
Donor modification of thermally activated delayed fluorescence photosensitizers for organocatalyzed atom transfer radical polymerization†
Alexander M.
Polgar
,
Shine H.
Huang
and
Zachary M.
Hudson
 *
*
Department of Chemistry, The University of British Columbia, 2036 Main Mall, Vancouver, British Columbia V6T 1Z1, Canada. E-mail: zhudson@chem.ubc.ca; Fax: +1-604-822-2847; Tel: +1-604-822-3266
First published on 24th June 2022
Abstract
Thermally activated delayed fluorescence (TADF) photosensitizers based on 9,10-dihydro-9,9-dimethylacridine/2,4,6-triphenylpyrimidine conjugates with strong visible absorption, large excited state reduction potentials, and long-lived triplet excited states were successfully employed in the organocatalyzed atom transfer radical polymerization (O-ATRP) of methacrylic monomers. A donor-modification strategy dramatically improved the stability of the photocatalyst radical cations, while retaining their high oxidizing strengths, a key requirement for controlled O-ATRP. Time-resolved photoluminescence studies of the catalysts support initiation by electron transfer from both singlet and triplet states, with functionalized donors producing higher driving forces for photoinduced electron transfer. A donor-modified TADF photocatalyst was found for the synthesis of methacrylic polymers with Đ below 1.3 at catalyst loadings down to 50 ppm. This catalyst was also successfully applied in block copolymer synthesis, while the unfunctionalized analogue yields entirely uncontrolled polymerization.
1. Introduction
Controlled radical polymerizations such as nitroxide-mediated polymerization (NMP),1 radical addition–fragmentation chain transfer (RAFT),2 and atom-transfer radical polymerization (ATRP)3 have empowered chemists to synthesize polymers of well-defined size, composition, and topology without the need for specialized equipment.4 In ATRP, polymerization is mediated by a metal complex (commonly Cu) which reversibly deactivates propagating radical chains through an inner-sphere single electron transfer.5 Initially, very high loadings (>1000 ppm) of Cu were required to achieve sufficient rates of polymerization, raising issues with the applicability of the technique in biomedicine, health/beauty products, and electronics.6 Lower Cu loadings (10–50 ppm) have since been realized by the addition of supplemental initiators or reducing agents to the polymerization to continuously regenerate the CuI activator.6–9Yagci and others have demonstrated the suitability of photoredox catalysis to mediate Cu ATRP with reduced catalyst loadings.10–13 The excited states of photosensitizers are powerful electron donors that can regenerate CuI through an oxidative quenching process, with a sacrificial electron donor to complete the catalytic cycle. In 2012, Hawker demonstrated photo-mediated ATRP with an iridium photocatalyst (PC) that reduces alkyl halide ATRP initiators from its excited state (PC*), generating a highly oxidizing photocatalyst radical cation (PC˙+) that controls polymerization by deactivating the propagating radical.14 This obviated the need for both a CuI activator and a sacrificial electron donor. Purely organic O-ATRP followed from the same group, using 10-phenylphenothiazine as a potent excited state reductant.15 Further development of O-ATRP has seen various phen(ox)azine derivatives and heteroatom-doped aromatic hydrocarbons find use as visible-light activated photocatalysts, which may be applied at reduced loadings, over a wide monomer scope, and under irradiation from sunlight.16–21 As a result of these innovations, photoinduced electron transfer (PET) has become a key feature of metal-free reversible deactivation radical polymerizations.22–26
In 2017, Huang et al. demonstrated O-ATRP using thermally activated delayed fluorescence (TADF) emitters as photocatalysts.27 TADF is widely deployed in fields ranging from organic electronics,28 photocatalysis,29 biological imaging,30 and chemical sensing31,32 due to a unique ability to interconvert singlet and triplet excited states. TADF emitters are commonly designed around a twisted donor–π–acceptor (D–π–A) motif to minimize overlap between the electron density of the HOMO and LUMO. This reduces the electronic exchange energy between electrons in the excited biradical state. Consequently, there is a small energetic gap between the first excited singlet (S1) and triplet (T1) states that may be overcome by thermal energy in a process termed reverse intersystem crossing (RISC). The result is a cycling between S1 and T1 that prolongs the excited state lifetime and facilitates singlet and triplet energy/electron transfer.33
The benefits of TADF emitters for O-ATRP were clearly demonstrated by Singh et al. in 2018.34 Through computational screening, and detailed structure–activity relationships, the authors identified the TADF emitter 2,4,5,6-tetrakis(diphenylamino)-1,3-benzenedicarbonitrile (4DPIPN) as an optimal photocatalyst for the polymerization of methyl methacrylate (MMA) at sub-ppm catalyst loadings. Due to the prolonged excited state lifetime of TADF emitters, the effective concentration of the excited state activator species in solution is higher than for a comparable concentration of a fluorescent photosensitizer.14 At the low catalyst loadings afforded by TADF catalysis, the polymer formed might be used without catalyst removal – greatly simplifying the procedure compared to conventional Cu-based ATRP.
The donor–acceptor nature of TADF has advantages for achieving controlled O-ATRP with an expanded monomer scope. As shown in Fig. 1, the charge transfer (CT) excited state of a TADF emitter with a D–π–A design can be approximated as a biradical with an oxidized donor (D˙+) and reduced acceptor (A˙−). In the activation step of O-ATRP, the excited state undergoes oxidative quenching with an alkyl halide initiator. The driving force for this step is proportional to the reduction potential of the ground-state acceptor. Singh et al. made use of this fact, using the weak acceptor diphenylsulfone, with a large negative ground-state reduction potential (E°(A/A˙−) = −2.3 V vs. SCE) for the controlled O-ATRP of styrene (E°(RBr/R˙ + Br−) ∼ −1.5 V; R = α-alkyl benzyl).34 Moreover, if electron transfer from the excited triplet state is dominant in an O-ATRP, the small ΔEST of TADF PCs allows less excitation energy to be lost via ISC – a unique feature of TADF photosensitizers.
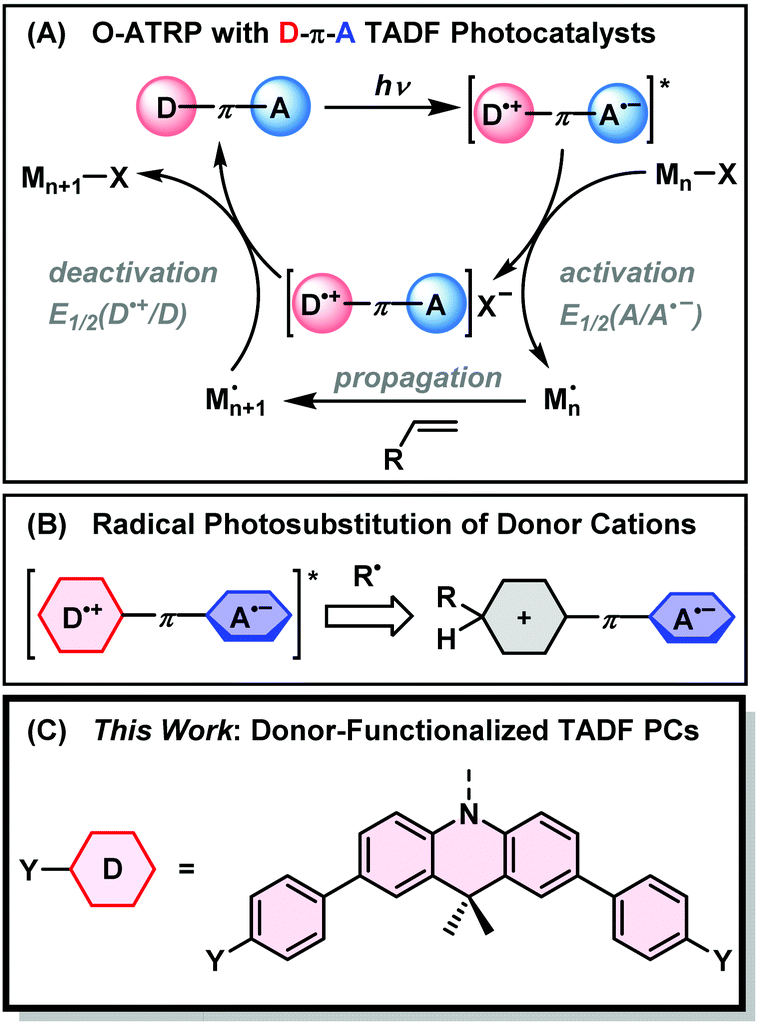 | ||
| Fig. 1 (A) Mechanism of organocatalyzed atom-transfer radical polymerization of olefinic monomers using D–π–A type photocatalysts. (B) Deactivation of charge transfer excited states by radical substitution at the donor. (C) Functionalized 9,10-dihydro-9,9-dimethylacridine donors used in this work. | ||
Matyjaszewski argued that deactivation of the propagating radical occurs through a termolecular encounter involving an associative electron transfer (AET) with the photocatalyst radical cation.35 Weaker donors, with higher oxidation potentials (E°(D˙+/D)), therefore provide a larger driving force for this step, and should yield greater control over polymer molecular weight and dispersity. TADF photocatalysts are unique in the ability to independently tune the driving force for activation and deactivation through choice of donor and acceptor moieties. The ideal TADF photocatalyst needs to balance the opposing demands of fast activation and deactivation (requiring weak donors and acceptors) with intense visible light absorption from a charge transfer excited state – which conversely relies on using stronger donors and acceptors.
Using electron donors and acceptors in photoredox catalysis comes with the drawback of unwanted excited state side reactivity.36 Exposed positions on the aromatic rings of catalysts with electron donor moieties can abstract radicals from the polymerization (Fig. 1B).27,37–39 This lowers the initiator efficiency of the reaction and can also deactivate the catalyst towards further photocatalytic cycling by changing its light-absorbing and electrochemical properties. König has shown that the acceptor is also vulnerable to radical photosubstitution chemistry in cyanobenzene-based catalysts such as 4DPIPN.40,41 Weak C–S and C–P bonds are also subject to cleavage in sulfone- or phosphine-oxide-based acceptors.42 Degradation of the acceptor might be mitigated by instead using electron-deficient nitrogen-based heterocycles, as any degradation of the acceptor would occur at a lower rate due to the disruption of aromaticity.
Here, we propose the use of donor-modified TADF emitters as photocatalysts for O-ATRP. Our design is based around 2,4,6-triphenylpyrimidine acceptors, having a high ground state reduction potential (E°(A/A˙−) = −1.7 V vs. SCE), and 9,10-dihyro-9,9-dimethylacridine donors with strongly oxidizing radical cations (E°(D˙+/D) ≥ 0.7 V) in the donor–acceptor conjugate. To limit excited state side-reactivity and improve the visible light absorption of the CT excited state, methylbenzene and methoxybenzene groups are appended to the para-positions of 9,10-dihyro-9,9-dimethylacridine (Fig. 1C). The donor-modified photocatalysts significantly outperform the catalyst without para-substitution, allowing for the polymerization of MMA and benzyl methacrylate (BnMA) with dispersities down to 1.27 at 50 ppm catalyst loading. Time-resolved fluorescence studies reveal rapid and near-quantitative electron transfer from the triplet state of the photocatalyst, with increased rates for the donor-modified catalysts that have higher driving forces for photoinduced electron transfer. The chain-end fidelity of the polymerization was corroborated by the synthesis of a block copolymer with high chain re-initiation efficiency. Overall, this work demonstrates the promise of molecular engineering for TADF emitters in photoredox-mediated controlled radical polymerizations.
2. Experimental
2.1 Instrumentation
Stern–Volmer quenching experiments were carried out in reagent grade (≥99%) N,N-dimethylacetamide (DMAc), dried over 3 Å molecular sieves and subjected to three freeze–pump–thaw cycles prior to storage in a nitrogen-filled glovebox. Prior to use, DMAc was filtered through a short column of activated neutral alumina. Appropriate amounts of photocatalyst and initiator stock solutions were combined to give 100 μM of the photocatalyst and 10–200 mM of the initiator. The solution was transferred to a quartz cuvette and sealed with a custom-made screw-cap with an O-ring seal to prevent oxygen quenching. Prompt and delayed fluorescence lifetimes were modelled by exponential reconvolution fitting with the instrument response function by optimizing the χ2 value of the fit.
2.2 Chemical synthesis
Details on the synthesis and NMR/HR-MS characterization of the photocatalysts used in this study can be found in the ESI.†2.3 Photopolymerization procedure
Photopolymerizations were conducted in a nitrogen-filled glovebox. The light source was a 435 ± 10 nm LED strip (DIY LED U-HOME SMD2835-60led) arranged along the inside of a 6-inch diameter crystallization dish. The total light output is estimated at 1 W (200 LEDs × 5 mW per LED). The setup was continuously cooled using a fan and the glovebox atmosphere is recirculated through a cold-water heat exchanger to maintain an average temperature between 28 and 30 °C over the course of the polymerization. The polymerizations were conducted in Teflon-septum-capped 1-dram vials, which were positioned 2 inches away from the edge of the crystallization dish. The dish was set on a magnetic stir plate set to 400 rpm. A photograph of the setup and the spectrum of the LED source are shown in Fig. S1 of the ESI.†The solvent used for photopolymerizations was DMAc (99% ACS Reagent Grade), which was degassed by three freeze–pump–thaw cycles and stored over 3 Å molecular sieves inside the glovebox. Directly before use in polymerizations, the solvent was passed through a short column of activated neutral alumina. All monomers were degassed by three freeze–pump–thaw cycles prior to use and passed through activated neutral alumina to remove inhibitor.
In a typical procedure, 100 μL of the monomer were added to a 1-dram vial containing a magnetic stir bar. Stock solutions of the initiator and photocatalyst were prepared at 50 and 2.5 or 0.5 mg mL−1 in DMAc, respectively. Appropriate amounts of the stock solutions were added to give theoretical DP of 200 (MMA) or 100 (BnMA) and catalyst loadings of 500 (2.5 mg mL−1 stock), 50, 25, and 10 ppm (0.5 mg mL−1 stock; ppm relative to monomer). DMAc was then added to each polymerization so that the total volume of solvent was 250 μL. The LED was then turned on and reaction kinetics were monitored by periodically removing 15 μL aliquots from the reaction, diluting in CDCl3 and analyzing by NMR the relative intensity of the monomer vs. polymer peaks. Polymers were isolated by pipetting the reaction mixture into ethanol and collecting the precipitate by vacuum filtration.
2.4 Density functional theory
Density functional theory calculations were carried out in Gaussian 16 rev B.01. All photocatalysts were optimized at the B3LYP/6-31G(d) level of theory in their ground singlet (S0), excited triplet (T1) and radical cation doublet (D+) electronic states.44,45 Frequency analysis verified that all structures were at local minima. Ground and excited state oxidation potentials were computed from the Gibbs free energies. ΔG(PC˙+/PC) = G(D˙+) − G(S0); ΔG*(PC˙+/3PC*) = G(D˙+) − G(T1). Free energies were converted into electrode potentials by taking a value of −100.5 kcal mol−1 for the standard hydrogen electrode.46 Finally, a value of 0.24 V vs. SHE was assumed for the SCE.43 Gas-phase B3LYP/6-31G(d) provided good agreement between theoretical and measured ground and excited state electrode potentials, within 0.3 eV. Additional diffuse or polarization functions or an implicit solvent model (DMAc) worsened the agreement between theory and experiment.Time-dependent DFT was performed at the ωB97XD/6-311++G(d,p) level of theory using a conductor-like polarizable continuum (CPCM) solvent model, with toluene as the solvent.47 The range-separation parameter ω was chosen through comparison of the theoretical charge transfer transition energy with experiment. The vertical singlet–triplet gap was determined using the Tamm–Dancoff approximation (TDA) at the optimized S0 geometry as the difference between the energy of the lowest excited singlet and lowest excited triplet state.48 TDA was employed to avoid underestimation of the triplet energy.49,50
3. Results and discussion
3.1 Catalyst design
Three D–π–A–π–D triads of 9,10-dihydro-9,9-dimethylacridine (DMA) and 2,4,6-triphenylpyrimdine (Pym) were synthesized (Section S1 of the ESI†), with the donor either bearing hydrogen atoms (PymDMA), methylphenyl groups (PymDMDTA), or methoxyphenyl (PymDMDMA) groups para- to the nitrogen atom (Fig. 2). Evaluation of their catalytic properties began by simulating their energetics in the singlet ground (S0), excited triplet (T1) and cationic doublet (D+) states at the B3LYP/6-31G(d) level of theory (Fig. 2B). The Gibbs free energies of the T1 state are highest for PymDMA at 58.9 kcal mol−1 above the ground state and lowest for PymDMDMA, bearing electron-donating methoxy groups, at 56.4 kcal mol−1. The energies of the doublets vary more widely, with PymDMA having a high-energy radical cation at 135.0 kcal mol−1, compared to 124.5 for PymDMDMA. The relative instability of the unmodified DMA radical cation can be expected to result in a lower driving force for photoinduced electron transfer.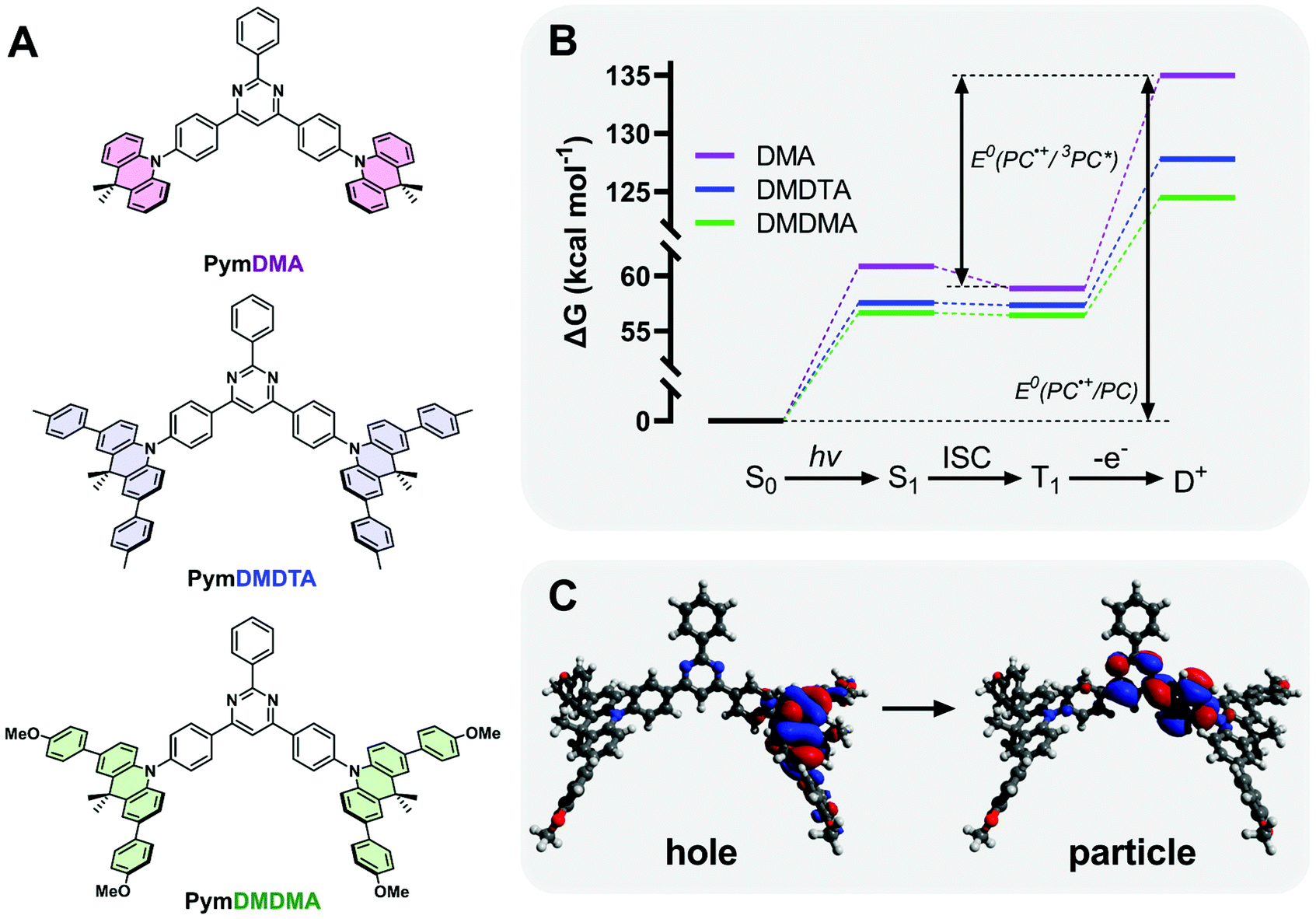 | ||
| Fig. 2 (A) Structure of the photocatalysts used in this study. (B) Calculated changes in free energy during the photocatalytic cycle at B3LYP/6-31G(d) level of theory. (C) Visualization of the particle–hole pair for the S1 transition of PymDMDMA at ωB97XD/6-311++g(d,p) level of theory with toluene CPCM. | ||
The computed triplet excited state reduction potentials E°(PC˙+/3PC*) are compiled in Table 1. PymDMA, as expected, has the lowest driving force for PET and PymDMDMA the highest. Nevertheless, all PCs are predicted to be sufficiently strong excited-state reductants to activate typical α-bromo ester ATRP initiators. Conversely, the unstable radical cation of PymDMA should provide the most rapid deactivation due to its high electron affinity E°(PC˙+/PC) = 1.25 V vs. SCE. Based on these results alone, PymDMA should provide the best control over O-ATRP, by limiting the concentration of propagating radicals through slow activation, and reducing their persistence time with fast deactivation.
| E°(PC˙+/PC) (V vs. SCE) | E°(PC˙+/1PC*) (V vs. SCE) | E°(PC˙+/3PC*) (V vs. SCE) | ω (Bohr−1) | ΔEST (meV) | |
|---|---|---|---|---|---|
| PymDMA | 1.25 | −1.39 | −1.30 | 0.1246 | 86 |
| PymDMDTA | 0.94 | −1.55 | −1.54 | 0.1005 | 9 |
| PymDMDMA | 0.80 | −1.66 | −1.65 | 0.1121 | 9 |
TDA-DFT was employed to calculate the singlet–triplet energy gap (ΔEST) and from this obtain an estimate of the energy of S1. To minimize the delocalization error in density functional theory,51 a range-separated hybrid functional ωB97XD, with variable amount of Hartree–Fock exchange, was used in conjunction with a diffuse basis set 6-311++G(d,p) to adequately describe charge transfer. Through optimization of the range-separation parameter, ω, the singlet–triplet gap of TADF emitters can be accurately predicted.52PymDMDTA and PymDMDMA have ΔEST below 10 meV, consistent with previous results showing that π-extended donors are effective for minimizing HOMO–LUMO overlap and ΔEST.53,54PymDMA has a higher ΔEST of 86 meV, which results in a higher-energy singlet state and corresponding greater driving force for singlet electron transfer.
The charge-transfer nature of the singlet excited states was confirmed through natural transition orbital (NTO) analysis (Fig. 2C). The first optically bright (f > 0) transition locates the electron density “hole” on the DMA donor, with small contributions from the para-aryl substituents. The “particle” is localized mainly on the pyrimidine acceptor and the phenyl bridge between the donor and acceptor. Some degree of hole–particle overlap is desirable for increased absorptivity of the lowest-energy charge transfer state, and this is observed in all cases on the phenyl bridge. Owing to their low ΔEST and strong excited-state charge-transfer nature, these materials are expected to be effective mediators of reverse intersystem crossing for efficient TADF.
3.2 Optical and electrochemical characterization
UV-vis absorption spectroscopy of all catalysts in toluene (Fig. 3A) shows moderately strong charge-transfer bands (εmax ∼ 4–6 × 103 M−1 cm−1) between 350 and 450 nm. As predicted by DFT, PymDMA has the highest-energy excitation to S1, peaking at 378 nm with the band extending to 440 nm. The donor-modified derivatives have bathochromically shifted absorptions, with the charge-transfer band extending to 460 nm. All three have sufficient absorption in the visible range, due to the moderate donor and acceptor strengths of DMA and Pym, respectively.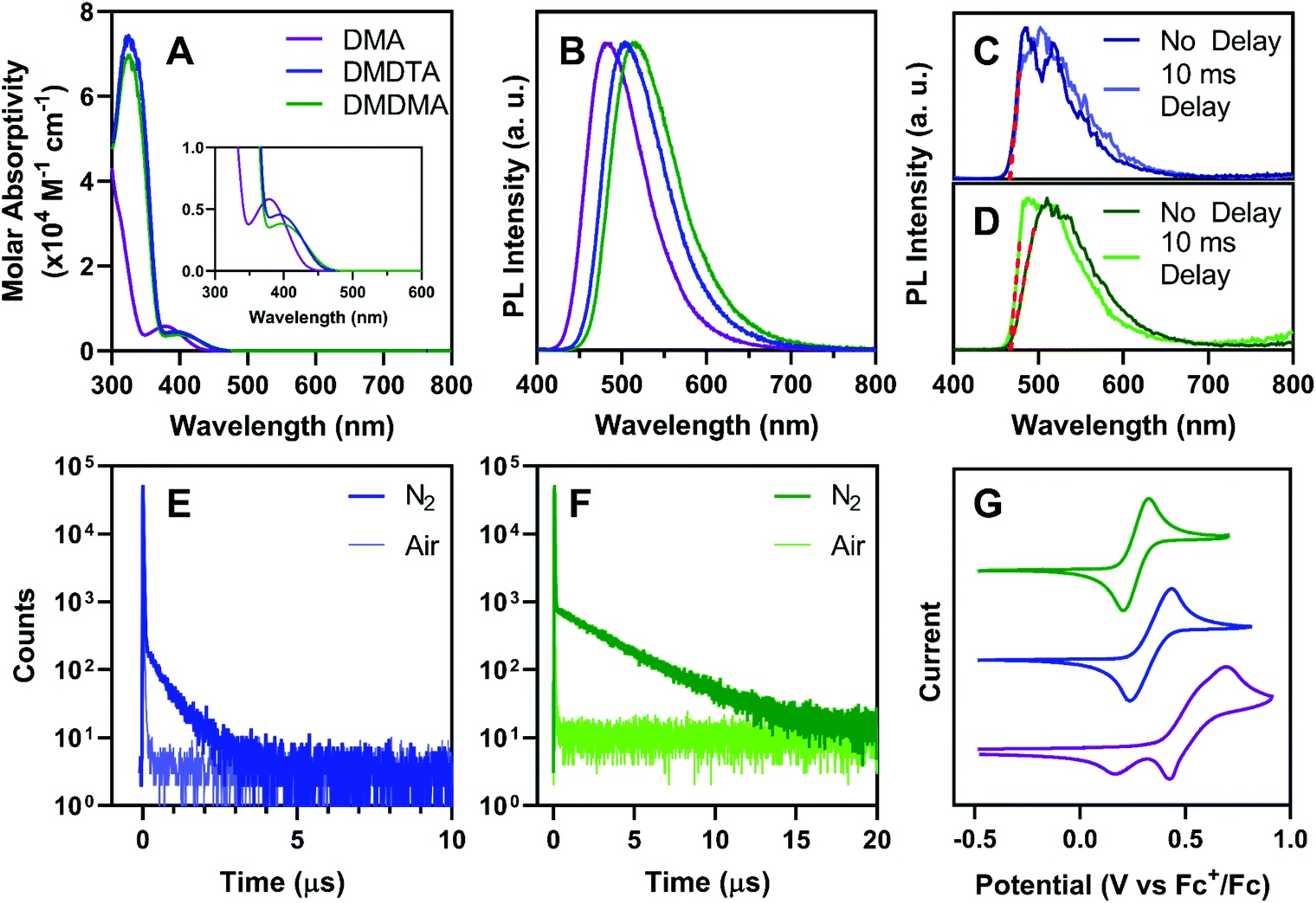 | ||
| Fig. 3 (A) UV-vis spectroscopy of all photocatalysts at 50 μg mL−1 in toluene. (B) Fluorescence spectroscopy of photocatalysts at 2 μg mL−1 in toluene, excited at 380 nm. Comparison of prompt fluorescence and phosphorescence of (C) PymDMDTA and (D) PymDMDMA in 2-MeTHF (2 μg mL−1) at 77 K, excited at 365 nm. Transient photoluminescence decays of (E) PymDMDTA and (F) PymDMDMA in degassed or aerated toluene (2 μg mL−1), excited at 313 nm. (G) Cyclic voltammetry of all photocatalysts at 4 mM in chloroform with 200 mM (nBu4N)PF6 supporting electrolyte. The scan rate was 20 mV s−1 and the first scan is presented. | ||
Bright photoluminescence is observed in aerated toluene (Fig. 3B) which increases 4–5 fold when the solutions are sparged with N2 (Fig. S2†). The relatively broad and featureless bands are typical of emission from charge-transfer excited states. The emission peak is bathochromically shifted as electron donating groups are appended to the donor. This indicates that donor-modification can enhance donor strengths, as has been shown elsewhere using carbazole as the “parent” donor.53,54 Time-resolved emission spectra taken at 77 K in 2-methyltetrahydrofuran (2-MeTHF) reveal no difference in the onsets of prompt fluorescence and phosphorescence after a 10 ms delay (Fig. 3C and D). This indicates a near-degeneracy of the S1 and T1 states, consistent with DFT, which is necessary to facilitate RISC.
Time-correlated single-photon counting (TCSPC) further supports the TADF mechanism of emission for donor-modified derivatives. Microsecond-timescale emission is observed in N2-sparged toluene, which disappears upon aerating the solution (Fig. 3E and F; see also Fig. S3†). Temperature-dependent photoluminescence decays of PymDMDTA and PymDMDMA show the expected increase in delayed emission as the temperature increases from 77 K to 298 K (Fig. S4†). PymDMA is a known TADF emitter with a ΔEST of 0.19 eV.55,56 The delayed fluorescence of PymDMA was remeasured in toluene and a lifetime of 29.6 μs was obtained (Fig. S5†), in reasonable agreement with a previous result of 20.7 μs in DPEPO film.55,56
The stability of the radical cation produced in O-ATRP was studied by cyclic voltammetry. Matyjaszewski and others have shown that radical cation stability is a key determining factor in controlling O-ATRP, as the persistence of PC˙+ in solution is directly correlated to the rate of deactivation.35PymDMA undergoes irreversible oxidation, producing a secondary species that is reduced at lower cathodic potentials (Fig. 3G). In contrast, PymDMDTA and PymDMDMA both show reversible oxidations at lower potentials than PymDMA, agreeing with the DFT-predicted values within 0.3 eV. Excellent reversibility is maintained over five successive cycles and is further attested to by the linearity in plots of peak anodic current versus the square root of the scan rate (Fig. S6 and S7†). The results are congruent with those of Miyake and co-workers,18 suggesting that addition to the radical cation of DMA occurs through the para-position, and may be prevented through core modifications.
The excited state reduction potentials were estimated using E°(PC˙+/1PC*) = E°(PC˙+/PC) − E0,0, where E0,0 is the relaxed S1 energy, estimated from the onset of fluorescence in 2-methyltetrahydrofuran at 77 K. In all cases, reduction occurs between −1.6 and −1.8 V, in good agreement with DFT. The donor-modified derivatives are stronger excited-state reductants by ∼0.1 V than PymDMA, which should lend them faster rates of activation. The ground-state reduction potential of 2,4,6-triphenylpyrimidine was measured at −1.7 V vs. SCE in N,N-dimethylformamide (Fig. S8†), validating the model of PET from the Pym˙− radical anion formed by photoinduced intramolecular charge transfer.
The measured photophysical and electrochemical parameters in degassed toluene and chloroform, respectively, are summarized in Table 2. Owing to the relatively low emission efficiency of PymDMDMA, the yield of intersystem crossing and rate of reverse intersystem crossing were calculated using an exact analysis provided by Adachi and co-workers, which does not rely on a priori assumptions about emission efficiency, other than the assumption of no radiative T1 decay.57 From this analysis, PymDMDMA is likely the most active catalyst for O-ATRP, with a high intersystem crossing yield, moderate rate of reverse intersystem crossing, strong driving force for photoelectron transfer and good visible light absorption. PymDMA should offer the best control due to its slower activation and larger driving force for deactivation, however its unstable radical cation will likely prevent the establishment of an ATRP equilibrium.
| E°(PC˙+/PC) (V vs. SCE) | E°(PC˙+/1PC*) (V vs. SCE) | λ abs (nm); ε (M−1 cm−1) | λ em (nm); φPL | τ p (ns) | τ d (μs) | φ ISC | k RISC (×106 s−1) | |
|---|---|---|---|---|---|---|---|---|
| a Wavelength of maximum absorption and molar extinction coefficient at λabs. b Wavelength of maximum emission and photoluminescence quantum yield. c Prompt fluorescence lifetime. d Delayed fluorescence lifetime. e Intersystem crossing yield. f Rate of reverse intersystem crossing. | ||||||||
| PymDMA | 0.96 | −1.67 | 378; 5800 | 483; 0.85 | 16 | 30 | 0.39 | 0.05 |
| PymDMDTA | 0.74 | −1.76 | 395; 4500 | 503; 0.73 | 15 | 0.59 | 0.22 | 1.14 |
| PymDMDMA | 0.67 | −1.79 | 398; 3900 | 517; 0.47 | 18 | 3.2 | 0.65 | 0.56 |
3.3 Kinetics of electron transfer
To mimic typical O-ATRP conditions, the photophysical properties of the PCs were examined in deoxygenated DMAc. The intramolecular charge transfer nature of the PC excited states results in bathochromic shifting of the emission maxima to ∼600 nm, compared to toluene. The photoluminescence quantum yields are reduced to between 9% for PymDMA and 0.9% for PymDMDMA, indicative of enhanced non-radiative relaxation of the excited states. This is accompanied by a reduction in the prompt and delayed fluorescence lifetimes of the PCs in DMAc, relative to toluene (Table 3). Stern–Volmer quenching experiments were then carried out for each of the PCs, using diethyl 2-bromo-2-methylmalonate (DBMM) and degassed DMAc as typical O-ATRP initiator and solvent, respectively (Fig. 4). Adding the initiator results in a progressive decrease in the delayed fluorescence lifetime, which can be fit to a linear increase in τ0/τ with increasing DBMM concentration (where τ0 is the lifetime in absence of the initiator and τ is the lifetime at a given [DBMM]).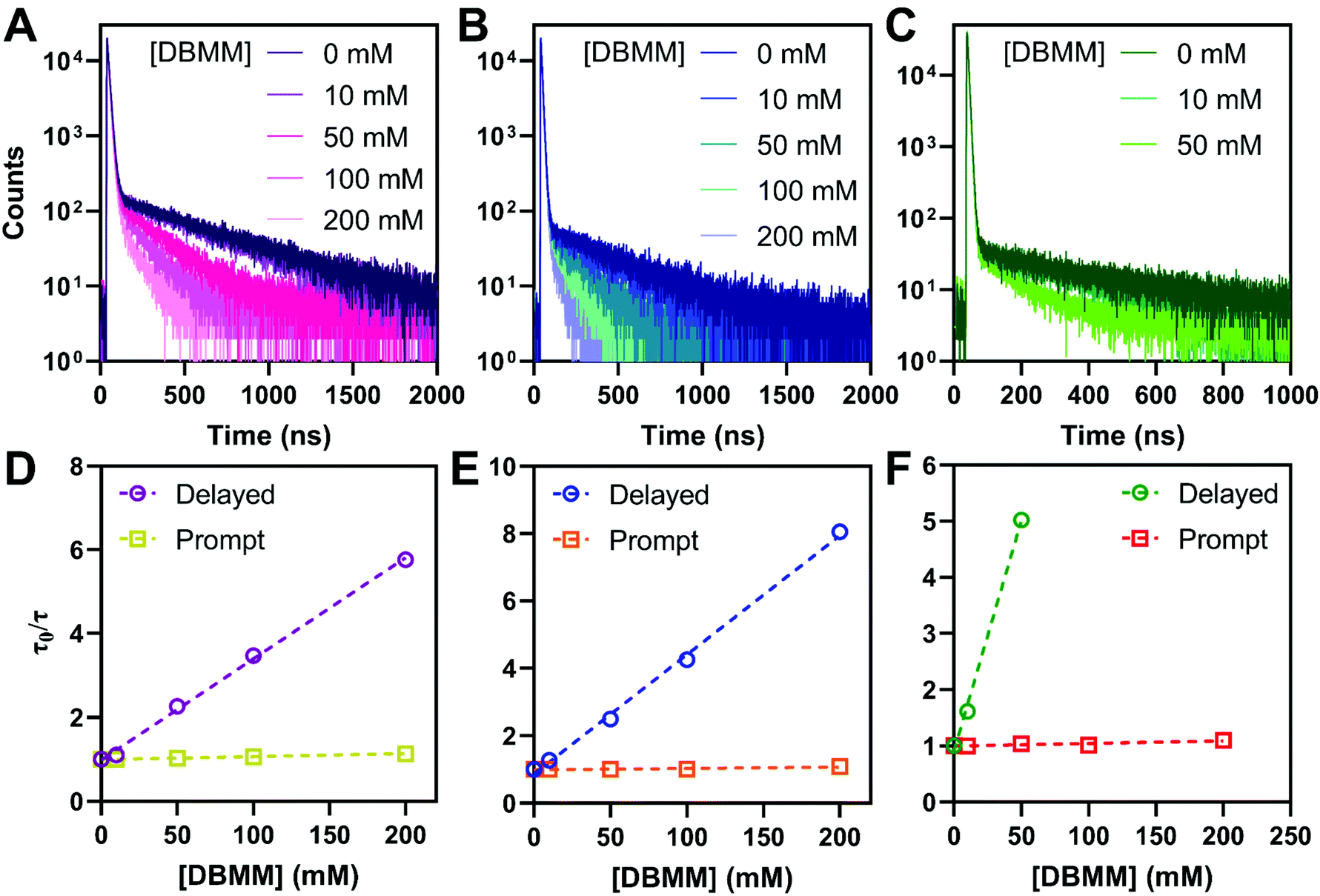 | ||
| Fig. 4 Transient photoluminescence decay of (A) PymDMA, (B) PymDMDTA, and (C) PymDMDMA dissolved in DMAc (100 μM) with the presence of varying amounts of DBMM initiator. Excited at 380 nm and emission collected at 600 nm. Stern–Volmer plots of the prompt and delayed fluorescence lifetimes as a function of DBMM concentration for (D) PymDMA, (E) PymDMDTA, and (F) PymDMDMA. | ||
| φ p,0 | φ d,0 | τ p,0 (ns) | τ d,0 (ns) | φ ISC | k RISC (×106 s−1) | k Tnr (×106 s−1) | k SET (×106 M−1 s−1) | k TET (×106 M−1 s−1) | |
|---|---|---|---|---|---|---|---|---|---|
| a Quantum yield of prompt fluorescence at zero quencher. b Quantum yield of delayed fluorescence at zero quencher. | |||||||||
| PymDMA | 0.067 | 0.0019 | 13 | 536 | 0.57 | 1.5 | 1.8 | 54 | 80 |
| PymDMDTA | 0.035 | 0.004 | 8.4 | 430 | 0.54 | 1.5 | 2.3 | 49 | 130 |
| PymDMDMA | 0.008 | 0.001 | 4.4 | 579 | 0.53 | 0.98 | 1.7 | 105 | 220 |
On the nanosecond timescale, a slight reduction in the prompt fluorescence lifetime is also observed (Fig. S9†). The effect is most pronounced for PymDMA, for which the lifetime decreases from 13.0 ns in the absence of initiator to 11.4 at 200 mM DBMM. This suggests, as has been argued elsewhere, that electron transfer from both singlet and triplet excited states is operative for initiator activation.58,59 The molar ratio of photocatalyst to initiator used in the experiments ranges between 0.1 and 0.005, comparable to common O-ATRP conditions. Somewhat higher concentrations of photocatalyst than are typical were necessary to account for the low intensity of the emission in DMAc.
The Stern–Volmer eqn (1) is used to extract the rate of singlet electron transfer (kSET) from the prompt fluorescence data.
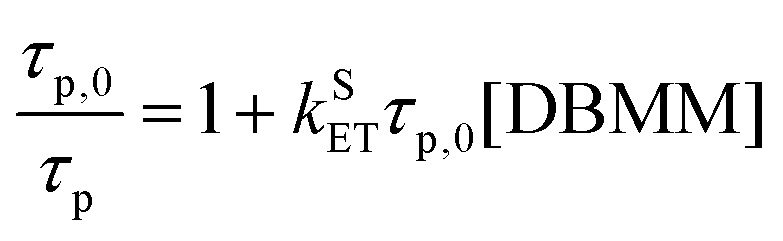 | (1) |
PymDMDMA has the most rapid singlet electron transfer at 1 × 108 M−1 s−1 (Table 3). This is still well below the diffusion limit (∼1010 M−1 s−1) indicating that electron transfer is under activation control for this system. Despite the higher driving force for PET in PymDMDTA, it has a slightly lower kSET than PymDMA. Given the structural similarity of the catalysts, this is surprising, but may reflect a larger reorganization energy for outer-sphere electron transfer in the more sterically encumbered photocatalysts. From DFT, structural reorganization upon the T1 → D+ transition (T1 and S1 are assumed to be structurally similar due to their near-degeneracy) is more significant in donor-modified derivatives since the rotation of the pendant aryl groups relative to DMA stabilizes the cation through charge delocalization. For PymDMDMA, the average equilibrium dihedral angle between DMA and methoxyphenyl groups decreases from 36.1° in T1 to 31.6° in D+. A comparable decrease is observed upon oxidation of PymDMDTA.
The rate of electron transfer from the triplet state (kTET) is similarly obtained by considering the rate at which the delayed fluorescence is quenched by DBMM. We begin by assuming the majority of excitons are deactivated after a single pass through the triplet state, which is reasonable given that >90% of all excitons are deactivated by fast non-radiative decay even in the absence of DBMM. The intensity ratio of delayed fluorescence to prompt fluorescence has been previously taken as a measure of the average number of cycles through T1,60 and is below 1 for all three PCs. The quantum yield of delayed fluorescence (φd) is then given by the product of the efficiencies of prompt fluorescence (φp), ISC (φISC), and RISC (φRISC), as an exciton that results in delayed fluorescence will first undergo ISC, then RISC before radiative relaxation from S1.
| φd = φpφISCφRISC | (2) |
Here, the analysis is simplified by assuming that φp and φISC are not significantly altered by the addition of quencher, which is reasonable given the slow rate of change in the prompt lifetime as a function of [DBMM] (see Fig. S9†). In this case, the ratio φd,0/φd can then be written:
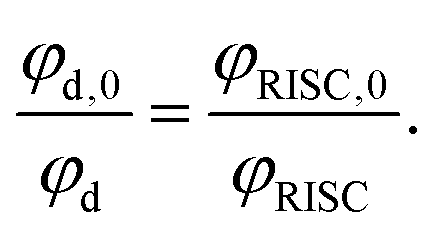 | (3) |
Because of the low fluorescence quantum yields in these systems, the lifetimes of delayed fluorescence are a more sensitive measure of excited state kinetics than φd. Assuming the rate constant of delayed fluorescence is unchanged by the DBMM concentration (kd,0 = kd), τd and φd are directly proportional over the range of [DBMM] tested (φd = kdτd),61–63 which then yields:
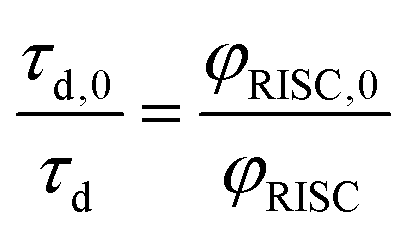 | (4) |
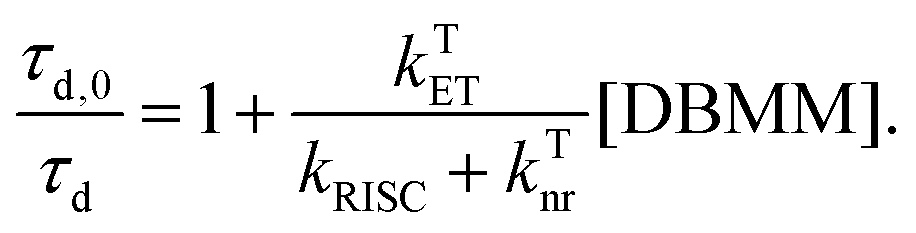 | (5) |
Using this equation, the rates of triplet electron transfer are estimated to be 1.5–3× faster than singlet electron transfer. The trend in triplet electron transfer rates tracks well with the excited state reduction potentials. Coupled with the large ISC yields in DMAc and the low efficiency of back electron transfer in triplet manifolds,64 these results suggest dominant triplet state activation over the range of initiator concentrations tested.
3.4 O-ATRP performance
The activity of the TADF photocatalysts was evaluated for the polymerization of methyl methacrylate (MMA, DPtheo = 200) and benzyl methacrylate (BnMA, DPtheo = 100). Control experiments were performed without catalyst, or with PymDMDMA as catalyst in the absence of initiator, or in the absence of light (Table 4). In the absence of photocatalysts, MMA and BnMA both form high molecular weight, high dispersity (Đ = Mw/Mn) polymers (Fig. S10†). The initiator efficiencies (I* = Mn,theo/Mn,exp) are expectedly low. Similar results are obtained in absence of an initiator. In the dark, no polymerization is observed as the catalysts are not sufficiently strong reductants in their ground states to activate DBMM.65| Run | PC | Monomer | [M]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[I]:[PC] :[I]:[PC] |
Conv.a (%) | M n (kDa) | I*c | Đ |
|---|---|---|---|---|---|---|---|
| a Conversion after 16 (8*) hours irradiation, determined through integration of polymer versus monomer signals in the 1H NMR. b Determined for the crude polymer after 16 (8*) hours by gel permeation chromatography. c I* = Mn,theo/Mn,exp. | |||||||
| No PC | None | MMA | 200:1:0 |
12 | 5090 | 0.00 | 2.14 |
| No PC | None | BnMA | 100:1:0 |
57 | 308 | 0.03 | 2.48 |
| No I | PymDMDMA | MMA | 200:0:0.1 |
13 | 3450 | 0.00 | 2.24 |
| No I | PymDMDMA | BnMA | 100:0:0.05 |
51 | 1570 | 0.00 | 2.67 |
| Dark | PymDMDMA | MMA | 200:1:0.1 |
0 | — | — | — |
| Dark | PymDMDMA | BnMA | 100:1:0.05 |
0 | — | — | — |
| 1 | PymDMA | MMA | 200:1:0.1 |
66 | 18.8 | 0.72 | 3.11 |
| 2 | PymDMDTA | MMA | 200:1:0.1 |
75 | 17.4 | 0.88 | 2.06 |
| 3 | PymDMDMA | MMA | 200:1:0.1 |
89 | 15.5 | 1.16 | 1.75 |
| 4 | PymDMDMA | MMA | 100:1:0.005 |
67* | 6.3* | 0.91 | 1.29 |
| 5 | PymDMDMA | MMA | 200:1:0.01 |
68* | 12.2* | 1.14 | 1.27 |
| 6 | PymDMDMA | MMA | 500:1:0.025 |
60* | 28.2* | 0.93 | 1.51 |
| 7 | PymDMDMA | MMA | 1000:1:0.05 |
55* | 42.2* | 0.77 | 1.70 |
| 8 | PymDMDMA | MMA | 200:1:0.005 |
72* | 13.2* | 1.11 | 1.58 |
| 9 | PymDMDMA | MMA | 200:1:0.002 |
56* | 14.7* | 0.78 | 1.81 |
| 10 | Phenox A0202 | MMA | 200:1:0.01 |
62* | 12.4* | 1.02 | 1.51 |
| 11 | Phenox A0202 | MMA | 200:1:0.005 |
65* | 14.4* | 0.92 | 1.86 |
| 12 | Phenox A0202 | MMA | 200:1:0.002 |
57* | 22.9* | 0.51 | 1.88 |
| 13 | PymDMA | BnMA | 100:1:0.05 |
81 | 15.8 | 0.90 | 2.04 |
| 14 | PymDMDTA | BnMA | 100:1:0.05 |
94 | 15.0 | 1.09 | 1.52 |
| 15 | PymDMDMA | BnMA | 100:1:0.05 |
99 | 15.3 | 1.14 | 1.31 |
The kinetics of polymerization with MMA and BnMA were studied at catalyst loadings of 500 ppm (Fig. 5, see also Fig. S11 and S12†). Hawker has shown that photosensitizers with long-lived excited states for O-ATRP have poor control over polymer dispersity at high catalyst loadings, owing to the persistence of the activator species (3PC*) in solution increasing the concentration of propagating radicals.14 500 ppm catalyst loading was chosen to mitigate these effects. In the polymerization of both methacrylic monomers, first-order kinetic behaviour is shown in plots of ln([M0]/[M]) versus time for PymDMDMA over 8 hours and PymDMDTA over 6 hours. For donor-unmodified PymDMA, a non-linear plot indicates the rapid onset of termination reactions, likely due to decomposition of its unstable radical cation. The dispersity of the polymer formed with PymDMA as the photocatalyst quickly increases at conversions greater than 40%, implying that termination processes outpace initiation.
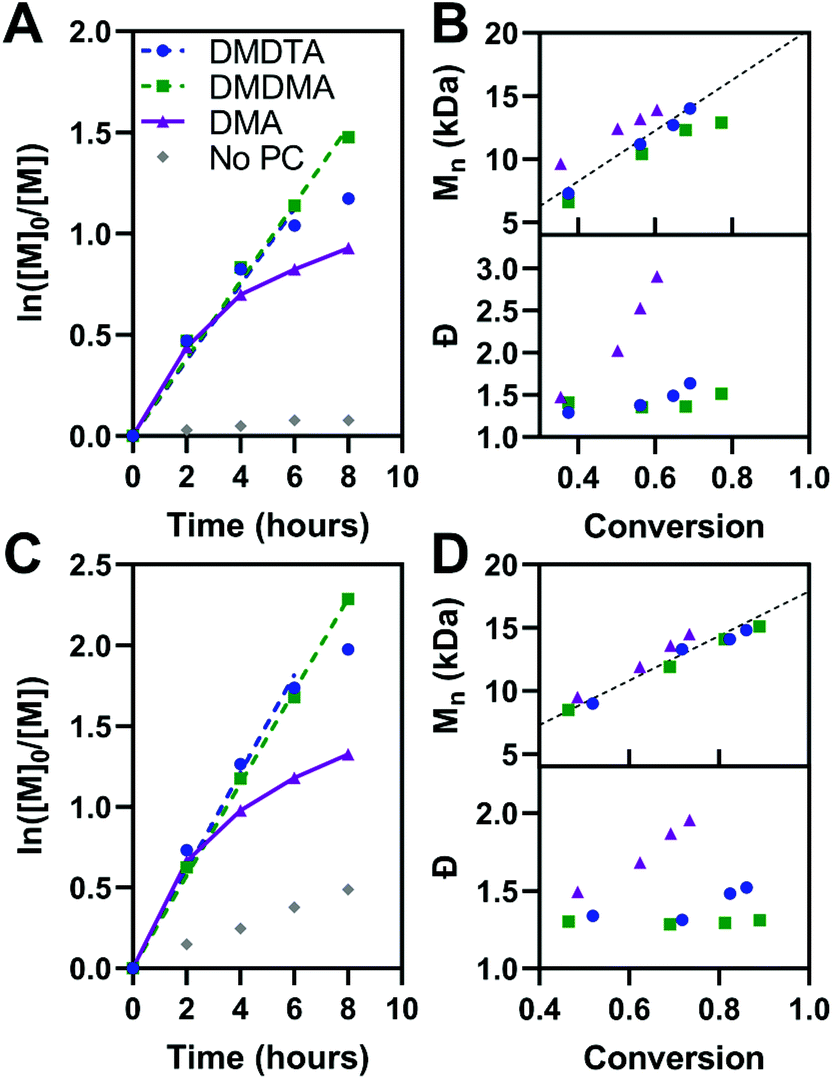 | ||
| Fig. 5 Polymerization kinetics and plots of molecular weight and dispersity as a function of conversion with different PCs at 500 ppm for (A and B) MMA and (C and D) BnMA. The theoretical molecular weight is given by the dashed black lines in (B) and (D). | ||
Donor-modified PymDMDMA and PymDMDTA polymerize MMA (target molecular weight = 20 kDa) with dispersities below 1.4 up to 75 and 70% conversion, respectively. Further conversion is associated with a steep increase in dispersity (Table 4, entries 2 and 3). Conversely, PymDMDMA can polymerize BnMA to 99% conversion with a final dispersity of 1.31 in 16 hours. The syntheses of acrylic polymers of methyl, n-butyl, and benzyl acrylate were attempted using PymDMDMA and PymDMDTA; in all cases poor control over molecular weight was obtained with initiator efficiencies below 0.6 and dispersities above 1.8 after 2 hours (>60% conversion).
The lower control over polymerizations mediated by PymDMA compared to its donor-modified derivatives is thought to originate from alkylation of the donor radical cation in the presence of DBMM. When a DMAc solution of PymDMA is irradiated with 435 nm light in the presence of 20 equivalents of DBMM, addition of DBMM to the DMA core is observed over 8 hours. In the 1H NMR spectrum (Fig. S11†), the meta- and para-hydrogen (relative to nitrogen) signals of the DMA donor are transformed from two doublets of doublets into one doublet (with additional hyperfine 4J coupling), consistent with replacement of either the meta- or para-hydrogen. By contrast, the 1H NMR spectra of PymDMDMA and PymDMDMTA are largely unchanged under 435 nm irradiation with DBMM over the same period of time (Fig. S12 and S13†), suggesting it is the para-hydrogen that is exchanged in the alkylation of PymDMA. Not only does this mode of reactivity lower the initiator efficiency of the polymerization, but it also provides a potential termination mechanism from propagating radicals combining with DMA radical cations, which will cause an increase in the polymer dispersity.
The activity of PymDMDMA at low catalyst loadings was evaluated and compared to the phenoxazine-based O-ATRP catalyst Phenox A0202 reported by Miyake (Fig. S17†).19 At 50 ppm, PymDMDMA performs better in the polymerization of MMA than at 500 ppm loading, with a lower dispersity of 1.27 and good initiator efficiency (Table 4, entry 4). It also has comparatively lower dispersity than Phenox A0202 at the same loading (Table 4, entry 10). At 50 ppm of PymDMDMA, higher target masses of 50 and 100 kDa result in an increase in dispersity compared to the 20 kDa target polymer (Table 4, entries 6 and 7 and Fig. S18†). Decreasing the catalyst loading below 50 ppm is associated with an increase in dispersity for both catalysts, indicating that the deactivator species is not able to achieve a sufficient concentration. At 10 ppm, both catalysts produce PMMA with dispersities over 1.8 and initiator efficiencies below 0.78 after 8 hours (entries 9 and 12).
To evaluate the chain-end retention provided by O-ATRP with PymDMDMA, a PMMA-b-p(BnMA) block copolymer was synthesized. 10 kDa PMMA was first prepared using 100 ppm PymDMDMA and halting the polymerization at 50% conversion to give macroinitiator with dispersity 1.18 after precipitation in ethanol. Chain extension with p(BnMA) was then attempted with 500 ppm PymDMDMA. After 8 hours, the conversion of BnMA was 93% by 1H NMR, similar to the polymerization using DBMM initiator. The 1H NMR spectrum of the precipitated polymer contains distinct resonances at δ 7.30 and 4.90 ppm of p(BnMA) and 3.62 ppm for the methyl ester of PMMA. In the DOSY spectrum of the block copolymer, all three resonances diffuse at the same level, with a narrow dispersion of diffusion coefficients between 3.6 and 3.9 × 10−10 m2 s−1 (Fig. 6A), suggesting covalent linkage of PMMA and p(BnMA) blocks. Integration of the 1H NMR spectrum (Fig. S19†) yields an excess of BnMA compared to theory (DP 119 compared to 93), suggesting that as much as 22% of macroinitiator chains are not re-initiated. While a clear separation between macroinitiator and block copolymer traces is observed by GPC (Fig. 6B), significant shouldering at low molecular weight is responsible for an increase in dispersity upon block formation. This is likely due to the presence of dead macroinitiator chains, as O-ATRP is not a truly living process and some degree of termination is to be expected.
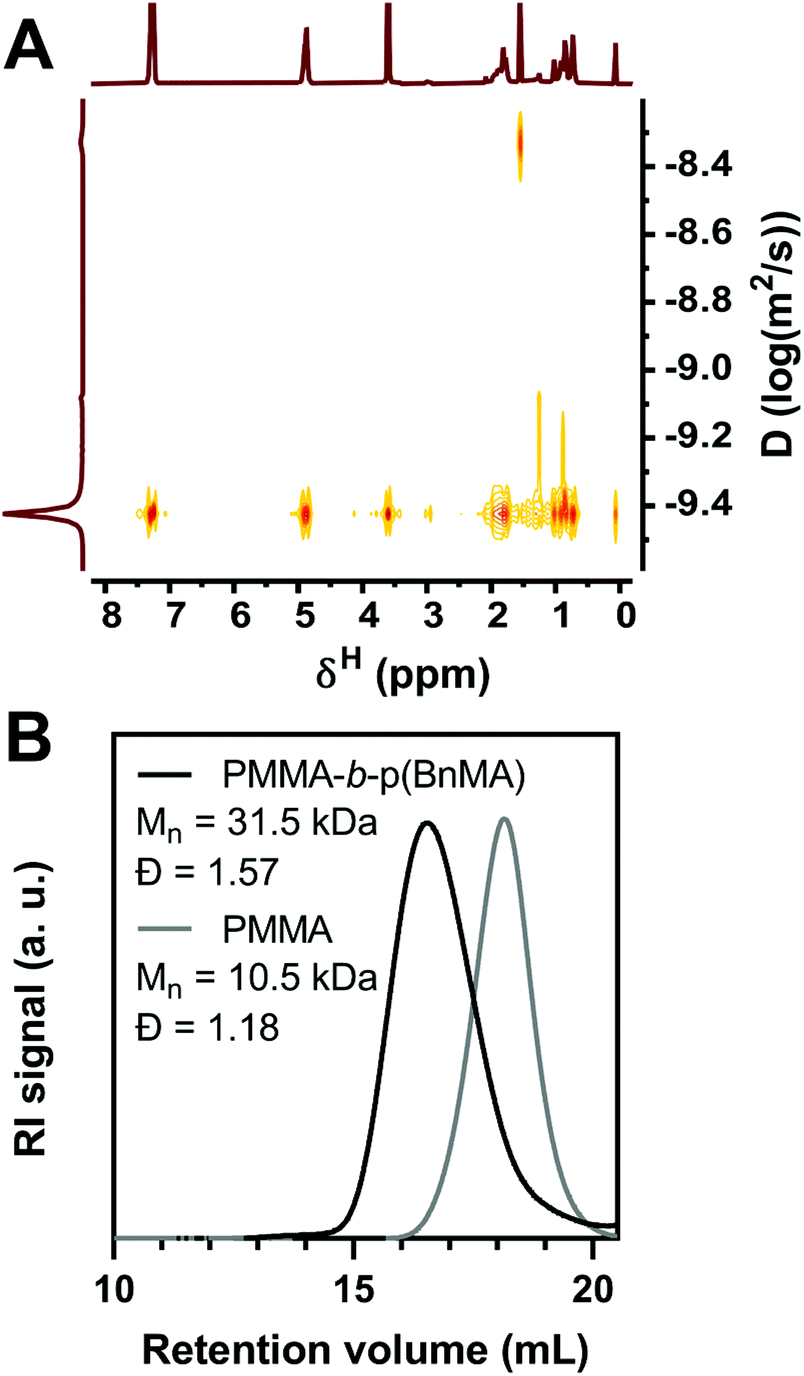 | ||
| Fig. 6 (A) DOSY spectrum of PMMA-b-p(BnMA) in CDCl3. (B) RI Chromatogram of PMMA macroinitiator and PMMA-b-p(BnMA). | ||
4. Conclusions
The promise of TADF for metal-free photocatalytic transformations relies on a strong understanding of structure–activity relationships. In this study, modification of the donor group used in TADF was investigated with respect to the ability to participate in dissociative and associative electron transfer for visible-light promoted O-ATRP. While the stability of the radical cation is found to be the strongest determining factor for controlled polymerization, significant differences amongst donor-modified catalysts are related to variation in the rates of photoinduced electron transfer. Our results are consistent with previous studies that posit both singlet and triplet electron transfer as initiation mechanisms, with the reduced singlet–triplet energy gap in TADF emitters benefitting both the yield and driving force of triplet electron transfer. The ability to predict these driving forces based on theoretical modelling is promising, as computations can inform the types of donors, acceptors, and their modifications to design the most efficient catalysts.The results of this study should be applicable to any of the donors commonly used in TADF. Carbazole, a widely used donor, is known to have irreversible oxidative chemistry which may hinder its applications in photoreductive catalysis.66 Donors with reversible oxidations, such as phenothiazine, phenoxazine, and phenazine, are still susceptible to turnover-limiting photosubstitution chemistry in the absence of appropriate donor-modification strategies. We anticipate that the widespread adoption of TADF photocatalysts in organic synthesis will greatly benefit from the development of more robust donor and acceptor fragments with increased potential for high conversions, reduced side-reactivity and catalyst recyclability.
Author contributions
All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We wish to thank Dr Maria Ezhova for performing the DOSY NMR study. The authors thank the Natural Sciences and Engineering Council of Canada (NSERC) for financial support. A. M. P. and S. H. H. thank NSERC for a Canada Graduate Scholarship and Undergraduate Student Research Award, respectively. Z. M. H. gratefully acknowledges support from the Canada Research Chairs Program.Notes and references
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- W. A. Braunecker and K. Matyjaszewski, Prog. Polym. Sci., 2007, 32, 93–146 CrossRef CAS.
- J. S. Wang and K. Matyjaszewski, J. Am. Chem. Soc., 1995, 117, 5614–5615 CrossRef CAS.
- K. Matyjaszewski, Adv. Mater., 2018, 30, 1706441 CrossRef PubMed.
- W. Jakubowski and K. Matyjaszewski, Angew. Chem., Int. Ed., 2006, 45, 4482–4486 CrossRef CAS PubMed.
- A. Anastasaki, V. Nikolaou, G. Nurumbetov, P. Wilson, K. Kempe, J. F. Quinn, T. P. Davis, M. R. Whittaker and D. M. Haddleton, Chem. Rev., 2016, 116, 835–877 CrossRef CAS PubMed.
- K. Matyjaszewski, W. Jakubowski, K. Min, W. Tang, J. Huang, W. A. Braunecker and N. V. Tsarevsky, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15309–15314 CrossRef CAS PubMed.
- M. A. Tasdelen, M. Uygun and Y. Yagci, Macromol. Chem. Phys., 2010, 211, 2271–2275 CrossRef CAS.
- M. A. Tasdelen, M. Ciftci and Y. Yagci, Macromol. Chem. Phys., 2012, 213, 1391–1396 CrossRef CAS.
- Q. Yang, F. Dumur, F. Morlet-Savary, J. Poly and J. Lalevée, Macromolecules, 2015, 48, 1972–1980 CrossRef CAS.
- S. Dadashi-Silab, K. Kim, F. Lorandi, G. Szczepaniak, S. Kramer, L. Peteanu and K. Matyjaszewski, ACS Macro Lett., 2022, 11, 376–381 CrossRef CAS PubMed.
- B. P. Fors and C. J. Hawker, Angew. Chem., Int. Ed., 2012, 51, 8850–8853 CrossRef CAS PubMed.
- N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. Read de Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096–16101 CrossRef CAS PubMed.
- Q. Ma, J. Song, X. Zhang, Y. Jiang, L. Ji and S. Liao, Nat. Commun., 2021, 12, 429 CrossRef CAS PubMed.
- J. C. Theriot, C. H. Lim, H. Yang, M. D. Ryan, C. B. Musgrave and G. M. Miyake, Science, 2016, 352, 1082–1086 CrossRef CAS PubMed.
- B. L. Buss, C. Lim and G. M. Miyake, Angew. Chem., Int. Ed., 2020, 59, 3209–3217 CrossRef CAS PubMed.
- B. G. McCarthy, R. M. Pearson, C. H. Lim, S. M. Sartor, N. H. Damrauer and G. M. Miyake, J. Am. Chem. Soc., 2018, 140, 5088–5101 CrossRef CAS PubMed.
- A. Allushi, S. Jockusch, G. Yilmaz and Y. Yagci, Macromolecules, 2016, 49, 7785–7792 CrossRef CAS.
- X. Liu, L. Zhang, Z. Cheng and X. Zhu, Polym. Chem., 2016, 7, 689–700 RSC.
- C. Wu, N. Corrigan, C.-H. Lim, W. Liu, G. M. Miyake and C. Boyer, Chem. Rev., 2022, 122, 5476–5518 CrossRef CAS PubMed.
- P. Lu, V. K. Kensy, R. L. Tritt, D. T. Seidenkranz and A. J. Boydston, Acc. Chem. Res., 2020, 53, 2325–2335 CrossRef CAS PubMed.
- C. Wu, K. Jung, Y. Ma, W. Liu and C. Boyer, Nat. Commun., 2021, 12, 478 CrossRef CAS PubMed.
- N. Corrigan, S. Shanmugam, J. Xu and C. Boyer, Chem. Soc. Rev., 2016, 45, 6165–6212 RSC.
- E. E. Stache, V. Kottisch and B. P. Fors, J. Am. Chem. Soc., 2020, 142, 4581–4585 CrossRef CAS PubMed.
- Z. Huang, Y. Gu, X. Liu, L. Zhang, Z. Cheng and X. Zhu, Macromol. Rapid Commun., 2017, 38, 1600461a CrossRef PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- M. A. Bryden and E. Zysman-Colman, Chem. Soc. Rev., 2021, 50, 7587–7680 RSC.
- F. Fang, L. Zhu, M. Li, Y. Song, M. Sun, D. Zhao and J. Zhang, Adv. Sci., 2021, 2102970 CrossRef CAS PubMed.
- N. R. Paisley, C. M. Tonge and Z. M. Hudson, Front. Chem., 2020, 8, 229 CrossRef CAS PubMed.
- F. Ni, N. Li, L. Zhan and C. Yang, Adv. Opt. Mater., 2020, 8, 1902187 CrossRef CAS.
- A. M. Polgar and Z. M. Hudson, Chem. Commun., 2021, 57, 10675–10688 RSC.
- V. K. Singh, C. Yu, S. Badgujar, Y. Kim, Y. Kwon, D. Kim, J. Lee, T. Akhter, G. Thangavel, L. S. Park, J. Lee, P. C. Nandajan, R. Wannemacher, B. Milián-Medina, L. Lüer, K. S. Kim, J. Gierschner and M. S. Kwon, Nat. Catal., 2018, 1, 794–804 CrossRef CAS.
- X. Pan, C. Fang, M. Fantin, N. Malhotra, W. Y. So, L. A. Peteanu, A. A. Isse, A. Gennaro, P. Liu and K. Matyjaszewski, J. Am. Chem. Soc., 2016, 138, 2411–2425 CrossRef CAS PubMed.
- J. J. Devery, J. J. Douglas, J. D. Nguyen, K. P. Cole, R. A. Flowers and C. R. J. Stephenson, Chem. Sci., 2015, 6, 537–541 RSC.
- B. McCarthy, S. Sartor, J. Cole, N. Damrauer and G. M. Miyake, Macromolecules, 2020, 53, 9208–9219 CrossRef CAS PubMed.
- Z. Zhang, W. Chen, Y. Zhang, Y. Wang, Y. Tian, L. Fang and X. Ba, Macromolecules, 2021, 54, 4633–4640 CrossRef CAS.
- D. A. Corbin, K. O. Puffer, K. A. Chism, J. P. Cole, J. C. Theriot, B. G. McCarthy, B. L. Buss, C. H. Lim, S. R. Lincoln, B. S. Newell and G. M. Miyake, Macromolecules, 2021, 54, 4507–4516 CrossRef CAS PubMed.
- S. Grotjahn and B. König, Org. Lett., 2021, 23, 3146–3150 CrossRef CAS PubMed.
- D. Kong, M. Munch, Q. Qiqige, C. J. C. Cooze, B. H. Rotstein and R. J. Lundgren, J. Am. Chem. Soc., 2021, 143, 2200–2206 CrossRef CAS PubMed.
- N. Lin, J. Qiao, L. Duan, L. Wang and Y. Qiu, J. Phys. Chem. C, 2014, 118, 7569–7578 CrossRef CAS.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. Winget, C. J. Cramer and D. G. Truhlar, Theor. Chem. Acc., 2004, 112, 217–227 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- S. Hirata and M. Head-Gordon, Chem. Phys. Lett., 1999, 314, 291–299 CrossRef CAS.
- M. J. G. Peach, M. J. Williamson and D. J. Tozer, J. Chem. Theory Comput., 2011, 7, 3578–3585 CrossRef CAS PubMed.
- N. T. Maitra, J. Chem. Phys., 2005, 122, 234104 CrossRef PubMed.
- P. Mori-Sánchez, A. J. Cohen and W. Yang, Phys. Rev. Lett., 2008, 100, 146401 CrossRef PubMed.
- H. Sun, C. Zhong and J.-L. Brédas, J. Chem. Theory Comput., 2015, 11, 3851–3858 CrossRef CAS PubMed.
- S. Hirata, Y. Sakai, K. Masui, H. Tanaka, S. Y. Lee, H. Nomura, N. Nakamura, M. Yasumatsu, H. Nakanotani, Q. Zhang, K. Shizu, H. Miyazaki and C. Adachi, Nat. Mater., 2015, 14, 330–336 CrossRef CAS PubMed.
- H. Kaji, H. Suzuki, T. Fukushima, K. Shizu, K. Suzuki, S. Kubo, T. Komino, H. Oiwa, F. Suzuki, A. Wakamiya, Y. Murata and C. Adachi, Nat. Commun., 2015, 6, 8476 CrossRef CAS PubMed.
- R. Komatsu, H. Sasabe, Y. Seino, K. Nakao and J. Kido, J. Mater. Chem. C, 2016, 4, 2274–2278 RSC.
- R. Komatsu, H. Sasabe, K. Nakao, Y. Hayasaka, T. Ohsawa and J. Kido, Adv. Opt. Mater., 2017, 5, 1600675 CrossRef.
- Y. Tsuchiya, S. Diesing, F. Bencheikh, Y. Wada, P. L. dos Santos, H. Kaji, E. Zysman-Colman, I. D. W. Samuel and C. Adachi, J. Phys. Chem. A, 2021, 125, 8074–8089 CrossRef CAS PubMed.
- D. Koyama, H. J. A. Dale and A. J. Orr-Ewing, J. Am. Chem. Soc., 2018, 140, 1285–1293 CrossRef CAS PubMed.
- A. Bhattacherjee, M. Sneha, L. Lewis-Borrell, G. Amoruso, T. A. A. Oliver, J. Tyler, I. P. Clark and A. J. Orr-Ewing, J. Am. Chem. Soc., 2021, 143, 3613–3627 CrossRef CAS PubMed.
- C. Baleizão and M. N. Berberan-Santos, J. Chem. Phys., 2007, 126, 204510 CrossRef PubMed.
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253–258 CrossRef CAS.
- F. B. Dias, Philos. Trans. R. Soc., A, 2015, 373, 20140447 CrossRef PubMed.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- N. D. Dolinski, Z. A. Page, E. H. Discekici, D. Meis, I. H. Lee, G. R. Jones, R. Whitfield, X. Pan, B. G. McCarthy, S. Shanmugam, V. Kottisch, B. P. Fors, C. Boyer, G. M. Miyake, K. Matyjaszewski, D. M. Haddleton, J. R. de Alaniz, A. Anastasaki and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 268–273 CrossRef CAS PubMed.
- K. Karon and M. Lapkowski, J. Solid State Electrochem., 2015, 19, 2601–2610 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py00470d |
| This journal is © The Royal Society of Chemistry 2022 |