
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible tuning of luminescence and magnetism in a structurally flexible erbium–anilato MOF†
Noemi
Monni
abd,
José J.
Baldoví
 b,
Víctor
García-López
b,
Mariangela
Oggianu
ad,
Enzo
Cadoni
a,
Francesco
Quochi
*cd,
Miguel
Clemente-León
*b,
Maria Laura
Mercuri
*ad and
Eugenio
Coronado
b
b,
Víctor
García-López
b,
Mariangela
Oggianu
ad,
Enzo
Cadoni
a,
Francesco
Quochi
*cd,
Miguel
Clemente-León
*b,
Maria Laura
Mercuri
*ad and
Eugenio
Coronado
b
aDipartimento di Scienze Chimiche e Geologiche, Università degli Studi di Cagliari, Complesso Universitario di Monserrato, 09042 Monserrato, Italy. E-mail: mercuri@unica.it
bInstituto de Ciencia Molecular, Universitat de València, Catedrático José Beltrán 2, 46980 Paterna, Spain. E-mail: miguel.clemente@uv.es
cDipartimento di Fisica, Università degli Studi di Cagliari, Complesso Universitario di Monserrato, 09042 Monserrato, Italy. E-mail: quochi@unica.it
dConsorzio Interuniversitario Nazionale per la Scienza e Tecnologia dei Materiali, INSTM, Via Giusti 9, 50121 Firenze, Italy
First published on 10th May 2022
Abstract
By combining 3,6-N-ditriazolyl-2,5-dihydroxy-1,4-benzoquinone (H2trz2An) with NIR-emitting ErIII ions, two different 3D neutral polymorphic frameworks (1a and 1b), differing in the number of uncoordinated water molecules, formulated as [Er2(trz2An)3(H2O)4]n·xH2O (x = 10, a; x = 7, b), have been obtained. The structure of 1a shows layers with (6,3) topology forming six-membered rings with distorted hexagonal cavities along the bc plane. These 2D layers are interconnected through the N4 atoms of the two pendant arms of the trz2An linkers, leading to a 3D framework, where neighboring layers are eclipsed along the a axis, with hexagonal channels filled with water molecules. In 1b, layers with (6,3) topology in the [101] plane are present, each ErIII ion being connected to three other ErIII ions through bis-bidentate trz2An linkers, forming rectangular six-membered cavities. 1a and 1b are multifunctional materials showing coexistence of NIR emission and field-induced slow relaxation of the magnetization. Remarkably, 1a is a flexible MOF, showing a reversible structural phase transition involving shrinkage/expansion from a distorted hexagonal 2D framework to a distorted 3,6-brickwall rectangular 3D structure in [Er2(trz2An)3(H2O)2]n·2H2O (1a_des). This transition is triggered by a dehydration/hydration process under mild conditions (vacuum/heating to 360 K). The partially dehydrated compound shows a sizeable change in the emission properties and an improvement of the magnetic blocking temperature with respect to the hydrated compound, mainly related to the loss of one water coordination molecule. Theoretical calculations support the experimental findings, indicating that the slight improvement observed in the magnetic properties has its origin in the change of the ligand field around the ErIII ion due to the loss of a water molecule.
Introduction
Metal–organic frameworks (MOFs), formed by organic molecules (linkers) and metal ions (nodes), are attracting considerable attention in materials science thanks to their fascinating architectures1,2 and the richness of their chemical and physical properties. Noteworthily, some of them possess structural flexibility,3 a unique feature that depends on the nature of metal–ligand interactions, which in turn, is influenced by both organic ligands and metal ions/clusters. As a direct consequence, flexible MOFs show dynamic properties such as opening/closing of pores, expansion/shrinkage of the framework and/or a reversible change of physical properties, induced by different types of stimuli such as light, electrical potential, pressure or inclusion of guest molecules.4–6 Despite the plethora of MOFs reported so far, those showing structural flexibility and dynamic behaviour are rare, in spite of their great potential in several applications, including storage, sensing and biomedicine. More importantly, this uncommon feature makes MOFs a challenging platform for designing multiple functionalities with the possibility to tune the physical properties upon reversible structural changes in response to external stimuli.7,8 Lanthanide based MOFs (LnIII-MOFs) in particular show peculiar luminescence properties with emission in the Vis to NIR range.9 In fact, LnIII-MOFs can enable strong light absorption by the organic linkers and efficient lanthanide emission sensitization via the so-called antenna effect, thereby circumventing the inherently low optical absorption rates due to the LnIII forbidden f–f transitions. In addition, LnIII ions possess large inherent magnetic anisotropy, i.e. large unquenched orbital angular moments, which impart strong magnetic anisotropy. Their interesting magnetic behaviour makes them attractive candidates to build single-ion magnets (SIMs),10 a class of mononuclear metal complexes exhibiting slow magnetic relaxation of molecular origin. Controlled spatial organization of SIMs to build large-scale ordered arrays can be achieved by assembling LnIII ions into a 3D framework affording the so-called SIM-MOF.11,12 Among the organic linkers which can either act as a powerful antenna or isolate magnetically the LnIII metal ions, 3,6-disubstituted (X)-2,5-dihydroxybenzoquinone derivatives with X = H, F, Cl, Br, I or CN (i.e. anilates) have recently been studied as building blocks for LnIII-based frameworks,13–16 leading to several bi-dimensional (2D) and a few three-dimensional (3D) lattices17–20 with interesting properties such as luminescence, SIM behaviour, gas/solvent adsorption/desorption and solvent exchange.21 These bridging ligands couple (antiferro)magnetically the transition metal centers but they provide a good magnetic isolation with LnIII metal ions as a result of the negligible overlap with the 4f orbitals.21 Additionally, redox-activity of the benzoquinone core has been shown to be a challenging strategy to tune (enhancing) magnetism and conductivity in benzoquinone-based 3D/2D frameworks and related nanostructures.22,23 Interestingly, Pontilliart et al. reported the first simultaneous reversible redox- and solvato-magnetic switching, observed in a dinuclear DyIII single-molecule magnet (SMM) complex based on a tetrathiafulvalene triad, through a reversible dehydration/rehydration process of the DyIII coordination sphere. Redox-activity plays a crucial role in the modulation of the DyIII magnetic relaxion through the addition of one water molecule. A significant modulation of SMM behaviour due to a hydration/dehydration process has been also observed in the Na9[Er(W5O18)2]·35H2O polyoxometalate.24,25 Previous attempts to tune the magnetic and optical properties of LnIII-anilato-based frameworks, based on the exchange of coordinated and non-coordinated solvent molecules, were reported as well, even though not accompanied by drastic structural changes.16 In the present work, the 3,6-N-ditriazolyl-2,5-dihydroxy-1,4-benzoquinone (H2trz2An) anilato derivative (Scheme 1)26 has been used to construct 3D MOFs, thanks to the coordinative properties of the N4 atoms of the two triazole pendant arms at the 3,6 positions of the anilato moiety. Indeed, it has afforded a MnII 3D coordination polymer27 and ultramicroporous CoII 3D MOFs, featuring an excellent capability in CO2 capture and separation.28 By resorting to LnIII ions – in particular, the NIR emitting luminescent ErIII ion – two 3D ErIII-based frameworks formulated as [Er2(trz2An)3(H2O)4]n·10H2O (1a, porous) and [Er2(trz2An)3(H2O)4]n·7H2O (1b, non-porous), have been obtained. These polymorphs were characterized by single-crystal X-ray diffraction (SC-XRD), luminescence spectroscopy and magnetometry, showing that both compounds present a field-induced slow relaxation of magnetization. Interestingly, all techniques proved a reversible structural flexibility and shrinkage/expansion dynamic behaviour of the 1a framework that originate from a change in the coordination environment of ErIII ions, in response to the removal of coordinative water molecules. Noteworthily, such a structural flexibility induces important and reversible structural changes after partial dehydration, thus allowing for a tuning of their SIM and luminescence properties, which can be rationalized by means of effective electrostatic ligand field calculations.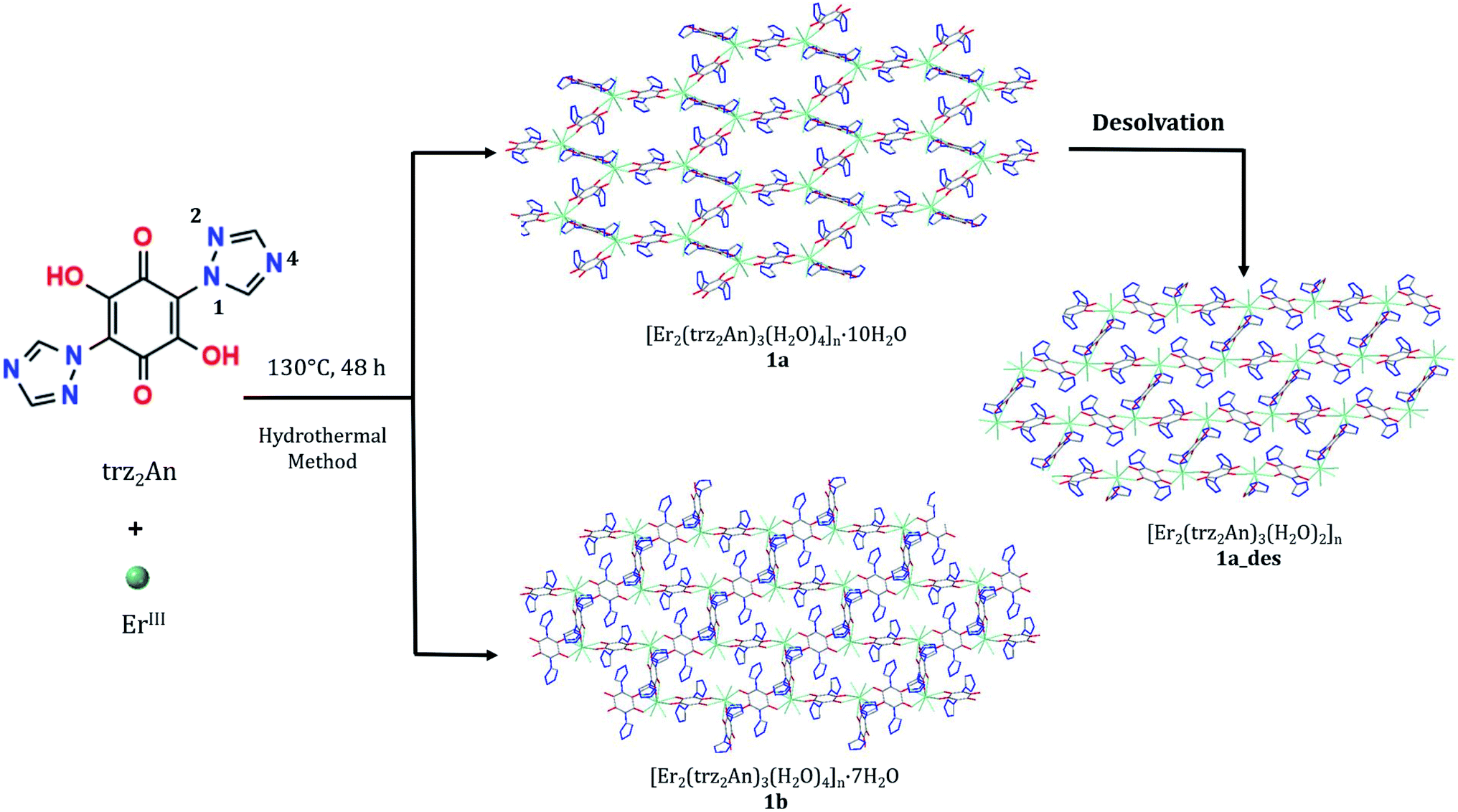 | ||
| Scheme 1 Synthetic strategy for 1a, 1a_des and 1b. | ||
Results and discussion
Synthesis
By combining Er(NO3)3·6H2O with H2trz2An in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, [Er2(trz2An)3(H2O)4]n·10H2O (1a) and [Er2(trz2An)3(H2O)4]n·7H2O (1b) were obtained through a hydrothermal method (Scheme 1). Isostructural compounds are also obtained with TbIII, DyIII and HoIII which will be published in due course. The synthetic protocol has been optimized for the ErIII derivative because it presents multifunctionality. Two types of crystals were observed in each hydrothermal batch: dark red block crystals (1a) and orange prismatic crystals (1b) (Fig. S1a†). Due to the difference in density, it was possible to separate both types of crystals by using a CH2Cl2/CH2Br2 solvent mixture. Thus, when the crystals were suspended in the CH2Cl2/CH2Br2 solution, crystals of 1a started floating, while crystals of 1b lied at the bottom (Fig. S1b†). The lower density of 1a agrees with the more porous structure of this compound, which is formed by interconnected hexagonal pores, in contrast to 1b with rectangular pores of smaller size (vide infra). In an attempt to decrease the number of solvate water molecules of 1a in a controlled way, by heating or applying vacuum, a drastic and reversible structural change was observed. By contrast, the 1b structure remains unaltered under the same aforementioned conditions. The removal of some of water molecules present in the channels and one coordinated water molecule in 1a results in a new crystalline phase, [Er2(trz2An)3(H2O)2]n·2H2O (1a_des), with a less porous structure formed by distorted rectangular cavities (vide infra).
:1 ratio, [Er2(trz2An)3(H2O)4]n·10H2O (1a) and [Er2(trz2An)3(H2O)4]n·7H2O (1b) were obtained through a hydrothermal method (Scheme 1). Isostructural compounds are also obtained with TbIII, DyIII and HoIII which will be published in due course. The synthetic protocol has been optimized for the ErIII derivative because it presents multifunctionality. Two types of crystals were observed in each hydrothermal batch: dark red block crystals (1a) and orange prismatic crystals (1b) (Fig. S1a†). Due to the difference in density, it was possible to separate both types of crystals by using a CH2Cl2/CH2Br2 solvent mixture. Thus, when the crystals were suspended in the CH2Cl2/CH2Br2 solution, crystals of 1a started floating, while crystals of 1b lied at the bottom (Fig. S1b†). The lower density of 1a agrees with the more porous structure of this compound, which is formed by interconnected hexagonal pores, in contrast to 1b with rectangular pores of smaller size (vide infra). In an attempt to decrease the number of solvate water molecules of 1a in a controlled way, by heating or applying vacuum, a drastic and reversible structural change was observed. By contrast, the 1b structure remains unaltered under the same aforementioned conditions. The removal of some of water molecules present in the channels and one coordinated water molecule in 1a results in a new crystalline phase, [Er2(trz2An)3(H2O)2]n·2H2O (1a_des), with a less porous structure formed by distorted rectangular cavities (vide infra).
Crystal structure
The structures of 1a and 1b were obtained at 120 K by SC-XRD. Both compounds crystallize in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . They are 3D neutral coordination frameworks formed by ErIII ions connected by trz2An bridging linkers. In 1a, the ErIII ion is ennea-coordinated with a {NO8} coordination sphere arising from two oxygens of three bidentate trz2An anilates, the N4 atom of one trz2An and two coordinated water molecules. The geometry of the first coordination sphere is closest to a spherical capped square antiprism (CSAPR-9) according to continuous SHAPE analysis (Fig. 1a and Table S9†).29 ErIII ions are linked to three neighboring ions through the oxygens of three trz2An anilates which coordinate in the bis-bidentate mode. This leads to layers with (6,3) topology forming six-membered rings with distorted hexagonal cavities along the bc plane (Fig. S2a†). Each cavity contains two anilato rings almost parallel to the layer (face-on) and four anilato rings almost perpendicular to it (edge-on). The largest Er–Er–Er angle in the hexagon is 134.4°, while the Er–Er distances of the diagonals of the hexagons are 12.8, 13.4 and 21.9 Å (Table S4†). These distances and angles are similar to those found in LnIII–anilato-based 2D compounds with an ennea-coordinated ligand field for the LnIII and spherical tricapped trigonal prism (TCTPR-9) geometry.30,31 The C–O and C–C bond lengths across the five-membered chelate ring for trz2An ligands also present typical values of anilato dianions (see Table S3†).30 A remarkable difference with respect to previous LnIII anilato-based 2D compounds is that these 2D layers are interconnected through the N4 atoms of the two 1,2,4-triazole substituted pendant arms of one of the three trz2An linkers coordinated to each ErIII (face-on linkers) (Fig. 1c). This leads to a porous 3D framework, where neighboring layers are eclipsed along the a axis with hexagonal channels filled with water molecules (Fig. 1b). The remaining N4 atoms of the 1,2,4-triazole substituted pendant arms of the other two trz2An linkers are hydrogen-bonded to coordinating water molecules.
. They are 3D neutral coordination frameworks formed by ErIII ions connected by trz2An bridging linkers. In 1a, the ErIII ion is ennea-coordinated with a {NO8} coordination sphere arising from two oxygens of three bidentate trz2An anilates, the N4 atom of one trz2An and two coordinated water molecules. The geometry of the first coordination sphere is closest to a spherical capped square antiprism (CSAPR-9) according to continuous SHAPE analysis (Fig. 1a and Table S9†).29 ErIII ions are linked to three neighboring ions through the oxygens of three trz2An anilates which coordinate in the bis-bidentate mode. This leads to layers with (6,3) topology forming six-membered rings with distorted hexagonal cavities along the bc plane (Fig. S2a†). Each cavity contains two anilato rings almost parallel to the layer (face-on) and four anilato rings almost perpendicular to it (edge-on). The largest Er–Er–Er angle in the hexagon is 134.4°, while the Er–Er distances of the diagonals of the hexagons are 12.8, 13.4 and 21.9 Å (Table S4†). These distances and angles are similar to those found in LnIII–anilato-based 2D compounds with an ennea-coordinated ligand field for the LnIII and spherical tricapped trigonal prism (TCTPR-9) geometry.30,31 The C–O and C–C bond lengths across the five-membered chelate ring for trz2An ligands also present typical values of anilato dianions (see Table S3†).30 A remarkable difference with respect to previous LnIII anilato-based 2D compounds is that these 2D layers are interconnected through the N4 atoms of the two 1,2,4-triazole substituted pendant arms of one of the three trz2An linkers coordinated to each ErIII (face-on linkers) (Fig. 1c). This leads to a porous 3D framework, where neighboring layers are eclipsed along the a axis with hexagonal channels filled with water molecules (Fig. 1b). The remaining N4 atoms of the 1,2,4-triazole substituted pendant arms of the other two trz2An linkers are hydrogen-bonded to coordinating water molecules.
 | ||
| Fig. 1 (a) Spherical capped square antiprismatic coordination geometry of the ErIII center in 1a. (b) View of 1a along the bc plane, showing hexagonal cavities. (c) View of 1a along the ac plane, showing layers bridged by trz2An linkers. The black, blue, red, and green spheres represent the C, N, O, and ErIII atoms, respectively. Hydrogen atoms and water molecules inside the pores are omitted for clarity. | ||
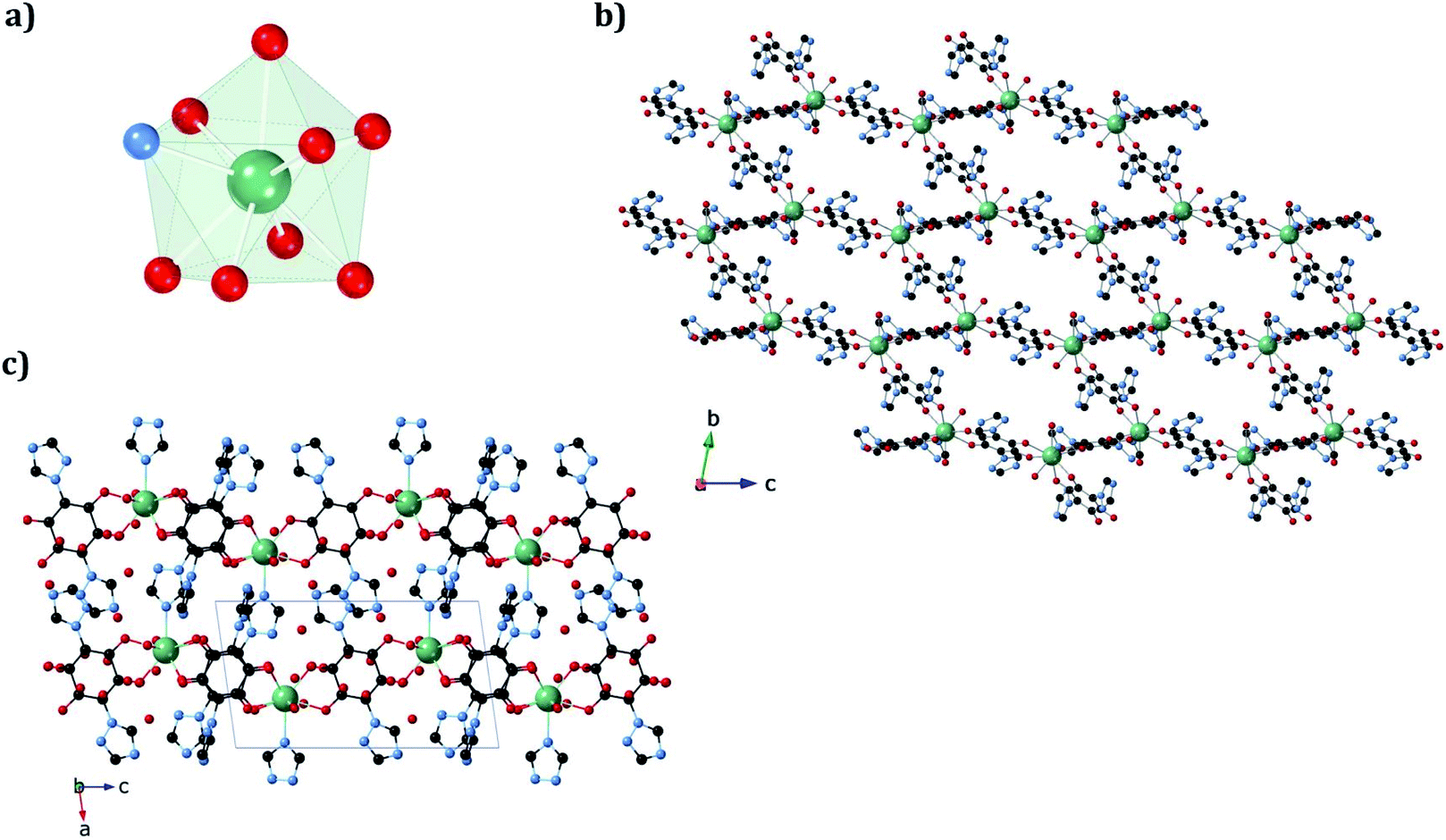
In 1b, the ErIII ion is ennea-coordinated as well, showing a {NO8} coordination sphere with six oxygens of three bidentate trz2An linkers, two coordinated water molecules and the N4 atom of one trz2An ligand. The coordination geometry is nearest to a spherical tricapped trigonal prism (TCTPR-9) according to continuous SHAPE analysis29 (Fig. 2a and Table S9†). The structure shows layers with (6,3) topology in the [101] plane, in which each ErIII ion is connected to three other ErIII ions through bis-bidentate trz2An linkers forming rectangular six-membered cavities, which adopt a brick-wall structure where the long axes of all the rectangles are parallel (Fig. S2b†). Thus, the two largest Er–Er–Er angles are close to 180° (172.5°) (see Table S6†). On the other hand, ErIII nodes of the same layer are almost coplanar and are connected to ErIII nodes of other layers, through the N4 atom of the two pending triazolyl arms of one of the three trz2An linkers (Fig. 2c), which are placed at the shortest side of the rectangular cavities, leading to a 3D framework. Finally, the C–O and C–C bond lengths across the five-membered anilato ring for trz2An linkers exhibit, as in 1a, typical values of anilato dianions (see Table S5†).30 The powder X-ray diffraction (PXRD) patterns of both 1a and 1b polycrystalline samples are fully consistent with the calculated ones (Fig. S3 and S4†), confirming the homogeneity and purity of the bulk sample. These polycrystalline samples were placed under vacuum in a Schlenk line and then sealed in a glove box in order to study if the vacuum could induce changes in the crystal structure. A structural change was observed in 1a leading to a new phase, 1a_des, where a loss of water molecules was found. Instead, no structural change was observed for 1b, as clearly shown in Fig. S5.† Remarkably, when 1a_des was left in air for two days, the sample is rehydrated forming the 1a phase as shown by PXRD measurements (Fig. 3). This proves that the dehydration/hydration process and the induced structural change are fully reversible.
 | ||
| Fig. 2 (a) Spherical tricapped trigonal prismatic coordination geometry of the ErIII center in 1b. (b) View along the bc plane, showing rectangular cavities. (c) View along the ac plane of 1b, showing layers bridged by trz2An linkers. The black, blue, red, and green spheres represent the C, N, O, and ErIII atoms, respectively. Hydrogen atoms and water molecules inside the pores are omitted for clarity. | ||
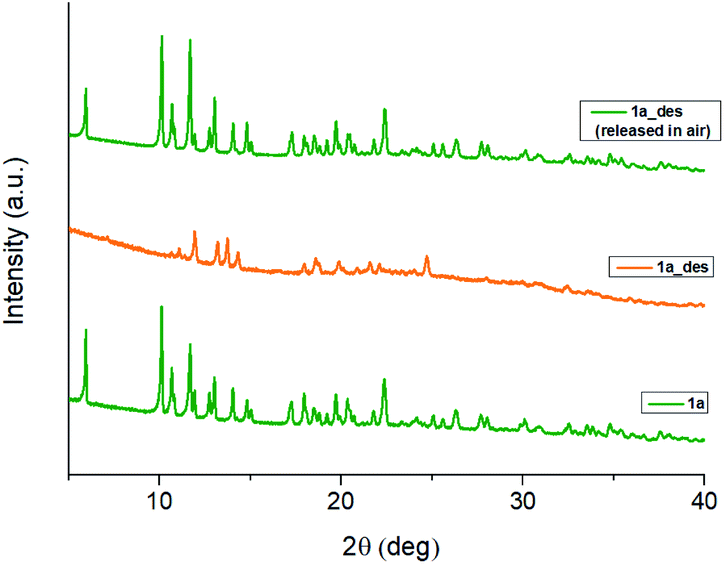 | ||
| Fig. 3 PXRD patterns of 1a (green), 1a_des (orange) and 1a_des left in air 2 days after vacuum treatment (green top), highlighting the full reversibility of the structural change induced by dehydration. | ||
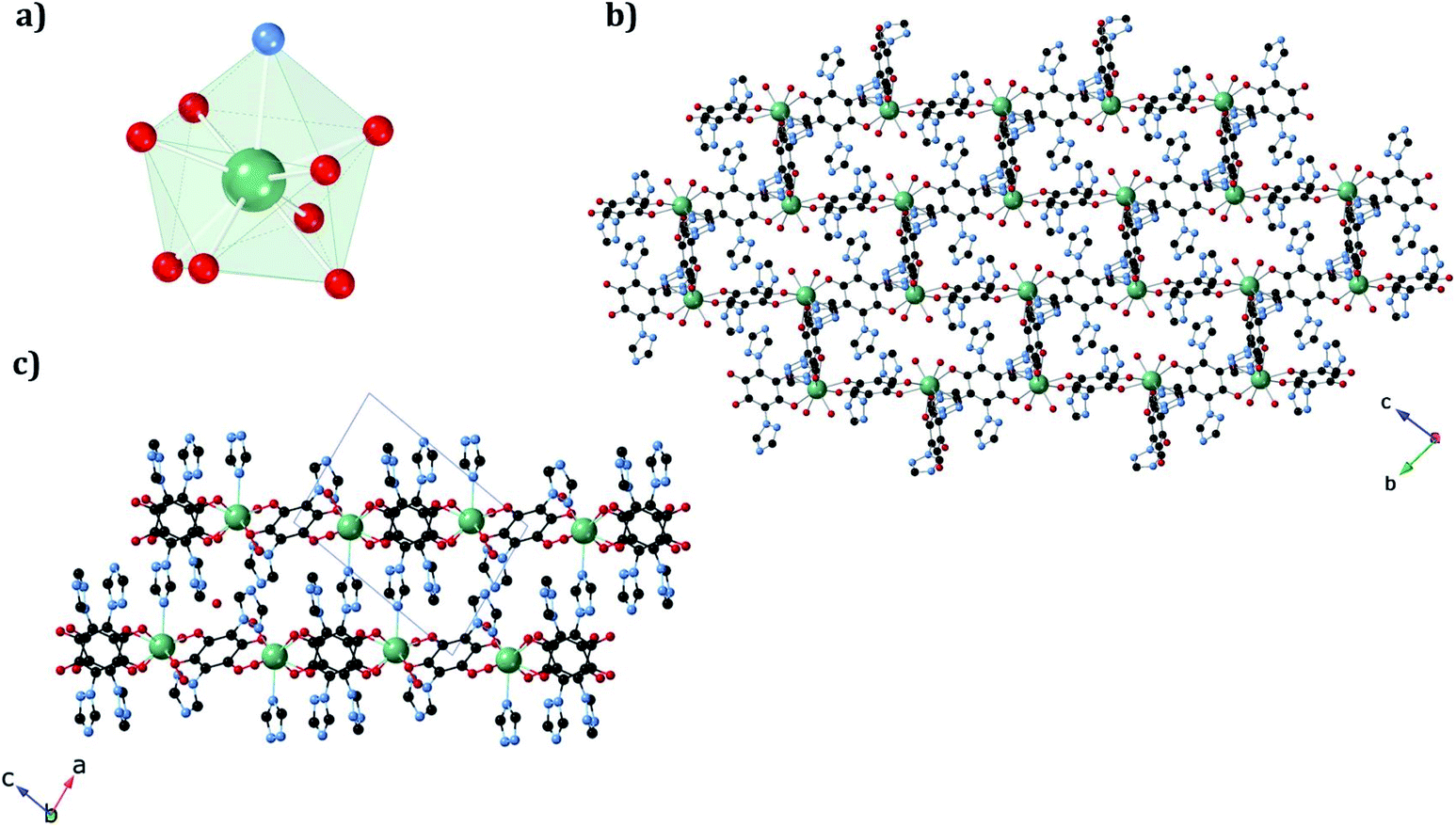
The structure of the new dehydrated phase, 1a_des, was investigated by SC-XRD. A vacuum was applied to a single crystal of 1a, which was then transferred to a diffractometer at 120 K. A similar structure was obtained if a single crystal of 1a was heated at 360 K in the diffractometer with a small difference in the water pore content. The ErIII ion in 1a_des (under vacuum/heating) shows a {NO7} coordination sphere arising from six oxygens of three bidentate trz2An linkers, the N4 atom of one trz2An linker and one coordinated water molecule, different from the {NO8} one observed in 1a. This is due to the loss of one water coordination molecule. The coordination geometry is close to a triangular dodecahedron (TDD-8) (Fig. 4a and Table S10†), according to continuous SHAPE analysis.29 Er–O bond lengths are in the range of 2.266(16)–2.430(16) Å for 1a_des after vacuum (2.28(2)–2.442(13) Å for 1a_des at 360 K), while Er–N bond lengths are 2.46(2) Å for 1a_des after vacuum (2.464(18) Å for 1a_des at 360 K). Again, the C–O and C–C bond lengths across the five-membered anilato ring for trz2An linkers exhibit typical values of anilato dianions,30 1.22(3)–1.26(2) and 1.57(3)–1.60(3) Å for 1a_des after vacuum (1.22(3)–1.27(2) and 1.52(3)–1.54(3) Å for 1a_des at 360 K) (see Table S7†). As in the 1a case, the structure shows layers with (6,3) topology in the bc plane, but interestingly, they exhibit a drastic change from a distorted hexagonal 2D network to a distorted 3,6-brickwall rectangular structure. Thus, the six-membered rectangular cavities of the layers contain four face-on and two edge-on anilato rings in 1a_des, instead of the two face-on and four edge-on ones found in 1a (Fig. S2b†). Remarkably, the 3D framework is maintained thanks to the N4 atom of the two pending triazolyl arms of one of the three trz2An linkers located in the shortest side of the rectangular cavity (face-on linker), indicating that the connectivity is the same as that of 1a. Neighboring layers are eclipsed along the a axis, leading to rectangular channels (Fig. 4b). These channels contain one crystallographically independent water solvate molecule forming a hydrogen bond with the coordinated water molecule. By heating the sample at 360 K, the solvate water molecule shows a disorder, and it was modeled with an occupancy of 0.5. This could be related to a more complete dehydration of the sample. As shown by PXRD, the phase of 1a_des is consistent with the calculated one from the CIF (Fig. S6†).
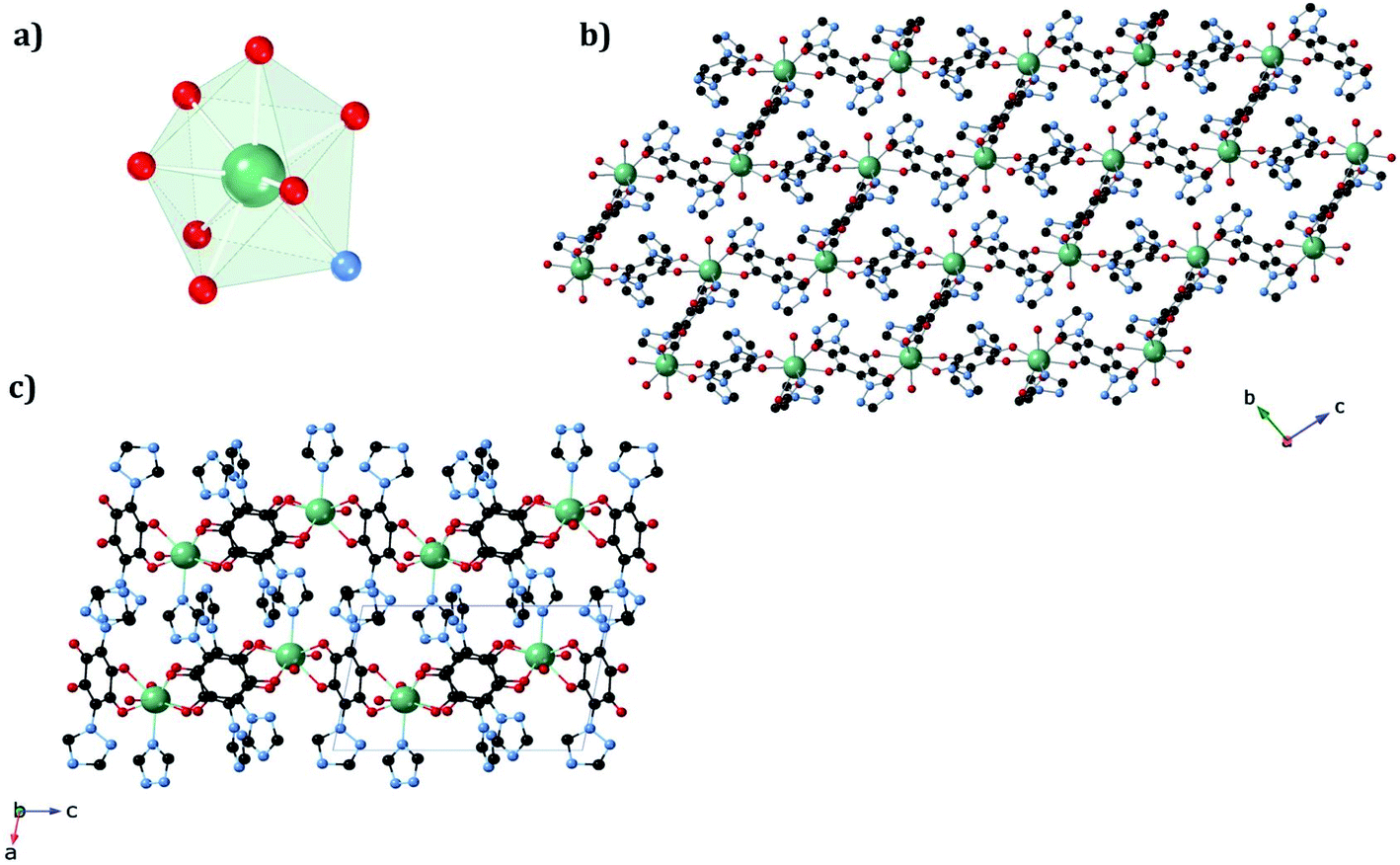 | ||
| Fig. 4 (a) Triangular dodecahedron coordination geometry of the ErIII ions in 1a_des. (b) View along the bc plane of the 1a_des framework, showing shrunk rectangular cavities. (c) View along the ac plane of 1a_des, showing layers bridged by trz2An linkers. The black, blue, red, and green spheres represent the C, N, O, and ErIII atoms, respectively. Hydrogen atoms and water molecules inside the pores are omitted for clarity. | ||
In conclusion, a structural phase transition from a hexagonal cavity to a rectangular one is observed when the 1a phase is dehydrated under vacuum or by heating the sample. This transition is fully reversible, as observed when the dehydrated phase (1a_des) is rehydrated.
Magnetic properties
DC magnetic susceptibility measurements of 1a, 1b, and 1a_des were carried out in the temperature range of 2–300 K using an applied magnetic field of 0.1 T (Fig. 5 and S10†). In the case of 1a, it was necessary to perform the measurements in a closed holder and in contact with H2O to prevent its dehydration and subsequent formation of 1a_des due to the vacuum of the squid chamber (∼2–3 mbar). We measure a χMT product at 300 K of 11.43 (1a), 11.36 (1b) and 11.58 (1a_des) cm3 K mol−1, which is close to the value expected for ErIII (11.475 cm3 K mol−1; 4I15/2), thus indicating that most of the ligand field split energy levels of the ground multiplet are populated at room temperature. χMT shows a slow and steady decrease upon cooling from 300 K to 50 K, which becomes sharper at lower temperatures reaching a value of 5.2, 5.5 and 6.1 cm−3 K mol−1 for 1a, 1b and 1a_des, respectively at 2 K. The decrease at low temperature is characteristic of the depopulation of these levels. The field dependence of magnetization was also measured in the 2–8 K temperature range by varying the magnetic field up to 5 T (Fig. S11 and S12†).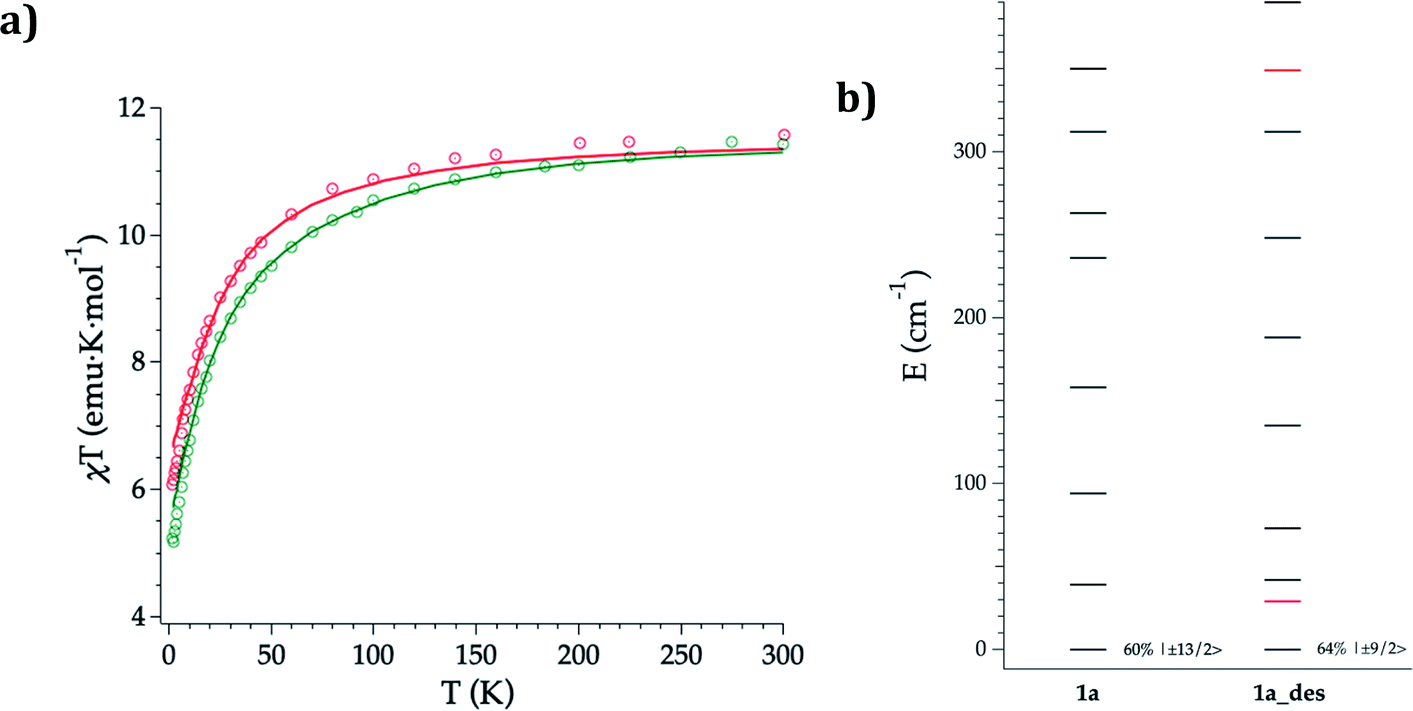 | ||
| Fig. 5 Experimental (symbols), fitted (solid line) temperature-dependence of the magnetic susceptibility from 2 to 300 K of 1a (green) and 1a_des (red) (a); calculated (black) and experimental (red) ground-J multiplet energy levels (b). Further details are provided in Table S13.† | ||
To further understand the observed magnetic behaviour of both phases, theoretical calculations were carried out using the SIMPRE computational package.32 The static magnetic susceptibility was successfully fitted by using the radial effective charge (REC) model33 (Dr = 0.98 Å and Zi = 0.161 for oxygens, and Dr = 1.20 Å and Zi = 0.05 for nitrogen) (Fig. 5 and S10†). This allowed the prediction of the magnetization curves, which are also in good agreement with the experimental data (Fig. S11 and S12†). The diagonalization of the crystal field Hamiltonian results in a ground state wave function mainly composed of 60% of the MJ = |±13/2〉 microstate in the easy axis direction for 1a and 1b, which is congruent with the observed slow relaxation of the magnetization. The first excited doublet is located at about 39 cm−1 and is mainly composed of the MJ = |±11/2〉 microstate (37%), mixed with |±3/2〉 and |±1/2〉 for 1a, while it is located at about 32 cm−1 and is mainly composed of |±15/2〉 and |±11/2〉 (63% between both of them) for 1b. The energy levels are approximately equispaced, and the total crystal field splitting reaches 355 cm−1. In the case of 1a_des, the loss of a water molecule that reduces the coordination number results in a drastic change of the ground state wave function, which is now described by a mixture between |±9/2〉 (64%), |±11/2〉 (14%) and |±7/2〉 (11%) microstates, while the first excited energy level is located at 42 cm−1 (see Tables S13 and S14†).
The dynamic magnetic properties were studied by susceptibility measurements performed with an alternating magnetic field (AC susceptibility). In the absence of a magnetic field, no signal in the out-of-phase molar susceptibility (χ′′) was observed. When magnetic DC fields of 0.09 T were applied, strong frequency-dependent peaks in both the in-phase molar susceptibility (χ′) and χ′′ appear with clear maxima of χ′′ below 2.2 K for 1a (Fig. S13†). On the other hand, the variable-frequency AC data of 1a at 2 K show a maximum at ca. 5800 Hz in χ′′ (see Fig. 6). This is a clear indication that 1a presents a field-induced slow relaxation of magnetization. Interestingly, when 1a is dehydrated to give 1a_des, the χ′′ maxima are shifted to higher temperatures (3.6 K for 1a_des and 2.2 K for 1a at a frequency of 10000 Hz under an applied DC field of 0.09 T) and at lower frequencies in the variable-frequency AC measurements (5800 Hz for 1a and 170 Hz for 1a_des at 2 K) as shown in Fig. 6 and 7. When the same desolvated sample is measured again in contact with H2O, the initial magnetic behavior of 1a is restored in a reversible way. This result confirms that the structural changes of 1a after dehydration modify the magnetic properties, in agreement with the decrease of the mixing with lower microstates found from the theoretical calculations for 1a_des. Thus, although the ground state of 1a_des is dominated by a lower MJ value than for 1a, importantly, most of the wave function is described by high MJ values (88% between |±11/2〉, |±9/2〉 and |±7/2〉), thus resulting in a more negligible contribution from those with lower MJ values. This situation is more favourable to reduce fast spin relaxation through quantum tunneling of the magnetization (QTM) at low temperatures in 1a_des. In contrast, in 1a, the non-negligible contribution of nonaxial crystal-field terms increases the coupling to the degenerate Kramers doublets in high-order perturbation theory34 leading to poorer SMM properties. Another interesting aspect is that the loss of a water molecule erases its molecular vibrations, preventing them from coupling with the ligand field split energy levels, thus suppressing this possible vibration-induced magnetic relaxation channel.35 It is likely that the combination of both effects (a reduction in QTM and molecular vibrations) is responsible for the slight increase in the blocking temperature of 1a after dehydration, as dynamic magnetic measurements indicate. Interestingly, the modulation of SIM properties has not been observed in the isostructural Dy-MOF.36
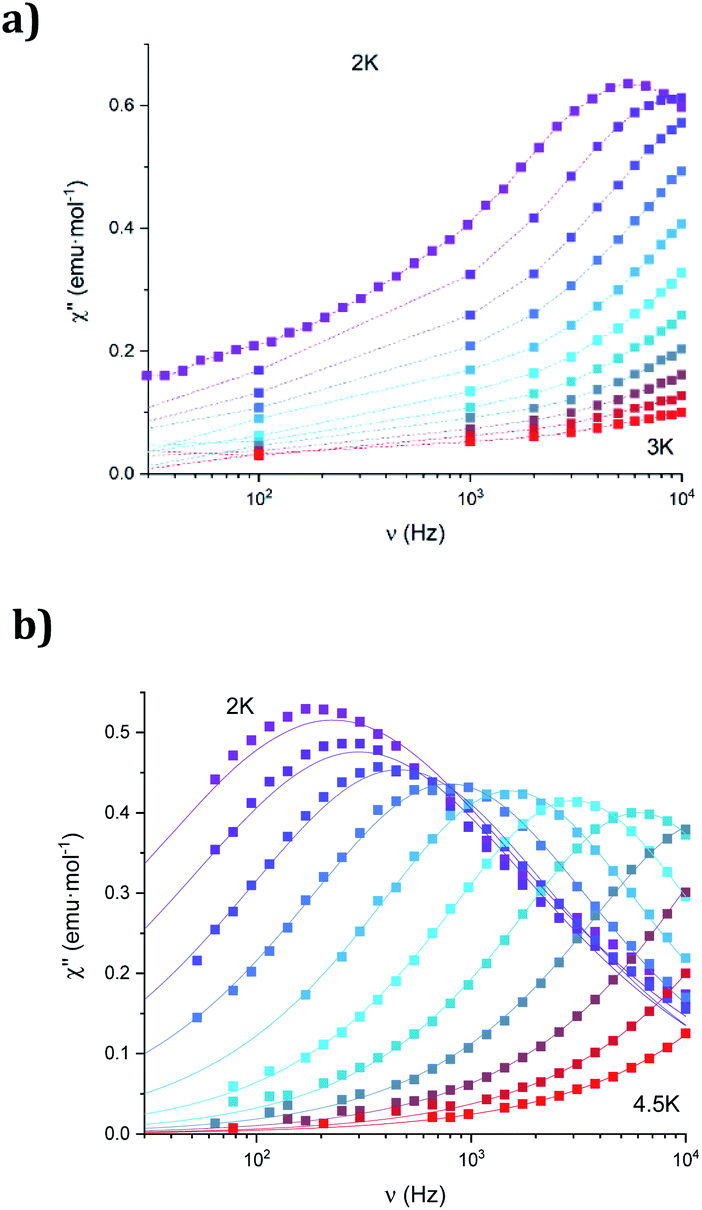 | ||
| Fig. 6 Frequency dependence of the out-of-phase component, χ′′, of 1a (a) and 1a_des (b) in an applied DC field of 0.09 T at the different temperatures. Solid lines represent the best fits to the Debye model. | ||
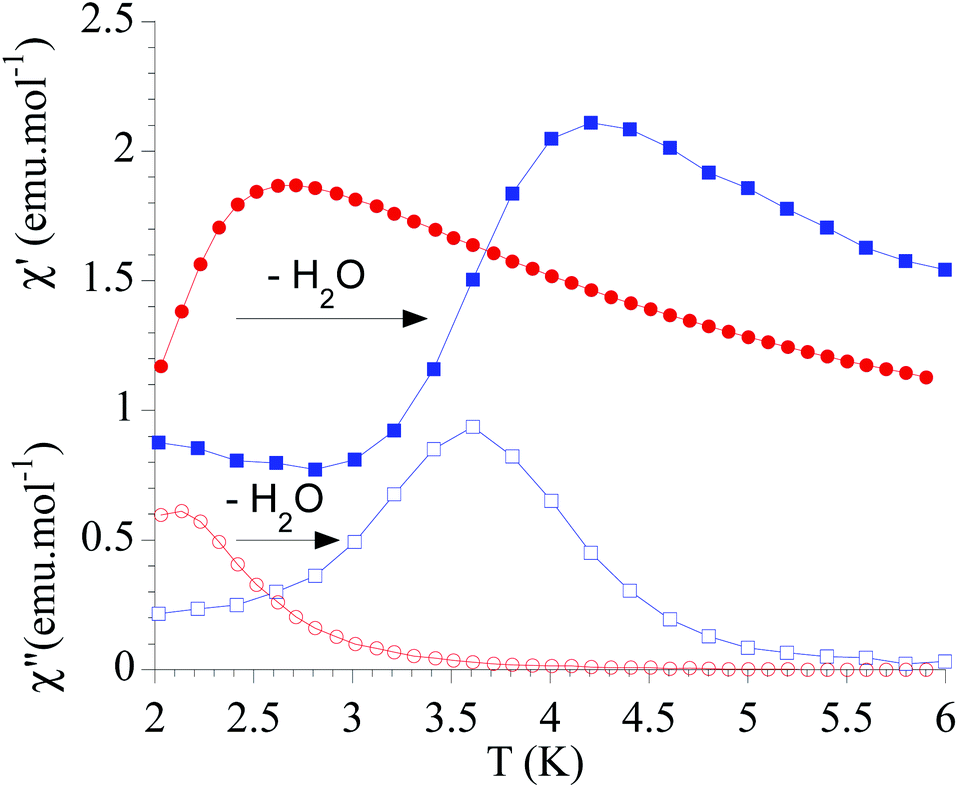 | ||
| Fig. 7 Temperature dependence of χ′ and χ′′ of 1a (full and empty red circles, respectively) and 1a_des (full and empty blue squares, respectively) in an applied DC field of 0.09 T at 10000 Hz. | ||
The Cole–Cole plots (χ′′ vs. χ′) of 1a_des confirm the presence of a single relaxation process (see Fig. S15†). Thus, at fixed temperatures between 2.0 and 3.5 K, semi-circular plots were obtained and fitted using a generalized Debye model, yielding the α parameter in the range of 0.17–0.40. This indicates moderately broad distributions of the relaxation processes. In the case of 1a, only one temperature could be studied due to the lower temperature of the maxima in χ′′ (see Fig. S15†). Again, the Cole–Cole plot at this temperature suggests the presence of a single relaxation process (α = 0.31). The relaxation times (τ) of 1a_des at the different temperatures were determined from the Debye model. They were fitted to the Arrhenius expression for a thermally activated process (Orbach, τ = τ0exp(Ueff/kBT)). However, the plots of τ vs. 1/T deviate from linearity at low temperatures, indicating the coexistence of multiple relaxation pathways as observed in other anilate-based LnIII complexes (Fig. S16†). We, therefore, applied the general model where the first, second, third and fourth terms include quantum tunneling, direct, Raman and Orbach relaxation processes, respectively.
| τ−1 = τQTM−1 + AH2T + CTn + τ0−1exp(−Ueff/KBT) |
Accurate fittings were obtained using Raman and Orbach relaxation processes with values comparable to those found in other anilate-based LnIII compounds with C = 16(7) s−1 K−5.3, τ0 = 5.0(5) × 10−10 s and Ueff = 39(2) K for 1a_des (Fig. S17†).16,20,37,38 The calculated value of n (5.3(3)) is smaller than the ideal value of 9 found for Raman processes. This suggests that these Raman-like relaxations are attributed to acoustic and optical vibrations.39 Finally, temperature dependence of the τ values of 1a was not fitted because of the lack of data points.
The AC susceptibility measurements of 1b show strong frequency-dependent peaks in both χ′ and χ′′ with the clear maxima of χ′′ below 4.2 K when a magnetic DC field of 0.1 T was applied (Fig. S18†). The variable-frequency AC data at different temperatures show the maxima of χ′′ at 160 Hz at 2 K and ca. 6000 Hz at 4 K (see Fig. S19†). Therefore, they indicate a field-induced slow relaxation of magnetization in agreement with theoretical calculations. The Cole–Cole plots (χ′′ vs. χ′) confirm the presence of a single relaxation process (see Fig. S20†). Thus, at fixed temperatures between 2.0 and 4.0 K, semi-circular plots were obtained and fitted using a generalized Debye model, yielding the α parameter in the range of 0.12–0.21. This indicates narrower distributions of the relaxation processes than in the case of 1a_des. Temperature dependence of the τ values of 1b was fitted using Raman and Orbach relaxation processes with values comparable to those found in 1a_des and other anilate-based LnIII compounds with C = 11(5) s−1 K−5.6, n = 5.6(4), τ0 = 2.0(5) × 10−9 s and Ueff = 42(3) K (Fig. S17†).16,20,35,36
Photoluminescence properties
Photoluminescence (PL) was excited using short laser pulses at a wavelength of 355 nm and analysed both spectrally and temporally. Near UV laser irradiation photoexcites the trz2An linker dianion in all compounds. ErIII emission sensitization then occurs via the optical antenna effect through a ligand-to-metal energy relaxation pathway. As clearly seen in Fig. 8a, ligand-centred emission of 1a and 1b measured under standard conditions appeared considerably redshifted and quenched with respect to that of the ligand dianion trz2An in the (Ph4P)2trz2An reference compound. This was rationalized as the consequence of strong singlet/triplet intersystem mixing (heavy-atom effect), ensuing a ligand-centred excited state that quickly relaxes toward the LnIII ion by ligand-to-metal energy transfer.40 In fact, as deduced by three exponential fitting of the PL decay profile and amplitude averaging of the time constants, the bare excited-state lifetime of trz2An measured in (Ph4P)2(trz2An) lies in the time scale of hundreds of picoseconds (∼460 ps, Fig. 8b), whereas the ligand-centred emission of 1a is nearly resolution-limited, hinting towards a quantitative ligand-to-metal energy sensitization with nearly unity efficiency. Similar considerations hold for 1b, where a monoexponential PL lifetime of ∼30 ps was observed. At room temperature (295 K), 1a and 1a_des exhibited different ErIII-centred (4I13/2 → 4I15/2) emission spectra, readily interpreted as a consequence of the different coordination environment experienced by ErIII in the two compounds (Fig. 9a). Cooling 1a_des under vacuum conditions down to 77 K resulted in higher spectral definition of the crystal-field-split 4I13/2 → 4I15/2 optical transitions (Fig. 9b). Up to 13 transitions could be clearly detected and fairly well fitted using a multiple Gaussian fit procedure, thereby demonstrating a non-negligible presence of hot bands (starting from higher energy states of the 4I13/2 multiplet) still at 77 K.41 A direct comparison between the emission spectra at 295 and 77 K allowed for a tentative identification of the zero-phonon transition, which was observed at 1529 nm. In turn, this enabled a direct comparison between the observed transition wavelengths and those inferred from the theoretically calculated energy spectrum of the 4I15/2 multiplet. Despite the strong vibrational quenching of the ErIII emission from coordinated water molecules, effectively reducing the ErIII excited-state lifetime from milliseconds to tens of nanoseconds,421a and 1a_des could be quite clearly distinguished on the basis of the decay profiles of the ErIII-centred NIR PL, the amplitude-averaged lifetime (extracted from biexponential fits) being 55 ns in 1a and 86 ns in 1a_des (Fig. 9c). This was ascribed to the different quenching produced by two coordinated water molecules in 1a and only one water molecule in 1a_des. Upon cooling at 77 K, the PL lifetime of 1a_des increased to 107 ns possibly due to partial suppression of vibrational quenching. The PL properties of 1a could not be measured at 77 K because the experimental technique used for cryogenic measurements involves 1a dehydration and consequent phase transition to 1a_des prior to cooling. As shown in Fig. 9d, 1b displayed practically the same room-temperature ErIII-centred NIR emission spectrum, both in air and under vacuum, in agreement with the results of PXRD measurements and confirming the existence of only one phase. The decay profile of the NIR PL showed a much lower sensitivity to environmental conditions, which was in part expected since in 1b the vacuum does not modify the number of water molecules coordinated to the ErIII ions (Fig. 9f). Comparison of the 295 and 77 K emission spectra (Fig. 9e) led to the identification of the emission peak centred at 1524 nm as the most probable zero-phonon transition in 1b, allowing for comparison of experiment and theory just as for 1a_des.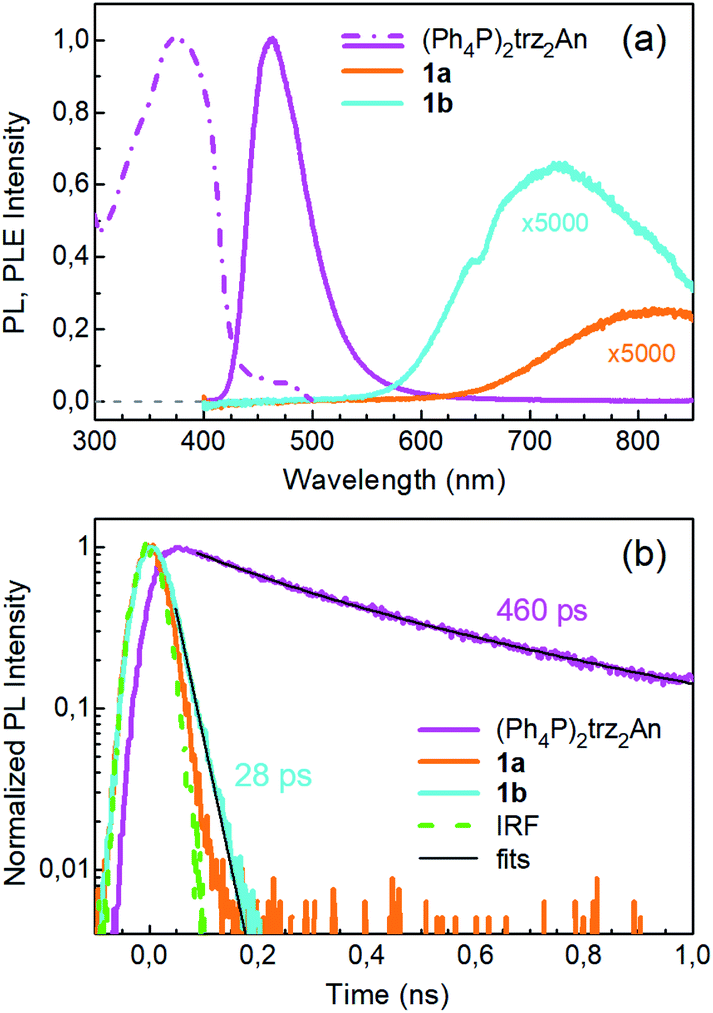 | ||
| Fig. 8 Ligand-centred PL properties of 1a, 1b and the reference (Ph4P)2(trz2An) linker, under 355 nm pulsed laser irradiance. (a) Ligand-centred PL spectra acquired under standard conditions and normalized to the peak of the spectrum of (Ph4P)2trz2An. The PL intensities of 1a and 1b are multiplied by a factor of 5000. The dashed-dotted line is the normalized PL excitation (PLE) spectrum of (Ph4P)2trz2An. The excitation spectra of the strongly quenched ligand emissions of 1a and 1b could not be clearly detected and are not shown. (b) Spectrally-integrated ligand-centred PL decay transients and the impulse response function (IRF) of the detection setup. The fit of a three exponential decay curve to the (Ph4P)2trz2An decay is shown, and the amplitude-averaged lifetime is indicated. Monoexponential fit of the 1b decay and the lifetime are reported as well. | ||
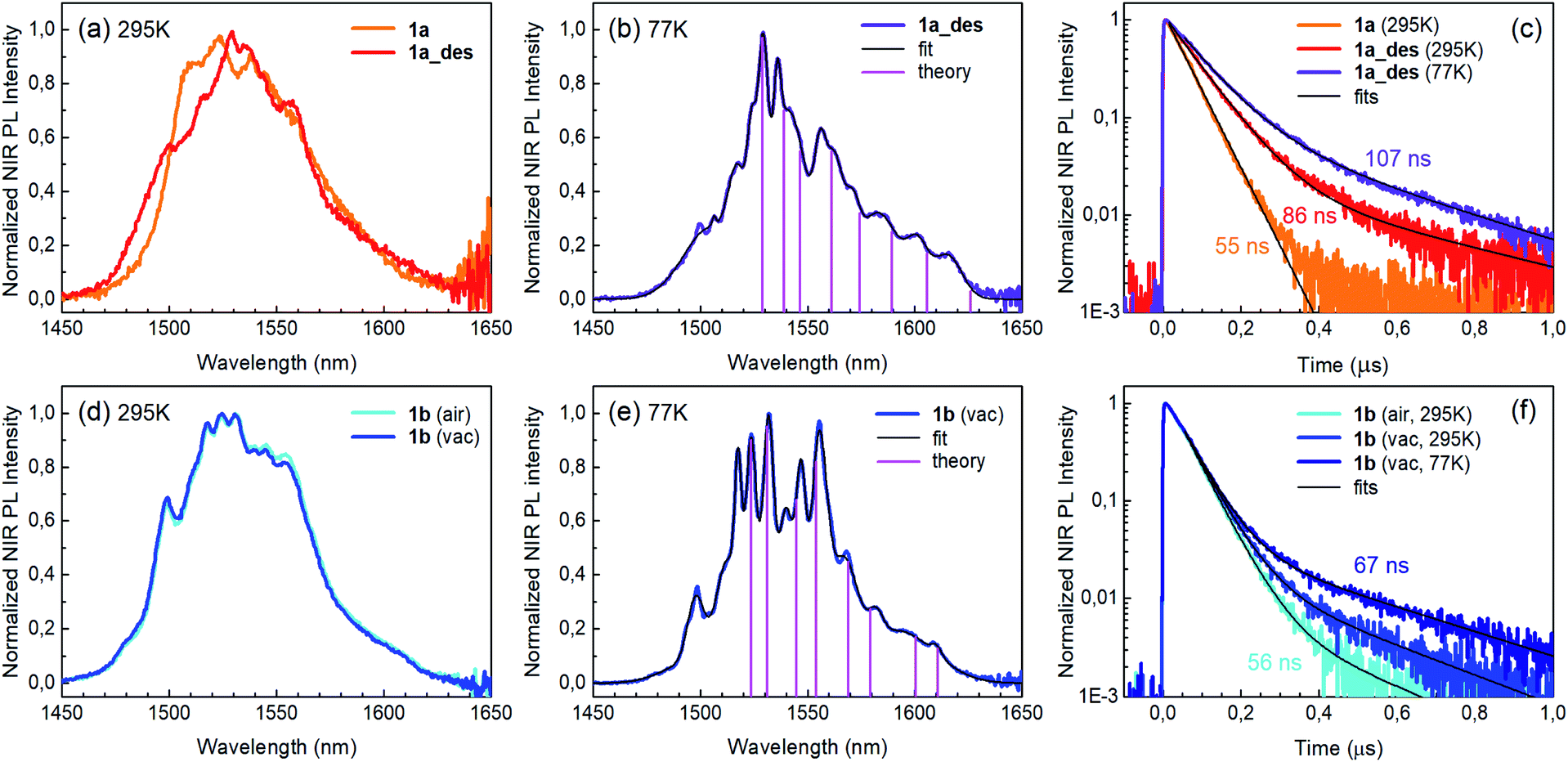 | ||
| Fig. 9 ErIII-centred NIR PL properties of 1a, 1a_des and 1b. (a) PL spectra of 1a and 1a_des at room temperature. (b) Low-temperature (77 K) spectrum of 1a_des with multiple Gaussian fit. Vertical bars represent the radiative transitions from the 4I13/2 lowest level to the ground (4I15/2) multiplet levels estimated from theoretical calculations. (c) PL decay transients for 1a and 1a_des at room temperature and at 77 K. Biexponential decay fits and amplitude-averaged lifetimes are shown as well. (d)–(f) Same as (a)–(c) but for 1b. | ||
Reversible phase transition from 1a to 1a_des was confirmed spectroscopically by performing several air/vacuum (rehydration/dehydration) cycles, where highly reproducible emission spectra and decay transients were obtained (see Fig. S21†). Overall, theoretical calculations of the ground (4I15/2) multiplet spectra turned out to be consistent with the low-temperature emission spectra, with regard to both the energy splitting between the first two levels and the whole energy spread within the multiplet. The agreement is quite impressive for 1b, as clearly seen in Fig. 9e.
Conclusions
The H2trz2An anilato derivative, bearing a triazole pendant arm at the 3,6 positions of the anilato moiety, has been used for the first time in combination with ErIII nodes to afford 1a and 1b 3D frameworks formed by 2D layers with (6,3) topology linked through the anilato ligands. A remarkable difference with respect to previous LnIII anilato-based compounds is that these 2D layers are interconnected through the N4 atoms of the two 1,2,4-triazole substituted pendant arms of trz2An, leading to formation of 3D frameworks. 1a and 1b constitute unique examples of multifunctional frameworks as they display NIR emission and field-induced slow relaxation of the magnetization. To the best of our knowledge, this is the first report of an anilato-based ErIII-MOF that shows SIM behaviour. Remarkably, the flexibility and porosity of the 1a MOF enable drastic structural changes after partial dehydration, under very mild conditions (vacuum or heating to 360 K), to a less porous 3D structure in 1a_des, different from 1b (vide supra). 1a_des shows an improvement of the magnetic blocking temperature and a modulation of the PL emission spectrum and lifetime, with respect to the hydrated compound, which are mainly related to the loss of one water coordination molecule. Full reversibility of the observed phase transition from 1a to 1a_des was demonstrated through several air/vacuum (rehydration/dehydration) cycles. The milder conditions required to achieve the switching from 1a to 1a_des and the fact that it affects both luminescent and SIM properties (multifunctionality) in a 3D extended architecture as the flexible anilato-based ErIII-MOF, herein reported, constitutes a significant advance with respect to LnIII-based discrete complexes (vide supra) showing an interesting solvato-switching of the SIM properties.24,25 Interestingly, the structure, as well as the luminescent and magnetic properties of 1a, are fully regenerated by simply exposing the crystals of 1a_des to air, which constitutes an additional advantage compared to some of the aforementioned molecular complexes.25Theoretical calculations indicate that the slight improvement of SIM properties is mainly due to the decrease of mixing with lower microstates, which reduces quantum tunneling of magnetization at low temperatures. The present work contributes to the ongoing research on flexible MOFs and sheds light on the important role of luminescence spectroscopy and magnetometry as probes for mechanism investigation of flexible LnIII-MOFs.
Author contributions
MLM and NM designed the MOF. MLM, ECa and MCL supervised the research project, with the contribution of ECo and FQ. NM synthesized and characterized the MOF under the supervision of MLM and MCL (structural and magnetic characterization) and with the help of VGL and MO. JJB performed the theoretical calculations and analyzed the magnetic data. FQ performed luminescent experiments and analyzed the data. MLM, MCL and FQ wrote the manuscript. All authors contributed to the final interpretation of the experimental results and critically revised the manuscript. All authors have read and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Fondazione di Sardegna – Convenzione triennale tra la Fondazione di Sardegna e gli Atenei Sardi, Regione Sardegna – L.R. 7/2007 Annualità 2018 – DGR 28/21 del 17.05.2015, through Projects F74I19000940007 and F74I19000920007, are acknowledged for the post-doctoral fellowship of MO. MUR (Ministry of Education, University, Research) and UNICA-UNISS Consortium PhD Course on Chemical Sciences and Technologies is also acknowledged for financing the PhD grant of NM. CESA (Centro d’Eccellenza per la Sostenibilità Ambientale, accordo di programma RAS-UNICA-IGEA-AUSI, project number E58C16000080003) is acknowledged for the PhD grant of MO. The CeSAR (Centro Servizi d'Ateneo per la Ricerca) core facility of the University of Cagliari is acknowledged for the use of the Ultrafast Optical Spectroscopy Laboratory. J. J. B. is thankful for support from the Plan Gen-T of Excellence of the Generalitat Valenciana (CDEIGENT/2019/022). Financial support from the EU (ERC Advanced Grant MOL-2D 788222), the Spanish MCIN (Grants PID2020-117264GB-I00 and PID2020-117152RB-I00 funded by MCIN/AEI/10.13039/501100011033 and Unidad de Excelencia María de Maeztu CEX2019-000919-M) and the Generalitat Valenciana (PROMETEO program) is acknowledged. We all thank A. Soriano-Portillo, J. M. Martínez-Agudo and G. Agustí for PXRD and magnetic measurements.Notes and references
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- H. C. J. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 43, 5415–5418 RSC.
- Z. Chang, D. H. Yang, J. Xu, T. L. Hu and X. H. Bu, Adv. Mater., 2015, 27, 5432–5441 CrossRef CAS PubMed.
- A. Ghoufi, K. Benhamed, L. Boukli-Hacene and G. Maurin, ACS Cent. Sci., 2017, 3, 394–398 CrossRef CAS PubMed.
- A. Ghoufi, A. Subercaze, Q. Ma, P. G. Yot, Y. Ke, I. Puente-Orench, T. Devic, V. Guillerm, C. Zhong, C. Serre, G. Férey and G. Maurin, J. Phys. Chem. C, 2012, 116, 13289–13295 CrossRef CAS.
- J. Y. Kim, L. Zhang, R. Balderas-Xicohténcatl, J. Park, M. Hirscher, H. R. Moon and H. Oh, J. Am. Chem. Soc., 2017, 139, 17743–17746 CrossRef CAS PubMed.
- F. X. Coudert, Chem. Mater., 2015, 27, 1905–1916 CrossRef CAS.
- S. M. Hyun, J. H. Lee, G. Y. Jung, Y. K. Kim, T. K. Kim, S. Jeoung, S. K. Kwak, D. Moon and H. R. Moon, Inorg. Chem., 2016, 55, 1920–1925 CrossRef CAS PubMed.
- S. V. Eliseeva and J. C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC.
- L. Sorace, C. Benelli and D. Gatteschi, Chem. Soc. Rev., 2011, 40, 3092–3104 RSC.
- L. H. G. Kalinke, D. Cangussu, M. Mon, R. Bruno, E. Tiburcio, F. Lloret, D. Armentano, E. Pardo and J. Ferrando-Soria, Inorg. Chem., 2019, 58, 14498–14506 CrossRef CAS PubMed.
- J. J. Baldoví, E. Coronado, A. Gaita-Ariño, C. Gamer, M. Giménez-Marqués and G. Mínguez Espallargas, Chem.–Eur. J., 2014, 20, 10695–10702 CrossRef PubMed.
- S. Ashoka Sahadevan, N. Monni, A. Abhervé, D. Marongiu, V. Sarritzu, N. Sestu, M. Saba, A. Mura, G. Bongiovanni, C. Cannas, F. Quochi, N. Avarvari and M. L. Mercuri, Chem. Mater., 2018, 30, 6575–6586 CrossRef CAS.
- S. Ashoka Sahadevan, N. Monni, M. Oggianu, A. Abhervé, D. Marongiu, M. Saba, A. Mura, G. Bongiovanni, V. Mameli, C. Cannas, N. Avarvari, F. Quochi and M. L. Mercuri, ACS Appl. Nano Mater., 2020, 3, 94–104 CrossRef CAS.
- S. Benmansour, A. Hernández-Paredes, M. Bayona-Andrés and C. J. Gómez-García, Molecules, 2021, 26, 1–21 CrossRef PubMed.
- S. Benmansour, A. Hernández-Paredes, A. Mondal, G. López Martínez, J. Canet-Ferrer, S. Konar and C. J. Gómez-García, Chem. Commun., 2020, 56, 9862–9865 RSC.
- K. Nakabayashi and S. Ohkoshi, Inorg. Chem., 2009, 48, 8647–8649 CrossRef CAS PubMed.
- B. F. Abrahams, J. Coleiro, K. Ha, B. F. Hoskins, S. D. Orchard and R. Robson, J. Chem. Soc., Dalton Trans., 2002, 2, 1586–1594 RSC.
- K. Bondaruk and C. Hua, Cryst. Growth Des., 2019, 19, 3338–3347 CrossRef CAS.
- A. Mondal, S. Roy and S. Konar, Chem.–Eur. J., 2020, 26, 8774–8783 CrossRef CAS PubMed.
- S. Benmansour and C. J. Gómez-García, Magnetochemistry, 2020, 6, 1–44 CrossRef.
- N. Monni, M. Sanna Angotzi, M. Oggianu, S. Ashoka Sahadevan and M. L. Mercuri, J. Mater. Chem. C, 2022, 10, 1548–1572 RSC.
- N. Monni, M. Oggianu, S. A. Sahadevan and M. L. Mercuri, Magnetochemistry, 2021, 7, 1–12 CrossRef.
- J. Flores Gonzalez, V. Montigaud, V. Dorcet, K. Bernot, B. Le Guennic, F. Pointillart and O. Cador, Chem.–Eur. J., 2021, 27, 10160–10168 CrossRef CAS PubMed.
- F. Pointillart, J. Flores Gonzalez, V. Montigaud, L. Tesi, V. Cherkasov, B. Le Guennic, O. Cador, L. Ouahab, R. Sessoli and V. Kuropatov, Inorg. Chem. Front., 2020, 7, 2322–2334 RSC.
- W. Gauβ, H. Heitzer and S. Petersen, Justus Liebigs Ann. Chem., 1973, 764, 131–144 CrossRef.
- C. J. Kingsbury, B. F. Abrahams and R. Robson, CCDC 1568063: Experimental Crystal Structure Determination, 2017.
- N. Monni, E. Andres-Garcia, K. Caamaño, V. García-López, J. M. Clemente Juan, M. Giménez-Marqués, M. Oggianu, E. Cadoni, G. Mínguez Espallargas, M. Clemente Leon, M. L. Mercuri and E. Coronado, J. Mater. Chem. A, 2021, 9, 25189–25195 RSC.
- M. Llunell, D. Casanova, J. Cirera, P. Alemany and S. Alvarez, SHAPE, version 2.1, University of Barcelona, Spain, 2013 Search PubMed.
- M. P. Van Koeverden, B. F. Abrahams, D. M. D'Alessandro, P. W. Doheny, C. Hua, T. A. Hudson, G. N. L. Jameson, K. S. Murray, W. Phonsri, R. Robson and A. L. Sutton, Chem. Mater., 2020, 32, 7551–7563 CrossRef CAS.
- S. Benmansour, I. Pérez-Herráez, C. Cerezo-Navarrete, G. López-Martínez, C. Martínez Hernández and C. J. Gómez-García, Dalton Trans., 2018, 47, 6729–6741 RSC.
- J. J. Baldoví, S. Cardona-Serra, J. M. Clemente-Juan, E. Coronado, A. Gaita-Ariño and A. Palii, J. Comput. Chem., 2013, 34, 1961–1967 CrossRef PubMed.
- J. J. Baldoví, J. J. Borrás-Almenar, J. M. Clemente-Juan, E. Coronado and A. Gaita-Ariño, Dalton Trans., 2012, 41, 13705–13710 RSC.
- K. Hymas and A. Soncini, Phys. Rev. B, 2021, 104, 205306 CrossRef CAS.
- L. Escalera-Moreno, J. J. Baldoví, A. Gaita-Ariño and E. Coronado, Chem. Sci., 2018, 9, 3265–3275 RSC.
- N. Monni, J. J. Baldoví, V. García-López, M. Oggianu, E. Cadoni, M. L. Mercuri, M. Clemente-León and E. Coronado, 2022, Manuscript in Preparation.
- W. R. Reed, M. A. Dunstan, R. W. Gable, W. Phonsri, K. S. Murray, R. A. Mole and C. Boskovic, Dalton Trans., 2019, 48, 15635–15645 RSC.
- P. Zhang, M. Perfetti, M. Kern, P. P. Hallmen, L. Ungur, S. Lenz, M. R. Ringenberg, W. Frey, H. Stoll, G. Rauhut and J. Van Slageren, Chem. Sci., 2018, 9, 1221–1230 RSC.
- R. Ishikawa, S. Michiwaki, T. Noda, K. Katoh, M. Yamashita and S. Kawata, Magnetochemistry, 2019, 5, 30 CrossRef CAS.
- F. Quochi, M. Saba, F. Artizzu, M. L. Mercuri, P. Deplano, A. Mura and G. Bongiovanni, J. Phys. Chem. Lett., 2010, 1, 2733–2737 CrossRef CAS.
- R. Marin, G. Brunet and M. Murugesu, Angew. Chem., Int. Ed., 2021, 60, 1728–1746 CrossRef CAS PubMed.
- F. Artizzu, L. Marchiò, M. L. Mercuri, L. Pilia, A. Serpe, F. Quochi, R. Orrù, F. Cordella, M. Saba, A. Mura, G. Bongiovanni and P. Deplano, Adv. Funct. Mater., 2007, 17, 2365–2376 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2130639–2130642. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc00769j |
| This journal is © The Royal Society of Chemistry 2022 |