
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecularly imprinted polymers in diagnostics: accessing analytes in biofluids
Yasemin L.
Mustafa
 ab,
Antonios
Keirouz
ab and
Hannah S.
Leese
*ab
ab,
Antonios
Keirouz
ab and
Hannah S.
Leese
*ab
aMaterials for Health Lab, Department of Chemical Engineering, University of Bath, Bath, BA2 7AY, UK. E-mail: h.s.leese@bath.ac.uk
bCentre for Biosensors, Bioelectronics and Biodevices (C3Bio), University of Bath, Bath, BA2 7AY, UK
First published on 22nd June 2022
Abstract
Bio-applied molecularly imprinted polymers (MIPs) are biomimetic materials with tailor-made synthetic recognition sites, mimicking biological counterparts known for their sensitive and selective analyte detection. MIPs, specifically designed for biomarker analysis within biofluids, have the potential to significantly aid patient diagnostics at the point-of-care, enabling self-health monitoring and management. Recent research in this field, facilitated by the hybridisation of materials science and biology, has developed and utilised a variety of different polymerisation synthesis methods tailored to the bio-application of MIPs. This review evaluates the principles of molecular imprinting for disease diagnostics, including recent progress in integrated MIP-sensor technologies for high-affinity analyte detection in complex biofluids from serum and saliva to cerebrospinal fluid, sweat, urine, nasopharyngeal fluid, and tears. The work highlights the state-of-the-art in the progression of MIP-sensor technologies’ translation into commercially available sensors and their potential contribution to disease detection systems in healthcare settings.
 Hannah S. Leese | Hannah S. Leese is assistant professor in the Department of Chemical Engineering at the University of Bath. She received her Ph.D. in Chemical Engineering (2013) and held post-doctoral research associate positions at Imperial College London (2013–2017) and the University of Manchester (2017–2018). Together, Hannah's team, the Materials for Health Lab, engineer advanced materials for minimally invasive point-of-care diagnostics for the early detection of disease – including molecularly imprinted polymers as sensor elements in devices, hydrogel-based microneedle technologies for drug delivery and biomarker uptake, devices for early detection of ovarian cancer and biomimetic protein-based fibres for wound dressing textiles. |
Introduction
Until recently, medical diagnostics have primarily relied on laboratory verification, often providing delayed results due to laborious processes, in addition to requiring specialised equipment and medically trained personnel.1–3 However, the rising demand for medical testing devices at the point-of-care, combined with the continuous interconnecting of medicine and digital technologies, has facilitated the fabrication of devices with high selectivity, specificity, and rapid response times.4 The evolvement of such devices has been internationally recognised as a focal point during the COVID-19 pandemic, highlighting further, the urgent call to advance accurate, rapid, and reliable medical diagnosis and treatment.5–9Point-of-care devices can negate issues associated with current clinical instruments, instead, offering fast results, small sample volumes, easy transportability, and minimal technical training. Commercially available point-of-care biosensors, such as the FreeStyle Libre 2 (Abbott, USA), Accutrend® Plus system (Roche, Switzerland), and CoaguChek® Pro II (Roche, Switzerland), are sensor-based biomarker monitoring systems for targeted diseases such as diabetes, cardiovascular conditions and blood coagulation, respectively (see Table 1).10–12 These biosensors have capitalised on biological recognition elements such as antibodies or enzymes to function as the molecular recognition element for specific target analyte(s) (e.g., glucose, lactate, cholesterol, and triglyceride) and provide a benchmark for MIP-based biosensors to reach the clinic.10–12 Furthermore, besides bearing high economic costs, antibodies have sensitive physicochemical properties, such as, sensitivity to temperature fluctuations and requiring storage conditions below −20 °C.13 Failure to abide by the storage requirements significantly affects their storage life and, more importantly, the affinity of these molecules with that of the target analyte. The latter is likely to alter their performance as a biosensor, leading to result ambiguity. Therefore, other means of development have considered changing traditional molecular biology practices by obtaining the selectivity of natural antibodies via synthetic routes, e.g., soft lithographic processes, systematic evolution of ligands by exponential enrichment, and molecular imprinting.14
| Specifications | Prototypes | |||||
|---|---|---|---|---|---|---|
| FreeStyle Libre 2 | Accutrend® Plus System | CoaguChek® Pro II | ||||
| a From the date of manufacturing. | ||||||
| Manufacturer | Abbott, USA | Roche Diagnostics, Switzerland | Roche Diagnostics, Switzerland | |||
| Price (£) | 58 | 271 | 1279 | |||
| Targeted disease | Diabetes | Cardiovascular | Blood coagulation | |||
| Sample material | Interstitial fluid | Capillary blood | Capillary, venous or arterial blood | |||
| Analytes | Glucose | Cholesterol | Glucose | Lactate | Triglyceride | Thromboplastin |
| Sample volume (μL) | — | 15–40 | 15–50 | 15–50 | 10–40 | ≥8 |
| Measuring time (sec) | 126–144 | 180 | 12 | 60 | Max. 174 | 9.6–96 |
| Memory capacity | 8 h | 100 values | 2000 results | |||
| Operating temperature | 10–45 °C | 18–35 °C | 18–35 °C | |||
| Advantages | Data capture in <4 cm range | Determination of 4 different analytes on one platform | Touchscreen | |||
| Suitable for 4–12 years | Test strips have a storage life of up to 18 monthsa | Several built-in quality-control functions (e.g., test strip temperature check and quality control) | ||||
| Easily attached to the body | Can store up to 100 different measurements with date and time | Save power mode | ||||
| Max. 14 day wear | Great precision and accuracy across the measuring range | Translatable into 13 different languages | ||||
| Readings stored every 15 min | Portable hand-held device | WLAN and unique QR code connectivity option | ||||
| Rapid data share | Large display – easy to read | |||||
| Excellent accuracy | ||||||
| No test strips are necessary | ||||||
| Stores up to 90 days of data | ||||||
| Portable hand-held device | ||||||
| Large display – easy to read | ||||||
| Water-resistant | ||||||
| Minimally invasive | ||||||
| Discreet and convenient | ||||||
| Limitations | 60 min warm-up period | Requires a lancing device | Additional components required | |||
| Finger pricks required (if readings do not match symptoms/expectations) | Additional components required | Requires a lancing device | ||||
| Sensor app and website compatibility is selective | Invasive | Invasive | ||||
| Daily patterns need at least 5 days of glucose data | Painful | Painful | ||||
| Scanning is required every 8 h | Potential blood contamination | Potential blood contamination | ||||
| Data transfer is dependent upon mobile connectivity | Expensive | Expensive | ||||
| Automatic data upload requires a wireless internet connection or mobile data connection | ||||||
The field of molecular imprinting historically dates to the 1940s following Pauling's hypothesis that the occurrence of antibodies in human cells is a consequence of the presence of antigens.15 Pauling attributed the high specificity of the antibody–antigen complex to the behaviour of the antigen, performing as a template, commanding the geometry of the antibody.15 It is argued that the catalyst of this discovery resulted from Polyakov's development in understanding the importance of solvent selection in 1931, specifically its effect on the pore structure of synthesised silica hydrogels.16 And since, a variety of synthetic approaches have been generated, giving rise to molecularly imprinted polymers (MIPs) which demonstrate selectivity likened to those of natural antibodies, thereby earning the titles of ‘artificial receptors’, ‘plastic antibodies’ and ‘antibody mimics’, leading to the development of molecularly imprinted technologies (MITs).17–19
MIPs have the potential to address many of the current challenges associated with antibody-based diagnostics, including complicated manufacture and handling, long-term stability, and loss of performance in organic media.17,20–27 MIPs are reasonably inert materials that can be exploited as affordable artificial receptors for biological sensor purposes owed to their selective, specific, biologically stable, and easily tailored (e.g., surface chemistry modifications and/or signalling functionalities) nature in combination with their exceptional physicochemical abilities and extensive shelf-life.17,20–25,27 Thus, exploitation of these material properties could facilitate improved biomarker detection and analysis compared to clinical biological receptors.28
To date, the field of MIPs has been employed in various applications;28–30 however, this review will evaluate the principles of molecular imprinting for biosensing and evaluate their effectiveness when detecting analytes in biofluids specifically; serum, saliva, cerebrospinal fluid, sweat, urine, nasopharyngeal fluid and tears. Recent progress in integrated MIP sensor technologies for high-affinity analyte detection from complex biofluids will be critically assessed, highlighting state-of-the-art for the progression into disease detection systems in healthcare settings and point-of-need. Throughout this review, the term biosensor will also be applied to sensing devices whereby the recognition element is comprised of a synthetic MIP species. Although this may differ from the International Union of Pure and Applied Chemistry (IUPAC) definition of traditional biosensor terminology, the vast majority of the literature covered in this review considers “biosensors” to include MIP-based sensing devices for the detection of biological analytes.
Mechanisms of molecularly imprinted polymers
MIP synthesis (Fig. 1) is typically achieved via the selection of suitable functional monomer(s) that form specific stable interactions with a template molecule (target analyte).31,32 The formation of a stable template–monomer complex is critical for the recognition capability of imprinted materials.33 The spatial arrangement of selected monomers around the template molecule is secured via polymerisation, aided by a crosslinking agent (dependent upon the type of molecular interactions selected), forming a three-dimensional polymeric network.34 Interactions within the polymer matrix, between the newly shaped polymer backbone and template, results in the installation of molecular recognition.26 These high-affinity binding sites are revealed subsequent to template extraction due to the formation of cavities complementary in shape, size, and chemical functionality to that of the template and closely related functional analogues.35,36 Molecular imprinting is classified according to the relationship between the MIP and the non-imprinted polymer (NIP) species. NIP synthesis follows the identical protocol employed to produce MIPs while omitting the inclusion of the template molecule. Upon synthesis, NIPs present a three-dimensional network of non-target-specific cavities, where non-specific binding may still occur on the surface thus, acting as a comparative control to selectivity and specificity. In turn, this enables the calculation of quantifiable values for the assessment of both the imprinting factor (IF) and the specificity adsorption (α) ratio for sensitivity and specificity determination, respectively.37 Collectively, these values measure MIP performance, which is largely a consequence of preparative techniques, amongst other factors, including chemical composition (e.g., solvent choice), polymer morphology, template–monomer complexation and their interactive forces.38–40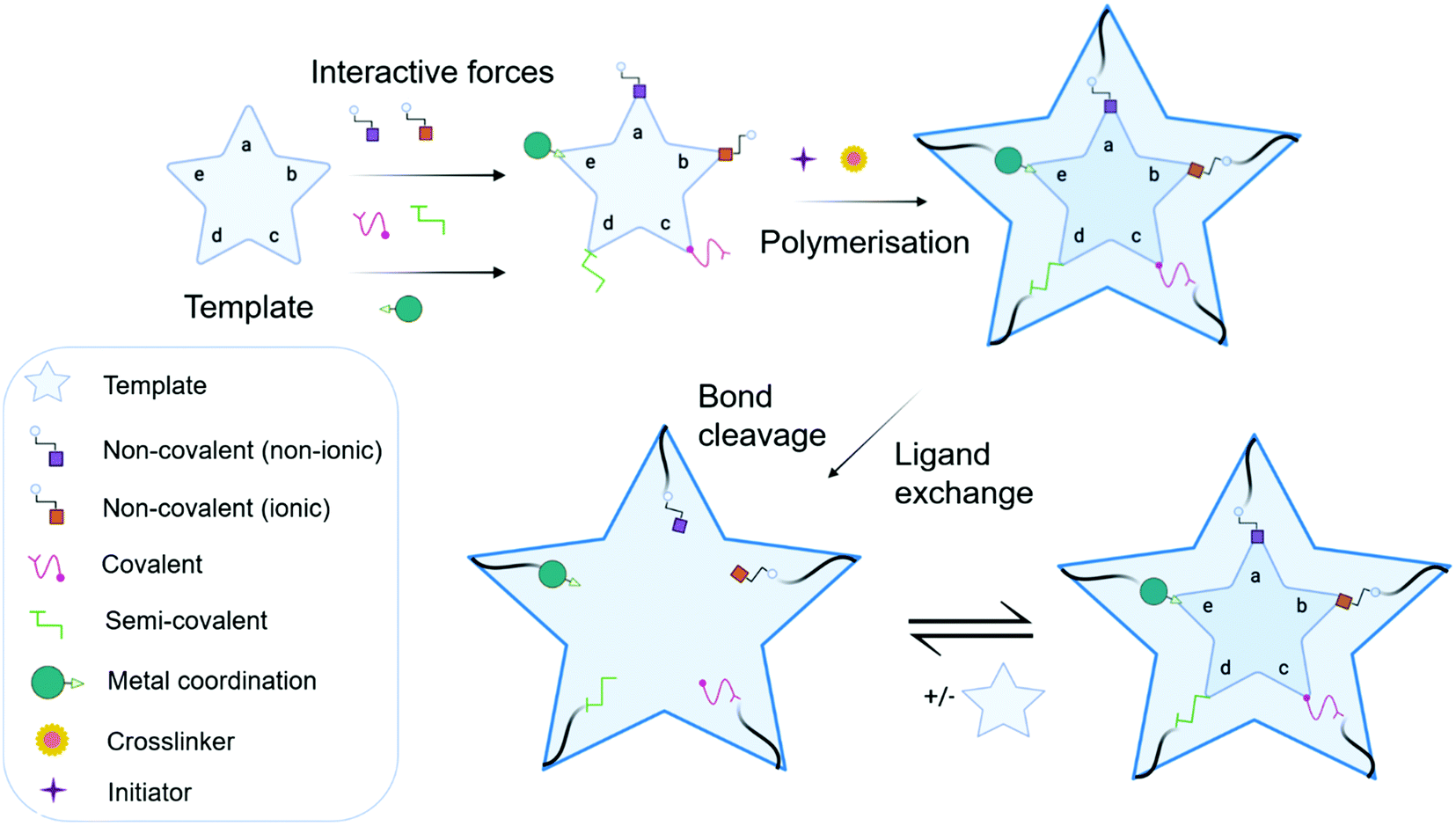 | ||
| Fig. 1 Molecular imprinting process depicting potential interactions within the template–monomer complex for a MIP synthesised via chemical polymerisation, where a, b, c, d, and e, represent non-covalent (non-ionic), non-covalent (ionic), covalent, semi-covalent, and metal coordination, respectively. NB. Interactions depend upon the selected template and monomer(s); therefore, the described interactions are not necessarily all simultaneously at play. | ||
The stability of the template–monomer complex and the energy required for template extraction is governed by the type of intermolecular interactions. For MIPs, molecular interactions are classified as either covalent or non-covalent, contingent on the available functional groups of both template molecules and functional monomer(s) in a suitable solvent.41 Typically, non-covalent imprinting (Fig. 1a and b) is stipulated by the self-assembly of the template molecule and functional monomer(s) solubilised in a suitable solvent via non-covalent interactions (e.g., hydrogen bonding, π–π stacking interactions, or electrostatic forces), followed by the addition of a crosslinking agent to initiate polymerisation.42,43 Covalent imprinting (Fig. 1c) describes a polymerisable and cleavable template–monomer composite, where the template is covalently bound to the functional monomer(s). In the latter, polymerisation is aided by a crosslinker, after which the template is cleaved off via acidic/basic hydrolysis, generating an imprinted species prepared for template rebinding through covalent forces.44,45 The final approach, semi-covalent imprinting (Fig. 1d and e), exploits both non-covalent and covalent interactions.25 Like the covalent approach, a template-functional monomer composite is formed, followed by the removal of the template molecule via hydrolysis, fabricating cavities for template rebinding through non-covalent bonding interactions.25
Commonly, non-covalent interactions are exploited for MIP synthesis, and this is primarily attributed to intermolecular forces, such as hydrogen bonding, encouraging interactions like that of biological recognition systems (e.g., receptor–ligand, enzyme–substrate, and antibody–antigen complexes).46 Additionally, the ease of template–monomer complex formation, template dissociation and capability to utilise an extensive range of different monomers, has led to non-covalent interactions being widely applied for MIP synthesis.20 Nevertheless, non-covalent imprinting suffers from limitations in the form of heterogeneous distribution of binding sites and non-specific binding, resultant of excess amounts of functional monomer(s) for pre-polymerisation complex stabilisation.43 Additionally, there have been reports suggestive of the covalent imprinting approach being superior due to the generation of uniformly accessible imprinted cavities as there is a more homogeneous distribution of binding sites and high selectivity throughout the MIP.47 However, despite these advantages, covalent imprinting is not widely adopted, attributed to time-consuming template rebinding, limiting its application as a biosensor.43 Furthermore, covalent imprinting necessitates additional fabrication steps for template–monomer composite stabilisation via covalent bond formation. In particular, covalent forces are restricted to diols, ketones, aldehydes, carboxylic acids, and amines.45 Semi-covalent interactions, like covalent interactions, can produce polymers with high selectivity; however, polymers formed via these interactions can suffer from non-specific binding due to uncleaved template molecules, occupying binding sites.37
The success of these antibody mimics has progressed their application for the employment of sensory components, detecting bioanalytes (e.g., electrolytes, metabolites, amino acids, proteins, and hormones) located within bodily fluids.48–50 These bioanalyte-rich media, including but not limited to, saliva, tears, and sweat offer essential information regarding patient health.51 By considering alternative biofluids, other than the current gold standard – arterial blood – enables biomarkers to be readily accessed in a minimally invasive manner, laying the foundations for their evolution from bench to bedside.28–30,52
Methodologies to produce molecularly imprinted polymers with example bio-applications
Today, molecular imprinting is broadly employed for various applications, such as biosensing, chromatographic separation and solid-phase extraction, among others.52–55 Traditionally, MIPs are formed via free-radical reactions employing preparation methods such as bulk, precipitation, emulsion, and suspension techniques (Fig. 2a–d).56 The degree of imprinting within a polymeric network is defined as the IF efficiency and can be calculated as shown by eqn (1), where the equilibrium binding capacity (Q), i.e., the amount of analyte bound by MIP and NIP species, denoted by QMIP and QNIP, respectively, is first determined. Q (eqn (2)) describes the divergence between the initial amount of target analyte in the introduced solution mixture relative to the amount in the supernatant, where Ci, Cf, m and V define the initial concentration of the target analyte in the introduced mixture, the equilibrium concentration of the target analyte in the solution mixture, the mass of the polymer and volume of the analyte solution mixture, respectively.37,57–59 Additionally, Q, can also be used to determine the specificity adsorption ratio (a), which describes the degree of selectivity of MIP species in relation to the NIP species, as shown by eqn (3).57IF and α values exceeding 1 are indicative of a good degree of molecular imprinting and target analyte specificity, respectively.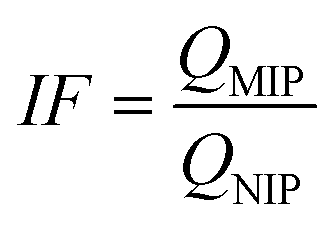 | (1) |
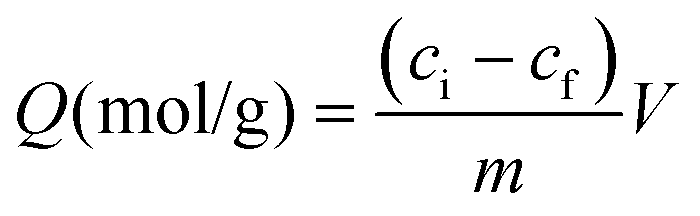 | (2) |
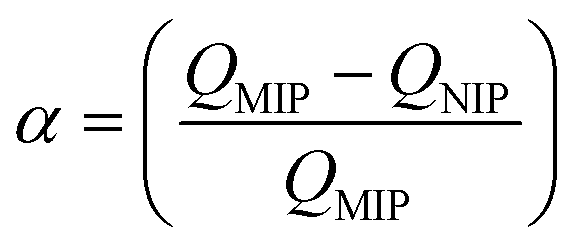 | (3) |
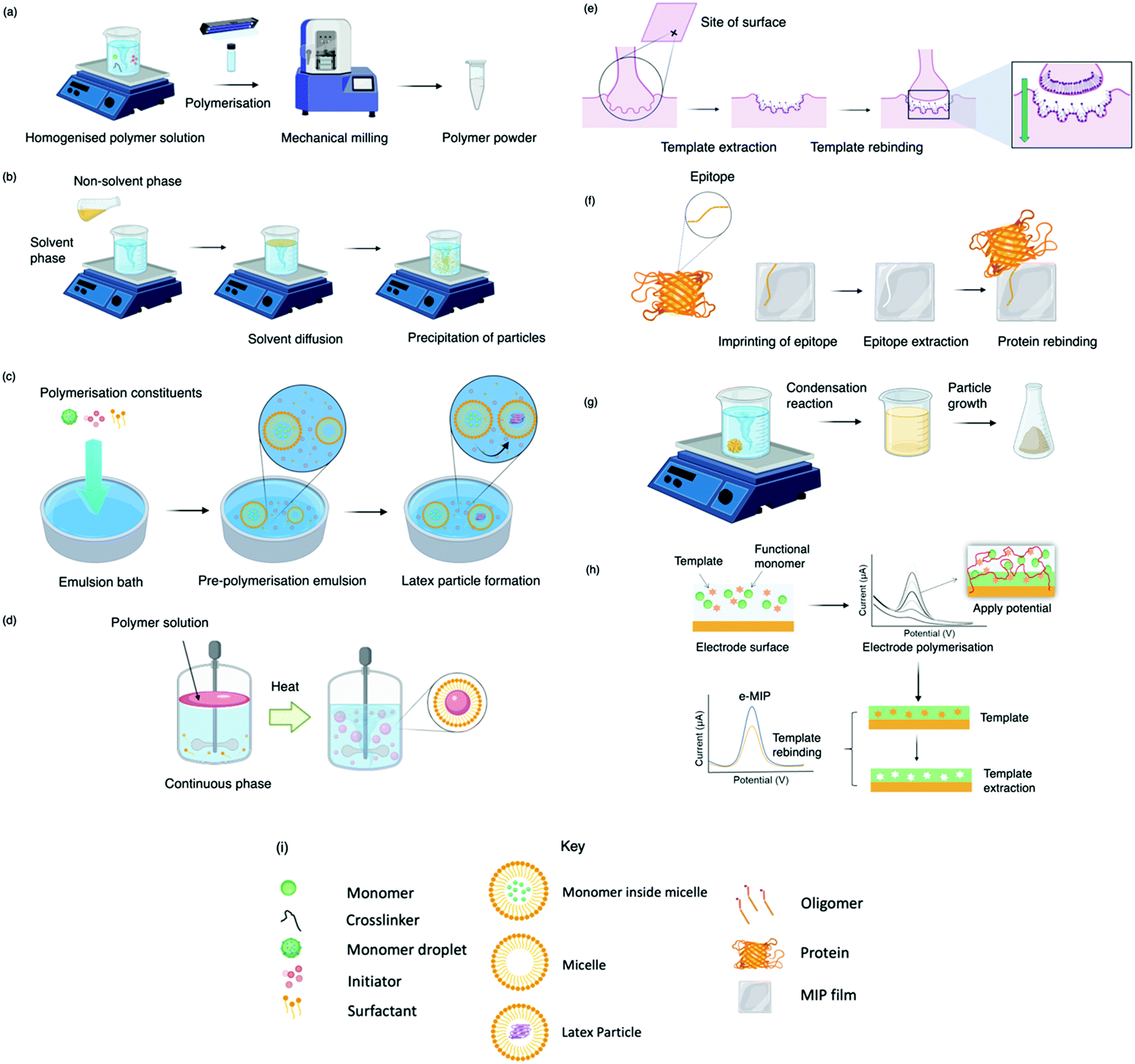 | ||
| Fig. 2 Illustrative diagram depicting a range of different polymerisation techniques including: (a) bulk, (b) precipitation, (c) emulsion, (d) suspension, (e) surface, (f) epitope, (g) sol–gel, (h) electropolymerisation, and (i) each technique's associated key. | ||
Bulk polymerisation (Fig. 2a) proceeds with the initiation of a soluble radical initiator in the presence of soluble monomer(s) (functional monomer and crosslinker) upon thermal or ultraviolet radiation exposure.60,61 Precipitation polymerisation (Fig. 2b) describes a homogeneous polymer solution consisting of soluble monomer(s) and initiator in a continuous phase.60,61 Upon initiation, a heterogeneous mixture is formed in which the insoluble polymer precipitates out.62 Olcer et al. utilised bulk and precipitation polymerisation to prepare a highly selective and reusable (up to five times) MIP to detect endocrine-disrupting chemicals, specifically, ibuprofen in drinking and tap water samples.60 In the study, ibuprofen, methacrylic acid, trimethylolpropane trimethacrylate were employed as template molecule, functional monomer, and crosslinker in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8:20 molar ratio, respectively, with 4,4-azobis(4-cyanovaleric acid) and acetonitrile as initiator and porogen. Precipitation polymerisation was more successful in formulating a MIP with higher template affinity compared to the bulk-synthesised MIP.60 The imprinted polymer prepared via precipitated means showed an approximately two-fold increase in sorption capacity compared to its control (non-imprinted polymer, NIP), calculated at 0.0387 mmol g−1 and 0.0235 mmol g−1, respectively at 50 mg L−1. Selectivity was demonstrated in the presence of compounds sharing structural likeness to that of ibuprofen, namely naproxen and ketoprofen (Fig. 3(a-i)). The success of the precipitation technique over the bulk technique was attributed to the MIP's spherical nature (Fig. 3(a-ii)), providing greater surface area compared to the monolithic MIP (Fig. (3(a-iii))), thus allowing for more selective cavity sites at the surface of the polymer, arguably the principal contribution to sorption performance.60
:8:20 molar ratio, respectively, with 4,4-azobis(4-cyanovaleric acid) and acetonitrile as initiator and porogen. Precipitation polymerisation was more successful in formulating a MIP with higher template affinity compared to the bulk-synthesised MIP.60 The imprinted polymer prepared via precipitated means showed an approximately two-fold increase in sorption capacity compared to its control (non-imprinted polymer, NIP), calculated at 0.0387 mmol g−1 and 0.0235 mmol g−1, respectively at 50 mg L−1. Selectivity was demonstrated in the presence of compounds sharing structural likeness to that of ibuprofen, namely naproxen and ketoprofen (Fig. 3(a-i)). The success of the precipitation technique over the bulk technique was attributed to the MIP's spherical nature (Fig. 3(a-ii)), providing greater surface area compared to the monolithic MIP (Fig. (3(a-iii))), thus allowing for more selective cavity sites at the surface of the polymer, arguably the principal contribution to sorption performance.60
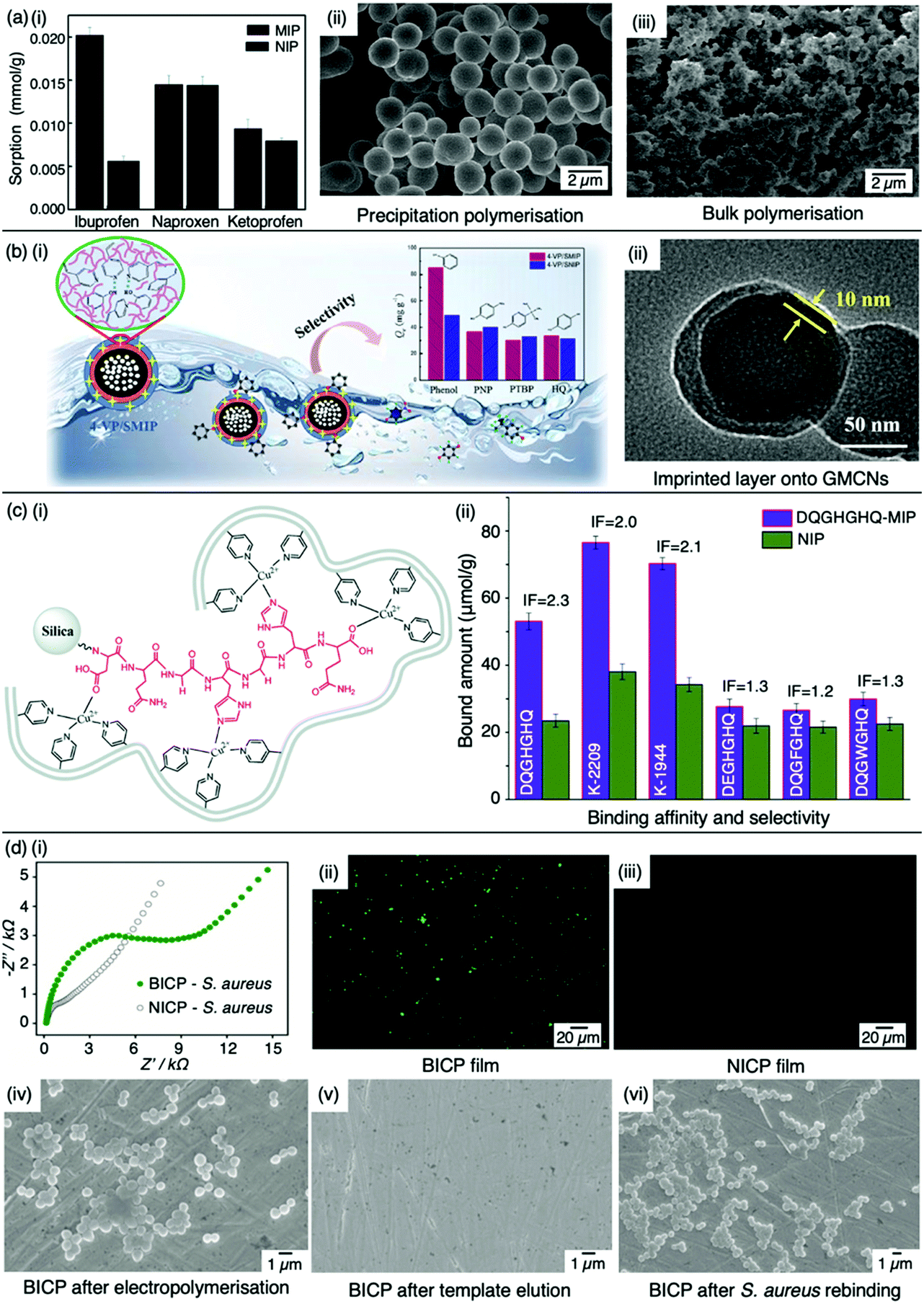 | ||
| Fig. 3 Molecular imprinting through different synthesis routes. (a) Comparison of precipitation vs bulk polymerised MIPs. (i) Sensitivity and selectivity of ibuprofen MIPs prepared via means of precipitation in the presence of structurally related compounds; (ii) scanning electron microscopy (SEM) image of MIP prepared via precipitation polymerisation, and; (iii) SEM image of MIP prepared via bulk polymerisation. Reproduced with permission from ref. 60, Copyright 2017 Royal Society of Chemistry. (b) Bulk polymerised MIPs presenting glucose-derived microporous carbon nanospheres. (i) Schematic illustration of phenol extraction from wastewater using a highly selective surface imprinted MIP; (ii) transmission electron microscopy image of 4-vinyl pyridine surface imprinted MIP. Reproduced with permission from ref. 87, Copyright 2020 Elsevier. (c) Epitope imprinted silica-conjugated MIP peptides. (i) Polymeric mixture of epitope-containing peptides secured via polymerisation; (ii) competitive binding of epitope-MIP with structurally similar peptide interferants. Reproduced with permission from ref. 88, Copyright 2016 Royal Society of Chemistry. (d) Bacteria-imprinted conducting polymer film. (i) Comparative electrochemical response of bacteria imprinted conductive polymer (BICP) sensor and corresponding non-imprinted conductive polymer (NICP) as control; (ii and iii) fluorescence images of BICP and corresponding NICP films after staphylococcus aureus retention; (iv–vi) scanning electron microscopy images of varying polymer films modified gold electrodes. Reproduced with permission from ref. 89, Copyright 2021 Elsevier. | ||
Despite bulk polymerisation being considered a simplistic approach, some synthesis protocols can require mechanical milling and, if necessary molecular sieving to provide uniform particle shapes and sizes.63,64 However, milling could reduce MIP capturing capacities by damaging or distorting imprinted interaction sites.65 Regardless of the limited control over the physical form of the obtained imprinted species, many MIP publications still employ the bulk polymerisation approach, possibly due to reproducibility and simplicity with respect to the preparation and formulation of this polymerisation process.60,61,63,66 Bulk polymerisation limitations have led to the development of alternative approaches, including in situ prepared monoliths, molecularly imprinted monoliths, MIP membranes, and MIP beads.22,67–71 Precipitation polymerisation (Fig. 2b) is credited to the processes improved heat control and direct preparation of large-scale production.72 However, precipitation polymerisation requires a greater volume of porogen and more rigorous reaction control compared to bulk polymerisation.72–74
Emulsion polymerisation (Fig. 2c) describes the dispersion of monomer(s) and surfactant(s) in an emulsion of either water-in-oil or oil-in-water. Polymer spheres are formed via the generation of free radicals by an initiator (water- or oil-soluble – dependent upon polymerisation type).75 In a recent study, Zhao et al. established a MIP to purify solanesol, an essential pharmaceutical intermediate and an organic substance to produce anti-ulcer and anti-cancer drugs via emulsion polymerisation. In this study, the effects of varying quantities of MIP chemical composition were investigated for MIP functionality performance purposes. Zhao et al. successfully synthesised spherical solanesol MIP (SS-MIP) microspheres, with particle sizes ranging between 50–500 mm, using solanesol, methyl methacrylate, ethylene glycol dimethacrylate and potassium persulfate, performing as template species, functional monomer, and crosslinker, in a 1:8:30 molar ratio, respectively. SS-MIP performance was demonstrated by maximum absorption retention (Qmax) of 56.97 mmol g−1 and reported an IF of 2.51.76 Although the fabricated SS-MIP showed good adsorption properties, more research is required to validate the performance of this means of polymerisation, including, but not limited to: (a) improved protocols for the formation of uniformly shaped and sized SS-MIPs, (b) investigation into the recognition sensitivity and specificity of SS-MIPs, and (c) study of SS-MIP applications.76 Ma et al., however, successfully demonstrated a large-scale applicatory function of SS-MIPs for high-purity solanesol extraction from tobacco leaves using chromatographic methods via suspension polymerisation, where methacrylic acid, ethylene glycol dimethacrylate, and 2,2′-azobisisoheptonitrile were used as functional monomer, crosslinker and initiator, respectively. The synthesised spherical SS-MIPs (particle diameter 250–350 mm) exhibited a Qmax of 107.3 mmol g−1 and IF of 3.9.77
In suspension polymerisation (Fig. 2d), a dispersed heterogeneous mixture of droplets is formed that undergoes polymerisation, ultimately forming polymer spheres.78 Gomes et al. produced polyphenol MIPs via precipitation and suspension polymerisation to determine the most superior process for morphology and performance, using polydatin, a precursor of resveratrol available in vegetable extracts, as a template.69 Polydatin is an important detectable target due to its anti-oxidant and anti-inflammatory capabilities, rendering it a valuable chemical for disease treatment.79 Gomes et al. synthesised a range of MIPs for the amphiphilic polydatin template through a parametric study that assessed a variety of functional monomers, crosslinkers, and solvents for the assessment of non-covalent interactions (including hydrophobic/hydrophilic interactions) between the template–monomer complex and their effects on imprinting efficiency. For natural extracts, hydrophilic/hydrophobic interactions are inevitable; thus, the adjustment of solvent polarity is critical for the isolation of different polyphenols.69 In this study, polydatin, 4-vinylpyridine, 2,2-azobisisobutyronitrile, water/methanol, and sorbitan mono-oleate (Span 80) as template species, functional monomer, initiator, solvent, and surfactant, respectively, presented a Qmax of ∼300 mmol g−1, greater than literature reported values.80–84
Some of the previously discussed polymerisation techniques can suffer from poor imprinted cavity access due to embedded binding sites, incomplete template removal and slow mass transfer rates. Surface imprinting (Fig. 2e), on the other hand, generates materials with large surface areas (surface-to-volume ratio) and high porosity owed to their distribution of externally available binding sites. Arranging cavity sites in this way is favourable for removing and rebinding the target analyte as imprinted cavities are readily exposed.85,86 Moreover, these surfaces are controllable, show low migration resistance towards selective adsorption, bypassing the template embedding phenomenon, resulting in greater adsorption capacities.69
Recent studies have shown surface or outer layer modifications of MIPs to be a popular method of molecular imprinting, utilising specific carriers such as silica, polystyrene microspheres, quantum dots, metal–organic frameworks, magnetic nanoparticles, and carbon nanomaterials.87,90–93 Qu et al. recently used surface molecular imprinting to fabricate a highly selective MIP prepared on glucose-derived microporous carbon nanospheres for the removal of phenol (Fig. 3(b-i)), a highly toxic substance found in wastewater which can affect the cardiovascular and central nervous system.94,95 Glucose-derived microporous carbon nanospheres were employed as support materials due to their rich pore structures and surface located oxygenic functional groups, where active layers were stabilised via silane coupling agents. An optimal polymeric mixture was synthesised using phenol, 4-vinylpyridine, ethylene glycol dimethacrylate, 2-methylpropionitrile, and toluene as template molecule, functional monomer, crosslinker, initiator, and solvent system in a 1:3:1.8:0.07 ratio, respectively. The assembly of this surface imprinted crosslinked polymer (Fig. 3(b-ii)) displayed a Qmax of 85.72 mg g−1. The successful isolation of phenol from wastewater was displayed by the relative selectivity factors (a) of phenol versus three interfering molecules; hydroquinone, p-nitrophenol and p-tert-butylphenol, valued at 8.38, 7.96 and 6.67, respectively, highlighting the selective capabilities of this synthesised MIP.94
Surface imprinting technology was also employed by Liu et al. to develop highly sensitive and specific magnetic MIPs (MMIPs). MMIPs with an average particle size of 2 μm were prepared via graphite-like carbon nitride–iron oxide nanoparticles as support matrices for the adsorption of atrazine, a herbicide with known carcinogenic effects. Adsorption isotherms demonstrated that MMIPs (1.82 mmol g−1) formulated by atrazine, methacrylic acid, ethylene glycol dimethacrylate, 2,2-azobisisobutyronitrile, and chloroform as template molecule, functional monomer, crosslinker, initiator, and solvent system, respectively, showed a greater adsorption capacity compared to its control counterpart (MNIP, 0.875 mmol g−1) at 80 mmol L−1. Additionally, adsorption of atrazine molecules was greater when compared to structural analogues (ametryn, atratin, hexazinoneas), demonstrating high selectivity towards atrazine pesticides for the detection of atrazine from complex substances.96
Epitope imprinting (Fig. 2f) exploits short characteristic regions of biomolecules (e.g., peptides) performing as templates.97 Epitope MIPs (EMIPs) can overcome concerns surrounding the imprinting of bulky macromolecular structures, often burdened with diffusion issues within the highly crosslinked three-dimensional polymeric network and conformational changes due to harsh polymerisation conditions and environments.97,98 EMIPs encourage the use of conventional monomers, facilitating improved template extraction procedures, and are more cost-effective as they avoid the need for an entire biomolecule, making it a valuable technique for the application of protein studies.99,100 Like traditional imprinting methods, the epitope approach can be utilised via bulk or surface imprinting, depending upon the desired template–monomer complex interactions.101 Template immobilisation onto a supporting matrix is critical for successful EMIP formation, where studies have reported the preparation of templates via boronate affinity, physical adsorption, metal ion chelation, and covalent bonding.98,102,103
Xing et al. developed a novel controllable oriented surface imprinting approach, utilising boronate affinity-anchored epitopes to imprint protein templates.98 This study was centred around the 2-microglobulin (b2M) protein sequence, where abnormal concentration levels are associated with diseases such as multiple myeloma.104 The C-terminus nonapeptide, attached with a lysine, was selected as the template epitope, with 2,4-difluoro-3-formyl-phenylboronic acid acting as the coordinating ligand. Consequential of boronate affinity, the glycated epitope was immobilised onto boronic acid-functionalised magnetic nanoparticles, coated with multiple silylating agents, facilitating template–monomer interactions via polycondensation reactions in a monomer ratio of 10:10:20:60, respectively.98 The synthesised glycated epitope-imprinted magnetic nanoparticles (average diameter 150 nm) presented an imprinting efficiency percentage and IF value of 54.2% and 5.8, respectively. The study further demonstrated the successful removal of b2M-epitope and protein using the synthesised glycated b2M EMIP, through fast equilibrium kinetics (∼20 min), attributed to easily accessible surface imprinted cavities. Additionally, b2M protein showed greater selectivity, attributed to the highest IF calculated at 6.5, compared to competitive analogous proteins ribonuclease A, ribonuclease B, horseradish peroxidase, and bovine serum albumin. Moreover, the imprinted species demonstrated long-term storage capabilities (ca. three months) with a minimal reduction in sensitivity performance (13.4% decrease) and withstood a total of six consecutive activities of rebinding.99
Tang et al. opted for the covalent approach to prepare two biomarker peptides (K-2209 and K-1944), reflective of gastric and liver cancer diagnosis, using heptapeptide (DQGHGHQ) as the epitope template.102 Porous silica was retained as a sacrificial substrate for template molecule immobilisation.105 Spherical imprinted particles were synthesised via hierarchical imprinting polymerisation, utilising the metal coordination interaction between copper(II) oxide, the template species, and 4-vinyl pyridine (performing as both functional monomer and coordinating ligand) for the formation of binding sites (Fig. 3(c-i)).88 The optimal pre-polymerisation mixture was formed of template, aqueous copper acetate, 4-vinyl pyridine, ethylene glycol dimethacrylate (crosslinker), and 2,2-azobisisobutyronitrile (initiator) in a 1:6:12:30 molar ratio, respectively, dissolved in acetonitrile. DQGHGHQ-MIP exhibited a 71–88% recovery performance for the K-1944 and K-2209. This study demonstrated the potential of epitope imprinting to aid biomarker screening for cancer diagnosis through individual and competitive batch binding experiments. The individual experiments highlighted the highest IF (2.2) for the DQGHGHQ-MIP, followed by K-2209 (2.0) and K-1944 (2.0), and competitive binding results exhibited a ca. 1.7-fold selectivity towards the DQGHGHQ-MIP apropos to its competitive peptide counterparts (DQGWGHQ, DQGFGHQ and DEGHGHQ) (Fig. 3(c-ii)).102
The sol–gel technique (Fig. 2g) offers high solvent and thermal stability, material homogeneity at the molecular level, and a one-pot fabrication process.58 Commonly, sol–gel imprinting utilises tetra-methyl or -ethyl orthosilicate as a precursor to introduce the template species into the polymeric framework.58 The polymeric mixture undergoes hydrolysis followed by polycondensation reactions, generating crosslinked polymeric gels.106 Sol–gel imprinting has attractive characteristics, including simple preparative techniques, controllable porosity and surface area, and easily tailored chemical functionality for enhanced selectivity.58 Unlike radical polymerisation, where polymer network formation has a dependency on both temperature and the choice of organic solvent (e.g., chloroform), this technique can be performed at room temperature with environmentally friendly solvents (e.g., water).107 For instance, Guoning et al. used Tween® 20, a non-ionic water-soluble surfactant, to encourage mild hydrolysis to develop a surfactant-mediated sol–gel system to fabricate protein MIP layers, utilising 3-(methacryloxy)propyltrimethoxysilane as crosslinker.108 Human serum albumin, one of the most abundant proteins present in blood plasma, was selected as the target protein as this biological analyte can correspond to ailments such as kidney disease.109,110 The authors formed a homogenous protein–polymer mixture, with no protein deformation within the organic solvent system (methanol and toluene) being reported. MIP layers were stabilised onto iron(II, III) oxide nanospheres to encourage rapid magnetic separation and immobilisation. Finally, ovalbumin was employed as a protein blocking agent to improve MIP selectivity by reducing the effects of non-specific binding. This method of molecular imprinting demonstrated a limit of detection (LoD) of 0.3 μg mL−1 and recovery ranges of 85.4–104.5%, where the authors stated the significance of the selective species with the potential to replace their biological counterparts (e.g., antibodies).108 Considering healthy human serum albumin levels found in urine samples are ∼30 mg mL−1, the authors have proposed a very sensitive means to detect the onset of kidney disease.111
Finally, MIPs can be produced via electropolymerisation (Fig. 2h). Established in the late ‘90s, electropolymerisation rapidly emerged as a strategy to develop thin MIP films via in situ spatial confinement of polymer layers directly onto the surface of an electrode.112,113 This form of polymerisation is initiated by the oxidation (voltage- or current-induced) of a specific monomer in an electrochemical cell, facilitating polymer growth.114 Electropolymerisation enables precise control over film thickness and shows good compatibility with aqueous media.115
Malitesta et al. prepared and characterised the first electrosynthesised MIP using glucose and o-phenylenediamine as template molecule and functional monomer, respectively.116 This pioneering study utilised o-phenylenediamine, a known enzyme-entrapping membrane previously employed to fabricate biosensors, to confirm the feasibility of an electropolymerised MIP via the exploitation of a neutral template.117 Direct analytical communication between the transducer element, quartz crystal microbalance and synthetic MIP species was established.116 These promising findings encouraged Özcan et al. to fabricate an electropolymerised MIP via a modified pencil graphite electrode to detect paracetamol (a regularly used analgesic and antipyretic drug) in clinical and pharmaceutical samples.118,119 Polypyrrole (pyrrole, monomer) films were processed via cyclic voltammetry deposition in the presence of lithium perchlorate (supporting electrolyte) with (MIP synthesis) or without (NIP synthesis) paracetamol.119 Differential pulse voltammetry was used to evaluate MIP and NIP performance, with a LoD of 7.9 × 10−7 M (3σ), highlighting the imprinted species sensitivity towards paracetamol. Moreover, this MIP demonstrated a constant and reproducible response (1.3%, relative standard deviation (RSD) (n = 6)), even when in the presence of competitive species (e.g., dopamine, phenacetin, ascorbic acid, phenol and D-glucose), highlighting the MIP sensor's selectivity towards paracetamol.119
More recently, Wang et al. designed a MIP electrochemical sensor using the conductive monomer 3-thiophenacetic acid for rapid sensitive, and label-free detection of Staphylococcus aureus (S. aureus), a Gram-positive pathogen responsible for a wide range of clinical infections.89,120 The conductive MIP film was deposited on the surface of a gold electrode, negating the use of toxic organic solvents or a crosslinking agent. Optimised conditions exhibited a significantly low LoD and very fast kinetic recognition valued at 2 CFU mL−1 and 10 min, respectively. This impedimetric sensor verified good MIP sensitivity and selectivity with respect to electrical impedance spectroscopy responses compared to NIP species (Fig. 3(d-i)) and structural analogues, including Gram-positive Listeria monocytogenes and Gram-negative Escherichia coli (E. coli) O157 and Salmonella Paratyphi B. Additionally, fluorescence imaging was utilised to assess the presence of S. aureus on the surface of the bacteria-imprinted conductive polymer film, reinforcing successfully installed S. aureus-specific recognition cavities, where target bacteria was visibly detectable on the MIP species (Fig. 3(d-ii)) and evidently absent on the NIP species (Fig. 3(d-iii)). Moreover, SEM images captured different polymer film-modified electrodes during the process of bacterial imprinting and recognition, verifying that S. aureus had successfully been embedded on the poly-3-thiophenacetic acid matrices (Fig. 3(d-iv–vi)). Ultimately, Wang et al.'s chosen polymerisation techniques have paved the way for potential bacteria-imprinted conductive polymer films for pathogen detection in applications such as food and water safety. The discussed polymerisation methods are summarised in Table 2.89
| MIP Preparation Techniques | Strengths | Drawbacks | Ref. | |
|---|---|---|---|---|
| Radical | Bulk | – Fast preparation methods without the use of sophisticated equipment | – MIPs require mechanical grinding and sieving - could destroy installed cavities | 56, 60 and 126 |
| – Synthesis of large batches | – Large volumes of porogen are obligatory | |||
| – Affordable synthesis | ||||
| – Polymer mixture is prepared in liquid form | ||||
| – Additional solvent is not required | ||||
| – MIP particle sizes can be readily adjusted | ||||
| Emulsion | – Addition of surfactant(s) or stabiliser(s) unrequired | – Emulsifying agent, hydrophobic monomer(s), and hydrophilic initiator is mandatory | 56, 76 and 126 | |
| – Forms high yield of monodispersed particles | ||||
| – Polymers are suitable for aqueous environments | ||||
| Suspension | – Simple synthesis | – Polymeric mixtures involve both aqueous and organic phases, but many monomers are not water-soluble | 56, 69 and 126 | |
| – Forms uniform MIP microspheres | – Large range between produced particle sizes (up to several hundreds of micrometres) | |||
| – Forms highly porous MIP films | – Initiator and monomer must be hydrophobic | |||
| – Requires surfactant(s) and stabiliser(s) | ||||
| Precipitation | – Quick and simple | – Only successful if polymeric chains are of the appropriate size to reach insolubility within the polymeric reaction mixture | 56, 61 and 126 | |
| – High yields of uniformly shaped MIP beads | – Requires large quantities of template | |||
| – Individual polymeric chains are cultivated into microspheres | ||||
| – Polymeric mixtures do not require porogens | ||||
| Sol–gel | – Reaction solvent(s) are environmentally friendly | – MIPs have low sensitivity | 56, 58 and 126 | |
| – Pore sizes can be readily controlled | – Polymerisation suffers from slow kinetics | |||
| – MIPs have excellent thermal and mechanical stability | – Solvent polarity, mechanical stirring, and polymerisation temperature affect the size of the particles | |||
| Epitope Imprinting | – Templates are small - can easily be removed | – Suitable protein regions must be screened | 56, 97 and 126 | |
| – Reduction in structural complications improves specificity and selectivity | – Surface charges need consideration | |||
| – Synthesised with organic solvents | ||||
| – Affordable | ||||
| Surface Imprinting | – Less template required | – Fewer cavities are formulated - decreased sensitivity | 56, 126 and 127 | |
| – Easier template extraction | ||||
| – Installed cavities are more accessible to template molecules providing favourable binding kinetics | ||||
| Electropolymerisation | – Direct polymerisation onto the electrode surface | – Recognition performance is governed by film thickness, choice of monomer(s), electrode potential, among others | 56, 115, 126 and 128 | |
| – Choice of monomer(s) are limited | ||||
Following polymerisation completion, the template requires removal from the three-dimensional polymer network to finalise the molecular imprinting process. Template removal is often achieved via washing methods using various solvents from aqueous-based to alcohols, and less commonly, by exposing the final product to elevated temperatures or electrochemically.121–123 Optimal template removal conditions should follow a simplistic approach, operating within a reasonable timeframe and using a minimal amount of environmentally friendly solvent(s).124 It is important to note that the NIP usually does not undergo the template removal step, as would be expected. However, for complete comparative analysis, both the MIP and NIP should be exposed to consistent chemical conditions throughout their syntheses. In addition, evidence has suggested that skipping the template extraction protocol for the NIP has led to unrealistic and overstated selectivity factors to that of the MIP.125
Development of MIPs as biosensors for point-of-care diagnostics
Molecular imprinting techniques have seen an expansion in target species, including cells and microorganisms.129–131 For example, the selective identification of microorganisms (e.g., pathogenic bacterial strains) in biological media via artificial MIP biosensing systems has gained widespread attention as a new class of sensing materials that permits for processing-free whole-cell bacterial detection.129 Traditionally, bacterial detection relies on cultures and antibiotic susceptibility tests, immunological assays, genome sequencing, and biochemical testing, often associated with laborious, expensive, and time-consuming practices.132,133 Recent work has described how utilising MIPs can provide a unique approach for the identification of bacterial pathogens.134–139Shen et al. integrated MIT to fabricate bacteria imprinted polymer (BIP) beads for microbiological disease detection via Pickering emulsion polymerisation.140 BIP beads were synthesised in phosphate-buffered saline and prepared for two different groups of bacteria, rod-shaped E. coli (E-BIP) and spherical-shaped Micrococcus luteus (M. luteus) (M-BIP), to determine whether whole-cell recognition performance of imprinted receptors was affected by cell shape. Prior to template extraction, polymer bead surfaces indicated the presence of E. coli (Fig. 4(a and b)); however, post-template extraction, only custom-made imprinted cavities were visibly present on the surface (Fig. 4(c and d)). Both E-BIP and M-BIP beads demonstrated favourable binding of the analogous template cells, indicative of preferential BIP bead selectivity between rod- and spherical-shaped bacteria cells (Fig. 4(e–h)).140 Thus, this study highlighted a practical way to fabricate bacterial recognition sites via molecular imprinting, developing possibilities towards biosensors for real-time examination of whole-cell bacteria species in biofluids.
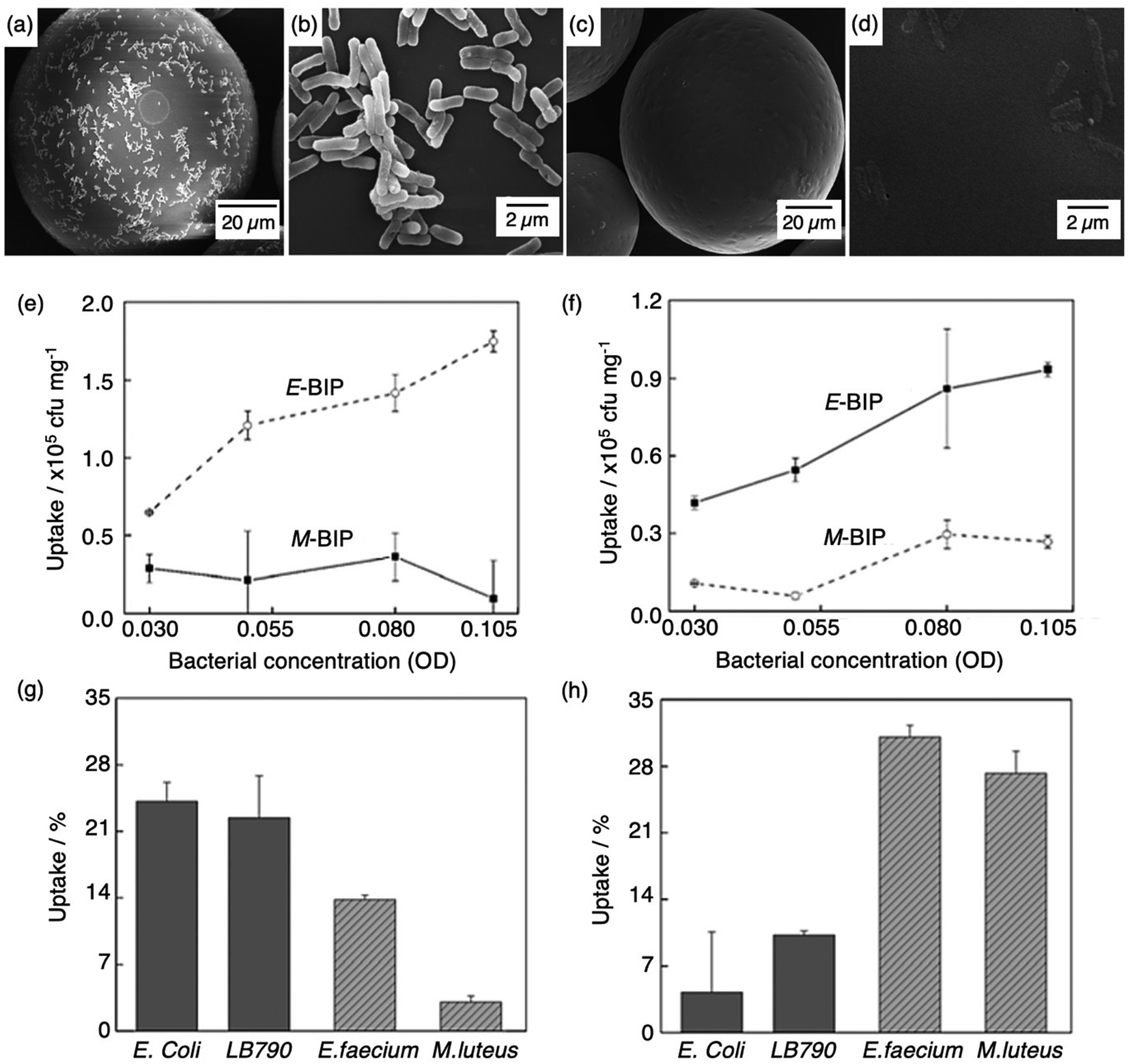 | ||
| Fig. 4 Bacterial imprinting at Pickering emulsion interfaces. (a and b) SEM images of E. coli imprinted polymer beads before template extraction and (c and d) after template extraction; (e and f) sensitivity and selectivity of E-BIP and M-BIP demonstrated by E. coli and M. luteus uptake, and (g and h) uptake of different cells. Reproduced with permission from ref. 140, Copyright 2014 John Wiley & Sons. †Escherichia coli (E. coli), Lactobacillus sakei (LB790), Enterococcus faecium (E. faecium), Micrococcus luteus (M. luteus). | ||
Roushani et al. introduced a new diagnostic method for the detection of Acinetobacter baumannii (A. baumannii), one of the most challenging agents of nosocomial infections worldwide.141 MIP synthesis was achieved via electropolymerisation of the template (A. baumannii) and the functional monomer (dopamine) on the surface of a glassy carbon electrode. Electrochemical properties were used to characterise the performance of the MIP and analogous NIP modified electrode, including cyclic voltammetry and impedance spectroscopy, in a hexacyanoferrate (operating as the electrochemical probe) redox system. This sensor presented a linear range of 102–107 CFU L−1 with a 30 CFU L−1 LoD in human blood serum samples, the first of its kind.141
MIPs have also been integrated with quartz crystal microbalances for facile, cost-effective, high-resolution, and label-free mass sensing, owing to this device's ability to measure very small mass changes on a quartz crystal resonator in real-time. This type of sensor has been extensively utilised for analytical purposes, attributed to the technique's high sensitivity and on-line acquisition aptitude.142,143 Quartz crystal microbalances are generally composed of a quartz disc incorporated with electrodes, and in terms of MIP sensors, they function by measuring the disturbances in frequency resonance resultant of target analyte adsorption/binding based on the Sauerbrey equation.144 Therefore, changes in resonance energy are proportional to interactions between the target analyte and imprinted cavity recognition sites.145
Tokoname et al. used a quartz crystal microbalance to recognise the rod-shaped bacilliform bacteria, where sensor performance was improved by integrating MIP technology with dielectrophoresis. The target bacterial species was precisely printed on the surface of a polypyrrole film, and the corresponding imprinted cavities were created by extracting the target template upon overoxidation. The integration of both technologies allowed real-time and selective detection of bacilli from apple juice samples with a LoD as low as 103 CFU mL−1 within 180 s, exclusive of any pre-treatment. These cavities showed high selectivity and were capable of successfully differentiating competitive rod-shaped bacterial species, including Acinetobacter calcoaceticus, E. coli, and Serratia marcescens, highlighting a simple and rapid bacterial detection method for potential future clinical point-of-care testing.146
For the detection of smaller biomolecules, Wang et al. integrated dual-emission fluorescent MIPs (DE-MIPs) into a facile test strip for the visual detection of dopamine via colorimetric analysis.147 Dopamine sensing is vital for the diagnosis, prevention, and management of neurological disorders including, but not limited to, Parkinson's disease, schizophrenia, and Huntington's disease.148–150 Dopamine selective DE-MIPs were prepared via the combination of two types of quantum dots with red and blue colour emissions using imprinting technology.56,151 The blue quantum dots were implanted into silica nanocores, preserving continuous fluorescent intensity, whilst the red quantum dots were hybridised into the imprinted polymer shell, encouraging dopamine interaction and stimulating fluorescence quenching during dopamine rebinding (Fig. 5a). DE-MIPs were layered onto a filter paper, formulating a dopamine test strip (Fig. 5b). Serum samples of 10 mL demonstrated dopamine detection within 180 s with a low LoD of 100–150 nM, showing sensitive detection to physiological dopamine levels. Thus the authors successfully prepared a facile and efficient strategy for rapid, visual, and on-site detection of physiologically important species available in biofluids.147
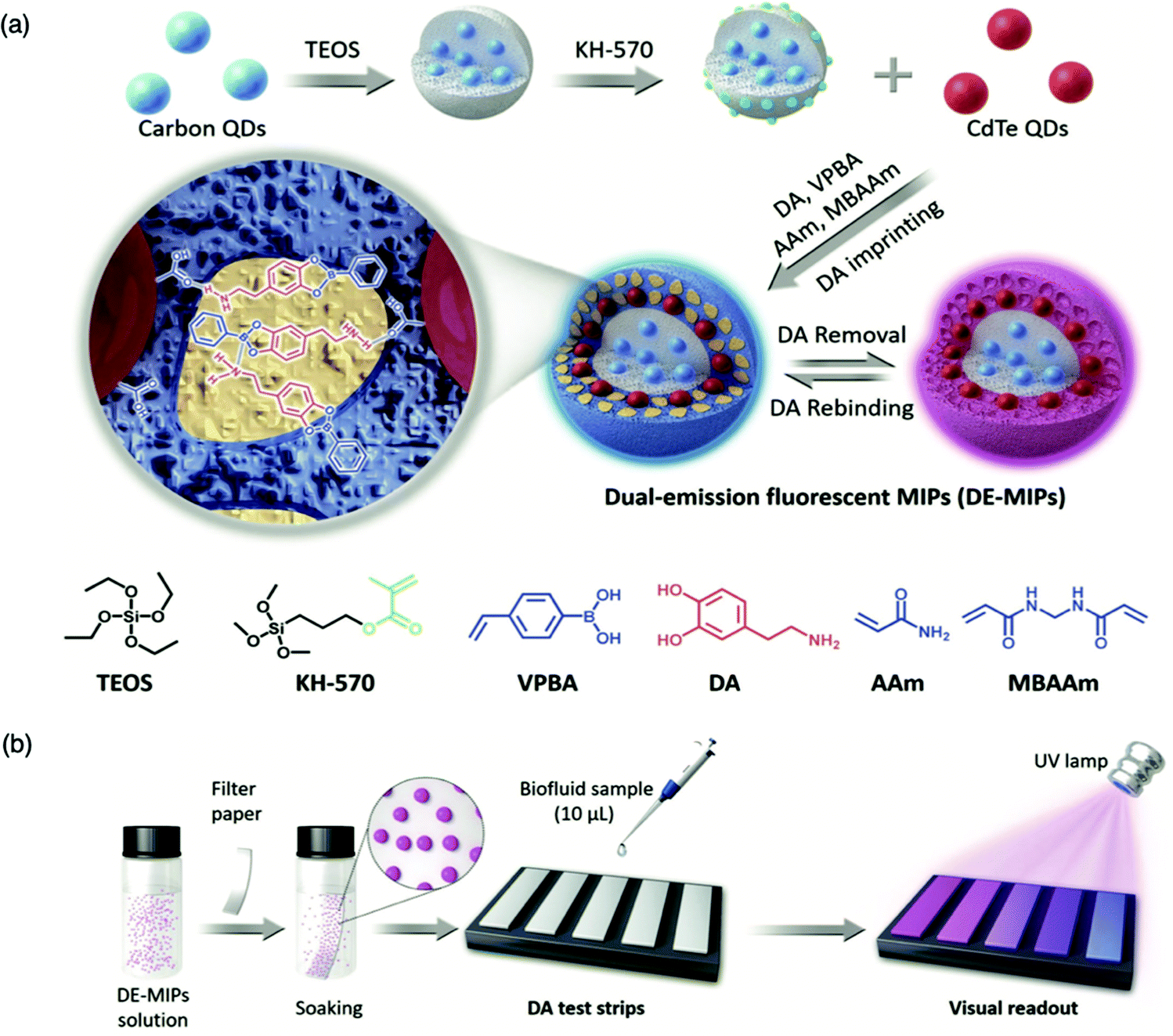 | ||
| Fig. 5 Molecularly imprinted fluorescent test strip for dopamine detection in biofluids. (a) Production of dual-emission fluorescent molecularly imprinted polymer nanoparticles (DE-MIPs) with specific dopamine recognition, and (b) DE-MIPs-coated filter paper as an easy dopamine test strip optical detection. Reproduced with permission from ref. 147, Copyright 2019 John Wiley & Sons. | ||
In addition, surface plasmon resonance, a non-invasive and label-free detection, is another technique that can be employed to develop MIPs into point-of-care biosensors.153 Surface plasmon resonance sensors are simple to function, provide real-time responses with high sensitivity and can be easily adapted for automation.152 With respect to MIP-based plasmon resonance sensors, the binding interaction event that occurs between the analyte and imprinted receptor causes variation of the plasmonic wave at the metal-dielectric interface, producing a detectable signal.153,154
Cenci et al. synthesised a library of MIP nanoparticles (diameter 20–50 nm) targeting the N-terminus of the iron regulating hormone, Hepiciden-25, via precipitation polymerisation.155 Sensitive and selective sensor response to the target hormone in spiked serum samples was detected within 180 s, using biotinylated MIP nanoparticles immobilised to a NeutrAvidin™ surface plasmon resonance sensor chip. Linearity was observed with the logarithm of Hepcidin-25 concentration in the range 7.2–720 pM, and LoD was calculated at 5 pM.157 Recently reported micro-MIP-based surface plasmon resonance, at best, reached nanomolar sensitivities.158–160 However, Cenci et al. highlighted biomarker detection on the picomolar level, credited to utilising completely polymeric MIP nanosized recognition elements integrated into surface plasmon resonance. Ultimately, these results demonstrate a viable approach for MIP integration into surface plasmon resonance for the identification of undetected peptides and proteins that have a fundamental role in pathogenesis but manage to elude existing means of detection.155
Garcia-Cruz et al. presented a generic electrochemical sensor platform, for a range of different targets, based on electro-responsive molecularly imprinted nanoparticles (e-nanoMIPs) for the potential employment of point-of-care diagnostics in the clinic.156 This technology was applied to measure the concentration of targets including, glucose, trypsin, paracetamol, C4-homoserine lactone, and tetrahydrocannabinol, for the detection/monitoring of disease (e.g., diabetes), biological molecules (e.g., digestive enzyme), and drug conjugates for the design of affordable, robust, and disposable sensors.156 The e-nanoMIP fabrication procedure involved controlled polymerisation of the monomer mixture (e.g., methacrylic acid, ethylene glycol dimethacrylate, and trimethylolpropane trimethacrylate) in the presence of a specific target analyte immobilised onto an activated solid phase support (glass beads). A polymerisable ferrocene derivative was added to modify the standard composition of the prepared MIP.156 Electrode modification relied upon well-established carbodiimide coupling of e-nanoMIP deposition onto the surface of a screen-printed gold electrode coated with self-assembled monolayers of alkanethiol.157 Experimentally, all sensors demonstrated an electrochemical response time of 7 min, as shown by differential pulse voltammetry.156,157 All e-nanoMIPs displayed a proportional increase in current response with respect to target analyte concentration (performance summary of each sensor is presented in Table 3), significant of sensitive and selective target detection. In essence, Garcia-Cruz et al. have developed a sensor fabrication process that can be exploited to assemble affordable, disposable, and transportable devices for microvolume sampling of a variety of target analytes present in biological media.156,158
| Target | Glucosea | C4-homoserine lactonea | Paracetamolb | Tetrahydrocannabinolb | Trypsinb |
|---|---|---|---|---|---|
| a Tested in phosphate buffer saline. b Tested in human plasma. | |||||
| Sensitivity | 5.6 mA mM−1 | 42 mA nM−1 | 10.1 mA mM−1 | 7.2 mA mM−1 | 0.25 mA nM−1 |
| LoD | 0.4 mM | 0.1 nM | 82 nM | 50 nM | 0.2 nM |
| Linear range | 0.8–50 mM | 6.2–800 nM | 100–1000 mM | 0.1–1000 mM | 6.5–100 nM |
Hong et al. utilised protein molecularly imprinting to fabricate a biomimetic sensor, exploiting the affinity of the C-reactive protein (CRP), an inflammatory protein, towards its natural ligand (phosphorylcholine).13 The molecularly imprinted protein was synthesised in the presence of O-4-nitropehnylphosphorylcholine, polyethylene glycol 400 dimethacrylate and 2,2′-dimethoxy-2-phenyl-acetophenone, performing as functional monomer, crosslinker and initiator in a 1:4640:3.4 ratio, respectively, facilitating the formation of immuno-like membranes for the development of a microfluidic biochip for the rapid detection of the target protein (CRP). In this work, a point-of-care device for the separation and sensing of CRP from blood serum based on an immune-like polymer membrane (Fig. 6) was developed using well-orientated molecularly imprinted nanocavities.13 The polymer membrane was integrated within a plastic microfluidic chip containing an enclosed interdigitated electrode array (Fig. 6a). The loading of human blood serum samples into the microfluidic biochip and the subsequent capture of the target protein by the protein imprinted recognition sites of the immuno-like membrane (Fig. 6b) was followed by the loading of sodium dodecyl sulphate and CRP release from the immuno-like membrane (Fig. 6c). This was proceeded by the final delivery of sodium dodecyl sulphate with the target protein to the electrodes (Fig. 6d) for electronic sensing based upon the rate of decay of the applied electric signal due to impedance changes. After initial incubation with spiked serum samples, CRP was detectable within 110 s. CRP was identifiable as low as 10 mg L−1. Although this level surpasses the low to high-risk categories recommended for cardiovascular disease detection and is likely to be most beneficial for high-risk categories only, it is apparent from the dynamic response of the decaying signal that the approach of molecularly imprinted CRP selective cavities has the potential to detect lower CRP concentrations and form the basis for an improved clinically applicable biochip for point-of-care-testing.159,160 Ultimately, the proposed approach is an adaptive technological platform, with the potential to facilitate cost-effective mass production for the application of a range of protein biomarkers.13
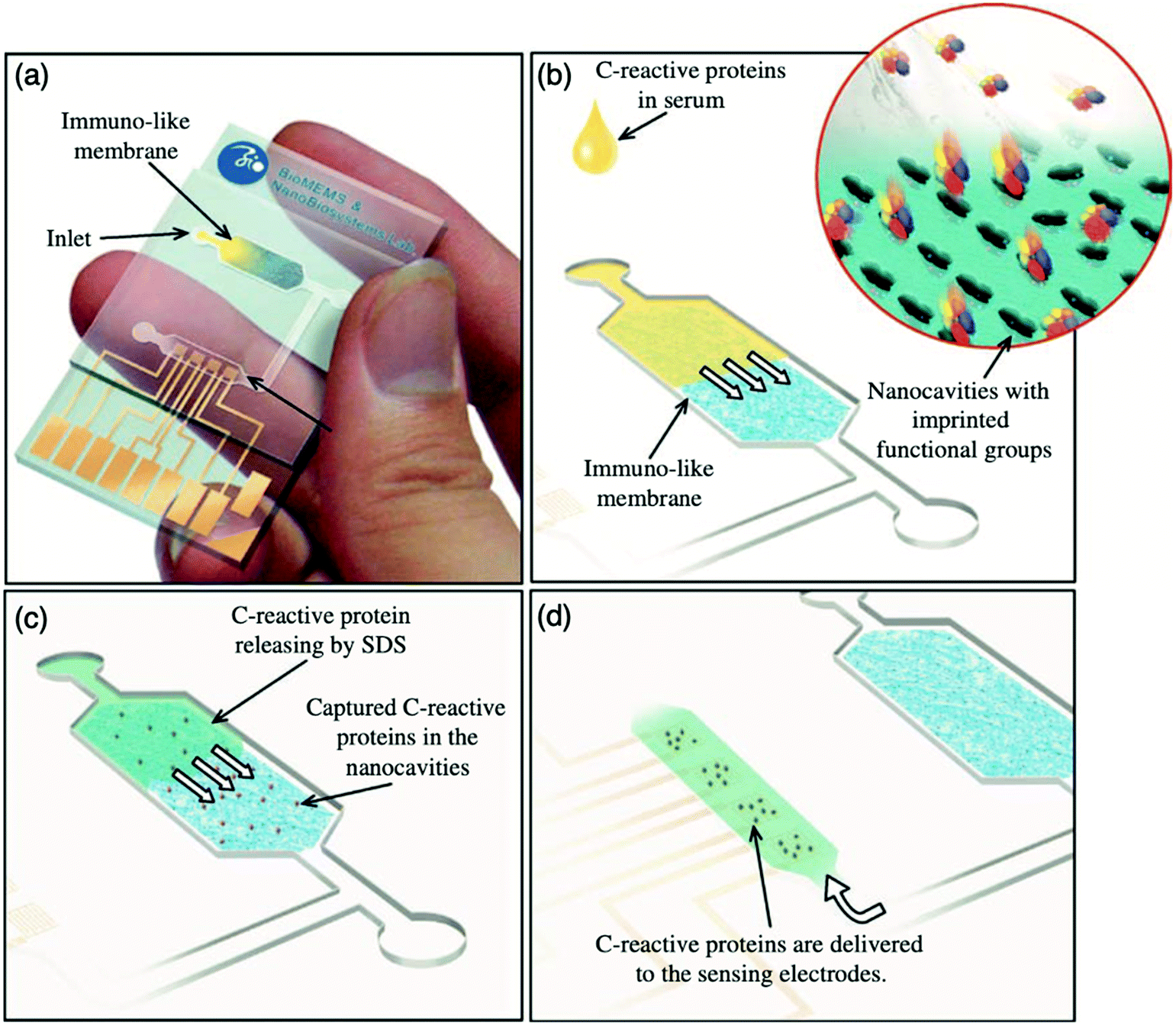 | ||
| Fig. 6 Point-of-care sensing platform based on immuno-like membranes with molecularly aligned nanocavities for CRP detection. (a) Prepared membranes with aligned molecular nano-recognition for point-of-care protein biosensing. Immuno-like membrane in microfluidic biochip; (b) human blood serum sample loading and capturing of CRP; (c) loading of sodium dodecyl sulphate and releasing of CRP from the immuno-like membrane, and delivery of sodium dodecyl sulphate with CRP to the electrodes for electronic sensing. Reproduced with permission from ref. 13, Copyright 2013 Elsevier. | ||
MIP-based biosensor capabilities towards detecting biological analytes within physiological fluids
Physiological media hosts a variety of biomarkers, and the biomarker concentration levels can give a detailed reflection of the status of human health.161 The following section will discuss developments of MIP biosensors in their capability towards detecting biological analytes within a range of different biofluids.Serum
Blood serum, the liquid left after blood clotting, exhibits many of the same solid components as plasma and is a popular biological fluid for disease detection, credited to its abundance of biochemical markers estimated at 250.161 In turn, several recent works have focused on the detection of biomarker analysis within blood serum for the diagnosis of diabetes mellitus, liver cancer, human immunodeficiency virus, breast cancer, hypertryptophanemia, coronary heart disease, and many others.134,162–165 You et al. utilised clinical blood serum samples for the detection of the amyloid-β oligomer, peptides responsible for the amyloid plaques present in Alzheimer brain lesions, via the fabrication of a novel electrochemical biosensor, where molecular imprinting was integrated with an aptamer-based sandwich assay as the sensor recognition unit.166 Amyloid-β oligomer-specific aptamer was assembled onto the surface of the Ag/SiO2 NPs via exploitation of the silver–thiol bond, establishing a Ag/SiO2–aptamer bioconjugate. The molecularly imprinted layer was prepared with amyloid-β oligomer, methacrylic acid, divinylbenzene, and 2,2′-azo-bis-(2,4-dimethyl)valeronitrile performing as target analyte, functional monomer, crosslinker, and initiator, respectively.166 Sensitivity was evaluated by measuring sensor response towards two variations of amyloid-β monomers, amyloid-β oligomers, and amyloid-β fibrils under identical conditions. Both variations of the amyloid-β monomers and amyloid-β fibrils showed a significantly weaker current response compared to that of the amyloid-β oligomers, credited to the specific recognition capabilities of the imprinted cavities installed via MIP and aptamer sandwich integration. Further investigation highlighted that different concentrations of the amyloid-β 1–42 oligomer showed a strong linear correlation ranging between 5 pg mL−1–10 ng mL−1 (R2 = 0.997), where the LoD was calculated at 1.22 pg mL−1.166 Thus, the response of the biosensor correlates to an amyloid-β oligomer concentration range within the detection range found in physiological serum of Alzheimer's disease patients (1.35–12.5 ng mL−1).167The preparation of 12 amyloid-β 1–42 MIP biosensors under identical conditions demonstrated a relative standard deviation (RSD) value of 7.7% for the detection of 1 ng mL−1 of amyloid-β 1–42 oligomer, indicating that the MIP could be reproduced satisfactorily. Stability was monitored by measuring the electrochemical signal of the sensor every week, where ∼91.2% of the original signal intensity was retained after 28 days. Recovery of the amyloid-β 1–42 oligomer from spiked healthy human blood serum samples ranged between 93–107.7% with acceptable RSDs ranging from 2.5 to 9.8%, verifying the applicability of the fabricated MIP biosensor for amyloid-β1–42 oligomer determination in clinical samples for early Alzheimer's disease diagnosis.171
Jaiswal et al. reported a MIP-based electrochemical sensor developed via a layer-by-layer approach, capable of quantitatively differentiating between ultra-trace levels of D- and L-serine in blood serum and cerebrospinal fluid of clinical patients.168 Serine, has a crucial role in the functioning of the central nervous system, and is a biomarker of psychiatric disorders (e.g., schizophrenia).169 Imprinted polymeric films were comprised of acrylamide, copper(II) oxide, 2,2′-bipyridine, and chloroform, acting as functional monomer, catalyst, ligand, and initiator, respectively. Layer-by-layer assembly was achieved by grafting synthesised imprinted polymeric films via spin coating on the surface of pencil graphite electrodes. Jaiswal et al. prepared a highly sensitive biosensor, with respect to other biosensors evaluating the same target analyte, inferred by a 0.24 ng mL−1 and 0.25 ng mL−1 LoD for D-serine (0.83–20.63 ng mL−1 LoD range) and L-serine (0.87–20.45 LoD range), respectively (inclusive of both biological fluids), utilised for sequential analysis of isomeric target analytes using a single electrode.170–175 The authors reported that the artificial recognition component, (i.e., the MIP), presented an IF of 25.61, absent of any regeneration limitations, cross-reactivity, and false positives, highlighting its capability to improve medical diagnostics for the correct management of administered medications and facility admittance.168
Luo et al. developed a magnetic surface molecularly imprinted-resonance light scattering sensor for the rapid and highly sensitive detection of the Japanese encephalitis virus (JEV), a mosquito-transmitted virus that can cause inflammation of the brain.176,177 The surface imprinted polymer was prepared on silicon-coated iron oxide microspheres and polymerisation was performed using aminopropyltriethoxysilane and tetraethyl orthosilicate as functional monomer and crosslinker to immobilise the target virus (JEV) and secure the three-dimensional polymeric network, respectively. The sensitive and selective detection of JEV was initially determined in Britton–Robinson buffer. Under optimised conditions, the LoD was observed at 1.3 pM (3.0%, RSD).176 In comparison to previously reported MIP optical sensors for the detection of JEV, the detection limit (1.3 pM) is clinically acceptable, and sensor response time (20 min) is significantly faster.176–179 The response of the magnetic MIPs towards JEV in the presence of other viruses, including Hepatitis A virus, dimensionally different Rabies virus, and Simian vacuolating virus 40, using an initial virus concentration of 75 ng mL−1, showed that the imprinted JEV could be selectively detected with low nonspecific adsorption when in the presence of virus interferents. The feasibility of the prepared MIP-sensor for the detection of JEV in clinically relevant samples was examined in spiked (10–90 ng mL−1) healthy human serum samples. Sensing performance was ascribed by recovery rates ranging between 98.0–110.2%, suggestive of the designed approach being a promising means to detect the target virus in patient samples.176
To address drawbacks of using antibodies for cancer biomarker recognition, Tawfik et al. utilised optical methods in the form of fluorescent molecularly imprinted conjugated polythiophenes to develop a simple and affordable enzyme-free amplification assay for the detection of α-fetoprotein, a biomarker of liver cancer, undetectable among healthy individuals, within blood serum samples.185 This strategy relied upon dual emission fluorescent molecularly imprinted conjugated polymers (MICPs) fabricated with specific α-fetoprotein and carcinoembryonic antigen affinity and enhanced fluorescence features (Fig. 7). Signal amplification was generated by exploiting two varying conjugated polymers green- and yellow-emitting polythiophenes, where photoluminescence quantum yield was observed at 35% and 55%, respectively. Both imprinted species were synthesised via ultraviolet chemical polymerisation (λ = 365 nm), where conjugated polythiophenes were solubilised in 4-vinylphenylboronic acid in the presence of the target analyte, polyethylene glycol dimethacrylate (crosslinker), and 1-hydroxycyclohexyl phenyl ketone (photo initiator).180 Initial α-fetoprotein sensitivity parameters were examined in phosphate-buffered saline, where a LoD of 2.50 pg mL−1 and 1.20 pg mL−1 for green- and yellow-emitting fluorescent MIPs, respectively, was recorded.180 Comparatively, the obtained LoD values showed improvement with regard to state-of-the-art α-fetoprotein biosensors.181–189
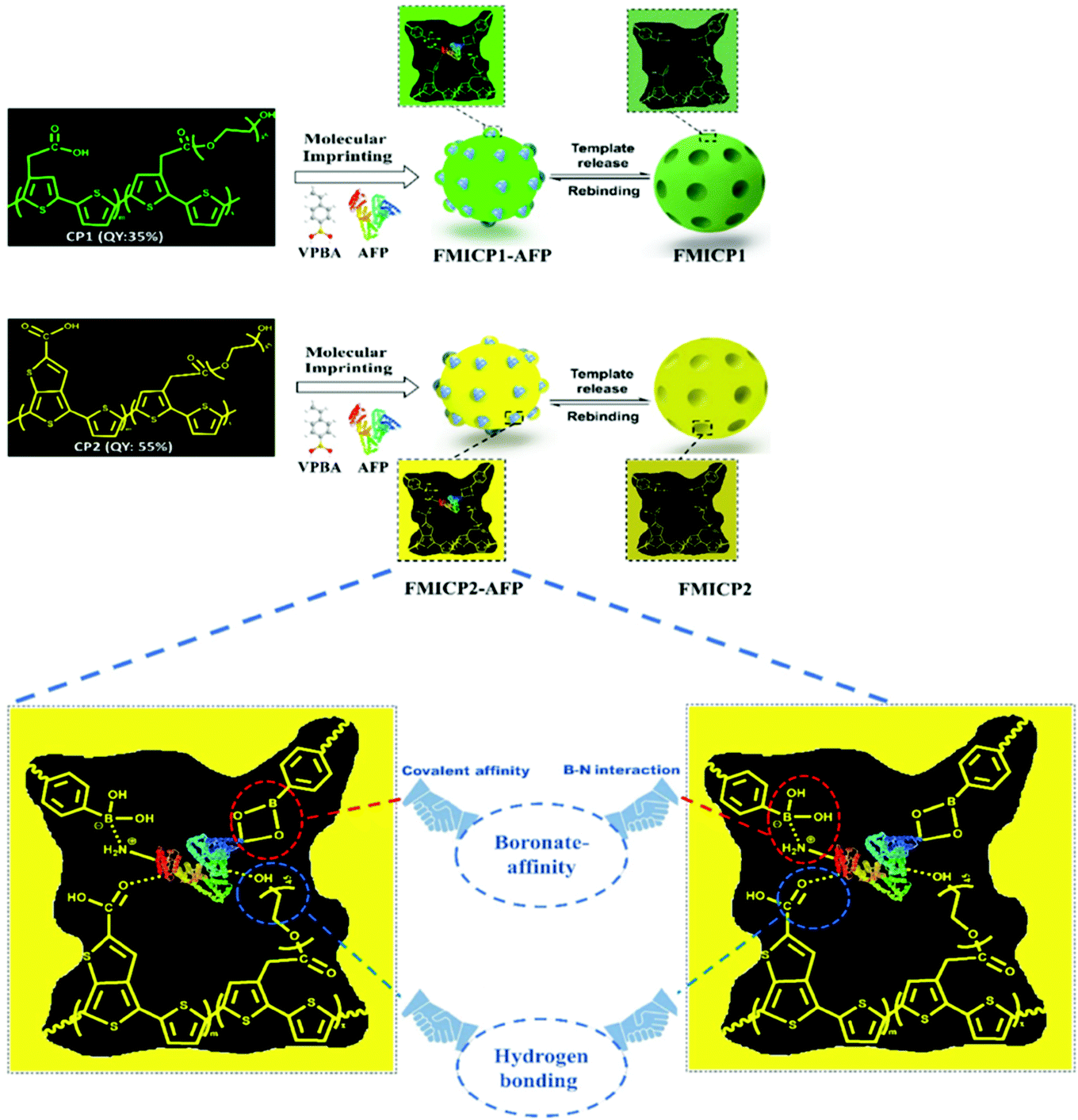 | ||
| Fig. 7 A dual emission non-ionic molecular imprinted conjugated polythiophene-based paper device for α-fetoprotein detection. Mechanistic insight into dual-emission conjugated polymers linked with boronate-affinity molecular imprinting technique. Reproduced with permission from ref. 180, Copyright 2020 Elsevier. | ||
The designed sensor's feasibility was surveyed in multiple human physiological fluids. Traces of α-fetoprotein in human blood serum were analysed, where LoDs for green- and yellow-emitting MICPs were valued at 5.0 pg mL−1 and 2.2 pg mL−1, respectively. For saliva samples, LoDs were calculated at 2.1 pg mL−1 and 1.1 pg mL−1 for green- and yellow-emitting MICPs, respectively. Attained values agreed with early α-fetoprotein detection in clinical samples, where the onset for positive results is 5 ng mL−1. In addition, target recovery from α-fetoprotein spiked blood serum varied between 98.9–110% (1.56–3.65%, RSD) and 98–105% (1.88–3.66%, RSD) for green- and yellow-emitting MICPs, respectively. For saliva samples, recovery ranges included 96.90–110% (1.91–4.21%, RSD) and 97.30–110% (2.43–3.66%, RSD) for green- and yellow-emitting MICPs, respectively.185
As a proof of principle, the designed MIP sensors were used for α-fetoprotein analysis of clinical blood serum provided by liver cancer patients. The quantified α-fetoprotein concentrations from both MIP biosensor assays were concurrent to those probed by enzyme-linked immunosorbent assay (ELISA) analysis, reflected by high linear correlations (R2 = 0.985 and R2 = 0.987, respectively). Therefore, Tawfik et al. has unveiled a pioneering step to advance universally accessible, affordable, portable, point-of-care cancer diagnostics using optical MIP biosensors in physiological samples, which will be of significant benefit in resource-limited clinical settings.180
Saliva
Unlike serum, saliva sampling is non-invasive, with significantly less risk of subsequent infection (from possible needle contamination) and overall cost.190 Furthermore, salivary biomarker detection is becoming a popular method for diagnostics, credited to its accessibility of biological analytes associated with disease, common to those found in human blood.134,137,191–195 Diouf et al. capitalised on this biofluid via the development of a MIP electrochemical sensor for the sensitive detection of glucose in saliva.196 Healthy physiological salivary and blood glucose levels range between 0.05–21.61 mg dL−1 and 70–120 mg dL−1, respectively.196 Despite the very selective, specific and sensitive nature of enzymatic glucose sensors, their applications are hindered by their complexity, lack of stability, limited operational conditions (e.g., temperature, pH, and humidity sensitivity, refer to Table 1), and heavy reliance on blood samples from finger-pricking.197,198 Despite these issues, there are minimal reports focused on non-enzymatic biosensors for the direct detection of glucose; therefore, there is a gap in the current clinical commercial market for the development of a non-invasive and effective glucometer for continuous, accurate, and rapid measurements.199In response to the current commercial limitations, Diouf et al. fabricated a non-enzymatic MIP sensor based on electrochemical polymerisation of the functional monomer acrylamide crosslinked by N,N′-methylene bis-acrylamide in the presence of glucose onto a gold surface printed electrode to determine glucose concentrations in real saliva samples (Fig. 8).196 Electrochemical detection of glucose within a working range of 0.5–50 μg mL−1 was achieved by introducing the working electrode into a known concentration of glucose. A ferricyanide redox probe was utilised to monitor decreasing current in response to increasing glucose concentration, explained by the binding of glucose molecules with that of MIP recognition cavities - impeding electron transport to the redox probe. Differential pulse voltammetry and electrical impedance spectrometry were utilised to determine the LoD, reported at 0.59 μg mL−1 and 1.6 μg mL−1, respectively. Notably, the NIP species showed a negligible change of signal, signifying that observed responses directly resulted from the immobilised glucose within imprinted glucose cavities.196 The selectivity of this glucose MIP sensor was measured using lactose and sucrose, two interfering analytes, naturally coexisting in biological saliva samples, with a similar structure to that of glucose. No significant response was obtained from either interferant. Thus, the proposed glucose MIP sensor showed a remarkable selectivity towards glucose molecules only. Additionally, the reproducibility (3.4%, RSD), repeatability (4.0%, RSD), and stability (retained 85% of its initial response) of the MIP sensor were also investigated, where stability was monitored at regular time intervals over a three-month period.196 The fabricated glucose MIP sensor was applied to saliva samples of six healthy volunteers. Sampling procedures involved mouth rinsing followed by storage at 4 °C. Saliva glucose levels were compared to that of finger-prick blood. Salivary glucose and fasting blood glucose values ranged between 0.22–0.86 mg dL−1 and 94–147 mg dL−1, respectively. Consequently, Diouf et al. have established an inexpensive, simple, and effective sensing platform for non-enzymatic glucose detection, assembling a promising tool for the future evolution of accurate and reliable non-invasive diabetes mellitus diagnosis using MIT. To further validate the obtained results within this study, additional data should be collected from saliva samples of diabetic patients.201
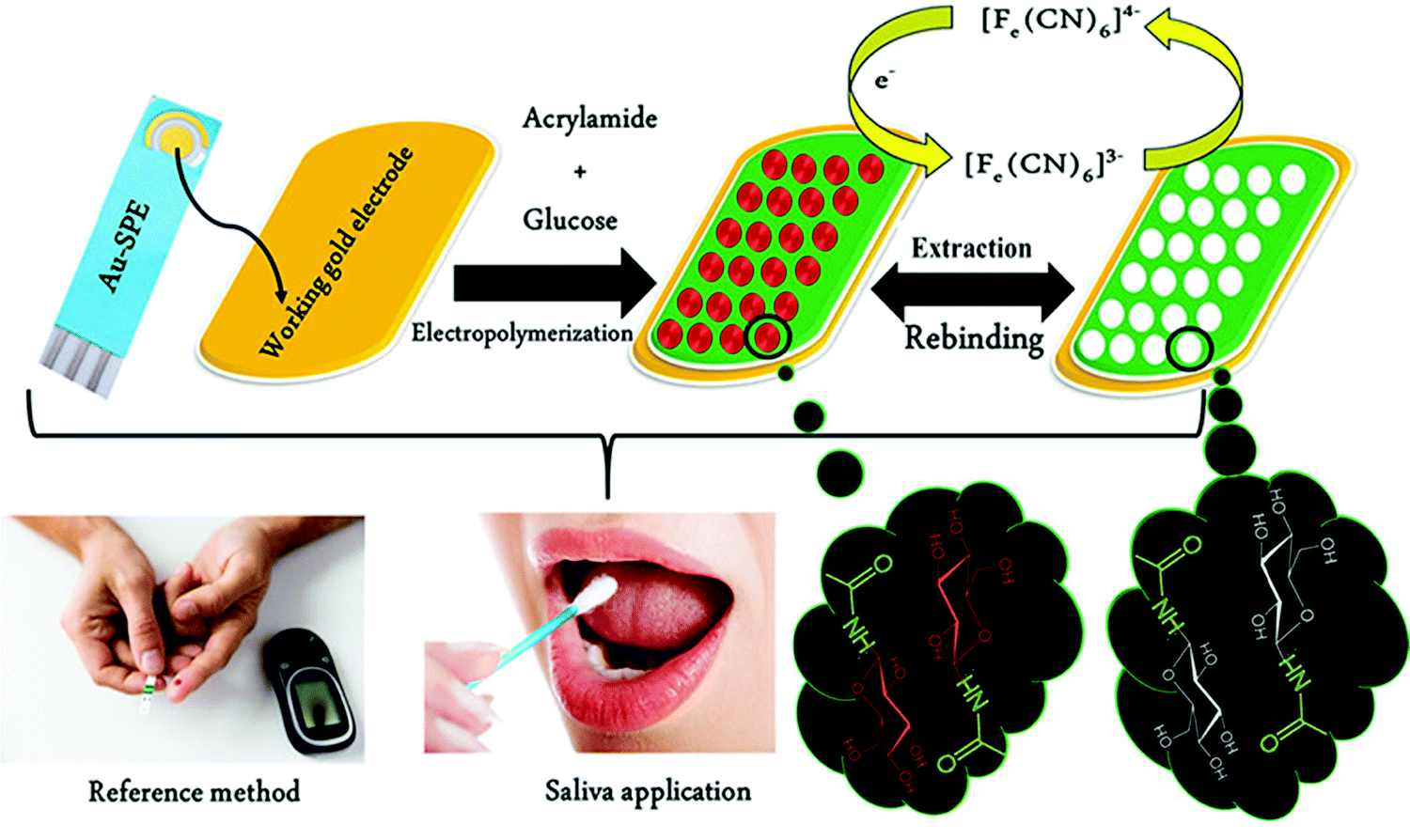 | ||
| Fig. 8 Fabrication of a MIP-based screen-printed gold electrode (Au-SPE) for the diagnosis of diabetes mellitus prepared in the presence of glucose as the template, where the binding sites of molecular interactions have been highlighted. Traditional methods of finger-prick blood sampling have been presented as a comparison to emphasise the ease and pain-free means of saliva sampling. Reproduced with permission from ref. 196, Copyright 2019 Elsevier. | ||
Salivary biomarker analysis was also adopted by Parnianchi et al., who recently developed, for the first time, a highly sensitive and selective electrochemical sensor integrated with a MIP for the detection of bilirubin.200 Bilirubin is a metabolite of the catalytic degradation of heme in haemoglobin and can be utilised as a biomarker of anaemia (low levels) and liver disease (high levels).201–203 Due to the electrochemical applications of multi-walled carbon nanotubes, these structures were used to modify the electrodes prior to o-phenylenediamine (functional monomer) electropolymerisation in the presence and absence of bilirubin for MIP and NIP electrode preparation, respectively.200,204
Preceding biosensor employment for the detection of bilirubin in real human samples, the sensing platform was first characterised using ferricyanide as an electrochemical probe. Analyses highlighted that when the target analyte was removed from the polymer film, there was a significant decrease in electron transfer resistance, credited to the successful imprinting of bilirubin cavities performing as electron transfer channels – reducing resistance. Ultimately, modified MIP electrodes showed a wide linear range of 12.08–91.81 fM with a 7.8 fM LoD.200 Sensor selectivity was investigated via structurally similar analogues to bilirubin, including progesterone, testosterone, dopamine, uric acid, and ascorbic acid. Each structure was examined three times and signified negligible cavity adsorption compared to the bilirubin template, valuing MIP sensor selectivity at 1.05 μA fM−1.200 Furthermore, this MIP electrode signified reasonable operational stability (i.e., it can be used several times), attributed to a minimal 5% signal loss over a period of 10 days, which is in agreement with other reported studies.205–208 Following optimisation testing, the selective detection of bilirubin in human saliva of neonates and adults, was analysed. Samples relating to healthy adult saliva were prepared in different concentrations ranging from dilution factors of 0–1000. MIP sensor performance was examined by the addition of standard bilirubin in the femtomolar range (12.08–91.81 fM), where clinically reasonable responses were observed as identified by an average bilirubin recovery of 97.41% with an RSD of 3.84 (n = 4). However, detection of bilirubin from the samples relating to infants with symptoms of jaundice using the developed MIP sensor showed a decrease in target analyte recovery (95.9% with an RSD of 6.0% (n = 4)) when compared to results obtained using high-performance liquid chromatography (HPLC).200
Cerebrospinal fluid
In contrast to saliva, cerebrospinal fluid sampling is a more invasive technique. Cerebrospinal fluid is an invaluable clinical specimen that offers critical information regarding diseases, infections, and severe conditions affecting the spinal cord and brain.209–211 Ji et al. fabricated a MIP monolith using a micropipette tip, prepared via epitope imprinting, for the highly selective extraction of cholecystokinin neuropeptides (Fig. 9).212 Cholecystokinin neuropeptides are active within the physiological and pathophysiological processes of the central nervous system, contributing to mental health disorders (e.g., schizophrenia) and epilepsy; therefore, the development of accurate analytical techniques within the fluid is vital.213,214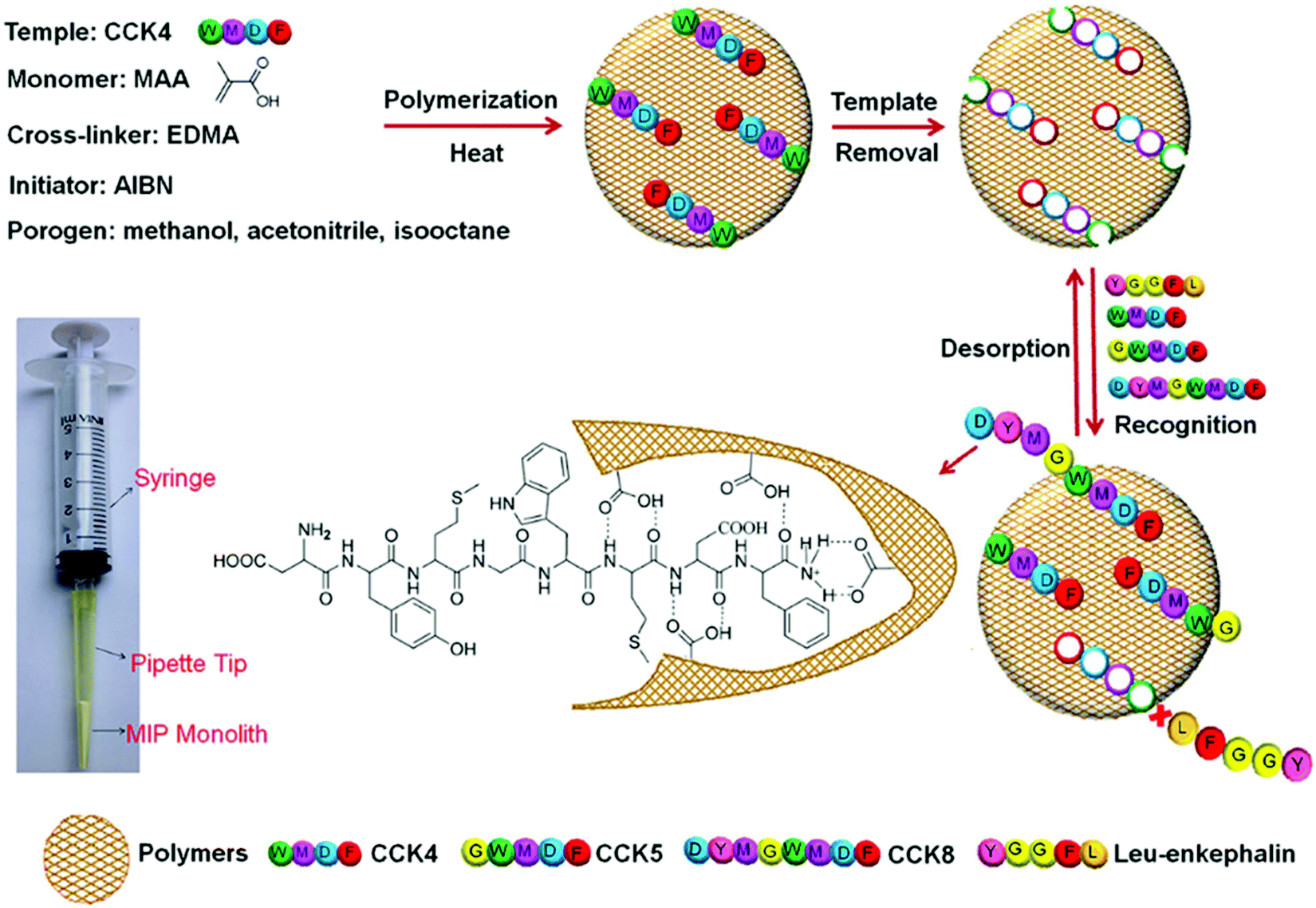 | ||
| Fig. 9 Schematic depiction of the MIP monolith fabrication protocol for the selective recognition of cholecystokinin (CCK) neuropeptides in cerebrospinal fluid. The monomer and template interactions are highlighted. A micropipette tip connected to a syringe is utilised to perform the molecularly imprinted solid-phase microextraction (MI-μ-SPE) for the detection of CCK neuropeptides via the epitope imprinting approach. Reproduced with permission from ref. 212, Copyright 2015 John Wiley & Sons. | ||
The designed MIP monolith exhibited specific recognition capability, high cholecystokinin neuropeptides (CCK) recapture capacity, and excellent reusability. Initially, in situ polymerisation occurred, where methacrylic acid, ethylene glycol dimethacrylate, and 2,2′-azobisisobutyronitrile were applied as functional monomer, crosslinker, and initiator, respectively, in the presence of the template CCK4. Template extraction revealed imprinted cavities that could selectively recognise CCK4 and structurally similar CCKs (i.e., species that shared the same imprinted amino acid sequences), including CCK5 and CCK8. In addition, three oligopeptides, including tetrapeptide, Leu-enkephalin and Met-enkephalin, were introduced to interfere with the specific binding observed for the CCK neuropeptides for sensor selectivity evaluation. Nonetheless, the MIP monolith demonstrated a significant molecular affinity and selectivity enrichment capability for CCK neuropeptides only.212 Sensor applicability was determined by monitoring CCK neuropeptide in clinical human cerebrospinal fluid samples. Formulated calibration curves for CCK4 (R2 = 0.981) and CCK8 (R2 = 0.973) signified LoDs calculated at 1.2 and 5.5 pM, respectively. Linearity and target recoveries (73.9–90.4%) were within clinically acceptable limits. The feasibility of the prepared MIP monolith sensor has shown that with the combination of sample purification and enrichment, it was possible to detect CCK neuropeptides in cerebrospinal fluid at physiological levels.212
Luliński et al. developed a MIP to detect tryptamine within cerebrospinal fluid to monitor cardiovascular pathologies (e.g., myocardial infarction, hypertension). The sensor was fabricated via molecularly imprinted solid-phase extraction (MISPE) and HPLC combined with fluorometric signal technology. The tryptamine-imprinted microscale sorbent (stationary phase), performing as sensor recognition, was synthesised via both bulk and precipitation polymerisation, where 4-vinylbenzoic acid and ethylene glycol dimethacrylate were employed as functional monomer and crosslinker, respectively. Static (sensitivity determination) and dynamic experiments (selectivity determination) revealed an IF of 15.4 and 18.62, respectively. Tryptamine determination in the presence of serotonin and L-tryptophan was validated using a complex matrix of bovine serum albumin yielding the recoveries of tryptamine that ranged between 98.7 and 107.0% (1.1–3.7%, RSD).215 Human cerebrospinal fluid (post-mortem) was then tested to determine the practicality of the MIP sensor in the presence of competitors. First, the elution fraction obtained from pure cerebrospinal fluid was spiked with standards of tyramine (1.31), serotonin (IF = 1.21), and L-tryptophan (IF = 1.16) to identify the presence of eluent interferants. The Qmax of tryptamine (186 ± 19 mmol g−1) is greater than that of the interfering species (15.9 ± 1.6, 39.9 ± 4.0, and 117 ± 12 mmol g−1, respectively), indicative of MIP target adsorption. The reported values are explained by structural analogy to that of tryptamine, as the interferants all share a similar indole ring with an ethylamine aliphatic chain. Nevertheless, the MIP sensor clearly demonstrates tryptamine selectivity irrespective of the present interferants. All signals were achieved in <15 min. Neat human cerebrospinal fluid spiked with the internal standard (5-methoxy-tryptamine) was also tested for extraction processes to quantify the concentration of tryptamine in the eluent. All signals were achieved in <25 min. Ultimately, MISPE techniques demonstrated a very low tryptamine LoD (19.9 nmol L−1) in bovine serum albumin, and tryptamine was also quantified in human cerebrospinal fluid (553 ± 45 nmol L−1 (n = 3)), even in the presence of competitive structural equivalents. Thus, the authors have developed a novel analytical procedure to determine tryptamine in real human samples for a better-magnified insight into cardiovascular-related ailments.215
Sweat
Sweat has a less complicated matrix than other physiological fluids, resulting in the secretion of metabolites in an acidic and electrolyte-rich fluid.216 The minimally invasive nature and easy collection of this fluid due to several accessible sampling sites enables sweat to be used in applications for continuous analysis.217 Recently, Mugo et al. utilised this media to develop a molecularly imprinted cortisol selective biosensor, fabricated via the layer-by-layer assembly for elevated cortisol detection, implicated in various stress-related conditions (e.g., post-traumatic stress disorder and bipolar disorder).218,219 As such, cortisol can be a valuable biomarker for diagnosing physiological conditions related to anxiety, depression, and mental health.220 The sensor layers comprised a stretchable polydimethylsiloxane base with carbon nanotube-cellulose nanocrystals (CNC/CNT) conductive nanoporous nanofilms. The MIP was fabricated via surface imprinting using glycidyl methacrylate (functional monomer) and ethylene glycol dimethacrylate (crosslinker) where is was deposited onto the CNC/CNT, forming the cortisol biomimetic receptor.218Sensitivity of the MIP was shown by a 180 s sensor response, with a 2.0 ± 0.4 ng mL−1 LoD (2.6%, RSD) to cortisol in phosphate buffer, attributed to the inherent receptor-specific cavities engrained in the MIP film.221 Increasing cortisol concentrations yielded a linear calibration (R2 = 0.92) with a high calibration sensitivity four orders of magnitude higher than the NIP, acting as a control. Moreover, the MIP cortisol sensor dynamic range was determined to be 6–60 ng mL−1, falling within the physiological cortisol range (8–50 ng mL−1) in human sweat, thus confirming the feasibility of the prepared sensor for cortisol detection in biological sweat.217 Although former reported cortisol biometric sensors have displayed lower LoD values; these sensors rely on antibodies.222–224 Specificity of the imprinted cortisol sensor was determined via analysis of analogous interfering chemical species, similar to those already present in human sweat. The interfering species tested included epinephrine, methoxyxprogestone, β-estradiol and glucose. The MIP sensor exhibited high specificity towards cortisol with a measurable difference in voltammetric readings, compared to the introduction of the other interfering species.218 Biosensor feasibility was evaluated for ex situ analysis of sweat samples. Following triplicate measurements, cortisol concentration was determined to be 33 ± 2 ng mL−1 (5.4%, RSD), which falls within the normal clinical range. The MIP sensor's cortisol content was also validated by HPLC, resulting in a very similar concentration of 29 ± 0.5 ng mL−1 (5%, RSD).218
In a recent study, Tang et al. developed a touch-based MIP biosensor (Fig. 10a) for selective cortisol detection through natural perspiration. Instead of sweat sampling via stimulative methods (e.g., exercise), this sensor was designed to measure fingertip sweat cortisol through touch via highly selective binding to the cortisol-imprinted electropolymerised polypyrrole coating, where Prussian blue was embedded as the redox probe. The high density of eccrine sweat glands present in the fingers can produce high sweat volumes, which can subsequently be collected by the highly porous, permeable, and sweat absorbing polyvinyl alcohol hydrogel. Thus, this MIP-based fingertip cortisol biosensor has generated a natural and practical sweat sampling technique, providing a stressless, label-free, low-cost point-of-care testing platform for mental health management and monitoring, exemplifying a potential step-change in the disciplines of wearable biosensor devices and personalised healthcare.225
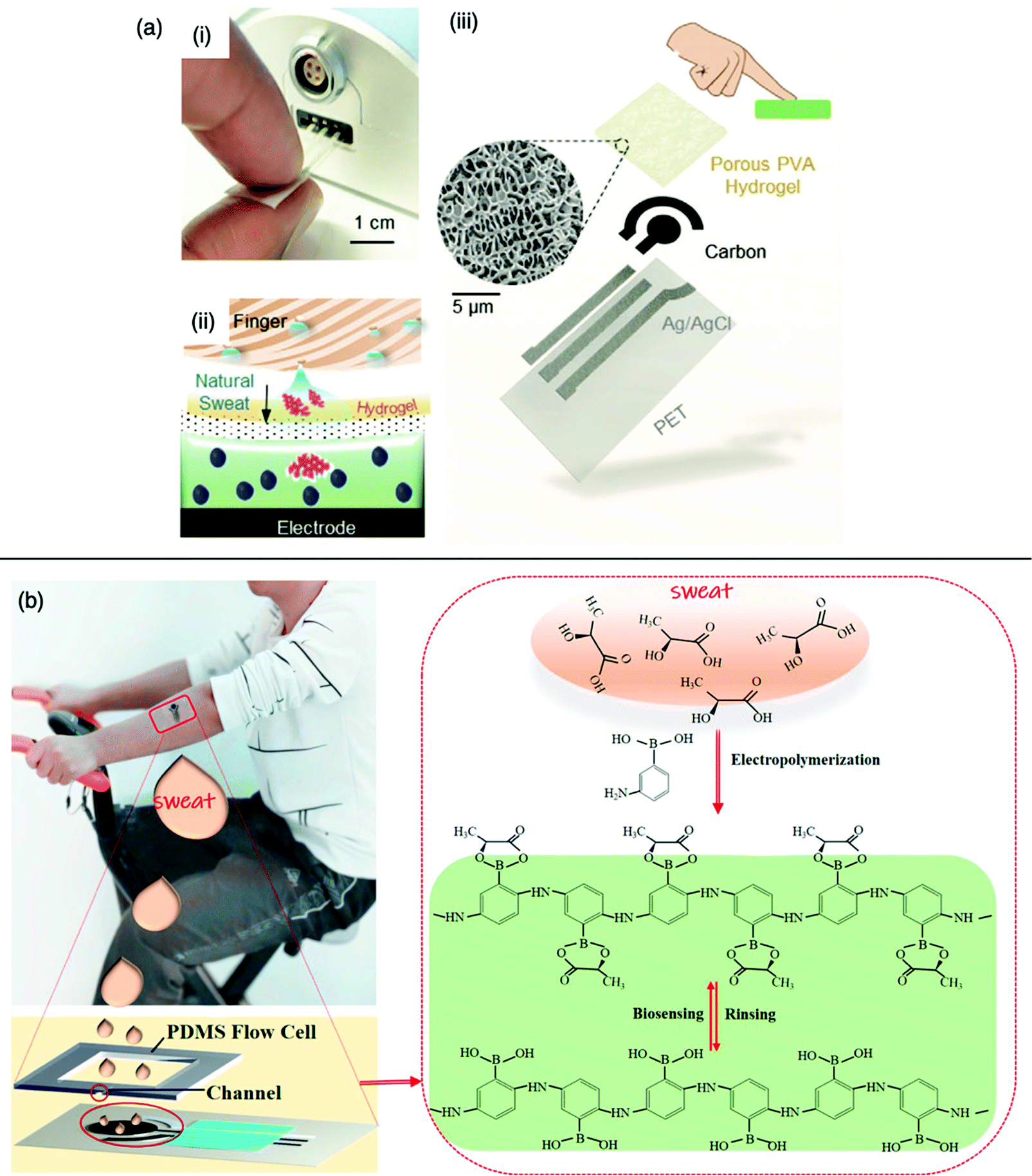 | ||
| Fig. 10 Molecularly imprinted sweat sensors for healthcare monitoring. (a) Cortisol-imprinted electropolymerised polypyrrole-based sensor for the rapid and stressless detection of cortisol (i) The touch-based cortisol biosensor demonstrating the single-touch application; (ii) sensing mechanism illustration describing the cortisol accumulation from finger sweat diffusing through the hydrogel onto the MIP electrode; and (iii) a structural depiction of the sensor with cryogenic scanning electron microscopy image of the porous hydrogel. Reproduced with permission from ref. 225, Copyright 2021 John Wiley & Sons. (b) Molecularly imprinted silver (Ag) nanowires for the detection of lactate. Application of the designed screen-printed three-electrode biosensor chip to a human volunteer for the feasible determination of sweat sensing during exercise exertion, where the binding principle of the MIP biosensor fabrication and sensing process is highlighted. Reproduced with permission from ref. 226, Copyright 2020 Elsevier. | ||
Zhang et al. developed a wearable electrochemical biosensor by incorporating silver nanowires and a MIP electropolymerised in the presence of 3-aminophenulboronic acid (functional monomer) on a screen-printed electrode for the non-invasive monitoring of lactate in human sweat.226 During exercise, sweat lactate concentrations can ascend to 25 mM; however, a severe imbalance can lead to anaerobic metabolism (e.g., pulmonary embolism or haemorrhagic shock).227,228 The MIP sensor exhibited high sensitivity and specificity for the detection of lactate in phosphate buffer saline from 10−6–10−1 M, significant of a 22 μM LoD. Furthermore, the sensors had high stability and reproducibility with a sensitivity recovery of 99.8% ± 1.7% seven months after being stored in the dark at room temperature. Glucose, urea, pyruvic acid, uric acid, sodium chloride, calcium chloride and ammonium hydroxide were among the many substances utilised to measure the sensor's selectivity, primarily due to their structural resemblance to the metabolic substances found in human sweat. Generally, the sensor response showed good selectivity towards the target and negligible response to the interferants. Urea, pyruvic acid and uric acid give the highest current changes around 3 × 10−7 A, which is only 1.4% of the specific response for lactate (14 mM) binding (2.2 × 10−5 A).226 Epidermal lactate measurements were performed on six healthy volunteers. A thin film of polydimethylsiloxane with a sweat cell was utilised for the upwards attachment of the electrode biosensor to the volunteer's skin, enabling sweat overflow after cell saturation (Fig. 10b). Differential pulse voltammetry responses were recorded every five minutes for a period of 30 minutes during exercise. After 10 minutes of exercise, skin moisture became evident, and the differential pulse voltammetry current began to show a reduction, resulting in sweat lactate build-up. After 30 minutes of exercise, a sweat lactate concentration of 14 mM was measured. The same MIP silver nanowires biosensor was then applied to the remainder of the volunteers, producing parallel current responses validating the MIP device reconcilability. Ultimately, the described epidermal biosensor showed a higher sensitivity for lactate concentrations through an easily accessible medium, as opposed to many other lactate biosensors, offering on-body monitoring of perspiration lactate for real-time monitoring of human health conditions.231
Urine
Urine can also provide real-time monitoring for biomarker studies, as highlighted by multiple reports based on the construction of MIP-based biosensors for urine analysis.138,211,229,230 Zhang et al. produced a MIP-based electrochemical biosensor supported by a dual-signal technique to evaluate human serum albumin in real urine samples.231 Urine albumin excretion, a bulky negatively charged protein, can indicate early-stage chronic nephritis, diabetes, and endothelial dysfunction.232 Sensor fabrication was achieved by modifying a glassy electrode substrate with gold nanoparticles and polythionine-methylene blue, displaying a MIP film synthesised via electropolymerisation using o-phenylenediamine and hydroquinone as functional monomers. Sensitivity was observed within the linear range of 10−10–10−4 g L−1 (R2 = 0.995) by simultaneously decreasing substrate and solution probe peak currents, indicative of human serum albumin occupied imprinted cavities (3 × 10−11 g L−1, LoD).231 Notably, the designed sensor demonstrated lower LoD values when compared with other published MIP-sensors specific for human serum albumin detection.210,229,231,233–236 The biosensor exhibited reasonable sensor selectivity when tested with a range of competitive substances, including glycine, glutamate, cysteine, tryptophan, histidine, dopamine, ascorbic acid, haemoglobin, and bovine serum albumin. The biosensor also demonstrated reproducibility (4.4%, RSD (n = 5)) and stability at 95.3% of initial signal retention. The MIP sensor applicability was evaluated by measuring target protein concentrations from urine samples of an unwell patient and a healthy volunteer, where serum albumin recovery varied between 90–104% (3.0–3.5%, RSD) and 95–105% (2.2–3.9%, RSD), respectively. These values concur with the immunoturbidimetry assay results from the hospital record, suggesting that the proposed biosensor proposed promising results for clinical assays.231Martins et al., like Tawfik et al., developed a paper-based biosensor, encouraging improved affordability, sustainability, and reliability of support materials for the design of point-of-care diagnostic devices. A label-free detection method involving direct MIP assembly on a paper platform, integrated with a conductive carbon ink for suitable electrochemical transduction, was incentivised for the recognition of 3-nitrotyrosine in urine. 3-Nitrotyrosine is an important marker for oxidative stress, responsible for neurodegenerative disorders (e.g., Alzheimer's and Parkinson's disease). The MIP film was deposited on the surface of the carbon-coated electrode through bulk polymerisation, followed by electropolymerisation using phenol as a functional monomer (Fig. 11).237 3-Nitrotyrosine concentrations were monitored via the immersion of the MIP-based device in spiked human urine samples. A linear correlation for both the MIP and NIP (control) species was observed for 3-nitrotyrosine concentrations ranging between 5 μM–1 mM; however, the MIP sensor demonstrated greater linear sensitivity (R2 = 0.994), reproducibility (smaller error bars) and an LoD of 22.3 nM (1–2%, RSD (n = 3)).237 In comparison to previously reported works involving MIPs coupled with electrochemical sensing that were amplified using nanomaterials, this reported LoD is clinically viable.242
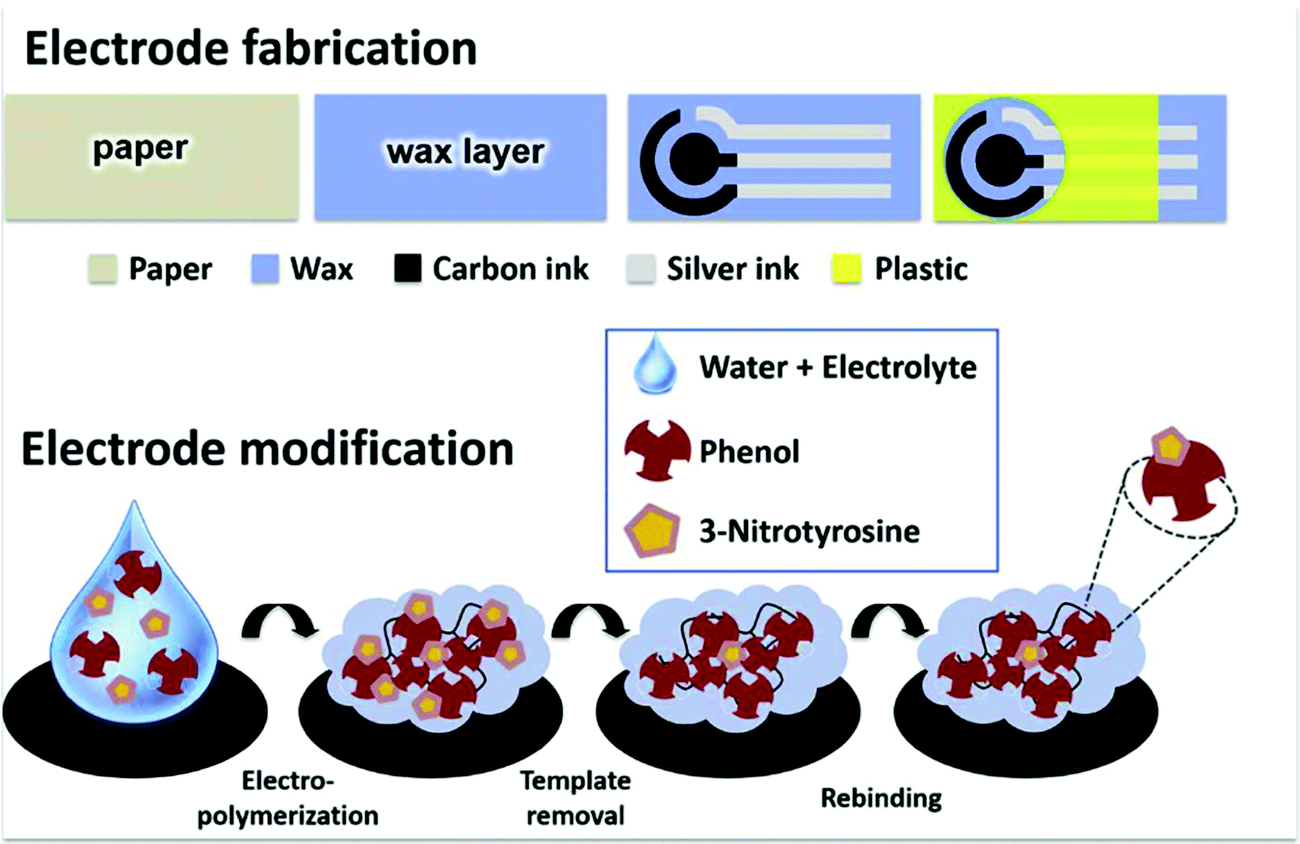 | ||
| Fig. 11 Illustrative depiction of a paper-based electrode. Electrode modification occurs via molecular imprinting of the 3-nitrotyrosine template, formulating a sensor film receptive of the template molecule. Reproduced with permission from ref. 237, Copyright 2020 Elsevier. | ||
Nasopharyngeal fluid
The COVID-19 pandemic has highlighted the importance of point-of-care testing using safely accessible physiological media, such as nasopharyngeal fluid. Although remarkable progress has already been achieved, there is still great demand for simple and rapid diagnostic tools for the early detection of the coronavirus disease. Molecular assays, i.e., real-time polymerase chain reaction (RT-PCR), have become the gold standard for accurate severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) detection.238 Unfortunately, due to the time-consuming analysis, instrumentation costs, and the need for medically trained personnel, the use of RT-PCR can be limited.239–241 Consequently, there is still an urgent need for more affordable, portable and sensitive devices for COVID-19 diagnostics.Commercially available point-of-care diagnostic tools for the detection of the SARS-CoV-2-specific antigens, such as the qualitative lateral flow immunochromatographic assays, utilise the antibody-based detection principle, producing results within 15–30 min.242 Alternatively, point-of-care diagnostic tools based on electrochemical sensing platforms have also been reported.243,244 Nevertheless, current forms of coronavirus detecting systems suffer from special storage systems to preserve their shelf life due to the environmental sensitivity of the biological materials.245
Ayankojo and co-authors have recently developed a MIP-based electrochemical sensor for the quantitative detection of the SARS-CoV-2 spike protein subunit S1 (ncovS1) found in the nasopharyngeal fluid of coronavirus patients for the rapid detection of coronavirus disease.245 The authors exploited the covalent interaction between 1,2-diols of the highly glycosylated protein and the boronic acid group of 3-aminophenyl-boronic acid (functional monomer). A disposable thin-film gold electrode chip was modified with a MIP film with selectivity toward the ncovS1, acting as the recognition element (Fig. 12a and b).245
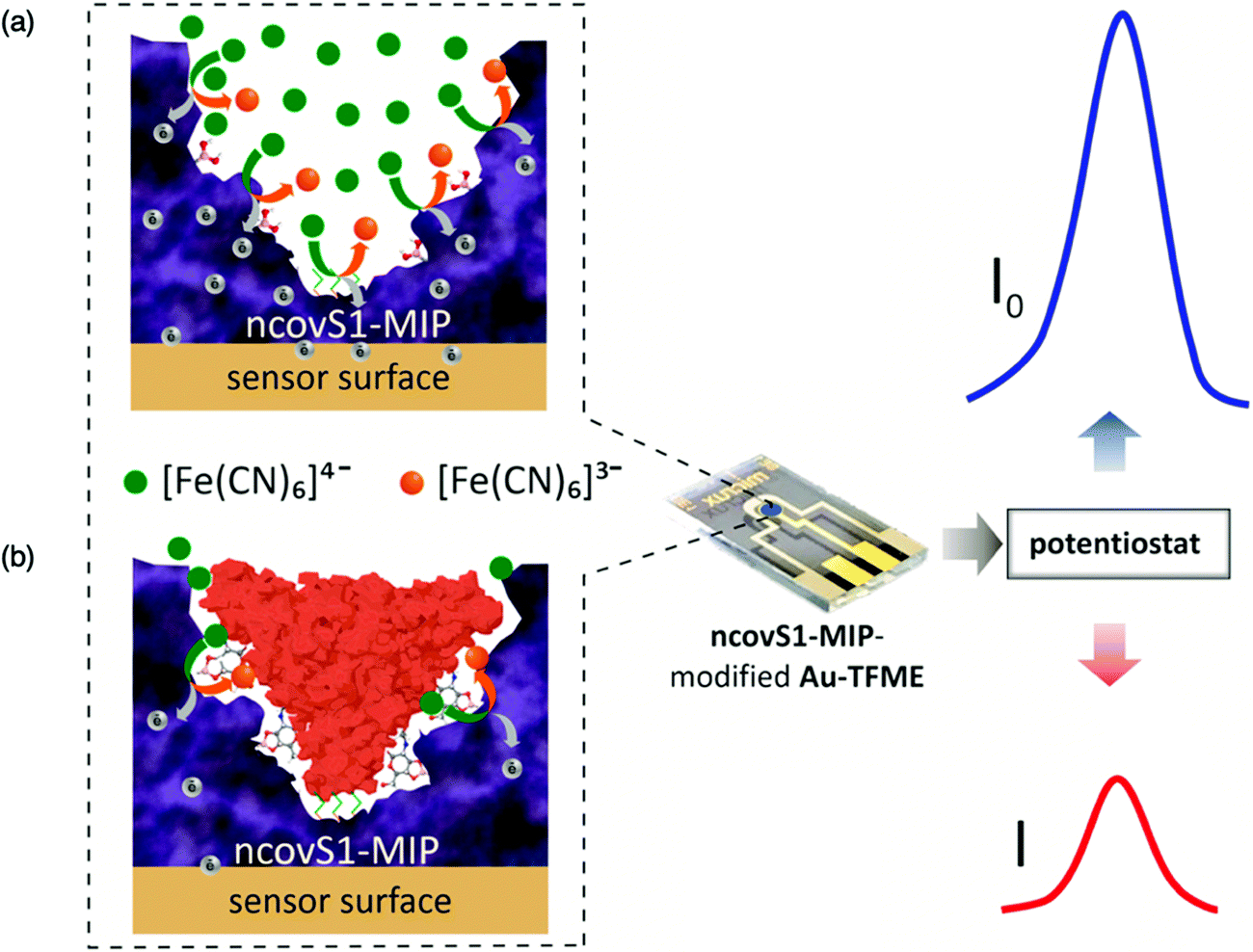 | ||
| Fig. 12 The operating principles of the developed coronavirus detecting device for the sensitive and selective detection of ncovS1 and ncovNP, respectively. (a) Redox probe readily carrying the charge through the ncovS1-MIP producing current (I0); (b) the rebound ncovS1 blocks pathways for the redox probe to carry the charge through ncovS1-MIP, leading to a concentration-dependent contraction in the recorded current (I). Reproduced with permission from ref. 245, Copyright 2022 Elsevier. | ||
Clinical samples consisted of nasopharyngeal specimens of three negative and five positive coronavirus patients in sample preservation solution, where their coronavirus status was previously confirmed via RT-PCR. The fabricated sensor displayed a reaction and measurement time of 15 and 5 min, respectively. Device sensitivity towards ncovS1 detection in nasopharyngeal samples showed a LoD of 4.8 pg mL−1 (linear range of 0–30 pg mL−1).245 When compared to other electrochemical sensor platforms for SARS-CoV-2 detection reported in the literature, the ncovS1 sensor presented a clinically reasonable performance with improved rapid results output.244,246–249
The selectivity of the sensor was determined by spiking negative coronavirus samples with varying concentrations of either ncovS1, SARS-CoV-2 nucleo-protein (ncovNP) or their mixture. The responses induced on the sensor by the increasing concentration of ncovNP were below the LoD, indicating no recognition for ncovNP, whereas increasing responses above the LoD were seen towards ncovS1. Furthermore, the responses induced by the mixture of both proteins in a 1:10 ratio are comparable to that from ncovS1, thus, indicating that the presence of ncovNP in the sample does not interfere, to any significant extent, with the sensor-specific recognition of ncovS1 thereby enabling its accurate analysis.245
The proposed strategy has established its suitability as a potential diagnostic tool for clinical assessment of SARS-CoV-2, demonstrating a substantial quantitative advantage apropos to the LoD over commercially available lateral-flow immunochromatography-based SARS-CoV-2 antigen tests through easily obtainable nasopharyngeal sampling.
Similarly, Raziq et al. report a MIP-based electrochemical sensor for the rapid detection of the SARS-CoV-2 nucleocapsid protein (ncovNP). This device is comprised of a disposable sensor chip in the form of a gold thin-film electrode (Au-TFE) interfaced with a MIP selective for ncovNP connected to a portable potentiostat, measuring the reduction in charge transfer intensity carried by the hexacyanoferrate redox probe via the thin ncovNP-MIP film to the Au-TFE.250 Fabrication of the sensor consisted of modifying an Au-TFE with a ncovNP-MIP film via electrochemical surface imprinting, utilising poly-m-phenylenediamine as functional monomer – rationalised through computational modelling.251 The performance of the resulting ncovNP sensor was initially assessed in lysis buffer, signifying a linear response up to 111 fM with an LoD of 15 fM. The rebinding time of ncovNP at the sensor's surface was also optimised to 15 min incubation in ncovNP-containing samples, as demonstrated by sensor saturation and response equilibrium.250
Sensor selectivity was assessed by evaluating its ability to distinguish between ncovNP and interfering proteins such as a subunit of SARS-Cov-2 spike protein, S1 (75 kDa, pI 6.0); hepatitis C virus surface viral antigen, E2 HCV (47 kDa, pI 8.2); Cluster of Differentiation 48 protein, CD48 (22 kDa, pI 9.3); and bovine serum albumin, BSA (66 kDa, pI 4.7). Target protein selection was established by protein size, isoelectric point molecular weight, and potential to be found in real biological samples. Nevertheless, the results highlighted that the response of the ncovNP sensor was greater towards the target protein as compared to the responses towards the interfering proteins.255
The clinical diagnostic feasibility of the ncovNP sensor was studied by analysing the nasopharyngeal swab specimens of four COVID-19 negative and four COVID-19 positive patients. The presence or absence of the viral infection in the clinical samples was confirmed with RT-PCR method. Initially, the sensor was calibrated using COVID-19 negative samples spiked with known concentrations of ncovNP. The sensor showed a pseudo-linear response versus ncovNP concentration in the range of 0.22–333 fM with a LoD of 27 fM. In addition, the sensor demonstrated appreciable selectivity towards ncovNP, since its response was almost insensitive to the addition of S1 in the COVID-19 negative sample but raised immediately after ncovNP was spiked. These results indicate the sensor's promising capability to respond towards ncovNP without significant disturbance towards the accurate determination of COVID-19 positive samples, where other proteins of SARS-CoV-2 are presented.250 Finally, sensor stability was inspected via the preparation of 12 sensors, which were tested on a weekly basis in the lysis buffer diluted COVID-19 negative samples spiked with ncovNP at a concentration of 66.6 fM. The results confirmed that the response of the as-prepared sensors remained unchanged for up to 9 weeks of storage, revealing excellent long-term stability.250 The presented strategy presents a new route for the development of express COVID-19 diagnostic tools, relying on an entirely different approach as compared to the currently available SARS-CoV-2 antigen tests. Thus, this method could symbolise a valuable alternative to a portable diagnostic platform for the rapid screening of COVID-19.250
Tears
Takeuchi et al. successfully demonstrated the use of tears as a physiological database for target biomarkers, enabling the predictive determination of cancer aggression at the onset. The developed molecular imprinting-based dynamic moulding approach, produced antibody-conjugated signalling nanocavities capable of cancer-related small extracellular vesicle (sEVs) recognition.252 The current status quo of in vivo sEV detection relies upon the collection of blood samples, micro-ribonucleic acids, and proteins embedded inside and/or outside sEVs.253 The analysis proceeds via RT-PCR, ELISA, Western blotting, and mass spectroscopy.254 These analytical methods usually involve time-consuming and tedious pre-treatments, such as ultracentrifugation and/or size-exclusion chromatography.252However, tears have been reported to contain sEVs and can be collected easily in a non-invasive manner using the Schirmer tear test strip by placing a filter paper inside the lower lid of the eye for tear extraction (Fig. 13).255,256 Tears provide a means of convenient and self-collecting sampling at the point-of-care. Thus, Takeuchi et al. fabricated a non-invasive, rapid, and sensitive platform for sensing intact sEVs within the tears sampled from breast cancer patients. The synthesis of sEV-binding nanocavities began with the conjugation of silica nanoparticles to hexahistidine peptide chains and free thiol groups as dynamic moulds. Thiol groups were then coupled with methacrylic acid (functional monomer) via disulphide exchange reaction with 2-(2-pyridyl)dithioethyl methacrylamide to yield methacrylamide-coupled hexahistidine-tagged silica nanoparticles, enabling copolymerisation with the polymer matrix.252 Subsequently, surface-initiated atom transfer radical polymerisation was performed to form a layer of 2-methacryloyloxyethyl phosphorylcholine-based biocompatible polymer matrix, whereby the formed coating was intended to minimise non-specific binding to the substrate.257
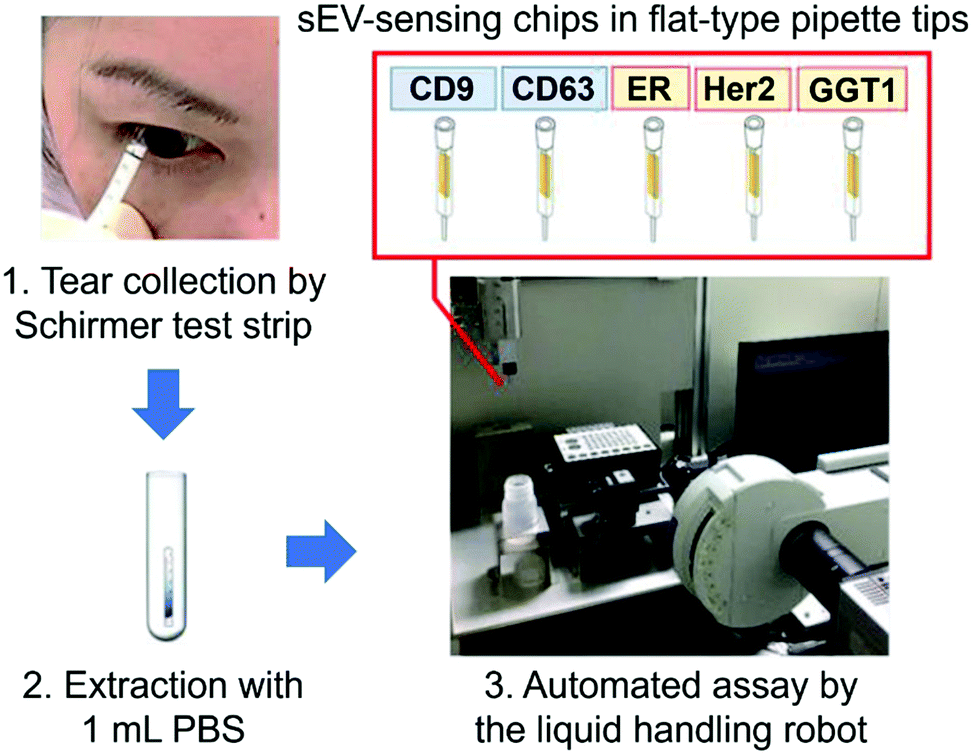 | ||
| Fig. 13 Detection of breast cancer-related small extracellular vesicles (sEV) available in tear samples using the Schirmer tear test strip. Reproduced with permission from ref. 252, Copyright 2020 ACS Publications. | ||
Tears collected from five breast cancer patients and five healthy volunteers were used to assess the efficiency of the sEV sensor chip towards cancer detection. Extracted tear sEVs were analysed exclusive of any pre-treatment, ensuring that those analysed were intact. The LoD was calculated at 1.2 × 10−17 M (linear range of 0–1.0 × 10−16 M), highlighting the comparability of the device in question to previously reported pre-treatment-free methods.258–263 The authors attributed the sensor's success to the highly dense sEV-sensing nanocavities, antibody orientation, and preferable round shape for small extracellular vesicle binding, enabling small extracellular vesicles to form multipoint interactions with two or more antibodies (anti-CD9).252
The developed sensor exhibited selectivity when successfully differentiating between healthy donors and breast cancer patients, as well as between samples collected before and after total mastectomy. Their nano-processing strategy can be easily repurposed for the specific detection of other types of cancer by changing the conjugated antibodies, thereby facilitating the establishment of liquid biopsy for early cancer diagnosis. Sensor development was further enhanced via the construction of a differential antibody array for breast cancer-related sEV detection. This involved the conjugation of five antibodies: anti-CD9, anti-CD63, anti-oestrogen receptor, antihuman epidermal growth factor receptor 2, and anti-GGT1 on each small extracellular vesicle-sensing chip. Moreover, the sensor showed an analysis time of 5 min, significantly less than that of ELISA-based strategies.257
Here we have explored the use of molecular imprinting technologies as the sensor components in diagnostic tools for the detection of analytes in a wide range of biofluids for the detection of a variety of biomarkers utilised to diagnose disease. The inherent and versatile ability of MIPs to identify pathological biomarkers, ranging from small molecules to whole-cell organisms and viruses was further explored through their ability to detect disease-specific biomarkers in biofluids such as serum, saliva, cerebrospinal fluid, sweat, urine, nasopharyngeal fluid, and tears, signifying MIPs’ potential role in the future of precision diagnostics. Recent progress in the field of MIP-biosensing exemplifies their clinically relevant sensitivity and specificity that can be attained by integrating molecular imprinted species with well-established nanomaterials and “intelligent” interfaces capable of manipulating and extracting analytes of interest from complex biological media. These cost-effective, highly stable, long shelf-life, artificial sensing elements can provide new avenues for developing non-invasive biosensing platforms for the rapid, accurate, and quantifiable detection of health-relevant biomarkers at point-of-care. A highlighted selection of the MIP-based biosensors targeting biofluid-specific biomarkers and their corresponding detectable diseases discussed in this review, is summarised in Table 4.
| Biological analyte | Biological media | Disease detection | Sensor | Linear ranges | Limit of detection | Ref. |
|---|---|---|---|---|---|---|
| Cerebrospinal fluid (CSF), matrix-assisted laser desorption/ionisation-time of flight mass spectroscopy (MALDI-ToF MS), organic electrochemical transistors (OECTs), lossy mode resonance (LMR), surface plasmon resonance (SPR), human immunodeficiency virus (HIV), human serum albumin (HSA), quartz crystal microbalance (QCM), solid-phase extraction (SPE), field-effect transistor (FET), and reflectometric interferometric spectroscopy (RIS). | ||||||
| Amyloid-β | Serum | Alzheimer's disease | Voltammetric | 5 pg mL−1–10 ng mL−1 | 1.22 pg mL−1 | 166 |
| D-/L-Aspartic acid | CSF | Neurological | Voltammetric | 1.73–1.79 ng mL−1 | 1.79 ng mL−1 | 136 |
| Bilirubin | Saliva | Liver function | Voltammetric and impedimetric | 12.08–91.81 fM | 7.8 fM | 200 |
| Carcinoembryonic antigen | Serum | Cancer | Fluorometric | 0.01–100 ng mL−1 | 3.0 pg mL−1 (green) | 264 |
| 1.3 pg mL−1 (yellow) | ||||||
| Cholecystokinin neuropeptides | CSF | Central nervous system | MALDI-ToF MS | 0.02–2.0 μg mL−1 | 1.2 and 5.5 pM | 212 |
| Cortisol | Saliva | Cardiovascular disease | Colorimetric | 2.50–20.0 ng mL−1 | 1.02 ng mL−1 | 193 |
| Cortisol | Saliva | Cardiovascular disease | Impedimetric | 0.50–64.0 nM | 0.14 nM | 265 |
| Cortisol | Sweat | Cardiovascular disease | OECTs | 0.10–1.0 μM | 2.68 μA dec−1 | 135 |
| Cortisol | Saliva | Stress | LMR | 0–1.0 × 10−6 g mL−1 | 2.59 × 10−14 g mL−1 | 266 |
| Cortisol | Sweat | Stress | Capacitive | 10–66 ng mL−1 | 2.0 ng mL−1 (±0.4) | 218 |
| Cortisol | Artificial sweat | Stress | Amperometric | 10 × 10−9–1 × 10−6 M | 0.2 × 10−9 M | 225 |
| Cholesterol | Serum | Coronary heart disease | Voltammetric | 1.0 × 10−18–10−13 M | 3.30 × 10−19 M | 165 |
| Cytochrome C | Urine | Apoptosis | Fluorometric | 0.20–60.0 μM | 0.11 μM | 267 |
| L-Dopa | Serum | Parkinson's disease | Electrochemical | 0.40–100 μM | 1.20 × 10−2 μM | 268 |
| Dopamine | Artificial CSF | Schizophrenia | Voltammetric | 0.30–100 μM | 0.10 μM | 209 |
| Dopamine | Urine | Parkinson's disease | Optical | 8.0–200.0 ng mL−1 | 1.50 ng mL−1 | 269 |
| Estriol | Serum | Breast cancer | Amperometric | 0.10–20.0 μg mL−1 | 2.0 × 10−3 μg mL−1 | 163 |
| Ferritin | Plasma | Kidney damage | Voltammetric | 120.0–360.0 mg dm−3 | 10.7 mg dm−3 | 229 |
| α-Fetoprotein | Serum | Cancer | Fluorometric | 0.01–100 ng mL−1 | 5.0 pg mL−1 (green) | 264 |
| 2.2 pg mL−1 (yellow) | ||||||
| α-Fetoprotein | Saliva | Cancer | Fluorometric | 0.01–100 ng mL−1 | 5.1 pg mL−1 (green) | 264 |
| 1.1 pg mL−1 (yellow) | ||||||
| Galectin-3 | Serum | Heart failure | SPR | 10.0–50.0 ng mL−1 | 2.0 ng mL−1 | 270 |
| Glucose | Saliva | Diabetes | Voltammetric and imedimetric | 0.5–50 μg mL−1 | 0.59 μg mL−1 and 1.6 μg mL−1 | 196 |
| Glucose | Saliva | Diabetes | Potentiometric | 3.20 × 10−7–1.0 × 10−3 M | 1.9 × 10−7 M (± 0.15) | 137 |
| Glucose | Synthetic tears | Diabetes | Fluorometric | 3.0–16.0 mM | 10.0 μg mL−1 | 271 |
| Haemoglobin | Urine | Anaemia | Potentiometric | 1.0–10.0 μg mL−1 | 1.0 μg mL−1 | 138 |
| HIV | Serum | HIV | Electrochemiluminescence | 0.30–3.0 nM | 3.0 × 10−7 nM | 164 |
| HIV-p24 | Serum | HIV-p24 | Voltammetric | 1.0 × 10−4–2 ng cm−3 | 8.3 × 10−5 ng cm−3 | 162 |
| HAS | Serum | Coronary heart disease | QCM | 5.0 × 10−5– × 10−4 g L−1 | 2.60 × 10−5 g L−1 | 236 |
| HAS | Serum | Coronary heart disease | Voltammetric | 8.0 × 10−4–2.0 × 10−2 g L−1 | 1.60 × 10−5 g L−1 | 234 |
| HAS | Serum | Coronary heart disease | Fluorometric | 1.70 × 10−2–3.0 × 10−1 g L−1 | 2.90 × 10−3 g L−1 | 233 |
| HAS | Serum | Coronary heart disease | Voltammetric | 1.99 × 10−6–30.91 × 10−6 g L−1 | 4.20 × 10−6 g L−1 | 210 |
| HAS | Plasma | Coronary heart disease | Voltammetric | 1.0 × 10−10–10−4 g L−1 | 2.0 × 10−11 g L−1 | 235 |
| HAS | Urine | Diabetes | Voltammetric | 1.0 × 10−10–10−4 g L−1 | 3.0 × 10−11 g L−1 | 231 |
| HAS | Urine | Kidney damage | Voltammetric | 0.02–0.10 g L−1 | 3.70 g L−1 | 229 |
| Insulin | Serum | Diabetes | MALDI-ToF MS | 23.6 ± 2.27 μM | 0.5 ng mL−1 | 134 |
| Interleukin-8 | Saliva | Oral cancer | Voltammetric | 0.10–10.0 pM | 4.0 × 10−2 pM | 194 |
| Japanese encephalitis virus | Serum | Japanese encephalitis virus | Resonance light scattering | 0–25 pM | 1.3 pM | 272 |
| Lactate | Sweat | Anerobic metabolism | Voltammetric | 1.0 × 10−6–10−1 M | 0.22 μM | 226 |
| Lactate | Sweat | Cell acidosis | Voltammetric | 1.0 × 10−6–10−1 M | 2.20 × 10−7 M | 226 |
| Lysozyme | Synthetic saliva | Chronic disease | Impedimetric | 2.20–292 mg L−1 | 0.9 mg L−1 and 2.1 mg L−1 | 273 |
| Lysozyme | Saliva and urine | Chronic disease | Fluorometric | 1.0 × 10−7–×10−6 M | 1.02 × 10−8 M | 274 |
| Melatonin | Urine | Urinary tract infection | Voltammetric | 1.75–2.11 pg mL−1 | 20.0 pg mL−1 | 230 |
| Melatonin | Plasma and urine | Parkinson's disease | Amperometry | 5.20 × 10−2–100 μm L−1 | 6.0 × 10−9 μM | 275 |
| Myoglobin | Serum | Myocardial infarction | Spectrophotometrically | 0.50–3.50 mg mL−1 | 623.0 mg g−1 | 276 |
| Norepinephrine | Urine | Bronchial asthma | Electrochemical | 0.50–80.0 μM | 0.10 μM | 277 |
| 3-Nitrotyrosine | Serum and urine | Alzheimer's disease | Electrochemical | 0.20–50.0 μM | 0.05 μM | 278 |
| 3-Nitrotyrosine | Serum | Atherosclerosis | Fluorometric | 5.0 × 10−2–1.85 μM | 1.70 × 10−2 μM | 279 |
| 3-Nitrotyrosine | Urine | Neurodegenerative disorders | HPLC with SPE | 2.50–55.0 μg mL−1 | 0.70 μg mL−1 | 280 |
| 3-Nitrotyrosine | Serum | Neurodegenerative disorders | SPR | 0.50–1.0 × 103 pM | 0.135 pM | 281 |
| 3-Nitrotyrosine | Urine | Oxidative stress | Voltammetric | 10 pg mL−1–1 μg mL−1 | 1.13 pg mL−1 | 237 |
| ncovNP | Nasopharyngeal fluid | COVID-19 | Electrochemical | 0.22–333 fM | 27 fM | 250 |
| ncovS1 | Nasopharyngeal fluid | COVID-19 | Electrochemical | 0–30 pg mL−1 | 4.8 pg mL−1 | 245 |
| Oxidised glutathione | Plasma | Cardiovascular disease | Amperometric | 0–86 × 10−7 M | 1.86 × 10−9 M | 282 |
| Phycocyanin | Urine | Cyanobacterial blooms | Fluorometric | 7.5 × 10−2 μM | 0.80–8.0 μM | 283 |
| D-/L-Pyroglutamic acid | Urine | Infection | Voltammetric | 1.80–173.6 ng mL−1 | 5.43 × 103–5.24 × 105 ng mL−1 | 211 |
| CSF | 1.50–180 ng mL−1 | 2.52 × 103–3.03 × 105 ng mL−1 | ||||
| Plasma | 1.30–153.8 ng mL−1 | 1.08 × 103–1.28 × 105 ng mL−1 | ||||
| Sarcosine | Urine | Cancer | RIS | 0.25–3.0 mM | 4.50 × 10−5 nM | 284 |
| D-/L-Serine | Serum | Schizophrenia | Voltammetric | 0.83–20.63 ng mL−1 (D-) | 0.24 ng mL−1 (D-) | 168 |
| 0.87–20–45 ng mL−1 (L-) | 0.25 ng mL−1 (L-) | |||||
| Tryptamine | CSF | Cardiovascular disease | Fluorometric | 0.3–1.5μmol L−1 | 1.46 μmol L−1 | 285 |
| Tryptophan | Serum | Schizophrenia | Voltammetric | 0.01–4.0 μM | 8.0 × 10−13 μM | 286 |
| 4.0–20.0 μM | ||||||
| 20.0–100.0 μM | ||||||
| Tryrosine | Serum | Parkinson's disease | Voltammetric | 0.1–400.0 μM | 4.60 × 10−2 μM | 287 |
| Uric acid | Urine | Hyperuricemia | SPR | 0.50–40.0 mg L−1 | 0.25 mg L−1 | 288 |
| Uric acid | Urine | Hyperpiesia | Voltammetric | 0.1–100.0 μM | 3.20 × 10−3 μM | 287 |
| Zika virus | Saliva | Zika virus | Potentiometric | 2.4 × 10−1–5.3 × 106 PFU | 10.0 PFU mL−1 | 289 |
Progress towards commercialising MIP-based biosensors
The Freestyle Libre 2, Accutrend® Plus system, and CoaguChek® Pro II (Table 1), are current benchmark examples of commercially available devices where biological materials have been employed as recognition elements for exclusive analyte detection. Throughout this review, from Diouf et al. MIP sensor for glucose detection, to Zhang et al. development of a wearable MIP sensor for the non-invasive monitoring of lactate in human sweat, the capability and potential of MITs to be replacements of their biological equivalents for the development of personalised medical care has been demonstrated. To date, along with the substantial advancements in developing MIP composites capable of detecting biomarkers in a wide range of biofluids, the field has seen some initial steps towards translating MIP technologies into commercially available products. One non-randomised clinical investigation for a MIP-based medical device aimed to quantifiably monitoring the effect of chemotherapy in colorectal cancer patients via the detection of transfer ribonucleic acid markers has been conducted in urine samples. The MIP-based sensor platform was designed to detect adenosine monophosphate by measuring surface acoustic waves in a microfluidic channel.290 Commercially, among the companies that have emerged, MIP Technologies, based in Lund, Sweden, is an ISO 9001:2008 certified company specialising in the design and production of custom-made polymeric separation materials through two commercialised products; Resna® (bulk resins) and Affinilute™ MIP Columns (solid-phase extraction columns) for separation.291 Allergy Amulet has marketed a point-of-care device to detect allergens at “clinically relevant thresholds” via MIP electrochemical sensing. The MIP detection element of the device was based on the electrochemical oxidation via differential pulse voltammetry of o-phenylenediamine (functional monomer) onto screen-printed electrodes under nitrogen atmosphere.292,293 MIP Diagnostics, based in Bedford, U.K., has been exploring the commercialisation of MIT through their “nanoMIPs” platform promoted for its use in the development of lateral flow devices and ELISA assays. Nanoparticle MIPs and nanoMIPs, are produced via bulk polymerisation using styrene as functional monomer, ethylene glycol dimethacrylate and trimethylolpropane trimethacrylate as crosslinkers, N-diethyldithiocarbamic acid benzyl ester as interferer, and pentaerythritol-tetrakis-(3-mercaptopropionate) as chain transfer agent, dissolved in acetonitrile using glass beads and UV polymerised. Depending on the application, the nanoMIPs have been subsequently conjugated onto sensor chips via coupling using carbodiimide crosslinker chemistry or through activation of the nanoMIP particles using NaOH, silanisation of the sensor chip's surface and subsequent immobilisation, among others.294–297 Their current research and development products (Troponin I, ProBNP and NT-ProBNP) rely on the nanoMIPs as the affinity reagent. As the biosensors market is anticipated to exceed $35 billion by 2026, following an increased demand for point-of-care healthcare monitoring and wearables, we foresee subsequent expansion to the commercialisation interest of MIT, allowing for growth opportunities in the research and development of smart MIP-based biodevices with a drive-in sustainable synthesis approaches.298Summary and future outlook
The function of MIPs performing as the synthetic recognition components within biosensors is motivated by improving healthcare technologies. However, to commercially compete with their biological counterparts further developments are required to accelerate the performance of MIP sensors in the application of clinical samples. Although the expansion of MIP-based point-of-care devices or other wearable sensors could still benefit from further development, the direct results obtained from using these simple biomarker detection techniques provide an accurate, rapid, convenient, reliable, affordable, and sustainable approach for on-site disease diagnostics. MIP biosensors’ high selectivity and sensitivity, miniaturised into portable devices, will inspire universally affordable and reliable biofluid detection.The synthetic affinity acquired through the “molecular memory” of the imprinted cavities towards a target species has signified the importance of molecular imprinting approaches, concerning the ability of such recognition matrices to identify specific analytes in complex mediums and their subsequent importance in diagnostics. This review has further demonstrated the ability of MIPs acting as sensing elements to present the required stability akin to selectivity and performance to that of enzymatic and antibody-based diagnostic counterparts via summarising the current status of the most up-to-date molecularly imprinted biosensor devices. The rudiments of MIP design, including the fundamental elements (e.g., functional monomer(s), crosslinker, initiator etc.), preparative methods, and characterisation techniques, have been evaluated, highlighting innovative and/or improved processes, such as bulk, emulsion, suspension, precipitation, and electropolymerisation, in addition to, sol–gel, surface, and epitope imprinting processes.
Integrating MIPs with nanocomposites to detect and purify biomarkers out of small volumes of biofluids has been critical for developing sensing platforms. As can be seen by the literature covered, incorporating smart technologies, and miniaturising the sensing element of such devices are important aspects of future commercialisation.
From small molecules to the advancement of facile methodologies, such as electropolymerisation, capable of imprinting macromolecules, whole microorganisms, and viruses, molecular imprinting has effectively expanded its ability to extend toward the detection of a wide range of biomarkers.
Until now, MIPs for biosensing has predominately focused on imprinting single species followed by optimisation, selectivity assessment in the presence of structurally homologous species and improved performance. Nonetheless, early disease detection predominately relies on the simultaneous identification of several biomarkers coexistent in the same media. The design and expansion of strategies towards the development of multiplexed sensing platforms or arrays of disease-relevant biomarkers can further expand the clinical significance of MIP-biosensors.
While lab-scale molecular imprinting has significantly progressed and there are many positive indicators of its future role in medical diagnostics, sustainable approaches to their production by employing green chemistry principles, is an essential element that needs to be addressed. As the principles of green chemistry are becoming more relevant to several scientific fields, molecular imprinting is destined to follow. At present, there has been disproportionate and insufficient research focusing on the use of less toxic and green chemicals that are less harmful to the users and non-polluting to the environment. Through green synthesis, the selection of green and renewable sourced reagents, reducing the number of reagents required for the processes, and the engagement of safer analytical methods, strategies focusing on sustainability and a “toxic-free” environment can be attained, contributing to both healthcare and environmental sustainability.
Conflicts of interest
The authors have no conflicts of interest to declare.Acknowledgements
The authors gratefully acknowledge financial support from the Engineering and Physical Sciences Research Council Grant EP/V010859/1 and the Royal Society Research Grant RSG\R1\201185. Fig. 1 and 2 and ToC were created with BioRender.com.References
- J. L. J. M. Müskens, R. B. Kool, S. A. van Dulmen and G. P. Westert, BMJ Qual. Saf., 2022, 31, 54–63 CrossRef PubMed.
- M.-S. Ong, F. Magrabi and E. Coiera, BMC Health Serv. Res., 2018, 18, 1–8 CrossRef PubMed.
- I. M. Lubin, J. R. Astles, S. Shahangian, B. Madison, R. Parry, R. L. Schmidt and M. L. Rubinstein, Diagnosis, 2021, 8, 281–294 CrossRef PubMed.
- J. Tu, R. M. Torrente-Rodríguez, M. Wang and W. Gao, Adv. Funct. Mater., 2020, 30, 1906713 CrossRef CAS.
- T. Pinheiro, A. R. Cardoso, C. E. A. Sousa, A. C. Marques, A. P. M. Tavares, A. M. Matos, M. T. Cruz, F. T. C. Moreira, R. Martins and E. Fortunato, ACS Omega, 2021, 6, 29268–29290 CrossRef CAS PubMed.
- S. A. Abid, A. A. Muneer, I. M. S. Al-Kadmy, A. A. Sattar, A. M. Beshbishy, G. E.-S. Batiha and H. F. Hetta, Life Sci., 2021, 273, 119117 CrossRef CAS PubMed.
- R. Samson, G. R. Navale and M. S. Dharne, 3 Biotech., 2020, 10, 1–9 CrossRef PubMed.
- A. Parihar, P. Ranjan, S. K. Sanghi, A. K. Srivastava and R. Khan, ACS Appl. Bio Mater., 2020, 3, 7326–7343 CrossRef CAS PubMed.
- L. Xu, D. Li, S. Ramadan, Y. Li and N. Klein, Biosens. Bioelectron., 2020, 170, 112673 CrossRef CAS PubMed.
- https://www.freestylelibre.co.uk, FreeStyle Libre.
- https://diagnostics.roche.com/global/en/products/instruments/accutrend-plus, Accutrend® Plus System.
- https://diagnostics.roche.com/gb/en/products/instruments/coaguchek-pro-ii, CoaguChek® Pro II.
- C.-C. Hong, C.-P. Chen, J.-C. Horng and S.-Y. Chen, Biosens. Bioelectron., 2013, 50, 425–430 CrossRef CAS PubMed.
- C. Lorenz, F. von Pelchrzim and R. Schroeder, Nat. Protoc., 2006, 1, 2204–2212 CrossRef CAS PubMed.
- L. Pauling, J. Am. Chem. Soc., 1940, 62, 2643–2657 CrossRef CAS.
- M. V. Polyakov, Zh. Fiz. Khim., 1931, 2, 799–805 Search PubMed.
- F. Canfarotta, A. Poma, A. Guerreiro and S. Piletsky, Nat. Protoc., 2016, 11, 443–455 CrossRef CAS PubMed.
- L. Ye, Y. Yu and K. Mosbach, Analyst, 2001, 126, 760–765 RSC.
- J. Pan, W. Chen, Y. Ma and G. Pan, Chem. Soc. Rev., 2018, 47, 5574–5587 RSC.
- G. Vasapollo, R. Del Sole, L. Mergola, M. R. Lazzoi, A. Scardino, S. Scorrano and G. Mele, Int. J. Mol. Sci., 2011, 12, 5908–5945 CrossRef CAS PubMed.
- E. V. Piletska, A. R. Guerreiro, M. J. Whitcombe and S. A. Piletsky, Macromolecules, 2009, 42, 4921–4928 CrossRef CAS.
- A. Poma, A. P. F. Turner and S. A. Piletsky, Trends Biotechnol., 2010, 28, 629–637 CrossRef CAS PubMed.
- G. Wulff, Angew. Chem., Int. Ed. Engl., 1995, 34, 1812–1832 CrossRef CAS.
- S. Yan, Y. Fang and Z. Gao, Biosens. Bioelectron., 2007, 22, 1087–1091 CrossRef CAS PubMed.
- C. Alexander, H. S. Andersson, L. I. Andersson, R. J. Ansell, N. Kirsch, I. A. Nicholls, J. O’Mahony and M. J. Whitcombe, J. Mol. Recognit., 2006, 19, 106–180 CrossRef CAS PubMed.
- E. Reville, E. Sylvester, S. Benware, S. Negi and E. B. Berda, Polym. Chem., 2022, 13, 3387–3411 RSC.
- L. Ye and K. Mosbach, Chem. Mater., 2008, 20, 859–868 CrossRef CAS.
- Z. El-Schich, Y. Zhang, M. Feith, S. Beyer, L. Sternbæk, L. Ohlsson, M. Stollenwerk and A. G. Wingren, Biotechniques, 2020, 69, 406–419 CrossRef CAS PubMed.
- D. Refaat, M. G. Aggour, A. A. Farghali, R. Mahajan, J. G. Wiklander, I. A. Nicholls and S. A. Piletsky, Int. J. Mol. Sci., 2019, 20, 6304 CrossRef CAS PubMed.
- M. Komiyama, T. Mori and K. Ariga, Bull. Chem. Soc. Jpn., 2018, 91, 1075–1111 CrossRef CAS.
- Y. Taguchi, E. Takano and T. Takeuchi, Langmuir, 2012, 28, 7083–7088 CrossRef CAS.
- K. Yoshimatsu, K. Reimhult, A. Krozer, K. Mosbach, K. Sode and L. Ye, Anal. Chim. Acta, 2007, 584, 112–121 CrossRef CAS PubMed.
- Y. Ge and A. P. F. Turner, Eur. J. Chem., 2009, 15, 8100–8107 CrossRef CAS.
- J. Haginaka, K. Nishimura, T. Kimachi, K. Inamoto, Y. Takemoto and Y. Kobayashi, Talanta, 2019, 205, 120149 CrossRef CAS PubMed.
- J. J. BelBruno, Chem. Rev., 2018, 119, 94–119 CrossRef PubMed.
- J. Xu, H. Miao and L. Zou, Angew. Chem., Int. Ed., 2021, 60, 24526–24533 CrossRef CAS PubMed.
- E. N. Ndunda, J. Mol. Recognit., 2020, 33, e2855 CrossRef CAS PubMed.
- B. Sellergren, Molecularly imprinted polymers: man-made mimics of antibodies and their application in analytical chemistry, Elsevier, 2000 Search PubMed.
- B. Mattiasson and L. Ye, Molecularly imprinted polymers in biotechnology, Springer, 2015, vol. 150 Search PubMed.
- L. Ye and K. Mosbach, React. Funct. Polym., 2001, 48, 149–157 CrossRef CAS.
- K. Haupt, P. X. Medina Rangel and B. T. S. Bui, Chem. Rev., 2020, 120, 9554–9582 CrossRef CAS.
- R. Arshady and K. Mosbach, Die Makromol. Chem., 2003, 182, 687–692 CrossRef.
- T. Takeuchi and J. Matsui, Acta Polym., 1996, 47, 471–480 CrossRef CAS.
- G. Wulff, A. Sarhan and K. Zabrocki, Tetrahedron Lett., 1973, 14, 4329–4332 CrossRef.
- K. Mosbach and O. Ramström, Bio/Technology, 1996, 14, 163–170 CAS.
- J. O’Mahony, A. Molinelli, K. Nolan, M. R. Smyth and B. Mizaikoff, Biosens. Bioelectron., 2006, 21, 1383–1392 CrossRef.
- S. N. N. S. Hashim, R. I. Boysen, L. J. Schwarz, B. Danylec and M. T. W. Hearn, J. Chromatogr. A, 2014, 1359, 35–43 CrossRef CAS PubMed.
- https://www.niehs.nih.gov/health/topics/science/biomarkers/index.cfm, National Institute of Environmental Health Sciences.
- S. Emaminejad, W. Gao, E. Wu, Z. A. Davies, H. Y. Y. Nyein, S. Challa, S. P. Ryan, H. M. Fahad, K. Chen and Z. Shahpar, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 4625–4630 CrossRef CAS PubMed.
- W. Gao, H. Y. Y. Nyein, Z. Shahpar, H. M. Fahad, K. Chen, S. Emaminejad, Y. Gao, L.-C. Tai, H. Ota and E. Wu, ACS Sens., 2016, 1, 866–874 CrossRef CAS.
- J. Heikenfeld, A. Jajack, B. Feldman, S. W. Granger, S. Gaitonde, G. Begtrup and B. A. Katchman, Nat. Biotechnol., 2019, 37, 407–419 CrossRef CAS PubMed.
- A. Martin-Esteban, Fresenius’ J. Anal. Chem., 2001, 370, 795–802 CrossRef CAS PubMed.
- W.-C. Lee, C.-H. Cheng, H.-H. Pan, T.-H. Chung and C.-C. Hwang, Anal. Bioanal. Chem., 2008, 390, 1101–1109 CrossRef CAS PubMed.
- G. Liu, Y. She, S. Hong, J. Wang and D. Xu, Appl. Sci., 2018, 8, 560 CrossRef.
- R. J. Ansell, J. K. L. Kuah, D. Wang, C. E. Jackson, K. D. Bartle and A. A. Clifford, J. Chromatogr. A, 2012, 1264, 117–123 CrossRef CAS.
- L. Chen, X. Wang, W. Lu, X. Wu and J. Li, Chem. Soc. Rev., 2016, 45, 2137–2211 RSC.
- L. M. Madikizela, S. S. Zunngu, N. Y. Mlunguza, N. T. Tavengwa, P. S. Mdluli and L. Chimuka, Water SA, 2018, 44, 406–418 CrossRef CAS.
- Y. Yang, Q. Li, G. Fang and S. Wang, RSC Adv., 2016, 6, 54510–54517 RSC.
- N. Sohrabi, R. Mohammadi, H. R. Ghassemzadeh and S. S. S. Heris, Microchem. J., 2022, 175, 107087 CrossRef CAS.
- Y. A. Olcer, M. Demirkurt, M. M. Demir and A. E. Eroglu, RSC Adv., 2017, 7, 31441–31447 RSC.
- R. Pratiwi, S. Megantara, D. Rahayu, I. Pitaloka and A. N. Hasanah, J. Young Pharm., 2019, 11, 12 CrossRef CAS.
- S. Pardeshi and S. K. Singh, RSC Adv., 2016, 6, 23525–23536 RSC.
- W. Siripairoj, A. Kaewchada and A. Jaree, J. Taiwan Inst. Chem. Eng., 2014, 45, 338–346 CrossRef CAS.
- K. Ji, X. Luo, L. He, S. Liao, L. Hu, J. Han, C. Chen, Y. Liu and N. Tan, J. Pharm. Biomed. Anal., 2020, 180, 113036 CrossRef CAS PubMed.
- V. B. Kandimalla and H. Ju, Anal. Bioanal. Chem., 2004, 380, 587–605 CrossRef CAS PubMed.
- E. Daniels, Y. L. Mustafa, C. Herdes and H. S. Leese, ACS Appl. Bio Mater., 2021, 4, 7243–7253 CrossRef CAS PubMed.
- M. Yoshikawa, K. Tharpa and S.-O. Dima, Chem. Rev., 2016, 116, 11500–11528 CrossRef CAS PubMed.
- X. Shen, C. Xu and L. Ye, Soft Matter, 2012, 8, 7169–7176 RSC.
- C. Gomes, G. Sadoyan, R. Dias and M. R. P. F. N. Costa, Processes, 2017, 5, 72 CrossRef.
- J. Peng, D. Xiao, H. He, H. Zhao, C. Wang, T. Shi and K. Shi, J. Sep. Sci., 2016, 39, 383–390 CrossRef CAS PubMed.
- M. Díaz-Álvarez, E. Turiel and A. Martín-Esteban, J. Chromatogr. A, 2016, 1469, 1–7 CrossRef.
- A. Beltran, R. M. Marcé, P. A. G. Cormack and F. Borrull, J. Chromatogr. A, 2009, 1216, 2248–2253 CrossRef CAS PubMed.
- Y. Liu, K. Hoshina and J. Haginaka, Talanta, 2010, 80, 1713–1718 CrossRef CAS PubMed.
- T. Alizadeh, Anal. Chim. Acta, 2010, 669, 94–101 CrossRef CAS PubMed.
- K. H. J. Buschow, M. C. Flemings and R. Cahn, The Encyclopedia of Materials: Science and Technology, Pergamon Imprint, 2001 Search PubMed.
- G. Zhao, J. Liu, M. Liu, X. Han, Y. Peng, X. Tian, J. Liu and S. Zhang, Appl. Sci., 2020, 10, 2868 CrossRef CAS.
- X. Ma, Z. Meng, L. Qiu, J. Chen, Y. Guo, D. Yi, T. Ji, H. Jia and M. Xue, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2016, 1020, 1–5 CrossRef CAS.
- E. Vivaldo-Lima, P. E. Wood, A. E. Hamielec and A. Penlidis, Ind. Eng. Chem. Res., 1997, 36, 939–965 CrossRef CAS.
- R. R. Watson, Polyphenols in plants: isolation, purification and extract preparation, Academic Press, 2018 Search PubMed.
- C. Gomes, G. Sadoyan, R. Dias and M. R. P. F. N. Costa, Processes, 2017, 5, 72 CrossRef.
- L. J. Schwarz, M. K. Potdar, B. Danylec, R. I. Boysen and M. T. W. Hearn, Anal. Methods, 2015, 7, 150–154 RSC.
- H. Cao, J. B. Xiao and M. Xu, Macromol. Res., 2006, 14, 324–330 CrossRef CAS.
- S. Ma, X. Zhuang, H. Wang, H. Liu, J. Li and X. Dong, Anal. Lett., 2007, 40, 321–333 CrossRef CAS.
- F.-F. Chen, X.-Y. Xie and Y.-P. Shi, J. Chromatogr. A, 2013, 1300, 112–118 CrossRef CAS PubMed.
- Y. Li, H.-H. Yang, Q.-H. You, Z.-X. Zhuang and X.-R. Wang, Anal. Chem., 2006, 78, 317–320 CrossRef CAS.
- S. R. Carter and S. Rimmer, Adv. Funct. Mater., 2004, 14, 553–561 CrossRef CAS.
- J. Yu, X. Wang, Q. Kang, J. Li, D. Shen and L. Chen, Environ. Sci.: Nano, 2017, 4, 493–502 RSC.
- M. M. Titirici, A. J. Hall and B. Sellergren, Chem. Mater., 2002, 14, 21–23 CrossRef CAS.
- R. Wang, L. Wang, J. Yan, D. Luan, J. Wu and X. Bian, Talanta, 2021, 226, 122135 CrossRef CAS PubMed.
- M. Zhang, H. T. Zhao, X. Yang, W. T. Zhang, J. Wang, G. Y. Liu, H. Zhang and A. J. Dong, RSC Adv., 2016, 6, 3714–3722 RSC.
- F. Ning, T. Qiu, Q. Wang, H. Peng, Y. Li, X. Wu, Z. Zhang, L. Chen and H. Xiong, Food Chem., 2017, 221, 1797–1804 CrossRef CAS PubMed.
- H. Dai, D. Xiao, H. He, H. Li, D. Yuan and C. Zhang, Microchim. Acta, 2015, 182, 893–908 CrossRef CAS.
- Y. Wang, J. Zhou, B. Zhang, L. Tian, Z. Ali and Q. Zhang, Chem. Eng. J., 2017, 327, 932–940 CrossRef CAS.
- Y. Qu, L. Qin, X. Liu and Y. Yang, Chemosphere, 2020, 251, 126376 CrossRef CAS.
- J. Cheng, P. R. Chang, P. Zheng and X. Ma, Ind. Eng. Chem. Res., 2014, 53, 1415–1421 CrossRef CAS.
- G. Liu, X. Yang, T. Li, Y. She, S. Wang, J. Wang, M. Zhang, F. Jin, M. Jin and H. Shao, Mater. Lett., 2015, 160, 472–475 CrossRef CAS.
- X. Wang, G. Chen, P. Zhang and Q. Jia, Anal. Methods, 2021, 13, 1660–1671 RSC.
- R. Xing, Y. Ma, Y. Wang, Y. Wen and Z. Liu, Chem. Sci., 2019, 10, 1831–1835 RSC.
- A. Rachkov and N. Minoura, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2001, 1544, 255–266 CrossRef CAS.
- A. Rachkov and N. Minoura, J. Chromatogr. A, 2000, 889, 111–118 CrossRef CAS.
- K. Yang, S. Li, L. Liu, Y. Chen, W. Zhou, J. Pei, Z. Liang and Y. Zhang, Adv. Mater., 2019, 31, 1902048 CrossRef CAS PubMed.
- A. Tang, L. Duan, M. Liu and X. Dong, J. Mater. Chem. B, 2016, 4, 7464–7471 RSC.
- S. Li, K. Yang, B. Zhao, X. Li, L. Liu, Y. Chen, L. Zhang and Y. Zhang, J. Mater. Chem. B, 2016, 4, 2739 RSC.
- S. Gatto, G. Ball, F. Onida, H. M. Kantarjian, E. H. Estey and M. Beran, Blood, 2003, 102, 1622–1625 CrossRef CAS PubMed.
- E. Yilmaz, O. Ramström, P. Möller, D. Sanchez and K. Mosbach, J. Mater. Chem., 2002, 12, 1577–1581 RSC.
- K. Kajihara, J. Asian Ceram. Soc., 2013, 1, 121–133 CrossRef.
- A. Florea, O. Hosu, B. Ciui and C. Cristea, Molecularly imprinted polymer-based sensors for biomedical and environmental applications, Scrievener Publishing, NJ, USA, 2016 Search PubMed.
- C. Guoning, S. Hua, L. Wang, H. Qianqian, C. Xia, Z. Hongge, L. Zhimin, C. Chun and F. Qiang, J. Pharm. Biomed. Anal., 2020, 190, 113511 CrossRef CAS PubMed.
- R. Rolla, D. Vay, E. Mottaran, M. Parodi, N. Traverso, S. Aricó, M. Sartori, G. Bellomo, L. W. Klassen and G. M. Thiele, Hepatology, 2000, 31, 878–884 CrossRef CAS PubMed.
- S. Sang, Y. Li, X. Guo, B. Zhang, X. Xue, K. Zhuo, C. Zhao, W. Zhang and Z. Yuan, Biosens. Bioelectron., 2019, 141, 111399 CrossRef CAS PubMed.
- W.-F. Chang, S.-Y. Huang, R.-H. Lee and Y.-C. Liu, J. Polym. Res., 2014, 21, 1–7 CAS.
- M. C. Blanco-López, M. J. Lobo-Castañón, A. J. Miranda-Ordieres and P. Tunon-Blanco, TrAC, Trends Anal. Chem., 2004, 23, 36–48 CrossRef.
- S. A. Piletsky and A. P. F. Turner, Electroanalysis, 2002, 14, 317–323 CrossRef CAS.
- G. Fomo, T. Waryo, U. Feleni, P. Baker and E. Iwuoha, Electrochemical Polymerization, ed. Jafar Mazumder, M. A., Sheardown, H., Al-Ahmed, A., 2019, pp. 105–131 Search PubMed.
- A. Herrera-Chacón, X. Cetó and M. del Valle, Anal. Bioanal. Chem., 2021, 413, 6117–6140 CrossRef PubMed.
- C. Malitesta, I. Losito and P. G. Zambonin, Anal. Chem., 1999, 71, 1366–1370 CrossRef CAS PubMed.
- C. Malitesta, F. Palmisano, L. Torsi and P. G. Zambonin, Anal. Chem., 1990, 62, 2735–2740 CrossRef CAS PubMed.
- R. N. Goyal and S. P. Singh, Electrochim. Acta, 2006, 51, 3008–3012 CrossRef CAS.
- L. Özcan and Y. Şahin, Sens. Actuators, B, 2007, 127, 362–369 CrossRef.
- F. Gu, W. He, S. Xiao, S. Wang, X. Li, Q. Zeng, Y. Ni and L. Han, Sci. Rep., 2020, 10, 1–8 CrossRef PubMed.
- A. Ramanaviciene and A. Ramanavicius, Biosens. Bioelectron., 2004, 20, 1076–1082 CrossRef CAS PubMed.
- G. Ceolin, Á. Orbán, V. Kocsis, R. E. Gyurcsányi, I. Kézsmárki and V. Horváth, J. Mater. Sci., 2013, 48, 5209–5218 CrossRef CAS.
- G. Evtugyn, A. Porfireva, A. Ivanov, O. Konovalova and T. Hianik, Electroanalysis, 2009, 21, 1272–1277 CrossRef CAS.
- J. Kronholm, K. Hartonen and M.-L. Riekkola, TrAC, Trends Anal. Chem., 2007, 26, 396–412 CrossRef CAS.
- G.-Q. Fu, H. Yu and J. Zhu, Biomaterials, 2008, 29, 2138–2142 CrossRef CAS PubMed.
- A. Adumitraăchioaie, M. Tertiş, A. Cernat, R. Săndulescu and C. Cristea, Int. J. Electrochem. Sci., 2018, 13, 2556–2576 CrossRef.
- M. Gao, Y. Gao, G. Chen, X. Huang, X. Xu, J. Lv, J. Wang, D. Xu and G. Liu, Front. Chem., 2020, 8, 1142 Search PubMed.
- P. Jolly, V. Tamboli, R. L. Harniman, P. Estrela, C. J. Allender and J. L. Bowen, Biosens. Bioelectron., 2016, 75, 188–195 CrossRef CAS PubMed.
- N. Idil and B. Mattiasson, Sensors, 2017, 17, 708 CrossRef.
- M. Jia, Z. Zhang, J. Li, X. Ma, L. Chen and X. Yang, TrAC, Trends Anal. Chem., 2018, 106, 190–201 CrossRef CAS.
- S. A. Zaidi, Crit. Rev. Anal. Chem., 2021, 51, 609–618 CAS.
- A. Ahmed, J. V. Rushworth, N. A. Hirst and P. A. Millner, Clin. Microbiol. Rev., 2014, 27, 631–646 CrossRef CAS PubMed.
- O. Lazcka, F. J. del Campo and F. X. Munoz, Biosens. Bioelectron., 2007, 22, 1205–1217 CrossRef CAS PubMed.
- Z. Wang, X. Fang, N. Sun and C. Deng, Anal. Chim. Acta, 2020, 1128, 1–10 CrossRef CAS PubMed.
- O. Parlak, S. T. Keene, A. Marais, V. F. Curto and A. Salleo, Sci. Adv., 2018, 4, eaar2904 CrossRef CAS PubMed.
- B. B. Prasad, A. Srivastava and M. P. Tiwari, Mater. Sci. Eng., C, 2013, 33, 4071–4080 CrossRef CAS.
- D.-M. Kim, J.-M. Moon, W.-C. Lee, J.-H. Yoon, C. S. Choi and Y.-B. Shim, Biosens. Bioelectron., 2017, 91, 276–283 CrossRef CAS PubMed.
- T. S. Anirudhan and S. Alexander, Eur. Polym. J., 2017, 97, 84–93 CrossRef CAS.
- D. Ivnitski, I. Abdel-Hamid, P. Atanasov and E. Wilkins, Biosens. Bioelectron., 1999, 14, 599–624 CrossRef CAS.
- X. Shen, J. Svensson Bonde, T. Kamra, L. Bülow, J. C. Leo, D. Linke and L. Ye, Angew. Chem., Int. Ed., 2014, 53, 10687–10690 CrossRef CAS PubMed.
- M. Roushani, M. Sarabaegi and A. Rostamzad, J. Iran. Chem. Soc., 2020, 17, 2407–2413 CrossRef CAS.
- S. Arif, S. Qudsia, S. Urooj, N. Chaudry, A. Arshad and S. Andleeb, Biosens. Bioelectron., 2015, 65, 62–70 CrossRef CAS PubMed.
- J. Y. Chen, L. S. Penn and J. Xi, Biosens. Bioelectron., 2018, 99, 593–602 CrossRef CAS PubMed.
- J. Kankare, Langmuir, 2002, 18, 7092–7094 CrossRef CAS.
- C. Cheubong, A. Yoshida, Y. Mizukawa, N. Hayakawa, M. Takai, T. Morishita, Y. Kitayama, H. Sunayama and T. Takeuchi, Anal. Chem., 2020, 92, 6401–6407 CrossRef CAS PubMed.
- S. Tokonami, Y. Nakadoi, M. Takahashi, M. Ikemizu, T. Kadoma, K. Saimatsu, L. Q. Dung, H. Shiigi and T. Nagaoka, Anal. Chem., 2013, 85, 4925–4929 CrossRef CAS PubMed.
- J. Wang, J. Dai, Y. Xu, X. Dai, Y. Zhang, W. Shi, B. Sellergren and G. Pan, Small, 2019, 15, 1803913 CrossRef.
- D. L. Robinson, A. Hermans, A. T. Seipel and R. M. Wightman, Chem. Rev., 2008, 108, 2554–2584 CrossRef CAS PubMed.
- S. Kruss, M. P. Landry, E. Vander Ende, B. M. A. Lima, N. F. Reuel, J. Zhang, J. Nelson, B. Mu, A. Hilmer and M. Strano, J. Am. Chem. Soc., 2014, 136, 713–724 CrossRef CAS PubMed.
- T. Patriarchi, J. R. Cho, K. Merten, M. W. Howe, A. Marley, W.-H. Xiong, R. W. Folk, G. J. Broussard, R. Liang and M. J. Jang, Science, 2018, 360, eaat4422 CrossRef PubMed.
- B. Kong, A. Zhu, Y. Luo, Y. Tian, Y. Yu and G. Shi, Angew. Chem., Int. Ed., 2011, 50, 1837–1840 CrossRef CAS PubMed.
- D. Michel, F. Xiao and K. Alameh, Sens. Actuators, B, 2017, 246, 258–261 CrossRef CAS.
- H. Šípová and J. Homola, Anal. Chim. Acta, 2013, 773, 9–23 CrossRef.
- K. B. Levin, O. Dym, S. Albeck, S. Magdassi, A. H. Keeble, C. Kleanthous and D. S. Tawfik, Nat. Struct. Mol. Biol., 2009, 16, 1049–1055 CrossRef CAS PubMed.
- L. Cenci, E. Andreetto, A. Vestri, M. Bovi, M. Barozzi, E. Iacob, M. Busato, A. Castagna, D. Girelli and A. M. Bossi, J. Nanobiotechnol., 2015, 13, 1–15 CrossRef.
- A. Garcia-Cruz, O. S. Ahmad, K. Alanazi, E. Piletska and S. A. Piletsky, Microsyst. Nanoeng., 2020, 6, 1–9 CrossRef PubMed.
- T. Kamra, S. Chaudhary, C. Xu, L. Montelius, J. Schnadt and L. Ye, J. Colloid Interface Sci., 2016, 461, 1–8 CrossRef CAS PubMed.
- A. L. Campaña, S. L. Florez, M. J. Noguera, O. P. Fuentes, P. Ruiz Puentes, J. C. Cruz and J. F. Osma, Biosensors, 2019, 9, 41 CrossRef.
- N. L. C. Members, G. L. Myers, R. H. M. Christenson, M. Cushman, C. M. Ballantyne, G. R. Cooper, C. M. Pfeiffer, S. M. Grundy, D. R. Labarthe and D. Levy, Clin Chem, 2009, 55, 378–384 Search PubMed.
- P. S. Jellinger, D. A. Smith, A. E. Mehta, O. Ganda, Y. Handelsman, H. W. Rodbard, M. D. Shepherd, J. A. Seibel, R. Kreisberg and R. Goldberg, Endocr. Pract., 2012, 18, 1–78 CrossRef PubMed.
- X. Zhang, B. Yao, Q. Hu, Y. Hong, A. Wallace, K. Reynolds, C. Ramsey, A. Maeder, R. Reed and Y. Tang, Mater. Chem. Front., 2020, 4, 2548–2570 RSC.
- Y. Ma, X.-L. Shen, Q. Zeng, H.-S. Wang and L.-S. Wang, Talanta, 2017, 164, 121–127 CrossRef CAS.
- X. Xin, S. Sun, M. Wang and R. Jia, Ionics, 2020, 26, 2633–2641 CrossRef CAS.
- B. Babamiri, A. Salimi and R. Hallaj, Biosens. Bioelectron., 2018, 117, 332–339 CrossRef CAS PubMed.
- H. Yang, L. Li, Y. Ding, D. Ye, Y. Wang, S. Cui and L. Liao, Biosens. Bioelectron., 2017, 92, 748–754 CrossRef CAS.
- M. You, S. Yang, Y. An, F. Zhang and P. He, J. Electroanal. Chem., 2020, 862, 114017 CrossRef CAS.
- I. W. Hamley, Chem. Rev., 2012, 112, 5147–5192 CrossRef CAS PubMed.
- S. Jaiswal, R. Singh, K. Singh, S. Fatma and B. B. Prasad, Biosens. Bioelectron., 2019, 124, 176–183 CrossRef PubMed.
- M.-A. B. MacKay, M. Kravtsenyuk, R. Thomas, N. D. Mitchell, S. M. Dursun and G. B. Baker, Front. Psychiatry, 2019, 10 DOI:10.3389/fpsyt.2019.00025.
- A. Kugimiya and E. Matsuzaki, Appl. Biochem. Biotechnol., 2014, 174, 2527–2536 CrossRef CAS PubMed.
- M. Yaqoob and A. Nabi, Talanta, 2001, 55, 1181–1186 CrossRef CAS.
- S. Li, Q. Yu, X. Lu and S. Zhao, J. Sep. Sci., 2009, 32, 282–287 CrossRef CAS PubMed.
- M. Saha and S. Das, J. Nanostruct. Chem., 2014, 4, 1–9 Search PubMed.
- A. K. Singh and M. Singh, J. Electroanal. Chem., 2016, 780, 169–175 CrossRef CAS.
- M. Roushani, M. Shamsipur and S. M. Pourmortazavi, J. Appl. Electrochem., 2012, 42, 1005–1011 CrossRef CAS.
- L. Luo, J. Yang, K. Liang, C. Chen, X. Chen and C. Cai, Talanta, 2019, 202, 21–26 CrossRef CAS PubMed.
- K. He, C. Chen, C. Liang, C. Liu, B. Yang, X. Chen and C. Cai, Sens. Actuators, B, 2016, 233, 607–614 CrossRef CAS.
- W. Feng, C. Liang, H. Gong and C. Cai, New J. Chem., 2018, 42, 3503–3508 RSC.
- C. Liang, H. Wang, K. He, C. Chen, X. Chen, H. Gong and C. Cai, Talanta, 2016, 160, 360–366 CrossRef CAS.
- S. M. Tawfik, M. R. Elmasry, M. Sharipov, S. Azizov, C. H. Lee and Y. I. Lee, Biosens. Bioelectron., 2020, 160, 112211 CrossRef CAS.
- G. Zhao, Y. Wang, X. Li, Q. Yue, X. Dong, B. Du, W. Cao and Q. Wei, Anal. Chem., 2019, 91, 1989–1996 CrossRef CAS.
- N. Xia, X. Wang, B. Zhou, Y. Wu, W. Mao and L. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 19303–19311 CrossRef CAS.
- L. Guerrini, R. Arenal, B. Mannini, F. Chiti, R. Pini, P. Matteini and R. A. Alvarez-Puebla, ACS Appl. Mater. Interfaces, 2015, 7, 9420–9428 CrossRef CAS PubMed.
- T. Yang, S. Hong, T. O’Malley, R. A. Sperling, D. M. Walsh and D. J. Selkoe, Alzheimer's Dement., 2013, 9, 99–112 CrossRef PubMed.
- L. Zhu, J. Zhang, F. Wang, Y. Wang, L. Lu, C. Feng, Z. Xu and W. Zhang, Biosens. Bioelectron., 2016, 78, 206–212 CrossRef CAS PubMed.
- Y. Yu, L. Zhang, C. Li, X. Sun, D. Tang and G. Shi, Angew. Chem., 2014, 126, 13046–13049 CrossRef.
- M. K. Kang, J. Lee, A. H. Nguyen and S. J. Sim, Biosens. Bioelectron., 2015, 72, 197–204 CrossRef CAS PubMed.
- H.-L. Shuai, X. Wu, K.-J. Huang and Z.-B. Zhai, Biosens. Bioelectron., 2017, 94, 616–625 CrossRef CAS.
- L. Liu, F. Zhao, F. Ma, L. Zhang, S. Yang and N. Xia, Biosens. Bioelectron., 2013, 49, 231–235 CrossRef CAS PubMed.
- F. G. Bellagambi, T. Lomonaco, P. Salvo, F. Vivaldi, M. Hangouët, S. Ghimenti, D. Biagini, F. Di Francesco, R. Fuoco and A. Errachid, TrAC, Trends Anal. Chem., 2020, 124, 115781 CrossRef CAS.
- E. Lamy and M. Mau, J. Proteomics, 2012, 75, 4251–4258 CrossRef CAS PubMed.
- C.-K. Yeh, N. J. Christodoulides, P. N. Floriano, C. S. Miller, J. L. Ebersole, S. E. Weigum, J. McDevitt and S. W. Redding, Tex. Dent. J., 2010, 127, 651 Search PubMed.
- G. Spano, S. Cavalera, F. di Nardo, C. Giovannoli, L. Anfossi and C. Baggiani, Anal. Methods, 2019, 11, 2320–2326 RSC.
- P. Tang, H. Zhang, J. Huo and X. Lin, Anal. Methods, 2015, 7, 7784–7791 RSC.
- A. S. Panchbhai, J. Oral Maxillofac. Res., 2012, 3, e3 Search PubMed.
- A. Diouf, B. Bouchikhi and N. el Bari, Mater. Sci. Eng., C, 2019, 98, 1196–1209 CrossRef CAS.
- J. Xu, Q. Sheng, Y. Shen and J. Zheng, Colloids Surf., A, 2017, 529, 113–118 CrossRef CAS.
- C. Hu, D.-P. Yang, F. Zhu, F. Jiang, S. Shen and J. Zhang, ACS Appl. Mater. Interfaces, 2014, 6, 4170–4178 CrossRef CAS.
- A. Ciudin, C. Hernández and R. Simó, Curr. Diabetes Rev., 2012, 8, 48–54 CrossRef CAS PubMed.
- F. Parnianchi, S. Kashanian, M. Nazari, C. Santoro, P. Bollella and K. Varmira, Microchem. J., 2021, 168, 106367 CrossRef CAS.
- Ç. Çiçek, F. Yılmaz, E. Özgür, H. Yavuz and A. Denizli, Chemosensors, 2016, 4, 21 CrossRef.
- C. Song, Y. Li, B. Wang, Y. Hong, C. Xue, Q. Li, E. Shen and D. Cui, Colloids Surf., B, 2021, 197, 111430 CrossRef CAS PubMed.
- W. Xiao, D. Zhi, Q. Pan, Y. Liang, F. Zhou and Z. Chen, Anal. Methods, 2020, 12, 5691–5698 RSC.
- I. Taurino, V. Van Hoof, G. De Micheli and S. Carrara, Thin Solid Films, 2013, 548, 546–550 CrossRef CAS.
- M. L. Yola, C. Göde and N. Atar, Electrochim. Acta, 2017, 246, 135–140 CrossRef CAS.
- A.-H. Wu and M.-J. Syu, Biosens. Bioelectron., 2006, 21, 2345–2353 CrossRef CAS PubMed.
- M.-J. Syu, T.-C. Chiu, C.-Y. Lai and Y.-S. Chang, Biosens. Bioelectron., 2006, 22, 550–557 CrossRef CAS PubMed.
- C.-Y. Huang, M.-J. Syu, Y.-S. Chang, C.-H. Chang, T.-C. Chou and B.-D. Liu, Biosens. Bioelectron., 2007, 22, 1694–1699 CrossRef CAS PubMed.
- J. Yang, Y. Hu and Y. Li, Biosens. Bioelectron., 2019, 135, 224–230 CrossRef CAS PubMed.
- B. B. Prasad, A. Prasad and M. P. Tiwari, Biosens. Bioelectron., 2013, 39, 236–243 CrossRef CAS PubMed.
- B. B. Prasad and I. Pandey, Sens. Actuators, B, 2013, 186, 407–416 CrossRef CAS.
- X. Ji, D. Li and H. Li, Biomed. Chromatogr., 2015, 29, 1280–1289 CrossRef CAS PubMed.
- M. E. Bowers, D. C. Choi and K. J. Ressler, Physiol. Behav., 2012, 107, 699–710 CrossRef CAS PubMed.
- A. L. LaCrosse and M. Foster Olive, CNS Neurol. Disord.: Drug Targets, 2013, 12, 619–632 CrossRef CAS PubMed.
- P. Luliński, M. Bamburowicz-Klimkowska, M. Dana and D. Maciejewska, J. Sep. Sci., 2017, 40, 1824–1833 CrossRef PubMed.
- M. Chung, G. Fortunato and N. Radacsi, J. R. Soc., Interface, 2019, 16, 20190217 CrossRef CAS.
- M. Jia, W. M. Chew, Y. Feinstein, P. Skeath and E. M. Sternberg, Analyst, 2016, 141, 2053–2060 RSC.
- S. M. Mugo and J. Alberkant, Anal. Bioanal. Chem., 2020, 412, 1825–1833 CrossRef CAS PubMed.
- J. A. Martin, J. L. Chávez, Y. Chushak, R. R. Chapleau, J. Hagen and N. Kelley-Loughnane, Anal. Bioanal. Chem., 2014, 406, 4637–4647 CrossRef CAS PubMed.
- R. C. Kessler, P. Berglund, O. Demler, R. Jin, K. R. Merikangas and E. E. Walters, Arch. Gen. Psychiatry, 2005, 62, 593–602 CrossRef PubMed.
- N. V. Zaryanov, V. N. Nikitina, E. V. Karpova, E. E. Karyakina and A. A. Karyakin, Anal. Chem., 2017, 89, 11198–11202 CrossRef CAS PubMed.
- M. Sekar, M. Pandiaraj, S. Bhansali, N. Ponpandian and C. Viswanathan, Sci. Rep., 2019, 9, 403 CrossRef CAS.
- K. S. Kim, S. R. Lim, S.-E. Kim, J. Y. Lee, C.-H. Chung, W.-S. Choe and P. J. Yoo, Sens. Actuators, B, 2017, 242, 1121–1128 CrossRef CAS.
- B. Sun, Y. Gou, Y. Ma, X. Zheng, R. Bai, A. A. A. Abdelmoaty and F. Hu, Biosens. Bioelectron., 2017, 88, 55–62 CrossRef CAS PubMed.
- W. Tang, L. Yin, J. R. Sempionatto, J. Moon, H. Teymourian and J. Wang, Adv. Mater., 2021, 33, 2008465 CrossRef CAS PubMed.
- Q. Zhang, D. Jiang, C. Xu, Y. Ge, X. Liu, Q. Wei, L. Huang, X. Ren, C. Wang and Y. Wang, Sens. Actuators, B, 2020, 320, 128325 CrossRef CAS.
- W. Jia, A. J. Bandodkar, G. Valdés-Ramírez, J. R. Windmiller, Z. Yang, J. Ramírez, G. Chan and J. Wang, Anal. Chem., 2013, 85, 6553–6560 CrossRef CAS PubMed.
- P. J. Derbyshire, H. Barr, F. Davis and S. P. J. Higson, J. Physiol. Sci., 2012, 62, 429–440 CrossRef CAS PubMed.
- Z. Stojanovic, J. Erdőssy, K. Keltai, F. W. Scheller and R. E. Gyurcsányi, Anal. Chim. Acta, 2017, 977, 1–9 CrossRef CAS PubMed.
- M.-H. Lee, D. O’Hare, Y.-L. Chen, Y.-C. Chang, C.-H. Yang, B.-D. Liu and H.-Y. Lin, Biomicrofluidics, 2014, 8, 54115 CrossRef.
- G. Zhang, Y. Yu, M. Guo, B. Lin and L. Zhang, Sens. Actuators, B, 2019, 288, 564–570 CrossRef CAS.
- C. D. Stehouwer, M. A. Gall, J. W. Twisk, E. Knudsen, J. J. Emeis and H. H. Parving, Diabetes, 2002, 51, 1157–1165 CrossRef CAS PubMed.
- Y.-Z. Wang, D.-Y. Li, X.-W. He, W.-Y. Li and Y.-K. Zhang, Microchim. Acta, 2015, 182, 1465–1472 CrossRef CAS.
- M. Cieplak, K. Szwabinska, M. Sosnowska, B. K. C. Chandra, P. Borowicz, K. Noworyta, F. D’Souza and W. Kutner, Biosens. Bioelectron., 2015, 74, 960–966 CrossRef CAS PubMed.
- J. Xia, X. Cao, Z. Wang, M. Yang, F. Zhang, B. Lu, F. Li, L. Xia, Y. Li and Y. Xia, Sens. Actuators, B, 2016, 225, 305–311 CrossRef CAS.
- X.-T. Ma, X.-W. He, W.-Y. Li and Y.-K. Zhang, Sens. Actuators, B, 2017, 246, 879–886 CrossRef CAS.
- G. V. Martins, A. C. Marques, E. Fortunato and M. G. F. Sales, Sens. Bio-Sens. Res., 2020, 28, 100333 CrossRef.
- H. Jayakody, D. Rowland, C. Pereira, R. Blackwell, T. Lasota, M. Laverick, L. Tisi, H. S. Leese and A. D. S. Walsham, Sci. Rep., 2022, 12, 1–8 CrossRef PubMed.
- C. H. Hernando, J. A. Á. Serra, M. J. E. Saco, S. Martínez-Nadal and C. V. Cerén, Anales de Pediatria, Elsevier, 2020, vol. 93, p. 264 Search PubMed.
- K. Hong, W. Cao, Z. Liu, L. Lin, X. Zhou, Y. Zeng, Y. Wei, L. Chen, X. Liu and Y. Han, Emerging Microbes Infect., 2020, 9, 2315–2321 CrossRef CAS PubMed.
- A. Wajnberg, M. Mansour, E. Leven, N. M. Bouvier, G. Patel, A. Firpo-Betancourt, R. Mendu, J. Jhang, S. Arinsburg and M. Gitman, Lancet Microbe, 2020, 1, e283–e289 CrossRef CAS PubMed.
- Find©, https://www.finddx.org/test-directory/), (accessed March 29, 2022).
- R. R. X. Lim and A. Bonanni, TrAC, Trends Anal. Chem., 2020, 133, 116081 CrossRef PubMed.
- G. Seo, G. Lee, M. J. Kim, S.-H. Baek, M. Choi, K. B. Ku, C.-S. Lee, S. Jun, D. Park, H. G. Kim, S.-J. Kim, J.-O. Lee, B. T. Kim, E. C. Park and S. Il Kim, ACS Nano, 2020, 14, 5135–5142 CrossRef CAS.
- A. G. Ayankojo, R. Boroznjak, J. Reut, A. Öpik and V. Syritski, Sens. Actuators, B, 2022, 353, 131160 CrossRef CAS PubMed.
- Y. Li, Z. Peng, N. J. Holl, M. R. Hassan, J. M. Pappas, C. Wei, O. H. Izadi, Y. Wang, X. Dong and C. Wang, ACS Omega, 2021, 6, 6643–6653 CrossRef CAS PubMed.
- S. Eissa and M. Zourob, Anal. Chem., 2020, 93, 1826–1833 CrossRef PubMed.
- B. Mojsoska, S. Larsen, D. A. Olsen, J. S. Madsen, I. Brandslund and F. A. Alatraktchi, Sensors, 2021, 21, 390 CrossRef CAS PubMed.
- L. Fabiani, M. Saroglia, G. Galatà, R. de Santis, S. Fillo, V. Luca, G. Faggioni, N. D’Amore, E. Regalbuto and P. Salvatori, Biosens. Bioelectron., 2021, 171, 112686 CrossRef CAS PubMed.
- A. Raziq, A. Kidakova, R. Boroznjak, J. Reut, A. Öpik and V. Syritski, Biosens. Bioelectron., 2021, 178, 113029 CrossRef CAS PubMed.
- R. Boroznjak, J. Reut, A. Tretjakov, A. Lomaka, A. Öpik and V. Syritski, J. Mol. Recognit., 2017, 30, e2635 CrossRef.
- T. Takeuchi, K. Mori, H. Sunayama, E. Takano, Y. Kitayama, T. Shimizu, Y. Hirose, S. Inubushi, R. Sasaki and H. Tanino, J. Am. Chem. Soc., 2020, 142, 6617–6624 CrossRef CAS PubMed.
- R. J. Berckmans, R. Lacroix, C. M. Hau, A. Sturk and R. Nieuwland, J. Extracell. Vesicles, 2019, 8, 1688936 CrossRef CAS PubMed.
- S. U. Wang, A. Khan, R. Huang, S. Ye, K. Di, T. Xiong and Z. Li, Biosens. Bioelectron., 2020, 154, 112056 CrossRef CAS PubMed.
- L. A. Aqrawi, H. K. Galtung, B. Vestad, R. Øvstebø, B. Thiede, S. Rusthen, A. Young, E. M. Guerreiro, T. P. Utheim and X. Chen, Arthritis Res. Ther., 2017, 19, 1–15 CrossRef PubMed.
- K. Mori, M. Hirase, T. Morishige, E. Takano, H. Sunayama, Y. Kitayama, S. Inubushi, R. Sasaki, M. Yashiro and T. Takeuchi, Angew. Chem., 2019, 131, 1626–1629 CrossRef.
- K. Ishihara, T. Ueda and N. Nakabayashi, Polym. J., 1990, 22, 355–360 CrossRef CAS.
- H. Im, H. Shao, Y. Il Park, V. M. Peterson, C. M. Castro, R. Weissleder and H. Lee, Nat. Biotechnol., 2014, 32, 490–495 CrossRef CAS PubMed.
- H. Shao, J. Chung, L. Balaj, A. Charest, D. D. Bigner, B. S. Carter, F. H. Hochberg, X. O. Breakefield, R. Weissleder and H. Lee, Nat. Med., 2012, 18, 1835–1840 CrossRef CAS.
- P. Zhang, X. Zhou, M. He, Y. Shang, A. L. Tetlow, A. K. Godwin and Y. Zeng, Nat. Biomed. Eng., 2019, 3, 438–451 CrossRef CAS PubMed.
- Y. Yoshioka, N. Kosaka, Y. Konishi, H. Ohta, H. Okamoto, H. Sonoda, R. Nonaka, H. Yamamoto, H. Ishii and M. Mori, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- G. Qiu, A. Thakur, C. Xu, S. Ng, Y. Lee and C. L. Wu, Adv. Funct. Mater., 2019, 29, 1806761 CrossRef.
- Y. Zhang, F. Wang, H. Zhang, H. Wang and Y. Liu, Anal. Chem., 2019, 91, 12100–12107 CrossRef CAS PubMed.
- S. M. Tawfik, M. R. Elmasry, M. Sharipov, S. Azizov, C. H. Lee and Y. I. Lee, Biosens. Bioelectron., 2020, 160, 112211 CrossRef CAS PubMed.
- H. D. Ertuğrul Uygun, Z. O. Uygun, E. Canbay, N. S. F. Gi Rgi and E. Sezer, Talanta, 2020, 206, 120225 CrossRef PubMed.
- S. P. Usha, A. M. Shrivastav and B. D. Gupta, Biosens. Bioelectron., 2017, 87, 178–186 CrossRef CAS PubMed.
- Y.-J. Yan, X.-W. He, W.-Y. Li and Y.-K. Zhang, Biosens. Bioelectron., 2017, 91, 253–261 CrossRef CAS PubMed.
- L. Lin, H.-T. Lian, X.-Y. Sun, Y.-M. Yu and B. Liu, Anal. Methods, 2015, 7, 1387–1394 RSC.
- H. Duan, L. Li, X. Wang, Y. Wang, J. Li and C. Luo, Spectrochim. Acta, Part A, 2015, 139, 374–379 CrossRef CAS.
- E. N. Primo, M. J. Kogan, H. E. Verdejo, S. Bollo, M. D. Rubianes and G. A. Rivas, ACS Appl. Mater. Interfaces, 2018, 10, 23501–23508 CrossRef CAS PubMed.
- S. Manju, P. R. Hari and K. Sreenivasan, Biosens. Bioelectron., 2010, 26, 894–897 CrossRef CAS PubMed.
- L. Luo, J. Yang, K. Liang, C. Chen, X. Chen and C. Cai, Talanta, 2019, 202, 21–26 CrossRef CAS PubMed.
- T. Di Giulio, E. Mazzotta and C. Malitesta, Biosensors, 2020, 11, 3 CrossRef PubMed.
- X. Zhang, S. Yang, R. Jiang, L. Sun, S. Pang and A. Luo, Sens. Actuators, B, 2018, 254, 1078–1086 CrossRef CAS.
- P. Gupta and R. N. Goyal, RSC Adv., 2015, 5, 40444–40454 RSC.
- R. Keçili, Int. J. Anal. Chem., 2018, 2018, 4359892 Search PubMed.
- J. Chen, H. Huang, Y. Zeng, H. Tang and L. Li, Biosens. Bioelectron., 2015, 65, 366–374 CrossRef CAS.
- S. Wang, G. Sun, Z. Chen, Y. Liang, Q. Zhou, Y. Pan and H. Zhai, Electrochim. Acta, 2018, 259, 893–902 CrossRef CAS.
- R. Jalili and M. Amjadi, Sens. Actuators, B, 2018, 255, 1072–1078 CrossRef CAS.
- L. Mergola, S. Scorrano, R. Del Sole, M. R. Lazzoi and G. Vasapollo, Biosens. Bioelectron., 2013, 40, 336–341 CrossRef CAS PubMed.
- S. P. Ng, G. Qiu, N. Ding, X. Lu and C.-M. L. Wu, Biosens. Bioelectron., 2017, 89, 468–476 CrossRef CAS PubMed.
- H. Hai, X. An and J. Li, Anal. Methods, 2015, 7, 2210–2214 RSC.
- X. Wang, J. Yu, J. Li, Q. Kang, D. Shen and L. Chen, Sens. Actuators, B, 2018, 255, 268–274 CrossRef CAS.
- S. E. Diltemiz and O. Uslu, Biotechnol. Prog., 2015, 31, 55–61 CrossRef CAS.
- P. Luliński, M. Bamburowicz-Klimkowska, M. Dana and D. Maciejewska, J. Sep. Sci., 2017, 40, 1824–1833 CrossRef PubMed.
- Y. Tian, P. Deng, Y. Wu, Z. Ding, G. Li, J. Liu and Q. He, Biomolecules, 2019, 9, 294 CrossRef CAS PubMed.
- W. Zheng, M. Zhao, W. Liu, S. Yu, L. Niu, G. Li, H. Li and W. Liu, J. Electroanal. Chem., 2018, 813, 75–82 CrossRef CAS.
- A. Göçenoğlu Sarıkaya, B. Osman, T. Çam and A. Denizli, Sens. Actuators, B, 2017, 251, 763–772 CrossRef.
- V. Ricotta, Y. Yu, N. Clayton, Y.-C. Chuang, Y. Wang, S. Mueller, K. Levon, M. Simon and M. Rafailovich, Analyst, 2019, 144, 4266–4280 RSC.
- L. A. Agrofoglio, A. Krstulja, C. De Schutter, P. Favetta, R. Delépée, V. Roy, C. Dejous, H. Hallil, J.-L. Lachaud and N. Lebal, IRBM, 2014, 35, 66–71 CrossRef.
- MIP Technologies, http://www.miptechnologies.com/, (accessed March 29, 2022).
- Allergy Amulet, https://www.allergyamulet.com/, (accessed May 29, 2022).
- M. Sundhoro, S. R. Agnihotra, B. Amberger, K. Augustus, N. D. Khan, A. Barnes, J. BelBruno and L. Mendecki, Food Chem., 2021, 344, 128648 CrossRef CAS.
- R. Rapini, F. Canfarotta, E. Mazzotta, C. Malitesta, G. Marrazza, S. Piletsky and E. Piletska, Analyst, 2019, 144, 7290–7295 RSC.
- MIP Diagnostics, https://www.mip-dx.com/, (accessed March 29, 2022).
- Z. Altintas, A. Guerreiro, S. A. Piletsky and I. E. Tothill, Sens. Actuators, B, 2015, 213, 305–313 CrossRef CAS.
- K. Smolinska-Kempisty, A. Guerreiro, J. Czulak and S. Piletsky, Sens. Actuators, B, 2019, 301, 126967 CrossRef CAS.
- MarketsandMarkets™, https://www.marketsandmarkets.com/Market-Reports/biosensors-market-798.html, (accessed March 29, 2022).
| This journal is © The Royal Society of Chemistry 2022 |