
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical generation and utilization of alkoxy radicals
Albara A. M. A.
El Gehani
,
Hussain A.
Maashi
,
James
Harnedy
and
Louis C.
Morrill
 *
*
Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: MorrillLC@cardiff.ac.uk
First published on 6th March 2023
Abstract
This highlight summarises electrochemical approaches for the generation and utilization of alkoxy radicals, predominantly focusing on recent advances (2012–present). The application of electrochemically generated alkoxy radicals in a diverse range of transformations is described, including discussion on reaction mechanisms, scope and limitations, in addition to highlighting future challenges in this burgeoning area of sustainable synthesis.
 From left to right: James Harnedy, Hussain A. Maashi, Albara A. M. A. El Gehani and Louis C. Morrill | James Harnedy obtained a Masters in Chemistry at the University of Nottingham (United Kingdom) and is now pursuing a PhD at Cardiff University. Hussain A. Maashi obtained a Bachelors in Chemistry at Jazan University (Saudi Arabia) and a Masters in Chemistry at Western Illinois University (United States). He is now pursuing a PhD at Cardiff University. Albara A. M. A. El Gehani obtained a Masters in Chemistry at Heriot-Watt University (United Kingdom) and is now pursuing a PhD at Cardiff University. James, Hussain and Albara are researching towards the development of new synthetic methodology enabled by electrochemistry. Louis Morrill received his PhD from the University of St Andrews in 2014 under the direction of Prof. Andrew Smith and undertook postdoctoral research at UC Berkeley with Prof. Richmond Sarpong. In June 2015, he initiated his independent research career at Cardiff University. Research in the group is focused on inventing new reactions in organic chemistry and developing sustainable catalytic methodologies for synthesis. |
1. Introduction
Radical chemistry has a rich and storied history, which describes the application of various radical intermediates across a diverse range of useful and elegant transformations.1 Alkoxy radicals are an important class of oxygen-centered radicals, which consist of an alkyl group bound to an electrophilic oxygen radical center.2 They are particularly high energy species due to the lack the stabilization provided by mesomeric effects and spin density delocalization found in other O-centered radicals, such as aryloxy radicals. Despite this, alkoxy radicals exhibit well defined yet diverse reactivity, including β-scission processes,3 hydrogen atom transfers (HATs),4 and addition to π-systems (Scheme 1).5 Although competition can exist between these reactivity modes, the desired pathway can often be favoured through a combination of substrate design and judicious choice of reaction conditions. As such, these privileged intermediates have been successfully employed across a plethora of powerful transformations spanning selective C–H functionalization, C–C bond activation, and heterocycle synthesis to access valuable products, including within complex molecule synthesis.6 Traditional methods for alkoxy radical generation involve the homolysis of weak oxygen–heteroatom bonds within pre-functionalized radical precursors in combination with radical initiators and/or thermal or UV photochemical activation, which can limit functional group tolerance. Photoredox catalysis has enabled alkoxy radical generation under milder conditions (i.e., low intensity irradiation with visible light),7 employing various radical precursors including peroxides, N-alkoxyphthalimides, N-alkoxypyridiniums, N-alkoxybenzimidazoles, N-alkoxytriazoliums and oxyimino acids. Recent advances in transition metal catalysis and photoredox catalysis have enabled alkoxy radical intermediates to be accessed directly from unprotected aliphatic alcohols.2 However, many of these approaches require the use of stoichiometric oxidants (e.g., K2S2O8 or hypervalent iodine reagents) and/or precious metal (photo)catalysts. As such, the development of alternative, more sustainable, approaches for the generation of alkoxy radicals, which also offer opportunities for the discovery of new reactivity, is an important and timely pursuit.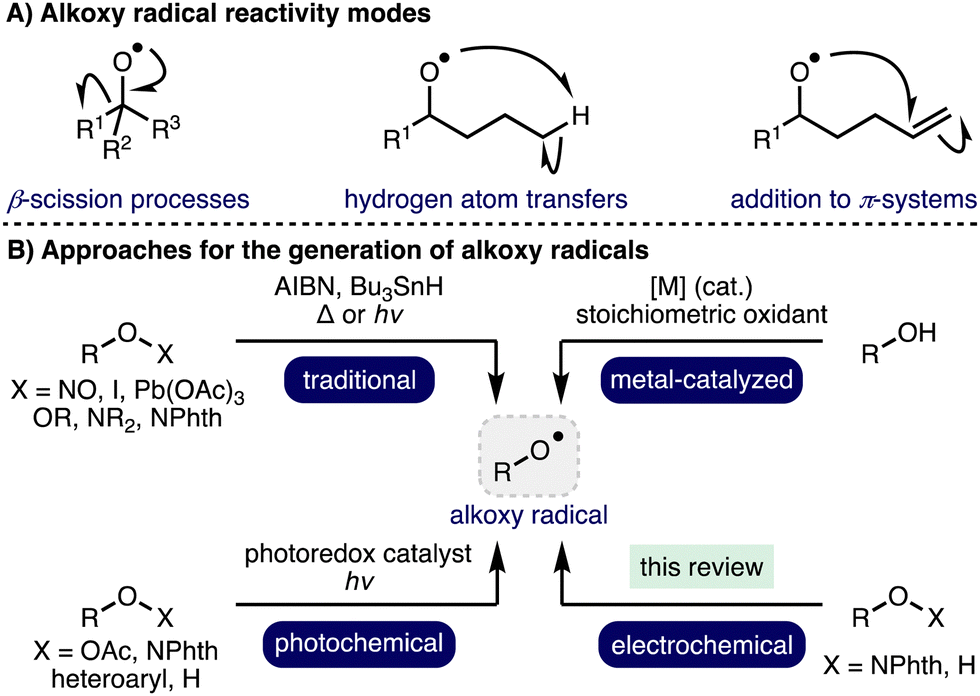 | ||
| Scheme 1 Alkoxy radical reactivity modes and generation methods. | ||
Organic electrochemistry represents one of the cleanest possible chemical processing technologies,8,9 which has recently undergone a renaissance due to the increasing global challenge to develop more sustainable synthetic methods to produce chemicals for society in combination with the increasing availability of standardized batch and flow electrochemical reactors.10,11 By careful tuning of electrochemical parameters, specific single electron transfer processes can be targeted, accessing powerful radical intermediates. In recent years, there has been a significant increase in reports that describe the development of electrochemical methods for the generation and utilization of alkoxy radicals. This highlight will summarise these developments and showcase the application of electrochemical generated alkoxy radical intermediates in a diverse range of electrosynthetic transformations, predominantly focusing on recent advances (2012–present).
2. β-Scission processes
The thermodynamically controlled homolytic cleavage of a β-C–C bond within alkoxy radicals is known as β-scission.3 This process leads to the generation of a carbonyl functional group and a C-centered radical, which is a powerful method for the selective activation of strong C–C σ-bonds.In 1996, Nikishin and co-workers reported the electrochemical oxidative ring opening of 1-methylcyclobutanol.12 When Mn(OAc)2·4H2O was employed in conjunction with LiCl, 5-chloropentan-2-one was produced in 43% yield. In 2019, Browne, Morrill, and co-workers expanded upon this methodology, demonstrating the Mn-mediated deconstructive chlorination of a broad range of cyclopropanols and cyclobutanols, which provided access to the correponding distally chlorinated ketones in up to 97% yield (Scheme 2).13 The process was scaled up employing flow electrochemistry in combination with inline purification technology to access products on gram scale. Cyclic voltammetry was used to probe the reaction mechanism, which was reported to proceed via the electrochemical generation of a reactive Mn(III)X2Cl species. It was proposed that this species facilitates the formation of the alkoxy radical and also acts as a chlorine atom source for the chlorination of the distal C-centred radical formed after alkoxy radical β-scission. An alternative mechanistic pathway was also suggested involving the formation of a Mn(III)-bound alkoxide intermediate, which can undergo reversible β-scission, rather than a free alkoxy radical intermediate.
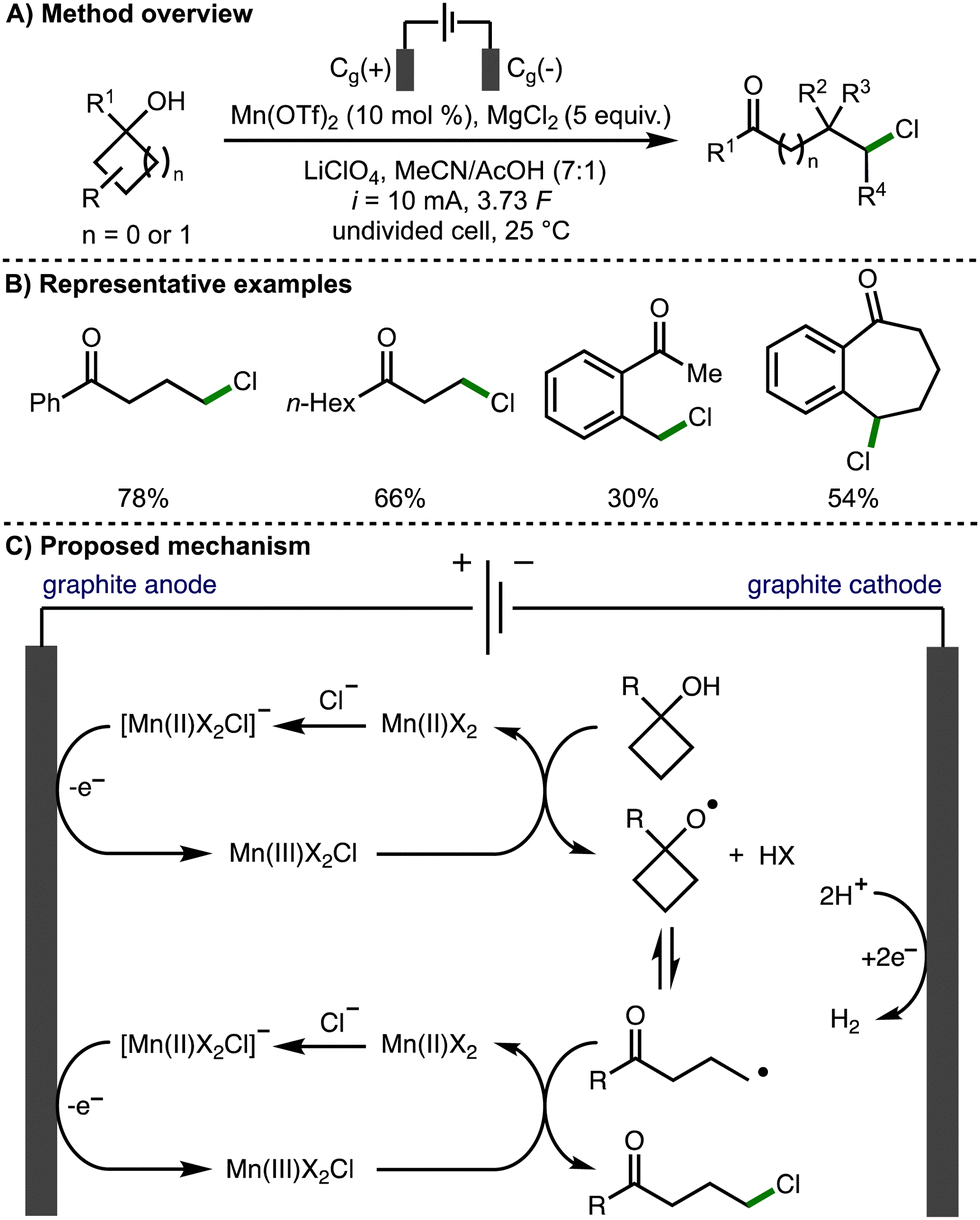 | ||
| Scheme 2 Manganese-catalyzed electrochemical deconstructive chlorination of cycloalkanols via alkoxy radicals (Browne and Morrill). | ||
In 2021, Parsons and co-workers reported the regioselective electrochemical cyclobutanol ring expansion to synthetically useful 1-tetralones (Scheme 3).14 The authors reported the formation of a range of substituted tetralones including products bearing electron-donating substituents and halogens, allowing for further synthetic elaboration. Desirable heteroaromatic products were also reported including fused indole, benzofuran and benzothiophene compounds. The authors proposed that the cyclobutanol substrates can undergo direct anodic oxidation to form a protonated alkoxy radical intermediate. Alternatively, indirect anodic oxidation can proceed in the presence of Mn(OTf)2, which results in the formation of a Mn(II)-alkoxide complex that can be oxidised to the corresponding Mn(III)-alkoxide species prior to undergoing inner sphere electron transfer to form the allkoxy radical and regenerate the Mn(II) catalyst. Subsequent alkoxy radical β-scission leads to the ring opening of the cyclobutanol, forming a distal C-centred radical, which undergoes intramolecular addition to the aromatic system to form a new 6-membered ring. Further anodic oxidation of the resultant α-oxygen radical and deprotonation affords the observed functionalised 1-tetralone products.
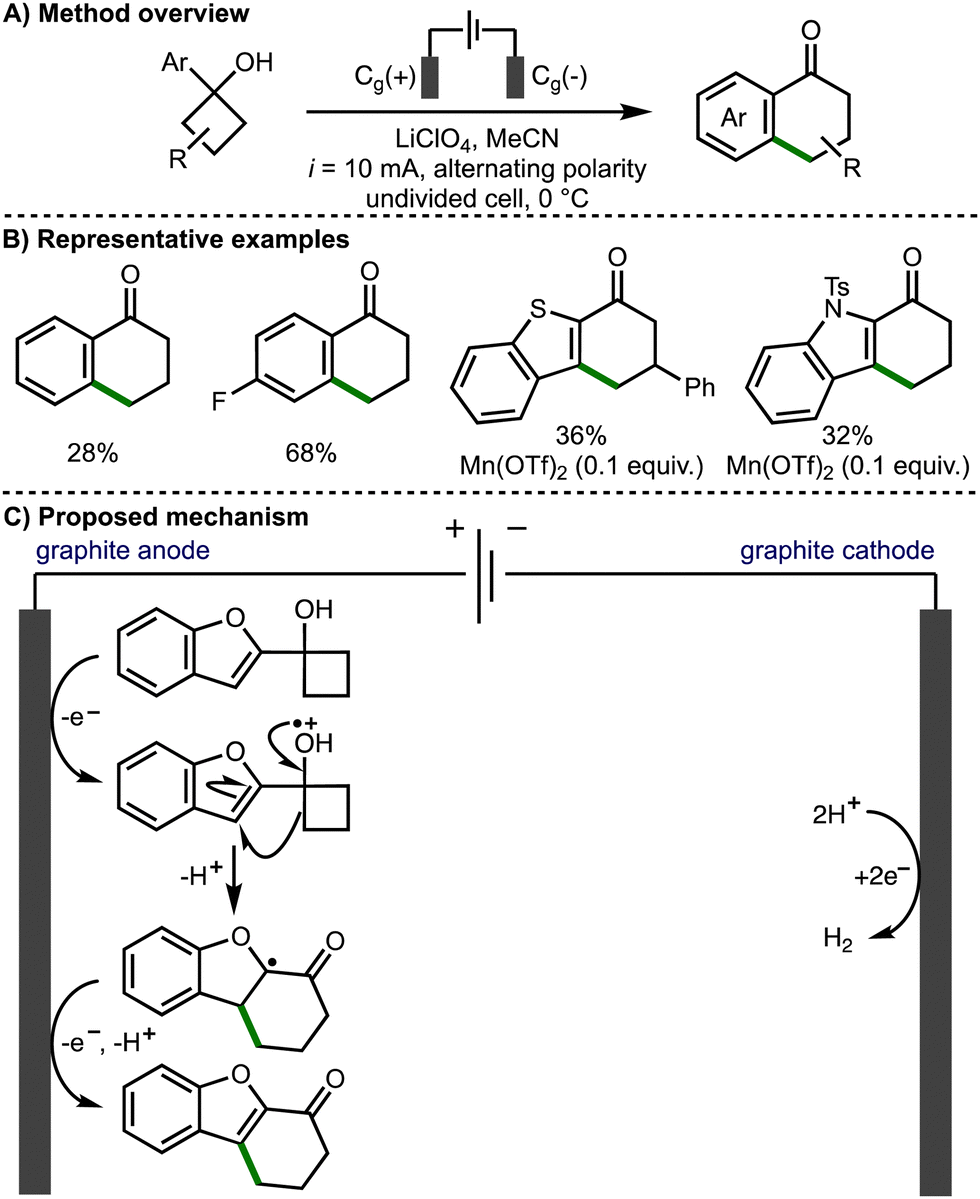 | ||
| Scheme 3 Regioselective electrochemical cyclobutanol ring expansion to 1-tetralones (Parsons). | ||
In 2021, Jirgensons and co-workers reported the electrochemical preparation of functionalised α,β-unsaturated esters from furfurylated ethylene glycols or amino alcohols, via the electro-oxidative fragmentation of electrochemically prepared spirocycles (Scheme 4).15 Based on deuterium labelling studies, the authors propose that the reaction mechanism proceeds via direct anodic oxidation of the spirocycle to the radical cation followed by methanolysis to generate an alkoxy radical intermediate and subsequent β-scission to form an α-heteroatom-stabilised C-centered radical. Further anodic oxidation of this radical intermediate leads to formation of the corresponding oxocarbenium/iminium species, which is intercepted by methanol to form the observed products.
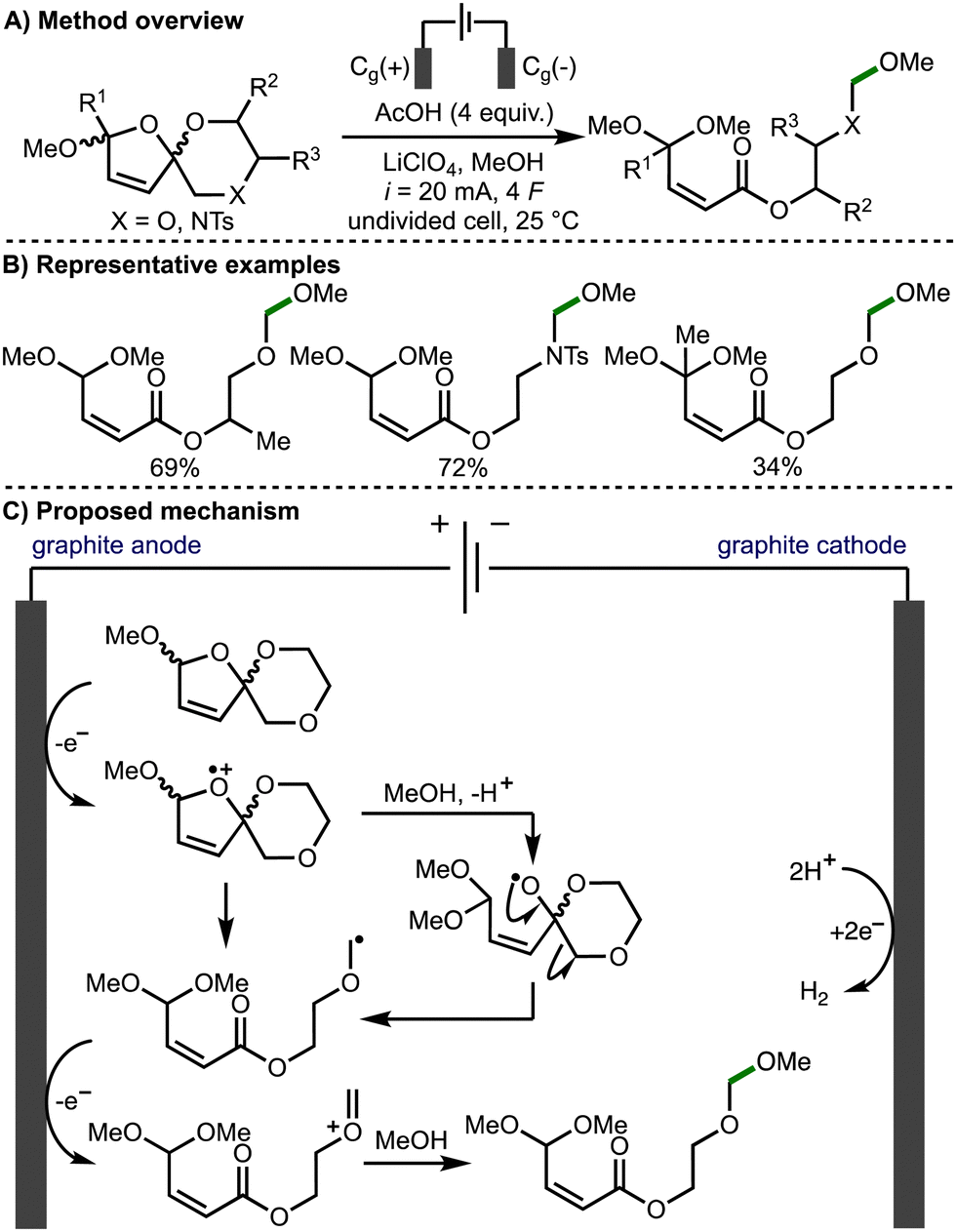 | ||
| Scheme 4 Torii-type electrosynthesis of α,β-unsaturated esters from furfurylated ethylene glycols and amino alcohols (Jirgensons). | ||
Also in 2021, Onomura and co-workers reported the electrophotochemical deconstructive bromination of tert-cycloalkanols (Scheme 5).16 The methodology was not limited to strained cycloalkanols and was successfully demonstrated using 5-, 6-, and 7-membered rings. However, it was limited to the formation of distally brominated ketone products. The scope of suitable tertiary cycloalkanols was shown to be broad with a variety substituents tolerated, and the method successfully performed on gram scale. The reaction was reported to proceed via the formation of a bromo cation species by electrochemical oxidation of a bromide anion. Hypobromite formation and light-promoted homolysis of the weak O–Br bond generated the alkoxy radical intermediate. Subsequent β-scission generates a distal C-centred radical, which undergoes a radical–radical coupling with a bromine radical to afford the observed product.
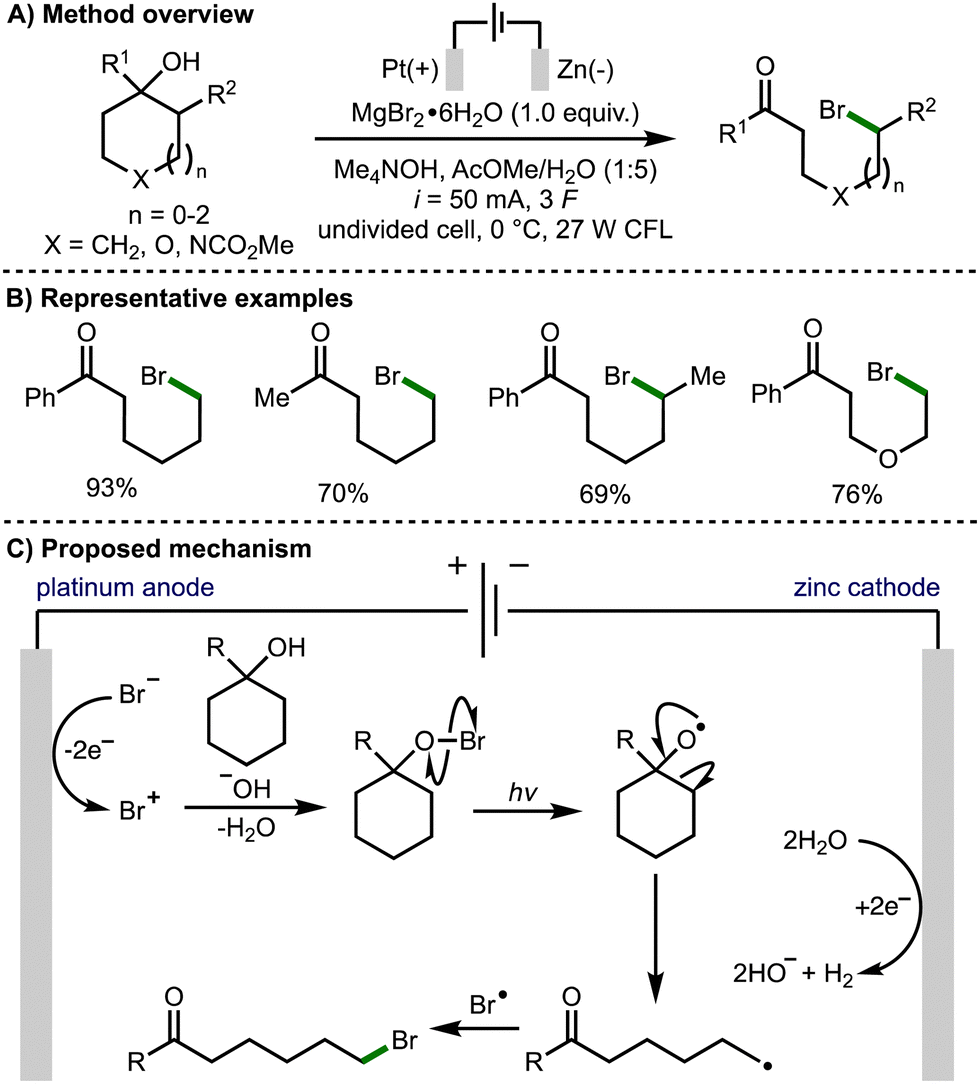 | ||
| Scheme 5 Electrophotochemical ring-opening bromination of tert-cycloalkanols (Onomura). | ||
In 2022, Morrill and co-workers reported an electrochemical method to access alkoxy radical intermediates via proton-coupled electron transfer (PCET) (Scheme 6).17 The transformation was successful on 4- to 12-membered tertiary cycloalkanols bearing an electron rich aryl ring. This electrochemical transformation employed a triarylamine redox mediator, which upon anodic oxidation (Eox = 0.91 V vs. Fc/Fc+) to the corresponding radical cation, can oxidise the aryl ring within the cycloalkanol. Proton-coupled electron transfer involving deprotonation of the alcohol with the Brønsted base and quenching of the arene radical cation results in formation of an alkoxy radical intermediate. Subsequent β-scission generates a distal C-centered radical, which is trapped with the SOMOphile, bromotrichloromethane in this case, to form a new C–Br bond. The authors also demonstrated the orthoganol derivatization of the products via a Ni-catalyzed ball-milling enabled cross-electrophile coupling of the C(sp3)−Br functionality, in addition to Baeyer–Villiger oxidation of the 4-methoxyphenyl ketone functionality to access the corresponding ester.
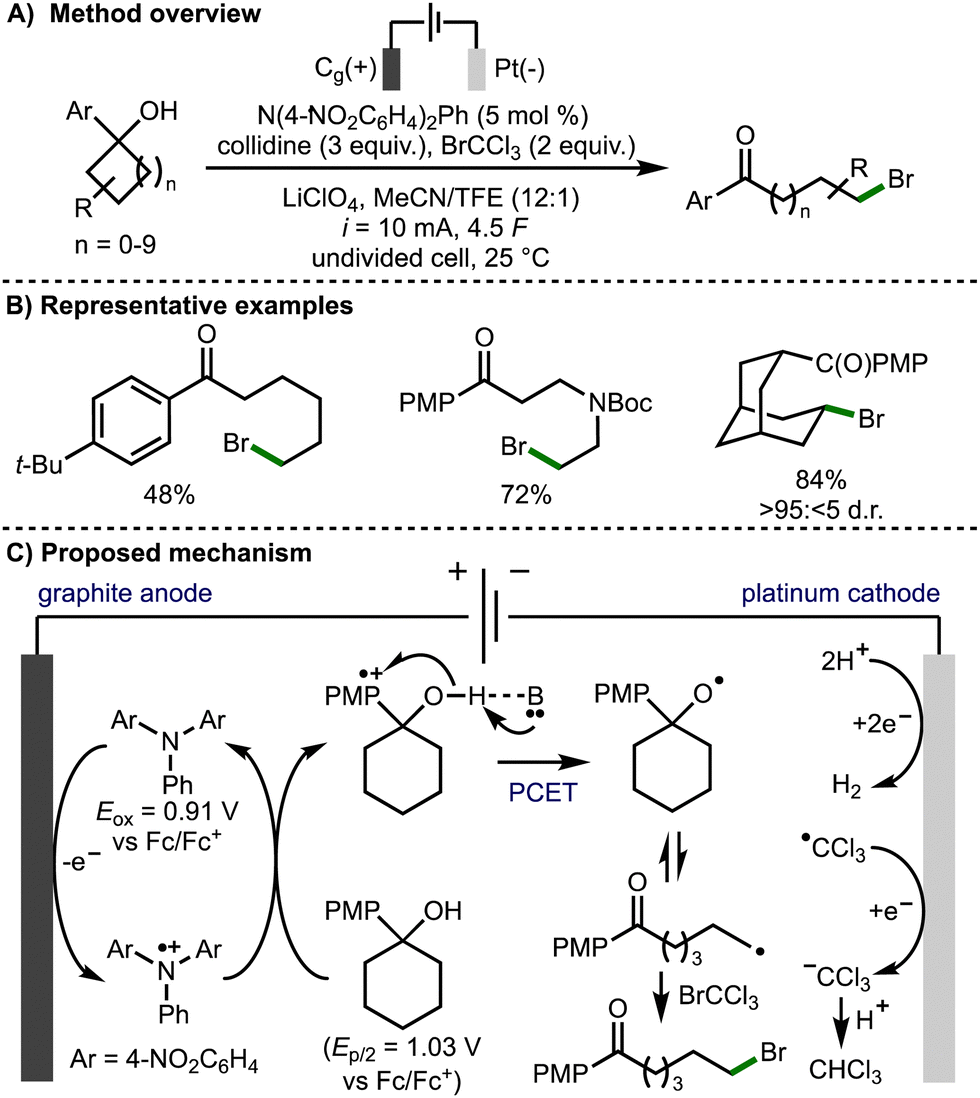 | ||
| Scheme 6 Electrochemical deconstructive functionalisation of cycloalkanols via alkoxy radicals enabled by proton-coupled electron transfer (Morrill). | ||
In 2022, Guo, Xia, and co-workers reported an efficient approach to the electrochemical generation of alkoxy radicals, by direct oxidation of the alkoxide, which was generated in situ (Scheme 7).18 The authors proposed a mechanism that begins with deprotonation of the tertiary cycloalkanol with potassium carbonate. The intermediate alkoxide is then proposed to oxidise at the surface of the graphite anode to form the alkoxy radical. After β-scission the distal C-centred radical undergoes further anodic oxidation to form a primary carbocation, which rearranges to form a more stable internal secondary carbocation. Subsequent deprotonation generates an enone intermediate, which is intercepted by addition of the nucleophile via Michael addition to generate the observed β-functionalised ketone products. The transformation was shown to have a broad scope tolerating a large range of tertiary cyclobutanols, secondary cyclobutanol, 1-phenylcyclopropanol and 1-phenylcyclopentanol. A range of N-, O-, C- and P-centred nucleophiles were also employed giving a variety of β-functionalised ketone products. The authors noted that the reaction was not tolerant of rings larger than 5-membered, and that only the β-functionalised carbonyl produces were accessible in useful yields.
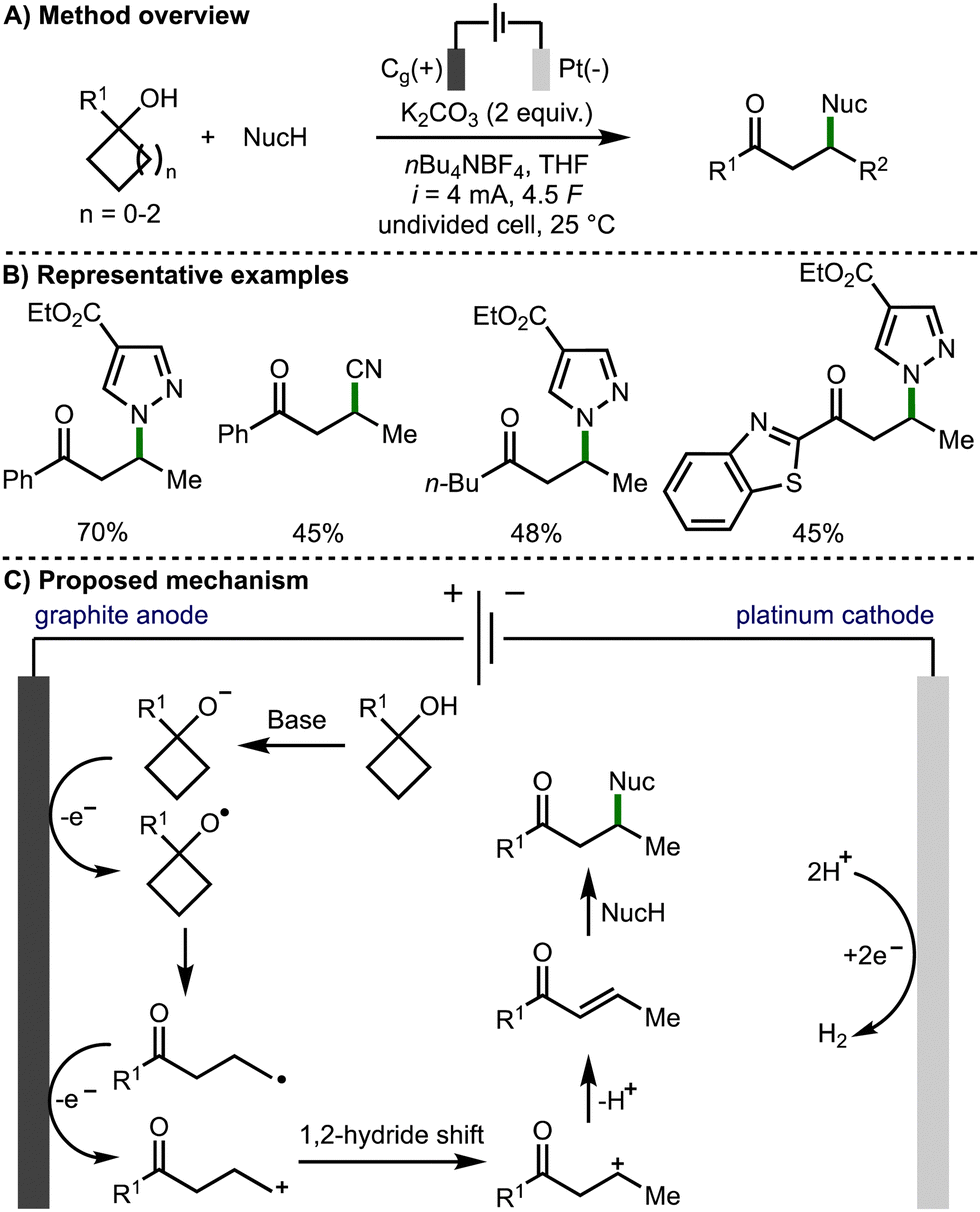 | ||
| Scheme 7 Electrochemical synthesis of β-functionalised ketones via ring opening of cycloalkanols (Guo and Xia). | ||
In 2022, Huang, Lei, and co-workers reported the electrophotochemical Ce-catalyzed ring opening functionalization of a broad range of tertiary cycloalkanols (Scheme 8).19 The transformation was applicable on a broad selection of functionalised tertiary alcohols with a range of aryl, heteroaryl and alkyl substitution at the 1-position tolerated. The developed method allowed for the formation of distally functionalized ketones products, with the formation of new C–CN, C–C, C–S and C–oxime bonds. The methodology employed CeCl3 as the catalyst and tetraethylammonium chloride as the electrolyte and chloride anion source. The combination of these reagents forms a [CeIIICl6]3− anion, which is oxidized at the graphite anode to a Ce(IV) species. This Ce(IV) complex undergoes ligand exchange with the tertiary alcohol substrate to form the Ce(IV)-alkoxide. The alkoxy radical is then formed by the photoinduced ligand-to-metal charge transfer (LMCT) homolysis of the cerium-bound alkoxide. Following β-scission, the distal C-centred radical is trapped by a SOMOphile, with the majority of those demonstrated being aryl sulfonyl compounds. Other SOMOphiles shown include 1,1-diphenylethylene for alkenylation, N-chlorosuccinamide for chlorination, and isoquinoline for heteroarylation.
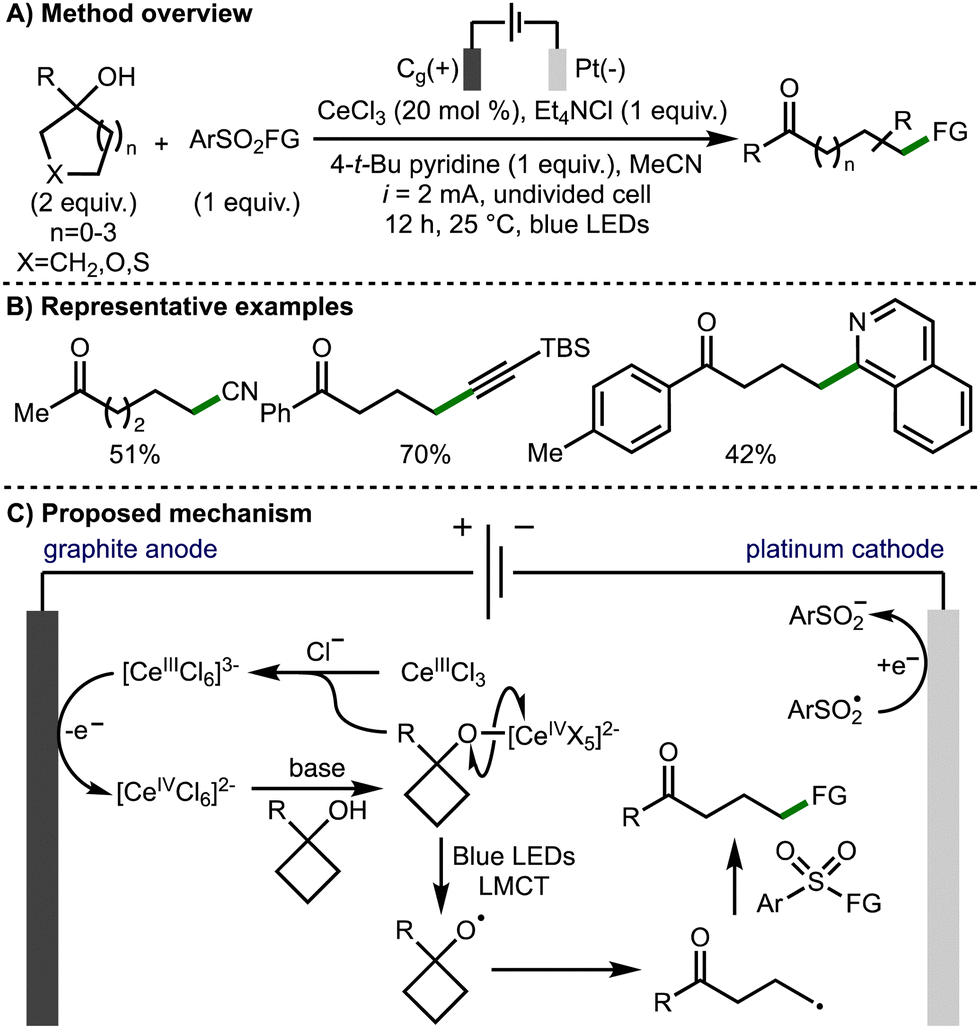 | ||
| Scheme 8 Electrophotochemical Ce-catalyzed ring-opening functionalization of cycloalkanols (Huang and Lei). | ||
Also in 2022, Jin, Liu, and co-workers reported the electrochemical oxidative cleavage of 1,2-diols to produce the corresponding carbonyl compounds (Scheme 9).20 For example, it was found that benzopinacol was converted to benzophenone in 97% isolated yield when n-hexylpyridinium tetrafluoroborate (HPyBF4) was employed as the supporting electrolyte in EtOH using galvanostatic conditions (i = 4 mA), a carbon plate anode and a nickel plate cathode in an undivided cell at room temperature. Various aryl-containing diol substrates were tolerated and the transformation was successfully demonstrated on gram scale. The authors proposed a reaction mechanism involving direct anodic oxidation of the diol to give an alkoxy radical intermediate. Subsequent alkoxy radical β-scission generated the ketone product and an α-hydroxy radical, which was proposed to undergo hydrogen atom transfer to form a second ketone product.
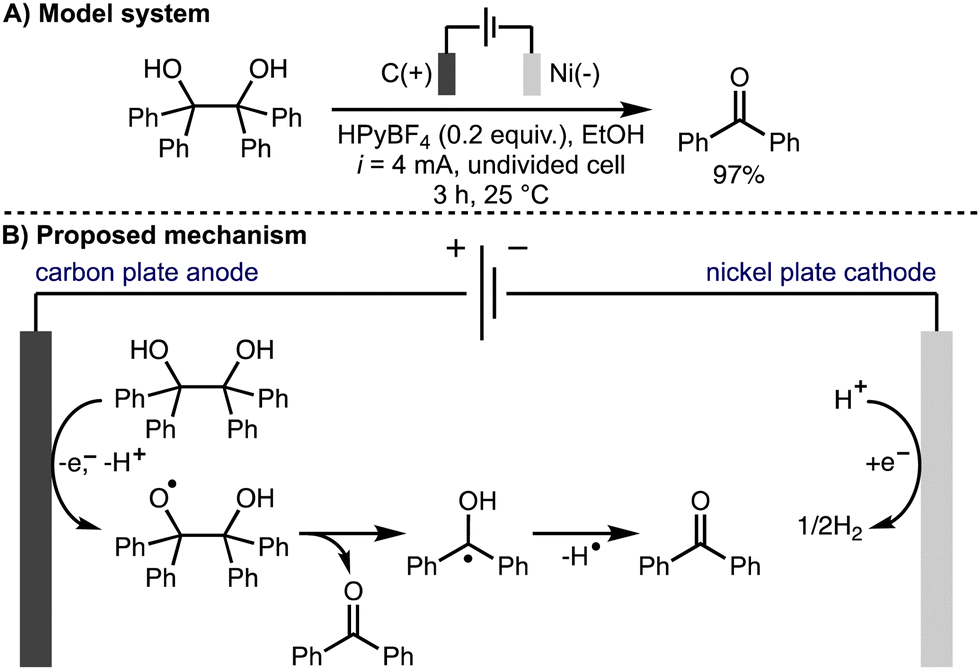 | ||
| Scheme 9 Oxidative cleavage of C–C bonds of 1,2-diols to ketones (Jin and Liu). | ||
3. Hydrogen atom transfer processes
The considerable difference in the bond dissociation free energy between RO–H bonds (BDFE ≈ 105 kcal mol−1) and most inactivated C–H bonds (BDFE ≈ 98–102 kcal mol−1) results in thermodynamically favoured hydrogen atom transfer events involving alkoxy radicals. As such, HAT processes involving alkoxy radicals, which can be intermolecular or intramolecular, are a powerful approach for site selective functionalization reactions.43.1 Intramolecular hydrogen atom transfer
In 2000, Markó reported an electrochemical method for spiroketal formation from ω-hydroxy-tetrahydropyrans (Scheme 10).21 A total of five examples were reported affording the corresponding spiroketals in good yields. The mechanism for this transformation was proposed to involve oxidation of an in situ generated alkoxide to form an alkoxy radical at the Pt anode. Intramolecular hydrogen atom transfer would generate a nucleophilic α-oxygen C-centered radical that is prone to a further single electron oxidation to give an oxocarbenium ion. Subsequent intramolecular nucleophilic attack by the pendant hydroxyl functionality formed the observed spiroketal products.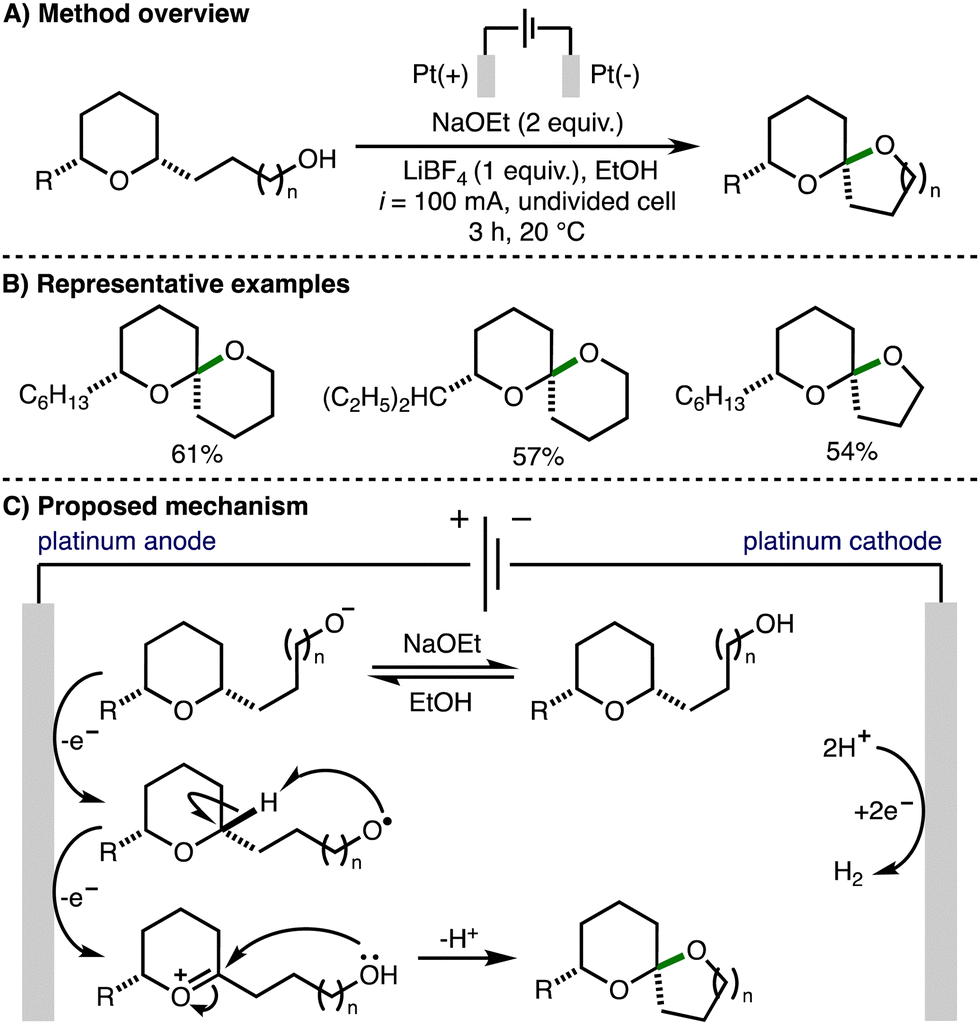 | ||
| Scheme 10 Electrochemical oxidative cyclization of ω-hydroxy-tetrahydropyrans to spiroketals (Markó). | ||
3.2 Intermolecular hydrogen atom transfer
In 2004, Griesbach, Comninellis, and co-workers reported the anodic methoxylation of p-tert-butyltoluene in MeOH-H2SO4-H2O solution using an electrochemical flow cell equipped with a boron doped diamond (BDD) anode.22 The authors proposed anodic generation of methoxy radicals, followed by intermolecular hydrogen atom transfer, and radical–radical coupling to form p-tert-butylbenzyl methyl ether. Further oxidation to p-tert-butylbenzaldehyde dimethyl acetal was proposed to occur via the same mechanism. In 2006, the same authors employed a similar approach for the electrochemical formation of trimethylorthoformate from formaldehyde dimethylacetal.23In 2012, Waldvogel and co-workers reported the anodic phenol-arene C–C cross-coupling using BDD anodes (Scheme 11).24 A selection of substituted phenols and electron-rich arenes underwent selective C–C cross-coupling, which provided access to a range of nonsymmetrical biaryls. The authors proposed initial formation of methoxy or hydroxy radicals on the BDD anode surface, which have increased lifetime in the presence of hexafluoroisopropanol. Subsequent intermolecular HAT with phenol would generate a phenoxy radical, which is trapped by excess arene to give the cross-coupled product after a second oxidation step.
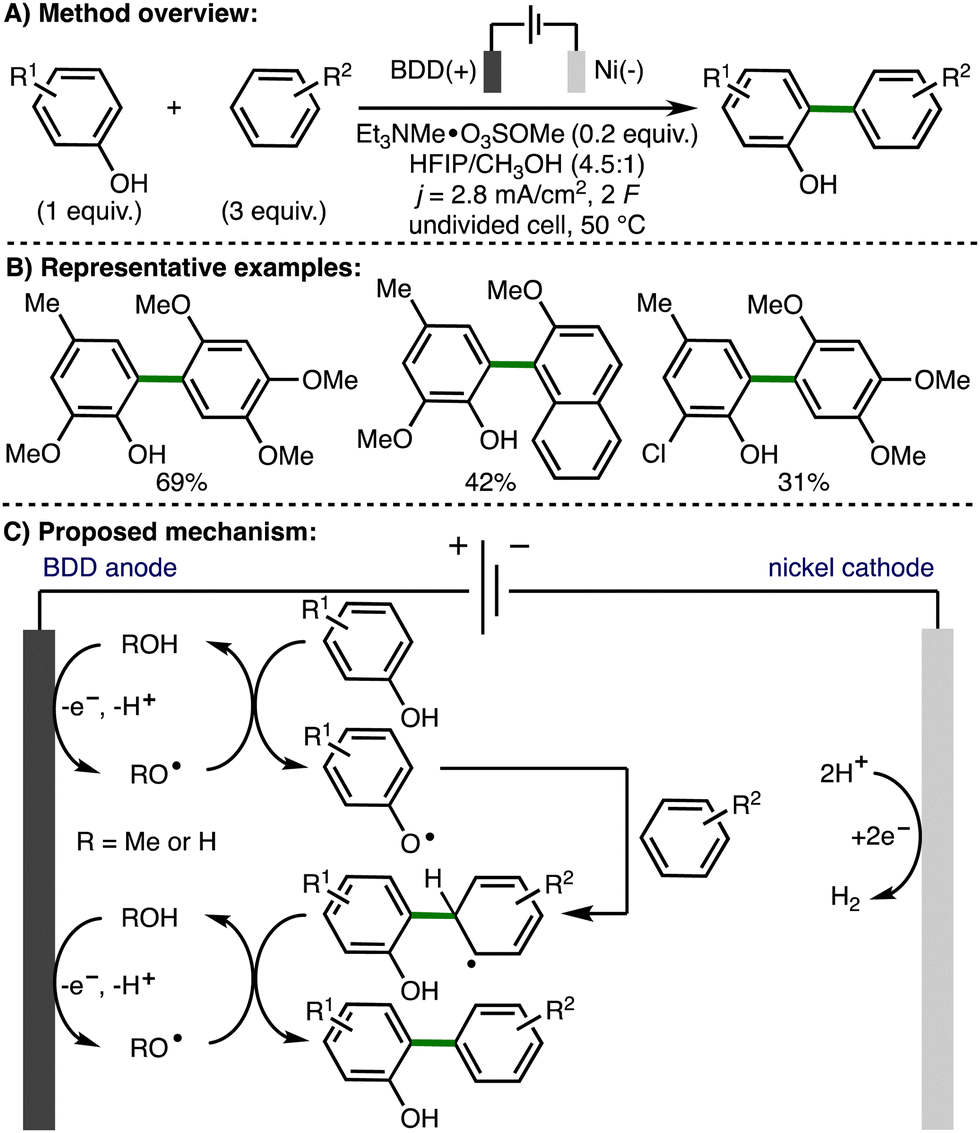 | ||
| Scheme 11 Anodic phenol-arene C–C cross-coupling using BDD anodes (Waldvogel). | ||
In 2013, Suryanarayanan and co-workers reported the regioselective anodic α-methoxylation of 2-oxazolidinone on boron doped diamond in acidic methanol (Scheme 12),25 forming 4-methoxy-2-oxazolidinone in up to 88% yield when a high current density (50 mA cm−2) was employed. Electron spin resonance (ESR) spectroscopic analysis of the radical species generated upon anodic oxidation of methanol in the presence of 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) provided support for the formation of methoxy radicals due to observation of signals that corresponded to the methoxy radical spin adduct. A linear sweep voltammogram of 0.1 M H2SO4/MeOH medium recorded in the presence of 2-oxazolidinone (0.01 M) using a BDD anode revealed a broad oxidation wave at approximately 1.5 V vs. SCE. Based on these observations, at high current density, the authors proposed a reaction mechanism involving anodic generation of methoxy radicals followed by regioselective intermolecular HAT with 2-oxazolidinone to produce a C-centered radical, which reacts further with a methoxy radical via radical–radical coupling to produce the observed product. At lower current density, the reaction mechanism was proposed to proceed via anodic oxidation of 2-oxazolidione to the corresponding radical cation, followed by deprotonation and radical–radical coupling.
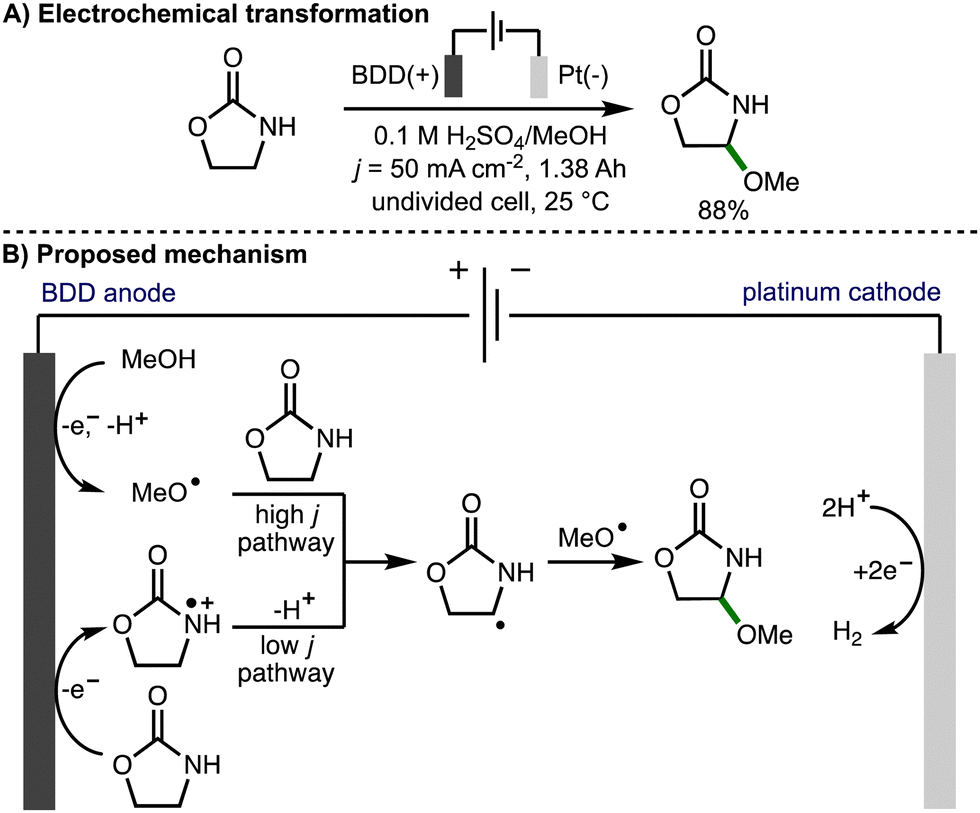 | ||
| Scheme 12 Regioselective anode α-methoxylation of 2-oxazolidinone on boron doped diamond in acidic methanol medium (Suryanarayanan). | ||
In 2021, Xu, Zeng, and co-workers reported the electrophotocatalytic generation of methoxy radicals and applied this to the formation of Si-functionalized benzimidazo-fused isoquinolinones (Scheme 13).26 The direct electrooxidation of trialkylsilanes is challenging due to their high oxidation potentials, and their use in redox chemistry is further limited by the high bond dissociation free energies of Si–H and α-Si–C–H bonds (BDFE > 92 kcal mol−1). To overcome these challenges, a strategy that combined electro-oxidation, photoinduced ligand-to-metal charge transfer (LMCT) and methoxyl radical mediated intermolecular hydrogen atom transfer was employed for the selective generation of silyl radicals. The proposed mechanism of this transformation initiates with anodic generation of a Ce(IV)Cln species, which undergoes ligand exchange with methanol to generate Ce(IV)OMeCln−1. Homolysis via photoinduced LMCT generates methoxy radicals, which can undergo intermolecular HAT with trialkylsilanes for the formation of silyl radicals. The silyl radical undergoes regioselective addition to the acrylamide functionality within the substrate, followed by cyclization, Ce(IV)-mediated oxidation, and deprotonation to deliver the observed Si-functionalized heterocyclic products.
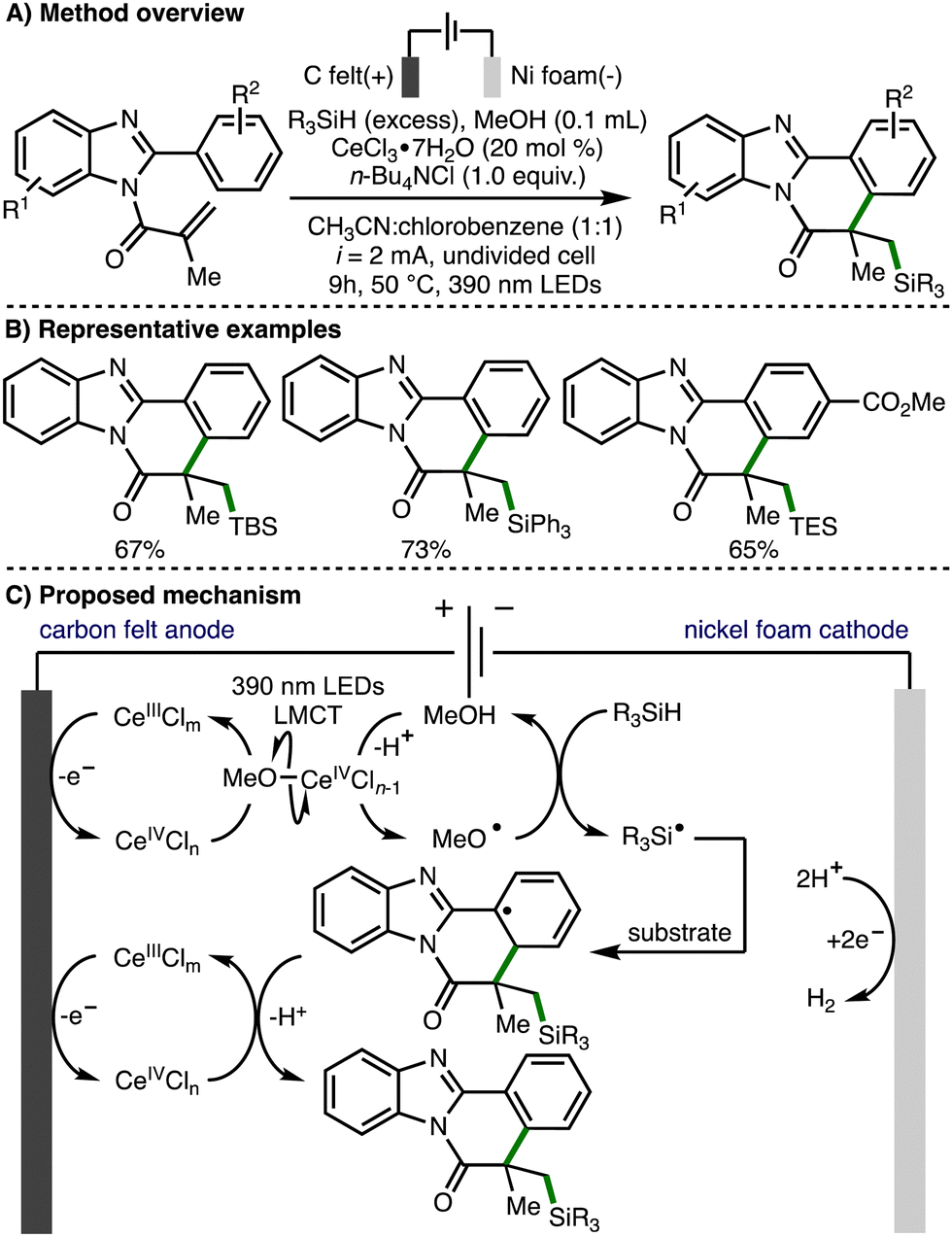 | ||
| Scheme 13 Electrophotocatalytic Si–H activation governed by polarity-matching effects (Xu and Zeng). | ||
In 2022, Xu, Zeng and co-workers extended this approach towards the activation of C–H bonds and reported the electrophotocatalytic alkylation of N-heteroarenes with unactivated alkanes (Scheme 14).27 In this transformation, intermolecular HAT between methoxy radicals and alkanes generates C-centered radicals, which add to various N-heteroarenes under acidic conditions to afford the observed alkylated N-heteroaromatic products. The same authors recently applied the same method to the synthesis of alkylated benzimidazo-fused isoquinolinones.28
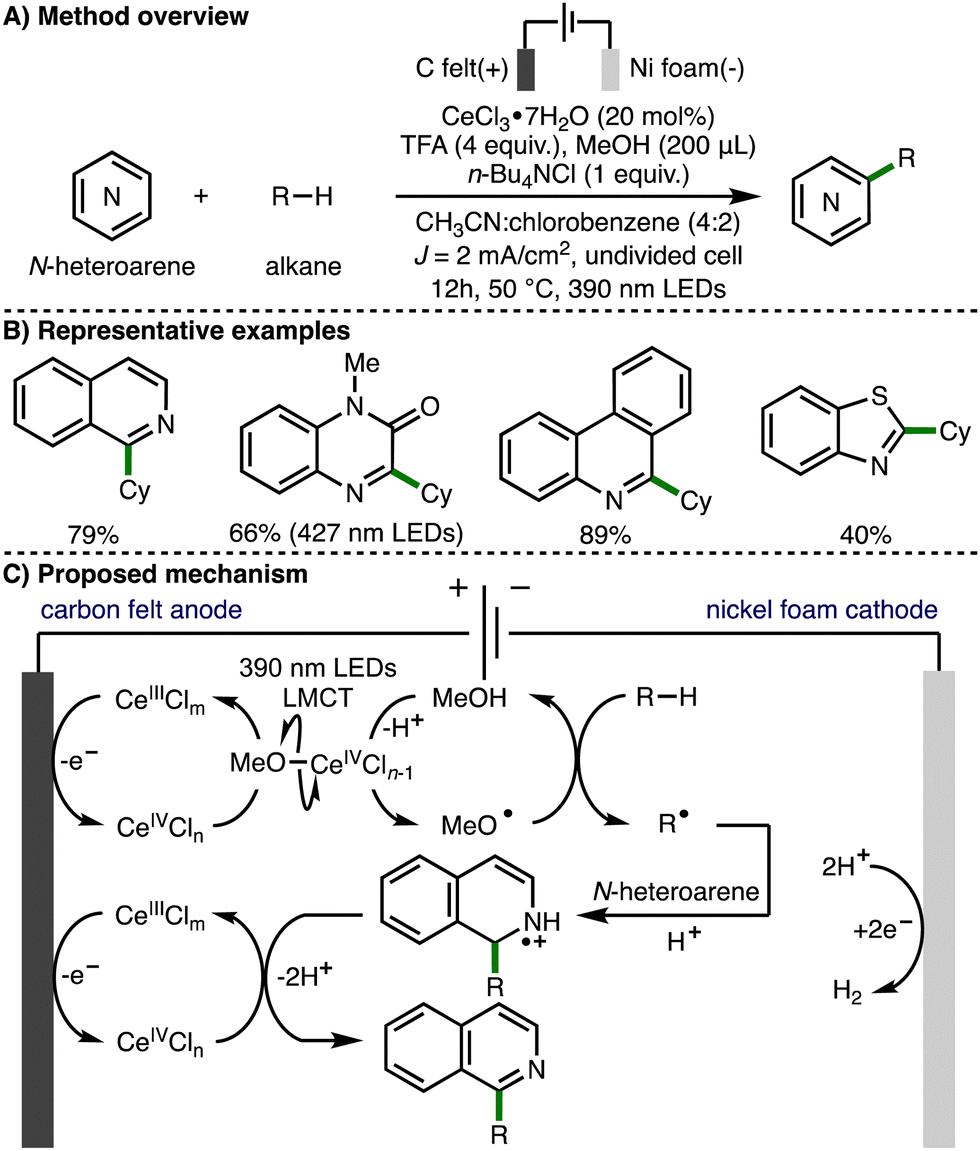 | ||
| Scheme 14 Electrophotocatalytic C–H functionalization of N-heteroarenes with unactivated alkanes under external oxidant-free conditions (Xu and Zeng). | ||
Also in 2022, Huang and co-workers reported the regioselective sulfoximido-oxygenation of alkenes with a variety of NH-sulfoximines and alcohols, obtaining the corresponding products in high yields (Scheme 15).29 The authors proposed that the reaction mechanism predominantly proceeds via anodic generation of an N-centred sulfoximidoly radical, which undergoes regioselective addition to the alkene, followed by anodic oxidation and trapping of the resulting carbocation with methanol to generate the observed products. However, cyclic voltammetry experiments revealed a MeOH oxidation peak of in the presence of K3PO4 (Ep = 1.29 V vs. SCE), which overlapped with the oxidation potential of the NH-sulfoximine. As such, the authors also proposed an alternative minor pathway for the generation of N-centered radicals, which involved initial anodic oxidation of methoxide to generate methoxy radicals, which could undergo an intermolecular HAT with the NH-sulfoximine to generate the reactive N-centred radical.
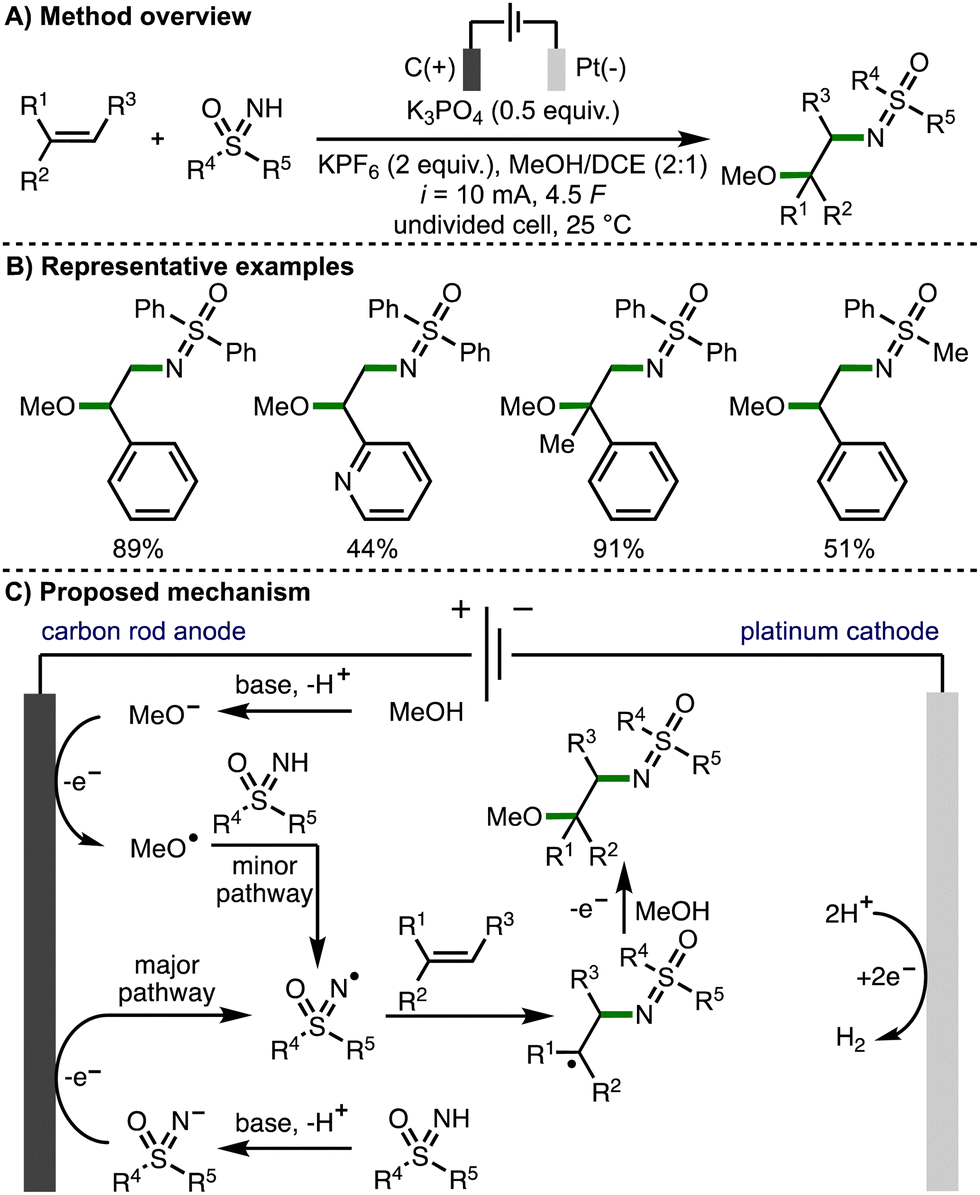 | ||
| Scheme 15 Electrochemically enabled sulfoximido-oxygenation of alkenes with NH-sulfoximines and alcohols (Huang). | ||
All the examples described previously involve the direct or indirect anodic generation of alkoxy radicals and their subsequent utilization in synthesis. With regards to the cathodic electrochemical generation of alkoxy radicals, in 2017, Terent’ev and co-workers investigated the electrochemical behaviour or N-oxyphthalimides (Scheme 16).30 Density functional theory (DFT) calculations were used to demonstrate that the favoured decomposition pathway of the radical anion generated upon single electron reduction of N-ethoxyphthalimide is the formation of the ethoxy radical and the phthalimide anion (ΔG = −17.9 kcal mol−1). This was supported by cyclic voltammetry analysis, which showed that N-ethoxyphthalimide has a reduction potential (Ered) of −1.80 V (vs. Fc/Fc+) with formation of the phthalimide anion observed.
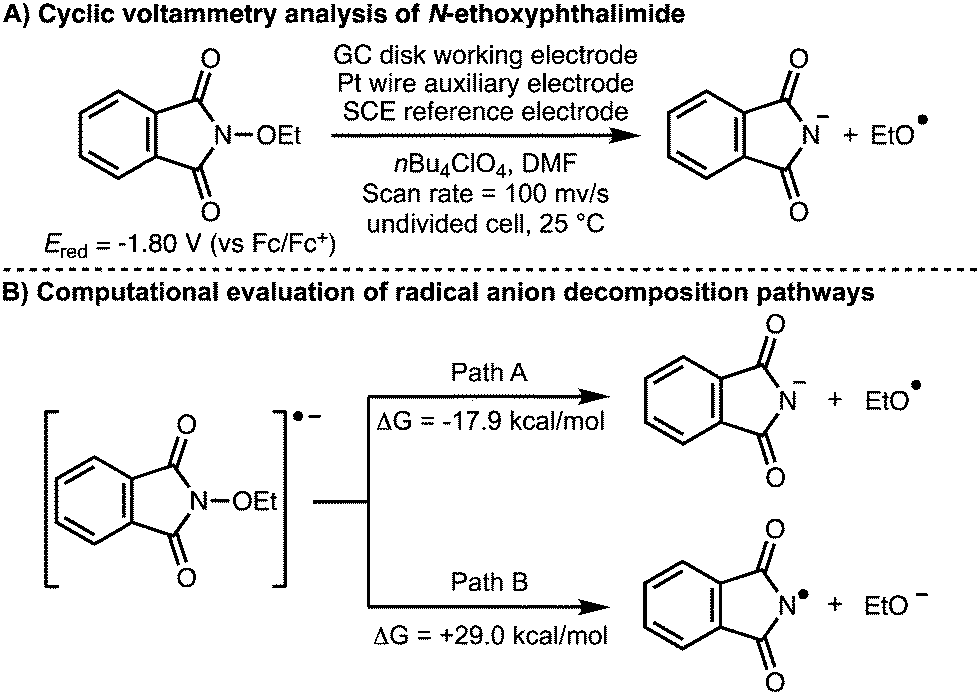 | ||
| Scheme 16 Electrochemical behaviour of N-oxyphthalimides (Terent'ev). | ||
In 2023, Fermi, Bertuzzi, Bandini, and co-workers reported the electrochemical C(sp3)–H functionalization of ethers via hydrogen atom transfer (Scheme 17).31 Under optimized electrochemical reaction conditions, it was found that a variety of ethers could undergo selective allylation using Morita–Baylis–Hillman acetates in the presence of redox active carbonates. A sacrificial zinc anode was used in combination with a graphite cathode, with either constant voltage electrolysis (CVE) or constant current electrolysis (CCE) employed depending upon the reagents used. The approach was extended to ether alkylation via Giese-type additions to vinyl sulfones and acrylates. The authors proposed a reaction mechanism involving cathodic reduction of the carbonate to generate the phthalimide anion and t-BuOCO2˙. This radical could undergo direct HAT with the ether substrate to generate an α-oxy radical (path a), or it could first extrude carbon dioxide to generate a t-butoxyl radical that could participate in HAT (path b). The α-oxy radical then undergoes regioselective addition to the MBH acetate, followed by cathodic reduction and loss of acetate anion to form the observed products.
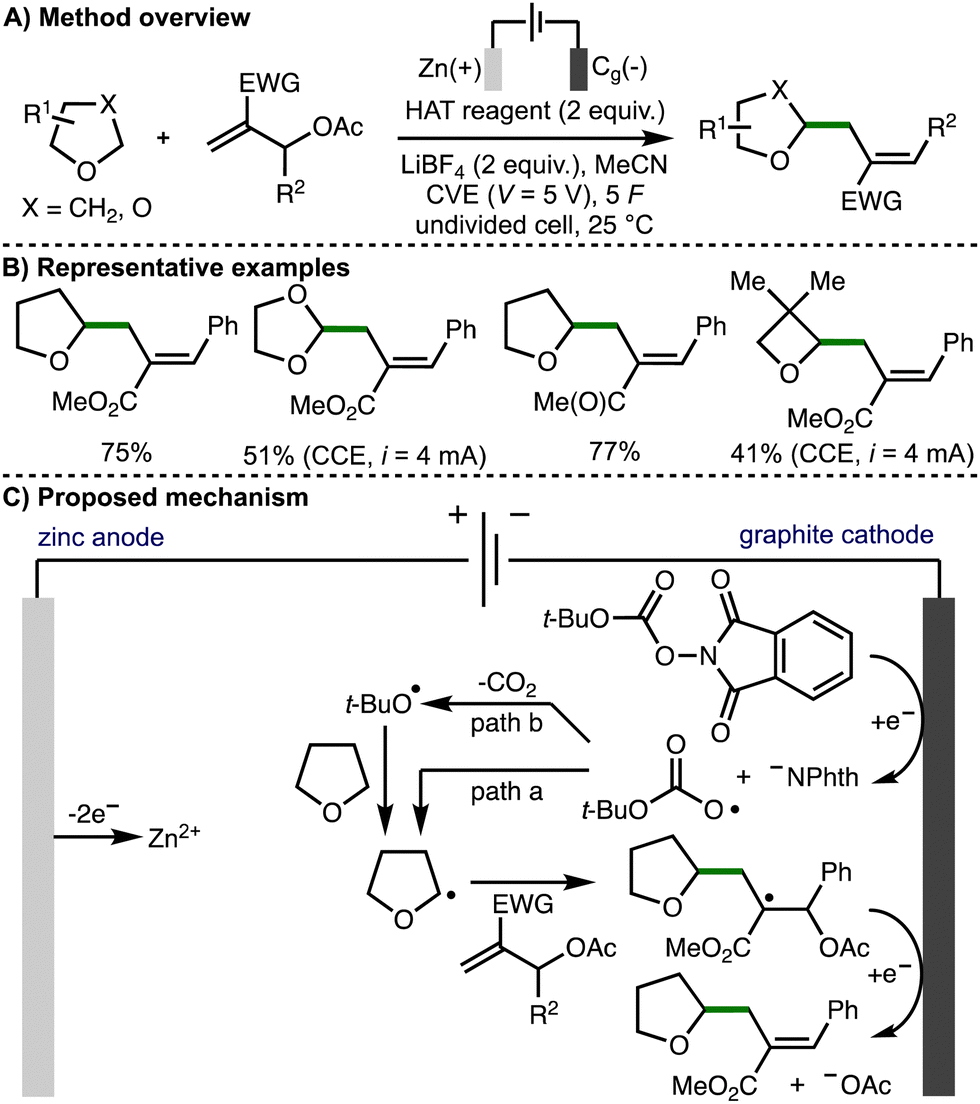 | ||
| Scheme 17 Electrochemical C(sp3)–H functionalization of ethers via hydrogen-atom transfer by means of cathodic reduction (Fermi, Bertuzzi and Bandini). | ||
4. Addition to π-systems
The addition of electrophilic alkoxy radicals to electron-rich C–C double bonds is a kinetically fast process and a powerful method for the construction of new C–O bonds.5 Building upon pioneering early contributions from Belleau,32 Tsutsumi,33,34 and Brettle,35,36 in 1981, Swenton and co-workers reported the methoxylation of electron rich naphthalenes,37 which was proposed to involve the addition of anodically generated methoxy radicals to aromatic radical cations. The same approach was subsequently employed by others for the methoxylation of isatin,38 4-methoxyanilines,39 dimethoxybenzenes,40 and electron-deficient aromatic rings.41In 2012, Einaga, Nishiyama and co-workers undertook a mechanistic study to determine if radical intermediates are generated in electrochemical oxidation reactions using methanol in conjunction with BDD, Pt, and glassy carbon anodes (Scheme 18).42 The dearomative dimethoxylation of a substituted dimethoxybenzene derivative in basic MeOH solution was selected for this investigation. It was found that the desired product was formed in quantitive yields when Pt net or BDD plate anodes were used, whereas the corresponding aldehyde was produced in 50% yield when a glassy carbon anode was employed. ESR spectra of product mixtures were obtained from anodic oxidations of methanolic solutions containing radical trapping agent 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) using BDD, Pt, and glassy carbon anodes. When BDD and Pt anodes were employed, the resulting spectra were indicative of nitroxyl radicals generated by reaction of DMPO with methoxy radicals. Furthermore, the relative intensities revealed that methoxy radicals are most efficiently generated using BDD anodes, followed by Pt, with negligable methoxy radical generation using glassy carbon. As such, using BDD or Pt anodes, it was proposed that the reaction mechanism proceeds through the EECrCp pathway previously proposed by Swenton,37 which involves anodic oxidation of the aromatic substrate and methoxide to generate the corresponding aromatic radical cation and methoxy radicals, respectively. Subsequent addition of the methoxy radical to the aromatic radical cation, followed by methoxide addition provides the observed dearomatized product. In 2016, the same authors employed this approach for the synthesis of a cyclohexadienone derivative possessing an α-D-glucopyranosyl moiety.43
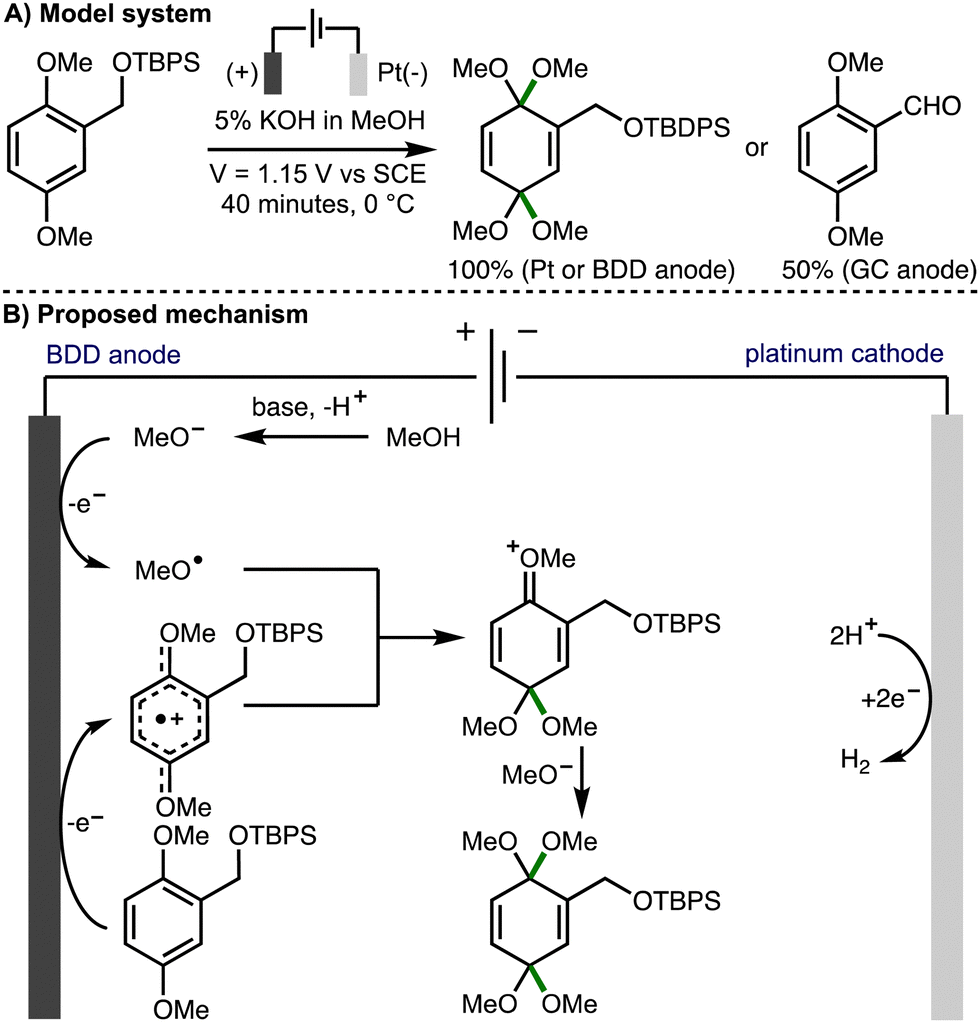 | ||
| Scheme 18 Anodic oxidation on a boron doped diamond electrode mediated by methoxy radicals (Einaga and Nishiyama). | ||
5. Conclusion and outlook
This highlight summarises electrochemical approaches for the generation and utilization of alkoxy radicals, predominantly focusing on recent advances (2012–present). The most significant advances in this area have been highlighted, and categorized according to the operative alkoxy radical reactivity mode, namely β-scission, hydrogen atom transfer, and addition to π-systems. A diverse array of electrochemical transformations have been covered, including deconstructive functionalization, heterocycle synthesis, biaryl formation, C–H functionalization, alkene difunctionalization, and dearomatization processes. Despite a sharp increase in the number of publications in this area over the past decade, many opportunities remain. These include, but are not limited to: (1) the development of novel electrochemical strategies for the direct or indirect anodic generation of alkoxy radicals, which address limitations (e.g., substrate scope) associated with existing approaches; (2) diversifying the range of C-centered radical functionalizations possible to include various bond formations; (3) the application of the developed methodologies towards the synthesis and/or derivitization of biologically important molecules; (4) the development of enantioselective processes; (5) further utilization of the cathodic generation of alkoxy radicals in synthesis; (6) employment of flow electrochemistry to improve efficiency and sustainability metrics; (7) the use of various electroanalytical tools for detailed analysis of electrochemical reaction mechanisms. It is anticipated that many new and exciting electrochemical transformations involving alkoxy radicals will be discovered to address these and other challenges in the coming years.Author contributions
Albara A. M. A. El Gehani, Hussain A. Maashi and James Harnedy contributed equally to this work.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the School of Chemistry, Cardiff University for generous support, in addition to AstraZeneca and the EPSRC (A. A. M. A. E. G. EP/R513003/1), and the Saudi Arabia cultural mission in the UK and the Department of Chemistry, University of Bisha, Saudi Arabia (H. A. M.), for PhD studentships.References
- M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714 CrossRef CAS PubMed.
- E. Tsui, H. Wang and R. R. Knowles, Chem. Sci., 2020, 11, 11124–11141 RSC.
- S. P. Morcillo, Angew. Chem., Int. Ed., 2019, 58, 14044–14054 CrossRef CAS PubMed.
- M. Salamone and M. Bietti, Acc. Chem. Res., 2015, 48, 2895–2903 CrossRef CAS PubMed.
- J. Hartung, T. Gottwald and K. Špehar, Synthesis, 2002, 1469–1498 CrossRef CAS.
- S. Yamashita, A. Naruko, Y. Nakazawa, L. Zhao, Y. Hayashi and M. Hirama, Angew. Chem., Int. Ed., 2015, 54, 8538–8541 CrossRef CAS PubMed.
- L. Chang, Q. An, L. Duan, K. Feng and Z. Zuo, Chem. Rev., 2022, 122, 2429–2486 CrossRef CAS PubMed.
- C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed.
- L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Chem. Soc. Rev., 2021, 50, 7941–8002 RSC.
- M. Yan, Y. Kawamata and P. S. Baran, Angew. Chem., Int. Ed., 2018, 57, 4149–4155 CrossRef CAS PubMed.
- C. Gütz, B. Klöckner and S. R. Waldvogel, Org. Process Res. Dev., 2016, 20, 26–32 CrossRef.
- N. I. Kapustina, L. L. Sokova and G. I. Nikishin, Russ. Chem. Bull., 1996, 45, 1246–1248 CrossRef.
- B. D. W. Allen, M. D. Hareram, A. C. Seastram, T. McBride, T. Wirth, D. L. Browne and L. C. Morrill, Org. Lett., 2019, 21, 9241–9246 CrossRef CAS PubMed.
- A. Petti, P. Natho, K. Lam and P. J. Parsons, Eur. J. Org. Chem., 2021, 854–858 CrossRef CAS.
- M. Darzina, A. Lielpetere and A. Jirgensons, Eur. J. Org. Chem., 2021, 4224–4228 CrossRef CAS.
- K. Yamamoto, H. Toguchi, M. Kuriyama, S. Watanabe, F. Iwasaki and O. Onomura, J. Org. Chem., 2021, 86, 16177–16186 CrossRef CAS PubMed.
- M. D. Hareram, A. A. M. A. El Gehani, J. Harnedy, A. C. Seastram, A. C. Jones, M. Burns, T. Wirth, D. L. Browne and L. Morrill, Org. Lett., 2022, 24, 3890–3895 CrossRef CAS PubMed.
- L. Zhao, Q. Zhong, J. Tian, M. Luo, C. Yang, L. Guo and W. Xia, Org. Lett., 2022, 24, 4421–4426 CrossRef CAS PubMed.
- Z. Yang, D. Yang, J. Zhang, C. Tan, J. Li, S. Wang, H. Zhang, Z. Huang and A. Lei, J. Am. Chem. Soc., 2022, 144, 13895–13902 CrossRef CAS PubMed.
- R. Wang, P. Sun, W. Jin, Y. Zhang, B. Wang, Y. Xia, F. Xue, A. Abdukader and C. Liu, Org. Chem. Front., 2022, 9, 2664–2670 RSC.
- I. E. Marko, Tetrahedron Lett., 2000, 41, 4383–4387 CrossRef CAS.
- D. Zollinger, U. Griesbach, H. Pütter and C. Comninellis, Electrochem. Commun., 2004, 6, 600–604 CrossRef CAS.
- R. Fardel, U. Griesbach, H. Pütter and C. Comninellis, J. Appl. Electrochem., 2006, 36, 249–253 CrossRef CAS.
- A. Kirste, B. Elsler, G. Schnakenburg and S. R. Waldvogel, J. Am. Chem. Soc., 2012, 134, 3571–3576 CrossRef CAS PubMed.
- K. R. Saravanan, V. Selvamani, K. Kulangiappar, D. Velayutham and V. Suryanarayanan, Electrochem. Commun., 2013, 28, 31–33 CrossRef CAS.
- Y. Jiang, K. Xu and C. Zeng, CCS Chem., 2022, 4, 1796–1805 CrossRef CAS.
- Z. Tan, X. He, K. Xu and C. Zeng, ChemSusChem, 2022, 15, e202102360 CAS.
- Z. Tan, Y. Jiang, K. Xu and C. Zeng, J. Catal., 2023, 417, 473–480 CrossRef CAS.
- J.-L. Wan and J.-M. Huang, Org. Lett., 2022, 24, 8914–8919 CrossRef CAS PubMed.
- M. A. Syroeshkin, I. B. Krylov, A. M. Hughes, I. V. Alabugin, D. V. Nasybullina, M. Y. Sharipov, V. P. Gultyai and A. O. Terent'ev, J. Phys. Org. Chem., 2017, 30, e3744 CrossRef.
- L. Rapisarda, A. Fermi, P. Ceroni, R. Giovanelli, G. Bertuzzi and M. Bandini, Chem. Commun., 2023, 59, 2664–2667 RSC.
- B. Belleau and N. L. Weinberg, J. Am. Chem. Soc., 1963, 85, 2525–2526 CrossRef CAS.
- T. Inoue, K. Koyama, T. Matsuoka, K. Matsuoka and S. Tsutsumi, Tetrahedron Lett., 1963, 4, 1409–1411 CrossRef.
- T. Inoue, K. Koyama, T. Matsuoaka and S. Tsutsumi, Bull. Chem. Soc. Jpn., 1967, 40, 162–168 CrossRef CAS.
- A. J. Baggaiey and R. Brettle, J. Chem. Soc. C, 1968, 2055–2059 RSC.
- R. Brettle and J. R. Sutton, J. Chem. Soc., Perkin Trans. 1, 1975, 1947–1954 RSC.
- M. G. Dolson and J. S. Swenton, J. Am. Chem. Soc., 1981, 103, 2361–2371 CrossRef CAS.
- A. Niedźwiecka-Kornaś, E. Bojarska, J. Kamińskib and Z. Kazimierczuk, Z. Naturforsch. B, 1998, 53, 620–624 CrossRef.
- M. C. Carreño and M. Ribagorda, J. Org. Chem., 2000, 65, 1231–1234 CrossRef PubMed.
- T. Saitoh, E. Suzuki, A. Takasugi, R. Obata, Y. Ishikawa, K. Umezawa and S. Nishiyama, Bioorg. Med. Chem. Lett., 2009, 19, 5383–5386 CrossRef CAS PubMed.
- S. Kumar, E-J. Chem., 2011, 8, 846–850 CrossRef CAS.
- T. Sumi, T. Saitoh, K. Natsui, T. Yamamoto, M. Atobe, Y. Einaga and S. Nishiyama, Angew. Chem., Int. Ed., 2012, 51, 5443–5446 CrossRef CAS PubMed.
- S. Yajima, T. Saitoh, K. Kawa, K. Nakamura, H. Nagase, Y. Einaga and S. Nishiyama, Tetrahedron, 2016, 72, 8428–8435 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |