
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iron-catalyzed graphitization for the synthesis of nanostructured graphitic carbons
R. D.
Hunter
a,
J.
Ramírez-Rico
 b and
Z.
Schnepp
*a
b and
Z.
Schnepp
*a
aSchool of Chemistry, University of Birmingham, Birmingham, B152TT, UK. E-mail: z.schnepp@bham.ac.uk
bDpto. Fisica de la Materia Condensada and Instituto de Ciencia de Materiales de Sevilla, Universidad de Sevilla-CSIC, 41092 Sevilla, Spain
First published on 7th February 2022
Abstract
Carbons are versatile and diverse materials that have numerous applications across energy and environmental sciences. Carbons with a graphitic structure are particularly appealing due to their high chemical stability, large surface areas and high thermal and electronic conductivity. Numerous methods exist to produce nanostructured graphitic carbons but some of these can be energy-intensive and/or have problems with scalability. One option that is being increasingly explored is the process of iron-catalyzed graphitization. This simply involves the pyrolysis of carbon-rich precursors in the presence of an iron catalyst and has been used to produce carbons with a wide range of structures and properties. This review will examine the current field of iron-catalyzed graphitization, with a focus on molecular organic or biomass precursors. Bio-derived precursors are particularly attractive as a potential option for sustainable production of graphitic carbons. We start with a brief introduction to some key carbon structures, the current applications in which they are employed and some of the key methods that have been developed to produce nanostructured graphitic carbons. We will then review the history of catalytic graphitization before evaluating the wide range of conditions and precursors that have been employed in catalytic graphitization. Finally, this review will investigate the current challenges facing iron-catalyzed graphitization, looking particularly at the limitations of the current understanding of the mechanistic aspects of graphitization, with a view to outlining where research in this field might progress.
 R. D. Hunter | Robert Hunter is currently studying for a PhD in the School of Chemistry at the University of Birmingham in the group of Dr Zoe Schnepp. His research is focused on the synthesis of porous graphitic carbons by iron-catalyzed graphitization of biomass-derived precursors. His work involves the use of both experimental and theoretical techniques to study mechanistic aspects of the graphitization process. |
 J. Ramírez-Rico | Joaquin Ramirez-Rico is a professor of Physics at the University of Sevilla, in Spain. He is the scientific director of the X-ray facility and heads a research group working on methods to convert biomass and natural resources into advanced materials for structural and functional applications. He was a Postdoctoral Fellow in Northwestern University and specialized in synchrotron radiation analysis for in situ characterization of materials. His current interests are in carbon materials for electrochemical energy storage and wastewater treatment. |
 Z. Schnepp | Zoe Schnepp is an Associate Professor in chemistry in the School of Chemistry at the University of Birmingham. Her research group is focussed on the synthesis of nanomaterials from biomass precursors, including ceramic nanostructures and porous carbons. |
1. Introduction
Carbon has been at the heart of human technological development since the ancient discovery that metals could be extracted from rocks by heating them with charcoal (smelting). In the modern era, carbons find broad applications in energy and environmental applications as well as in pigments and as fillers for elastomers.1 The value of carbon materials can be evidenced by the fact that graphite is now classed as a critical material,2 due to its importance in refractory materials and battery technology.3 Many governments now recognise the need for future carbon materials to be sustainable, both for environmental concerns and economic security.One class of carbon materials that is receiving increasing attention is that of nanostructured graphitic carbons. These boast a wide variety of useful properties that include high chemical stability, large accessible surface areas and high thermal and electronic conductivity. This makes graphitic carbon materials valuable in technologies such as batteries,4 fuel cells5 and in separation/purification science.6 Numerous techniques have been developed to produce complex graphitic nanostructures, e.g., chemical vapour deposition,7 arc discharge,8 or laser ablation9 for the synthesis of carbon nanotubes. However, many of these methods are energy intensive and/or difficult to scale up. Therefore, there has been an international drive to develop cheaper, scalable, and more sustainable routes to nanostructured graphitic carbons.10
A promising method for the synthesis of nanostructured graphitic carbons is catalytic graphitization. Catalytic graphitization is broadly defined as the “transformation of non-graphitic carbon into graphite by heat treatment in the presence of certain metals or minerals”.11 The process normally occurs at a much lower temperature than graphitization without a catalyst, which makes it attractive from both an environmental and economic perspective. Various transition metals have been used to promote catalytic graphitization, but iron is particularly appealing, due to its abundance and low toxicity. Iron-catalyzed graphitization can be considered to include processes such as the production of graphite within blast furnaces or the chemical vapour deposition synthesis of carbon nanotubes. We will discuss these briefly, but the bulk of this article will describe the procedure of pyrolyzing mixtures of iron and organic precursors in an inert atmosphere to produce a range of nanostructured graphitic materials (Fig. 1). The simple conditions and wide range of potential organic precursors available for this process (including raw biomass) make it particularly appealing in terms of sustainability. We will compare the many examples of iron-catalyzed graphitization later in the review, but it is useful to start with a brief overview of carbon chemistry.
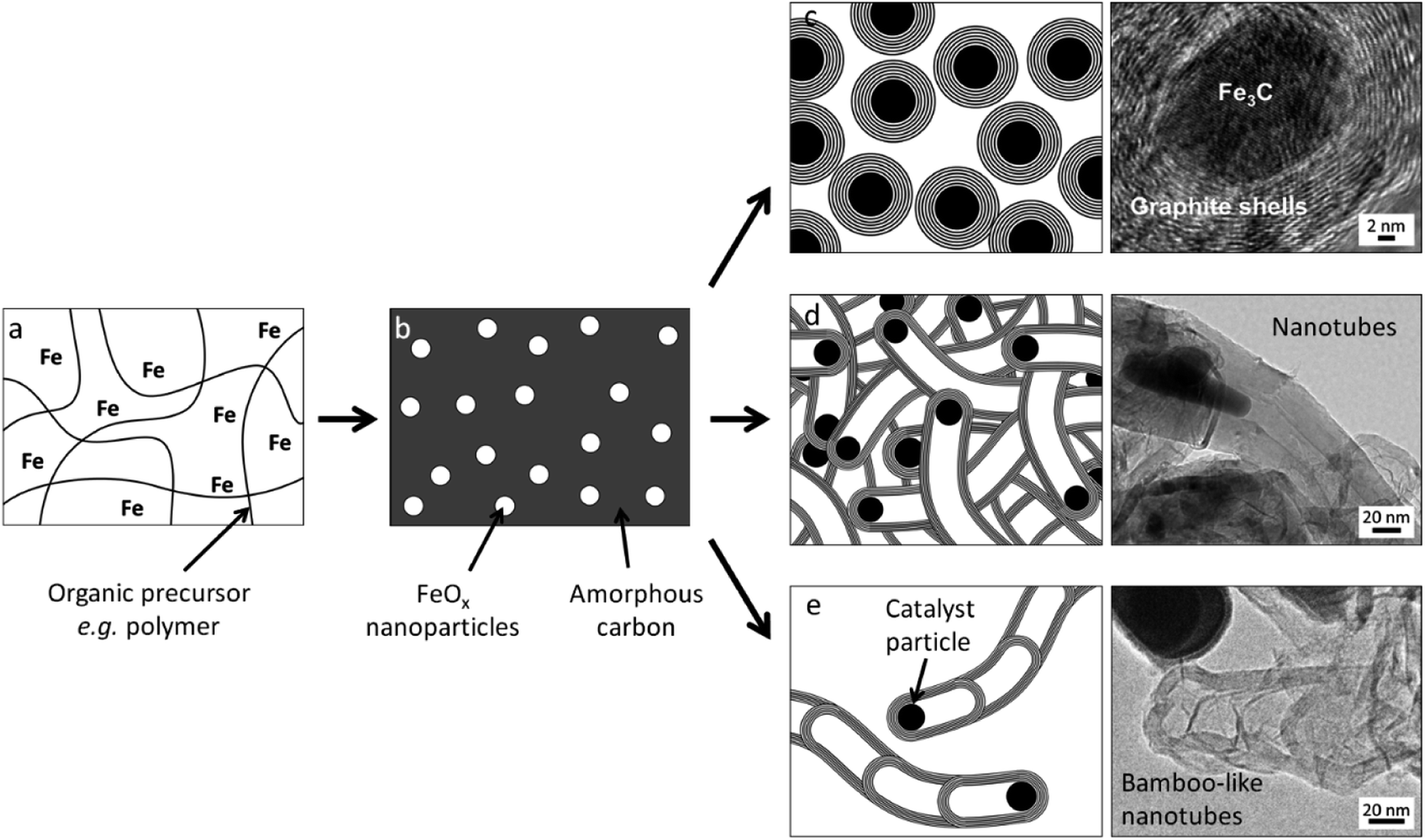 | ||
| Fig. 1 Schematic of a typical iron-catalyzed graphitization process of pyrolyzing (a) an organic/iron mixture to initially produce (b) amorphous carbon and iron oxide nanoparticles then a variety of graphitic nanostructures such as (c) shells, (d) nanotubes and (e) bamboo-like nanotubes. Images reproduced with permissions from ref. 145 and 160. | ||
2. Carbon materials
2.1 Defining carbon structures
Defining (and naming) carbon materials is made challenging by the complexity and wide range of structures that are possible. Heating any organic material (synthetic or biological) in a low-oxygen atmosphere will generate a carbon-rich solid, with the chemistry and structure depending on the heating conditions and the nature of the precursor. Most of these carbon materials do not have a perfectly regular structure like that of diamond or graphite but instead are comprised of a mixture of features with significant variability and disorder. Considerable effort has gone into characterizing different types of carbon materials, for example through 13C NMR spectroscopy,12 Raman spectroscopy,13 X-ray diffraction,14,15 transmission electron microscopy,16 total scattering,17 electron energy loss spectroscopy (EELS) and X-ray photoelectron spectroscopy.18 These techniques seek to probe features such as hybridization of the carbon atoms, the presence of sheet-like structures, interlayer spacing, orientation of stacked sheets, and sheet curvature caused by pentagonal or heptagonal rings of carbon. These materials are not just academic curiosities. Carbon blacks and activated carbons, for example, are widely used for air and water purification, as pigments in inks and as fillers in elastomers and plastics.19 More recently, new carbon materials are also being developed for high-tech energy applications such as anodes for lithium and sodium batteries, where understanding of the fine structure is critical to optimizing their properties.20–22Amorphous carbons are defined as containing a mixture of sp2 and sp3 hybridised carbon atoms and display no long-range crystalline order throughout their structure.23 The surface of amorphous carbon typically consists of many reactive, dangling bonds,24 and may also contain heteroatoms such as hydrogen, oxygen, nitrogen, or boron throughout the carbon network, depending on the method of synthesis. In contrast, graphitic carbons are composed of sp2 hybridized carbon atoms arranged in hexagonal layers with long-range order that can be detected by diffraction methods. Strictly speaking, the term ‘graphitic’ should be reserved for carbon materials where the individual graphene layers are packed in an ABAB vertical arrangement.25 However, many other carbon materials are commonly referred to as ‘graphitic’ if they have sheet-like structures arranged with an interplanar spacing similar to that of graphite. These can include multiwalled carbon nanotubes (Fig. 2a),26 ‘nanoscrolls’,27 bamboo-like carbon nanotubes (Fig. 2b),28,29 which have a segmented structure, and onion-like carbons (Fig. 2c),30–32 which consist of roughly spherical concentric layers of graphitic carbon surrounding a central ‘core’ that can either be hollow or contain a nanoparticle catalyst. Graphitic carbons have various appealing properties, including high electronic conductivity and chemical and thermal stability.33 Another term that is commonly used in describing some carbon materials is ‘turbostratic’, derived from “turbo” (rotated) and “strata” (layer). Turbostratic carbons contain stacked sheets of sp2-bonded carbon like graphite, but the sheets may exhibit random translation of the graphene layers along with rotational disorder, resulting in areas of larger interlayer spacing.34 This can be observed in X-ray diffraction patterns as a broadening and/or a small shift to a lower 2θ value of the characteristic (002) peak.
 | ||
| Fig. 2 Transmission electron microscope (TEM) images of (a) multiwalled carbon nanotube, (b) bamboo-like carbon nanotubes and (c) onion-like graphitic carbon shell. Images modified with permission from ref. 26, 29 and 31. | ||
An important factor in describing the chemistry of carbonization and carbon materials is the fact that some carbons resist graphitization. This was discovered in the early twentieth century and led to a seminal paper by Rosalind Franklin, who coined the terms ‘graphitizing’ and ‘non-graphitizing’ to describe different types of carbon.35 She observed that some organic materials, such as polyvinylchloride and pitch, could be converted to crystalline graphite at >2200 °C whereas others, such as polyvinylidene chloride and sugar retain a porous isotropic structure even up to 3000 °C. Graphitizing and non-graphitizing carbons are commonly called ‘soft’ and ‘hard’ carbons, terms that relate to their observed physical properties. Understanding of the structure of hard and soft carbons has evolved with the development of increasingly sophisticated analytical techniques. The structures proposed by Franklin involved small crystallites of graphite, each of which contains several small graphene layers. In graphitizing carbons, these crystallites are approximately parallel to each other, facilitating graphitization (Fig. 3a). In non-graphitizing carbons, she proposed that the individual crystallites were crosslinked in a random orientation (Fig. 3b). More recent studies, using aberration-corrected TEM, 13C NMR and total scattering, provide evidence for a ‘fullerene-like’ structure for microporous carbons, involving curved sheets of graphene arising from non-hexagonal carbon rings (Fig. 3c and d).36–39 The structures of the different types of carbon are important as they may have implications for the process of catalytic graphitization as well as helping us to describe and characterize some of the materials produced by catalytic graphitization.
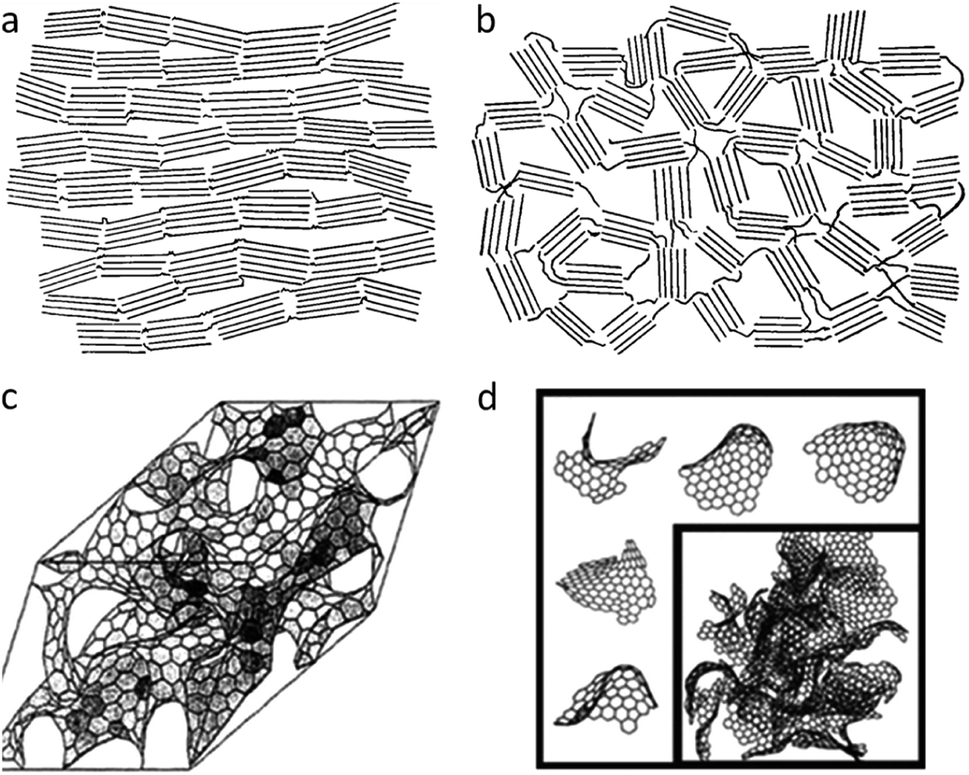 | ||
| Fig. 3 Structures of (a) graphitizing and (b) non-graphitizing carbons proposed by R. Franklin. Also (c) curved graphitic sheet and (d) fullerene-like structural models of amorphous carbon. Images modified with permission from ref. 35, 38 and 39. | ||
2.2 Applications of carbon materials
As noted above, there are numerous applications for carbon materials and some of these represent large industries, for example the production of carbon black as a filler to improve the wear resistance of tyres.40 Other significant industrial examples include carbon fibres for lightweight composites41 and activated carbon for purification and environmental remediation.42 It is outside of the scope of this article to cover all the applications of carbons and there are numerous reviews on the subject.43–45 Instead, we will focus on several key applications that are the motivation of much of the work in the field of catalytic graphitization. These applications exploit the many useful properties of carbons produced by catalytic graphitization, such as porosity, electronic conductivity, chemical stability, and tuneable surface chemistry. Another driving force in this area is the potential for catalytic graphitization as a sustainable route to carbons since iron can produce graphitic structures at relatively low temperature (∼800 °C).One of the biggest areas of interest for carbons produced by catalytic graphitization is as electrocatalysts for the oxygen reduction reaction (ORR) in proton exchange membrane fuel cells.46 The ORR occurs at the cathode, where oxygen is reduced by electrons and protons to produce water. This process commonly uses a noble-metal catalyst such as platinum, decorated on porous carbons. However, the cost and scarcity of noble-metals has led to a drive to find noble-metal free alternatives. Numerous authors have turned to functionalised carbons, many of which are produced by pyrolysis of organic species. This simple process can lead to a wide range of carbons, and it is possible to introduce various chemical and structural features to optimise catalytic activity. This can be achieved through the addition of iron species and by incorporating nitrogen functionality via the organic precursor or additives.47 There are various potential ORR-active sites in Fe/N-doped carbons, such as Fe–Nx, N/C, and Fe3C@C48 and many of the materials rely on the beneficial properties of graphitic nanostructures (such as high electronic conductivity and thermal/chemical stability) that are produced during synthesis.
Pure graphite remains the material of choice for lithium-ion battery anodes as it displays excellent stability and reversibility.49 However, natural and synthetic graphite both have limitations in terms of energy density, cost, and sustainability so there are considerable efforts to develop alternatives.50 Catalytic graphitization offers a promising alternative and has been used to convert biomass into graphitic carbon materials that can be used in lithium-ion batteries.51 The appeal of this method is that enables a relatively low processing temperature and the opportunity to use a renewable feedstock. Catalytic graphitization also has the advantage of being able to convert non-graphitizing carbons into graphitic structures. There are only a handful of examples of iron-catalyzed graphitization being used to produce materials for lithium-ion battery anodes but given the wide interest in using biomass as a precursor for this application,52 it is likely to become a much larger area of research in the future.53 It should be noted that there are also many efforts to produce nanostructured graphitic carbons for other energy storage applications such as sodium-ion batteries and supercapacitors.54 The structural and chemical requirements for these applications are different and there are now many examples of carbons for energy storage produced by pyrolysis or catalytic graphitization of a range of precursors.55,56
A final promising application for nanostructured graphitic carbons is for use as adsorbents for separation or purification. As mentioned above, activated carbons are already used widely in industry for removal of various impurities or contaminants from air or water. Recently, many authors have investigated graphitic nanostructures such as carbon nanotubes and graphene for purification and separation applications.57,58 There are various ways that graphitic features are believed to facilitate adsorption of contaminants, including π–π interactions with aromatic functionality of organic pollutants59 or binding of heavy metal ions to defect sites or to tube ends.60 Indeed, carbon nanostructures with significant sp2 character such as nanotubes or graphene have been shown to have specific advantages over activated carbon.61–63 However, the high cost of these specialist carbon materials limits their range of applications, particularly in environmental remediation.64 Iron-catalyzed graphitization may offer an economical and sustainable solution, given the potential to transform bio-derived materials65 or even waste biomass66 into complex carbons with promising adsorption properties.
2.3 Production of carbon materials
This review focuses on the simple method of pyrolyzing organic molecules or materials with an iron precursor to generate graphitic carbon nanostructures. However, it is useful to give a brief overview of other approaches to synthesizing carbon materials as studying them can offer insight into the mechanism of iron-catalyzed graphitization. The first known production of carbon materials dates to the paleolithic era, where early humans used charcoal for cave paintings.67 Charcoal was made in simple earth-covered kilns, where wood is slowly burned with a limited supply of air. The development of charcoal is tied to the discovery of metal smelting.68 Charcoal fires burn much hotter than wood fires, which are limited by the release of significant amounts of water and volatiles. The heat and reductive environment of a charcoal fire is what facilitates the extraction of metals from their ores. Charcoal has endured as a useful material to humankind due to its ease of production and high purity compared to coal and coke.69 Many modern carbon materials are produced using technology that is fundamentally very similar to that of ancient charcoal kilns i.e. pyrolysis of organic matter in a low-air environment. However, modern furnaces enable much better control over heating conditions and purity of atmosphere. The main factor affecting the properties of the resulting carbon is the nature of the organic precursor. For example, carbon black is a fine powder with high surface area and electronic conductivity that is typically produced by the reaction of hydrocarbon fuels with a limited supply of air.19 In contrast, biochars are produced by the pyrolysis of biomass in the absence of oxygen and tend to maintain the macrostructure of the biomass source.70 Biochars have reasonably high porosity but carbons that are produced by pyrolysis of solid materials can gain higher porosity through ‘activation’. This can be achieved through chemical (e.g. phosphoric acid, sodium hydroxide or zinc chloride) or physical (e.g. steam) treatment of the char.71Carbon black and activated carbon are two of the most industrially important carbon materials but there have been substantial efforts to develop routes to more specialist carbon materials such as fullerenes or nanotubes. One of the earliest examples is arc discharge. In this method, an electrical arc between two electrodes in a non-conductive gas is used to generate a plasma. The high-temperature plasma vaporizes solid graphite from the anode, which then deposits onto the cooled cathode as structures such as carbon nanotubes72 and fullerenes.73 The graphite anode can be doped with various metals such as nickel, iron or cobalt, which act as catalysts for the growth of nanotubes and can facilitate growth of specific structures such as single or double-walled carbon nanotubes.74 Arc-discharge continues to be an area of interest for many researchers75 and there is still much to be learned about graphitization in this system, but that is outside the scope of this review. Another technique for growing carbon nanotubes is laser ablation, which was developed to create more controllable conditions. Laser ablation was first reported by Guo et al.,9 and uses a pulsed laser beam to vaporize a graphite target with embedded metal catalyst particles. Like arc discharge, the resulting carbon vapour is deposited onto a cooled substrate in the form of nanotubes and fullerenes.76
Arc-discharge and laser ablation both require a lot of energy and are difficult to scale up. As a result, an alternative method called chemical vapour deposition (CVD) was developed to facilitate the industrial manufacture of carbon nanotubes. In addition to the lower energy requirements, CVD has the advantage of not requiring pure graphite as a reagent. Instead, a gaseous hydrocarbon such as acetylene flows over a two-dimensional substrate. The substrate is coated in nanoparticles of a metal catalyst (typically iron, cobalt, or nickel) and when the hydrocarbon gas is heated, it decomposes and dissolves into the catalyst. Carbon nanostructures grow from the catalyst nanoparticles by various mechanisms such as float growth or base growth (Fig. 4). Compared to arc discharge and laser ablation, CVD can produce a relatively high purity product in a high yield. By changing various experimental parameters such as reaction temperature or the chemical nature of the catalyst or substrate, it is possible to influence the structure of the final product and CVD methods are widely used in industrial manufacture of carbon nanostructures. It is worth noting that many of the methods discussed in this section employ transition metal catalysts such as nickel, cobalt or iron. Considerable effort has gone into elucidating the mechanism by which these catalysts produce carbon nanostructures. While the conditions of iron-catalyzed graphitization are very different from those of arc discharge, laser ablation and CVD, there is much we can learn from the extensive mechanistic studies of these processes, and we will return to this later in the review.
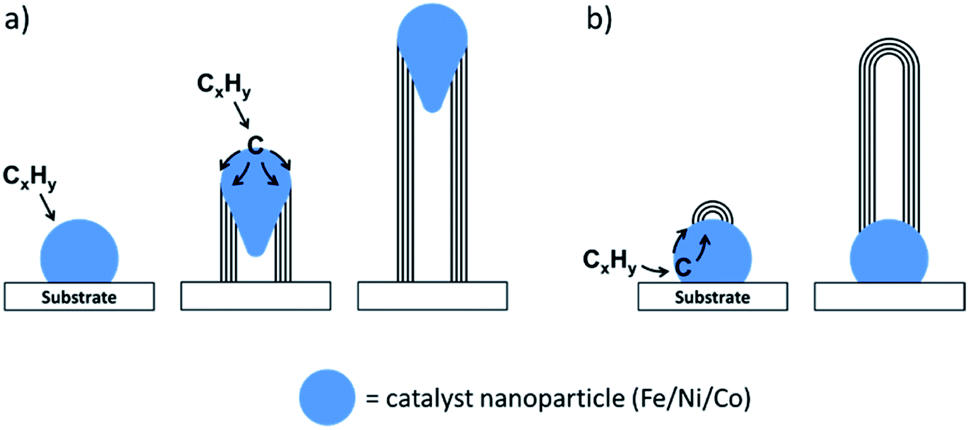 | ||
| Fig. 4 (a) Float growth and (b) base growth mechanisms of carbon nanotube growth in chemical vapour deposition, driven by decomposition of a hydrocarbon gas and dissolution of carbon into a catalyst particle. | ||
3. A brief history of catalytic graphitization
Humankind has been exploiting catalytic graphitization for thousands of years, long before the technology evolved for us to understand the chemistry. For example, pottery from an ancient settlement called Keeladi in India has a durable black coating that was found to be made up of multi-walled carbon nanotubes.77 These are believed to have been formed during firing from carbonization of plant matter alongside naturally occurring catalytic iron species. In another example, ancient Damascus steel blades, renowned for their strength and sharpness, have shown a complex microstructure of carbon nanotubes and iron carbide nanowires.78 Catalytic graphitization by iron may even occur naturally throughout the universe, with evidence that iron has driven the formation of graphitic structures found on meteorites.79In more recent years, there have been more deliberate efforts to understand and exploit the process of catalytic graphitization. Many of these have stemmed from an interest in improving the efficiency of graphite production by reducing the typical synthesis temperature of >3000 °C.80 The ability of ‘impurities’ to drive catalytic graphitization at much lower temperatures was discovered by Acheson81 in 1896. Since then, many different elements and alloys have been shown to promote the transformation of amorphous carbon to graphitic carbon.82 Early attempts involved the pyrolysis of mixtures of metal powders with amorphous carbons.83 The differences in the type of carbons formed (graphitic or turbostratic/graphitic) were linked to two different graphitization mechanisms. The first of these was proposed to be formation and subsequent decomposition of metal carbides and the second was suggested to involve dissolution of carbon into the catalyst, followed by reprecipitation.84 The latter mechanism was believed to be driven by formation of a solution that is saturated with respect to disordered carbon but supersaturated with respect to graphite.85,86 The precise composition of the carbon precursor was found to affect the degree of graphitization, but importantly, it was shown that even ‘non-graphitizable’ carbons could be graphitized at relatively low temperatures (<1400 °C).87
The specific phenomenon of iron-catalyzed graphitization has been studied in considerable detail since the early reports of catalytic graphitization. This is partly due to the importance of graphitization within the iron and steel industry. For example, in blast furnaces, iron ore is heated with a porous carbon such as coke, which acts as a reducing agent and energy source and is also crucial in maintaining the permeability of the reactor contents for upward flowing gases.88 For environmental reasons, there is a need to minimize coke consumption, which has driven investigations into coke degradation and transformation within blast furnaces.89 One mechanism by which this occurs is the graphitization of coke carbon. A model study used to probe this system combined coke with a fine iron powder (particle size <5 μm). Heating the mixture resulted in the formation of graphitic carbon above 1200 °C.90 The process is believed to result from a dissolution–precipitation mechanism within molten iron catalyst particles.
Another observation that prompted study into iron-catalyzed graphitization was the unwanted deposition of carbon on metals exposed to carbon monoxide. For example, the conversion of carbon monoxide to solid carbon on iron surfaces was observed to cause deactivation of Fischer–Tropsch catalysts,91 damage to brickwork in blast furnaces,92 and carburization in heat exchangers of nuclear reactors.93 While carbon deposition can be problematic in these circumstances, it was recognised that the same process may enable industrial production of useful carbons. In studying carbon deposition, many authors observed tube like filaments, with small crystals of iron or iron carbide (Fe3C), indicating that the iron had catalyzed the filament formation. Work quickly moved to the deliberate synthesis of these tubular filaments from decomposition of carbon monoxide94 or benzene95 over iron catalysts. This research led to development of CVD synthesis of carbon nanotubes,96 as discussed above.
Alongside development of gaseous precursors for catalytic graphitization, there has been increasing interest in iron-catalyzed graphitization of non-gaseous organic precursors. This typically involves mixing solvated iron precursors such as ferrocene, iron nitrate, iron acetate or iron chloride with a solid or dissolved organic material.83,97,98 The mixture is then pyrolyzed in an inert atmosphere to drive thermal decomposition of the organic matter to amorphous carbon. Alongside this, the iron precursor also decomposes. In many cases this results firstly in iron oxide nanoparticles, which are then transformed by carbothermal reduction to Fe or Fe3C. These particles then drive graphitization, which is believed to occur either by dissolution/reprecipitation of carbon or formation/decomposition of carbides. Due to the relatively small size of the catalytic iron nanoparticles, the graphitization process occurs at the relatively low temperature of 800 °C or 900 °C. In many instances, tubular nanostructures are formed, suggesting that the iron-containing catalyst particles move through the amorphous carbon matrix during graphitization, perhaps driven by dissolution and reprecipitation of carbon. Multiple variations of this procedure now exist, using a wide range of organic precursors and these will form the basis of the next sections of this review. We will then return to the question of the mechanism of graphitization.
4. Organic precursors used in iron-catalyzed graphitization
This section will present the main categories of organic precursors that have been used in iron-catalyzed graphitization. A few examples from the literature will be discussed to highlight the diversity of the method, focusing particularly on those in which effort has been made to explain or influence the formation of the graphitic carbon product.4.1 Small organic molecules
The simplest organic precursors used in iron-catalyzed graphitization are small molecules such as sucrose and glucose. These sugars are highly soluble in water so can be combined with aqueous iron salts such as iron(III) nitrate to produce a homogeneous solution or gel. This is then dried and pyrolyzed to yield graphitic carbon structures (Fig. 5a). An example of this can be seen in the work of Yang et al., who synthesized graphitic shell-like structures containing metal particles by pyrolyzing a mixture of sucrose and iron(III) nitrate above 700 °C (Fig. 6a).99 The metal nanoparticles can be removed by acid-washing to leave graphitic “capsules” (Fig. 6b), alongside a smaller number of graphitic nanotubes, highlighting the possibility of the formation of different nanostructures within the same product. Sevilla et al. found that a combination of iron nitrate and glucose could be used to produce filamentous multi-walled nanotubes, suggesting that the catalyst in this system is highly mobile (Fig. 6c and d).100 It is difficult to know whether sucrose and glucose themselves influence the process of graphitization as the metal![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :organic ratio and heating conditions are quite different in these two papers. Also, the sucrose was mixed with iron nitrate in water, whereas the glucose system employed ethanol as a solvent. It seems most likely, however, that the metal:organic ratio is the dominant reason for the difference in these systems. The ‘shell’ like structures were produced from a system containing 3 millimoles of iron per gram of sucrose and the nanotubes came from mixtures of 0.4 or 0.8 millimoles of iron per gram of glucose. The glucose system would therefore have a lot more amorphous carbon available, which could allow for substantial movement of the catalyst particles during catalytic graphitization before all the carbon is consumed. In a further example using aqueous glucose and iron nitrate, shell-like structures were observed rather than nanotubes.101 Again, it is difficult to conclude anything about the mechanism as the authors also used NaCl as a sacrificial template but the ratio of 1.7 millimoles of iron per gram of glucose is similar to the levels used by Yang et al. to produce shell-like structures. This lends credence to the argument that the metal:organic ratio is most important in dictating the type of structures formed in these systems. Another study that used sucrose, glucose and urea with iron acetylacetonate showed that all organic precursors resulted in carbons that show a mixture of shell-like and short tubular structures.102 Some differences were observed in the iron/iron carbide composition and the graphite peak height in XRD but the suggestion overall is that iron:organic ratio has a more significant impact on graphitization than the chemistry of the different small-molecule precursors.
:organic ratio and heating conditions are quite different in these two papers. Also, the sucrose was mixed with iron nitrate in water, whereas the glucose system employed ethanol as a solvent. It seems most likely, however, that the metal:organic ratio is the dominant reason for the difference in these systems. The ‘shell’ like structures were produced from a system containing 3 millimoles of iron per gram of sucrose and the nanotubes came from mixtures of 0.4 or 0.8 millimoles of iron per gram of glucose. The glucose system would therefore have a lot more amorphous carbon available, which could allow for substantial movement of the catalyst particles during catalytic graphitization before all the carbon is consumed. In a further example using aqueous glucose and iron nitrate, shell-like structures were observed rather than nanotubes.101 Again, it is difficult to conclude anything about the mechanism as the authors also used NaCl as a sacrificial template but the ratio of 1.7 millimoles of iron per gram of glucose is similar to the levels used by Yang et al. to produce shell-like structures. This lends credence to the argument that the metal:organic ratio is most important in dictating the type of structures formed in these systems. Another study that used sucrose, glucose and urea with iron acetylacetonate showed that all organic precursors resulted in carbons that show a mixture of shell-like and short tubular structures.102 Some differences were observed in the iron/iron carbide composition and the graphite peak height in XRD but the suggestion overall is that iron:organic ratio has a more significant impact on graphitization than the chemistry of the different small-molecule precursors.
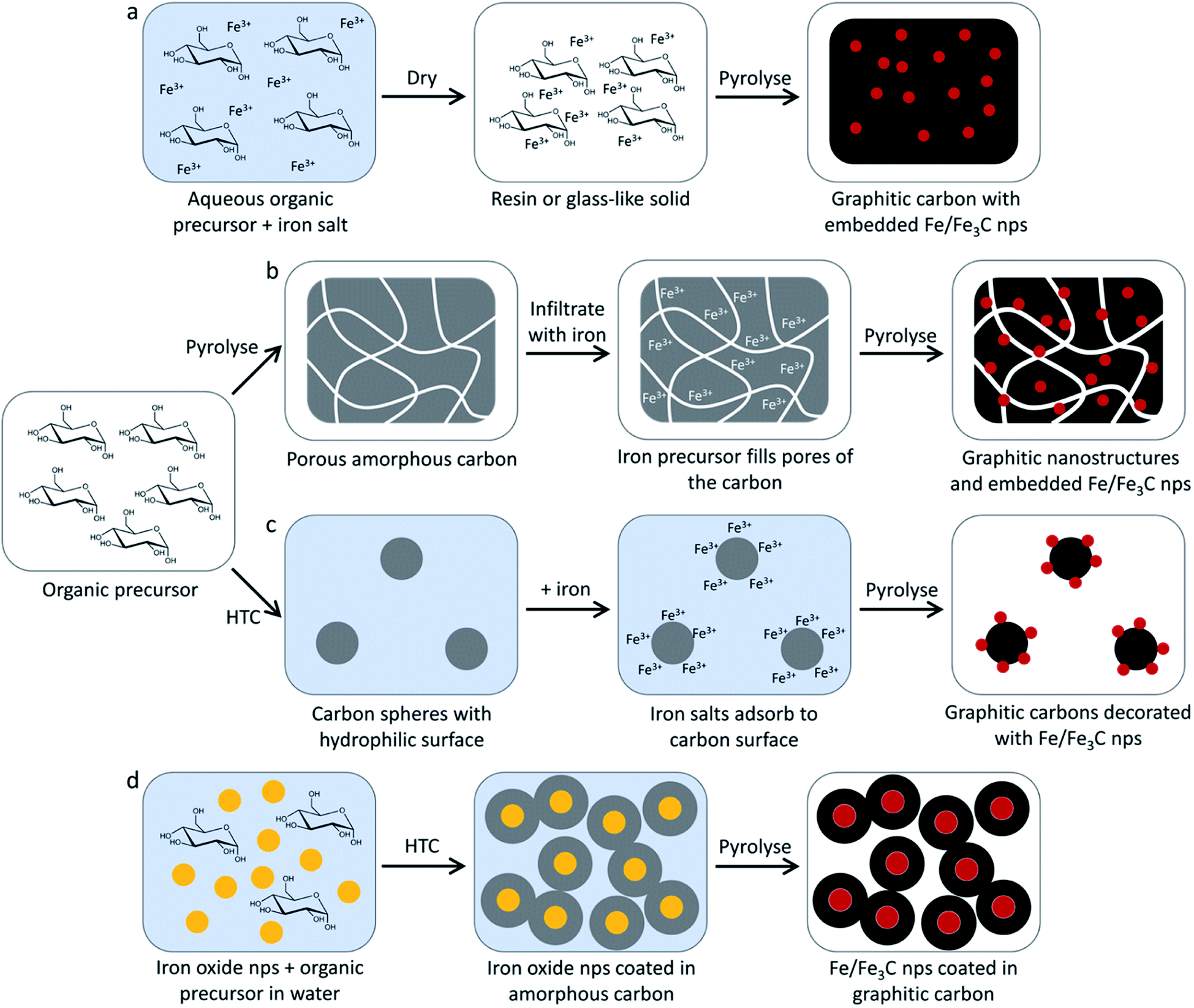 | ||
| Fig. 5 Schematic showing some of the methods that have been used to prepare nanostructured graphitic carbons from small organic molecule precursors such as glucose. These include (a) simple pyrolysis of a mixture of the organic precursor with an iron salt, (b) pyrolysis of the organic precursor followed by infiltration with an iron salt and a second pyrolysis step, (c) hydrothermal carbonization (HTC) followed by adsorption of iron species onto the resulting carbon spheres and (d) hydrothermal carbonization around iron oxide nanoparticles, followed by pyrolysis. | ||
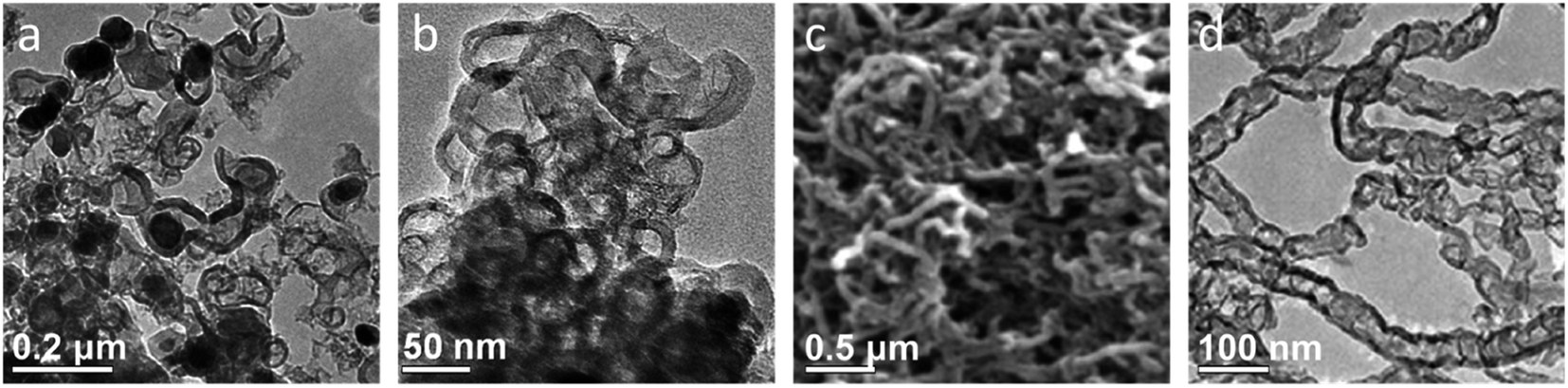 | ||
| Fig. 6 TEM images of (a) graphitic nanostructures with embedded iron nanoparticles produced by pyrolyzing a mixture of sucrose and iron nitrate and (b) the same sample after acid-washing. (c) Scanning electron microscope (SEM) and (d) TEM images of graphitic nanotubes produced from glucose and iron nitrate (after acid washing to remove iron catalyst particles). Figures modified with permission from ref. 99 and 100. | ||
Sugars have also been used to produce carbons where graphitic nanostructures are combined with macrostructural features such as pores or spherical shapes. The simplest approach to this is to initially produce an amorphous carbon and then introduce iron before pyrolyzing (Fig. 5b). This can be achieved just by pyrolyzing a sugar such as glucose to produce a porous amorphous carbon,103 but more elaborate structures can be formed by templating sugars with amphiphilic copolymers before carbonizing them.104 In the copolymer-templated example, the resulting amorphous carbon was then decorated with iron oxide nanoparticles by hydrothermal treatment and then pyrolyzed a second time to drive graphitization. Hydrothermal chemistry can also be used to produce the amorphous carbon precursor e.g. via hydrothermal carbonization of sucrose solutions to generate carbon spheres. The surface of these can be impregnated with iron nitrate, followed by pyrolysis to generate carbon spheres with a highly graphitic surface (Fig. 5c).105 Hydrothermal carbonization can also be used to deposit carbon from glucose around iron oxide nanoparticles before pyrolysis to drive graphitization (Fig. 5d). This has the advantage of maintaining a relatively consistent catalyst particle size.106 It is not particularly instructive to continue discussing all examples of iron-catalyzed graphitization that employ small molecules. A selection of examples from the literature are listed in Table 1 and it can easily be seen that a wide range of precursors and conditions have been used, even within this small area focusing on small molecules. It is clear from these examples that there is scope for tailoring graphitic nanostructures even in very simple systems but there is a need for systematic investigation.
| Product description | Organic precursor | Iron source | Temp. (°C) | Notes | Catalyst |
|---|---|---|---|---|---|
| Graphitic tubes/shells108 | Glucose | Fe(NO3)3 | 800 | BET surface area 343 m2 g−1 | Fe3C |
| Graphite103 | Glucose | Fe powder | 1200 | Glucose pyrolysed to form hard carbon before grinding with iron powder | |
| Carbon-encapsulated magnetic nanoparticles, core–shell structure106 | Glucose | Fe3O4 nanoparticles | 700–850 | BET surface area 134–202 m2 g−1 | |
| Graphene-like carbon shell encapsulating iron carbide nanoparticles107 | Glucose | Fe(NO3)3 | 800 | Potassium nitrate added as a promoter, BET surface area 238 m2 g−1 | Fe3C |
| Mesoporous graphitic carbon108 | Glucose | Fe(NO3)3 | 800 | Comparison with starch and cellulose BET surface area 343 m2 g−1 | Fe3C |
| Graphitic mesoporous carbon, carbon nanotubes99 | Sucrose | Fe(NO3)3 | 700/800 | BET surface area 198 m2 g−1 | |
| Nanoporous graphitic carbon, wormlike porous structure104 | Sucrose | Fe(NO3)3 | 600–900 | Initial hydrothermal treatment, F123 copolymer template, BET surface area 329 m2 g−1 | |
| Graphitic nanoribbons105 | Sucrose | Fe(NO3)3 | 800 | Initial hydrothermal treatment | |
| Hollow carbon nanospheres109 | Sucrose | Iron oxide nanoparticles | 450 | Very broad peak in XRD | Fe3C |
| Fe3C or Fe nanoparticles with graphitic shells102 | Glucose, sucrose, or urea | Iron acetylacetonate | 800 | BET surface area 40–240 m2 g−1 | Fe3C or Fe |
| Graphite encapsulated iron carbide/iron nanosheet composites110 | Glucose and glycine | Fe(NO3)3 | 700–1000 | BET surface area 75–260 m2 g−1 | Fe3C then Fe |
| Iron particles with graphitic carbon shells111 | Glucose + dicyandiamide or urea | K3[Fe(CN)6] | 800/900 | NaCl template, dicyandiamide or urea for N-doping | Fe/Fe3C |
| Iron and nitrogen doped carbon nanostructures112 | Glycine | FeCl2 | 900 | Additional template of silica beads, BET surface area 740 m2 g−1 | |
| Iron-doped porous carbon, graphene sheets, particles wrapped by graphitic carbon113 | L-Histidine | Fe2O3 and FeCl3 | 1000 then 1000 | Ball milling and acid wash after first pyrolysis, BET surface area 200–315 m2 g−1 | |
| Metallic nanoparticles in graphitic shells114 | Citric acid | Fe(NO3)3 | 800 | Initial formation of metal citrate gel at 120 °C | |
| Iron carbide encapsulated in graphitic layers115 | 1,8-Diaminonaphthalene | FeCl3 | 700–1000 | Initial polymerisation step, BET surface area 510–920 m2 g−1 | Fe3C |
4.2 Synthetic polymers
A large range of synthetic polymers have been used as precursors for graphitic carbons and these are summarized in Table 2. As with small organic molecule precursors, the simplest way to produce graphitic carbons from synthetic polymers is to produce a homogeneous mixture of the polymer with an iron compound and pyrolyze in an inert atmosphere. For example, Huang et al. produced bamboo-like hollow carbon fibres from an aqueous mixture of polyethylene glycol and iron sulfate.116 Interestingly, these authors found that the carbon nanofibre yield decreased with a high iron loading, suggesting that catalyst particle size may be important in regulating graphitization. Rather than mixing an iron precursor directly with the polymer, it is also possible to mix the iron precursor with a monomer and then initiate polymerization, before pyrolyzing the resulting material. This method has been used to produce graphitic carbon nanostructures from polyfurfuryl alcohol. It should be noted that the structures produced in these systems are turbostratic (i.e. no regular stacking between layers) rather than truly graphitic, as indicated by an absence of hkl diffraction in selected area electron diffraction (SAED) studies.97 This is probably true of most of the examples in this section but is typically not stated by authors.| Product description | Organic precursor | Iron source | Temp. (°C) | Comments |
|---|---|---|---|---|
| Turbostratic carbon shells and tubes surrounding Fe nanoparticles97 | Furfuryl alcohol | Ferrocene | 450–820 | Initial polymerization step to polyfurfuryl alcohol |
| Turbostratic carbon, shell-like structures125 | Furfuryl alcohol | Ferrocene | 700 | BET surface area 200 m2 g−1 |
| Graphitic mesoporous carbons121 | Phenolic resin | Fe(NO3)3 | 900 | Silica xerogel template, BET surface area 1010 m2 g−1 |
| Graphitic mesoporous carbon117 | Phenolic resin | Fe(NO3)3 or ferrocene | 700 | BET surface areas of 607 m2 g−1 and 248 m2 g−1 |
| Onion-like carbon126 | Phenol/formaldehyde | Ferrocene | 1000 | Different mixing methods compared |
| Microporous carbons, graphitic layers118 | Phenol/formaldehyde | Ferrocene | 1000 | Mechanical mixing compared to solution mixing, BET surface areas 216–632 m2 g−1 |
| Mesoporous carbon122 | Phenol/formaldehyde resin | Fe(NO3)3 | 900 | SBA-15 silica nanocast BET surface area 670 m2 g−1 |
| Mesoporous graphite-like carbon123 | Phenol/formaldehyde | Ammonium iron citrate | 700 | Pluronic P123 as templating agent |
| Ordered mesoporous carbons with partially graphitized network127 | Phenol/formaldehyde | FeCl3/FeSO4 | 800 | Initial hydrothermal treatment and copolymer P123 |
| Graphitic mesoporous carbon128 | Resorcinol/formaldehyde | Iron citrate | 900 | Silica sol template, surface area depends on iron content |
| Highly ordered Fe-containing mesoporous carbon124 | Resorcinol/formaldehyde | Fe(NO3)3 | 800 | Triblock copolymer pluronic F127 templating agent |
| Metal-doped carbon aerogels129 | Resorcinol/formaldehyde | Iron acetate | 900 | BET surface area 461 m2 g−1 |
| Graphitic carbon spheres130 | Resorcinol/formaldehyde spheres | Prussian blue | 1000 | BET surface area 381 m2 g−1 |
| Macroporous monolithic graphitic carbon119 | Resorcinol/formaldehyde xerogel | FeCl3 | 1000 | Spinodal decomposition during polymerization introduces macroporosity, BET surface area 465 m2 g−1 |
| Onion-like or nanocapsule-like graphitic carbon131 | Resorcinol/formaldehyde | Iron(II) acetate | 1100 | Two step pyrolysis to minimise cracking |
| Monolithic porous graphitic carbons132 | Resorcinol, furfural xerogel | FeCl3 | 1050 | BET surface areas up to 400 m2 g−1 |
| Carbon nanotubes and shells120 | Polyethylene/polyvinyl alcohol | Iron hydroxide needles | 750 | |
| Carbon nanofibers with bamboo-like hollow fibril morphology116 | Polyethylene glycol | FeSO4 | 750 | Graphitization believed to be facilitated by sulfur dissolution into the catalyst particles |
| Carbon nanotubes133 | Polypropylene | Fe nanoparticles | 700 | Dissolution of precursor in xylene |
| Multilayer graphitic nanosheets/nanoshells134 | Poly(4-ethylstyrene-co-divinylbenzene) | Iron(II) acetylacetonate | 850 |
A common type of synthetic polymer used for catalytic graphitization is phenolic resins. These are thermosetting polymers that are generally synthesized from the reaction of phenols with formaldehyde and are commonly used as industrial adhesives. The type of structures and porosity of graphitic carbons produced from phenolic resins depends on the type of iron precursor (e.g. iron nitrate or ferrocene)117 and also the method of mixing, with mechanical mixing of solid precursors favouring micropores and homogeneous mixing of solvated precursors favouring mesopores.118 This is due to the homogeneously-mixed sample displaying more graphitic features, compared to the micropores of amorphous carbon. Organic gels formed from the reaction between resorcinol and formaldehyde have also been successfully graphitized by iron-based catalysts. Again, the polymerization is performed in the presence of the iron precursor, allowing a homogeneous mixture of the two components. An interesting example by Hasegawa et al. uses resorcinol, iron chloride and formaldehyde in an ethanol/water solvent.119 The iron chloride acts as an acid catalyst to initiate polymerization and as the reaction proceeds, the polymer network becomes more hydrophobic, leading to spinodal decomposition and the formation of a macroporous structure. The pore size can be controlled simply by varying the ethanol:water ratio and the macroporosity is maintained in the graphitic product after pyrolysis (Fig. 7a).
 | ||
| Fig. 7 (a) SEM image of macroporous carbon produced from resorcinol-formaldehyde resin, (b) TEM image of iron hydroxide needles with amorphous carbon coating and (c) the same sample after further pyrolysis, showing empty needle-shaped carbon shells. Also shown are TEM images of (d) Fe-doped SBA-15 mesoporous silica and (e) carbon templated from that silica and f) ordered mesoporous carbon from Pluronic F127-templating of resorcinol formaldehyde with iron nanoparticle circled. Figures modified with permissions from ref. 119, 120, 122 and 124. | ||
Another route to introduce porosity into graphitic carbons produced from synthetic polymers is to use hard or soft templates. These can be sacrificial or can themselves form part of the carbon product or the catalyst. Maksimova et al.,120 embedded needle-shaped iron hydroxide nanoparticles in polyvinylalcohol and polyethylene before pyrolysis under nitrogen. TEM images of samples heated to 600 °C showed needle-like iron oxide crystals (from iron hydroxide decomposition) coated in a layer of amorphous carbon (Fig. 7b). Pyrolysis to 750 °C results in carbothermal reduction of the iron oxide to iron and a markedly different structure (Fig. 7c) where the carbon has retained the needle-like structure but the catalyst particles have moved out of the carbon shells to form much larger, rounded particles. This suggests that the catalyst phase is highly mobile and the authors propose that this indicates a melting transition of the catalyst particles. Sevilla et al. produced mesoporous carbons by impregnating silica xerogels with a solution of phenolic resin in methanol, followed by pyrolysis and etching of the silica template with HF.121 The resulting mesoporous carbons were then filled with ethanolic iron nitrate and pyrolyzed to 400–900 °C before removing the metal catalyst particles by acid washing. The combination of graphitic features and porosity from the silica template led to remarkably high surface areas and bimodal porosity. Interestingly, these authors found that a lower pyrolysis temperature (≤600 °C) favoured graphitization, perhaps because high temperatures drove sintering of the catalyst particles. In a similar method, Li et al. synthesized a mesoporous silica template (Fig. 7d) with embedded iron oxide particles that was infiltrated with phenolic resin, pyrolyzed at 900 °C and washed with HF to generate a mesoporous graphitic carbon (Fig. 7e).122
The challenge with using silica as a template is the need for harsh treatment (HF or strong NaOH) to remove the silica. An alternative approach is to use soft templates which can be combusted during the pyrolysis process. For example, Wang et al. used Pluronic P123 to introduce mesoporosity into a phenolic resin.123 Pyrolysis without iron produced a carbon with regular porosity and a high surface area of 800 m2 g−1. However, much of this ordered porosity appears to be lost if an iron precursor is also included in the phenolic resin, with the formation of graphitic nanostructures and a lower BET surface area. Another system showed more successful retention of ordered mesoporosity. Li et al. employed a triblock copolymer Pluronic F127 to produce resorcinol-formaldehyde resins with ordered mesoporosity.124 They discovered that a high iron content led to loss of the ordered porosity but that it could be maintained at a low iron:organic ratio (Fig. 7f), with metallic iron nanoparticles dispersed throughout the structure.
4.3 Biopolymers
Polymers derived from biomass (often called biopolymers) offer an attractive alternative to synthetic polymers as they are renewable and could therefore offer a sustainable route to materials. Many biopolymers may also be waste products of industrial processes so producing graphitic carbons may be a way to add value to unwanted materials. In this section, we will consider both soluble and insoluble biopolymers as it is instructive to do so, and some may ‘swell’ rather than truly dissolve. In the following section we will discuss raw biomass. A summary of biopolymer-derived graphitic carbons can be found in Table 3.| Product description | Organic precursor | Iron source | Temp. (°C) | Comments |
|---|---|---|---|---|
| Turbostratic carbon with a ribbon morphology146 | Microcrystalline cellulose spheres | Fe(NO3)3 | 800 | Detailed mechanism study |
| Graphitic carbon shells and tube-like structures147 | Cellulose filter paper | Fe(NO3)3 | 800 | In situ TEM images of graphitization |
| Iron or iron carbide nanoparticles embedded in graphitic carbon matrix148 | Cellulose | Iron oxide nanoparticles | Up to 800 | Initial hydrothermal treatment |
| Mesoporous graphitic carbon149 | Microcrystalline cellulose spheres | Fe(NO3)3 | 800 | Various metals compared |
| Carbon encapsulated iron carbide nanoparticles150 | Cellulose | Fe3O4 nanoparticles from FeCl2/FeCl3 | 800–1600 | Thicker graphitic shell at higher temperature |
| Mesoporous graphite-containing carbon composites151 | Cellulose | FeCl3/Fe(NO3)3 | 500–1000 | Various cellulose precursors |
| Mesoporous graphitic carbon108 | Cellulose fibres | Fe(NO3)3 | 800 | Compared to glucose and starch, BET surface area 358 m2 g−1 |
| Porous graphitic carbon152 | Cellulose | Fe(NO3)3 | 850 | Liquid, gaseous and solid products characterized |
| Microporous or mesoporous carbon108 | Potato starch | Fe(NO3)3 | 800 | Graphitization very slow compared to cellulose or glucose |
| Graphitic carbon nanostructures100 | Starch | Fe(NO3)3 | 900 | Comparison of starch, glucose and sucrose |
| Graphitic-carbon-encapsulated iron nanoparticles141 | Kraft lignin | Fe(NO3)3 | 700–1000 | Larger particles believed to create thicker graphitic shells |
| Graphene-encapsulate iron particles, multilayer graphene sheets/flakes, core–shell structure139 | Kraft lignin | Fe(NO3)3 | 1000 | Lignin dissolved in THF |
| Graphene-encapsulated iron particles140 | Kraft lignin | Fe(NO3)3 | 1000 | Different iron:lignin ratios |
| Graphene-encapsulated iron nanoparticles153 | Kraft lignin | Fe(NO3)3 | 1000 | Effect of pyrolysis gases investigated: Ar, CO2, H2, CH4 |
| Carbon-encapsulated iron nanoparticles and carbon tubules154 | Kraft lignin | Fe nanoparticles or Fe(NO3)3 | 1000 | Comparison of solid and aqueous iron precursors |
| Core shell structures155 | Lignin, cellulose and hemicellulose | Fe(NO3)3 | 1000 | Biomass pyrolyzed before Fe addition and second pyrolysis step. Limited graphitization for lignin |
| Porous carbon sheets143 | Agar | Fe(NO3)3 | 800 | Al(NO3)3 as templating agent |
| Graphitic mesoporous capsules, graphitized carbon156 | Gelatin | Fe(NO3)3 | 800 | Mg(NO3)3 to prevent sintering |
| Sponge-like graphitic carbon142 | Chitosan | FeCl3, FeCl2 or (Fe(Phen)3Cl2) | 900 | Freeze drying of chitosan gel introduces macroporosity |
| N-doped carbon with a high degree of graphitization157 | Chitosan | FeCl3 | 800–1000 | Graphitic N functionality |
The most abundant biopolymer found in nature is cellulose, which is the main component in the cell walls of green plants. Cellulose is a polysaccharide consisting of linear chains of β(1→4) linked glucose units. Cellulose molecules align to form strong microfibrils, with individual cellulose polymers bound together by hydrogen bonds and it is networks and arrays of these microfibrils that provides much of the mechanical strength of plants. Cellulose can be extracted from plant material (or from cellulose-producing bacteria) but is very difficult to solubilise, requiring specialist conditions such as ionic liquids135 or concentrated NaOH at controlled temperatures136 to be dissolved. All the examples of iron-catalyzed graphitization of cellulose therefore use pure solid forms of cellulose such as fibres or microcrystalline powders.
A range of iron precursors have been used for catalytic graphitization of cellulose. Hoekstra et al. pyrolyzed microcrystalline cellulose spheres impregnated with three trivalent iron salts: iron(III) nitrate, ammonium iron(III) citrate and iron(III) chloride.65 The study showed several interesting results. X-ray diffraction of all three systems showed similar compositions in the resulting carbons, with peaks for Fe and Fe3C and a broad peak corresponding to the interlayer plane spacing of graphite. However, only the iron nitrate and ammonium iron citrate showed the shell-like and tube-like graphitic nanostructures characteristic of most examples of catalytic graphitization. The sample prepared from FeCl3 showed much less evidence of graphitic nanostructures in TEM images and appeared to have a high amorphous carbon content. This observation was supported by porosimetry data, which showed mesoporosity in both the iron nitrate and ammonium iron citrate systems but only microporosity in the FeCl3 system (Fig. 8a, d, g). Investigation of the structure of the materials and the mechanism of formation offered insight into the reason for this disparity. Temperature-dependent X-ray diffraction data (Fig. 8b, e and h) showed that all samples initially formed iron oxide (Fe3O4) but for iron nitrate and ammonium iron citrate the peaks are very broad indicating nanoparticles. In contrast, the Fe3O4 peaks for the sample prepared from FeCl3 were sharp, indicating large crystallites. In the nitrate and ammonium iron citrate systems, the magnetite nanoparticles are reduced to wüstite (FeO) nanoparticles (again indicated by very broad diffraction peaks) before reduction to Fe/Fe3C. In contrast, the sharp magnetite peaks in the FeCl3 system are transformed to sharp wüstite peaks before reduction to Fe/Fe3C. SEM images support these observations, with carbons prepared from iron nitrate and ammonium iron citrate containing small nanoparticles of iron, indicated by the bright spots on the backscattered electron images (Fig. 8c, f, i). The carbon derived from FeCl3, however, shows very large iron particles, which appear to be located mainly on the carbon surface. The authors demonstrate that the reason for the very large particles is the volatility of the FeCl3 precursor driving evaporation of FeCl3 from the loaded microcrystalline cellulose spheres and deposition of large particles on the surface. The work provides further evidence that graphitization is closely linked to catalyst particle size. Interestingly, the iron chloride also influences the carbonization process and particularly the formation of micropores because of its Lewis acid character, which ‘activates’ the carbon by promoting dehydration rather than depolymerization reactions.137,138
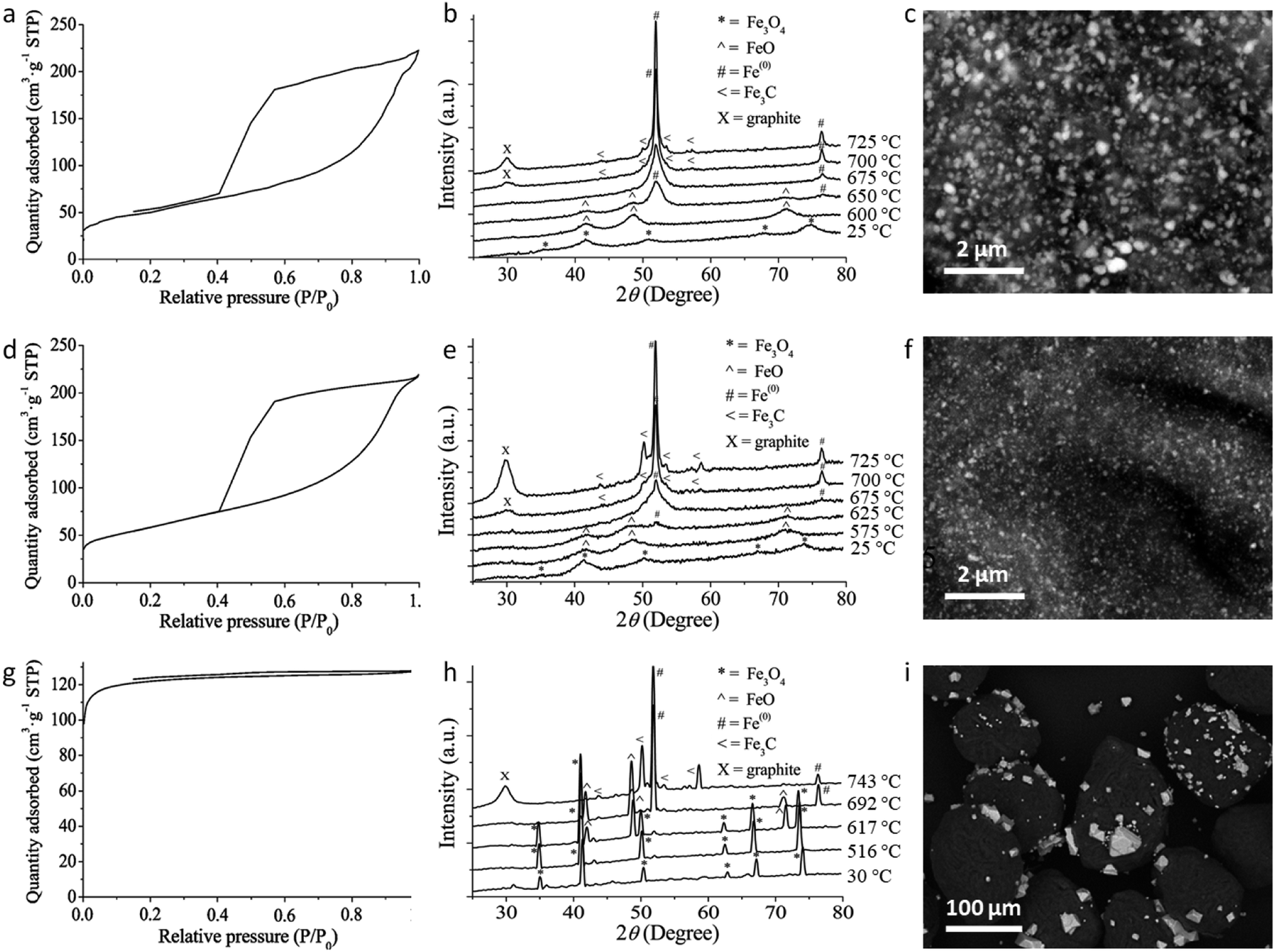 | ||
| Fig. 8 Nitrogen adsorption/desorption isotherms, temperature dependent X-ray diffraction and SEM (backscattered electron) images of carbons produced from microcrystalline cellulose and (a–c) iron nitrate, (d–f) ammonium iron citrate and (g–i) iron(III) chloride. NB: temperature-dependent XRD was performed on samples preheated to 500 °C. Figures modified with permission from ref. 65. | ||
Another study compared the graphitization of cellulose, starch and glucose, using iron nitrate as the iron precursor.108 Cellulose and starch are both composed of polymers that are based on glucose monomers but while glucose is fully water-soluble, cellulose is insoluble and starch only swells to form gels. Pyrolysis of each of these precursors with aqueous iron nitrate resulted in carbons with very different structures, where cellulose and glucose carbons were mesoporous and the starch carbon was primarily microporous and with a much broader graphite XRD peak. Further study of the system showed that a mesoporous graphitic carbon could be formed from starch, but only by extending the pyrolysis time (Fig. 9). Small-angle X-ray scattering data indicated that the iron carbide particles in the starch system grow very slowly, as can be inferred by the broad peaks in X-ray diffraction data. The authors postulated that the complex gel-structure of starch granules allows intercalation of the Fe3+ precursor within the constituent amylose and amylopectin molecules (Fig. 10). The thermal stability of starch means that the organic network persists for a long time around the iron precursor as it gradually agglomerates to form iron oxide nanoparticles. Smaller iron oxide nanoparticles would in turn lead to small iron carbide nanoparticles. Glucose, being a small molecule, decomposes much earlier, which would facilitate mass transport of iron species through the developing carbon. In the insoluble cellulose system, the iron is dispersed over the surface, again facilitating mass transport and particle growth. The implication from this data is that there is a critical size of Fe3C particle that must be reached before graphitization can occur. This is something that has also been suggested by other authors.116,118
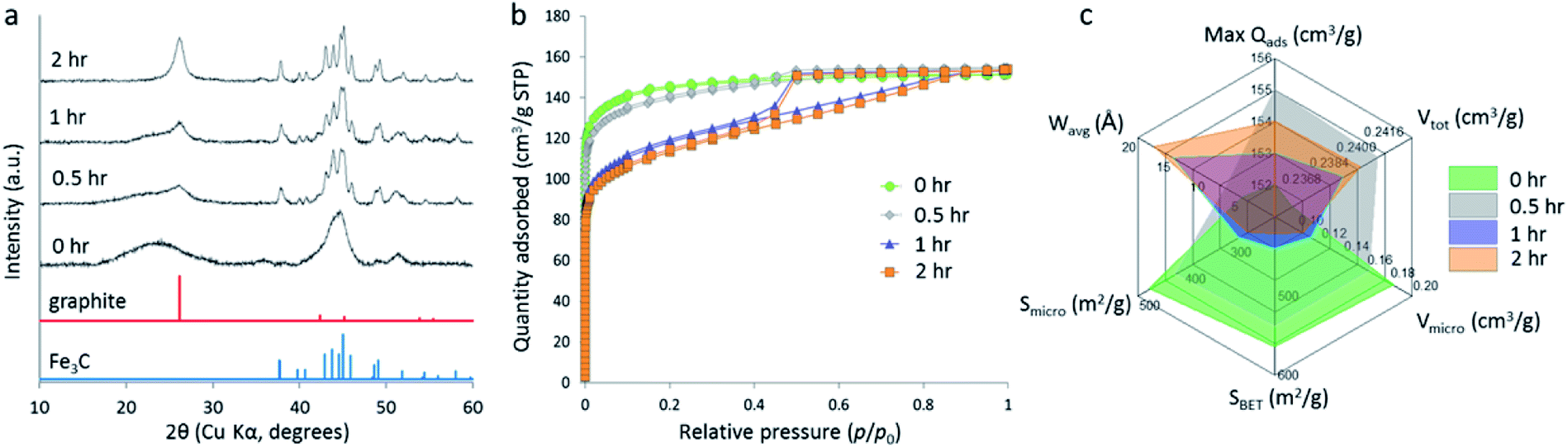 | ||
| Fig. 9 (a) XRD data, (b) N2 adsorption/desorption isotherms and (c) porosimetry data for carbons prepared by pyrolyzing starch and Fe(NO3)3 to 800 °C for various hold-times. Figure reproduced with permission from ref. 108. | ||
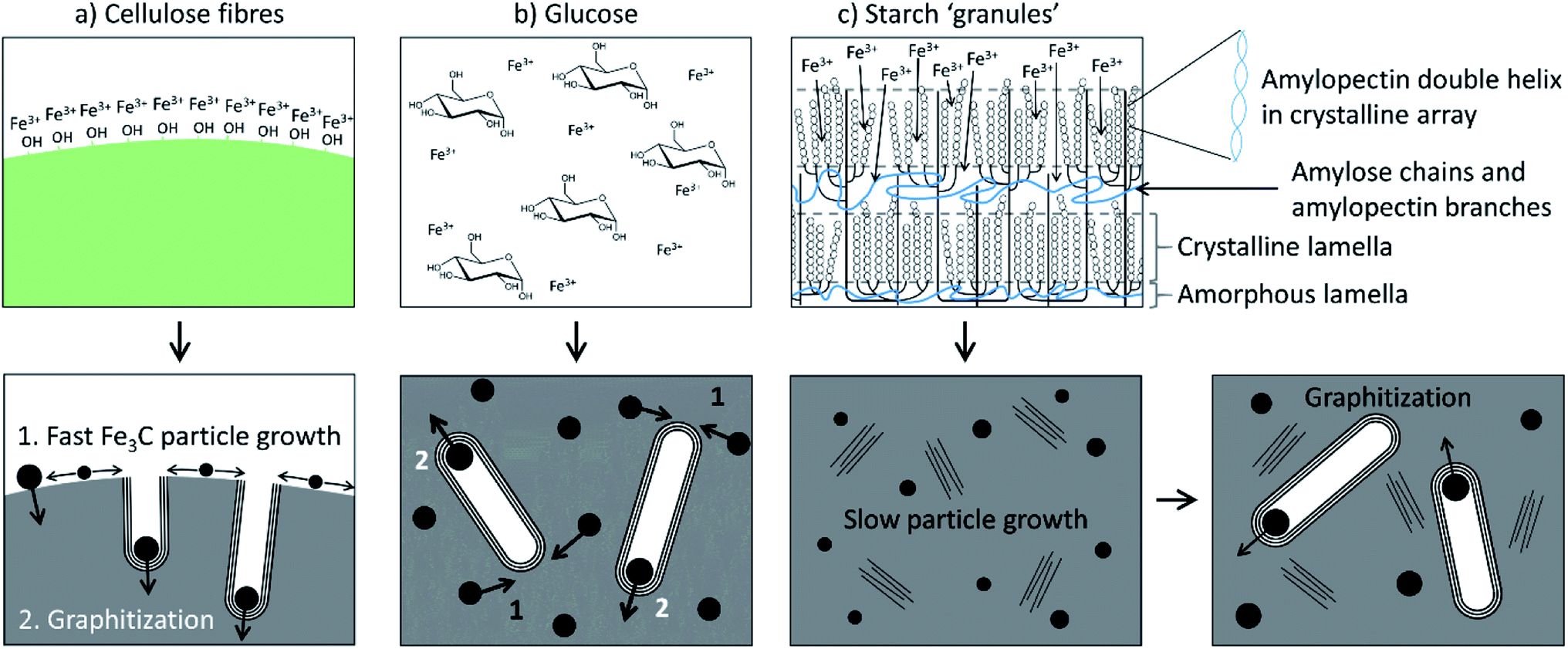 | ||
| Fig. 10 Schematic of proposed iron-catalyzed graphitization mechanisms for (a) cellulose, (b) glucose and (c) starch. Figure reproduced with permission from ref. 108. | ||
The other major component of many plant-based biomass sources is lignin. Lignin is an abundant polyaromatic molecule and a waste product of the Kraft wood pulping process, making it a popular candidate for graphitization. The challenge of working with lignin is that it is insoluble in water. To address this, several authors report graphitization of lignin by first dissolving lignin in tetrahydrofuran (THF) and then adding aqueous iron nitrate to maximise homogeneity between the lignin and iron.139,140 After drying the mixtures and subjecting them to pyrolysis under argon, shell-like encapsulated metal nanoparticles were found in the carbon products.140 Similar shell-like nanostructures were reported by Zhang et al., but rather than solubilizing the lignin in THF, the authors added aqueous iron nitrate to lignin powder.141 This resulted in a large polydispersity in the catalyst particles, reflecting the fact that the iron would just coat the lignin particles rather than be dispersed throughout the lignin molecules. Interestingly, the authors found some evidence that the number of graphene layers around a catalyst particle may be correlated to the diameter of that particle.
There are many other biopolymers produced by both plants and animals and several of these have been employed in the production of carbons by iron-catalyzed graphitization. Most of these examples involve polymers that are soluble in hot water, producing gels that can trap aqueous iron precursors. The gels can then be dried in an oven to produce a dense material or freeze dried to give a sponge-like structure which is retained during pyrolysis.142 An advantage of many biopolymers is that they contain functional groups which can facilitate dispersion of an ionic iron precursor. For example, agar is derived from seaweed and consists of two polysaccharides (agarose and agaropectin) which contain hydroxyl groups that can coordinate to aqueous Fe3+ to produce a precursor for catalytic graphitization.143 Chitosan is another polysaccharide that been used to generate porous graphitic carbons. The advantage of chitosan is that each saccharide monomer contains an amine group, which can facilitate doping of nitrogen into carbons during pyrolysis.142 A final example is gelatin, which is a polypeptide derived from collagen. Gelatin forms sticky liquids and resins when combined with metal nitrates and these expand to form foams on drying.144 Like the freeze-dried chitosan sponge, the foam-like structure of the gelatin–metal nitrate mixtures is also maintained during pyrolysis and if gelatin is combined with iron nitrate, the resulting carbon foam contains iron carbide nanoparticles coated in ‘onion-like’ layers of graphitic carbon.145
4.4 Raw biomass
There is considerable interest in the conversion of raw biomass into functional carbon materials for various applications and there are some excellent reviews of this specific field.20,158 For catalytic graphitization using iron-precursors, one of the most interesting features of products derived from biomass is that they maintain their macrostructure during pyrolysis (Fig. 11a). This is not just aesthetically attractive. Biological materials encompass a wide range of complex and highly evolved structures and capturing these in a functional material can give unique properties.159 For example, the interconnected pore structure of wood is maintained during iron-catalyzed graphitization, leading to advantageous properties for the resulting carbon in supercapacitors.54 While normal (iron-free) pyrolysis will also maintain the macrostructure of biomass, the addition of iron catalysts introduces mesoporosity through the formation of graphitic nanostructures. Doping biomass with iron salts leads to the formation of Fe or Fe3C nanoparticles during pyrolysis. These nanoparticle catalysts drive graphitization, as in examples earlier in this review. However, in raw biomass, the catalyst nanoparticles often appear to be highly mobile, driving conversion of the biomass-derived amorphous carbon into a dense network of graphitic carbon nanotubes (Fig. 11b–d).160 The fact that the Fe or Fe3C nanoparticles produce the graphitic nanostructures means that the size of the catalyst particles directly impacts the porosity of the resulting carbon. This can be observed in nitrogen porosimetry data of graphitic carbons produced from Fe(NO3)3/sawdust at various ratios (Fig. 11e), where a higher Fe-content leads to larger pores. This can also be observed in TEM images, (Fig. 11f/g) which show much smaller particles at a lower Fe/sawdust ratio. Interestingly, the degree of graphitization does not seem to be affected by a significant drop in iron content, possibly indicating that the smaller Fe/Fe3C catalyst particles travel further through the amorphous carbon matrix. The broad graphitic peak in X-ray diffraction patterns of these samples suggests that the graphitic carbon nanotubes have a high level of disorder, resembling a turbostratic rather than a truly graphitic carbon. In a similar system, Gomez-Martin et al. showed that pyrolysis of FeCl3-doped wood samples to higher temperatures (1000–2000 °C) leads to a much sharper graphitic peak, indicating more regular graphitic structure.51 There is a drop in the BET surface area at higher temperatures (200 m2 g−1 at 850 °C to 31 m2 g−1 at 2000 °C) but this is still higher than a control sample (no Fe) pyrolyzed to 2000 °C, showing that some of the porosity can be maintained. This is reflected in TEM images, which still identify graphitic nanostructures in FeCl3-doped wood pyrolyzed to 2000 °C.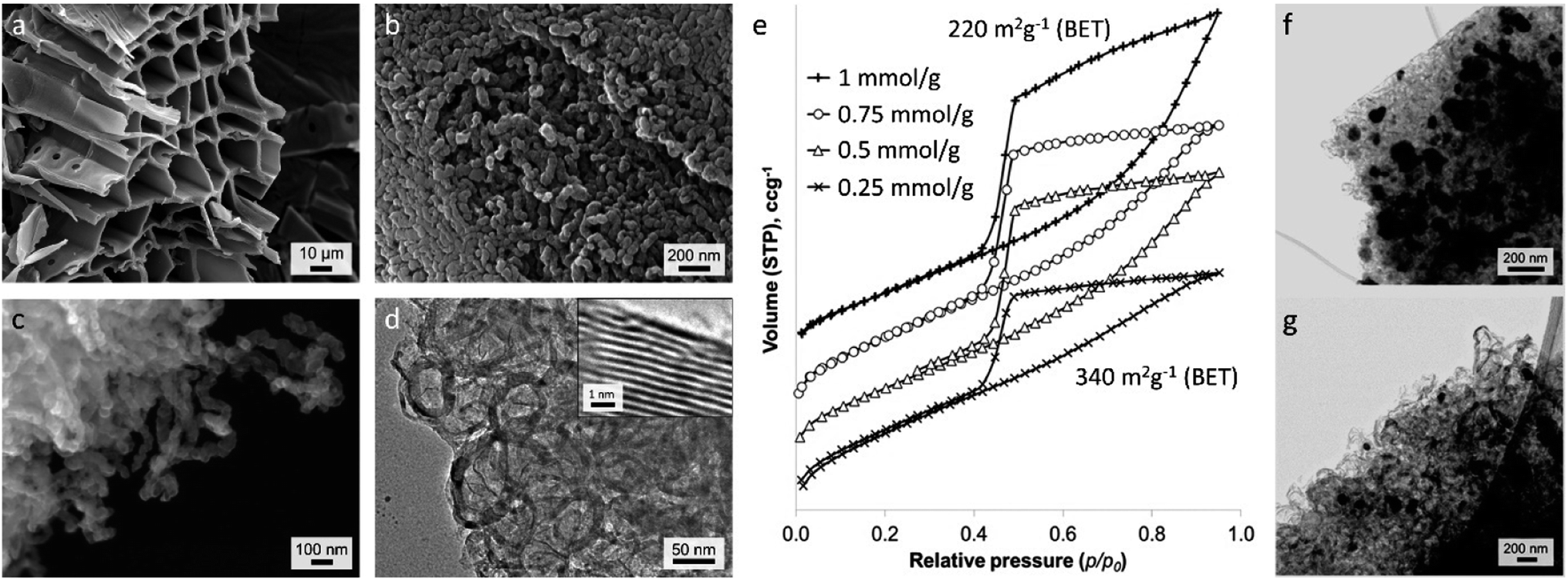 | ||
| Fig. 11 (a–c) SEM and (d) TEM images of carbon produced from iron-catalyzed graphitization of sawdust. (e) N2 adsorption–desorption isotherms (offset along y-axis) of graphitic carbons produced from Fe(NO3)3 and sawdust at various Fe/biomass ratios and TEM images of samples prepared from (f) 1 mmol and (g) 0.5 mmol Fe(NO3)3 per gram of sawdust. Images modified with permission from ref. 160. | ||
A factor that must be considered in iron-catalyzed graphitization of biomass is the ability of the iron salt to penetrate the biomass structure. Many authors mill biomass to a fine powder before infiltration with an iron salt to maximize coating of the biological material in iron. Hunter et al. demonstrated this systematically, showing that milling a wide range of biomass precursors before iron-catalyzed graphitization led to porous graphitic carbons with consistent adsorptive properties. Another consideration is the nature of the iron precursor. Liu et al. describe an intriguing process using FeCl3 as a catalyst for carbon nanotube production from sawdust.161 Their process involves a fast pyrolysis step, where powdered sawdust is loaded with an iron salt, dried, flushed with N2 and then inserted into a preheated quartz tubular reactor (Fig. 12a).162 The resulting carbon powder was covered in a dense mat of graphitic nanotubes (Fig. 12b). The authors demonstrate that the rapid heating leads to degradation of the cellulosic precursor into volatile low molecular weight hydrocarbons. These dissolve into surface Fe or Fe3C nanoparticles to drive the formation of graphitic nanotubes in a process analogous to chemical vapour deposition. Interestingly, Fe(NO3)3 and Fe2(SO4)3 catalysts did not produce the surface mat of graphitic nanotubes (Fig. 12c) and neither did CuCl2 or NiCl2, indicating that the combined catalytic effect of iron and chloride is required for this phenomenon to occur. Zhang et al. reported similar structures from the fast pyrolysis of FeCl3-treated rice husks.163 It is possible that the unique behaviour of FeCl3 derives from the Lewis acid character of this salt, which is known to change the decomposition pathway of cellulosic materials.164–166 There are many other examples of raw biomass being used to produce graphitic carbons and it is difficult to draw further conclusions due to the large range of iron precursors and pyrolysis conditions used. For the benefit of the reader, these are summarized in Table 4.
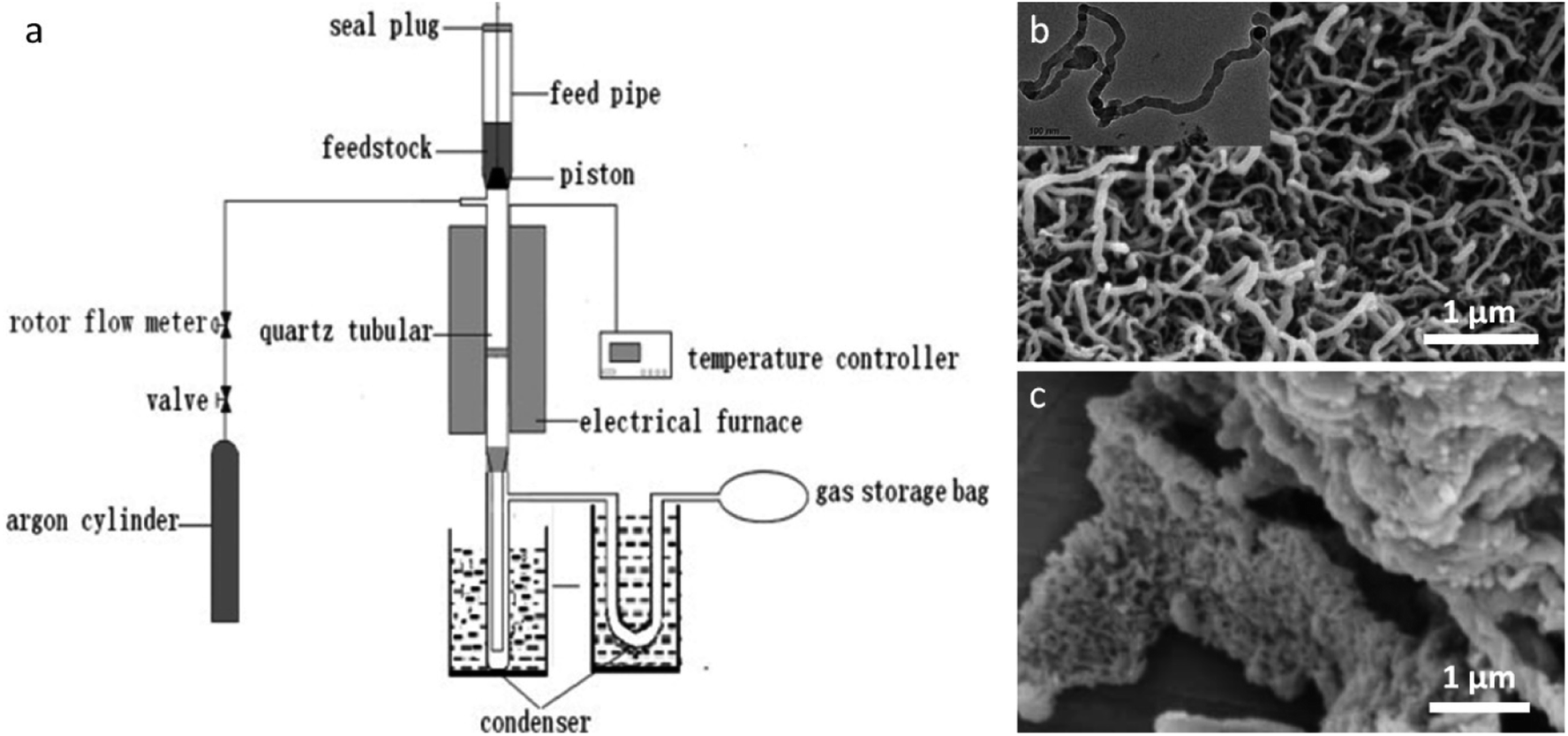 | ||
| Fig. 12 (a) Diagram of setup for fast pyrolysis and SEM images of samples prepared by fast pyrolysis of sawdust powder with (b) FeCl3 (inset TEM image) and c) Fe(NO3)3. Figures modified with permission from ref. 161 and 162. | ||
| Product description | Organic precursor | Iron source | Temp. (°C) | Comments |
|---|---|---|---|---|
| Continuous and bamboo-like graphitic nanotubes160 | Softwood sawdust | Fe(NO3)3 | 800 | Porosity depends on iron:biomass ratio |
| Nanofibers/mesoporous carbon composites161 | Sawdust | FeCl3 | 600–800 | Fast pyrolysis process, comparison of Fe(NO3)3 and Fe2(SO4)3 |
| Graphitic carbon nanostructures167 | Pine wood sawdust | Fe(NO3)3 | 900/1000 | Comparison to Ni |
| Onion-like structure, curved graphitic shells168 | Beech wood | FeCl3 | 1000/1300 | Impregnation with FeCl3 solution in isopropanol |
| Porous graphitic carbons54 | Beech wood | FeCl3 | 1000–1600 | Slow pyrolysis to 500 °C to reduce cracking |
| Onion-like graphitic shells51 | MDF wood | FeCl3 | 850–2000 | Slow pyrolysis to 500 °C to reduce cracking |
| Mesoporous graphitic carbons66 | Bamboo, nut shells, grasses, wood | Fe(NO3)3 | 800 | Mechanical milling increases graphitization for hard biomass |
| Carbon microfibres with iron nanoparticles169 | Bamboo | Fe(NO3)3 | 800 | Hydrothermal pre-treatment of bamboo in NaOH |
| Porous graphitic carbon170 | Bamboo | K2FeO4 | 800 | Bamboo pyrolyzed to 400 °C before infiltration with iron precursor |
| Porous graphene-like nanosheets171 | Coconut shell | FeCl3 | 900 | ZnCl2 used for simultaneous activation and graphitization, BET surface area 1874 m2 g−1 |
| Porous graphitic carbon172 | Coconut shell | Fe(NO3)3 | 1000 | Coconut shell milled to a powder before infiltration with Fe(NO3)3 |
| Magnetic nanofibers/porous carbon composites163 | Rice husk | FeCl3 | 600 | Various pretreatments of rice husk including hydrothermal and NaOH |
| Fe/N-doped carbon173 | Soy bean milk | FeCl3 | 600–1000 | BET surface areas 879–1164 m2 g−1 |
| Onion-like graphitic carbon174 | Cotton | Iron(III) acac | 650 | DMF solvent |
| Nanoporous carbon@carbon fibre composites175 | Cotton | FeCl3 | 500–600 | MOF precursor, initial activation step |
| Mesoporous carbon/iron carbide nanocomposite176 | Cotton fabric | Fe(NO3)3 | 800 | BET surface area 154–410 m2 g−1 |
| Nitrogen-doped porous graphitic carbon177 | Water hyacinth | Fe(NO3)3 | 700 | Dopamine hydrochloride as N source |
| Hierarchically porous carbon nanosheets178 | Moringa Oleifera stems | FeCl3 | 800 | ZnCl2 as activation catalyst |
| Graphitic core–shell structures179 | Miscanthus grass powder | Fe(NO3)3 | 900 | Graphitization enhanced with cobalt |
| Magnetic carbon nanocages180 | Pine tree resin | Fe(NO3)3 | 1000 | Fe3C catalyst |
| Hierarchical porous graphitic carbon181 | Chopsticks | Fe(NO3)3 | 850 | Potassium oxalate as activating agent |
| Worm-like structures, carbon nano-capsule182 | Chinese chestnuts | Fe(NO3)3 | 400–800 | Gas and liquid byproducts also characterized |
| Graphitized carbon nanosheets183 | Citrus grandis skins | FeCl3 | 1200 | Biomass milled, ZnCl2 cocatalyst |
| Carbon-shell coated iron nanoparticles184 | Coffee grounds | Fe(NO3)3 | 800 | Coffee grounds washed before infiltration with Fe(NO3)3 |
| Mesoporous graphitic carbon185 | Chestnut shell, bamboo, poplar, cotton, lotus | Fe(NO3)3 | 800 | Different iron loading and pyrolysis conditions investigated |
| ‘Graphite-shell-chains’186 | Wood, coffee, tofu residue, cotton | Fe(NO3)3 | 850 | Electron microscopy shows tube-like structures |
| Graphitic structures187 | Oil palm frond | Fe(NO3)3 | 1000–1400 | Silica also added |
| Graphitized porous carbon188 | Phoenix tree leaves | K2FeO4 | 650–950 | Pyrolyzed biomass (400 °C) mixed with K2FeO4 powder |
| Carbon shells/tubes189 | Oryza sativa pulp | FeCl3 | 800 | NaOH pretreatment |
| Porous graphitic carbon microtubes190 | Willow catkins | K4Fe(CN)6 | 900 | FeCl2 comparison |
4.5 Organometallics
The examples discussed in the preceding sections have all used a separate organic and metal precursor. An alternative method is to use a single reactant containing both components, i.e., an organometallic complex containing iron (Table 5). This route has been less widely explored but there are some examples of direct pyrolysis of iron complexes such as iron gluconate.191 One challenge with organometallic precursors is their lack of thermal stability or even volatility. Some authors have resolved this by polymerizing the precursor, e.g., the catalytic crosslinking of ferrocene.192 Another approach is to exploit the volatility of the precursor. For example, Leonhardt et al. used one furnace to drive sublimation of ferrocene before passing the vapour into a second furnace at a much higher temperature, where carbon nanostructures were deposited onto a substrate.193 A similar approach has been used to produce graphitic nanocages by bubbling a mixture of nitrogen and carbon-rich gas such as acetylene through liquid iron pentacarbonyl and passing the resulting vapour through a tube furnace.194,195 High pressure reactors have also been used for catalytic graphitization of ferrocene.196 A final note on the use of organometallic prearccursors is that they can be readily templated. Lee et al. mixed iron phthalocyanine with mesoporous silica (SBA-15) in a pestle and mortar. Pyrolysis resulted in sublimation and decomposition of the iron phthalocyanine and deposition of graphitic carbon and iron nanoparticles through the silica structure.197 The silica can subsequently be removed with HF or NaOH and the resulting carbon retains the high porosity and surface area (877 m2 g−1) of the template.| Product description | Precursor | Temp. (°C) | Comments |
|---|---|---|---|
| Mesoporous graphitic carbon197 | Iron phthalocyanine | 900 | Mesoporous silica template, BET surface area 877 m2 g−1 |
| Multi-walled carbon nanotubes198 | Iron phthalocyanine | 850 | Initial vaporization at 650 °C, then flow of gas into second high T furnace with Ar |
| Graphitic carbon nanostructures, nanocapsule/nanopipes191 | Iron(II) gluconate | 900–1000 | KMnO3 post-treatment to remove iron species and amorphous carbon |
| Graphitic porous carbons with 3D nanonetwork192 | Ferrocene | 700–900 | Initial reflux in CCl4/AlCl3 to drive crosslinking of cyclopentadienyl rings |
| Fe-filled carbon nanotubes193 | Ferrocene | 860–920 | Initial sublimation of ferrocene at 150 °C, then flow of gas into second high T furnace with Ar |
| Graphitic nanocages | Fe(CO)5, acetylene and ammonia | 750–1050 | Nitrogen doping in product |
| Graphitic nanocages | Fe(CO)5 and ethanol | 900 | Vertical tube furnace with three heating zones |
| Carbon-encapsulated iron carbide nanoparticles196 | Ferrocene | 600–1600 | High pressure, mechanism study |
| Core@shell nanocomposites199 | Ferrocene | 900 | High pressure |
5. Mechanism of catalytic graphitization
One of the current drawbacks of catalytic graphitization is the limited understanding of the mechanism. As seen in the sections above, there are many examples of graphitic nanostructures synthesized by pyrolysis of organic and iron precursors. The choice of precursors and synthesis conditions vary greatly between different studies and can all significantly affect the chemistry and structure of the graphitic carbon product. As a result, it is difficult to directly compare different systems or predict which may result in graphitic carbon materials with enhanced properties. Future development of the method of catalytic graphitization requires a more detailed understanding of the mechanism. While there are few studies on the mechanism of catalytic graphitization in pyrolysis, there has been considerable effort to understand the process of CVD synthesis of carbon nanotubes.200,201 The processes are fundamentally different. CVD synthesis of carbon nanotubes involves decomposition of a hydrocarbon gas and dissolution of carbon into a catalyst particle. In contrast, catalytic graphitization involves decomposition of an organic precursor into amorphous carbon and graphitization of that solid carbon by a catalyst particle. However, it may be possible to gain insight into the mechanism of catalytic graphitization by studying CVD chemistry.5.1 Insights from CVD synthesis of carbon nanotubes: nucleation and growth
As noted in section 2.3, CVD synthesis of carbon nanotubes can occur by float growth or base growth. In both situations, the first step is the initial nucleation of solid carbon from the catalyst particle. A popular theory for nucleation of carbon nanotubes on catalyst particles is known as the “yarmulke” mechanism, first proposed by Dai et al.202 The yarmulke mechanism involves the initial formation of a graphene ‘cap’ over the catalyst nanoparticle, which is stabilized by binding of the edges to the catalyst nanoparticle. Computer simulations have been widely used to probe the precipitation of carbon from saturated FexCy catalyst nanoparticles and the formation of the graphene cap as the first step to SWCNTs.203 As new carbon is introduced to the catalyst particle, the cap lifts of the catalyst and elongates to form a SWCNT (Fig. 13a). Successful detachment of the cap and thus nanotube growth is dependent on multiple factors such as temperature and work of adhesion of the graphitic cap on the nanoparticle, which is governed by the interfacial energy and thus linked to chemical and physical properties of the catalyst particle.204 Cap lift-off and nanotube growth has been observed experimentally using in situ environmental TEM (Fig. 13b).205,206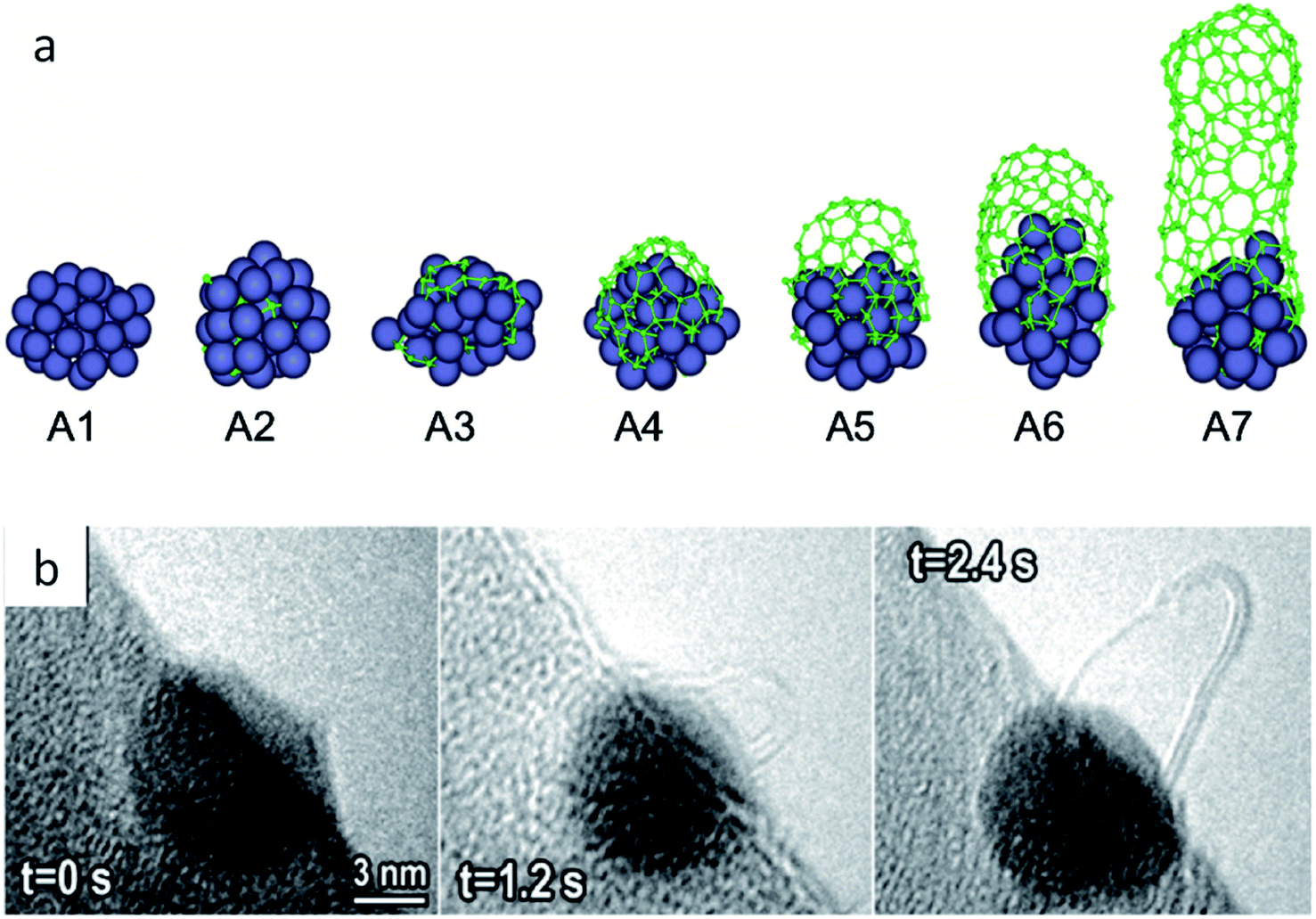 | ||
| Fig. 13 (a) Molecular dynamics simulation of carbon cap formation and lift-off and (b) still images taken from environmental TEM video footage of carbon nanotube growth from an iron catalyst. Images modified with permission from ref. 204 and 205. | ||
Simulations have also shown the possibility of cap formation followed by growth to form a continuous shell around the catalyst particle.207 This is believed to occur when there is not enough energy to overcome the interfacial energy between the cap and the catalyst and correlates to experimental observations of graphitic shells coating catalyst particles in CVD. Factors that appear to contribute to shell formation include particle size (very small particles are not encapsulated), catalyst phase (Fe or Fe3C) and temperature (encapsulated particles tend to be found in the lower temperature regions of CVD ovens). Interestingly, there is evidence that the number of ‘shells’ of graphite around a catalyst particle is linked to the diameter of that particle. Given that the addition of carbons to the edge of an existing graphene sheet is energetically more favourable than nucleation of a new sheet, it is believed that the first shell is completed before a new shell nucleates underneath. This would lead to exhaustion of the carbon supply in the catalyst, which could explain experimental evidence that encapsulated particles in CVD synthesis tend to be formed of Fe rather than Fe3C.208 Catalytic graphitization leads to similar ‘encapsulated’ and tube-like structures, suggesting that there may be parallels with the processes of CVD. However, while some examples of catalytic graphitization produce mainly shells and others mainly tubes, there are also plenty of examples where a mixture of products is formed.
Another feature of catalyst diameter is that it influences the diameter of the resulting carbon nanotube. This is believed to be due to cap lift-off occurring after the cap reaches a similar diameter to the catalyst cluster.209 Cheung et al. demonstrated experimentally that monodisperse iron nanoparticles could be used to produce carbon nanotubes with controlled diameter via CVD.210 These authors also note that an efficient carbon supply is required to drive the growth of large-diameter nanotubes from large catalyst particles. On the other end of the scale, catalyst clusters that are too small (<20 atoms) have been shown in simulations to produce poor quality carbon nanotubes, due to inconsistencies in binding between the cluster and the dangling bonds of the nanotube.211 This reflects some experimental observations that nanoparticles must reach a critical size before they become catalytically active in CVD.212 A similar level of control of nanotube diameter has also been seen in catalytic graphitization. If catalyst particles are smaller, they produce nanotubes and shells of smaller diameter, which is reflected in the porosity of the carbon material.65,160 A point to note is that many authors have observed Fe or Fe3C nanoparticles to be ‘liquid-like’ during the graphitization process and very quickly agglomerate to form larger particles. Controlling this process may be a route to a wider range of graphitic products.
5.2 Insights from CVD synthesis of carbon nanotubes: types of growth
Nanotube growth in CVD can proceed in two different ways depending on specific reaction conditions. One possibility is tip-growth, otherwise known as float-growth, in which the catalyst nanoparticle detaches from the substrate and leaves a carbon nanotube trail between itself and the substrate (Fig. 4).213 Alternatively, nanotube formation can proceed via base-growth, in which the catalyst nanoparticle remains in contact with the substrate and the nanotube grows outwards from the nanoparticle.214 The dominating mechanism is dependent on the interactions between the catalyst nanoparticle and the substrate, demonstrated by the work of Wang et al. in which iron was used as a catalyst and the nature of the substrate was varied.215 They suggested that if the interfacial energy between the catalyst particle and the substrate is greater than the surface energy of the substrate itself, tip-growth would dominate, which they observed with a silica substrate. Conversely, with a tantalum substrate, the iron/tantalum interfacial energy is lower than the surface energy of pure tantalum so base growth was the dominant pathway. While non-carbon interfaces are generally not relevant to iron-catalyzed graphitization, it will still be important to consider the different interfacial energies that may contribute to catalyst movement.A second important observation from CVD nanotube growth is that of continuous and bamboo-like carbon nanotubes. As the name suggests, bamboo-like carbon nanotubes resemble the structure of bamboo, with regular compartment-like graphitic structures (Fig. 14a).216 This contrasts with the straight channel formed in normal carbon nanotubes (Fig. 14b/c). Bamboo-like carbon nanotubes are believed to form by periodic nucleation steps, where a series of graphitic layers form across the trailing edge of the catalyst particle during float growth (Fig. 14d). The catalyst particle then detaches from the graphitic layers and moves forward before stopping to generate another nucleation point.217 Simulations suggest that bamboo-like carbon nanotubes form in conditions of higher carbon concentration,218 which correlates to an in situ TEM study which identified Fe3C as the catalyst for bamboo-like carbon nanotubes and Fe as active for continuous carbon nanotubes.216
 | ||
| Fig. 14 TEM images of (a) bamboo-like and (b and c) continuous carbon nanotubes and (d) in situ growth of a bamboo-like carbon nanotube. Figures modified with permission from ref. 216 and 217. | ||
Continuous and ‘bamboo-like’ nanotubes have also been identified in many reports of catalytic graphitization, sometimes within the same sample.160 Given that many reports of catalytic graphitization identify both Fe3C and Fe in X-ray diffraction data, it is possible that the different materials drive two different types of graphitization. Some interesting insight comes from work by Ichihashi et al., who grew amorphous carbon ‘nanopillars’ by electron-beam-induced chemical vapor deposition on an iron-doped carbon substrate.219 On heating the sample to 650 °C inside a transmission electron microscope, they observed the formation of iron nanoparticles in the substrate followed by ‘liquid-like’ movement of particles outwards along the carbon pillars, leaving a graphitic ‘trail’ behind (Fig. 15). In one case, they observed the liquid-like particle moving at constant speed along the carbon nanopillar to form a multi-walled nanotube. In another case, the catalyst particle moved more slowly, and the tail of the particle periodically paused for several hundred milliseconds, forming graphene caps within the nanotube. Crucially, the nanoparticle catalysts never moved back along the pillar once graphitization was complete, indicating that graphitic carbon does not re-dissolve in the catalyst once it has formed. The authors do not propose a reason for the different behaviour, but they do suggest that the driving force for movement is due to the difference in solubility of amorphous carbon in iron compared to graphitic carbon in iron. A difference in solubility of amorphous carbon in Fe compared to Fe3C could potentially then be a reason for slower or faster movement of a catalyst particle through amorphous carbon.
 | ||
| Fig. 15 Images from an environmental TEM video showing movement of an iron catalyst particle along a carbon ‘nanopillar’. Figure modified with permission from ref. 219. | ||
5.3 The chemical nature of the catalyst
In CVD synthesis and in catalytic graphitization, the chemical nature of the catalyst is a matter of debate. The high activity of iron-based catalyst particles is commonly credited to the high solubility of carbon in iron at temperatures of around 700–800 °C. One of the proposed mechanisms by which graphitization occurs is dissolution-precipitation. Here, the iron particle dissolves carbon atoms until it reaches a point of supersaturation, at which point graphitic carbon is precipitated from the particle. This hypothesis has been used to explain why transition metals with a low carbon solubility such as copper show poor catalytic activity for graphitization, while metals with high carbon solubility such as iron and nickel show high activity.139 An alternative theory suggests that the formation of metastable carbides is crucial to graphitization. These decompose into a more thermodynamically stable metal species and precipitate graphitic carbon in the process. There is evidence for Fe220 and Fe3C221 being the active catalyst for CVD synthesis of carbon nanotubes and other authors have shown that both phases can be active catalysts, but for different graphitic products.216 Fe and Fe3C are both phases that are commonly identified in in situ X-ray diffraction studies of iron-catalyzed graphitization, often within the same sample.146,149,222,223 This raises the possibility that both dissolution-precipitation and carbide decomposition are potential mechanisms in catalytic graphitization. However, other pathways may also be possible. For example, a study by Yan et al. proposed that as iron carbide is stable at the temperatures involved in catalytic graphitization, it is unlikely that the formation of graphitic carbon is due to metal carbide decomposition. Instead, they suggested that the iron carbide and pure iron species may both act as dissolution-precipitation catalysts, contributing to a high catalytic activity in iron species.139An important point to note is that the crystal phases present during catalytic graphitization may change as the sample is cooled. For example, in situ synchrotron XRD data from Gomez-Martin et al. showed that the onset of graphitization of a wood precursor corresponded to the appearance of an Fe3C phase.222 During cooling, the Fe3C phase transforms to γ-Fe then α-Fe, triggering a second carbon precipitation step. This change in the crystalline composition on cooling highlights the importance of studying graphitization in situ. Systems that are only studied after removal from the furnace will offer limited information on the mechanism by which they were formed.
Another question that is linked to the chemical nature of the catalyst is how carbon is transported within the catalyst particles. Again, much of the evidence so far on this phenomenon comes from studies of CVD processes. For example, Yoshida et al. collected in situ environmental TEM footage of MWCNTs growing from Fe3C nanoparticles.221 All the cylinder layers of the nanotube were observed to grow at the same rate, regardless of diameter, which strongly suggests migration of carbon atoms through the bulk of the catalyst particle. Other examples propose diffusion of carbon atoms around the surface of the catalyst nanoparticles. Again, there is a possibility that different mechanisms operate in different systems. In the complex world of iron-catalyzed graphitization, there may even be multiple mechanisms operating within the same sample.
The mechanism of iron-catalyzed graphitization can be further complicated when considering doping, or the presence of impurities within the system. Doping of Fe–C materials with heteroatoms has been an intense field of study as it has led to improved performance in various applications. For example, N-doped carbons that contain iron species are known to be highly active in the oxygen reduction reaction224,225 and N-doped carbons are also of interest in lithium-ion batteries.226 There is also considerable interest in phosphorus227 and sulfur228-doped carbons. Given that iron can form alloys with nitrogen, phosphorus and sulfur, it is reasonable to assume that the presence of these heteroatoms will influence the chemistry of Fe–C graphitization catalysts, whether or not the heteroatoms become integrated into the resulting graphitic carbon. While there are few studies on the effect of heteroatom doping on graphitization catalysts, it is clear that the presence of these species affects graphitization. For example, sulfur doping was shown to affect the number of layers in carbon nanotubes grown by CVD.229 Sulfur has also been used to prevent agglomeration of iron catalyst particles during CVD, which in turn controls the diameter of the carbon nanotubes.230 It is possible that iron sulfide (FeS) or iron nitride (Fe3N) may act as graphitization catalysts in their own right. For example, there have been reports of graphitic structures forming around iron sulfide nanoparticles.231 A final point that is worth noting is that different nitrogen-containing precursors can lead to different types of nitrogen feature in the resulting graphitic carbon, which suggests that the precursor can affect the mechanism of graphitization.232
5.4 The physical nature of the catalyst
Determining the physical state of the catalyst particle during graphitization is also challenging. A commonly cited theory describing the physical state of the catalyst in CVD processes is the vapour-liquid-solid mechanism first proposed by Wagner and Ellis to explain the growth of silicon whiskers.233 This mechanism suggests that the catalyst particle must be in a liquid state to promote growth. However, the melting temperature of bulk iron is 1538 °C,234 much higher than typical reaction temperatures in CVD and catalytic graphitization. Despite this, many in situ environmental TEM studies have observed ‘liquid like’ behaviour.235 Harutyunyan et al. proposed that the formation of a liquid phase is essential for growth of carbon nanotubes in CVD and the formation of a solid particle hinders growth.236 Other authors, however, show evidence that the catalyst particle remains in the solid state (demonstrated by the presence of lattice fringes in in situ TEM images) and that the ‘liquid-like’ behaviour is due to the constant fluctuation of the catalyst nanoparticle phase.221 While many of the environmental TEM studies have probed CVD processes, similar liquid-like behaviour has been observed in the iron-catalyzed graphitization of cellulose (Fig. 16). The catalyst particle was observed to move through the solid carbon matrix with the front edge of the particle creeping slowly forwards and dissolving the amorphous matrix. The trailing edge of the particle moves in a very different way and stops periodically to allow build-up of graphitic layers before suddenly detaching and moving forward very quickly. This implies that the interfacial tension between the catalyst and the graphene surface is very different to the interfacial tension between the catalyst and the amorphous carbon.147 The video footage showed that the particle stopped moving once the amorphous carbon was exhausted. Interestingly, the experiment also showed that some particles remained stationary throughout the experiment, which may indicate that some particles became encapsulated in graphite or that they were composed of an inactive phase.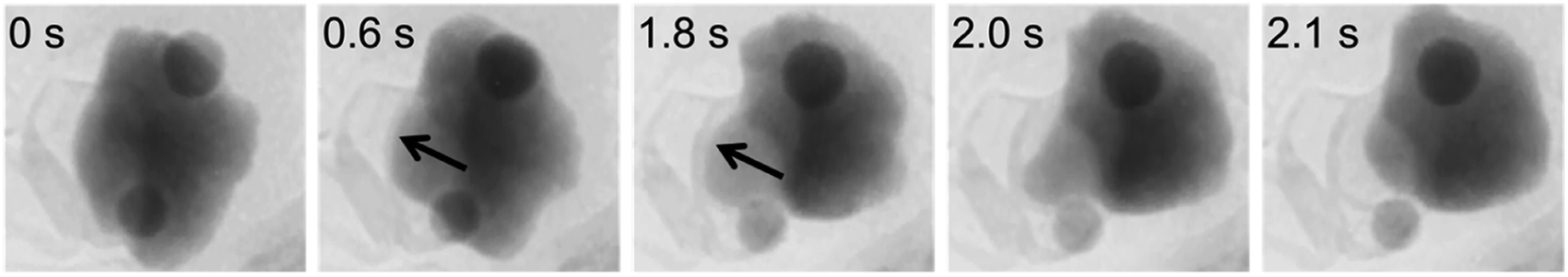 | ||
| Fig. 16 Still images from in situ ETEM footage of an iron-rich particle moving through a carbon matrix. Arrows show the layers of graphite forming on the trailing edge of the catalyst. Images modified with permission from ref. 147. | ||
Computer simulations have been used to offer insight into the physical properties of the graphitization catalyst. Methods such as molecular dynamics have shown that the melting temperature of metal nanoparticles can be considerably lower than the corresponding bulk material, raising the possibility that graphitization catalysts may be in a liquid or liquid-like state.237 One model, carried out by Ding et al., suggests that both states are possible.238 Their study modelled the growth of single-walled carbon nanotubes on both solid and liquid iron particles and found that the two routes had similar growth mechanisms. The main difference was that the main diffusion pathway of the carbon atoms in liquid nanoparticles was through the bulk, while surface diffusion dominated in solid nanoparticles. In real systems, it may be that both routes take place at the same time, because catalyst particles are unlikely to be completely uniform in size. It is possible that the smaller particles are in a liquid state and the larger particles in a solid state at the same temperature. The study by Ding et al. not only suggests that this is plausible, but that the resulting nanostructure may be the same in both scenarios. A further factor to consider is that some metals, including iron, are known to undergo surface melting. This may provide an explanation for the liquid-like behaviour of the catalyst particles in some systems. Ding et al. have used molecular dynamics to demonstrate that surface melting occurs at temperatures below the melting temperature in both free239 and supported iron nanoclusters,240 with the depth of the surface melt increasing with temperature until the cluster undergoes complete melting. Therefore, as well as both solid and liquid states, it is possible that the nanoparticles occupy ‘in-between’ states in the process of graphitization. Modelling can never take into consideration all the factors of an experimental study. One important consideration when considering the size effect is the introduction of a surface, which is applicable to both CVD and catalytic graphitization processes. While stronger interactions caused by a lower contact angle can decrease the melting temperature,241 favourable epitaxy between the nanoparticle and the substrate can raise the melting temperature by hundreds of degrees.242 This implies that the suggested variation in melting temperature may not be a purely size-based argument. There are many more challenges and discoveries remaining in this field.
5.5 In situ methods for studying the mechanism of graphitization
The need to understand the mechanism of catalytic graphitization has required the development of novel in situ methods. By following the graphitization reaction as a function of time and temperature, it is possible to determine the extent of graphitization and the presence of phase transformations or reactions between carbon and the catalyst. Two in situ methods, X-ray diffraction and transmission electron microscopy, have received the most attention. In the case of X-ray diffraction, one of the earliest accounts is from Fitzer and Weisenburger, who devised a special heating stage to follow graphitization of coke (without any catalyst) using a laboratory diffractometer.243 More recent papers from Hoekstra et al. studied the in situ pyrolysis and graphitization of microcrystalline cellulose spheres with copper, nickel, cobalt and iron.65,146,149 These studies used a commercially available heating stage on a laboratory X-ray diffractometer to identify the crystalline phases present during pyrolysis and confirmed the presence of iron oxide intermediates and both Fe3C and Fe during the graphitization step. The availability of synchrotron sources has enabled experiments with better time resolution. These experiments are technically challenging, due to the need to hold the samples in a quartz capillary in the synchrotron beam, while heating and maintaining an inert atmosphere. Some studies used nitrogen gas flow through223 or around244 the capillary while another partially carbonized the sample before sealing it within a quartz capillary.222 In all these studies, a hot air blower was used to heat the sample to the required temperature (Fig. 17). Despite the very different set-up to a laboratory furnace, the capillaries appear to replicate the conditions of a laboratory furnace effectively.245 There are many other synchrotron techniques that have been used to study carbon materials or graphitization by other metals. For example, synchrotron based XPS was employed to study the growth of carbon nanotubes using ZrO2 nanoparticles as catalysts in a low-pressure CVD system. While Zr metal can catalyze graphitization at very high temperatures,246 Steiner et al. found that the catalytic effect for nanotube growth of ZrO2 took place without carbothermal reduction, with no visible changes in the Zr 3d XPS spectrum during synthesis.247 Other authors have used in situ synchrotron small-angle X-ray scattering (SAXS) to probe the (catalyst-free) pore structure of polyacrylonitride-based carbon fibres during graphitization.248 Undoubtedly, similar methods will be applied to the question of iron-catalyzed graphitization in the future. | ||
| Fig. 17 Schematic of an experimental setup used to study pyrolysis of a gelatin/Fe(NO3)3 mixture in situ viewed from (a) the top and (b) the side. Figure reproduced with permission from ref. 223. | ||
In situ or environmental TEM has also been used to study graphitization of carbon by iron and other metals. The challenge in these experiments again is replicating the laboratory furnace environment as closely as possible. Since electron microscopes operate under a high vacuum, it is necessary to be cautious when interpreting results from in situ TEM experiments as the materials may behave differently in the vacuum compared to the N2 or Ar atmosphere of the laboratory furnace. It is also important to consider how the high energy electron beam may be interacting with the sample. The earliest example of in situ TEM investigations of graphitization come from Krivoruchko et al.235 The authors dispersed iron hydroxide particles over an amorphous carbon film on a TEM grid. Heating the sample inside the microscope led first to reduction of the iron hydroxide and then movement of the resulting ‘liquid-like’ Fe particles across the carbon film, producing a trail of graphitic carbon. Glatzel et al., used a similar approach but dispersed a sample of cellulose/Fe(NO3)3 on a TEM grid before heating in situ and recording video footage.147 They observed very similar ‘liquid-like’ movement as the Fe/Fe3C nanoparticles graphitized the sample.
6. Other graphitization catalysts
Although iron is the most studied catalyst for graphitization, many other elements are known to drive graphitization, with transition metals and especially iron, cobalt and nickel being the most efficient. From these three, the highest catalytic activity is normally found for iron, followed by cobalt and then nickel, as evidenced by the interplanar distance of the (002) planes and Raman intensity ratio between D and G bands of the resulting graphitic carbon.139,167,191,249–251 The reason for this order is not clear, although some authors argue that is related to the number of electron vacancies in their d-shell orbitals.82,139 Group VIII metals have a d-shell occupied by 6–10 electrons, with electron configurations of [Ar] 3d64s2 for iron, [Ar] 3d74s2 for cobalt and [Ar] 3d84s2 for nickel, and the energy levels of their electronic configuration would only slightly change by accepting electrons from carbon, allowing for the formation of covalent bonds and the dissolution of carbon by the metal. According to this criterion, the catalytic activity of transition metals should have the order iron > cobalt > nickel, in agreement with experimental evidence.An additional factor for explaining the high catalytic graphitization efficiency of iron may be related to carbon solubility at high temperatures and the ability to form metal carbides. While iron can form a stable carbide with carbon, nickel and cobalt carbides are metastable, due to their weaker carbon bonds, with the order of the enthalpy being: Fe–C < Co–C < Ni–C.252 The maximum solubility of carbon in nickel is about 0.6 wt% at 1327 °C, while the maximum solubility of carbon in cobalt is 0.9 wt% at 1320 °C. For iron, the maximum solubility of carbon is 2.06 wt% at 1153 °C for the austenite (fcc) phase. This drops sharply to 0.02 wt% at 723 °C, where the transition to ferrite (bcc) iron occurs, due to the smaller interstitial positions in the ferrite lattice.253 According to the maximum solubility, the order is again iron > cobalt > nickel for the catalytic activity of these three metals. The abrupt decrease in solubility upon the austenite to ferrite transition may also help explain the higher efficiency of iron as a graphitization catalyst from a classic solution-precipitation view.
Other elements have shown to be active catalysts for graphitization, as reviewed by Ōya and Marsh.82 Ōya and Ōtani studied the graphitization of formaldehyde derived carbon at 2600–3000 °C by 10 wt% of Mg, Si, Ca, Cu, and Ge, which formed only graphitic carbon, and Al, Ti, V, Cr, Mn, Fe, Co, Ni, Mo and W, which formed both graphitic and turbostratic carbon.83 In addition, B was found to markedly accelerate homogeneous graphitization, while Zn, Sn, Sb, Pb and Bi had no catalytic effect. Yokokawa et al. studied the graphitization of furfuryl alcohol-derived carbon at 1400–2300 °C by copper compounds and copper metal.87 Weisweiler et al. used monolithic glass-like carbon crucibles filled with different metals and found Ni, Co, Fe, Pt, Mo, Cr and B to be highly effective in catalyzing graphitization, while Ag, Mg, Zn, Cd, Ge, Sn, Pb, Sb, Bi, Se, Te and Pd showed no reaction.85 More recent works have studied the catalytic graphitization of rare earth elements,254 yttrium,255 manganese121 and magnesium.256
7. Conclusions and perspective
Iron-catalyzed graphitization has the potential to be a scalable and economical route to carbons with complex graphitic structures. Natural graphite is listed by many governments as a critical material and there is a pressing need to identify routes to synthetic graphite materials. The appeal of iron-catalyzed graphitization is the simplicity. Organic matter is combined with an iron precursor and pyrolyzed in an inert atmosphere. For example, a complex network of graphitic nanotubes can be produced by heating a mixture of sawdust with iron nitrate. The method has proven to be extremely flexible, and authors have utilised a wide range of organic precursors and additional templates to produce carbons with diverse properties. One challenge that now needs to be addressed is how to translate this fascinating system into real-world applications. Iron-catalyzed graphitization appears to be scalable and flexible but there are very few examples of systematic or large-scale studies of this process. Without these, it is difficult to understand exactly how different precursors and heating conditions can be tuned to optimize the graphitic carbons towards certain applications. The few systematic studies that do exist have shown that it is possible to achieve dramatic variations in carbon structure through simple changes in precursors.65,108Another challenge in iron-catalyzed graphitization is understanding the mechanism. It is still not clear why some organic precursors produce graphitic shells while others generate graphitic nanotubes. It is possible that the different precursors or conditions lead to different catalytic routes. This may relate to the chemical nature of the catalyst (Fe or Fe3C) or to its physical state. When the catalyst does become mobile and produce graphitic nanotubes, it is not known why this movement is apparently random. If the movement of the catalyst particles could be understood and then controlled, it may be possible to further optimize the properties of the resulting carbons by introducing directional order between the nanotubes. On a simpler level, it is also important to gain a further understanding of how catalyst size influences the graphitization process and thus the porosity of the resulting materials. It is clear from all the examples in this paper that there is still much to be discovered in the field of iron-catalyzed graphitization. Increasingly sophisticated simulations, and synchrotron and electron microscopy experiments will undoubtedly offer insight into the nature of the catalytic process. However, there is also a lot of scope in simple, systematic experimental studies of different precursors and conditions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the University of Birmingham (RDH), Leverhulme Trust under Grant No. RPG-2020-076 (ZS) and Consejería de Economía, Conocimiento, Empresas y Universidad, Junta de Andalucia, Spain, under Grant. No P20/01186 (JRR).References
- D. D. L. Chung, Carbon Materials Science and Applications, World Scientific Publishing Co., Singapore, 2019 Search PubMed.
- Study on the EU's list of Critical Raw Materials, 2020, https://ec.europa.eu/docsroom/documents/42883/attachments/1/translations/en/renditions/native, accessed November 2021 Search PubMed.
- G. J. Simandl, S. Paradis and C. Akam, in Symposium on Strategic and Critical Materials Proceedings, ed. G. J. Simandl and M. Neetz, British Columbia Geological Survey Paper, British Columbia Ministry of Energy and Mines, Victoria, British Columbia, 2015, vol. 2015-3, pp. 163–171 Search PubMed.
- X. Hu, X. Sun, S. J. Yoo, B. Evanko, F. Fan, S. Cai, C. Zheng, W. Hu and G. D. Stucky, Nano Energy, 2019, 56, 828–839 CrossRef CAS.
- Y. Hu, J. O. Jensen, W. Zhang, L. N. Cleemann, W. Xing, N. J. Bjerrum and Q. Li, Angew. Chem., Int. Ed., 2014, 53, 3675–3679 CrossRef CAS PubMed.
- B. Li, L. Yang, C. Q. Wang, Q. P. Zhang, Q. C. Liu, Y. D. Li and R. Xhao, Chemosphere, 2017, 175, 332–340 CrossRef CAS PubMed.
- M. Meyyappan, J. Phys. D: Appl. Phys., 2009, 42, 213001 CrossRef.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- T. Guo, P. Nikolaev, A. Thess, D. T. Colbert and R. E. Smalley, Chem. Phys. Lett., 1995, 243, 49–54 CrossRef CAS.
- M. M. Titirici, R. J. White, N. Brun, V. L. Budarin, D. S. Su, F. del Monte, J. H. Clark and M. J. MacLachlan, Chem. Soc. Rev., 2015, 44, 250–290 RSC.
- IUPAC Gold Book, https://goldbook.iupac.org/html/C/C00894.html, accessed May 2021 Search PubMed.
- H. Darmstadt, C. Roy, S. Kaliaguine, G. Xu, M. Auger, A. Tuel and V. Ramaswamy, Carbon, 2000, 38, 1279–1287 CrossRef CAS.
- A. C. Ferrari and J. Robertson, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 14095 CrossRef CAS.
- F. R. Feret, Analyst, 1998, 123, 595–600 RSC.
- A. N. Popova, Coke Chem., 2017, 60, 361–365 CrossRef.
- P. J. F. Harris, Z. Liu and K. Suenaga, J. Phys.: Condens. Matter, 2008, 20, 362201 CrossRef.
- J. M. Stratford, P. K. Allan, O. Pecher, P. A. Chater and C. P. Grey, Chem. Commun., 2016, 52, 12430–12433 RSC.
- A. S. Marriott, A. J. Hunt, E. Bergström, K. Wilson, V. L. Budarin, J. Thomas-Oates, J. H. Clark and R. Brydson, Carbon, 2014, 67, 514–524 CrossRef CAS.
- J.-B. Donnet, R. C. Bansal and M.-J. Wang, Carbon Black, Marcel Dekker, Inc., New York, 1993 Search PubMed.
- W. Long, B. Fang, A. Ignaszak, Z. Wu, Y.-J. Wang and D. Wilkinson, Chem. Soc. Rev., 2017, 46, 7176–7190 RSC.
- X. Dou, I. Hasa, D. Saurel, C. Vaalma, L. Wu, D. Buchholz, D. Bresser, S. Komaba and S. Passerini, Mater. Today, 2019, 23, 87–104 CrossRef CAS.
- Y. Li, Y.-S. Hu, H. Li, L. Chen and X. Huang, J. Mater. Chem. A, 2016, 4, 96–104 CAS.
- E. Fitzer, K.-H. Köchling, H. P. Boehm and H. Marsh, Pure Appl. Chem., 1995, 67, 473–506 Search PubMed.
- H. Pan, M. Pruski, B. C. Gerstein, F. Li and J. S. Lannin, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 6741–6745 CrossRef CAS PubMed.
- M. Monthioux, Carbon, 2020, 160, 405–406 CrossRef CAS.
- A. Ghemes, J. Muramatsu, Y. Minami, M. Okada, Y. Inoue and H. Mimura, Journal of Advanced Research in Physics, 2012, 3, 011207 Search PubMed.
- X. Xie, L. Ju, X. Feng, Y. Sun, R. Zhou, K. Liu, S. Fan, Q. Li and K. Jiang, Nano Lett., 2009, 9, 2565–2570 CrossRef CAS PubMed.
- Y. Saito and T. Yoshikawa, J. Cryst. Growth, 1993, 134, 154–156 CrossRef CAS.
- W. Fan, Z. Li, C. You, X. Zong, X. Tian, S. Miao, T. Shu, C. Li and S. Liao, Nano Energy, 2017, 37, 187–194 CrossRef CAS.
- D. Pech, M. Brunet, H. Durou, P. Huang, V. Mochalin, Y. Gogotsi, P.-L. Taberna and P. Simon, Nat. Nanotechnol., 2010, 5, 651–654 CrossRef CAS PubMed.
- W. Lian, H. Song, X. Chen, L. Li, J. Huo, M. Zhao and G. Wang, Carbon, 2008, 46, 525–530 CrossRef CAS.
- M. E. Plonska-Brzezinska, ChemNanoMat, 2019, 5, 568–580 CrossRef CAS.
- R. Van Noorden, Nature, 2011, 469, 14–16 CrossRef CAS PubMed.
- A. Bianco, H.-M. Cheng, T. Enoki, Y. Gogotsi, R. H. Hurt, N. Koratkar, T. Kyotani, M. Monthioux, C. R. Park, J. M. D. Tascon and J. Zhang, Carbon, 2013, 65, 1–6 CrossRef CAS.
- R. E. Franklin, Proc. R. Soc. London, Ser. A, 1951, 209, 196–218 CAS.
- P. J. F. Harris, J. Mater. Sci., 2013, 48, 565–577 CrossRef CAS.
- A. C. Forse, C. Merlet, P. K. Allan, E. K. Humphreys, J. M. Griffin, M. Aslan, M. Zeiger, V. Pressesr, Y. Gogotsi and C. P. Grey, Chem. Mater., 2015, 27, 6848–6857 CrossRef CAS.
- S. J. Townsend, T. J. Lenosky, D. A. Muller, C. S. Nichols and V. Elser, Phys. Rev. Lett., 1992, 69, 921–924 CrossRef CAS PubMed.
- A. P. Terzyk, S. Furmaniak, P. J. F. Harris, P. A. Gauden, J. Włoch, P. Kowalczyk and G. Rychlicki, Phys. Chem. Chem. Phys., 2007, 9, 5919–5927 RSC.
- A. R. Payne and R. E. Whittaker, Composites, 1970, 1, 203–214 CrossRef CAS.
- D. D. L. Chung, Carbon Fiber Composites, Butterworth-Heinemann, Newton, MA, 1994 Search PubMed.
- H. Marsh and F. Rodríguez-Reinoso, Activated Carbon, Elsevier, Oxford, 2006 Search PubMed.
- M. R. Benzigar, S. N. Talapaneni, S. Joseph, K. Ramadass, G. Singh, J. Scaranto, U. Ravon, K. Al-Bahily and A. Vinu, Chem. Soc. Rev., 2018, 47, 2680–2721 RSC.
- L. Wang and X. Hu, Chem.–Asian J., 2018, 13, 1518–1529 CrossRef CAS PubMed.
- M. M. Sabzehmeidani, S. Mahnaee, M. Ghaedi, H. Heidari and V. A. L. Roy, Mater. Adv., 2021, 2, 598–627 RSC.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- C. W. B. Bezerra, L. Zhang, K. Lee, H. Liu, A. L. B. Marques, E. P. Marques, H. Wang and J. Zhang, Electrochim. Acta, 2008, 53, 4937–4951 CrossRef CAS.
- H. Tan, J. Tang, J. Kim, Y. V. Kaneti, Y.-M. Kang, Y. Sugahara and Y. Yamauchi, J. Mater. Chem. A, 2019, 7, 1380–1393 RSC.
- Y. Nishi, Chem. Rec., 2001, 1, 406–413 CrossRef CAS PubMed.
- J. Asenbauer, T. Eisenmann, M. Kuenzel, A. Kazzazi, Z. Chen and D. Bresser, Sustainable Energy Fuels, 2020, 4, 5387–5416 RSC.
- A. Gomez-Martin, J. Martinez-Fernandez, M. Ruttert, A. Heckmann, M. Winter, T. Placke and J. Ramirez-Rico, ChemSusChem, 2018, 11, 2776–2787 CrossRef CAS PubMed.
- W.-J. Liu, H. Jiang and H.-Q. Yu, Energy Environ. Sci., 2019, 12, 1751–1779 RSC.
- C. Liedel, ChemSusChem, 2020, 13, 2110–2141 CrossRef CAS PubMed.
- A. Gutiérrez-Pardo, J. Ramírez-Rico, R. Cabezas-Rodríguez and J. Martínez-Fernández, J. Power Sources, 2015, 278, 18–26 CrossRef.
- Y. Li, J. Hu, Z. Wang, K. Yang, W. Huang, B. Cao, Z. Li, W. Zhang and F. Pan, ACS Appl. Mater. Interfaces, 2019, 11, 24164–24171 CrossRef CAS PubMed.
- N. Wang, Q. Liu, B. Sun, J. Gu, B. Yu, W. Zhang and D. Zhang, Sci. Rep., 2018, 8, 9934 CrossRef PubMed.
- V. Gupta and T. A. Saleh, Environ. Sci. Pollut. Res., 2013, 20, 2828–2843 CrossRef CAS PubMed.
- J. Xu, Z. Cao, Y. Zhang, Z. Yuan, Z. Lou, X. Xu and X. Wang, Chemosphere, 2018, 195, 351–364 CrossRef CAS PubMed.
- W. Chen, L. Duan and D. Zhu, Environ. Sci. Technol., 2007, 41, 8295–8300 CrossRef CAS PubMed.
- H. J. Wang, A. L. Zhou, F. Peng, H. Yu and J. Yang, J. Colloid Interface Sci., 2007, 316, 277–283 CrossRef CAS PubMed.
- S. A. C. Carabineiro, T. Thavorn-amornsri, M. F. R. Pereira, P. Serp and J. L. Figueiredo, Catal. Today, 2012, 186, 29–34 CrossRef CAS.
- L. Ji, W. Chen, L. Duan and D. Zhu, Environ. Sci. Technol., 2009, 43, 2322–2327 CrossRef CAS PubMed.
- M. J. Sweetman, S. May, N. Mebberson, P. Pendleton, K. Vasilev, S. E. Plush and J. D. Hayball, J. Carbon Res., 2017, 3, 18 CrossRef.
- S. C. Smith and D. F. Rodrigues, Carbon, 2015, 91, 122–143 CrossRef CAS.
- J. Hoekstra, A. M. Beale, F. Soulimani, M. Versluijs-Helder, D. van de Kleut, M. Koelewijn, J. W. Geus and L. W. Jenneskens, Carbon, 2016, 107, 248–260 CrossRef CAS.
- R. D. Hunter, J. Davies, S. J. A. Hérou, A. Kulak and Z. Schnepp, Philos. Trans. R. Soc., A, 2021, 379, 20200336 CrossRef CAS PubMed.
- H. Valladas, Meas. Sci. Technol., 2003, 14, 1487–1492 CrossRef CAS.
- P. Harris, Interdiscip. Sci. Rev., 1999, 24, 301–306 CrossRef.
- M. J. Antal and M. Grønli, Ind. Eng. Chem. Res., 2003, 42, 1619–1640 CrossRef CAS.
- J. Lehmann and S. Joseph, Biochar for Environmental Management Science and Technology, Earthscan, London, 2009 Search PubMed.
- Z. Heidarinejad, M. H. Dehghani, M. Heidari, G. Javedan, I. Ali and M. Sillanpää, Environ. Chem. Lett., 2020, 18, 393–415 CrossRef CAS.
- N. Arora and N. N. Sharma, Diamond Relat. Mater., 2014, 50, 135–150 CrossRef CAS.
- N. I. Alekseyev and G. A. Dyuzhev, Carbon, 2003, 41, 1343–1348 CrossRef CAS.
- Y. Saito, T. Nakahira and S. Uemura, J. Phys. Chem. B, 2003, 107, 931–934 CrossRef CAS.
- R. Hu, T. Furukawa, X. Wang and M. Nagatsu, Appl. Surf. Sci., 2017, 416, 731–741 CrossRef CAS.
- J. Prasek, J. Drbohlavova, J. Chomoucka, J. Hubalek, O. Jasek, V. Adam and R. Kisek, J. Mater. Chem., 2011, 21, 15872–15884 RSC.
- M. Kokarneswaran, P. Selvaraj, T. Ashokan, S. Perumal, P. Sellappan, K. D. Murugan, S. Ramalingam, N. Mohan and V. Chandrasekaran, Sci. Rep., 2020, 10, 19786 CrossRef CAS PubMed.
- M. Reibold, P. Paufler, A. A. Levin, W. Kochmann, N. Pätzke and D. C. Meyer, Nature, 2006, 444, 286 CrossRef CAS PubMed.
- E. Charon, J.-N. Rouzaud and J. Aléon, Carbon, 2014, 66, 178–190 CrossRef CAS.
- H. Marsh and A. P. Warburton, J. Appl. Chem., 1970, 20, 133–142 CrossRef CAS.
- E. G. Acheson, US Pat., 568323 (1896), 616974 (1899) and 645285 (1900) Search PubMed.
- A. Oya and H. Marsh, J. Mater. Sci., 1982, 17, 309–322 CrossRef CAS.
- A. Oya and S. Otani, Carbon, 1979, 17, 131–137 CrossRef CAS.
- S. M. Irving and P. L. Walker, Carbon, 1967, 5, 399–400 CrossRef CAS.
- W. Weisweiler, N. Subramanian and B. Terwiesch, Carbon, 1971, 9, 755–761 CrossRef CAS.
- T. Britt and P. C. Pistorius, Metall. Mater. Trans. B, 2021, 52, 1–5 CrossRef CAS.
- C. Yokokawa, K. Hosokawa and Y. Takegami, Carbon, 1966, 4, 459–465 CrossRef CAS.
- S. Gupta, V. Sahajwalla, J. Burgo, P. Chaubal and T. Youmans, Metall. Mater. Trans. B, 2005, 36, 385–394 CrossRef.
- K. Li, H. Li, M. Sun, J. Zhang, H. Zhang, S. Ren and M. Barati, Energy Fuels, 2019, 33, 10941–10952 CrossRef CAS.
- H. Li, H. Zhang, K. Li, J. Zhang, M. Sun and B. Su, Fuel, 2020, 279, 118531 CrossRef CAS.
- P. L. Walker, J. F. Rakszawski and G. R. Imperial, J. Phys. Chem., 1959, 63, 133–140 CrossRef CAS.
- W. R. Davis, R. Slawson and G. R. Rigby, Nature, 1953, 171, 756 CrossRef CAS.
- W. R. Ruston, M. Warzee, J. Hennaut and J. Waty, Carbon, 1969, 7, 47–57 CrossRef CAS.
- H. P. Boehm, Carbon, 1973, 11, 583–590 CrossRef.
- A. Oberlin and M. Endo, J. Cryst. Growth, 1976, 32, 335–349 CrossRef CAS.
- M. José-Yacamán, M. Miki-Yoshida, L. Rendón and J. G. Santiesteban, Appl. Phys. Lett., 1993, 62, 657 CrossRef.
- H. Marsh, D. Crawford and D. W. Taylor, Carbon, 1983, 21, 81–87 CrossRef CAS.
- J. Ozaki, M. Mitsui and Y. Nishiyama, Carbon, 1998, 36, 131–135 CrossRef CAS.
- J. Yang and S. Zuo, Diamond Relat. Mater., 2019, 95, 1–4 CrossRef CAS.
- M. Sevilla, C. Sanchís, T. Valdés-Solís, E. Morallón and A. B. Fuertes, Carbon, 2008, 46, 931–939 CrossRef CAS.
- L. Chen, Z. Wang, C. He, N. Zhao, C. Shi, E. Liu and J. Li, ACS Appl. Mater. Interfaces, 2013, 5, 9537–9545 CrossRef CAS PubMed.
- C. Defilippi, M. O. A. Mukadam, S. A. Nicolae, M. R. Lees and C. Giordano, Materials, 2019, 12, 323 CrossRef CAS PubMed.
- M. N. Obrovac, X. Zhao, L. T. Burke and R. A. Dunlap, Electrochem. Commun., 2015, 60, 221–224 CrossRef CAS.
- M. J. Xie, J. Yang, J. Y. Liang, X. F. Guo and W. P. Ding, Carbon, 2014, 77, 215–225 CrossRef CAS.
- J. Hoekstra, A. M. Beale, F. Soulimani, M. Versluijs-Helder, J. W. Geus and L. W. Jenneskens, New J. Chem., 2015, 39, 6593–6601 RSC.
- H. Niu, Y. Wang, X. Zhang, Z. Meng and Y. Cai, ACS Appl. Mater. Interfaces, 2012, 4, 286–295 CrossRef CAS PubMed.
- Z. Tian, C. Wang, J. Yue, X. Zhang and L. Ma, Catal. Sci. Technol., 2019, 9, 2728–2741 RSC.
- R. D. Hunter, J. L. Rowlandson, G. J. Smales, B. R. Pauw, V. P. Ting, A. Kulak and Z. Schnepp, Mater. Adv., 2020, 1, 3281–3291 RSC.
- R. Atchudan, S. Perumal, T. N. J. I. Edison and Y. R. Lee, Mater. Lett., 2016, 166, 145–149 CrossRef CAS.
- Z. Cao, M. Qin, C. Zuo, Y. Gu and B. Jia, J. Colloid Interface Sci., 2017, 491, 55–63 CrossRef CAS PubMed.
- S. Juvanen, A. Sarapuu, S. Vlassov, M. Kook, V. Kisand, M. Käärik, A. Treshchalov, J. Aruväli, J. Kozlova, A. Tamm, J. Leis and K. Tammeveski, ChemElectroChem, 2021, 8, 2288–2297 CrossRef CAS.
- I.-A. Choi, D.-H. Kwak, S.-B. Han, J.-Y. Park, H.-S. Park, K.-B. Ma, D.-H. Kim, J.-E. Won and K.-W. Park, Appl. Catal., B, 2017, 211, 235–244 CrossRef CAS.
- J. J. Shi, X. M. Hu, M. R. Madsen, P. Lamagni, E. T. Bjerglund, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, ACS Appl. Nano Mater., 2018, 1, 3608–3615 CrossRef CAS.
- H. Khani, N. S. Grundish, D. O. Wipf and J. B. Goodenough, Adv. Energy Mater., 2020, 10, 1903215 CrossRef CAS.
- M. Xiao, J. Zhu, L. Feng, C. Liu and W. Xing, Adv. Mater., 2015, 27, 2521–2527 CrossRef CAS PubMed.
- C. W. Huang, L. C. Hsu and Y. Y. Li, Nanotechnology, 2006, 17, 4629–4634 CrossRef CAS PubMed.
- Z. Tang, Y. Song, X. He and J. Yang, Mater. Lett., 2012, 89, 330–332 CrossRef CAS.
- K. Inomata and Y. Otake, Microporous Mesoporous Mater., 2011, 143, 60–65 CrossRef CAS.
- G. Hasegawa, K. Kanamori and K. Nakanishi, Mater. Lett., 2012, 76, 1–4 CrossRef CAS.
- N. I. Maksimova, O. P. Krivoruchko, G. Mestl, V. I. Zaikovskii, A. L. Chuvilin, A. N. Salanov and E. B. Burgina, J. Mol. Catal. A: Chem., 2000, 158, 301–307 CrossRef CAS.
- M. Sevilla and A. B. Fuertes, Carbon, 2006, 44, 468–478 CrossRef CAS.
- J. J. Li, Y. Liang, B. J. Dou, C. Y. Ma, R. J. Lu, Z. P. Hao, Q. Xie, Z. Q. Luan and K. Li, Mater. Chem. Phys., 2013, 138, 484–489 CrossRef CAS.
- A. M. Wang, J. W. Ren, B. F. Shi, G. Z. Lu and Y. Q. Wang, Microporous Mesoporous Mater., 2012, 151, 287–292 CrossRef CAS.
- J. S. Li, J. Gu, H. J. Li, Y. Liang, Y. X. Hao, X. Y. Sun and L. J. Wang, Microporous Mesoporous Mater., 2010, 128, 144–149 CrossRef CAS.
- J. I. Ozaki, K. Nozawa, K. Yamada, Y. Uchiyama, Y. Yoshimoto, A. Furuichi, T. Yokoyama, A. Oya, L. J. Brown and J. D. Cashion, J. Appl. Electrochem., 2006, 36, 239–247 CrossRef CAS.
- C. G. Renda, R. Bertholdo, T. Venancio, A. P. Luz, V. C. Pandolfelli and A. A. Lucas, Ceram. Int., 2019, 45, 12196–12204 CrossRef CAS.
- B. Zhang, C. Liu, W. Kong and C. Qi, Frontiers of Materials Science, 2016, 10, 147–156 CrossRef.
- J. Qi, L. H. Jiang, Q. W. Tang, S. Zhu, S. L. Wang, B. L. Yi and C. Q. Sun, Carbon, 2012, 50, 2824–2831 CrossRef CAS.
- A. Abdelwahab, J. Castelo-Quibén, J. F. Vivo-Vilches, M. Pérez-Cadenas, F. J. Maldonado-Hódar, F. Carrasco-Marin and A. F. Pérez-Cadenas, Nanomaterials, 2018, 8, 266 CrossRef PubMed.
- A. Chen, Y. Yu, T. Xing, R. Wang, Y. Zhang and Q. Li, J. Mater. Sci., 2015, 50, 5578–5582 CrossRef CAS.
- M. M. Gaikwad, M. Kakunuri and C. S. Sharma, Mater. Today Commun., 2019, 20, 100569 CrossRef CAS.
- W. Kicinski, M. Norek and M. Bystrzejewski, J. Phys. Chem. Solids, 2013, 74, 101–109 CrossRef CAS.
- Y.-H. Chung and S. Jou, Mater. Chem. Phys., 2005, 92, 256–259 CrossRef CAS.
- F. Cesano, M. M. Rahman, F. Bardelli, A. Damin and D. Scarano, ChemistrySelect, 2016, 1, 2536–2541 CrossRef CAS.
- S. Zhu, Y. Wu, Q. Chen, Z. Yu, C. Wang, S. Jin, Y. Ding and G. Wu, Green Chem., 2006, 8, 325–327 RSC.
- M. Egal, T. Budtova and P. Navard, Biomacromolecules, 2007, 8, 2282–2287 CrossRef CAS PubMed.
- A. Huidobro, A. C. Pastor and F. Rodríguez-Reinoso, Carbon, 2001, 39, 389–398 CrossRef CAS.
- A. Solak and P. Rutkowski, J. Mater. Cycles Waste Manage., 2014, 16, 491–499 CrossRef CAS.
- Q. C. Yan, J. H. Li, X. F. Zhang, E. Hassan, C. J. Wang, J. L. Zhang and Z. Y. Cai, J. Nanopart. Res., 2018, 20, 223 CrossRef.
- Q. G. Yan, J. H. Li, X. F. Zhang, J. L. Zhang and Z. Y. Cai, Nanomater. Nanotechnol., 2018, 8, 1–12 Search PubMed.
- X. F. Zhang, Q. G. Yan, J. H. Li, I. W. Chu, H. Toghiani, Z. Y. Cai and J. L. Zhang, Polymers, 2018, 10, 183 CrossRef PubMed.
- G. Daniel, T. Kosmala, F. Brombin, M. Mazzucato, A. Facchin, M. C. Dalconi, D. Badocco, P. Pastore, G. Granozzi and C. Durante, Catalysts, 2021, 11, 390 CrossRef CAS.
- S. W. Zhang, M. Y. Zeng, J. X. Li, J. Li, J. Z. Xu and X. K. Wang, J. Mater. Chem. A, 2014, 2, 4391–4397 RSC.
- A. E. Danks, M. J. Hollamby, B. Hammouda, D. C. Fletcher, F. Johnston-Banks, S. E. Rogers and Z. Schnepp, J. Mater. Chem. A, 2017, 5, 11644–11651 RSC.
- Z. Schnepp, Y. Zhang, M. J. Hollamby, B. R. Pauw, M. Tanaka, Y. Matsushita and Y. Sakka, J. Mater. Chem. A, 2013, 1, 13576–13581 RSC.
- J. Hoekstra, A. M. Beale, F. Soulimani, M. Versluijs-Helder, J. W. Geus and L. W. Jenneskens, J. Phys. Chem. C, 2015, 119, 10653–10661 CrossRef CAS.
- S. Glatzel, Z. Schnepp and C. Giordano, Angew. Chem., Int. Ed., 2013, 52, 2355–2358 CrossRef CAS PubMed.
- K. Lotz, A. Wutscher, H. Dudder, C. M. Berger, C. Russo, K. Mukherjee, G. Schwaab, M. Havenith and M. Muhler, ACS Omega, 2019, 4, 4448–4460 CrossRef CAS PubMed.
- J. Hoekstra, M. Versluijs-Helder, E. J. Vlietstra, J. W. Geus and L. W. Jenneskens, ChemSusChem, 2015, 8, 985–989 CrossRef CAS PubMed.
- S. Shi, W. Che, K. Liang, C. Xia and D. Zhang, J. Anal. Appl. Pyrolysis, 2015, 115, 1–6 CrossRef CAS.
- A. N. Prusov, S. M. Prusova, A. G. Zakharov, A. V. Bazanov and V. K. Ivanov, Fibre Chem., 2018, 50, 154–160 CrossRef CAS.
- S. Xia, N. Cai, W. Lu, H. Zhou, H. Xiao, X. Chen, Y. Chen, H. Yang, X. Wang, S. Wang and H. Chen, J. Cleaner Prod., 2021, 329, 129735 CrossRef CAS.
- Q. Yan, X. Zhang, J. Li, E. Hassan, C. Wang, J. Zhang and Z. Cai, J. Mater. Sci., 2018, 53, 8020–8029 CrossRef CAS.
- X. Zhang, Q. Yan, J. Li, J. Zhang and Z. Cai, Materials, 2018, 11, 139 CrossRef PubMed.
- S. T. Neeli and H. Ramsurn, Carbon, 2018, 134, 480–490 CrossRef CAS.
- Z. Schnepp, Y. Zhang, M. J. Hollamby, B. R. Pauw, M. Tanaka, Y. Matsushita and Y. Sakka, J. Mater. Chem. A, 2013, 1, 13576–13581 RSC.
- J. Zhao, Y. Liu, X. Quan, S. Chen, H. Yu and H. Zhao, Appl. Surf. Sci., 2017, 396, 986–993 CrossRef CAS.
- Q. Chen, X. Tan, Y. Liu, S. Liu, M. Li, Y. Gu, P. Zhang, S. Ye, Z. Yang and Y. Yang, J. Mater. Chem. A, 2020, 8, 5773–5811 RSC.
- S. Sotiropoulou, Y. Sierra-Sastre, S. S. Mark and C. A. Batt, Chem. Mater., 2008, 20, 821–834 CrossRef CAS.
- E. Thompson, A. E. Danks, L. Bourgeois and Z. Schnepp, Green Chem., 2015, 17, 551–556 RSC.
- W.-J. Liu, K. Tian, Y.-R. He, H. Jiang and H.-Q. Yu, Environ. Sci. Technol., 2014, 48, 13951–13959 CrossRef CAS PubMed.
- W.-J. Liu, F.-X. Zeng, H. Jiang and H.-Q. Yu, Bioresour. Technol., 2011, 102, 3471–3479 CrossRef CAS PubMed.
- S. Zhang, Y. Su, S. Zhu, H. Zhang and Q. Zhang, J. Anal. Appl. Pyrolysis, 2018, 135, 22–31 CrossRef CAS.
- Z. Xu, Y. Zhou, Z. Sun, D. Zhang, Y. Huang, S. Gu and W. Chen, Chemosphere, 2020, 241, 125120 CrossRef CAS PubMed.
- Z. Xu, Z. Sun, Y. Zhou, W. Chen, T. Zhang, Y. Huang and D. Zhang, Colloids Surf., A, 2019, 582, 123934 CrossRef CAS.
- J. Bedia, M. Peñas-Garzón, A. Gómez-Avilés, J. J. Rodriguez and C. Belver, J. Carbon Res., 2020, 6, 21 CrossRef CAS.
- M. Sevilla, C. Sanchís, T. Valdés-Solís, E. Morallón and A. B. Fuertes, J. Phys. Chem. C, 2007, 111, 9749–9756 CrossRef CAS.
- A. Gomez-Martin, A. Gutierrez-Pardo, J. Martinez-Fernandez and J. Ramirez-Rico, Fuel Process. Technol., 2020, 199, 106279 CrossRef CAS.
- L. Ma, Y. Xu, Y. Liu, H. Zhang, J. Yao, N. Li, C. M. Li, W. Zhou and J. Jiang, ACS Sustainable Chem. Eng., 2019, 7, 17919–17928 CrossRef CAS.
- Y. Gong, D. Li, C. Luo, Q. Fu and C. Pan, Green Chem., 2017, 19, 4132–4140 RSC.
- L. Sun, C. Tian, M. Li, X. Meng, L. Wang, R. Wang, J. Yin and H. Fu, J. Mater. Chem. A, 2013, 1, 6462–6470 RSC.
- Q. Liu, J. Gu, W. Zhang, Y. Miyamoto, Z. Chen and D. Zhang, J. Mater. Chem., 2012, 22, 21183–21188 RSC.
- Y. Liu, J. Ruan, S. Sang, Z. Zhou and Q. Wu, Electrochim. Acta, 2016, 215, 388–397 CrossRef CAS.
- F. Wu, R. Huang, D. Mu, B. Wu and Y. Chen, Electrochim. Acta, 2016, 187, 508–516 CrossRef CAS.
- X. Li, E. Cui, Z. Xiang, L. Yu, J. Xiong, F. Pan and W. Lu, Colloids Surf., A, 2020, 819, 152952 CAS.
- L. Chen, T. Ji, L. Mu, Y. Shi, L. Brisbin, Z. Guo, M. A. Khan, D. P. Young and J. Zhu, RSC Adv., 2016, 6, 2259–2269 RSC.
- Z. Yan, C. Dai, M. Zhang, X. Lu, X. Zhao and J. Xie, Int. J. Hydrogen Energy, 2019, 44, 4090–4101 CrossRef CAS.
- Y. Cai, Y. Luo, H. Dong, X. Zhao, Y. Xiao, Y. Liang, H. Hu, Y. Liu and M. Zheng, J. Power Sources, 2017, 353, 260–269 CrossRef CAS.
- I. Major, J. M. Pin, E. Behazin, A. Rodriguez-Uribe, M. Misra and A. Mohanty, Green Chem., 2018, 20, 2269–2278 RSC.
- E. Petala, Y. Georgiou, V. Kostas, K. Dimos, M. A. Karakassides, Y. Deligiannakis, C. Aparicio, J. Tuček and R. Zbořil, ACS Sustainable Chem. Eng., 2017, 5, 5782–5792 CrossRef CAS.
- X. Zhang, H. Li, K. Zhang, Q. Wang, B. Qin, Q. Cao and L. Jin, J. Electrochem. Soc., 2018, 165, A2084–A2092 CrossRef CAS.
- S. Xia, K. Li, H. Xiao, N. Cai, Z. Dong, C. Xu, Y. Chen, H. Yang, X. Tu and H. Chen, Bioresour. Technol., 2019, 287, 121444 CrossRef CAS PubMed.
- W. Tian, Q. Gao, Y. Tan and Z. Li, Carbon, 2017, 119, 287–295 CrossRef CAS.
- M. A. Ahsan, A. R. Puente Santiago, A. Rodriguez, V. Maturano-Rojas, B. Alvarado-Tenorio, R. Bernal and J. C. Noveron, J. Cleaner Prod., 2020, 275, 124141 CrossRef CAS.
- S. Xia, N. Cai, J. Wu, H. Xiao, J. Hu, X. Chen, Y. Chen, H. Yang, X. Wang and H. Chen, Fuel Process. Technol., 2020, 209, 106543 CrossRef CAS.
- K. Suzuki, Y. Saito, N. Okazaki and T. Suzuki, Sci. Rep., 2020, 10, 12131 CrossRef CAS PubMed.
- A. S. Kamal, N. H. Jabarullah and R. Othman, Mater. Today: Proc., 2020, 31, 211–216 Search PubMed.
- J. He, D. Zhang, Y. Wang, J. Zhang, B. Yang, H. Shi, K. Wang and Y. Wang, Appl. Surf. Sci., 2020, 515, 146020 CrossRef CAS.
- B. S. Purwasasmita, F. Tafwidli and R. Septawendar, J. Aust. Ceram. Soc., 2013, 49, 119–126 CAS.
- X. Zhang, K. Zhang, H. Li, Q. Cao, L. Jin and P. Li, J. Power Sources, 2017, 344, 176–184 CrossRef CAS.
- M. Sevilla, C. Salinas Martínez-de Lecca, T. Valdés-Solís, E. Morallón and A. B. Fuertes, Phys. Chem. Chem. Phys., 2008, 10, 1433–1442 RSC.
- Z. H. Li, H. Y. Zhang, H. P. Zhu, L. Q. Li and H. Y. Liao, J. Mater. Sci., 2016, 51, 5676–5684 CrossRef CAS.
- A. Leonhardt, M. Ritschel, D. Elefant, N. Mattern, K. Biedermann, S. Hampel, C. Müller, T. Gemming and B. Büchner, J. Appl. Phys., 2005, 98, 074315 CrossRef.
- Z. M. Sheng, M. H. Hu, Z. Y. Dai, C. Y. Hong, C. K. Chang, Q. Z. Chen and X. J. Chang, Microporous Mesoporous Mater., 2016, 234, 224–229 CrossRef CAS.
- J. N. Wang, L. Zhang, J. J. Niu, F. Yu, Z. M. Sheng, Y. Z. Zhao, H. Chang and C. Pak, Chem. Mater., 2007, 19, 453–459 CrossRef CAS.
- A. O. Baskakov, I. S. Lyubutin, S. S. Starchikov, V. A. Davydov, L. F. Kulikova, T. B. Egorova and V. N. Agafonov, Inorg. Chem., 2018, 57, 14895–14903 CrossRef CAS PubMed.
- K. T. Lee, X. Ji, M. Rault and L. F. Nazar, Angew. Chem., Int. Ed., 2009, 48, 5661–5665 CrossRef CAS PubMed.
- J. Song, M. Sun, Q. Chen, J. Wnag, G. Zhang and Z. Xue, J. Phys. D: Appl. Phys., 2004, 37, 5–9 CrossRef CAS.
- S. S. Starchikov, V. A. Zayakhanov, A. L. Vasiliev, S. Lyubutin, A. O. Baskakov, Y. A. Nikiforova, K. O. Funtov, M. V. Lyubutina, L. F. Kulikova, V. N. Agafonov and V. A. Davydov, Carbon, 2021, 178, 708–717 CrossRef CAS.
- V. Jourdain and C. Bichara, Carbon, 2013, 58, 2–39 CrossRef CAS.
- J.-P. Tessonier and D. S. Su, ChemSusChem, 2011, 4, 824–847 CrossRef PubMed.
- H. Dai, A. G. Rinzler, P. Nikolaev, A. Thess, D. T. Colbert and R. E. Smalley, Chem. Phys. Lett., 1996, 260, 471–475 CrossRef CAS.
- A. J. Page, Y. Ohta, S. Irle and K. Morokuma, Acc. Chem. Res., 2010, 43, 1375–1385 CrossRef CAS PubMed.
- M. A. Ribas, F. Ding, P. B. Balbuena and B. I. Yakobson, J. Chem. Phys., 2009, 131, 224501 CrossRef PubMed.
- C. T. Wirth, B. C. Bayer, A. D. Gamalski, S. Esconjauregui, R. S. Weatherup, C. Ducati, C. Baehtz, J. Robertson and S. Hofmann, Chem. Mater., 2012, 24, 4633–4640 CrossRef CAS.
- S. Hofmann, R. Sharma, C. Ducati, G. Du, C. Mattevi, C. Cepek, M. Cantoro, S. Pisana, A. Parvez, F. Cervantes-Sodi, A. C. Ferrari, R. Dunin-Borkowski, S. Lizzit, L. Petaccia, A. Goldoni and J. Robertson, Nano Lett., 2007, 7, 602–608 CrossRef CAS PubMed.
- F. Ding, A. Rosen, E. E. B. Campbell, L. K. L. Falk and K. Bolton, J. Phys. Chem. B, 2006, 110, 7666–7670 CrossRef CAS PubMed.
- Z. D. Zhang, J. G. Zheng, I. Skorvanek, G. H. Wen, J. Kovac, F. W. Wang, J. L. Yu, Z. J. Li, X. L. Dong, S. R. Jin, W. Liu and X. X. Zhang, J. Phys.: Condens. Matter, 2001, 13, 1921–1929 CrossRef CAS.
- F. Ding, K. Bolton and A. Rosén, J. Phys. Chem. B, 2004, 108, 17369–17377 CrossRef CAS.
- C. L. Cheung, A. Kurtz, H. Park and C. M. Lieber, J. Phys. Chem. B, 2002, 106, 2429–2433 CrossRef CAS.
- F. Ding, A. Rosen and K. Bolton, J. Chem. Phys., 2004, 121, 2775–2779 CrossRef CAS PubMed.
- Z. Yu, D. Chen, B. Tøtdal and A. Holmen, Catal. Today, 2005, 100, 261–267 CrossRef CAS.
- S. Hofmann, G. Csanyi, A. C. Ferrari, M. C. Payne and J. Robertson, Phys. Rev. Lett., 2005, 95, 036101 CrossRef CAS PubMed.
- A. A. Puretzky, D. B. Geohegan, S. Jesse, I. N. Ivanov and G. Eres, Appl. Phys. A: Mater. Sci. Process., 2005, 81, 223–240 CrossRef CAS.
- Y. Wang, B. Li, P. S. Ho, Z. Yao and L. Shi, Appl. Phys. Lett., 2006, 89, 183113 CrossRef.
- Z. He, J.-L. Maurice, A. Gohier, C. S. Lee, D. Pribat and C. S. Cojocaru, Chem. Mater., 2011, 23, 5379–5387 CrossRef CAS.
- M. Lin, J. P. Y. Tan, C. Boothroyd, K. P. Loh, E. S. Tok and Y.-L. Foo, Nano Lett., 2007, 7, 2234–2238 CrossRef CAS PubMed.
- F. Ding, K. Bolton and A. Rosén, J. Electron. Mater., 2006, 35, 207–210 CrossRef CAS.
- T. Ichihashi, M. Ishida, Y. Ochiai and J. Fujita, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.–Process., Meas., Phenom., 2004, 22, 3221–3223 CrossRef CAS.
- C. T. Wirth, S. Hofmann and J. Robertson, Diamond Relat. Mater., 2009, 18, 940–945 CrossRef CAS.
- H. Yoshida, S. Takeda, T. Uchiyama, H. Kohno and Y. Homma, Nano Lett., 2008, 8, 2082–2086 CrossRef CAS PubMed.
- A. Gomez-Martin, Z. Schnepp and J. Ramirez-Rico, Chem. Mater., 2021, 33, 3087–3097 CrossRef CAS.
- Z. Schnepp, A. E. Danks, M. J. Hollamby, B. R. Pauw, C. A. Murray and C. C. Tang, Chem. Mater., 2015, 27, 5094–5099 CrossRef CAS.
- A. Mehmood, J. Pampel, G. Ali, H. Y. Ha, F. Ruiz-Zepeda and T.-P. Fellinger, Adv. Energy Mater., 2018, 8, 1701771 CrossRef.
- D. Menga, J. L. Low, Y.-S. Li, I. Arčon, B. Koyotürk, F. Wagner, F. Ruiz-Zepeda, M. Gaberšček, B. Paulus and T.-P. Fellinger, J. Am. Chem. Soc., 2021, 143, 18010–18019 CrossRef CAS PubMed.
- J. Wu, Z. Pan, Y. Zhang, B. Wang and H. Peng, J. Mater. Chem. A, 2018, 6, 12932–12944 RSC.
- G. Li, J. Yu, W. Yu, L. Yang, X. Zhang, X. Liu, H. Liu and W. Zhou, Small, 2020, 16, 2001980 CrossRef CAS PubMed.
- S. S. Shah, S. M. A. Nayem, N. Sultana, A. J. S. Ahammad and M. A. Aziz, ChemSusChem, 2022, 15, e202101282 Search PubMed.
- J. Wei, H. Zhu, Y. Jia, Q. Shu, C. Li, K. Wang, B. Wei, Y. Zhu, Z. Wang, J. Luo, W. Liu and D. Wu, Carbon, 2007, 45, 2152–2158 CrossRef CAS.
- S.-H. Lee, J. Park, H.-R. Kim, J. Lee and K.-H. Lee, RSC Adv., 2015, 5, 41894–41900 RSC.
- Z. M. Sheng, N. N. Li, Q. M. Xu, C. Y. Hong, S. Y. Wu, C. K. Chang, S. Han and C. M. Li, Sustainable Energy Fuels, 2021, 5, 4080–4086 RSC.
- Z. M. Sheng, C. Y. Hong, N. N. Li, Q. Z. Chen, R. P. Jia, D. Y. Zhang and S. Han, Electrochim. Acta, 2018, 259, 1104–1109 CrossRef CAS.
- R. S. Wagner and W. C. Ellis, Appl. Phys. Lett., 1964, 4, 89–90 CrossRef CAS.
- G. J. Long and H. P. Leighly, J. Chem. Educ., 1982, 59, 948–953 CrossRef CAS.
- O. P. Krivoruchko and V. I. Zaikovskii, Mendeleev Commun., 1998, 3, 97–99 CrossRef.
- A. R. Harutyunyan, T. Tokune and E. Mora, Appl. Phys. Lett., 2005, 87, 051919 CrossRef.
- M. Takagi, J. Phys. Soc. Jpn., 1954, 9, 359–363 CrossRef.
- F. Ding, A. Rosen and K. Bolton, Carbon, 2005, 43, 2215–2217 CrossRef CAS.
- F. Ding, K. Bolton and A. Rosen, Eur. Phys. J. D, 2005, 34, 275–277 CrossRef CAS.
- F. Ding, A. Rosen, S. Curtarolo and K. Bolton, Appl. Phys. Lett., 2006, 88, 133110 CrossRef.
- F. Ding, A. Rosen, S. Curtarolo and K. Bolton, Appl. Phys. Lett., 2006, 88, 133110 CrossRef.
- D. Schebarchov and S. C. Hendy, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 085407 CrossRef.
- E. Fitzer and S. Weisenburger, Carbon, 1974, 12, 657–666 CrossRef CAS.
- M. S. Chambers, D. S. Keeble, D. Fletcher, J. A. Hriljac and Z. Schnepp, Inorg. Chem., 2021, 60, 7062–7069 CrossRef CAS PubMed.
- M. S. Chambers, R. D. Hunter, M. J. Hollamby, B. R. Pauw, A. J. Smith, T. Snow, A. E. Danks and Z. Schnepp, Inorg. Chem., 2021 Search PubMed , submitted..
- H. Marsh and A. P. Warburton, Carbon, 1976, 14, 47–52 CrossRef CAS.
- S. A. Steiner, T. F. Baumann, B. C. Bayer, R. Blume, M. A. Worsley, W. J. MoberlyChan, E. L. Shaw, R. Schlögl, A. J. Hart, S. Hofmann and B. L. Wardle, J. Am. Chem. Soc., 2009, 131, 12144–12154 CrossRef CAS PubMed.
- P. Xiao, Y. Gong, D. Li and Z. Li, Microporous Mesoporous Mater., 2021, 323, 111201 CrossRef CAS.
- F. J. Maldonado-Hódar, C. Moreno-Castilla, J. Rivera-Utrilla, Y. Hanzawa and Y. Yamada, Langmuir, 2000, 16, 4367–4373 CrossRef.
- C. J. Thambiliyagodage, S. Ulrich, P. T. Araujo and M. G. Bakker, Carbon, 2018, 134, 452–463 CrossRef CAS.
- M. Sevilla and A. B. Fuertes, Carbon, 2013, 56, 155–166 CrossRef CAS.
- F. Yu, J. N. Wang, Z. M. Sheng and L. F. Su, Carbon, 2005, 43, 3018–3021 CrossRef CAS.
- H. Baker, ASM Handbook: Alloy Phase Diagrams, 1998, vol. 3 Search PubMed.
- R. Wang, G. Lu, W. Qiao and J. Yu, Langmuir, 2016, 32, 8583–8592 CrossRef CAS PubMed.
- S. Yi, Z. Fan, C. Wu and J. Chen, Carbon, 2008, 46, 378–380 CrossRef CAS.
- L. Zhao, X. Zhao, L. T. Burke, J. C. Bennett, R. A. Dunlap and M. N. Obrovac, ChemSusChem, 2017, 10, 3409–3418 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |