
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCloneable inorganic nanoparticles
Alexander R.
Hendricks
 a,
Bradley F.
Guilliams
a,
Rachel S.
Cohen
a,
Tony
Tien
a,
Gavin A.
McEwen
a,
Kanda M.
Borgognoni
b and
Christopher J.
Ackerson
*a
a,
Bradley F.
Guilliams
a,
Rachel S.
Cohen
a,
Tony
Tien
a,
Gavin A.
McEwen
a,
Kanda M.
Borgognoni
b and
Christopher J.
Ackerson
*a
aDepartment of Chemistry, Colorado State University, Fort Collins, CO 80523, USA. E-mail: chris.ackerson@colostate.edu
bNational Institute of Environmental Health Sciences, Durham, NC 27709, USA
First published on 2nd June 2023
Abstract
When a defined protein/peptide (or combinations thereof) control and define the synthesis of an inorganic nanoparticle, the result is a cloneable NanoParticle (cNP). This is because the protein sequence/structure/function is encoded in DNA, and therefore the physicochemical properties of the nanoparticle are also encoded in DNA. Thus the cloneable nanoparticle paradigm can be considered as an extension of the central dogma of molecular biology (e.g. DNA → mRNA → protein → cNP); modifications to the DNA encoding a cNP can modify the resulting properties of the cNP. Inorganic ion oxidoreductases (e.g., mercuric reductase, tellurite reductase, etc.) can select and reduce specific inorganic oxyanions and coordination complexes, creating zerovalent precipitates. Other proteins/peptides (often genetically concatenated to the parent oxidoreductase) serve as ligands, directing the size, shape, crystal structure and other properties of the nanoparticle. The DNA encoding a cNP can be recombinantly transferred into any organism. Ideally, this enables recombinant production of cNPs with the same defined physiochemical properties. Such cNPs are of interest for applications ranging from molecular imaging, bio-remediation, catalysis, and biomining. In this Feature Article we detail and define the cNP concept, and retrace the story of our creation of a cloneable Se NanoParticle (cSeNP). We also describe our more preliminary work that we expect to result in cloneable semiconductor quantum dots, cloneable Te nanoparticles, and other cNP formulations. We highlight the application of cNPs in cellular electron microscopy and compare this approach to other cloneable imaging contrast approaches.
 Alexander R. Hendricks | Alex Hendricks is about to complete his chemistry PhD under Prof. Chris Ackerson at Colorado State University. Prior to CSU, Alex earned a B.S. in Chemistry from the University of Minnesota. His research has mainly focused on engineering the catalytic activity of metal reductases. Outside of the lab, Alex dedicates his time to creating art, climbing, running, cooking, consulting, reading, and story writing. |
 Bradley F. Guilliams | Bradley Guilliams obtained B.S. degrees in Chemistry and Biology in 2019 and an M.S. degree in Chemistry in 2020 from Western Carolina University with Prof. Scott Huffman and Prof. Brian Byrd. He is currently a chemistry PhD candidate under Prof. Chris Ackerson at Colorado State University. His research centers on cryo-electron tomography of metal reductases as contrast tools to enhance intracellular imaging. Outside of the lab, Bradley spends his time backcountry skiing, climbing, cooking, and riding bikes. |
 Rachel S. Cohen | Rachel Cohen is a chemistry graduate student at Colorado State University working under Prof. Chris Ackerson and Prof. Chris Snow. She obtained a B.S. in Chemistry in 2021 from the University of Massachusetts, Amherst. Her current research is focused in computational design of metal reductases, particularly in the context of the cloneable nanoparticle project. |
 Tony Tien | Tony Tien is a Bioengineering graduate student at Colorado State University working under Prof. Chris Ackerson. He obtained his B.S. in Chemical Engineering from Colorado School of Mines in 2021 where he worked on developing fluorescence-based analyte nanosensors with Prof. Kevin Cash. His current research focuses on transforming cloneable nanoparticles into new model systems as well as investigating the role of peptides for nanoparticle size control. |
 Gavin A. McEwen | Gavin McEwen earned a B.S. in Chemistry from Dartmouth College and is currently a chemistry PhD student working under Prof. Chris Ackerson at Colorado State University. His research focuses on developing and studying peptides for greater control over the size and morphology of cloneable selenium nanoparticles. Outside of the lab, Gavin enjoys the plentiful outdoor recreation available in Colorado. |
 Kanda M. Borgognoni | Kanda Borgognoni received her B.S. in Biochemistry from Loyola University New Orleans in 2016. As an undergraduate, she conducted research with Dr Joelle Underwood at Loyola and Dr Robert Siggins at LSU Health Sciences Center. She joined Dr Chris Ackerson's research group as a graduate student at Colorado State University. She received her Ph. D. in 2021 where her thesis work involved developing in situ cloneable nanoparticle methodologies for imaging applications. As a postdoctoral researcher at the NIEHS, she is currently working with Dr Robin E. Stanley to define the mechanisms of ribosome biogenesis in vivo using cryo-electron tomography. |
 Christopher J. Ackerson | Christopher Ackerson is a Colorado native and Professor of Chemistry at Colorado State University. He started at CSU as an Assistant Professor in 2009. He earned a B.S. in Biochemistry (with honors) from the University of Texas at Austin and a PhD in Biophysics from Stanford University under supervision of Professor Roger Kornberg. Prof. Ackerson's research focuses on developing inorganic contrast agents, such as gold nanoclusters and cloneable nanoparticles, for use in biological imaging. Outside of science, Prof. Ackerson enjoys running, bicycling, raising 4 children and playing competitive pinball. |
Introduction
Soluble inorganic nanoparticles (iNPs) typically comprise an inorganic core passivated by an organic ligand shell. In vitro synthetic methodology for producing iNPs of well-defined elemental composition, size, shape and crystal structure is now relatively mature; rules and guidelines exist that form starting points for synthesis of novel iNPs, based on prior work. Biogenic nanoparticles (bNPs) are those produced with biomolecules, such as crude cell extracts or sugars. These tend to be less well-controlled, relying on serendipitous interactions between biomolecules and iNP precursors; well-developed frameworks for predictive bNP synthesis do not yet exist.A relatively new concept in nanoparticle synthesis is that of a cloneable nanoparticle (cNP), which can be considered a sub-type of bNP. When physicochemical properties of an iNP, such as elemental composition, size, shape, crystal-structure, allotrope, optical properties, and magnetism, are encoded in DNA, the iNP is a “cloneable NP” (cNP). These cNPs are synthesized by proteins, usually inside cells. Protein sequence, and therefore structure and function, are genetically encoded. Therefore, nanoparticles whose physicochemical properties are controlled by proteins during synthesis have their properties encoded in DNA. This framing implies that the DNA encoding any cNP can be recombinantly transferred into any organism, endowing that organism with the ability to make the cNP.
cNPs as defined in this article are made in living cells. cNPs do occur naturally. Magnetosomes,1,2 ferritins,3–5 and DNA-binding protein from starved cells (Dps)6,7 are examples of naturally occurring cNPs. Each of these examples makes an iron oxide nanoparticle of defined size, shape, elemental composition, and magnetism.
bNPs (and therefore cNPs) attract interest for a diverse array of applications. Our primary interest in cNP engineering is to make cloneable contrast in images of biological systems generated with electrons, X-rays, light and/or magnets (i.e., electron microscopes, CT-scanners, optical microscopes, and MRI imagers). Our secondary interest is using cNPs for environmental bioremediation of metal(loid) contaminations.
There are many ongoing studies developing bNPs for uses including catalysis,8 solar photoconversion,9 antimicrobial therapy,10,11 and theranostic (i.e., combined therapeutics and diagnostics) agents.12,13 The applications of biogenic NPs listed here are at preliminary stages. It can be expected that the greater synthetic control of NP properties in biological contexts will become more desirable or necessary as implementations advance. cNPs may enable such means of improving synthetic control in many of the currently proposed uses of biogenic NPs.
The cNP synthesis paradigm
We have developed a modular cNP platform that links genotype (DNA) to phenotype (cNP composition). This platform adapts in vitro NP synthesis (Fig. 1, top panel) approaches to use cellularly available reactants (Fig. 1, bottom panel).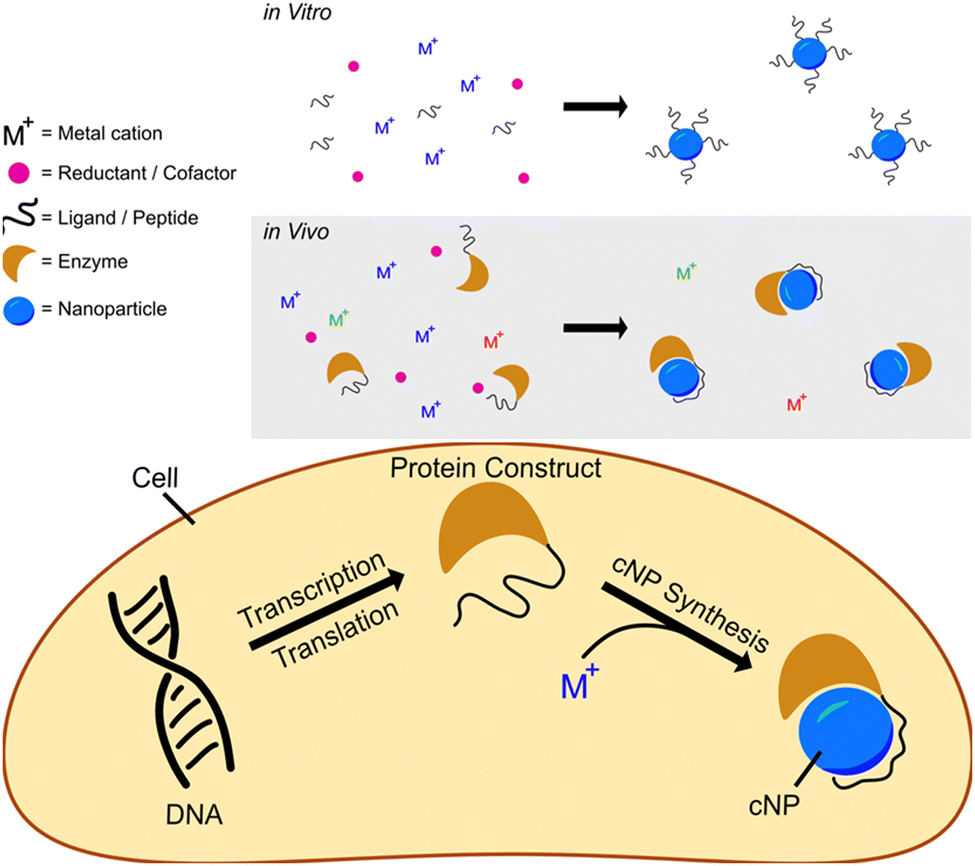 | ||
| Fig. 1 Schematics showing reductive nanoparticle synthesis approaches in vitro (top panel) and in vivo (bottom panel). | ||
In vitro reductive iNP synthesis usually begins with three chemical components: inorganic cations, reductants, and ligands. In this synthetic approach, reductants change the oxidation states of cations from soluble (e.g., aqueous) to insoluble (particulate, solid) states. Very often, the reduced particulate oxidation state is zerovalent. Ligands present during synthesis cap growing inorganic solids at nanoparticulate size, suppressing growth into macroscopic bulk solid14 and directing particle size, shape, fluorescence, etc.
In adapting this paradigm to cellular environments, we similarly use three distinct chemical components, with the additional use of inorganic ion oxidoreductases. In this context, bioavailable metal(loid) cations or coordination complexes act as the metal(loid) source. Inorganic ion oxidoreductases select the ‘desired’ ion (coordination complex) from a background that contains all essential elements (Se, Zn, Fe, etc). Enzyme cofactors (NADH, NADPH, glutaredoxin, etc.) act in concert with enzymes as the reductant. Peptides or proteins that bind to and/or control iNP properties can be concatenated to or coexpressed with the oxidoreductase and act as ligands in cNP synthesis; in this role, they offer the prospect of size, shape, and crystal structure definition. The combination of oxidoreductases and iNP binding peptides/proteins results in synthetic control of cNP synthesis inside cells rivalling the synthetic control that is possible with well-developed in vitro methods.
cNP composition parameters
cNPs require metal(loids) coordination-complex precursors that are at least transitively stable inside cells (e.g., not easily reduced by ubiquitous reducing molecules such as sugars and aromatic or thiol-containing amino acids). Such metal(loid)s include essential elements such as Fe, Co, Zn, Se, W and Mo. In addition, many non-essential metal(loids), such as Bi (a pharmaceutical component in Pepto Bismol) or Au (a pharmaceutical component of rheumatoid arthritis drugs such as Auranofin) might also form the basis for a cNP. Some metal(loids) regarded as highly toxic, such as, can even form the basis for a cNP, if the toxicity can be appropriately modulated. In the case of arsenicals, the metal complex arsphenamine (Salvarsan) is a treatment for syphilis.15For metal(loid) ions to be cNP reactants, they must be enzymatically reducible by enzymes – therefore the redox potential of the metal(loid) precursor must be in a range accessible to biological reductants such as NADPH or glutaredoxin. (i.e., their oxidation state must be modulated by the enzyme). The redox potential of a metal(loid) ion is significantly impacted by ligation. The impact of ligation on redox potential exerts effects through multiple pathways, including ligand lability/inertness, the electron transfer (tunnelling) properties of the ligand, and the metal(loid) oxidation state favored by the ligand (influenced by ligand geometry). As an example of how ligation changes redox potentials, consider the reduction of Cu2+to Cu0. The reduction potential of aquated Cu2+ in the reaction [Cu(H2O)6]2+ + 2e → Cu(s) + 6H2O(l) is +0.337 V versus S.H.E., whereas [Cu(NH3)4]2+ + 2e → Cu(s) + 4NH4 is at −0.05 V.16
Many intracellular metal ions are glutathione-ligated, in part due to the ∼mM concentration of glutathione (GSH) inside most cells, combined with the high affinity of thiols for many metal(loids).17 Many metal(loid) oxidoreductases are thus unsurprisingly related to glutathione reductase (GSHR). GSHR,18,19 mercuric reductase,20 tellurite reductase,21 and glutathione reductase-like metalloid reductase (GRLMR)22 all belong to the class I FAD dependent oxidoreductase enzyme family. Functional differences among enzymes in this family mainly comprise substrate selection—where substrates are different metal(loids) ligated by GSH.
Enzymatic metal(loid) ion reduction is a relatively common cellular feature. Many enzymatic systems may therefore possess cNP chemistry; however, most enzymes described to date lack inherent means of enforcing uniform size/shape/elemental composition. A great deal of iNP binding peptides have, in contrast, been described for their ability to exert strong influence on NP size/shape/elemental composition yet lack intrinsic means of reducing metal(loid) cations. Thus, the combination of iNP-binding peptides with metal(loid) ion reductases represents a platform or paradigm for cNP synthesis.
iNP binding dodecapeptides were first isolated from phage display libraries.23 So-far, peptides that bind to a about 100 iNPs of differing size/shape/elemental composition are now described in the literature. A subset of iNP binding peptides can also assist in the synthesis of iNPs. For instance, A3 peptide in the presence of Au3+ salts and a reductant (which can include the buffer HEPES serving as reductant) makes 10 nm AuNPs. The Ge8 peptide, in the presence of AgNO3 and H2O2, makes chiral, curved silver nanowires.24
These synthesis-assisting peptides do not constitute cNPs on their own; while they are encoded in DNA and promote NP formation, they do not change the oxidation states of the ions (and therefore solubility). Thus, they do not represent a self-contained cNP system. Notably, some of the early literature on iNP binding peptides confuses this issue, as early papers did not recognize that buffer salts can donate electrons too, and reduce many metal ions in the absence of peptides.25,26
Many proteins have also been identified that bind to inorganic nanoparticles and might therefore similarly control their shape and size during synthesis. Broadly speaking, there is existing literature on proteins that form a ‘corona’ around iNPs.27,28 For selenium nanoparticle(s) (SeNPs) a proteomic study identified about 50 proteins that are found in the SeNP corona,29 for SeNPs made in cells or exposed to cell extracts.
By analogy to what is now known about library isolated peptides in the context of bNP synthesis, we expect that some naturally occurring proteins can also direct cNP size and shape. That suite of proteins may include those that confer an adaptive advantage to their host (in the context of metal toxicity) and those that evolved for other functional endpoints but serendipitously control cNP size/shape.
Above we outlined cellular equivalents of reductants and ligands that can synthesize well-defined cNPs. This combination of cellular reductants and ligands can be encoded in a single chimeric DNA construct. This results in a chimeric protein that incorporates all chemical activities required to make a cNP.
Because a single protein (polypeptide chain) controls all functions, mutations to the DNA encoding the protein can subsequently change properties of the cNP. This allows the adaptation of recently developed and powerful directed evolution methods for the purpose of cellular nanoparticle synthesis.30
Cloneable contrast
Our primary motivation in developing the cNP platform is to develop cloneable imaging contrast. Cloneable contrast imaging agents, such as green fluorescent protein (GFP), were first described and adopted for fluorescence microscopy.31,32 What makes GFP useful in imaging is that the DNA encoding GFP can be easily ‘cloned’ or concatenated to the DNA encoding any protein of interest. The protein of interest now ‘lights up’ in fluorescence microscopy and is distinctly visible against the cellular background, where nothing else fluoresces notably at the GFP emission wavelength of 509 nm. Meanwhile, the available toolbox of fluorescent proteins has grown to the extent that multi-color (multiplexed imaging) studies are routine—e.g., dynamic protein interactions with several players can be observed.33GFP represents a ‘benchmark’ for cloneable imaging contrast. The properties of GFP that make it so useful are: (1) it works in both live and fixed cells; (2) it is entirely self-contained in a single DNA segment—requiring nothing further such as exogenous building blocks or upstream cellular engineering (3) There is 1 GFP for each protein that it is concatenated to. This allows straightforward quantification of protein copy number through quantification of fluorescence signal.
Cloneable contrast agents that function in electron, magnet and X-ray-based imaging modalities are extensively explored, but not yet widely adopted by biologists using these imaging methods. Such modalities are nevertheless important, as they allow tissue imaging in magnification and penetration domains outside what visible-light microscopy is capable of. Cloneable contrast in these imaging modalities may or may not use approaches that create cNPs. Approaches that use ferritin, singlet-oxygen generation proteins, and phytochelatins—such as metallothionein—are outlined below. So far, no cloneable contrast approach for EM is widely used because there are severe limitations associated with each method proposed to date:
Ferritin is a ∼400 kDa protein constituted from 24 polypeptide units. Under iron-rich conditions, ferritins can store up to ∼4500 iron atoms as a highly regulated mineral core, which can be easily observable in certain imaging contexts such as EM or correlative light and electron microscopy (CLEM).3,4Ferritin presents two substantial challenges for widespread use as a contrast agent. First, its multimericity can heavily convolute labelling strategies. Second, its comparatively large size casts a shadow in images, obscuring potentially important areas within images. The size may also impact function of ferritin-protein conjugates by slowing diffusion kinetics of concatemerized protein(s) of interest.
Certain proteins can locally deposit diaminobenzidine (DAB) polymer, such as mini singlet oxygen generator34 (miniSOG) and engineered ascorbate peroxidase35 (APEX2). In these cases, radical oxygen is enzymatically produced, which subsequently activates DAB to form osmiophilic polymers. These polymers then preferentially absorb electron dense osmium stains proximal to the point of polymer origination. While osmiophilic DAB polymers can be a highly sensitive method of enhancing contrast, they diffuse distally from their point of origination (in some cases to nearly 200 nm), substantially reducing the resolution at which interpretations of images (collected at much higher resolution) can be made. DAB polymerization is moreover infeasible in living cells or in frozen-hydrated cells, requiring fixation which introduces imaging artefacts.
Metallothioneins are small (0.5–14 kDa) cysteine-rich proteins which stoichiometrically bind metal atoms. Previous works have demonstrated that metallothionein can bind a wide array of metal atoms, including Au, Pt, Ag, Pb, Cd, and As. Metallothioneins have been implemented as cloneable contrast tags.36 However, they are barely visible above the background in electron micrographs. This can also be exacerbated by competition from natively expressed metallothionein, which upregulates in the presence of high intracellular metal concentration.
Inorganic nanoparticles are known to have properties that can produce contrast in EM, visible-light, X-ray, and magnetic imaging modalities. An appropriately formulated cNP may therefore be able to produce contrast in each of these imaging modalities simultaneously, while overcoming many of the limitations of existing cloneable contrast agents.
This feature article outlines our development of a cNP platform, with some applications highlighted. The most ‘mature cNP’ in our lab is comprised of ∼5 nm Selenium NPs—making it a cloneable selenium nanoparticle (cSeNP.)
We summarize a set of 4 papers from our group, which in concert describe the development of this cSeNP platform. The reductase is Glutathione Reductase-Like Metalloid Reductase (GRLMR), which was isolated from Pseudomonas moraviensis subspecies stanleyae—among the most Se tolerant microorganisms ever described. This species may show promise for bioremediation of selenium contamination—an area where previous work has been done collecting biogenically produced Se(0) from wastewater.37,38
Transmission electron microscopy (TEM) and electron tomography of the P. moraviensis stanleyae shows ∼50 nm diameter intracellular SeNPs. There is no apparent membrane or protein structure surrounding these SeNPs. GRLMR can be transferred among species, endowing other species with enhanced Se tolerance.
GRLMR alone does not control SeNP size. To introduce size control, we isolated a peptide that binds to SeNPs from a phage display library. Concatenation of this peptide to GRLMR at the DNA level results in a construct that consistently makes 35 nm diameter SeNPs in vitro and ∼5 nm diameter SeNPs in vivo. Furthermore, we show that inclusion of transition metals such as Cadmium in the cell cultures that make cSeNPs allows instead the production of CdSe semiconductor quantum dots.
There are ‘less mature’ cNPs in the pipeline in our lab. For instance, we have isolated a tellurium reductase from environmental isolates, and this may form the basis for a cloneable tellurium nanoparticle in future work.
While not a cNP, we have examined metallothionein as a cloneable contrast agent, concluding that it must be present in high copy number to be visible in EM.
We have also made some substantial and unpublished investigation of the capsule-forming and Fe-concentrating protein Dps. Here we will outline our approach and the shortcomings of that approach which precluded prior publication.
Summary of cNP developments
Development of the cSeNP is described in 4 manuscripts, beginning with a manuscript describing the isolation of a selenium hyperaccumulating bacterium, followed by a paper describing the isolation of a selenium reducing enzyme and characterization of the SeNP products of that enzyme. A subsequent paper showed that the Se tolerance can be recombinantly transferred to other species, facilitating recombinant SeNP production. A fourth paper isolates a SeNP binding peptide and shows how incorporation of this peptide with the selenium reductase significantly alters enzyme activity and results in size-controlled, 5 nm diameter, red SeNPs which remain attached to the enzyme that synthesizes them, representing a cSeNP.Pseudomonas moraviensis subsp. stanleyae, a bacterial endophyte of hyperaccumulator Stanleya pinnata, is capable of efficient selenite reduction to elemental selenium under aerobic conditions38
Our work on the cSeNP began with isolation and characterization of a bacterium specialized for high Se conditions.38 In this paper, we report Pseudomonas moraviensis subspecies stanleyae, a bacterial endophyte that was isolated from the root tissue of Se hyperaccumulator Stanleyae pinnata (colloquial name Prince's Plume) found growing in the naturally seleniferous soil in the Pine Ridge Natural area of Colorado, USA.The bacterium was characterized phylogenetically by fatty acid methyl ester analysis and multi locus sequence analysis. The sequence analysis showed 97.3% nucleotide identity to P. moraviensis. This subspecies was therefore dubbed Pseudomonas moraviensis stanleyae, taking the name of the plant in which it was found.
Because P. moraviensis stanleyae was found as an endophyte in a selenium hyperaccumulating plant, it was hypothesized that the bacterium is a selenium specialist (possibly working in symbiosis with the plant in which it lives). The bacterial strain was grown in various concentrations of SeO42− (selenate) and SeO32− (selenite). When grown on selenite supplemented LB-agar, the bacterial colonies appeared bright red—consistent with production of red-allotrope SeNPs (Fig. 2, panel A). When examined by TEM, rod shaped bacteria coated with SeNPs were apparent (Fig. 2, panel B.) Overall, we observed that the bacterium could tolerate up to 120 mM of selenite and 150 mM selenate, making this the most selenium tolerant organism so-far described.
 | ||
| Fig. 2 Panel a shows P. moraviensis stanleyae growing on SeO32− supplemented agar. The red color is indicative of red Se(0) formation as a result of selenite reduction. Panel b shows a TEM image of P. moraviensis stanleyae grown in the presence of selenite. The bacteria appear coated with SeNPs. Figure adapted from (38) with permission. | ||
Growth kinetics in the presence of high concentrations of Se oxyanions showed that the growth of P. moraviensis stanleyae was not significantly impeded by 10 mM selenate—but no red Se(0) was observed with selenate. In the presence of selenite, growth was somewhat slower, and red selenite appeared after about 10 hours (Fig. 3).
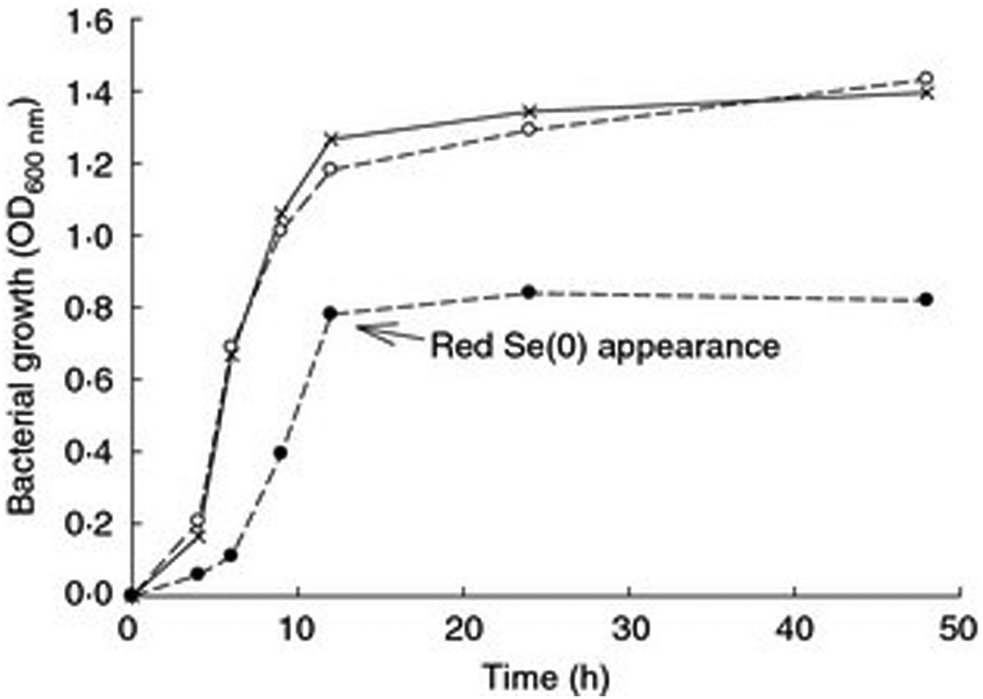 | ||
| Fig. 3 Aerobic growth of P. moraviensis stanleyae in Luria Broth media without Se oxyanions (x), and in the presence of 10 mM selenite (solid circles) and 10 mM SeO42− (open circles). Figure adapted from (38) with permission. | ||
The observation that P. moraviensis stanleyae converted SeO32− to Se(0) while SeO42− was not reduced was also made by observing the aerobic reduction of each selenium oxyanion. Fig. 4 shows the concentration of SeO32− and SeO42− as determined by a microchip capillary electrophoresis method that we developed for this purpose.39 This figure shows that SeO42− concentrations persisted near their original 10 mM concentrations in the culture for as long as they were measured (up to 50 hours). SeO32− concentration, however, diminished over time due to its reduction to red Se(0). Combined, these observations suggest that the mechanism for SeO32− tolerance involves conversion of SeO32− to Se(0), whereas SeO42− tolerance must be due to other mechanisms that do not reduce selenium oxyanions (such as efflux pumping). Overall, this paper identifies the most Se tolerant organism described to date, capable of surviving in liquid culture supplemented with up to 150 mM SeO42−. Subsequent work determines the molecular mechanism of SeO32− reduction.
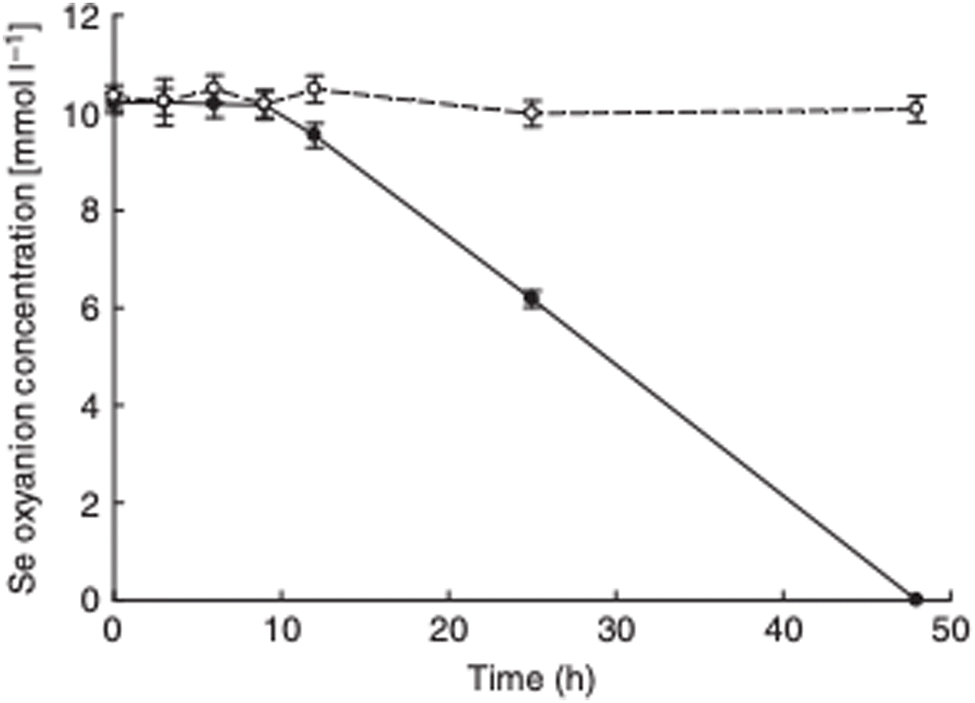 | ||
| Fig. 4 Aerobic reduction of SeO42− (open circles) and SeO32− (closed circles) by P. moraviensis stanleyae as a function of time. Figure adapted from (38) with permission. | ||
Progress toward cloneable inorganic nanoparticles40
We continued the characterization of P. moraviensis stanleyae.40 This paper focuses on electron microscopy characterization of P. moraviensis stanleyae and unraveling the mechanism of SeO32− reduction.The initial EM imaging of P. moraviensis stanleyae (Fig. 2) left several microscopic aspects of the system ambiguous. From these images, it is unclear whether SeNPs are present inside cells as well as outside cells. This initial data also lacks elemental imaging, which could confirm that the punctate inclusions are in fact comprised of Se, as inferred.
In the present paper, P. moraviensis stanleyae cells were grown in the presence of SeO32− and cells were then imaged by scanning electron microscopy (SEM) as glutaraldehyde-fixed dehydrated cells. The SEM based imaging allows energy dispersive X-ray spectroscopy (EDS) elemental analysis of the cells.
SEM imaging revealed capsule-shaped cells, most of which contained one or more dark inclusions. EDS mapping showed that these inclusions were Se rich, suggesting that these are SeNPs. The putative SeNPs were circular with an average diameter of 107 ± 35 nm (for 50 observations). These dark inclusions were not observed in control experiments where P. moraviensis stanleyae was grown in the absence of Se supplementation.
To this point, it was unclear if SeNPs were present inside or outside of the observed cells. Electron tomography is an imaging modality that allows 3D reconstructions of cells, which can resolve questions about particle location. P. moraviensis stanleyae was again grown with selenite supplementation and was preserved for electron tomographic image acquisition by freeze substitution. In this method, cells are vitrified by high pressure freezing, then the water in the cells is substituted for acetone. Following acetone substitution, the cells can be fixed and plastic embedded. This approach is recognized as providing the highest fidelity preservation of cellular ultrastructure outside of imaging of cells that are vitrified and imaged as frozen-hydrated samples (which is technically difficult to do for cells that are as thick as P. moraviensis stanleyae.)
The results of electron tomographic reconstruction of P. moraviensis stanleyae are shown in Fig. 5. Fig. 5 panel A shows a 2D image of a P. moraviensis stanleyae cell, which contains two very clear large inclusions, assumed to be SeNPs. Panels B and C show a 3D reconstruction of the cell. The reconstruction is segmented so that the membrane is shown in green and putative SeNPs are shown in red. The reconstruction reveals 3 additional particles that were not apparent in the 2D image (possibly because they were distant from the focal plane and/or too small to be visible in the 2D image. It is common for 3D reconstructions to reveal details that are not visible in any of the 2D images used to perform the reconstruction.
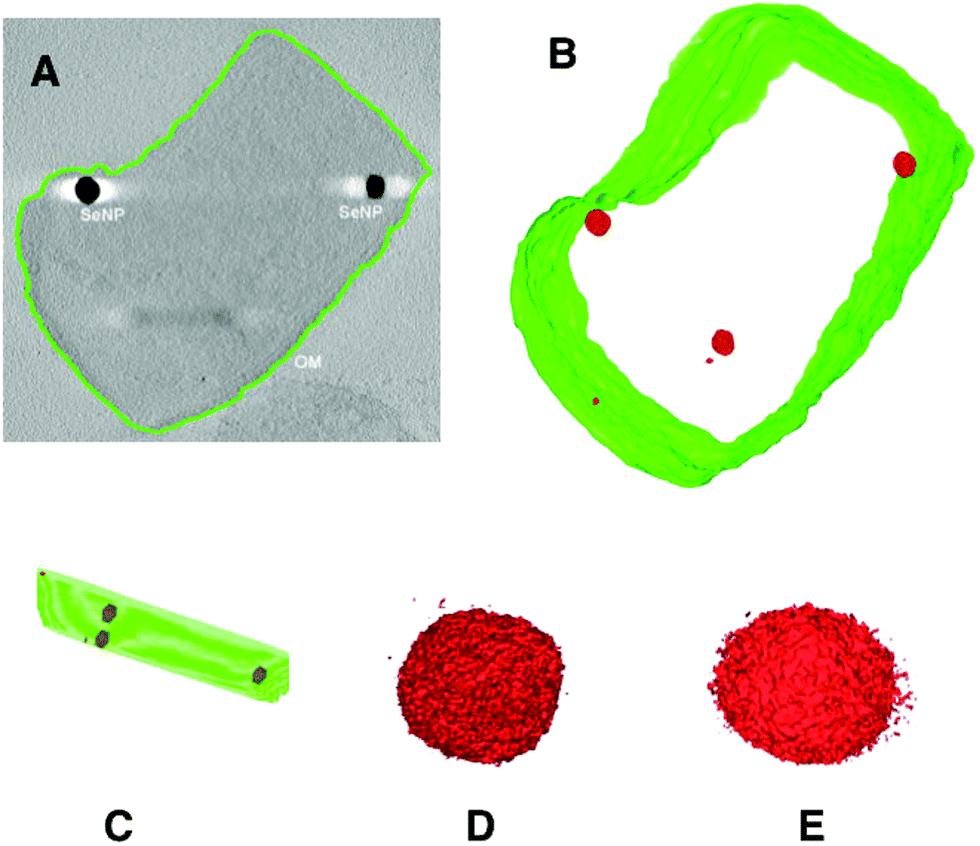 | ||
| Fig. 5 Electron tomographic reconstruction of P. moraviensis stanleyae grown in SeO32− media. The reconstruction was segmented to reveal the outer membrane and SeNPs (panels A–C). Panels D and E show two of the SeNPs at higher magnification. Figure adapted (40) with permission. | ||
Panels B and C of Fig. 5 show unambiguously that the putative SeNPs are present intracellularly. Furthermore, there is no evidence for membrane encapsulation, which is observed for other inorganic intracellular NPs, such as magnetite within membrane encapsulated magnetosomes.41 Panels D and E of Fig. 5 show 2 of the SeNPs at greater magnification. The individual particles are irregular in shape, although overall they are approximately spherical. The irregularity in shape suggests that there is no structured protein coat surrounding these particles, as is the case with other intracellular NPs such as ferritin and Dps.
Most inorganic clusters and nanoparticles require an organic ligand shell to be stable; in the absence of a ligand shell, inorganic nanoparticles fuse, ultimately forming macroscopic material. Selenium, however, is one of a handful of elements for which ‘naked’ or un-ligated clusters and nanoparticles are stable. The reasoning for this is that Se (as well as, Bi and Sb, among others) can self-satisfy its valence bonding conditions by bonding to other Se atoms.42 This aspect of Se chemistry suggests that the SeNPs inside P. moraviensis stanleyae might not have or need a well-defined biological coating, such as those found around iron oxides. It is probable that the SeNPs do maintain a so-called ‘protein corona’, but that this forms as a result of direct exposure of the surface of SeNPs to bacterial cytoplasm.
To gain insight into the mechanism of intracellular SeNP formation, we assayed all soluble proteins of P. moraviensis stanleyae for SeO32− reduction capability. This assay involved fractionating all the soluble proteins from P. moraviensis stanleyae on a non-denaturing gel, which is expected to leave enzyme function intact. Non-denaturing gels containing all soluble proteins were incubated with SeO32− salts and various enzyme cofactors, including NADH and NADPH. With the combination of NADPH and SeO32−, we observed a set of red bands in the gel, presumably comprised of Se(0) precipitates made by NADPH dependent enzymatic reduction of SeO32− (Fig. 6). The bands did not appear in the absence of NADPH and appeared very faintly in the presence of NADH.
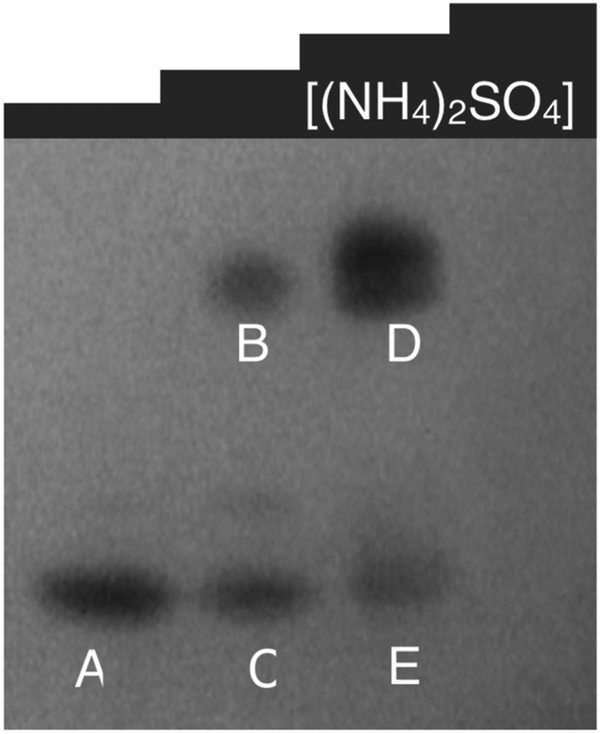 | ||
| Fig. 6 Non-denaturing gel of soluble proteins from P. moraviensis stanleyae, stained with NADPH and SeO32−. The lanes correspond to fractions eluted from a hydrophobic interaction column at various concentrations of ammonium sulfate. Bands appear where NADPH dependent SeO32− activity is spatially localized. Figure adapted from (40) with permission. | ||
To identify the NADPH dependent enzyme(s) responsible for SeO32− precipitation, we excised each band from the gel and subjected it to proteomic mass spectrometry analysis. This analysis revealed a total of 7 NADPH dependent enzymes out of 113 total proteins present in the combination of all 5 lanes. The NADPH dependent enzymes were Nitrite and sulfite reductase, isocitrate dehydrogenase, glutathione reductase, Methylenetetrahydrofolate reductase, ketoacyl-ACP reductase, Thiol peroxidase and 4-Aminobutyrate aminotransferase.
Of these enzymes, we hypothesized that a glutathione reductase-like enzyme was primarily responsible for SeNP particle formation, because glutathione reductase (GSHR) belongs to the same enzyme family as mercuric reductase, and GSHR has also been shown to reduce Au3+ ions.43 To address this hypothesis, we acquired commercially produced GSHR from Sigma-Aldrich, and tested it for SeO32− reductase activity. We observed that GSHR produced spherical SeNPs of an average diameter of 61 ± 37 nm in vitro, when both SeO32− and NADPH were present. Notably, we also observed that GSHR reduced TeO32− as well.
Combined, this paper shows that GSHR can reduce SeO32− intracellularly in an NADPH dependent process. The enzymatic product is SeNPs, of relatively high polydispersity. The SeNPs are thought to be ‘naked’ or stable without a defined and monolayer-like ligand shell. Overall, this paper postulates that GSHR or similar inorganic ion reductases form a promising basis for cNPs as enzymes that make electron dense intracellular nanoparticles from essential elements. A full adaptation of such reductases should include better SeNP size control, retention of the SeNP at the enzyme, and enzymes that reduce their metal substrates faster and more effectively than ‘background’ reduction by, for example, native GSHR.
Metalloid reductase of Pseudomonas moraviensis stanleyae conveys nanoparticle mediated metalloid tolerance22
Under the hypothesis that the GSHR from P. moraviensis stanleyae is specialized for Se reduction, we proceeded with study of the P. moraviensis stanleyae GSHR. In the paper, we began by sequencing the genome of P. moraviensis stanleyae. From the whole genome sequence, we identified the DNA encoding the most closely related enzyme to GSHR from this organism. This sequence was cloned into a protein expression vector, and the GSHR-like protein from P. moraviensis stanleyae was expressed recombinantly in Escherichia coli.The substrate selectivity of the P. moraviensis stanleyae GSHR-like protein was determined for oxidized glutathione (GSSG), selenite, and selenodiglutathione (GS-Se-SG). For comparison, commercially obtained baker's yeast GSHR was analyzed with the same substrates.
Enzyme assays for the GSHR-like enzyme and GSHR are shown in Fig. 7. Michaelis–Menten analysis of this data revealed that the P. moraviensis stanleyae GSHR-like enzyme prefers GS-Se-SG as a substrate over GS-SG; the KM for GSSG was determined to be 8.22 mM for GSSG and 336 μM for GS-Se-SG. For baker's yeast GSHR, the KM values for each substrate were similar (103 μM and 133 μM for GSSG and GS-Se-SG respectively)—showing a slightly greater affinity for GSSG substrates. Other investigations of substrate selectivity for baker's yeast GSHR found similar KM values.44
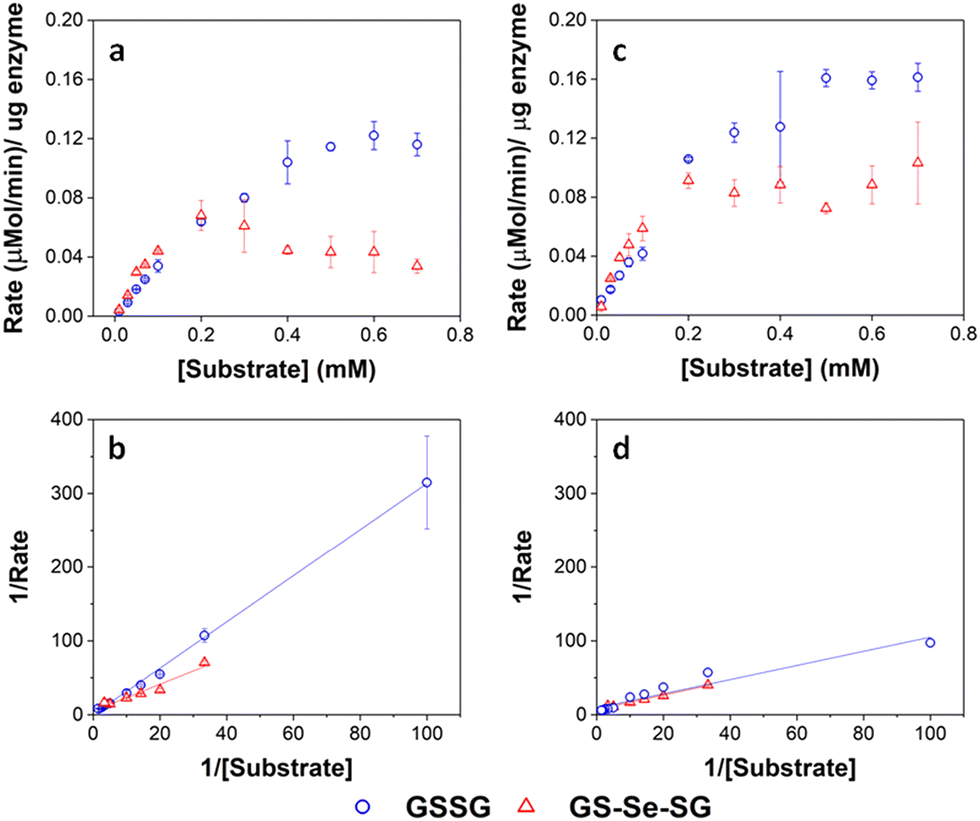 | ||
| Fig. 7 Enzyme kinetic assays for GRLMR (panel a) and GSHR (panel c). Lineweaver–Burke plots for GRLMR (panel b) and GSHR (panel d). The drop in rate at high concentrations of GS-Se-SG is attributed to SeNP formation interfering with the optical assay. GSSG indicated by blue circles, GS-Se-SG indicated by red triangles. Figure adapted from (22) with permission. Further permissions related to the material excerpted should be directed to the ACS. | ||
The 25-fold difference in KM values for the enzyme from P. moraviensis stanleyae suggests that the enzyme is adapted for reducing GS-Se-SG. Therefore, we named this enzyme “Glutathione Reductase-Like Metalloid Reductase” (GRLMR) due to its substrate preference.
Another difference between GRLMR and GSHR enzymes is the lack of a substrate binding pocket cysteine or methionine residue on GRLMR that might serve as a locus for glutathionylation. Baker's yeast GSHR, a canonical GSHR enzyme, can be glutathionated at C239, resulting in inhibition of the enzyme.45 Similarly, E. coli and Homo sapiens GSHR contain a methionine residue in this position, which might also be glutathionylated. This is thought to be a regulatory mechanism. In GRLMR, homology models show that this residue is a serine residue, which cannot be glutathionylated (Fig. 8). The absence of GSH based inhibition of GRLMR also suggests that this enzyme is distinct from canonical GSHR enzymes.
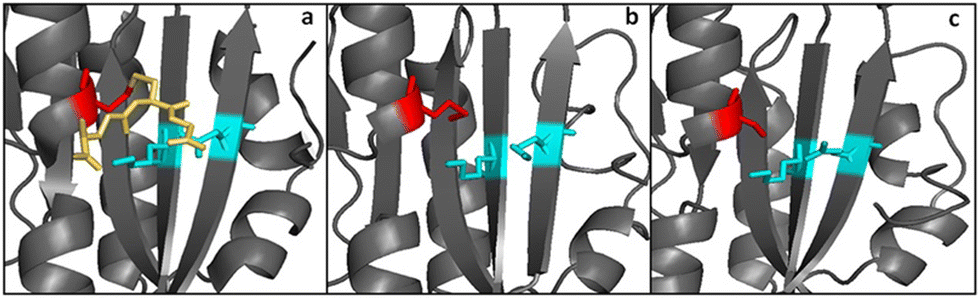 | ||
| Fig. 8 Panels a and b show the substrate binding region from yeast and E. coli GSHR crystal structures. Panel c shows the same region for a homology model of GRLMR. The residue shown in red is glutathionylated in panel a. In panel c, the red-colored residue is not capable of being glutathionylated. Figure adapted from ACS Omega 2018, 3, 14902–14909. | ||
Expression of GRLMR in E. coli confers selenium recombinant selenium tolerance to its host species. In the presence of mM concentrations of selenite, GRLMR creates nanoparticles to impart selenium tolerance for the cells (Fig. 9). In our testing, we defined selenium tolerance as the amount of selenium required to inhibit 90% of cell growth (IC90). By testing for the IC90 of SeO32− in laboratory strains of Escherichia coli, cells with the GRLMR gene had an IC90 of 21.3 ± 9.8 mM, whereas without GRLMR, the IC90 was 1.89 ± 0.46 mM. We observed a similar increase in Se tolerance for the SS320 strain of E. coli. The ten-fold higher Se tolerance when GRLMR was expressed was interpreted as imparting Se tolerance. Notably, GRLMR expressing cells grew poorly unless they were supplemented with 1 μM selenite. This was interpreted as GRLMR scavenging Se to the point that cells were Se deficient.
 | ||
| Fig. 9 Panels a–d show SEM images of fixed BL21 E. coli. Panels a and c show cells without the GRLMR plasmid, whereas panels b and d show cells that include the plasmids that are expressing GRLMR. Panels c and d show EDS overlay, where areas that are mapped to Se are colored red. Figure adapted from (22) with permission. Further permissions related to the material excerpted should be directed to the ACS. | ||
For our ultimate goal of creating cNPs useful in EM imaging of cells, it is critical that the enzyme that synthesizes the SeNP also bind to the SeNP that it has made. We observe that about 10% of GRLMR enzymes are associated with SeNPs in pull-down assays (where the mass of the SeNP allows SeNPs and associated proteins to be collected by centrifugation). This small association makes sense—an efficient enzyme is likely to turn over its product and not be associated with the product. In fact, we observe that ∼20% of baker's yeast GSHR is SeNP associated after SeNP synthesis. The smaller fraction of GRLMR associated with SeNPs is additional evidence for the specialization of this enzyme for SeNP production.
Overall, the GRLMR enzyme accomplishes reduction of a cellularly ubiquitous and essential element, Selenium. However, the SeNPs are quite polydisperse in size and do not remain attached to the enzyme that made them. To make a cSeNP, the GRLMR enzyme needs modification.
Metalloid reductase activity modified by a fused Se0 binding peptide46
In the paper, we describe the isolation and activity of a SeNP binding peptide. When concatenated to GRLMR, this peptide changes the activity of the enzyme so that it controls SeNP size and SeNPs remain largely associated with the enzyme.A SeNP binding peptide (SeBP) was isolated from the New England Biolabs PhD 12-mer phage display library (NEB PhD Kit) after three rounds of selection versus 8.4 ± 2.7 nm SeNPs, whose size was confirmed via TEM. Following three rounds of selection, the output phage peptide library was found to be dominated by two sequences (Table 1). The sequence that appeared at the highest frequency (LTPHKHHKHLHA) was designated as SeBP and was used for future experiments.
| Peptide sequence | Frequency | pI | Polystyrene | GRLMR | SiNP | |
|---|---|---|---|---|---|---|
| Wild type | N/A | N/A | 3.9 × 104 | 1.5 × 105 | 1.8 × 104 | |
| SeBP | LTPHKHHKHLHA | 19/33 | 9.37 | 3.7 × 104 | 2.6 × 105 | 3.0 × 105 |
| SeBP2 | GPHHMHHHRTHH | 7/33 | 10.47 | 1.1 × 109 | 2.1 × 109 | 7.9 × 109 |
| SeBP3 | WPRHHWHTNYMR | 1/33 | 11.15 | 3.2 × 108 | 3.9 × 108 | 1.3 × 108 |
| SeBP4 | GWHSPHAHWRVK | 1/33 | 10.61 | 4.4 × 104 | 3.3 × 105 | 3.7 × 105 |
| SeBP5 | THYNPLRINPIT | 1/33 | 9.95 | 9.8 × 103 | 6.8 × 105 | 1.8 × 104 |
| SeBP6 | KVHIMHFHHHSL | 1/33 | 9.08 | N/A | N/A | N/A |
| SeBP7 | HSWSTIKRIETM | 1/33 | 9.07 | N/A | N/A | N/A |
| SeBP8 | WPHLQHHKATSR | 1/33 | 10.61 | N/A | N/A | N/A |
| SeBP9 | HDRMTKSSFSPP | 1/33 | 9.07 | N/A | N/A | N/A |
Subsequent experiments explored the effect of concatenating SeBP to GRLMR (GRLMR-SeBP) versus GRLMR alone in terms of nanoparticle size, enzyme-nanoparticle binding, and enzyme activity. It was discovered that GRLMR-SeBP exerted control over the size of nanoparticles formed. SEM images of nanoparticles formed using 40 μM, 1 mM, 5 mM, and 10 mM SeO32− revealed that GRLMR-SeBP formed smaller and less polydisperse sized particles (Fig. 10). For GRLMR, particle size varied with SeO32− concentrations such that at low concentrations SeNPs were smaller, and at high concentrations particles were larger. However, the GRLMR-SeBP construct synthesized ∼35 nm diameter particles independent of SeO32− concentration at the physiologically relevant SeO32− concentrations of 40 μM to 5 mM. This demonstrates that the SeBP exerts remarkable control over SeNP size.
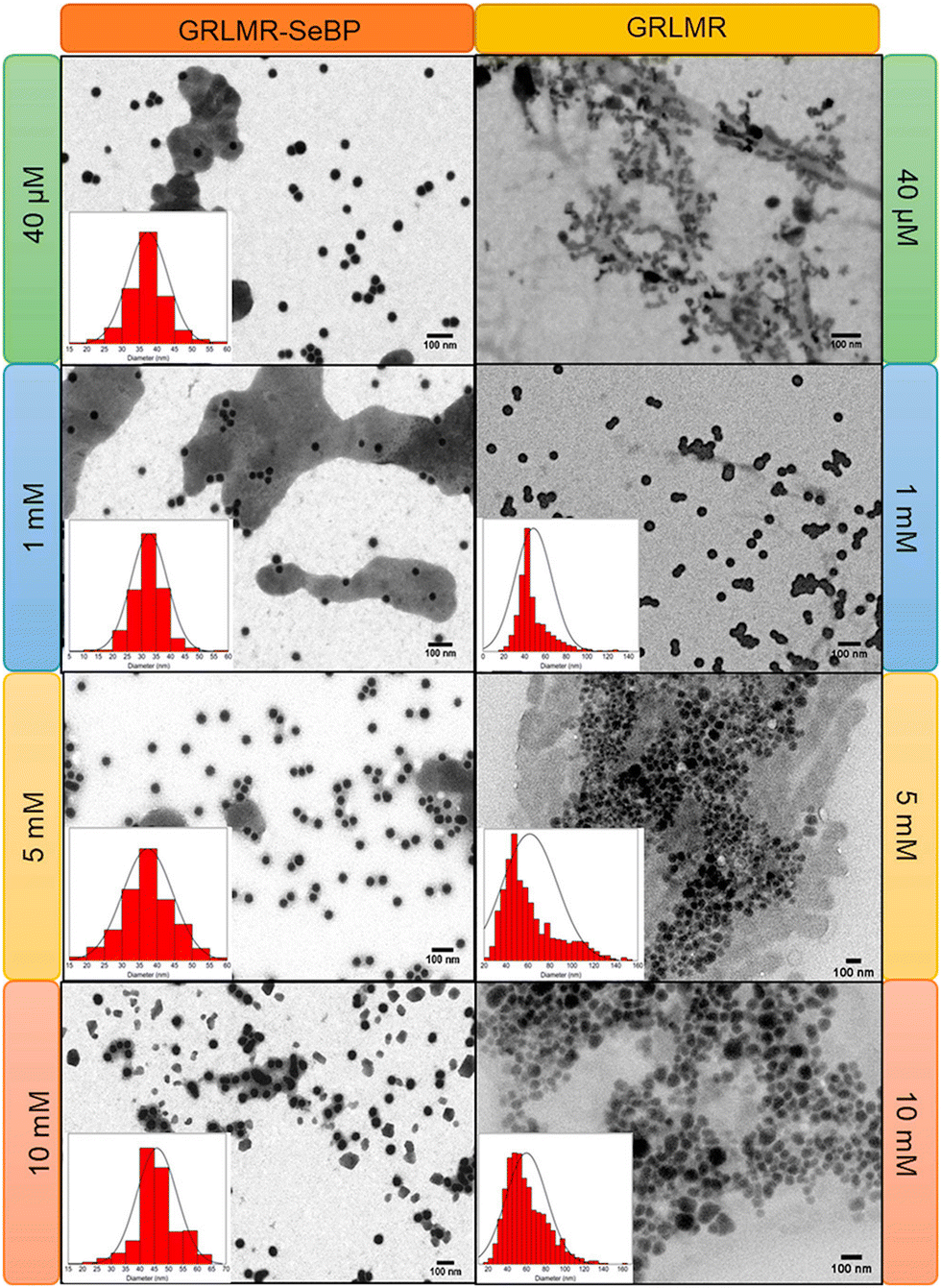 | ||
| Fig. 10 SEM images of nanoparticles formed using GRLMR-SeBP or GRLMR at indicated concentrations of SeO32−. A histogram of the size distributions is shown where it could be determined. Figure adapted from (46) with permission. Copyright 2020 American Chemical Society. | ||
To compare the abilities of GRLMR-SeBP versus GRLMR to bind SeNPs, two different centrifugation assays were performed. In the first centrifugation assay, SeNPs were enzymatically synthesized, and centrifugation was then used to remove SeNPs from solution along with any associated enzyme. The percentage of bound enzyme was determined by measuring the protein concentration in the soluble fraction after precipitation. It was found that 83.1 ± 1.0% of GRLMR-SeBP was bound to SeNPs, while 14.4 ± 2.6% of GRLMR was bound to SeNPs (Fig. 11). This demonstrates that concatenation of SeBP to GRLMR improves nanoparticle binding affinity.
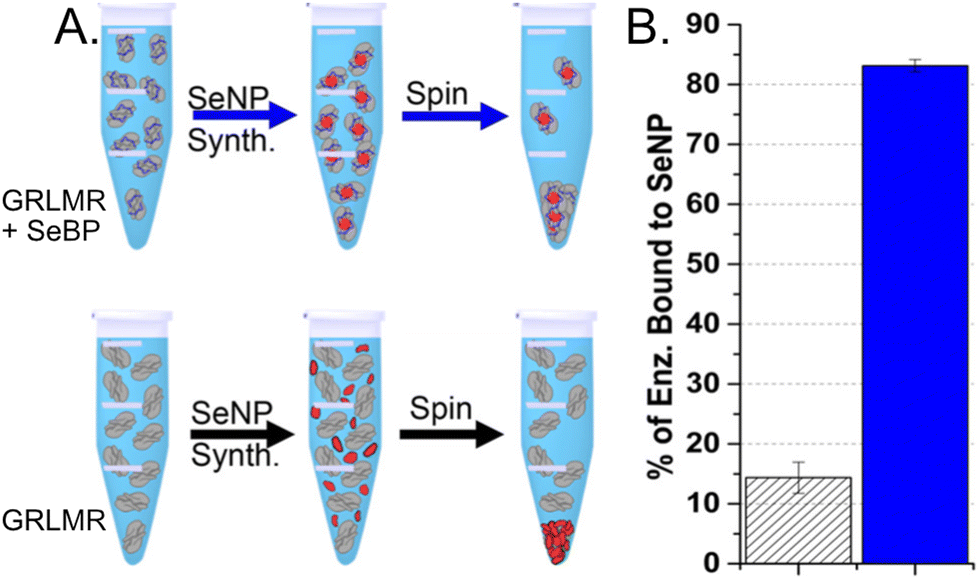 | ||
| Fig. 11 (a) Schematic of the centrifugation assay used to assess enzyme-nanoparticle binding for GRLMR-SeBP (top) and GRLMR (bottom). (b) Percentage of enzyme that remained bound to nanoparticles following centrifugation for GRLMR (diagonal stripe) versus GRLMR-SeBP (blue). Figure adapted from (46) with permission. Copyright 2020 American Chemical Society. | ||
In the second centrifugation assay, SeNPs were abiotically synthesized using NaBH4 reduction; the percentage of bound enzyme was again determined by measuring the protein concentration in the soluble fraction after precipitation. GRLMR-SeBP preferentially binds smaller SeNPs (30 nm or 50 nm) compared to larger SeNPs (125 nm or 900 nm). This indicates that there is an intrinsic preference of GRLMR-SeBP to bind SeNPs that are close to the typical SeNP size formed by enzymatic reduction. These results demonstrate that GRLMR-SeBP forms smaller and more monodisperse SeNPs than GRLMR. The results also demonstrate that concatenating SeBP to GRLMR improves the binding affinity for smaller SeNPs.
Raman spectroscopy was used to confirm enzyme conjugation by monitoring established histidine-metal binding modes and peptide backbone conformation in both free and SeNP bound states. The interpretation of Raman spectra suggested that the SeBP peptide forms covalent bonds to histidine residues via mono- and bivalent conjugation, which is supported by observing conformational shifts in its backbone signature from an unordered structure to a β-sheet configuration. Finally, the enzymatic activities of GRLMR versus GRLMR-SeBP were measured by spectroscopically monitoring NADPH depletion during enzymatic reactions. It was found that the KM is significantly lower for GRLMR-SeBP than GRLMR alone when HNaSeO3 was used as the substrate (0.22 ± 0.06 mM versus 1.92 ± 1.28 mM). Additionally, the kcat was significantly higher for GRLMR-SeBP than GRLMR when using HNaSeO3 as the substrate (40 ± 2 min−1versus 23 ± 9 min−1). When GSSG was used as the substrate, the measured KM or kcat values were not significantly different for GRLMR versus GRLMR-SeBP (Fig. 12). These results indicate that concatenating SeBP to GRLMR leads to a more favourable enzyme-substrate complex and faster enzyme activity when selenite is used as the substrate. A possible reason for the increased enzyme-substrate stability and faster enzyme activity is that SeBP preconcentrates selenite near the active site. This could be due to the introduction of five positively charged histidine residues near the active site.
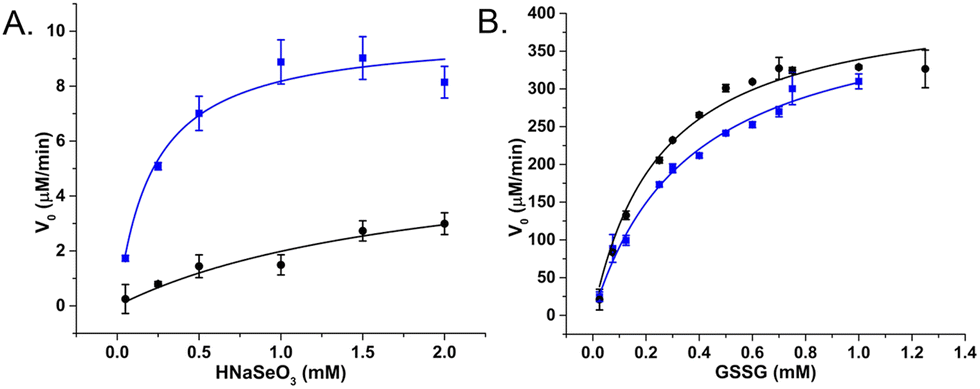 | ||
| Fig. 12 V 0 plotted against substrate concentration of GRLMR (black) and GRLMR-SeBP (blue) using (a) HNaSeO3 as a substrate, (b) GSSG as a substrate. Figure adapted from (46) with permission. Copyright 2020 American Chemical Society. | ||
Overall, the GRLMR-SeBP construct appears to constitute a cloneable SeNP (cSeNP). This construct selects the element (Se) from a background containing many metal(loid) ions; the construct reduces the Se in an NADPH dependent process to form Se precipitates of the red allotrope of Se(0). The SeBP arrests the particle size at a consistent size. Therefore, this construct—which is encoded in DNA—makes 35 nm diameter, spherical red Se nanoparticles that remain associated with the construct.
Assessment of molecular imaging with this cSeNP construct is ongoing and the cSeNP appears effective—localizing SeNPs to the expected intracellular positions of the proteins to which cSeNP is genetically concatenated.
Motivation for “multi-color” cNPs
Above is the ‘story of a cSeNP’. For our goal of creating cNPs for imaging applications, multiplexed, simultaneous tagging of different proteins with distinguishable tags is a longer-term goal. Fluorescent proteins that were originally isolated as green (e.g., GFP) were subsequently evolved in laboratories to emit different colors (red, yellow, etc.).33 The multiple colors of fluorescent protein now enables multiplexed imaging of multiple proteins of interest, each labeled with a different color of fluorescent protein. Similarly, multiple cNP proteins, each producing a distinguishable cNP, comprise a panel of cNPs that enable multiplexed tagging of proteins of interest.Inorganic NPs can be distinguished in EM on the basis of their elemental composition, size, and/or shape. For other modalities like X-ray imaging, elemental composition of the cNP may be the chief means of distinguishing different cNPs. The basis for distinguishing different elements in EM and X-ray imaging modalities may be as simple as elemental electron density (for instance, it is straightforward to distinguish Au and Ag NPs using TEM, where Au NPs simply have notably higher contrast). However, more sophisticated spectroscopic methods such as EDS and electron energy loss spectroscopy (EELS) can also map elemental compositions in TEM.47,48 In X-ray imaging, X-ray fluorescence methods may allow elemental mapping (although the resolution may be low) and tuning the X-ray wavelength to absorption edges of particular elements can also distinguish different elements, although variable wavelength X-rays require specialized X-ray sources such as synchrotrons.
In this context, we are working to make size/shape or elementally distinct cNPs. Most of our work to-date has been toward making elementally distinct cNPs.
Identification of a TeO32− reductase/mycothione reductase from Rhodococcus erythropolis PR421
In this paper, we began by collecting environmental samples from abandoned mine sites in Clear Creek County, Colorado, USA. Mining in this region dates to the 1851 Colorado Gold Rush, and most mines have not been active for over a century. Many of these mines leach mine runoff that carries substantial metal contamination.The environmental sampling was facilitated by the Clear Creek Watershed Foundation—a nonprofit organization dedicated to the cleanup of mine sites in Clear Creek County, whose mine runoff contaminates Clear Creek with toxic metals.
Here, we worked under the hypothesis that microorganisms that survive in normally lethal concentrations of toxic metals may reduce these metals to zerovalent form as a means of detoxification. One of the isolates from this sample collection was able to grow on LB-Agar that contained normally lethal concentrations of Fe(II), Cu(II), AsO32−, SeO32−, TeO32−, Cd(II) and Zn(II).
To discover any inorganic oxidoreductases involved in this remarkable set of metal tolerances, we followed the same procedure that we used to isolate GRLMR from the Se specialized P. moraviensis stanleyae. This involves first fractionating all soluble proteins on one or more non-denaturing gels, then staining gels with different metal salts and enzyme cofactors, and finally performing proteomic mass spectrometry analysis on any bands that appear from combinations of enzyme cofactors and metal salts to identify enzymes that may have precipitated the metal salts.
Fig. 13 shows the results of this screening for metal(loid) reductase activity in this isolate. Among the inorganic ions tested, only TeO32− was enzymatically reduced in-gel (Fig. 13, panel A). Proteomic mass spectrometry revealed 13 proteins associated with the precipitate, 4 of which are NAD(P)H dependent enzymes (Fig. 13, panel B). Of those enzymes, we judged the mycothione reductase to be of the greatest interest because it belongs to the same class I pyridine nucleotide-disulphide oxidoreductase enzyme family as GRLMR and mercuric reductase. Notably, mycothione reductase fills a similar role as glutathione reductase; in mycothione containing organisms, mycothione is used to establish redox balance—whereas in most organisms, GSH accomplishes this.
 | ||
| Fig. 13 (a) Native PAGE gel sections incubated in indicated metal(loid) ions and NADPH. The arrow indicates reduced insoluble TE0 particulate. Cu(II) was precipitated in gel with and without protein. (b) Proteins associated with the excised Te(0) by LC-MS/MS. Entries in italics indicate the presence of an NAD(P)H binding domain. Figure adapted from (21) with permission. | ||
Mass spectrometry identified the organism as Rhodococcus erythropolis PR4. The genome of this organism was already sequenced and available in published sequence databases. We used the existing genomic information to determine the sequence of the mycothione reductase, and then expressed and purified this enzyme recombinantly from BL21 E. coli.
In in vitro experiments, the mycothione reductase was found to have significant selectivity for tellurite over selenite. This was surprising because the oxyanions are very similar, and the reduction potential for selenite makes it easier to reduce than tellurite. The standard reduction of potential for the reaction TeO32− + 3H2O + 4e− ↔ Te + 6OH− is −0.57 V vs. Hydrogen. The corresponding reduction of SeO32− is −0.366 V. Thus, SeO32− is the preferred substrate for reduction in the absence of a mechanism for substrate selectivity.
This selectivity for Te over Se was quantified in assays in which both TeO32− and SeO32− were present in different molar ratios. Fig. 14 shows that in equimolar concentrations of Te and Se, the enzymatic precipitant incorporated 7 tellurium atoms for each selenium atom as determined by inductively coupled plasma mass spectrometry analysis. Even when selenium was present in a 10-fold excess to tellurium, the enzymatic products incorporated tellurium in an almost 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio.
:1 ratio.
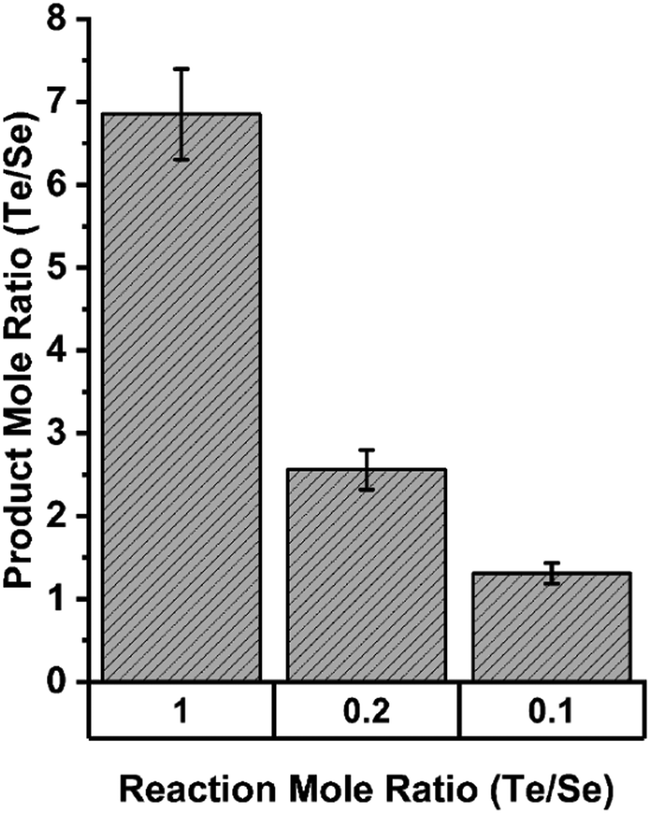 | ||
| Fig. 14 Elemental composition of precipitates resulting from enzymatic reactions in indicated ratios of tellurite and selenite as determined by ICP MS. Figure adapted from (21) with permission. | ||
This selectivity was found to depend on pH. Assays done at pH values of 6, 7, 8 and 9 showed the greatest discrimination between tellurium and selenium at pH 6, with decreasing selectivity as pH rises (Fig. 15).
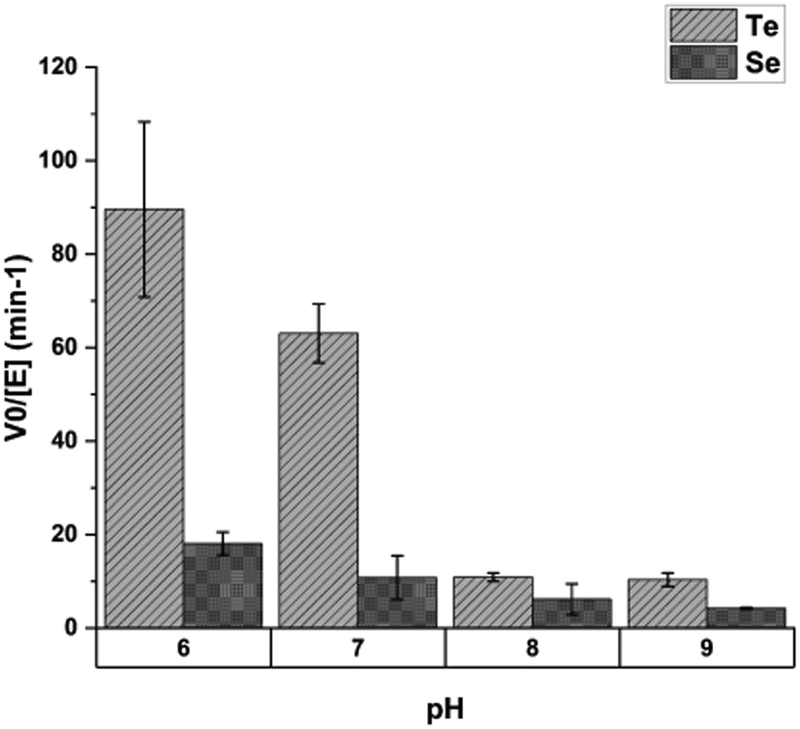 | ||
| Fig. 15 The effect of pH on mycothione reductase substrate selectivity; Y-axis shows initial velocity of mycothione reductase with tellurite or selenite. Each substrate was present at 2 mM. Figure adapted from (21) with permission. | ||
In this paper, we postulated a mechanism for selectivity of tellurium over selenium by the mycothione reductase which we now view as flawed. Here, we postulate what we believe to be a more plausible mechanism for enzymatic preference of Te over Se.
A homology model for the structure of mycothione reductase was generated with Phyre2. This homology model was subsequently aligned with GRLMR in Pymol to show mutations which may contribute to the observed substrate specificity.
Several notable mutations in mycothione reductase are located in an apparent cavity that is oppositional to the putative substrate entry channel of GRLMR. Four of the seven mutations within this region result in aromatic residues for mycothione reductase, with several of these residues being Histidine. This set of aromatic residues may contract the immediately surrounding region through π–π stacking interactions; this contraction may expand the substrate entry channel, accommodating of TeO32−, which is a larger substrate than SeO32−. The protonation of the histidine residues at lower pH values is implicated in this structural rearrangement by the pH dependence of the observed selectivity.
In summary, we isolated a Class I pyridine nucleotide-disulphide oxidoreductase that is specialized for tellurite reduction. This is a curious finding because tellurite reduction is energetically more difficult than selenite reduction, and selenite and tellurite are otherwise very similar oxyanions. This enzyme currently represents a lower priority for our adaptation to a cNP purpose because of the toxicity of tellurium. However, it is attractive for adaptation because mycothione is not present in most cells, meaning TeO32− will not be competing with mycothione as a substrate. (In our testing, the mycothione reductase did not act upon oxidized glutathione in any detectable manner.)
Enzyme-catalyzed in situ synthesis of temporally and spatially distinct CdSe quantum dots in biological backgrounds49
Metal chalcogenide semiconductor nanoparticles are often referred to as quantum dots. This naming reflects the observation that electrons within semiconductor nanoparticles behave like a quantum particle in a box. Quantum dots can be very bright luminescent emitters with quantum yields over 90%. This makes quantum dots attractive as potential luminescent labels in optical or fluorescence microscopy.Many semiconductor nanoparticles are made as metal-selenides, for example, CdSe. In this paper, we found that when Cd2+ ions are included in the synthesis of SeNPs by GRLMR, what results are luminescent CdSe nanoparticle quantum dots, instead of the non-emissive SeNPs that we made previously. Therefore, GRLMR joins cystathionine γ-lyase from Stenotrophomonas maltophilia50,51 and nitrate reductase from Fusarium oxysporum52 in a category of enzymes that generate active monomers to help mediate metal nanoparticle growth.
In prior papers that identified enzymes that synthesize quantum dots, a general theme is that the enzymes generate dianionic chalcogenides (e.g., S2− from cysteine). These dianionic chalcogenides then crystalize with dicationic metals (e.g., Cd2+). The resulting nanocrystals comprise quantum dots.
Following this rationale, we hypothesized that GRLMR might reduce GS-Se-SG (where Se's oxidation state is +2) to Se2−. The Se2−ions could then crystallize with any Cd2+ forming fluorescent semiconductor nanocrystals. This rationale is summarized in Fig. 16. In this rationale, the presence of dissolved oxygen oxidizes Se2− to Se(0) which forms the usual observed SeNP product of GRLMR. Evidence for this hypothesis arises from the observation that the QDs only form when oxygen is excluded from GRLMR catalyzed SeNP synthesis in the presence of Cd2+.
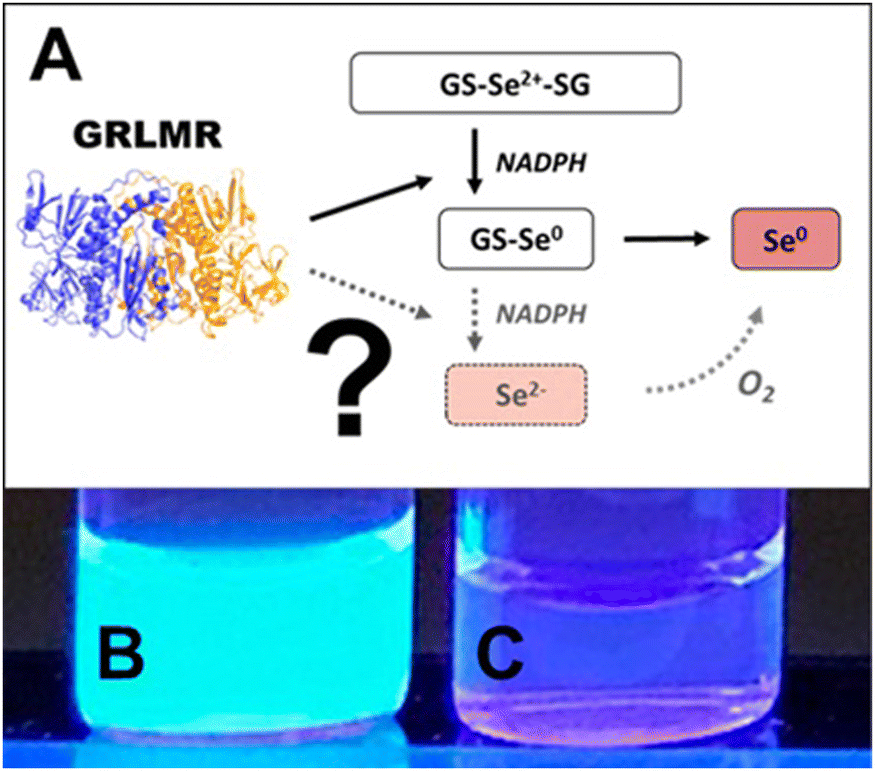 | ||
| Fig. 16 (A) Sketch of suggested selenite reduction pathway via GRLMR. Fluorescence can be visualized through UV illumination of particles formed by reduction of Se2+ to Se2− by GRLMR with Cd2+ present in (B) air-free conditions and (C) in atmosphere. Figure adapted from (49) with permission. | ||
Much of this paper describes mechanistic work showing that while GSH can also reduce SeO32− to Se(0) and Se2−, the rate of this process is slow compared to the enzymatic process. Overall, this paper shows that GRLMR can be used to form CdSe semiconductor quantum dots. This is appealing because of the possibility of making cNPs that are both electron dense and fluorescent. Such a tool is useful in the emerging field of CLEM—where fluorescence microscopy images are used to determine which parts of a sample should be examined at high resolution by EM, and which parts are of less interest.
Other approaches to cloneable contrast in EM
In addition to the cNP approach, we have attempted to form cloneable contrast agents from the naturally occurring metal handling proteins Dps and metallothionein. The Dps approach failed completely and has never been published. The metallothionein approach functions in the limited circumstances of high-copy number proteins in fixed cells, as outlined below.Metallothionein as a cloneable tag for protein localization by electron microscopy of cells53
We explored the use of metallothionein as a cloneable contrast tag. Metallothioneins are small cysteine rich proteins belonging to the broader class of phytochelatins. Such proteins are naturally expressed during heavy metal stress to most cells. The cysteine-rich proteins chelate metal ions, helping to sequester and reduce toxic effects of metals. There is some prior history of investigation of metallothionein as a cloneable contrast agent by others.Previous studies demonstrated by mass-spectrophotometric measurement that metallothionein can capture enough gold atoms to theoretically be visible in cellular electron microscopy. Metallothionein can achieve this function when conjugated to a protein.36 These studies increased the appeal of metallothionein as a cloneable contrast tag but did not demonstrate success in imaging.
We do not define cloneable contrast tags formed by metallothionein as a biogenic cNP for three reasons. First, these tags are essentially inorganic coordination complexes—which are not classically ‘particulate’—even as they may appear as such in an electron microscope. Second, the chemistries developed that allow successful imaging of metallothionein are incompatible with living cells; more specifically, the chemistries developed here use Aun+ salts, which are quite reactive with living cells. Finally, a cNP can be modified by altering the DNA encoding the cNP. In an approach where the underlying chemistry is stoichiometric binding of metal ions to cysteine, the only way to modify the construct to modify metal activity is adding or removing cysteine residues to the protein. This manipulation is only able to modify the number of metal ions coordinated—i.e., changing the DNA cannot dramatically modify materials properties such as elemental composition, crystal structure, size, or shape.
Nonetheless, metallothionein is appealing for potential applications as a cloneable contrast tag, whose electron scattering properties could be enhanced post-fixation and before EM imaging.
In this study, we attempted to label several proteins, which provided an assessment of how well metallothionein performed as a cloneable contrast tag. Metallothioneins range in weight from 0.5–14 kDa, which satisfies the requirement of a small-size tag. To assess the ability of metallothionein to label proteins with a high copy number, wild-type desmin (a filamenting protein) was tagged with a single metallothionein and expressed in bacteria. After desmin was freeze-substituted and treated with aurothiomalate, the metallothionein-labeled desmin showed staining with small particles. Furthermore, this type of staining was not seen in the wild-type desmin, which indicated that the staining was due to metallothionein and that the staining resulted in a good signal to noise ratio (Fig. 17).
 | ||
| Fig. 17 Tomographic slices of (A) wild-type desmin (B) metallothionein-labeled desmin. Scale bars = 100 nm. Figure adapted from (53) with permission. Copyright 2015 Royal Microscopical Society. | ||
To assess the ability of metallothionein to localize a tagged protein in the complex environment of a cell, an SPC42 protein product was tagged with two copies of metallothionein. SPC42 is a component of the yeast spindle pole body and is located approximately at the plane of the nuclear envelope. It was found that following the same procedure as followed for desmin did not yield adequate signal. Instead, use of a Nanoprobes gold enhancement kit was required (Fig. 18). To this point, sections were stained with aurothiomalate after freeze substitution. Therefore, staining during the process of freeze substitution was assessed next.
 | ||
| Fig. 18 (A) Diagram of spc42; the metallothionein tag was added to the C-terminus. (B) Tomographic slices of wild-type spc42. Tomographic slices of Spc42-2x metallothionein (C) incubated with aurothiomalate alone, and (D) first stained with aurothiomalate followed by enhancement with the Nanoprobes gold enhancement kit. Scale bars = 100 nm. Figure adapted from (53) with permission. Copyright 2015 Royal Microscopical Society. | ||
It was found that the best signal was achieved by first staining sections with Au(III)Cl3 during freeze-substitution, then treating samples with diglyme, and finally enhancing using a silver enhancement protocol.54 Control samples not tagged with metallothionein did not show staining, regardless of whether they underwent the diglyme and silver enhancement protocol. However, metallothionein tagged samples that underwent staining during freeze-substitution, diglyme treatment, and silver enhancement showed strong labeling, which allowed single particles to be detected (Fig. 19). The studies using desmin and spc42 provide evidence that metallothionein can reveal the location of proteins with many copies localized to a small area.
 | ||
| Fig. 19 Staining of wild-type spc42 during freeze substitution (A) without diglyme treatment and silver enhancement and (B) with diglyme treatment and silver enhancement. Staining is not observed in either condition. Staining of Spc42-2xMTH sections (C) without diglyme and silver enhancement and (D) with diglyme treatment and silver enhancement, which allowed for the detection of particles (D, inset). Scale bars = 100 nm. Figure adapted from (53) with permission. Copyright 2015 Royal Microscopical Society. | ||
To assess the capabilities of metallothionein as a tag for low-copy number proteins, protein components of the nuclear-pore complex were tagged with metallothionein and stained. No staining of the nuclear pore complex was observed, although many staining and enhancement protocols were attempted.
Despite unsuccessful attempts to stain components of the NPC, the successful applications of metallothionein to stain both desmin and spc42 demonstrates the potential of metallothionein as a cloneable contrast tag in fixed cells.
Overall, this paper shows that metallothionein can be effective to localize high copy number proteins that are concentrated in specific areas, such as filamentous or ultrastructural proteins. For low copy number or isolated proteins, metallothionein does not appear effective. These approaches with metallothionein also require that cells are fixed before metal incubation and are therefore somewhat limited compared to the cNP platform approach, which can use essential elements to form high contrast particles in live cells.
Dps as a cloneable tag
Iron oxide encapsulating proteins such as ferritin and Dps create iron oxides that can be identified in electron microscopy. Ferritin has been investigated as a cloneable tag.3 Ferritin is comprised of 24 protein subunits that assemble into a capsule which can contain an iron oxide core of up to ∼8 nm in diameter. Concatenating a ferritin subunit to a protein of interest at the DNA level can cause a ferritin capsule to become attached to that protein of interest. This approach has not become widely used because several proteins of interest can become integrated into a single ferritin capsule. This cross-linking, as well as the sheer size of ferritin, are likely to cause artifactual observations around the localization of the protein of interest.An improvement on this approach may be to use Dps, which is sometimes described as a mini-ferritin. Dps is a capsule protein comprised of 12 subunits, which can contain an iron oxide mineral up to ∼4 nm in diameter, which can be visualized in cells. A ‘single-chain’ Dps—where up to 12 Dps protomers are linked with a flexible linker sequence at the DNA level is in principle possible.
Our work on Dps involved making the DNA that encodes Dps from 12 Dps protomers, each linked by a flexible linker. Despite many attempts, we were unable to express functional Dps as a 12-mer, 6-mer, 4-mer or 3-mer construct. For now, we hypothesize the linker that we chose may have been incompatible with DPS folding and therefore function. We hypothesize that different linkers might allow DPS protomers to be linked into functional units, using modern computational protein folding assessment tools to identify successful linkers.
Outlook and future directions for cNPs
As a field, synthetic chemistry aims to develop and improve methods for creation of well-defined molecules and materials. The synthetic chemistry sub-field of organic synthesis is particularly impressive in this regard, with well-developed methods for discrete addition or removal of atoms to defined locations in complex molecules. Cyclization reactions, solid phase synthesis and enantioselective control, among other tools, facilitate the precision synthesis of carbon-rich products including pharmaceuticals, oligonucleotides, and plastics.Outside of organic synthesis, precise ‘magic number’ nanoclusters are now routinely made with atomic precision and resulting defined structure, up to about ∼500 metal atoms. These nanoclusters are already used as extrinsic biological contrast agents, catalysts, and their continued synthetic exploration allows advancement of fundamental knowledge of cluster physics. Improved control over NP synthesis in biological contexts is a logical (and destined) progression.
Research on biogenic NPs appears to be currently in a “gold rush” era, with sharply growing numbers of active researchers and publications (Fig. 20). These publications include those that address fundamental aspects of nanoparticle chemistry, new synthetic routes, and a wide variety of potential applications. The research area is presently highly interdisciplinary, with contributions coming from chemists, physicists, geologists, and microbiologists.
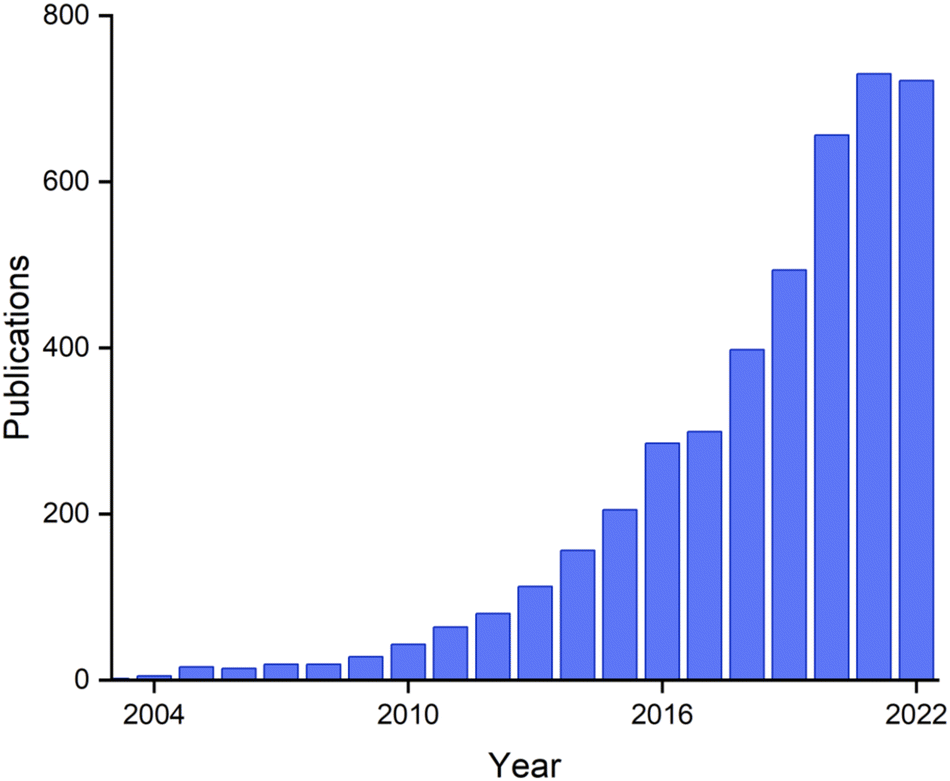 | ||
| Fig. 20 Papers on the topic of ‘biogenic nanoparticles’ per year, from Web of Science. | ||
As the field of biogenic nanoparticles continues to develop, we anticipate an increasing emphasis on synthetic precision, by analogy to the development of other synthetic chemistry sub-fields. We postulate that the cNP platform approach, which mixes and matches well-defined metal oxidoreductases and proteins and peptides that enforce NP shape, provides a robust approach to improving biogenic NP precision and design. Biological evolution over eons has produced many examples of atomically precise/defined molecules. For bio-organic synthesis, biology often allows synthetic organic transformations that are difficult or impossible to achieve in vitro. It's an ambition of ours to push cNP synthesis to achieve similar levels of atomic precision in nanomaterials synthesis, using well-defined biological approaches.
One way to conceptualize this approach is as an extension of the central dogma of molecular biology. This states that DNA contains all the information to create an atomically precise protein. To convert DNA information into protein function, the DNA is transcribed into RNA, which is then translated into functional proteins.
Extending this dogma to cNP synthesis, we suggest that DNA (information) can be transcribed into mRNA (information transfer) which is then translated into protein (translating information into function). The protein then creates a cNP, translating function into discrete synthetic product. This approach suggests the possibility of next-generation nanoscale materials where the synthetic information is not contained in journal-article text, but rather in DNA sequence.
Much in the same way that unnatural amino acid incorporation has expanded the “chemical space” that biology is allowed to partake in, so too will cNPs open the door to exploring new interfaces of nanoscale inorganic nanomaterials and biosystems. As we approach this paradigm, we ask ourselves: what new and exciting things will be discovered?
cNP synthesis does have intrinsic limitations. Not every metal(loid) on the periodic table, for instance, is compatible with redox reactions that are biologically achievable. Elements with redox potentials outside the range of −1.0 V to 1.0 V relative to SHE will either be too favorable for controlled reduction or too unfavorable for any redox chemistry to occur at all. In the case of ‘uncontrolled’ reduction—oxidation of ubiquitous biomolecules such as sugars, tyrosine and tryptophan, and nucleobases by easily reduced elements is likely to be implicated. Such elements are likely to be reduced in nonspecific ways within biological matrices. Pourbaix diagrams describe expected redox potential versus pH and can help identify elements that are good candidates for inclusion in cNPs.55,56
For our target applications in imaging, electron-rich (e.g., higher-Z) elements are the most attractive, as they provide greater contrast relative to the less electron rich elements of C, O, N, H and P that comprise most of the biological matrix. Based on these considerations, Zn, As, Te, Fe, Cd, Se, Bi and Co ions are of highest interest for cNP incorporation because of relatively favorable redox potentials and Z-values. We consider comparably toxic elements as lower priority choices. Although the cNP machinery may confer tolerance to elements such as Cd and Te, their intrinsic toxicity at small concentrations presents complications that are less pronounced for biologically essential elements such as Zn, Fe and Se, as well as elements that are generally less toxic such as Bi (a component in pharmaceuticals such as Pepto-Bismol). Bismuth is particularly attractive for our future work as it is the most electron-rich non-radioactive element on the periodic table.
Many metal(loid) oxidoreductases are comparatively large proteins. Class I pyridine nucleotide-disulphide oxidoreductases, including GRLMR, are typically ∼100 kDa homodimers—before they are saddled with an attached inorganic nanoparticle.19–21,57 For comparison, widely used fluorescent proteins like GFP are ∼27 kDa monomers. GFP is well known to alter the biological activity of many of the proteins that it is concatenated to, potentially resulting in artefactual observations.58,59 It's hypothesized that larger ‘tags’ are more likely to alter biological activity of concatenated proteins; large size can hinder POI diffusion kinetics and position contrasting markers distally to the point of interest, whereas multimericity can convolute labelling efficiency. Therefore, one of our future directions in cNP synthesis is to develop smaller inorganic oxidoreductases.
We are investigating approaches for making GRLMR a functional monomer of decreased mass. Other metal(loid) oxidoreductase enzymes may be of interest as starting points for further engineering. For instance, a 27 kDa NADH-dependent cytochrome b5 reductase was recently isolated from a strain of Muchor racemosus;60,61 similar to GRLMR, the enzyme conveys nanoparticle-mediated metal tolerance to silver and palladium when cloned into laboratory microorganisms. Alternatively, arsenate reductase62 may be amenable to engineering processes that alter its substrate specificity to metals other pnictides such as Bismuth.
It is possible that newfound and promising enzymatic systems may possess kinetics or substrate specificity that nullify their otherwise ideal characteristics (e.g., mass, stability, metal(loid) reduction, monomericity, etc.). Given the discrete genetic nature of cNPs, we offer the prospect of using directed evolution as a means of manipulating enzyme function to better serve the needs of cloneable contrast. Doing so will necessitate the development of robust selection methods for metal reductases, which is a relatively unexplored area.
Conclusions
In this feature-article, we have described the development of a cNP platform technology. Our main goal is to develop cNP based cloneable contrast agents for biological imaging. It is conceivable that our approach may one day result in a single cloneable contrast agent that allows imaging in many modalities (optical, fluorescent, electron, X-ray, magnetic imaging). We are also interested, to a lesser extent, in using cNP approaches for bioremediation of toxic metals.The platform is not limited to applications in imaging and bioremediation. Rather, we expect that many applications explored for biogenic NPs such as biomining and solar photoconversion might one day benefit from the increased precision of synthetic product made possible with the well-defined components of this platform and their intrinsic linkage to laboratory evolvable DNA sequences. The intracellular aspect of cNP synthesis further offers the prospect of integrating cNPs with other cellular processes to offer the prospect of cellular engineering to accomplish yet unimagined goals.
Author contributions
A. R. H., R. S. C., G. A. M., T. T., K. M. B., and C. J. A. wrote the first draft of the manuscript. A. R. H., R. S. C., G. A. M., B. F. G., K. M. B., and C. J. A. revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge assistance from David Holm, director of the Clear Creek Watershed Foundation. Charleston Ducote designed the table of contents figure. CJA acknowledges NIH award R01 GM137139 for support of this work.Notes and references
- A. Dieudonné, D. Pignol and S. Prévéral, Magnetosomes: Biogenic Iron Nanoparticles Produced by Environmental Bacteria, Appl. Microbiol. Biotechnol., 2019, 103(9), 3637–3649, DOI:10.1007/s00253-019-09728-9.
- C. Jogler, W. Lin, A. Meyerdierks, M. Kube, E. Katzmann, C. Flies, Y. Pan, R. Amann, R. Reinhardt and D. Schüler, Toward Cloning of the Magnetotactic Metagenome: Identification of Magnetosome Island Gene Clusters in Uncultivated Magnetotactic Bacteria from Different Aquatic Sediments, Appl. Environ. Microbiol., 2009, 75(12), 3972–3979, DOI:10.1128/AEM.02701-08.
- Q. Wang, C. P. Mercogliano and J. Löwe, A Ferritin-Based Label for Cellular Electron Cryotomography, Structure, 2011, 19(2), 147–154, DOI:10.1016/j.str.2010.12.002.
- N. I. Clarke and S. J. Royle, FerriTag Is a New Genetically-Encoded Inducible Tag for Correlative Light-Electron Microscopy, Nat. Commun., 2018, 9(1), 2604, DOI:10.1038/s41467-018-04993-0.
- B. Jiang, L. Fang, K. Wu, X. Yan and K. Fan, Ferritins as Natural and Artificial Nanozymes for Theranostics, Theranostics, 2020, 10(2), 687–706, DOI:10.7150/thno.39827.
- V. O. Karas, I. Westerlaken and A. S. Meyer, The DNA-Binding Protein from Starved Cells (Dps) Utilizes Dual Functions To Defend Cells against Multiple Stresses, J. Bacteriol., 2015, 197(19), 3206–3215, DOI:10.1128/JB.00475-15.
- K. Orban and S. E. Finkel, Dps Is a Universally Conserved Dual-Action DNA-Binding and Ferritin Protein, J. Bacteriol., 2022, 204(5), e00036, DOI:10.1128/jb.00036-22.
- M. J. Capeness, V. Echavarri-Bravo and L. E. Horsfall, Production of Biogenic Nanoparticles for the Reduction of 4-Nitrophenol and Oxidative Laccase-Like Reactions, Front. Microbiol., 2019, 10, 997, DOI:10.3389/fmicb.2019.00997.
- S. Y. Sawant, M. S. Sayed, T. H. Han, M. R. Karim, J.-J. Shim and M. H. Cho, Bio-Synthesis of Finely Distributed Ag Nanoparticle-Decorated TiO2 Nanorods for Sunlight-Induced Photoelectrochemical Water Splitting, J. Ind. Eng. Chem., 2019, 69, 48–56, DOI:10.1016/j.jiec.2018.09.002.
- P. Singh, A. Garg, S. Pandit, V. Mokkapati and I. Mijakovic, Antimicrobial Effects of Biogenic Nanoparticles, Nanomaterials, 2018, 8(12), 1009, DOI:10.3390/nano8121009.
- A. M. Fayaz, K. Balaji, M. Girilal, R. Yadav, P. T. Kalaichelvan and R. Venketesan, Biogenic Synthesis of Silver Nanoparticles and Their Synergistic Effect with Antibiotics: A Study against Gram-positive and Gram-negative Bacteria, Nanomedicine, 2010, 6(1), 103–109, DOI:10.1016/j.nano.2009.04.006.
- M. C. Zambonino, E. M. Quizhpe, L. Mouheb, A. Rahman, S. N. Agathos and S. A. Dahoumane, Biogenic Selenium Nanoparticles in Biomedical Sciences: Properties, Current Trends, Novel Opportunities and Emerging Challenges in Theranostic Nanomedicine, Nanomaterials, 2023, 13(3), 424, DOI:10.3390/nano13030424.
- V. Nayak, K. R. Singh, R. Verma, M. D. Pandey, J. Singh and R. Pratap Singh, Recent Advancements of Biogenic Iron Nanoparticles in Cancer Theranostics, Mater. Lett., 2022, 313, 131769, DOI:10.1016/j.matlet.2022.131769.
- A. Heuer-Jungemann, N. Feliu, I. Bakaimi, M. Hamaly, A. Alkilany, I. Chakraborty, A. Masood, M. F. Casula, A. Kostopoulou, E. Oh, K. Susumu, M. H. Stewart, I. L. Medintz, E. Stratakis, W. J. Parak and A. G. Kanaras, The Role of Ligands in the Chemical Synthesis and Applications of Inorganic Nanoparticles, Chem. Rev., 2019, 119(8), 4819–4880, DOI:10.1021/acs.chemrev.8b00733.
- S. P. Fricker, Metal Based Drugs: From Serendipity to Design, Dalton Trans., 2007, 4903, 10.1039/b705551j.
- K. L. Haas and K. J. Franz, Application of Metal Coordination Chemistry To Explore and Manipulate Cell Biology, Chem. Rev., 2009, 109(10), 4921–4960, DOI:10.1021/cr900134a.
- M. Jozefczak, T. Remans, J. Vangronsveld and A. Cuypers, Glutathione Is a Key Player in Metal-Induced Oxidative Stress Defenses, Int. J. Mol. Sci., 2012, 13(3), 3145–3175, DOI:10.3390/ijms13033145.
- P. R. E. Mittl and G. E. Schulz, Structure of Glutathione Reductase from Escherichia Coli at 1.86 Å Resolution: Comparison with the Enzyme from Human Erythrocytes, Protein Sci., 2008, 3(5), 799–809, DOI:10.1002/pro.5560030509.
- I. Carlberg and B. Mannervik, in [59] Glutathione Reductase, Methods in Enzymology, Elsevier, vol. 113, 1985, pp. 484–490 DOI:10.1016/S0076-6879(85)13062-4.
- B. Fox and C. T. Walsh, Mercuric Reductase. Purification and Characterization of a Transposon-Encoded Flavoprotein Containing an Oxidation-Reduction-Active Disulfide, J. Biol. Chem., 1982, 257(5), 2498–2503, DOI:10.1016/S0021-9258(18)34951-2.
- Z. J. Butz, A. Hendricks, K. Borgognoni and C. J. Ackerson, Identification of a TeO32− Reductase/Mycothione Reductase from Rhodococcus Erythropolis PR4, FEMS Microbiol. Ecol., 2020, 97(1), fiaa220, DOI:10.1093/femsec/fiaa220.
- R. Nemeth, M. Neubert, T. Ni and C. J. Ackerson, The Metalloid Reductase of Pseudomonas Moravenis Stanleyae Conveys Nanoparticle Mediated Metalloid Tolerance, ACS Omega, 2018, 3(11), 14902–14909, DOI:10.1021/acsomega.8b00826.
- S. R. Whaley, D. S. English, E. L. Hu, P. F. Barbara and A. M. Belcher, Selection of Peptides with Semiconductor Binding Specificity for Directed Nanocrystal Assembly, Nature, 2000, 405(6787), 665–668, DOI:10.1038/35015043.
- C. J. Carter, C. J. Ackerson and D. L. Feldheim, Unusual Reactivity of a Silver Mineralizing Peptide, ACS Nano, 2010, 4(7), 3883–3888, DOI:10.1021/nn100630v.
- K. Chandra, K. S. B. Culver, S. E. Werner, R. C. Lee and T. W. Odom, Manipulating the Anisotropic Structure of Gold Nanostars Using Good's Buffers, Chem. Mater., 2016, 28(18), 6763–6769, DOI:10.1021/acs.chemmater.6b03242.
- J. Xie, J. Y. Lee and D. I. C. Wang, Seedless, Surfactantless, High-Yield Synthesis of Branched Gold Nanocrystals in HEPES Buffer Solution, Chem. Mater., 2007, 19(11), 2823–2830, DOI:10.1021/cm0700100.
- C. K. Payne, A Protein Corona Primer for Physical Chemists, J. Chem. Phys., 2019, 151(13), 130901, DOI:10.1063/1.5120178.
- P. Pino, B. del; Pelaz, Q. Zhang, P. Maffre, G. U. Nienhaus and W. J. Parak, Protein Corona Formation around Nanoparticles – from the Past to the Future, Mater. Horiz., 2014, 1(3), 301–313, 10.1039/C3MH00106G.
- M. Lenz, B. Kolvenbach, B. Gygax, S. Moes and P. F. X. Corvini, Shedding Light on Selenium Biomineralization: Proteins Associated with Bionanominerals, Appl. Environ. Microbiol., 2011, 77(13), 4676–4680, DOI:10.1128/AEM.01713-10.
- K. Chen and F. H. Arnold, Engineering New Catalytic Activities in Enzymes, Nat Catal., 2020, 3(3), 203–213, DOI:10.1038/s41929-019-0385-5.
- O. Shimomura, Structure of the Chromophore of Aequorea Green Fluorescent Protein, FEBS Lett., 1979, 104(2), 220–222, DOI:10.1016/0014-5793(79)80818-2.
- M. Chalfie, Y. Tu, G. Euskirchen, W. Ward and D. Prasher, Green Fluorescent Protein as a Marker for Gene Expression, Science, 1994, 263(5148), 802–805, DOI:10.1126/science.8303295.
- D. M. Chudakov, M. V. Matz, S. Lukyanov and K. A. Lukyanov, Fluorescent Proteins and Their Applications in Imaging Living Cells and Tissues, Physiol. Rev., 2010, 90(3), 1103–1163, DOI:10.1152/physrev.00038.2009.
- X. Shu, V. Lev-Ram, T. J. Deerinck, Y. Qi, E. B. Ramko, M. W. Davidson, Y. Jin, M. H. Ellisman and R. Y. Tsien, A Genetically Encoded Tag for Correlated Light and Electron Microscopy of Intact Cells, Tissues, and Organisms, PLoS Biol., 2011, 9(4), e1001041, DOI:10.1371/journal.pbio.1001041.
- S. S. Lam, J. D. Martell, K. J. Kamer, T. J. Deerinck, M. H. Ellisman, V. K. Mootha and A. Y. Ting, Directed Evolution of APEX2 for Electron Microscopy and Proximity Labeling, Nat. Methods, 2015, 12(1), 51–54, DOI:10.1038/nmeth.3179.
- C. P. Mercogliano and D. J. DeRosier, Concatenated Metallothionein as a Clonable Gold Label for Electron Microscopy, J. Struct. Biol., 2007, 160(1), 70–82, DOI:10.1016/j.jsb.2007.06.010.
- L. C. Staicu, E. D. van Hullebusch and C. Ackerson, Editorial: Microbial Biominerals: Toward New Functions and Resource Recovery, Front. Microbiol., 2021, 12, 796374, DOI:10.3389/fmicb.2021.796374.
- L. C. Staicu, C. J. Ackerson, P. Cornelis, L. Ye, R. L. Berendsen, W. J. Hunter, S. D. Noblitt, C. S. Henry, J. J. Cappa, R. L. Montenieri, A. O. Wong, L. Musilova, M. Sura-de Jong, E. D. van Hullebusch, P. N. L. Lens, R. J. B. Reynolds and E. A. H. Pilon-Smits, Pseudomonas Moraviensis Subsp. Stanleyae, a Bacterial Endophyte of Hyperaccumulator Stanleya Pinnata, Is Capable of Efficient Selenite Reduction to Elemental Selenium under Aerobic Conditions, J. Appl. Microbiol., 2015, 119(2), 400–410, DOI:10.1111/jam.12842.
- S. D. Noblitt, L. C. Staicu, C. J. Ackerson and C. S. Henry, Sensitive, Selective Analysis of Selenium Oxoanions Using Microchip Electrophoresis with Contact Conductivity Detection, Anal. Chem., 2014, 86(16), 8425–8432, DOI:10.1021/ac502013k.
- T. W. Ni, L. C. Staicu, R. S. Nemeth, C. L. Schwartz, D. Crawford, J. D. Seligman, W. J. Hunter, E. A. H. Pilon-Smits and C. J. Ackerson, Progress toward Clonable Inorganic Nanoparticles, Nanoscale, 2015, 7(41), 17320–17327, 10.1039/C5NR04097C.
- S. Kashyap, T. J. Woehl, X. Liu, S. K. Mallapragada and T. Prozorov, Nucleation of Iron Oxide Nanoparticles Mediated by Mms6 Protein in Situ, ACS Nano, 2014, 8(9), 9097–9106, DOI:10.1021/nn502551y.
- D. M. P. Mingos and D. J. Wales, Prentice Hall advanced reference series, Introduction to Cluster Chemistry, Prentice Hall, Englewood Cliffs, N.J, 1990 Search PubMed.
- D. Scott, M. Toney and M. Muzikár, Harnessing the Mechanism of Glutathione Reductase for Synthesis of Active Site Bound Metallic Nanoparticles and Electrical Connection to Electrodes, J. Am. Chem. Soc., 2008, 130(3), 865–874, DOI:10.1021/ja074660g.
- R. D. Mavis and E. Stellwagen, Purification and Subunit Structure of Glutathione Reductase from Bakers’ Yeast, J. Biol. Chem., 1968, 243(4), 809–814, DOI:10.1016/S0021-9258(19)81737-4.
- J. Yu and C.-Z. Zhou, Crystal Structure of Glutathione Reductase Glr1 from the Yeast Saccharomyces Cerevisiae, Proteins, 2007, 68(4), 972–979, DOI:10.1002/prot.21354.
- Z. J. Butz, K. Borgognoni, R. Nemeth, Z. N. Nilsson and C. J. Ackerson, Metalloid Reductase Activity Modified by a Fused Se 0 Binding Peptide, ACS Chem. Biol., 2020, 15(7), 1987–1995, DOI:10.1021/acschembio.0c00387.
- S. R. Adams, M. R. Mackey, R. Ramachandra, S. F. Palida Lemieux, P. Steinbach, E. A. Bushong, M. T. Butko, B. N. G. Giepmans, M. H. Ellisman and R. Y. Tsien, Multicolor Electron Microscopy for Simultaneous Visualization of Multiple Molecular Species, Cell Chem. Biol., 2016, 23(11), 1417–1427, DOI:10.1016/j.chembiol.2016.10.006.
- M. Scotuzzi, J. Kuipers, D. I. Wensveen, P. de Boer, K. W. Hagen, J. P. Hoogenboom and B. N. G. Giepmans, Multi-Color Electron Microscopy by Element-Guided Identification of Cells, Organelles and Molecules, Sci. Rep., 2017, 7(1), 45970, DOI:10.1038/srep45970.
- R. A. Riskowski, R. S. Nemeth, K. Borgognoni and C. J. Ackerson, Enzyme-Catalyzed in Situ Synthesis of Temporally and Spatially Distinct CdSe Quantum Dots in Biological Backgrounds, J. Phys. Chem. C, 2019, 123(44), 27187–27195, DOI:10.1021/acs.jpcc.9b05519.
- R. Dunleavy, L. Lu, C. J. Kiely, S. McIntosh and B. W. Berger, Single-Enzyme Biomineralization of Cadmium Sulfide Nanocrystals with Controlled Optical Properties, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(19), 5275–5280, DOI:10.1073/pnas.1523633113.
- Y. Wang, H. Chen, Z. Huang, M. Yang, H. Yu, M. Peng, Z. Yang and S. Chen, Structural Characterization of Cystathionine γ-Lyase SmCSE Enables Aqueous Metal Quantum Dot Biosynthesis, Int. J. Biol. Macromol., 2021, 174, 42–51, DOI:10.1016/j.ijbiomac.2021.01.141.
- S. Hamedi, M. Ghaseminezhad, S. Shokrollahzadeh and S. A. Shojaosadati, Controlled Biosynthesis of Silver Nanoparticles Using Nitrate Reductase Enzyme Induction of Filamentous Fungus and Their Antibacterial Evaluation, Artif. Cells, Nanomed., Biotechnol., 2017, 45(8), 1588–1596, DOI:10.1080/21691401.2016.1267011.
- M. K. Morphew, E. T. O’Toole, C. L. Page, M. Pagratis, J. Meehl, T. Giddings, J. M. Gardner, C. Ackerson, S. L. Jaspersen, M. Winey, A. Hoenger and J. R. Mcintosh, Metallothionein as a Clonable Tag for Protein Localization by Electron Microscopy of Cells: metallothionein for protein localization in cells, J. Microsc., 2015, 260(1), 20–29, DOI:10.1111/jmi.12262.
- M. Morphew, W. He, P. J. Bjorkman and J. R. McINTOSH, Silver Enhancement of Nanogold Particles during Freeze Substitution for Electron Microscopy: enhancement of nanogold during freeze substitution, J. Microsc., 2008, 230(2), 263–267, DOI:10.1111/j.1365-2818.2008.01983.x.
- Y. Choi and S. Y. Lee, Biosynthesis of Inorganic Nanomaterials Using Microbial Cells and Bacteriophages, Nat. Rev. Chem., 2020, 4(12), 638–656, DOI:10.1038/s41570-020-00221-w.
- Y. Choi, T. J. Park, D. C. Lee and S. Y. Lee, Recombinant Escherichia Coli as a Biofactory for Various Single- and Multi-Element Nanomaterials, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(23), 5944–5949, DOI:10.1073/pnas.1804543115.
- D. J. Carothers, G. Pons and M. S. Patel, Dihydrolipoamide Dehydrogenase: Functional Similarities and Divergent Evolution of the Pyridine Nucleotide-Disulfide Oxidoreductases, Arch. Biochem. Biophys., 1989, 268(2), 409–425, DOI:10.1016/0003-9861(89)90309-3.
- M. T. Swulius and G. J. Jensen, The Helical MreB Cytoskeleton in Escherichia Coli MC1000/PLE7 Is an Artifact of the N-Terminal Yellow Fluorescent Protein Tag, J. Bacteriol., 2012, 194(23), 6382–6386, DOI:10.1128/JB.00505-12.
- Z. Huang, C. Zhang, S. Chen, F. Ye and X.-H. Xing, Active Inclusion Bodies of Acid Phosphatase PhoC: Aggregation Induced by GFP Fusion and Activities Modulated by Linker Flexibility, Microb. Cell Fact., 2013, 12(1), 25, DOI:10.1186/1475-2859-12-25.
- S. A. Mirzaei, M. T. Yazdi and Z. Sepehrizadeh, Secretory Expression and Purification of a Soluble NADH Cytochrome B5 Reductase Enzyme from Mucor Racemosus in Pichia Pastoris Based on Codon Usage Adaptation, Biotechnol. Lett., 2010, 32(11), 1705–1711, DOI:10.1007/s10529-010-0348-z.
- N. A. Setayesh, Z. Sepehrizadeh, E. Jaberi and M. T. Yazdi, Cloning, Molecular Characterization and Expression of a CDNA Encoding a Functional NADH-Cytochrome B5 Reductase from Mucor Racemosus PTCC 5305 in E. Coli, Biol. Res., 2009, 42(2), 137–146, DOI:10.4067/S0716-97602009000200001.
- R. Mukhopadhyay and B. P. Rosen, Arsenate Reductases in Prokaryotes and Eukaryotes, Environ. Health Perspect., 2002, 110(5), 745–748, DOI:10.1289/ehp.02110s5745.
| This journal is © The Royal Society of Chemistry 2023 |