
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Next steps for solvent-based CO2 capture; integration of capture, conversion, and mineralisation
David J.
Heldebrant
 *ab,
Jotheeswari
Kothandaraman
a,
Niall Mac
Dowell
c and
Lynn
Brickett
d
*ab,
Jotheeswari
Kothandaraman
a,
Niall Mac
Dowell
c and
Lynn
Brickett
d
aPacific Northwest National Laboratory, Richland, WA, USA. E-mail: david.heldebrant@pnnl.gov
bWashington State University, Pullman, WA, USA
cImperial College London, London, England
dUS Department of Energy, Office of Fossil Energy, USA
First published on 19th May 2022
Abstract
In this perspective, we detail how solvent-based carbon capture integrated with conversion can be an important element in a net-zero emission economy. Carbon capture and utilization (CCU) is a promising approach for at-scale production of green CO2-derived fuels, chemicals and materials. The challenge is that CO2-derived materials and products have yet to reach market competitiveness because costs are significantly higher than those from conventional means. We present here the key to making CO2-derived products more efficiently and cheaper, integration of solvent-based CO2 capture and conversion. We present the fundamentals and benefits of integration within a changing energy landscape (i.e., CO2 from point source emissions transitioning to CO2 from the atmosphere), and how integration could lead to lower costs and higher efficiency, but more importantly how CO2 altered in solution can offer new reactive pathways to produce products that cannot be made today. We discuss how solvents are the key to integration, and how solvents can adapt to differing needs for capture, conversion and mineralisation in the near, intermediate and long term. We close with a brief outlook of this emerging field of study, and identify critical needs to achieve success, including establishing a green-premium for fuels, chemicals, and materials produced in this manner.
Introduction
As the world continues to drive toward a net zero economy, fossil fuels will still be needed to supplement renewables during this transition to net-zero emissions. In this context, it is generally understood that CO2 capture and storage (CCS) technology has the potential to play a significant role in meeting climate targets.1,2 Presently, the biggest barrier to carbon capture technologies is the high total cost of capture, which comprises a combination of both energy demand and capital costs. In the past 20 years, there have been great gains in energy efficiency, though reductions in CAPEX remain challenging due to limited number of commercial systems in operation. To date, the total costs of capture are cheapest for concentrated gas streams such as coal-derived flue gas or cement kilns, spanning $47–80 (USD) per tonne CO2.3 Notably, there are escalating costs from capturing CO2 as the concentration becomes increasingly dilute from natural gas combined cycle (NGCC) flue gas ($80–100 (USD) per tonne CO2)4 and even higher costs and complexity for highly dilute streams, e.g. negative emission technologies (NETs) such as direct air capture (DAC) costs $150–1000 (USD) per tonne CO2.5,6Solvent-based processes are the most mature CCS technologies owing to their significant commercial deployment for the purification of natural gas.7 While not the lowest energy carbon capture and separation (CCS) approach, solvents can achieve 60% thermodynamic efficiency, while having the lowest total costs of capture ($45–47.1 (USD) per tonne CO2).8 Most importantly solvents are the only technologies that can currently be manufactured at the requisite scale9 for point-sources such as coal and natural gas powerplants, cement kilns and steel furnaces.
In the past decade, solvent-based processes have undergone numerous changes in formulation and process to increase efficiency while reducing costs for point-source emissions. The field has seen a shift from simple strippers to more efficient configurations such as, lean-vapor compression with absorber intercooling or two-stage flash regeneration as a means to recoup heat, whereas the latter also is much cheaper and also bypasses the energy-intensive first stage of CO2 compression.4,10–14 Similarly, solvent formulations have shifted from first-generation aqueous alkanolamines to more complex second-generation aqueous amines, and now into third-generation water-lean solvents that are faster at absorbing CO2 and are more energy efficient to regenerate.7,15 Ultimately, leading solvent technologies that project to be 19% cheaper than Shell's CANSOLV technology.16
While attractive from a cost, ease of manufacture, and timeliness perspective, solvent-based processes should not be considered a panacea for CCS. This is because solvents have been primarily designed to mitigate point-source emissions but not those of legacy emissions using Negative Emission Technologies (NETs). In the near term, it may make sense to focus CCU applications on fixed point sources, thus contributing to the near-term avoidance of emissions, and the development improved capture technologies. In the longer term, however, it will be important to obtain non-lithospheric carbon as an industrial feedstock, and in this context, the further development of direct air capture (DAC) technologies will be key. Solvents can, therefore, be considered the cornerstone of a foundation on which many carbon capture technologies will be built to achieve the global goal of deep decarbonisation. Thus, our focus should be primarily on deployment so we can initiate large-scale emission reductions, but also so subsequent technologies and later stage NET approaches can benefit from the CO2 transport and storage infrastructure that will be established in the process, as well as markets for CO2, and CO2-containing materials.
The driver to increased deployment of CCU today is either to introduce regulatory requirements or to provide industry an economic incentive to utilise captured CO2. Outside of a few markets of enhanced oil recovery (EOR) and niche CO2 use for agriculture, there are limited markets for CO2 at meaningful scales. Thus, one way to reverse this reality is to provide more economic incentives for CCU, which could come from one of or a combination of tax credits, such as a modification from the well-known 45Q credit, or policy drivers like buying “green” mandates. There are dozens of materials that can be made from CO2,17 and whilst their markets are small relative to the scales required for whole-economy net zero, there remains potential for a meaningful contribution.
Owing to their unique ability to both cost-effectively capture CO2 at industrial scale and act as a medium for chemical conversion, solvent-based processes are the prime medium for integrated capture and conversion. Starting with the 2019 paper from the National Academy of Sciences on transforming separation science,18 there have been reviews, workshops, and Faraday transactions that thought leaders have begun to set the stage for reactive separations related to CCU.2,19–21 We build on these initial efforts and thoughts, and offer a more detailed definition of integrated capture and conversion, and identifying what is needed to make it more tangible. In this contribution, we identify the needs and priorities for capture and conversion in solvent-based processes in the near, intermediate, and long-term focusing on how solvents can lay the foundation on which timely and potentially profitable deep decarbonization can be built.
Discussion
Nature's integrated capture and conversion
Over millennia, natural systems have had time to perfect integrated capture and conversion of gas molecules. There are a handful of natural examples of systems (e.g. hemoglobin, carboxylic anhydrase) that capture and chemically convert atmospheric gases. Here, for completeness, we briefly describe examples that mirror our carbon-based energy systems.We first highlight aerobic respiration, which involves O2 being absorbed, activated, and then transported to be used as a chemical oxidant. Respiration is analogous to the combustion of fossil fuels to release energy. O2 is absorbed in the lungs, being chemically coordinated to Fe2+ in heme groups found at the active site hemoglobin in red blood cells. This resulting iron superoxide (captured and activated O2) is circulated in the blood stream to muscle cells where O2 is consumed during chemical oxidation of fuel. This reaction provides energy for locomotion and heat. Unlike our combustion of fuels, in respiration, after O2 is consumed, the chemical process continues in reverse, removing 20–25% of the body's waste CO2via coordinated transport by the same heme group in hemoglobin. The waste CO2 is expunged by the lungs during exhalation, in a continuous process of inhalation and exhalation.
Plants capture, activate, and then convert CO2 into glucose in the Calvin cycle. CO2 is first absorbed in the chloroplasts, being first acted upon by the enzyme ribulose-1,5-bisphosphate carboxylase-oxygenase (RuBisCO). In RuBisCo, the active site includes the Lysine moiety which chemically fixates CO2 as a carbamate (captured CO2) that is stabilized by a Mg2+ in the active site. The captured/activated CO2 is then transcarboxylated to ribulose 1,5, biphosphate, making two molecules of glycerate-3-phosphate, which then are converted to glucose in later stages of the Calvin cycle.
Humans also perform capture and conversion of CO2 in metabolic pathways such as gluconeogenesis, fatty acid synthesis, and amino acid catabolism. In our bodies, CO2 is first hydrolyzed by the enzyme carbonic anhydrase, making a bicarbonate which is first transcarboxylated to the biotin co-factor with the aid of adenosine triphosphate by any of the four carboxylase enzymes. Once the CO2-biotin carboxylate adduct is formed, the activated CO2 is subsequently reacted with metabolites such as pyruvate to make fuels such as glucose and fatty acids. Conversely, extremophiles such as cyanobacteria achieve glycogenesis by means of a slightly different approach due to the absence of light to provide the energy source. In the deep ocean, bacteria capture CO2 and H2S from deep-sea vents, and produce glucose by using H2S as a chemical reductant to reduce CO2 captured as a carbamate.22,23 This chemotrophic capture and conversion of CO2 is driven by the deep-sea vents, because of the immense heat providing the driving force for the chemical reactions.
We point to these systems as examples, as there are already energy technologies that emulate these biological processes. Aerobic respiration can be considered an analogue for chemical looping combustion, and glucogenesis by organisms such as algae is the fundamental process behind algal biofuel production. As these types of technologies exist for performing chemical energy storage and release of chemical energy during combustion, we can see a possibility/reality where solvent-based carbon capture and conversion are integrated to emulate natural systems. From this concept, we can envision a modular multi-product chemical manufacturing facility (Fig. 1) similar to Shell's Pearl GTL plant,24 albeit making value-added products from CO2 instead of natural gas.
 | ||
| Fig. 1 Our vision of a 21st-century point source manufacturing center from CO2. | ||
Why integrate capture and conversion?
While many reactions of CO2 have been reported in the literature, there is an unwritten assumption that CO2 is free. In reality, CO2 typically has to be recovered from a range of sources of varying dilution, and each of the CO2 absorption, desorption, compression, and transportation unit operations come at a cost. Reactive separation of CO2via conversion in the capture solvent is a logical approach to reduce the cost and energy demands requisite for CO2 capture. Natural systems are integrated because it makes sense from an energy perspective, though there are other potential benefits of doing so. Integration also provides improved thermodynamic efficiency, the ability to alter or bypass limiting chemical equilibria, the use of new drivers to increase the chemical potential for separation and conversions (much like reactive distillations), and most importantly, more favorable economics due to reduced redundancy. We further expand on potential drivers, detailing how each can be a motivator for integration.Integration can be used to enhance CO2's reactivity for conversion
As we aim to process (hopefully) many millions of tonnes of CO2 a year, the energy demand and rates greatly influence the CAPEX and OPEX of any utilization approach, and any means to reduce either, will improve the prospects of commercial viability. To date, approaches to utilize CO2 have focused on optimizing and refining every variable available except the most obvious one, the CO2 itself.CO2 is a very stable molecule, thermodynamically and kinetically speaking. Revisiting Valence Shell Electron Pair Repulsion (VSEPR) theory, CO2 (gas) is sp hybridized, and strong overlap of the bonding orbitals limits reactivity. Reactions that involve the central carbon (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C*O) require a nucleophile to deposit paired electrons in a small and highly shielded sp antibonding orbital (Fig. 2, left). As a result, the activation energy (Eact) for most reactions of CO2 are high, and, consequently, reaction rates are slow. This high activation energy is why reductions of CO2 in its native state require catalysts that function at high temperatures (>300 °C) to achieve an appreciable rate.
C*O) require a nucleophile to deposit paired electrons in a small and highly shielded sp antibonding orbital (Fig. 2, left). As a result, the activation energy (Eact) for most reactions of CO2 are high, and, consequently, reaction rates are slow. This high activation energy is why reductions of CO2 in its native state require catalysts that function at high temperatures (>300 °C) to achieve an appreciable rate.
 | ||
| Fig. 2 On orbitals and nucleophile attack/availability. | ||
Conversely, chemically captured CO2 is trigonal planar sp2 hybridized anionic carboxylates. This hybridization is nearly universal in solution (e.g. as bicarbonate in sea water, or carbamates in aqueous amines) and in amine-functionalized solid sorbents that capture CO2 as carbamates. This molecular geometry is sterically and electronically more favorable for nucleophilic attack because the p* antibonding orbital is perpendicular to the plane of the molecule (Fig. 2, right), making it less shielded and more accessible. Further, when CO2 is reduced or attacked by a nucleophile, the resulting CO2-containing intermediate or species are commonly trigonal planar (sp2). This is in part why the majority of the natural systems, e.g., RuBisCO, described above capture, transport, or react CO2 in an activated sp2 state (e.g., biotin-carboxylates, bicarbonate).
Anionic carboxylates are negatively charged, but they can be reduced in catalytic processes. Anionic carboxylates readily coordinate to cationic metal complexes and heterogeneous catalysts. Once the carboxylates are coordinated to metals, they become charge-neutral, enabling reduction. It has recently been demonstrated that alkylcarbonates are more reactive toward ruthenium hydride complexes than gas-phase CO2 under comparable conditions via an inner-sphere reduction.25 It is likely that carbamates share a similar reactivity. We have also shown coordination of CO2-containing ions to heterogeneous catalyst interfaces at temperatures comparable to CO2 release from solvents (∼120 °C),26 suggesting durable heterogeneous catalysts could be employed for integrated capture and conversion approaches. For the recent reviews related to integrated capture and conversion, see ref. 27–29.
The capture solvent can aid conversion
A benefit of performing a conversion reaction inside a carbon capture solvent allows us to proceed at ambient pressures of CO2. In most reported catalytic hydrogenations or conversions, higher pressures are applied to provide a high concentration of CO2 in solution. For example, the mole fraction of physically dissolved CO2 in water and organics are ∼0.00044 and >0.01, respectively, at 40 °C under 1 atm CO2.30–35 In particular, CO2 pressures of 15–30 bar CO2 are common concentrations for conversions because CO2 is a non-polar molecule whereas the conventional solvents used are polar. Conversely, as the CO2 gas is already chemically reacted with the capture solvent, high CO2 pressure is not needed when the conversions are performed in a capture solvent. The CO2-loaded carbon capture solvents (after treating a high [CO2] flue gas) entail CO2-rich loadings of approximately 5 wt%.4,36 From a systems perspective, the ability to react at lower pressure negates the cost and duty of CO2 compressors to achieve sufficient concentrations of CO2 in solution.The rate of chemical reactions can be greatly influenced by solvent effects. Chemically selective solvent-based carbon capture is a reaction between a weak acid (CO2) and a strong base (amines). As like all acid–base reactions, proton transfer and charge solvation are the primary reaction steps, which are favored in polar protic or polar aprotic solvents such as water, propylene carbonate, glycols, sulfolane, and, more recently, ILs.37,38 Solvents in general, favor the reaction because they are able to provide charge solvation and facilitate proton transfers. Solvents are employed in many organic chemistry reactions and thermocatalytic conversions because they solvate catalysts in addition to stabilizing high-energy transition states (i.e., rehybridization of CO2), in turn lowering the activation energy of a reaction (Eact) few of kcal mol−1,39 and thereby allowing reactions to proceed more readily. As carbon capture solvents are polar protic, or polar aprotic media, these solvent effects provide a lever to control reactivity that remains unavailable to gas-phase catalytic processes.
Changing speciation allows for bypassing limiting chemical equilibria, enabling lower T and P reactions
The reactions of CO2 in gas phase are very different from the condensed-phase chemistry. For instance, in the conventional gas-phase CO2 hydrogenation to methanol reaction, the typical reaction intermediates are formate (HCOOad), acetal (OCH2Oad), and methoxy (OCH3ad),40,41 whereas in the condensed phase, inside a carbon capture solvent, the intermediates involved are carbamate (–NCOO−) or carbonate (–OCOO−), formate (HCOO−), and formamide (–NCHO)/formate ester (–OCHO).25,26,42–46 Depending on the catalyst, solvent, and reaction conditions (T, P, diluent, additives etc.), we can selectively form one of these value-added chemicals/intermediates—formate (or formic acid), methanol, and methane.The CO2 hydrogenation to formic acid is strongly endergonic 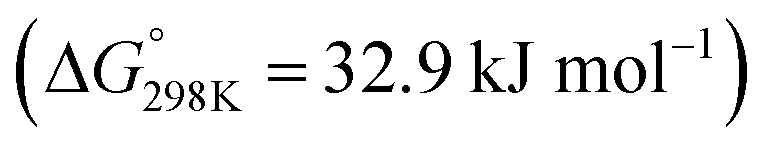 . However, in the presence of an ionic liquid or organic/inorganic bases, the reaction becomes exergonic, moving equilibrium toward formic acid and formate.39 A base is typically used to drive the formation of formate upon CO2 hydrogenation. Then, the formic acid is separated from the base by ion exchange or thermal cleavage. Coincidentally, the CO2 capture solvents typically contain amine units, which can act as a base to promote the formation of ammonium formate. The hydrogenation of captured CO2 to formate has been shown by us and others in both aqueous and water-lean solvents.25,42,45,47,48
. However, in the presence of an ionic liquid or organic/inorganic bases, the reaction becomes exergonic, moving equilibrium toward formic acid and formate.39 A base is typically used to drive the formation of formate upon CO2 hydrogenation. Then, the formic acid is separated from the base by ion exchange or thermal cleavage. Coincidentally, the CO2 capture solvents typically contain amine units, which can act as a base to promote the formation of ammonium formate. The hydrogenation of captured CO2 to formate has been shown by us and others in both aqueous and water-lean solvents.25,42,45,47,48
Low-temperature methanol synthesis from CO2 has been gaining lot of attention recently.26,27,43,49–52 The methanol formation from CO2/H2 is an exothermic process, and, based on the entropy, low temperature will give higher conversion to methanol, but due to slower reaction kinetics, the methanol synthesis is typically performed at high pressures and temperatures (>200 °C). The presence of alcohols and bases were shown to favor the formation of methanol at low temperatures (<170 °C) by reacting with formate to make formamides and formate esters to drive reaction in solution phase.
C1 products like methanol or methane made from the hydrogenation of CO2 generates quantitative amounts of water as a byproduct. The use of low-water or water-lean capture solvent is an attractive option compared to aqueous solvents because the excess water in aqueous solvent can reverse the reaction. The use of a water-lean capture solvent also offers higher concentrations of dissolved CO2 as compared to aqueous solvents, lowering the temperature and pressures required for synthesis, thereby significantly suppressing the reverse water gas shift (CO2 + H2 → CO + H2O).
Heat from conversion can be recovered to drive regeneration of the solvent or reused elsewhere
Solvent-based carbon capture is an exothermic reaction, driven by the large enthalpic driver between the heat of protonation from (most commonly, but not limited to) carbamic acid (made from amines and CO2) and the amine. Mathias has identified an optimal heat of solution for capture of CO2 from point sources to span between −60 to 85 kJ mol−1, where the thermodynamics are optimal for the separation from coal-derived flue gases.36,53,54 The regeneration of the solvent (release of CO2) is endothermic, requiring an equally strong if not stronger enthalpic driver to force the regeneration. Solvents have been regenerated by many approaches, including but not limited to electrochemical, pH, or thermal means, with the latter being the most common.7,36,55–58 In each case, reactivation of the solvent requires a substantive amount of energy (2.0–3.3 GJ per tonne CO2) to reactivate the sorbent and deliver a relatively pure CO2 stream.Conversely, most value-added products that could be made from CO2 are made from high-energy reagents like metal oxides, silicates, epoxides, or reductants such as e− or H2. The most common conversion of CO2 is reduction to chemicals or fuels such as formate (or formic acid), formamides, methanol, or methane, all of which are downhill energetically due to strong exotherm driven by the production of water as a byproduct in most of these cases. These most common products made from reduced CO2, entail reaction enthalpies of −84 kJ mol−1 (ammonium formate), 33 kJ mol−1 (dimethyl formamide), −49 kJ mol−1 (methanol), and −165 kJ mol−1 (methane). Similarly, mineralisation of CO2 is highly exothermic, with enthalpies for mineralisation of −118 kJ mol−1 and −179 kJ mol−1 for MgCO3 and CaCO3 respectively.
It is desirable to optimize thermodynamics of both (capture and conversion) processes to minimize energy losses. Integration of the capture and conversion allows this to happen because the scale of the endotherm in regeneration of the solvent is comparable to the exothermic conversion. Thus, from a systems perspective, depending on the availability and quality of the heat of the conversion reaction, we could and should aim to offset the regeneration of the solvent (Fig. 3) or repurpose the heat elsewhere in an integrated CO2 to chemicals plant. We concede that there is a large difference in scale between conversion and capture, in that there is not enough reagent to consume all captured CO2, and high-energy reagents like minerals and H2 require energy-intensive processes to produce. We posit that integration could be viable for slipstreams that can scaled to the availability of reagents and quality of heat to drive the solvent regeneration (or other processes) via chemical conversion.
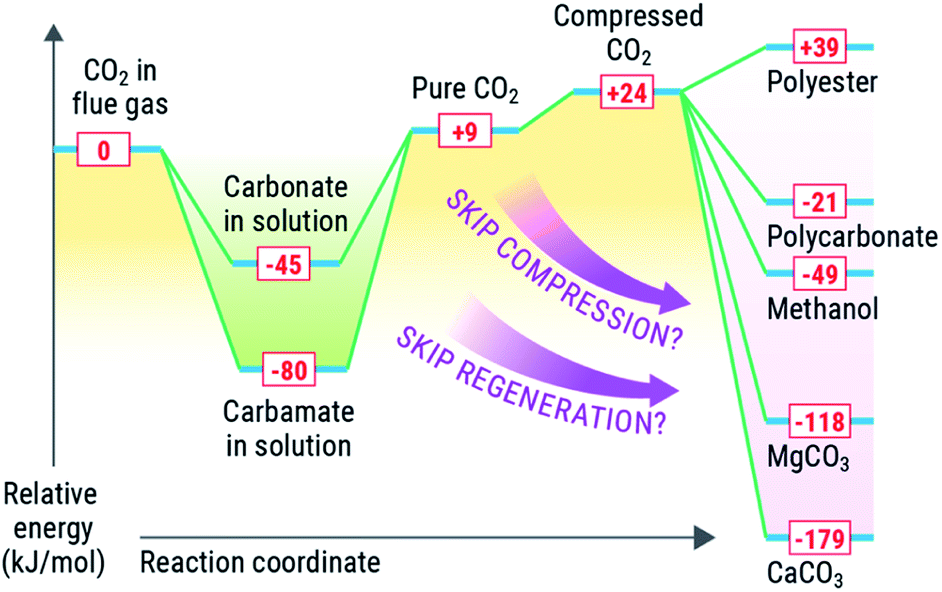 | ||
| Fig. 3 Conceptual energy comparison for capture and conversion of CO2. | ||
Removing units of operation reduces energy and capital costs
As mentioned previously, a capture process has an absorber, heat exchangers, pumps, and heaters, all of which will be in a conversion process as well. Also, from a capital perspective, utilizing systems and reagents for multiple steps removes the redundancy, enabling a cheaper integrated system as compared to disparate systems. Similarly, if a smaller, modular capture and conversion unit were made, the system could be configured without a compressor, potentially saving money and energy in the process.Also, from a systems perspective, converting CO2 into condensed-phase products saves energy because this approach can bypasses the need for the high-compression energy to compress CO2 to a supercritical state for transportation in pipelines. In solvent-based processes, CO2 is released from a solvent at ∼1.8 bar at 120 °C, though more recent approaches by Rochelle have shown sizable energy savings if CO2 is thermally compressed and released at 6 bar, which allows solvents like piperazine (PZ) to bypass the first (and most energy-intensive) stage of compression.13,59 The compression energy of CO2 according to the US DOE Case 12B baseline is about 32 MW,4 not counting the energies associated with transportation, which of course are variable. If the slipstream conversion can make CO2 into a product that exists in the condensed phase, this energy demand could be eliminated for compressor work, potentially reducing its size and cost.
Selling CO2-derived products can pay for the initial separation
CCS has been deployed in limited cases such as (Port Charles, Aquistore BD3) because there are limited market incentives to cover the costs of capture ≥$47.1 (USD) per tonne CO2 and $20 (USD) for transportation and storage. Selling CO2-derived products could off-set some costs associated with carbon capture, thereby providing enough incentive to encourage commercialization.Presently, there are a few target large-volume economically profitable and scalable chemicals that could achieve approximately 0.3 to 0.6 Gt CO2 per year reductions by the year 2050, with breakeven costs of ∼$80 to $320 (USD) per tonne of CO2.60 Urea (140 Mt CO2 per year breakeven at $100 (USD) per tonne), and polycarbonate polyols (10–50 Mt per year breakeven at $2600 (USD) per year) could be initial targets. It is noteworthy that, under the revisions proposed to the 45Q tax credit, incentives to capture and geologically sequester CO2 were proposed in the range of $85–200 (USD) per tonne – not dissimilar to the range required to make some CO2 capture and conversion projects economically viable. We further observe that these and other economic assessments assume decoupled capture and conversion, with the conversion proceeding with on a nearly pure CO2 stream. Integrated capture and conversion systems could reduce energy demands, and selling prices, potentially lowering breakeven points for urea, polyols, and potentially fuels. Further, breakeven points could become lower for other chemicals that are not presently profitable or scalable, giving us a larger market for CO2-containing products and thus a larger potential to avoid greenhouse gas emissions. It will, however, be important to recognise that the fate of this CO2 will be its emission to atmosphere, and in a net zero paradigm, these emissions must be accounted for and compensated by the permanent removal of carbon dioxide removal from the atmosphere, such as bioenergy with CCS (BECCS), DAC, enhanced weathering (EW) or another equivalent pathway. Thus, in the longer term, CO2 used in the production of fuels, and platform chemicals will need to come from the atmosphere, or other equivalently sustainable source.
Bringing it all together, an integrated capture and conversion example
Combining all of the advantages described earlier, we recently showed economic and energetic benefits of an integrated CO2 capture and conversion process to make CO2-neutral synthetic natural gas. In this process, the CO2 is first captured using a single-component water-lean solvent, 2-EEMPA (total costs of capture at $47.1 (USD) per tonne CO2).8,61 The CO2-rich solvent is mixed with H2 and hydrogenated in the presence of a commercial Ru/Al2O3 catalyst at 120 °C which is ∼3 °C higher than the solvent's reported regeneration temperature. The energies associated with CO2 compression are lessened as the reaction is performed in the condensed phase, and similarly the exothermic hydrogenation is used to partially offset the solvent regeneration enthalpy. In this process >90% of the CO2 is hydrogenated, producing a mixture of hydrocarbons, mostly methane at temperatures which are less than half of conventional gas-phase Sabatier reactions. The technoeconomic assessment (TEA) of the integrated process from CO2 captured from a coal-fired powerplant reported a reduction in the capital cost and minimum synthetic natural gas selling price by 32% and 12%, respectively.61 Further, the integration of the two processes enabled an increase in thermal efficiency by 5% as compared to separate capture and conversion. The improvement in thermal efficiency, reduction in capital costs and product selling price (compared to conventional non-integrated process) all point to integration as a key driver that could provide comparable benefits to other CCU systems.Where should integrated solvent-based CCUS focus?
With the case laid out for integrated capture and conversion, we first note that there are three time periods of impact, each with differing needs. Here we highlight areas of need with respect to chemistry, catalysts, processes, and markets for each of these time periods.In the near term, finding a way to further incentivize the separation is critical
Improvements in CO2 capture technologies in the near term (<10 years) are needed to enable the energy sector to deploy carbon capture on point-source generators (power plants). Thus, near-term efforts should focus on integrated conversion of CO2 into a range of value-added products, that can further develop CCU at meaningful scales and establish a marketplace for CO2-containing materials.Here research approaches should assess the viability of integration of capture and conversion of CO2 into economical and scalable chemicals like urea and polyols, and carbon-neutral energy-carriers such as methanol, methane, formic acid and dimethylether, similarly to what has been shown for methane.61 Combined theoretical and experimental R&D efforts should assess coupled chemical processes as a means to conjoin two chemical processes thereby bypassing the energies associated with regeneration of the capture medium and compression of CO2 (Fig. 1). These integrated reactive separation approaches allow us to design new condensed-phase catalytic systems that bypass limiting chemical equilibria of conventional high temperature gas-phase reductions of CO2 into energy-carriers.
True integration of capture and conversion requires a solvent that can perform both capture and conversion. There are many notable groups doing work on thermocatalytic conversions of CO2 providing insight on the basic reactivity, though we would define these “amine-promoted” conversions of CO2 as they use additives that are not carbon capture solvents.26,43–45,47,62–64 All of these approaches utilize gas-phase catalysis, or the use of chemicals or promoters that have not yet been validated as viable solvents for CO2 capture. Polyamines, volatile secondary or tertiary amines or solvents and promoters like tetrahydrofuran or ethanol are either too viscous, too energy inefficient, or too volatile to be used in capture technologies.
The first criteria for a viable capture and conversion solvent should be the capability of absorbing a sufficient amount of CO2 from a stream such as coal-derived flue gas or a cement kiln.4 The maximum uptake under equilibrium that any solvent could achieve is the equilibrium partial pressure of CO2 (P*) over the liquid at a given temperature. As post-combustion processes absorb at approximately 40 °C, they are configured to achieve >90% capture of 1.4 kPa CO2, the P* needs to be ∼1.4 kPa to perform the separation. Mathias's work provides critical information on the thermodynamics of capture (Fig. 4).53 He identifies that the minimum enthalpy of solution that could perform this separation is ∼−60 kJ mol−1 with a favored “Goldie Locks” range of −65 to 85 kJ mol−1. This range clearly shows why viable post-combustion solvents are chemical sorbents because physical solvents such as Rectisol and Selexol are too weak to capture 90% CO2 at 40 °C. We would also like to point out that if a solvent cannot work for concentrated streams like coal or natural gas exhaust, conditions in DAC are significantly more challenging because the P* of CO2 in air is a meager 0.04 kPa, meaning solvents need to be even stronger or have other drivers (e.g. precipitation) to capture CO2 any meaningful amount of CO2. Thus, one area of need is for more data on the P* of common solvents or promoters used in CO2 conversion to identify which, if any, of these co-solvents or promoters are capable of the initial separation.
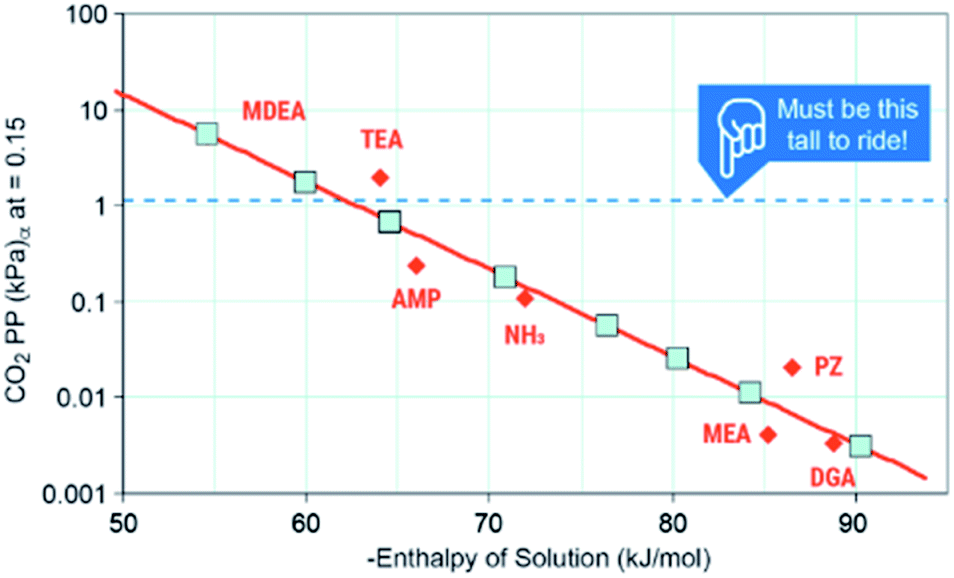 | ||
| Fig. 4 Solvent ranges of viability. Image adapted with data from Mathias, Int. J. Greenh. Gas Control, 2013, 19, 262–270.53 | ||
The second criteria is that the solvent or co-solvent cannot be volatile or viscous. As we have detailed before, solvents are designed to minimize evaporative losses in the absorption side of the process.36 Coal-derived power plants and other exhaust streams flow millions of pounds of gas an hour over a liquid in the absorber, meaning any volatile solvent such as triethyl amine or ethanol or co-solvent like THF will evaporate in the absorber column. Second, the rheological properties of the solvent need to be considered, notably that viscous ionic liquids or polymeric amines are prone to become quite viscous after CO2 complexation, which will cause greatly reduced rates of mass transfer, increasing the size and cost of the absorption unit.
Last, integrating capture and conversion requires a minimum level of TEA against a plausible reference case, such as those set by the U.S. Department of Energy's cost and performance baseline or comparable targets. Industrial benchmarks like 5 M monoethanolamine (MEA) or Shell's Cansolv have reboiler duties of 3.3 and 2.4–2.5 GJ per tonne CO2 and projected costs below $66 and $50 (USD) per tonne respectively.4 If a solvent used for capture does not meet or exceed these benchmarks, then they should not be considered targets in an integrated approach. We should instead focus on chemical conversions in validated post-combustion solvents.
There are only a few integrated capture and conversion approaches using what we define as “viable” post-combustion carbon capture solvents. Thermocatalytically, we have shown that CO2 could be made into methane in a single-component water-lean solvent 2-EEMPA.61 Similarly, Leitner and Francio have shown hydrogenation of CO2 hydrogenation using a 20 wt% MEA solution and homogenous catalyst in biphasic systems.65 Conversely, Sargent has shown an elegant electrocatalytic approach to convert CO2 into syngas in 5 M MEA, a process that was a finalist for the carbon XPRIZE.66 We have also shown the ability to coproduce two value-added chemicals—propylene glycol and methanol—at the same time with no waste and 100% atom efficiency, helping us utilize all the CO2 and avoid producing waste.50
In the intermediate term we need to expand solvents to capture CO2 from a greater number of point sources and reconstitute CO2 into materials or products that are CO2-negative
CCUS in the intermediate term (10–20 years), we assume continued growth of renewable energy to meet global energy needs, though there will still be some enduring point sources such as coal-, natural gas-, and biomass-fired power plants, as well as cement kilns, and steel plants that will continue to emit concentrated CO2 streams. Here, solvent-based capture will first need to be adapted for treatment of these remaining concentrated exhaust sources.Similar to the near term, utilization will require continued monetization of CO2, which is critical to achieve widespread deployment of CCU technology. Here, it makes little sense to continue to make C1 products identified in the near term, as those markets should already be established, and those products are CO2-neutral at best. To achieve tangible NETs, we will need new integrated CCU technologies that can produce a multitude of large-volume CO2-containing chemicals and materials that do not emit CO2 (e.g., polymers, composites). When we define NET's, we abide by the guidelines set forth by Ramirez.67 These materials represent sizable markets and potential CO2 sinks, though the valorization can be further driven by tax incentives. One example, the revised 45Q tax credit in the US, which provides $85 (USD) per tonne if the CO2 is “permanently sequestered”, noting that further clarification on the definition of “permanent” in this context will be required.
CO 2 utilization must follow the principles of green chemistry and be atom and energy efficient.2,68,69 This means, that our products need to contain the entire CO2 molecule, not fragments or materials that contain 1–3 atoms of the CO2. Similarly, we should avoid products and reactions that require high-energy C–O bond breakage because that adds even more energy demands to CCUS approaches.
Presently, there are a handful of chemicals that could utilize CO2 in its entirety. There have been many reviews that cover the size and scale of available markets though there is a disconnect in the commercial viability of the products we can presently make that consume the entire CO2 molecule. Reactions that utilize the entire CO2 molecule include carboxylation, cyclization, and polymerization with epoxides. CO2 polymerizations to make poly and cyclic carbonates, and reductions into fuels involve electrophilic attack of the central carbon in CO2 by strong nucleophiles. There are several approaches to make cyclic and polymeric carbonates from gaseous CO2, though there are only a few examples of integrated capture and conversion to materials.70 We laud the pioneering groups (Inoue, Darrensbourgh, Coates, and others)71,72 who are leading the field, but the desirable polycarbonates cannot yet be made with the requisite polydispersity index and molecular weight at a low enough cost. Further, polycarbonates made from CO2 have inadequate mechanical strength and low glass transition temperatures and are prone to thermal degradation and hydrolysis (releasing CO2 after a few years), thus limiting the application scope.
We suggest the focus in the long term should be attempting to make chemically durable, large-volume commodity materials that contain CO2 equivalents in their linkage that cannot yet be made from CO2. Viable targets include but are not limited to polyurethanes (NC(O)O) and polyesters (OC(O)O). Polyurethanes are valuable materials in adhesives, coatings, and foam insulation, with millions of tons a year produced and market sizes in the billions of USD.73 Similarly, polyesters are used ubiquitously as bottles for the beverage industry, and fabrics for the textiles industry with millions of tons a year and market sizes billions of dollars per year.74,75 Industrially, polyurethanes are made by polyaddition between different diisocyanates and diols (or polyols). The polyols used for the polyurethane synthesis can be polyether, polyester, polycarbonate, acrylic polyols, or polybutadiene polyols.76 The ability to use a wide range of polyols, isocyanates, and additives for polyurethane synthesis to achieve different properties makes them suitable for many applications. The polyether polycarbonate synthesized from epoxide and CO2 is used to make polyurethane flexible foams.77,78 It should be noted that these flexible foams containing up to 20% CO2 are already commercialized by the German polymer manufacturer Covestro and used in mattresses and upholstery furniture.79
On the other hand, isocyanates, the other raw material used for polyurethane synthesis, are synthesized by treating the corresponding amines with phosgene. Using CO2 as a C1 source for isocyanate synthesis instead of phosgene (which is highly toxic and prepared from CO and Cl2), is attractive from an economical and sustainability standpoint, but are still in exploratory stages.80 Alternatively, an isocyanate-free route to directly incorporate CO2 into polyurethane linkage in fewer steps is also in its infancy.81
Conversely, polyesters are conventionally made by a polycondensation reaction between dicarboxylic acids and diols or by ring opening polymerization (ROP) of lactones. Direct use of CO2 as a comonomer for copolymerization with diynes and terpolymerization with diynes and dihalides has also been explored in addition to indirect approaches where the CO2-derived polymerizable building blocks (mostly lactones) are used to produce polyesters. The direct polymerization of ethylene (or olefins) and CO2 is the straightforward approach to produce polyester, but it remains elusive mainly because of the high activation energy for the CO2 insertion into the growing polymer, the facile homopolymerization of ethylene competes with the alternating CO2/ethylene copolymerization.82–85
In the long term, the question is, can the captured CO2 inside the solvent reduce the free-energy barriers for desired reactions of CO2? Reducing our dependence on fossil resources will require an atom-economical, energy-efficient, cost-effective, industrially scalable, and environmentally friendly approach to utilize CO2 as a monomer in polyurethane and polyester synthesis. It should be noted that catalysts will also play a crucial role to achieve these transformations.
In addition, achieving near-term needs, methanol can be a CO2-sourced building block to produce various polymers. Conversion of methanol to olefins and subsequent polymerization or epoxidation can provide polyethylene, polypropylene, polyesters, polycarbonates, and others. This will allow us to incorporate most of the CO2 into polymers and to rely less on the fossil resources for hydrocarbons.
Our inability to make polyesters and polyurethanes is in part due to our antediluvian approach of converting CO2 either via addition and elimination reactions that rely on nucleophilic attack of the weakly electrophilic central carbon, or electrophilic attack on the weakly nucleophilic oxygens. Revisiting the orbital hybridization discussion above, if we are attempting to polymerize or co-polymerize CO2 in its linear SP hybridized state, we are limited in the types of reagents or catalysts that we can utilize. To date, the field has seen slow and steady improvements mostly driven by Edisonian research approaches. To date, the field has focused on altering catalysts, reagents, chelants, solvents, temperature, and pressure, but there is only so much that we can bend, distort, and chelate, with our current reagent pool. The lone variable left to be sufficiently altered is the CO2 itself. Captured CO2 are almost universally anionic sp2 hybridized carboxylates. This hybridization offers more electrophilic and nucleophilic active sites than sp hybridized CO2, so we question why we as a community should not assess reactivities of these different chemical species and bonds? For this reason, we should avoid being biased as we strive to develop CO2-containing materials. We should instead focus our efforts on the harder challenges of designing new reactivities that deviate from rudimentary approaches that rely on electrophilic and nucleophilic attack.
Similarly, catalysts will be an important element of this mid-term strategy, and they will need to be reinvented to operate in the condensed phase acting on captured CO2 in solution. Presently, the majority of catalytic approaches to convert CO2 operate on the assumption of outer-sphere reactions (reduction or transcarboxylation) where the CO2 does not directly (or is assumed not to) coordinate to the active metal. This greatly limits us as we strive to design new and more efficient catalysts. CO2 captured in a solvent will be in the form of anionic carboxylates which will directly coordinate to cationic metal sites, so our focus should be on designing catalysts or processes that operate via inner-sphere mechanisms in the condensed phase. Performing reactions in the capture solvent could be done at lower temperatures and pressures while also achieving the economic and energetic reasons identified above. Thus, our thinking on catalyst design and operation needs to align with (or unlock) the new reactive approaches for CO2 described earlier. Also note, the catalyst needs to be tolerant of the SOx and NOx in the flue gas.
Depending on the product of interest, the choice of the nature of the catalyst i.e. heterogenous or homogenous, used will also be critical, mostly from the separation standpoint. In the case of solid products such as polymers, homogenous catalysts will facilitate easy separation of the solid products from the catalyst and capture solvent. On the other hand, in the case of volatile liquid products such as methanol, the use the heterogenous catalyst will ease the separation of the catalyst from the liquid products and capture solvent. Then, the liquids products can be separated from the capture solvent via distillation. Similarly, in the case of gaseous products such as methane, the use of heterogenous catalyst will still work best for the separation of the product and capture solvent.
In the longer term, solvent-based technologies will need to adapt to permanent NETs as the world prioritizes deep decarbonization
Long term (>20 years), large-scale carbon capture and utilization will have been de-risked, and commercialized, with the global markets for CO2 and its products will have been established.There are many NETs identified as a part of a multipronged approach, including but not limited to enhanced weathering, seawater capture, afforestation, reforestation, and DAC, with the latter being the current trend in the CCUS community. The vision for DAC is the fabrication and deployment of millions of “artificial trees” powered by renewables to capture CO2 from air, coupled with CO2 utilization, though preferably sequestration. The inherent volatility of solvents and high energy demand for thermal-swing regeneration make solvents less attractive for integrated CO2 capture and conversion from dilute streams as in DAC. Similarly, afforestation and reforestation approaches are not amenable to integration with solvent-based capture processes. Solvents are unlikely to provide value to sub-surface sequestration as this step involves pumping almost pure CO2 into reservoirs.
Solvents could, however, provide the means to expedite the glacial rate of mineral carbonation. Mineral carbonation, i.e., mineralisation of gaseous CO2 occurs with alkaline earth metals (e.g., Ca, Mg), that are present in naturally occurring silicate and oxide minerals. There is a plethora of data on the viability, cost, and economics of the millions of metric tonnes of global mine tailings that could mineralize gigatons of CO2.86–91 Mineralisation is a spontaneous process that is downhill energetically,92 though mineralisation is not a panacea. While favorable enthalpically, the high kinetic barriers of these mineralisation reactions limit the rate of carbonation to a glacial pace. This is in part due to the mineralisation being impeded by the formation of passivating surface layers of carbonate.90
Recent efforts have focused on means to promote enhanced weathering (mineralisation), by mechanical, thermal, and chemical means. Mineralisation reactions are greatly influenced by surface area of the mineral that is reacting with the gaseous CO2. Currently, the easiest (and most inefficient) way to increase the rate of mineralisation is mechanical grinding of the rock to increase its available surface area. Second, the rate of mineralisation can be enhanced with elevated temperature and pressure, a process which also requires additional energy input. Third, mineralisation is best performed in aqueous solution, with recent sequestration studies showing that mineralisation is greatly enhanced by carbonating hot water.93 Mineralisation can be further expedited by the addition of chemical additives such as acids and bases to digest the passivating carbonate interface.
Solvent-enhanced weathering is a textbook example of an integrated capture and conversion approach. The primary benefit of integration is the exploitation of the strong exotherm of mineralisation (−118 kJ mol−1 to −179 kJ mol−1 for MgCO3 and CaCO3 respectively) to provide the regeneration enthalpy of the solvent (<85 kJ mol−1).92,94 Additionally, solvents can serve as a transport medium, providing a highly concentrated CO2 source to promote mineralisation. Solvents also consist of water and bases that are known to enhance the rates of mineralisation by digesting the passivating carbonate interfaces.
While this may sound like fiction, this concept has already been demonstrated at the laboratory scale. We highlight the work of the groups of Gadikota, Liu, Park, and Bourgeois, showing the viability and improved performance of integration of solvent-based capture and mineralisation approaches (Fig. 5).94–98 Gadikota recently showed the effectiveness of MEA, sodium glycinate (NaGly), 2-amino-2-methylpropanol (AMP), and water-lean solvents such as 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), on the carbon mineralisation of CaO, CaSiO3, and MgO.99 Liu demonstrated the viability of capture using CO2-rich solutions of MEA, PZ, diethanolamine (DEA), AMP, N-methyldiethanolamine (MDEA) to perform subsequent mineralisation and precipitation of calcium carbonate as a means to regenerate the solvents.96 Bourgeois and Leclaire expanded integrated solvent-enhanced mineralisation to magnesium silicates, demonstrating enhanced rate and yield of mineral carbonation of MgSiO4 using aqueous solutions with polyamines.97 These works are of note because they further demonstrate that solvents can enhance the rate and degree of sequestration by mineralisation with silicates, and additional economic incentives by selling MgCO3, SiO aggregates, and valuable ores recovered from these processes. Integrated approaches like these demonstrate the feasibility and sizable befits of integrated capture and mineralisation.
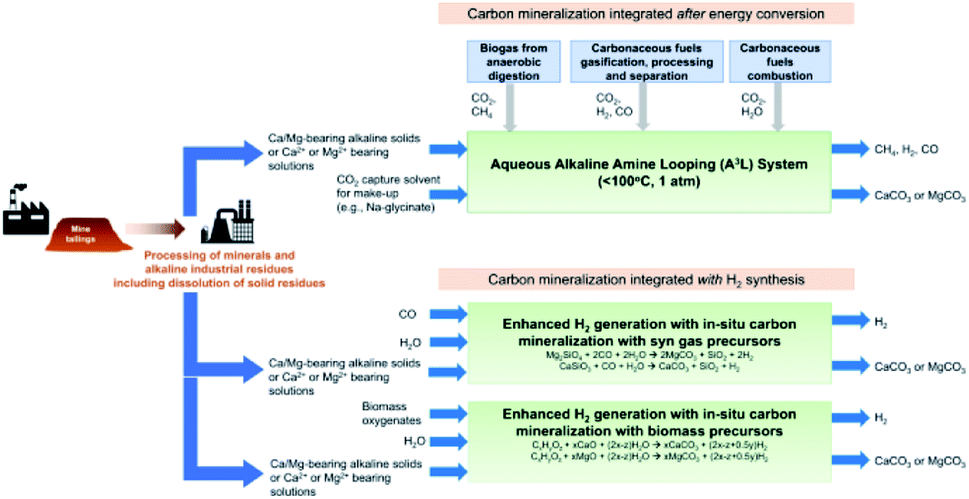 | ||
| Fig. 5 Conceptual solvent-based capture and concurrent mineralisation. Image from Gadikota et al., 2021.100 | ||
With benefits come challenges. Integrated solvent-based capture and utilization will require significant R&D efforts to ensure long-term viability of this type of approach. The first challenge will be ensuring chemical tolerance of the solvents to these substrates and products. A second challenge will be sufficient and economical recovery of solvent from particles to ensure the costs of solvent loss are negligible. Another challenge will be scaling and matching the rates and scales of capture and mineralisation streams to ensure one process does not dwarf the other. It should be noted that these processes will entail significant logistical hurdles to clear in order to deliver millions of tons substrates (likely by rail), and where to ship and store the carbonaceous products.
The answer to this question may come from the economic drivers for mineralisation. The present costs of transport and sequestration of CO2 are approximately $20 (USD) per tonne CO2, in addition to a cost of capture target of $30 (USD) per tonne CO2, which at present is economically unfeasible to make a profit without incentives or a market for mineralisation. Sequestration in the US could provide a revised $85 (USD) per tonne CO2 45Q, mineralisation could be profitable at a reasonable $25 (USD) per tonne. As stated above, these margins are yet enough to entice broad commercialization, but could be made to be if other markets and incentives can be made available via an integrated capture and mineralisation approach. Bourgeois makes a compelling case that precipitated carbonates and silicates can be recovered and sold as binding agents, mineral aggregates, and concrete for commercial construction applications providing a further economic incentive for capture and permanent sequestration.101 If materials like this could be sold as concrete constitutes, there could be a sizable reduction in the carbon footprint of the construction industry, which Liu estimates to be more than 4 billion tonnes on CO2.96
Needs and outlook
Our vision is to use solvent-based processes to convert point-source emission sites to factories that manufacture a myriad of CO2-containing products adaptable to market needs. We are at the turning point where we can continue to use 20th century monolithic capture and conversion infrastructure or we can begin the transition to a new 21st century paradigm of integrated solvent-based carbon capture and conversion technologies (Fig. 1).To make this vision a reality, there are many areas of need. First, more studies of known reactivity of CO2 in viable carbon capture solvents is needed to identify viable solvents, catalysts, reagents and products that could impact the three time periods identified above, including studies of solvent durability and lifetime for utilization reactions. Second, we have to expand the product base, which can only occur from the design and realization of new reactivities of captured CO2. Also, one can envision further integration approaches where the processes are in a single unit, where utilization can be used to influence VLE in the absorber, and thus further drive the absorption. Third, processes should assess modular microchannel reactors that can run in parallel for varied products. Fourth, recent advances in additive manufacturing opens new doors for advanced heat integration enabling more efficient integrated systems. Lastly, comprehensive TEA and life cycle analysis (LCA) studies will be needed to qualify and quantify the full range of benefits of integration in order to produce policy-relevant evidence to co-create the legislative landscape to enable the at-scale deployment of these technologies.
Conclusions
In this contribution, we have made a case for how solvent-based carbon capture can be integrated with conversion as a key element of the transition to a net-zero emission economy. Carbon dioxide utilization has always been a lofty target for at-scale production of green CO2-derived fuels, chemicals and materials, as these materials would command enough revenue to pay for capture, with further incentives from tax credits. The key to making CO2-derived materials and products market competitive is by integration of solvent-based CO2 capture and conversion. We have envisioned how solvents could be adapted for the differing needs for capture, conversion, and mineralisation in the near, intermediate, and long term. We have identified white space and research needs in this emerging field to achieve success. Ultimately, integration of capture and conversion represents promising approach to achieve global net-zero emissions.Author contributions
Dr Heldebrant provided conceptualization, supervision, writing. Dr Kothandaraman, Professor Mac Dowell and Ms Brickett provided investigation and writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division.Notes and references
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- J. Leclaire and D. J. Heldebrant, Green Chem., 2018, 20, 5058–5081 RSC.
- https://www.iea.org/commentaries/is-carbon-capture-too-expensive .
- R. E. James, D. Kearins, M. Turner, M. Woods, N. Kuehn and A. Zoelle, Cost and Performance Baseline for Fossil Energy Plants Volume 1: Bituminous Coal and Natural Gas to Electricity, NETL-PUB-22638, 2019 Search PubMed.
- https://www.iea.org/reports/ccus-in-clean-energy-transitions .
- D. W. Keith, G. Holmes, D. St. Angelo and K. Heidel, Joule, 2018, 2, 1573–1594 CrossRef CAS.
- D. J. Heldebrant and J. Kothandaraman, in Carbon capture and storage,, ed. Mai Bui and N. M. Dowell, The Royal Society of Chemistry, 2020, pp. 36–68, DOI: 10.1039/9781788012744-00036.
- R. F. Zheng, D. Barpaga, P. M. Mathias, D. Malhotra, P. K. Koech, Y. Jiang, M. Bhakta, M. Lail, A. V. Rayer, G. A. Whyatt, C. J. Freeman, A. J. Zwoster, K. K. Weitz and D. J. Heldebrant, Energy Environ. Sci., 2020, 13, 4106–4113 RSC.
- J. E. Bara, Greenhouse Gases: Sci. Technol., 2012, 2, 162–171 CrossRef CAS.
- J. R. Scherffius, S. Reddy, J. P. Klumpyan and A. Armpriester, Energy Proc., 2013, 37, 6553–6561 CrossRef CAS.
- Z. Amrollahi, I. S. Ertesvag and O. Bolland, Int. J. Greenh. Gas Control, 2011, 5, 1393–1405 CrossRef CAS.
- Y. J. Lin and G. T. Rochelle, 12th International Conference on Greenhouse Gas Control Technologies, Ghgt-12, 2014, vol. 63, pp. 1504–1513 Search PubMed.
- Y. J. Lin, E. Chen and G. T. Rochelle, Faraday Discuss., 2016, 192, 37–58 RSC.
- M. S. Walters, R. H. Dunia, T. F. Edgar and G. T. Rochelle, Energy Proc., 2013, 37, 2133–2144 CrossRef CAS.
- D. J. Heldebrant, P. K. Koech, V. A. Glezakou, R. Rousseau, D. Malhotra and D. C. Cantu, Chem. Rev., 2017, 117, 9594–9624 CrossRef CAS PubMed.
- Y. Jiang, P. M. Mathias, C. J. Freeman, J. A. Swisher, R. F. Zheng, G. A. Whyatt and D. J. Heldebrant, Int. J. Greenh. Gas Control, 2021, 106 Search PubMed.
- C. Maeda, Y. Miyazaki and T. Ema, Catal. Sci. Technol., 2014, 4, 1482–1497 RSC.
- National Academies of Sciences, Engineering, and Medicine, A Research Agenda for Transforming Separation Science, The National Academies Press, Washington, DC, 2019 Search PubMed.
- W. Leitner and M. Schmitz, Faraday Discuss., 2021, 230, 413–426 RSC.
- NREL, Reactive CO₂ Capture: Process Integration for the New Carbon Economy Workshop Proceedings, 2020, https://www.nrel.gov/bioenergy/workshop-reactive-co2-capture-2020-proceedings.html Search PubMed.
- National Academies of Sciences, Engineering, and Medicine, Negative Emissions Technologies and Reliable Sequestration: A Research Agenda, The National Academies Press, Washington, DC, 2019 Search PubMed.
- H. Felbeck and G. N. Somero, Trends Biochem. Sci., 1982, 7, 201–204 CrossRef CAS.
- C. M. Cavanaugh, S. L. Gardiner, M. L. Jones, H. W. Jannasch and J. B. Waterbury, Science, 1981, 213, 340–342 CrossRef CAS PubMed.
- https://www.shell.com/about-us/major-projects/pearl-gtlhttps://www.shell.com/about-us/major-projects/pearl-gtl.html.
- D. B. Lao, B. R. Galan, J. C. Linehan and D. J. Heldebrant, Green Chem., 2016, 18, 4871–4874 RSC.
- J. Kothandaraman, R. A. Dagle, V. L. Dagle, S. D. Davidson, E. D. Walter, S. D. Burton, D. W. Hoyt and D. J. Heldebrant, Catal. Sci. Technol., 2018, 8, 5098–5103 RSC.
- S. Kar, A. Goeppert and G. K. S. Prakash, Acc. Chem. Res., 2019, 52, 2892–2903 CrossRef CAS PubMed.
- J. W. Maina, J. M. Pringle, J. M. Razal, S. Nunes, L. Vega, F. Gallucci and L. F. Dumee, ChemSusChem, 2021, 14, 1805–1820 CrossRef CAS PubMed.
- L. Li, X. Chen, Z. Chen, R. Gao, H. Yu, T. Yuan, Z. Liu and M. Maeder, Greenhouse Gases: Sci. Technol., 2021, 11, 807–823 CrossRef CAS.
- X. Li, Y. Jiang, G. Han and D. Deng, J. Chem. Eng. Data, 2016, 61, 1254–1261 CrossRef CAS.
- A. Ostonen, E. Sapei, P. Uusi-Kyyny, A. Klemela and V. Alopaeus, Fluid Ph. Equilibria, 2014, 374, 25–36 CrossRef CAS.
- J. J. Carroll, J. D. Slupsky and A. E. Mather, J. Phys. Chem. Ref. Data, 1991, 20, 1201–1209 CrossRef CAS.
- P. M. Mathias, F. Zheng, D. J. Heldebrant, A. Zwoster, G. Whyatt, C. M. Freeman, M. D. Bearden and P. Koech, ChemSusChem, 2015, 8, 3617–3625 CrossRef CAS PubMed.
- Z. Yuan, M. R. Eden and R. Gani, Ind. Eng. Chem. Res., 2015, 55, 3383–3419 CrossRef.
- G. A. Whyatt, A. Zwoster, F. Zheng, R. J. Perry, B. R. Wood, I. Spiry, C. J. Freeman and D. J. Heldebrant, Ind. Eng. Chem. Res., 2017, 56, 4830–4836 CrossRef CAS.
- D. J. Heldebrant, P. K. Koech, V. Glezakou, R. Rousseau, D. Malhotra and D. C. Cantu, Chem. Rev., 2017, 117, 9594–9624 CrossRef CAS PubMed.
- T. N. Borhani and M. Wang, Renew. Sustain. Energy Rev., 2019, 114 Search PubMed.
- K. A. Mumford, Y. Wu, K. H. Smith and G. W. Stevens, Front. Chem. Sci. Eng., 2015, 9, 125–141 CrossRef CAS.
- Y. Yasaka, C. Wakai, N. Matubayasi and M. Nakahara, J. Phys. Chem. A, 2010, 114, 3510–3515 CrossRef CAS PubMed.
- M. D. Porosoff, B. Yan and J. G. Chen, Energy Environ. Sci., 2016, 9, 62–73 RSC.
- S. Kattel, P. J. Ramirez, J. G. Chen, J. A. Rodriguez and P. Liu, Science, 2017, 355, 1296–1299 CrossRef CAS PubMed.
- M. Yadav, J. C. Linehan, A. J. Karkamkar, E. van der Eide and D. J. Heldebrant, Inorg. Chem., 2014, 53, 9849–9854 CrossRef CAS PubMed.
- N. M. Rezayee, C. A. Huff and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 1028–1031 CrossRef CAS PubMed.
- J. Kothandaraman, A. Goeppert, M. Czaun, G. A. Olah and G. K. Prakash, J. Am. Chem. Soc., 2016, 138, 778–781 CrossRef CAS PubMed.
- J. Kothandaraman, A. Goeppert, M. Czaun, G. A. Olah and G. K. S. Prakash, Green Chem., 2016, 18, 5831–5838 RSC.
- S. Wesselbaum, T. Vom Stein, J. Klankermayer and W. Leitner, Angew. Chem., Int. Ed., 2012, 51, 7499–7502 CrossRef CAS PubMed.
- Y. N. Li, L. N. He, A. H. Liu, X. D. Lang, Z. Z. Yang, B. Yu and C. R. Luan, Green Chem., 2013, 15, 2825–2829 RSC.
- D. Wei, H. Junge and M. Beller, Chem. Sci., 2021, 12, 6020–6024 RSC.
- J. Kothandaraman and D. J. Heldebrant, Green Chem., 2020, 22, 828–834 RSC.
- J. Kothandaraman and D. J. Heldebrant, RSC Adv., 2021, 10, 42557–42563 RSC.
- J. Kothandaraman, A. Goeppert, M. Czaun, G. A. Olah and G. K. S. Prakash, J. Am. Chem. Soc., 2016, 138, 778–781 CrossRef CAS PubMed.
- S. Wesselbaum, V. Moha, M. Meuresch, S. Brosinski, K. M. Thenert, J. Kothe, T. V. Stein, U. Englert, M. Holscher, J. Klankermayer and W. Leitner, Chem. Sci., 2015, 6, 693–704 RSC.
- P. M. Mathias, S. Reddy, A. Smith and K. Afshar, Int. J. Greenh. Gas Control, 2013, 19, 262–270 CrossRef CAS.
- P. M. Mathias, Fluid Phase Equilib., 2014, 362, 102–107 CrossRef CAS.
- Y. Liu, H. Z. Ye, K. M. Diederichsen, T. Van Voorhis and T. A. Hatton, Nat. Commun., 2020, 11, 2278 CrossRef CAS PubMed.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- S. Jin, M. Wu, R. G. Gordon, M. J. Aziz and D. G. Kwabi, Energy Environ. Sci., 2020, 13, 3706–3722 RSC.
- S. E. Renfrew, D. E. Starr and P. Strasser, ACS Catal., 2020, 10, 13058–13074 CrossRef CAS.
- M. S. Walters, R. H. Dunia, T. F. Edgar and G. T. Rochelle, Energy Proc., 2013, 37, 2133–2144 CrossRef CAS.
- C. Hepburn, E. Adlen, J. Beddington, E. A. Carter, S. Fuss, N. Mac Dowell, J. C. Minx, P. Smith and C. K. Williams, Nature, 2019, 575, 87–97 CrossRef CAS PubMed.
- J. Kothandaraman, J. Saavedra Lopez, Y. Jiang, E. D. Walter, S. D. Burton, R. A. Dagle and D. J. Heldebrant, ChemSusChem, 2021, 14, 4812–4819 CrossRef CAS PubMed.
- J. Kothandaraman and D. J. Heldebrant, RSC Adv., 2020, 10, 42557–42563 RSC.
- Z. Z. Yang, L. N. He, Y. N. Zhao, B. Li and B. Yu, Energy Environ. Sci., 2011, 4, 3971–3975 RSC.
- J. R. Khusnutdinova, J. A. Garg and D. Milstein, ACS Catal., 2015, 5, 2416–2422 CrossRef CAS.
- M. Scott, B. Blas Molinos, C. Westhues, G. Francio and W. Leitner, ChemSusChem, 2017, 10, 1085–1093 CrossRef CAS PubMed.
- G. Lee, Y. G. C. Li, J. Y. Kim, T. Peng, D. H. Nam, A. S. Rasouli, F. W. Li, M. C. Luo, A. H. Ip, Y. C. Joo and E. H. Sargent, Nat. Energy, 2021, 6, 46–53 CrossRef CAS.
- S. E. Tanzer and A. Ramírez, Energy Environ. Sci., 2019, 12, 1210–1218 RSC.
- P. T. Anastas and J. B. Zimmerman, Environ. Sci. Technol., 2003, 37, 94A–101A CrossRef PubMed.
- J. C. W. P. T. Anastas, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- J. Kothandaraman, J. Zhang, V.-A. Glezakou, M. T. Mock and D. J. Heldebrant, J. CO2 Util., 2019, 32, 196–201 CrossRef CAS.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388–2410 CrossRef CAS PubMed.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS PubMed.
- https://www.researchandmarkets.com/reports/4118824/20polyurethane-market-size-share-and-trends .
- https://www.360marketupdates.com/global-polyester-fiber-market-17940105 .
- https://www.gminsights.com/industry-analysis/polyester-fiber-market .
- J. O. Akindoyo, M. D. H. Beg, S. Ghazali, M. R. Islam, N. Jeyaratnam and A. R. Yuvaraj, RSC Adv., 2016, 6, 114453–114482 RSC.
- N. von der Assen and A. Bardow, Green Chem., 2014, 16, 3272–3280 RSC.
- E. Orgilés-Calpena, F. Arán-Aís, A. M. Torró-Palau, E. Montiel-Parreño and C. Orgilés-Barceló, Int. J. Adhesion Adhes., 2016, 67, 63–68 CrossRef.
- https://www.covestro.com/en/sustainability/flagship-solutions/co2-as-a-raw-material .
- W. Leitner, G. Franciò, M. Scott, C. Westhues, J. Langanke, M. Lansing, C. Hussong and E. Erdkamp, Chem. Ing. Tech., 2018, 90, 1504–1512 CrossRef CAS.
- L. Maisonneuve, O. Lamarzelle, E. Rix, E. Grau and H. Cramail, Chem. Rev., 2015, 115, 12407–12439 CrossRef CAS PubMed.
- C. J. Price, B. J. E. Reich and S. A. Miller, Macromolecules, 2006, 39, 2751–2756 CrossRef CAS.
- R. Nakano, S. Ito and K. Nozaki, Nat. Chem., 2014, 6, 325–331 CrossRef CAS PubMed.
- V. Moha, D. Cozzula, M. Holscher, W. Leitner and T. E. Muller, ChemSusChem, 2016, 9, 1614–1622 CrossRef CAS PubMed.
- B. Grignard, S. Gennen, C. Jerome, A. W. Kleij and C. Detrembleur, Chem. Soc. Rev., 2019, 48, 4466–4514 RSC.
- K. S. Lackner, C. H. Wendt, D. P. Butt, E. L. Joyce and D. H. Sharp, Energy, 1995, 20, 1153–1170 CrossRef CAS.
- J. M. Matter and P. B. Kelemen, Nat. Geosci., 2009, 2, 837–841 CrossRef CAS.
- J. M. Bielicki, M. F. Pollak, J. P. Fitts, C. A. Peters and E. J. Wilson, Int. J. Greenh. Gas Control, 2014, 20, 272–284 CrossRef CAS.
- P. M. Haugan, Geophys. Res. Lett., 2004, 31 Search PubMed.
- P. Kelemen, S. M. Benson, H. Pilorgé, P. Psarras and J. Wilcox, Front. Clim., 2019, 1 Search PubMed.
- E. C. La Plante, I. Mehdipour, I. Shortt, K. Yang, D. Simonetti, M. Bauchy and G. N. Sant, ACS Sustain. Chem. Eng., 2021, 9, 10727–10739 CrossRef CAS.
- S. Lin, T. Kiga, Y. Wang and K. Nakayama, Energy Proc., 2011, 4, 356–361 CrossRef CAS.
- J. M. Matter, M. Stute, S. O. Snaebjornsdottir, E. H. Oelkers, S. R. Gislason, E. S. Aradottir, B. Sigfusson, I. Gunnarsson, H. Sigurdardottir, E. Gunnlaugsson, G. Axelsson, H. A. Alfredsson, D. Wolff-Boenisch, K. Mesfin, D. Fernandez de la Reguera Taya, J. Hall, K. Dideriksen and W. S. Broecker, Science, 2016, 352, 1312–1314 CrossRef CAS PubMed.
- S. Hong, G. Sim, S. Moon and Y. Park, Energy Fuels, 2020, 34, 3532–3539 CrossRef CAS.
- M. Liu and G. Gadikota, Fuel, 2020, 275 Search PubMed.
- W. Liu, L. Teng, S. Rohani, Z. Qin, B. Zhao, C. C. Xu, S. Ren, Q. Liu and B. Liang, Chem. Eng. J., 2021, 416 Search PubMed.
- C. Julcour-Lebigue, F. Bourgeois, L. Cassayre, J. Leclaire, S. Touzé, M. Cyr and F. Bailly, Design of a hybrid leaching process for mineral carbonation of magnesium silicates: learnings and issues raised from combined experimental and geochemical modelling approaches, International Conference on Accelerated Carbonation for Environmental and Material Engineering (ACEME), Newcastle, Australia, 11 March 2018–14 March 2018 Search PubMed.
- M. Liu and G. Gadikota, Energy Fuels, 2018, 33, 1722–1733 CrossRef.
- M. Liu, A. Hohenshil and G. Gadikota, Energy Fuels, 2021, 35, 8051–8068 CrossRef CAS.
- G. Gadikota, Commun. Chem., 2021, 4, 21 CrossRef.
- F. Bourgeois, P. Laniesse, M. Cyr and C. Julcour, Front. Energy Res., 2020, 8, 113 CrossRef.
| This journal is © The Royal Society of Chemistry 2022 |