
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Swift C–C bond insertion by a 12-electron palladium(0) surrogate†
Kevin
Breitwieser
 a,
Fabian
Dankert
a,
Annette
Grünwald
ab,
Paula R.
Mayer
a,
Frank W.
Heinemann
b and
Dominik
Munz
*ab
a,
Fabian
Dankert
a,
Annette
Grünwald
ab,
Paula R.
Mayer
a,
Frank W.
Heinemann
b and
Dominik
Munz
*ab
aCoordination Chemistry, Saarland University, Campus C4.1, Saarbrücken D-66123, Germany. E-mail: dominik.munz@uni-saarland.de
bFriedrich-Alexander-Universität Erlangen-Nürnberg, Inorganic and General Chemistry, Egerlandstr. 1, Erlangen D-91058, Germany
First published on 14th September 2023
Abstract
The selective activation of C–C bonds holds vast promise for catalysis. So far, research has been primarily directed at rhodium and nickel under harsh reaction conditions. Herein, we report C–C insertion reactions of a 12-electron palladium(0) surrogate stabilized by a cyclic(alkyl)(amino) carbene (CAAC) ligand. Benzonitrile (1), biphenylene (2), benzocyclobutenone (3), and naphtho[b]cyclopropene (4) were studied. These substrates allow elucidation of the effect of ring strain as well as hybridization encompassing sp3, sp2 and sp hybridized carbon atoms. All reactions proceed quantitatively at or below room temperature. This work therefore outlines perspectives for mild C–C bond functionalization catalysis.
The functionalization of C–C bonds allows for the modification of organic molecules without the requirement for specific functional groups.1 However, the elegance of this reaction-type is compromised by the strong and sterically shielded C–C bond, which renders the insertion step difficult. The most common transition metals used for catalytic transformations of C–C bonds are arguably rhodium and nickel. This is exemplified by the Rh-catalyzed cyclobutene ring opening2 and the Ni-catalyzed transfer hydrocyanation.3 However, also here, harsh reaction conditions, namely refluxing toluene or xylene solvents, are needed to break the C–C bond. These reactions may even take several days if not weeks to achieve high conversions.4,5 As such, mild pathways to isolable, yet strained group-10 metallacycles are scarce as highlighted by Moret et al. for a nickelacyclobutane.6
So far, only a handful of C–C insertion products have been reported for platinum, and even fewer for palladium. Jones and colleagues pioneered the field and used nickel(0), palladium(0), and platinum(0) phosphine complexes for the activation of biphenylene (Scheme 1a),7–12 as well as the photochemical activation of diphenylacetylene.13 The insertion of the palladium(0) complex into biphenylene proceeded in reasonable yield only upon heating to 68 °C for 14 days.10,12,14 Even for Ni(NHC)2, only a yield of 66% was obtained after stirring overnight.15,16 The oxidative addition of benzonitrile with a bond dissociation energy (BDE) of 555 kJ mol−1, which considerably exceeds the one of the Ph–F bond (BDE = 485 kJ mol−1),17 seems even more challenging.11,18 Thus, it requires in the case of palladium the addition of Lewis-acids such as BEt319 or silylium cations as studied by Gagné and coworkers (Scheme 1b).20,21
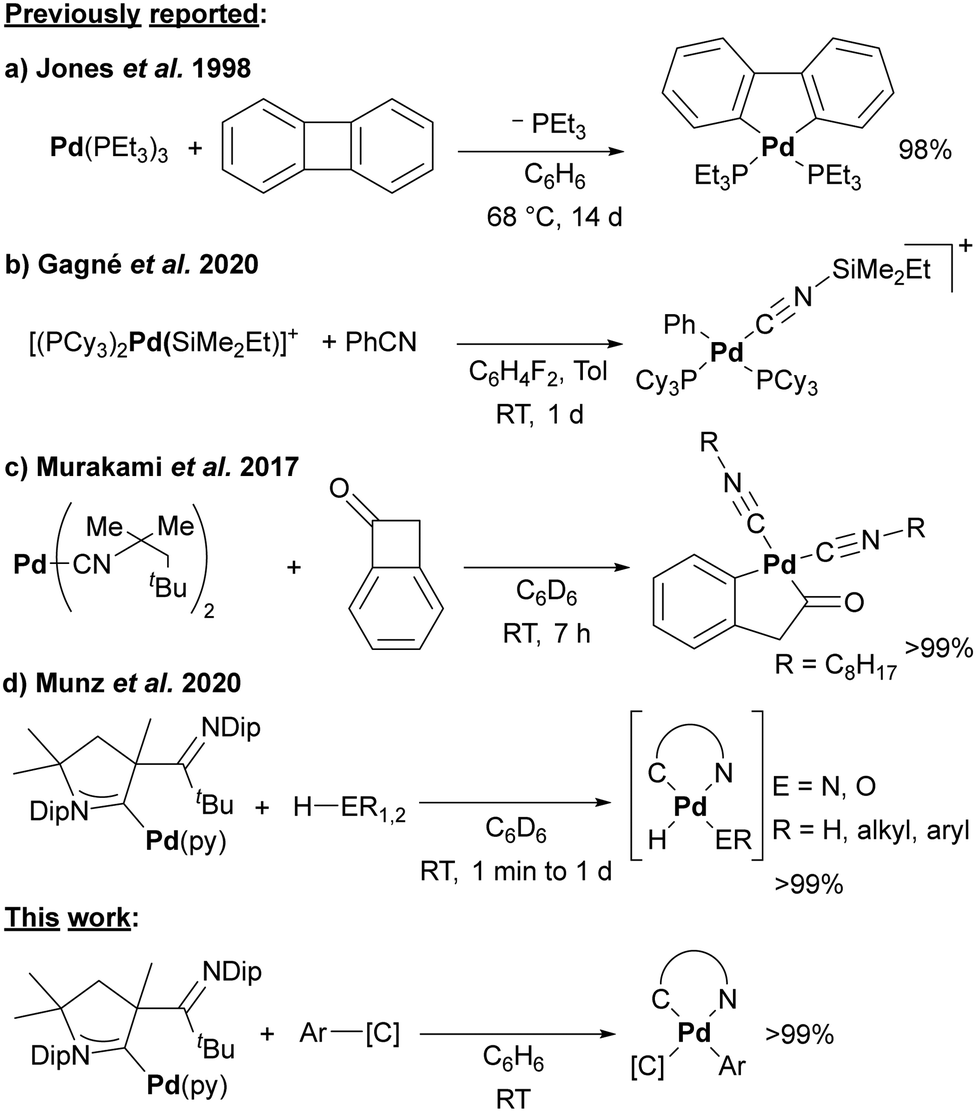 | ||
| Scheme 1 Insertion into C–C and H–E (E = N, O) bonds by palladium(0) complexes. | ||
The C–C insertion of benzocyclopropene and naphtho[b]cyclopropene with (PEt3)Ni(COD)22,23 and (PPh3)2Pt(C2H4)24 have been studied as well, yet the products obtained with palladium proved unstable. Notwithstanding this, Murakami and coworkers isolated the insertion product of benzocyclobutenone with a palladium(0) complex bearing sterically hindered isocyanide ligands (Scheme 1c).25 We reported a palladium(0) cyclic (alkyl)(amino) carbene (CAAC)26–30 complex (1) with a labile pyridine ligand, which renders this complex a surrogate for monocoordinated 12-electron palladium(0).27,31 This complex serves as precursor for exceedingly reactive nitrene complexes32–34 and homolytically cleaves O–H and N–H bonds, whereby the hemilabile imino group allows to trap the transient Pd-hydrido species (Scheme 1d).35 Thus, we reasoned that the softness of the palladium(0) metal center should also allow for the insertion into C–C bonds under unprecedented mild conditions.
Indeed, treating dark-red 1 in benzene with equimolar equivalents (or a small excess of 1.1 to 1.5 eq.) of either benzonitrile (2), benzocyclobutenone (3), biphenylene (4), or naphtho[b]cyclopropene (5) led to an instantaneous color change from red to yellow/orange, which is indicative for the formation of palladium(II) (Scheme 2). Further, yellow precipitates/crystals started to form immediately. Compounds 3 and 5 crystallized quantitatively within 3–24 hours, whereas 2 and 4 required the addition of pentane in the workup. Running the reactions in thawing benzene likewise led to an immediate color-change and quantitative conversion within <5 min according to the in situ1H spectroscopic analysis. An exception is 2, where ≈4 h are required (Fig. S6, ESI†). As expected, all these reactions take considerably longer in coordinating pyridine-d5, as illustrated by a reaction time of ≈4 days for 2. Thereby, the in situ1H-NMR spectroscopic analysis (see Fig. S7 for C6D6, ESI†) reveals an intermediate, which we propose to be the η2-coordinate π-complex.35
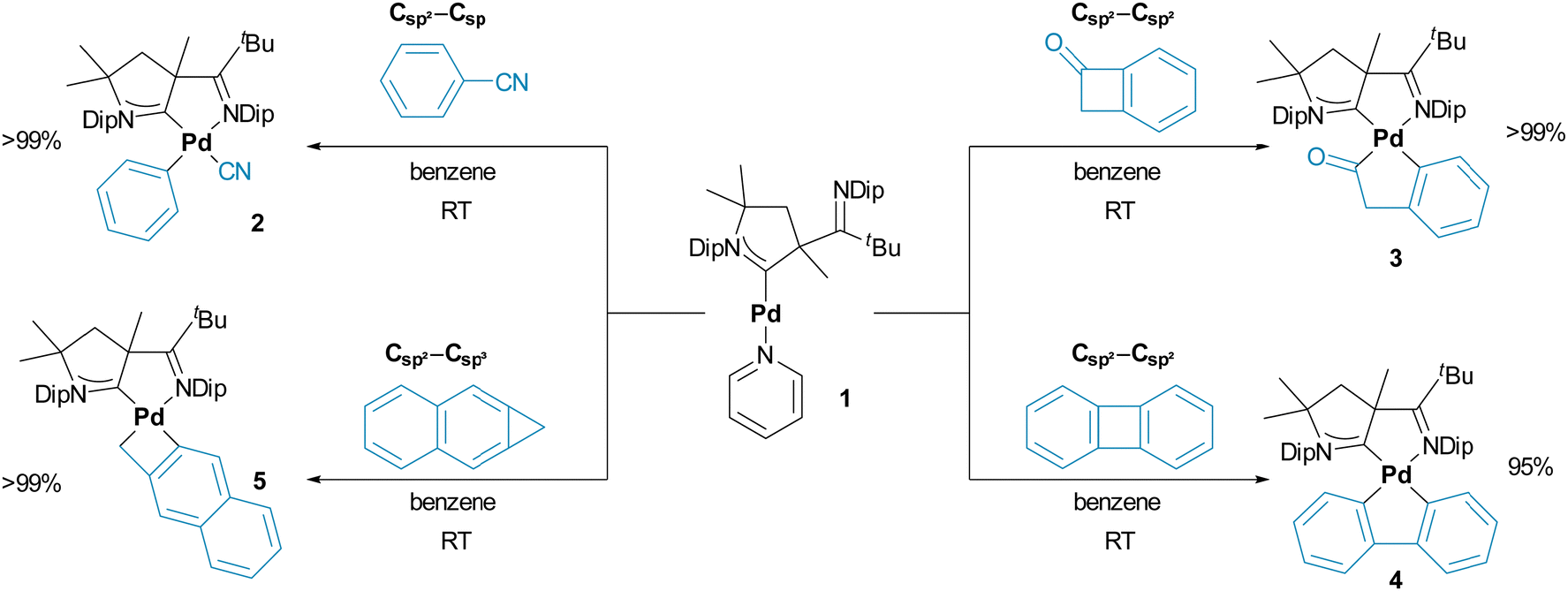 | ||
| Scheme 2 Insertion of complex 1 into Csp2–Csp, Csp2–Csp2 and Csp2–Csp3 bonds. | ||
Single crystals suitable for XRD analysis were obtained directly from the reaction in case of 3 and 5, via slow evaporation of a concentrated solution of 2 in pyridine, or through diffusion techniques (pentane/pyridine) at −30 °C for 4 (Fig. 1). Comparing the bond lengths of the Pd–CAr bonds between the four complexes reveals remarkable trans influences. For 4, the Pd–C bond trans to the CAAC is exceedingly long with 2.085(4) Å. In contrast, the other Pd–Ar bond is much shorter with 1.990(4) Å, and thus in the common range for palladium(II) phenyl complexes. Likewise, an elongated Pd–Ar bond of 2.085(2) Å was found in single crystals of 3. Likely due to the constraints of the four-membered ring, this bond is moderately shorter (2.049(7) Å) in 5, yet still unusually long in respect to common palladium phenyl compounds. The torsion angles between the CAAC and the aryl rings are also noteworthy. While this angle is large for 4 (N1–C1–C37–C42, 76.3°) and 3 (N2–C1–C36–C35, 39.3°), it is more acute for 5 (N2–C1–C38–C39, 18.9°). As a result, 5 features better spatial overlap between the aromatic π-system of the aryl ligand and the metal ion as well as the vacant p(z)-orbital of the carbene, which results in a short Pd–Ccarbene bond of only 2.011(7) Å.
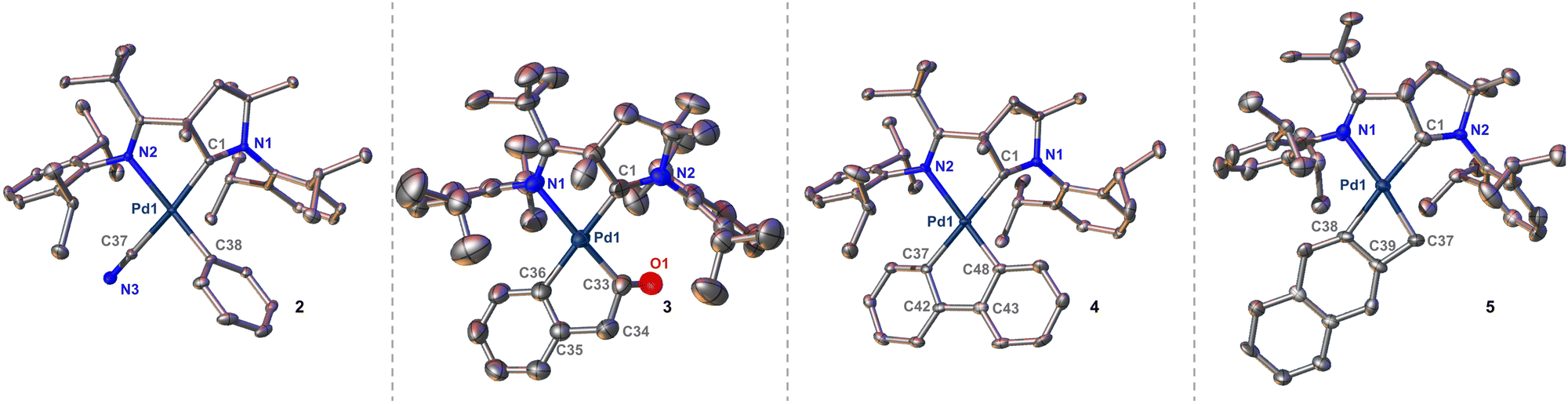 | ||
| Fig. 1 Solid state structures of complexes 2, 3, 4 and 5. Thermal ellipsoids are shown at 50% probability. Hydrogen atoms and co-crystallized solvent molecules are omitted for clarity. Selected bond lengths [Å] and (torsion) angles [°]: 2: Pd1–N2, 2.1846(14); Pd1–C1, 1.9847(17); Pd1–C37, 2.0182(18); Pd1–C38, 2.0107(17). 3: Pd1–N1, 2.2460(15); Pd1–C1, 2.0565(18); Pd1–C33, 1.970(2); Pd1–C36, 2.0859(19); N2–C1–C36–C35, 39.3. 4: Pd1–N2, 2.210(4); Pd1–C1, 2.044(4); Pd1–C37, 2.085(4); Pd1–C48, 1.990(4); N1–C1–C37–C42, 76.3. 5: Pd1–N1, 2.175(6); Pd1–C1, 2.011(7); Pd1–C37, 2.062(6); Pd1–C38, 2.049(7); N2–C1–C38–C39, 18.9. | ||
In the case of 2, this bond is even shorter with 1.9847(17) Å due to the trans-influence of the cyanido ligand. The palladacycles 4 and 5 are almost planar with the sum of the internal angles being close to the ideal (4, 538.5° vs. 540°; 5, 359.2° vs. 360°). In contrast, palladacycle 3 is significantly bent (522.2° vs. 540°) and the carbonyl group is twisted out of plane with a short distance of approximately 3.1 Å to the mean plane of the phenyl ring of the diisopropylphenyl group. Small inter-ligand distances are also found for the phenylene groups in 3 and 4. These palladacycle–CAAC interactions parallel the stability of complexes 3, 4 and 5. While 5 is perfectly stable in pyridine, solutions of 4 turn colorless within 2 h, thereby depositing insoluble needles. SC-XRD analysis identified these crystals as the di(pyridine) complex 6 (Fig. 2). In case of 3, this transformation requires heating to 80 °C overnight (ESI†). Substitution reactions of CAACs are very unusual, and PdII(CAAC) complexes are usually inert towards strong nucleophiles, including phosphines and other carbene-ligands.36,37 We therefore propose that the loss of the CAAC ligand is due to steric congestion.
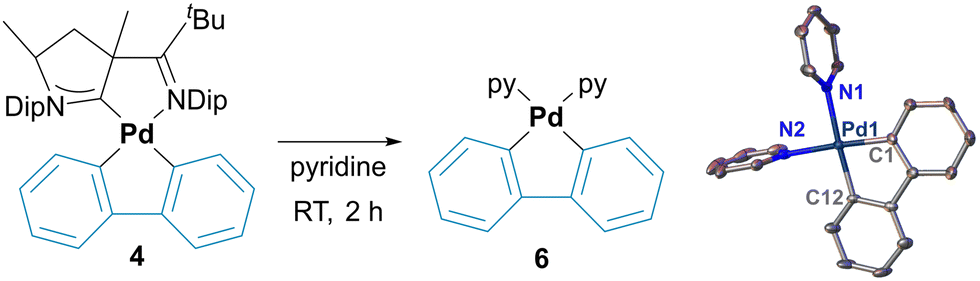 | ||
| Fig. 2 Solid state structures for the di(pyridine) complexes 6. Thermal ellipsoids at 50% probability. Hydrogen atoms and co-crystallized solvent molecules are omitted for clarity. Selected bond lengths [Å]: Pd1–N1, 2.127(3); Pd1–N2, 2.128(3); Pd1–C1, 2.002(3); Pd1–C12, 2.002(3). | ||
The ease of C–C activation may by understood by the interplay of ring strain and carbon atom hybridization. No ring strain is required for the oxidative addition into the Csp2–Csp bond of benzonitrile (2) despite the high BDE of 555 kJ mol−1.11 Although lower s-character of the C–C bond reduces the BDE (Ph–Ph, BDE = 418 kJ mol−1),17 it renders these compounds kinetically more stable (vide infra). As such, ring strain is required (3, computed at 116 kJ mol−1, Table S2; 4,38 222 kJ mol−1) for the oxidative addition. In the case of 3, selective insertion into C(O)–Csp2 was obtained, although the C(O)–Csp3 bond (CH3C(O)–Et, BDE = 349 kJ mol−1vs. CH3C(O)–Ph, BDE = 413 kJ mol−1)39 is weaker. Compound 5 showcases that even Csp3–Csp2 bonds (Ph–Et, BDE = 427.6 kJ mol−1)39 may be activated in the presence of substantial ring-strain (computed at 291 kJ mol−1, Table S2 (ESI†); literature values benzocyclopropenes, 285–300 kJ mol−1).40 Indeed, we did not observe a room-temperature reaction with benzocyclobutene, which features a Csp3–Csp2 bond with a considerably reduced strain of 136 kJ mol−1.40
It is intriguing to note that the observed reactivity aligns with computational predictions by Ananikov, Musaev, and Morokuma.41 Based on the calculation of oxidative addition transitions states for CH3–CH3, Ph–Ph and HC2–C2H with Pd(PMe3)2, the authors proposed that low barriers, viz. fast reactions, are due to enhanced s-character in the transition state. The BDEs of the Pd–R and C–C bonds, which both follow the trend HC2 > Ph > CH3, play a secondary role in the Bell–Evans–Polanyi (Hammond's postulate, respectively) sense. It was furthermore shown that ground state inhibition through the formation of π-complexes in the case of olefins and especially alkynes somehow compromises this general trend. This matches perfectly with the slow formation of 2 pending the formation of an intermediate (vide supra). We furthermore would like to highlight that the reactivity trend also coincides with the oxidative addition of organic (pseudo)halides, which has been attributed to both the higher stability of the forming Pd–C bonds as well as π-backbonding.42
In summary, we present the oxidative addition of biphenylene, benzonitrile, naphto[b]cyclopropene, and benzocyclobutenone to a palladium(0) complex. These insertion reactions proceed swiftly in (cold) benzene and, hence, under unprecedented mild conditions. Further, we report that complex 4 undergoes the unusual substitution of the CAAC ligand in pyridine. The ease of C–C bond insertion correlates with ring-strain and hybridization. Current efforts are targeting catalysis with stronger C–C bonds at elevated temperatures.
K.B.; investigation (synthesis and characterization 3, 5 and 6, computations), formal analysis, visualization, writing – original draft. A.G.; investigation (synthesis 2 and 4). P.M.; characterization 4 and determination of all reaction times (investigation). F.D., crystallographic analysis 3, 5 (investigation), contribution to writing – original draft; F.W.H. crystallographic analysis 2, 4, 6 (investigation). D.M.; conceptualization, funding acquisition, project administration, supervision, writing – review and editing.
This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 Research and Innovation Program (grant no. 948185). Instrumentation and technical assistance for the XRD analysis of 2 and 4 were provided by the Service Center X-ray Diffraction, with financial support from Saarland University and the German Science Foundation (DFG) (Project INST 256/506-1). We thank Peter Chen for providing a sample of naphtho[b]cyclopropene and N. Marigo for the preparation of starting materials. We also thank the Friedrich-Alexander-Universität Erlangen-Nürnberg (FAU) for generous financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For reviews, see: (a) B. Rybtchinski and D. Milstein, Angew. Chem., Int. Ed., 1999, 38, 870–883 CrossRef; (b) F. Chen, T. Wang and N. Jiao, Chem. Rev., 2014, 114, 8613–8661 CrossRef CAS PubMed; (c) L. Souillart and N. Cramer, Chem. Rev., 2015, 115, 9410–9464 CrossRef CAS PubMed; (d) M. Murakami and N. Ishida, J. Am. Chem. Soc., 2016, 138, 13759–13769 CrossRef CAS PubMed; (e) T. Kondo, Eur. J. Org. Chem., 2016, 1232–1242 CrossRef CAS; (f) G. Fumagalli, S. Stanton and J. F. Bower, Chem. Rev., 2017, 117, 9404–9432 CrossRef CAS PubMed; (g) P. H. Chen, B. A. Billett, T. Tsukamoto and G. Dong, ACS Catal., 2017, 7, 1340–1360 CrossRef CAS PubMed; (h) R. Vicente, Chem. Rev., 2021, 121, 162–226 CrossRef CAS PubMed; (i) A. P. Y. Chan and A. G. Sergeev, Coord. Chem. Rev., 2020, 413, 213213 CrossRef CAS; (j) G. Dong, C–C Bond Activation, Top. Curr. Chem., 346, Springer, Berlin, Heidelberg, 2014 CrossRef; (k) M. Murakami and N. Chatani, Cleavage of Carbon-Carbon Single Bonds by Transition Metals, Wiley-VCH, Weinheim, Germany, 2015; for highlights, see: CrossRef; (l) S. Hu, T. Shima and Z. Hou, Nature, 2014, 512, 413–415 CrossRef CAS PubMed; (m) A. Sattler and G. Parkin, Nature, 2010, 463, 523–526 CrossRef CAS PubMed; (n) M. Jakoobi, Y. Tian, R. Boulatov and A. G. Sergeev, J. Am. Chem. Soc., 2019, 141, 6048–6053 CrossRef CAS PubMed; (o) M. Jakoobi, N. Halcovitch, G. F. Whitehead and A. G. Sergeev, Angew. Chem., Int. Ed., 2017, 56, 3266–3269 CrossRef CAS PubMed.
- T. Seiser and N. Cramer, J. Am. Chem. Soc., 2010, 132, 5340–5341 CrossRef CAS PubMed.
- X. Fang, P. Yu and B. Morandi, Science, 2016, 351, 832–836 CrossRef CAS PubMed.
- C. Perthuisot, B. L. Edelbach, D. L. Zubris, N. Simhai, C. N. Iverson, C. Müller, T. Satoh and W. D. Jones, J. Mol. Catal. A: Chem., 2002, 189, 157–168 CrossRef CAS.
- B. D. Swartz, W. W. Brennessel and W. D. Jones, Organometallics, 2011, 30, 1523–1529 CrossRef CAS.
- M. L. G. Sansores-Paredes, S. van der Voort, M. Lutz and M.-E. Moret, Angew. Chem., Int. Ed., 2021, 60, 26518–26522 CrossRef CAS PubMed.
- B. L. Edelbach, R. J. Lachicotte and W. D. Jones, Organometallics, 1999, 18, 4040–4049 CrossRef CAS.
- N. Simhai, C. N. Iverson, B. L. Edelbach and W. D. Jones, Organometallics, 2001, 20, 2759–2766 CrossRef CAS.
- C. Müller, R. J. Lachicotte and W. D. Jones, Organometallics, 2002, 21, 1975–1981 CrossRef.
- J. J. Garcia and W. D. Jones, Organometallics, 2000, 19, 5544–5545 CrossRef CAS.
- J. J. Garcia, N. M. Brunkan and W. D. Jones, J. Am. Chem. Soc., 2002, 124, 9547–9555 CrossRef CAS PubMed.
- B. L. Edelbach, R. J. Lachicotte and W. D. Jones, J. Am. Chem. Soc., 1998, 120, 2843–2853 CrossRef CAS.
- C. Müller, C. N. Iverson, R. J. Lachicotte and W. D. Jones, J. Am. Chem. Soc., 2001, 123, 9718–9719 CrossRef PubMed.
- B. L. Edelbach, D. A. Vicic, R. J. Lachicotte and W. D. Jones, Organometallics, 1998, 17, 4784–4794 CrossRef CAS.
- T. Schaub and U. Radius, Chem. – Eur. J., 2005, 11, 5024–5030 CrossRef CAS PubMed.
- T. Schaub, M. Backes and U. Radius, Organometallics, 2006, 25, 4196–4206 CrossRef CAS.
- J. A. Dean, Lange's Handbook of Chemistry, McGraw-Hill, INC., New York St. Louis San Francisco Auckland Bogotá Caracas Lisbon London Madrid Mexico Milan Montreal New Delhi Paris San Juan São Paulo Singapore Sydney Tokyo Toronto, 15th edn, 1999.
- S. Lachaize, D. C. Gallegos, J. J. Antonio, A. C. Atesin, T. A. Ateşin and W. D. Jones, Organometallics, 2023, 42, 2134–2147 CrossRef CAS.
- L. Munjanja, C. Torres-López, W. W. Brennessel and W. D. Jones, Organometallics, 2016, 35, 2010–2013 CrossRef CAS.
- A. L. Wierschen, J. Lowe, N. Romano, S. J. Lee and M. R. Gagné, Organometallics, 2020, 39, 1258–1268 CrossRef CAS.
- A. L. Wierschen, N. Romano, S. J. Lee and M. R. Gagne, J. Am. Chem. Soc., 2019, 141, 16024–16032 CrossRef CAS PubMed.
- R. Neidlein, A. Rufińska, H. Schwager and G. Wilke, Angew. Chem., Int. Ed. Engl., 1986, 25, 640–642 CrossRef.
- C. Krüger, K. Laakmann, G. Schroth, H. Schwager and G. Wilke, Chem. Ber., 2006, 120, 471–475 CrossRef.
- P. J. Stang, L. Song and B. Halton, J. Organomet. Chem., 1990, 388, 215–220 CrossRef CAS.
- S. Okumura, F. Sun, N. Ishida and M. Murakami, J. Am. Chem. Soc., 2017, 139, 12414–12417 CrossRef CAS PubMed.
- V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef CAS PubMed.
- J. Chu, D. Munz, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 7884–7887 CrossRef CAS PubMed.
- M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef CAS PubMed.
- F. Vermersch, L. Oliveira, J. Hunter, M. Soleilhavoup, R. Jazzar and G. Bertrand, J. Org. Chem., 2022, 87, 3511–3518 CrossRef CAS PubMed.
- K. Breitwieser and D. Munz, Adv. Organomet. Chem., 2022, 78, 79–132 CrossRef CAS.
- D. Munz, J. Chu, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2016, 55, 12886–12890 CrossRef CAS PubMed.
- A. Grünwald, B. Goswami, K. Breitwieser, B. Morgenstern, M. Gimferrer, F. W. Heinemann, D. M. Momper, C. W. M. Kay and D. Munz, J. Am. Chem. Soc., 2022, 144, 8897–8901 CrossRef PubMed.
- S. J. Goodner, A. Grünwald, F. W. Heinemann and D. Munz, Aust. J. Chem., 2019, 72, 900–903 CrossRef CAS.
- A. Grünwald, N. Orth, A. Scheurer, F. W. Heinemann, A. Pöthig and D. Munz, Angew. Chem., Int. Ed., 2018, 57, 16228–16232 CrossRef PubMed.
- A. Grünwald, F. W. Heinemann and D. Munz, Angew. Chem., Int. Ed., 2020, 59, 21088–21095 CrossRef PubMed.
- E. Tomás-Mendivil, M. M. Hansmann, C. M. Weinstein, R. Jazzar, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2017, 139, 7753–7756 CrossRef PubMed.
- N. Marigo, B. Morgenstern, A. Biffis and D. Munz, Organometallics, 2023, 42, 1567–1572 CrossRef CAS PubMed.
- I. Novak, J. Phys. Chem. A, 2008, 112, 2503–2506 CrossRef CAS PubMed.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- R. Benassi, S. Ianelli, M. Nardelli and F. Taddei, J. Chem. Soc., Perkin Trans. 2, 1991, 1381–1386 RSC.
- V. P. Ananikov, D. G. Musaev and K. Morokuma, Organometallics, 2005, 24, 715–723 CrossRef CAS.
- A. Ariafard and Z. Lin, Organometallics, 2006, 25, 4030–4033 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2288556–2288560. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cc03964a |
| This journal is © The Royal Society of Chemistry 2023 |