
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRedox-triggerable firefly luciferin-bioinspired hydrogels as injectable and cell-encapsulating matrices†
Minye
Jin
abc,
Alisa
Gläser
a and
Julieta I.
Paez
 *ac
*ac
aINM – Leibniz Institute for New Materials, Campus D2-2, 66123, Saarbrücken, Germany. E-mail: j.i.paez@utwente.nl
bChemistry Department, Saarland University, 66123, Saarbrücken, Germany
cDevelopmental Bioengineering, University of Twente, Drienerlolaan 5, 7522 NB, Enschede, The Netherlands
First published on 15th August 2022
Abstract
Stimuli-responsive hydrogels are smart materials that respond to variations caused by external stimuli and that are currently exploited for biomedical applications such as biosensing, drug delivery and tissue engineering. The development of stimuli-responsive hydrogels with defined user control is relevant to realize materials with advanced properties. Recently, our group reported firefly luciferin-inspired hydrogel matrices for 3D cell culture. This platform exhibited advantages like rapid gelation rate and tunability of mechanical and biological properties. However, this first molecular design did not allow fine control of the gelation onset, which restricts application as a cell-encapsulating matrice with injectable and processable properties. In this article, we endow the firefly luciferin-inspired hydrogels with redox-triggering capability, to overcome the limitations of the previous system and to widen its application range. We achieve this goal by introducing protected macromers as hydrogel polymeric precursors that can be activated in the presence of a mild reductant, to trigger gel formation in situ with a high degree of control. We demonstrate that the regulation of molecular parameters (e.g., structure of the protecting group, reductant type) and environmental parameters (e.g., pH, temperature) of the deprotection reaction can be exploited to modulate materials properties. This redox-triggerable system enables precise control over gelation onset and kinetics, thus facilitating its utilization as an injectable hydrogel without negatively impacting its cytocompatibility. Our findings expand the current toolkit of chemically-based stimuli-responsive hydrogels.
1. Introduction
Hydrogels are porous hydrophilic crosslinked networks. Due to their high water content and tunable biophysical and biochemical properties, hydrogels are used as extracellular matrix mimics in therapeutics delivery, in vitro disease modelling, tissue gluing and tissue engineering.1–3 In particular, stimuli-responsive hydrogels have attracted interest as smart materials for biomedical applications. Stimuli-responsive hydrogels that experience changes in their equilibrium swelling,4 undergo sol–gel5 or gel–sol transition,6 or can modify their bioactivity7 in response to an applied stimulus (also called a “trigger”) have been reported. Diverse physical (e.g., temperature,8,9 light,6,10 electrical, magnetic and ultrasound fields11–13) and chemical (e.g., pH,14,15 enzymes16 and small molecules17,18) triggers have been used to tune materials properties on demand. These smart biomaterials have been successfully implemented in a wide range of applications including biosensing, cell and drug delivery and as scaffolds for tissue engineering.19,20Among chemically-responsive hydrogels, those systems that can be triggered or actuated by biocompatible redox reactions are promising for biomedical applications because of the easy access and low cost of redox triggers. For instance, redox-responsive functional groups in the hydrogel can react with mild oxidants or reductants, leading to either the formation or the cleavage of covalent bonds, which induces changes in materials properties.21 One representative redox reaction applied for preparation of biocompatible systems is the catechol to quinone oxidation that is used to trigger sol–gel transition.6 Another example is the thiol/disulfide reactive pair. The thiol to disulfide oxidation can be applied to trigger sol–gel transition or to increase the crosslinking density,22,23 while the disulfide to thiol reduction is typically used for promoting gel–sol transition or to decrease the crosslinking density in the material.24,25 These strategies have mostly been implemented to modulate mechanical properties on demand24 and to promote controlled drug release26 from the hydrogel matrix. However, one aspect that remains relatively unexplored in this field is the use of mild redox reactions to precisely trigger the gelation onset as well as to control the rate of the gelation process, without compromising materials cytocompatibility. When implemented in the context of cell encapsulation, these redox-triggerable matrices with flexible control over gelation onset and rate are envisioned as versatile hydrogel platforms with tunable properties. These smart hydrogels have application potential as injectable matrices and printable hydrogel inks.
Recently, our group reported soft biomaterials whereby the crosslinking chemistry was inspired by the biochemical reactions occurring in fireflies and that lead to the formation of luciferin in vivo.27 Firefly luciferin-inspired polyethylene glycol (PEG)-based hydrogel for 3D cell culture were presented. These hydrogels were crosslinked via the so-called luciferin click ligation, which involves a condensation reaction between cyanobenzothiazole (CBT) and cysteine (Cys) groups, to form a luciferin-like adduct. The resulting materials, termed as CBT–Cys hydrogels, exhibited efficient gelation rate under physiological conditions, high homogeneity at the microscale, good cytocompatibility and tunable mechanical strength within physiologically relevant values. Upon stem cell encapsulation and by regulating hydrogel's bioactivity through incorporation of biochemical cues, cell–materials interaction was modulated. Moreover, the gelation rate of the system was regulated within the seconds range by adjusting pH within the close-to-physiological range without impacting the final mechanical strength.27
Despite the mentioned advantages, the CBT–Cys system presented some limitations regarding user's control over system's properties. First, it did not allow for control of the gelation onset since the crosslinking reaction starts as soon as CBT and Cys precursors are mixed. This could complicate its application as in situ curing injectable matrices where too fast gelation rate could lead to clogging of the syringe during injection. Second, even at pH 6.6, quick gel formation was observed (i.e., gelation time remained consistently <1 min). This short gelation time proved ideal for homogeneous encapsulation of cells in a culture plate,27 but might be too short for other biomedical scenarios requiring slower gelation rate (e.g., within minutes) such as those involving injectability and in situ crosslinking for therapeutic delivery, tissue adhesion and tissue engineering.28,29 Therefore, gaining user control on gelation onset and rate would increase the application potential of this platform. Third, we and others30 found that under physiological media (e.g., in buffer pH 7.4) Cys precursors undergo oxidation of Cys groups to form disulfide bridges, which inactivates the reactivity of Cys groups towards CBT moieties over time. Consequently, Cys precursors had to be freshly prepared before gel formulation. Increasing chemical stability of Cys precursors would allow stock solutions of this macromer to be prepared at one time point and used later, which is convenient for applications involving hydrogel fabrication in combination with processing technologies. In this line, regulating the reactivity and stability of the Cys precursor could be key to solve these issues. We envisioned that the introduction of redox-triggers to the luciferin-inspired gels will enable to overcome all the mentioned limitations of the existing system, thus facilitating this system's application as injectable matrices with tunable properties.
In this article, we investigate whether the incorporation of redox-responsive moieties onto the Cys precursor of luciferin-inspired hydrogels could impart the system with redox-triggerable gelation onset. To this end, the PEG-Cys precursor that contained terminal 1,2-aminothiol motifs was chemically modified with a disulfide protecting group at the thiol rest to obtain a PEG-Cys(SR) precursor, therefore impeding the crosslinking reaction with PEG-CBT in aqueous conditions (shown in Fig. 1a, upper path). Upon the addition of a mild reductant (i.e., the redox trigger), the protecting group was cleaved and free PEG-Cys was generated in situ, which in the presence of PEG-CBT promoted hydrogelation via luciferin click ligation (Fig. 1a, lower path). Herein, we demonstrate that tuning of the disulfide cleavage reaction at the molecular level enables to tightly modulate important materials properties such as gelation onset and kinetics, as well as mechanical strength of derived materials. Tunability of materials properties is possible by tailoring intrinsic and extrinsic parameters of the deprotection reaction. Specifically, we studied the influence of the molecular structure of the protecting group of the PEG-Cys(SR) precursor, the reductant type used, and environmental cues such as pH and temperature; on the course of the gelation process. Working conditions for flexible preparation of cell-encapsulating hydrogels were found, while high viability of embedded stem cells was preserved. Finally, it is demonstrated that these redox-triggerable hydrogels can be formulated as injectable matrices with potential for the development of in-bath crosslinkable inks. These redox-triggerable bioinspired hydrogels with high user's control on materials properties complement and expand the current palette of chemically-based stimuli-responsive systems. These smart biomaterials are expected to pave the way for innovative applications in the biomedical field.
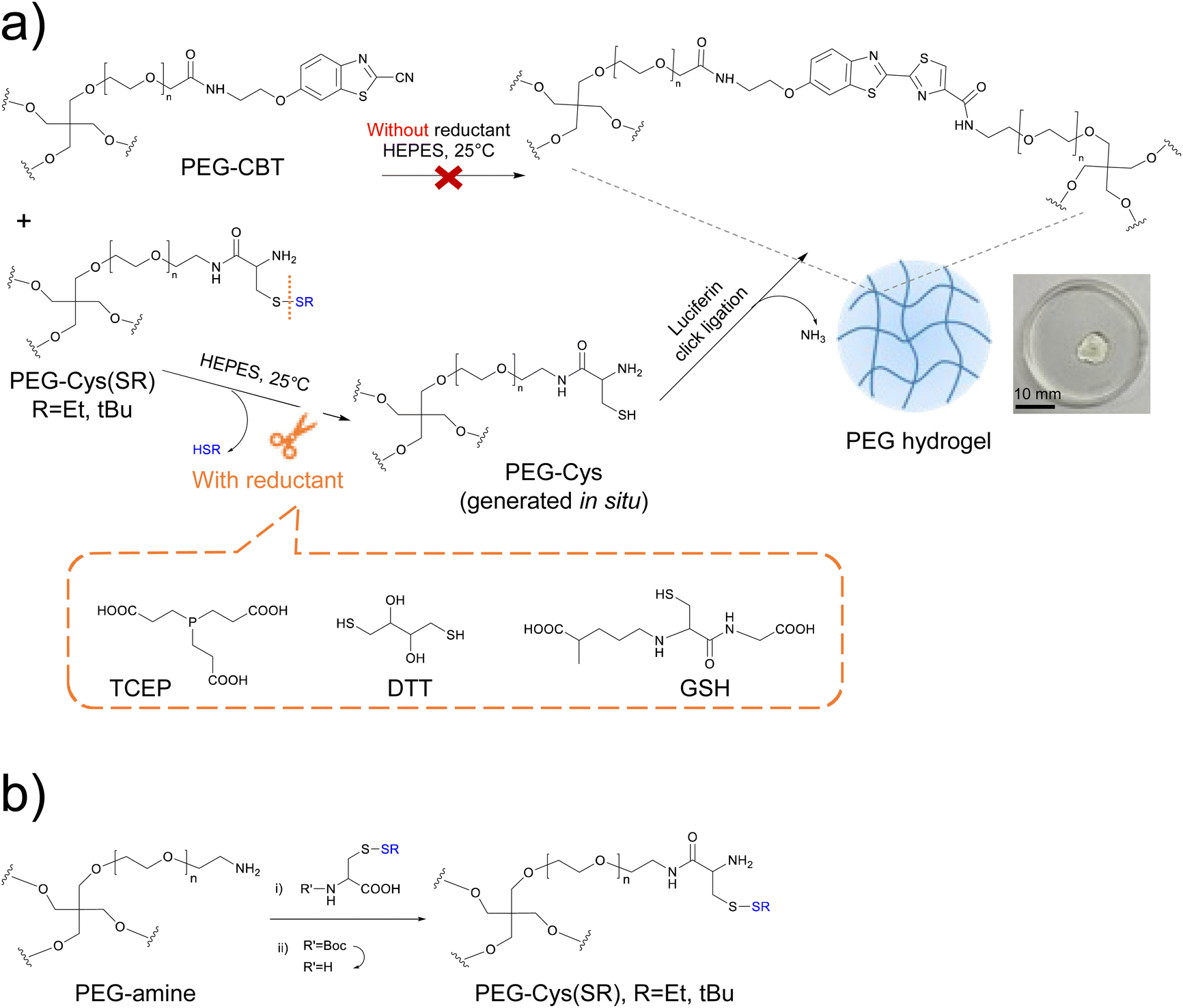 | ||
Fig. 1 (a) Schematics of the redox-triggered firefly luciferin-bioinspired crosslinking, used in this work as a strategy for on demand gelation. In the absence of a reductant, mixing PEG-CBT and PEG-Cys(SR) precursors does not lead to the formation of a hydrogel, because the thiol residue of Cys is protected (upper path). In contrast, the addition of a reductant cleaves the protecting group from PEG-Cys(SR) and generates PEG-Cys in situ, which in presence of PEG-CBT precursor forms a hydrogel (lower path). The reductants used in this work, TCEP, DTT and GSH, are shown. A photography of a swollen PEG-Cys(SEt) gel at 3.3 wt% is included. Scale bar = 10 mm. (b) Synthesis of PEG-Cys(SR) macromers. Reagents and conditions: (i) HBTU, HOBT, DIPEA, dry DMF, room temp., 2 d; (ii) TFA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TIS:water (95:2.5:2.5), room temp., 1.5 h. :TIS:water (95:2.5:2.5), room temp., 1.5 h. | ||
2. Results and discussion
Design and synthesis of PEG-Cys(SR) macromers for redox-triggerable gelation
We envisioned that thiol-protected PEG-Cys precursors will be key for the design of redox-triggerable hydrogels via luciferin click ligation crosslinking. We considered 4-arm PEG-Cys(SR) macromers presenting different protecting groups at the thiol residue (R = Et, tBu) and diverse molar masses (10 and 20 kDa). Different protecting groups (in this case, alkyl disulfides) are expected to show diverse cleavage rate in the presence of a reductant, thus enabling control over gelation kinetics. Additionally, the choice of precursor's molar mass was done to enable tuning of important properties of derived materials (such as crosslinking degree and gelation rate), while leading to hydrogels that are adequate for cell encapsulation applications.27,31Control of the disulfide cleavage reaction at the molecular level was expected to enable fine tuning of materials properties at the micro/macroscopic level. According to reported work, the rate of disulfide cleavage depends on the chemical structure of the disulfide and on the specific reductant used.32–34 Regarding the structure of the disulfide protecting group, factors such as the steric hindrance of the lateral chain, the redox potential of the parent disulfide and the pKa of the thiol leaving group are deemed important. Concerning the reductant type, parameters such as the redox potential of the reductant, the specific reaction mechanism taking place and the chemical orthogonality between the reductant and the CBT moiety,27 are all aspects that could play a role in deprotection kinetics. In general, a faster deprotection is attributed to a smaller steric size of alkyl disulfide,35 to a lower pKa of the thiol leaving group,36 and to the use of a stronger reductant. In our study, Cys precursor derivatives with different protecting groups were synthesized: PEG-Cys(SR), whereby R = Et or tBu. With smaller steric hinderance and lower pKa of thiol leaving group (HSEt = 10.6 vs. HStBu = 11.2), we anticipated faster deprotection rate and thus faster gelation rate for the system derived from the PEG-Cys(SEt) precursor. As for the reductant, we used dithiothreitol (DTT), tris-(2-carboxyethyl)-phosphine (TCEP) and glutathione (GSH) (Fig. 1a), which are widely used in the chemical biology laboratory.37 Note that, additionally, GSH exists in vivo and is used by living cells to regulate numerous redox processes.38 Besides the distinct reduction potential of these three reductants,39–41 it is important to mention the specific mechanism of disulfide cleavage that occurs in each case. TCEP, a phosphine-based reductant, cleaves disulfide bonds via an irreversible mechanism that forms thiols and phosphine oxide.42 In contrast, GSH and DTT present thiol groups and cleave disulfide bonds through a disulfide–thiol exchange mechanism.38,39 Moreover, while the –COOH side groups of TCEP are foreseen chemically orthogonal to CBT–Cys reaction, the thiol groups present in GSH and DTT might chemically interfere with the luciferin coupling as they can transiently react with CBT groups to form thioimidate adducts.43 This could eventually delay the gelation speed.
The PEG-Cys(SR) macromers were successfully synthesized in two steps from a commercial precursor PEG-amine (see scheme in Fig. 1b). Cys amino acid variants, presenting Boc protecting group at the amine rest and disulfide protecting groups at the thiol rest, had their free –COOH group activated with HBTU/HOBT mixture and were subsequently coupled to PEG-amine. The intermediate Boc-containing macromers showed a high substitution degree (>90%), as revealed by 1H-NMR spectroscopy. In a second step, the Boc group was removed in TFA:TIS:water (95:2.5:2.5) deprotection cocktail. Under these mild acidic conditions that include TIS as scavenger, the Boc group was selectively cleaved while the disulfide bond that protects the thiol rest remained stable.44,45 Successful Boc cleavage was proven by 1H-NMR, by observing the disappearance of the signal of the Boc group at 1.42 ppm. After purification by dialysis and lyophilization, PEG-Cys(SEt) and PEG-Cys(StBu) macromers were obtained in 90% and 87% yield, respectively. Furthermore, PEG-CBT macromers (10 and 20 kDa) were prepared according to our previously published protocol.27 Detailed description of the synthesis, purification, and characterization of the PEG macromers can be found in the ESI.†
PEG-Cys(SR) macromers show long-term stability upon storage in aqueous solution
The aqueous stability of hydrogel precursors is relevant for the prospective storage of these compounds in solution, which could prove beneficial for the usage of these solutions in applications that require pre-mixing of precursors (e.g., injectable matrices and hydrogel inks in processing technologies).Previously, we had demonstrated that PEG-CBT macromers are stable in aqueous solution for at least 1 month,27 but we had not investigated the stability of PEG-Cys macromers. We noticed that solutions of PEG-Cys precursor had to be freshly prepared to prevent the inactivation of the macromer's reactivity, apparently due to oxidation of thiols to disulfides. To get a deeper insight into the stability of PEG-Cys under physiologically relevant medium, 1H NMR spectroscopy experiments were conducted. PEG-Cys precursor solution at 1 mM (20 mg mL−1) concentration was prepared in deuterated phosphate buffer saline (d-PBS) at pH 7.4 and at room temperature, and its 1H NMR spectra were recorded over time up to 4 weeks. During this time, the solutions were stored in a closed NMR tube and under ambient conditions (i.e., at room temperature and exposed to light from the laboratory). After 1 week of ageing, PEG-Cys evidenced spectral changes in the aliphatic region that are consistent with the formation of disulfide bonds (Fig. S1a†). Namely, the doublet at 2.9 ppm corresponding to the methylene of the free Cys group decreased in intensity and a new multiplet attributed to the methylene of the formed disulfide appeared at 3.2–3.0 ppm. This is in line with spectral changes previously reported for small Cys variants measured under similar conditions.30 In our studies, PEG-Cys precursor showed complete conversion of free thiols to disulfides within 2 weeks of ageing. Furthermore, we investigated the reactivity of the aged precursor, by assessing its ability to form hydrogels when mixed with PEG-CBT precursor. A 4-weeks aged PEG-Cys solution proved unable to form hydrogels upon mixing with PEG-CBT (see Fig. S2,† left panel).
Next, we studied the stability of the PEG-Cys(SR) macromers by 1H NMR under same conditions. Both PEG-Cys(SEt) and PEG-Cys(StBu) compounds demonstrated high stability in aqueous conditions as no significant spectral change was observed after at least 12 weeks of ageing, within the investigated conditions (Fig. S1b and c†). Subsequently, 4-weeks aged PEG-Cys(SR) precursor solution was mixed with PEG-CBT precursor in presence of 1 eq. reductant and formation of hydrogels was observed in both cases (Fig. S2,† central and right panels). Preliminary rheological characterization demonstrated that hydrogels formed from aged PEG-Cys(SR) precursors showed similar gelation kinetics and final mechanical strength than gels prepared from fresh precursor solutions (Fig. S2a†). This indicates that aged PEG-Cys(SR) precursors remain chemically reactive. Altogether, these studies demonstrate the excellent long-term stability of PEG-Cys(SR) precursors under physiologically relevant conditions that, in relation to the existing PEG-Cys precursor, is advantageous for storage and pre-mixing of reactive macromer solutions. In the next section, a systematic investigation of the formation of redox-triggered hydrogels is presented.
Choice of protecting group and reductant type allows modulation of gelation onset and rate without affecting gel's final mechanical strength
Redox-triggered hydrogels were fabricated in 20 mM HEPES buffer at pH 8.0 under mild reductive conditions using TCEP as reductant. First, 10 kDa PEG-CBT and PEG-Cys(StBu) precursor solutions at 3.3 wt% concentration were mixed at 1:1 volume ratio, and no hydrogel formation was observed (see Fig. S3†), confirming that masking the thiol residue of the Cys group effectively prevents hydrogelation. When 1 equivalent of TCEP reductant solution was added to obtain a reactive mixture of final 5 wt% polymer concentration and CBT:Cys(SEt):TCEP (1:1:1) molar ratio; a hydrogel formed in 19 s, as revealed by a macroscopic test (Table 1). This proves that the addition of TCEP triggers the disulfide cleavage of PEG-Cys(SEt) precursor, thus unmasking the thiol moiety and triggering the formation of a hydrogel through luciferin click ligation. The formed hydrogels looked transparent and homogeneous to the naked eye and showed the pale-yellow color27 that is characteristic of newly formed luciferin-like crosslinks (see picture of a hydrogel in insert of Fig. 1a).
| Hydrogel | CBT–Cys(SEt) | CBT–Cys(StBu) | CBT–Cys (control) |
|---|---|---|---|
| Conditions: 20 kDa, 5 wt% polymer concentration, in HEPES buffer at pH 8.0 containing 20 mM TCEP, T = 25 °C, n = 3. | |||
| Gelation time | 18.6 ± 0.6 s | 198.6 ± 7.7 s | 17.3 ± 0.6 s |
Furthermore, when the other protected precursor, PEG-Cys(StBu), was pre-mixed with PEG-CBT followed by the addition of TCEP, a hydrogel formed in ∼200 s (Table 1), which is about 10-fold slower than when PEG-Cys(SEt) was used. This indicates that, as expected, a bulkier protecting group at the thiol rest35 and a higher pKa of the thiol leaving group36 lead to slower disulfide cleavage, thus delaying the unmasking of the precursor and consequently decreasing the gelation rate. This finding validates the molecular design of the redox-triggerable Cys(SR) precursors. For comparison, a hydrogel obtained from mixing PEG-CBT and PEG-Cys (i.e., without protecting groups at the thiol rest of Cys) was prepared as control under the same conditions, observing a gelation time of 18 s. Interestingly, the gelation rates obtained from either of the two precursors, PEG-Cys(SEt) or PEG-Cys, are similar; suggesting that the unmasking of PEG-Cys(SEt) is very efficient under these conditions (or that, at least, it is faster than the Cys–CBT coupling). To get a deeper insight on the unmasking reaction of PEG-Cys(SEt) with TCEP under physiologically relevant media, we characterized the disulfide cleavage at the molecular level under conditions that mimic gel preparation shown above. The H-Cys(SEt)-OH amino acid was synthesized, dissolved at 50 mM concentration in d-PBS buffer pH 7.4, mixed with TCEP at (1:1) molar ratio, and the disulfide cleavage reaction was monitored by 1H NMR spectroscopy at 25 °C. Complete disulfide cleavage (∼100% conversion) was observed within 5 min of reaction (Fig. S4†). Unfortunately, it was not possible to follow the unmasking process at shorter reaction times due to limitations of our NMR experiment, meaning that it is possible that the unmasking reaction was complete at time <5 min. In comparison, under similar experimental conditions, the CBT–Cys coupling reaction between small molecules has been reported to reach ∼80% conversion within 45 min.46 Thus, our NMR result indicates that the unmasking of PEG-Cys(SEt) precursor by TCEP under the studied conditions is noticeably faster than CBT–Cys coupling, which explains the similar gelation times shown above for gels derived from either PEG-Cys(SEt) or PEG-Cys precursors.
To further investigate the impact of the protecting group of the PEG-Cys(SR) variants on the gelation kinetics and mechanical strength of the hydrogels, the gelation of these materials was studied by oscillatory shear rheometry. In the following, the different hydrogels are denoted CBT–Cys(SR) when they are derived from PEG-Cys(SR) variants (R = Et, tBu) and denoted CBT–Cys when they are prepared from PEG-Cys precursor (control material).
A solution containing PEG-CBT and PEG-Cys(SR) was mixed directly on the bottom plate of the rheometer, followed by addition of TCEP solution and quick mixing, and the evolution of storage modulus (G′) and loss modulus (G′′) was monitored over time at 25 °C (Fig. 2). At 5 wt% concentration, formation of CBT–Cys(SEt) hydrogel was observed in the beginning of the experiment, indicated by G′ > G′′. This evidences an efficient curing, with gelation time <1 min (note that 1 min is the approx. time required for sample preparation and measurement setting), in good agreement with the values obtained by the macroscopic test shown on Table 1. In comparison, CBT–Cys(StBu) gels prepared under same conditions formed in ∼3 min. During the curing process, G′ increased and reached G′ = 1334 Pa vs. G′ = 1191 Pa after 10 min, for the Et and tBu variants, respectively (Fig. 2a). In a separate experiment, CBT–Cys(SR) gels of same composition were prepared in molds, let cure for 120 min, swelled in buffer until equilibrium (∼24 h), and the final mechanical strength of the gels after swelling was measured. CBT–Cys(SEt) and CBT–Cys(StBu) gels showed G′ of 2202 and 2052 Pa, respectively; and the obtained G′ values proved not significantly different indicating similar crosslinking density in both CBT–Cys(SEt) and CBT–Cys(StBu) gels (Fig. 2b). Overall, the observed trend in the gelation rate CBT–Cys(Et) > CBT–Cys(StBu) seems to reflect the rate of deprotection of the Cys variants and demonstrates that the structure of the protecting group of the Cys precursor can be used to regulate the gelation time of derived materials from a few seconds to minutes.
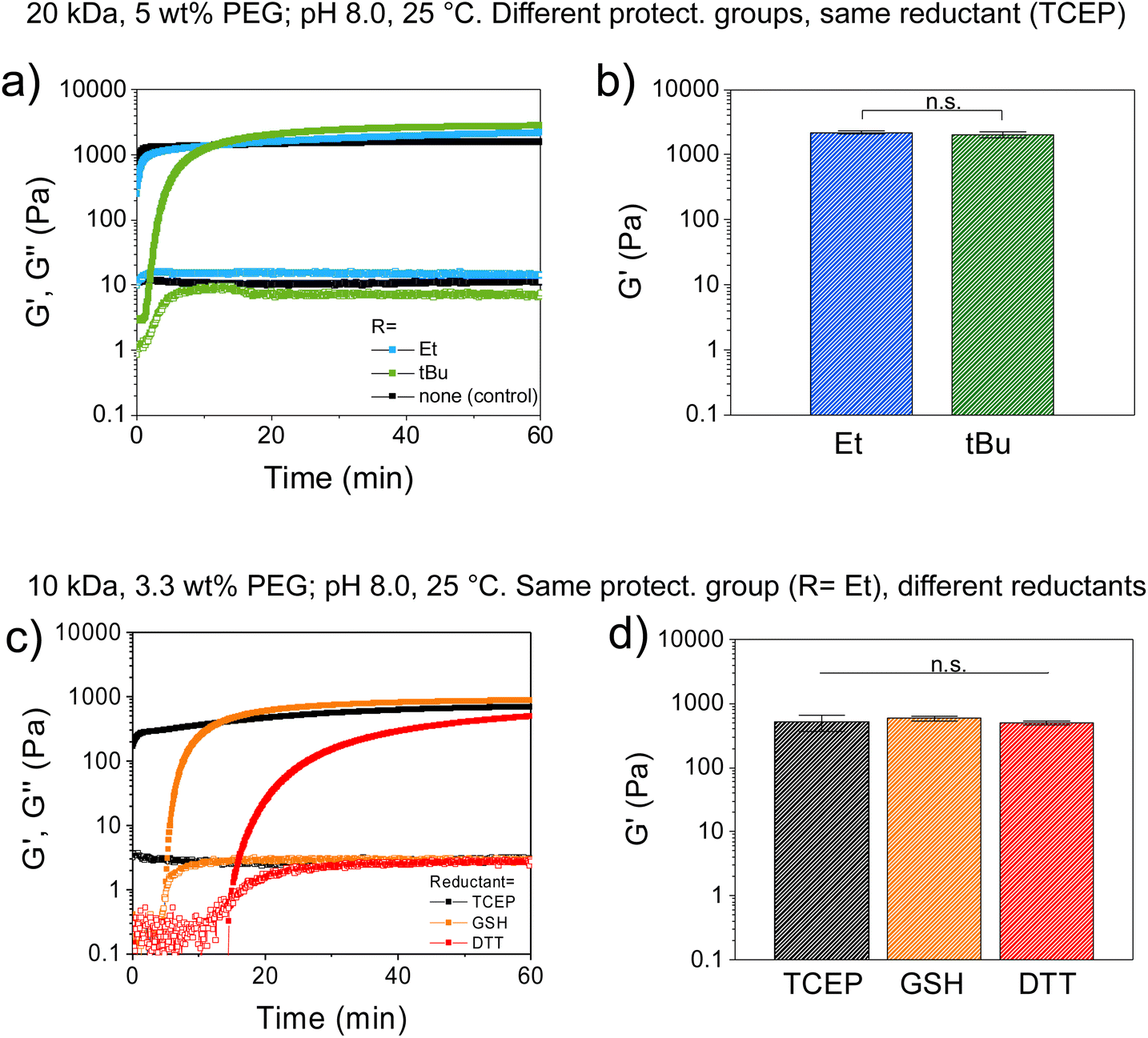 | ||
| Fig. 2 Study of gelation kinetics and mechanical strength of redox-triggered hydrogels by shear oscillatory rheometry, at varying Cys protecting group and reductant type. Shear storage (G′) and loss (G′′) moduli were followed as a function of time and denoted as closed and open symbols, respectively. (a and b) Effect of different Cys-protecting groups of PEG-Cys(SR) precursor, on the gelation of CBT–Cys(SR) gels under constant reductant type (TCEP), before (a) and after (b) swelling. A CBT–Cys gel, prepared from unprotected PEG-Cys is shown as control. (c and d) Effect of reductant type (TCEP, GSH or DTT) on the gelation of CBT–Cys(SEt) gels, before (c) and after (d) swelling (i.e., at constant protecting group). Gel composition: (a and b) 20 kDa PEGs, 5 wt% polymer content, in HEPES buffer, at (1:1) reductant:Cys(SR) molar ratio, T = 25 °C. (c and d) 10 kDa PEGs, 3.3 wt% polymer content, in HEPES, at (1:1) reductant:Cys(SEt) molar ratio, T = 25 °C. The reductant used is indicated in each case. In all cases, data are plotted as mean ± SD, n = 3. In (b) & (d), statistical significance was analyzed by ANOVA followed by the post hoc Tukey test (*p < 0.05 set for statistical significance level; n.s. = not significant). | ||
Next, we investigated the effect of the reductant type on the course of the gelation process. CBT–Cys(SEt) hydrogels were prepared at 3.3 wt% concentration, using TCEP, DTT or GSH as reductants, while keeping constant the CBT:Cys(SEt):reductant (1:1:1) molar ratio. In this case, gelation time points measured by rheology were <1 min, 5 min and 15 min, for TCEP, GSH and DTT, respectively. Moreover, G′ evolved 90% in 5 min, 10 min and 60 min for gels containing TCEP, GSH, or DTT, respectively (Fig. 2c). Final G′ values measured after swelling were 792, 977 and 842 Pa for TCEP, GSH and DTT systems, respectively, and these values were found not significantly different (Fig. 2d). This indicates that gelation rate followed the trend TCEP > GSH > DTT, while no significant difference in gel's mechanical strength was found after the swelling process. Interestingly, these gelation kinetics results did not completely agree with kinetic data of reduction of small aryldisulfides in Tris buffer (100 mM, pH 8.2, 25 °C), where reported cleavage rate followed the trend: TCEP > DTT > GSH;33 neither completely follow the trend of the reduction potential, where DTT (−0.33 V),47 TCEP (−0.29 V),40 and GSH (−0.26 V).41 According to their mere relative reductive strength, it was expected that DTT would be the stronger reductant, leading to the most efficient disulfide cleavage and to the fastest redox-triggering; whereas our rheological characterization results suggest that DTT is the least efficient of the reductants. This could indicate that in the reduction reaction using DTT, other molecular effects such as competing side reactions might play a role in the redox-triggering process, thus the kinetics of disulfide cleavage does not solely depend on reduction potential of the reductant. In line with previous work, we hypothesize that redox-triggering by DTT is slower than expected because this reductant is not chemically orthogonal to the system since its thiol groups can engage in CBT-thiol reversible coupling.46 However, it is known that in presence of free Cys groups, CBT–Cys coupling prevails over CBT-thiol coupling.46 In our system, DTT (a dithiol), is presumed to act both as a reductant and as a transient crosslinker, thus leading to redox-triggered CBT–Cys crosslinked networks with slower gelation rate than expected from its mere reductive character.27
Collectively, our findings prove that in CBT–Cys(SR) gels the structure of the protecting group of the Cys precursor and the choice of reductant type, enable fine control of gelation onset and adjustment of the gelation rate without affecting final gel mechanics. The same final stiffness indicates same crosslinking density and similar conversion of crosslinking in the network. These results are surprising since they are in contrast to reported work on analogous PEG hydrogels crosslinked via thiol-vinylsulfone, thiol-methylsulfonyl or thiol-maleimide coupling reactions, where generally a slower gelation kinetics resulted in stiffer hydrogels.48 It had been hypothesized that a slower curing kinetics allowed higher reaction conversion in the network and rendered a network with fewer defects.48 The reason for the difference between such reported work and our system is currently unknown. We speculate that in our redox-triggerable hydrogels, the fact that two reactions are needed for network formation (precursor unmasking followed by network crosslinking), instead of only crosslinking as in the mentioned report, influences gelation rate but not the final conversion of the network.
Furthermore, CBT–Cys(StBu) hydrogels prepared under same conditions evidenced gelation times generally slower than CBT–Cys(SEt) analogues and their gelation times also depended on the reductant used. At 3.3 wt% polymer concentration, gelation times for CBT–Cys(StBu) system of 27 min and 24 h were observed for TCEP and DTT, respectively; while no gel formed in the presence of GSH (Table 2). A faster gelation rate TCEP > DTT agrees with the trend observed above, whereas the fact that GSH is not able to trigger gel formation indicates that the disulfide cleavage becomes unfavorable under these conditions. We hypothesize that the redox potential of GSH half-reaction is off-range for the RSStBu half-reaction and/or that the steric hindrance of RSStBu rest is too big for effective disulfide cleavage by GSH. Moreover, adjustment of polymer content in the gel formulation can also be used to regulate materials properties, whereby a higher polymer content leads to faster gelation. Table 2 summarizes the gelation time of various formulations of CBT–Cys(SR) hydrogels. Overall, the specific combination of the molecular structure of the protecting group and the reductant type determines gelation onset and kinetics, with gelation times spanning from seconds to minutes, or even to hours range. This redox-responsive system with on-demand crosslinking provides flexible working conditions for gel preparation.
| Hydrogel | Final polymer content | CBT:Cys(SR):reductant molar ratio |
Reductant | Gelation time |
|---|---|---|---|---|
| Conditions: 10 kDa PEG macromers, in 20 mM HEPES pH 8.0.a T = 25 °C.b T = 37 °C. | ||||
| CBT–Cys(SEt)a | 3.3 wt% | 1:1:1 |
TCEP | 25 s |
| GSH | 8 min | |||
| DTT | 14 min | |||
| CBT–Cys(StBu)a | 3.3 wt% | 1:1:1 |
TCEP | 27 min |
| GSH | no gel | |||
| DTT | 24 h | |||
| 6.6 wt% | 1:1:1 |
TCEP | 2.3 min | |
| 1.7 minb | ||||
| DTT | 2.5 h | |||
Adjustment of environmental conditions within close-to-physiological range can be used to regulate materials properties
We investigated the possibility to adjust environmental cues, such as pH and temperature, within close-to-physiological range as a means of modulating gelation rate of CBT–Cys(SR) systems. To this end, CBT–Cys(SR) hydrogels were prepared at 5 wt% using TCEP as reductant, in buffer of varying pH (pH = 8 or 7) and at varying temperature (T = 25 or 37 °C); and rheological characterization was performed. Close-to-physiological range was chosen since our final goal is to implement the redox-triggered hydrogel platform for encapsulation of living cells within injectable matrices for biomedical applications. Time-sweep experiments revealed a slower gelation rate when pH decreased from 8 to 7. CBT–Cys(SEt) gel formed in 30 s and 42 s (∼1.4-fold slower), while CBT–Cys(StBu) gel formed in 1.7 and 7 min (∼4.1-fold slower), when the pH decreased from 8 to 7, respectively (Fig. 3a). This effect of pH on gelation kinetics could be attributed to two possibilities: (i) the disulfide cleavage by TCEP is less efficient at lower pH, or (ii) after disulfide cleavage and exposure of free Cys group, the rate of crosslinking is slower at lower pH, because of the smaller thiolate:thiol ratio. It is hypothesized that the second possibility dominates, in line with previous work on CBT–Cys gels,27 and considering that TCEP has been reported to cleave disulfide bonds efficiently over a broad pH range from 9.0 to 1.5.49
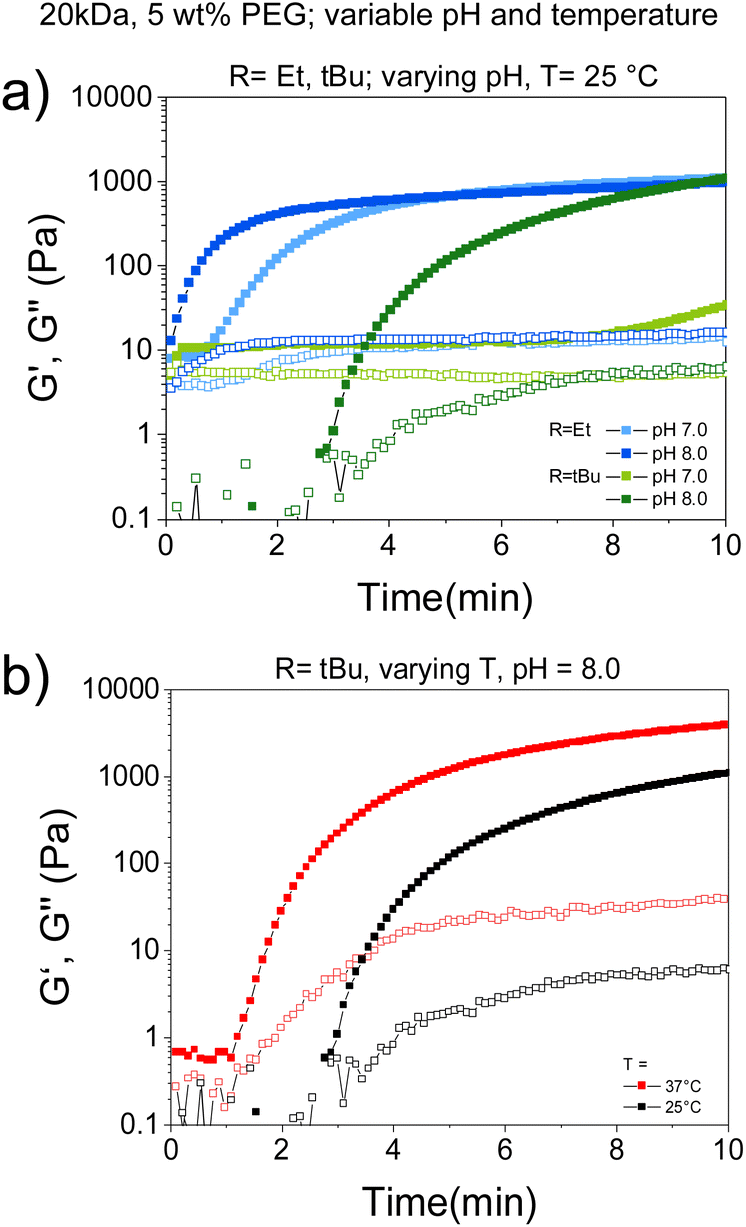 | ||
| Fig. 3 Study of the gelation kinetics and mechanical strength of TCEP-triggered CBT–Cys(SR) hydrogels by shear oscillatory rheometry at varying pH and temperature. (a) Effect of pH on gelation of CBT–Cys(SR) systems. (b) Effect of temperature on the gelation of CBT–Cys(StBu) system. Gel composition: 20 kDa PEGs, 5 wt% polymer content, in HEPES, at (1:1) TCEP:Cys(SR) molar ratio. Specific pH and temperature are indicated in each case. In all cases, data are plotted as mean ± SD, n = 3. | ||
To check whether different gelation rate regulated by pH additionally influences the final mechanical strength of the materials, gels at different pH values were prepared and G′ after equilibrium swelling were compared. G′ ranged 1710–2202 Pa and no significant difference was obtained across different pH values and protecting groups (Fig. S5†). These results are in good agreement with a previous report on CBT–Cys system,27 and prove the possibility of pH-regulating the gelation rate of redox-triggered CBT–Cys(SR) hydrogels, without affecting final gel mechanics.
Moreover, the effect of temperature on gelation rate was studied in CBT–Cys(StBu) system. In this case, gelation time decreased from 3 min to 1 min (3-fold faster) when the curing temperature increased from 25 to 37 °C (Fig. 3b and Table 2). Importantly, temperature adjustment also impacted gel's mechanical strength: G′ = 1072 Pa vs. G′ = 3879 Pa when curing was performed at 25 and 37 °C, respectively. This evidences that the increase of temperature also impacted the crosslinking degree of the formed networks, presumably due to increased conversion, in good agreement with previous reports of other hydrogels which crosslinking was based on thiol-mediated nucleophilic coupling reactions.48,50 Altogether, gelation rate of CBT–Cys(SR) gels can be tuned between seconds, minutes, and hours, depending on the specific choice of molecular and environmental parameters of the CBT–Cys(SR) system.
Redox-triggered hydrogels are cytocompatible
To assess the feasibility of applying the redox-triggered CBT–Cys(SR) gels for cell-encapsulation, the cytocompatibility of these systems was studied. A procedure schematically shown in Fig. 4a was followed. PEG-CBT precursor was biofunctionalized with the cell-adhesive cyclo(RGDfK(C)) peptide and then pre-mixed with the PEG–Cys(SR) precursor and human mesenchymal stem cells (hMSC) suspension. To this mixture, TCEP reductant solution was added to trigger the crosslinking process that led to the obtention of hydrogels with embedded cells. The final gel composition was 3 wt% PEG and 0.03 wt% (0.46 mM) cyclo(RGDfK(C)) peptide. The latter is within the range (0.05–3.5 mM) of previously reported cell-encapsulating PEG gels.51–53 In our experiments, such concentration of RGD peptide corresponds to 1/8 of total Cys groups while PEG-Cys(SR) precursor provides remaining 7/8 of Cys groups after deprotection. This concentration of RGD peptide allows to reach a hydrogel with convenient balance of mechanical strength and cell-adhesiveness to support cell survival. Although at this composition gelation time was 20 s and 1 min for CBT–Cys(SEt) vs. CBT–Cys(StBu) gels, respectively, hydrogels were left curing for 15 min in incubator. In these gels, luciferin click ligation is used both for biofunctionalization of PEG chains and for gelation.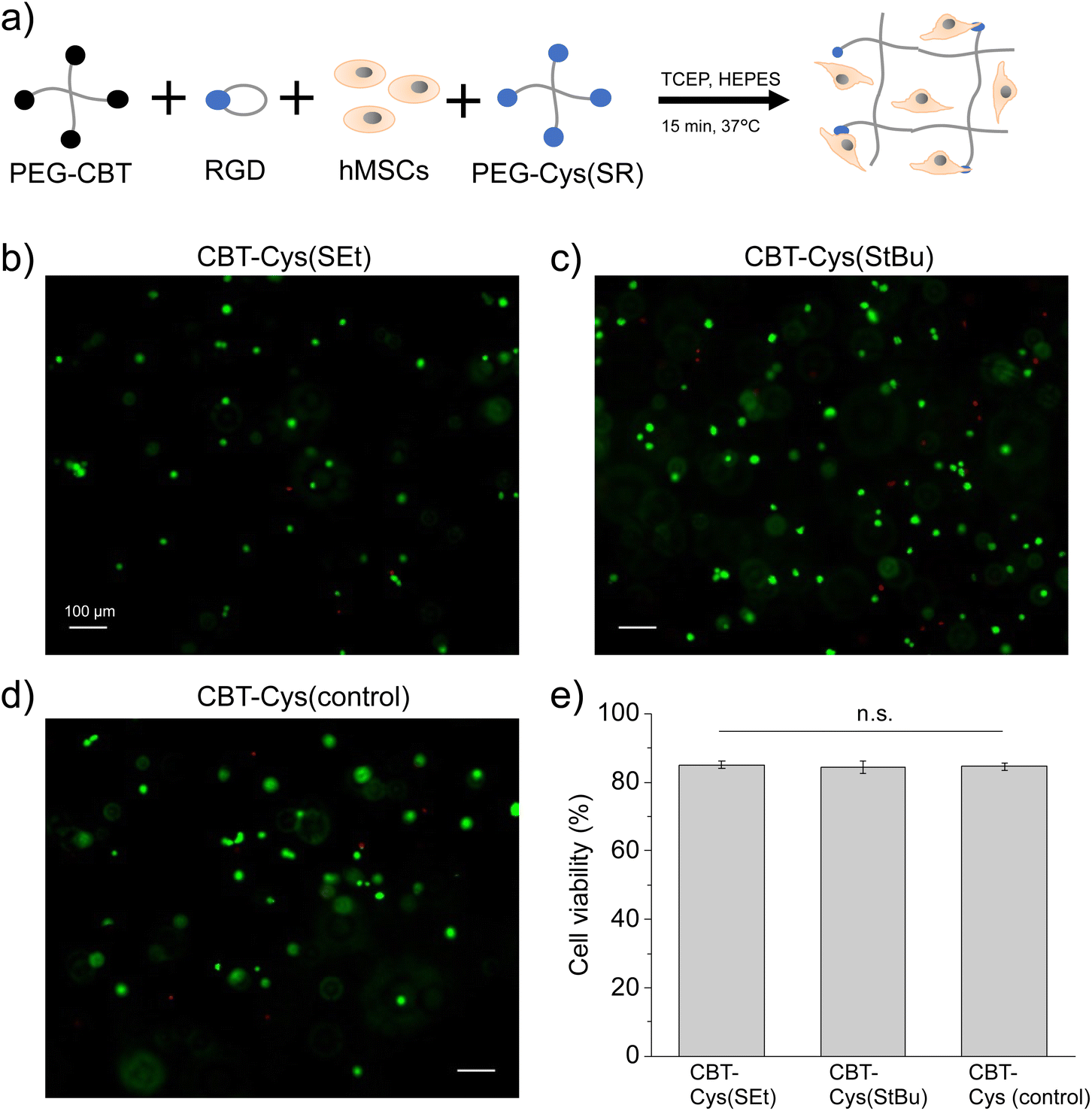 | ||
| Fig. 4 Cytocompatibility study of CBT–Cys(SR) hydrogels biofunctionalized with cell-adhesive cyclo(RGDfK(C)) peptide. (a) Scheme of procedure followed for cell encapsulation, including biofunctionalization of PEG-CBT with RGD followed by redox-triggered crosslinking and cell encapsulation. (b–d) Representative fluorescence images showing post-encapsulation hMSCs survival after live (green)/dead (red) staining at 1 day of culture, and (e) corresponding quantification of cell viability, at varying protecting groups CBT–Cys(SR) (b) R = Et, (c) R = tBu. (d) A CBT–Cys control gel is shown for comparison. Scale bars: 100 μm. Statistical analysis was performed by ANOVA followed by post hoc Tukey test (*p < 0.05 used for statistical significance; n.s. = not significant). Gel composition: 20 kDa, 3 wt% PEGs, 0.03 wt% (0.46 mM) cyclo(RGDfK(C)), starting cell density per gel was 5000 cells. | ||
Embedded cells were cultured for 1 day, live/dead assay was performed, and cell viability was quantified (Fig. 4b–e). Cells encapsulated in CBT–Cys(SR) hydrogels maintained high viability (>83%), similar to the value observed in CBT–Cys control gels and in good agreement with other established hydrogel systems for cell encapsulation, such as the thiol-vinylsulfone materials demonstrated in previous reports.27,48 At 3 days post-encapsulation, embedded cells kept high viability (>83%) and showed homogeneous density distribution across the gels (see Fig. S6†). These results prove that the use of TCEP as redox-trigger of gelation does not negatively impact cell survival, at least within the tested conditions (here, TCEP concentration was 2.8 mM). This demonstrates the good cytocompatibility of CBT–Cys(SR) hydrogels and the possibility of reaching materials with homogeneous cell density.
Redox-triggering can be exploited for formulating injectable and in-bath crosslinkable hydrogel matrices
Injectable matrices for therapeutic delivery and tissue repair, either alone or in combination with processing and scaffolding technologies (e.g., extrusion-based printing), are becoming increasingly important for tissue engineering and regenerative medicine.54,55 These scenarios entail the delivery of low viscous (pre)mixed precursor solutions through a fine needle or nozzle to the application site, followed by an on-demand quick gelation step for mechanical stabilization that is typically triggered by the action of an external stimulus.56,57 We foresaw that the redox-triggerable firefly luciferin-inspired hydrogel platform could be easily adapted towards injectable and in-bath crosslinking scenarios, thus combining the advantage of tunable materials properties with the ease of processing such matrices.To prove this concept, an experiment to test injectability and in situ in-bath crosslinking of our materials was designed (Fig. 5a). 10 wt% PEG-CBT and PEG-Cys(SEt) precursors were pre-mixed and colored with green food dye for visualization purposes. The precursors mixture was injected via micropipette into a reductant bath that contained TCEP and buffer/glycerol equivolume combination. Note that glycerol was added to adjust the viscosity of the bath to better support the aimed crosslinked material. The precursors mixture could be injected through the fine pipette tip without clogging into the bath, and formation of a hydrogel was observed within 10 s (Fig. 5b and Video S1 in ESI†). After in-bath crosslinking and removal of the reductant bath, a hydrogel with good mechanical stability was obtained (Fig. 5c). A control experiment was performed, in which PEG precursors were injected into a bath that contained buffer/glycerol but lacked the reductant. In this case, the precursors mixture rapidly dissolved in the bath and no hydrogel was formed (Fig. 5d and Video S2 in ESI†), demonstrating that only the presence of reductant can trigger in-bath gelation.
 | ||
| Fig. 5 Demonstration of the use of redox-triggered gelation for developing injectable and in-bath crosslinkable hydrogel matrices. A solution mixture containing PEG-CBT and PEG-Cys(SEt) precursors was injected into a reductant bath, and CBT–Cys(SEt) gelation was triggered in situ. (a) Schematics of experimental setup. (b) Crosslinking of CBT–Cys(SEt) hydrogel in the reductant bath (TCEP was used). (c) Hydrogel after in-bath crosslinking followed by removal of the reductant bath. (d) Negative control: injecting precursors mixture in a bath without TCEP did not lead to the formation of a hydrogel. Instead, PEGs solution diluted in the bath. Precursors solutions: 10 kDa, 10 wt% PEG-CBT and 10 wt% PEG-Cys(SEt), (1:1) volume ratio in HEPES pH 8.0 (containing green food dye for clear visualization). Reductant bath: HEPES:glycerol (1:1) volume ratio, 20 mM TCEP. Scale bar: 1 cm. (b–d) Snapshots of videos recorded during the experiments. The full videos can be found as ESI.† | ||
As demonstrated in a previous section of this article, the good stability of aqueous solutions of PEG-Cys(SR) precursors facilitates the preparation of stock solutions, which can be pre-mixed with PEG-CBT precursor at a convenient time before processing the mixture for on-demand crosslinking. We anticipate that the redox-triggered hydrogel platform will be advantageous for the development of injectable matrices for drug delivery as well as bioinks for extrusion-based 3D bioprinting.
3. Conclusions
A novel redox-triggered firefly luciferin-inspired hydrogel platform was developed in this work. PEG-Cys(SR) precursors demonstrated good long-term stability upon storage in physiologically-relevant aqueous conditions, which is expected to facilitate applicability of this platform in those settings requiring pre-mixing of reactive precursors. The CBT–Cys(SR) system allows flexible in situ gelation, whereby gelation onset and rate can be fine modulated by adjustment of molecular parameters (e.g., protecting group structure and reductant type) and environmental parameters (e.g., pH and temperature) of the deprotection reaction. Depending on the specific choice of conditions, gelation times spanning from seconds to minutes to hours could be easily achieved. Furthermore, stem cells encapsulated in biofunctionalized CBT–Cys(SR) hydrogels showed high viability after 1–3 days of encapsulation, demonstrating the good cytocompatibility of these systems. With redox-triggered gelation, these smart hydrogels are envisioned as injectable matrices for drug delivery and tissue engineering as well as inks for extrusion-based printing of soft constructs for regenerative medicine. These bioinspired materials with upgraded tunability expand the toolkit of stimuli-responsive materials.Author contributions
M. J. and A. G. synthesized macromer precursors and performed rheological characterization experiments. M. J. performed microscopy and cell experiments. M. J. and J. I. P. wrote the initial manuscript. J. I. P. conceived and supervised the project, and acquired funding. All authors contributed with data analysis, provided critical feedback on the manuscript and approved its final version.Conflicts of interest
There are no conflicts of financial interest to declare.Acknowledgements
The authors thank financial support from the Deutsche Forschungsgemeinschaft (DFG, project no. 422041745) awarded to J. I. P. The authors thank Dr Gülistan Koçer (INM) for supporting cell experiments and Prof. A. del Campo (INM) for mentoring and support.References
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1880 CrossRef CAS PubMed.
- D. Seliktar, Science, 2012, 336, 1124–1128 CrossRef CAS PubMed.
- I. Willner, Acc. Chem. Res., 2017, 50, 657–658 CrossRef CAS PubMed.
- F. Gao, Y. Zhang, Y. Li, B. Xu, Z. Cao and W. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 8956–8966 CrossRef CAS PubMed.
- J. A. Burdick, D. Frankel, W. S. Dernell and K. S. Anseth, Biomaterials, 2003, 24, 1613–1620 CrossRef CAS PubMed.
- M. Villiou, J. I. Paez and A. del Campo, ACS Appl. Mater. Interfaces, 2020, 12, 37862–37872 CrossRef CAS PubMed.
- A. Farrukh, J. I. Paez and A. del Campo, Adv. Funct. Mater., 2019, 29, 1807734 CrossRef.
- L. Klouda, Eur. J. Pharm. Biopharm., 2015, 97, 338–349 CrossRef CAS PubMed.
- L. Klouda and A. G. Mikos, Eur. J. Pharm. Biopharm., 2008, 68, 34–45 CrossRef CAS PubMed.
- I. Tomatsu, K. Peng and A. Kros, Adv. Drug Delivery Rev., 2011, 63, 1257–1266 CrossRef CAS PubMed.
- Z. Liu, J. Liu, X. Cui, X. Wang, L. Zhang and P. Tang, Front. Chem., 2020, 8, 124 CrossRef CAS PubMed.
- M. Antman-Passig and O. Shefi, Nano Lett., 2016, 16, 2567–2573 CrossRef CAS PubMed.
- S. Kennedy, J. Hu, C. Kearney, H. Skaat, L. Gu, M. Gentili, H. Vandenburgh and D. Mooney, Biomaterials, 2016, 75, 91–101 CrossRef CAS PubMed.
- D. Schmaljohann, Adv. Drug Delivery Rev., 2006, 58, 1655–1670 CrossRef CAS PubMed.
- N. A. Peppas and D. S. Van Blarcom, J. Controlled Release, 2016, 240, 142–150 CrossRef CAS PubMed.
- R. Y. Tam, L. J. Smith and M. S. Shoichet, Acc. Chem. Res., 2017, 50, 703–713 CrossRef CAS PubMed.
- M. D. Konieczynska and M. W. Grinstaff, Acc. Chem. Res., 2017, 50, 151–160 CrossRef CAS PubMed.
- V. Yesilyurt, M. J. Webber, E. A. Appel, C. Godwin, R. Langer and D. G. Anderson, Adv. Mater., 2016, 28, 86–91 CrossRef CAS PubMed.
- H. L. Lim, Y. Hwang, M. Kar and S. Varghese, Biomater. Sci., 2014, 2, 603–618 RSC.
- N. Eslahi, M. Abdorahim and A. Simchi, Biomacromolecules, 2016, 17, 3441–3463 CrossRef CAS PubMed.
- X. Sui, X. Feng, M. A. Hempenius and G. J. Vancso, J. Mater. Chem. B, 2013, 1, 1658–1672 RSC.
- B. D. Fairbanks, S. P. Singh, C. N. Bowman and K. S. Anseth, Macromolecules, 2011, 44, 2444–2450 CrossRef CAS PubMed.
- M. E. Bracchi, G. Dura and D. A. Fulton, Polym. Chem., 2019, 10, 1258–1267 RSC.
- Z. Gao, B. Golland, G. Tronci and P. D. Thornton, J. Mater. Chem. B, 2019, 7, 7494–7501 RSC.
- L. Wu, S. Di Cio, H. S. Azevedo and J. E. Gautrot, Biomacromolecules, 2020, 21, 4663–4672 CrossRef CAS PubMed.
- M. Huo, J. Yuan, L. Tao and Y. Wei, Polym. Chem., 2014, 5, 1519–1528 RSC.
- M. Jin, G. Koçer and J. I. Paez, ACS Appl. Mater. Interfaces, 2022, 14, 5017–5032 CrossRef CAS PubMed.
- A. K. Mahanta and P. Maiti, ACS Appl. Bio Mater., 2019, 2, 5415–5426 CrossRef CAS PubMed.
- J. I. Paez, O. Ustahüseyin, C. Serrano, X.-A. Ton, Z. Shafiq, G. K. Auernhammer, M. d'Ischia and A. del Campo, Biomacromolecules, 2015, 16, 3811–3818 CrossRef CAS PubMed.
- D. Bermejo-Velasco, A. Azémar, O. P. Oommen, J. Hilborn and O. P. Varghese, Biomacromolecules, 2019, 20, 1412–1420 CrossRef CAS PubMed.
- J. Feng, X.-A. Ton, S. Zhao, J. I. Paez and A. Del Campo, Biomimetics, 2017, 2, 23 CrossRef PubMed.
- G. Liang, H. Ren and J. Rao, Nat. Chem., 2010, 2, 54–60 CrossRef CAS PubMed.
- P. K. Pullela, T. Chiku, M. J. Carvan and D. S. Sem, Anal. Biochem., 2006, 352, 265–273 CrossRef CAS PubMed.
- M. Kar, Y.-R. Vernon Shih, D. O. Velez, P. Cabrales and S. Varghese, Biomaterials, 2016, 77, 186–197 CrossRef CAS PubMed.
- S. E. Boiadjiev and D. A. Lightner, J. Am. Chem. Soc., 2000, 122, 11328–11339 CrossRef CAS.
- T. J. Bechtel and E. Weerapana, Proteomics, 2017, 17, 1600391 CrossRef PubMed.
- E. B. Getz, M. Xiao, T. Chakrabarty, R. Cooke and P. R. Selvin, Anal. Biochem., 1999, 273, 73–80 CrossRef CAS PubMed.
- V. Ribas, C. García-Ruiz and J. C. Fernández-Checa, Front. Pharmacol., 2014, 5, 151 Search PubMed.
- W. W. Cleland, Biochemistry, 1964, 3, 480–482 CrossRef CAS PubMed.
- E. A. Dergousova, I. Y. Petrushanko, E. A. Klimanova, V. A. Mitkevich, R. H. Ziganshin, O. D. Lopina and A. A. Makarov, Biomolecules, 2017, 7, 18 CrossRef PubMed.
- K. Khazim, D. Giustarini, R. Rossi, D. Verkaik, J. E. Cornell, S. E. D. Cunningham, M. Mohammad, K. Trochta, C. Lorenzo, F. Folli, S. Bansal and P. Fanti, Transl. Res., 2013, 162, 16–25 CrossRef CAS PubMed.
- J. A. Burns, J. C. Butler, J. R. Moran and G. M. Whitesides, J. Org. Chem., 1991, 56, 2648–2650 CrossRef CAS.
- Z. Zheng, P. Chen, G. Li, Y. Zhu, Z. Shi, Y. Luo, C. Zhao, Z. Fu, X. Cui, C. Ji, F. Wang, G. Huang and G. Liang, Chem. Sci., 2017, 8, 214–222 RSC.
- A. Isidro-Llobet, M. Álvarez and F. Albericio, Chem. Rev., 2009, 109, 2455–2504 CrossRef CAS PubMed.
- E. J. Ste.Marie and R. J. Hondal, J. Pept. Sci., 2018, 24, e3130 CrossRef PubMed.
- H. Ren, F. Xiao, K. Zhan, Y.-P. Kim, H. Xie, Z. Xia and J. Rao, Angew. Chem., Int. Ed., 2009, 48, 9658–9662 CrossRef CAS PubMed.
- V. Domkin and A. Chabes, Sci. Rep., 2014, 4, 5539 CrossRef CAS PubMed.
- J. I. Paez, A. Farrukh, R. Valbuena-Mendoza, M. K. Wlodarczyk-Biegun and A. Del Campo, ACS Appl. Mater. Interfaces, 2020, 12, 8062–8072 CrossRef CAS PubMed.
- J. C. Han and G. Y. Han, Anal. Biochem., 1994, 220, 5–10 CrossRef CAS PubMed.
- N. J. Darling, Y.-S. Hung, S. Sharma and T. Segura, Biomaterials, 2016, 101, 199–206 CrossRef CAS PubMed.
- K. Bott, Z. Upton, K. Schrobback, M. Ehrbar, J. A. Hubbell, M. P. Lutolf and S. C. Rizzi, Biomaterials, 2010, 31, 8454–8464 CrossRef CAS PubMed.
- C. Adelöw, T. Segura, J. A. Hubbell and P. Frey, Biomaterials, 2008, 29, 314–326 CrossRef PubMed.
- J. J. Moon, J. E. Saik, R. A. Poché, J. E. Leslie-Barbick, S.-H. Lee, A. A. Smith, M. E. Dickinson and J. L. West, Biomaterials, 2010, 31, 3840–3847 CrossRef CAS PubMed.
- J. H. Lee, Biomater. Res., 2018, 22, 27 CrossRef PubMed.
- E. Lepowsky, M. Muradoglu and S. Tasoglu, Bioprinting, 2018, 11, e00034 CrossRef.
- I. T. Ozbolat and M. Hospodiuk, Biomaterials, 2016, 76, 321–343 CrossRef CAS PubMed.
- L. Yu and J. Ding, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, rheological characterization and cell culture as well as complete experimental section. See DOI: https://doi.org/10.1039/d2py00481j |
| This journal is © The Royal Society of Chemistry 2022 |