
Delivery of gold nanoparticle-conjugated M2e influenza vaccine in mice using coated microneedles
Lazar D.
Nesovic†
,
Carsen J.
Roach†
,
Gaurav
Joshi
and
Harvinder Singh
Gill
 *
*
Department of Chemical Engineering Texas Tech University, 8th and Canton, Lubbock, Texas 79409, USA. E-mail: harvinder.gill@ttu.edu
First published on 23rd June 2023
Abstract
As a prospective influenza vaccination platform, a microneedle patch offers advantages such as self-administration and reduction of needle-phobia-associated vaccination avoidance. In an effort to design a broadly protective influenza vaccine we have previously developed a vaccine formulation containing the highly conserved ectodomain sequence of the M2 influenza protein (M2e) attached to the surface of gold nanoparticles (AuNPs) with CpG as a soluble adjuvant (AuNP-M2e + sCpG). Our previous studies have used the intranasal route for vaccination and demonstrated broad protection from this vaccine. Here we asked the question whether the same formulation can be effective when administered to mice using microneedles. We demonstrate that the microneedles can be coated with AuNP-M2e + sCpG formulation, and the AuNPs from the coating can be readily resuspended without aggregation. The AuNPs were delivered with high efficiency into murine skin, and the AuNPs cleared the skin within 12 h of microneedle treatment. After vaccination, strong M2e-specific humoral and cellular responses were stimulated, and the vaccinated mice were 100% protected following a lethal challenge with influenza A/PR/8/34 (H1N1).
1. Introduction
Immunization campaigns across the world continue to be plagued with a multitude of complex issues,1 which severely diminish the impact of vaccines. Cost of administration2 and the need for a cold storage infrastructure3 are commonly recognized as two important bottlenecks in vaccination campaigns. In addition, the need for trained health care workers who can promptly administer vaccines using needles and syringes is an another obstacle that must be addressed during vaccination campaigns. For example, the measles and rubella vaccine must be administered within 6 h after reconstitution,4 and if the vaccine is not in the form of a single-dose, then healthcare workers sometimes hesitate to reconstitute it for fear of wasting it, and ask the parents of the child to come back a different day to receive the vaccine. Such events are called ‘missed opportunities’. The coronavirus pandemic caused by COVID-19 is another example where significant disparities in vaccine distribution in low-and middle-income countries were observed due to challenges in supplying and delivering the vaccine throughout the world.5To overcome these bottlenecks related to vaccine administration, the vaccine innovation prioritization strategy (VIPS) alliance was formed, which through a multi-year, in-depth discussion with different stakeholders concluded in 2020 that microneedle (MN) patches are a promising platform for the next generation of vaccination systems. MN arrays in which the vaccine is formulated in a solid form have been shown to be thermally stable,6–8 do not cause long-term damage to skin,9,10 and are perceived as easy to administer,11,12 and painless.13
With this in mind, we set out to establish the ability of coated MNs to deliver an M2e-based broadly protective influenza vaccine. Influenza is a highly contagious, viral respiratory disease, which causes yearly epidemics, poses a continuous threat of pandemics, and results in an estimated average of close to 400![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths annually.14 Hemagglutinin (HA), which is the prominent antigen in current influenza vaccines is subject to point mutations,15,16 therefore, new vaccines must be formulated every year. To address this challenge, significant effort has been put into developing a broadly protective (sometimes also known as universal) influenza vaccine. All three influenza virus surface proteins, namely, HA, neuraminidase (NA), and matrix-2 (M2) protein are being evaluated to make a broadly protective influenza vaccine.17 The M2 protein whose 23-amino acid-long extracellular domain (M2e) has remained remarkably conserved since the 1918 pandemic is one antigen being developed.18 In our previous studies, we have demonstrated that intranasal delivery of a consensus M2e peptide attached to the surface of gold nanoparticles (AuNPs) in combination with CpG in a soluble form (sCpG) induces robust anti-M2e immunoglobulin response and confers 100% survival against lethal challenges with influenza A/PR/8/34 (H1N1), A/California/04/2009 (H1N1), and a highly pathogenic avian influenza strain A/Vietnam/1203/2004 (H5N1).19–21 Additionally, we recently demonstrated that the AuNP-M2e + sCpG formulation is thermally stable when stored in lyophilized form for 3 months at 4 °C or 37 °C, or for 2 weeks at 50 °C, and that it also confers protective immunity in ferrets.22
000 deaths annually.14 Hemagglutinin (HA), which is the prominent antigen in current influenza vaccines is subject to point mutations,15,16 therefore, new vaccines must be formulated every year. To address this challenge, significant effort has been put into developing a broadly protective (sometimes also known as universal) influenza vaccine. All three influenza virus surface proteins, namely, HA, neuraminidase (NA), and matrix-2 (M2) protein are being evaluated to make a broadly protective influenza vaccine.17 The M2 protein whose 23-amino acid-long extracellular domain (M2e) has remained remarkably conserved since the 1918 pandemic is one antigen being developed.18 In our previous studies, we have demonstrated that intranasal delivery of a consensus M2e peptide attached to the surface of gold nanoparticles (AuNPs) in combination with CpG in a soluble form (sCpG) induces robust anti-M2e immunoglobulin response and confers 100% survival against lethal challenges with influenza A/PR/8/34 (H1N1), A/California/04/2009 (H1N1), and a highly pathogenic avian influenza strain A/Vietnam/1203/2004 (H5N1).19–21 Additionally, we recently demonstrated that the AuNP-M2e + sCpG formulation is thermally stable when stored in lyophilized form for 3 months at 4 °C or 37 °C, or for 2 weeks at 50 °C, and that it also confers protective immunity in ferrets.22
We postulated that by coupling this already thermally-stable influenza vaccine formulation with MNs, it may be feasible to synergistically harness the benefits of MNs as a vaccine delivery system. Therefore, in the present study, we sought to investigate the ability to coat MNs with the AuNP-M2e + sCpG vaccine and to determine whether these coated MNs can stimulate protective immunity in mice.
2. Materials and methods
2.1 Chemicals, proteins, cells, & antibodies
Tween 20 was purchased from Fisher Scientific (PA, USA). Gold(III) chloride hydrate, trisodium citrate dihydrate, phosphate-citrate buffer tablets, trypsin (TPCK-treated) and concanavalin A from Canavalia ensiformis were purchased from Sigma-Aldrich (St Louis, MO, USA). Phosphate-buffered saline (PBS) was bought from Mediatech, Inc. (Manassas, VA, USA). Bovine serum albumin (BSA), fetal bovine serum (FBS), O-phenylenediamine (OPD), Gibco RPMI 1640 (1×), Gibco MEM (1×), and Gibco antibiotic–antimycotic (100×) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Isoflurane (NDC 14043-704-06) was obtained from Patterson Veterinary (Greeley, CO, USA). Milli-Q (Millipore, MA, USA) water with a resistance of 18.2 MΩ cm was used in all experiments. Falcon 70 μm cell strainers were purchased from Corning (Corning, NY, USA). Carboxymethylcellulose (CMC) disodium salt (CAS 9004-32-4) was purchased from CarboMer, Inc. (San Diego, CA, USA). Lutrol F-68 NF (CAS 9003-116-6) was purchased from BASF (Mt. Olive, NJ, USA). Chicken red blood cells with a concentration of 5% were purchased from Lampire Biological Laboratory (Pipersville, PA, USA). M2e (acetylated-SLLTEVETPIRNEWGSRSNDSSDC-amidated; molecular weight: 2736 Da) was chemically synthesized by AAPPTec (LLC, KY, USA). Madin–Darby Canine Kidney (MDCK) cells were purchased from ATCC (Manassas, VA, USA). Horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG, IgG1, and IgG2a antibodies were purchased from Southern Biotech (Birmingham, AL, USA). Cytokine ELISA kits for IL-6, IL-17, IFN-γ, and TNF-α were purchased from R&D Systems (Minneapolis, MN, USA). CpG oligodeoxynucleotide (ODN) 1826 (5′-TCCATGACGTTCCTGACGTT-3′) with phosphorothioate backbone was synthesized by Integrated DNA Technologies (Coralville, IA, USA) and is recognized by toll-like receptor 9 (TLR9). Notation sCpG refers to the fact that CpG is present in the formulation in free soluble form.2.2 Animals
Female BALB/c mice aged 6–8 weeks were purchased from Charles River Laboratories (Wilmington, MA, USA). All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Texas Tech University and Experiments were approved by the Animal Ethics Committee of Texas Tech University.2.3 Synthesis of AuNPs
AuNPs were synthesized by the Turkevich method, as described previously.19 Briefly, 300 ml of water was brought to a boil and 1 ml of water containing 120 mg of gold(III) chloride hydrate was added. After 1 minute, 10 ml of water containing 350 mg trisodium citrate dihydrate was added. The reaction mixture changed to black upon addition of sodium citrate, then purple shortly after. The reaction was continued for an additional 15 min with stirring at 100 °C. Synthesized AuNPs were kept overnight at room temperature and then stored at 4 °C until further use. UV-visible (vis) spectral analysis was performed on a Cary 300 UV-vis spectrophotometer (Agilent Technologies, Inc., CA, USA).2.4 AuNP characterization
AuNP size and morphology was analyzed using Hitachi (MI, USA) H-8100 transmission electron microscope. Transmission electron micrographs (TEMs) were obtained by evaporating 10 μl of sample on a 300-mesh copper grid coated with carbon film.2.5 Vaccine formulation and microneedle coating
Both M2e and CpG were originally received in lyophilized form and were dissolved in endotoxin-free water. The AuNP-M2e + sCpG formulation was prepared as follows. 1 μl tween 20 was added to 1 ml of synthesized AuNPs, containing 224 μg AuNPs, and vortexed. The mixture was centrifuged at 17000 RCF for 25 min at 4 °C and 932 μl of supernatant was discarded. To the remaining AuNPs, 12 μl of 1 mM M2e was added and mixed overnight with gentle shaking at 4 °C. The next day, 20 μl of 4 mg ml−1 CpG was added and the mixture was gently shaken for 2 h at 4 °C. Following this, AuNP-M2e + sCpG formulation was lyophilized. The microneedle coating solution was prepared as follows. After lyophilization, the vaccine formulation was resuspended in an aqueous solution containing two coating excipients—CMC (1% (w/v)) which acts as a viscosity enhancer and helps to form thicker coatings, and Lutrol F68 (0.5% (w/v)) which acts as a surfactant and aids in complete wetting of the MN surface to allow for a uniform coating along the MN shaft. This coating solution was then housed in a pipette tip and microneedle patches were dip coated using an automated x–y–z motion controller-based coating station developed in house.23–27 Similarly, the M2e + sCpG formulation without AuNPs was prepared by mixing equal volumes of a solution containing sCpG and M2e, and a solution containing 2% CMC (w/v) and 1% Lutrol F68 (w/v). MNs were dip coated using M2e + sCpG to serve as control patches. Upon coating MNs with AuNP-M2e + sCpG formulation, MNs were dissolved in water, and the coated mass of both M2e and CpG was confirmed via NanoDrop (ThermoFisher Scientific).
2.6 MN fabrication and coating stability
MN arrays were made from 304 grade stainless steel and each array contained a total of 57 needle tips, each 700 μm long. Coating of MNs was carried out by a dip coating method as described previously.28 In short, the vaccine formulations were placed in an orifice and coated onto MNs via dip-coating method that utilizes a coating station assembled in-house. To inspect the uniformity of coatings on MNs, a stereomicroscope (Olympus SZX16, Olympus America Inc.) was used. A MN patch coated with AuNP-M2e + sCpG was dissolved in water, and the resulting solution's absorption spectra was compared to that of a freshly prepared vaccine formulation.2.7 Scanning electron microscopy
Coated MN arrays were sputter-coated with iridium on Q150T Plus turbomolecular pumped coater (Quorum Technologies, United Kingdom) and visualized via a scanning electron microscope (SEM) (Zeiss Crossbeam 540, Germany). Energy-dispersive X-ray spectroscopy was performed via AZtec software (Oxford Instruments, United Kingdom).2.8 In vivo measurement of delivery efficiency
The delivery efficiency of coated MNs was calculated as described previously with modifications.24 In brief, ten MN patches were coated with the AuNP-M2e + sCpG formulation. Subsequently, each coated MN patch (n = 10) was separately suspended in 1 ml of water and the AuNPs in dissolved coatings were quantified using UV-Vis against a standard curve to determine the initial amount of AuNPs coated on MNs (M1). A new set of coated MNs (n = 10) was inserted into murine skin in vivo and held in via thumb pressure for 5 min. MNs were then removed from skin and submerged in 700 μl water to determine the amount of AuNPs left on the patch (M2). The area of skin treated with coated MNs was then gently wiped with a pre-soaked cotton swab (n = 10), and the swab submerged in water to quantify the amount of AuNPs left on the skin (M3). The delivery efficiency was then calculated as (M1–M2–M3)/M1 × 100%. The percentage of AuNPs left on the MN patch or skin was calculated as (M2/M1) × 100% or (M3/M1) × 100%, respectively.2.9 Mice immunizations and treatment groups
24 h prior to immunization, mice were anesthetized with isoflurane using a Euthanex EZ-150C classic anesthesia machine and their backs were shaved followed by treatment with Nair hair removal cream to remove all hair from the immunization region. On immunization day, mice were anesthetized with isoflurane and MNs were pressed into the skin and manually held for 5 min. In terms of treatment groups, the following strategy was employed – to reproduce the amount of vaccine that was intranasally administered to the mice in our previous study,21 mice were administered 60 μg AuNPs, 8.2 μg M2e, and 20 μg sCpG per individual mouse, in the form of two coated-MN patches administered one immediately after the other (2-patch) (Table 1). Two patches were used instead of a single patch because when a single MN patch was coated with 60 μg AuNPs, 8.2 μg M2e, and 20 μg sCpG the coating layer was thick, and this led to a large fraction of the coating to deposit on the skin surface during insertion, leading to low delivery efficiency. Hence, two patches each containing 30 μg AuNPs, 4.1 μg M2e, and 10 μg sCpG were administered in succession. To study the dose effect, another group of mice received only a single patch containing half the dose, i.e., 30 μg AuNPs, 4.1 μg M2e, and 10 μg sCpG per individual mouse (1-patch) (Table 1). To determine whether absence of AuNPs decreases the immunogenicity of M2e when delivered into skin via coated MNs, a separate group of mice received MN vaccination consisting of only M2e (4.1 μg) and soluble CpG (10 μg) (1-patch-NoAuNPs) (Table 1). Finally, a naïve group of mice received no MNs, and only underwent the influenza virus challenge on day 42 (Table 1). All the vaccine formulations were administered on day 1 and repeated (boosted) on day 21.| Group no. | Experimental group | No. of MN patches administered | M2e (μg) | Soluble CpG (μg) | AuNPs (μg) |
|---|---|---|---|---|---|
| 1 | Naïve | — | — | — | — |
| 2 | 1-Patch-NoAuNPs | 1 | 4.1 | 10 | — |
| 3 | 1-Patch | 1 | 4.1 | 10 | 30 |
| 4 | 2-Patch | 2 | 8.2 | 20 | 60 |
2.10 Serum antibody measurements
Blood was collected on study days 0, 21, and 42 through submandibular bleeding. The collected blood was equilibrated at room temperature to allow for clotting to occur. Following this, samples were centrifuged at 3000 rcf for 20 minutes at 4 °C and the supernatant was collected and stored at −80 °C until further use.2.11 Serum ELISA
Serum antibodies were measured via ELISA as described previously.24 The final titer was defined as the reciprocal of the highest dilution exhibiting positive values above the cut-off value, determined with a confidence limit above 99.9% according to the method described by Frey et al.292.12 Cytokine ELISA
Mice were euthanized on day 42 and the spleens were collected. Single cell suspensions were obtained from spleens through mechanical dissociation by passing tissue pieces through a 70 μm filter into the 24-well plate, and subsequently supplemented with RPMI media containing 10% (v/v) FBS and 1% (v/v) antibiotic–antimycotic (Thermo Fisher Scientific (Waltham, MA, USA). Isolated splenocytes were cultured in 24-well plates for 72 h at 37 °C with either M2e, concanavalin A (positive control), or the RPMI culture media (negative control). After 72 h of culture, the cells were centrifuged at 500 rcf for 5 min. The supernatants were collected and analyzed for the presence of IL-6, IL-17, IFN-γ, and TNF-α using R&D systems quantikine ELISA kits.2.13 Bone marrow ELISA
Following euthanasia on day 42, the femurs of mice were removed and soaked in 70% ethanol and water for 5 min. Next, 500 μl of RPMI media was twice pushed through the interior of the bone using a 29-gauge needle and collected separately. Samples were incubated at 37 °C for 96 h. Following this, samples were centrifuged at 500 rcf for 5 min and the supernatants were analyzed by ELISA for the presence of M2e-specific antibodies.2.14 Bronchoalveolar lavage
Mice euthanized on day 42 were analyzed for the presence of M2e-specific antibodies in the respiratory tract as follows. An 18-gauge catheter (EXELINT International, Redondo Beach, CA) was inserted into the trachea and 1 ml of PBS was pushed into the lungs. Immediately following this, the PBS was withdrawn from the lungs and stored at −80 °C until analysis using ELISA.2.15 Virus challenge
Mice were anesthetized with isoflurane and 30 μl of the influenza A (H1N1-A/PR/8/34) virus was administered intranasally at a dose of approximately 3 × 50% lethal dose (LD50) on day 42.2.16 Determining influenza A (H1N1-A/PR/8/34) viral titer by TCID50 assay
Mice were euthanized 72 h post influenza challenge and their lungs were collected in 10% (w/v) MEM, which contained 1% (v/v) antibiotic–antimycotic. The lungs were homogenized using Qiagen 5 mm stainless steel beads and a Qiagen TissueLyser LT. Next, homogenized lung samples were centrifuged at 5000 rcf for 10 minutes at 4 °C and the supernatants were stored at −80 °C until further analysis.Separately, MDCK cells were suspended in MEM media containing 10% (v/v) FBS and 1% (v/v) antibiotic–antimycotic. Next, MDCK cells were added to a 96-well plate with 50000 cells per well and incubated overnight at 37 °C. Media was removed from plates and adhered cells were washed twice with PBS. Lung supernatant was serially diluted 10× with 2 μg ml−1 TPCK-treated trypsin in MEM with 1% (v/v) antibiotic–antimycotic, added to each well, and cultured at 37 °C for 72 h.
Following the 72 h incubation period, 50 μl of supernatant was collected from each plate and combined with 50 μl of 0.5% chicken RBCs in PBS in a conical 96-well plate. Plates were incubated for 1 h at room temperature. The absence of virus was confirmed in wells in which RBCs could be seen settling at the bottom of the well, indicating agglutination did not occur. The observed RBC agglutination titers were used to calculate the median tissue culture infectious dose TCID50, or the dilution of the virus required to infect 50% of cell samples. The log10(TCID50) values were calculated using Reed–Muench method.30
2.17 Statistical analysis
Statistical analyses were performed on GraphPad Prism 9 for Windows (CA, USA). Multifactorial ANOVA with post hoc Tukey analysis or paired t-test were used for all comparisons.3. Results
3.1. Characterization of MN coatings
The MN coatings comprise AuNPs, M2e and CpG. Fig. 1A describes the association between the different species. M2e is attached to the AuNP surface through a thiol–Au bond, with thiol group being contributed by a cysteine residue in M2e. M2e that is not attached to AuNPs is not removed from the formulation because a previous study has demonstrated its critical role in stimulating a protective immune response.20 The adjuvant CpG is not attached to AuNPs. The ability of AuNPs to form a colloidal suspension was assessed using UV-Vis (Fig. 1B). The UV-vis absorption spectra peaks for the bare AuNPs, freshly prepared vaccine formulation (AuNPs + M2e + sCPg), and AuNPs + M2e + sCpG obtained from dissolving coated MNs was 518 nm, 523 nm, and 526 nm, respectively (Fig. 1C). SEM image of a coated MN (Fig. 1D) illustrates the coating uniformity. The energy-dispersive X-ray spectroscopy (EDS) identified that gold (Au) was uniformly distributed in the MN coatings (Fig. 1E). EDS for iron (Fe), which is highly abundant in grade 304 stainless steel showed presence of Fe in the uncoated portion of MN shaft but its signal was occluded by the coatings (Fig. 1F). Composition analysis of coated MNs revealed gold to be the most abundant element in the coated region (49.4 ± 0.3 wt%) (Fig. 1G), while no gold was identified in the uncoated region of the MN (Fig. 1H). MNs coated with AuNPs + M2e + CpG (Fig. 1I and J) or M2e + sCpG (Fig. 1K and L) exhibited uniform coatings.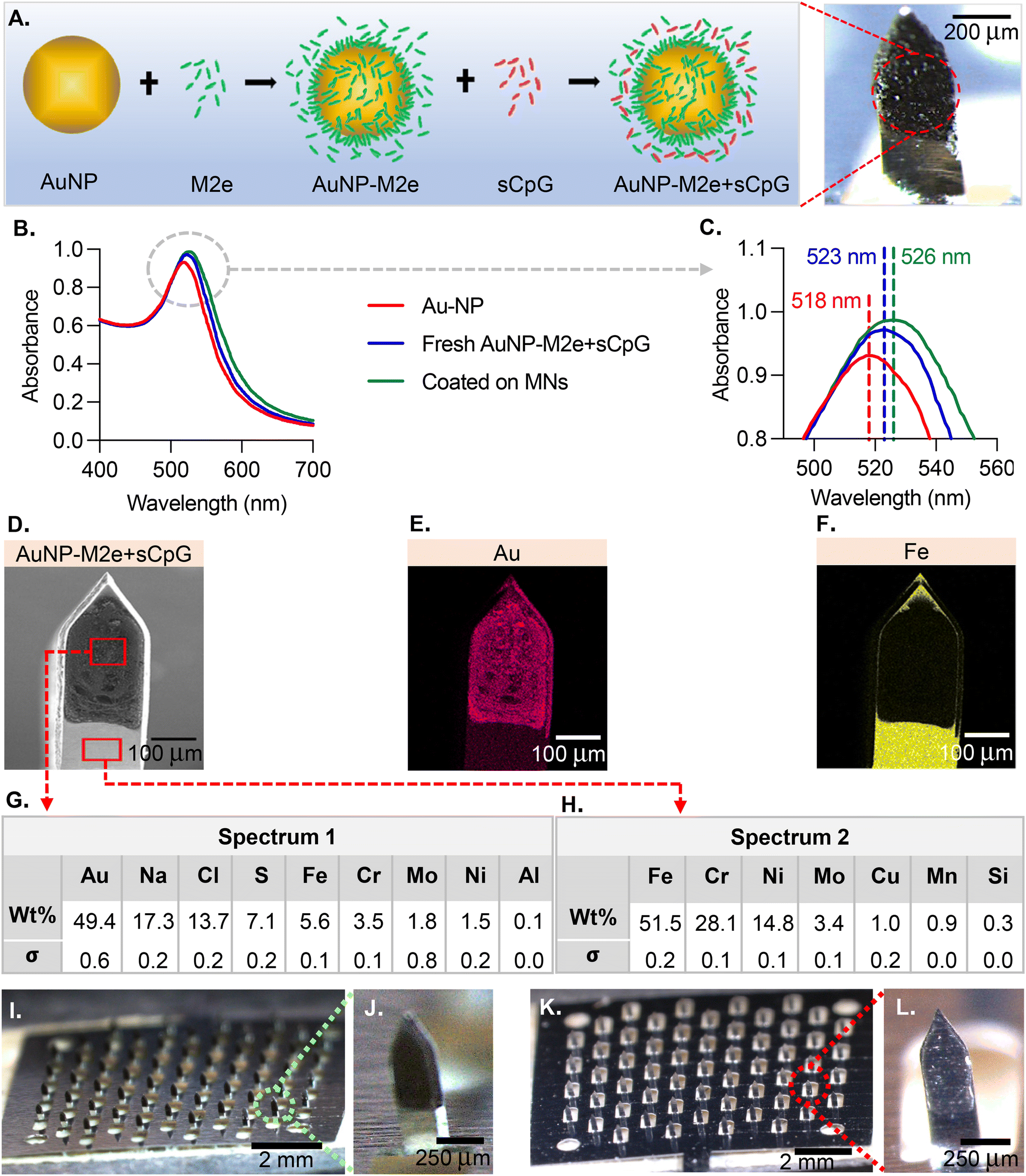 | ||
| Fig. 1 Vaccine formulation design and stability. (A) Vaccine design scheme. To enhance its poor immunogenicity, M2e is conjugated to the surface of a AuNP. By keeping M2e in excess in the aqueous AuNP solution, full surface coverage of M2e is ensured on the surface of AuNPs. CpG is also included in the formulation in soluble form (sCpG) as an adjuvant that targets toll-like receptor 9 (TLR9) and initiates induction of T helper 1 immune response. The finalized vaccine formulation is then resuspended in coating excipient solution (1% CMC (w/v) and 0.5% F68 (w/v)) and coated onto a MN patch. (B) Absorption spectra of bare AuNPs, a freshly prepared vaccine formulation, and a lyophilized formulation coated on MNs. (C) A close up of maximum absorption peaks. (D) SEM image of a single MN coated with AuNP-M2e + sCpG formulation. (E) Energy-dispersive X-ray spectroscopy (EDS) image showing Au composition of a coated MN. (F) EDS image showing Fe composition of a coated MN. (G) Distribution of elements in the coated region of a MN (spectrum 1). (H) Distribution of elements in the uncoated region of a MN (spectrum 2). (I) Digital image of a stainless-steel MN patch housing 57 MNs coated with a formulation of AuNP-M2e + sCpG. (J) Zoomed-in image of a single MN coated with the AuNP-M2e + sCpG formulation. (K) Digital image of a MN patch coated with M2e + sCpG formulation. (L) Zoomed-in image of a single MN coated with M2e + sCpG formulation. | ||
3.2. Delivery efficiency of coated MNs into murine skin
Based on analysis of MNs coated with AuNP-M2e + sCpG, the average amount of AuNPs coated on MNs was 31.0 μg (±2.1 standard deviation (SD)), the amount left on the patch post-insertion into murine skin was 3.2 μg (±0.4 SD), and the amount left on the skin was 1.4 μg (±0.3 SD) (Fig. 2A). Coated AuNPs were delivered into the skin at 85.4% (±1.4 SD) efficiency (Fig. 2B), which is consistent with previous studies reporting 72% to 91% delivery of coated material into skin.23–25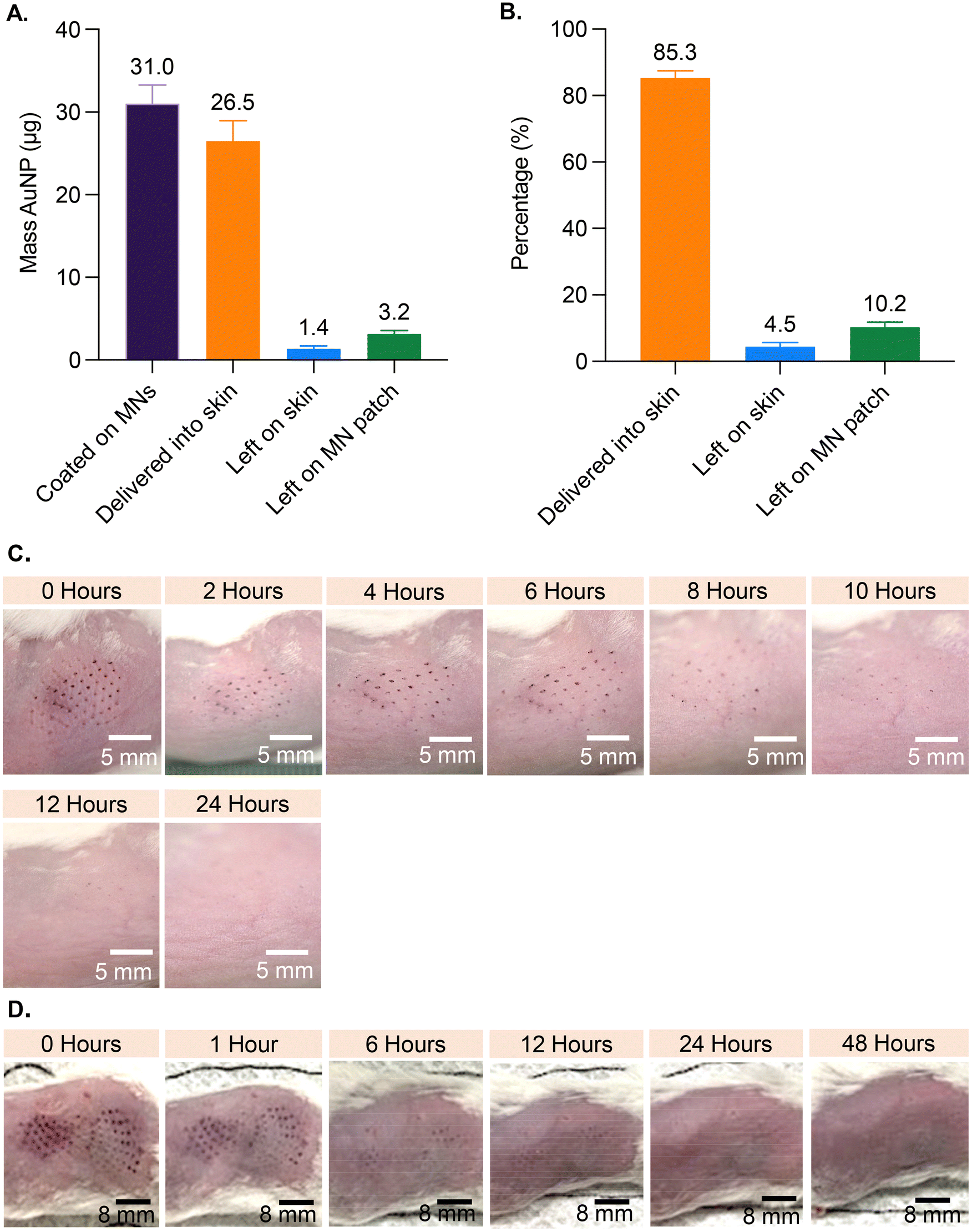 | ||
| Fig. 2 Delivery efficiency and appearance of skin post MN treatment. MN patches were coated with AuNP-M2e + sCpG and applied to murine skin (N = 10 mice, with each mouse receiving one coated patch): (A) quantification of AuNPs, and (B) delivery efficiency of the coatings. Data is presented as mean + standard deviation. Digital images showing appearance of skin after MN treatment. Treated mice received (C) one patch, or (D) two patches: one immediately after the other. | ||
3.3. Skin appearance and state after insertion of coated MNs into murine skin
To investigate the effect of inserting AuNP-coated MNs into skin, the appearance of treated murine skin was monitored at various timepoints for up to 96 h post vaccination (Fig. 2). Mice in the 1-patch group exhibited mild erythema at the application site, which completely resolved within 8 h of treatment, and showed no symptoms of bleeding (Fig. 2C). Mice in the 2-patch group displayed erythema at the MN insertion site, which fully resolved within 6 to 12 h of treatment (Fig. 2D). No permanent skin tissue alterations were observed at the MN-treated sites regardless of the number of MN patches or the amount of AuNPs and vaccine applied on to the skin. Post MN treatment, the skin contained spots in the form of purple dots that mimicked the pattern of MNs on the patch (Fig. 2C and D). The AuNP dots faded over time and fully disappeared within 24 to 48 h (Fig. 2C and D).3.4. Serum anti-M2e IgG antibody response
In the current study, all the treatment groups (Table 1) received formulations via coated MNs on days 0 and 21 (booster dose) (Fig. 3A). After two doses of MN vaccination, M2e-specific IgG, IgG1 and IgG2a in serum increased significantly (Fig. 3B–D) for both 1-patch and 2-patch groups, as compared to both naïve and 1-patch-NoAuNPs groups (p < 0.0001). Exclusion of AuNPs from the coating formulation was detrimental and the immune response was no different from the unimmunized naïve mice (p > 0.999) (Fig. 3B). The 2-patch group consistently showed significantly higher levels of anti-M2e IgG (p < 0.0001), IgG1 (p = 0.02), and IgG2a (p < 0.0001) as compared to 1-patch group (Fig. 3B–D). Of note, the booster dose (day 21 vaccination) significantly increased the M2e-specific antibody response, as witnessed by significantly higher serum levels of IgG, IgG1, and IgG2a on day 42 as compared to day 21 in 1-patch and 2-patch groups.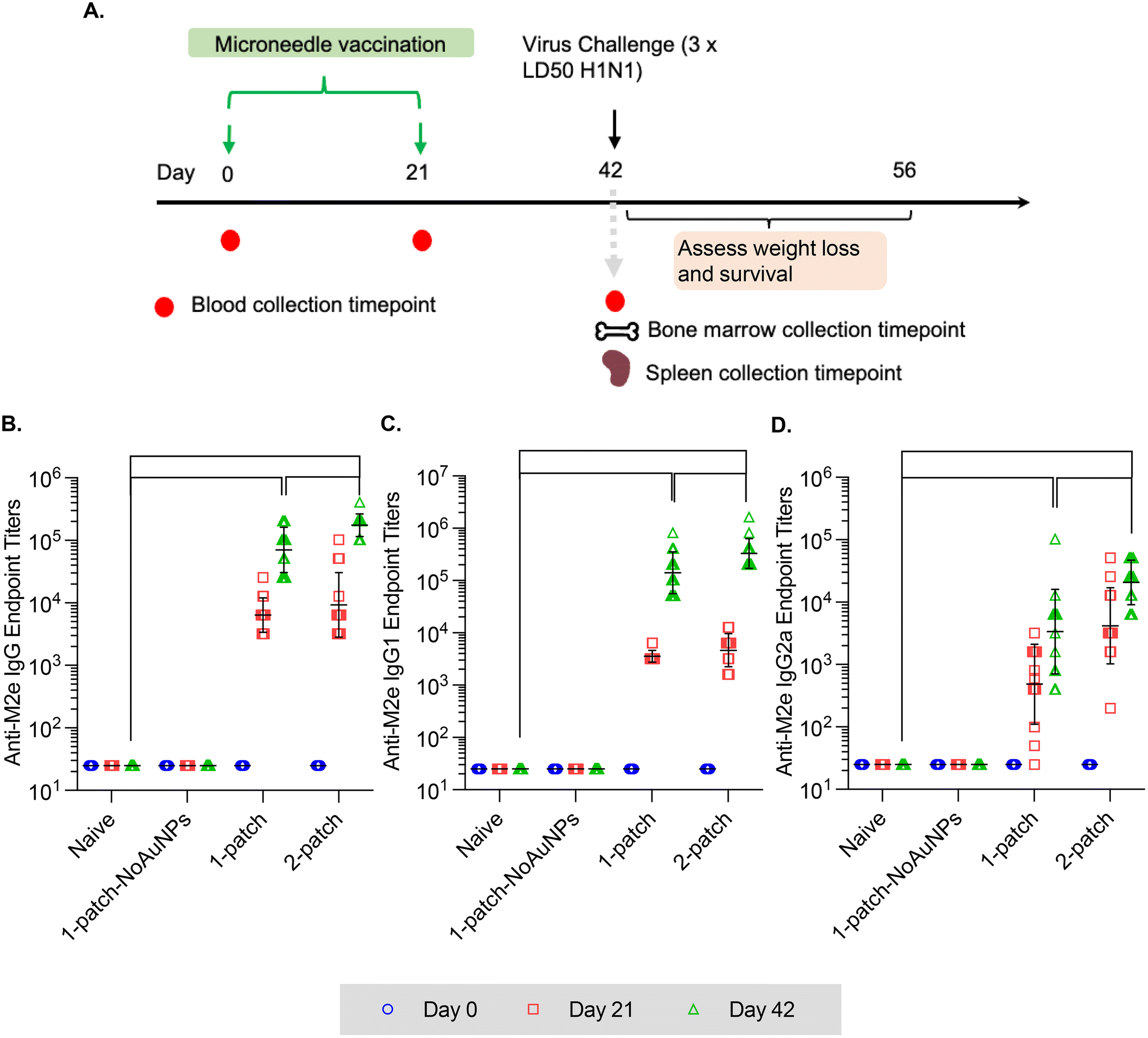 | ||
| Fig. 3 Anti-M2e IgG serum titers following a two-dose vaccination regimen with coated MNs. (A) Immunization schedule. Mice were immunized on days 0 and 21, and intranasally challenged with 3 × 50% lethal dose (LD50) dose of influenza virus A/PR/8/34 (H1N1) on day 42. Blood was collected on days 0, 21 and 42; bone marrow and spleen were collected on day 42. (B) Anti-M2e IgG response in murine serum. (C) Anti-M2e IgG1 response in murine serum. (D) Anti-M2e IgG2a response in murine serum. N = 13 in each group. p < 0.05, *; p < 0.01, **; p < 0.001, ***; p < 0.0001, ****. Each symbol represents an animal. Data is presented as geometric mean ± geometric standard deviation. | ||
3.5. Protection against virus challenge
To evaluate the ability of coated MN vaccination to confer protection in immunized mice against lethal influenza challenge, the mice were intranasally challenged with approximately 3 × 50% lethal dose (LD50) of H1N1-A/PR/8/34 influenza virus, and their bodyweight and survivability was recorded for up to two weeks post-virus exposure. Mice vaccinated with only M2e + sCpG (1-patch-NoAuNPs group) as well as the naïve (untreated) mice demonstrated similar weight loss and mortality, with both groups demonstrating abrupt weight loss starting on day 4 post-challenge, ultimately resulting in 0% survival by day 10 post-challenge (0/11 mice survived in either group) (Fig. 4A and B). A 100% survival (10/10 mice) and a significantly smaller weight loss was observed in both 1-patch and 2-patch groups (Fig. 4A and B). Both groups demonstrated the peak weight loss on day 7 post-challenge, following which they began to recover, ultimately regaining 96.4% ± 2.2% and 94.8% ± 1.9% of weight in 1-patch and 2-patch groups, respectively (Fig. 4B). We further sought to evaluate the protective effect of MN-based vaccination by examining lung virus titers. As seen in Fig. 4C, both 1-patch and 2-patch groups displayed significantly lower log10(TCID50) influenza virus titers as compared to naïve and 1-patch-NoAuNPs groups, thereby demonstrating that vaccination with MNs confers enhanced level of protection against lethal influenza challenge.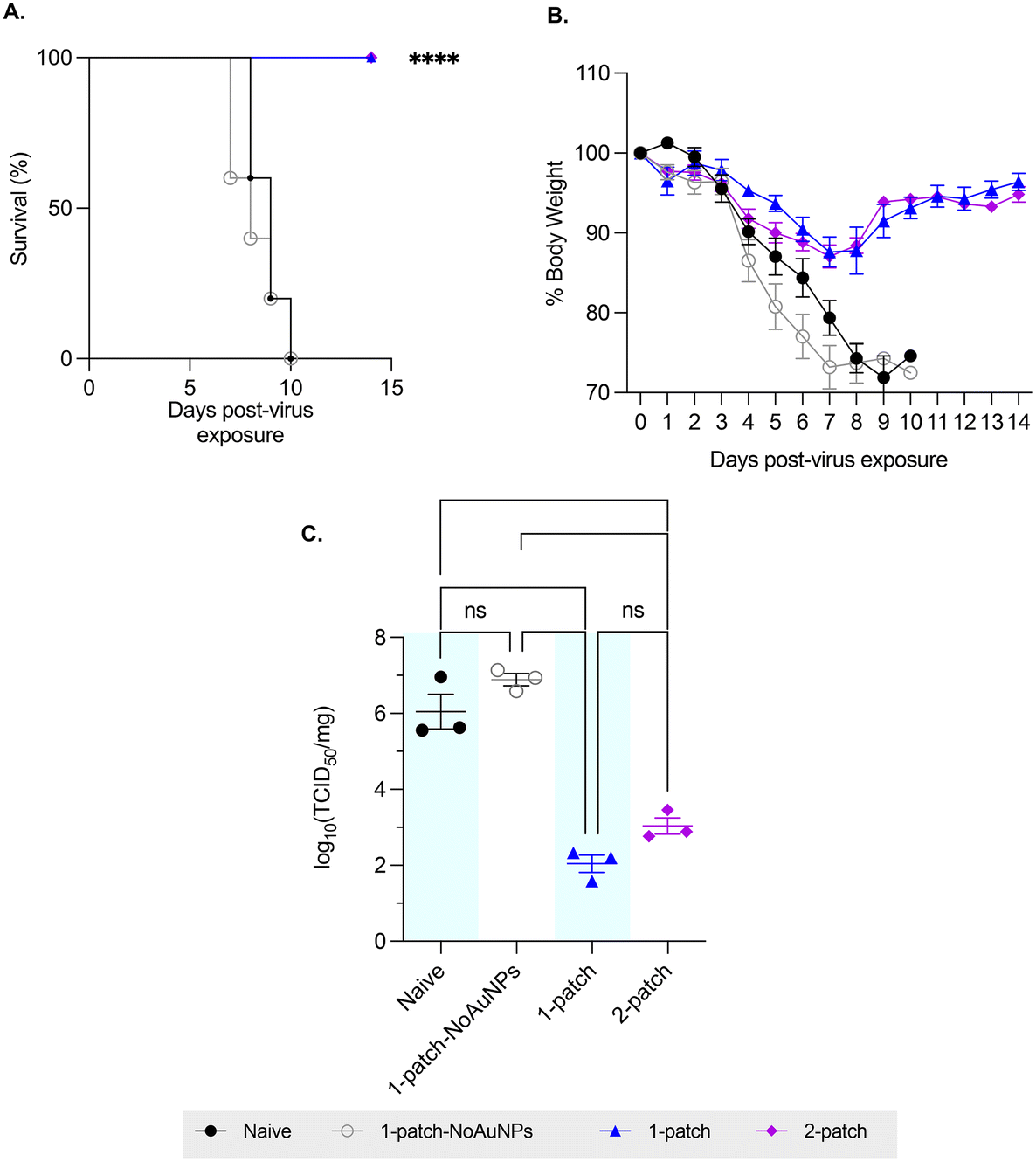 | ||
| Fig. 4 Effect of challenge with A/PR/8/34 (H1N1) influenza virus. Mice were challenged with 3× LD50 of H1N1-A/PR/8/34. (A) Survival percentages (N = 5 per group). (B) Average body weights (N = 5 per group). (C) Lung virus load on day 3 post-challenge ((N = 3 per group). Each symbol represents an animal. Data is represented as mean ± SEM. p < 0.05, *; p < 0.01, **; p < 0.001, ***; p < 0.0001, ****. | ||
3.6. Bone marrow anti-M2e IgG antibody response
Bone marrow plasma cells are known to maintain a long-term humoral immunity following vaccination.31 Accordingly, we examined the levels of M2e-specific IgG antibodies in bone marrow culture supernatants of vaccinated mice on day 42. The naïve and 1-patch-NoAuNPs groups had undetectable levels of anti-M2e IgG, IgG1 and IgG2a antibodies, while 1-patch and 2-patch groups produced anti-M2e IgG, IgG1 and IgG2a, at levels significantly higher (p < 0.01) than both 1-patch-NoAuNPs and the naïve groups (Fig. 5A–C). There was no difference between 1-patch and 2-patch groups except the anti-M2e IgG2a levels, which were significantly higher in the 2-patch groups (p < 0.05).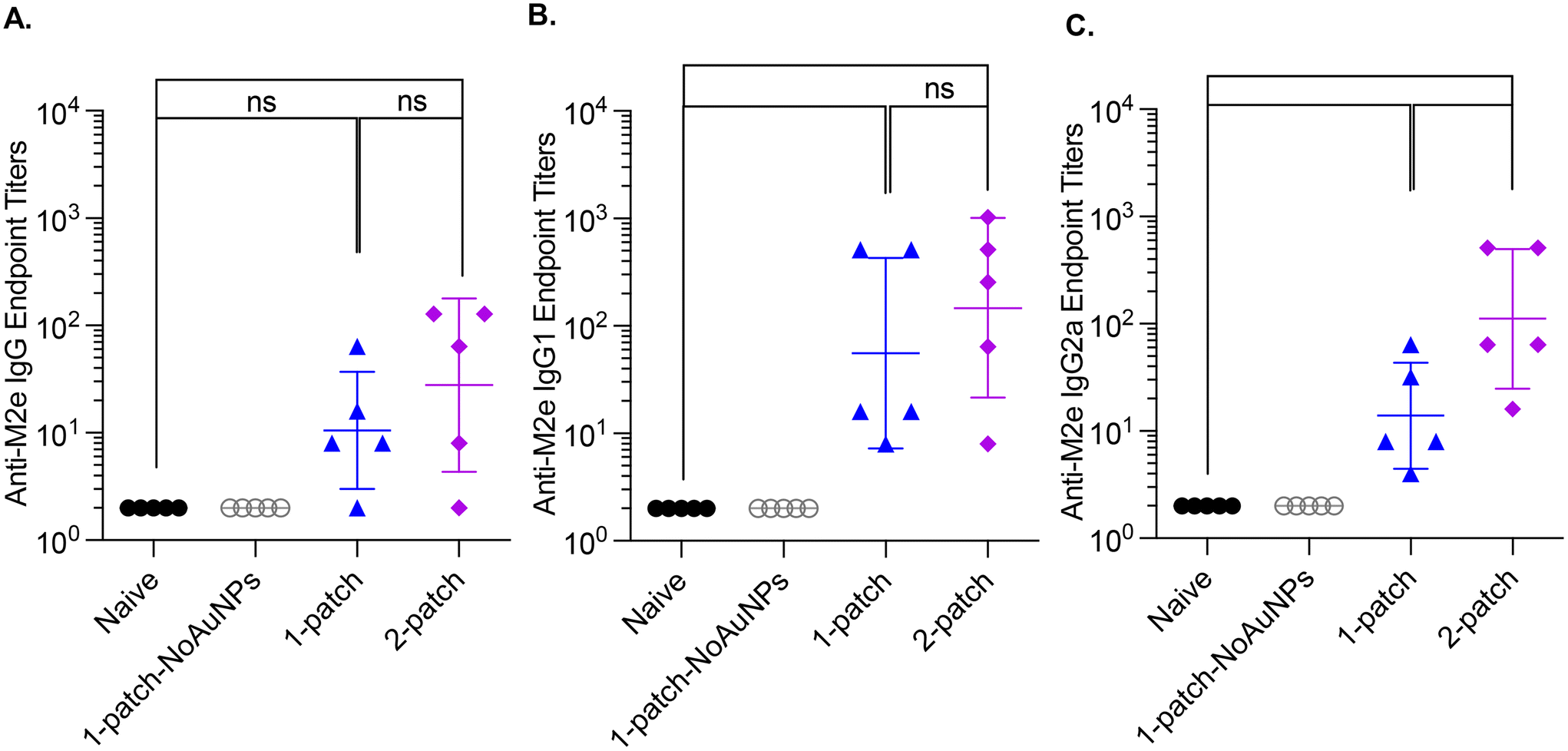 | ||
| Fig. 5 Anti-M2e IgG bone marrow titers following a two-dose vaccination regimen with coated MNs. Mice were vaccinated on day 0 and 21, femurs were collected post-euthanasia on day 42, flushed twice with 500 ml RPMI to collect cells, which were then cultured. Cell culture supernatants were then analyzed for presence of anti-M2e antibodies. (A) Anti-M2e IgG response. (B) Anti-M2e IgG1 response. (C) Anti-M2e IgG2a response. N = 5 in each group. p < 0.05, *; p < 0.01, **; p < 0.001, ***; p < 0.0001, ****. Each symbol represents an animal. Data is represented as geometric mean ± geometric standard deviation. | ||
3.7. Bronchoalveolar lavage anti-M2e IgG antibody response
To evaluate the effect of vaccination with coated MNs on generation of humoral immunity in the lungs of immunized mice, bronchoalveolar lavage (BAL) fluid was collected from influenza-challenged mice post-euthanasia and assessed for levels of M2e-specific IgG antibodies. Both 1-patch and 2-patch groups exhibited significantly higher levels of IgG, IgG1, and IgG2a than naïve and 1-patch-NoAuNPs groups (p < 0.0001) (Fig. 6A–C). While BAL levels of IgG1 and IgG2a exhibited no significant difference between 1-patch and 2-patch groups (Fig. 6B and C), the overall IgG levels were significantly higher in 2-patch group as compared to 1-patch group (p = 0.0408) (Fig. 6A). Interestingly, no M2e-speciffic IgA response was observed in BAL fluid of MN-immunized mice (Fig. 6D).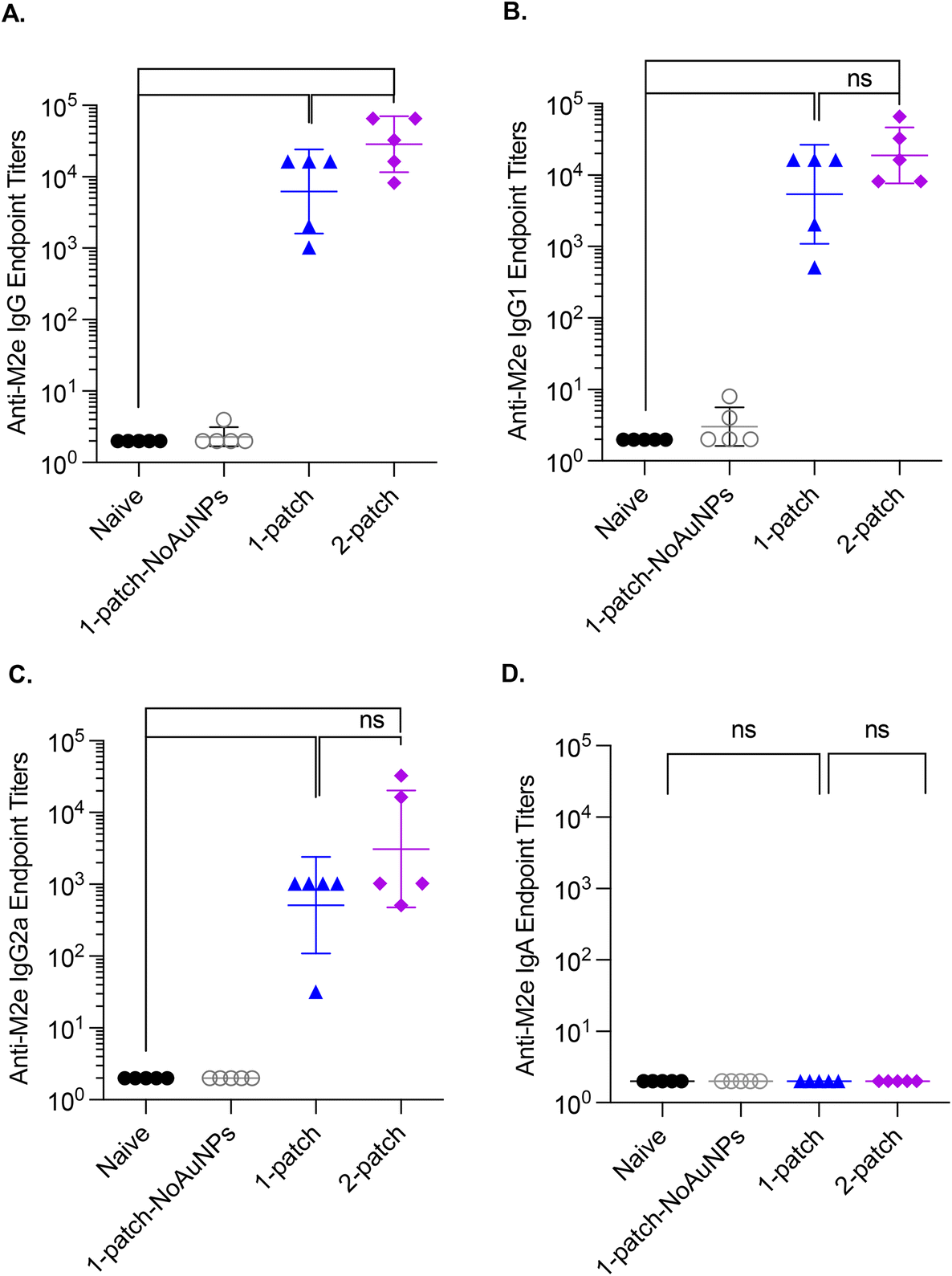 | ||
| Fig. 6 Anti-M2e IgG bronchoalveolar lavage fluid titers following a two-dose vaccination regimen with coated MNs. Mice were vaccinated on day 0 and 21, BAL fluid samples were collected post-euthanasia on day 42, and analyzed for presence of anti-M2e antibodies. (A) Anti-M2e IgG response. (B) Anti-M2e IgG1 response. (C) Anti-M2e IgG2a response. (D) Anti-M2e IgA response. N = 5 in each group. P < 0.05, *; p < 0.01, **; p < 0.001, ***; p < 0.0001, ****. Each symbol represents an animal. Data is represented as geometric mean ± geometric standard deviation. | ||
3.8. Splenocyte cytokine profile
To better characterize the cellular immune response generated by coated MN vaccination, ex vivo splenocyte cytokine production was measured after M2e restimulation. The splenocytes of mice immunized with only M2e + sCpG (1-patch-NoAuNPs group) exhibited no significant increase in cytokine levels for any of the cytokines, as compared to naïve mice (Fig. 7A–D). Further, no significant difference in IL-6, IL-17, IFN-γ, and TNF-α levels was observed between 1-patch and 2-patch groups (Fig. 7A–D). Only the 2-patch group displayed a noticeable, although not significant, increase in IL-6 compared to naïve group (p = 0.01051) (Fig. 7A). Both 1-patch and 2-patch groups exhibited greater IL-17 response compared to naïve and 1-patch-NoAuNPs groups (Fig. 7B), while a significant increase in IFN-γ was observed in the 2-patch group as compared to both naïve and 1-patch-NoAuNPs group (p = 0.0143 and p = 0.0214, respectively) (Fig. 7C). Likewise, both 1-patch and 2-patch groups exhibited significantly higher levels of TNF-α as compared to naïve and 1-patch-NoAuNPs groups (Fig. 7D).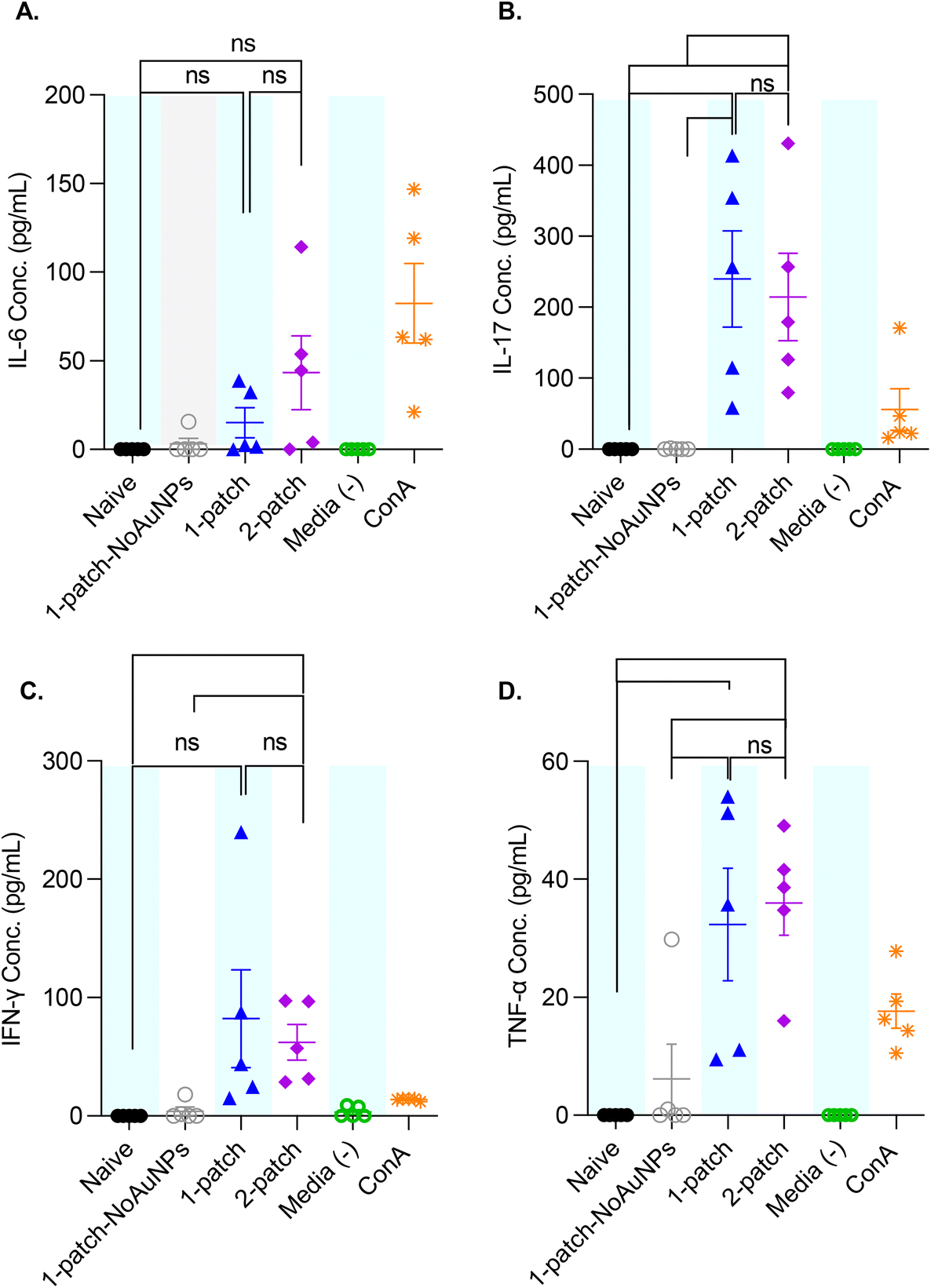 | ||
| Fig. 7 Anti-M2e IgG titers of splenocyte culture supernatants following a two-dose vaccination regimen with MNs. Mice were vaccinated on day 0 and 21, spleens were collected post-euthanasia on day 42, and spleen culture supernatants analyzed for presence of (A) IL-6, (B) IL-17, (C) IFN-γ and (D) TNF-α. Cell culture media alone, and kit-provided control were used as negative and positive controls, respectively. N = 5 in each group. p < 0.05, *; p < 0.01, **; p < 0.001, ***; p < 0.0001, ****. Data comparison was performed via paired t-test. Each symbol represents an animal. Data is represented as mean ± SEM. | ||
4. Discussion
In this study, an influenza vaccine formulation comprising AuNPs as a carrier of the antigen (M2e) was delivered into the superficial mouse skin using coated MNs. The adjuvant in the formulation, CpG (toll like receptor-9: TLR-9 agonist) was not attached to AuNPs but was simply mixed. A 100% survival of the immunized mice was observed against a lethal influenza infection.M2e is highly conserved and has been investigated as an antigen to make a broadly protective influenza vaccine. However, because M2e is poorly immunogenic various recombinant and synthetic approaches have been employed to attach M2e to different carriers to enhance its immunogenicity.32–38 We chose to utilize AuNPs as M2e carriers due to their many attractive features such as inert core, low cytotoxicity,39,40 tight nanoparticle size control,41 inherent adjuvanticity,42,43 and ease of conjugation with peptides via thiol group.44 In our formulation, M2e is added in an amount greater than what is required to fully cover the AuNP surfaces. This additional M2e, which exists in free form was found to be critical in enhancing immune protection – when it was removed from the formulation, the immune response decreased.20 CpG, which is a TLR-9 agonist was also found to be a critical component of the formulation for providing a strong immune response and protecting animals against lethal influenza virus infection.19 CpG promotes Th1-dominant cellular and humoral immune response characterized by secretion of IFN-γ and IgG2a antibodies in mice.45 When delivered into murine skin, CpG can stimulate skin-resident Langerhans cells to uptake the concurrently administered antigen and present it to CD4+ T cells for induction of Th1 response.46,47
One of the uniqueness and a challenge in this study was the need to coat MNs with inorganic colloidal AuNPs and then ensure their removal from the MNs without causing their aggregation because particle aggregation can increase the colloidal size and negatively impact vaccine immunogenicity. Another problem associated with coating particles on MNs using the dip coating approach is that colloidal particles can settle over time, which can impact coating reproducibility. In the past, as proof-of-principle, latex beads (10 μm in diameter) and barium sulfate particles (1 μm in diameter) were dip coated on MNs, however, characterization was not performed to demonstrate reproducibility in achieving consistent particle loading across different MN batches.48 Another class of colloids that have been widely coated on microneedles include the bacteria, inactivated/live viruses and virus-like particles, which are all natural colloids.49 These natural colloids can often be concentrated in an aqueous formulation to help perform the MN dip coating operation. The hydrophilic protein corona that naturally exists around the virus-like particles, viruses and bacteria is an important determinant of their colloidal stability, however, various additives have also been used to both stabilize the proteins in the corona and to prevent aggregation.50 Recently, rather than dip coating microparticles onto MNs, particles were attached to MN surfaces by air-spraying dry microparticles onto MNs precoated with an adhesive layer.51 This approach could work under certain circumstances, but in our case due to limited amount of active material available this was not a viable approach, and importantly, spraying the MNs can cause particle attachment to other parts of the MN array besides the sharp MN structures.
In our case, the vaccine formulation that we wanted to deliver using MNs contained 60 μg AuNPs, 8.2 μg M2e, and 20 μg sCpG. To coat this large amount on a patch containing 57 MNs, we required a highly concentrated coating liquid. Typically, to concentrate viruses, bacteria and virus-like particles, a centrifugation spin cycle with aspiration of the supernatant and resuspension of the solid pellet in a lower volume can be employed to achieve a concentrated solution. Alternatively, centrifugal filters with an appropriate molecular weight cutoff can be used. However, in our case, the vaccine formulation contains CpG and some M2e in free form (i.e., not attached to AuNPs), which can be lost during the centrifugation step, but these are essential for stimulating a strong immune response. Thus, these centrifugation approaches were not suitable. In our previous study22 we have shown that this vaccine formulation can be lyophilized and readily resuspended in water without aggregation. Therefore, we decided to also lyophilize the formulation but instead of using water, we resuspended it in the coating liquid consisting of 1% (w/v) carboxymethyl cellulose (CMC) as a viscosity enhancer and 0.5% (w/v) pluronic F-68 as a surfactant. Indeed, as determined via UV-VIS spectroscopy, we were able to fully resuspend the AuNPs in this coating solution. Not only did the AuNPs remain stable and highly dispersed even when the liquid was allowed to sit overnight (data not shown), but when the MNs were dip coated using this coating solution, AuNPs were uniformly distributed along the entire MN shaft (as demonstrated via EDS imaging). The stability of the AuNPs could be attributed to M2e, which is attached to the AuNPs surfaces. In addition, although we used CMC as a viscosity enhancer, it has also been shown to act as a AuNP stabilizer.52–54 Similarly, pluronic block copolymers (including pluronic F-68 used in this study) have also been shown to interact with pre-synthesized AuNPs to prevent their aggregation by enhancing steric repulsion between AuNPs.55
Cellular response upon vaccination can provide necessary protection against potential virus infection. We encouragingly found IFN-γ, TNF-α, IL-6 and IL-17 to all be elevated in mice immunized with AuNP-M2e + sCpG formulation. In terms of antiviral properties, IFN-γ can control expression of receptors responsible for virus entry and block virus replication.56 Further, a recent study found healthy individuals to have increased numbers of IFN-γ-producing natural killer (NK) cells post influenza vaccination.57 A slight increase of IFN-γ observed in our study is hence encouraging and expected to be induced by the presence of CpG in our vaccine formulation.58 IL-6 has been shown to increase B-cell mediated production of IgG antibody, whose systemic induction is crucial in preventing influenza infection.59 IL-17 and TNF-α were likewise reported to both play important roles in protection against influenza virus.60,61
We observed a robust dose-effect of our MN vaccination platform, as MN immunization with both half and full vaccine doses of AuNP-M2e + sCpG resulted in 100% survival. A possible shortcoming of our study lies in the fact that we had to utilize two coated MN patches (instead of one) to deliver a full dose of our vaccine formulation. When we attempted to coat a full dose of vaccine onto a single MN patch, the resulting coatings were extremely thick and the majority of the coating wiped off onto the skin surface during insertion, thereby leading to low delivery efficiency. To overcome this issue, we used two patches instead. In future studies, a possible solution may be to increase the number of MNs on the patch to accommodate the higher load.
While we tested the protective efficacy of our vaccine against the A/PR/8/34 (H1N1) strain, future studies warrant investigation of AuNP-M2e + sCpG vaccine efficacy against other influenza A strains. In addition, it will be important to test the efficacy of MNs in an animal model that more-closely resembles human patients, in terms of skin properties and lung physiology. While we have already demonstrated the stability of our AuNP-M2e + sCpG formulation at various temperatures, it will be important to examine the stability of our vaccine when coated on MNs and stored under similar conditions. In conclusion, our results collectively encourage further evaluation of the AuNP-M2e + sCpG-coated-MNs as a potential broadly protective influenza A vaccine platform.
Conflicts of interest
Harvinder Singh Gill is a co-inventor on a patent related to the development of pollen grains for oral vaccines. This potential conflict of interest has been disclosed and is managed by Texas Tech University.Acknowledgements
This research was supported by the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH) under award number R01AI137846. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.We thank Dr Bo Zhao of Texas Tech University College of Arts and Sciences Microscopy for help in SEM and EDS imaging.
References
- E. Zerhouni, Cell, 2019, 179, 13–17 CrossRef CAS PubMed.
- A. Portnoy, S. Ozawa, S. Grewal, B. A. Norman, J. Rajgopal, K. M. Gorham, L. A. Haidari, S. T. Brown and B. Y. Lee, Vaccine, 2015, 33, A99–A108 CrossRef PubMed.
- D. Chen and D. Zehrung, J. Pharm. Sci., 2013, 102, 29–33 CrossRef CAS PubMed.
- J. L. Goodson and P. A. Rota, Drug Delivery Transl. Res., 2022, 12, 959–967 CrossRef PubMed.
- R. Forman, S. Shah, P. Jeurissen, M. Jit and E. Mossialos, Health Policy, 2021, 125, 553–567 CrossRef PubMed.
- M. J. Mistilis, A. S. Bommarius and M. R. Prausnitz, J. Pharm. Sci., 2015, 104, 740–749 CrossRef CAS PubMed.
- M. J. Mistilis, J. C. Joyce, E. S. Esser, I. Skountzou, R. W. Compans, A. S. Bommarius and M. R. Prausnitz, Drug Delivery Transl. Res., 2017, 7, 195–205 CrossRef CAS PubMed.
- T. T. Nguyen, Y. Oh, Y. Kim, Y. Shin, S.-K. Baek and J.-H. Park, Hum. Vaccines Immunother., 2021, 17, 316–327 CrossRef CAS PubMed.
- S. A. Coulman, J. C. Birchall, A. Alex, M. Pearton, B. Hofer, C. O'Mahony, W. Drexler and B. Považay, Pharm. Res., 2011, 28, 66–81 CrossRef CAS PubMed.
- M. Haq, E. Smith, D. N. John, M. Kalavala, C. Edwards, A. Anstey, A. Morrissey and J. C. Birchall, Biomed. Microdevices, 2009, 11, 35–47 CrossRef CAS PubMed.
- M. Mvundura, C. Frivold, A. J. Osborne, P. Soni, J. Robertson, S. Kumar, J. Anena, A. Gueye, M. Menozzi-Arnaud and B. Giersing, Vaccine, 2021, 39, 7195–7207 CrossRef PubMed.
- H. Amani, M.-A. Shahbazi, C. D'Amico, F. Fontana, S. Abbaszadeh and H. A. Santos, J. Controlled Release, 2021, 330, 185–217 CrossRef CAS PubMed.
- H. S. Gill, D. D. Denson, B. A. Burris and M. R. Prausnitz, Clin. J. Pain, 2008, 24, 585 CrossRef PubMed.
- J. Paget, P. Spreeuwenberg, V. Charu, R. J. Taylor, A. D. Iuliano, J. Bresee, L. Simonsen and C. Viboud, J. Global Health, 2019, 9(2), 020421 CrossRef PubMed.
- K. Subbarao, B. R. Murphy and A. S. Fauci, Immunity, 2006, 24, 5–9 CrossRef CAS PubMed.
- H. Kim, R. G. Webster and R. J. Webby, Viral Immunol., 2018, 31, 174–183 CrossRef CAS PubMed.
- Y. H. Jang and B. L. Seong, Front. Cell. Infect. Microbiol., 2019, 9, 344 CrossRef CAS PubMed.
- W. Fiers, M. De Filette, K. El Bakkouri, B. Schepens, K. Roose, M. Schotsaert, A. Birkett and X. Saelens, Vaccine, 2009, 27, 6280–6283 CrossRef CAS PubMed.
- W. Tao, K. S. Ziemer and H. S. Gill, Nanomedicine, 2014, 9, 237–251 CrossRef CAS PubMed.
- W. Tao and H. S. Gill, Vaccine, 2015, 33, 2307–2315 CrossRef CAS PubMed.
- W. Tao, B. L. Hurst, A. K. Shakya, M. J. Uddin, R. S. Ingrole, M. Hernandez-Sanabria, R. P. Arya, L. Bimler, S. Paust and E. B. Tarbet, Antiviral Res., 2017, 141, 62–72 CrossRef CAS PubMed.
- R. S. Ingrole, W. Tao, G. Joshi and H. S. Gill, Vaccine, 2021, 39, 4800–4809 CrossRef CAS PubMed.
- A. K. Shakya, C. H. Lee and H. S. Gill, J. Controlled Release, 2017, 265, 75–82 CrossRef CAS PubMed.
- A. K. Shakya, R. S. Ingrole, G. Joshi, M. J. Uddin, S. Anvari, C. M. Davis and H. S. Gill, J. Controlled Release, 2019, 314, 38–47 CrossRef CAS PubMed.
- A. K. Shakya, C. H. Lee and H. S. Gill, Mol. Pharm., 2020, 17, 3033–3042 CrossRef CAS PubMed.
- A. K. Shakya, C. H. Lee and H. S. Gill, J. Allergy Clin. Immunol., 2018, 142, 2007–2011 CrossRef CAS PubMed.
- A. K. Shakya, C. H. Lee, M. J. Uddin and H. S. Gill, Mol. Pharm., 2018, 15, 5437–5443 CrossRef CAS PubMed.
- H. S. Gill and M. R. Prausnitz, Pharm. Res., 2007, 24, 1369–1380 CrossRef CAS PubMed.
- A. Frey, J. Di Canzio and D. Zurakowski, J. Immunol. Methods, 1998, 221, 35–41 CrossRef CAS PubMed.
- L. J. Reed and H. Muench, Am. J. Epidemiol., 1938, 27, 493–497 CrossRef.
- C. W. Davis, K. J. Jackson, M. M. McCausland, J. Darce, C. Chang, S. L. Linderman, C. Chennareddy, R. Gerkin, S. J. Brown and J. Wrammert, Science, 2020, 370, 237–241 CrossRef CAS PubMed.
- S. Neirynck, T. Deroo, X. Saelens, P. Vanlandschoot, W. M. Jou and W. Fiers, Nat. Med., 1999, 5, 1157–1163 CrossRef CAS PubMed.
- R. S. Ingrole, W. Tao, J. N. Tripathy and H. S. Gill, Nano LIFE, 2014, 4, 1450004 CrossRef PubMed.
- L. I. Ibañez, K. Roose, M. De Filette, M. Schotsaert, J. De Sloovere, S. Roels, C. Pollard, B. Schepens, J. Grooten and W. Fiers, PLoS One, 2013, 8, e59081 CrossRef PubMed.
- J. Fan, X. Liang, M. S. Horton, H. C. Perry, M. P. Citron, G. J. Heidecker, T.-M. Fu, J. Joyce, C. T. Przysiecki and P. M. Keller, Vaccine, 2004, 22, 2993–3003 CrossRef CAS PubMed.
- A. Seth, F. K. Ritchie, N. Wibowo, L. H. Lua and A. P. Middelberg, PLoS One, 2015, 10, e0117203 CrossRef PubMed.
- W. A. Ernst, H. J. Kim, T. M. Tumpey, A. D. Jansen, W. Tai, D. V. Cramer, J. P. Adler-Moore and G. Fujii, Vaccine, 2006, 24, 5158–5168 CrossRef CAS PubMed.
- J. W. Huleatt, V. Nakaar, P. Desai, Y. Huang, D. Hewitt, A. Jacobs, J. Tang, W. McDonald, L. Song and R. K. Evans, Vaccine, 2008, 26, 201–214 CrossRef CAS PubMed.
- Y. Pan, S. Neuss, A. Leifert, M. Fischler, F. Wen, U. Simon, G. Schmid, W. Brandau and W. Jahnen-Dechent, Small, 2007, 3, 1941–1949 CrossRef CAS PubMed.
- E. E. Connor, J. Mwamuka, A. Gole, C. J. Murphy and M. D. Wyatt, Small, 2005, 1, 325–327 CrossRef CAS PubMed.
- B. Duncan, C. Kim and V. M. Rotello, J. Controlled Release, 2010, 148, 122–127 CrossRef CAS PubMed.
- N. G. Bastús, E. Sánchez-Tilló, S. Pujals, C. Farrera, M. J. Kogan, E. Giralt, A. Celada, J. Lloberas and V. Puntes, Mol. Immunol., 2009, 46, 743–748 CrossRef PubMed.
- H. K. Patra, S. Banerjee, U. Chaudhuri, P. Lahiri and A. K. Dasgupta, Nanomedicine, 2007, 3, 111–119 CrossRef CAS PubMed.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- C. L. B. Millan, R. Weeratna, A. M. Krieg, C.-A. Siegrist and H. L. Davis, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 15553–15558 CrossRef CAS PubMed.
- K. Sugita, K. Kabashima, K. Atarashi, T. Shimauchi, M. Kobayashi and Y. Tokura, Clin. Exp. Immunol., 2007, 147, 176–183 CrossRef CAS PubMed.
- T. Jakob, P. S. Walker, A. M. Krieg, M. C. Udey and J. C. Vogel, J. Immunol., 1998, 161, 3042–3049 CrossRef CAS.
- H. S. Gill and M. R. Prausnitz, J. Controlled Release, 2007, 117, 227–237 CrossRef CAS PubMed.
- R. S. Ingrole and H. S. Gill, J. Pharmacol. Exp. Ther., 2019, 370, 555–569 CrossRef CAS PubMed.
- Y.-C. Kim, F.-S. Quan, R. W. Compans, S.-M. Kang and M. R. Prausnitz, J. Controlled Release, 2010, 142, 187–195 CrossRef CAS PubMed.
- J.-E. Choi, H.-R. Cha, S. Kim, J. S. Kim, M.-J. Kim, H. W. Chung, S.-K. Baek, J. M. Lee and J.-H. Park, J. Controlled Release, 2022, 351, 1003–1016 CrossRef CAS PubMed.
- A. Caires, H. Mansur, A. Mansur, S. Carvalho, Z. Lobato and J. Dos Reis, Colloids Surf., B, 2019, 177, 377–388 CrossRef CAS PubMed.
- M. M. Khalaf, F. F. El-Senduny, H. M. Abd El-Lateef, H. Elsawy, A. H. Tantawy and S. Shaaban, J. Mol. Liq., 2021, 340, 117202 CrossRef CAS.
- H. E. Emam and H. B. Ahmed, Int. J. Biol. Macromol., 2018, 111, 999–1009 CrossRef CAS PubMed.
- S. Y. Kim, J. C. Ha and Y. M. Lee, J. Controlled Release, 2000, 65, 345–358 CrossRef CAS PubMed.
- M. G. Katze, Y. He and M. Gale, Nat. Rev. Immunol., 2002, 2, 675–687 CrossRef CAS PubMed.
- B. R. Long, J. Michaelsson, C. P. Loo, W. M. Ballan, B.-A. N. Vu, F. M. Hecht, L. L. Lanier, J. M. Chapman and D. F. Nixon, Clin. Vaccine Immunol., 2008, 15, 120–130 CrossRef CAS PubMed.
- C. Bode, G. Zhao, F. Steinhagen, T. Kinjo and D. M. Klinman, Expert Rev. Vaccines, 2011, 10, 499–511 CrossRef CAS PubMed.
- R. Yang, A. R. Masters, K. A. Fortner, D. P. Champagne, N. Yanguas-Casás, D. J. Silberger, C. T. Weaver, L. Haynes and M. Rincon, J. Exp. Med., 2016, 213, 2281–2291 CrossRef CAS PubMed.
- W.-T. Ma, X.-T. Yao, Q. Peng and D.-K. Chen, Open Biol., 2019, 9, 190109 CrossRef CAS PubMed.
- S. H. Seo and R. G. Webster, J. Virol., 2002, 76, 1071–1076 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |