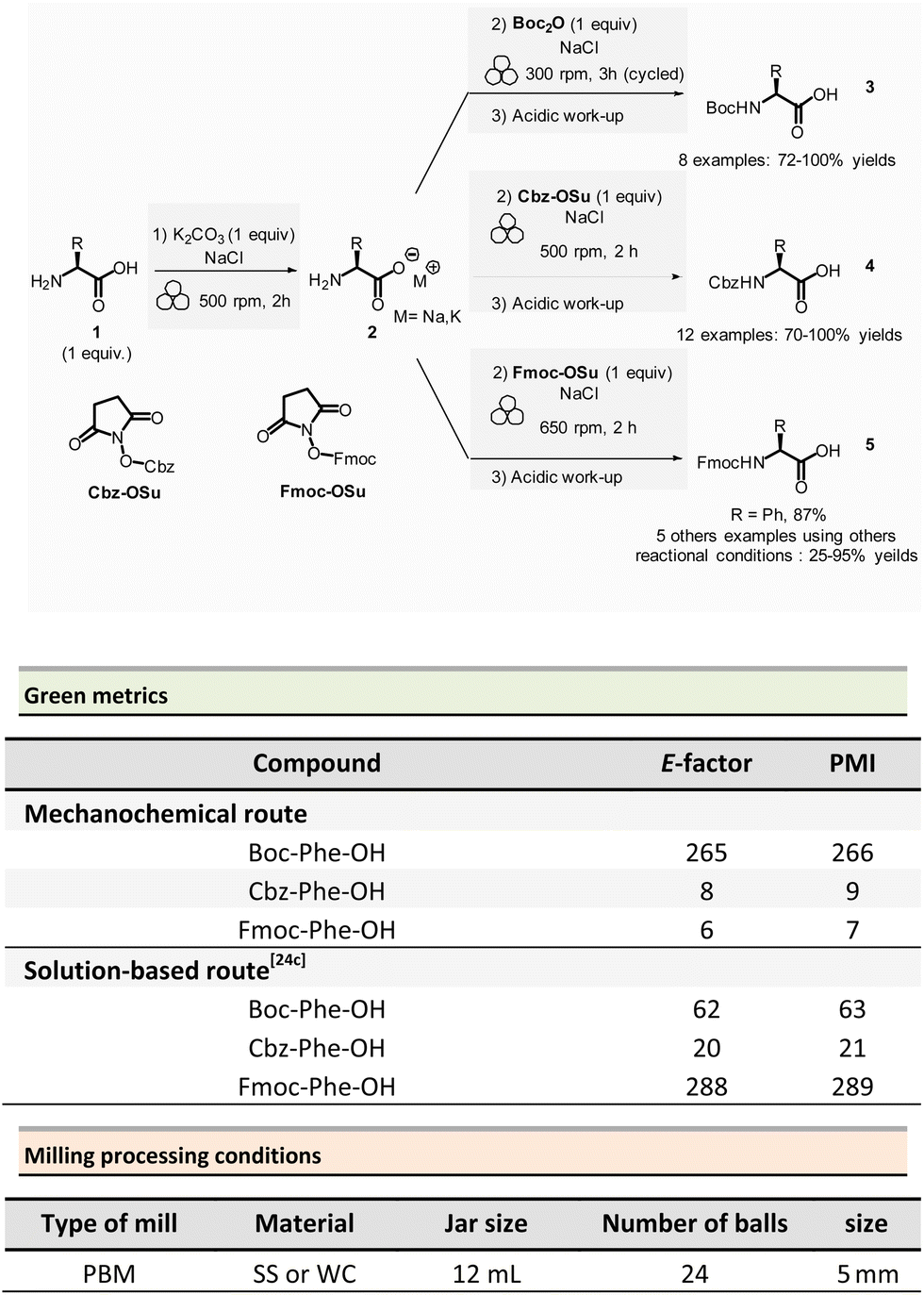 | ||
| Scheme 1 Amino acids protection with Boc, Cbz, and Fmoc groups by mechanochemistry. | ||
Following a simplified work-up, pure N-protected amino acids 3-4 were isolated by precipitation in water. For Fmoc protecting group, several reaction conditions were employed for preparation of short series of amino acids 5. When (L)-Fmoc-Phe-OH amino acid 1 was used as reagent, the same stainless-steel jars were used compared to Boc and CBz groups, and the reactions afforded after 2 h at 650 rpm (second step) and work-up the protected amino acid 5 in 87% yield. Adapting material and reaction conditions, five other amino acids were also obtained in 25–95% yields. These Fmoc amino acids were produced using a stainless steel (12 mL or 20 mL) or a tungsten carbide (50 mL) jar, containing 24 to 80 balls, and grinding at 500 rpm for first step (2 h) and 500, 650 or 750 rpm (one case in cycled mode) for second step (2–3 h).
In this case, the reaction outcome depended on the milling rotation speed. In particular, the yield was doubled at higher milling speed (650 rpm) and the Fmoc amino acids were obtained with full selectivity (i.e., no formation dibenzofulvene by-product was observed, even in traces, despite the presence of a base into the reaction medium). Moreover, the reaction outcome (yields and selectivity) was not influenced by the nature of the milling media used, the same results being obtained materials having different density and hardness (e.g., stainless-steel vs. tungsten carbide) were used.
This example paved also the way towards the unprecedented introduction of green chemistry metrics applied to a mechanochemical syntheses, in order to compare their greenness versus the corresponding solution-based methods.
The environmental factor (E-factor) determined for the N-protected amino acids Cbz-Phe-OH, and Fmoc-Phe-OH were lower than the corresponding solution-based reactions (Scheme 1). In contrast, the E-factor score for Boc-Phe-OH, was better in solution for solvent-based Boc-protection due to the liquid–liquid extraction work-up required for mechanochemical synthesis (265 for mechanochemistry vs. 62 in solution).
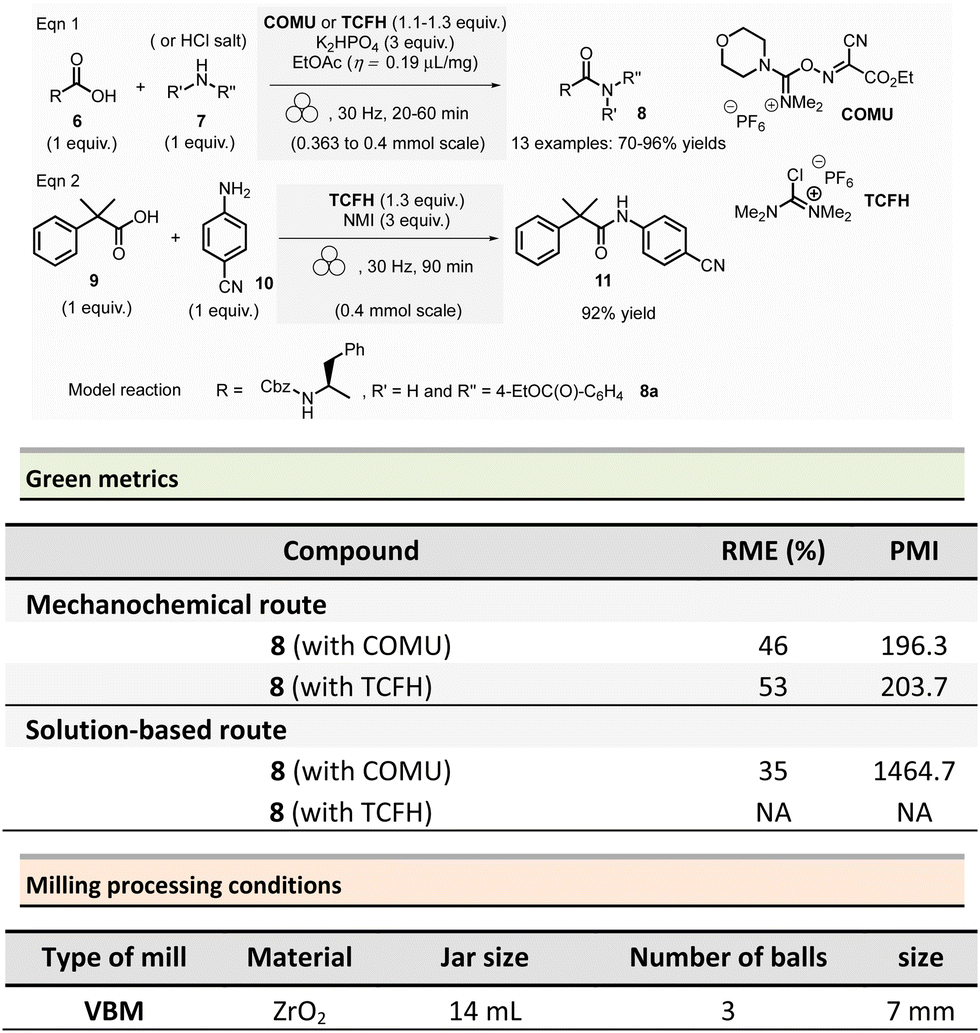
Aav et al. described amidations using uronium-type coupling reagents (COMU and TCFH) via mechanochemical activation (Scheme 2).65 Typically, the coupling reactions of carboxylic acids derivatives 6 with protected amino acids, N-Boc-piperazine or aniline derivatives 7 were performed in a ZrO2-coated jar (14 mL) containing three ZrO2 balls (7 mm ∅). The reaction needed a slight excess of COMU (1.1 equiv.) or TCFH reagents (1.1–1.3 equiv.), a large significant excess quantity of base (K2HPO4, 3 equiv.), under liquid-assisted grinding (LAG)34 conditions, using a small amount of not harmful ethyl acetate (η = 0.19 μL mg−1). This procedure gave a ranging of 70–96% yields in amides 8.
 | ||
| Scheme 2 Mechanical synthesis of amides using uronium-type reagents. | ||
Reaction times ranged from 20–60 min at 30 Hz employing a VBM. Notably, the coupling of hindered carboxylic acid 9 with poor nucleophilic amine 10 in the presence of TCFH/1-methylimidazole (NMI) was also efficient; however, it required a longer milling time (90 min at 30 Hz). The TCFH/NMI system under LAG conditions (EtOAc) was successfully applied for the polyamidation of the six carboxylic acid functions of biotin[6]uril with 80% of yield and 99% of purity (detected by HPLC).
Better yields were obtained with both COMU/K2HPO4 and TCFH/K2HPO4 under mechanochemical activation for the model reaction (Scheme 2, eqn (1)) (86–92% vs. 70–96%).66 The isolation of the reaction products was readily performed by filtration and water wash for the mechanochemical process, whereas column chromatography was required in conventional solution approaches. Green metrics such as reaction mass efficiency (RME) and product mass intensity (PMI) showed unambiguously that mechanochemical procedures outperformed solution-based method. By mechanochemistry, RME was 46% and 53%, respectively, for COMU and TCFH activating agents, compared to 35% for a solution-based procedure using COMU. However, RME was not calculated for the corresponding solution-based process involving TCFH. PMI (COMU/K2HPO4) was also substantially lowered by over 7-fold (196.3 vs. 1464.7 in solution), underlining that mechanochemical reaction produced less waste.
The only notable drawback concerning this reaction is linked to the reproductive toxicity of tetramethylurea produced as a byproduct of TCFH-activated reaction.
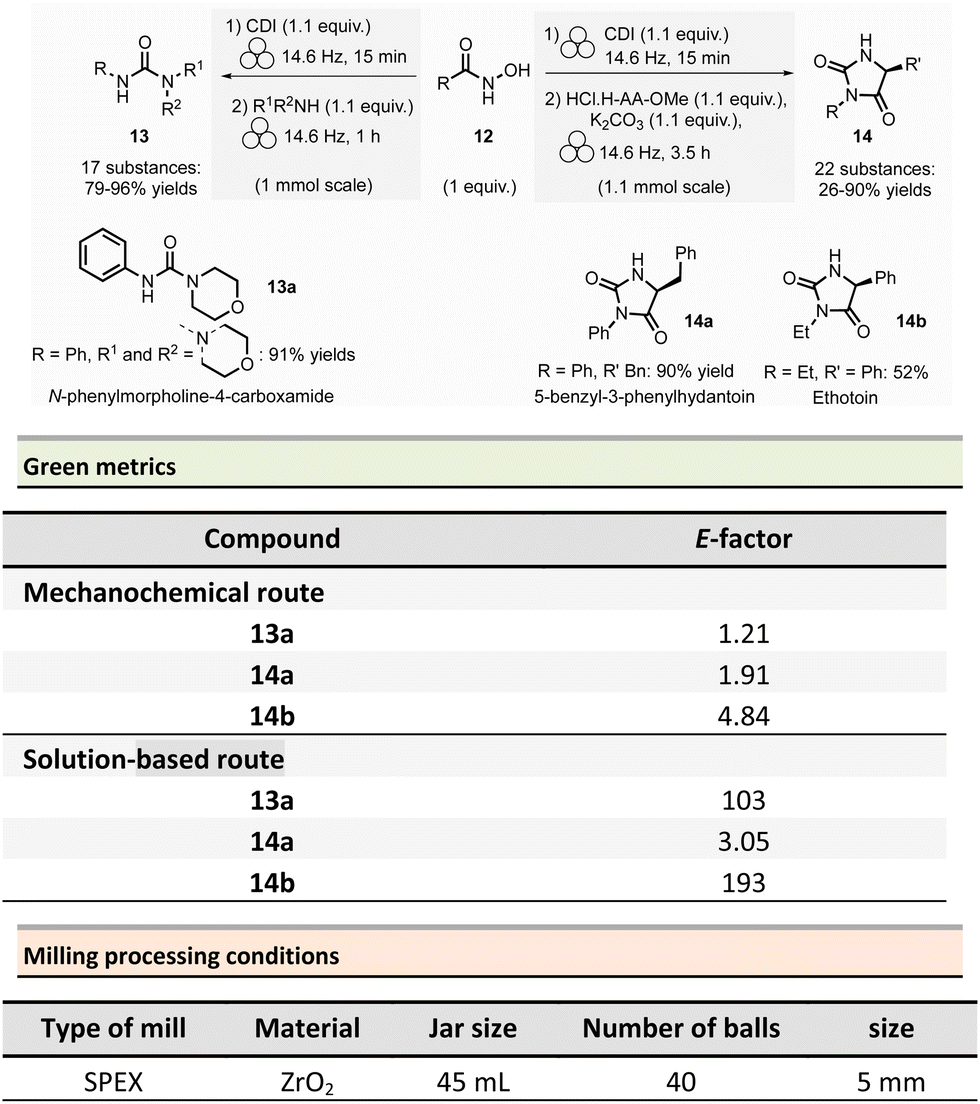
Non-symmetrical ureas and hydantoins (including pharmaceutical ingredient ethotoin)67 were mechanochemically synthesized in 2019 by Colacino, Porcheddu, et al. (Scheme 3).68,69 The one-pot/two-step sequence, employs hydroxamic acid 12 as starting material. First, activation of hydroxamic acid by 1,1-carbonyldiimidazole (1.1 mmol), followed by a subsequent Lossen transposition afforded in situ the reactive isocyanate. Reactions were performed using a SPEX shaker mill. Milling hydroxamic acid (1.0 mmol) and CDI (1.1 mmol) into a ZrO2 jar (45 mL) containing 40 ZrO2 balls (5 mm ∅) at 14.6 Hz for 15 min. In a second step, amine (1.1 mmol) was added, and the reaction mixture was ground for one more hour. In contrast to solution-based approaches, the reaction did not require the presence of a base to occur. Finally, the trituration of the resulting solid with a 15% w/w citric acid aqueous solution, followed by filtration and drying under vacuum with P2O5, produced the pure ureas 13. Seventeen ureas 13 were obtained using this methodology in yields ranging from 79% to 96%. When amino esters were used instead of amines in the second step, hydantoins species were obtained in 26% to 90% yields after 3.5 h of milling.
 | ||
| Scheme 3 Urea and hydantoin mechanochemical synthesis through Lossen rearrangement. | ||
This mechanochemical procedure avoided using toxic isocyanates, alkyl halides or dialkylsulfates as reagents and DMF or DMAc as solvents providing a safer and greener approach to these species. It is worth noticing that this method allowed to selectively prepare: (i) N-methylated hydantoins not accessible by conventional solvent-based procedures due to safety reasons (e.g., use of flammable and harmful methylisocyanate), (ii) N-phenyl substituted hydantoins, which are not accessible by other mechanochemical procedures70 and (iii) long chain N-alkylated hydantoins directly from hydroxamic acids obtained directly from commercially available reactants (e.g., carboxylic acids),71 which bypasses the steps required to obtain non commercially available isocyanates. Consequently, the E-factor was better than traditional solution-based procedures.72–74 For instance, for N-phenylmorpholine-4-carboxamide 13a, the E-factor (without work-up) was 1.21 (91% yield) vs. 103 (99% yield) in solution.
The E-factor remains favourable towards mechanochemical procedures for 5-benzyl-3-phenylhydantoin 14a (1.91 with 90% yield vs. 3.05 with 79% yield, in solution), while for ethotoin 14b, the E-factor was 4.84 (52% yield) vs. 193 (65% yield) for mechanochemical vs. solutions-based approaches, which requires a column chromatography purification step.
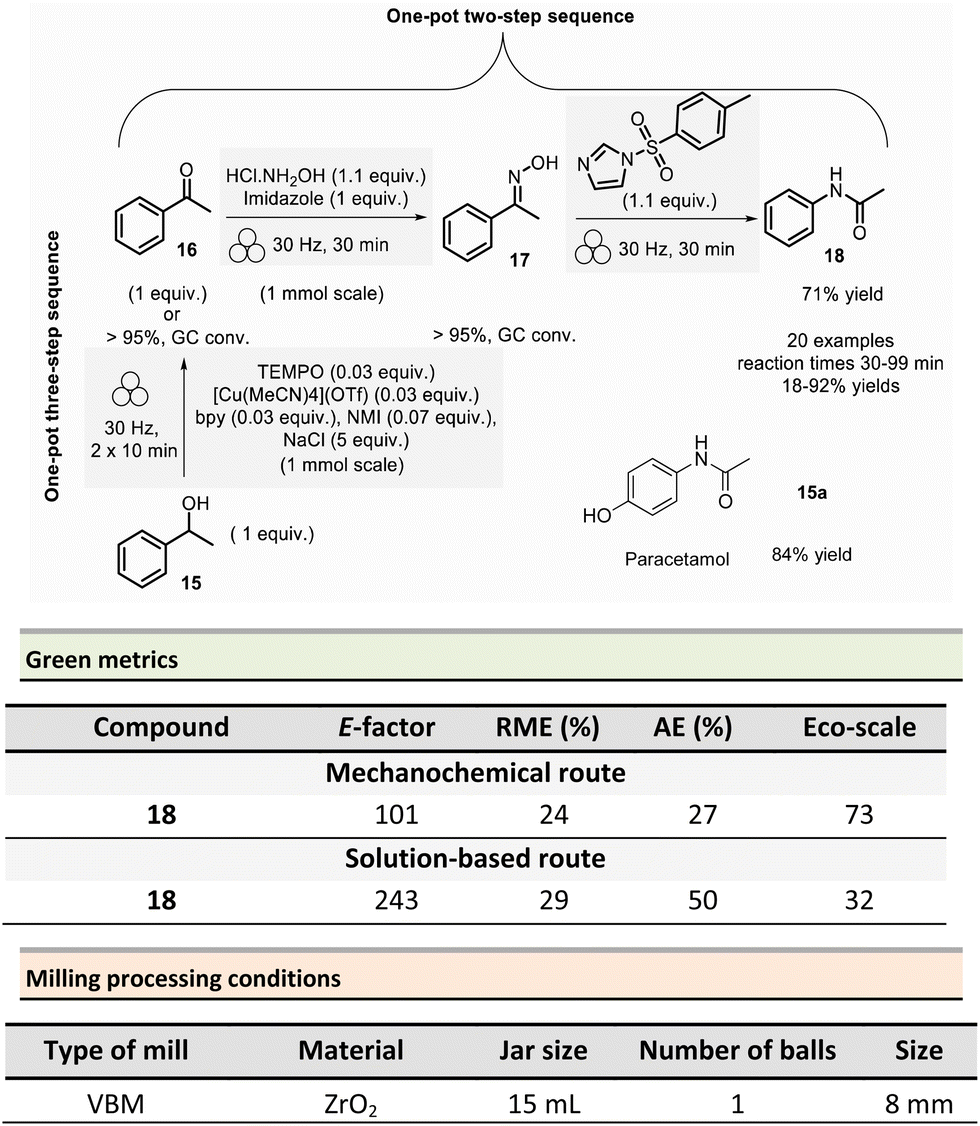
In 2021, Mocci et al. developed a mechanochemical procedure for the Beckmann rearrangement reaction (BKR) to access amides from in situ synthesized oximes.75 The BKR generally requires strong acids, harsh conditions, and hazardous reagents. Even though milder conditions could be employed, toxic coupling reagents (e.g., cyanuric chloride, BOPCl) could not be avoided. As a representative example, N-phenylacetamide 18 was obtained in a one-pot/two-step synthesis using a VBM. The first step was performed on 1.0 mmol scale by milling acetophenone 16 (1 equiv.), hydroxylamine hydrochloride (1.1 equiv.), and imidazole (1 equiv.) at 30 Hz for 30 min in a ZrO2 jar (15 mL) with one ZrO2 ball (8 mm ∅). To the in situ formed oxime intermediate 17, p-toluenesulfonylimidazole (1.1 mmol) was added, and the mixture was then milled at 30 Hz for further 30 min to afford N-phenylacetamide 18 (Scheme 4).
 | ||
| Scheme 4 N-Phenylacetamide preparation by mechanochemical Beckmann rearrangement. | ||
To eliminate the imidazolium tosylate by-product, the reaction crude was triturated with water, 10% w/w citric acid aqueous solution, and 10% w/w potassium carbonate aqueous solution, filtered off, and dried in vacuo over Na2SO4. Furthermore, varying the ketones and the reaction times (30–99 min), allowed a large scope of N-acetyl, N-aryl, or N-alkyl amides to be produced in 18–92% yields. It is worth highlighting that this methodology enables an affordable route to the active pharmaceutical ingredient (API) paracetamol 18a in 84% yield from 4-(1-hydroxyethyl)phenol, as illustrated so far in Scheme 4 for the obtention of phenylacetamide 18. The first step consisted in the oxidation of 1-phenylethanol 15 (1.0 mmol scale) into acetophenone using a combination of 2,2,6,6-tetramethylpiperidine 1-oxyl (TEMPO)/air as oxidant, [Cu(MeCN)4]OTf as the catalyst, N-methylimidazole and 2,2′-bipyridyl (bpy) as ligands, and sodium chloride as a milling agent used to avoid paste-like mixture. Interestingly, grinding the mixture twice for 10 min at 30 Hz, allowed the formation of acetophenone in yields over 95% determined by gas chromatography (GC). Subsequent steps remained unchanged, and phenylacetamide 18 was isolated in 71% overall yield (Scheme 4).
Green metrics were calculated for the mechanochemical preparation of phenylacetamide 18 according to the one-pot, two-step sequence and compared to a similar solution-based process.76 In solution, a mixture of acetophenone (1 equiv.), NH2OH·HCl (1.6 equiv.), and sodium acetate (2 equiv.) was refluxed for 1 h in EtOH/H2O (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).77 Then, the newly obtained acetophenone oxime (1 equiv.), was reacted with p-toluenesulfonyl chloride (0.02 equiv.), and ZnCl2 (0.02 equiv.) in dry MeCN during 1 h at reflux.76 The yields were comparable for the two procedures (91% vs. 86% in solution). However, AE and RME were better in solution, with 50% for AE and 29% for RME, compared to the mechanochemical method (27% for AE and 24% for RME). Green metrics favour solution-based procedures due to the only waste produced being acetic acid, sodium chloride, and water. Whereas the mechanochemical method produced imidazolium chloride and 4-methylbenzenesulfonate imidazolium waste. Finally, the mechanochemical procedure displayed an E-factor of 101 and an Eco-scale score of 73, while solution-based techniques scored 243 and 32, respectively.
:1).77 Then, the newly obtained acetophenone oxime (1 equiv.), was reacted with p-toluenesulfonyl chloride (0.02 equiv.), and ZnCl2 (0.02 equiv.) in dry MeCN during 1 h at reflux.76 The yields were comparable for the two procedures (91% vs. 86% in solution). However, AE and RME were better in solution, with 50% for AE and 29% for RME, compared to the mechanochemical method (27% for AE and 24% for RME). Green metrics favour solution-based procedures due to the only waste produced being acetic acid, sodium chloride, and water. Whereas the mechanochemical method produced imidazolium chloride and 4-methylbenzenesulfonate imidazolium waste. Finally, the mechanochemical procedure displayed an E-factor of 101 and an Eco-scale score of 73, while solution-based techniques scored 243 and 32, respectively.
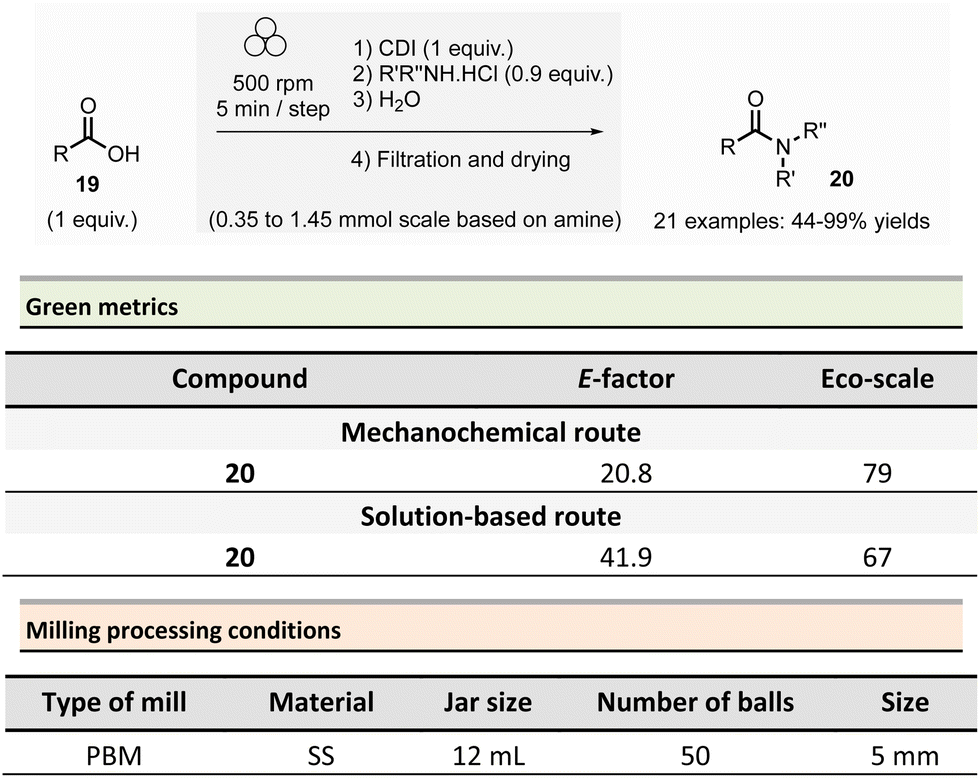
In 2012, Métro et al. developed a mechanochemical acylation of nucleophiles (mostly amines) using N,N-carbonyldiimidazole (CDI) as activating agent.78 Twenty-one amides 20 were obtained from carboxylic acid derivatives 19 in good to almost quantitative yields (44–99%) (Scheme 5). Typically, during the two-step optimized procedure, the carboxylic acid activation was accomplished with CDI under 5 min at 500 rpm in a PBM (using stainless-steel grinding media with 50 balls of 5 mm diameter). Then, amine hydrochlorides were added, and the mixture was milled for a further 5 min at 500 rpm. The by-products were imidazole hydrochloride, carbon dioxide, and a small amount of unreacted carboxylic acid starting material. Typically, the amides were readily purified by aqueous work-up under grinding conditions (5 min, 500 rpm), filtration, washing with deionized water, and drying under vacuum. Using the developed approach, teriflunomide, an active metabolite of Leflunomide approved by the FDA for treating multiple sclerosis, was also synthesized with 81% yield. In addition, this methodology was also extended to the formation of C–O, C–S, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds.
C bonds.
 | ||
| Scheme 5 CDI-mediated amidation by mechanochemistry. | ||
Careful characterization by ICP-MS or gravimetric analysis of the final products showed ppm traces of metal impurities (Fe, Zr, Cr, Y, Si) which originated from the degradation of the milling balls and jar during the grinding process.79 Metal contamination was drastically diminished by optimizing reaction times and milling media used by assessing the use of zirconium oxide, agate, or PTFE milling media. The studies showed that stainless-steel was the most suitable grinding media for this reaction since it took less time, gave the best yield, and metal particle contaminants could be removed by simple filtration without the need for harmful EtOAc.
Evaluation of the environmental impact of this solvent-free preparation of N-benzylbenzamide with CDI was also compared to other classical solution procedures using (N,N′-diisopropylbenzyl-amine-2-boronic acid (IBA), N,N′-dicyclohexylcarbodiimide (DCC),80 thionyl chloride (SOCl2),81N,N′-carbodiimidazole (CDI)82 in THF and sulfated tungstate.83 Among all species tested, the CDI reagent furnished the best yield with 92% for both reactions in solution and without solvent. The E-factor score calculated for mechanical milling was 20.8, which is half of that calculated for CDI in THF (41.9). None of the other coupling reagents had better metrics. Finally, with an Eco-scale score of 79, the mechanical method was excellent compared to all the different reactions conditions in solution (Eco-scale between 44 and 67).
Browne et al. developed an original and efficient ball milling method for direct amide bond formation using methyl or ethyl esters as starting materials (from reagents 21 and 24),84 circumventing the need of any activating reagent, usually needed when using carboxylic acids as starting materials. The solvent-free reaction was carried out using an amine (from 22 and 25), 1.2 equivalent of ester, and a substoichiometric amount of potassium tert-butoxide (0.85 mol%) as a base. Typically, 1.0 mmol scale reactions were milled at 30 Hz for 1 or 2 h (depending on substrates) using a stainless-steel milling jar (14 mL) with one stainless-steel ball (4 g) (Scheme 6). The only by-product of the reaction was the corresponding primary alcohol (i.e., methanol or ethanol). Using this method, several dozens of amides 23 and 26 were synthesized in moderate to high yield (11% to 99%). The reaction substrate scope comprises aromatic, alkyl, alkenyl, and heteroaromatic esters as electrophiles, and primary, secondary or cyclic amines as nucleophiles. Ammonium salts can also be used as starting materials. However, an excess of base (i.e., 1.85 equivalent) was required. Moreover, this methodology was successfully applied to synthesizing five relevant pharmaceuticals and agropharmaceuticals species (i.e., CL-82198 MMP13 inhibitor, Lidocaine, Coramine, Fenfuram and Moclobemide) in 47 to 92% yields. Notably, this procedure was also upscaled ten-fold for the synthesis of Moclobemide, by increasing the size of the milling reactor and the number of balls, affording similar yields. Using the model reaction reported in Scheme 6, eqn (1) (ethyl benzoate and morpholine), mechanical methods afforded complete amidation, whereas only 8% of the target amide was obtained in solution. This reaction's AE was consistently higher (55%) than those performed using traditional solution methods. For instance, in the best solution conditions (phosgene/Et3N), an AE of 47% was determined. Even if the difference in AE is not so high, the mechanochemical method has the advantage of avoiding toxic and harmful chemicals (e.g. phosgene). Moreover, concerning PMI, the mechanochemical amide bond formation was greener than the solution counterpart (PMI = 1.94 vs. a similar solution reaction reported by Yoon and coworkers, PMI = 59.28).85
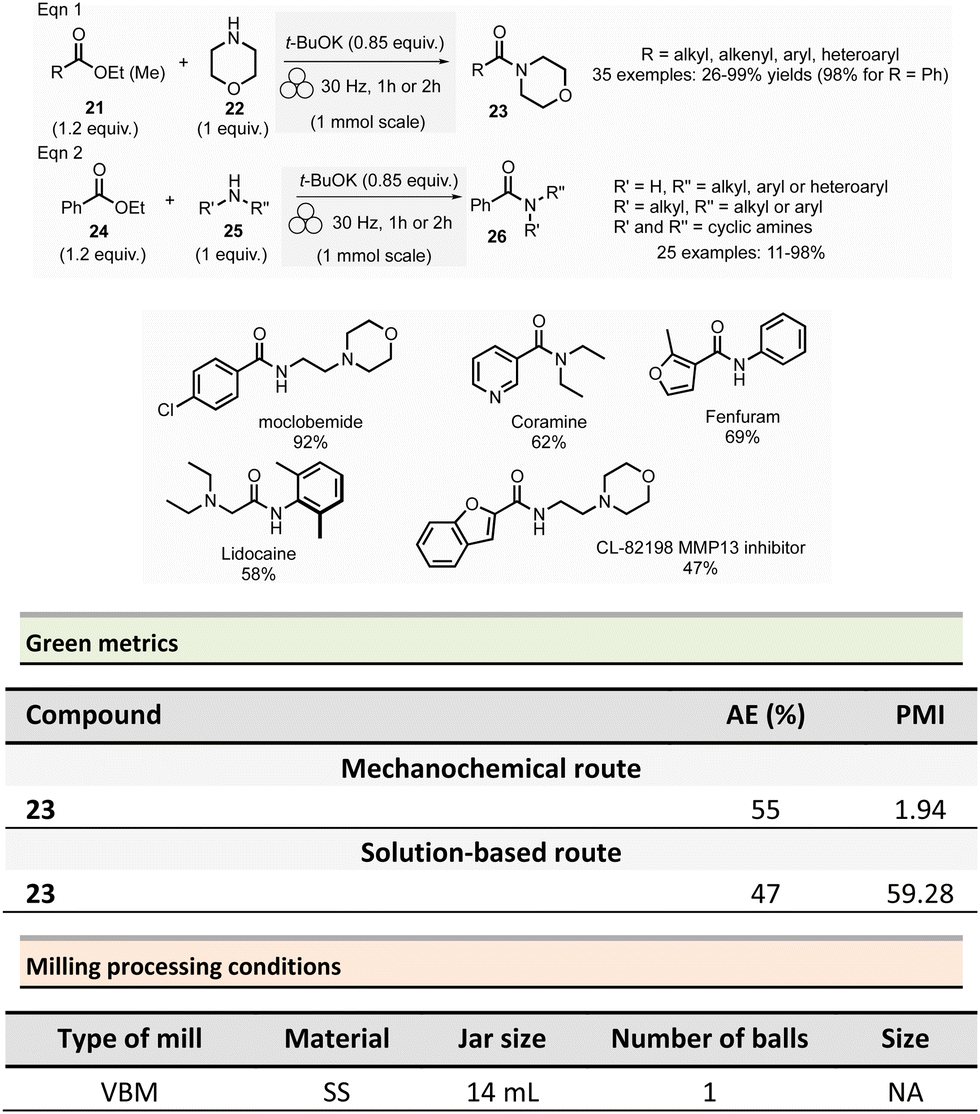 | ||
| Scheme 6 Direct amide bond formation from ester in mechanochemistry. | ||
In 2022, Wadouachi et al. reported the one-pot/two-step mechanochemical synthesis of four potential surfactants (4S)-N-alkyl-4,5-bis-sulfooxypentanamides 29 from bio-based (S)-γ-hydroxymethyl-γ-butyrolactone 27 (2H-HBO) in a one-pot two-step process (Scheme 7).86 This short sequence consisted of an aminolysis of 2H-HBO 27 followed by a disulfation reaction using a PBM. The optimized conditions consisted of grinding 2H-HBO (1 equiv.) with alkylamine (1 equiv.) in a ZrO2 jar (20 mL), charged with eighty ZrO2 balls (5 mm ∅) under an inert argon atmosphere. The rotation was at 400 rpm in reverse mode (regular reversal of rotation direction) for 47 min (8 cycles of 5 min with 1 min rest between each cycle). The disulfation was accomplished by adding SO3·pyridine complex (4 equiv.) and a drop of EtOAc (η = 0.6 μL mg−1) into the jar under argon. The complete conversion occurred after 95 min (16 cycles of 5 min with 1 min rest) at 400 rpm or 500 rpm in reverse mode. The mixture was then milled again for 11 min (3 cycles of 3 min with 1 min pause between cycles) in the presence of NaHCO3 (8 equiv.). The final disulfate 29 was recovered after an aqueous work-up and purification step using reverse phase chromatography in good yields (77%, 62%, and 79% for dodecylamine, tetradecylamine, and hexadecylamine and octadecylamine, respectively).
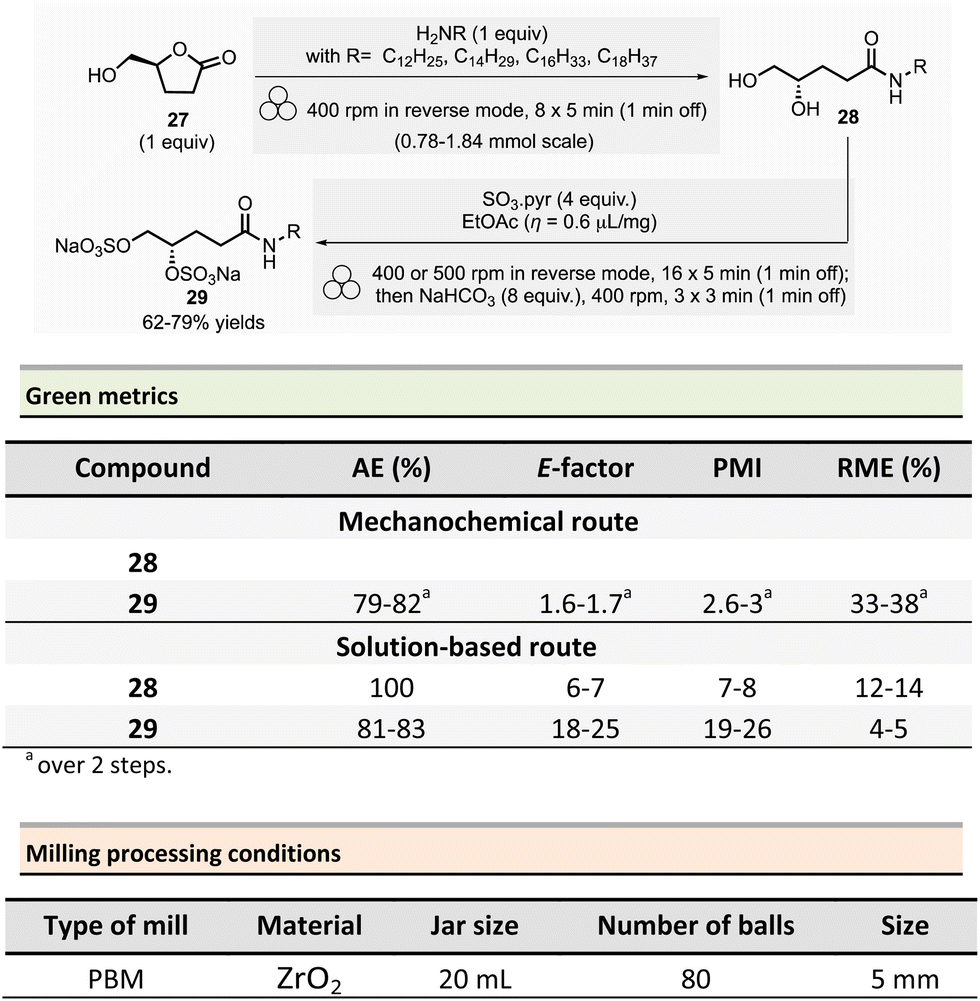 | ||
| Scheme 7 Bis-sulfoxypentanamide synthesis with one-pot/two-step procedure by ball-milling. | ||
It is worth to note that sulfation of oligoglucuronanes usually requires heating in DMF for prolonged time, with improved results in term of reaction kinetic when microwave-assisted heating is used, allowing to reduce the amounts of by-products due to the decomposition of the carbohydrates. Contrarily, the reaction conducted by mechanochemical activation does not require any external heating to occur, and the by-products are absent or only formed in traces.
To assess the environmental footprint of the mechanochemical strategy, authors also prepared the same four surfactants according to the following a two-step solution procedure: (i) (step 1) alkylamine (1.05 equiv.), 2H-HBO 27 (1 equiv.) in 2-propanol (c ≈ 0.1 mM) were stirred at 50 °C for 48 h, followed by purification by recrystallization (71–83% yield of amide 28). (ii) (step 2) 28 was then reacted with SO3·Pyr (4 equiv.) in dry pyridine (c ≈ 0.1 mM) at room temperature for about 48 h, then MeOH (10 equiv.) was added, and the mixture was stirred for additional 30 min at room temperature, in the presence of NaHCO3 (8 equiv.). The crude was dissolved in water and purified on reverse column chromatography (59–73% yields).
The green metrics, without work-up, calculated for the surfactants highlighted the superior green performance of the mechanochemical procedure. The AE was comparable for both methods, with 81–83% for step 2 of the solution-based process vs. 79–82% for the mechanochemical one. On the other hand, the E-factor is less favourable for conventional solution procedures, being 6–7 for step 1 and 18–25 for step 2 vs. 1.6–1.7 by mechanochemistry. Along the same trend, PMI was 7–8 for step 1 and 19–26 for step 2 vs. 2.6–2.7 for the mechanochemical sequence. Both metrics confirmed that, in addition to the experimental simplicity of the set-up, mechanochemical synthesis provides better green metrics. Finally, RME continued to show the same trend with 12–14% for step 1 and 4–5% for step 2 vs. 33–38% when using the reported ball milling strategy.
In 2017, Colacino et al. published a new ball-milling method for the synthesis of dipeptides and tripeptides using N-protected α-aminoacyl benzotriazoles 30 as building-blocks (Fmoc-AA1-Bt, Boc-AA1-Bt, or Cbz-Phe-Bt) and amino esters hydrochloride 31 (HCl·H2N-AA2-OtBu or HCl·H-Phe-NH2) in the presence of Hünig base (N,N-diisopropylethylamine, DIPEA) (Scheme 8).87N-Acylbenzotriazoles 30 are air- and water-stable reagents while remaining more reactive than the corresponding N-acylimidazoles.
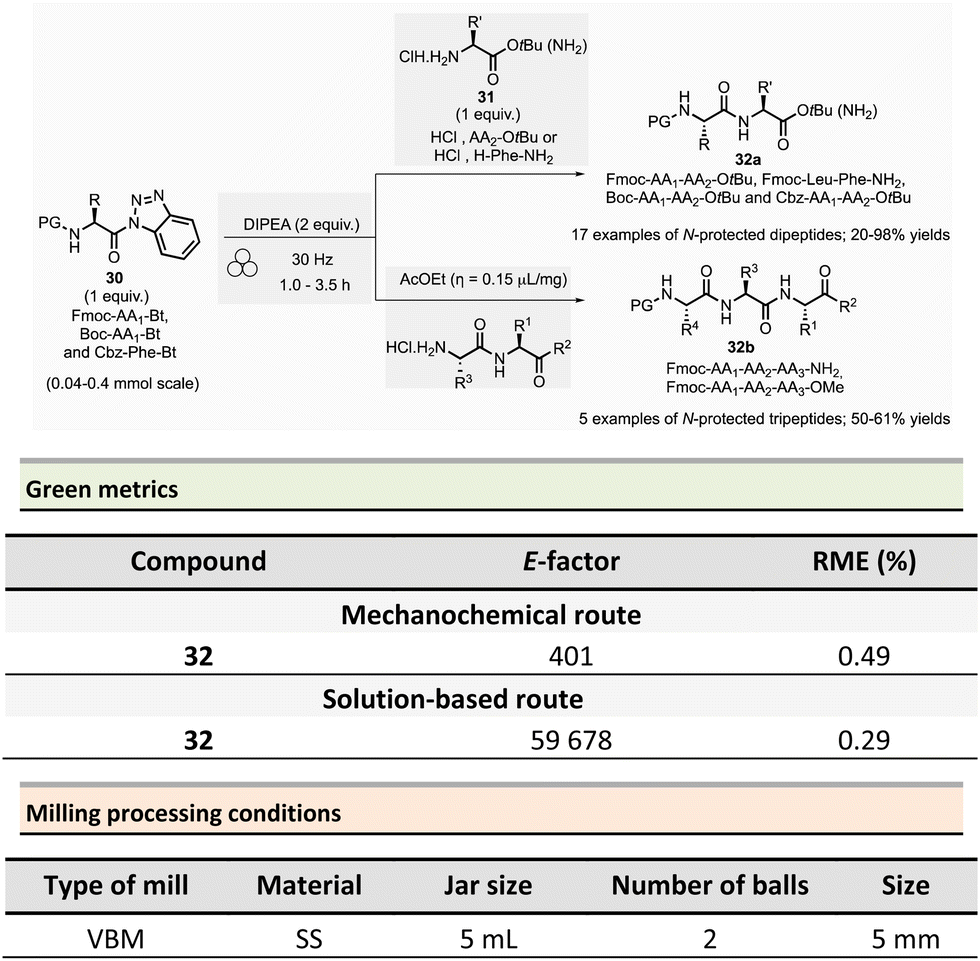 | ||
| Scheme 8 Protected dipeptides formation from N-acylbenzotriazoles by mechanochemistry. | ||
The typical mechanical procedure for synthesizing protected dipeptides was carried out in a stainless-steel jar with 2 stainless-steel balls (5 mm ∅) at 30 Hz using a VBM for 1 to 3.5 h. After precipitation, by adding water to the reaction mixture, and filtration, the protected dipeptides 32a (PG-AA1-AA2-OtBu) were readily isolated in 20 to 98% yields. In addition, LAG (ethyl acetate, η = 0.15 μL mg−1) was beneficial for preparing five protected tripeptides 32b (Fmoc-AA1-AA2-AA3-OtBu and Fmoc-AA1-AA2-AA3-NH2) in yields ranging from 50 to 61%. The LAG procedure was also used to synthesize a more complex biotinylated peptide (Biotin-Ahx-RGDfV-NH2 linear peptide).
Comparative green metrics based on the synthesis of three different peptides demonstrated the environmental advantage of mechanochemistry versus the standard solution-based procedures.88 For N-protected dipeptide Fmoc-Leu-Phe-NH2, both reaction time and yield were improved by mechanochemistry compared to the solution-based process (1h vs. 24 h and 91% vs. 55% yield). The E-factor was once more in favour of mechanochemical activation (401 vs. 59678) due to the recovery of the final product in solution required an additional HPLC purification step, negatively impacting the E-factor value. Likewise, the reaction mass efficiency (RME) was better for mechanochemistry (0.49) than for the reaction in solution (0.29). In addition to eco-friendly parameters, the added value of this mechanochemical activation lays in a lower production cost calculated to produce 1 g of dipeptide (54€ vs. 298€ in solution), with an evident economic advantage in view of a synthesis in larger scales, where costs are usually multiplied.
Mechanochemical methods were also successful in the formation of C–N bonds (e.g., imine synthesis, N-acyl hydrazones, aminals, etc.).
In 2018, Colacino, Porcheddu, et al. synthesized hydrazone-based APIs, namely nitrofurantoin 35a and dantrolene 35b, by ball-milling.89 Nitrofurantoin was successfully obtained using a VBM at a 0.84 mmol scale from the equimolar reaction of 1-aminohydantoin hydrochloride 33 and 5-nitro-2-furfural 34a. The mixture was milled at 30 Hz for 30 min in a stainless-steel jar (5 mL) with 2 balls (5 mm ∅). Complete conversion of starting materials was observed, and a simple work-up consisting of adding water, followed by filtration and drying, afforded pure nitrofurantoin 35a in 85% yield (Scheme 9). Moreover, no base was needed to generate the free reactive amine during the reaction.
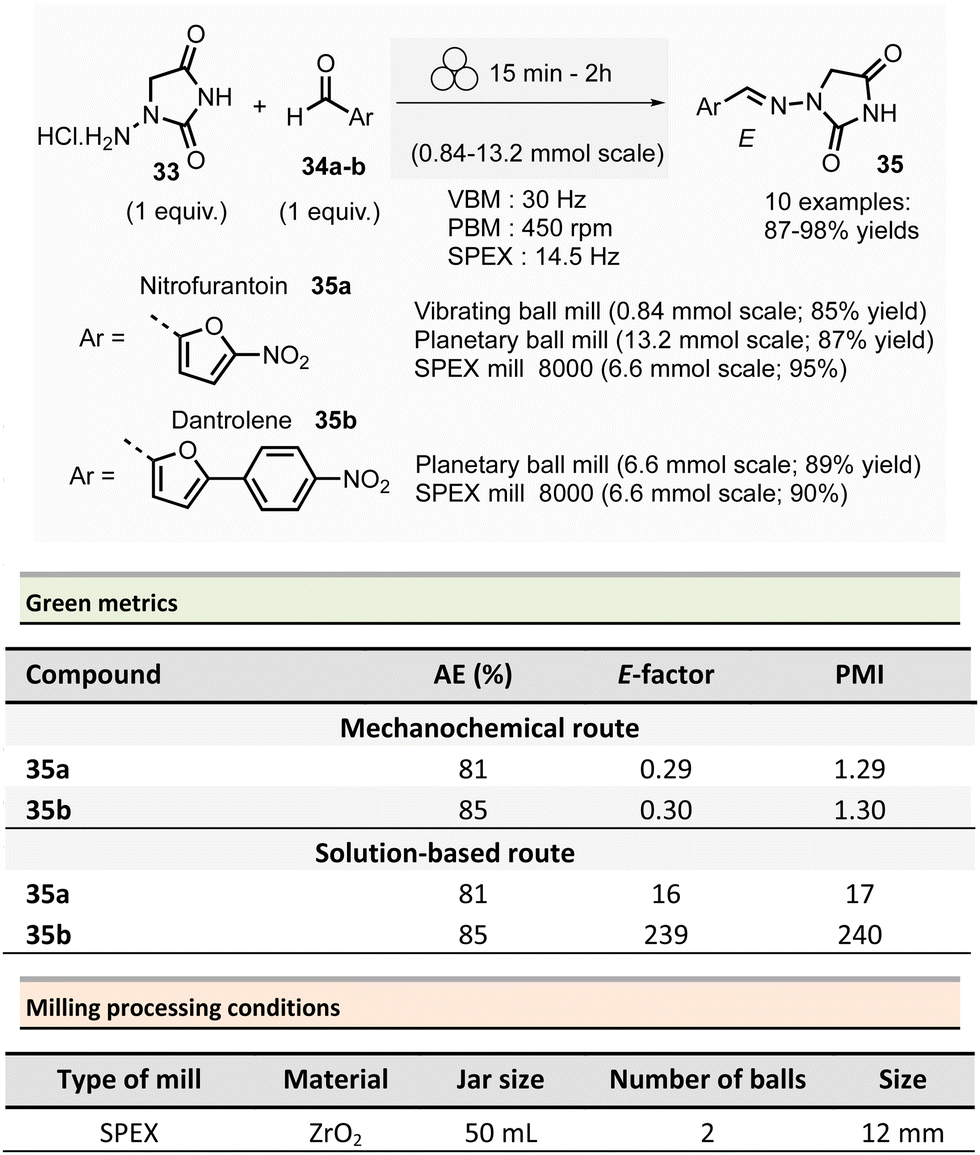 | ||
| Scheme 9 Hydrazones synthesis by ball-milling, applied to the mechanochemical preparation of active pharmaceutical ingredients nitrofurantoin 35a and dantrolene 35b. | ||
To assess the influence of different milling apparatuses, the same reaction was also conducted on a larger scale and it furnished 87% yield in 2 h using a PBM (Schemes 9, 13.2 mmol scale, zirconium oxide jar and balls, 600 rpm). When using a SPEX mill, 95% yield was reached in only 15 min (Scheme 9, on a 6.6 mmol scale, using zirconium oxide jar and balls). It is worth noticing that, independently on the milling apparatus used (VBM. PBM or SPEX), the use of stainless steel or zirconium oxide milling media led to the same results, while the reaction was incomplete when agate jars were used, clearly indicating, in this case, that the hardness and the density of the milling media are some of process parameters to be taken into account when optimising a mechanochemical process. In the latter, complete conversion was obtained using the reactants equimolar amounts, allowing the recovery of pure nitrofurantoin 35a directly by “scratching it out the powder” from the jar. In the case of the synthesis of dantrolene 35b (from reagents 33 and 34b), the reaction needed 2 h with both PBM and SPEX mills to afford 89% and 90% yields, respectively (Scheme 9). Seven hydrazones were prepared using the PBM protocol developed in excellent yields (87–96%) and recovered by precipitation in water. When 2-hydroxybenzaldehyde was used as the substrate, a VBM (1.32 mmol, 30 Hz, 2 h) was preferred (instead of the previous PBM protocol), providing the corresponding hydrazone with the best results (98% yield).
When considering the green metrics for nitrofurantoin 35a and dantrolene 35b, yields (both ≥90%) and AE (81% and 85% respectively, for 35a and 35b) were comparable for both mechanochemical and solution-based process, however, the E-factor was better for the mechanochemical process (for nitrofurantoin 35a: 0.29 vs. 16 and for dantrolene 35b: 0.30 vs. 239 by mechanochemistry vs. in solution respectively). Consequently, also, the PMI (E-factor + 1) resulted sensibly lower for mechanochemistry (1.29 and 1.30 for 35a and 35b respectively) compared to solution-based reactions (17 and 240 35a and 35b respectively).90–92 Moreover, the mechanochemical strategy avoids using toxic solvents (DMF, ACN), excess reagents, corrosive highly concentrated solutions of strong acids, and bases for synthesis and workup. Also, there is no need for pH adjustments or heating–cooling thermal cycles – which strongly reduces the environmental impact and production cost and improves the process's safety. For example, only considering the costs of reagents and solvents, the calculated price for 1 g of dantrolene 35b by mechanochemistry was calculated to 54.7€, compared to the 133.9€ needed for a solution-based reaction.
To evaluate the greenness of this reaction, Colacino et al. pioneered the use of DOZN 2.0 tool for nitrofurantoin synthesis, to quantitatively assess a mechanochemical process against the 12 principles of green chemistry (vide supra).93 The aggregated scores obtained using different mechanochemical devices both in batch (SPEX) and continuous (TSE) processes were compared with the solution counterparts (Fig. 4). For the mechanochemical processes, the best aggregate scores were obtained when using SPEX (0.05) and TSE (0.06) in accordance with their associated shorter reaction times, better yields, no need for work-up, and improved resources use (e.g., no excess of reagents). These methods outperformed VBM and PBM, which display aggregate scores of (1.64 and 0.67, respectively) which required trituration with water to recover the final product. Another essential feature is productivity, TSE apparatus is 2.33 more productive than SPEX (ca. 1.5 g)89 over a period of 15 min, delivering ca. 3.5 g94 of nitrofurantoin 35a.
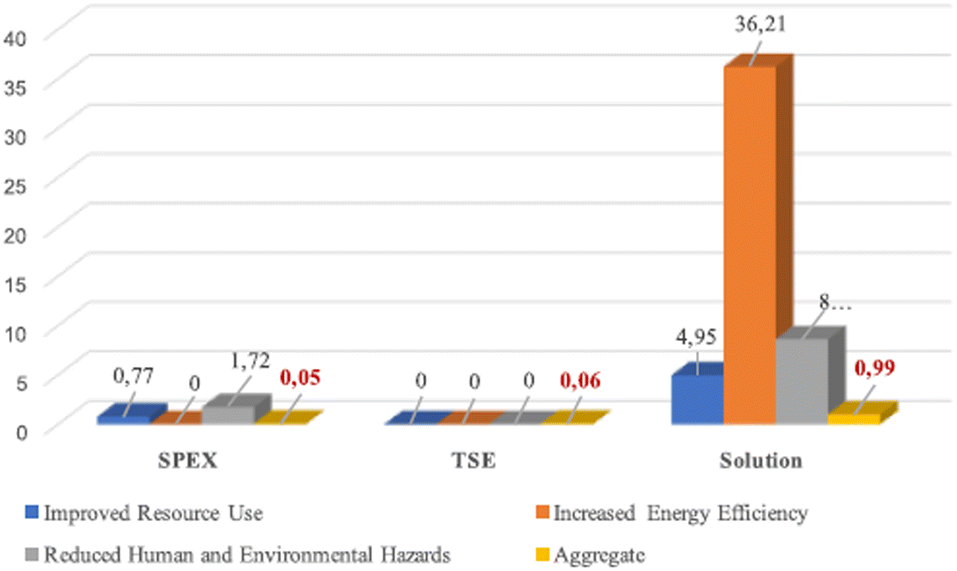 | ||
| Fig. 4 Greenness comparison between SPEX, TSE, and solution synthesis in a batch of nitrofurantoin 35a using DOZN 2.0 tool. Adapted with permission of the American Chemical Society from ref. 93. | ||
When comparing mechanochemical procedures to solution-based counterparts,92 better scores were obtained by mechanochemistry for 8 out of the 12 principles of green chemistry. The major difference in energy efficiency is due to the need to heat the solvent during batch synthesis. In the end, regarding the space-time yields (STY), the productivity is higher using TSE (68000 kg m−3 day−1) compared to solvent-batch synthesis (430 kg m−3 day−1) and also higher compared to estimated continuous flow synthesis for fine chemicals (4000 kg m−3 day−1).95
The Saha group developed the synthesis of 2,3-dihydroquinazolin-4(1H)-ones 38 in the presence of Brønsted acid catalyst either using a mortar-pestle or a tumbler ball mill (Scheme 10)20 In optimized conditions, for a 0.73 mmol scale, equimolar amounts of anthranilamide 36 and benzaldehyde 37 were ground in the presence of para-toluenesulfonic acid (10 mol%) for 3 min in a mortar. After simple trituration in water, pure product 38 was collected with a 95% yield.
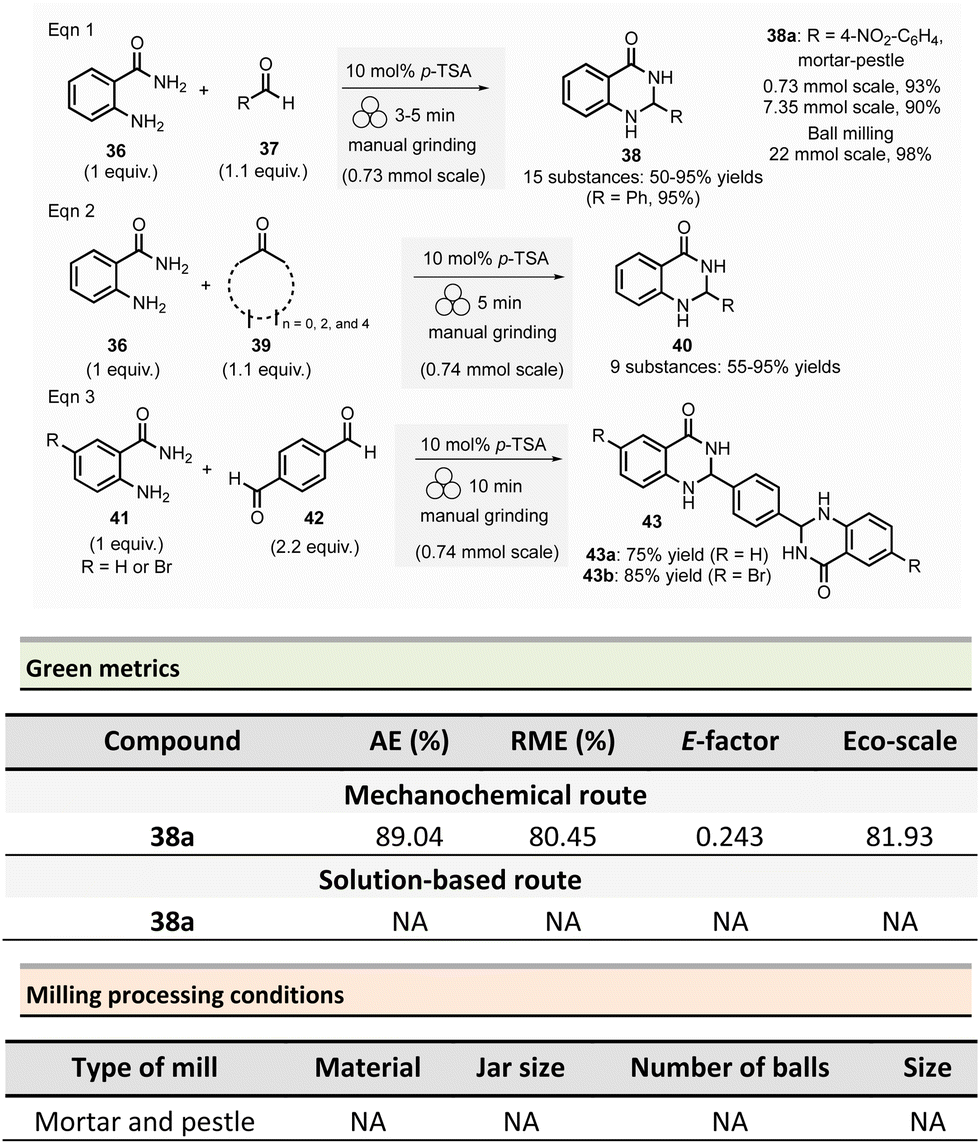 | ||
| Scheme 10 Dihydroquinazolin-4(1H)-ones and bis-quinazolinones formation by ball-milling. | ||
This methodology was extended to aromatic aldehydes, and by varying the reaction time from 3 to 15 min a library of fifteen 2,3-dihydroquinazolin-4(1H)-ones 38 with yields ranging from 50 to 95% was synthesized. Moreover, a gram-scale synthesis using anthranilamide and p-nitrobenzaldehyde afforded 90% yields (which is close to the 93% yield obtained for 0.1 g scale) after 10 min of grinding (Scheme 10).
The extension of this methodology to ketones at a 0.74 mmol scale permitted the formation of nine dihydroquinazolinones 40 in 55% to 95% yields. Similarly, the mechanosynthesis of two bis-dihydroquinazolinones from either 4-bromoanthranilamide or anthranilamide with terephthaldehyde afforded the desired products in good yields (75% and 85%, respectively).
In order to improve efficiency and productivity, a tumbler ball milling apparatus was used for the multigram synthesis of 2-(4-nitrophenyl)-2,3-dihydroquinazolinone 38a. The best reaction conditions were obtained using anthranilide (3.0 g, 0.013 mol) and 4-nitrobenzaldehyde (3.65 g, 0.024 mol) in a stainless-steel reactor vessel (200 mL) loaded with ninety stainless-balls (7.9 mm ∅), and 4 h grinding at 40 rpm. The corresponding dihydroquinazoline 38a was isolated in almost quantitative yields (98%).
For the model reaction (R = 4-NO2-C6H4) performed on a gram scale (7.35 mmol) employing a mortar and pestle, the calculated green metrics demonstrated the environmentally friendly nature of the process (AE = 89.04%, RME = 80.45%, E-factor of 0.243 and, an excellent Eco-scale of 81.93). Nevertheless, no comparison with solution-based reaction was mentioned by the authors.
Borchardt et al. described an efficient and eco-friendly ball-milling synthesis of HAT(CN)6 (46, hexaazatriphenylenehexacarbonitrile)96 (Scheme 11).97,98 The optimized two-step sequence required first to react hexaketocyclohexane octahydrate 44 (1.3 mmol, 1.0 equiv.) with an excess of diaminomaleonitrile 45 (7.84 equiv.) in the presence of water (η = 0.1 μL mg−1, LAG conditions) using a ZrO2 milling vial (10 mL) charged of two ZrO2 mill balls (10 mm ∅).
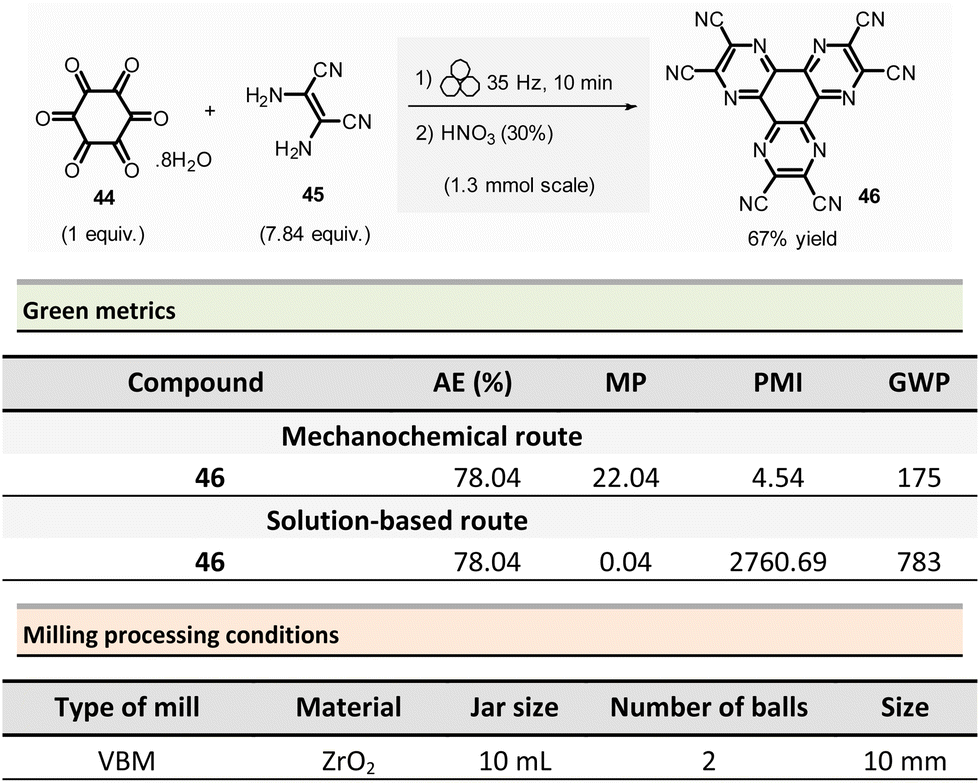 | ||
| Scheme 11 Mechanochemical synthesis of hexaazatriphenylenehexacarbonitrile HAT(CN)6, 46. | ||
The mixture was ground at 35 Hz for 10 min, then the mixture was treated in a glass flask with nitric acid (30%) at 110 °C for 1 h. After a simple work-up, the pure HAT-CN was isolated in 67% yield compared to the 50% yield obtained by traditional solution-based methods.
Concerning green metrics, while AE was equal for both mechanochemistry and solution (78.04%), PMI and MP (mass productivity) were better for the mechanical process (PMI: 4.54 vs. 2760.69 and MP: 22.04 vs. 0.04, respectively). Moreover, the LAG-mechanochemical reaction occurred sensibly faster (10 min vs. 420 min). The authors assessed the environmental impact of the mechanochemical process by calculating its global warming potential (GWP). To the best of our knowledge, the use of this metrics is unprecedented in the field, not commonly measured by chemists, but preferentially used by process engineers. The global warming potential (GWP) measures how much energy the emissions of 1 ton of a gas will absorb over a given period, relative to the emissions of 1 ton of carbon dioxide (CO2). This reaction displayed a more favourable GWP 175 vs. 783 CO2 equivalents than in solution.
Mechanosynthesis of various salen 48 and salophen 49 ligands and their complexes, including metals such as Zn, Ni, Pd, Cu, Co, and Mn(Cl), has been accomplished at both laboratory- and multigram-scales using ball mills and twin screw extruders (TSE).99–103 Nevertheless, in 2021, García et al. reported the mechanosynthesis of four Br–salen or Br–salophen complexes with aluminium or indium on a small scale (0.1–1 g) using a VBM.104 The four complexes were readily produced using a two-step sequence, which started by condensation of bromosalicylaldehyde 47 (2 equiv.) with either 1,2-phenylenediamine (1 equiv.) or ethylenediamine (1 equiv.). The resulting Br–salen 48 and Br–salophen 49 ligands were isolated in 92% and 98% yields, respectively (Scheme 12, eqn (1)). The complexes were prepared by subsequent treatment of Br–salen or Br–salophen with either AlCl3 or InCl3. The mixture was milled for 4 h at 30 Hz. After washing the crude with water and a drying step, the complexes were obtained in good to excellent yields (70–97%) (Scheme 12, eqn (1)).
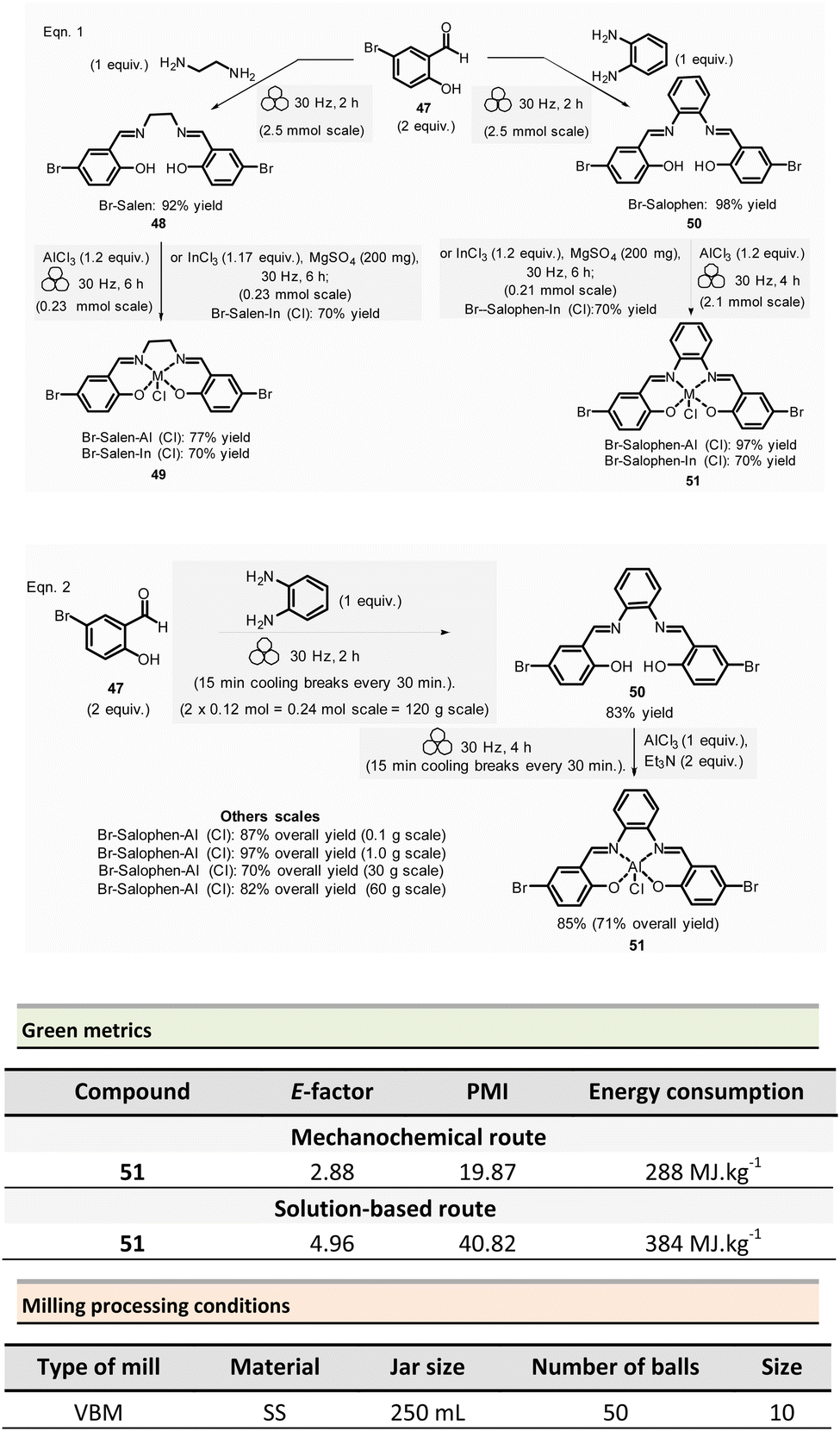 | ||
| Scheme 12 Mechanochemical preparation of Br–salen and Br–salophen ligands and their corresponding complexes. | ||
The large-scale synthesis (30–120 g) of Br–salophen–Al(Cl) complex 51 in a PBM was also developed. For 0.24 mol scale (120 g), the reaction consisted in grinding for 2 h with 15 min cooling breaks every 30 min at 30 Hz, a mixture of 5-bromosalicylaldehyde 47 (2 equiv.) and 1,2-phenylenediamine (1 equiv.) in two stainless-steel jars (250 mL) containing stainless-steel balls (balls mass = 500 g overall). The purpose of the cooling breaks during the cycled milling (8 cycles of 15 min with 30 minutes breaks in between two cycles) avoids any possible thermal activation of the reaction or decomposition of the reactants, due to the heat that might be produced if a continuous milling for 2 h was achieved. The Br–salophen 49 was recovered after simple water removal by drying in 83% yield. Further, complex Br–salophen–Al 51 synthesis was achieved on ≈ a 0.1 mol scale by treatment of Br–salophen 49 (1 equiv.) with aluminium chloride (1 equiv.) and triethylamine (2 equiv.) charged in an oven-dried stainless-steel milling jar. The mixture was ground at 30 Hz in 4 cycles of 30 min each with a 15 min pause between them. After washing the reaction crude with water, the final product was recovered by filtration in 71% overall yield (after drying).
Comparison of E-factors calculated for Br–salophen–Al (Cl) 51 at five different scales (0.1 g, 1 g, 30 g, 60 g, and 120 g) emphasized the effectiveness of mechanosynthesis over conventional solution-based method (Scheme 12, eqn (2)). For instance, on 240 mmol scale, E-factor was of 2.88 vs. 4.96 with solvent. Furthermore, the authors mentioned that when the reaction was done without an HCl trapping agent (i.e., in the absence of triethylamine base) the E-factor would have been be only 0.65 for mechanosynthesis and 2.81 in solution.
PMI values were also favourable towards mechanosynthesis, with lower PMI values than the corresponding solution-based synthesis. On a larger scale (0.24 mol), the calculated PMI value for the solventless reaction was 19.87 (1.65 without Et3N) vs. 40.82 (23.35 without Et3N) in the solvent procedure.
Energy consumption and costs for the five scales reported (0.1 g, 1 g, 30 g, 60 g, and 120 g) were lower than in solution for all mechanochemical procedures. To perform these calculations, as a rough measure, the energy consumption assessment was approximated to the maximum power consumption (as stated in the apparatus technical specifications) during the milling process and negligible power consumption when idle. Whereas for the solution-based methodology, the hot plate was presumed to be at maximum power while heating and negligible power when only stirring. The obtained energy consumption was translated into industrial production cost using US electricity prices from official sources.105
For example, at 0.24 mol scale, the energy consumption for ball-milling using a PBM was 288 MJ kg−1, whereas utilising a hotplate stirrer, the value rose to 384 MJ kg−1. The calculated energy consumption was 25% lower, translating into lower production costs (average cost of 5.18 USD kg−1 and 6.87 USD kg−1, respectively, for milling- and solution-based processes). Based on these results, it could be anticipated that the difference could be even more favourable at a larger scale as solvent costs were not considered.
In 2022, the same group reported the synthesis of four sterically hindered fluorescent salen and salophen complexes.106 The optimized preparation of 3,5-di-tert-butyl functionalized salen 53 and salophen 55, ligands and their respective aluminium and indium complexes were obtained on a 2.5 mmol scale (Scheme 13). For this purpose, 3,5-di-tert-butylsalicylaldehyde 52 (2 equiv.) and ethylene diamine (1 equiv.) were ball-milled in a stainless milling media (one 10 mL jar loaded with one 10 mm ∅ ball at 30 Hz for 2 h) affording the target salen ligand 53 in 93% yield. For salophen ligand 55 (from 1,2-phenylene diamine), a catalytic amount of acetic acid (70 μL) was added to enable ligand formation in a 90% yield. Their corresponding salen and salophen metal complexes were obtained in 70% to 88% yields, respectively, by reacting the corresponding ligand (1 equiv.) with AlCl3 or InCl3 (1.5 equiv.) and MgSO4 (≈13 equiv.) at 30 Hz for 6 h. The green metrics calculated for t-Bu-salen 53 and t-Bu-salophen 55 ligands and their respective complexes showed lower E-factors than in solution (0.4 and 4.03 vs. 2.05 and 5.19 in solution, for salen and salophen complexes, respectively). For instance, for the t-Bu–salen–Al (Cl) complex, the E-factors were 3.02 for the mechanochemical synthesis and 4.76 for its preparation in solution.106 Mechanochemistry displayed the same trend for PMI (1.02 to 1.66) versus 3.17 to 28.9 for solution-based reactions. For the t-Bu-salen–Al (Cl) complex, PMI was 1.13 for mechanochemistry vs. 39.7 in solution. In all cases, RMEs were more favourable mechanochemical procedures than their solution counterparts (60–89% vs. 43–53%, respectively). Then, for the t-Bu–salen–In (Cl) complex, the RME was 60% in mechanosynthesis and 49% for traditional solvent-based procedures.
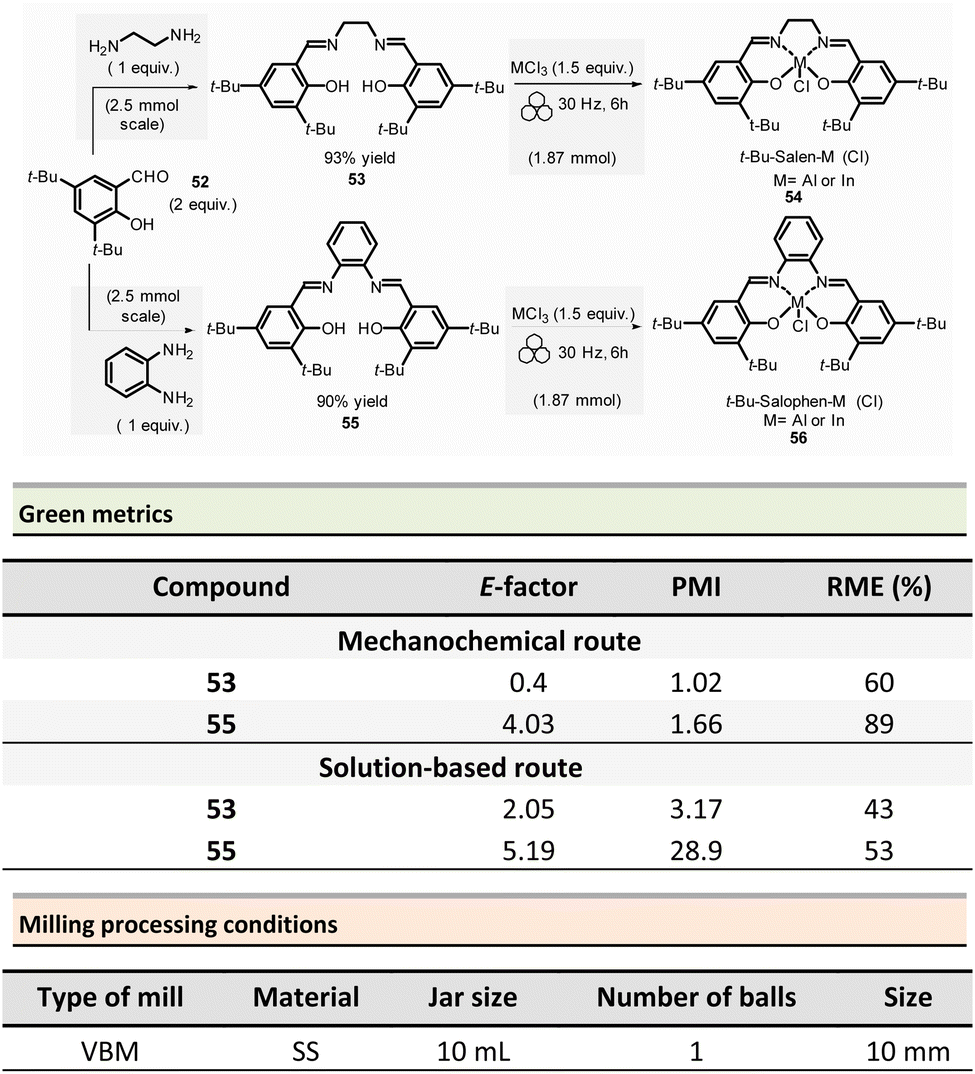 | ||
| Scheme 13 Mechanochemical t-Bu–salophen complexes formation with aluminium and indium metals. | ||
Energy efficiency costs were also calculated at different scales (from 0.1 g to 120.0 g) for the four complexes (t-Bu–salen–M(Cl) and t-Bu–salophen–M(Cl), M = Al and In). For each scale, estimated energy consumption and approximative electricity cost were systematically lower than in the solution. For the larger scale (120 g), the estimated energy consumption was 384 MJ kg−1 for mechanical activation, whereas the value was higher (512 MJ kg−1) in solution. The predicted energy savings was 25%, making the ball-milling production cheaper than conventional “wet” routes (10.4€ kg−1vs. 13.8€ kg−1, respectively). It is worth highlighting that the significant energy and cost difference calculated for the 0.1 g-scale, with a 16-fold production cost for conventional laboratory scales (18681.0€ kg−1 and 1,167.3€ kg−1 for solution vs. mechanochemistry).
Multicomponent reactions and synthesis of heterocycles
This section reports the mechanochemical preparations of heterocycles and compares them with similar solution-based reactions. Multicomponent reactions (MCR) are also included in this section.107–109 MCR presents an intrinsically high atom economy and is often environmentally benign. A synergistic combination of solvent-free processes with multicomponent transformations would be relevant for synthesizing a wide range of organic compounds, including APIs.110 Some of these reactions were recently reviewed with in the contexts of pharmaceuticals,111 and several mechanochemical MCR were already described.112–115 However, here we reported the ones where green metrics have been calculated.Coumarine 59 synthesis by Pechmann condensation was recently revisited by Ranu team.116 This original reaction, published in 1883,117 consisted of the condensation of a phenol derivative with a β-ketoester catalyzed by Brønsted acids (such as sulfuric acid). Despite several advantageous modifications, which included the use of milder acids, this process still requires a large quantity of acid, as well as having a limited substrate scope.
Ranu et al. proposed a catalytic and solvent-free milling process that avoids hazardous solvents, excess acid, and high temperatures. The optimized model reaction (Scheme 14, eqn (1)) was carried out at 5.0 mmol scale by mixing phloroglucinol 57, ethyl acetoacetate 58 (1.1 equiv.), and methylsulfonic acid as catalyst (10 mol%) in a stainless-steel jar (10 mL) containing ten stainless-steel balls (5 mm ∅) in a PBM rotating at 500 rpm for 2 h. The coumarin 59 was recovered in 87% yield after dilution of the residue in ethanol, followed by crystallization. At a 25 mmol scale, the yield rose to 91%. Employing this methodology, thirty-two coumarines were prepared in yields ranging from 50 to 93% from a wide range of phenol derivatives and β-ketoesters. Furthermore, an extension of this technique enables access to seven pyrano-annulated indoles (48–86%). For the model reaction presented in Scheme 14 (eqn (2)), only one pyranoindole regioisomer 62 was formed. Eco-scale scores for the synthesized coumarins and pyranoindoles varied from 71 to 90.5 and 68.5 to 83, respectively. More specifically, for the mechanosynthesis of 5,7-dihydroxy-4-methyl-2H-chromen-2-one 59 (Scheme 14, eqn (1)), the Eco-scale score and E-factor obtained were 89.5 and 0.67, respectively, whereas in solution the reported values were 87.5 and 0.76, respectively.118
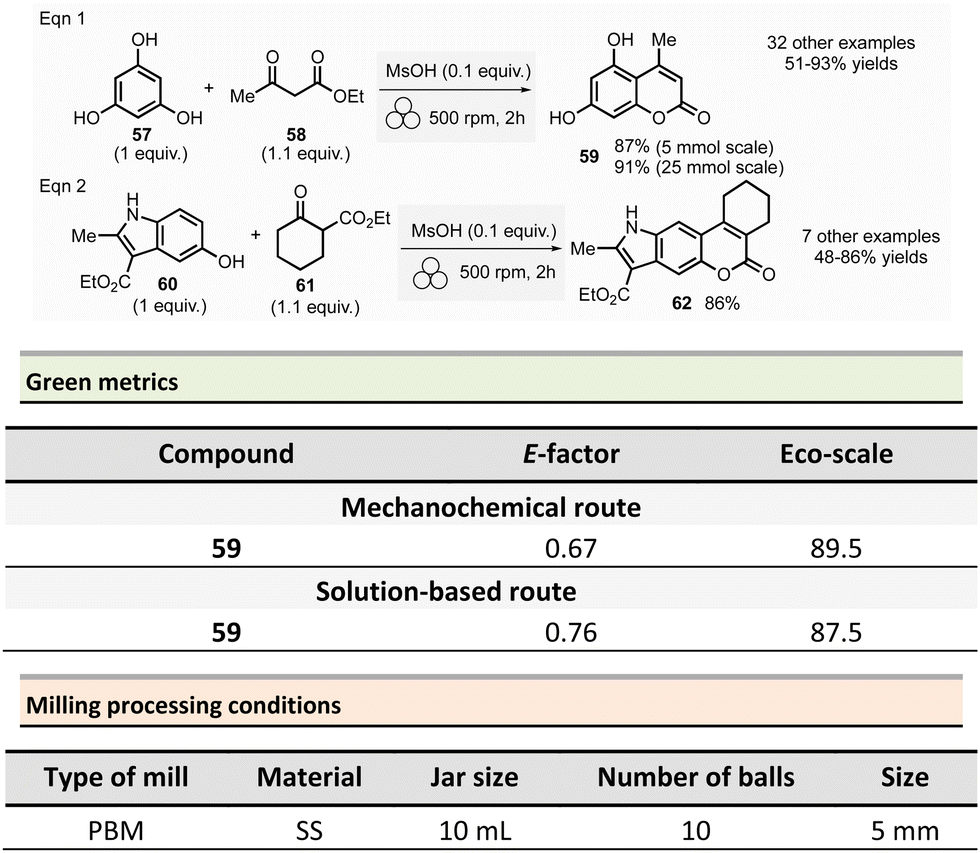 | ||
| Scheme 14 Coumarines and pyranoindoles mechanochemical synthesis via Pechmann condensation. | ||
The eco-friendly advantage (i.e., better metrics) displayed by the mechanochemical approach was attributed to the higher yields obtained (87% vs. 81%), after a work-up consisting of a precipitation in water of the product, followed by a filtration and a crystallisation in EtOH.
In 2022, Porcheddu et al. described an indole and indoline synthesis by Fischer and interrupted Fischer indolization using a ball-milling strategy to avoid the harsh and harmful conditions generally required for these reactions (Scheme 15)119 Traditional indole synthetic procedures required the presence of strong acids or Lewis acids,120 high temperatures, and toxic solvents. More eco-friendly methods were also developed using EtOH and/or water solvents. However, they either required large quantities of p-toluene sulfonic acid (6 equiv.),121 or the use of toxic ionic liquids bearing sulfonic acid groups.122–124
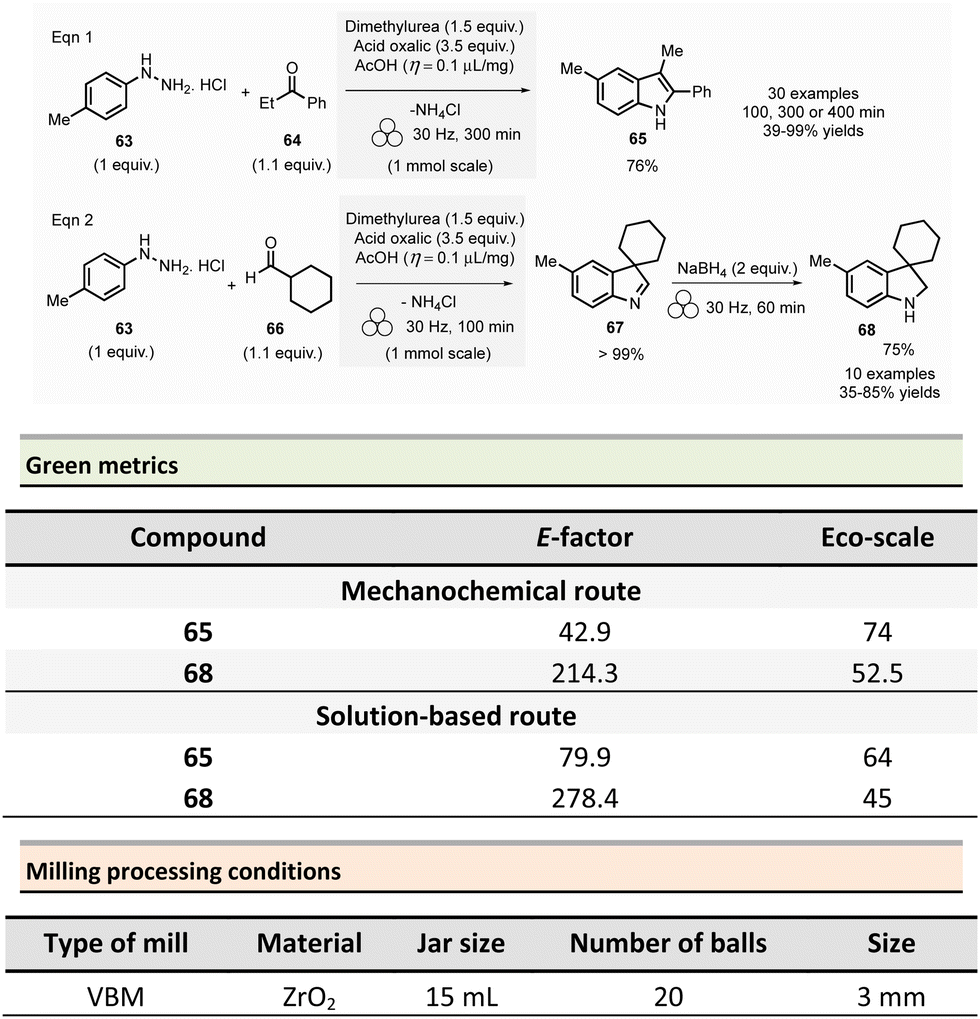 | ||
| Scheme 15 Indole by Fischer mechanochemical synthesis and indolines by interrupted Fischer indolisation. | ||
To address these issues, an environmentally sustainable and optimized approach based on mechanochemical methodologies was developed (Scheme 15). This procedure, conducted at 1.0 mmol scale, consisted in milling 4-methylphenylhydrazine hydrochloride 63 (1 equiv.), propiophenone 64 (1.1 equiv.), oxalic acid (3.5 equiv.), and dimethylurea (1.5 equiv.) under LAG conditions (acetic acid, η = 0.1 μL mg−1). The reaction was carried out in a 15 mL ZrO2 milling jar containing 20 milling balls (∅ = 3 mm, balls mass = 6.5 g overall) for 300 min at 30 Hz. After water addition and filtration, the pure product was recovered in 76% yield. The authors also studied the recyclability of the dimethylureas/oxalic acid mixture. This mixture was successfully reused four times with only a slight yield reduction (from 76% for 1st batch to 70% for 4th batch). By optimizing reaction time (i.e., 100, 300, or 400 min), thirty indoles were obtained by either reacting various arylhydrazines hydrochloride with ketones or mixing 4-methylphenylhydrazine hydrochloride with aldehydes or ketones in yields ranging from 39% to 99%. Moreover, the developed methodology was successfully extended to indolines 68. Interrupted Fischer indole reaction of 4-methylphenylhydrazine hydrochloride 63 with cyclohexane carboxaldehyde 66 furnished almost quantitatively indolenine 67 (Scheme 15, eqn (2)). Further, indolenine 67 was mechanically reduced by the addition of sodium tetraborohydride (30 Hz, 60 min) to produce the corresponding indoline 68. Using this procedure, ten other indolines were synthesized in yields ranging from 35 to 85%.
The green metrics of this process were compared to solution-phase reactions. In solution, indoles 65 were was obtained by reacting phenylhydrazine hydrochloride and cyclohexanone using acidic clay conditions.125 Indoline 68 were obtained by reacting 4-phenylhydrazine hydrochloride 63 and cyclohexane carboxaldehyde 66 in the presence of acetic acid and sodium triacetoxyborohydride.126 For the synthesis of indole and indoline in solution, Eco-scale values were 64 and 45, respectively, and the E-factor reached 79.9 and 278.4. A comparison of Eco-scale scores obtained for the mechanochemical synthesis (74 for indole and 52.5 for indoline syntheses), indicates that ball-milling protocols are greener than solution-based procedures. Even, E-factor values were undoubtedly in favour of mechanical activation, with lower values of 42.9 for indole and 214.3 for indoline than in solution. In 2007, a one-step microwave protocol was developed to produce porphyrins reacting pyrrole and aldehyde in the presence of propionic acid in nitrobenzene at 120 °C for 10 min (20% yield for R = p-methoxyphenyl).127 Conventional heating in solution for the synthesis of porphyrin 71 (Scheme 16, R = 4-MeOC6H4) resulted in low yields (i.e., 20%).
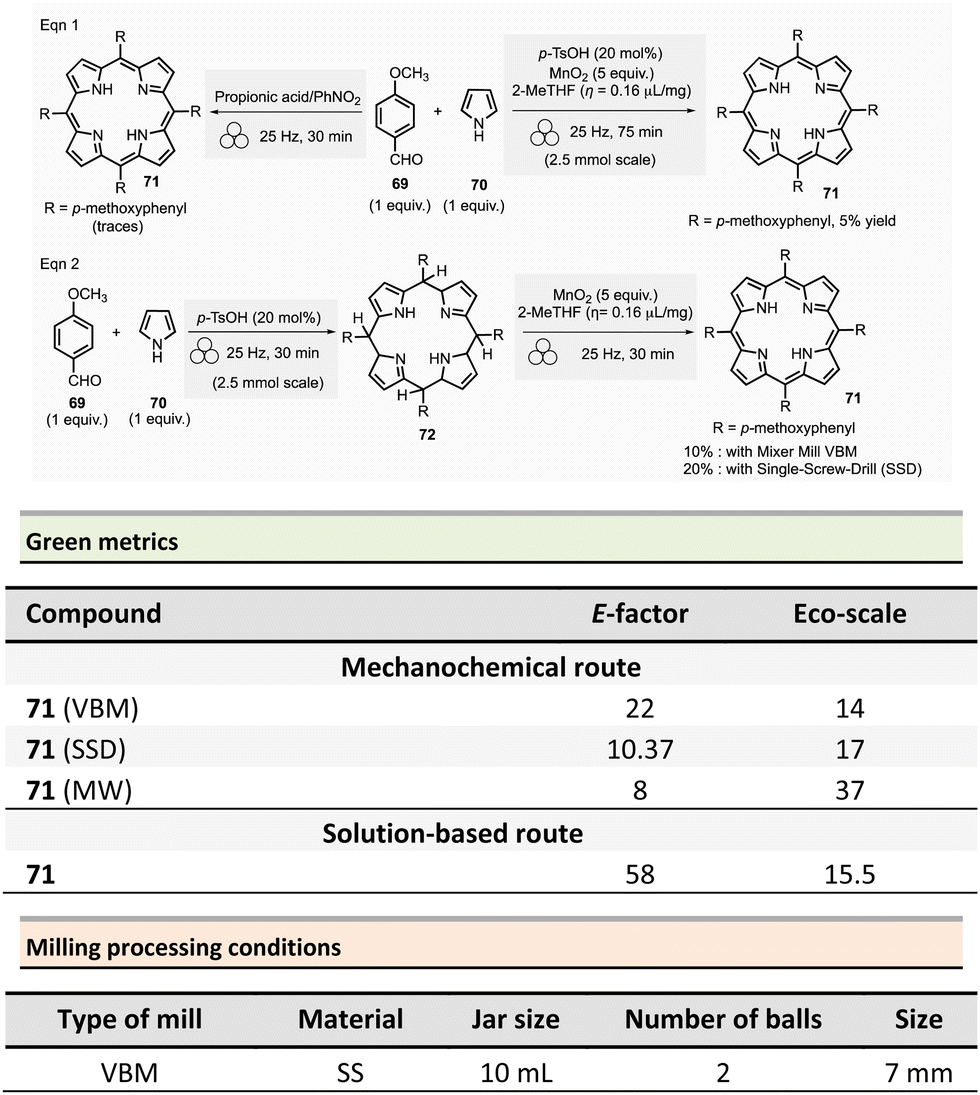 | ||
| Scheme 16 One and two-step porphyrin syntheses by mechanochemistry. | ||
In 2019 and 2020, Pineiro et al. revisited the synthesis of substituted meso-porphyrins using mechanochemistry.21,128 While neat mechanical activation using a VBM resulted in only traces of porphyrin 71,128 grinding a mixture of pyrrole 70 (1 equiv.), 4-methoxybenzaldehyde 69 (1 equiv.), catalytic amounts of p-toluenesulfonic acid (p-TsOH, 20 mol%) and MnO2 (5 equiv.) under LAG conditions (2-MeTHF, η = 0.16 μL mg−1) in a stainless-steel grinding jar containing two stainless-steel balls (7 mm ∅) afforded porphyrin in 5% yield after 75 min at 25 Hz.128
An alternative two-step mechanical synthesis was also evaluated by the same group and revealed to be better for overall yield in porphyrin (10%) than one-pot synthesis. The first step consisted of preparing a porphyrinogen scaffold, which was subsequently oxidized into the porphyrin during the second step. The first reaction used 20 mol% of p-toluenesulfonic acid and the same equipment as the one-step procedure at 25 Hz for 30 min. Porphyrinogen oxidation was achieved by grinding at 25 Hz for 30 min at room temperature the mixture of porphyrinogen, MnO2 (5 equiv.), and 2-MeTHF (η = 0.16 μL mg−1), yielding 10% of the targeted porphyrin. However, the same reaction in solution appeared to be more efficient, furnishing the porphyrin 71 with a 19% yield.128 In any case, four other porphyrins (R = aryls) were mechanochemically synthesized by a two-step strategy in low yields ranging from 7% to 27%.128
The same group reported in 2020, a new automated mechanic-stirrer device adapted for mechanosynthesis. This new tool comprises a stainless-steel cylindrical reactor (4.33 mL) equipped with a stainless-steel mobile single-screw drill (SSD device) rotating at 250 rpm. The SSD device's rotational movement grinds the reactants placed in the stainless-steel cylindrical reactor (Fig. 1). The authors claimed that this new device combines the mortar's simplicity and an automated apparatus's reproducibility. This new equipment was thus used to form porphyrin 71 (R = 4-MeOC6H4), and it proved to be practically effective, giving the same yield than solution-based methodologies, i.e., 20% of yield (Scheme 16, eqn (2)).21 Notably, the modest yield obtained is comparable to those observed for the best solution-based methodologies.
For the two-step procedures (Scheme 16, eqn (2)), the Eco-scale values were comparable (R = 4-MeOC6H4, 2-MeTHF, MnO2, 10% yield) with values of 14 and 17 for the reaction carried out using the VBM and the SSD setup, respectively, while the solution counterpart (i.e., 2-MeTHF/MnO2) was similar with a value of 15.5. However, E-factor values were better for mechanical milling, with values of 22 and 10.37 for VBM and SSD. In contrast, solution-based methods displayed an E-factor of 58. On the other hand, the microwave one-step procedure with a small amount of water (200 °C, 10 min; 14% yield) had the lowest footprint of all the reported syntheses with E-factor and Eco-scale calculated values of 8 and 37, respectively.
In 2021, Pineiro et al. prepared a series of metalloporphyrins from their corresponding metal-free porphyrins by sono- or mechanochemical activations.129 Generally speaking, conventional approaches to metalloporphirins metalloporphyrins involve the complexation of a metal salt by porphyrin in solution. The major drawbacks of this methodology derive from the use of hazardous solvents (e.g., DMF, CHCl3/MeOH) and the need for a significant excess of metal salts required.
A safer procedure was proposed by Pineiro et al. by grinding the porphyrin (R = 3,4-(MeO)2C6H3, 50 mg) with 5 or 10 equivalents of the corresponding salt (i.e., Zn(II), Cu(II), Co(II), Mn(III), Pd(II), Pt(II)) in the presence of NaOH (0,5 or 10 equiv.) in a stainless-steel jar (10 mL) containing two stainless-steel balls (7 mm ∅) at 25 Hz for 30–300 min (Scheme 17). Furthermore, a liquid–liquid extraction with ethyl acetate/water, followed by drying and solvent removal, furnished the corresponding metalloporphyrins in yields ranging from 30% for Mn(OAc)2·4H2O to 97% for Zn(OAc)2·2H2O. Using a similar mechanochemical route, a selection of hydrophobic porphyrins 73 [i.e., R = C6H5, 3,5-Cl2-C6H3, 3-HO-4-MeO-C6H3, 3,4,5-(MeO)3-C6H2)] and hydrophilic porphyrins 75 [i.e., R =, 3,4-(MeO)2-C6H3, 3,5-(MeO)2-C6H3, 3-HO-C6H4 (3-HOTPP), 3-NO2-C6H4, and methylpyridinium (TMePyP)] afforded the corresponding copper-complexes 74 and 76 in yields ranging from 70 to 90%.
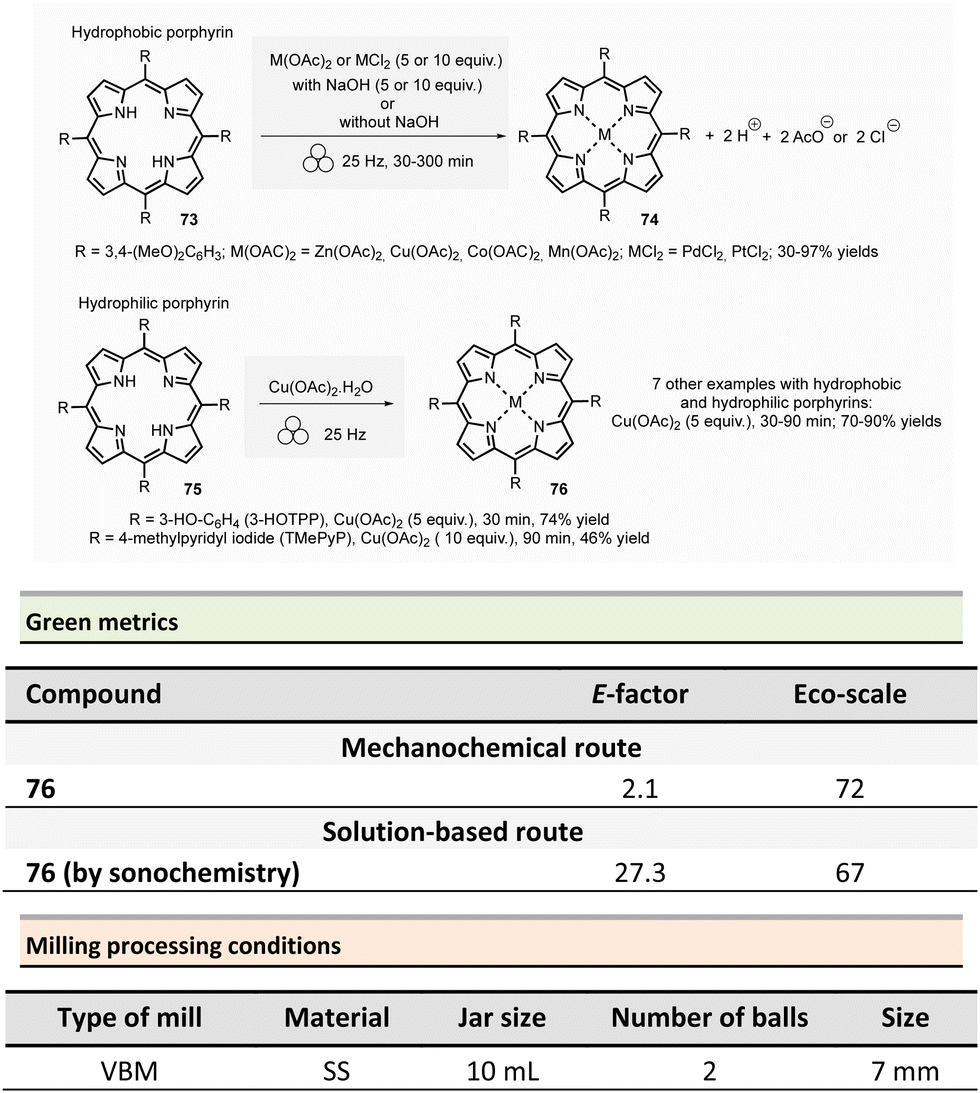 | ||
| Scheme 17 Metalloporphyrin synthesis by mechanochemistry. | ||
On the other hand, sonochemistry was preferentially used for water-soluble porphyrins counterparts [i.e., R = 3-HO-C6H4, (3-HOTPP), 4-HO2C-C6H4, 4-HSO3-C6H4 and methylpyridyl iodide (TMePyP)]. When these reactions were carried out under ultrasound conditions with 1 equivalent of metal salt [Zn(II), Cu(II) and Mn(II)] dissolved in an alkaline solution (NaOH, 2 M), porphyrin complexes were obtained in 32 to 85% yields. Specifically, in the case of Cu(II) salt, 2 equivalents were needed to afford a quantitative yield.
Concerning green metrics, the stoichiometric amount of copper acetate used during the sonochemical route helps explain the better atom economy displayed compared to mechanochemistry with porphyrins 3-HOTPP and TmePyP – which required a significant excess. However, E-factor values obtained for sonochemistry (27.3) and mechanochemistry (2.1), as well as Eco-scale scores calculated for sonochemistry (67) and mechanochemistry (72), unequivocally reflect the greenness of the mechanical methodologies.
The SSD device's rotational movement grinds the reactants placed in the stainless-steel cylindrical reactor (Fig. 1). This new device combines the mortar's simplicity and an automated apparatus's reproducibility.21 The apparatus was successfully employed for di- and tri-component reactions to prepare several chalcones 78 (71% to 99% yields) (Scheme 18, eqn (1)), 3,4-dihydropyrimidinones (55–98%) (Scheme 18, eqn (2)), 4,6-diaryldihydropyrimidinones and 4,6-diaryldihydropyrimidinethiones (Scheme 18, eqn (3), 47–96% yields) and 5-(4-iodophenyl)-1,3-diphenyl-1H-pyrazoline (Scheme 18, eqn (4), 42% yield). Almost all reactions performed with this custom custom-made SSD device (except for pyrazoline formation) afforded equal or superior yields than other mechanochemical- or solution-based methods.128,130–139 For diphenyl chalcone 78, the E-factor score was significantly better using the SSD device (0.17) than any green procedures (solvent-free procedure ground with mortar and pestle) previously reported (E-factor: 0.39 to 0.51).130–132 In contrast, the Eco-scale (74.5) was slightly worse than the solvent-free protocol (grinding with mortar and pestle) developed by Shan et al. (Eco-scale score of 78).131
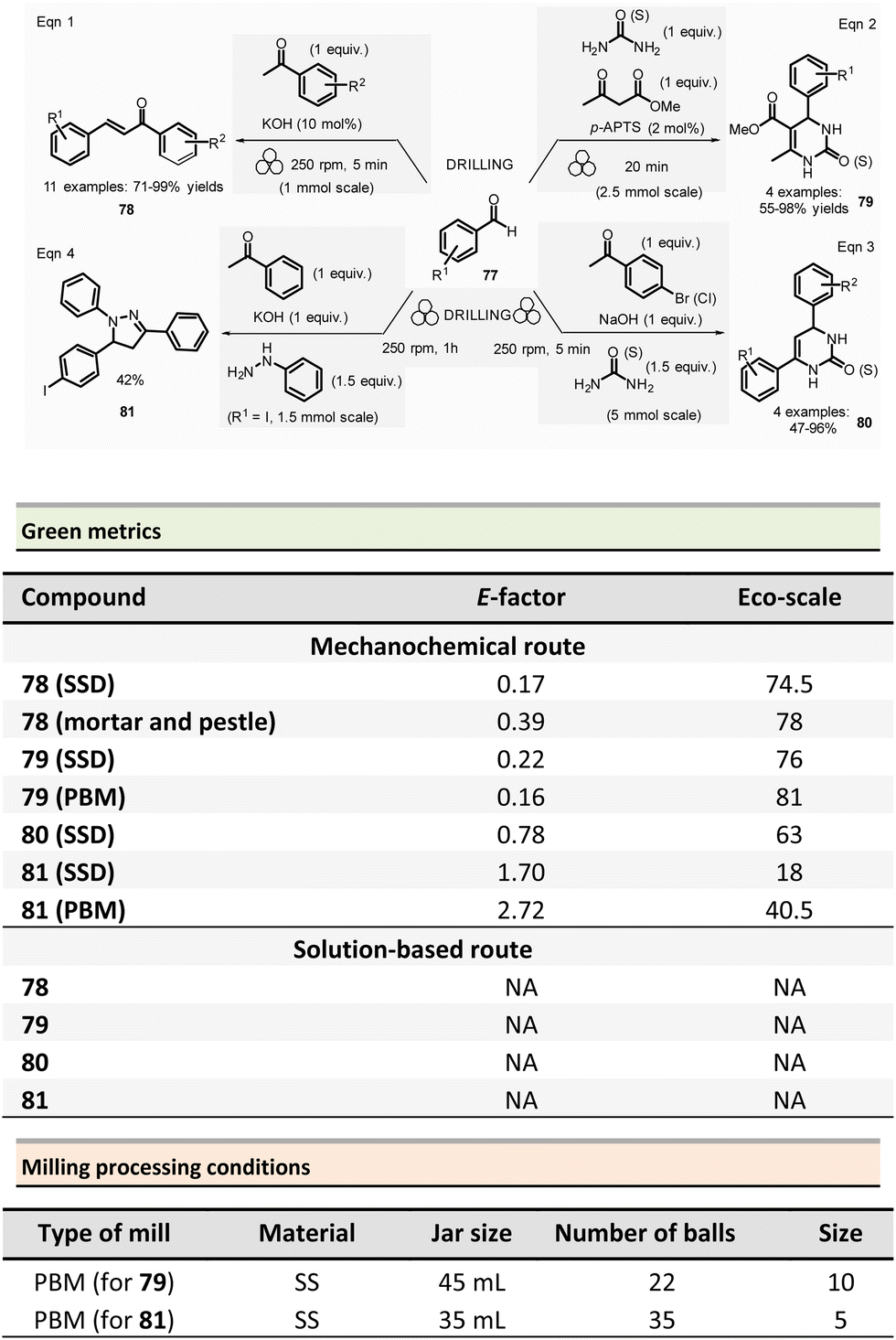 | ||
| Scheme 18 Mechanochemical synthesis of chalcones, dihydropyrimidinones (or thiones), and a 1H-pyrazoline. | ||
The SSD device was also used for the Biginelli synthesis of 3,4-dihydropyrimidine-2-(1H)-thione 79 by reacting methyl acetoacetate, benzaldehyde and urea. The E-factor and Eco-scale scores were equal to 0.22 and 76, respectively. However, an already already-existing protocol by M'Hamed et al. using solvent-free and ball mill strategy was better in terms of E-factor (0.16) and Eco-scale (81).133E-Factor and Eco-scale for the SSD preparation of 4,6-diphenyldihydropyrimidinone 80 were 0.78 and 63, respectively, which are environmentally favourable when compared to performing the in acetonitrile under basic or acidic conditions.135–137 The SSD procedure still remained relevant when compared to solvent-free microwave reaction under acidic conditions for 80.136 For 5-(4-iodophenyl)-1,3-diphenyl-1H-pyrazoline 81, the E-factor (1.70) was slightly more favourable for the SSD device than the high-speed ball milling counterpart (2.72),138 but the Eco-scale was more favourable for ball milling (40.5) compared to 18 by SSD device).
In 2022, Blazquez-Barbadillo et al. synthesized unsymmetrical 1,4-diaryl-1,4-dihydropyridines 85 by a one-pot, two-step mechanochemical reaction (Scheme 19).115 The reaction is ball-milled in a PBM for 2 h for the first step with 1 equivalent of aldehyde 83 and 1 equivalent of aromatic amine 82 to produce intermediate E-imine 84. Subsequently, an equimolar amount of β-ketoester, catalyst, and ethanol are added, and the reaction vessel and ball-milled for additional 2 hours. The final unsymmetrical 1,4-diaryl-1,4-dihydropyridines 85 were purified by silica gel column chromatography, also used in the solution-based process. The greenness of the reaction was compared to the solution based-procedure using DOZN 2.0 tool.16
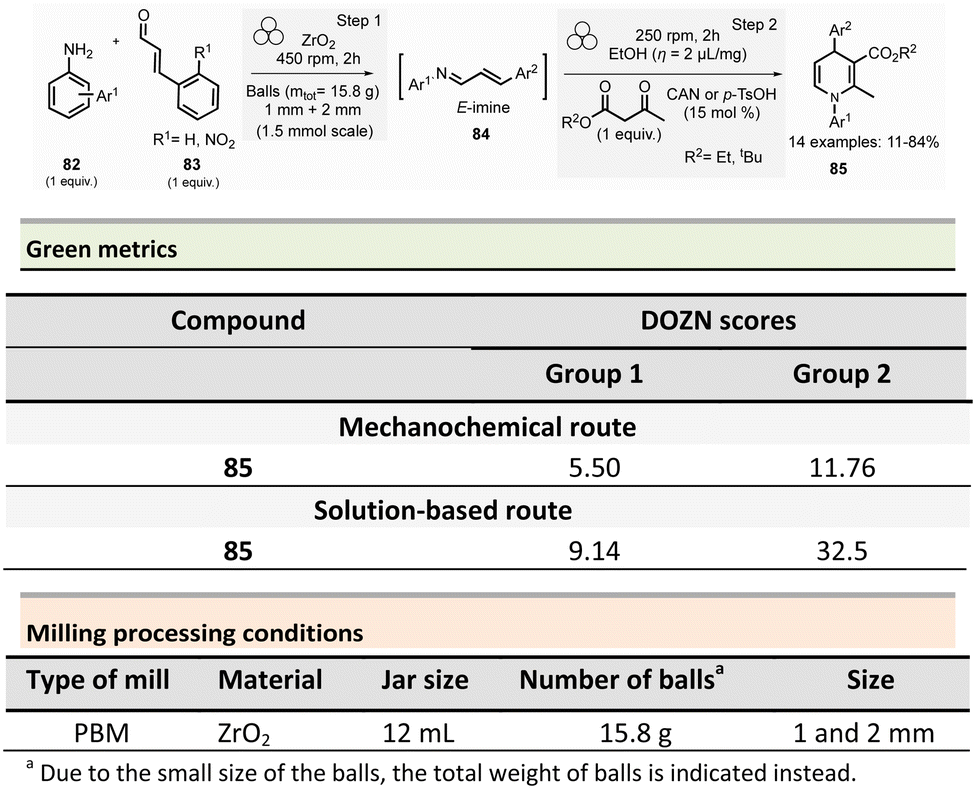 | ||
| Scheme 19 1,4-Dihydropyridine synthesis by ball-milling. | ||
An aggregate score of 1 was obtained for both methods. However, the individual scores for the subgroups of principles showed better resource use (group 1) and a better reduced human and environmental hazards (group 3) (5.50 vs. 9.14 and 11.76 vs. 32.5, respectively) for the ball-milling method compared to solution-based procedures.
Jang et al. described an MCR mechanical synthesis of 2-aminobenzimidazoles and pyrimidines derivatives using ZnO nanoparticles (ZnO NPs) as the catalyst.140 The most effective ZnO NPs catalyst was prepared via a sol–gel method employing a specific directing agent (Scheme 20). Under optimal reaction conditions at room temperature and under an inert argon atmosphere, the condensation reaction of equimolar amounts of 2-aminobenzimidazole 86 (3.0 mmol), 2-nitrobenzaldehyde 87 (3.0 mmol), and ethyl acetoacetate 88 (3.0 mmol), catalysed by ZnO NPs (0.4 mol%), occurs in a tungsten carbide jar containing 20 milling balls (5 mm ∅) at 600 rpm for 40 min. The corresponding benzoimidazopyrimidine 89 was isolated in 82% yield by recrystallization from a water/acetone mixture. Interestingly, the catalyst was recycled up to 5 times without any efficiency loss. This method produces a higher yield, in a shorter reaction time, with a more straightforward purification step than other protocols reported in the literature. When condensations were led with different alkyl-, aryl- or heteroarylaldehydes from 2-aminobenzimidazole and ethyl acetoacetate, pyrimidines were isolated in range of 73–87% yield (16 examples). When 2-aminobenzothiazole were employed as starting material, 5 other pyrimidines were produced in 72–82% yields, and finally, when urea or thiourea were used, 4 other compounds were obtained in 77–80% yields. It is noticed that a large-scale reaction (60 mmol) was also performed between 2-aminobenzimidazole, benzaldehyde and ethyl acetoacetate, and an equivalent yield to the smaller scales was observed.
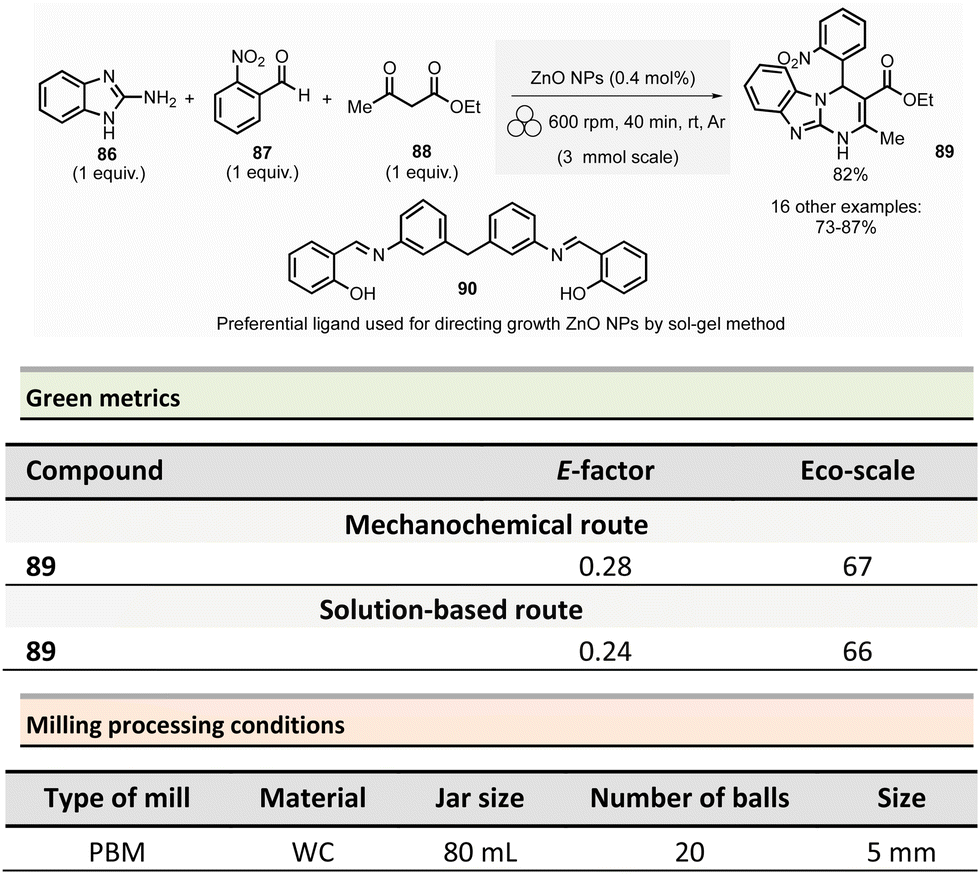 | ||
| Scheme 20 Model reaction for benzoimidazopyrimidine synthesis by mechanochemistry. | ||
For the reaction presented in Scheme 20, an Eco-scale score of 67 and an E-factor of 0.28 were obtained. These values showed to be better141–144 or comparable (66 and 0.24)145 to other methodologies reported with or without solvents. In addition, Jang et al. method140 is better than Liu's method145 in solution due to both reduced reaction time (40 min vs. to 3 h) and temperature (room temperature vs. 100 °C, respectively).
Baltas et al. developed a mechanochemical route to annulated 1,2,4-triazoles 94 using a one-pot, two-step strategy (Scheme 21).146 For this purpose, equimolar amounts of 1-hydrazinophthalazine hydrochloride 91 and 3,4-dimethoxybenzaldehyde 92 absorbed on silica were ground with sodium acetate producing the corresponding hydrazine 93.
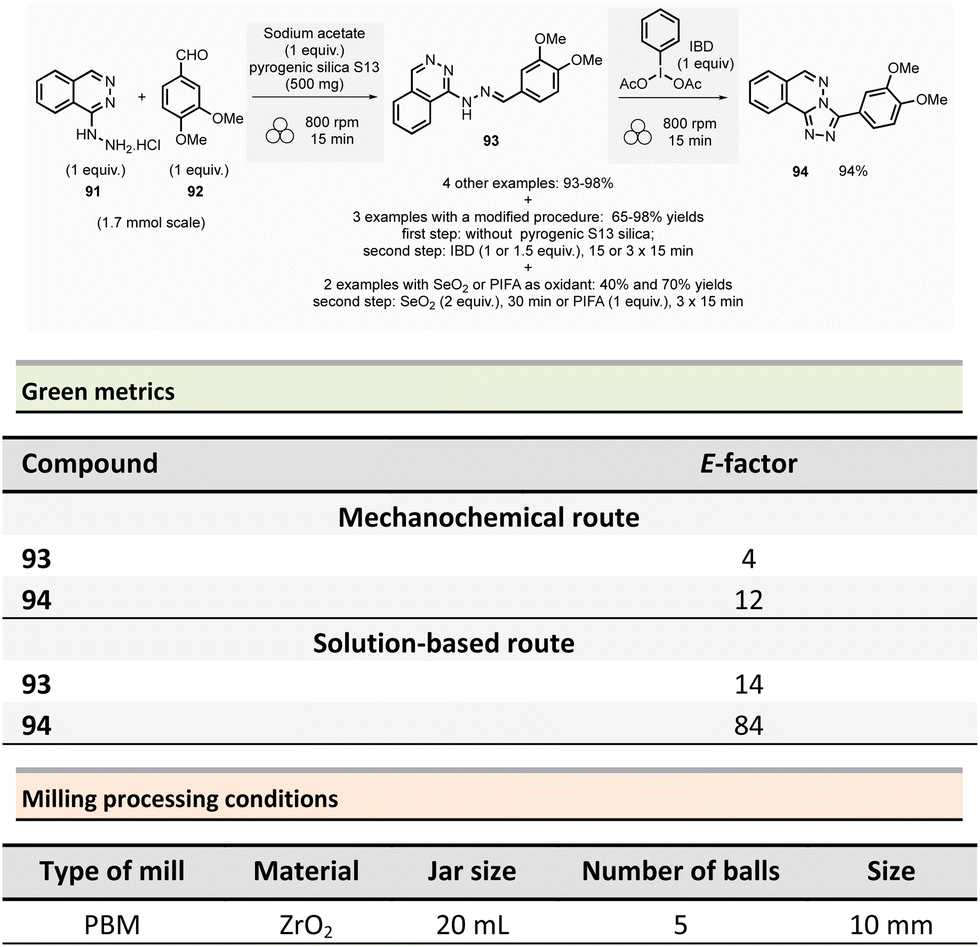 | ||
| Scheme 21 One-pot two-step sequence for annulated 1,2,4-triazole by mechanochemistry. | ||
This reaction was conducted on a 1.7 mmol scale with zirconium oxide jars (20 mL) containing five ZrO2 balls (10 mm ∅) and milled at 800 rpm for 15 min in a PBM. Moreover, addingiodobenzene diacetate (IBD, 1 equiv.) as an oxidizing agent afforded the intramolecular cyclization product after milling at 800 rpm for 15 min. The annulated triazole 94 was obtained in 94% yield. A small library of triazoles was synthesized employing the same strategy on four nonphenolic aromatic aldehydes (yields 93–98%), or using a modified procedure for three heteroaryl carboxaldehydes (conditions for the 1st step: without pyrogenic silica, 800 rpm, 3 × 15 min, 2nd step: IBD = 1 or 1.5 equiv., 800 rpm, 3 × 15 min; yields 65–98%). Selenium oxide or PIFA were also used as an oxidant in the reaction with p-hydroxy-benzaldehyde or vanillin, leading to the corresponding annulated 1,2,4-triazoles 94 in 40% or 70% yield (conditions first step: pyrogenic silica, 800 rpm, 15 min, and second step: SeO2 = 2 equiv., 800 rpm, 30 min). Based on these results, the use of an oxidant based on hypervalent iodine salts (e.g., IBD) not only delivered a better yield of 1,2,4-triazoles 94, but displayed a lower toxicity profile compared to the use of SeO2.
Green metrics for each mechanochemical step were calculated and compared for the mechanochemical synthesis (Scheme 21, conditions used in the 1st step: without pyrogenic silica and 2 × 15 min, and in the 2nd-step 3 × 15 min) and solution methods (conditions in the 1st step: EtOH, AcONa, reflux, 1 h and in the 2nd-step: IBD, CH2Cl2, 4 h). The E-factor calculated for the mechanochemical synthesis was 4 for the 1st-step and 12 for the 2nd-step, whereas for the solution-based protocol were higher (14 and 84, respectively).
Sharma et al. developed a 3-component reaction leading to fused pyrano-spirooxindoles via manual mortar and pestle grinding.147 Equimolar amounts of isatin 95 and malononitrile were first ground for 10 min, followed by the addition of dimedone and then a further 15 min grinding (Scheme 22). Using malonitrile or ethyl cyanoacetate with several isatin derivatives or acetanaphthalenequinone, and different cyclic 1,3-diketones, a series of sixteen amino-2-oxospiro[indoline-3,4′-pyran]-3′-carbonitriles were produced in excellent yields (87–96%). After filtration and washing with water, pure pyrano-spirooxindole 96 was obtained in 94% yield. From the model reaction shown in Scheme 22 (5.0 mmol scale), an excellent E-factor (0.054), as well as good atom economy (95%), reaction mass efficiency (95%), and carbon economy (94.91%), were calculated. However, no comparison with other methodologies was provided by the authors.
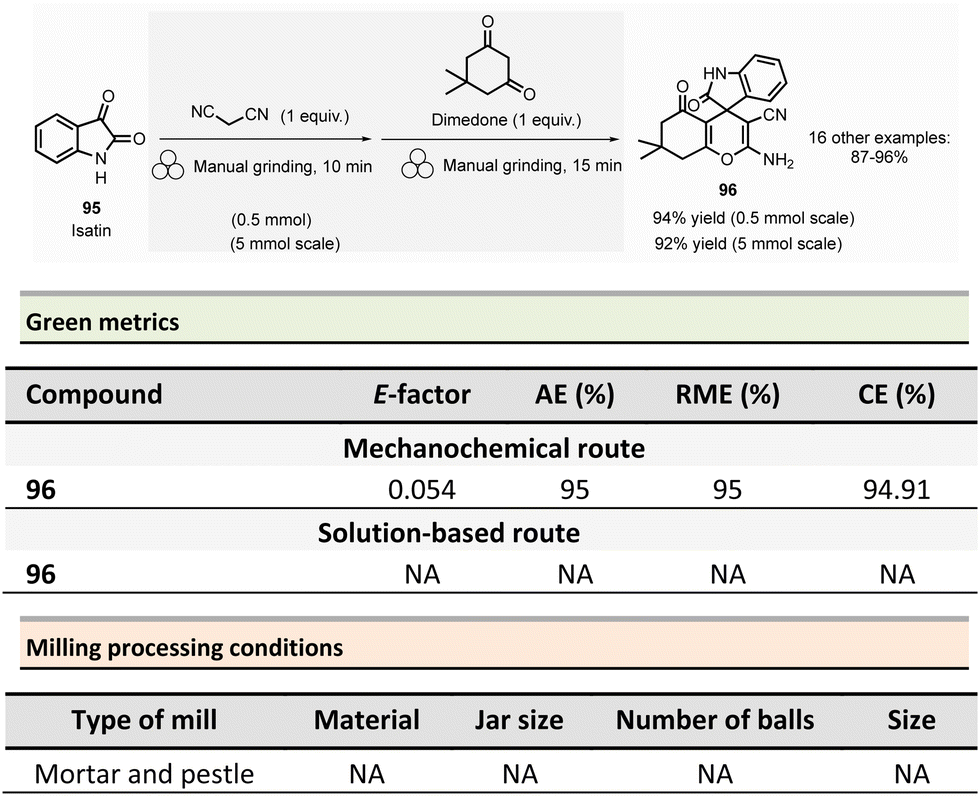 | ||
| Scheme 22 Model reaction for 2-amino-2-oxospiro[indoline-3,4′-pyran]-3′-carbonitrile synthesis by manual grinding. | ||
Catalytic processes mediated by transition metals or acids
Bolm et al. described a new solvent-free method for N-sulfenylations of sulfoximines and sulfonimidamides by disulfides mediated by silver oxide.148 The typical reaction (Scheme 23, eqn (1)) consisted in grinding equimolar amounts of S-methyl S-phenyl sulfoximine 97 (1 equiv.) with diphenylsulfide 98 (1 equiv.), in the presence of silver oxide (0.5 equiv.) and silica gel (60 mg), in a stainless-steel milling jar (10 mL) loaded with one stainless-steel ball (10 mm ∅) for 90 min at 30 Hz under atmospheric conditions. The resulting phenylthioimino sulfanone 99 was isolated in high yields (92%). On a larger scale (5.0 mmol), and modifying the reaction conditions (30 Hz for 10 min followed by heating for 30 min in an oven at 80 °C), the corresponding sulfenyl product 99 was obtained in 85% yield after purification by column chromatography. The authors extended this methodology (0.2 mmol scale, 30 Hz for 90 min) to a wide range sulfoximines and sulphides substrates. Twenty-seven N-sulfenylation products were obtained in yields ranging from 45 to 92%. Sulfonimidamides 100 were also tested with success on diphenylsulfide 98, leading to a library of ten compounds 101 in yields ranging from 79% to 89% yields (Scheme 23, eqn (2)). The efficiency of the N-sulfenylation of S-methyl S-phenyl sulfoximine with diphenylsulfide was compared to the reaction in solution (dichloroethane, 80 °C, 7 h, air atmosphere, 90% yield). An E-factor of 2.3 was obtained by mechanical milling (30 Hz, 90 min), unambiguously highlighting the superiority of ball-milling over the solution-based strategy (E-factor 27.6). A further advantage could be attributed to mechanochemistry is its efficiency for N-sulfenylation of S-methyl S-phenyl sulfoximine with dialkyl disulfides – not achieved in solutions, as previously mentioned.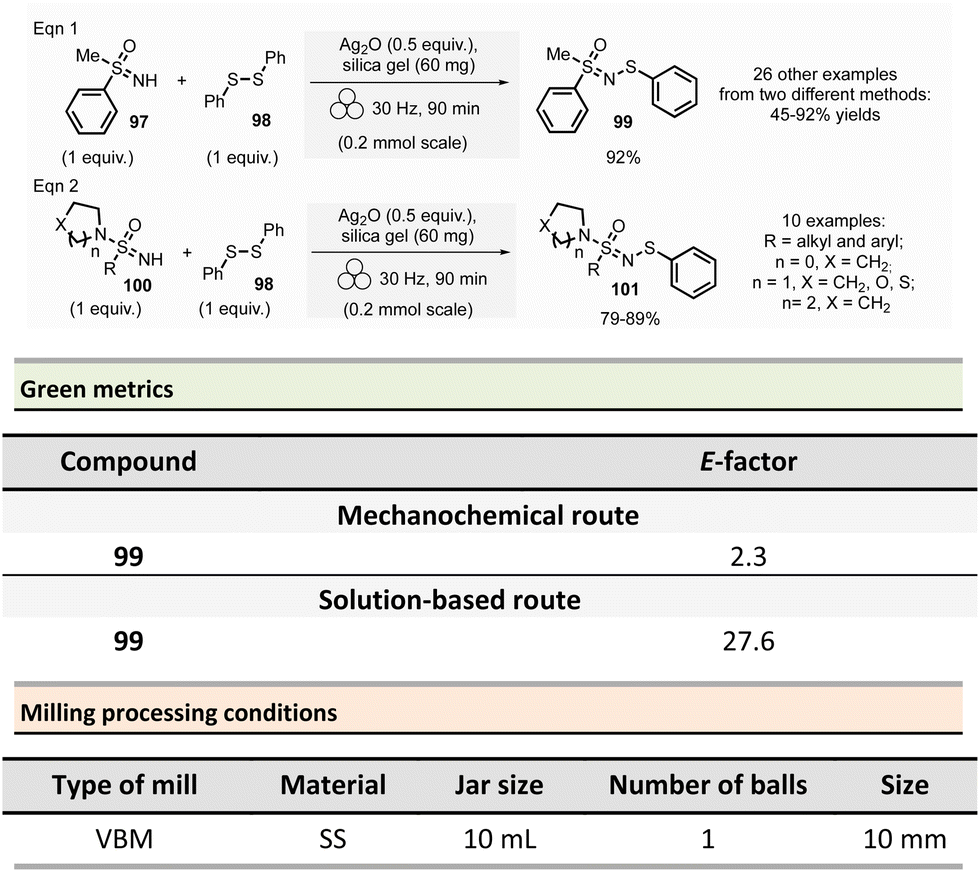 | ||
| Scheme 23 N-Sulfenylations of sulfoximines and sulfonimidamides by mechanochemistry. | ||
In 2022, Bolm et al. developed a regioselective chlorosulfoximidation of allenes by ball milling.12 Surprisingly, the reaction appeared to be catalyzed by traces of metal coming from the stainless-steel jar. The traces of metal generate a sulfoximidoyl radical, which subsequently adds to allene. The typical reaction procedure consisted in grinding sulfoximidoyl chloride 102 (1.5 equiv.), phenylallene 103 (1 equiv.), and silica (7 equiv.) in a stainless-steel milling jar (10 mL) containing 10 stainless-steel balls (5 mm ∅) at 25 Hz for 198 min under argon (Scheme 24, eqn (1)). The chlorosulfonylalkene 104 was obtained after column chromatography in 77% yield. Fifteen compounds were synthesized by mixing N-tosyl arylsulfoximidoyl chlorides, with mono- or disubstituted allenes. Notably, all the reactions performed were fully regioselective, leading to a single isomer in 46 to 82% yields. The obtained chlorosulfonylalkenes 104 were subsequently reacted with a series of heteroatom-based nucleophiles (sodium toluenesulfinate, benzylamine allylamine, and sodium azide). Reactions were performed on a 0.1 mmol scale with a stainless-steel milling jar (5 mL) containing one stainless-steel ball (7 mm ∅) at 25 Hz for 10 min under an air atmosphere. The corresponding substituted products 105 were obtained in excellent yields (80–92%).
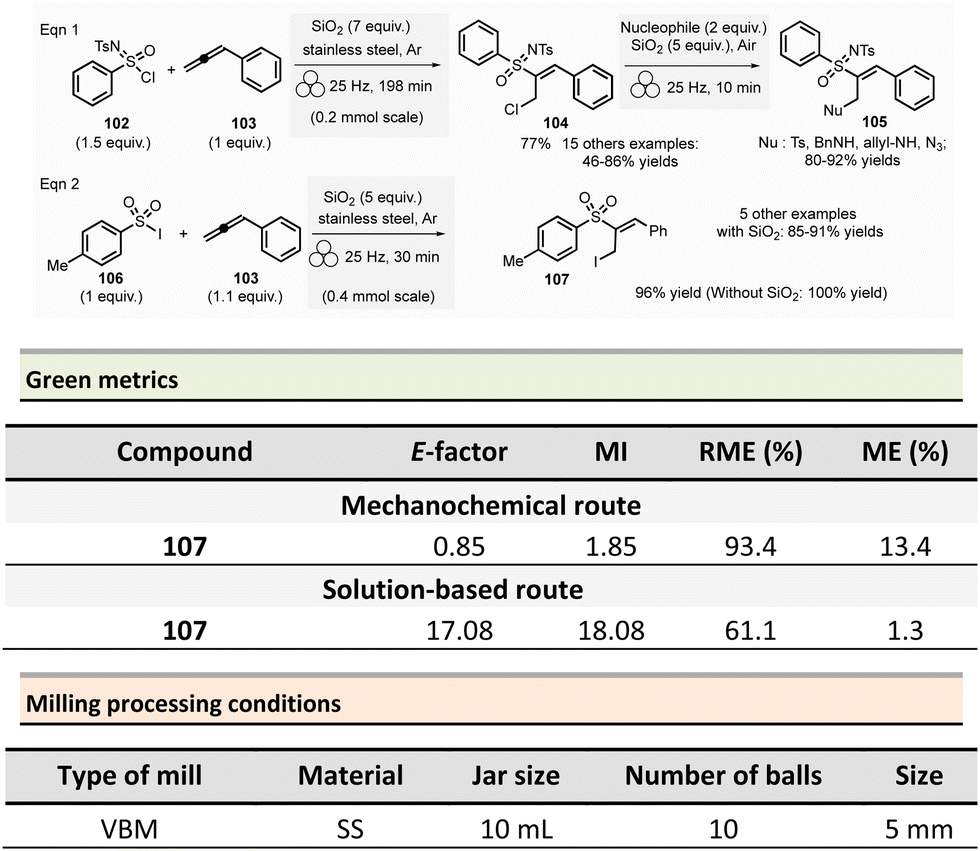 | ||
| Scheme 24 Chlorosulfoximidation of phenylallene followed by subsequent nucleophilic substitutions, and iodosulfonations of arylallenes by mechanochemistry. | ||
Notably, p-toluenesulfonyl iodide 106 was also able to react with phenylallene 103 (Scheme 24, eqn (2)). The reaction was carried out in a stainless-steel milling jar (10 mL) containing ten stainless-steel balls (5 mm ∅) at 25 Hz for 30 min under argon. The iodovinylsulfonyl derivative obtained 107 was isolated in 96% yield or even quantitatively in the absence of silica. Five other alkenes were also obtained from aryl allenes in the presence of silica (85–91%).
Comparison of green metrics (Scheme 24, eqn (2)) was favourable to mechanochemistry with respect to solution methods (E-factor: 0.85 vs. 17.08, mass intensity: 1.85 vs. 18.08, RME: 93.4% vs. 61.1%, molar efficiency: 13.4% vs. 1.3%).149 These parameters were even better when milling in the absence of silica (100% yield). In such conditions, an E-factor of 0.03, a mass intensity of 1.03, a RME of 97.2% and a molar efficiency of 47.6% were obtained. An extra underlining advantage was the absence of purification.
The same team also published a palladium-catalyzed oxidative procedure for the esterification of alcohols by ball-milling.150 The previous green protocols for this reaction generally used heterogeneous catalysis or microwave irradiation,151–153 which required high temperature, high pressure, organic solvents, and excess alcohol.
Bolm et al. also developed a new mechanochemical method for the self-esterification of alcohols using Pd(OAc)2 (5 mol%) and xantphos (5 mol%) as the catalytic system, in the presence of benzyl chloride (2 equiv.) and K3PO4 (3 equiv.) as a base. The best yields were observed when the reaction was carried out in a 0.8 mmol scale milling of compound 108 for two hours at 30 Hz using a stainless-steel milling jar (5 mL) loaded with two milling balls (10 mm ∅). In this manner, from benzyl alcohol (108) seven esters 109 were obtained in yields ranging from 38% (4-NO2C6H4CH2OH) to 87% (4-MeO-3-FC6H3CH2OH) after a column chromatography). The method is versatile because it is possible to obtain mixed esters starting from two different alcohols. In the best reaction conditions, at 0.2 mmol scale, one stainless-steel ball was used to react [1,1′-biphenyl]-4-yl methanol derivative 110 (1 equiv.), 2-methyl-propan-1-ol 111 (3 equiv.) and K3PO4 (5 equiv.) using the same amounts of catalyst, ligand, and oxidant. After purification on column chromatography, these conditions gave the desired ester 112 (78% yield). In addition, chromatographic purification was required to eliminate two side-products – [1,1′-biphenyl]-4-yl-methyl-[1,1′-biphenyl]-4-carboxylate (4%) and isobutyl isobutyrate (20%), still present in the reaction mixture. Applying this procedure, seventeen mixed esters were obtained starting from various benzyl alcohols and primary or secondary alkyl alcohols (35–92% yields).
The metrics for the mechanochemical self-esterification were compared with those for solution-based protocols: with values of AE (45%) and RME (38%) for the ball-milling method (Scheme 25, eqn (1)). In solution, the AE was 76%, and the RME was 64%. For this specific method, the green metrics are better for the solution-based process, due to the greener nature of the oxidant used (O2), replaced by two equivalents of benzyl chloride in the ball-milling reaction.154 Nevertheless, the E-factor and the molar efficiency (5.4 and 10%, respectively) are better than in solution (23.7 and 2%, respectively) due to the absence of solvent during the ball-milling process. The Eco-scales for both mechanochemistry and in solution were comparable (59 vs. 56). For the cross-esterification (Scheme 25, eqn (2)), the AE was better in solution than in ball-milling (79% vs. 50%).155 Nevertheless, RME (24% vs. 30%), E-factor (30.7 vs. 7.9), molar efficiency (1 vs. 7), and Eco-scale (31 vs. 60) are favourable to the mechanochemical routes.
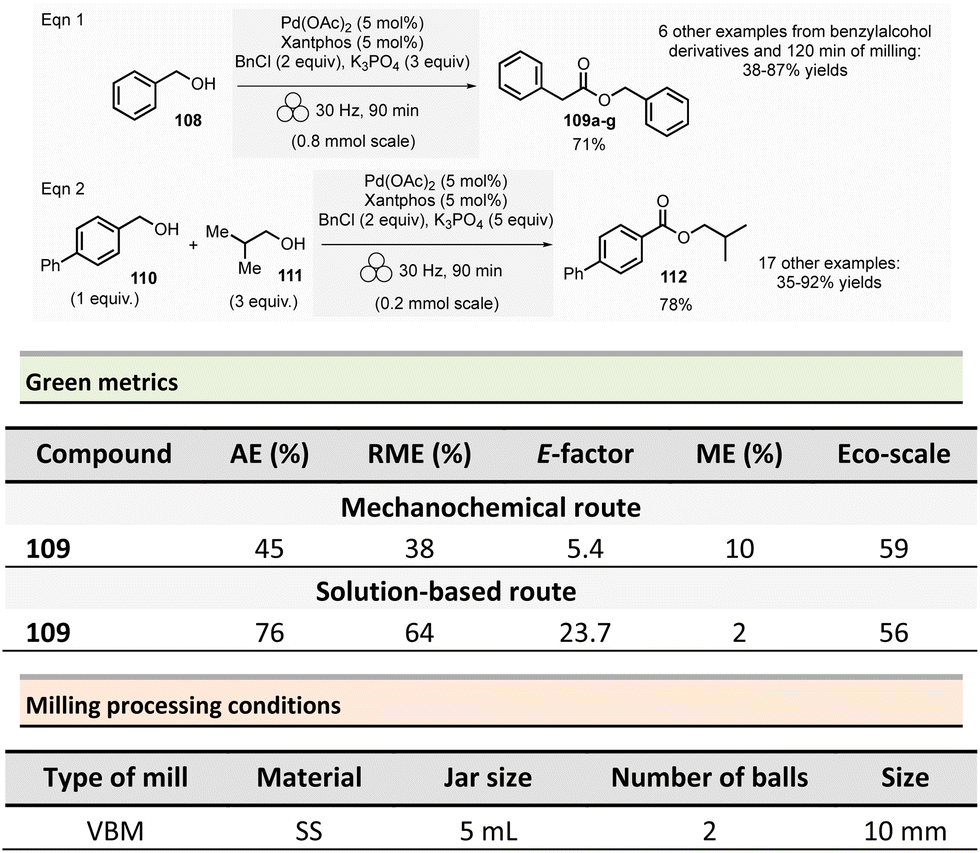 | ||
| Scheme 25 Palladium-catalyzed oxidative self-esterification and cross-esterification by mechanochemistry. | ||
Guo et al. reported the mechanochemical cross-coupling reaction of 2-mercatobenzothiazoles and bromoacetophenone derivatives. The catalytic response is mediated by a transition metal-N-Heterocyclic carbene (metal-NHC) complex ([NiLBr]PF6) 118 (Scheme 26, eqn (1)).156 For instance, the [NiLBr]PF6 complex was readily obtained using a four-step mechanochemical synthesis. The benzimidazole 113 and 2-chloromethylpyridine 114 were first ground in the presence of NaOH and water (η = 0.01 μL mg−1) to produce N-pyridylmethylbenzimidazole 115 (95% yield). This compound was then treated with dibromomethane under milling conditions to afford benzimidazolium salt 116 ([H2L]Br2, 38% yield).
 | ||
| Scheme 26 Metal-NHC catalyzed alkylation of 2-mercaptobenzothiazole (by manual grinding or ball-milling). | ||
Then, [H2L]Br2 was reacted by manual grinding (agate mortar and pestle) with Ni(OAc)2 under LAG conditions (η = 0.71 μL mg−1) [MeCN/H2O (1:1) or MeCN/MeOH (1:1)] to afford [NiL]Br2·2H2O and [NiL]Br2·MeOH 117 almost quantitatively. Lastly, to the obtained 1:1 mixture of complexes, an anion exchange reaction was performed by manual mixing with ammonium hexafluorophosphate affording the catalyst [NiLBr]PF6118 in 97% after recrystallization.
Catalyst 118 (1.8 mol%) was then used for the reaction of 2-bromoacetophenone 120 (1 equiv.) with 2-mercaptobenzothiazole 119 (1 equiv.) under manual grinding (agate mortar) in order to avoid potential catalytic activity of balls which are used under ball milling process. The use of 30 μL of methanol in LAG conditions (η = 0.08 μL mg−1) enabled 93% yield (61% with 1.2 mol% of catalyst) of the desired product 121 after purification by silica gel chromatography (Scheme 26, eqn (2)). By comparison, only 25% yield was obtained in solution at room temperature with 1.2 mol% of catalyst loading. Eight other benzothiazolylthioketones 121a-i were synthesized in low to good yields (16–88%). Although for the reaction presented in Scheme 26 (eqn (2)), the E-factor was 0.520, and the Eco-scale score equals 76.5, no comparison was made with the solution-based protocol.
In 2021, Barcellos et al. published a simple and efficient one-step mechanochemical preparation of copper oxide(II) nanoparticles (CuO NPs), which were later employed to catalyze the reduction of nitroarenes.157 Several other mechanochemical routes to CuO NPs were previously reported using diverse copper sources (e.g., Cu(OAc)2·2H2O,158,159 CuSO4·5H2O,160 CuCl2·2H2O,160 and Cu(OH)2161). After calcination at 400 °C and 500 °C, the NPs obtained from Cu(OAc)2·2H2O presented an average size of 75 nm and 86 nm, respectively. Smaller CuO NPs (7 nm to 34 nm) were obtained by grinding CuSO4·5H2O and CuCl2·2H2O at slow rotating (290–300 rpm) for 1–3 h in the presence of sodium hydroxide/sodium chloride. A similar particle size was observed by grinding sodium chloride with Cu(OH)2. Finally, Barcellos et al. managed the synthesis of ultra-small and quasi-spherical CuO NPs (7.84 ± 2.08 nm) in gram-scale by a straightforward and fast eco-friendly one-pot protocol (Scheme 27, eqn (1)). The reaction was carried out by milling Cu(OH)2 (122, 1 equiv.) and sodium chloride (2 equiv.) acting as a grinding agent and avoiding paste-like mixture, for 20 min at high-speed (1000 rpm) using high-energy tungsten carbide mill jars containing tungsten carbide balls (3 and 4 mm ∅, 1:1 w/w). The quasi-spherical nanoparticles of copper oxide 123 were recovered after repeated water treatment and drying steps with 88% yield.
 | ||
| Scheme 27 Mechanochemical preparation of CuO nanoparticles and nitroarene reductions mediated by CuONPs in water. | ||
Green metrics assessment for the NPs synthesized from different copper salts was also performed. Comparing the atom economy (AE), Cu(OH)2 turned out to be the best metal precursor as per the Barcellos’ procedure, with an AE of 81.54%, followed by CuCl2 (59.17%). Considering a yield of 88%, the real atom economy (RAE) was 71.8%. Moreover, for the same reaction, the E-factor was equal to 2.05 when water was not considered. The “complete” E-factor reached 38.63 when water is taken into consideration.
If NaCl could be recycled during the process, the E-factor would have been only 0.39. Among all the methods described in the literature, mechanical preparations of CuONPs from Cu(OAc)2/urea,159 and Cu(OAc)2/ammonium oxalate158 followed by calcination at 500 °C and 400 °C resulted greener with an E-factors of 2.26 and 2.92 vs. 38.63, respectively. Nevertheless, such E-factor calculations were performed with an estimated yield of 99%. The mass of oxygen during the calcination step was also not considered, which can skew the comparison between these different protocols. It can be noticed that the solution-based procedure (Cu(NO3)2·3H2O162) displayed comparable E-factor values (2.58 and 28.04 for E-factor and complete E-factor, respectively). Finally, the catalytic efficiency of the nanoparticles prepared by Barcellos et al. was determined by reduction of nitroarenes 124, using 8 mol% of catalyst and 2.5 to 4 equivalents of NaBH4 in water at 70 °C for 10–60 min. For all reactions, aniline derivatives 125 were produced in excellent yields (92–98%). Moreover, using nitrobenzene as substrate, CuONPs catalyst could be reused five times without significant loss of efficacy.
In an original article, Singh et al. described the synthesis of fourteen bis-coumarins in 90 to 95% yields by mechanical ball-milling. The reaction consisted of a double condensation of 4-hydroxycoumarin 126 with various aryl or heteroaryl aldehydes 127 with a zwitterionic liquid coated CuO as catalyst (Scheme 28).163
 | ||
| Scheme 28 Model reaction of bis-coumarine synthesis by mechanochemistry. | ||
As a representative example, 4-hydroxycoumarin 126 (2 equiv.), 4-hydroxybenzaldehyde 127 (1 equiv.), and the catalyst (0.5 mol%) were milled at 600 rpm for 3 h in a grinding jar containing 45 tungsten carbide balls (5 mm ∅). A simple water and methanol washing afforded the bis coumarin 128 in 90% yield (95% from benzaldehyde). Most of the methods describing the condensation reaction of 4-hydroxycoumarin 126 with 4-hydroxybenzaldehyde in solution gave lower yields (66–93%),164,165 except for work by Su et al. where a higher yield (97%) was obtained by performing the reaction under reflux in ethanol.166 However, this reaction required a longer reaction time (24 h) when compared to the mechanochemical route (3 h). In addition, the catalyst employed during the mechanocatalytic reaction was reused 10 times without any loss of efficiency. When the mechanochemical reaction was carried out at a 10 mmol scale, the Eco-scale score was equal to 64 and the E-factor to 0.18.
Gon Kim et al. developed the direct aryl C–H amidation of acyl and carbamoyl azides mediated by an iridium(III) catalyst (Scheme 29).167 Acyl azides are thermally unstable and prone to Curtius rearrangement into isocyanates at 50 °C. To avoid the formation of isocyanate, the C–H activation was attempted at room temperature.168,169 In their preliminary work, Gon Kim et al. investigated the stability of 4-nitrobenzoyl azide under solvent-free milling (1 h, 30 Hz) using different milling apparatuses and conditions. The energy transferred to the reaction system had to be controlled to limit the formation of isocyanate. Their studies highlighted that a Teflon jar (2.2 g cm−3) with stainless-steel balls was the best-performing milling media. In comparison, ZrO2 (5.7 g cm−3), stainless-steel (7.9 g cm−3), or tungsten carbide (15.6 g cm−3) jars and balls produced 7 to 21% of the undesired Curtius rearrangement. Further, C–H amidation optimization was carried out at 0.1 mmol scale by reaction of p-nitrobenzoyl azide 129 (1.8 equiv.) and t-butylbenzamide 130 (1 equiv.) (Scheme 29, eqn (1)). The best conditions found for this reaction consisted in grinding both substrates with the catalyst (i.e., [Cp*IrCl2]2, 5 mol%), and a combination of silver salts (AgNTf2, 20 mol%, and AgOAc 20 mol%). The mixture was milled in a Teflon jar (10 mL) with one stainless-steel ball (10 mm ∅) for 10 min at 30 Hz producing the desired bis amide 131 in 93% yield. A gram-scale synthesis afforded desired product 131 after 20 min of grinding, followed by recrystallization in ethyl acetate in 71% yield (Scheme 29, eqn (1)). A combination of various benzoyl azides 134 and benzamides 130 or 8-methylquinoline 132 in the presence of 5 to 10 mol% of iridium catalyst was studied either by ball-milling or in solution (1,2-dichlorethane). Eleven other amides were formed. Yields were better by mechanical stirring or very close to those obtained in 1,2-dichloroethane (Scheme 29, eqn (1) and (2)).
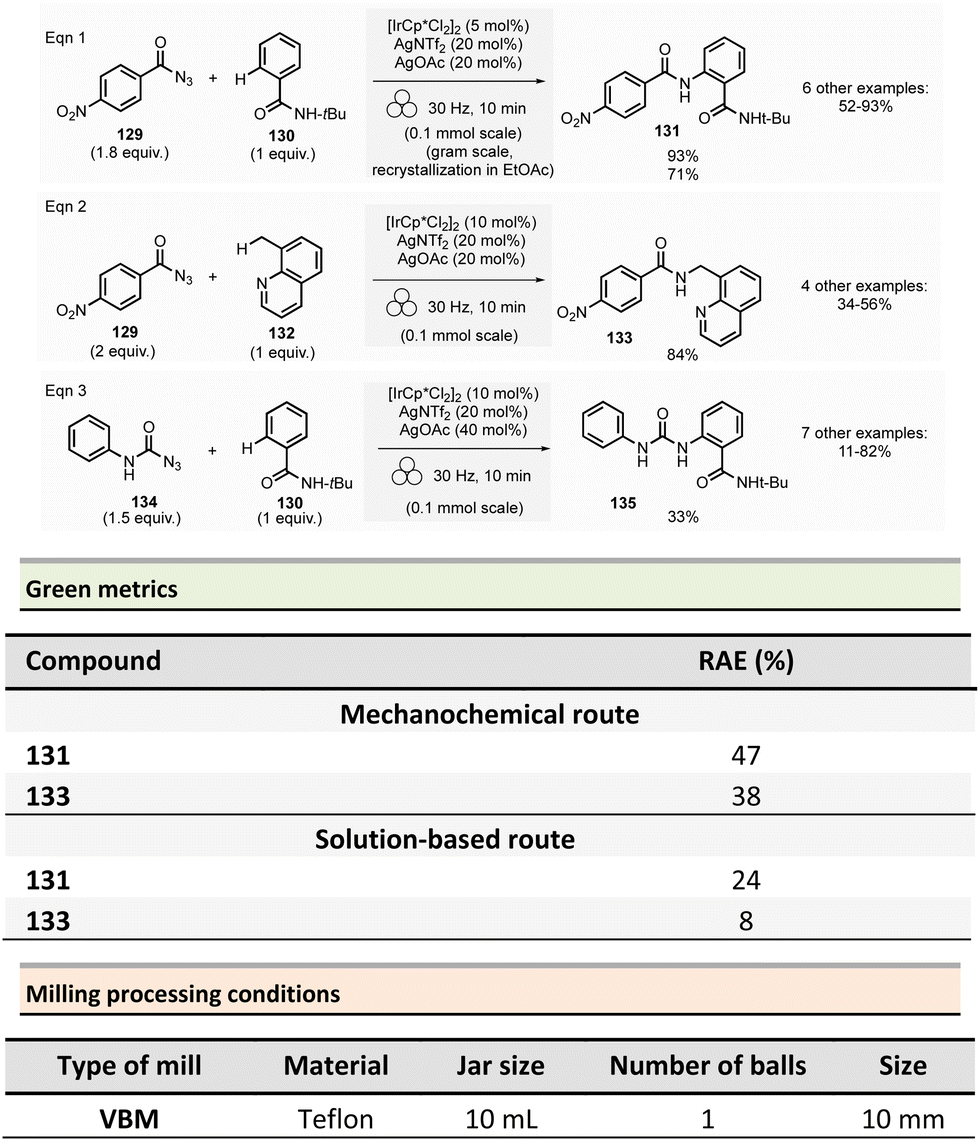 | ||
| Scheme 29 C–H amidation of acyl and carbamoyl azides mediated by Ir(III) catalyst by mechanochemistry. | ||
Authors also extended this C–H amidation to carbamoyl azides by both mechanical grinding and solution phase reactions. Eight ureas were obtained in 11 to 82% yields. Notably, in all cases, yields were in favour of mechanochemistry. The authors calculated the RAE for the first two reactions (Scheme 29, eqn (1) and (2)). For N-(tert-butyl)-2-(4-nitrobenzamido)benzamide 131, the RAE was better for the ball-mill method (47%) compared to the solution-based process (24%). For 4-nitro-N-(quinolin-8-ylmethyl)benzamide 133 synthesis, RAE was almost 5-fold better in mechanochemistry than in solution (i.e., 38% vs. 8%, respectively).
Bis-indolylquinones are substances having multiple pharmacological activities. Menéndez et al. proposed an original synthesis of mono-indolylquinones or bis-symmetrical and unsymmetrical indolylquinones by mechanochemistry.170 Mono-indolylquinones 138 were prepared, by grinding with a high speed ball-milling (HSBM), indole 136 (1 equiv.), 2,5-chloro- or 2,5-dibromoquinone 137 (1 equiv.), Fetizon reagent (Ag2CO3 on Celite, 2 equiv.) used as an oxidant in the presence of p-TsOH (1 equiv.) acting as Brønsted acid for 1 h at 20 Hz in a zirconium oxide jar (20 mL) containing one zirconium ball (20 mm ∅) (Scheme 30, eqn (1)). After purification by precipitation, the final products 139 (X = Cl or Br) were recovered in 96% and 80% yields, respectively (Scheme 30, eqn (1)). An extension of this reaction afforded 12 other indolylquinones 138 isolated in 42% to 98% yields.
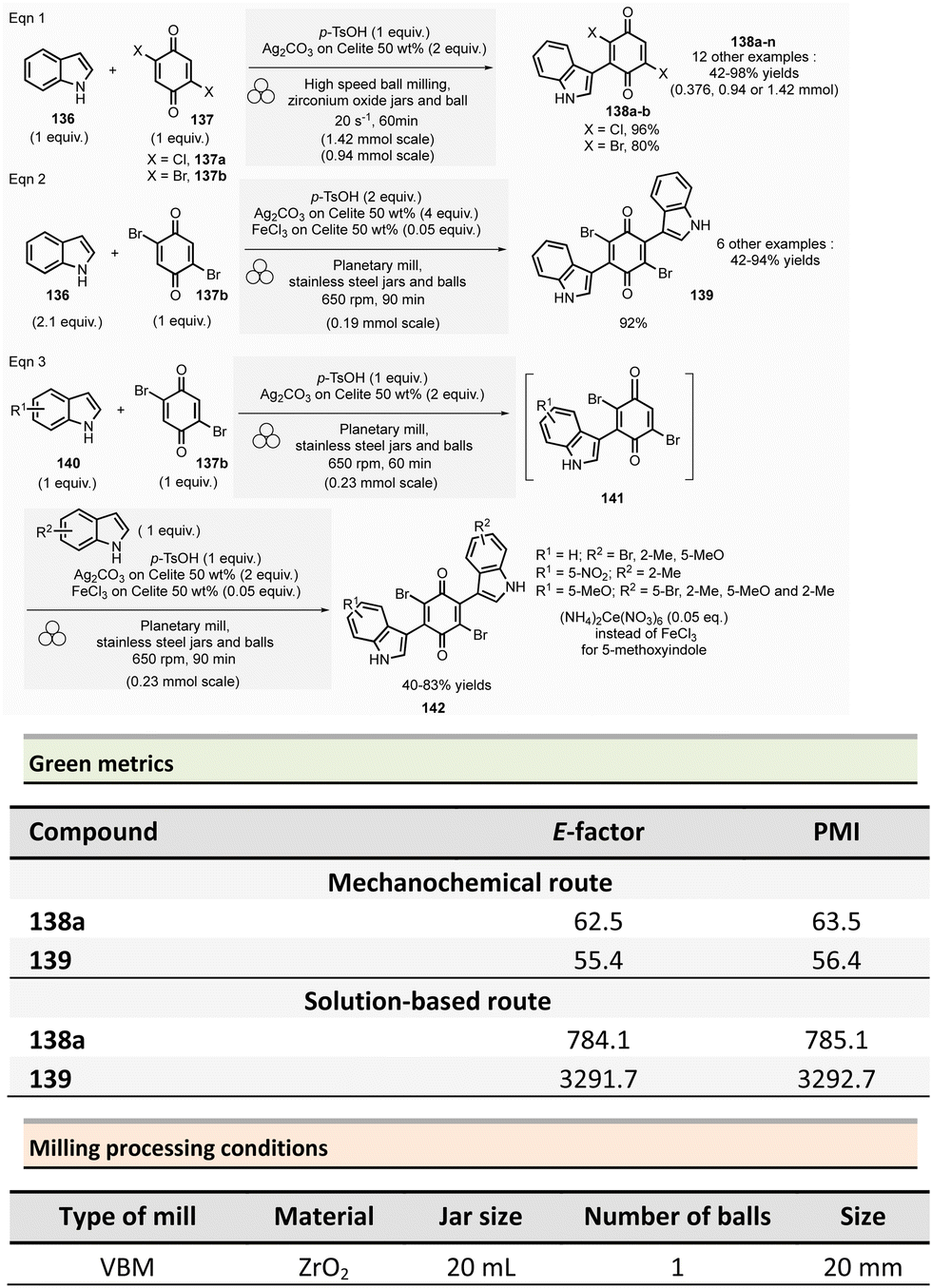 | ||
| Scheme 30 Mechanochemical preparation of indolylquinones. | ||
Interestingly, this methodology allowed the preparation of symmetrical bis-indolylquinones 139 using a PBM (instead of HSBM due to unsatisfying results) rotating at 650 rpm for 90 min with changes in rotation direction every 2 min (reverse rotation). In a stainless-steel jar (12 mL) filled with thirty balls (5 mm ∅), indole 136 (2 equiv.) was ground with p-TsOH (2 equiv.), Fetizon reagent (4 equiv.) and FeCl3 (5 mol%) on Celite (or CAN 5 mol% for 5-methoxyindole) (Scheme 30, eqn (2)).
The corresponding bis-indolylquinone 139 was synthesized in 92% yield. Six other products were prepared in 42% (from 5-bromo-1H-indole) to 94% yield (for 5-methoxy-2-methyl-1H-indole). Consecutively unsymmetrical bis-indolylquinones 142 were prepared using a one-pot, two-step synthetic route. The first step occurred in the conditions described above (650 rpm for 1 h) with indoles 136 (1 equiv.), Fetizon reagent (2 equiv.) and p-TsOH (1 equiv.). After completion of the reaction, the second step was launched by the addition of the second indole 136 (1 equiv.), p-TsOH (1 equiv.), Fetizon reagent (2 equiv.) and FeCl3 (5 mol%) on Celite. The mixture was ground for 90 min. After a work-up, unsymmetrical bis-indolylquinones 142 were isolated in 40% to 83% yields.
All the mechanical reactions took place in shorter times than in solution (26–62 h). Furthermore, green metrics for mono-indolylquinone 138a (X = Cl, Scheme 30, eqn (1)), the E-factor, and PMI showed to be better (62.5 and 63.5, respectively) compared to solution-based reaction (784.1 and 785.1).171 In the same way, for the synthesis of bis-indolylquinone 139 (Scheme 30, eqn (2)), the difference between mechanochemistry (E-factor; 55.4, and PMI: 56.4) and the reaction in solution (E-factor: 3291.7, and PMI: 3292.7) was also in favourable to mechanical milling.172
In 2020, Malvestiti et al. developed a mechanical protocol for the thiocyanation of aryl compounds via C–H functionalization.173 Ball-mill reactions were all performed on a 0.2 mmol scale, without solvent, in short reaction times. The best conditions found for thiocyanation of ortho- and meta-substituted anilines were to grind together anilines (1 equiv.), ammonium thiocyanate (1.5 equiv.), and ammonium persulfate (1.5 equiv.) in the presence of silica (150 mg) in a stainless-steel jar (5 mL) with two balls (7 mm ∅) at 25 Hz for 1 h (Scheme 31, eqn (1)). When the reaction was performed in a Teflon jar a lower yield of 72% was obtained for 144.
 | ||
| Scheme 31 Thiocyanation of aniline and phenol derivatives by mechanochemistry. | ||
Starting from 2-nitroaniline 143, the final product 144 was recovered in 92% yield. Next, the reaction was extended to seven anilines leading to the corresponding thiocyanate derivatives in 45% to 92% yields. The mechanochemical thiocyanation was fully regioselective, and only the para-amino thiocyanates 144 were observed. In the presence of a reactive neighbouring group, a further reaction occurred. Then 3-aminophenol afforded 6-aminobenzooxathiol-2-one in low yield (15%). For para-substituted anilines 145, the thiocyanation cannot occur in para-position (Scheme 31, eqn (2)).
Consequently, the 2-aminobenzothiazoles 146 were isolated in 18–71% yields. The methodology was also applied to phenol derivatives (Scheme 31, eqn (3)).
Ortho- and meta-substituted phenols 147 led preferentially to para-thiocyanation products 148 in 8% to 94% yields (R = H, para-thiocyanation afforded 96% yield). The nature of the second group strongly influences the overall yields. As expected, electron-withdrawing groups have a detrimental effect, whereas the presence of an electron-donating one appeared favourable. By contrast with para-substituted anilines, para-substituted phenols gave benzooxathiol-2-ones 150 only in low yields (14–30%) (Scheme 31, eqn (4)). The mechanochemical thiocyanation was also extended to others arenes such as N,N-dimethylaniline, anisole, 1,2,3- and 1,3,5-trimethoxybenzene, thioanisole, 1-naphthol, and indole. The resulting thiocyanates were obtained in low to excellent yields (33–89%).
The green metrics were calculated for aniline as substrate, only considering the reaction without purification steps. AE values for ball-mill and solution reactions were comparable (0.38 vs. 0.34), while the yield was better for the solvent process (67% vs. 90%).174 For the other parameters, ball-milling was significantly more favourable. The inverse of the stoichiometric factor (1/SF) was equal to 0.72 in mechanical milling, whereas it was only 0.59 for the conventional reaction. The MRP and RME values were 0.42 and 0.077 versus 0.23 and 0.042 in solution. E-Factor exhibited the same tendency (12.0 vs. 22). These metrics stated the importance of avoiding both solvent use and reagents excess.
Yu et al. described the mechanochemical aryl radical formation by homolytic cleavage of aryldiazoniums.175 The C–H (hetero)arylation of 1H- and 2H-indazoles, N-methyl-3-methylindole, benzothiazole, 2-methylthiophene, phenyl derivatives was developed. Typically, the reaction was performed on 0.3 mmol scale (Scheme 32, eqn (1)): 4-methoxyphenyldiazonium tetrafluoroborate 151 (2.5 equiv.), 2-phenyl-2H-indazole 152 (1.0 equiv.), NaCl (1.0 g) acting as a grinding agent and avoiding paste-like mixture, and one drop of EtOAc (η = 0.06 μL mg−1) or CH2Cl2 (for 4-nitroaryldiazonium) were ground at 30 Hz for 30 min in a stainless-steel jar (15 mL) containing one stainless-steel ball (14 mm ∅). Ethyl acetate as LAG additive was necessary for the reaction.
 | ||
| Scheme 32 C–H (hetero)arylations, C–H sulfenylations, cascade- and HAT-additions using aryldiazonium tetrafluoroborates by ball-milling. | ||
The mixture was removed from the jar and purified by column chromatography to afford indazole 153 an 84% yield. Similarly, twenty others 1H and 2H indazoles were obtained in yields ranging from 24% to 87% (Scheme 32, eqn (1)). When 1,3,5-trimethoxybenzene, 1,3,5-trimethylbenzene, 1,4-dimethylbenzene, N-methyl-3-methylindole, benzothiazole, and 2-methylthiophene were used as substrates, modified conditions were applied. With arenes, two equivalents of aryldiazonium salt were added stepwise (1 equivalent each time) and ball-milled in the presence of NaCl (0.5 g each time) and CH2Cl2 as LAG additive (η = 0.12 μL mg−1) added in two equivalent portions for each cycle (30 minutes twice, Scheme 32, eqn (2)). In the case of heteroarenes (eqn (3)), it was necessary to have 4 or 5 equiv. and no LAG additive for the homocoupling of heteroarenes. Besides, a simplified purification (liquid–liquid extraction) was adopted for arene coupling instead of the required column chromatography for heteroarenes. Final products were recovered in 56 to 70% yields for arenes (5 substances, Scheme 32, eqn (2)) and 32–87% yields for heteroarenes (4 substances, Scheme 32, eqn (3)).
Similarly, aryl- and heteroarylthioethers or arylpinacol boronate esters were obtained by transformation of heteroaryl- or aryldiazonium tetrafluoroborates, with dialkyl- or diarylsulfide or bis(pinacolato)diboron substrates in the presence of NaCl (Scheme 32, eqn (4)).
For instance, on a 0.5 mmol scale, milling methoxyphenyldiazonium tetrafluoroborate (1 equiv.) with diphenyldisulfide (1.25 equiv.) and NaCl (1.0 g) at 30 Hz for 2 h, afforded, after column chromatography, the corresponding diarylthioether in 82% yield (Scheme 32, eqn (3)). Eight other thioethers were obtained in yields ranging from 37% to 64% and two aryl pinacolboronate esters in 52% yield when Ar = 4-ClC6H4- and 62% yield with Ar = 4-MeOC6H4-. As a representative example (4-methoxyphenyl)(phenyl)sulfane was scaled-up to an 8.0 mmol-scale milling for 3 h affording 62% yield. For such reactions, sodium chloride acted as an activator. Most halogen salts (NaCl, KCl, NaBr) were effective, while NaBF4 and neutral alumina did not give the target products. The homolytic fragmentation was attributed to the relative instability of in situ formed aryl diazonium chlorides. The authors also demonstrated that the excess NaCl could be recycled and reused at least five times without significant yield loss (78% yield after the fifth time).
Yu et al.175 also reported that 1,3,5-trimethoxybenzene or indole derivatives could undergo a C–H cascade addition starting from aryldiazoniums, and styrene derivatives in the presence of NaCl (Scheme 32, eqn (5)). Direct hydrogen atom transfer (HAT) addition of aryl radical was also efficient on position 3 of indoles starting from 4-hydroxystyrene derivatives (Scheme 32, eqn (6)). Finally, the aryl radical reaction promoted by NaCl was also extended to prepare five APIs (including dantrolene).
Green metrics were calculated for 2,3-diphenyl-2H-indazole synthesized by mechanochemistry and in solution (Scheme 32, eqn (1)).176 An AE of 58.7% was found instead of 57.6% in solution, an E-factor of 3.5 vs. 22.1, a RME of 22.1% vs. 4.3% and a good Eco-scale score of 75 vs. 46 in solution. A similar trend was observed for (4-methoxyphenyl)(phenyl)-sulfane 157 (Scheme 32, eqn (4)). Indeed, a better AE of 39.1% was calculated in the solventless process compared to 4.9% for the reaction done in solution.177,178 The E-factor value obtained was excellent, with 2.1 vs. 25.8 in solution. Even RME was better, with 32% compared to that one in solution, which was 3.7%. Finally, Eco-scale score was 71 in mechanochemistry and only 51 for solution-based reactions.
Generally speaking, especially for metal-catalysed reactions, the removal of the catalyst at the end of the process is a need. In this regard, any post-synthetic treatment, by liquid–liquid extraction or worst, when a purification by column chromatography is required, will negatively affect the green metrics and the entire environmental footprint of the mechanochemical process. This aspect has to be carefully evaluated with the purpose to design/conceive mechanochemical syntheses leading to a straightforward recovery of the final product, keeping at the minimum the number of operations required during the work-up procedures.
Miscellaneous
Borchardt's group published a synthesis of porous organic polymers (POPs) using ball-milling techniques. These POPs were synthesized by applying a Friedel–Crafts alkylation between 1,3,5-triphenylbenzene 164 (TPB) and an excess of a cross-linking reagent (CH2Cl2 or CHCl3) in the presence of AlCl3 (Scheme 33).179 Similarly to the solution-based method, the use of harmful CH2Cl2 or CHCl3 can not be avoided here, being linking agents. The advantage of conducting the synthesis by ball-milling limits the amount used, contrarily to the corresponding solution-based method, where they also act as solvents, and thus used in very large excess. The typical reaction consisted in grinding under an inert atmosphere, TPB 164 (1 equiv., 1.63 mmol) with CH2Cl2 or CHCl3 (6 equiv.) and AlCl3 (24 equiv.) for 1 h (CHCl2) or 0.5 h (CHCl3) at 30 Hz in a ZrO2 jar (50 mL) with twenty-two ZrO2 balls (10 mm ∅), following by washing the resulting solid with water and acetone to remove the AlCl3 excess and the residual starting material. A flexible and a rigid polymer were obtained in 95% (CH2Cl2) and quantitative yields (CHCl3), respectively.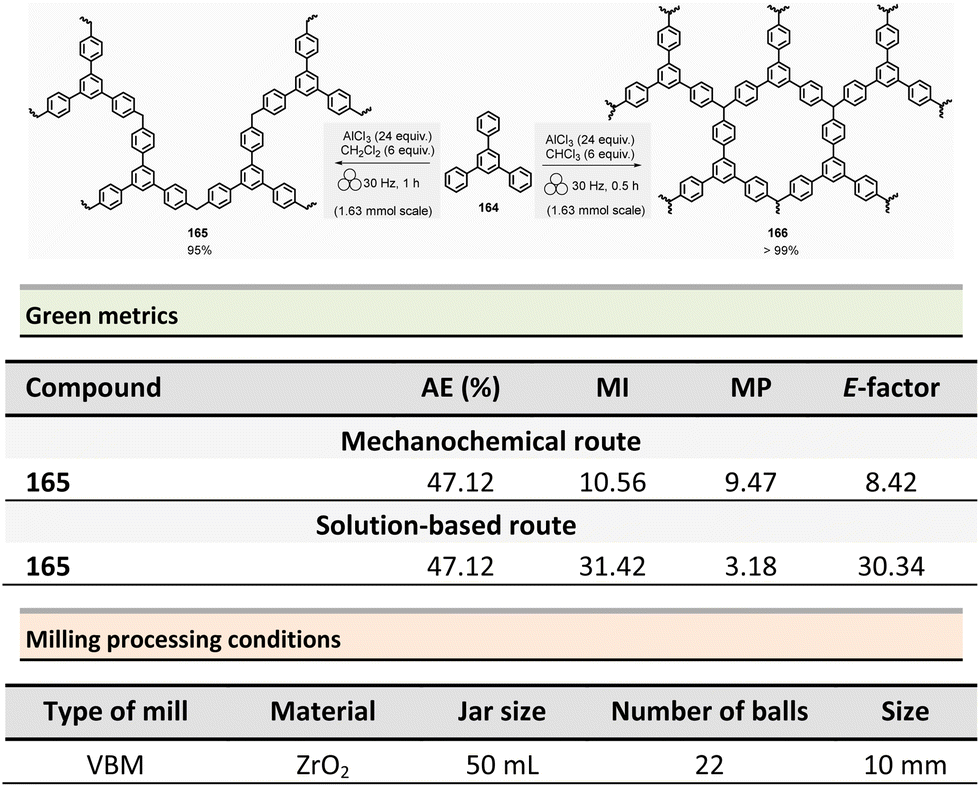 | ||
| Scheme 33 Porous organic polymers (POPs) preparation by mechanochemistry. | ||
The solution-based reaction required a longer time (48 h) and a cleaning of the polymer using a Soxhlet extractor for 24 h.180 Both procedures highlight that mechanical milling is advantageous by reducing the reaction time and the amount of cross-linking agent necessary. Regarding their properties, both polymers adsorb CO (4.37 mmol CO2 per g for CH2Cl2-based polymer and 4.74 mmol CO2 per g for CHCl3-derivative.
These values were very close to those observed for the polymers synthesized in solution (4.35 mmol and 4.71 mmol CO2 per g for CH2Cl2- and CHCl3-based polymers, respectively). Additionally, selectivity between N2/CO2 was calculated using ideal adsorption solution theory (IAST) method displaying 73.98 (90/10) for CH2Cl2 and 93.81 (90/10) for CHCl3.181 Authors demonstrated that the specific surface areas (SSABET) of CH2Cl2-based POPs depended on the number of equivalents of cross-linking agents used. A value of 1220 m2 g−1 was observed for six equivalents and 1670 m2 g−1 with 15 equivalents. This latter value was very close to SSABET observed in solution with CH2Cl2 (1685 m2 g−1). By contrast, SSABET of CHCl3-POPs remained not affected (1280 m2 g−1 for six equivalents and 1270 m2 g−1 with 15 equivalents).
Regarding the green metrics, without considering the work-up, AE values revealed the same in solution and for mechanical reactions (47.12% for CH2Cl2 and 37.01% for CHCl3). For CH2Cl2, MI was equal to 10.56 vs. 31.42 (ball milling vs. solution), MP 9.47 vs. 3.18 and E-factor 8.42 vs. 30.34. These results highlighted that mechanochemical milling was more environmentally friendly than the solution methods. Accounting for the work-up, E-factor for mechanochemistry reached 295.68, which is higher than in solution (278.27). This detriment could be compensated if the water used for the work-up in mechanochemistry could be reused. In this ideal case, E-factor would become lower for the mechanochemical process (134.87) than the procedure carried out in solution. The other green metrics (without work-up) for the CHCl3 cross-linked polymer followed the same trend. However, if work-up is included, a slight advantage was given to mechanochemistry with an E-factor of 261.89 vs. 297.94 for the solution process.
In 2014, James Mack compared Eco-scale values between mechanochemistry and the corresponding reaction in solution to answer how much greener mechanochemical reactions are. For this purpose, a supported Wittig reaction was developed, and the merits of each approach were determined (Scheme 34).182 In solution, benzyl triphenylphosphonium bromide 169 was prepared by mixing benzylbromide 168 (1 equiv.) with triphenylphosphine 167 (1.5 equiv.) in refluxing toluene for 3 h. The phosphonium salt was then isolated by filtration and then dried. Then, the phosphonium salt, benzaldehyde (1 equiv.), and NaOH (excess) were diluted in CH2Cl2/H2O (1:1), and the mixture was refluxed for 30 min. The stilbene was then recovered pure after a work-up and column chromatography in 55% yield with a 56:44 E/Z ratio. In contrast, in mechanochemistry, the preparation of stilbene was performed according to a one-pot, two-step sequence. The phosphonium salt was first prepared by combining benzyl bromide with a polymer-supported triphenylphosphine (1.64 mmol of PhCH2Br per gram of polymer) in a stainless-steel jar with a stainless-steel milling ball (5 mm ∅). The mechanical milling was then carried out on a SPEX shaker mill for 2 h. Subsequently, cesium carbonate (2.63 mmol per gram of resin), ethanol (2 mL per gram of resin), and benzaldehyde (1.54 mmol per gram of polymer) were added in the jar, and the mixture was milled for 2 h. After addition of ethyl acetate, filtration, and removal of solvent, the pure stilbene 170 was obtained in 73% yield with a 54:46 E/Z ratio.
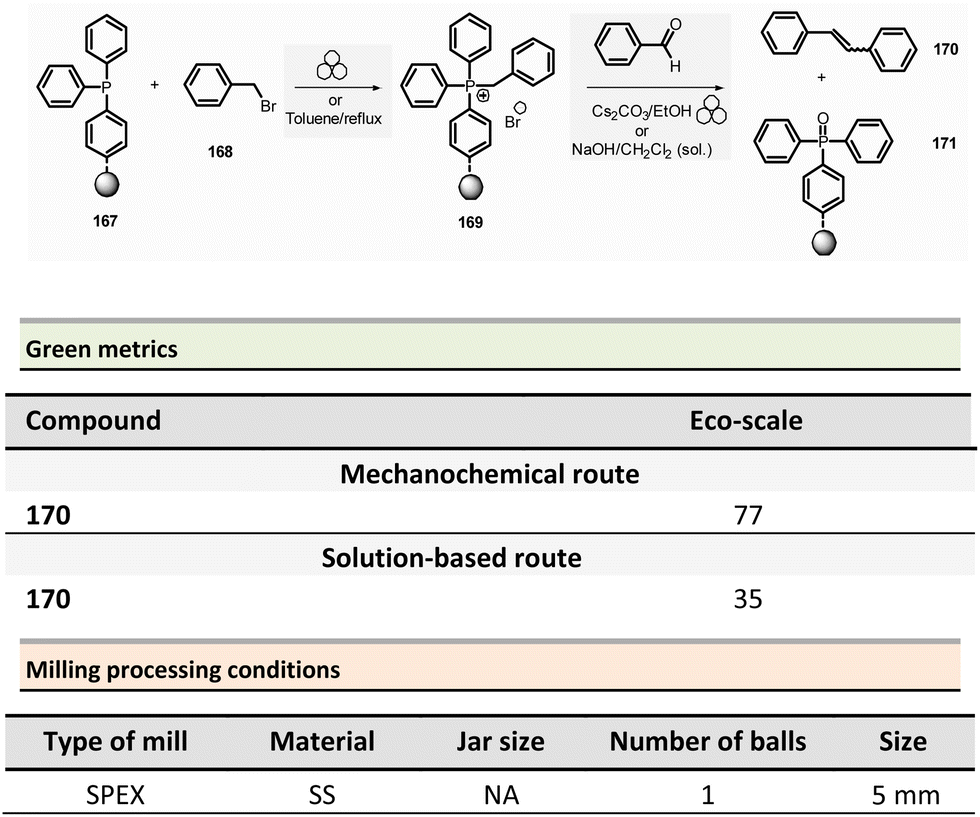 | ||
| Scheme 34 Witting reaction by mechanochemistry. | ||
The mechanochemical procedure displayed an Eco-scale of 77, which is consistent with a green reaction. By contrast, the Eco-scale for solution-based strategy was much lower (35). For the authors, the better Eco-scale score exhibited by mechanochemistry was directly linked to strong penalties of the solution-based process, i.e. a lower yield, use of less safe solvents (cyclohexane used for the chromatography) and reactants (NaOH). In addition, the work-up, specifically the purification by column chromatography, has a detrimental impact on the metrics. Another advantage in favour of mechanochemistry not considered by Eco-scale is the reaction set-up time. Mechanochemical reactions are often faster to set- up than solution counterparts. This Wittig reaction required 5 hours and 11 min for the overall procedure by mechanochemistry compared to the 7 hours and 37 min needed in solution.
Life cycle assessment in mechanochemistry
In 2022, Spatari et al. reported the first example of life cycle environmental impact for the production of the API nitrofurantoin 35a by TSE (i.e., continuous flow mechanochemistry) and compared the obtained metrics with the solvent-batch synthesis.183 In the mechanochemical process, no solvent was used, along with no excess of reagents, leading to fewer resources consumed and less waste/s produced. Many APIs are synthesized by batch processes in solution, leading to high energy consumption and consequently a high release of CO2 (from 10 to more than 1000 kg of CO2 equiv. per kg of API).184Life cycle assessment (LCA) in the chemical field can evaluate the sustainability of a reaction considering resource consumption, environmental impact, and effects on human health. Prior to the LCA for the nitrofurantoin synthesis, a life cycle inventory (LCI) was conducted for both the TSE process and solvent-batch synthesis (Scheme 35).
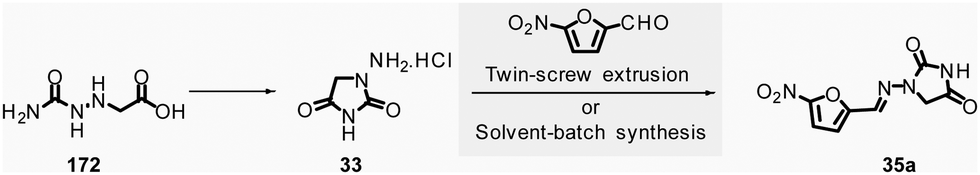 | ||
| Scheme 35 Model reaction for life cycle assessment of continuous flow mechanochemical synthesis of nitrofurantoin 35a. | ||
The LCI included the 2-semicarbazidoacetic acid 172 and the aminohydantoin hydrochloride 33 in the inventory. 5-Nitrofurfural was not considered since it is absent from the ecoinvent database,185 and the same ratio is used in both methods. Life cycle impact assessment (LCIA), like global warming, terrestrial and ecotoxicity (freshwater), ionizing radiation, human non-carcinogenic toxicity, and fossil resource scarcity, were also taken into account. The rate of synthesis by TSE is 0.23 g min−1, and the production of 1 kg of nitrofurantoin required 6.61 kW h. The unique by-product is hydrochloric acid which is trapped by a scrubber. For the production of 1 kg of nitrofurantoin, lower PMI (1.2 vs. 25), lower wastewater (<0.01 vs. 19), and lower cost (4.5$ vs. 37.6$) are obtained by the TSE process compared to the solvent-batch synthesis.186–188 The LCIA metrics followed the same trends, which are ten times smaller by TSE process than solvent-batch synthesis. We can notice a more significant energy consumption (mainly electricity) by TSE compared to the batch protocol in solution, which does not affect the life cycle score. The use of eight equivalents of 1-aminohydantoin hydrochloride in solution significantly impacts LCA. It is also noticed that toxic ammonia and hydrazine used to produce 2-semicarbazidoacetic acid 172 negatively impacts terrestrial and freshwater ecotoxicity.
Based on an annual need in the US of 4323 ± 301 kg of nitrofurantoin per year, a reduction from 2624 (solvent-batch synthesis) to 330 tons of CO2 (by TSE process) has been estimated. In addition, avoiding solvent use allows for reduces terrestrial ecotoxicity from 120140 tons of toxic emissions to 14850 tons. Ultimately, these reductions in environmental footprint also appear in terms of operating costs, from $162000 for solvent-batch synthesis to $19000 by TSE.
Industrial implementation
Since most industrial applications of mechanochemistry are protected by patents, detailed process information and their green metrics are difficult to obtain. However, the number of patents on comprising mechanochemical methodologies is rapidly increasing189 illustrating an acceleration in industrial adoption (Fig. 5).190,191 | ||
| Fig. 5 Number of patents involving mechanochemistry over time. Google patent search accessed on 19th July 2023. | ||
Conclusions and outreach
In conclusion, there is no perfect and universal parameter to assess a chemical process's overall sustainability. Combining several complementary parameters is necessary to fully embrace the complexity of this subject. In this respect and concerning mechanochemical procedures, widespread green indicators suggested it has an edge over mainstream solution-based methodologies by exhibiting a lower environmental footprint in most cases. According to the green metrics discussed throughout this review, this difference can be primarily attributed to three distinctive factors: (i) the generalized absence of bulk solvents, (ii) precise control over the stoichiometry (i.e., using agents in a stoichiometrically rather than in excess), and (iii) more selective reactions enabling simplified work-up procedures.In addition, emerging large-scale methods and tools, such as TSE, despite their sporadic use in organic synthesis, have the potential to be disruptive technology within the chemical industry.39,51,190,191 The trust of industrial chemists and chemical engineers needs to be gained to achieve the required technology readiness level for their implantation. This can only be enabled by (i) steadily growing a broader panel of mechanochemical organic and inorganic reactions, (ii) expanding the current pool of knowledge and know-how within industrially relevant fields, (iii) promoting the combination of existing well-established methodologies such as metal-catalysis, photocatalysis, etc. with ball-milling to reach even greener reactions, and (iv) increasing the awareness and training in mechanochemistry for the future generations of researchers and chemical professionals. This is not a chimaera, and it is already happening, as witnessed by the ongoing research and training activities developed within the European Programme COST Action CA18112 ‘Mechanochemistry for Sustainable Industry’192–194 All this, in turn, will create the synergy required for greater use of mechanochemistry by industries that would benefit the environment.192
We hope the key examples highlighted in this review serve as a pitstop for academic and industrial chemists to fully consider mechanochemical technologies in their reaction and/or process design (or redesign).
We believe implementing environmentally promising technologies – such as mechanochemistry, inter alia – would lead to a more sustainable future and, ultimately, the survival of our specie.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This article is based upon work from COST Action CA18112 Mechanochemistry for Sustainable Industry,51,192–194 supported by COST (European Cooperation in Science and Technology). COST (European Cooperation in Science and Technology) is a funding agency for research and innovation networks. Our Actions help connect research initiatives across Europe and enable scientists to grow their ideas by sharing them with their peers. This boosts their research, career and innovation (https://www.cost.eu). E. C., N. F. and D. V. are grateful to Région Occitanie (France) for the Pre-Maturation 2020 – MECH-API grant (ESR_PRE-MAT – 00262). E. C. is grateful to Campus France and French-Estonian cooperation Programme Hubert Curien France-Estonia (PHC PARROT 2021-2023). E. C. is grateful to European Union's Horizon 2021–2027 Research and Innovation Programme for the generous funding under grant agreement no. 101057286 (IMPACTIVE).195 F. G. would like to thank the support of Fundación para el Fomento en Asturias de la Investigación Científica Aplicada y la Tecnología (FICYT) through the Margarita Salas Senior Program (AYUD/2021/59709) and the Ministerio de Ciencia e Innovación through the project PID2021-127407NB-I00. A. P. would like to thank the support of MIUR Italy, PRIN 2017 project (grant number: 2017B7MMJ5_001) “MultIFunctional poLymer cOmposites based on groWn matERials (MIFLOWER) and Fondazione di Sardegna (FdS, F72F20000230007).Notes and references
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, Oxford, 1998 Search PubMed.
- W. C. Clark and A. G. Harley, Annu. Rev. Environ. Resour., 2020, 45, 331–386 CrossRef.
- B. Trost, Science, 1991, 254, 1471–1477 CrossRef CAS PubMed.
- W. Wang, J. Lü, L. Zhang and Z. Li, Front. Chem. Sci. Eng., 2011, 5, 349–354 CrossRef.
- R. Sheldon, A. I. Arends and U. Hanefeld, Green Chem. and Catalysis, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed.
- C. Jimenez-Gonzalez, C. S. Ponder, Q. B. Broxterman and J. B. Manley, Org. Process Res. Dev., 2011, 15, 912–917 CrossRef CAS.
- A. D. Curzons, D. N. Mortimer, D. J. C. Constable and V. L. Cunningham, Green Chem., 2001, 3, 1–6 RSC.
- F. G. Calvo-Flores, ChemSusChem, 2009, 2, 905–919 CrossRef CAS PubMed.
- J. Andraos, Org. Process Res. Dev., 2005, 9, 149–163 CrossRef CAS.
- J. Andraos and M. Sayed, J. Chem. Educ., 2007, 84, 1004–1010 CrossRef CAS.
- D. J. C. Constable, A. D. Curzons and V. L. Cunningham, Green Chem., 2002, 4, 521–527 RSC.
- D. Kong, M. M. Amer and C. Bolm, Green Chem., 2022, 24, 3125–3129 RSC.
- K. Van Aken, L. Strekowski and L. Patiny, Beilstein J. Org. Chem., 2006, 2, 1–7 Search PubMed.
- D. Kralisch, D. Ott and D. Gericke, Green Chem., 2015, 17, 123–145 RSC.
- A. G. Parvatker and M. J. Eckelman, ACS Sustainable Chem. Eng., 2019, 7, 350–367 CrossRef CAS.
- A. DeVierno Kreuder, T. House-Knight, J. Whitford, E. Ponnusamy, P. Miller, N. Jesse, R. Rodenborn, S. Sayag, M. Gebel, I. Aped, I. Sharfstein, E. Manaster, I. Ergaz, A. Harris and L. Nelowet Grice, ACS Sustainable Chem. Eng., 2017, 5, 2927–2935 CrossRef CAS.
- F. Gomollón-Bel, Chem. Int., 2019, 41, 12–17 Search PubMed.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- M. Pérez-Venegas and E. Juaristi, ACS Sustainable Chem. Eng., 2020, 8, 8881–8893 CrossRef.
- G. Yashwantrao, V. P. Jejurkar, R. Kshatriya and S. Saha, ACS Sustainable Chem. Eng., 2019, 7, 13551–13558 CrossRef CAS.
- C. Gomes, C. S. Vinagreiro, L. Damas, G. Aquino, J. Quaresma, C. Chaves, J. Pimenta, J. Campos, M. Pereira and M. Pineiro, ACS Omega, 2020, 5, 10868–10877 CrossRef CAS PubMed.
- D. Tan and F. García, Chem. Soc. Rev., 2019, 48, 2274–2292 RSC.
- J. F. Reynes, V. Isoni and F. García, Angew. Chem., Int. Ed., 2023, 202300819 Search PubMed.
- F. Effaty, L. Gonnet, S. G. Koenig, K. Nagapudi, X. Ottenwaelder and T. Friščić, Chem. Commun., 2023, 59, 1010–1013 RSC.
- G. Báti, D. Csókás, T. Yong, S. M. Tam, R. R. S. Shi, R. D. Webster, I. Pápai, F. García and M. C. Stuparu, Angew. Chem., Int. Ed., 2020, 59, 21620–21626 CrossRef PubMed.
- J. Andersen, J. Brunemann and J. Mack, React. Chem. Eng., 2019, 4, 1229–1236 RSC.
- R. R. A. Bolt, S. E. Raby-Buck, K. Ingram, J. A. Leitch and D. L. Browne, Angew. Chem., Int. Ed., 2022, 61, 202210508 CrossRef PubMed.
- N. Cindro, M. Tireli, B. Karadeniz, T. Mrla and K. Užarević, ACS Sustainable Chem. Eng., 2019, 7, 16301–16309 CrossRef CAS.
- J. Mack and M. Shumba, Green Chem., 2007, 9, 328–330 RSC.
- J. F. Reynes and F. Garcia, Angew. Chem., Int. Ed., 2023, 62, 202215094 CrossRef PubMed.
- A. A. L. Michalchuk, The Thermodynamics and Kinetics of Mechanochemical Reactions – An Experimental Approach, in Mechanochemistry and Emerging Technologies for Sustainable Chemical Manufacturing, ed. E. Colacino and F. Garcia, CRC Press, 2023, ch. 3, pp. 59–91 DOI:10.1201/9781003178187.
- For kinetics of mechanochemical reaction, please read: M. Carta, L. Vugrin, G. Miletić, M. Juribašić Kulcsar, P. C. Ricci, I. Halasz and F. Delogu, Angew. Chem., Int. Ed., 2023, e202308046 CAS , ahead of print.
- V. Martinez, T. Stolar, B. Karadeniz, I. Brekalo and K. Uzarevic, Nat. Rev. Chem., 2023, 7, 51–65 CrossRef CAS PubMed.
- D. Hasa and W. Jones, Adv. Drug Delivery Rev., 2017, 117, 147–161 CrossRef CAS PubMed.
- C. Wang, C. Yue, A. Smith and J. Mack, J. Organomet. Chem., 2022, 976, 122430 CrossRef CAS.
- B. P. Hutchings, D. E. Crawford, L. Gao, P. Hu and S. L. James, Angew. Chem., Int. Ed., 2017, 56, 15252–15256 CrossRef CAS PubMed.
- J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS PubMed.
- N. R. Rightmire and T. P. Hanusa, Dalton Trans., 2016, 45, 2352–2362 RSC.
- E. Colacino, V. Isoni, D. Crawford and F. García, Trends Chem., 2021, 3, 335–339 CrossRef CAS.
- R. R. A. Bolt, J. A. Leitch, A. C. Jones, W. I. Nicholson and D. L. Browne, Chem. Soc. Rev., 2022, 51, 4243–4260 RSC.
- K. J. Ardila-Fierro and J. G. Hernández, ChemSusChem, 2021, 14, 2145–2162 CrossRef CAS PubMed.
- F. Roschangar and J. Colberg, in Green Techniques for Organic Synthesis and Medicinal Chemistry, ed. W. Zhang and B. W. Cue, John Wiley & Sons, Ltd, Chichester, UK, 2018, pp. 1–19 Search PubMed.
- For a collection of articles on environmental metrics, please see: https://www.sciencedirect.com/journal/current-opinion-in-green-and-sustainable-chemistry/special-issue/108G6G89CDK, accessed december 16, 2022.
- J. Andersen and J. Mack, Green Chem., 2018, 20, 1435–1443 RSC.
- D. Tan and T. Friščić, Eur. J. Org. Chem., 2018, 18–33 CrossRef CAS.
- F. Cuccu, L. Luca, F. Delogu, E. Colacino, N. Solin, R. Mocci and A. Porcheddu, ChemSusChem, 2022, 15, 202200362 CrossRef PubMed.
- D. Virieux, F. Delogu, A. Porcheddu, F. García and E. Colacino, J. Org. Chem., 2021, 86, 13885–13894 CrossRef CAS PubMed.
- J. G. Hernández and C. Bolm, J. Org. Chem., 2017, 82, 4007–4019 CrossRef PubMed.
- A. Porcheddu, E. Colacino, L. Luca and F. Delogu, ACS Catal., 2020, 10, 8344–8394 CrossRef CAS.
- S. Hwang, S. Grätz and L. Borchardt, Chem. Commun., 2022, 58, 1661–1671 RSC.
- X.-Z. Lim, Chem. Eng. News, 2020, 98 Search PubMed , online issue. https://cen.acs.org/pharmaceuticals/process-chemistry/Interlocking-screws-crank-pharmaceuticals/98/i38?utm_source=Synthesis&utm_medium=Synthesis&utm_campaign=CENRS, accessed December 16, 2022.
- F. Leon and F. Garcia, in Comprehensive Coordination Chemistry, ed. E. C. Constable, G. Parkin and L. Que, Jr., Elsevier, Amsterdam, 3rd edn, 2021, vol. 9, ch. 9.19, pp. 620–679 Search PubMed.
- A. A. Gečiauskaitė and F. García, Beilstein J. Org. Chem., 2017, 13, 2068–2077 CrossRef PubMed.
- T. Stolar and K. Užarević, CrystEngComm, 2020, 22, 4511–4525 RSC.
- T. Friščić and W. Jones, Cryst. Growth Des., 2009, 9, 1621–1637 CrossRef.
- J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337–2347 RSC.
- A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, J. Comb. Chem., 1999, 1, 55–68 CrossRef CAS PubMed.
- P. Ertl, E. Altmann and J. M. McKenna, J. Med. Chem., 2020, 63, 8408–8418 CrossRef CAS PubMed.
- M. C. Bryan, C. Dalton, A. Díaz-Rodríguez, J. Doerfler, O. D. Engl, P. Ferguson, A. G. Molina, Z. S. Han, J. Hosford, G. P. Howell, M. Hutchby, W. Li, R. H. Munday, A. Navarro, M. Parmentier, J. Pawlas, P. F. Richardson, W. J. I. Smith, A. Steven, B. S. Takale, J. A. Terrett, D. S. Treitler and M. Zeng, Org. Process Res. Dev., 2022, 26, 251–262 CrossRef CAS.
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer, Jr, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411–420 RSC.
- M. C. Bryan, P. J. Dunn, D. Entwistle, F. Gallou, S. G. Koenig, J. D. Hayler, M. R. Hickey, S. Hughes, M. E. Kopach, G. Moine, P. Richardson, F. Roschangar, A. Steven and F. J. Weiberth, Green Chem., 2018, 20, 5082–5103 RSC.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- R. M. de Figueiredo, J.-S. Suppo and J.-M. Campagne, Chem. Rev., 2016, 116, 12029–12122 CrossRef CAS PubMed.
- L. Konnert, A. Gauliard, F. Lamaty, J. Martinez and E. Colacino, ACS Sustainable Chem. Eng., 2013, 1, 1186–1191 CrossRef CAS.
- T. Dalidovich, K. A. Mishra, T. Shalima, M. Kudrjašova, D. G. Kananovich and R. Aav, ACS Sustainable Chem. Eng., 2020, 8, 15703–15715 CrossRef CAS.
- A. El-Faham and F. Albericio, J. Pept. Sci., 2010, 16, 6–9 CrossRef CAS PubMed.
- L. Konnert, F. Lamaty, J. Martinez and E. Colacino, Chem. Rev., 2017, 117, 13757–13809 CrossRef CAS PubMed.
- A. Porcheddu, F. Delogu, L. De Luca and E. Colacino, ACS Sustainable Chem. Eng., 2019, 7, 12044–12051 CAS.
- D. Virieux, F. Delogu, A. Porcheddu, F. García and E. Colacino, J. Org. Chem., 2021, 86, 13885–13894 CrossRef CAS PubMed.
- L. Konnert, M. Dimassi, L. Gonnet, F. Lamaty, J. Martinez and E. Colacino, RSC Adv., 2016, 6, 36978–36986 RSC.
- R. Mocci, L. D. Luca, F. Delogu and A. Porcheddu, Adv. Synth. Catal., 2016, 358, 3135–3144 CrossRef CAS.
- L. Mistry, K. Mapesa, T. W. Bousfield and J. E. Camp, Green Chem., 2017, 19, 2123–2128 RSC.
- Y. Chen, L. Su, X. Yang, W. Pan and H. Fang, Tetrahedron, 2015, 71, 9234–9239 CrossRef CAS.
- K. H. Dudley and D. L. Bius, J. Heterocycl. Chem., 1973, 10, 173–180 CrossRef CAS.
- R. Mocci, E. Colacino, L. D. Luca, C. Fattuoni, A. Porcheddu and F. Delogu, ACS Sustainable Chem. Eng., 2021, 9, 2100–2114 CrossRef CAS.
- H.-J. Pi, J.-D. Dong, N. An, W. Du and W.-P. Deng, Tetrahedron, 2009, 65, 7790–7793 CrossRef CAS.
- Y. Gao, J. Liu, Z. Li, T. Guo, S. Xu, H. Zhu, F. Wei, S. Chen, H. Gebru and K. Guo, J. Org. Chem., 2018, 83, 2040–2049 CrossRef CAS PubMed.
- T.-X. Métro, J. Bonnamour, T. Reidon, J. Sarpoulet, J. Martinez, F. Lamaty, T.-X. Métro, J. Bonnamour, T. Reidon, A. Duprez, J. Sarpoulet, J. Martinez and F. Lamaty, Chem. Commun., 2012, 48, 11781–11783 RSC.
- T.-X. Métro, J. Bonnamour, T. Reidon, A. Duprez, J. Sarpoulet, J. Martinez and F. Lamaty, Chem. – Eur. J., 2015, 21, 12787–12796 CrossRef PubMed.
- J. C. Sheehan and G. P. Hess, J. Am. Chem. Soc., 1955, 77, 1067–1068 CrossRef CAS.
- G. Chattopadhyay, S. Chakraborty and C. Saha, Synth. Commun., 2008, 38, 4068–4075 CrossRef CAS.
- G. W. Anderson, J. Am. Chem. Soc., 1960, 82, 4596–4600 CrossRef.
- P. S. Chaudhari, S. D. Salim, R. V. Sawant and K. G. Akamanchi, Green Chem., 2010, 12, 1707–1710 RSC.
- W. I. Nicholson, F. Barreteau, J. A. Leitch, R. Payne, I. Priestley, E. Godineau, C. Battilocchio and D. L. Browne, Angew. Chem., Int. Ed., 2021, 60, 21868–21874 CrossRef CAS PubMed.
- B. R. Kim, H.-G. Lee, S.-B. Kang, G. H. Sung, J.-J. Kim, J. K. Park, S.-G. Lee and Y.-J. Yoon, Synthesis, 2012, 42–50 CAS.
- C. Herrlé, S. Toumieux, M. Araujo, A. Peru, F. Allais and A. Wadouachi, Green Chem., 2022, 24, 5856–5861 RSC.
- L. Gonnet, T. Tintillier, N. Venturini, L. Konnert, J.-F. Hernandez, F. Lamaty, G. Laconde, J. Martinez and E. Colacino, ACS Sustainable Chem. Eng., 2017, 5, 2936–2941 CrossRef CAS.
- A. R. Katritzky, A. Singh, D. N. Haase and M. Yoshioka, ARKIVOC, 2008, 2009, 47–56 Search PubMed.
- E. Colacino, A. Porcheddu, I. Halasz, C. Charnay, F. Delogu, R. Guerra and J. Fullenwarth, Green Chem., 2018, 20, 2973–2977 RSC.
- T. Hosoya, H. Aoyama, T. Ikemoto, Y. Kihara, T. Hiramatsu, M. Endo and M. Suzuki, Bioorg. Med. Chem., 2003, 11, 663–673 CrossRef CAS PubMed.
- D. C. Fabry, Y. A. Ho, R. Zapf, W. Tremel, M. Panthöfer, M. Rueping and T. H. Rehm, Green Chem., 2017, 19, 1911–1918 RSC.
- X. Li and F. Zhang, Method for Preparing And Refining Nitrofurantoin, Chinese Pat., CN101450941 2009 Search PubMed.
- P. Sharma, C. Vetter, E. Ponnusamy and E. Colacino, ACS Sustainable Chem. Eng., 2022, 10, 5110–5116 CrossRef CAS.
- D. E. Crawford, A. Porcheddu, A. S. McCalmont, F. Delogu, S. L. James and E. Colacino, ACS Sustainable Chem. Eng., 2020, 8, 12230–12238 CrossRef CAS.
- M. Baumann, T. S. Moody, M. Smyth and S. Wharry, Org. Process Res. Dev., 2020, 24, 1802–1813 CrossRef CAS.
- W. Pickhardt, M. Wohlgemuth, S. Grätz and L. Borchardt, J. Org. Chem., 2021, 86, 14011–14015 CrossRef CAS PubMed.
- B. Kurpil, A. Savateev, V. Papaefthimiou, S. Zafeiratos, T. Heil, S. Özenler, D. Dontsova and M. Antonietti, Appl. Catal., B, 2017, 217, 622–628 CrossRef CAS.
- R. Walczak, B. Kurpil, A. Savateev, T. Heil, J. Schmidt, Q. Qin, M. Antonietti and M. Oschatz, Angew. Chem., Int. Ed., 2018, 57, 10765–10770 CrossRef CAS PubMed.
- M. Ferguson, N. Giri, X. Huang, D. Apperley and S. L. James, Green Chem., 2014, 16, 1374–1382 RSC.
- D. Crawford, J. Casaban, R. Haydon, N. Giri, T. McNally and S. L. James, Chem. Sci., 2015, 6, 1645–1649 RSC.
- P. Milbeo, F. Quintin, L. Moulat, C. Didierjean, J. Martinez, X. Bantreil, M. Calmès and F. Lamaty, Tetrahedron Lett., 2021, 63, 152706 CrossRef CAS.
- D. E. Crawford, C. K. Miskimmin, J. Cahir and S. L. James, Chem. Commun., 2017, 53, 13067–13070 RSC.
- L. Leoni, A. Carletta, L. Fusaro, J. Dubois, N. A. Tumanov, C. Aprile, J. Wouters and A. Dalla Cort, Molecules, 2019, 24, 2314–2324 CrossRef CAS PubMed.
- V. K. Singh, A. Chamberlain-Clay, H. C. Ong, F. León, G. Hum, M. Y. Par, P. Daley-Dee and F. García, ACS Sustainable Chem. Eng., 2021, 9, 1152–1160 CrossRef CAS.
- For electric power monthly price please see: https://www.eia.gov/electricity/monthly/epm_table_grapher.php?t=epmt_5_6_a, accessed december 16, 2022.
- F. Leon, C. Li, J. F. Reynes, V. K. Singh, X. Lian, H. C. Ong, G. Hum, H. Sun and F. García, Faraday Discuss., 2022 10.1039/D2FD00117A.
- A. Dömling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083–3135 CrossRef PubMed.
- B. H. Rotstein, S. Zaretsky, V. Rai and A. K. Yudin, Chem. Rev., 2014, 114, 8323–8359 CrossRef CAS PubMed.
- S. E. John, S. Gulati and N. Shankaraiah, Org. Chem. Front., 2021, 8, 4237–4287 RSC.
- M. S. Singh and S. Chowdhury, RSC Adv., 2012, 2, 4547–4592 RSC.
- S. Kar, H. Sanderson, K. Roy, E. Benfenati and J. Leszczynski, Chem. Rev., 2022, 122, 3637–3710 CrossRef CAS PubMed.
- M. Leonardi, M. Villacampa and J. C. Menéndez, Chem. Sci., 2018, 9, 2042–2064 RSC.
- C. Fiore, I. Sovic, S. Lukin, I. Halasz, K. Martina, F. Delogu, P. C. Ricci, A. Porcheddu, O. Shemchuk, D. Braga, J.-L. Pirat, D. Virieux and E. Colacino, ACS Sustainable Chem. Eng., 2020, 8, 18889–18902 CrossRef CAS.
- K. Martina, L. Rotolo, A. Porcheddu, F. Delogu, S. R. Bysouth, G. Cravotto and E. Colacino, Chem. Commun., 2018, 54, 551–554 RSC.
- C. Blazquez-Barbadillo, J. F. González, A. Porcheddu, D. Virieux, J. C. Menéndez and E. Colacino, Green Chem. Lett. Rev., 2022, 15, 881–892 CrossRef CAS.
- A. D. Sharapov, R. F. Fatykhov, I. A. Khalymbadzha, V. V. Sharutin, S. Santra, G. V. Zyryanov, O. N. Chupakhin and B. C. Ranu, Green Chem., 2022, 24, 2429–2437 RSC.
- H. Von Pechmann and C. Duisberg, Ber. Dtsch. Chem. Ges., 1883, 16, 2119–2128 CrossRef.
- T. Sugino and K. Tanaka, Chem. Lett., 2001, 110–111 CrossRef CAS.
- A. Porcheddu, R. Mocci, M. Brindisi, F. Cuccu, C. Fattuoni, F. Delogu, E. Colacino and M. V. D’Auria, Green Chem., 2022, 24, 4859–4869 RSC.
- D. F. Taber and P. K. Tirunahari, Tetrahedron, 2011, 67, 7195–7210 CrossRef CAS PubMed.
- Y.-K. Lim and C.-G. Cho, Tetrahedron Lett., 2004, 45, 1857–1859 CrossRef CAS.
- D.-Q. Xu, J. Wu, S.-P. Luo, J.-X. Zhang, J.-Y. Wu, X.-H. Du and Z.-Y. Xu, Green Chem., 2009, 11, 1239–1246 RSC.
- S. P. M. Ventura, A. M. M. Gonçalves, T. Sintra, J. L. Pereira, F. Gonçalves and J. A. P. Coutinho, Ecotoxicology, 2013, 22, 1–12 CrossRef CAS PubMed.
- T. P. Thuy Pham, C.-W. Cho and Y.-S. Yun, Water Res., 2010, 44, 352–372 CrossRef PubMed.
- A. Dhakshinamoorthy and K. Pitchumani, Appl. Catal., A, 2005, 292, 305–311 CrossRef CAS.
- K. G. Liu, J. R. Lo and A. J. Robichaud, Tetrahedron, 2010, 66, 573–577 CrossRef CAS.
- B. F. O. Nascimento, M. Pineiro, A. M. d’A, M. R. Rocha Gonsalves, M. Ramos Silva, A. Matos Beja and J. A. Paixão, J. Porphyrins Phthalocyanines, 2007, 11, 77–84 CrossRef CAS.
- C. Gomes, M. Peixoto and M. Pineiro, J. Porphyrins Phthalocyanines, 2019, 23, 889–897 CrossRef CAS.
- C. Gomes, M. Peixoto and M. Pineiro, Molecules, 2021, 26, 6652–6665 CrossRef CAS PubMed.
- D. R. Palleros, J. Chem. Educ., 2004, 81, 1345–1347 CrossRef CAS.
- Z. Shan, X. Luo, L. Hu and X. Hu, Sci. China: Chem., 2010, 53, 1095–1101 CrossRef CAS.
- N. M. Rateb and H. F. Zohdi, Synth. Commun., 2009, 39, 2789–2794 CrossRef CAS.
- M. Ould M’hamed, A. G. Alshammari and O. M. Lemine, Appl. Sci., 2016, 6, 431–436 CrossRef.
- A. K. Bose, S. Pednekar, S. N. Ganguly, G. Chakraborty and M. S. Manhas, Tetrahedron Lett., 2004, 45, 8351–8353 CrossRef CAS.
- Z.-T. Wang, L.-W. Xu, C.-G. Xia and H.-Q. Wang, Tetrahedron Lett., 2004, 45, 7951–7953 CrossRef CAS.
- B. Liang, X. Wang, J.-X. Wang and Z. Du, Tetrahedron, 2007, 63, 1981–1986 CrossRef CAS.
- G. Sabitha, K. B. Reddy, R. Srinivas and J. S. Yadav, Helv. Chim. Acta, 2005, 88, 2996–2999 CrossRef CAS.
- X. Zhu, Z. Li, C. Jin, L. Xu, Q. Wu and W. Su, Green Chem., 2009, 11, 163–165 RSC.
- C. A. Henriques, S. M. A. Pinto, G. L. B. Aquino, M. Pineiro, M. J. F. Calvete and M. M. Pereira, ChemSusChem, 2014, 7, 2821–2824 CrossRef CAS PubMed.
- T. Raj, H. Sharma, Mayank, A. Singh, T. Aree, N. Kaur, N. Singh and D. O. Jang, ACS Sustainable Chem. Eng., 2017, 5, 1468–1475 CrossRef CAS.
- A. Shaabani, A. Rahmati and S. Naderi, Bioorg. Med. Chem., 2005, 15, 5553–5557 CrossRef CAS PubMed.
- G. B. D. Rao, B. N. Acharya, S. K. Verma and M. P. Kaushik, Tetrahedron Lett., 2011, 52, 809–812 CrossRef CAS.
- P. K. Sahu, P. K. Sahu and D. D. Agarwal, RSC Adv., 2013, 3, 9854–9864 RSC.
- T. Raj, P. Saluja and N. Singh, Sens. Actuators, B, 2015, 206, 98–106 CrossRef CAS.
- J. Liu, M. Lei and L. Hu, Green Chem., 2012, 14, 840–846 RSC.
- L. Gonnet, C. André-Barrès, B. Guidetti, A. Chamayou, C. Menendez, M. Baron, R. Calvet and M. Baltas, ACS Sustainable Chem. Eng., 2020, 8, 3114–3125 CrossRef CAS.
- A. S. Hussen, A. P. Pandey and A. Sharma, ChemistrySelect, 2018, 3, 11505–11509 CrossRef CAS.
- D. Kong, D. Ma, P. Wu and C. Bolm, ACS Sustainable Chem. Eng., 2022, 10, 2863–2867 CrossRef CAS.
- N. Lu, Z. Zhang, N. Ma, C. Wu, G. Zhang, Q. Liu and T. Liu, Org. Lett., 2018, 20, 4318–4322 CrossRef CAS PubMed.
- P. van Bonn, D. Dreßler, F. Weitenhagen and C. Bolm, ACS Sustainable Chem. Eng., 2022, 10, 1361–1366 CrossRef CAS.
- M. Caporaso, G. Cravotto, S. Georgakopoulos, G. Heropoulos, K. Martina and S. Tagliapietra, Beilstein J. Org. Chem., 2014, 10, 1454–1461 CrossRef PubMed.
- R. V. Jagadeesh, H. Junge, M.-M. Pohl, J. Radnik, A. Brückner and M. Beller, J. Am. Chem. Soc., 2013, 135, 10776–10782 CrossRef CAS PubMed.
- J. Wang, F. Jiang, C. Tao, H. Yu, L. Ruhlmann and Y. Wei, Green Chem., 2021, 23, 2652–2657 RSC.
- S. Gowrisankar, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 5139–5143 CrossRef CAS PubMed.
- C. Liu, J. Wang, L. Meng, Y. Deng, Y. Li and A. Lei, Angew. Chem., Int. Ed., 2011, 50, 5144–5148 CrossRef CAS PubMed.
- X. Cui, X. Hao and F. Guo, Dalton Trans., 2022, 51, 4377–4385 RSC.
- A. Lucchesi Schio, M. R. Farias Soares, G. Machado and T. Barcellos, ACS Sustainable Chem. Eng., 2021, 9, 9661–9670 CrossRef CAS.
- B. Ameri, S. S. H. Davarani, R. Roshani, H. R. Moazami and A. Tadjarodi, J. Alloys Compd., 2017, 695, 114–123 CrossRef CAS.
- A. Tadjarodi and R. Roshani, Curr. Chem. Lett., 2014, 3, 215–220 CrossRef CAS.
- S. Moniri Javadhesari, S. Alipour, S. Mohammadnejad and M. R. Akbarpour, Mater. Sci. Eng., C, 2019, 105, 110011 CrossRef CAS PubMed.
- H. Ayask, J. Khaki and M. Haddad Sabzevar, JUFGNSM, 2015, 48, 37–44 Search PubMed.
- Udayabhanu, P. C. Nethravathi, M. A. Pavan Kumar, D. Suresh, K. Lingaraju, H. Rajanaika, H. Nagabhushana and S. C. Sharma, Mater. Sci. Semicond. Process., 2015, 33, 81–88 CrossRef CAS.
- M. Mayank, A. Singh, P. Raj, R. Kaur, A. Singh, N. Kaur and N. Singh, New J. Chem., 2017, 41, 3872–3881 RSC.
- D. Završnik, S. Muratović, D. Makuc, J. Plavec, M. Cetina, A. Nagl, E. D. Clercq, J. Balzarini and M. Mintas, Molecules, 2011, 16, 6023–6040 CrossRef PubMed.
- A. R. Karimi, M. Karimi and Z. Dalirnasab, New York.
- C.-X. Su, J.-F. Mouscadet, C.-C. Chiang, H.-J. Tsai and L.-Y. Hsu, Chem. Pharm. Bull., 2006, 54, 682–686 CrossRef CAS PubMed.
- K. Yoo, E. J. Hong, T. Q. Huynh, B.-S. Kim and J. G. Kim, ACS Sustainable Chem. Eng., 2021, 9, 8679–8685 CrossRef CAS.
- N. Cindro, M. Tireli, B. Karadeniz, T. Mrla and K. Užarević, ACS Sustainable Chem. Eng., 2019, 7, 16301–16309 CrossRef CAS.
- M. Tireli, M. Juribašić Kulcsár, N. Cindro, D. Gracin, N. Biliškov, M. Borovina, M. Ćurić, I. Halasz and K. Užarević, Chem. Commun., 2015, 51, 8058–8061 RSC.
- M. Piquero, C. Font, N. Gullón, P. López-Alvarado and J. C. Menéndez, ChemSusChem, 2021, 14, 4764–4775 CrossRef CAS PubMed.
- M. C. Pirrung, K. Park and Z. Li, Org. Lett., 2001, 3, 365–367 CrossRef CAS PubMed.
- M. C. Pirrung, L. Deng, Z. Li and K. Park, J. Org. Chem., 2002, 67, 8374–8388 CrossRef CAS PubMed.
- E. de Oliveira Lima Filho and I. Malvestiti, ACS Omega, 2020, 5, 33329–33339 CrossRef CAS PubMed.
- T. B. Mete, T. M. Khopade and R. G. Bhat, Tetrahedron Lett., 2017, 58, 415–418 CrossRef CAS.
- X. Yang, H. Wang, Y. Zhang, W. Su and J. Yu, Green Chem., 2022, 24, 4557–4565 RSC.
- K. C. C. Aganda, J. Kim and A. Lee, Org. Biomol. Chem., 2019, 17, 9698–9702 RSC.
- X. Li, J. Du, Y. Zhang, H. Chang, W. Gao and W. Wei, Org. Biomol. Chem., 2019, 17, 3048–3055 RSC.
- In the original work, these values were calculated taking into account the molecular weight of solvents (usually not accounted in the calculation of the atom economy, AE) and the number of equivalents of reactants.
- A. Krusenbaum, J. Geisler, F. J. L. Kraus, S. Grätz, M. V. Höfler, T. Gutmann and L. Borchardt, J. Polym. Sci., 2022, 60, 62–71 CrossRef CAS.
- V. Rozyyev, Y. Hong, M. S. Yavuz, D. Thirion and C. T. Yavuz, Adv. Energy Sustainability Res., 2021, 2, 2100064 CrossRef CAS.
- C. M. Simon, B. Smit and M. Haranczyk, Comput. Phys. Commun., 2016, 200, 364–380 CrossRef CAS.
- K. Leahy, A. M. Mack and J. Mack, ACS Symp. Ser., 2014, 1186, 129–137 CrossRef CAS.
- O. Galant, G. Cerfeda, A. S. McCalmont, S. L. James, A. Porcheddu, F. Delogu, D. E. Crawford, E. Colacino and S. Spatari, ACS Sustainable Chem. Eng., 2022, 10, 1430–1439 CrossRef CAS.
- A. G. Parvatker, H. Tunceroglu, J. D. Sherman, P. Coish, P. Anastas, J. B. Zimmerman and M. J. Eckelman, ACS Sustainable Chem. Eng., 2019, 7, 6580–6591 CrossRef CAS.
- G. Wernet, C. Bauer, B. Steubing, J. Reinhard, E. Moreno-Ruiz and B. Weidema, Int. J. Life Cycle Assess., 2016, 21, 1218–1230 CrossRef.
- F. Shuai, X. Wang and J. Zhang, Improved Method for Synthesis and Purification of Nitrofurantoin, Chinese Pat., CN108069944, 2016 Search PubMed.
- D. Jack, J. Pharm. Pharmacol., 1959, 11, 108T–114T CrossRef CAS.
- J. H. Coleman, W. Hayden, M. Donough and C. J. O’Keefe, Process of Preparing 2-Semicarbazidoacetic Acid, U.S. Pat., US2779786, 1957 Search PubMed.
- T. Bendele, Intellectual Property (IP) Strategy in Mechanochemistry, in Mechanochemistry and Emerging Technologies for Sustainable Chemical Manufacturing, ed. E. Colacino and F. Garcia, CRC Press, 2023, ch. 5, pp. 105–135 DOI:10.1201/9781003178187.
- F. Gomollón-Bel, Chem. Eng. News, 2022, 100, 21–22 Search PubMed.
- F. Gomollón-Bel, ACS Cent. Sci., 2022, 8, 1474–1476 CrossRef PubMed.
- For more information on COST Action CA18112 ‘Mechanochemistry for Sustainable Industry’: https://www.mechsustind.eu/, accessed November 21, 2022.
- J. G. Hernández, I. Halasz, D. E. Crawford, M. Krupicka, M. Baláž, V. André, L. Vella-Zarb, A. Niidu, F. García, L. Maini and E. Colacino, Eur. J. Org. Chem., 2020, 8–9 CrossRef.
- M. Baláž, L. Vella-Zarb, J. G. Hernández, I. Halasz, D. E. Crawford, M. Krupička, V. André, A. Niidu, F. García, L. Maini and E. Colacino, Mechanochemistry: a disruptive innovation for the industry of the future, Chem. Today, 2019, 37, 32–34 Search PubMed . https://www.teknoscienze.com/tks_article/mechanochemistry-a-disruptive-innovation-for-the-industry-of-the-future/.
- https://www.mechanochemistry.eu .
| This journal is © The Royal Society of Chemistry 2023 |