
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aminoalcohol derivatives by nickel-catalyzed enantioselective coupling of imines and dienol ethers†
Jae Yeon
Kim‡
 ,
Thomas Q.
Davies‡
and
Alois
Fürstner
*
,
Thomas Q.
Davies‡
and
Alois
Fürstner
*
Max-Planck-Institut für Kohlenforschung, 45470 Mülheim/Ruhr, Germany. E-mail: fuerstner@kofo.mpg.de
First published on 28th September 2023
Abstract
The reductive coupling of dienol ethers with N-tosylimines catalyzed by Ni(0) in the presence of a VAPOL-derived phosphoramidite ligand follows an unprecedented regiochemical course; it furnishes syn-configured 1,2-aminoalcohol derivatives in good chemical yields with up to 94% ee.
Our group has recently reported that the nickel-catalyzed reductive coupling of aldehydes with dienol ethers, when carried out in the presence of the VAPOL-derived phosphoramidite ligand L1, takes an unprecedented regiochemical course.1–4 Whereas classical “Tamaru–Mori”-type reactions are distinguished by C–C-bond formation at the diene terminus leading to products such as rac-2,5–9 Ni(0)/L1 compels the electron-rich diene 1 to react at the least nucleophilic and arguably most hindered C-atom carrying the oxygen substituent to give the mono-protected 1,2-diol derivative 3 in good yield and appreciable optical purity (Scheme 1).1 Shortly thereafter, we showed that this outcome is no singularity; rather, electron-deficient sorbate esters 4 (and related dienes) also follow an “inverse” course relative to all literature precedent5,10 in that they furnish compounds such as 6 rather than aldol-type products of type rac-5.2 In addition to the unorthodox connectivity pattern manifested in 6,11 the high level of enantioselectivity is noteworthy, since asymmetric Tamaru-type reactions in general were even recently called a “largely unresolved challenge”;12 indeed, only a few examples were known prior to our work.10,13–16 Although mechanistic studies17 into these transformations proved exceptionally challenging and the reasons for the observed reversal enforced by the ligand L1 therefore remain elusive, a more systematic exploration of this unorthodox yet arguably enabling reactivity pattern is warranted.
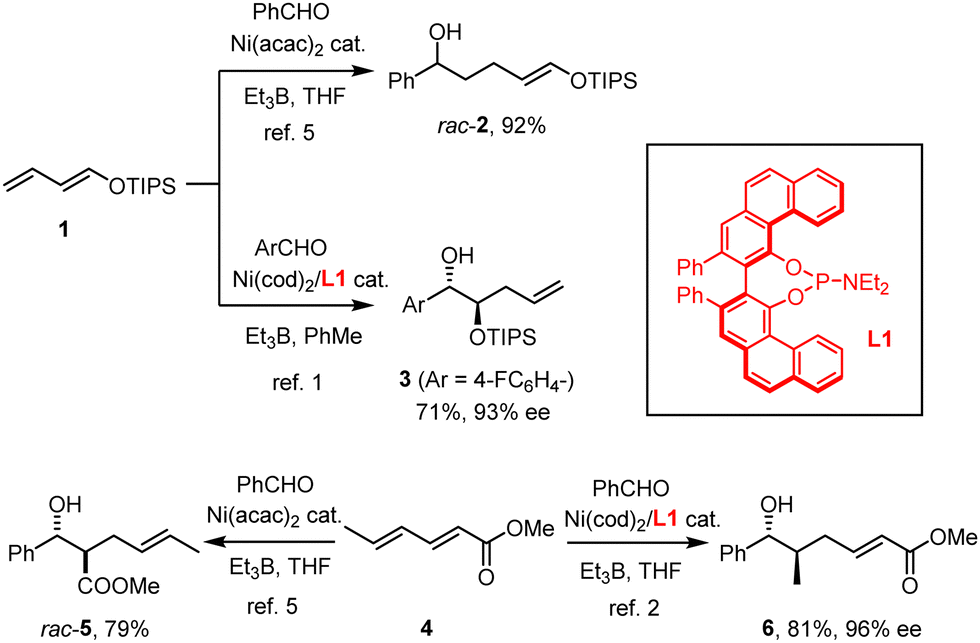 | ||
| Scheme 1 The VAPOL-derived phosphoramidite ligand L1 as a “game-changer” in Ni-catalyzed reductive coupling reactions. | ||
In this context, an extension to imines as electrophilic partners seemed particularly attractive for the prospect of gaining access to valuable vic-aminoalcohol derivatives. Under the standard conditions employed for the preparation of 3,1 the reaction of benzaldehyde N-tosylimine 8 with silyl dienol ether 1 (R = TBS) proceeded extremely slowly, not least because of the poor solubility of the substrate in toluene (Scheme 2(A)). To remedy the issue, amidosulfone 7 was tested as an imine surrogate,18 which led to an apparently homogeneous solution upon addition of di-isopropylethylamine to the mixture; this base gently releases imine 8 in solution yet is too bulky to quench the Et3B as the promoter of choice. The fact that product 9a was obtained in moderate yield but with excellent optical purity was deemed encouraging; surprisingly though, transformation into aziridine 10 and comparison with literature data19 suggested that the product was syn-configured (see below), whereas diols such as 3 derived from aldehydes under the same conditions had invariably been anti-configured.1 Unfortunately, however, the material contained substantial amounts of 11a, in which the double bond is shifted by one position (9a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :11a ≈ 4:1).
:11a ≈ 4:1).
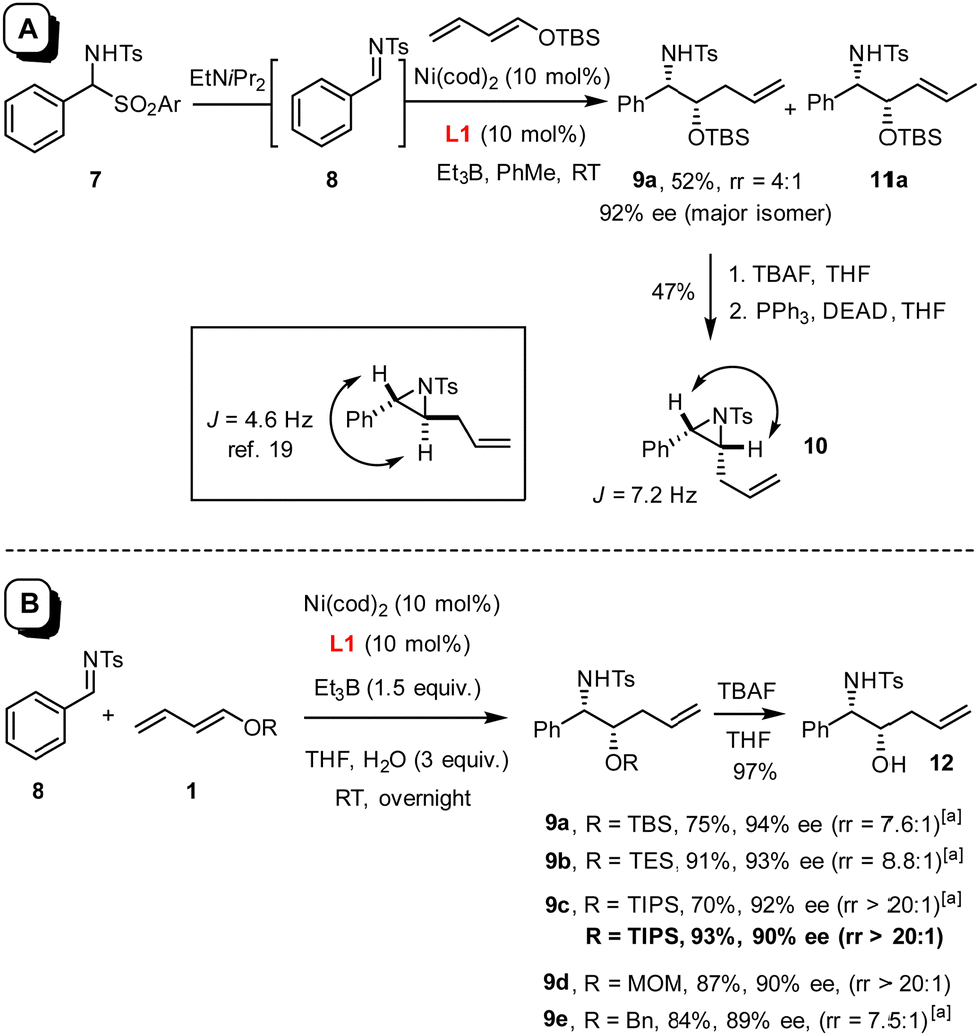 | ||
| Scheme 2 Panel A: lead finding (Ar = Ph); panel B: preparation of syn-configured aminoalcohol derivatives by reductive coupling under optimized conditions; a starting from sulfone 7 (Ar = p-tolyl) (rather than imine 8) in the presence of EtN(iPr)2 (1.2 equiv.). | ||
As this isomer is virtually impossible to remove by any method other than HPLC, its formation had to be suppressed from the outset. To this end, the solvent, the reaction temperature, the nickel source,§ the promoter,¶ the N-substituent on the imine,|| the O-substituent of the dienol ether were varied and numerous additives tested, without much success. It was not until an attempt was made to form an imine in situ from benzaldehyde and p-methoxybenzylamine that a key observation was made: although the desired product was almost racemic in this case, the rate of reaction was massively enhanced as compared to what was seen with preformed N-PMB imine.20 This comparison suggested that water generated in situ might play a critical role; therefore, further attempts at optimization focussed on this particular parameter.21 Indeed, addition of three equivalents of degassed H2O proved optimal, provided THF (rather than toluene) was used as the solvent (for details, see the ESI†). Under these conditions, product 9a (R = TBS) was obtained in 75% yield and 94% ee with a notably improved regioisomer ratio of ≈7.6:1; slightly better results were obtained for 9b bearing a smaller TES-group. Gratifyingly though, recourse to the more bulky TIPS dienol ether 1 (R = TIPS) furnished product 9c virtually as a single isomer; the regioisomeric alkene could not be detected by 1H NMR in the crude material (rr > 20:1). Moreover, the N-tosylimine 8 itself performed even better in this case than the sulfone surrogate 7 (Scheme 2(B)), which is an advantage in terms of atom economy. Cleavage of the TIPS group with TBAF afforded alcohol 12; its structure in the solid state confirmed the relative and absolute configuration as originally deduced based on the data recorded for the derived aziridine 10 (Fig. 1).22 The MOM-protected dienol ether proved similarly suitable for reductive coupling to give 9d exclusively, whereas the corresponding benzyl dienol ether resulted in an isomer mixture and was therefore not used any further.
 | ||
| Fig. 1 Structure of compound 12 in the solid state. | ||
Fig. 2 shows an assortment of aminoalcohol derivatives obtained by this new procedure from aromatic and heteroaromatic tosyl aldimines; their configuration was assigned in analogy to compound 12. Electron-rich as well as electron-deficient substrates proved suitable, although the latter tend to give better ee's. The fact that an aryl chloride and even a bromide as well as a thiophene ring remained intact is a remarkable virtue of this catalyst system based on Ni(0). Equally relevant is the compatibility with a pinacolboronate ester, which constitutes an excellent handle for downstream functionalization. It is also noteworthy that placement of a methyl substituent ortho to the aldimine did not prevent the reductive coupling from occurring. In cases such as 17, in which the optical purity was insufficient, recrystallization of the sample from tBuOMe/n-hexane allowed the issue to be addressed.
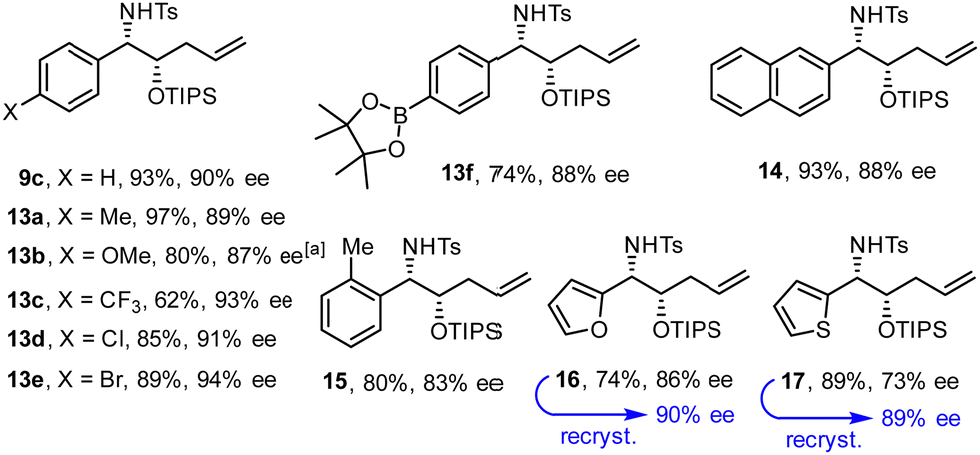 | ||
| Fig. 2 Aminoalcohol derivatives formed by reductive coupling of (hetero)aromatic tosyl aldimines with silyl dienol ether 1 (R = TIPS); for the conditions, see Scheme 2(B);a at 0 °C. | ||
N-Tosyl aldimines derived from aliphatic aldehydes also participate in reductive coupling with dienol ethers 1 (Scheme 3). In this case, only one equivalent of degassed water should be added to the reaction mixture to minimize formation of the corresponding internal alkene isomer. Although traces of this side product were always present in the crude material, chromatographic purification allowed this impurity to be removed after cleavage of the silyl ether. The ee's obtained with the aliphatic aldimines were in the range of 81–86%, but recrystallization proved again serviceable for solid materials.
 | ||
| Scheme 3 Reductive coupling of aliphatic tosyl aldimines with silyl dienolether 1 (R = TIPS). | ||
The reaction scales well. Thus, product 9c was formed in essentially the same yield and unchanged ee of 90% when the reductive coupling was performed on 1 mmol scale (Scheme 4(A)). Subsequent hydroboration/oxidation furnished alcohol 24; treatment of the derived mesylate with tBuOK in THF gave the disubstituted piperidine derivative 25, which is a valuable building block for the formation of non-peptidic neurokine NK1 receptor antagonists such as L-733,060 or CP-99,994.23 In this context, it is important to note that the tosyl group of 25 can be readily cleaved with magnesium powder in MeOH upon ultrasonication of the suspension in a laboratory cleaning bath.24 This method is so mild that the TIPS-ether survives, as demonstrated by the formation of compound 26, which was obtained with unaltered optical purity.
 | ||
| Scheme 4 Synthetic applications. Panel A: preparation of a building block for non-peptidic neurokine NK1 receptor antagonists; panel B: synthesis of the naturally occurring γ-amino acid statine. | ||
Scalability was also demonstrated for compound 28, which can be elaborated in only two steps into statine (30, Scheme 4(B)). This γ-amino acid is the essential constituent responsible for the activity of the potent aspartyl protease inhibitor pepstatin.25 To this end, the double bond of 28 was oxidatively cleaved with acidic permanganate and the sulfonamide of the resulting acid 29 deprotected with lithium naphthalenide in THF; the moderate yields are solely due to the very high polarity of the compounds.
In summary, this study extends our recent finding that Ni(0) allies with a VAPOL-derived phosphoramidite ligand to give a so far unique catalyst system that imposes unprecedented regioselectivity as well as appreciable enantioselectivity onto reductive coupling reactions of dienes with different electrophilic partners. Future studies in our laboratory will try to extend the portfolio of this novel type of transformation, optimize the attained level of induction by ligand tuning, and gain basic mechanistic understanding for how this specific nickel/ligand combination exerts its function. Pertinent results along these lines will be reported in due course.
Open Access funding provided by the Max Planck Society.
Conflicts of interest
There are no conflicts to declare.Notes and references
- T. Q. Davies, J. J. Murphy, M. Dousset and A. Fürstner, J. Am. Chem. Soc., 2021, 143, 13489–13494 CrossRef CAS PubMed.
- T. Q. Davies, J. Y. Kim and A. Fürstner, J. Am. Chem. Soc., 2022, 144, 18817–18822 CrossRef CAS PubMed.
- The inverted course was initially observed as a side reaction during an attempted application of nickel catalyzed reductive coupling to natural product synthesis, see: M. Heinrich, J. J. Murphy, M. K. Ilg, A. Letort, J. T. Flasz, P. Philipps and A. Fürstner, J. Am. Chem. Soc., 2020, 142, 6409–6422 CrossRef CAS PubMed.
- For VAPOL, see: (a) J. Bao, W. D. Wulff, J. B. Dominy, M. J. Fumo, E. B. Grant, A. C. Rob, M. C. Whitcomb, S.-M. Yeung, R. L. Ostrander and A. L. Rheingold, J. Am. Chem. Soc., 1996, 118, 3392–3405 CrossRef CAS; (b) Z. Ding, W. E. G. Osminski, H. Ren and W. D. Wulff, Org. Process Res. Dev., 2011, 15, 1089–1107 CrossRef CAS.
- M. Kimura, A. Ezoe, M. Mori, K. Iwata and Y. Tamaru, J. Am. Chem. Soc., 2006, 128, 8559–8568 CrossRef CAS PubMed.
- M. Kimura, A. Ezoe, K. Shibata and Y. Tamaru, J. Am. Chem. Soc., 1998, 120, 4033–4034 CrossRef CAS.
- M. Takimoto, Y. Hiraga, Y. Sato and M. Mori, Tetrahedron Lett., 1998, 39, 4543–4546 CrossRef CAS.
- M. Kimura and Y. Tamaru, Top. Curr. Chem., 2007, 279, 173–207 CrossRef CAS.
- For related reductive coupling reactions involving π-components other than dienes, see: (a) E. P. Jackson, H. A. Malik, G. J. Sormunen, R. D. Baxter, P. Liu, H. Wang, A.-R. Shareef and J. Montgomery, Acc. Chem. Res., 2015, 48, 1736–1745 CrossRef CAS PubMed; (b) E. A. Standley, S. Z. Tasker, K. L. Jensen and T. F. Jamison, Acc. Chem. Res., 2015, 48, 1503–1514 CrossRef CAS PubMed; (c) Nickel Catalysis in Organic Synthesis: Methods and Reactions, ed. S. Ogoshi, Wiley-VCH, Weinheim, 2020 Search PubMed.
- Y. Yang, S.-F. Zhu, H.-F. Duan, C.-Y. Zhou, L.-X. Wang and Q.-L. Zhou, J. Am. Chem. Soc., 2007, 129, 2248–2249 CrossRef CAS PubMed.
- We are aware of a single study in which the different termini of substituted 1,3-dienes could be addressed in reductive coupling reactions with formaldehyde using either a nickel- or a ruthenium catalyst, see: A. Köpfer, B. Sam, B. Breit and M. J. Krische, Chem. Sci., 2013, 4, 1876–1880 RSC.
- M. Holmes, L. A. Schwartz and M. J. Krische, Chem. Rev., 2018, 118, 6026–6052 CrossRef CAS PubMed.
- Y. Sato, N. Saito and M. Mori, J. Am. Chem. Soc., 2000, 122, 2371–2372 CrossRef CAS.
- Y. Sato, Y. Hinata, R. Seki, Y. Oonishi and N. Saito, Org. Lett., 2007, 9, 5597–5599 CrossRef CAS PubMed.
- N. Saito, A. Kobayashi and Y. Sato, Angew. Chem., Int. Ed., 2012, 51, 1228–1231 CrossRef CAS PubMed.
- For important recent advances, see: (a) J. S. Marcum and S. J. Meek, J. Am. Chem. Soc., 2022, 144, 19231–19237 CrossRef CAS PubMed; (b) J.-T. Ma, T. Zhang, B.-Y. Yao, L.-J. Xiao and Q.-L. Zhou, J. Am. Chem. Soc., 2023, 145, 19195–19201 CrossRef CAS PubMed.
- S. Ogoshi, K. Tonomori, M. Oka and H. Kurosawa, J. Am. Chem. Soc., 2006, 128, 7077–7086 CrossRef CAS PubMed.
- E. Marcantoni, A. Palmieri and M. Petrini, Org. Chem. Front., 2019, 6, 2142–2182 RSC.
- M. K. Ghorai, A. Kumar and D. P. Tiwari, J. Org. Chem., 2010, 75, 137–151 CrossRef CAS PubMed.
- The fact that in situ and ex situ generated imines behave differently in Tamaru–Mori-type reactions is precedented, see: M. Kimura, A. Miyachi, K. Kojima, S. Tanaka and Y. Tamaru, J. Am. Chem. Soc., 2004, 126, 14360–14361 CrossRef CAS PubMed.
- It is known that certain Tamaru–Mori-type reactions benefit from the presence of water in the medium for reasons that remain speculative, see: (a) M. Kimura, A. Ezoe, S. Tanaka and Y. Tamaru, Angew. Chem., Int. Ed., 2001, 40, 3600–3602 CrossRef CAS; (b) M. Kimura, M. Mori, N. Mukai, K. Kojima and Y. Tamaru, Chem. Commun., 2006, 2813–2815 RSC.
- For a stereocomplementary reductive coupling reaction to form anti-configured aminoalcohols, see: K. Spielmann, M. Xiang, L. A. Schwartz and M. J. Krische, J. Am. Chem. Soc., 2019, 141, 14136–14141 CrossRef CAS PubMed.
- G.-L. Li and G. Zhao, Org. Lett., 2006, 8, 633–636 CrossRef CAS PubMed.
- P. F. Koh, P. Wang, J. M. Huang and T. P. Loh, Chem. Commun., 2014, 50, 8324–8327 RSC.
- J. Marciniszyn, J. A. Hartsuck and J. Tang, J. Biol. Chem., 1976, 251, 7088–7094 CrossRef CAS PubMed.
- L. Nattmann and J. Cornella, Organometallics, 2020, 39, 3295–3300 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, characterization data, copies of NMR spectra of new compounds. CCDC 2294750. See DOI: https://doi.org/10.1039/d3cc04582j |
| ‡ J. Y. K. and T. Q. D. contributed equally. |
| § The use of the bench-stable Ni(0) stilbene complex [Ni(tBu-stb)3]26 instead of [Ni(cod)2] led to markedly lower yields of product, although this precatalyst had been very active in the reductive coupling of sorbate esters.2 |
| ¶ For reasons that are not entirely clear, Et3B worked much better than Et2Zn, although the latter has often been used in Tamaru–Mori-type reactions. |
| || PhCH = NTs and PhCH = NSO2C6H4CF3 gave good ee's; low ee's and/or low yields were obtained with PhCH = NSO2Me, PhCH = NSO2tBu, PhCH = NSO2Bn, PhCH = NBn, PhCH = NPMB, PhCH = N(2-pyridyl), PhCH = NP(O)Ph2 and PhCH = NP(O)(OEt)2. |
| This journal is © The Royal Society of Chemistry 2023 |