
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Deoxyfluorination tunes the aggregation of cellulose and chitin oligosaccharides and highlights the role of specific hydroxyl groups in the crystallization process†
Giulio
Fittolani
 ab,
Surusch
Djalali
ab,
Manishkumar A.
Chaube
a,
Theodore
Tyrikos-Ergas‡
ab,
Marlene C. S.
Dal Colle
ab,
Andrea
Grafmüller
c,
Peter H.
Seeberger
ab and
Martina
Delbianco
*a
ab,
Surusch
Djalali
ab,
Manishkumar A.
Chaube
a,
Theodore
Tyrikos-Ergas‡
ab,
Marlene C. S.
Dal Colle
ab,
Andrea
Grafmüller
c,
Peter H.
Seeberger
ab and
Martina
Delbianco
*a
aDepartment of Biomolecular Systems, Max Planck Institute of Colloids and Interfaces, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: martina.delbianco@mpikg.mpg.de
bDepartment of Chemistry and Biochemistry, Freie Universität Berlin, Arnimallee 22, 14195 Berlin, Germany
cDepartment of Theory and Biosystems, Max Planck Institute of Colloids and Interfaces, Am Mühlenberg 1, 14476 Potsdam, Germany
First published on 13th October 2022
Abstract
Cellulose and chitin are abundant structural polysaccharides exploited by nature in a large number of applications thanks to their crystallinity. Chemical modifications are commonly employed to tune polysaccharide physical and mechanical properties, but generate heterogeneous mixtures. Thus, the effect of such modifications is not well understood at the molecular level. In this work, we examined how deoxyfluorination (site and pattern) impact the solubility and aggregation of well-defined cellulose and chitin oligomers. While deoxyfluorination increased solubility in water and lowered the crystallinity of cellulose oligomers, chitin was much less affected by the modification. The OH/F substitution also highlighted the role of specific hydroxyl groups in the crystallization process. This work provides guidelines for the design of cellulose- and chitin-based materials. A similar approach can be imagined to prepare cellulose and chitin analogues capable of withstanding enzymatic degradation.
Introduction
Cellulose1 and chitin2 are naturally abundant structural polysaccharides. Plants, fungi, crustaceans, and insects rely on these polysaccharides for generating mechanically stable cell walls and bacteria exploit the fibrous nature of cellulose for creating dense and impenetrable biofilms.3,4 Cellulose and chitin are based on a similar backbone consisting of β-1,4 linked glucose (Glc) or N-acetyl glucosamine (GlcNAc) monosaccharide units, respectively (Fig. 1A). A dense network of inter- and intramolecular hydrogen bonds are responsible for the high crystallinity and insolubility of these two polysaccharides.5,6 Both polymers share the key intramolecular hydrogen bond between the OH-3 and O-5′ of the following residue. Additionally, cellulose crystal structures display intra- and inter-molecular hydrogen bond between OH-6' and OH-2,5,7,8 while in chitin the NHAc carbonyl group provides further inter-chain stabilization.6,9,10 Beside hydrogen bonds, additional stabilization is gained from hydrophobic interactions between the C–H rich faces of the monosaccharides.11 | ||
| Fig. 1 (A) Hydrogen bond pattern of cellulose and chitin. (B) Deoxyfluorinated analogues studied in this work. (C) Building blocks used for the AGA of deoxyfluorinated cellulose and chitin backbones. | ||
The crystalline character of cellulose and chitin is key in many applications. For example, cellulose and chitin nanocrystals, CNCs and ChNCs respectively, have raised interest for the fabrication of optical materials.12–16 While many aspects of cellulose and chitin crystals are established, details on the crystallization process and the precise role of each hydroxyl group is often a matter of debate.8,9 Furthermore, not all cellulose and chitin allomorphs have yet been described in detail.17,18 Top-down approaches rely on extraction from natural sources and do not grant access to polysaccharides with uniform dispersity, limiting our understanding of crystallization. Moreover, chemical modification protocols, aiming to tune cellulose and chitin crystallinity, lack control over the modification pattern.19,20 In contrast, bottom-up approaches guarantee precise saccharide sequences and patterns of modification, important determinants of the materials and biological properties.21–25
Perturbing the primary sequence of an oligosaccharide with non-natural residues offers a strategy to systematically tune the macroscopic properties of a material. In addition, this approach provides indirect information on the role of the replaced unit (or functional group). In this regard, fluorination of monosaccharides has drawn considerable attention.26 Fluorination is widely explored in medicinal and material chemistry,27–33 owing to the unique features of the F atom. Fluorine is a small, highly electronegative atom that can replace C–OH, C–H, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds with the chemically inert C–F bond.28,29 Despite the high polarization of the C–F bond, hydrogen bonds involving the fluorine atom are rare. Thus, deoxyfluorination offers a strategy to tune hydrogen bonding and interactions with water molecules with impact on conformation, aggregation propensity, and interactions with proteins.34,35 To date, most studies are focused on simple monosaccharides;34,36 limited attention has been given to the construction of more complex fluorinated oligosaccharides which require multistep syntheses, stereo-control, and harsh conditions.37–39
O bonds with the chemically inert C–F bond.28,29 Despite the high polarization of the C–F bond, hydrogen bonds involving the fluorine atom are rare. Thus, deoxyfluorination offers a strategy to tune hydrogen bonding and interactions with water molecules with impact on conformation, aggregation propensity, and interactions with proteins.34,35 To date, most studies are focused on simple monosaccharides;34,36 limited attention has been given to the construction of more complex fluorinated oligosaccharides which require multistep syntheses, stereo-control, and harsh conditions.37–39
Fluorinated cellulose40 and chitin41 oligomers, prepared by enzymatic synthesis, suggested that deoxyfluorination affects their crystallinity. Still, enzymatic synthesis does not allow for precise control over the pattern of substitution, degree of polymerization, and have a limited substrate scope.42 Here, we report the Automated Glycan Assembly (AGA) of a collection of cellulose and chitin oligomers incorporating deoxyfluorinated monosaccharides with precise pattern and site of modification (Fig. 1B and C). Molecular dynamics (MD) simulations served as a tool to interpret and rationalize the systematic characterization of these compounds performed by powder X-ray diffraction (XRD) and NMR spectroscopy. These insights will provide guidance for the future design of cellulose and chitin based materials.
Results and discussion
Building blocks synthesis
We targeted the synthesis of deoxyfluorinated cellulose and chitin oligomers as model compounds to understand the role of each hydroxyl group in the crystallization process. We previously reported that the 3F modification imparts higher flexibility to single cellulose oligomers by disrupting hydrogen bonds and preventing formation of crystalline domains.22,43 Here, we extend the study to new patterns and site of modification. BB1–5 were employed to construct the β-1,4 cellulose and chitin backbone using AGA (Fig. 1C). BB1 and BB4 were chosen to install the non-modified Glc and GlcNAc units, respectively, while BB2, BB3, and BB5 were synthesized to introduce the deoxyfluorinated residues. Each BB was equipped with a reactive anomeric leaving group, either a thioether or a dibutyl phosphate, permanent protecting groups (Bn, Bz, or TCA), and a temporary protecting group (Fmoc) to guarantee the regioselective step-wise elongation of β-1,4 glycosidic linkages. β-Stereoselectivity was ensured by participation of the C-2 Bz or N-TCA protecting groups. The synthesis of BB2 was performed following previously reported procedures.22 Deoxyfluorinated Glc and GlcNAc BBs (BB3 and BB5) were synthesized from commercially available starting materials, minimizing the number of synthetic steps.The installation of a fluorine atom at the C-6 position of Glc was envisioned through the regioselective deoxyfluorination of compound 1, readily accessible in four steps from a commercially available intermediate (see the ESI†). Regioselective tosylation of 1,44 followed by the nucleophilic substitution with CsF on intermediate 2, yielded only trace amounts of desired product 3a and a significant amount of the elimination side-product 3b (Fig. 2 and Table S1, entries 1 and 2†). The direct regioselective deoxyfluorination of 1 using DAST in the presence of pyridine resulted in the decomposition of the starting material (Table S1, entries 3 and 4†).45 When the reaction was performed in the absence of base, compound 3a was formed as a major product in modest 34% yield (Fig. 2 and Table S1, entries 5 and 6†). Increasing the reaction temperature only led to complex mixtures which complicated isolation (Table S1, entry 7†). The modest yield of the DAST-mediated deoxyfluorination of thioglycoside 1 were ascribed to the several side-reactions. Elimination, to give 3b, is a commonly observed side-reaction in nucleophilic substitution using the basic fluoride ion.46 A migration of the anomeric SEt moiety47 can generate 3c (observed as anomeric fluoride at ca. 130–140 ppm in 19F NMR). Lastly, DAST-mediated thioglycoside activation48–50 can produce reactive species capable of undergoing glycosylation with the nucleophiles present in solution or promote starting material decomposition, as observed when the reaction was performed at higher temperature. After the deoxyfluorination step, installation of Fmoc on 3a completed the synthesis of BB3 (Fig. 2).
 | ||
| Fig. 2 Synthesis of BB3. | ||
For the synthesis of the 6F-GlcNAc BB (BB5), different deoxyfluorination strategies were explored (Fig. 3). Regioselective deoxyfluorination using DAST on intermediate 4, bearing three hydroxyl groups, gave a complex mixture of products with only trace amounts of 5 (Fig. 3).51 Thus, triol 4 was converted into intermediate 6 in two steps, because an efficient deoxyfluorination procedure was reported for an analogous thioglycoside intermediate.41 The subsequent DAST-mediated deoxyfluorination of 6 yielded a complex mixture of side-products (Fig. 3). Next, inspired by the successful deoxyfluorination of the 3-O-Bn Glc intermediate 1, triol 4 was converted into diol 9 in three steps (Fig. 3). DAST-mediated deoxyfluorination, performed in a mixture of DCM and 1,4-dioxane, afforded the desired product 10 together with a complex mixture of side-products that complicated purification (Table S1, entry 1†). Furthermore, anomerization48–50 was observed (Table S2, entry 1†). To simplify purification and characterization, the reaction was performed in ACN as solvent. These conditions yielded the target compound 10 in 23% yield and minimized anomerization52 (Table S2, entry 2†). To complete the synthesis of BB5, Fmoc was installed and the thioether leaving group replaced by dibutyl phosphate (Fig. 3). The latter step aimed to increase the donor reactivity that could be beneficial when working with unreactive acceptors such as the GlcNAc-OH-4.52,53 During the installation of the dibutyl phosphate anomeric group, a trichloro oxazoline was also formed54 (Scheme S1†), but it could be easily converted into the desired BB5, adapting a reported procedure.55
 | ||
| Fig. 3 Synthesis of BB5. | ||
In both synthetic routes, the success of the C-6 deoxyfluorination of the thioglycoside intermediates proved highly dependent on the protecting group pattern, potentially modulating the rate of the multiple side reactions that can occur.41,45 While the implementation of a more stable anomeric group (e.g. O-based glycoside) could overcome the synthetic hurdles associated to the thioether leaving group, this operation would imply additional protecting group manipulations that might result in comparable overall yields. Thus, other routes were not explored.
Automated glycan assembly of F-cellulose and F-chitin
Fluorinated cellulose and chitin hexasaccharides were prepared by AGA, following cycles of glycosylation and Fmoc deprotection on solid supports L1 or L2 (Fig. 4). Solid-phase methanolysis of Bz esters (Module F, only for cellulose oligomers) was followed by cleavage from the solid support (Module G1) and hydrogenolysis of the Bn and TCA protecting groups (Module H1 or H2). A single final purification step afforded a collection of fluorinated cellulose and chitin oligomers (Module I). For details on the AGA modules we refer to the ESI.† | ||
| Fig. 4 AGA of fluorinated cellulose and chitin oligomers. Yields of isolated products after AGA, deprotection, and purification are reported in parentheses (see the ESI†). The oligomers A6, N6, AFAAFA and FAFAFA were synthesized according to previously reported protocols.22 The oligosaccharides are represented following the Symbol Nomenclature For Glycans (SNFG).56 | ||
The cellulose analogues were assembled including either the 3F- or the 6F-Glc units, indicated with a capital F and non-capital f, respectively. For both analogues, different substitution patterns were explored: alternating (FAFAFA), “random” (AFAAFA and AfAAfA), block (FFFAAA, and fffAAA), and fully substituted (FFFFFF and ffffff) (Fig. 4). Two 6F-chitin analogues with “random” (NNfNNNfN) and block (NfNfNfNNN) substitution patterns were synthesized (Fig. 4). Deoxyfluorination did not significantly affect the reactivity of the BB donors. The low reactivity of the OH-453 of the 6F-GlcNAc acceptors bound to the solid support was circumvented with a double cycle of glycosylation to minimize deletion sequences.
F-cellulose
To analyze the impact of the 3F- and 6F-Glc modifications on the macroscopic properties such as crystallinity, we relied on MD simulations and powder XRD. We previously observed that the 3F-Glc modification disrupted the intramolecular OH-3⋯O-5′ hydrogen bond in AFAAFA, enhancing the flexibility of the single molecule and disrupting the crystallinity typical of cellulose.22,43 The systematic analysis of all oligomers with different substitution patterns revealed that, while all analogues were more soluble in water (>1 mg mL−1) than the unmodified AAAAAA, the hexasaccharide with a block pattern (FFFAAA) displayed a cellulose II-type XRD profile, despite the high degree of substitution (Fig. 5A). In contrast, when the pattern was alternating (FAFAFA) an amorphous XRD profile was obtained. A very different behavior was observed for the hexasaccharide with a block pattern of 6F-Glc residues (fffAAA), which displayed again an amorphous XRD. These data indicate that the substitution site (6F versus 3F) is a key parameter in the aggregation process (Fig. 5A). This result is consistent with that observed in the Ramachandran plots extracted from single molecule atomistic MD simulations (Fig. 5B); while FFFAAA resembled AAAAAA, fffAAA showed a broader distribution, with a wider range of Ψ dihedral values populated (Fig. 5B, top). The radius of gyration plots confirmed the higher overall single molecule flexibility for the 6F substituted analogue fffAAA, exploring extended geometries as well as more compact conformations (Fig. 5B, bottom). This trend was observed for all the 3F and 6F analogues (see ESI, Section 4.3†). At high degree of substitution, both 3F and 6F oligomers resulted in amorphous XRDs (Fig. 5A).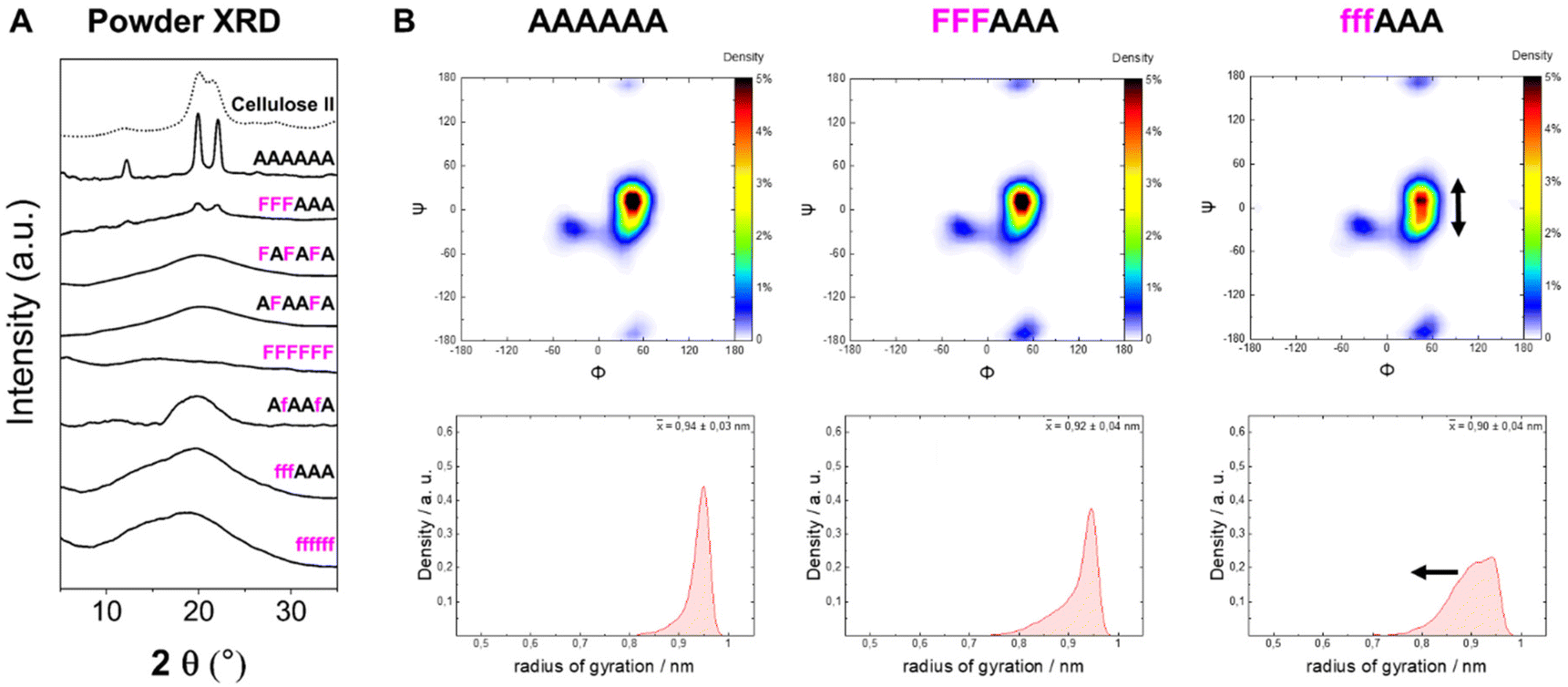 | ||
| Fig. 5 (A) Powder XRD profiles of 3F- and 6F-cellulose compared to cellulose II profile (dotted line). (B) Ramachandran plots for all glycosidic linkages combined (top) and radius of gyration plots (bottom) extracted from the MD simulations. The dihedral angles are defined as Φ = H1C1O4C4 and Ψ = C1O4C4H4 (further details are reported in Fig. S3 in the ESI†). | ||
The introduction of fluorine into the oligosaccharides enabled the use of the 19F NMR channel to perform structural studies.34,57 The 19F NMR spectrum of FFFAAA in deuterium oxide (D2O) indicated a different chemical environment surrounding the 3F moiety (i.e. internal versus external units, ca. 2.5 ppm difference), potentially due to significantly different interactions with solvent molecules (Fig. 6A). In contrast, fffAAA displayed a smaller chemical shift difference between internal and external residues (ca. 1 ppm, Fig. 6C). For the 6F substituted analogues, 19F NMR allowed for a qualitative analysis58 of the populations of the ω dihedral angle, using the calculated values for the three ω rotamers as reference59,60 (Fig. 6B). We measured the 3JH5–F6 values for external (27.2 Hz) and internal (25.8 Hz) residues (Fig. 6C), suggesting a lower populated gg rotamer in the external residue compared to internal ones. A complete estimation of the gg, gt, and tg populations required an additional experimental value (i.e.3JH6–H5) which could not be extracted due to severe spectral overlap in the 1H NMR. MD simulations supported this experimental observation, showing a similar trend of less populated gg conformations for external residues, both for AAAAAA (Fig. 6D, top) and for fffAAA (Fig. 6D, bottom). Furthermore, fluorine at the C-6 showed across all simulated structures a small rise of the tg rotamer (see the ESI, Section 4.6†), that could not be detected by NMR. This is surprising because the electronegative fluorine substituent should favor the gg rotamer due to the gauche effect.61 We speculate that these populations could be the results of the fluorine atom disrupting the organization of the surrounding water molecules62 (see the ESI†) or an artifact of the force field.
 | ||
| Fig. 6 (A) Fluorinated cellobiose repeating unit (top) and excerpt of the 19F NMR of FFFAAA in D2O (bottom). (B) Definition of gg, gt, and tg rotamers of the ω dihedral angle and previously reported calculated 3JH5–F6 values.59,60 (C) Excerpt of the 19F NMR of fffAAA in D2O and respective 3JH5–F6 values. (D) Predicted populations of the rotamers for the ω dihedral angles for external (left) and internal (right) residues of two cellulose analogues. | ||
Overall, these data suggest that the 6F substitution has a bigger impact than the 3F substitution on the crystallization of cellulose oligomers. This implies that the OH-6 plays a more important role than OH-3 in the crystallization process, potentially due to its higher exposure to solvent molecules, as indicated by NMR analysis of the 3F and 6F analogues. The higher exposure to water also suggest the prominent role of OH-6 in the formation of intermolecular hydrogen bonds, triggering aggregation.
F-chitin
In contrast to 6F-cellulose, the two 6F-chitin oligomers with ‘random’ (NNfNNNfN) and block (NfNfNfNNN) pattern were less soluble in water (<2 mg mL−1) than NNNNNN (13–17 mg mL−1 (ref. 22)). Both analogues retained the α-chitin-type XRD profile and the conformational behavior observed for the natural counterpart (NNNNNN)22 (Fig. 7A). High similarity in the Ramachandran and radius of gyration plots indicated comparable molecular conformations, with tendency to adopt an extended shape (Fig. 7B). We qualitatively analyzed the ω dihedral angle by measuring the 3JH5–F6 coupling constants for NfNfNfNNN. The smaller 3JH5–F6 for external residues was interpreted as a less populated gg rotamer compared to internal ones, as observed for the 6F-cellulose analogues (Fig. S6A and S6B†).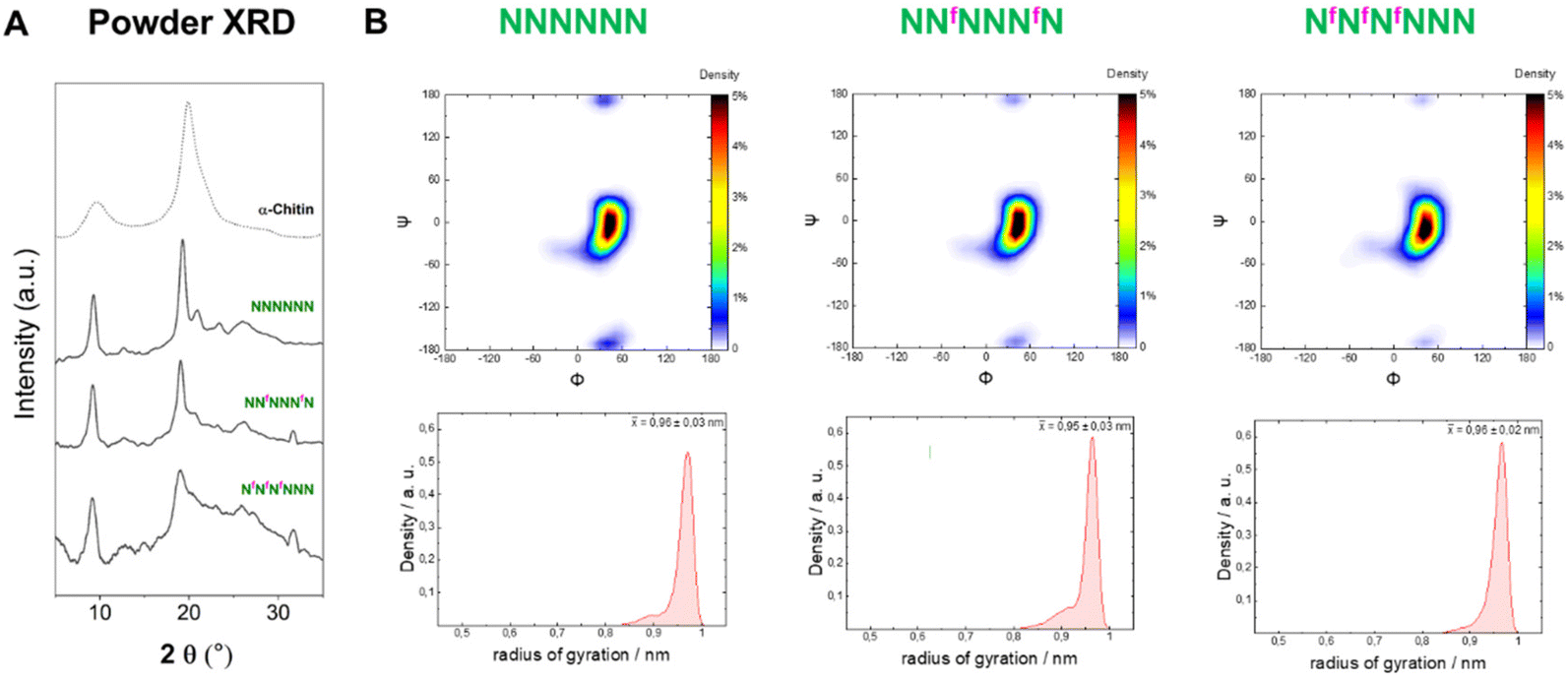 | ||
| Fig. 7 (A) Powder XRD profiles of 6F-chitin compared to α-chitin profile (dotted line). (B) Ramachandran plots for all glycosidic linkages combined (top) and radius of gyration plots (bottom) extracted from the MD simulations. | ||
From these results, it appears that chitin is more tolerant than cellulose to C-6 modifications, supporting the hypothesis that chitin crystallization is driven by other functionalities (e.g. the amide group that stabilizes inter-sheet hydrogen bonds10) rather than the OH-6. In the crystal structure of α-chitin the C-6 side chain can adopt a variety of orientations,6,10 supporting the observed high tolerance of chitin for C-6 modifications.
Conclusions
Seven new fluorinated oligomers of cellulose and chitin were assembled by AGA using non-natural 3F-Glc, 6F-Glc and 6F-GlcNAc BBs, granting precise control over the oligomer length and fluorination pattern. Their conformational behavior and macroscopic properties were studied by XRD analysis, NMR spectroscopy, and MD simulations. While, in general, deoxyfluorination of cellulose results in higher water solubility and lower crystallinity, we found that the pattern (block versus alternated) and site of modification (3F versus 6F) can be exploited to tune the sample crystallinity. In cellulose, the 6F modification impacted the molecular conformation more than the 3F substitution. In contrast, chitin conformation was much less affected by the 6F modification, with all the F-chitin retaining the native crystallinity.This work highlights deoxyfluorination as a valuable mean to tune the solubility and aggregation of cellulose and chitin as well as to pinpoint the importance of the replaced hydroxyl group in the crystallization process. In the future, deoxyfluorination can be imagined to increase the stability towards enzymatic degradation of cellulose or chitin-based materials.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We thank the Max Planck Society, the MPG-FhG Cooperation Project Glyco3Dysplay, the German Federal Ministry of Education and Research (BMBF, grant number 13XP5114), and the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation – SFB 1449 – 431232613; sub-project C2) for generous financial support. We thank Daniel Werner for assistance during XRD measurements. Open Access funding provided by the Max Planck Society.References
- Y. Nishiyama, J. Wood Sci., 2009, 55, 241–249 CrossRef CAS.
- M. Rinaudo, Prog. Polym. Sci., 2006, 31, 603–632 CrossRef CAS.
- D. J. Cosgrove, Nat. Rev. Mol. Cell Biol., 2005, 6, 850–861 CrossRef CAS PubMed.
- H.-C. Flemming, J. Wingender, U. Szewzyk, P. Steinberg, S. A. Rice and S. Kjelleberg, Nat. Rev. Microbiol., 2016, 14, 563–575 CrossRef CAS.
- Y. Nishiyama, J. Sugiyama, H. Chanzy and P. Langan, J. Am. Chem. Soc., 2003, 125, 14300–14306 CrossRef CAS PubMed.
- P. Sikorski, R. Hori and M. Wada, Biomacromolecules, 2009, 10, 1100–1105 CrossRef CAS.
- P. Langan, Y. Nishiyama and H. Chanzy, Biomacromolecules, 2001, 2, 410–416 CrossRef CAS PubMed.
- P. Chen, Y. Ogawa, Y. Nishiyama, M. Bergenstråhle-Wohlert and K. Mazeau, Cellulose, 2015, 22, 1485–1493 CrossRef CAS.
- Y. Ogawa, C. M. Lee, Y. Nishiyama and S. H. Kim, Macromolecules, 2016, 49, 7025–7031 CrossRef CAS.
- R. Minke and J. Blackwell, J. Mol. Biol., 1978, 120, 167–181 CrossRef CAS PubMed.
- B. Medronho, A. Romano, M. G. Miguel, L. Stigsson and B. Lindman, Cellulose, 2012, 19, 581–587 CrossRef CAS.
- B. Frka-Petesic and S. Vignolini, Nat. Photonics, 2019, 13, 365–367 CrossRef CAS PubMed.
- R. M. Parker, G. Guidetti, C. A. Williams, T. Zhao, A. Narkevicius, S. Vignolini and B. Frka-Petesic, Adv. Mater., 2018, 30, 1704477 CrossRef.
- R. M. Parker, B. Frka-Petesic, G. Guidetti, G. Kamita, G. Consani, C. Abell and S. Vignolini, ACS Nano, 2016, 10, 8443–8449 CrossRef CAS.
- A. Narkevicius, L. M. Steiner, R. M. Parker, Y. Ogawa, B. Frka-Petesic and S. Vignolini, Biomacromolecules, 2019, 20, 2830–2838 CrossRef CAS.
- A. Narkevicius, R. M. Parker, J. Ferrer-Orri, T. G. Parton, Z. Lu, G. T. Kerkhof, B. Frka-Petesic and S. Vignolini, Adv. Mater., 2022, 2203300 CrossRef CAS.
- M. Wada, L. Heux and J. Sugiyama, Biomacromolecules, 2004, 5, 1385–1391 CrossRef CAS.
- Y. Ogawa, J.-L. Putaux and Y. Nishiyama, Curr. Opin. Chem. Biol., 2022, 70, 102183 CrossRef CAS PubMed.
- S. C. Fox, B. Li, D. Xu and K. J. Edgar, Biomacromolecules, 2011, 12, 1956–1972 CrossRef CAS PubMed.
- H. C. Arca, L. I. Mosquera-Giraldo, V. Bi, D. Xu, L. S. Taylor and K. J. Edgar, Biomacromolecules, 2018, 19, 2351–2376 CrossRef CAS.
- G. Fittolani, D. Vargová, P. H. Seeberger, Y. Ogawa and M. Delbianco, J. Am. Chem. Soc., 2022, 144, 12469–12475 CrossRef CAS PubMed.
- Y. Yu, T. Tyrikos-Ergas, Y. Zhu, G. Fittolani, V. Bordoni, A. Singhal, R. J. Fair, A. Grafmüller, P. H. Seeberger and M. Delbianco, Angew. Chem., Int. Ed., 2019, 58, 13127–13132 CrossRef CAS PubMed.
- T. Tyrikos-Ergas, V. Bordoni, G. Fittolani, M. A. Chaube, A. Grafmüller, P. H. Seeberger and M. Delbianco, Chem. – Eur. J., 2021, 27, 2321–2325 CrossRef CAS.
- S. Basa, M. Nampally, T. Honorato, S. N. Das, A. R. Podile, N. E. El Gueddari and B. M. Moerschbacher, J. Am. Chem. Soc., 2020, 142, 1975–1986 CrossRef CAS PubMed.
- T. Tyrikos-Ergas, S. Gim, J. Huang, S. Pinzón Martín, D. Varón Silva, P. H. Seeberger and M. Delbianco, Nat. Commun., 2022, 13, 3954 CrossRef CAS.
- K. Huonnic and B. Linclau, Chem. Rev. DOI:10.1021/acs.chemrev.2c00086.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- J. C. Biffinger, H. W. Kim and S. G. DiMagno, ChemBioChem, 2004, 5, 622–627 CrossRef CAS PubMed.
- D. O′hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS.
- R. Hevey, Chem. – Eur. J., 2021, 27, 2240–2253 CrossRef CAS.
- N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS.
- M. Inoue, Y. Sumii and N. Shibata, ACS Omega, 2020, 5, 10633–10640 CrossRef CAS PubMed.
- B. Linclau, A. Ardá, N.-C. Reichardt, M. Sollogoub, L. Unione, S. P. Vincent and J. Jiménez-Barbero, Chem. Soc. Rev., 2020, 49, 3863–3888 RSC.
- J. D. Martínez, A. I. Manzano, E. Calviño, A. De Diego, B. Rodriguez de Francisco, C. Romanò, S. Oscarson, O. Millet, H.-J. Gabius, J. Jiménez-Barbero and F. J. Cañada, J. Org. Chem., 2020, 85, 16072–16081 CrossRef PubMed.
- V. Denavit, J. St-Gelais, T. Tremblay and D. Giguére, Chem. – Eur. J., 2019, 201901346 Search PubMed.
- M. L. Uhrig, B. Lantaño and A. Postigo, Org. Biomol. Chem., 2019, 17, 5173–5189 RSC.
- T. J. Kieser, N. Santschi, L. Nowack, A. Axer, G. Kehr, S. Albrecht and R. Gilmour, ACS Chem. Neurosci., 2020, 11, 2129–2136 CrossRef CAS.
- A. Axer, R. P. Jumde, S. Adam, A. Faust, M. Schäfers, M. Fobker, J. Koehnke, A. K. H. Hirsch and R. Gilmour, Chem. Sci., 2021, 12, 1286–1294 RSC.
- P. Andrade, J. C. Muñoz-García, G. Pergolizzi, V. Gabrielli, S. A. Nepogodiev, D. Iuga, L. Fábián, R. Nigmatullin, M. A. Johns, R. Harniman, S. J. Eichhorn, J. Angulo, Y. Z. Khimyak and R. A. Field, Chem. – Eur. J., 2021, 27, 1374–1382 CrossRef.
- A. Makino, M. Ohmae and S. Kobayashi, Macromol. Biosci., 2006, 6, 862–872 CrossRef CAS PubMed.
- T. Keenan, F. Parmeggiani, J. Malassis, C. Q. Fontenelle, J.-B. Vendeville, W. Offen, P. Both, K. Huang, A. Marchesi, A. Heyam, C. Young, S. J. Charnock, G. J. Davies, B. Linclau, S. L. Flitsch and M. A. Fascione, Cell Chem. Biol., 2020, 27, 1199–1206 CrossRef CAS PubMed.
- K. Anggara, Y. Zhu, G. Fittolani, Y. Yu, T. Tyrikos-Ergas, M. Delbianco, S. Rauschenbach, S. Abb, P. H. Seeberger and K. Kern, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, 1–6 CrossRef.
- Y. Zhu, T. Tyrikos-Ergas, K. Schiefelbein, A. Grafmüller, P. H. Seeberger and M. Delbianco, Org. Biomol. Chem., 2020, 18, 1349–1353 RSC.
- D. A. Williams, K. Pradhan, A. Paul, I. R. Olin, O. T. Tuck, K. D. Moulton, S. S. Kulkarni and D. H. Dube, Chem. Sci., 2020, 11, 1761–1774 RSC.
- P. A. Champagne, J. Desroches, J. D. Hamel, M. Vandamme and J. F. Paquin, Chem. Rev., 2015, 115, 9073–9174 CrossRef CAS PubMed.
- P.-C. Lin, A. K. Adak, S.-H. Ueng, L.-D. Huang, K.-T. Huang, J. A. Ho and C.-C. Lin, J. Org. Chem., 2009, 74, 4041–4048 CrossRef PubMed.
- K. C. Nicolaou, R. E. Dolle and D. P. Papahatjis, J. Am. Chem. Soc., 1984, 106, 4189–4192 CrossRef CAS.
- K. Suzuki, Y. Ito and O. Kanie, Carbohydr. Res., 2012, 359, 81–91 CrossRef CAS PubMed.
- I. Ohtsuka, T. Ako, R. Kato, S. Daikoku, S. Koroghi, T. Kanemitsu and O. Kanie, Carbohydr. Res., 2006, 341, 1476–1487 CrossRef CAS.
- P. J. Card and G. S. Reddy, J. Org. Chem., 1983, 48, 4734–4743 CrossRef CAS.
- P. R. Andreana and D. Crich, ACS Cent. Sci., 2021, 7, 1454–1462 CrossRef CAS.
- D. Crich and V. Dudkin, J. Am. Chem. Soc., 2001, 123, 6819–6825 CrossRef CAS.
- R. Enugala, L. C. R. Carvalho, M. J. Dias Pires and M. M. B. Marques, Chem. – Asian J., 2012, 7, 2482–2501 CrossRef CAS PubMed.
- T. J. Donohoe, J. G. Logan and D. D. P. Laffan, Org. Lett., 2003, 5, 4995–4998 CrossRef CAS PubMed.
- A. Varki, R. D. Cummings, M. Aebi, N. H. Packer, P. H. Seeberger, J. D. Esko, P. Stanley, G. Hart, A. Darvill, T. Kinoshita, J. J. Prestegard, R. L. Schnaar, H. H. Freeze, J. D. Marth, C. R. Bertozzi, M. E. Etzler, M. Frank, J. F. Vliegenthart, T. Lütteke, S. Perez, E. Bolton, P. Rudd, J. Paulson, M. Kanehisa, P. Toukach, K. F. Aoki-Kinoshita, A. Dell, H. Narimatsu, W. York, N. Taniguchi and S. Kornfeld, Glycobiology, 2015, 25, 1323–1324 CrossRef CAS.
- P. Valverde, J. I. Quintana, J. I. Santos, A. Ardá and J. Jiménez-Barbero, ACS Omega, 2019, 4, 13618–13630 CrossRef CAS PubMed.
- V. Denavit, D. Lainé, C. Bouzriba, E. Shanina, É. Gillon, S. Fortin, C. Rademacher, A. Imberty and D. Giguère, Chem. – Eur. J., 2019, 25, 4478–4490 CrossRef CAS PubMed.
- R. J. Abraham, E. J. Chambers and W. A. Thomas, Magn. Reson. Chem., 1992, 30, S60–S65 CrossRef CAS.
- L. Phillips and V. Wray, J. Chem. Soc. B, 1971, 1618 RSC.
- N. Santschi, N. Aiguabella, V. Lewe and R. Gilmour, J. Fluor. Chem., 2015, 179, 96–101 CrossRef CAS.
- R. E. Rosenberg, J. Phys. Chem. A, 2019, 123, 7651–7660 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ob01601j |
| ‡ Current affiliation: Department of Chemistry, University of Illinois, 61801 Urbana, Illinois, USA. |
| This journal is © The Royal Society of Chemistry 2022 |