
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light-induced protein labeling in live cells with aryl azides
Yixin
Zhang†
a,
Jiawei
Tan†
a and
Yiyun
Chen
 *abc
*abc
aState Key Laboratory of Bioorganic and Natural Products Chemistry Centre of Excellence in Molecular Synthesis, Shanghai Institute of Organic Chemistry, University of Chinese Academy of Sciences, Chinese Academy of Sciences, 345 Lingling Road, Shanghai 200032, P. R. China. E-mail: yiyunchen@sioc.ac.cn
bSchool of Physical Science and Technology ShanghaiTech University, 100 Haike Road, Shanghai 201210, P. R. China
cSchool of Chemistry and Material Sciences, Hangzhou Institute for Advanced Study, University of Chinese Academy of Sciences, 1 Sub-lane Xiangshan, Hangzhou 310024, P. R. China
First published on 27th January 2023
Abstract
Chemical labeling of proteins in live cells helps to probe their native functions in biological systems. Aryl azides are chemically inert under physiological conditions, but they are activated by certain external stimuli. Recently, photocatalytic live-cell applications of aryl azides by visible light irradiation have become a burgeoning new field in chemical biology. In this Feature Article, we focus on the recent progress of protein labeling in live cells with aryl azides induced by visible-light irradiation. Light irradiation activates aryl azides to generate highly reactive intermediates, which enables protein labeling for protein functionalization, crosslinking, and profiling. The activation mechanism of aryl azides by light irradiation is categorized as photolysis, energy-transfer, and electron-transfer. The extracellular and intracellular protein labeling applications in live cells with aryl azides induced by visible light are discussed, including recent advances in red-light-induced extracellular protein labeling.
 Yixin Zhang | Dr Yixin Zhang received her PhD degree in organic chemistry from Dalian Institute of Chemical Physics, Chinese Academy of Sciences in 2015. She then joined Prof. Yiyun Chen's group at the Shanghai Institute of Organic Chemistry as a research assistant. She is currently an associate research scientist focusing on light-induced biocompatible tools to study biology. |
 Jiawei Tan | Jiawei Tan is a PhD student under the guidance of Prof. Yiyun Chen at the Shanghai Institute of Organic Chemistry. He received his BSc degree in pharmaceutical sciences from China Pharmaceutical University. His research focuses on photocatalysis-induced protein cross-linking. |
 Yiyun Chen | Prof. Dr Yiyun Chen received his PhD degree in organic chemistry from Princeton University and did postdoctoral research on chemical biology at Harvard University and Howard Hughes Medical Institute. He joined the faculty team at the Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences in 2011. He currently leads a research team on biocompatible photochemistry as the principal investigator at the State Key Laboratory of Bio-Organic and Natural Products Chemistry, which focuses on organic photochemistry and photochemical biology. |
Introduction
Protein chemical labeling creates artificially modified proteins, immobilized enzymes, and antibody–drug conjugates, which are useful for native biological function probing, catalytic efficiency improvement, and selective delivery of cytotoxic drugs to target cancer cells.1 Chemical modification of proteins can be achieved by natural amino acid labeling or bioorthogonal labeling of unnatural groups in biocompatible conditions without impairing protein structures and functions.2,3Light has high spatiotemporal precision for external modulation. Aryl azides are chemically inert under physiological conditions, but they are activated by certain external stimuli.4 Light irradiation activates aryl azides to generate highly reactive intermediates, which enables protein labeling for protein functionalization, crosslinking, and profiling. Recently, photocatalytic live-cell applications of aryl azides by visible light irradiation have become a burgeoning new field in chemical biology (Fig. 1).5–7 In this feature article, we discuss the reaction pathways of aryl azides by photolysis and photocatalysis. The various biological applications of aryl azides protein labeling induced by light irradiation are discussed on the level of test tubes, extracellular membranes, and intracellular compartments, including recent advances in red-light-induced extracellular protein labeling.
 | ||
| Fig. 1 Chemical labeling of proteins with aryl azides induced by light irradiation for various biological applications. PC, photocatalyst. | ||
The reaction mechanism of light-induced aryl azide transformations
The photolysis pathways of aryl azides
It is known that the direct photoactivation of aryl azides under UV light (250–400 nm) irradiation forms the highly reactive singlet nitrene by releasing nitrogen gas (Fig. 2).8 These singlet nitrenes have a short lifetime of t1/2 ≈ 1–10 ns to quickly undergo three different reaction pathways: (i) they undergo X–H insertions by the nature of nitrenes where X = C, N, O, and S;9 (ii) they rearrange to benzazirines and subsequently ring-expand to seven-membered ketenimines (t1/2 = 5 ms to 1 s),10 in which the long-lived ketenimines are susceptible to hydrolysis11 and nucleophilic trapping;10 and (iii) they form the lower-energy triplet nitrenes with biradical nature (t1/2 = 300 ns) by intersystem crossing (ISC) (kISC = 3.2 × 106 s−1), after which they form anilines by H atom abstraction or dimerize to azobenzenes.12 Under certain light irradiation conditions, the triplet nitrenes cyclize to benzazirines and further rearrange to ketenimines.13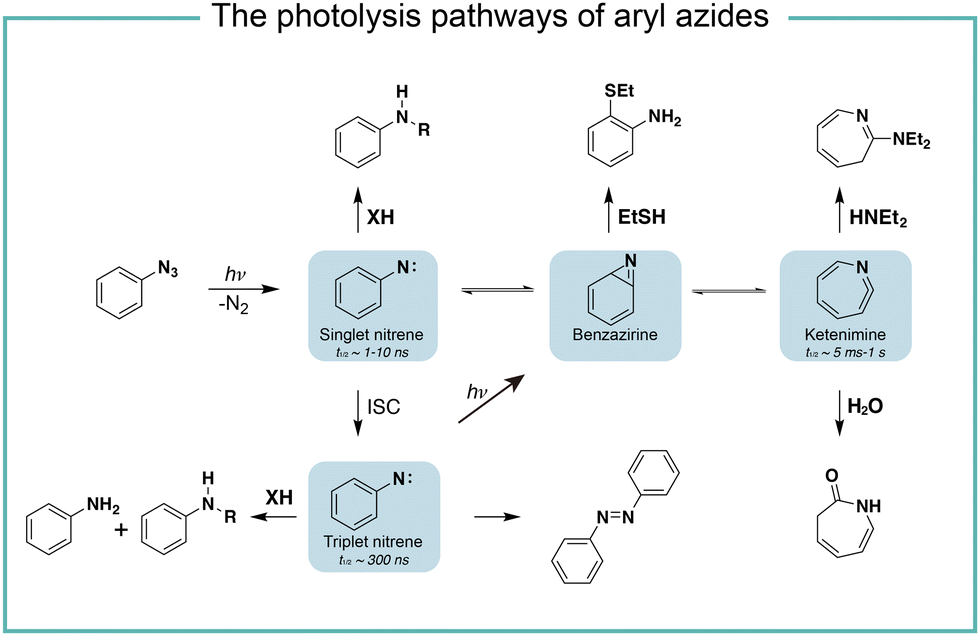 | ||
| Fig. 2 The photolysis pathways of aryl azides by direct light irradiation. ISC, intersystem crossing. | ||
The substituent groups on aryl azides greatly affect the ratio of different intermediates generated in the photolysis pathway.12 For example, the para-iodine-substituted aryl azides have a fast ISC rate (kISC = 7.2 × 107 s−1) due to the heavy atom effect, which facilitates the conversion of singlet nitrenes to triplet nitrenes and slows down the ring-expansion rearrangement. Similarly, aryl azides with a strong π donating para-methoxy substituent (kISC > 5.0 × 108 s−1) or para-dimethylamino substituent (kISC = 8.3 × 109 s−1) have fast ISC rates and also favour the triplet pathway rather than the ring-expansion rearrangement.14
The steric effect also affects the reaction pathways. The rate of singlet nitrene rearrangement to ketenimine is decreased in the case of polyfluorinated aryl azides. The ortho-fluorine substitution was believed to effectively inhibit ring expansion and led to a longer lifetime of singlet nitrenes.15 Recently, the computational calculations from Holland and coworkers indicated that no difference in the rearrangement barrier between ortho-fluorine substituted aryl azides and unsubstituted aryl azides. They hypothesized that the electronic complexity of the open-shell nitrene species contributed to the differences.16
The energy-transfer pathways of aryl azides
In photocatalytic reactions, the photocatalysts first absorb light irradiation to reach the excited state. Afterwards, the energy-transfer or electron-transfer from the excited photocatalyst to aryl azides initiates the subsequent transformations. In the energy-transfer pathway, the excited photosensitizer needs to have a matched triplet energy with aryl azides for the following photochemical transformation.17 In 1987, Raviv and Salomon reported that the energy-transfer process activated 5-iodonaphthalene-1-azide with 480 nm irradiation for membrane protein labeling, in which the undecylamine fluorescein was used as the photosensitizer.18 In 2021, we developed photocatalytic aryl azides protein labeling with organic dyes by the energy-transfer pathway and systematically investigated the reaction mechanism (Fig. 3).7 | ||
| Fig. 3 The photocatalysis pathway of aryl azides via the energy-transfer process. (a) The photocatalytic azide transformation induced by organic dyes; (b) the LC-MS/MS analysis of nucleophilic amino acid residues labeling on BSA proteins by aryl azides. | ||
The organic dyes acridine orange, fluorescein, and rhodamine 123 utilized in the study had a poor match of redox potential (Ered1/2 > −1.22 V, vs. SCE) with the reduction potential of azides (Eredp1/2 = −1.84 V, vs. SCE), suggesting that the single electron-transfer pathway was unlikely. These organic dyes had triplet energy greater than 1.9 eV and effectively converted aryl azides. In contrast, organic dyes with triplet energy of less than 1.9 eV did not convert aryl azides. In intermediate trapping experiments, the anilines, azepines, and thiol ether-substituted anilines were obtained and indicated the triplet nitrene, ketenimine, and benzazirine intermediates (Fig. 3a).19 In addition, the nucleophilic attacks at the ketenimine intermediates were observed by various nucleophilic residues through the LC-MS/MS analysis of photocatalytic protein labeling, whereas the less reactive amino acid residues such as Gln and Gly were not observed (Fig. 3b).
The electron-transfer pathways of aryl azides
When the oxidation potential of the excited state photocatalyst matches the reduction potential of aryl azides, the electron-transfer pathway activates aryl azides (Fig. 4). Liu and coworkers reported in 2011 the electron-transfer process for azide reduction induced by photocatalyst Ru(bpy)3Cl2 upon visible light irradiation.20 In the photoreduction of aryl azides in organic solvents, the trialkyl amine and Hantzsch ester were used as the sacrificial reductants for the azide reduction. In the neutral aqueous conditions, the ascorbates were used as the reductant. MacMillan and coworkers reported in 2022 a red-light-excited SnIV chlorin e6 catalyst for protein labeling with aryl azides and reductant NADH under red light irradiation.6 In both cases, the aryl azides were reduced to azide radical anions by the reduced photocatalyst in the ground state. After rapidly releasing molecular nitrogen and protonation, the azide radical anion is converted to the aminyl radical (Fig. 4a). In the case of Sn catalysis, the aminyl radical acts as a reactive intermediate for the protein proximity labeling. The generation and quenching of SnIII species by NADH and azide were suggested from the transient-absorption spectroscopy measurement. | ||
| Fig. 4 The photocatalysis pathway of aryl azides via the electron-transfer process. (a) The reductive pathway; (b) the redox-neutral pathway. RED, reductant. | ||
In the study of Rovis and coworkers in the same year, they reported the perfluorinated azide transformation to triplet nitrenes via a redox-neutral mechanism with photocatalyst Os((3,4,7,8-Me4phen))3(PF6)2 (Fig. 4b).21 The photoexcited catalyst Os* reduced perfluorinated azides by electron transfer to generate the reduced azide and the oxidized Os+. The reduced azide released molecular nitrogen to yield the nitrene, which was further oxidized by Os+ to afford triplet nitrenes. The computational modelling analysis of the aryl azides and the intermediate trapping experiments suggested the redox-neutral electron-transfer pathway for triplet nitrene formation. MacMillan and coworkers later used the transient absorption and spectro-electrochemistry studies to elucidate the electron-transfer mechanism for photocatalytic aryl azides activation under blue light with iridium photocatalysis.22 The generation of oxidized iridium was observed, agreeing with the electron-transfer pathway.
Protein labeling with aryl azides in test tubes
Aryl azides are chemically stable and easily synthesized, and have been extensively studied in protein labeling.23 The photolysis of aryl azides has been used to synthesize functionalized biomolecules such as fluorophore-tagged proteins, antibody–drug conjugates, and radiolabelled antibodies.5 Holland and co-workers synthesized the zirconium-89 radiolabeled monoclonal antibodies by the photochemical conjugation of aryl azides (Fig. 5a).24 The metal-binding chelate for Zr was linked with aryl azides to enable the one-pot conjugation in pH 8–8.5 buffered solution. The one-pot photochemical conjugation and radio labeling of 89Zr could be finished in less than 90 minutes, and was amenable to full automation.24 | ||
| Fig. 5 In vitro protein labeling via aryl azides photolysis. (a) The aryl azides promoted photo-radiosynthesis of 89Zr-radiolabeled monoclonal antibodies; (b) the aryl azides photoaffinity labeling of target proteins with structure and strategy optimization; (c) the incorporation strategies of aryl azides used in protein–protein photo-crosslinking; (d) the enzymatic incorporation of aryl azides in protein-RNA photo-crosslinking. mAbs, monoclonal antibodies; UAA, unnatural amino acid; aaRS, aminoacyl-tRNA synthetase; POI, protein of interest; Ecm1, encephalitozoon cuniculi mRNA cap (guanine N-7) methyltransferase; eIF4E, eukaryotic initiation factor 4E. | ||
Photoaffinity labeling with aryl azides is useful for target protein identification in complex biological environments.25,26 Typically, a photoaffinity labeling probe comprises a binding ligand as the bait, a photo-crosslinker for protein capture, and an affinity tag for imaging or target enrichment and isolation (Fig. 5b). The use of aryl azides for protein photoaffinity labeling as photo-crosslinking handles was first reported by Fleet in 1969.27 Aryl azides typically require 260–365 nm irradiation for photolysis and the nitrene insertion into desired targets generally occurs in low yields (<30%).28 With structural optimization, Geiger and coworkers red-shifted aryl azides to the 480 nm absorption with the m-nitro-substitution.29 Keana and coworkers later reported polyfluorinated aryl azides to increase the C–H and N–H insertion efficiency.30 To further improve the crosslinking efficiency of aryl azides, Li and coworkers performed strategy optimization by developing a DNA-programmed photoaffinity labeling method (DPAL) (Fig. 5b).31 In the DPAL probe, protein-binding ligands and multiple phenyl azide crosslinkers were attached to the complementary DNA strand. The crosslinkers improved the labeling efficiency by nearly 7-fold without affecting the labeling specificity and the target binding.
Aryl azides have also been used to study protein–protein interactions for understanding cellular functions and mechanisms in living systems. The photo-crosslinking with aryl azides may trap transient protein–protein interaction in a spatiotemporal-specific fashion by covalently linking low-affinity interacting macromolecules.32 Through genetic code expansion and enzymatic incorporation, the amino-acid-analogue bearing aryl azides can be introduced to the site of proteins with specificity (Fig. 5c). Schultz and coworkers first reported the addition of p-azido-L-phenylalanine on the dimer interface of glutathione-S-transferase by genetic code expansion, and the photochemical activation of aryl azides enabled the protein–protein interaction studies.33 Ting and coworkers engineered an aryl azide ligase to site-specifically incorporate fluorinated aryl azides on target proteins, which could study interacting proteins in cell lysates by UV-induced photo-crosslinking.34
The aryl azides can also be enzymatically transferred to nucleic acids to identify the RNA–protein interaction. Rentmeister and coworkers discovered that the S-adenosyl-L-methionine (SAM) analogues bearing aryl azides may act as a SAM surrogate for the wild-type methyltransferase. The aryl azides incorporation on the N7 position of the mRNA cap was highly efficient by cap N7 methyltransferase Ecm1 (Fig. 5d). The affinity of the modified 5′ caps was reflected by the photo-crosslinking of the aryl azide-modified mRNAs with a known cap interacting protein elF4E.35
Extracellular protein labeling with aryl azides
The short wavelength UV light irradiation may affect a living biological system negatively and there were isolated reports on photolytic protein labeling with aryl azides in live cells. Paulson and coworkers introduced azide-modified sialic acids (AAz-NeuAc) to glycosylated proteins by metabolic incorporation at the B cell surface (Fig. 6a).36 Under 254 nm UV irradiation for 20 min, the crosslinking of glycoproteins and coreceptors of their B-cell receptors CD22 was observed, which revealed the homomultimeric complexes of CD22 on the B cell surface.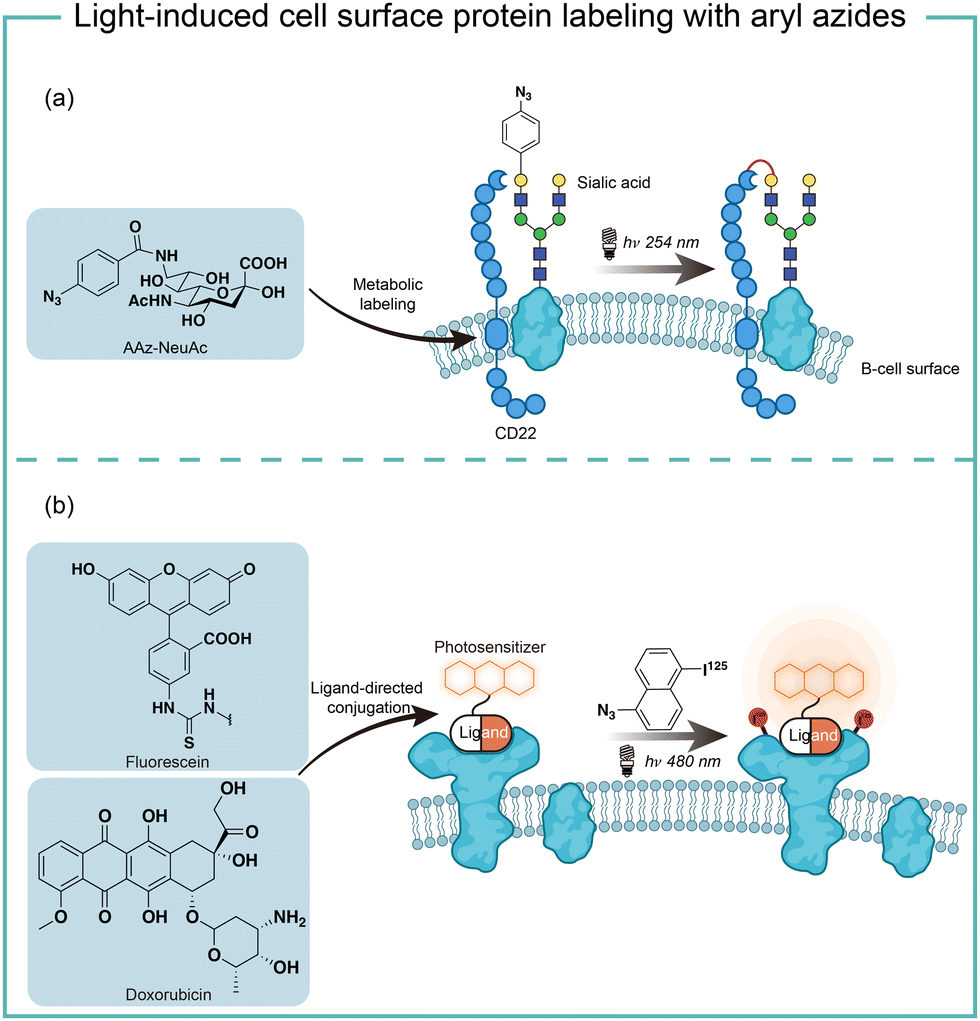 | ||
| Fig. 6 Cell surface protein labeling via aryl azides photolysis and photosensitization in live cells. (a) Metabolic incorporation of azide-modified sialic acid into cell-surface glycoproteins for in situ protein–protein photo-crosslinking. (b) Photocatalytic proximity protein labeling using ligand-directed photocatalysts. | ||
Besides metabolic incorporation, photocatalysts may be introduced as the ligand-conjugated form to target its surrounding proteins. Salomon and coworkers conjugated a fluorescein photocatalyst to lima bean lectin, which specifically interacted with receptor molecules on the rod outer segment and lymphocyte plasma membrane (Fig. 6b).37 Upon 480 nm light irradiation, the membrane receptor proteins surrounding fluorescein at extracted membranes were labelled by 5-iodonaphthalene-1-azides. When using free fluorescein or direct photolysis by UV irradiation, random labeling was observed. This approach was later utilized to explore the interaction of the cytotoxic drug doxorubicin with their transporter proteins in live cells.38 In that case, doxorubicin acts as both a cytotoxic drug and an effective photosensitizer. After binding to the functional multidrug transporter P170 on live drug-resistant cells, doxorubicin sensitized 5-iodonaphthalene-1-azides to label those proteins specifically. In contrast, these multiple membrane proteins were labelled in a nonspecific manner in either lysed cells or drug-sensitive cells. If the calcium channel blocker was added to reverse the drug resistance, the selective labeling was also not observed. These results suggested the P170 transporter association with doxorubicin drug resistance and indicated the advantages of photolabeling in live cells.
Intracellular protein labeling with aryl azides
Intracellular protein labeling in their native environment provides powerful tools in delineating cellular homeostasis and dysfunction. To overcome the phototoxicity of UV irradiation, Rhee and coworkers exploited 4-azido-N-ethyl-1,8-naphthalimide (AzNP) for photolytic proximity labeling in live cells with blue light (Fig. 7).39 Through the self labeling enzymes, AzNP was incorporated into various HaloTag-conjugated proteins of interest. The proximal proteins were crosslinked by the rapidly generated aryl nitrene species by blue light activation in live cells. Using the HaloTag-conjugated SARS-CoV-2 nucleocapsid (N) proteins and the AzNP ligand, this proximity photo-crosslinking method was utilized to identify the host interactome of SARS-CoV-2 N proteins. Multiple RNA-binding proteins in stress granules were exclusively enriched by the mass spectrometry analysis of the crosslinked proteins. This photo-crosslinking could be performed in cultured cells and mouse brain tissue, but the crosslinking efficiency was low in the tissue samples due to the low penetration of blue light.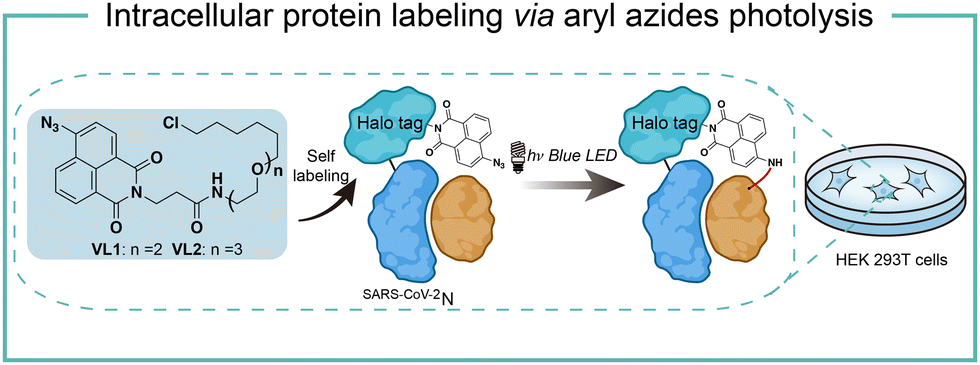 | ||
| Fig. 7 Intracellular protein labeling via aryl azides photolysis. | ||
In the case of photocatalysis, the activation of aryl azides is confined to the vicinity of photocatalysts, which distance-dependence allows spatial control in a cellular region of interest. Liu and coworkers discovered a Ru(II)-catalyzed azide reduction reaction to release amines under visible light irradiation from the DNA-encoded reaction discovery system. The reaction was highly chemoselective and compatible with various functional groups including alcohols, phenols, acids, alkenes, alkynes, aldehydes, and alkyl halides. Under mild aqueous conditions, this photocatalytic azide reduction reaction executed smoothly on nucleic acid and oligosaccharide substrates without affecting the enzyme RNaseA activity in the same solution (Fig. 8a).20 Winssinger and coworkers later applied this photocatalytic azide reduction in living systems (Fig. 8b).40,41 By using oligomeric protein templates and nucleic acid templates, the photocatalyst Ru(II) was brought pseudo-intramolecularly to the aryl azide-caged rhodamine. A clear turn-on fluorescent signal was observed after 30 min 455 nm LED light irradiation to indicate the rhodamine photo-release. The templated reaction rates in response to the protein oligomer were at least 30-fold faster than the untemplated reaction. The templated photocatalytic azide reaction was successfully used in miRNA imaging and protein interaction identification in live cells and vertebrates.
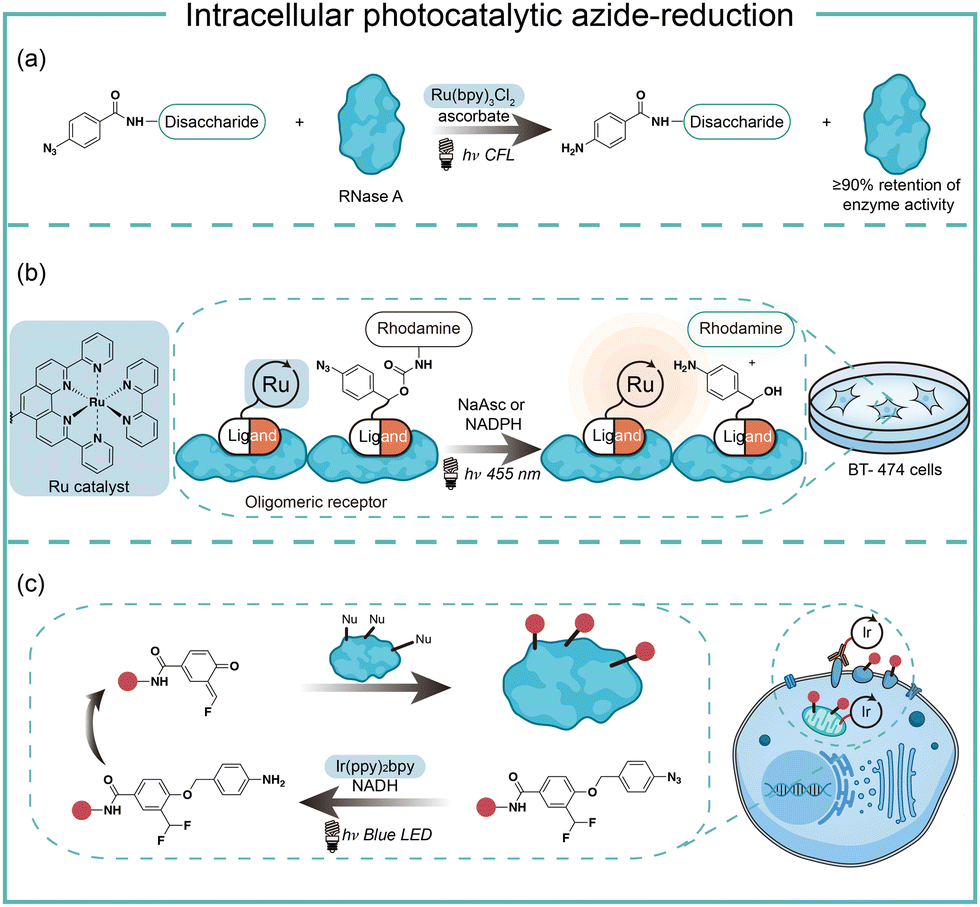 | ||
| Fig. 8 Intracellular photocatalytic azide-reduction via visible-light irradiation. (a) The biomolecule-compatible photocatalytic azide-reduction by the Ru catalyst. (b) The photocatalytic decaging of azides by the Ru catalyst for in vivo fluorophore release. (c) The photocatalytic decaging of azides by the Ir catalyst for in vivo protein labeling. | ||
By carefully choosing photocatalysts, Chen and coworkers discovered a cell-membrane permeable Ir(ppy)2bpy photocatalyst with mitochondria-targeting specificity.42 Upon blue LED light irradiation, the Ir(III) photocatalyst allowed rapid reduction of the azido benzyl-cage to release quinone methide as a highly reactive Michael acceptor for proximity protein labeling. These photocatalytic decaging and trapping reactions profiled the mitochondrial proteome in live HeLa cells as well as hard-to-transfect macrophage RAW264.7 cells, which observed 70% mitochondria specificity among all enriched proteins (Fig. 8c). The Ir(III) catalyst was further conjugated to antibodies for cell surface proteome profiling.43 The photocatalytic decaging occurred on cells targeted by the Ir(III)-antibody conjugate to label the neighbouring molecules with blue LED irradiation (18 mW cm−2).
Transition-metal-based photocatalysis has potential heavy-metal cytotoxicity,44 which may limit its biological applications. We employed organic dyes to catalyse the aryl azides protein labeling for intracellular applications (Fig. 9).7 Organic dyes have historically been used for intracellular imaging and tracing due to their native subcellular localization and excellent biocompatibility.45 We found that the commercially available organic dyes such as acridine orange (AO), fluorescein (Flu), and rhodamine 123 (Rh123) triggered the aryl azides protein labeling under low-energy blue or green LED light irradiation. The labeling reaction finished in 1 minute or even seconds when using high-energy green light irradiation. The addition of reactive oxygen species and metal ions at the concentrations above the physiological relevance did not affect the protein labeling.46 In contrast, cellular reductants such as glutathione and N-acetylcysteine effectively quenched the protein labeling, supporting the proximity labeling in the intracellular environment.47 These photocatalytic dyes also exhibited low quantum yields of singlet oxygen, suggesting their promising biocompatibility in live cells.
 | ||
| Fig. 9 Subcellular protein profiling via photocatalytic aryl azides activation with organic dyes. (a) Green-light-excited organic dye catalyst for aryl azides photoactivation and subcellular protein labeling. (b) The workflow and results of the dynamic mitochondrial proteome change mapping from rotenone treatment. AO, acridine orange; Rh123, rhodamine 123; Bio-PhN3, aryl azide-biotin probe. | ||
Successful demonstration of this photocatalytic azide conjugation reaction in the mitochondria of live cells has been shown through the mitochondrially localized dyes Rh123. Upon 515 nm low energy green LED light (2.9 mW cm−2) irradiation for 60 min, Rh123 mediated aryl azides labeling reactions in the mitochondria with excellent colocalization with mitochondrion dye TMRE (Fig. 9a). The labeled proteins mainly located in the mitochondrial matrix and inner membrane by mass spectrometry analysis, which was consistent with the Rh123 sub-mitochondrial localization. Afterwards, the mitochondria-specific labeling strategy was applied for dynamic mitochondrial proteome investigation for stress-response protein identification (Fig. 9b). With mild mitochondrial stress induced by 0.02 μM of rotenone, several potential mitochondrial stress-response proteins were identified.48 The AO-induced nucleus labeling was later realized with the nucleus-specific dye AO under a higher energy green LED irradiation in 1 min.49 These photocatalytic azide-promoted labeling methods offer precise protein labeling in subcellular compartments of live cells, which holds excellent potential for intracellular protein network investigations.
Red-light-induced extracellular protein labeling with aryl azides
To improve the poor tissue penetration from short-wavelength light irradiation, MacMillan and coworkers used the red-light absorbing catalysts SnIV chlorin e6 to activate aryl azides for protein labeling (Fig. 10a).6 Protein labeling was observed from the Sn photocatalyst and aryl azide probe with NADH as the reductant upon red light irradiation in minutes. In the optimized labeling conditions, the red-light-activated protein labeling demonstrated detectable biotinylation signals after penetrating several layers of tissue (>10 mm). They further conjugated the red-light-excitable Sn photocatalyst to a secondary antibody to selectively target EGFR (epidermal growth factor receptor) on the cell surface. Upon 660 nm wavelength light irradiation, the reactive aminyl radical intermediate selectively targeted EGFR and its neighbouring proteins. This method was further deployed to profile erythrocyte cell-surface proteins in the whole mouse blood and a total of 24 proteins were identified with mostly erythrocyte cell surface proteins. In contrast, the blue light activation failed to label the complex biological sample and confirmed the advantages of red light for tissue penetration. | ||
| Fig. 10 Red-light-induced extracellular protein profiling via photocatalytic aryl azides activation using an antibody conjugated metal catalyst. (a) Red-light-excited Sn photocatalyst for aryl azides photoactivation and EGFR microenvironment labeling; (b) red-light-excited Os photocatalyst for aryl azides photoactivation and EpCAM microenvironment labeling; (c) the proximity labeling radius of different photoactivation probes through the Ir-induced photoactivation. EGFR, epidermal growth factor receptor; EpCAM, epithelial cell adhesion molecule; FWHM, full width at half maximum. | ||
Another elegant example of red-light photocatalytic labeling of proteins was reported by Rovis and coworkers (Fig. 10b).21 The osmium photocatalyst was utilized for perfluoroaryl azide activation under red light irradiation to label proteins in aqueous buffer conditions. Minimal reactive oxygen species (ROS) were generated in the red-light mediated labeling process, indicating the osmium photocatalyst's compatibility for live-cell environment profiling. This azide activation process was further applied in the microenvironment mapping of targeted epithelial cell adhesion molecule (EpCAM) in different colon cancer cell systems. Several enriched membrane proteins with previously known and unknown association to EpCAM were identified by this method. They later compared this red-light photocatalytic system for mapping the EpCAM microenvironment with other proximity labeling methods. The proximal protein labeling region by nitrene was between the diazirine-based μMap system50 and peroxidase-based phenoxy radical platform.47
These results were consistent with the direct labelling measurement via super-resolution microscopy developed by MacMillan and coworkers (Fig. 10c).22 In their model, the labeling radius for the photocatalytic aryl azides activation was 119 ± 33 nm, which was larger than that of the diazirine at 54 ± 12 nm. The labeling radius was consistent with the slower quenching rate from triplet nitrenes compared to carbenes. The perfluoroaryl azides showed smaller labeling radius and increased reactivity compared to nonfluorinated congeners. The molecular diffusion coefficient of the intermediate dictated the spatial selectivity, and the labeling radius of aryl azides could be tuned by the PEG linker's length.
Conclusion and outlook
In conclusion, the photochemical transformations of aryl azides by photolysis and photocatalysis provide powerful tools for the covalent modification of natural proteins in live cells. Through chemical synthesis, genetic encoding, and enzymatic incorporation, aryl azides can be introduced into small molecules, DNA, RNA, and proteins for the photochemical labeling of proteins. When combined with proteomic tools, the efficient profiling of specific protein families and spatial proteomes in complex biological systems can be achieved. While the protein residue preference has been demonstrated by aryl azides photolabeling in some cases, the diversified reactive intermediates and the complex biological environment call for further investigations on the photochemical labeling of aryl azides (Fig. 11).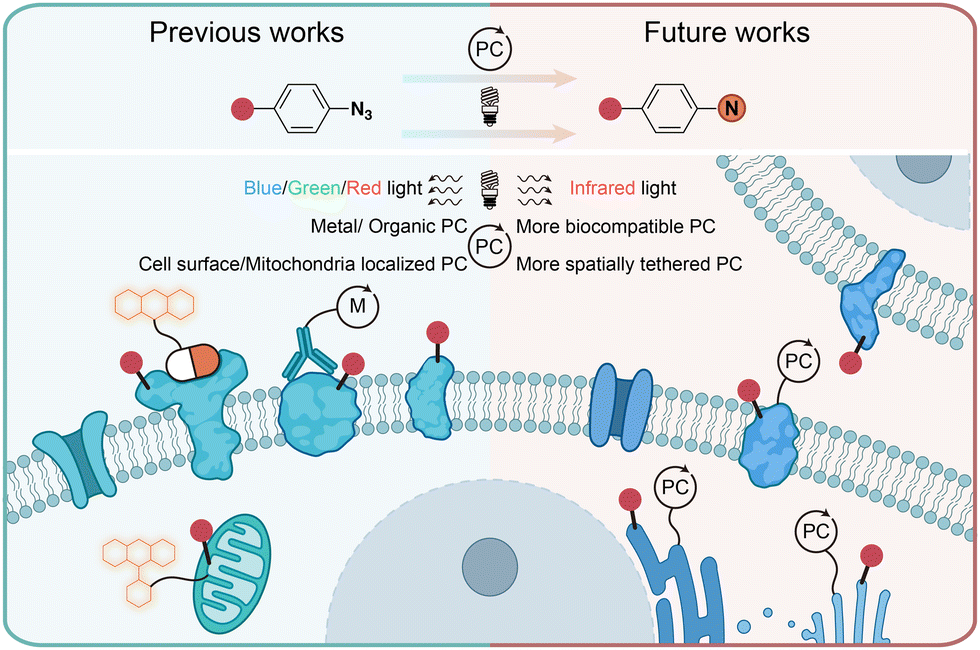 | ||
| Fig. 11 Current and outlook of light-induced protein labeling with aryl azides in live cells. M, metal. | ||
Photolytic azide labeling has been widely applied in protein conjugation, protein affinity labeling, and protein–protein crosslinking. However, most of the photolytic labeling is implemented in test tubes due to the phototoxicity and poor cell penetration of short wavelength irradiation. Encouragingly, the visible-light-induced photocatalytic azide labeling enables live cell applications. The photocatalytic labeling allows protein interaction profiling on the cell surface and the dynamic proteome studies in subcellular machinery. In contrast to enzymatic-based proximity labeling techniques51 such as APEX and BioID, the photocatalytic azide labeling provides high spatiotemporal resolution from external light irradiation. The localized photocatalysts afford the targeted probing for cell surface and mitochondria proteins. It is expected that improved spatial labeling requiring new spatially located photocatalysts, and the development of more biocompatible photocatalysts with longer wavelength absorption such as infrared light is the future direction to enable disease research and drug discovery.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support was provided by the National Natural Science Foundation of China 91753126, 22277133, 21622207, 21602242, CAS Interdisciplinary Innovation Team JCTD-2020-16, Program of Shanghai Academic/Technology Research Leader 21XD1424700, and Strategic Priority Research Program of the Chinese Academy of Sciences XDB20020200.References
- T. Tamura and I. Hamachi, J. Am. Chem. Soc., 2019, 141, 2782–2799 CrossRef CAS PubMed.
- K. Lang and J. W. Chin, Chem. Rev., 2014, 114, 4764–4806 CrossRef CAS PubMed.
- E. Basle, N. Joubert and M. Pucheault, Chem. Biol., 2010, 17, 213–227 CrossRef CAS PubMed.
- D. Y. Huang and G. B. Yan, Adv. Synth. Catal., 2017, 359, 1600–1619 CrossRef CAS.
- J. P. Holland, M. Gut, S. Klingler, R. Fay and A. Guillou, Chem. – Eur. J., 2020, 26, 33–48 CrossRef CAS PubMed.
- B. F. Buksh, S. D. Knutson, J. V. Oakley, N. B. Bissonnette, D. G. Oblinsky, M. P. Schwoerer, C. P. Seath, J. B. Geri, F. P. Rodriguez-Rivera, D. L. Parker, G. D. Scholes, A. Ploss and D. W. C. MacMillan, J. Am. Chem. Soc., 2022, 144, 6154–6162 CrossRef CAS PubMed.
- H. Wang, Y. Zhang, K. Zeng, J. Qiang, Y. Cao, Y. Li, Y. Fang, Y. Zhang and Y. Chen, JACS Au, 2021, 1, 1066–1075 CrossRef CAS PubMed.
- N. P. Gritsan, T. Yuzawa and M. S. Platz, J. Am. Chem. Soc., 1997, 119, 5059–5060 CrossRef CAS.
- M. S. Platz, Acc. Chem. Res., 1995, 28, 487–492 CrossRef CAS.
- Y. Z. Li, J. P. Kirby, M. W. George, M. Poliakoff and G. B. Schuster, J. Am. Chem. Soc., 1988, 110, 8092–8098 CrossRef CAS.
- F. R. Bou-Hamdan, F. Levesque, A. G. O'Brien and P. H. Seeberger, Beilstein J. Org. Chem., 2011, 7, 1124–1129 CrossRef CAS PubMed.
- N. P. Gritsan and M. S. Platz, Chem. Rev., 2006, 106, 3844–3867 CrossRef CAS PubMed.
- T. Schleif, J. Mieres-Perez, S. Henkel, E. Mendez-Vega, H. Inui, R. J. McMahon and W. Sander, J. Org. Chem., 2019, 84, 16013–16018 CrossRef CAS PubMed.
- N. P. Gritsan, D. Tigelaar and M. S. Platz, J. Phys. Chem. A, 1999, 103, 4465–4469 CrossRef CAS.
- W. L. Karney and W. T. Borden, J. Am. Chem. Soc., 1997, 119, 3347–3350 CrossRef CAS.
- D. F. Earley, A. Guillou, S. Klingler, R. Fay, M. Gut, F. d'Orchymont, S. Behmaneshfar, L. Reichert and J. P. Holland, JACS Au, 2022, 2, 646–664 CrossRef CAS PubMed.
- Q. Q. Zhou, Y. Q. Zou, L. Q. Lu and W. J. Xiao, Angew. Chem., Int. Ed., 2019, 58, 1586–1604 CrossRef CAS PubMed.
- Y. Raviv, Y. Salomon, C. Gitler and T. Bercovici, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 6103–6107 CrossRef CAS PubMed.
- W. T. Borden, N. P. Gritsan, C. M. Hadad, W. L. Karney, C. R. Kemnitz and M. S. Platz, Acc. Chem. Res., 2000, 33, 765–771 CrossRef CAS PubMed.
- Y. Chen, A. S. Kamlet, J. B. Steinman and D. R. Liu, Nat. Chem., 2011, 3, 146–153 CrossRef CAS PubMed.
- N. E. S. Tay, K. A. Ryu, J. L. Weber, A. K. Olow, D. C. Cabanero, D. R. Reichman, R. C. Oslund, O. O. Fadeyi and T. Rovis, Nat. Chem., 2023, 15, 101–109 CrossRef CAS PubMed.
- J. V. Oakley, B. F. Buksh, D. F. Fernandez, D. G. Oblinsky, C. P. Seath, J. B. Geri, G. D. Scholes and D. W. C. MacMillan, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2203027119 CrossRef CAS PubMed.
- J. Das, S. N. Patil, R. Awasthi, C. P. Narasimhulu and S. Trehan, Synthesis, 2005, 1801–1806 CrossRef CAS.
- A. Guillou, D. F. Earley, M. Patra and J. P. Holland, Nat. Protoc., 2020, 15, 3579–3594 CrossRef CAS PubMed.
- F. Kotzybahibert, I. Kapfer and M. Goeldner, Angew. Chem., Int. Ed. Engl., 1995, 34, 1296–1312 CrossRef CAS.
- E. Smith and I. Collins, Future Med. Chem., 2015, 7, 159–183 CrossRef CAS PubMed.
- G. W. J. Fleet, R. R. Porter and J. R. Knowles, Nature, 1969, 224, 511–512 CrossRef CAS.
- E. Karaj, S. H. Sindi and L. M. V. Tillekeratne, Bioorg. Med. Chem., 2022, 62, 116721 CrossRef CAS PubMed.
- M. W. Geiger, M. M. Elliot, V. D. Karacostas, T. J. Moricone, J. B. Salmon, V. L. Sideli and M. A. Stonge, Photochem. Photobiol., 1984, 40, 545–548 CrossRef CAS.
- J. F. W. Keana and S. X. Cai, J. Org. Chem., 1990, 55, 3640–3647 CrossRef CAS.
- G. Li, Y. Liu, X. Yu and X. Li, Bioconjugate Chem., 2014, 25, 1172–1180 CrossRef CAS PubMed.
- D. P. Murale, S. C. Hong, M. M. Haque and J. S. Lee, Proteome Sci., 2017, 15, 14 CrossRef PubMed.
- J. W. Chin, S. W. Santoro, A. B. Martin, D. S. King, L. Wang and P. G. Schultz, J. Am. Chem. Soc., 2002, 124, 9026–9027 CrossRef CAS PubMed.
- H. Baruah, S. Puthenveetil, Y. A. Choi, S. Shah and A. Y. Ting, Angew. Chem., Int. Ed., 2008, 47, 7018–7021 CrossRef CAS PubMed.
- F. Muttach, F. Masing, A. Studer and A. Rentmeister, Chem. – Eur. J., 2017, 23, 5988–5993 CrossRef CAS PubMed.
- S. Han, B. E. Collins, P. Bengtson and J. C. Paulson, Nat. Chem. Biol., 2005, 1, 93–97 CrossRef CAS PubMed.
- Y. Raviv, T. Bercovici, C. Gitler and Y. Salomon, Biochemistry, 1989, 28, 1313–1319 CrossRef CAS PubMed.
- Y. Raviv, H. B. Pollard, E. P. Bruggemann, I. Pastan and M. M. Gottesman, J. Biol. Chem., 1990, 265, 3975–3980 CrossRef CAS PubMed.
- P. K. Mishra, M. G. Kang, H. Lee, S. Kim, S. Choi, N. Sharma, C. M. Park, J. Ko, C. Lee, J. K. Seo and H. W. Rhee, Chem. Sci., 2022, 13, 955–966 RSC.
- K. K. Sadhu, T. Eierhoff, W. Roemer and N. Winssinger, J. Am. Chem. Soc., 2012, 134, 20013–20016 CrossRef CAS PubMed.
- L. Holtzer, I. Oleinich, M. Anzola, E. Lindberg, K. K. Sadhu, M. Gonzalez-Gaitan and N. Winssinger, ACS Cent. Sci., 2016, 2, 394–400 CrossRef CAS PubMed.
- Z. Huang, Z. Liu, X. Xie, R. Zeng, Z. Chen, L. Kong, X. Fan and P. R. Chen, J. Am. Chem. Soc., 2021, 143, 18714–18720 CrossRef CAS PubMed.
- Z. Q. Liu, X. Xie, Z. Y. Huang, F. Lin, S. B. Liu, Z. J. Chen, S. N. Qin, X. Y. Fan and P. R. Chen, Chem, 2022, 8, 2179–2191 CAS.
- L. Gourdon, K. Cariou and G. Gasser, Chem. Soc. Rev., 2022, 51, 1167–1195 RSC.
- K. M. Dean and A. E. Palmer, Nat. Chem. Biol., 2014, 10, 512–523 CrossRef CAS PubMed.
- C. C. Winterbourn, Nat. Chem. Biol., 2008, 4, 278–286 CrossRef CAS PubMed.
- H.-W. Rhee, P. Zou, N. D. Udeshi, J. D. Martell, V. K. Mootha, S. A. Carr and A. Y. Ting, Science, 2013, 339, 1328–1331 CrossRef CAS PubMed.
- N. Y. Li, K. Ragheb, G. Lawler, J. Sturgist, B. Rajwa, J. A. Melendez and J. P. Robinson, J. Biol. Chem., 2003, 278, 8516–8525 CrossRef CAS PubMed.
- A. Pierzynska-Mach, P. A. Janowski and J. W. Dobrucki, Cytometry A, 2014, 85A, 729–737 CrossRef CAS PubMed.
- J. B. Geri, J. V. Oakley, T. Reyes-Robles, T. Wang, S. J. McCarver, C. H. White, F. P. Rodriguez-Rivera, D. L. Parker, Jr., E. C. Hett, O. O. Fadeyi, R. C. Oslund and D. W. C. MacMillan, Science, 2020, 367, 1091–1097 CrossRef CAS PubMed.
- W. Qin, K. F. Cho, P. E. Cavanagh and A. Y. Ting, Nat. Methods, 2021, 18, 133–143 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2023 |