
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStrategies for the preparation of high-performance inorganic mixed-halide perovskite solar cells
Xin Liu
 *,
Jie Li,
Xumei Cui,
Xiao Wang
* and
Dingyu Yang
*
*,
Jie Li,
Xumei Cui,
Xiao Wang
* and
Dingyu Yang
*
a, College of Optoelectronic Engineering, Chengdu University of Information Technology, Chengdu 610225, China. E-mail: liuxin@cuit.edu.cn; wangxiao@cuit.edu.cn; yangdingyu@cuit.edu.cn
First published on 16th November 2022
Abstract
Inorganic halide perovskites have attracted significant attention in the field of photovoltaics (PV) in recent years due to their superior intrinsic thermal stability and excellent theoretical power conversion efficiency (PCE). CsPbI3 with a bandgap of ∼1.7 eV is considered to be the most potential candidate for PV application. However, bulk CsPbI3 films exhibit poor phase stability. The substitution of some iodide ions with bromide/chloride in CsPbI3 results in the formation of mixed-halide CsPbX3 perovskites, which exhibit a good balance between phase stability and efficiency. The halogen-tunable mixed-halide inorganic perovskites have a bandgap matching the sunlight region and show great potential for application in multi-junction tandem and semitransparent solar cells. Herein, the progress of mixed-halide CsPbX3 PSCs is systematically reviewed, including CsPbIxBryCl3−x−y- and CsPbIBr2-based IPSCs. In the case of CsPbIBr2 IPSCs, we introduce the low-temperature deposition of CsPbIBr2 films, doping methods for the preparation of high-quality CsPbIBr2 films and strategies for improving the performance of solar cells. Furthermore, the mechanism of crystallization/interface engineering for the preparation of high-quality CsPbIBr2 films and efficient solar cells devices is emphasized. Finally, the development direction of further improving the PV performance and commercialization of mixed-halide IPSCs are summarized and prospected.
 Xin Liu | Xin Liu is an Assistant Professor with the College of Optoelectronic Engineering, Chengdu University of Information Technology. She received her B.S. Degree from Hefei University of Technology in 2011 and PhD from South China University of Technology in 2016. From 2016 to 2017, she joined high-tech companies in China for research on organic light-emitting diodes. From 2017 to 2019, she was a Post-Doctoral Fellow with the University of Electronic Science and Technology of China. She joined Chengdu University of Information Technology in 2020 as an Assistant Professor. Her research interests include inorganic/organic semiconductor materials, thin-film photovoltaic devices and device physics. |
 Xiao Wang | Xiao Wang is an Assistant Professor with the College of Optoelectronic Engineering, Chengdu University of Information Technology. She received her B.S. Degree in 2012 and PhD in 2018 from the University of Electronic Science and Technology of China. During 2016–2018, she worked as a Visiting Scholar at the Centre for Organic Photonics & Electronics (COPE) at The University of Queensland. From June 2019 to June 2020, she was a Post-Doctoral Fellow at COPE. She joined Chengdu University of Information Technology in 2021 as an Assistant Professor. Her research interests include semiconductor and device physics, organic photovoltaics, and perovskite solar cells. |
 Dingyu Yang | Dingyu Yang is a Professor with the College of Optoelectronic Engineering, Chengdu University of Information Technology. He received his B.S. and M.S. Degree from Lanzhou University in 2000 and 2003, respectively, and PhD from Sichuan University in 2009. From October 2012 to February 2013, he worked as a Visiting Scholar at Nanyang Technological University. He joined Chengdu University of Information Technology in 2003. His research interests include optoelectronic detection technology and new energy technology. |
1 Introduction
Halide perovskites have received increasing attention in the past few years as bright new stars in optoelectronic devices. In the case of thin-film photovoltaic (PV) devices, halide perovskite materials exhibit excellent photophysical properties such as high extinction coefficient/carrier mobility, long carrier diffusion length, high defect tolerance, small exciton binding energy and tunable bandgaps.1–7 Strikingly, since Miyasaka et al. first adopted the halide perovskite material as a light absorber in a PV device and reported a power conversion efficiency (PCE) of 3.8% in 2009,8 organic–inorganic hybrid perovskite solar cells (OIH-PSCs) have obtained a PCE of more than 25%,9–16 which is comparable to that of the silicon-based solar cells currently dominating the PV field. The general chemical formula of organic–inorganic hybrid perovskite (OIHP) materials is ABX3, where A is a monovalent organic cation (methylammonium (CH3NH3+ and MA+), formamidinium (HC(NH2)2+ and FA+) or their mixture), B is a divalent cation (Pb2+ and Sn2+) and X is a halogen ion (I−, Br−, and Cl− or their mixture). However, due to the presence of volatile organic components, i.e., MA+ or FA+, OIH-PSCs undergo some compositional and structural degradation under persistent attack from heat, humidity, oxygen and light.17–23 Thus, to realize the commercialization of perovskite PV devices, the instability issues of OIH-PSCs have attracted great attention.24–26Inorganic cesium (Cs+) ions completely occupy the A-sites in the general chemical formula of ABX3, forming inorganic cesium lead halide perovskite materials (CsPbX3). Due to the lack of volatile and hygroscopic organic cations, these materials show robust resistance to high temperature and moisture.27–30 In 2015, Hodes's group revealed that organic components are not essential components to achieve efficient inorganic perovskite solar cells (IPSCs).31 They also demonstrated that the CsPbBr3 perovskite material has better thermal stability than organic–inorganic hybrid MAPbBr3 perovskite, and the CsPbBr3-based PV devices exhibit better thermal stability.32 In 2016, the PCE of mixed-halide IPSCs based on the first CsPbIBr2 and the first CsPbI2Br was 4.7%33 and close to 10%,34 respectively. Since then, the efficiency and stability of IPSCs have been rapidly improved by referring to the research experience of OIH-PSCs and the exploration of inorganic perovskite, as illustrated in Fig. 1a. However, compared with the Shockley–Queisser (SQ) efficiency limits and the requirements of practical PV applications,35,36 there is still much room to improve the efficiency and long-term stability of IPSCs (Fig. 1b).
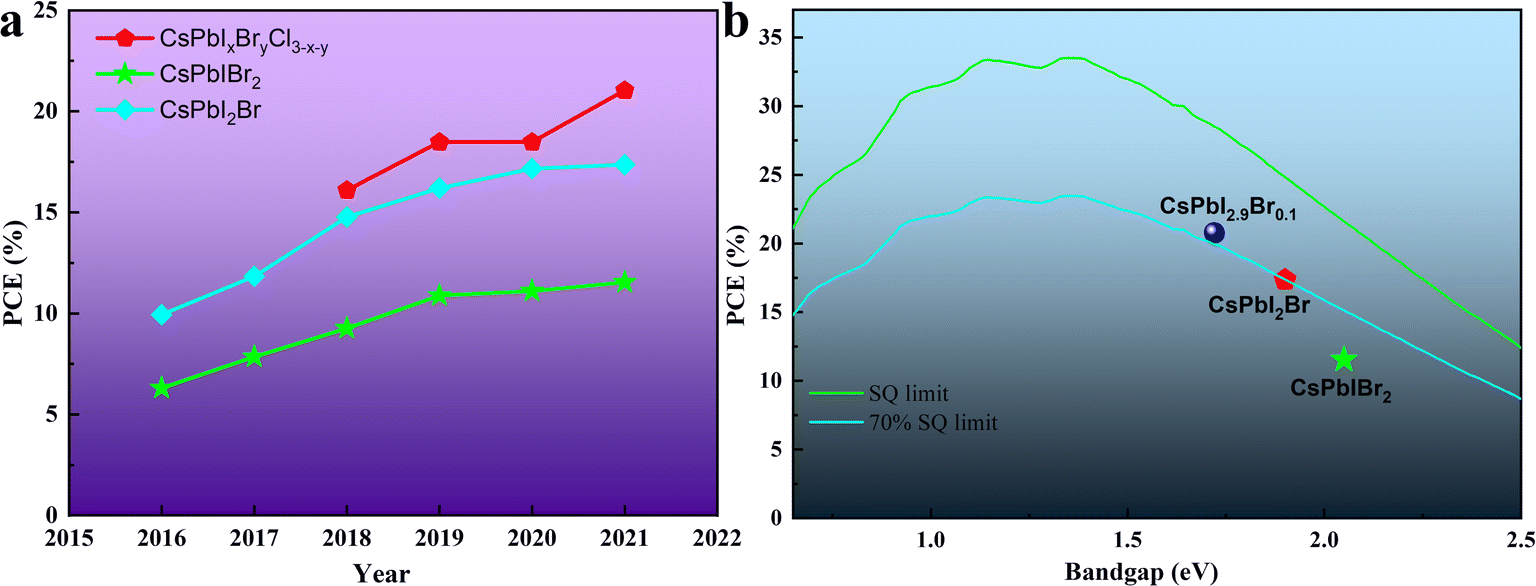 | ||
| Fig. 1 (a). Champion PCE of IPSCs with time. The devices are divided into 3 groups by changing the halogen compositions. (b). Champion PCE of different IPSCs compared with the SQ limit. | ||
At present, the commonly used light absorption layers in IPSCs include CsPbBr3, CsPbIBr2, CsPbI2Br and CsPbI3. CsPbI3 with a bandgap (Eg) of about 1.7 eV is considered the most potential candidate for PV application. Unfortunately, bulk CsPbI3 exhibits poor phase stability, which rapidly converts to a non-perovskite orthorhombic phase (Eg = 2.82 eV).37–40 The large bandgap of CsPbBr3 (Eg = 2.25 eV) limits the light collection, consequently reducing the device efficiency.41,42 Alternatively, the substitution of some of iodide ions with bromide/chloride ions in CsPbI3 to form mixed-halide CsPbX3 perovskites can provide a good balance between phase stability and efficiency. Mixed-halide inorganic perovskites such as CsPbIBr2 have attracted significant attention due to their increased Goldschmidt's tolerance factor t, thus stabilizing the black phase by partially substituting I− with the smaller Br−. Furthermore, cubic-phase CsPbIBr2 and CsPbI2Br perovskites show great potential in multi-junction tandem and semitransparent solar cells owing to their suitable optical bandgap of 2.05 eV43 and 1.92 eV,44 respectively. Importantly, mixed-halide inorganic perovskites can remain as a photoactive black phase at room temperature (RT), but inevitably convert to a non-perovskite phase when exposed to a humid environment. This phase transformation process is reversible when heated to 350 °C in a dry environment.45 To overcome the problems of environmental instability and achieve high-performance IPSCs, composition engineering and interface engineering play very important roles. For instance, Li+ doping can improve the optical, morphological and electronic properties of CsPbIBr2 films. Doping Mn2+ or Sn2+ in CsPbIBr2 can narrow its bandgap and enlarge its light response region. The introduction of the smaller F− in the X-site of CsPbI2Br can induce the formation of an α/δ-phase heterojunction, which facilitates efficient exciton dissociation and charge transport. Interfacial strategies between the perovskite film and the ETL or HTL can provide more suitable energy level alignments and passivate defects, and then effectively suppress the interfacial recombination.
In this review, as outlined in Fig. 2, we systematically summarize the reported mixed-halide CsPbX3 IPSCs. Firstly, the recent progress on the replacement of non-halogen or halogen anions in CsPbX3 IPSCs will be introduced. Then, we discuss and analyze the progress on CsPbIBr2-IPSCs. The section on CsPbIBr2-IPSCs covers the preparation of CsPbIBr2 films at low temperature, doping methods for the fabrication of high-quality CsPbIBr2 films and strategies for improving the PV performance of solar cells. The effective methods for the preparation of high-quality CsPbIBr2 films and the mechanism of crystallization/interface engineering for high-performance solar cells devices are emphasized. Finally, we present a summary and prospect on promising directions for further promoting the PV performance and realizing the commercialization of mixed-halide CsPbX3 solar cells.
 | ||
| Fig. 2 Summary and strategies for the preparation of high-performance mixed-halide CsPbX3 IPSCs. | ||
2 Basic properties of CsPbX3 perovskites
2.1 Crystal structure
Regarding crystal-based thin film optoelectronic devices, it is important to deeply understand their crystal structure to obtain high-performance halide perovskite PV devices with long-term stability. Halide perovskites are generally represented by the chemical formula ABX3, where the monovalent cation A-site such as MA+, FA+, and Cs+ occupies the corner positions (0, 0, 0), the bivalent cation B-site such as Pb2+, Sn2+, and Ge2+ is located at the central positions (1/2, 1/2, 1/2), and the anion X such as I−, Br− and Cl− is located at the center of the six planes (1/2, 1/2, 0), forming the crystal structure of three-dimensional (3D) perovskites with corner-sharing BX6 octahedra (Fig. 3a). According to the environmental conditions, CsPbX3 inorganic perovskite can form 4 types of crystal structures including cubic phase (α-, Pm3m), tetragonal phase (β-, P4/mbm), orthorhombic phase (γ-, Pbnm), and non-perovskite orthorhombic phase (δ-, Pnma).46 The optoelectronic properties and crystal stability of the CsPbX3 perovskite are significantly influenced by the different tilting angles of the PbX6 octahedra, and therefore the type of CsPbX3 perovskite phase is closely related to its PV properties and device long-term stability.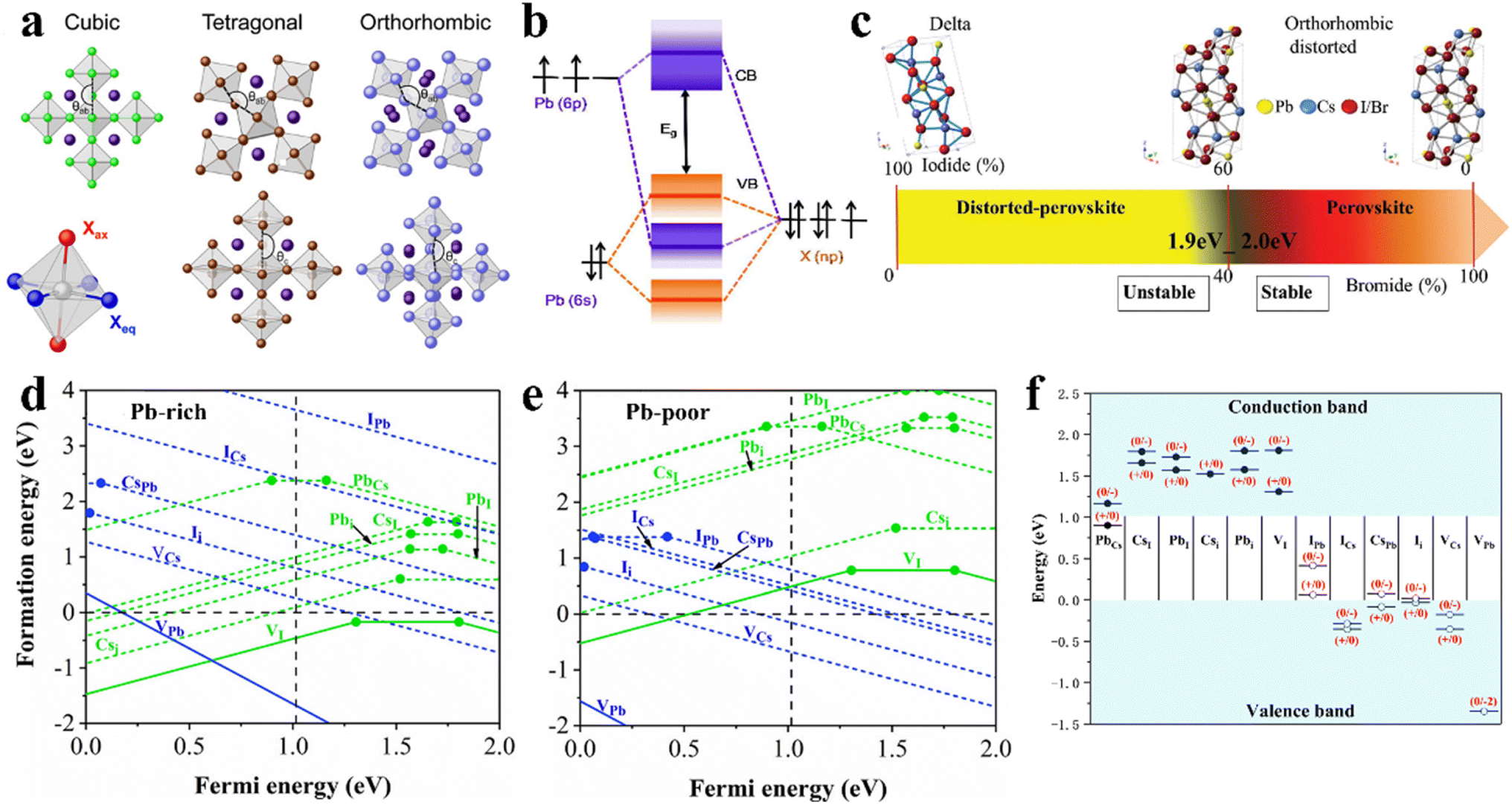 | ||
| Fig. 3 (a). Polyhedron models of crystal structures of cubic, tetragonal, and orthorhombic. Reprint with permission.67 Copyright 2018, the American Chemical Society. (b). Schematic representation of bonding/antibonding orbitals of APbX3 exhibiting the formation of the VB and CB. Reproduced with permission.52 Copyright 2016, the American Chemical Society. (c). CsPbX3 crystal structures as a function of the iodine/bromine ratio. Reproduced with permission.61 Copyright 2018, Wiley-VCH. Calculated defect formation energy as a function of the Fermi energy EF of Pb-rich (d) and Pb-poor (e) films of cubic CsPbI3. (f). Intrinsic point defect transition energy levels in cubic CsPbI3. (d–f) Reproduced with permission.65 Copyright 2017, the American Institute of Physics. | ||
The formation and geometric stability of the crystal structure of ABX3 compounds can be empirically determined using Goldschmidt's tolerance factor (t) and the octahedral factor (μ), which allow researchers to pre-screen the formation of suitable components of a stable perovskite lattice. To maintain the perovskite cubic crystal structure, 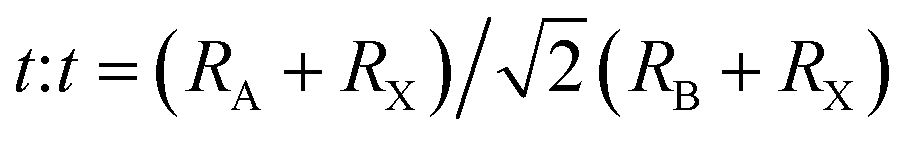 (ref. 47 and 48) and μ(RB/RX) (ref. 49) should be satisfied in the range of 0.9 to 1 and 0.4 to 0.9,50 where RA, RB, and RX are the ionic radii of the corresponding cation and anion, respectively. The BX6 framework can only contain certain ions to achieve the geometric stability of halide perovskites. A t factor below 0.9 leads to a distorted perovskite structure due to the tilting of the PbX6 octahedra. A t factor between 0.9 and 1.0 leads to the formation of symmetric cubic-phase perovskite. When t is greater than 1, a hexagonal structure with a face-sharing octahedron is formed. Meanwhile, with a t factor at the required lower or upper limit, a too small or large A cation generally leads to a non-perovskite phase. The μ assesses whether the B atoms will tend to octahedral coordination of the X atoms. When it is greater than 0.4, a stable BX6 octahedron can be formed. Thus, because the requirements of t and μ must be satisfied simultaneously, only a limited number of combinations of A, B, and X ion types can form 3D perovskites.
(ref. 47 and 48) and μ(RB/RX) (ref. 49) should be satisfied in the range of 0.9 to 1 and 0.4 to 0.9,50 where RA, RB, and RX are the ionic radii of the corresponding cation and anion, respectively. The BX6 framework can only contain certain ions to achieve the geometric stability of halide perovskites. A t factor below 0.9 leads to a distorted perovskite structure due to the tilting of the PbX6 octahedra. A t factor between 0.9 and 1.0 leads to the formation of symmetric cubic-phase perovskite. When t is greater than 1, a hexagonal structure with a face-sharing octahedron is formed. Meanwhile, with a t factor at the required lower or upper limit, a too small or large A cation generally leads to a non-perovskite phase. The μ assesses whether the B atoms will tend to octahedral coordination of the X atoms. When it is greater than 0.4, a stable BX6 octahedron can be formed. Thus, because the requirements of t and μ must be satisfied simultaneously, only a limited number of combinations of A, B, and X ion types can form 3D perovskites.
Cs+ has been identified as the preferred inorganic ion to substitute the organic MA+/FA+ in the perovskite structure. In the case of CsPbX3 perovskite compounds, Cs+ completely occupies the A-site. The photoactive black phase of CsPbI3 perovskite is easily converted to the undesirable yellow δ-CsPbI3 at room temperature (RT) due to the small ionic radius of Cs+, which makes it difficult to support the [PbI6]4− octahedron. The partial replacement of I− by Br− can reduce the size of the Cs-X coordination polyhedron, enabling Cs+ to retain the [PbX6]4− octahedron structure, and then adjust the values of t and μ to a more desirable range.51 Due to the partial replacement of I− with the smaller Br−, mixed-halogen inorganic perovskites such as CsPbI2Br and CsPbIBr2 exhibit excellent stability, and thus have attracted much attention. Furthermore, the bandgaps of the cubic-phase CsPbI2Br and CsPbIBr2 perovskites are 1.92 eV44 and 2.05 eV,43 respectively, showing great potential in tandem and semitransparent solar cells. More importantly, the mixed-halogen CsPbX3 perovskites can maintain a photoactive black phase at RT, but inevitably transform to a non-perovskite phase when exposed to a humid environment. Given that the distortion degree of the [PbI6]4− octahedron will seriously influence the transition between different phases, its distortion degree can be controlled by introducing steric hindrance or external disturbances, which will help CsPbX3 to remain in the desired black phase.
2.2 Optoelectronic properties of CsPbX3 perovskites
The excellent optical and electrical properties of semiconductor perovskites greatly depend on the nature of their electronic structures. For typical CsPbX3 perovskites, their conduction band minimum (CBM) is composed of a combination of lead 6p orbital and halogen np orbitals with the main contribution from Pb 6p (Fig. 3b), while their valence band maximum (VBM) mainly consists of mixed lead 6s orbitals and halogen np orbitals with the dominant contribution from X np.52 The contribution of the element to each band strongly depends on the perovskite stoichiometry. It is well known that the high efficiency of PSCs is related to direct their bandgap transition, high absorption coefficient, etc., while the optical absorption of perovskite materials is related to their electronic band structure.To gain deeper insight into the relationships between the structure and perovskite PV performance, theoretical investigations are conducive to acquiring a comprehensive understanding of CsPbX3 perovskites and serve as a guidance to accurately design and develop new component materials with advanced optoelectronic properties. Adopting PbI2 as an initial structural model, polymorphs of alkali metal lead halide perovskites with the cubic structure have been studied, indicating that while the Pb2+ 6s lone-pairs are stereochemically inert, the presence of proximal instabilities can have implications in the functional properties of these materials.53 Thus, the excitation and recombination of electrons and excitons are confined to the octahedron, similar to the widely studied OIHPs.54,55 According to density functional theory (DFT) and considering relativistic corrections and spin–orbit interactions, the electronic structures of cubic-phase CsPbCl3, CsPbBr3, and CsPbI3 perovskites have been calculated.56 It is evident that the energy band structures of CsPbX3 perovskites are not affected by their halide composition apart from the difference in their bandgap values, and thus all the CsPbX3 perovskites exhibit direct bandgaps, demonstrating numerous potential applications in the field of optoelectronics.57–60
A continuous change in halide composition from I to Br to Cl leads to systematic changes in the optical bandgap of halide perovskites (Fig. 3c).61 It is worth noting that the Urbach parameter (Eu), which is a measure of the sub-bandgap tail absorption and associated with static disorder in semiconductors, is abnormally low for halide perovskites. Most importantly, the optical absorption coefficients (α) of Pb-based halide perovskites are quite high and their absorption onsets are very sharp, indicating that only a very thin film can absorb all photons above their bandgap.
In CsPbX3 perovskites, data on the diffusion lengths and their related parameters (carriers mobilities and lifetimes) are limited. Obviously, perovskite films obtained under different conditions show different charge carrier lifetimes. Using the time-resolved microwave conductivity technique, Hutter et al. reported that the charge carrier lifetimes exceeded 10 μs in vapor-deposited CsPbI3, while the carrier lifetimes of spin-coated black-phase CsPbI3 films were less than 0.2 μs.62 For the same CsPbI3 perovskite fabricated by solution deposition, the lifetime of the film was over 20 ns.63 In addition, the lifetime value was around 14 ns for a solution-deposited CsPbI2Br film.64 Values of 2–7 μs were reported for CsPbBr3 macroscopic single crystals.
Given that defects can significantly change the electronic properties of perovskite materials, thus affecting the performance of PSCs, it is imperative to have a deep understanding of the defect characteristics to obtain efficient PV devices. In an ideal crystal structure, each atom is located in a specific position. However, due to the defective lattice arrangement at extended distances or the addition of foreign atoms, perovskite polycrystalline films grown and post-processed by solution processes at low temperature will inevitably have some defects.
The crystallographic defects in CsPbX3 show at least three types of different point defects. Fig. 3d and e show the formation energies of CsPbX3 intrinsic point defects calculated in Pb-rich and Pb-poor films.65,66 There are three types of intrinsic point defects, as follows: (1) vacancies (atoms missing in the lattice: VCs, VPb, and VI), (2) interstitials (atoms occupying the space between atoms in the lattice: Csi, Pbi, and Ii), and (3) anti-site substitutions (atoms occupying the wrong site in the lattice: CsPb, CsI, PbCs, PbI, ICs, and IPb), where AB means that A is replaced by B. According to the energy sites of the point defects in the bandgap, they can be divided into shallow and deep-level states. The activation energy of shallow-level defects is lower, while deep-level defects are far away from the CBM and VBM and close to the center of the bandgap, as shown in Fig. 3f.65 Shallow defects, such as VCs, VI, VPb, Csi, Ii, and CsPb, have a low formation energy, while deep defects, such as PbI, Pbi, IPb, and ICs, have a high formation energy. Electrons or holes are captured by deep defect states and are difficult to detrap.
3 CsPbX3 (X: non-halogen or halogen mixed ions)-based IPSCs
Vacuum thermal deposition is mature technology used in industry at present, which can easily prepare multilayer films in large areas, and the resultant films have good uniformity. However, the vacuum deposition method requires precise control of the stoichiometric ratios of the precursor, and it is usually difficult to control the crystal structure. Thus, the solution-processed methods are usually adapted to prepare CsPbX3 perovskite films owing to their simple preparation and low energy cost, especially given that most perovskite films only require low-temperature annealing. Generally, perovskite deposition can be divided into two solution processing methods, i.e., one-step and two-step sequential deposition. In a two-step process, PbX2 is first deposited on the substrate, and then coated with CsX, allowing the two precursors to react to form a CsPbX3 film. In a typical one-step process, the perovskite film is directly deposited from a solution containing all the precursors.The replacement of non-halogen or halogen anions in the X-site is an effective strategy to improve the structure stability of inorganic CsPbX3 perovskite. Thus far, halogen ions of Cl− and F− and non-halogen ions of Ac− and SCN− have been used to dope CsPbX3 to finely tune its crystal structure and phase stability. For example, Fu et al. introduced F− into the X-site of CsPbI2Br to adjust its phase heterostructure and t.68 It was proven that incorporating the smaller F− could induce the formation of an α/δ-phase heterojunction, which is beneficial for the efficient dissociation of excitons and charge transport. In another study, Ac− doping in the CsPbI2Br perovskite produced multiple benefits including lower trap densities, longer carrier lifetime, and fast charge transportation, thus resulting in a PCE of 15.56% and ultrahigh Voc of 1.30 V for CsPbI2−xBr(Ac)x-based IPSCs.69 Recently, using the one-step ultrasonic spray deposition method, Liu's group prepared Ac−-doped CsPbI2Br perovskite.70 Combining the vacuum extraction during processing, they obtained a CsPbI2Br film with improved quality, full coverage and long carrier lifetime. Also, a PCE of 10.06% was obtained for n–i–p IPSCs with good thermal stability due to the reduced defect density and nonradiative recombination loss. Table 1 summarizes the PV performance of non-halogen or mixed-halogen ion-based CsPbX3 IPSCs.
| Device architecture | Deposition method | Active area (cm2) | PCE (%) | Jsc (mA cm−2) | Voc (V) | FF (%) | Stability | Year (ref.) |
|---|---|---|---|---|---|---|---|---|
| FTO/c-TiO2/CsPbI2.85Br0.15 (@210 °C)/Spiro-OMeTAD/Au | One-step spin-coating | 0.09 | 18.43 | 20.64 | 1.09 | 82.3 | — | 2021 (ref. 78) |
| ITO/ZnO/CsPbI2.4Br0.6 (@275 °C)/Spiro-OMeTAD/Au | One-step spin-coating | — | 18.14 | 18.16 | 1.22 | 82.07 | — | 2021 (ref. 79) |
| FTO/c-TiO2/CsPbI2.33Br0.67 (@190 °C)/carbon | One-step spin-coating | 0.07 | 12.40 | 17.30 | 1.01 | 70.98 | Retained 84% of initial efficiency after stored in ambient environment with RH 15–20% for 200 h | 2021 (ref. 80) |
| ITO/P3CT-N/CsPbI2.8Br0.2 (@180 °C)/PCBM/Ag | One-step spin-coating | 0.09 | 13.14 | 18.78 | 1.00 | 70.0 | Maintained 80% of the initial PCE value exposed to an atmosphere with RH in the range of 40–60% for 6 h | 2022 (ref. 92) |
| ITO/ZnO/CsPbI2.4Br0.6:Cl/Spiro-OMeTAD/Au | One-step spin-coating | 0.0475 | 17.14 | 17.57 | 1.21 | 80.36 | — | 2021 (ref. 83) |
| FTO/c-TiO2/CsPbI2.85Br0.149Cl0.001 (@210 °C)/Spiro-OMeTAD/Au | One-step spin-coating | 0.09 | 19.65 | 19.94 | 1.23 | 80.11 | Maintained 91.2% of initial PCE value for over 30 days (RH 15–30%, at 10 °C) | 2021 (ref. 84) |
| FTO/SnO2/CsPbI2.5Br0.5 (@350 °C)/Spiro-OMeTAD/MoO3/Au | One-step spin-coating | 0.05 | 17.10 | 17.67 | 1.30 | 74.18 | Maintained 85% of initial PCE exposed to N2 atmosphere after 1500 h under continuous light illumination | 2020 (ref. 85) |
| FTO/SnO2/CsPb(I0.75Br0.25)3-0.5FAOAc (@280 °C)/Spiro-OMeTAD/Au | One-step spin-coating | 0.16 | 17.0 | 15.9 | 1.34 | 79.6 | — | 2020 (ref. 86) |
| FTO/c-TiO2/CsPbI2.84Br0.16-0.1CsTa (@180 °C)/Spiro-OMeTAD/Au | One-step spin-coating | 0.09 | 16.59 | 19.48 | 1.10 | 77.30 | Maintained 87.41% of the initial PCE after 500 h of storage under 80% RH at 80 °C | 2021 (ref. 87) |
| FTO/c-TiO2/CsPbI3−xBrx (@210 °C)/HA/Spiro-OMeTAD/Au | One-step spin-coating | 0.09 | 20.8 | 20.55 | 1.233 | 81.9 | — | 2021 (ref. 88) |
| ITO/SnO2/CsPbI1.8Br1.2 (@160 °C)/TACl/PBDB-T/MoO3/Au/ZnO/PFN/PM6:Y6/MoO3/Al | One-step spin-coating | 0.0988 | 21.04 | 13.36 | 2.05 | 76.82 | Retained 94% of initial PCE after 120 h of UV-light irradiation | 2022 (ref. 89) |
| FTO/SnO2/ZnO/CsPbI1.5Br1.5 (@200 °C)/Spiro-OMeTAD/Au | One-step spin-coating | 0.06 | 14.05 | 14.1 | 1.29 | 77.1 | — | 2021 (ref. 91) |
| FTO/TiO2/CsPbI2Br:1.0% Co(Ac)2 (@50 °C/@150 °C/@270 °C)/Spiro-OMeTAD/Ag/Au | One-step spin-coating (Ac− doped) | — | 15.04 | 15.43 | 1.21 | 80.46 | Retained 76% of initial PCE for over 50 days storage in N2 glovebox | 2020 (ref. 93) |
| ITO/SnO2/5% Pb(Ac)2:CsPbI2Br (@280 °C)/PTAA/Au | One-step ultrasonic spray (Ac− doped) | 0.09 | 10.06 | 13.99 | 1.12 | 65 | Retained 76% of the initial efficiency stored in N2 glovebox at 85 °C | 2021 (ref. 70) |
| FTO/c-TiO2/5% Pb(Ac)2:CsPbI2Br (@350 °C)/Spiro-OMeTAD/Ag | One-step spin-coating (Ac− doped) | 0.07 | 12 | 13.98 | 1.17 | 74 | Maintained ≈80% PCE after 30 days storage at T ≈ 20 °C and RH ≈ 20% | 2018 (ref. 94) |
| FTO/TiO2/4.5% Pb(Ac)2:CsPbI2Br (@35 °C/@120 °C/@165 °C)/Spiro-OMeTAD/Au | One-step spin-coating (Ac− doped) | 0.09 | 15.56 | 15.28 | 1.30 | 78.51 | Maintained 98% of initial PCE for 14 days storage in air (25 °C and 30% humidity) | 2019 (ref. 69) |
| ITO/SnO2/1% CsCl:CsPbI2Br (@45 °C/@110 °C/@180 °C)/Spiro-OMeTAD/Au | One-step spin-coating (Cl− doped) | 0.1 | 11.04 | 12.87 | 1.33 | 64 | Retained ∼80% of initial PCE after 360 h exposure in ambient air with RH ∼65% | 2021 (ref. 95) |
| FTO/c-TiO2/0.015 M Pb(DDTC)2:CsPbI2Br (@43 °C/@160 °C)/P3HT/Au | One-step spin-coating (DDTC− doped) | 0.0625 | 17.03 | 15.78 | 1.34 | 80.52 | Maintained > 98% of original PCE in ambient conditions with RH = 15 ± 3% for 1440 h | 2020 (ref. 96) |
| FTO/c-TiO2/m-TiO2/CsPbBrI1.78F0.22 (@150 °C)/Spiro-OMeTAD/Ag | One-step spin-coating (F− doped) | — | 10.26 | 14.94 | 1.01 | 68 | Retained 69.81% of the initial PCE after 10 days storage under 20% RH at RT | 2018 (ref. 68) |
| FTO/c-TiO2/m-TiO2/4-GBACl:CsPbI2Br (@50 °C/@160 °C)/Spiro-OMeTAD/Ag | One-step spin-coating (Pb-X framework) | 0.09 | 15.59 | 15.42 | 1.28 | 79 | Maintained 88% of initial PCE after 1200 h aging at 25 °C and 20% RH under ambient conditions | 2021 (ref. 97) |
| FTO/c-TiO2/CsGA0.04PbTh3Ac0.02 (@210 °C)/Spiro-OMeTAD/Au | One-step spin-coating (GCA passivation) | 0.09 | 19.37 | 20.14 | 1.17 | 82.1 | Retained 80% of initial PCE value after being stored for one month under ambient conditions without encapsulation | 2022 (ref. 81) |
| ITO/SnO2/β-CsPbI2.85(BrCl)0.15/PTB7-th BHJ (@210 °C)/Spiro-OMeTAD/Au | One-step spin-coating (DMAI-mediated) | 0.0832 | 19.0 | 20.7 | 1.12 | 81.8 | Maintained over 90% of the initial PCE after being stored in N2 glovebox for 6 months exposed to light illumination for 144 h | 2022 (ref. 82) |
3.1 CsPbIxCl3−x-based IPSCs
The regulation of the halide component by introducing Br and Cl has been proven to be a feasible route to improve the stability of IPSCs. For example, appropriate Cl doping in CsPbX3 can improve the stability of the black phase.71–73 Surface Cl-doped CsPbI3 IPSCs with PTACl passivation treatment exhibited a PCE of 19.03% with high stability.73 Dastidar et al. co-deposited colloidal NCs of pure CsPbCl3 and CsPbI3 to ensure high Cl doping levels and nanometer-scale mixed film, which exhibited improved stability in humid conditions compared to the undoped state.71 In another study, a stable black-phase γ-CsPbI3 was fabricated by doping Cl ions (Fig. 4a), in which the incorporation of Cl decreased the trap density and improved the electron and hole mobilities.72 Consequently, the Cl-treated γ-CsPbI3 IPSCs yielded a PCE as high as 16.07% and exhibited slight degradation after continuous light soaking or long-term exposure in dry air. A small amount of lead chloride (PbCl2) additive was introduced in the CsPbI3−xBrx perovskite precursor to suppress the recombination in the perovskite film.74 Consequently, CsPbI3−xBrx IPSCs with a bandgap of 1.77 eV exhibited an exciting PCE of 18.64% and Voc as high as 1.25 V with the Voc loss as low as 0.52 V, which showed excellent photostability with less than 6% efficiency drop under continuous 1 sun equivalent illumination over 1000 h.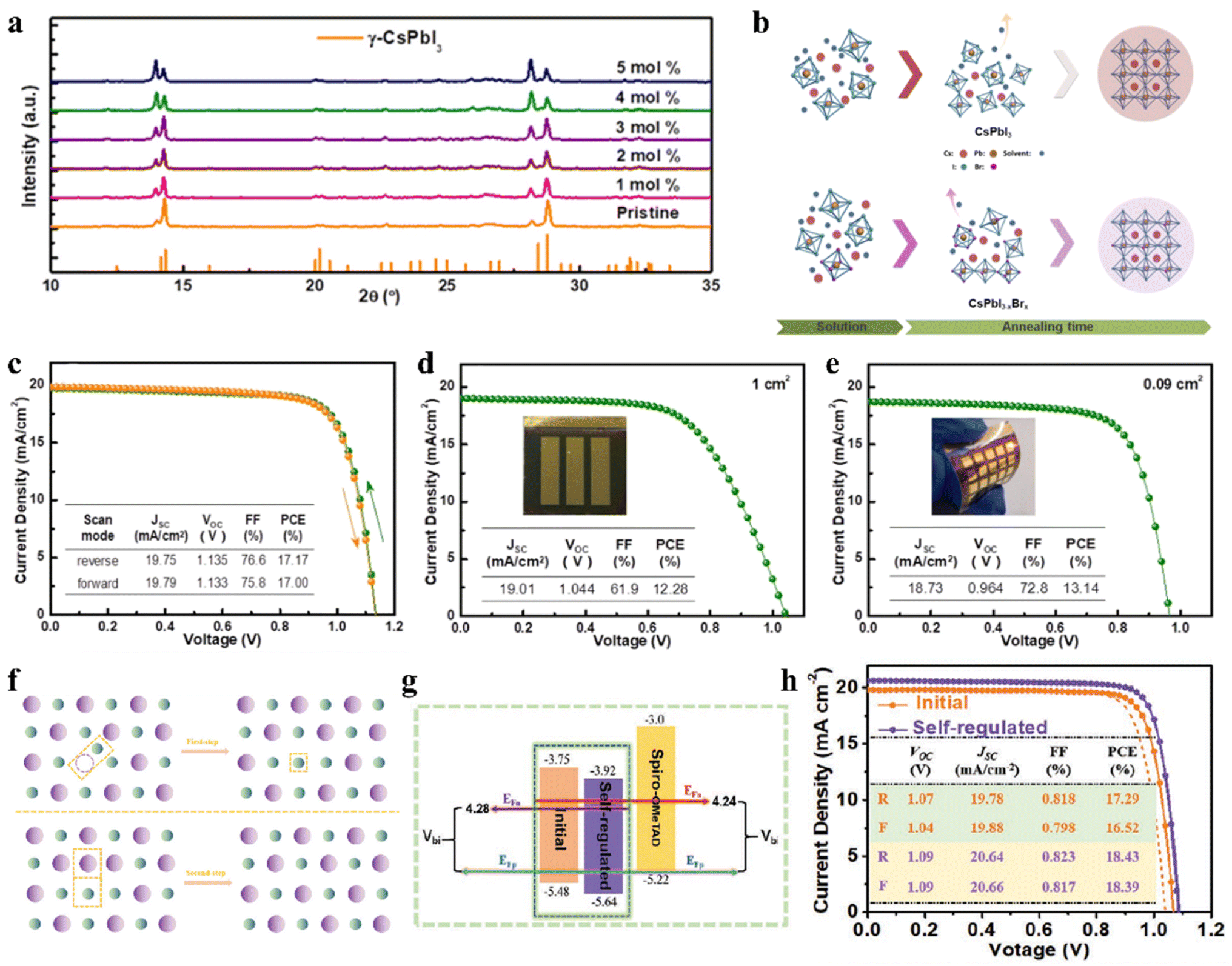 | ||
| Fig. 4 (a). XRD patterns of γ-CsPbI3:Clx films. Reproduced with permission.72 Copyright 2019, Elsevier. (b). Schematic diagram for Br-driven crystalline grain growth. J–V characteristics (c) under both the reverse and forward scan directions with an active area of 0.09 cm2; (d) on a glass substrate with an area of 1 cm2; and (e) on a PET/ITO/TiO2 substrate with an area of 0.09 cm2. (b–e) Reprinted with permission.76 Copyright 2019, the American Chemical Society. (f). Proposed schematic diagram of defect-regulation processes. (g). Schematic illustration of energy levels and Vbi. (h). J–V characteristics measured in the forward and reverse directions under AM1.5G illumination. (f–h) Reproduced with permission.78 Copyright 2021, Wiley-VCH. | ||
3.2 CsPbIyBr3−y-based IPSCs
The incorporation of small amount of Br can also adjust the t, while the mixing of I/Br can improve the stability of the CsPbI3 perovskite.75 By incorporating 5% Br ions in CsPbI3 (Fig. 4b), the mixed-halide CsPbI2.85Br0.15 IPSCs achieved a PCE of 17.17% and stabilized PCE of 16.83% with low Eloss of 0.58 eV and delivered a PCE of 12.28% and 13.14% for large areas (1 cm2) and flexible substrates, as shown in Fig. 4c–e, respectively.76 CsPb0.4Sn0.6I2.4Br0.6 perovskite with a narrow bandgap of 1.35 eV was developed and the corresponding IPSCs exhibited a PCE of 12.34%.77 The defect concentration of aged CsPbI3−xBrx polycrystalline films was about 2-orders of magnitude lower than that of the freshly synthesized films due to the self-digested anti-site defect pairs, in which the origin of the defect annihilation is intimately associated with the strain in the CsPbI3−xBrx film (Fig. 4f and g).78 The assembled CsPbI3−xBrx IPSCs acquired a PCE of 18.43% after self-regulation treatment, which was higher than that of the device with the fresh film (17.29%), as illustrated in Fig. 4h. Meanwhile, the hysteresis was suppressed owing to the reduced strain.Currently, Wu et al. systematically studied the working mechanism of the light soaking (LS) effect in CsPb(I1−xBrx)3 IPSCs.79 They found that LS can promote the migration of halogen ions, effectively giving rise to defect passivation. Based on these understandings, a PCE of 18.14% for CsPb(I0.8Br0.2)3 IPSCs was achieved by fine-tuning the amount of excessive PbI2 in the precursor. Composition engineering strategy was proposed to achieve high-quality perovskite films with a large grain size of over 1 μm and fabricate carbon-based IPSCs by incorporating a certain amount of bromide in the CsPbI3 perovskite (Fig. 5a).80 It was found that the incorporation of bromide induced a high-quality intermediate phase and contributed to the formation of a smooth perovskite film, thus leading to a longer carrier lifetime and lower band edge disorder. Finally, carbon electrode-based CsPbI2.33Br0.67 IPSCs (Fig. 5b and c) exhibited a PCE of 12.40% and retained 84% of their initial value after storage for 200 h in the ambient environment (RH 15–20%).
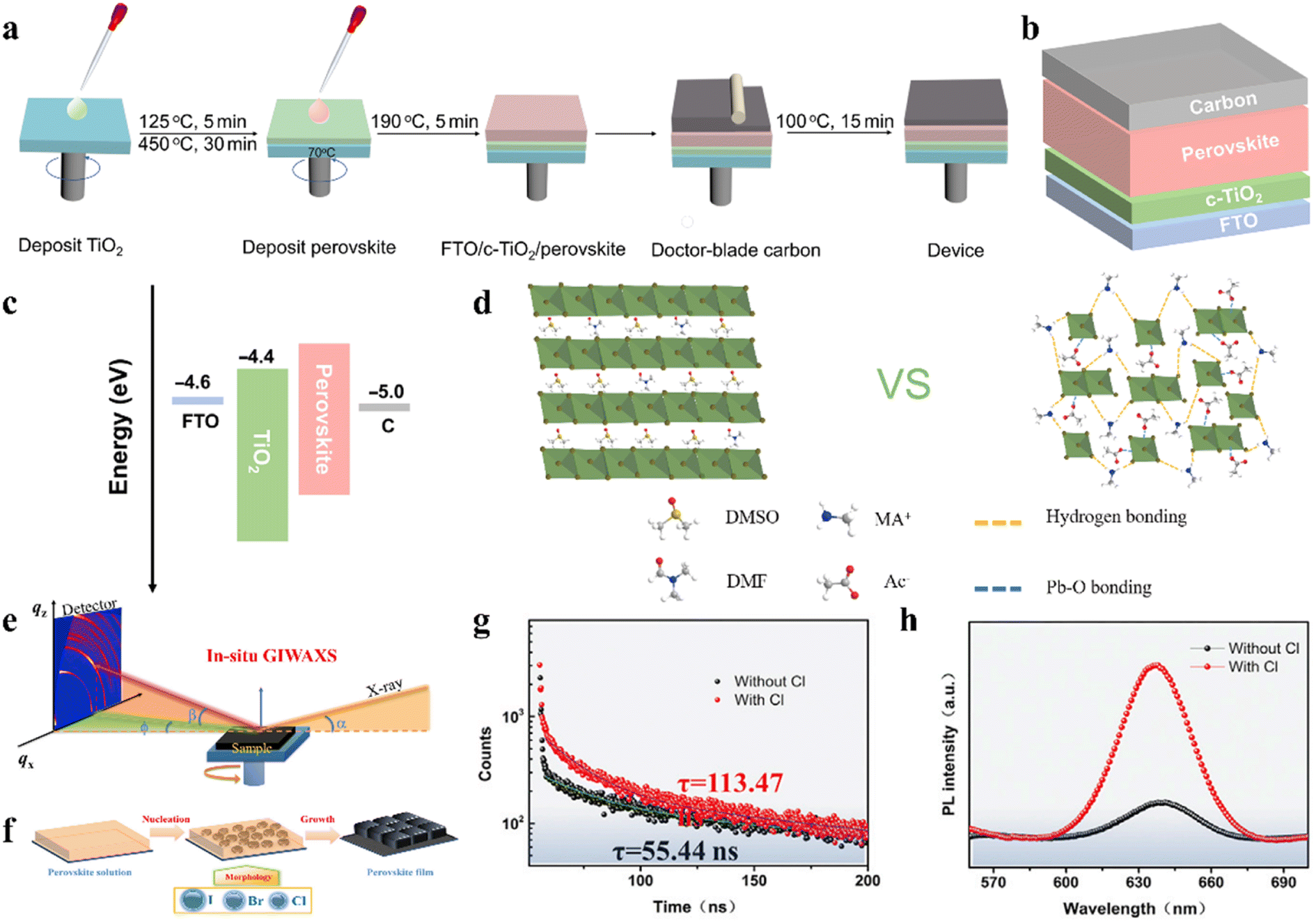 | ||
| Fig. 5 (a). Schematic illustration of device fabrication process. (b). Device configuration of HTM-free carbon-based IPSCs. (c). Energy level diagram of IPSCs. (a–c) Reproduced with permission.80 Copyright 2021, Wiley-VCH. (d). Schematic illustration of interaction in the precursor solutions. Reproduced with permission.85 Copyright 2020, Wiley-VCH. (e). Schematic of in situ GIWAXS characterization. (f). Schematic of perovskite film-formation procedures. (g). TRPL decay curves. h. Steady-state PL. (e–h) Reproduced with permission.83 Copyright 2021, Elsevier. | ||
3.3 CsPbIxBryCl3−x−y-based IPSCs
Several studies have demonstrated that the phase stability of CsPbX3 perovskites can be effectively improved by combining different halogens.81,82 To investigate the influence of ternary mixed-halides on the film crystallization mechanism and phase evolution of inorganic perovskite CsPbX3 (X = I, Br, Cl) under spin-coating, Ma et al. adopted the state-of-art in situ grazing-incidence wide-angle X-ray scattering (GIWAXS) technique, as shown in Fig. 5e.83 They found that the Br component not only can regulate the crystallization kinetics and affect the formation of the film, but also promote the photoactive phase formation and suppress the unwanted yellow phase transition, as shown by the mechanism presented in Fig. 5f. Moreover, the introduction of Cl can improve the crystallinity and orientational order of the bulk film, which helps to prolong the charge carrier lifetime and suppress the non-radiative recombination (Fig. 5g and h). Hence, with the assistance of Cl doping, the optimized composition CsPb(I0.8Br0.2)3:Cl IPSCs obtained a record high PCE of 17.14% with the small Voc Eloss of 0.6 eV. Another example reported a PCE of 19.65% with Voc of 1.23 V, corresponding to Eloss of 0.48 eV for triple halide-mixed CsPb(I2.85Br0.149Cl0.001) IPSCs deposited in the ambient atmosphere using an in situ hot oxygen cleansing strategy.84 It was found that the hot oxygen treatment not only effectively removed the organic residues, but also passivated the halogens vacancies to reduce the trap states and non-radiative recombination losses in the perovskite layer.The coordination interaction of the precursor solution plays a key role in regulating the crystallization of perovskites. For example, an effective interaction tailoring strategy was developed for the CsPbI3−xBrx perovskite by adopting the ionic liquid solvent MAAc.85 The results showed that oxygen with lone pair electrons (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) on Ac− had strong interaction with Pb2+ and the N–H⋯I hydrogen bonds, which enabled the formation of a stable perovskite precursor solution and allowed the high-quality production of pinhole-free, large grain size, flat inorganic perovskite films by retarding the crystallization (Fig. 5d). By controlling the ratio of I and Br, a series of IPSCs with a one-step, without the necessity for anti-solvent treatment, air-processing approach regardless of humidity, showed a PCE of 13.82% (CsPbI1.5Br1.5), 15.82% (CsPbI2Br), and 17.10% (CsPbI2.5Br0.5), respectively.
O) on Ac− had strong interaction with Pb2+ and the N–H⋯I hydrogen bonds, which enabled the formation of a stable perovskite precursor solution and allowed the high-quality production of pinhole-free, large grain size, flat inorganic perovskite films by retarding the crystallization (Fig. 5d). By controlling the ratio of I and Br, a series of IPSCs with a one-step, without the necessity for anti-solvent treatment, air-processing approach regardless of humidity, showed a PCE of 13.82% (CsPbI1.5Br1.5), 15.82% (CsPbI2Br), and 17.10% (CsPbI2.5Br0.5), respectively.
An intermediate-phase engineering strategy was developed to obtain robust interfacial contact by utilizing volatile organic salts, as shown by the mechanism in Fig. 6a.86 The introduction of organic cations doped in the perovskite lattice led to the formation of an organic–inorganic hybrid perovskite intermediate phase in the initial film and promoted high-quality interfacial contact through hydrogen bonding (Fig. 6b and c). In addition, CsPbI2.84Br0.16 films with small grain sizes were achieved by utilizing cesium trimethylacetate (CsTa) organic salt as an additive, in which the large steric hindrance effects of the Ta− anions efficiently prevented the tilt of the [PbI6]4− octahedra to inhibit the phase transition process from the corner-shared perovskite to the edge-shared non-perovskite structure.87 Furthermore, the Ta− groups firmly bonded onto the surface of the CsPbI2.84Br0.16 crystal at the X-site and increased the energy barrier for X-site vacancy generation (from 0.816 eV to 1.217 eV). Finally, the 0.1-CsTa HPbI3-prepared IPSCs exhibited a PCE of 16.59% and retained 80.88% of their initial efficiency after more than 1200 h in air (relative humidity (RH): 20%).
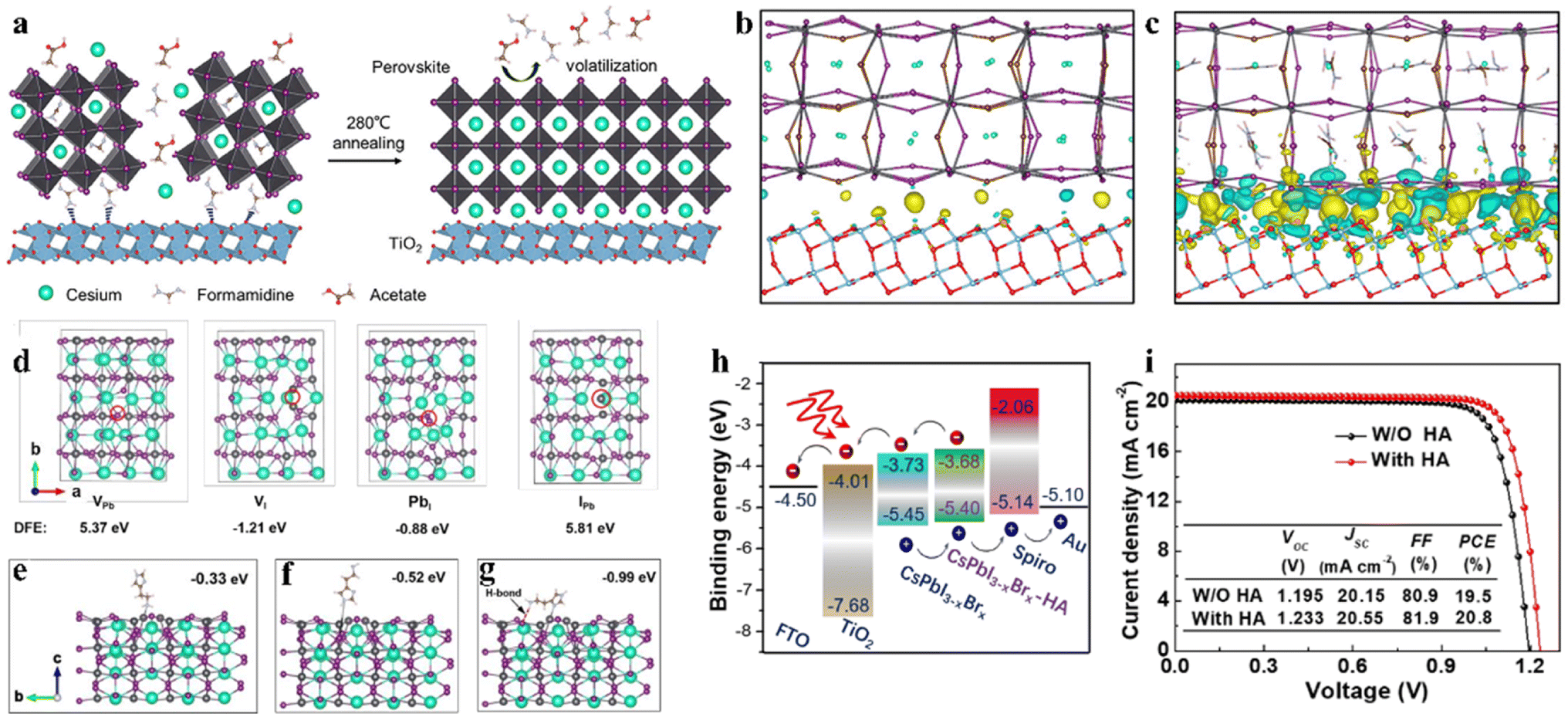 | ||
| Fig. 6 (a). Schematic illustration of the IPE process. The real-space distribution of difference charge density at the interfaces for (b) CsPb(I0.75Br0.25)3/TiO2 and (c) Cs0.5FA0.5Pb(I0.75Br0.25)3/TiO2. (a–c) Reproduced with permission.86 Copyright 2020, Elsevier. (d). Top view of the four types of surface defects and the corresponding DFT. Theoretical models of perovskite with molecular surface interaction of VI with HA: (e) –NH2 with VI; (f) imidazole ring with VI; (g) synergetic effect of both –NH2 and imidazole with VI. (h). Energy diagram of a complete IPSC passivated by HA. (i). J–V curves of the devices with and without HA passivation measured at 100 mW cm−2 irradiation in the reverse scan direction. (d–i) Reproduced with permission.88 Copyright 2021, Wiley-VCH. | ||
Interfacial modification has been proven to be an effective method for improving the perovskite phase stability, passivating defects and enhancing the performance of IPSCs. According to their DFT investigation, Gu et al. disclosed that the iodine vacancies (VI) on the surface of CsPbI3−xBrx perovskite films were the predominant defects trapping free charge carriers.88 To intentionally passivate VI in the perovskite films and prohibit the nonradiative recombination in devices, the histamine (HA) molecule was adopted, which could effectively interact with VI on the surface of the perovskite film via synergetic effects from both Lewis-base-acid reaction and hydrogen bond formation, and thus significantly reduced the number of uncoordinated Pb2+ and Pb clusters, as the mechanism illustrated in Fig. 6d–g. Moreover, the energy level position was also regulated to facilitate hole transfer at the heterojunction contact between the perovskite and HTL, as shown in Fig. 6h. Consequently, by optimizing the concentration of HA, the CsPbI3−xBrx IPSCs delivered an outstanding PCE of 20.8% (Fig. 6i) and stabilized value of 20.4%, corresponding to 72% of the SQ efficiency limit. Recently, a surface reconstruction (SR) strategy was developed by post-treating a CsPbI1.8Br1.2 film with the organic ammonium halide salt trimethylammonium chloride (TACl) to reduce the surface defect states (Fig. 7d).89 The repaired CsPbI1.8Br1.2 surface effectively inhibited nonradiative recombination and promoted hole transport, providing efficient charge recombination in the interconnecting layer in the two-terminal tandem SCs (2T-TSCs) (Fig. 7a and b). Consequently, the CsPbI1.8Br1.2 perovskite/organic 2T-TSCs yielded a PCE of 21.04% with an ultrahigh Voc of 2.05 V, as shown in Fig. 7c.
 | ||
| Fig. 7 (a). Left: device structure of all-inorganic perovskite/organic 2T TSC. Right: crystal structure of front all-inorganic perovskite CsPbI1.8Br1.2 and the molecular structures of the donor and acceptor materials in the rear cell. (b). Normalized absorption spectra of CsPbI1.8Br1.2 and PM6:Y6 films and the AM1.5G spectrum. (c). J–V curves of 2T TSC, front and rear solar cells under AM1.5G 100 mW cm−2 illumination. (d). Schematic illustration of the TACl and IPA synergistically induced SR processes. (a–d) Reproduced with permission.89 Copyright 2022, Wiley-VCH. (e). Pristine perovskite solution (left) and perovskite/FITC hybrid solution (right). (f). Schematic illustration of the interaction between FITC and perovskite. (g). Energy level diagram constructed from UV-vis and UPS measurements. (e–g) Reproduced with permission.91 Copyright 2021, Wiley-VCH. | ||
Besides, some other small molecules, such as the π-conjugated Lewis base 6TIC-4F, which contains a strong electron-donating core and 2 electron-withdrawing units, were dissolved in the anti-solvent to passivate uncoordinated defects on the surface/grain boundaries via the direct coordination of N atoms possessing lone pair electrons with the lead ion through the formation of Lewis adducts, thereby suppressing the non-radiative recombination and further increasing the PV performance.90 Later, Zhang et al. fabricated efficient and stable CsPbI1.5Br1.5 IPSCs (Fig. 7g) with a PCE of 14.05% and Voc of 1.29 V by incorporating an organic dye, i.e., fluorescein isothiocyanate (FITC), as a passivator in the perovskite precursor (Fig. 7e).91 The carboxyl and thiocyanate groups of FITC not only minimized the trap states by forming interactions with the uncoordinated Pb2+ ions, as illustrated by the mechanism in Fig. 7f, but also significantly increased the grain sizes and improved the crystallinity of the perovskite films during annealing.
4 CsPbIBr2-based IPSCs
Inorganic CsPbIBr2 perovskite as the light absorber has attracted tremendous attention due to its high-temperature stability at more than 460 °C and low phase transition temperature of about 100 °C. Ma et al. first studied CsPbIBr2 IPSCs and adopted a dual source thermal evaporation process to deposit the CsPbIBr2 perovskite light-absorber.33 The CsPbIBr2 films with an optical bandgap of 2.05 eV displayed a stable PL emission at 2.00 eV. No low-energy PL feature was observed in the CsPbIBr2 perovskite film, suggesting there is no halide segregation in the Cs mixed-halide perovskites, as depicted in Fig. 8a. The CsPbIBr2 IPSCs without the use of an HTL obtained a PCE of 4.7% under reverse scan and 3.7% under forward scan, showing a large hysteresis, as shown in Fig. 8b. Later, the same group deposited CsPbIBr2 films using the spray-assisted-solution method and achieved a PCE of 6.3% with negligible hysteresis.98 However, it was found that for the CsPb(BrxI1−x)3 family, for x > 0.4, the phase segregated into I-rich and Br-rich phases under illumination (Fig. 8c).44 Later, Li et al. observed phase segregation into an I-rich phase both at the GBs on the film surface and in the film bulk, in the form of clusters.99 A high density of mobile ions generated by phase segregation quickly moves along the GBs as ion migration “highways”, and finally piles up at the CsPbIBr2/TiO2 interface, resulting in the formation of larger injection barriers, which hamper electron extraction and lead to strong J–V hysteresis in the IPSCs (Fig. 8d–h). This explained why the planar CsPbIBr2 IPSCs exhibited severe hysteresis in the efficiency measurement, showing a PCE of up to 8.02% in the reverse scan and reduced PCE of 4.02% in the forward scan. Combining all these studies, it can be concluded that while phase separation may occur, it seems to depend on the particular films involved and how they are prepared. Table 2 summarizes the doping/low-temperature fabrication of CsPbIBr2 films and the PV performance of the corresponding IPSCs.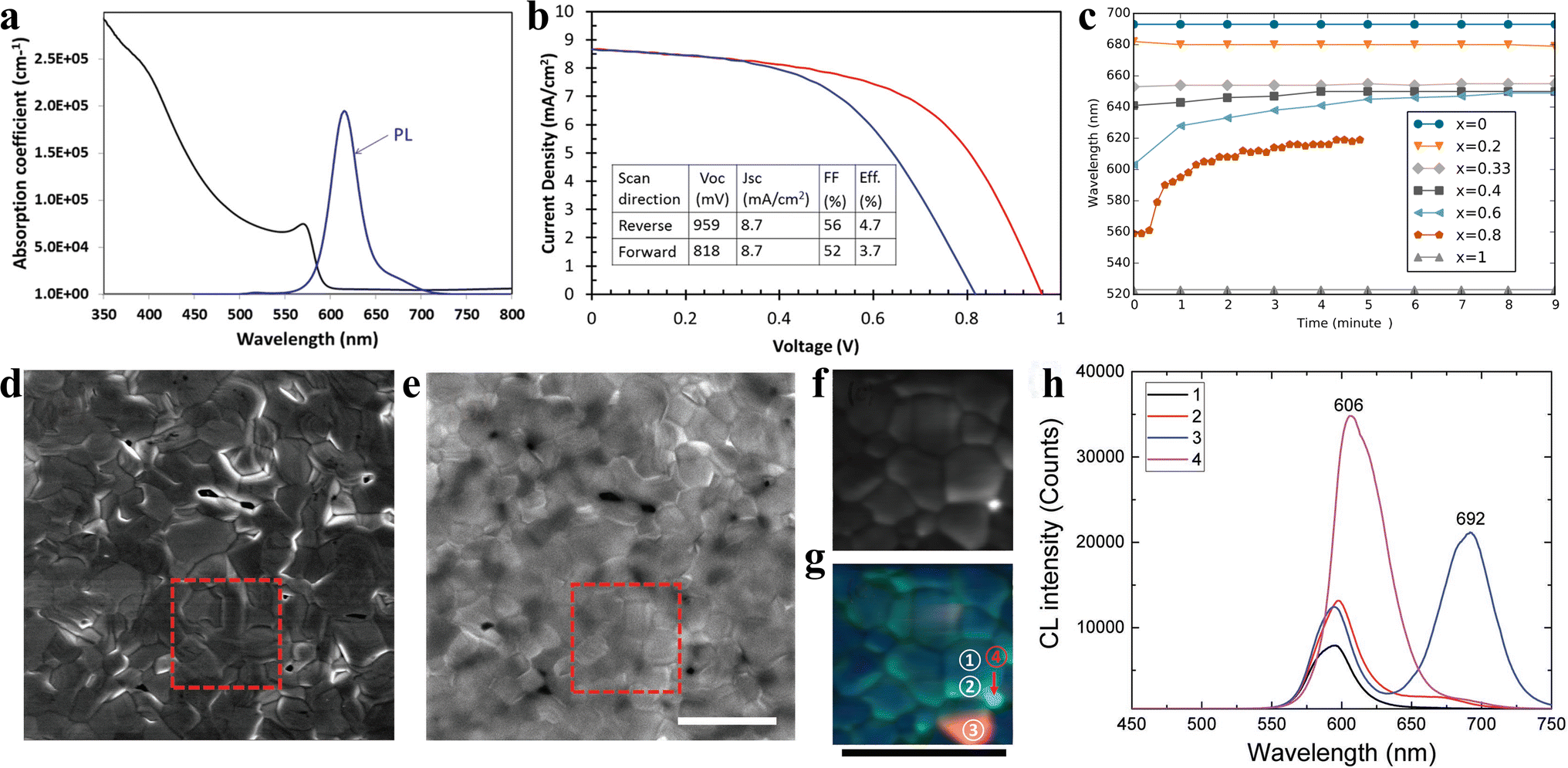 | ||
| Fig. 8 (a). Absorption coefficient and steady-state PL spectrum of CsPbIBr2 sample. (b). J–V curves of the best-performing CsPbIBr2 cell. (a and b) Reproduced with permission.33 Copyright 2016, Wiley-VCH. (c). PL peak position as a function of time for CsPb(BrxI1−x)3 materials under ∼l sun illumination. Reprinted with permission.44 Copyright 2016, the American Chemical Society. (d) Secondary electron SEM image and (e) CL PMT mapping of the CsPbIBr2 film (1024 × 1024 pixels (≈11 nm on a side); dwell time 50 μs per pixel) with the electron beam acceleration voltage of 5 kV and current of 799 pA. The square region in d was further studied by CL spectrum mapping with different spectral windows of 530–630 nm (pixel size of 40 nm × 40 nm; dwell time of 10 ms). (g). Superposition of (f). (h). CL spectra for the area inside a CsPbIBr2 grain (GI; region ① in (g)), grain boundary (GB; region ② in (g)), and I-rich phase (areas ③ and ④ in (g)). All the scale bars are 3 μm. (d–h) Reproduced with permission.99 Copyright 2017, Wiley-VCH. | ||
| Device architecture | Deposition method | Active area (cm2) | PCE (%) | Jsc (mA cm−2) | Voc (V) | FF (%) | Stability | Year (ref.) |
|---|---|---|---|---|---|---|---|---|
| FTO/c-TiO2/CsPbIBr2 (@250 °C)/Au | Dual source evaporation | 0.159 | 4.7 | 8.7 | 0.959 | 56 | — | 2016 (ref. 33) |
| FTO/bl-TiO2/mp-TiO2/CsPbIBr2 (@300 °C)/Spiro-OMeTAD/Au | Spray assisted two-step solution | 0.159 | 6.3 | 7.8 | 1.127 | 72 | — | 2016 (ref. 98) |
| FTO/In2S3/CsPbIBr2 (@160 °C)/Spiro-OMeTAD/Ag | Spin-coating | 0.11 | 5.59 | 7.76 | 1.09 | 65.94 | — | 2019 (ref. 100) |
| FTO/c-TiO2/CsPbIBr2 (@320 °C)/Spiro-OMeTAD/Au | A gas-assisted method | 0.16 | 8.02 | 9.69 | 1.227 | 67.4 | — | 2017 (ref. 99) |
| FTO/NiOx/CsPbIBr2 (@160 °C)/MoOx/Au | One-step solution | 0.09 | 5.52 | 10.56 | 0.85 | 62 | — | 2017 (ref. 101) |
| FTO/NiOx/CsPbIBr2 (@160 °C)/ZnO/Al | Single-step method | 0.04 | 5.08 | 8.53 | 0.97 | 61.4 | — | 2018 (ref. 118) |
| ITO/SnO2/CsPbIBr2 (@100 °C)/Spiro-OMeTAD/Au | Spin-coating (CB anti-solvent) | 0.16 | 9.5 | 11.52 | 1.19 | 69 | — | 2018 (ref. 119) |
| FTO/c-TiO2/m-TiO2/CsPbIBr2 (@350 °C)/carbon | Two-step solution | 0.09 | 7.36 | 13.15 | 0.99 | 57 | Decreased by 8% of initial PCE value | 2018 (ref. 111) |
| FTO/c-TiO2/m-TiO2/CsPb0.9Sn0.1IBr2 (@350 °C)/carbon | Two-step sequential solution-phase process | 0.09 | 11.33 | 14.30 | 1.26 | 63 | The encapsulated PSCs exhibited almost no degradation after being kept for >3 months at RT | 2017 (ref. 110) |
| ITO/SnO2/C60/CsPb0.75Sn0.25IBr2 (@150 °C)/Spiro-OMeTAD/Au | Spin-coating (CB anti-solvent) | 0.10 | 11.53 | 12.57 | 1.21 | 75.8 | Maintained over 90% of the initial PCE after 120 min illumination without encapsulation | 2018 (ref. 102) |
| ITO/SnO2/CsPb0.7Sn0.3IBr2 (@160 °C)/Spiro-OMeTAD/Au | One-step spin-coating | 0.06 | 14.1 | 15.5 | 1.18 | 76.7 | Retained over 75% of original PCE after 10 days exposure to air | 2022 (ref. 120) |
| FTO/c-TiO2/CsPbIBr2 (@100 °C)/carbon | One-step spin-coating | 0.09 | 6.55 | 9.11 | 1.142 | 63 | Retained 95% initial efficiency after 288 h of storage | 2018 (ref. 103) |
| ITO/NH4Cl–ZnO/CsPbIBr2 (@160 °C)/Spiro-OMeTAD/Ag | One-step spin-coating | 0.11 | 10.16 | 11.52 | 1.27 | 69.17 | Remained almost 70% of the initial value after storage for 800 h in a cabinet (RH: 15%, T: 25 °C) | 2020 (ref. 106) |
| FTO/SnO2/Cs0.99MA0.01PbIBr2 (@150 °C)/Spiro-OMeTAD/Au | One-step spin-coating | 0.06 | 10.47 | 11.94 | 1.21 | 72.5 | Retained 84% of initial PCE after 30 days | 2020 (ref. 104) |
| ITO/c-TiO2/CsPbIBr2 (@160 °C)/carbon | One-step spin-coating (Zn(Ac)2-doped) | 0.12 | 10.65 | 11.80 | 1.291 | 70 | Maintained 95% of starting PCE value after 30 days with continuous aging | 2022 (ref. 121) |
| ITO/SnO2/MgO/CsPbIBr2 (@160 °C)/Spiro-OMeTAD/Ag | One-step spin-coating | 0.11 | 11.04 | 11.70 | 1.36 | 69.35 | — | 2020 (ref. 107) |
| FTO/c-TiO2/CsPbIBr2 (@160 °C)/Spiro-OMeTAD/Au | One-step spin-coating | 0.09 | 10.78 | 11.63 | 1.25 | 74 | — | 2020 (ref. 105) |
| FTO/c-TiO2/Li–CsPbIBr2 (@280 °C)/CuPc/carbon | One-step spin-coating | 0.071 | 9.25 | 10.27 | 1.22 | 74 | Maintained 96% of initial PCE after being exposed to air with RH of 40% at 60 °C for one month | 2019 (ref. 112) |
| FTO/c-TiO2/CsPbIBr2 (@160 °C)/carbon | One-step spin-coating | 0.09 | 9.04 | 10.87 | 1.26 | 66 | Maintained 95% of initial PCE after 7d storage | 2020 (ref. 113) |
| FTO/c-TiO2/Rb or Ac co-doped CsPbIBr2 (@250 °C)/carbon | One-step spin-coating | 0.09 | 10.78 | 11.74 | 1.37 | 67 | Retained 98% of initial PCE after storage for 7 days | 2021 (ref. 114) |
| FTO/c-TiO2/CsI(PbBr2)0.95(CoCl2)0.05 (@250 °C)/Spiro-OMeTAD/Ag | One-step spin-coating | 0.07 | 10.43 | 12.48 | 1.25 | 66.88 | Maintained above 90% of initial PCE for 25 days in the air at 25 °C and RH = 20% without encapsulation | 2020 (ref. 115) |
| FTO/c-TiO2/CsPb0.99Zn0.01IBr2 (@250 °C)/Spiro-OMeTAD/Ag | One-step spin-coating | 0.07 | 10.51 | 11.92 | 1.28 | 69 | Maintained 91% of initial PCE after 30 days without encapsulation and stored at 25 °C under ambient conditions with RH of 20% | 2021 (ref. 117) |
| FTO/c-TiO2/CsPbIBr2-0.50% Cu (@225 °C)/Spiro-OMeTAD/Ag | One-step spin-coating | 0.0625 | 10.4 | 12.8 | 1.21 | 67.1 | Retained almost 75% of initial PCE when heated at 90 °C in ambient atmosphere with 30% humidity | 2020 (ref. 116) |
| ITO/ZnO/CsPbIBr2 (@140 °C)/Spiro-OMeTAD/Au | One-step spin-coating (vacuum-assisted low-temperature engineering) | 0.04 | 11.01 | 11.34 | 1.289 | 75.31 | Retained over 87% of initial PCE after being continuously heated at 80 °C in an inert atmosphere for 80 days without encapsulation | 2022 (ref. 122) |
| FTO/c-TiO2/CsPbIBr2 (@150 °C)/ZnPc/carbon | Two-step spin-coating (CB and IPA mixed anti-solvent) | 0.09 | 8.48 | 10.33 | 1.23 | 66.9 | Retained about 90% of initial efficiency after storage at 20% RH in air for 30 days | 2022 (ref. 123) |
4.1 Preparation of CsPbIBr2 films at low temperature
The high-temperature preparation of CsPbIBr2 is a major obstacle for practical applications and flexible devices. Hence, it is urgent to develop processes for the preparation of CsPbIBr2 perovskite layers at low temperature.100 A new device structure (FTO/NiOx/CsPbIBr2/MoOx/Au) of all-inorganic PSCs was developed by using a low-temperature stable-transition-film (STF) (Fig. 9a) to prepare a highly dense and pinhole-free CsPbIBr2 thin film with high crystalline quality (Fig. 9b).101 There was no phase separation in this film, but the hysteresis effect still occurred. The PCE of the device rapidly declined in the first 20 s, and gradually reached equilibrium within 100 s, which may be due to light-induced segregation. The low work function MoOx (4.3 eV) cathode buffer layer with ultra-thin thickness (4 nm) resulted in a decrease in the Schottky barrier, contact resistance and interface trap-state density (Fig. 9c), which increased the PCE of the IPSCs from 1.3% to 5.52%. Later, a series of CsPb1−xSnxIBr2 perovskite alloys was prepared through a one-step anti-solvent method at a lower annealing temperature (150 °C), which presented tunable bandgaps from 2.04 to 1.64 eV.102 Finally, the optimal CsPb0.75Sn0.25IBr2 perovskite with Eg = 1.78 eV exhibited a homogeneous and densely crystallized morphology.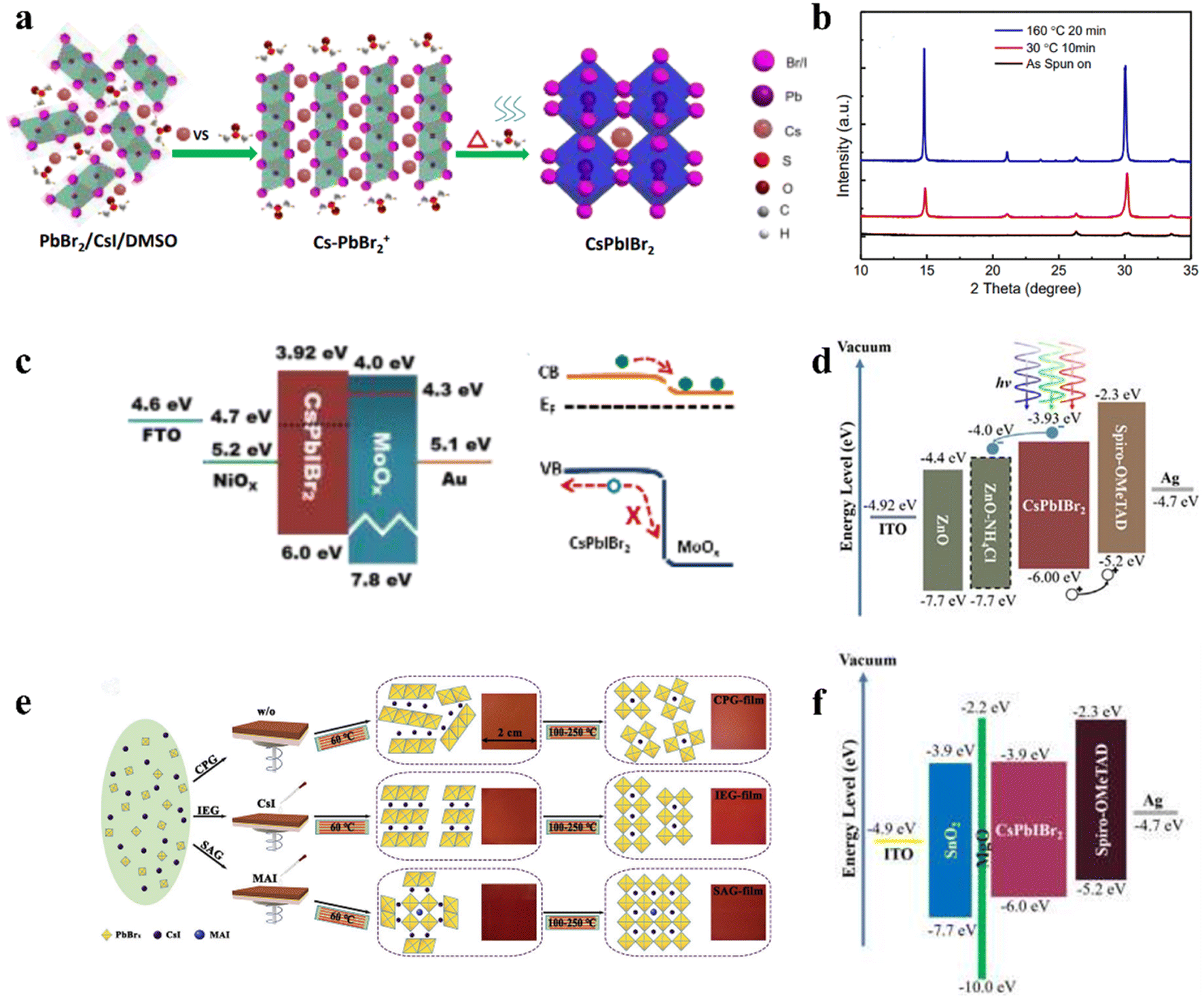 | ||
| Fig. 9 (a). CsPbIBr2 crystal growth mechanism. (b). XRD patterns of film structural evolution process. (c). Energy band diagram and surface energy bands bend downwards at the CsPbIBr2/MoOx interface. (a–c) Reproduced with permission.101 Copyright 2017, Elsevier. (d). Energy level diagrams of the device. Reproduced with permission.106 Copyright 2020, Wiley-VCH. (e). Schematic illustration of different fabrication methods for CsPbIBr2 films. Reproduced with permission.104 Copyright 2020, Wiley-VCH. (f). Energy band diagrams of the CsPbIBr2 solar cells with MgO passivation layer. Reproduced with permission.107 Copyright 2020, Wiley-VCH. | ||
Perovskite precursor engineering is an effective strategy to reduce the temperature for the fabrication of CsPbIBr2 thin films. For example, pure-phase and full-coverage CsPbIBr2 films could be obtained at a temperature as low as 100 °C by controlling the precursor solution aging time in one-step spin-coating method.103 The carbon-based IPSCs with these CsPbIBr2 films delivered a PCE of 6.55%. Later, a low-temperature seed-assisted growth (SAG) method was reported for high-quality perovskite films by treating the CsPbIBr2 precursor film with methylammonium halides (MAX, X = I, Br, and Cl), followed by annealing treatment, during which MA-perovskite seeds were formed and acted as nuclei for the growth of the CsPbIBr2 perovskite, as shown by the mechanism in Fig. 9e. The MABr-treated CsPbIBr2 perovskite (Pvsk-Br) processed at the low temperature of 150 °C showed an excellent surface morphology with micrometer-sized grains, resulting in long carrier lifetime and low trap density.104 In addition, the incorporation of n-butylammonium iodide (BAI) as an additive in the CsPbIBr2 precursor not only improved the crystallization and morphology of the perovskite layers to reduce the trap density and restrain the nonradiative recombination, but also decreased the annealing temperature.105 Consequently, the CsPbIBr2 IPSCs fabricated at 160 °C with an optimal BAI concentration of 0.1% exhibited a PCE of 10.78% and Voc of 1.25 V.
Simultaneous optimization of the perovskite layer and the ETL is an efficient way to reduce the energy losses and improve the Voc for high-performance CsPbIBr2 IPSCs. Introducing a trace of ammonium chloride (NH4Cl) into a sol–gel-derived ZnO as ETL could simultaneously improve the Voc, FF, and PCE of the CsPbIBr2 IPSCs.106 The NH4Cl-modified ZnO ETL exhibited a higher electron mobility and reduced work function, leading to a more suitable energy-level alignment between the perovskite and ETL, as shown in Fig. 9d. Finally, the CsPbIBr2 IPSCs with the configuration of ITO/NH4Cl-modified ZnO/CsPbIBr2/Spiro-OMeTAD/Ag under a low fabrication temperature of 160 °C achieved a PCE of 10.16% and outstanding Voc to 1.27 V. Later, the same group reported that the insertion of an ultrathin wide band MgO layer between the SnO2 ETL and CsPbIBr2 photo-absorber not only can passivate the undesirable recombination, and thereby enhance the Voc, but also provide a better substrate for CsPbIBr2 growth to reduce the interface δ-phase perovskite.107 Furthermore, the tunneling effect and better alignment effectively blocked holes and accelerated the movement of electrons to the electrode, as shown in Fig. 9f.
4.2 Doping strategies for high-quality CsPbIBr2 films
Under the guidance of t, compositional engineering of ABX3 is another way to modulate the crystallization of inorganic perovskite films.108,109 The introduction of different dopants can modify the surface of the perovskite crystal or incorporate them into the crystal lattice to replace one of the substituents. Doping CsPbIBr2 with Mn2+ or Sn2+ can narrow the bandgap and extend the light response region. A novel CsPb0.9Sn0.1IBr2 perovskite with an Eg of 1.79 eV (Fig. 10a) was prepared through a convenient two-step sequential solution-phase process in ambient air without the need for a glovebox or humidity control.110 Consequently, the CsPb0.9Sn0.1IBr2 IPSCs exhibited a PCE of 11.33% and Voc of 1.26 V, with a voltage loss of only 0.53 V (Fig. 10b), which is very low for a Br-rich perovskite material. The high Voc is mainly due to the better energy level matching between the VBM of the perovskite and the CBM of the ETL. Later, an Mn-doped CsPb1−xMnxI1+2xBr2−2x perovskite was synthesized in the ambient atmosphere without any humidity control.111 The Mn-doped films with appropriate dopant concentration showed better crystallinity and morphology, and a slightly decreased Eg (1.85 eV instead of 1.89 eV without Mn).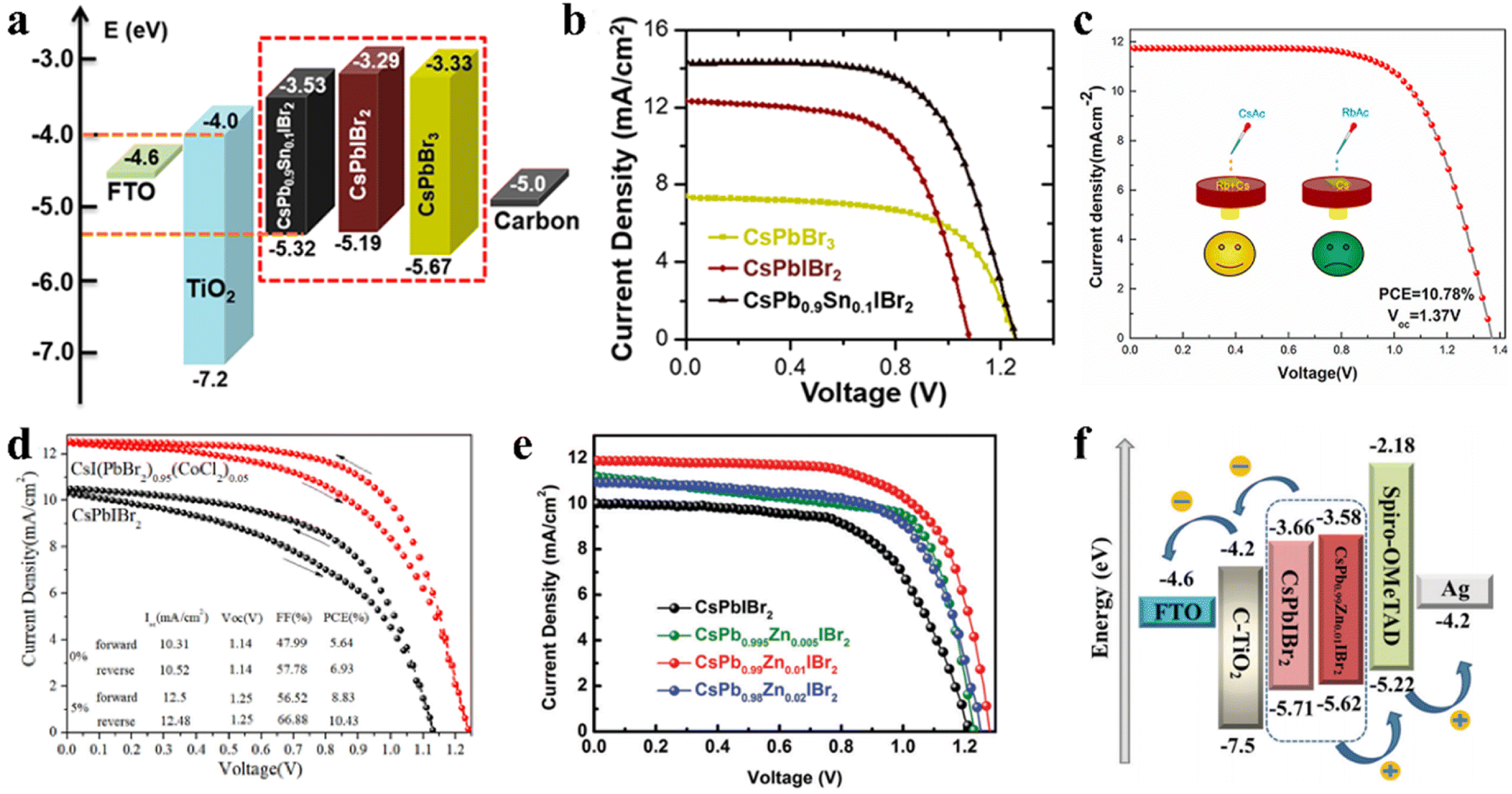 | ||
| Fig. 10 (a). Energy level diagrams of IPSCs. (b). J–V plots of IPSCs based on CsPbBr3, CsPbIBr2, and CsPb0.9Sn0.1IBr2, respectively. (a and b) Reprinted with permission.110 Copyright 2017, the American Chemical Society. (c). J–V curves of IPSCs with multi-source Rb/Ac-doped CsPbIBr2 films. Reproduced with permission.114 Copyright 2021, Elsevier. (d). J–V curves of optimal CsPbIBr2 and CsI(PbBr2)1−x(CoCl2)x devices under forward and reverse scans. Reproduced with permission.115 Copyright 2020, Elsevier. (e). J–V curves of the CsPb1−xZnxIBr2 (x = 0, 0.005, 0.01 and 0.02) IPSCs under simulated AM1.5G illumination. (f). Corresponding energy level diagrams with CsPbIBr2 and CsPb0.99Zn0.01IBr2 devices. (e and f) Reproduced with permission.117 Copyright 2021, The Royal Society of Chemistry. | ||
In addition, the Li doping strategy can improve the optical, morphological and electronic properties of CsPbIBr2 films.112 The Li-doped CsPbIBr2 films possess low trap-state densities and long carrier lifetime, contributing to a lower energy loss and a higher charge collection efficiency. In another study, by employing Zn substitution, carbon-based and HTL-free CsPbIBr2 IPSCs exhibited a PCE of 9.04% and 8.09% under low temperature annealing conditions (160 °C and 100 °C, respectively), as reported by Jiang et al.113 Later, they reported simultaneous cation/anion doping in a CsPbIBr2 film (Fig. 10c). The Rb/Ac co-doped CsPBIBr2 IPSCs exhibited a PCE of 10.78% with a large Voc of 1.37 V, originating from the long carrier lifetime and low recombination.114 To slow down the rapid formation and growth of CsPbIBr2 crystals, CoCl2 was used as a morphology controller.115 The slow crystallization resulted in low trap states and grain boundary in the CsPbIBr2 films, reducing the Eloss and enhancing the Voc by up to 1.25 V (Fig. 10d).
By doping an appropriate amount of Cu2+ (0.50 at%) in the CsPbIBr2 perovskite lattice, the high-quality CsPbIBr2 film showed increased crystallinity with expanded grain sizes, optimized energy level alignment, decreased trap density, and reduced charge recombination.116 Consequently, the CsPbIBr2-0.50% Cu-based device with the architecture of FTO/c-TiO2/CsPbIBr2-0.50% Cu/Spiro-OMeTAD/Ag exhibited a PCE of 10.4% and retained 75% of its initial PCE when heated at 90 °C in an ambient atmosphere with 30% humidity. In addition, Long et al. incorporated ZnBr2 in the CsPbIBr2 perovskite precursor and obtained CsPb1−xZnxIBr2 perovskite films using a one-step spin-coating method.117 Zn2+ doping not only can modulate the crystallization of the CsPbIBr2 perovskite film and improve the morphology to suppress charge recombination and decrease the trap states, but also regulate the energy band level of CsPbIBr2, which improved the built-in potential and Voc of the CsPbIBr2 IPSCs (Fig. 10e and f).
4.3 Strategies for improving the performance of CsPbIBr2-IPSCs
Interfacial recombination and nonradiative recombination in CsPbIBr2 IPSCs hinder the device performance. Accordingly, an effective way to suppress the interfacial recombination is to construct more suitable energy level alignments between the perovskite film and the ETL or HTL.124–128 For example, a surface modification strategy was proposed for the ETL/perovskite interface by employing SmBr3, wherein a gradient energy band is formed at the interface with an outstanding hole-blocking effect, as shown by the mechanism in Fig. 11a.129 SmBr3 interface modification could not only improve the charge extraction, but also suppressed the charge recombination occurring at the interface and the nonradiative recombination inside the perovskite material. In another study, band alignment engineering at the TiO2/CsPbIBr2 heterojunction by modifying TiO2 with CsBr clusters was reported.130 The CsBr modifier causes a beneficial increase in the CBM from −4.00 to −3.81 eV for the TiO2 ETL, thus promoting favorable band alignment at the heterojunction, suppressing recombination, and improving the extraction and transport of charge carriers. Table 3 summarizes several strategies for the preparation of high-quality CsPbIBr2 films and the PV performance of the corresponding IPSCs.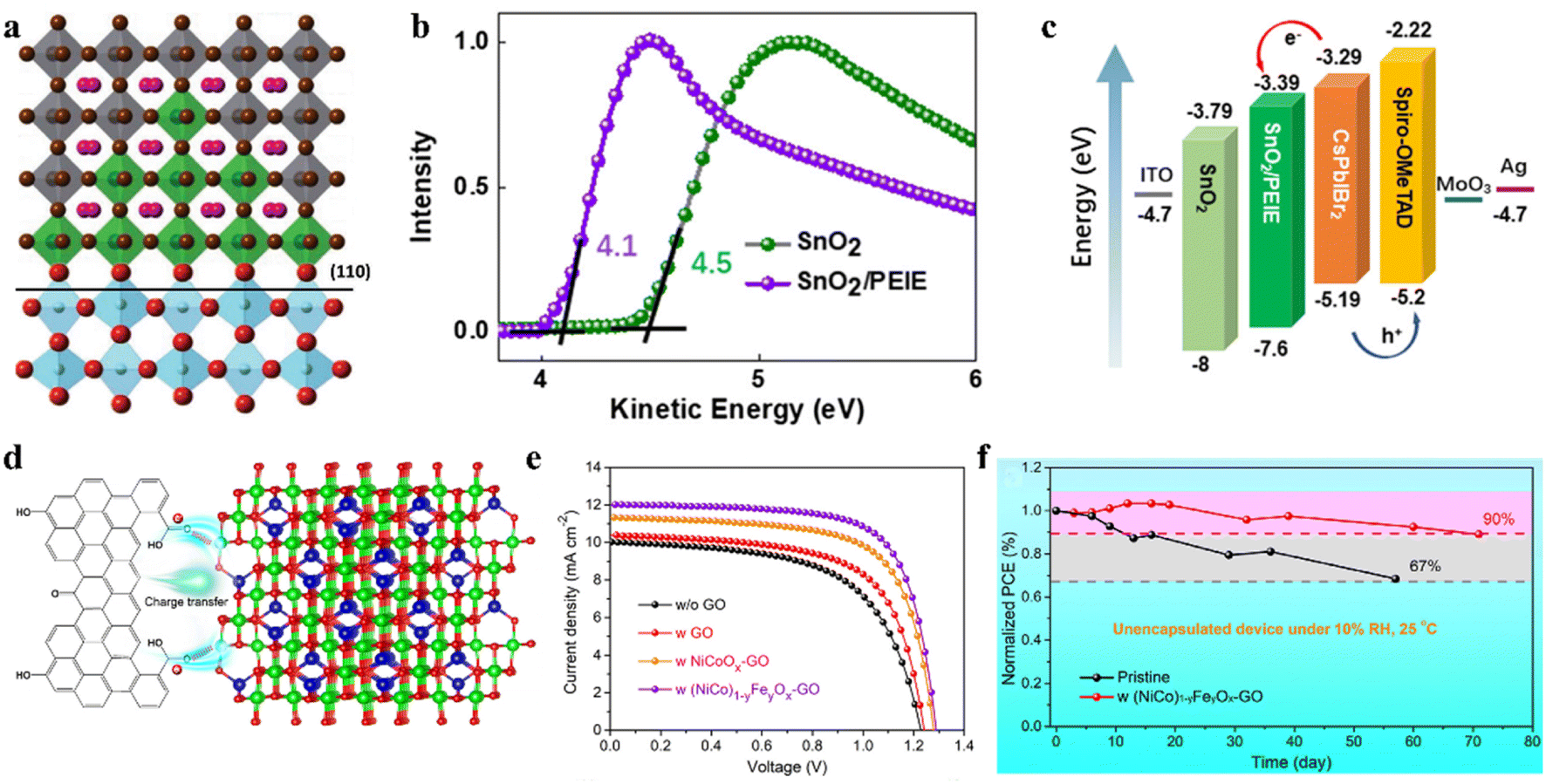 | ||
| Fig. 11 (a). Schematic diagram depicting the SIM on the (110) crystal plane of rutile-TiO2 (for octahedra, grey: PbI2Br4, green: SmBr6, blue: TiO6; for spheres, and pink: Cs). Reproduced with permission.129 Copyright 2018, Wiley-VCH. b. UPS spectra of SnO2 and SnO2/PEIE. (c). Energy-level diagram of IPSCs. (b and c) Reproduced with permission.132 Copyright 2021, Wiley-VCH. (d). Schematic diagram of charge transfer from GO to inorganic NPs. (e). Characteristic J–V curves of various solar cells. (f). Long-term stability of IPSCs. (d–f) Reproduced with permission.134 Copyright 2021, Wiley-VCH. | ||
| Device architecture | Deposition method | Active area (cm2) | PCE (%) | Jsc (mA cm−2) | Voc (V) | FF (%) | Stability | Year (ref.) |
|---|---|---|---|---|---|---|---|---|
| FTO/c-TiO2/CsPbIBr2 (@280 °C)/carbon | Spin-coating (CsI treatment) | 0.09 | 9.16 | 10.66 | 1.245 | 69 | Retained 90% over 60 days and 97% over 7 days of initial efficiency, stored controllably in ≈45% RH at 25 °C or 85 °C at zero humidity, respectively | 2018 (ref. 141) |
| FTO/NiOx/CsPbIBr2 (@160 °C)/ZnO/Al | Single-step method | 0.04 | 5.08 | 8.53 | 0.97 | 61.4 | — | 2018 (ref. 118) |
| FTO/c-TiO2/CsPbIBr2 (@280 °C)/carbon | One-step spin-coating (light-process) | 0.09 | 8.60 | 11.17 | 1.283 | 60 | — | 2019 (ref. 142) |
| FTO/c-TiO2/CsPbIBr2 (@120 °C)/Spiro-OMeTAD/Au | Spin-coating (DEE anti-solvent) | 0.10 | 9.17 | 10.24 | 1.20 | 74.6 | Maintained 90% of the initial PCE in 40% humidity ambient | 2019 (ref. 143) |
| ITO/SnO2/CsPbIBr2 (@280 °C)/Spiro-OMeTAD/Ag | One-step spin-coating (pre-heating process) | — | 9.86 | 10.69 | 1.267 | 71 | Retained ∼80% of initial efficiency over 72 h | 2019 (ref. 144) |
| FTO/c-TiO2(SmBr3)/CsPbIBr2 (@225 °C)/Spiro-OMeTAD/Au | One-step spin-coating | 0.09 | 10.88 | 12.75 | 1.17 | 73 | — | 2019 (ref. 129) |
| FTO/c-TiO2(CsBr)/CsPbIBr2 (@280 °C)/carbon | Two-step spin-coating | 0.09 | 10.71 | 11.80 | 1.261 | 72 | — | 2019 (ref. 130) |
| ITO/SnO2/CsPbIBr2 (@160 °C)/carbon | One-step spin-coating | 0.08 | 7.00 | 8.50 | 1.23 | 67 | Retained 95.5% of initial performance at 90 °C in air without encapsulation | 2019 (ref. 131) |
| FTO/c-TiO2/CsPbIBr2 (@160 °C)/Spiro-OMeTAD/Au | One-step spin-coating | 0.09 | 7.31 | 8.80 | 1.28 | 64.9 | — | 2018 (ref. 137) |
| FTO/c-TiO2/CsPbIBr2 (@260 °C)/NP-GO/carbon | One-step spin-coating | 0.09 | 10.95 | 12.03 | 1.29 | 70.58 | Retained 90% of initial PCE after aging in 10% RH air condition for 70 days without encapsulation | 2021 (ref. 134) |
| ITO/c-TiO2/CsPbIBr2 (@160 °C)/BHJ/carbon | One-step spin-coating | — | 11.54 | 11.79 | 1.31 | 74.47 | — | 2021 (ref. 135) |
| FTO/c-TiO2/CsPbIBr2 (@280 °C)/CsPbI3 QDs/Spiro-OMeTAD/Au | One-step spin-coating | — | 10.32 | 11.09 | 1.20 | 77.7 | Maintained 90% of initial PCE without encapsulation devices stored in air (RH: 25%, T: 25 °C) | 2021 (ref. 136) |
| FTO/c-TiO2/PEG:CsPbIBr2 (@200 °C)/Spiro-OMeTAD/Ag | One-step spin-coating | 0.078 | 11.10 | 12.25 | 1.21 | 74.82 | Retained over 90% of the initial PCE after 600 h storage in ambient condition without encapsulation | 2020 (ref. 138) |
| FTO/c-TiO2/CsPbIBr2 (@280 °C)/Spiro-OMeTAD/Au | One-step spin-coating | 0.16 | 10.1 | 12.11 | 1.13 | 74 | Retained 96% of initial PCE for 30 days under 40% RH | 2021 (ref. 139) |
| FTO/c-TiO2/CsPb(SO3)IBr2 (@225 °C)/Spiro-OMeTAD/Au | One-step spin-coating | 0.09 | 10.57 | 12.27 | 1.21 | 71 | Maintained over 80% of initial PCE after aging for 198 h in air without encapsulation | 2020 (ref. 140) |
| FTO/c-TiO2/CsPbIBr2 (@225 °C)/Spiro-OMeTAD/Au | One-step spin-coating | 0.09 | 10.04 | 11.35 | 1.23 | 72 | — | 2021 (ref. 145) |
| FTO/c-TiO2/CsPbIBr2 (@260 °C)/carbon | One-step spin-coating | 0.09 | 10.61 | 11.58 | 1.293 | 70.86 | Remained 56% of the initial PCE over 28 h with a humidity of 50% and a temperature of 25 °C | 2022 (ref. 146) |
| FTO/c-TiO2/CsPbIBr2 (@260 °C)/carbon | One-step spin-coating (SBTCl post-treatment) | 0.09 | 10.56 | 11.43 | 1.327 | 69.7 | After storage in 5% RH without encapsulation over 110 days and persistent light irradiation over 16 h in 50% RH condition | 2022 (ref. 147) |
| FTO/c-TiO2/CsPbIBr2 (@280 °C)/Spiro-OMeTAD/Au | One-step spin-coating (GuaSCN additive) | 0.04 | 10.90 | 12.05 | 1.23 | 73.71 | Retained ∼95% of initial value after being stored for over 600 h without encapsulation in air | 2022 (ref. 148) |
An interface engineering process was developed for SnO2 ETL surface passivation by employing an SnCl2 solution.131 Surface passivation of SnO2 can not only accelerate electron extraction from the perovskite film, but also effectively suppress the recombination at the interface between the CsPbIBr2 perovskite and SnO2 due to the higher recombination resistance. Later, an interfacial engineering strategy through the insertion of a thin polyethylenimine ethoxylated (PEIE) film between the SnO2 ETL and perovskite film was employed to reduce the energy loss in CsPbIBr2 IPSCs.132 The PEIE as a modifier showed positive effects on the device performance owing to several reasons, as follows: (1) the interactions between the amino groups of PEIE and CsPbIBr2 film can improve the crystallinity and enlarge the grain sizes of the perovskite during film formation. (2) The favorable energy-level alignment between SnO2/PEIE and CsPbIBr2 perovskite can maximize the device built-in potential (Fig. 11b). (3) The passivation effects of PEIE on the perovskite can alleviate the nonradiative recombination at the interface and enhance the charge extraction ability (Fig. 11c). Finally, the SnO2/PEIE-based CsPbIBr2 IPSCs showed a remarkable Voc of 1.29 V and PCE of 11.2%. Moreover, the SnO2/PEIE-based CsPbIBr2 IPSCs maintained over 80% of their initial value after continuous one sun illumination for 500 h. Besides, a CsBr dual-interface modification strategy was used to modify both surfaces of the CsPbIBr2 perovskite with the traditional configuration of FTO/TiO2/CsPbIBr2/Spiro-OMeTAD/Au.133 The TiO2/perovskite interface modification reduced the pinhole and trap-state densities, and regulation of perovskite/Spiro-OMeTAD produced a smoother surface and better crystallinity. Consequently, the synergistic effects of both modifications led to a PCE of 10.33% with a promising Voc of 1.24 V. In addition, the optimized CsPbIBr2 IPSCs retained 60% of their initial efficiency after 60 h of aging in the ambient atmosphere.
In addition to optimizing the ETL/perovskite interface, perovskite/carbon interfacial engineering can boost the performance of CsPbIBr2 IPSCs. For instance, inorganic (NiCo)1−yFeyOx nanoparticle-decorated graphene oxide (GO) was used as a hole collection layer in all-inorganic CsPbIBr2 PSCs with the architecture of FTO/c-TiO2/CsPbIBr2/NP-GO/carbon.134 The introduction of high-valance-state Fe3+ in NiCoOx induced the formation of more interstitial oxygen atoms and withdrew some electrons from the Ni2+/Co2+ ions. The particle electrons for the oxygen-containing groups in the GO surface spontaneously transferred to the inorganic NPs owing to their electropositivity to minimize the charge localization of GO, thus forming p-type-doped GO and an oriented dipole moment from GO to (NiCo)1−yFeyOx, as shown by the mechanism in Fig. 11d. Consequently, the NP-GO-tailored CsPbIBr2 IPSCs delivered a PCE of 10.95% and retained 90% of their initial efficiency after aging in 10% RH ambient conditions for 70 days owing to the self-encapsulation effect (Fig. 11e and f). In another study, a thin bulk-heterojunction (BHJ) layer (19 nm) consisting of poly(3-hexylthiophene-2,5-diyl) and [6,6]-phenyl methyl C61 butyric acid methyl ester (P3HT:PCBM) was integrated in CsPbIBr2 IPSCs with the configuration of ITO/TiO2/CsPbIBr2/BHJ/carbon.135 The introduction of the thin BHJ layer led to an expanded light absorption range, better charge transfer dynamics, suppressed interfacial energy loss in the CsPbIBr2/BHJ film and CsPbIBr2/BHJ/carbon, and improved long-term stability. The CsPbIBr2 IPSCs with an integrated BHJ layer showed a PCE of 11.54%.
QDs films can also be adopted as functional layers for PV devices with other bulk absorbers layers. For example, Sr-doped CsPbI3 QDs (Sr-CsPbI3 QDs) were introduced as an interfacial layer in CsPbIBr2 IPSCs to improve the device performance.136 The Sr-CsPbI3 QDs were synthesized by using SrCl2 as a co-precursor. The modification of the Sr-CsPbI3 QD interface not only optimized the charge transfer process and suppressed the interface recombination between the perovskite and HTL, but also restrained the nonradiative recombination in the CsPbIBr2 perovskite film.
The defect states at the grain boundaries and on the surface of the CsPbIBr2 polycrystalline film led to nonradiative carrier recombination, which reduced the Voc and final PCE of the corresponding PSCs. The combination of functional compounds in the perovskite precursor solution is considered to be an effective method to assist the formation of high-quality perovskite films. The intramolecular interactions between the perovskite precursors and these new compounds have important effects on the crystal dynamics of the perovskite. For example, high-quality CsPbIBr2 films were obtained by mixing a small amount of polyethylene glycol (PEG).137 PEG can not only improve coverage of the CsPbIBr2 perovskite film on the TiO2 layer, but also improve the wettability of the precursor solution. The self-assembled PEG network can slow down crystal growth and restrain the aggregation of the perovskite crystals during the process of perovskite phase formation, which effectively passivates the defect states at the grain boundaries and surface of the CsPbIBr2 bulk film. Later, the Lewis base PEG was also adopted as an additive to modify a CsPbIBr2 perovskite film. The PEG:CsPbIBr2 film exhibited suppressed non-radiative electron–hole recombination, a favorable energy band structure and less sensitivity to moisture, which originated from the reduced crystallization rate and strong interaction with Pb2+ (Fig. 12a). Ultimately, the device based on PEG:CsPbIBr2 delivered a PCE of 11.10%. Moreover, the PEG-modified device showed excellent long-term stability, retaining over 90% of its initial PCE after 600 h storage in ambient conditions without encapsulation, as shown in Fig. 12b.138 In addition, a high-quality CsPbIBr2 perovskite film was prepared by combining both substrate preheat treatment (SPT) and NH4PF6 precursor additive engineering.139 Sulfamic acid sodium salt (SAS) was also utilized as an additive to optimize the CsPbIBr2 perovskite film.140 SAS not only can regulate the crystallization process, resulting in a high-quality perovskite, but also possibly introduce an additional internal electric field effect, which favors electron transport and injection due to the inhomogeneous ion distribution.
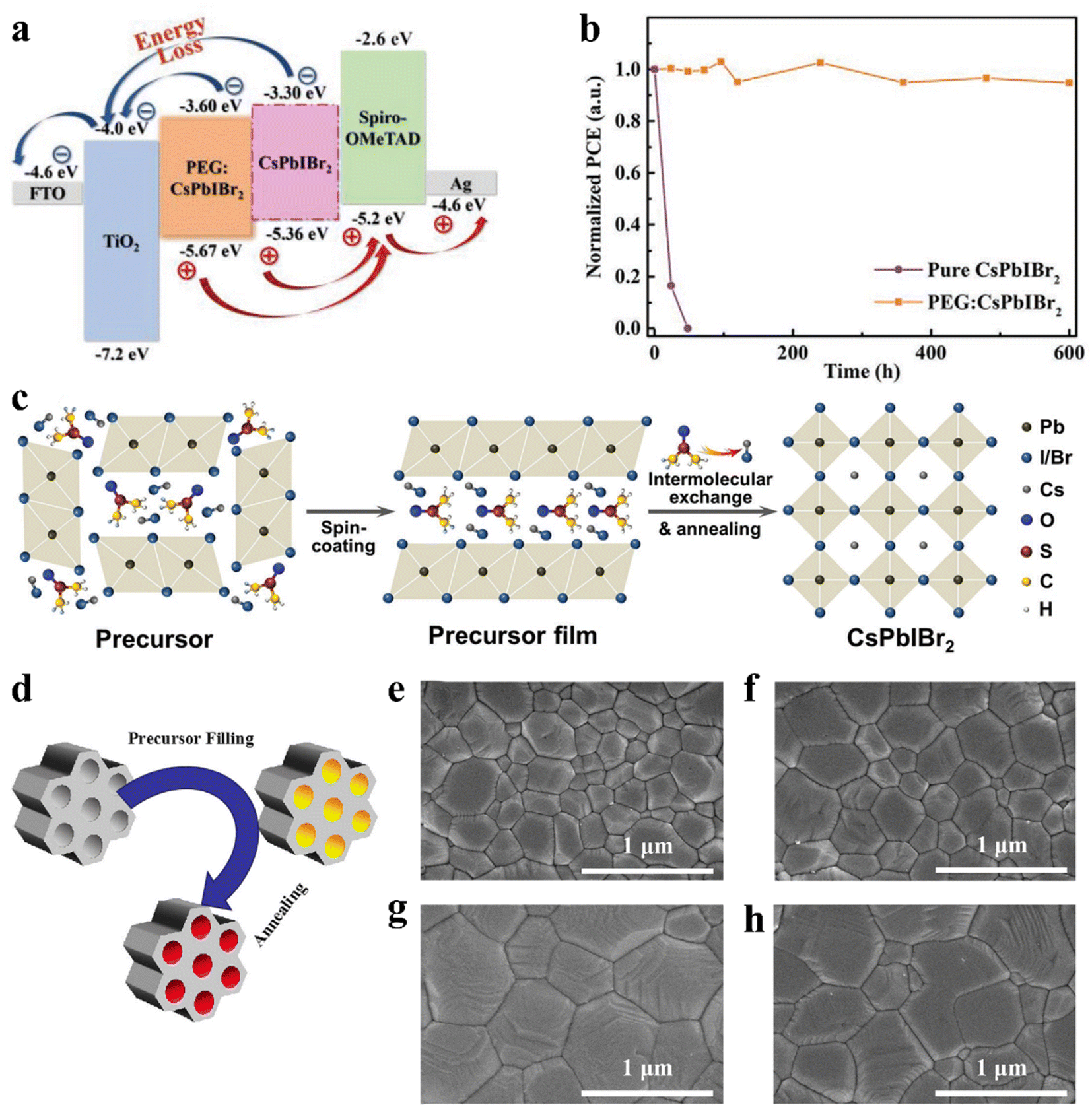 | ||
| Fig. 12 (a). Corresponding energy band diagrams of the cell. (b). Time-dependent (RH of 35% and temperature of 25 °C) of normalized PCE for the devices with and without PEG. (a and b) Reproduced with permission.138 Copyright 2020, Wiley-VCH. (c). Illustration of intermolecular exchange strategy. Reproduced with permission.141 Copyright 2018, Wiley-VCH. (d). Schematic diagram of the nanoconfined crystallization of CsPbIBr2 in ZrSBA-15. Top-view SEM images of various CsPbIBr2 perovskite films: (e). without ZrSBA-15; (f). with 0.01 wt% ZrSBA-15; g. with 0.1 wt% ZrSBA-15; and (h). with 1 wt% ZrSBA-15. (d–h) Reproduced with permission.145 Copyright 2021, Wiley-VCH. | ||
To improve the PV performance of CsPbIBr2 IPSCs, many other effective strategies have been extensively explored, such as post-treatment of the perovskite films, surface passivation and crystallization engineering. An intermolecular exchange strategy for CsPbIBr2 films was presented, wherein an optimized methanol solution of CsI was spin-coated on the CsPbIBr2 precursor film via the conventional one-step solution route (Fig. 12c).141 The resulting CsPbIBr2 films consisted of high crystallinity with few grain boundaries, which did not exhibit segregation compared to the same films without the CsI treatment exhibiting phase-segregation. A light-processing strategy was developed to produce a full-coverage, pure-phase CsPbIBr2 film.142 The CsPbIBr2 precursor films formed by the one-step spin-coating route were exposed in a simulated one-sun source for a duration of 60 min, followed by thermal annealing.
In another study, the anti-solvent and organic ion surface passivation strategies were adopted to precisely control the growth of CsPbIBr2 crystals.143 A high-quality CsPbIBr2 film was successfully obtained by introducing diethyl ether as the anti-solvent to improve the film coverage, crystallization, and homogeneous packing of the grains. Furthermore, guanidinium surface passivation can restrain the formation of the pinholes by assisting the secondary growth of the CsPbIBr2 film, which can suppress the formation of iodide vacancies and inactivation of the uncoordinated iodide species in the bulk and at the grain boundaries. Guo et al. reported a pre-heating-assisted one-step spin-coating method,144 where during spin-coating, the high-temperature substrate accelerates the volatilization of the solvent molecules, resulting in the complete coverage and higher crystallization of CsPbIBr2 films. By optimizing the substrate-preheating temperature, the IPSCs exhibited a PCE of 9.86% with a stabilized output of 8.78% and high Voc of 1.267 V. Besides, nanoconfined crystallization is considered a novel and effective strategy because of the absence of chemical reactions. 1D ordered mesoporous silica is introduced into inorganic perovskite precursors to facilely induce nanoconfined crystallization, as illustrated by the mechanism in Fig. 12d. Zr-doped SBA-15 (ZrSBA-15) nanoplatelets with suitable sizes were synthesized and added to the perovskite precursors to prepare 1D CsPbIBr2 perovskite monocrystals, facilitating charge transport and extraction.145 ZrSBA-15 is not only beneficial for the crystallization and morphology of the perovskite (Fig. 12e–h), but also reduces the defect density and improves the film stability.
5 Summary and prospect
Herein, the latest research progress of mixed-halide IPSCs was reviewed, including CsPbIxBryCl3−x−y- and CsPbIBr2-based IPSCs. Significant progress has been made in the preparation of stable and efficient mixed-halide IPSCs. The bandgaps and stability of mixed-halide perovskites are superior to that of pure halide inorganic perovskites. Compositional engineering has been widely used in mixed-halide IPSCs, which is beneficial to improve the phase stability and reduce the defect density.Crystallization and interface engineering have been extensively developed to obtain high-quality mixed-halide IPSCs. The combination of functional compounds in the perovskite precursor solution can assist in the formation of high-quality mixed-halide inorganic perovskite films. Intramolecular interactions between the perovskite precursors and these particular compounds have important effects on the crystal dynamics of the perovskite. Moreover, perovskite precursor engineering has been proven to be effective in reducing the temperature for the preparation of mixed-halide inorganic perovskite thin films. In addition, interfacial modification can effectively improve the perovskite phase stability, passivate defects and enhance the performance of mixed-halide IPSCs. Therefore, constructing more suitable energy level alignments between the perovskite film and the ETL or HTL can effectively suppress the interfacial recombination.
The efficiency of the reported mixed-halide IPSCs is significantly lower than that of organic–inorganic hybrid PSCs, and their stabilities are far from reaching commercial PV applications. Thus, effective strategies need to be selected and/or developed to further improve the PCE and stability for facilitating the PV application of mixed-halide IPSCs. At present, the research on large-scale modules is insufficient. For the manufacturing of large-area PV device modules, the low-temperature preparation of functional layers can simplify the fabrication process and reduce the industrialization cost. For the normal n–i–p structure, TiO2 and SnO2 prepared by low-temperature solution are better choices as the ETLs.
The mixed-halide inorganic perovskites have reasonable bandgaps and show great potential in semitransparent and tandem PV applications. Thus far, there is not enough research on multi-junction tandem and flexible PV applications. To maximize the light spectrum utilization, it is an inevitable choice to develop multi-junction tandem solar cells with mixed-halide inorganic perovskite as the top cells. Because the HTLs, such as undoped HTM and NiOx, can be processed at low temperatures, it is easier to prepare flexible PSCs adopting the inverted p–i–n architecture. Besides, graphene and its derivatives are expected to be utilized as charge transport materials or electrodes in flexible PSCs.
Mixed-halide inorganic perovskites have excellent thermal stability and promising theoretical efficiency. However, their stability lags behind the rapid growth in PCE, and thus becomes the next major challenge. To produce high-quality mixed-halide inorganic perovskite and efficient PV devices, effective strategies should be continuously explored. Once the stability of mixed-halide IPSCs is resolved, either in a single junction or multi-junction tandem with silicon cells, it will be a major development in the PV field.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Sichuan Science and Technology Program (No. 2022YFG0295, 2022NSFSC1200), Dazhou Science and Technology Project (No. 20YYJC0003).References
- J. H. Noh, S. H. Im, J. H. Heo, T. N. Mandal and S. Il Seok, Nano Lett., 2013, 13, 1764–1769 CrossRef CAS PubMed.
- S. D. Stranks, G. E. Eperon, G. Grancini, C. Menelaou, M. J. P. Alcocer, T. Leijtens, L. M. Herz, A. Petrozza and H. J. Snaith, Science, 2013, 342, 341 CrossRef CAS PubMed.
- Q. Lin, A. Armin, R. C. R. Nagiri, P. L. Burn and P. Meredith, Nat. Photonics, 2015, 9, 106 CrossRef CAS.
- W. Nie, H. Tsai, R. Asadpour, A. J. Neukirch, G. Gupta, J. J. Crochet, M. Chhowalla, S. Tretiak, M. A. Alam and H. Wang, Science, 2015, 347, 522 CrossRef CAS PubMed.
- Q. Dong, Y. Fang, Y. Shao, P. Mulligan, J. Qiu, L. Cao and J. Huang, Science, 2015, 347, 967 CrossRef CAS PubMed.
- D. W. DeQuilettes, S. M. Vorpahl, S. D. Stranks, H. Nagaoka, G. E. Eperon, M. E. Ziffer, H. J. Snaith and D. S. Ginger, Science, 2015, 348, 683 CrossRef CAS PubMed.
- A. K. Jena, A. Kulkarni and T. Miyasaka, Chem. Rev., 2019, 119, 3036 CrossRef CAS PubMed.
- T. Miyasaka, A. Kojima, K. Teshima and Y. Shirai, J. Am. Chem. Soc., 2009, 131, 6050 CrossRef PubMed.
- H. Min, M. Kim, S. U. Lee, H. Kim, G. Kim, K. Choi, J. H. Lee and S. Il Seok, Science, 2019, 366, 749 CrossRef CAS PubMed.
- H. Lu, Y. Liu, P. Ahlawat, A. Mishra, W. R. Tress, F. T. Eickemeyer, Y. Yang, F. Fu, Z. Wang, C. E. Avalos, B. I. Carlsen, A. Agarwalla, X. Zhang, X. Li, Y. Zhan, S. M. Zakeeruddin, L. Emsley, U. Rothlisberger, L. Zheng, A. Hagfeldt and M. Grätzel, Science, 2020, 370, eabb8985 CrossRef CAS PubMed.
- H. Min, M. Kim, S.-U. Lee, H. Kim, G. Kim, K. Choi, J. H. Lee and S. Il Seok, Science, 2020, 370, 749 CrossRef PubMed.
- G. Kim, H. Min, K. S. Lee, D. Y. Lee, S. M. Yoon and S. Il Seok, Science, 2020, 370, 108 CrossRef CAS PubMed.
- T. G. P. Jason, J. Yoo1, G. Seo, M. R. Chua, C. S. M. Yongli Lu, F. Rotermund, Y.-Ki Kim, N. JoongJeon, J. S. Correa-Baena, C. B. Juan-Pablo, V. Bulović, S. S. Shin and M. G. Bawendi1, Nature, 2021, 590, 587 CrossRef PubMed.
- J. Jeong, M. Kim, J. Seo, H. Lu, P. Ahlawat, A. Mishra, Y. Yang, M. A. Hope, F. T. Eickemeyer, M. Kim, Y. J. Yoon, I. W. Choi, B. P. Darwich, S. J. Choi, Y. Jo, J. H. Lee, B. Walker, S. M. Zakeeruddin, L. Emsley, U. Rothlisberger, A. Hagfeldt, D. S. Kim, M. Grätzel and J. Y. Kim, Nature, 2021, 592, 381 CrossRef CAS PubMed.
- W. Hui, L. Chao, H. Lu, F. Xia, Q. Wei, Z. Su, T. Niu, L. Tao, B. Du, D. Li, Y. Wang, H. Dong, S. Zuo, B. Li, W. Shi, X. Ran, P. Li, H. Zhang, Z. Wu, C. Ran, L. Song, G. Xing, X. Gao, J. Zhang, Y. Xia, Y. Chen and W. Huang, Science, 2021, 371, 1359 CrossRef CAS PubMed.
- https://www.nrel.gov/pv/assets/pdfs/best-research-cell-efficiencies-rev220630.pdf, n.d., DOI: https://www.nrel.gov/pv/assets/pdfs/best-research-cell-efficiencies-rev220630.pdf.
- T. Leijtens, G. E. Eperon, S. Pathak, A. Abate, M. M. Lee and H. J. Snaith, Nat. Commun., 2013, 4, 2885 CrossRef PubMed.
- T. Supasai, N. Rujisamphan, K. Ullrich, A. Chemseddine and T. Dittrich, Appl. Phys. Lett., 2013, 103, 183906 CrossRef.
- A. Dualeh, P. Gao, S. Il Seok, M. K. Nazeeruddin and M. Grätzel, Chem. Mater., 2014, 26, 6160 CrossRef CAS.
- B. Conings, J. Drijkoningen, N. Gauquelin, A. Babayigit, J. D'Haen, L. D'Olieslaeger, A. Ethirajan, J. Verbeeck, J. Manca, E. Mosconi, F. De Angelis and H. G. Boyen, Adv. Energy Mater., 2015, 5, 1500477 CrossRef.
- N. Aristidou, I. Sanchez-Molina, T. Chotchuangchutchaval, M. Brown, L. Martinez, T. Rath and S. A. Haque, Angew. Chem., Int. Ed., 2015, 54, 8208 CrossRef CAS PubMed.
- E. Smecca, Y. Numata, I. Deretzis, G. Pellegrino, S. Boninelli, T. Miyasaka, A. La Magna and A. Alberti, Phys. Chem. Chem. Phys., 2016, 18, 13413 RSC.
- N. Aristidou, C. Eames, I. Sanchez-Molina, X. Bu, J. Kosco, M. Saiful Islam and S. A. Haque, Nat. Commun., 2017, 8, 15218 CrossRef PubMed.
- Y. Rong, L. Liu, A. Mei, X. Li and H. Han, Adv. Energy Mater., 2015, 5, 1501066 CrossRef.
- T. A. Berhe, W. N. Su, C. H. Chen, C. J. Pan, J. H. Cheng, H. M. Chen, M. C. Tsai, L. Y. Chen, A. A. Dubale and B. J. Hwang, Energy Environ. Sci., 2016, 9, 323 RSC.
- N. H. Tiep, Z. Ku and H. J. Fan, Adv. Energy Mater., 2016, 6, 1501420 CrossRef.
- P. Yu, W. Zhang, F. Ren, J. Wang, H. Wang, R. Chen, S. Zhang, Y. Zhang, Z. Liu and W. Chen, J. Mater. Chem. C, 2022, 10, 4999 RSC.
- Y. Pan, Y. Zhang, W. Kang, N. Deng, Z. Yan, W. Sun, X. Kang and J. Ni, Mater. Adv., 2022, 3, 4053 RSC.
- Y. Yuan, G. Yan, R. Hong, Z. Liang and T. Kirchartz, Adv. Mater., 2022, 2108132 CrossRef CAS PubMed.
- J. Ma, M. Qin, P. Li, L. Han, Y. Zhang and Y. Song, Energy Environ. Sci., 2022, 15, 413 RSC.
- M. Kulbak, D. Cahen and G. Hodes, J. Phys. Chem. Lett., 2015, 6, 2452 CrossRef CAS PubMed.
- M. Kulbak, S. Gupta, N. Kedem, I. Levine, T. Bendikov, G. Hodes and D. Cahen, J. Phys. Chem. Lett., 2016, 7, 167 CrossRef CAS PubMed.
- Q. Ma, S. Huang, X. Wen, M. A. Green and A. W. Y. Ho-Baillie, Adv. Energy Mater., 2016, 6, 1502202 CrossRef.
- R. J. Sutton, G. E. Eperon, L. Miranda, E. S. Parrott, B. A. Kamino, J. B. Patel, M. T. Hörantner, M. B. Johnston, A. A. Haghighirad, D. T. Moore and H. J. Snaith, Adv. Energy Mater., 2016, 6, 1502458 CrossRef.
- Z. Guo, A. K. Jena, G. M. Kim and T. Miyasaka, Energy Environ. Sci., 2022, 15, 3171 RSC.
- Z. Guo, S. Zhao, N. Shibayama, A. Kumar Jena, I. Takei and T. Miyasaka, Adv. Funct. Mater., 2022, 32, 2207554 CrossRef CAS.
- Q. A. Akkerman, V. D'Innocenzo, S. Accornero, A. Scarpellini, A. Petrozza, M. Prato and L. Manna, J. Am. Chem. Soc., 2015, 137, 10276 CrossRef CAS PubMed.
- G. E. Eperon, R. J. Sutton, A. A. Haghighirad, H. J. Snaith, G. M. Paternò, A. Zampetti and F. Cacialli, J. Mater. Chem. A, 2015, 3, 19688 RSC.
- P. Luo, W. Xia, S. Zhou, L. Sun, J. Cheng, C. Xu and Y. Lu, J. Phys. Chem. Lett., 2016, 7, 3603 CrossRef CAS PubMed.
- Y. Hu, F. Bai, X. Liu, Q. Ji, X. Miao, T. Qiu and S. Zhang, ACS Energy Lett., 2017, 2, 2219 CrossRef CAS.
- H. Li, G. Tong, T. Chen, H. Zhu, G. Li, Y. Chang, L. Wang and Y. Jiang, J. Mater. Chem. A, 2018, 6, 14255 RSC.
- X. Liu, X. Tan, Z. Liu, H. Ye, B. Sun, T. Shi, Z. Tang and G. Liao, Nano Energy, 2019, 56, 184 CrossRef CAS.
- W. S. Subhani, K. Wang, M. Du and S. F. Liu, Nano Energy, 2019, 61, 165 CrossRef CAS.
- R. E. Beal, D. J. Slotcavage, T. Leijtens, A. R. Bowring, R. A. Belisle, W. H. Nguyen, G. F. Burkhard, E. T. Hoke and M. D. McGehee, J. Phys. Chem. Lett., 2016, 7, 746 CrossRef CAS PubMed.
- S. Mariotti, O. S. Hutter, L. J. Phillips, P. J. Yates, B. Kundu and K. Durose, ACS Appl. Mater. Interfaces, 2018, 10, 3750 CrossRef CAS PubMed.
- J. K. Sun, S. Huang, X. Z. Liu, Q. Xu, Q. H. Zhang, W. J. Jiang, D. J. Xue, J. C. Xu, J. Y. Ma, J. Ding, Q. Q. Ge, L. Gu, X. H. Fang, H. Z. Zhong, J. S. Hu and L. J. Wan, J. Am. Chem. Soc., 2018, 140, 11705 CrossRef CAS PubMed.
- P. K. Nielsen, C. Hemmingsen, S. U. Friis, J. Ladefoged and K. Olgaard, Peritoneal Dial. Int., 1995, 15, 18 CrossRef CAS.
- Z. Cheng and J. Lin, CrystEngComm, 2010, 12, 2646 RSC.
- C. Li, X. Lu, W. Ding, L. Feng, Y. Gao and Z. Guo, Acta Crystallogr., Sect. B: Struct. Sci., 2008, 64, 702 CrossRef CAS PubMed.
- W. Travis, E. N. K. Glover, H. Bronstein, D. O. Scanlon and R. G. Palgrave, Chem. Sci., 2016, 7, 4548 RSC.
- D. B. Straus, S. Guo, A. M. Abeykoon and R. J. Cava, Adv. Mater., 2020, 32, 2001069 CrossRef CAS PubMed.
- V. K. Ravi, G. B. Markad and A. Nag, ACS Energy Lett., 2016, 1, 665 CrossRef CAS.
- J. Brgoch, A. J. Lehner, M. Chabinyc and R. Seshadri, J. Phys. Chem. C, 2014, 118, 27721 CrossRef CAS.
- C. C. Stoumpos, C. D. Malliakas, J. A. Peters, Z. Liu, M. Sebastian, J. Im, T. C. Chasapis, A. C. Wibowo, D. Y. Chung, A. J. Freeman, B. W. Wessels and M. G. Kanatzidis, Cryst. Growth Des., 2013, 13, 2722 CrossRef CAS.
- A. Filippetti and A. Mattoni, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 125203 CrossRef.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692 CrossRef CAS PubMed.
- K. Heidrich, W. Schäfer, M. Schreiber, J. Söchtig, G. Trendel, J. Treusch, T. Grandke and H. J. Stolz, Phys. Rev. B, 1981, 24, 5642 CrossRef CAS.
- X. Li, Y. Wu, S. Zhang, B. Cai, Y. Gu, J. Song and H. Zeng, Adv. Funct. Mater., 2016, 26, 2435 CrossRef CAS.
- X. Li, F. Cao, D. Yu, J. Chen, Z. Sun, Y. Shen, Y. Zhu, L. Wang, Y. Wei, Y. Wu and H. Zeng, Small, 2017, 13, 1603996 CrossRef PubMed.
- Y. Wang and H. Sun, Small Methods, 2017, 1700252 Search PubMed.
- S. Sanchez, N. Christoph, B. Grobety, N. Phung, U. Steiner, M. Saliba and A. Abate, Adv. Energy Mater., 2018, 8, 1802060 CrossRef.
- E. M. Hutter, R. J. Sutton, S. Chandrashekar, M. Abdi-Jalebi, S. D. Stranks, H. J. Snaith and T. J. Savenije, ACS Energy Lett., 2017, 2, 1901 CrossRef CAS PubMed.
- S. Dastidar, S. Li, S. Y. Smolin, J. B. Baxter and A. T. Fafarman, ACS Energy Lett., 2017, 2, 2239 CrossRef CAS.
- J. K. Nam, S. U. Chai, W. Cha, Y. J. Choi, W. Kim, M. S. Jung, J. Kwon, D. Kim and J. H. Park, Nano Lett., 2017, 17, 2028 CrossRef CAS PubMed.
- Y. Li, C. Zhang, X. Zhang, D. Huang, Q. Shen, Y. Cheng and W. Huang, Appl. Phys. Lett., 2017, 111, 162106 CrossRef.
- Y. Huang, W. J. Yin and Y. He, J. Phys. Chem. C, 2018, 122, 1345 CrossRef CAS.
- R. J. Sutton, M. R. Filip, A. A. Haghighirad, N. Sakai, B. Wenger, F. Giustino and H. J. Snaith, ACS Energy Lett., 2018, 3, 1787 CrossRef CAS.
- L. Fu, Y. Zhang, B. Chang, B. Li, S. Zhou, L. Zhang and L. Yin, J. Mater. Chem. A, 2018, 6, 13263 RSC.
- H. Zhao, Y. Han, Z. Xu, C. Duan, S. Yang, S. Yuan, Z. Yang, Z. Liu and S. Liu, Adv. Energy Mater., 2019, 9, 1902279 CrossRef CAS.
- Y.-T. Yu, S.-H. Yang, L.-H. Chou, I. Osaka, X.-F. Wang and C.-L. Liu, ACS Appl. Energy Mater., 2021, 4, 5466 CrossRef CAS.
- S. Dastidar, D. A. Egger, L. Z. Tan, S. B. Cromer, A. D. Dillon, S. Liu, L. Kronik, A. M. Rappe and A. T. Fafarman, Nano Lett., 2016, 16, 3563 CrossRef CAS PubMed.
- K. Wang, Z. Jin, L. Liang, H. Bian, H. Wang, J. Feng, Q. Wang and S. Frank Liu, Nano Energy, 2019, 58, 175 CrossRef CAS.
- Y. Wang, X. Liu, T. Zhang, X. Wang, M. Kan, J. Shi and Y. Zhao, Angew. Chem., Int. Ed., 2019, 58, 16691 CrossRef CAS PubMed.
- Q. Ye, Y. Zhao, S. Mu, F. Ma, F. Gao, Z. Chu, Z. Yin, P. Gao, X. Zhang and J. You, Adv. Mater., 2019, 31, 1905143 CrossRef CAS PubMed.
- J. Lin, M. Lai, L. Dou, C. S. Kley, H. Chen, F. Peng, J. Sun, D. Lu, S. A. Hawks, C. Xie, F. Cui, A. P. Alivisatos, D. T. Limmer and P. Yang, Nat. Mater., 2018, 17, 261 CrossRef CAS PubMed.
- H. Wang, H. Bian, Z. Jin, H. Zhang, L. Liang, J. Wen, Q. Wang, L. Ding and S. F. Liu, Chem. Mater., 2019, 31, 6231 CrossRef CAS.
- S. Lee, J. Moon, J. Ryu, B. Parida, S. Yoon, D. G. Lee, J. S. Cho, S. Hayase and D. W. Kang, Nano Energy, 2020, 77, 105309 CrossRef CAS.
- Z. Yao, Z. Xu, W. Zhao, J. Zhang, H. Bian, Y. Fang, Y. Yang and S. Liu, Adv. Energy Mater., 2021, 11, 2100403 CrossRef CAS.
- X. Wu, J. Ma, M. Qin, X. Guo, Y. Li, Z. Qin, J. Xu and X. Lu, Adv. Funct. Mater., 2021, 31, 2101287 CrossRef CAS.
- X. Wu, F. Qi, F. Li, X. Deng, Z. Li, S. Wu, T. Liu, Y. Liu, J. Zhang and Z. Zhu, Energy Environ. Mater., 2021, 4, 95 CrossRef CAS.
- K. Wang, S. Ma, X. Xue, T. Li, S. Sha, X. Ren, J. Zhang, H. Lu, J. Ma, S. Guo, Y. Liu, J. Feng, A. Najar and S. Liu, Adv. Sci., 2022, 9, 2105103 CrossRef CAS PubMed.
- F. Wang, Z. Qiu, Y. Chen, Y. Zhang, Z. Huang, N. Li, X. Niu, H. Zai, Z. Guo, H. Liu and H. Zhou, Adv. Mater., 2022, 34, 2108357 CrossRef CAS PubMed.
- J. Ma, M. Qin, Y. Li, X. Wu, Z. Qin, Y. Wu, G. Fang and X. Lu, Matter, 2021, 4, 313 CrossRef CAS.
- K. Wang, C. Gao, Z. Xu, Q. Tian, X. Gu, L. Zhang, S. Zhang, K. Zhao and S. Liu, Adv. Funct. Mater., 2021, 31, 2101568 CrossRef CAS.
- X. Wang, X. Ran, X. Liu, H. Gu, S. Zuo, W. Hui, H. Lu, B. Sun, X. Gao, J. Zhang, Y. Xia, Y. Chen and W. Huang, Angew. Chem., Int. Ed., 2020, 59, 13354 CrossRef CAS PubMed.
- J. Zhang, Z. Wang, A. Mishra, M. Yu, M. Shasti, W. Tress, D. J. Kubicki, C. E. Avalos, H. Lu, Y. Liu, B. I. Carlsen, A. Agarwalla, Z. Wang, W. Xiang, L. Emsley, Z. Zhang, M. Grätzel, W. Guo and A. Hagfeldt, Joule, 2020, 4, 222 CrossRef CAS.
- H. Zhao, Y. Fu, Z. Li, S. Yang, B. Xu, X. Liu, J. Xu, S. Frank Liu and J. Yao, J. Mater. Chem. A, 2021, 9, 4922 RSC.
- X. Gu, W. Xiang, Q. Tian and S. Frank Liu, Angew. Chem., 2021, 133, 23348 CrossRef.
- W. Chen, D. Li, X. Chen, H. Chen, S. Liu, H. Yang, X. Li, Y. Shen, X. Ou, Y. Yang, L. Jiang, Y. Li and Y. Li, Adv. Funct. Mater., 2022, 32, 2109321 CrossRef CAS.
- J. Wang, J. Zhang, Y. Zhou, H. Liu, Q. Xue, X. Li, C. C. Chueh, H. L. Yip, Z. Zhu and A. K. Y. Jen, Nat. Commun., 2020, 11, 177 CrossRef CAS PubMed.
- W. Zhang, J. Xiong, J. Li and W. A. Daoud, Adv. Energy Mater., 2021, 11, 2003585 CrossRef CAS.
- Y. Zhao, K. Zhao, L. Wan, Y. Tan and Z. S. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 6906 CrossRef CAS PubMed.
- L. Ye, H. Wang, Y. Wei, P. Guo, X. Yang, Q. Ye and H. Wang, ACS Appl. Energy Mater., 2020, 3, 658 CrossRef CAS.
- Z. Zeng, J. Zhang, X. Gan, H. Sun, M. Shang, D. Hou, C. Lu, R. Chen, Y. Zhu and L. Han, Adv. Energy Mater., 2018, 8, 1801050 CrossRef.
- I. S. Jin, K. S. Kim and J. W. Jung, J. Power Sources, 2021, 512, 230481 CrossRef CAS.
- J. He, J. Liu, Y. Hou, Y. Wang, S. Yang and H. G. Yang, Nat. Commun., 2020, 11, 4237 CrossRef CAS PubMed.
- H. Li, X. Hao, B. Chang, Z. Li, L. Wang, L. Pan, X. Chen and L. Yin, ACS Appl. Mater. Interfaces, 2021, 13, 40489 CrossRef PubMed.
- C. F. J. Lau, X. Deng, Q. Ma, J. Zheng, J. S. Yun, M. A. Green, S. Huang and A. W. Y. Ho-Baillie, ACS Energy Lett., 2016, 1, 573 CrossRef CAS.
- W. Li, M. U. Rothmann, A. Liu, Z. Wang, Y. Zhang, A. R. Pascoe, J. Lu, L. Jiang, Y. Chen, F. Huang, Y. Peng, Q. Bao, J. Etheridge, U. Bach and Y. B. Cheng, Adv. Energy Mater., 2017, 1700946 CrossRef.
- B. Yang, M. Wang, X. Hu, T. Zhou and Z. Zang, Nano Energy, 2019, 57, 718 CrossRef CAS.
- C. Liu, W. Li, J. Chen, J. Fan, Y. Mai and R. E. I. Schropp, Nano Energy, 2017, 41, 75 CrossRef CAS.
- N. Li, Z. Zhu, J. Li, A. K. Y. Jen and L. Wang, Adv. Energy Mater., 2018, 1800525 CrossRef.
- W. Zhu, Q. Zhang, C. Zhang, Z. Zhang, D. Chen, Z. Lin, J. Chang, J. Zhang and Y. Hao, ACS Appl. Energy Mater., 2018, 1, 4991 CrossRef CAS.
- W. Zhang, J. Xiong, J. Li and W. A. Daoud, Small, 2020, 16, 2001535 CrossRef CAS PubMed.
- C. Zhang, K. Wang, Y. Wang, W. S. Subhani, X. Jiang, S. Wang, H. Bao, L. Liu, L. Wan and S. Liu, Sol. RRL, 2020, 4, 2000254 CrossRef CAS.
- H. Wang, S. Cao, B. Yang, H. Li, M. Wang, X. Hu, K. Sun and Z. Zang, Sol. RRL, 2020, 4, 1900363 CrossRef CAS.
- H. Wang, H. Li, S. Cao, M. Wang, J. Chen and Z. Zang, Sol. RRL, 2020, 4, 2000226 CrossRef CAS.
- A. K. Jena, A. Kulkarni, Y. Sanehira, M. Ikegami and T. Miyasaka, Chem. Mater., 2018, 30, 6668 CrossRef CAS.
- F. Ünlü, E. Jung, J. Haddad, A. Kulkarni, S. Öz, H. Choi, T. Fischer, S. Chakraborty, T. Kirchartz and S. Mathur, APL Mater., 2020, 8, 070901 CrossRef.
- J. Liang, P. Zhao, C. Wang, Y. Wang, Y. Hu, G. Zhu, L. Ma, J. Liu and Z. Jin, J. Am. Chem. Soc., 2017, 139, 14009 CrossRef CAS PubMed.
- J. Liang, Z. Liu, L. Qiu, Z. Hawash, L. Meng, Z. Wu, Y. Jiang, L. K. Ono and Y. Qi, Adv. Energy Mater., 2018, 8, 1800504 CrossRef.
- X. Tan, X. Liu, Z. Liu, B. Sun, J. Li, S. Xi, T. Shi, Z. Tang and G. Liao, Appl. Surf. Sci., 2019, 143990 Search PubMed.
- Y. Guo, F. Zhao, Z. Li, J. Tao, D. Zheng, J. Jiang and J. Chu, Org. Electron., 2020, 83, 105731 CrossRef CAS.
- Y. Guo, F. Zhao, X. Wang, J. Tao, D. Zheng, J. Jiang, Z. Hu and J. Chu, Sol. Energy Mater. Sol. Cells, 2021, 221, 110918 CrossRef CAS.
- H. Sun, L. Yu, H. Yuan, J. Zhang, X. Gan, Z. Hu and Y. Zhu, Electrochim. Acta, 2020, 349, 136162 CrossRef CAS.
- P. Liu, X. Yang, Y. Chen, H. Xiang, W. Wang, R. Ran, W. Zhou and Z. Shao, ACS Appl. Mater. Interfaces, 2020, 12, 23984 CrossRef CAS PubMed.
- Y. Long, C. Wang, X. Liu, J. Wang, S. Fu, J. Zhang, Z. Hu and Y. Zhu, J. Mater. Chem. C, 2021, 9, 2145 RSC.
- J. Lin, M. Lai, L. Dou, C. S. Kley, H. Chen, F. Peng, J. Sun, D. Lu, S. A. Hawks, C. Xie, F. Cui, A. P. Alivisatos, D. T. Limmer and P. Yang, Nat. Mater., 2018, 17, 261 CrossRef CAS PubMed.
- Y. Jiang, J. Yuan, Y. Ni, J. Yang, Y. Wang, T. Jiu, M. Yuan and J. Chen, Joule, 2018, 2, 1356 CrossRef.
- W. Zhang, H. Liu, X. Qi, Y. Yu, Y. Zhou, Y. Xia, J. Cui, Y. Shi, R. Chen and H. L. Wang, Adv. Sci., 2022, 2106054 CrossRef CAS PubMed.
- D. Wang, W. Li, T. Zhang, X. Liu, X. Jin, B. Xu, D. Li, Z. Huang, Q. Li, Z. Lan and J. Wu, ACS Appl. Energy Mater., 2022, 5, 2720 CrossRef CAS.
- J. Huang, S. He, W. Zhang, A. Saparbaev, Y. Wang, Y. Gao, L. Shang, G. Dong, L. Nurumbetova, G. Yue and Y. Tu, Sol. RRL, 2022, 6, 2100839 CrossRef CAS.
- J. Yang, H. Yu, S. Wu, C. Cai, J. Gao, X. Lu, X. Gao, L. Shui, S. Wu and J.-M. Liu, ACS Appl. Energy Mater., 2022, 5, 2881 CrossRef CAS.
- Z. Guo, A. K. Jena, I. Takei, G. M. Kim, M. A. Kamarudin, Y. Sanehira, A. Ishii, Y. Numata, S. Hayase and T. Miyasaka, J. Am. Chem. Soc., 2020, 142, 9725 CAS.
- S. Oz, K. A. Jena, A. Kulkarni, K. Mouri, T. Yokoyama, I. Takei, F. Unlu, S. Mathur and T. Miyasaka, ACS Energy Lett., 2020, 5, 1292 CrossRef CAS.
- Z. Guo, A. K. Jena, I. Takei, M. Ikegami, A. Ishii, Y. Numata, N. Shibayama and T. Miyasaka, Adv. Funct. Mater., 2021, 31, 2103614 CrossRef CAS.
- H. Zhong, W. Li, Y. Huang, D. Cao, C. Zhang, H. Bao, Z. Guo, L. Wan, X. Zhang, X. Zhang, Y. Li, X. Ren, X. Wang, D. Eder, K. Wang, S. F. Liu and S. Wang, ACS Appl. Mater. Interfaces, 2022, 14, 5183 CrossRef CAS PubMed.
- X. Zhao, T. Liu, Q. C. Burlingame, T. Liu, R. Holley, G. Cheng, N. Yao, F. Gao and Y. L. Loo, Science, 2022, 377, 307 CrossRef CAS PubMed.
- W. S. Subhani, K. Wang, M. Du, X. Wang and S. Frank Liu, Adv. Energy Mater., 2019, 1803785 CrossRef.
- W. Zhu, Z. Zhang, W. Chai, Q. Zhang, D. Chen, Z. Lin, J. Chang, J. Zhang, C. Zhang and Y. Hao, ChemSusChem, 2019, 12, 2318 CrossRef CAS PubMed.
- Z. Guo, S. Teo, Z. Xu, C. Zhang, Y. Kamata, S. Hayase and T. Ma, J. Mater. Chem. A, 2019, 7, 1227 RSC.
- J. Wang, X. Wu, Y. Liu, Q. Xue, H. L. Yip, A. K. Y. Jen and Z. Zhu, Energy Technol., 2021, 9, 2100562 CrossRef CAS.
- X. Jiang, W. S. Subhani, K. Wang, H. Wang, L. Duan, M. Du, S. Pang and S. Liu, Adv. Mater. Interfaces, 2021, 8, 2001994 CrossRef CAS.
- J. Du, J. Duan, X. Yang, Y. Duan, Q. Zhou and Q. Tang, Angew. Chem., Int. Ed., 2021, 60, 10608 CrossRef CAS PubMed.
- D. Wang, W. Li, R. Li, W. Sun, J. Wu and Z. Lan, Sol. RRL, 2021, 5, 2100375 CrossRef CAS.
- Y. Xu, Q. Wang, L. Zhang, M. Lyu, H. Lu, T. Bai, F. Liu, M. Wang and J. Zhu, Sol. RRL, 2021, 5, 2100669 CrossRef CAS.
- J. Lu, S. C. Chen and Q. Zheng, ACS Appl. Energy Mater., 2018, 1, 5872 CrossRef.
- Y. You, W. Tian, M. Wang, F. Cao, H. Sun and L. Li, Adv. Mater. Interfaces, 2020, 7, 2000537 CrossRef CAS.
- J. Pan, X. Zhang, Y. Zheng and W. Xiang, Sol. Energy Mater. Sol. Cells, 2021, 221, 110878 CrossRef CAS.
- Y. Wang, K. Wang, W. S. Subhani, C. Zhang, X. Jiang, S. Wang, H. Bao, L. Liu, L. Wan and S. Liu, Small, 2020, 16, 1907283 CrossRef CAS PubMed.
- W. Zhu, Q. Zhang, D. Chen, Z. Zhang, Z. Lin, J. Chang, J. Zhang, C. Zhang and Y. Hao, Adv. Energy Mater., 2018, 8, 1802080 CrossRef.
- Q. Zhang, W. Zhu, D. Chen, Z. Zhang, Z. Lin, J. Chang, J. Zhang, C. Zhang and Y. Hao, ACS Appl. Mater. Interfaces, 2019, 11, 2997 CrossRef CAS PubMed.
- B. Zhang, W. Bi, Y. Wu, C. Chen, H. Li, Z. Song, Q. Dai, L. Xu and H. Song, ACS Appl. Mater. Interfaces, 2019, 11, 33868 CrossRef CAS PubMed.
- Y. Guo, X. Yin, J. Liu and W. Que, J. Mater. Chem. A, 2019, 7, 19008 RSC.
- X. Jiang, K. Wang, H. Wang, L. Duan, M. Du, L. Wang, Y. Cao, L. Liu, S. Pang and S. Frank Liu, Small Sci., 2021, 1, 2000054 CrossRef CAS.
- Q. Zhang, J. Duan, Q. Guo, J. Zhang, D. Zheng, F. Yi, X. Yang, Y. Duan and Q. Tang, Angew. Chem., 2022, 134, e202116632 Search PubMed.
- F. Yan, P. Yang, J. Li, Q. Guo, Q. Zhang, J. Zhang, Y. Duan, J. Duan and Q. Tang, Chem. Eng. J., 2022, 430, 132781 CrossRef CAS.
- Q. Wang, Y. Xu, L. Zhang, A. Yang, T. Bai, F. Liu, M. Lyu and J. Zhu, ACS Appl. Energy Mater., 2022, 5, 3110 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |