
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ visualisation and analysis of dynamic single atom processes in heterogeneous catalysts
Pratibha L.
Gai
 *abc and
Edward D.
Boyes
*abd
*abc and
Edward D.
Boyes
*abd
aThe York Nanocentre, University of York, York YO10 5DD, UK. E-mail: pratibha.gai@york.ac.uk; ed.boyes@york.ac.uk
bDepartment of Physics, University of York, York YO10 5DD, UK
cDepartment of Chemistry, University of York, York YO10 5DD, UK
dDepartment of Electronic Engineering, University of York, York YO10 5DD, UK
First published on 18th November 2021
Abstract
Heterogeneous chemical reactions catalyzed over solid surfaces at operating temperatures are used to produce a vital part of energy, food, healthcare products, cleaner environments and chemicals. The reactions take place at the atomic level, with active structures forming under dynamic reaction conditions. Therefore, real time in situ visualisation and analysis of gas–solid catalyst reactions under realistic controlled working conditions are crucial to understanding the relation between the dynamic catalyst atomic structure and function for the rational development of improved catalysts and processes. Increasingly, dynamic single atoms and atom clusters are believed to be important in catalytic reactions leading to enhanced catalyst performance. However, dynamic chemical reaction mechanisms at the single atom level under extreme reaction conditions of gas and temperature are not well understood. The novel development of the analytical atomic resolution Environmental (Scanning) Transmission Electron Microscope (E(S)TEM) enables the visualisation and analysis of gas–solid catalyst reactions at the single atom level under controlled reaction conditions of gas environment and temperature. The recent development of the ESTEM advances the capability of the established ETEM, with detection of dynamic single atoms and the associated processes of selected heterogeneous catalysts in their working state. The data provide improved understanding of dynamic single atom processes and reaction mechanisms in heterogeneous catalyst, including the catalytic activity and deactivation at the atomic level and the chemistry underpinning important technological processes. The highlights include platinum/carbon systems of interest in fuel cells to meet energy demands and reduce environmental pollution, and the activation of supported cobalt practical catalysts in Fischer–Tropsch syntheses to produce fuels. Benefits of atomic resolution-E(S)TEM to science and technology include new knowledge of dynamic single atom processes in heterogeneous catalysts and improved processes.
Introduction
Chemical reactions catalysed by solid surfaces play a major role in catalysis for the production of energy sources, industrial chemicals, environmental emission controls and healthcare.1 The reactions take place at the atomic level. In situ visualisation and analysis of reacting catalysts at this level was not possible before, due to limitations of the instrumentation. A conventional electron microscope (EM) normally operates in high vacuum and at room temperature. Early in situ microscopy methods with ex situ reactor cells inserted into the EM had imaging at lower (nms) resolution and limited gas and analytical capabilities. Therefore, the nature of the dynamic gas–solid catalytic reaction at operating temperatures at the fundamental atomic level was unknown. Visualising and analysing chemical reaction properties on the atomic scale under reaction conditions of gas and temperature environments presented a formidable challenge globally. However, it is increasingly believed that single atoms and very small clusters can act as primary active sites in catalytic reactions; as well as providing a primary mechanism for catalyst deactivation.We have addressed the challenge in two stages: at atomic lattice resolution and then advancing to single atom sensitivity. We designed and developed the atomic resolution environmental transmission electron microscope (atomic resolution-ETEM) to visualise and analyse gas–solid catalyst reactions at the atomic level at working temperatures (Boyes and Gai),2 with applications in catalysis.3,4 This ETEM development has been featured.5
In this perspective, we highlight advances in in situ environmental scanning transmission electron microscopy (ESTEM) with single atom resolution developed in our laboratory at University of York, UK, for directly probing in real-time, dynamic gas–catalyst reactions at the single atom level under controlled conditions of a continuously flowing gas environment and at operating temperatures. The ESTEM supports direct real time visualisation and analysis of materials, especially dynamic catalysts, under conditions representative of technological of real-world chemical processes. It extends and complements the capabilities of the in situ atomic resolution-ETEM also supported on the same instrument.2
Following the pioneering development of STEM imaging of single atoms in vacuum by Crewe et al.,6 high angle annular fark field (HAADF) STEM imaging in the high vacuum environment of the EM has been widely used to characterise static catalytic materials.7–9 For atomic number (Z) contrast in HAADF imaging in the STEM, electrons scattered through high angles are collected, with the image intensity approximately proportional to Z2 where Z is the atomic number.6 The signal can provide high contrast images of heavier atoms and small particles on lighter supports which are typical structures of supported heterogeneous catalysts. Spherical aberration correctors have led to greatly improved resolution.10 Methods to identify single atoms are reported in the literature for high vacuum studies,6,11 where single atoms appear as white dots in HAADF images and are quantified by intensity profiles and calibration of intensities.6,12 Micro-electromechanical system (MEMS) heating holders13,14 such as from DENSsolutions13 provide temperature control.
However, reliable in situ visualisation and analysis of reacting single atoms in controlled gas and temperature environments in real-time have been lacking, leading to insufficient understanding of dynamic reaction mechanisms in chemical reactions at the single atom level. Here the development and applications of novel analytical in situ ESTEM at single atom resolution in controlled continuously flowing gas and temperature environments12 employed in heterogeneous gas–solid catalyst reactions and applications are presented, following the advances in AC-ETEM.15 In our gas-in-microscope design, outlined in the following sections, gas is introduced directly into the column of the electron microscope in both the new ESTEM,12,16–21 and in the longer established ETEM.2 ESTEM with dynamic Z-contrast imaging enables reliable detection of metal single atoms on supports, whereas the TEM image contrast is dominated by diffraction and phase effects which can make the visibility of supported single atoms and other atomic species difficult.19 Here we explore single atom dynamics in chemical reactions using heterogeneous catalysts in reacting gas and temperature environments for improved understanding of reaction pathways at the single atom level.
Development of analytical in situ ESTEM with single atom resolution under controlled reaction environments
Since gas–catalyst reactions at operating temperatures take place at the atomic level, we pioneered the atomic resolution-ETEM2 incorporating heating and analytical functionalities. The ETEM enables in situ visualisation and analysis of gas–solid reactions at the atomic lattice level in real time, under continuous controlled gas atmosphere and temperature reaction conditions. In the construction and development of the in situ atomic resolution-ETEM2, a new gas-in-microscope design of the open aperture in situ environmental cell (ECELL) reactor uses the EM sample chamber as the reactor and is thus integral to the electron microscope. Further, the objective lens pole-pieces in the ETEM incorporate radial holes constructed for the critical first stage of differential pumping with apertures added inside and keyed to the bores of the objective lens.2 It is thus a major change from the conventional vacuum methods and the earlier lower resolution environmental EM methods with ex situ reactor cells inserted into the EM. The ETEM development has opened a new field for studying gas–solid reactions and reaction mechanisms at working temperatures at the atomic level. The atomic-resolution ETEM development2 enables gas pressures of many mbars and temperatures up to 1000 °C, or higher. Liquid phase in situ microscopy of reactions between low-vapour pressure liquid, solid catalyst and gas at the sub-nanometer level to produce polymer intermediates and chemicals are also possible.22 The original atomic resolution-ETEM design and development of Boyes and Gai2 is used for production by leading EM manufacturers23 and is utilised by researchers internationally in academia and industry, leading to scientific and technological advances with significant real-world benefits.20,23,24Recently microelectromechanical systems (MEMS) have been incorporated into specialised holders to support temperature, gas and liquid environments. In the gas-cell holders, gas is normally contained between two silicon nitride windows of several nm thickness, and they can operate at a pressure of an atmosphere under static gas conditions, or with low flow rates. However, for reference, it may be noted that in many technological catalytic reactors gas pressures of many bars (up to 100 bars or more) are employed.20 EM gas-in-holder technologies13,14 are still evolving regarding resolution and analytical sensitivity.
For the applicability of STEM and electron diffraction in reaction environments, a new approach has been necessary to support full scientific goals.12,16 To visualise and analyse single atom dynamics reliably in supported catalyst systems during chemical reactions in situ and in real time, an Environmental Scanning TEM (ESTEM) with single atom resolution and key additional analytical capabilities has been developed with an open aperture, gas-in-microscope design and constructed.12,15–21 Dynamic in situ experiments in the electron microscope benefit from aberration correction (AC) to provide a series of unique single images, each at optimal and minimal defocus, with a continuous range of spatial frequencies, rather than requiring an impractical multi-image through-focal series.
The need to accommodate special specimen holders in an aberration corrected instrument has been one of the more important criteria driving our specification and modification of a double aberration-corrected (2AC) electron microscope, the JEOL 2200FS (2AC) FEG TEM/STEM operating at 200 kV, in the Nanocentre at University of York, to provide both the ETEM and for the first time ESTEM functionalities.12,16–18 Currently, the majority of in situ aberration corrected (AC) ETEMS use the earlier in situ atomic resolution-ETEM design and development of Boyes and Gai,2 described in the preceding sections. The development of the novel in situ AC ESTEM12,16–21 and its applications to nanoparticle catalysts25–29 are highlighted in the following sections.
Since aligning the sample into a zone axis orientation is a prerequisite for atomic resolution electron microscopy, an increased specimen tilt range is also important. Both the conditions benefit from the larger gap ARP/HRP/Midi objective lens polepiece. Parameters for ultra-high-resolution (UHR) and Midi polepieces are shown for comparison in Table 1. A Midi design was chosen for our double (TEM and STEM) Cs aberration corrected instrument to provide flexibility in stage space and manipulation.
| Polepiece type | Gap range | Uncorrected Cs | Uncorrected Cc | Cc with Cs Ac | Std hot stage? |
|---|---|---|---|---|---|
| UHR | 2.2–2.5 mm | 0.5–0.6 mm | 1.0–1.2 mm | 1.4 mm | No |
| Midi | 4.3–5.4 mm | 1.0–1.2 mm | 1.2–1.4 mm | 1.6 mm | Yes |
The ESTEM functionalities include controlled reaction conditions of continuously flowing gas environments and temperatures at operating temperatures of ≥1000 °C (controlled with a MEMS heating stage from DENSsolutions), dynamic ESTEM-HAADF Z-contrast imaging with single atom resolution, energy dispersive X-ray spectroscopy (EDX), electron energy loss spectroscopy (EELS) for chemical analysis and dynamic electron diffraction (ED) analyses of nanoparticle structures, during the course of reactions. The new permanently mounted environmental cell (ECELL, or gas reaction cell) developments retain the full performance of the original core instrument with single atom resolution.12,15–21 HAADF is also required to set up the early STEM corrector. The open-aperture gas-in-microscope ETEM and ESTEM instruments have differentially pumped column sections separated by fixed beamline apertures and a new gas tolerant pumping system. The use of open differential apertures removes the need for ‘gas-in-holder’ or other containment windows. Further, both the ESTEM and ETEM (referred to as E(S)TEM) capabilities have been incorporated in the same instrument for novel studies of nanoparticle catalysts with sub-Angstrom (<0.1 nm) resolution in continuous gas atmospheres at working temperatures.12,16–20 The method gains access to the sequencing of key intermediate phases and states which may be metastable with respect to temperature and reaction atmosphere. Minimally invasive electron (e)-beam conditions without contamination, catalyst activation only after loading into the EM reactor and in situ analysis, are required for the studies.
The advances allow for reliable environmental-STEM-HAADF (ESTEM-HAADF) imaging and analysis of reacting single atoms and reaction mechanisms at the single atom level. Rational design of novel reactions and nano-synthesis are possible with the in situ facilities. The structure, chemistry and function of dynamic catalysts are revealed while the sample is immersed in a flowing controlled gas atmosphere and held at the operating temperature. The new AC E(S)TEM system with single atom resolution operates in both ESTEM and ETEM modes, typically with <1 to >10 Pa gas pressures; in the latter case for a gas supply of ≫10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 monolayers per second (>0.1 mbar) measured directly at the sample position with inlet gas pressures up to a few mbar. The gas supply is adequate to flood the sample surface with gas coverage, fully sufficient to drive the chemistry12,16–21,25–30 and catalysts are regularly activated in situ; as they may be in practical applications. In this context, Surface Science defines ‘high pressure’ as >0.1Pa (>0.001 mbar).31 The gas environment-ESTEM is in contrast with the usual high vacuum STEM. In the atomic resolution-ETEM,2,23 many mbars of gas pressures are possible.
000 monolayers per second (>0.1 mbar) measured directly at the sample position with inlet gas pressures up to a few mbar. The gas supply is adequate to flood the sample surface with gas coverage, fully sufficient to drive the chemistry12,16–21,25–30 and catalysts are regularly activated in situ; as they may be in practical applications. In this context, Surface Science defines ‘high pressure’ as >0.1Pa (>0.001 mbar).31 The gas environment-ESTEM is in contrast with the usual high vacuum STEM. In the atomic resolution-ETEM,2,23 many mbars of gas pressures are possible.
In the ESTEM experiments minimally invasive low dose electron beam techniques are used throughout.12,16–21,24–29 Data on in situ experiments under low electron dose conditions are checked in parallel with in situ data from ‘blank’ calibration experiments in which the dynamic experiments are conducted without the electron beam (by blanking the beam) and the beam is switched on only to record the final reaction end point. In addition to providing direct and unparalleled insights into the dynamic evolution of structural changes and reaction mechanisms at the atomic level, the ESTEM and ETEM allow the detection, in real time, of surface as well as sub-surface structural phenomena. They include defect evolution and growth, access to metastable states, dynamic electron diffraction, changes in chemical composition and oxidation states. The aim is non-invasive characterisation under benign conditions without contamination. Under carefully controlled conditions, data from in situ ESTEM can be directly related to atomic structure–function (activity) relationships in technological processes.28,29 In catalysis, this correlation is crucial to optimising the synthesis and development of improved catalysts. Analyses of solid–state reaction mechanisms in the EM reactor further show that the mechanisms are consistent with those in technological catalytic reactors employing high gas pressures.30,32 Because of the small amounts of solid reactant in the microscope sample, measurements of reaction products are often carried out on larger samples in an ex situ micro-reactor operating under similar reaction environmental conditions to those used in the EM reactor for the nanostructural correlation28,29 although on-line methods are now being developed.
The images presented here are all high angle annular dark field (HAADF) images (unless stated otherwise), using the original JEOL STEM detector. The recording conditions for the STEM data were magnifications 8–12 MX with 512 and 1024 line frames and sampling at 0.015 or 0.03 nm; i.e. at 6× or 3× over sampling of the 0.1 nm digital imaging resolution element, using pixel dwell times of 19 or 38 μs (with 50 Hz line synchronisation). Some images were also recorded with faster frame integrations. The incident probe had a calibrated convergence semi-angle and the collection angles for HAADF imaging were 110 mrads and 170 mrads, respectively, for the inner and outer diameters of the detector. Short sequences of images recorded at reduced frame times and pixel counts were also assembled into a video to analyse and demonstrate the dynamic, and in some cases, competitive nature of the processes involved.
An outline schematic of the double aberration-corrected E(S)TEM development is shown in Fig. 1a. A schematic of the ESTEM functionalities is shown in Fig. 1b. An example of single atoms, clusters and nascent nanoparticles of carbon supported Pt catalysts is shown in the colorised image in Fig. 1c. The migratory single atoms on the support between the more substantial clusters have been imaged in controlled hydrogen gas and temperature environments.
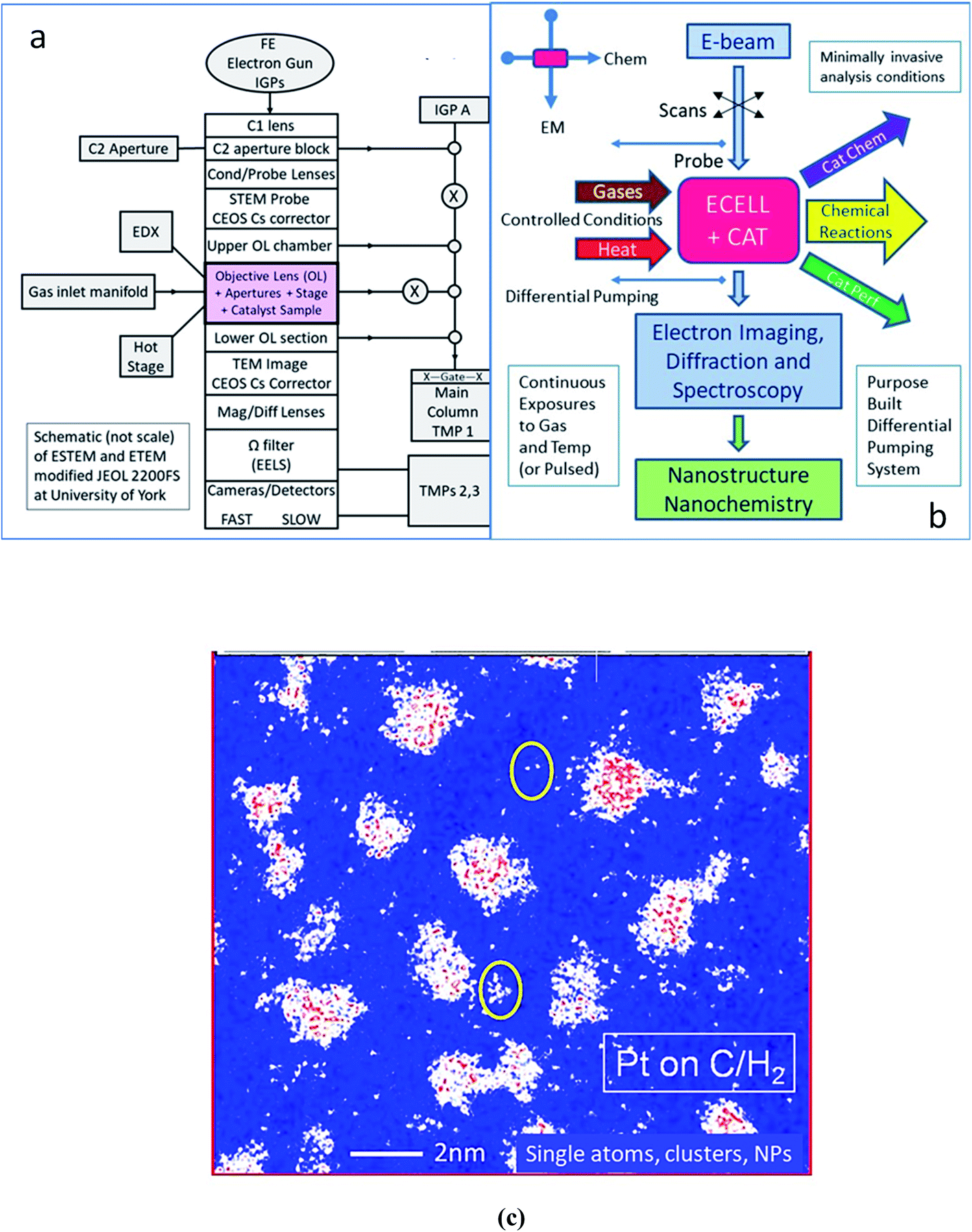 | ||
| Fig. 1 (a) A schematic of the double aberration corrected ESTEM/ETEM (E(S)TEM) development by Boyes and Gai with an integrated gas chemical reactor. Apertures and differential pumping with pressure zones sustain continuous gas pressures around the sample. (b) A schematic of the E(S)TEM functionalities. In the ESTEM and ETEM, the EM column shares a catalyst sample (CAT) with gas environment chemical reactor (ECELL). EC is the ECELL, with reaction providing insights into single atom dynamics with dynamic catalyst nanostructure, reaction pathways and chemical basis of performance. (c) The distribution of single atoms of platinum (white dots), Pt atom clusters and Pt nanoparticles in a model Pt/C catalyst in hydrogen in ESTEM-HAADF. Scale bar is 2 nm. | ||
Applications of analytical in situ ESTEM
Single atom dynamics in Pt/C nanoparticle catalysts in controlled redox environments
Supported noble metal nanoparticles are used widely as heterogeneous catalysts for chemical technologies for the production of fuels, fuel cells, chemicals, healthcare and in pollution control. They are important for world economic development and sustainability. As the nature of the active reaction sites of the metal nanoparticle, along with the processes by which the particle sinters and deactivates, have been shown to change both in magnitude and mechanism with the reaction environment,16–21,27–29,33 it is of fundamental importance to activate, react, observe and analyse nanoparticle systems in situ in real time under controlled continuous gas flow reaction conditions and operating temperatures. Single atom dynamics in controlled redox environments at operating temperatures in supported gold nanocatalysts using the ESTEM have also been reported.19 Sintering dynamics of single atoms under vacuum, air, Ar at suitable pressure can be examined.In situ studies of single atom dynamics in heterogeneous catalysts presented in the following sections are significant for the emerging field of single atom catalysts (SAC) where one of the objectives is to maintain the stability of an active population of single atoms, or very small clusters, on a substrate. Previous studies reported investigations of static SAC carried out in a high vacuum EM at room temperature.34,35 There are instructive reports on reaction processes over metal surfaces (especially over single crystal metal surfaces) using scanning probe methods (SPM), which are summarised in a recent review.36 In SPM methods tracking and analysis of dynamic single atoms between 3D nanoparticles, including any sub-surface contributions, in controlled continuously flowing gas environments at operating temperatures, are especially challenging. With the ESTEM, tracking and analysis of dynamic single atoms between 3D nanoparticles and their interactions have been performed in controlled reaction environments revealing atom movements and local substrate pinning site interactions.28,33
In the following sections we present examples of some recent applications that exploit the single atom-resolution ESTEM for understanding single atom processes in nanocatalysts in their functioning state. In supported metal nanocatalysts, insights into continuing populations of single atoms on the support between more established nanoparticles, or pre-particle rafts, under reaction conditions are required. These include applications of dynamic single atom catalysis, migrating agents of competitive growth catalysts, as well as deactivation and lower co-ordination rafts. With the ESTEM we can also identify adjuncts to established particle surfaces, not previously reliably visible.
Reduction-oxidation (redox) studies at the single atom level of Pt/C nano-catalysts, important in fuel cell applications, are presented in Fig. 2. The reduction is carried out in flowing hydrogen gas with pressure at the sample of 2 Pa, as a function of temperature, up to 500 °C. The sample is then cooled to 200 °C without the hydrogen gas and oxygen is introduced with the gas pressure of 1.9 Pa at the sample. Fig. 2a–c show a redox sequence of the nano-catalyst: (a) the catalyst at room temperature (RT) showing Pt single atoms and irregular (unfaceted) nanoparticles; (b) in hydrogen at 500 °C with increased particle faceting and (c) in oxygen at 200 °C. This sequence shows Pt single atoms and small clusters are visible at these temperatures and are consistent with the observations in the H2 reduction of platinum12 and gold nano-catalysts.19 The observations further indicate that the migration of loosely bound single atoms contribute to particle shape changes. It is shown that the minimisation of surface energy leads to the removal of under co-ordinated surface atoms in favour of facets, and such a process is proposed to impact the activity of nano-catalysts.12,16–21 The newly discovered single atom interaction mechanisms indicate that the primary role of irregular nanoparticles in reactions is to act as reservoirs and recipients of low co-ordinated atoms and clusters, and faceted particles generally act as spectators.12,16–20 These findings have important implications in the development and operation of single atoms and nanoparticle catalysts. Fig. 2c indicates that more Pt single atoms are present in oxygen gas (than in hydrogen). At 200 °C, the data suggest interactions between Pt atoms and the chemisorbed oxygen, as well as interactions with nanoparticles which exhibit irregular structures and less faceting (Fig. 2c).
 | ||
| Fig. 2 In situ ESTEM-HAADF images of single atom dynamics of Pt/C catalyst in redox reactions, in flowing gas and at operating temperatures: (a) RT; (b) in hydrogen gas at 500 °C; (c) in oxygen gas at 200 °C. The scale bar is 2 nm. | ||
Sintering dynamics and stability of Pt nanoparticle catalysts in chemical reactions at the single atom level
Atomic level deactivation mechanisms play a critical role in the loss of catalyst performance. Here we present real-time in situ visualisation and quantitative analysis of dynamic atom-by-atom sintering and stability of model and practical carbon supported Pt nanoparticle catalysts on carbon supports under controlled reaction conditions of temperature and continuously flowing gas to obtaining a deeper understanding of the mechanisms.The size of a metal particle is a key factor in its performance in catalytic applications.37,38 Based on dynamic correlations between increased catalytic activity and reduced surface atom coordination numbers28 and on static studies in EM high vacuum,34 there is an increasing recognition that single atoms and sub-nanometer (nm) clusters can lead to improved catalyst performance in chemical processes. In heterogeneous catalysis employing supported metal nanoparticles, particle growth (sintering) can cause quantitative deactivation by the loss of surface area,39 and qualitatively by changes in particle form with size and environment.
Nanoparticle deactivation due to sintering is generally explained in terms of two mechanisms: Ostwald ripening (OR), or particle migration and coalescence (PMC).39 So far, approaches to study sintering of nanoparticles have been at the nanoparticle level and address the sintering mechanisms in terms of particle size distributions (PSD).40–42 This has been implemented via individual particle size tracking, by fitting theoretical models to PSDs, using a mean-field approximation.39–41 In the mean-field model, the growth and decay of nanoparticles is described in terms of interface-controlled and diffusion-controlled ripening models. They include a mean-field approximation where the concentration of atoms (or atomic species) on the support is constant beyond the screening distance from the edge of the particle, with this mean-field concentration governing the atom exchange for particles in the ripening process.39 This approximation is elaborated in the literature41,42 as well as limitations of the mean-field model in understanding the OR mechanisms.39,41,42 The studies of PSDs in heterogeneous nanoparticle catalyst systems have indicated OR at low to moderate temperatures.40,41
However, since sintering of nanoparticles can take place at the single-atom level, it is crucial to have a deeper understanding of particle sintering mechanisms at that level. Here we present insights into the mechanisms of particle sintering at the single-atom level. Our results track the areal density of dynamic single atoms on the support between nanoparticles and attached to them; both as migrating species in the degradation of performance and as potential new independent active species. We demonstrate that the decay of smaller nanoparticles is initiated by a local lack of single atoms; while a post decay increase in single atom density indicates anchoring sites on the substrate before agglomeration to larger particles. The analyses unveil a relationship between the density and mobility of single atoms and the particle size. They are combined with practical catalysts important in technological processes. The findings generate new insights into nanoparticle sintering dynamics at the single atom level, important in the development of efficient supported nanoparticle systems for improved chemical processes and dynamic single atom catalysis.
Synthesis experiments and in situ ESTEM analysis of Pt/C nanoparticle catalysts
Pt/C system is of interest in a variety of chemical processes described in the preceding sections. We studied both model and technologically important practical (real-world) systems to obtaining insights into the behaviour of the nanoparticles as a function of temperature in hydrogen gas environments, with single atom resolution.To prepare model Pt nanoparticles on carbon, a JEOL JFC2300HR sputter coater was used to deposit 0.2 nm of Pt onto the 5 nm thick C window supports on DENS SH30 Wildfire S3 MEMS chips with internal temperature measurements, purchased from DENSsolutions. Before the EM experiments, the samples were heated in high vacuum at 600 °C to produce particles with radii in the range 0.5 to 1.5 nanometers (nm) and primarily in [110] crystallographic orientations. Practical (real-world) samples were prepared on 5 nm carbon coated MEMS chips supplied by DENSsolutions. Aqueous H2PtCl6 solution (3 μL, 0.1 mM) was deposited onto the transparent window followed by calcination in H2 gas in a tubular furnace (40 mL min−1 H2, 200 °C, 1 °C min−1, 1 h), forming polydisperse particles from about 1 nm to a few nm in diameter.
We first performed controlled dynamic sintering OR experiments on model Pt/C catalyst samples at 250 °C in 3Pa of H2 in the ESTEM. Several areas of the samples were examined to confirm the observations.
In the present investigations, a limited dose electron beam with careful calibration procedures and beam blanking were employed to avoid possible deleterious effects of the electron beam as described in the preceding sections, with an estimated integrated dose rate exposure averaged over the experiment up to about a few electrons per square Angstrom per second.20,29,33 The particles here were only exposed to the beam during data setup, interval examinations and actual data acquisition. The images were 1024 × 1024, with a pixel dwell time of 19.5 μs. For the in situ ESTEM studies, high-purity hydrogen gas (99.9995% from BOC, U.K.) was used with a pressure of 2–3 Pa, fully sufficient to drive the chemical reaction. The dynamic in situ ESTEM experiments track the development or degradation of nanoparticle nanostructures in real time and reliably track single atoms between 3D nanoparticles in working catalyst systems. These single atoms can act both as migrating species influencing the long-term performance degradation of catalysts and as potential active species in dynamic catalytic reactions and were previously largely invisible to direct studies. The dependence of the particle size on the single atom density during sintering or OR can now begin to be studied at the single atom resolution using the ESTEM.
Previous work in the literature had reported the effects of OR through particle size changes as described in the preceding sections, leading to an inferred understanding of reaction mechanisms involved.40–42 Other work has utilised scanning probe methods.36 The aim of our work is to obtain an improved understanding of the dynamic relationship between the effect (particle size change) and the cause involving single atom density and mobility, using a Pt/C nanoparticle catalyst system under controlled reaction conditions in a flowing gas environment at operating temperatures using the ESTEM. The science presented here leads to new knowledge in understanding of sintering in OR, with implications for an improved nanostructural design basis for materials and processes.
Results and discussion
Analysis of experimental results
To define the interaction range of nanoparticles and thus determine the local atomic concentration a method known as the Voronoi construction has been reported.43 This approach has been utilised in different microscopy methods by several researchers to provide a better understanding of sintering.41,43The application of the Voronoi approach43 is that assuming a local model, variations in the single atom density can be correlated to the particle size and decay. Local correlations, whereby single atoms are attributed to individual or groups of particles, have been explored with the Voronoi construction, including for the Pt/C system.33 The region of influence of a particle consists of any point that is closer to that particle than to any other. Nearest neighbours are defined as particles that share a Voronoi boundary. The accuracy of density measurements depends on single atoms detected being representative of the total population, limited by the detection efficiency, sample size and the migration rate.33 Additionally, parameters such as temperature and reaction gas pressure affect the number and detectability of single atoms and the electron beam scan rate dependence has been quantified.33 Relating the particle size to the single atom density under controlled reaction conditions in experiments requires an adequate sampling with large data sets to provide a representative overview of the dynamic processes. Further, constraints on the single atom detectability, the dependence on the migration speed of the atom versus the scan rate need to be considered.33 Despite the challenges, insights into single atom dynamics in both the model as well as practical systems can be obtained, described in the experimental studies and analyses in the following sections.
In the model system, a 23.5 nm × 23.5 nm area has been investigated for about 75 minutes to understand the relationship between the particle decay and single atom density (population). A dynamic ESTEM-HAADF image sequence of Pt/C catalyst in flowing hydrogen gas at 250 °C from the same sample area is shown in Fig. 3. It illustrates that the single atom density at (A) t = 10 min, is high. It decreases considerably at (B) t = 40 min and then increases again after particles 17 and 18 have decayed at (C) t = 70 min. Following the application of the Voronoi construction to the sample area, the decay of particles 17 and 18 is analysed by averaging single atom numbers over a larger area (the nearest neighbour Voronoi regions) as well as over the time.33 This improves the statistics sufficiently to enable quantitative time dependent single atom density observations.
 | ||
| Fig. 3 Dynamic in situ ESTEM-HAADF observations of single atom dynamics from the same local area of particles 17 and 18 imaged in 3 Pa H2 at 250 °C. Images are recorded at time intervals of (A) t = 10 min, (B) t = 40 min and (C) t = 70 min. A decrease in the single atom density is observed in (B), with an increased disorder of particles 17 and 18. (Particle 20 was not included in the Voronoi analysis, as it was not within the field of view of the microscope throughout the experiment. It is included here to illustrate the local environment of the particle 18). The dynamic observations reveal that as they are extinguished in (C) the local single atom density increases, with details given in Fig. 4. | ||
A clear correlation between the change in size of particles 17 and 18, and the density of single atoms in the nearest neighbour Voronoi cells is shown in Fig. 4. The dynamic in situ ESTEM observations have further revealed that the local single atom density begins to decrease prior to particle size changes, indicating the decay of particles 17 and 18 is driven by the absence of single atoms to replace those emitted by the particles in a competitive dynamic process. The data also present evidence of interactions between particles 17 and 18. Once the concentration of single atoms decreases below the rate of emission from particle 17, the particle size begins to decrease at t = 30 minutes, accompanied by an increase in the size of the particle 18. This indicates that it is emitting at a lower rate. When their sizes are approximately equal at t = 50 minutes, the particle 18 also begins to decay, which corresponds with a spike in the size of the particle 17 at t = 60 minutes. If for example, (a) emission from both particles is a function of the surface area, rather than only of the irregularity of the external surface, defined as an unfaceted or disordered exterior; and (b) the irregularity reduces the influence of the Gibbs–Thomson39 relationship on emissivity, smaller particles can survive in a lower single atom concentration due to their assumed lower emission. As described in the preceding sections analyses from several areas of the sample have been carried out in our experiments.
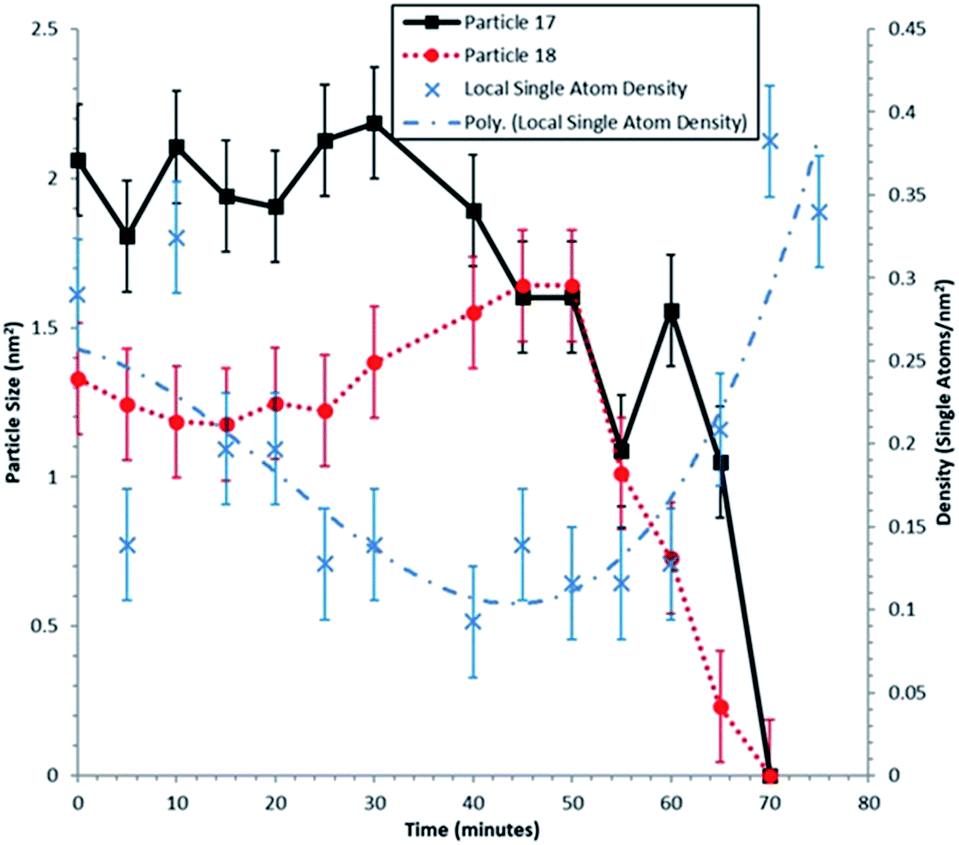 | ||
| Fig. 4 Sizes of particles 17 and 18 during experimental time frame of 75 minutes. The dynamic sampling rate in the in situ ESTEM studies is 1 acquisition per 5 minutes. The local single atom density is calculated from Voronoi cells neighbouring particles 17 and 18, the cells included are: 12, 13, 14, 15, 16, 17, 18, and 19. A third order polynomial has been fitted to the local single atom density data, indicated by the dot-dash line. Errors in the particle sizes are calculated by measuring the change in the particle size. The error in the single atom density is calculated by taking 5 successive images, counting the number of single atoms, then finding the corresponding single atom density variations. | ||
The dynamic observations have revealed that towards the end of the experiment at t = 60–70 minutes, the single atom density increases again. Particles 17 and 18 decay to r = 0 at t = 70 minutes. Thus, a reduction in the single atom density, once these net emitters have disappeared, would be expected. The increase in density over 4 observations (20 minutes) implies that the single Pt atoms are being anchored on the carbon support. Diffusion proceeds via a series of jumps between anchoring sites, with single atoms remaining stationary between hops. This is consistent with the conclusion reported in the literature44 in which Au atoms remain stable on the C support for ∼15 s before hopping ∼2 nm each time. The anchoring of migratory single atoms on stabilized active sites for reactions is an area of increasing interest, e.g. W-species on grain boundaries of zirconia18,45 and platinum on γ-Al2O3.46
Dynamic single atom density as a function of particle size
The initial change in the single atom density around particles 17 and 18 may be influenced by the changing particle structure of the nearest neighbours. Particles 17 and 18 are irregular in external profile and make up two of the smallest particles observed. However, their local single atom density appears to increase or stay constant over the experimental time frame.33 For understanding the relationship between particle size and single atom density, knowledge of the particle surface, especially the nature of irregular (i.e. unfaceted, non-equilibrium) external surfaces of the particle, is required. The decay of small nanoparticles with irregular external surfaces (17 and 18 illustrated in Fig. 3) indicates that they are inherently unstable and that the local single atom density can be used for an enhanced understanding of the nature of the nanoparticle surface. This may be challenging to achieve in practice when using an averaging technique on a sample of several hundred nanoparticles,33 as the density magnitude can be offset by the local particle size, the extent of surface faceting, particle geometry and the nanostructural geography of the neighbourhood in which it finds itself.The findings have revealed two distinct populations of nanoparticles, with irregular external surfaces at lower temperatures and more faceted exteriors at higher temperatures. The emissivity of particles with irregular external surfaces is found to be greater than that of facetted ones; consistent with previous reports.12,16–21 The results further elucidate that even after extended heating at 250 °C the surfaces of more established supported nanoparticles acquire or retain single atoms, or very small clusters, on otherwise regular surfaces. Fig. 3, acquired after heating for 70 minutes at 250 °C in hydrogen, is a notable example of this. These phenomena are not included in simulations of catalytic properties and nanoparticle sintering reported in the literature. The new insights also reveal that although many single atoms follow OR, some isolated single atoms continue after sintering, indicating the presence of pinning sites on the support in both OR and single atom catalysis.
Sintering dynamics in practical Pt/C nanoparticle catalyst system
It is important to compare the behaviour of model catalysts with practical systems of technological interest. We studied dynamic sintering of the practical catalysts as a function of hydrogen gas and temperature in the ESTEM. Images were recorded at 4 Mx magnification, with 2048 × 2048 pixels, dwell times of 19 μs and 50 Hz line synchronisation. Careful calibration procedures were followed throughout to avoid deleterious effects of the electron beam, as described in the preceding sections. The influences of the experimental conditions were investigated systematically by adjusting the beam on/off ratio, facilitated by the high stability of the MEMS stage employed, and gas pressures over the range 2–20 Pa.Suitable regions were identified for in situ reaction experiments and unreacted samples were imaged initially in vacuum. Samples were subsequently heated under vacuum to remove any surface contamination.33 The samples were then reacted in H2 gas to 300 °C, then ramped in 100 °C steps (30 °C min−1) over 80 minutes. Dynamic images were recorded in reaction environments at the operating temperature. (For comparison, in some experiments after 80 minutes at 700 °C the temperature was reduced to 300 °C for imaging).
Sintering and particle growth
The growth of the supported practical platinum nanoparticles as a function of time, and temperature in an environment of hydrogen gas using the ESTEM at the single atom resolution are presented in the following sections.A real time dynamic image sequence in hydrogen gas in ESTEM-HAADF is shown in Fig. 5. Nine particles are identified in the first image and the progression of the particle size is shown in Table 2. Through the sequence, small particles 4 and 8 are observed to shrink and eventually decay after 56 and 62 minutes, respectively, at 600 °C. The migration of single atoms is observed between the nanoparticles. The observations indicate that the decay of smaller particles is associated with the emission of single atoms and their reattachment, resulting in the growth of larger particles.
 | ||
| Fig. 5 Real time dynamic in situ ESTEM-HAADF images of a reaction sequence in H2 gas as a function of temperature and time, from the same region of practical Pt/C sample, illustrating single atom dynamics and the effect on the size and stability of the Pt nanoparticles on the support. Nine particles are identified in the first image shown at the top and are monitored in the following sequence. Images from left to right: top row: at 500 °C after 36 min, 41 min and 46 min; middle row at 600 °C, after 36 min, 41 min and 46 min; and bottom row at 600 °C after 51 min, 56 min and 62 min. Scale bar is 5 nm and applies to all the images. | ||
| Particle number | Particle area/nm2 | |||
|---|---|---|---|---|
| 500 °C, 51 min | 600 °C, 31 min | 600 °C, 56 min | 600 °C, 62 min | |
| 1 | 8.2 | 8.8 | 8.9 | 9.2 |
| 2 | 1.8 | 2.0 | 1.5 | 1.5 |
| 6 | 4.2 | 6.5 | 7.9 | 8.2 |
| 9 | 3.0 | 3.6 | 3.9 | 4.0 |
After 31 minutes at 600 °C, particle 6 has grown at the expense of particles 5 and 7. In the images recorded after 46 and 51 minutes the particles 3 and 6 coalesce, and in subsequent images rearrangement of the particle is observed. Thus, the sequence indicates Ostwald Ripening and decay of small particles/coalescence occurring at 600 °C.
Dynamic single atom distribution
Single atom species are observed to migrate on the support between the nanoparticles during reduction reaction in hydrogen gas, consistent with studies on model systems.19,20 The migration of single atoms and effects on the particle dynamics, stability and size during the reaction are illustrated in colorised dynamic image sequences in Fig. 6 as a function of temperature and time in hydrogen gas environment. The colour coding of these images is on a ‘pseudo-temperature’ scale to indicate sample thickness (or the projected atomic column length). Here the blue background is the thin carbon support film, for which the scattered intensity is minimal, and rising through the colour chart to red at the top end of the depicted intensity and eventual signal saturation. The aim here is to achieve improved reproductive contrast of single atoms and very small clusters against the carbon film background. | ||
| Fig. 6 (a) to (f): Real time dynamic in situ ESTEM-HAADF images of practical Pt nanoparticles on carbon, in hydrogen gas as a function of operating temperature, revealing single atom dynamics. The observations show the migration of single atoms and the effect on the nanoparticles during the reaction in hydrogen gas, as a function of the reaction temperature. Temperature is increased by 100 °C every 80 minutes. ((g) dynamic image in vacuum at 700 °C shown for comparison); (h) intensity profiles of regions 1 and 2 in (a) indicate the presence of Pt single atoms and clusters. | ||
The figure elucidates the migration of single atoms recorded from the same region of the sample: Fig. 6a–d illustrate the emission of single atoms from irregular (unfaceted) smaller particles and decay of the smaller particles: from left to right: (a) image recorded in vacuum at 300 °C; (b) in hydrogen at 300 °C; (c) in hydrogen at 400 °C; in hydrogen at 500 °C. Bottom row: from right to left (e) in hydrogen at 600 °C and (f) in hydrogen at 700 °C. (An image recorded in vacuum at 700 °C is also shown in (g) for comparison). Fig. 6(h) shows line intensity profiles displaying pronounced intensity in regions containing Pt single atoms and Pt atom clusters in (a), in comparison to the background intensity. Significantly, the dynamic observations reveal that single atoms are observed at elevated temperatures of 700 °C in H2 gas environment.
The decrease in the single atom concentration and adatom emission are attributed to the growth and increased faceting of the particles.33 The dynamic observations reveal that in general the well-established faceted (ordered) particles which show ordered crystalline lattice have fewer single atoms in close proximity than the more raft-like, irregular (disordered) particles. The findings are consistent with those in the model system.12,19
The dynamic in situ observations of practical Pt/C system have revealed the migration of single Pt atoms between Pt nanoparticles on the carbon support, with single atom population existing among more established nanoparticles and in continuous interchange with them over a considerable length scale in a competitive process, as observed for the model systems. Our findings show that single atoms continue to exist in hydrogen gas even at much higher temperatures, such as 700 °C for Pt, than previously anticipated. The observations of single atom dynamics are relevant to chemical processes employing Pt/C systems in industrial catalysis and open up opportunities for dynamic single atom studies in other complex practical catalysts. The in situ observations reveal that there is a continuing population of single atoms with important implications on catalyst reactivity, as well as providing the atom-by-atom deactivation mechanism.
Supported Co-based practical catalysts for Fischer–Tropsch process
Fischer–Tropsch (FT) catalysis is a major industrial process for converting syngas (hydrogen and CO) to transportation fuels and other hydrocarbons.47 The process generally uses oxide supported cobalt catalysts. Despite considerable literature on the FT process, the effect of pretreatment (activation) in hydrogen on the catalysts at the atomic level is not well understood. Effects of pretreatment of both dried, reduced (D) and dried calcined reduced (DC) supported real-world (practical) cobalt (Co) catalyst precursors on various supports such as SiO2, TiO2, ZrO2 and Al2O3 have been visualised and analysed using in situ E(S)TEM with single atom resolution, and complemented by chemical methods, including, Extended X-ray Absorption Fine Structure (EXAFS), X-ray Absorption Spectroscopy (XAS) and Diffuse Reflectance Infrared Fourier Transform Spectroscopy (DRIFTS).28 The methods have provided insights into the activation process in FT catalysis, including cobalt dispersion including Co single atoms, the presence of cobalt nanoparticles with face-centered cubic (fcc) and hexagonal close packed (hcp) phases, the dynamic atomic structure and the activity of the catalysts.The ESTEM is used for tracking single atom dynamics in the activation of cobalt/silica catalysts used for the FT process. In D samples, many Co single atoms, Co single atom dynamics, Co atom clusters and Co nanoparticles are revealed as shown in Fig. 7a and b, recorded at 2 min intervals, from the same area of the sample. In DC samples, the ESTEM reveals the presence of large Co-oxide agglomerates near Co nanoparticles and clusters (c and d), leading to the reduction of the catalyst surface area. The correlation of the nanostructure with the FT catalytic activity reveals higher activity for D, with the ratio of the relative activity for D/DC of about 1.6, indicating that the dynamic active species with Co single atoms, clusters and small NPs of Co play a pivotal role in the CO adsorption and enhancing the catalyst performance.28 The findings on the other supports have revealed that D samples are more active than the DC samples irrespective of the support employed, with no change to the selectivity values.28
 | ||
| Fig. 7 Dynamic ESTEM-HAADF image sequence as a function of time in hydrogen at 400 °C. (a) and (b) are from the same area of the sample near P: (a) practical D-Co/SiO2 catalyst, showing Co single atoms (about 100 pm), clusters (indicated at top thin arrow) and nanoparticles (NPs) and (b) tracking of the atoms, clusters and NPs. (Scale bar is 2 nm); (c) DC Co/SiO2-catalyst, showing primarily Co-oxide agglomerates (e.g. at A) and Co NPs; in (d) the square area in (c) is enlarged. | ||
Fig. 8 shows particle size distributions (PSD) in D-Co/SiO2 catalyst, showing mainly small particles. A histogram of the PSD shows counts vs. particle size with a peak at about 2.4 nm. Particle sizes in DC Co/SiO2 catalysts have shown that that they vary between about 0.2 nm to large agglomerates of 50 nm or more, as shown in Fig. 7c.
 | ||
| Fig. 8 Particle size distributions (PSD) in D-Co/SiO2 catalysts, showing mainly small particles. The PSD show counts vs. particle size with a peak at about 2.4 nm. | ||
Based on the ESTEM and chemical data, removal of the calcination process results in improved activity of the supported catalysts. The dynamic ESTEM observations have revealed more highly dispersed Co metal species (Fig. 7a and b), in reacting D samples, leading to smarter pretreatment synthesis methods with more active sites for CO adsorption in FT catalysis.
Conclusions
The development and applications of the novel analytical in situ environmental scanning transmission electron microscope (ESTEM) with single atom resolution and gas-in-microscope design (the highest resolution method currently available) to visualise and analyse single atom dynamics in heterogeneous catalysts in controlled environments of continuously flowing gases at operating temperatures in chemical reactions are presented. Applications of the quantitative in situ ESTEM include studies of sintering and redox mechanisms of industrially important nanoparticle catalysts. In dynamic ESTEM-HAADF studies of Pt/C catalyst systems, the newly discovered single atom interactions in hydrogen indicate that the primary role of irregular nanoparticles in the evolution of the nanostructure is to act as a reservoir of low co-ordinated atoms. The observations of single atom dynamics have important implications in the formulation of new mechanisms of wider applicability to understanding the development of the nanostructure and therefore the dependent properties in more detail. The findings of sintering dynamics at the single atom level have revealed important relationships between the single atom density and changes in the particle size in dynamic sintering of carbon-supported Pt atoms in hydrogen environments. The in situ observations elucidate the relationship of single atom density with a specific particle or its nearest neighbours and that the particle decay is initiated by a lack of local single atoms. A post-decay increase in the single atom density indicates that anchoring sites exist on the support with the diffusion of single atoms governing the OR. The findings provide a new level of detail in understanding the OR mechanism. The results have important implications in optimising the site stability of reacting single atoms in working nanoparticle catalyst systems and on their role in particle coarsening and deactivation. Dynamic activation of F-T catalysts has revealed the relationship between single atoms and clusters with the catalyst function.The ESTEM findings provide dynamic analysis and structural insights at the single atom level during the course of catalytic reactions, important in the development of efficient catalysts and processes. Challenges in studying catalysts in E(S)TEM include the efficiency of signals. Data acquisition using direct electron detection cameras will be helpful. Low dose protocols described in the preceding sections are required. The ESTEM is opening new opportunities for in situ visualisation and analysis of single atom dynamics in a wide range of catalytic reactions.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors acknowledge support from the UK Engineering and Physical Science Research Council (EPSRC, now UK Research and Innovation) for a research Grant EP/J0118058/1 to P. L. G. and E. D. B. They thank Thomas E. Martin, Robert W. Mitchell and David C. Lloyd for support and contributions.References
- R. A. van Santen, Heterogeneous Catalysis: from Principles to applications, ed. M. Beller, A. Renken and R. A. van Santen, Weinheim, Wiley-VCH Verlag, 2012, pp. 113–151 Search PubMed.
- E. D. Boyes and P. L. Gai, Ultramicroscopy, 1997, 67, 219–232 CrossRef CAS.
- P. L. Gai and K. Kourtakis, Science, 1995, 267, 661–663 CrossRef CAS PubMed.
- P. L. Gai, Adv. Mat., 1998, 10, 1259–1263 CrossRef CAS.
- J. Haggin, ACS Chem. Eng. News., 1995, 73, 39–41 CrossRef.
- A. V. Crewe, J. Wall and J. Langmore, Science, 1970, 168, 1338–1341 CrossRef CAS PubMed.
- M. M. Treacy, A. Howie and C. Wilson, Phil. Mag., 1978, 38, 569–572 CrossRef CAS.
- A. A. Herzing and A. F. Carley, Science, 2008, 321, 1331–1335 CrossRef CAS.
- J. Sanchez, M. Lopez-Haro, J. C. Hernando-Garrido, G. Blanco, M. Cauqui, I. Rodriguez, J. A. Perez-Omil and J. Calvino, J. Mat. Chem. A, 2019, 7, 8993–9003 RSC.
- M. Haider, S. Uhlemann, E. Schwan, H. Rose, B. Kabius and K. Urban, Nature, 1998, 392, 768–769 CrossRef CAS.
- P. E. Batson, N. Dalby and O. Krivanek, Nature, 2002, 418, 617–619 CrossRef CAS.
- E. D. Boyes, M. R. Ward, L. Lari and P. L. Gai, Ann. Phys., 2013, 525, 423–429 CrossRef CAS.
- www.denssolutions.com .
- www.protochips.com .
- P. L. Gai and E. D. Boyes, Microsc. Res. Tech., 2009, 72, 153–160 CrossRef CAS.
- E. D. Boyes and P. L. Gai, J. Phys.: Conf. Ser., 2014, 522, 012004 CrossRef CAS.
- P. L. Gai and E. D. Boyes, J. Phys.: Conf. Ser., 2010, 241, 010255–010260 CrossRef.
- E. D. Boyes and P. L. Gai, C. R. Physics., 2014, 15, 200–213 CrossRef CAS.
- P. L. Gai, L. Lari, M. R. Ward and E. D. Boyes, Chem. Phys. Lett., 2014, 592, 355–359 CrossRef CAS.
- E. D. Boyes, A. P. LaGrow, M. R. Ward, R. W. Mitchell and P. L. Gai, Acc. Chem. Res., 2020, 53, 390–399 CrossRef CAS PubMed.
- E. D. Boyes, A. P. LaGrow, M. R. Ward, R. W. Mitchell and P. L. Gai, Philos. Trans. R. Soc., 2020, 378, 20190605 CrossRef.
- P. L. Gai, Microsc. Microanal., 2002, 8, 21–28 CrossRef CAS.
- P. L. Gai, E. D. Boyes, P. Hansen, S. Helveg, S. Giorgio and C. Henry, MRS Bull., 2007, 32, 1044–1048 CrossRef CAS.
- L. Luo, M. Su, P. Yan and D. Schreiber, et al. , Nat. Mat., 2018, 17, 514–518 CrossRef CAS PubMed.
- A. P. LaGrow, D. C. Lloyd, P. L. Gai and E. D. Boyes, J. Am. Chem. Soc., 2017, 139, 179–185 CrossRef CAS.
- A. P. LaGrow, D. C. Lloyd, P. L. Gai and E. D. Boyes, Chem. Mater., 2018, 30, 197–203 CrossRef CAS.
- M. R. Ward, R. W. Mitchell, E. D. Boyes and P. L. Gai, J. Energy Chem., 2021, 57, 281–290 CrossRef.
- R. W. Mitchell, D. C. Lloyd, L. G. A. van de Water, P. R. Ellis, K. A. Metcalfe, C. Sibbald, L. H. Davies, D. I. Enache, G. J. Kelly, E. D. Boyes and P. L. Gai, ACS Catal., 2018, 8, 8816–8829 CrossRef CAS.
- P. L. Gai, K. Yoshida, M. R. Ward, M. Walsh, L. G. A. van de Water, R. T. Baker, M. J. Watson and E. D. Boyes, Catal. Sci. Technol., 2016, 6, 2214–2227 RSC.
- P. L. Gai and E. D. Boyes, Atomic resolution ETEM, in Handbook of Nanoscopy, ed. Van Tendeloo G., Van Dyck D. and Pennycook S., Wiley-VCH, Germany, 2012; 1, pp. 375–390 Search PubMed.
- G. Somorjai, R. York, D. Butcher and J. Park, Phys. Chem. Chem. Phys., 2007, 9, 3500–3513 RSC.
- M. S. Huberty, A. L. Wagner, A. McCormick and E. Cussle, AIChE J., 2012, 58, 3526 CrossRef CAS.
- T. E. Martin, R. W. Mitchell, E. D. Boyes and P. L. Gai, Philos. Trans. R. Soc., A, 2020, 378, 20190597 CrossRef CAS.
- X. F. Yang, A. Wang, B. Quiao, J. Li and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1745 CrossRef CAS PubMed.
- Z. Sun, S. Wang and W. Chen, J. Mat. Chem. A, 2021, 9, 5296–5319 RSC.
- P. Jiang, X. Bao and M. Salmeron, Acc. Chem. Res., 2015, 48, 1524–11531 CrossRef CAS PubMed.
- S. Mostafa, J. R. Croy, L. Li, J. Yang, A. Frenkel and B. Cuenya, J. Am. Chem. Soc., 2010, 132, 15714–15719 CrossRef CAS.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef CAS.
- P. Wynblatt and N. Gjostein, Prog. Solid State Chem., 1975, 9, 21–58 CrossRef CAS.
- P. Hansen, A. DeLaRiva, S. R. Challa and A. Datye, Acc. Chem. Res., 2013, 46, 1720–1730 CrossRef PubMed.
- S. B. Simonsen, I. Chorkendorff and M. Skoglundh, et al. , J. Catal., 2011, 281, 147–155 CrossRef CAS.
- S. R. Challa, A. Delariva and J. Sehsted, et al. , J. Am. Chem. Soc., 2011, 133, 20672–20675 CrossRef CAS.
- N. Bartelt, W. Theis and R. Tromp, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11741 CrossRef CAS.
- P. E. Batson, Microsc. Microanal., 2008, 14, 89–97 CrossRef CAS.
- N. Shihu, K. Yoshida, E. D. Boyes, D. Brown and P. L. Gai, Catal. Sci. Technol., 2011, 1, 413–423 RSC.
- J. Kwak, J. Hu, D. Mei, C. Yi, D. Kim and A. Peden, Science, 2009, 325, 1670–1673 CrossRef CAS PubMed.
- D. L. King and A. De Klerk, Overview of Feed-to-Liquid (XTL) Conversion, ACS Symp. Ser., 2011, 1084, 1–24 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |