
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
β-1,3-Glucan synthesis, novel supramolecular self-assembly, characterization and application†
Robert
Pylkkänen
 *ab,
Pezhman
Mohammadi
*b,
Ville
Liljeström
c,
Wojciech
Płaziński
de,
Grégory
Beaune
c,
Jaakko V. I.
Timonen
f and
Merja
Penttilä
ab
*ab,
Pezhman
Mohammadi
*b,
Ville
Liljeström
c,
Wojciech
Płaziński
de,
Grégory
Beaune
c,
Jaakko V. I.
Timonen
f and
Merja
Penttilä
ab
aDepartment of Bioproducts and Biosystems, School of Chemical Engineering, Aalto University, FI-00076 Aalto, Finland. E-mail: robert.pylkkanen@aalto.fi
bVTT Technical Research Centre of Finland, FI-02044 VTT, Finland
cNanomicroscopy Center, OtaNano, Aalto University, FI-00076 Aalto, Finland
dJerzy Haber Institute of Catalysis and Surface Chemistry, Polish Academy of Sciences, 30-239 Krakow, Poland
eDepartment of Biopharmacy, Faculty of Pharmacy, Medical University of Lublin, 20-093 Lublin, Poland
fDepartment of Applied Physics, School of Science, Aalto University, FI-00076 Aalto, Finland
First published on 4th October 2022
Abstract
β-1,3-Glucans are ubiquitously observed in various biological systems with diverse physio-ecological functions, yet their underlying assembly mechanism and multiscale complexation in vitro remains poorly understood. Here, we provide for the first-time evidence of unidentified β-1,3-glucan supramolecular complexation into intricate hierarchical architectures over several length scales. We mediated these unique assemblies using a recombinantly produced β-1,3-glucan phosphorylase (Ta1,3BGP) by fine-tuning solution conditions during particle nucleation and growth. We report a synthesis of interconnected parallel hexagonal lamellae composed of 8 nm thick sheets of highly expanded paracrystals. The architecture consists of β-1,3-glucan triple-helices with considerable inter–intra hydrogen bonding within, as well as in between adjacent triple-helices. The results extend our understanding of β-1,3-glucan molecular organization and shed light on different aspects of the crystallization processes of biomolecules into structures unseen by nature. The presented versatile synthesis yields new materials for diverse medical and industrial applications.
Introduction
Glucans are one of the most abundant organic polymers on Earth. They are inherently stiff molecules due to the bulkiness and rigidness of the glucose monomers they are composed of.1 The unique properties of glucans largely revolve around the specific glycosidic linkage connecting adjacent glucose monomers. In nature, glucans are utilized for a wide range of physiological processes. α-Linked glucans are commonly associated with intracellular energy storage (e.g., starch, α-1,4-glucan) and β-linked glucans with cell wall structure (e.g., cellulose, β-1,4-glucan). Both α- and β-linked glucans can also be found in microbial exopolysaccharides where they function in cell protection, cell adhesion to solid surfaces, and cell-to-cell interactions. A β-1,3-linkage introduces repeating twists to the polymer chain because the C1 and C3 carbon atoms of the glucose ring are not directly opposite each other. As a result of repeating twists, β-1,3-glucan chains have the natural tendency to form helical structures.In natural systems, linear and unbranched β-1,3-glucans can be found in the forms of callose2,3 (cell wall component of embryophytes, i.e., land plants), curdlan4–12 (exopolysaccharide of Gram-negative bacteria), and paramylon13–17 (intracellular energy storage in some protozoa species, such as euglenids and haptophytes). Extensive studies on extracted and purified naturally occurring β-1,3-glucans have revealed them forming triple-helical associations composed of parallel β-1,3-glucan chains with extensive hydrogen bonding within one triple helix and between adjacent helices in a hexagonal crystalline unit cell.18,19 Single-helical forms have also been reported.20
The regio- and stereoselective construction of glycosidic linkages is challenging due to each monosaccharide unit containing multiple hydroxyl groups with similar reactivity and each linkage acting as a stereogenic center.21 Despite this, the chemical synthesis of short β-1,3-gluco-oligosaccharides up to six glucose units in length has been reported.22,23 On the contrary, enzymes offer greater regio- and stereoselectivity and can be used to consistently synthesize linear unbranched β-1,3-glucan. For example, isolated membrane fractions of the oomycete Saprolegnia monoica and the plant blackberry (Rubus fruticosa) produce weakly crystalline β-1,3-glucan when incubated in the presence of UDP-glucose and cellobiose (activator) in vitro.24 In an alternative enzymatic approach, β-1,3-glucan chains have also been synthesized using mutants of hydrolytic enzymes25 together with activated (typically fluorinated) substrates, however the glycosyl fluoride donors are often unstable at higher temperatures26 meaning the reactions are limited to 30–37 °C.
Taking advantage of an emerging approach we produced relatively long β-1,3-glucan chains with high crystallinity using the reverse reaction of a phosphorylase enzyme. Phosphorylases have the capability of reversibly catalyzing the liberation of monosaccharide 1-phosphate from the nonreducing end of specific glycosides in the presence of inorganic phosphate.27,28 They have received more attention only recently, despite offering large potential in utilizing accessible and relatively stable monosaccharide 1-phosphates for stereo- and regio-selective synthesis of glycosidic linkages (e.g. commercial synthesis of 2-O-α-D-glucosyl glycerol29 and kilogram scale synthesis of lacto-N-biose).30 A thermostable β-1,3-glucan phosphorylase with an optimum temperature of 75 °C was recently characterized,31,32 presenting an opportunity to examine the effects of reaction temperature on β-1,3-glucan synthesis.
We hypothesized similar mechanisms that grant curdlan the unique ability to form thermo-reversible and thermo-irreversible gels33–42 could result in unique β-1,3-glucan assemblies that are dependent on the reaction temperature. Here we report for the first time the growth of β-1,3-glucan into single crystals with a macroscopic appearance of a hexagonally shaped lamellar structure with multidomain morphology and hierarchical chirality. Strikingly, the seemingly separate lamellar domains are all parts of a single crystal mediated through spiral growth through screw-dislocation which span the crystalline layer at both crystallographic and morphological levels.
Results and discussion
β-1,3-Glucan synthesis
Of the natural β-1,3-glucans, curdlan has a unique ability to form thermo-reversible and thermo-irreversible gels upon heat treatment. Thermo-reversible gels are formed when an aqueous curdlan solution is heated up to 55 °C and then cooled, while thermo-irreversible gels are formed when the solution is heated at above 80 °C. These transition temperatures are dependent on many factors, such as molecular weight, concentration, heating rate, dispersing method, and the addition of inorganic salts.34–37 A possible mechanism for this gel-forming ability has been suggested to be the disassociation of curdlan microfibrils at 60 °C due to disruption of hydrogen bonding and the subsequent reassociation of curdlan microfibrils at higher temperatures driven by hydrophobic interactions.Taking a similar approach, we set out to perform β-1,3-glucan synthesis reactions (Fig. 1a) at temperatures ranging from ambient temperature (25 °C) up to 80 °C using Thermosipho africanus β-1,3-glucan phosphorylase (Ta1,3BGP) that we produced recombinantly. Glycosyl acceptor and donor concentrations employed were typical for reverse reactions with phosphorylases, i.e., 50 mM glucose (acceptor) and 200 mM glucose 1-phosphate (donor).43,44 Insoluble β-1,3-glucan was produced in all temperatures (Fig. 1b). The aqueous solutions displayed brownish color in reaction temperatures above 50 °C, and this could be attributed to Maillard and/or caramelization reactions. However, the insoluble glucans had a pure white color after washing with water.
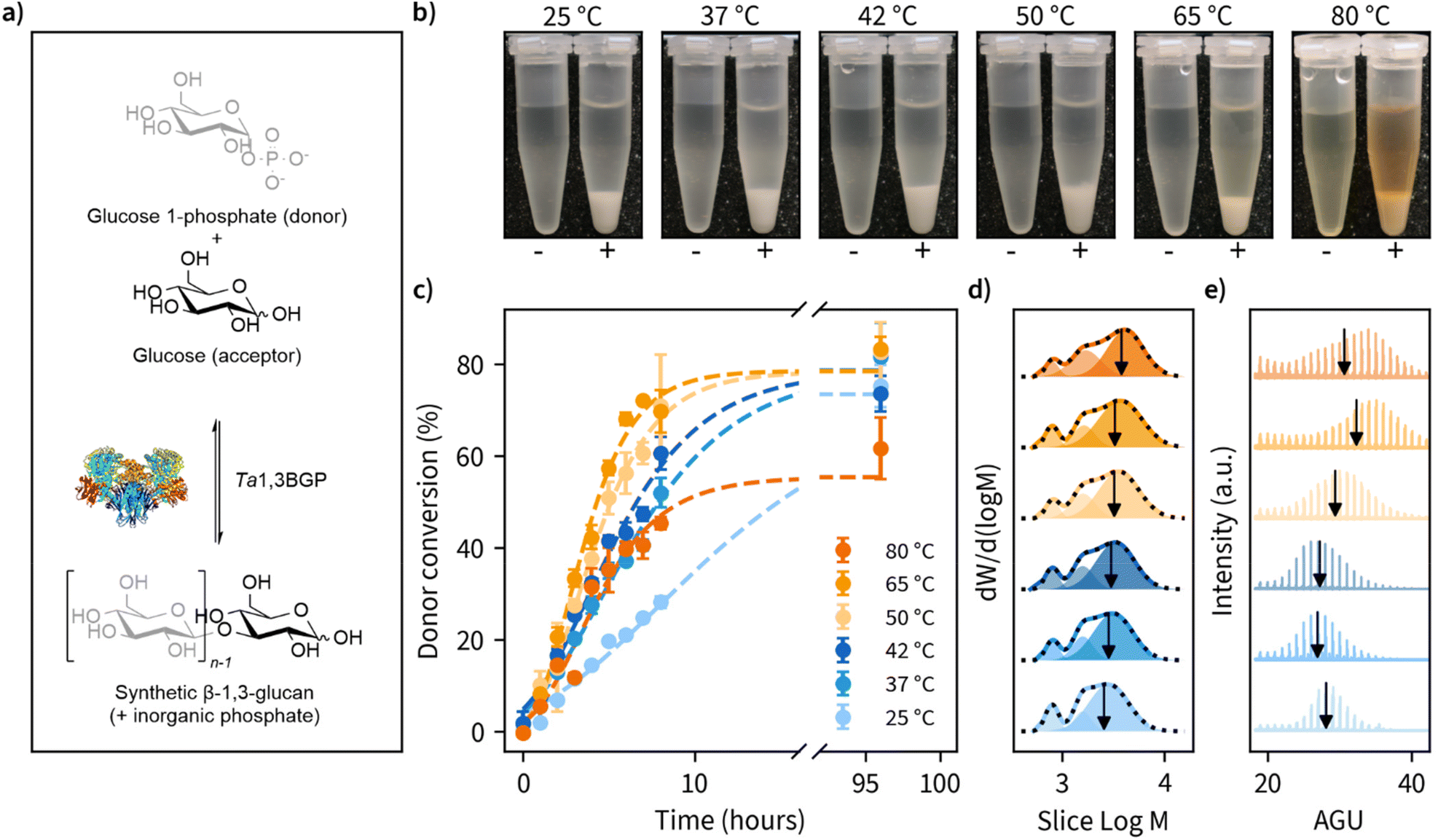 | ||
Fig. 1 Effect of reaction temperature on the enzymatic β-1,3-glucan synthesis reaction rates, product molar mass distributions and morphology. (a) Photographs of the β-1,3-glucans synthesized at different reaction temperatures illustrating the formation of insoluble precipitate when the glycosyl donor (α-D-glucose 1-phosphate) and glycosyl acceptor (glucose) are incubated in the presence Thermosipho africanus β-1,3-glucan phosphorylase (+) and lack of insoluble product when the enzyme is omitted (−). (b) Progression of glycosyl donor conversion from α-D-glucose 1-phosphate measured using a colorimetric assay to quantify the green complex formed between malachite green, molybdate and free orthophosphate. (c) Molar mass distributions of the reaction products measured using size-exclusion chromatography (SEC) showing the obtained multimodal distributions as lines and three individual fitted peaks as transparent fills. The cumulative sum of the fitted peaks is shown as a black dashed line. The calculated number average degree of polymerization (![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n) for the largest peak is indicated as a black vertical arrow. M is the molecular weight corresponding to averaged slice retention time and W is the normalized slice height used for molecular weight distribution calculation. (d) MALDI-ToF-MS spectra of the β-1,3-glucans synthesized at different temperatures with a peak-to-peak difference of one anhydroglucose unit (AGU, 162 Da) and the calculated n is indicated as a vertical line. (e) Representative SEM images of β-1,3-glucan morphology for when synthesis is performed at reaction temperatures up to 37 °C and (f) 42 °C and above. n) for the largest peak is indicated as a black vertical arrow. M is the molecular weight corresponding to averaged slice retention time and W is the normalized slice height used for molecular weight distribution calculation. (d) MALDI-ToF-MS spectra of the β-1,3-glucans synthesized at different temperatures with a peak-to-peak difference of one anhydroglucose unit (AGU, 162 Da) and the calculated n is indicated as a vertical line. (e) Representative SEM images of β-1,3-glucan morphology for when synthesis is performed at reaction temperatures up to 37 °C and (f) 42 °C and above. | ||
During the reactions, we determined glucose 1-phosphate (donor) conversion by measuring the liberation of free orthophosphate using a colorimetric assay (Fig. 1c). Reaction rates increased with reaction temperature up to 65 °C but decreased slightly moving from 65 to 80 °C. Donor conversion reached approximately 80% in all cases except 80 °C (approximately 60%). These results suggest prolonged incubations above 65 °C may result in decreased enzymatic activity, despite an optimum temperature of 75 °C reported for Ta1,3BGP.45 We also observed the spontaneous liberation of orthophosphate from glucose 1-phosphate in the absence of enzyme at temperatures of 80 °C and above, accounting for approximately 10% of the measured donor conversion at 80 °C. The detailed individual plots showing all replicate measurements and full timescale for these measurements can be found in Fig. S1.†
Ta1,3BGP has been reported to produce β-1,3-glucan chains up to at least 34 glucose units in length,31 but the effect of reaction temperature on chain length has not been studied. Furthermore, a report46 of low molecular weight (<20 glucose units) β-1,3-glucans being incapable of forming helical structures warranted further examination of the molar mass distributions (MMDs) of the reaction products. Fig. 1d illustrates the MMDs obtained using size-exclusion chromatography (SEC). The number average degree of polymerization (DP) for the obtained multimodal MMDs ranged from 11.2 glucose units (25 °C) to 13.7 glucose units (80 °C), with longer chains being produced at higher reaction temperatures. At first, we were surprised by the multimodality of these MMDs and re-examined them by dividing each distribution into three separate peaks. We found the two smallest molecular weight fractions to have approximately constant DP of 4.8 ± 0.0 and 9.5 ± 0.1, calculated between all reaction temperatures. Meanwhile, the fraction corresponding to the largest molecular weight showed an increase in DP from 15.7 (25 °C) up to 23.4 (80 °C). The detailed parameters obtained from distribution fitting are shown in Tables S2, S3 and the corresponding plots in Fig. S2.† One possible explanation for the observed multimodality is ‘alkaline peeling’ (β-elimination reaction) which β-1,3-glucans have tendency to undergo in dilute bases,47 such as the 0.1 M NaOH which was used as the eluent for SEC. Therefore, we decided to also estimate molar mass distributions using MALDI-ToF-MS, which additionally yields information about peaks corresponding to individual glucan chains (Fig. 1e). These results suggested slightly higher DP for the products. A DP ranging from 27.0 to 33.8, with masses for chains up to at least 34 visible agrees with an earlier report.31 Furthermore, a similar trend of larger molecular weight β-1,3-glucan production in higher reaction temperatures was also observed with MALDI-ToF-MS. The detailed analysis together with both unprocessed and processed MALDI-ToF-MS spectra can be found in Tables S2, S3 and Fig. S3, S4.† The summary of MMD measurements is given in Table 1.
| T (°C) | SEC, cumulative sum of all peaks | SEC, largest peak | MALDI-ToF-MS | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
n
|
w
|
DPn | PDI |
n
|
w
|
DPn | PDI |
n
|
w
|
DPn | PDI | |
| 25 | 1839 | 2560 | 11.2 | 1.39 | 2568 | 3132 | 15.7 | 1.22 | 4625 | 4649 | 28.4 | 1.01 |
| 37 | 1992 | 2760 | 12.2 | 1.39 | 2852 | 3457 | 17.5 | 1.21 | 4387 | 4422 | 27.0 | 1.01 |
| 42 | 1998 | 2860 | 12.2 | 1.43 | 3013 | 3625 | 18.5 | 1.20 | 4298 | 4363 | 26.4 | 1.02 |
| 50 | 2134 | 3135 | 13.1 | 1.47 | 3234 | 3959 | 19.9 | 1.22 | 4832 | 4882 | 29.7 | 1.01 |
| 65 | 2175 | 3287 | 13.3 | 1.51 | 3273 | 4134 | 20.1 | 1.26 | 5492 | 5526 | 33.8 | 1.01 |
| 80 | 2244 | 3393 | 13.7 | 1.51 | 3806 | 4491 | 23.4 | 1.18 | 5216 | 5254 | 32.1 | 1.01 |
Morphological characterization
Next, we examined the morphological characteristics of the produced insoluble β-1,3-glucans. We started with confocal microscopy, which enabled high-throughput investigation of products synthesized in different solution conditions in hydrated state. These examinations revealed a significant difference between non-ordered structures synthesized at temperatures up to 37 °C and ordered particles with approximately 10 μm diameter that were synthesized at 42 °C and above (Fig. 2a). Interestingly, at reaction temperatures of 65 °C and above, the ordered particles showed signs of disintegration. Given that the resolution of the confocal images was not sufficient to accurately describe particle morphology, we also employed high-resolution SEM for imaging (Fig. 2b). To avoid ambient drying artifacts and retain the morphological features of insoluble particles for imaging, we prepared the specimens by cryo-vitrification followed by rapid lyophilization. According to the images obtained via SEM, glucans synthesized in lower temperatures formed less-organized sheet-like structures, while higher reaction temperatures yielded well-ordered particles composed of stacked lamellar sheets with 10 μm diameter and a facetted appearance with hexagonal geometry. Furthermore, disruption of particle morphology at reaction temperatures above 65 °C was also evident. All these observations are in good agreement with our confocal microscopy images of particle morphology in wet state. More confocal microscope images of hydrated specimens can be found in Fig. S5–S8† and more SEM images at different magnifications in Fig. S9–S14.† | ||
| Fig. 2 Effect of reaction temperature on hexagonal β-1,3-glucan microparticle assembly and crystallinity. (a) Transmitted light images obtained with a confocal microscope of never-dried β-1,3-glucan particles synthesized at different temperatures. (b) SEM images of dried β-1,3-glucan particles synthesized at different reaction temperatures. (c) 2D synchrotron wide-angle X-ray scattering (WAXS) patterns of dried β-1,3-D-glucans prepared at different temperatures (λ = 0.82656 Å). Yellow arrows annotate the major diffraction rings corresponding to triple-helical β-1,3-glucan, and the corresponding Miller indices. (d) 1D WAXS profiles with major diffraction peaks indicated with arrows (λ = 1.5418 Å). (e) SEM images of different types of hexagonal assemblies observed for β-1,3-D-glucans synthesized at 50 °C. (f) Micropipette aspiration of hexagonal particles synthesized at 50 °C performed with a micropipette with a tip diameter of 10 μm and pressure ranging from 0 to −1000 mbar. | ||
Since the chain length of the particles (>20 glucose units) was sufficient for helix formation46 and the hexagonal shape suggested a hexagonal crystalline unit cell, we next measured the wide-angle X-ray scattering (WAXS) patterns for dry β-1,3-glucan particles (Fig. 2c and d). All major diffraction rings corresponding to triple-helical β-1,3-glucan were clearly visible, and, interestingly, reaction temperature did not seem to have a major contribution towards crystallinity or crystal form of the produced glucans. This suggests even the less ordered sheet-like structures synthesized at lower temperatures had triple-helical conformation. The unprocessed and processed 1D WAXS data can be found in Fig. S15, S16,† and the assignment of peaks to the corresponding Miller indices in Fig. S17.† It should be noted the broad 111 diffraction ring could have some contribution from the 120 and 201 diffraction rings. However, the contribution of these peaks remains unclear as they cannot be resolved well enough.
Regarding the uniformity of the particles, we observed several different well-ordered hexagonal microparticle assemblies, and some of these observations are highlighted in Fig. 2e. The most common structure consisted of stacked sheets with similar dimensions, but some other interesting shapes were also present. This includes self-enclosed circular assemblies of stacked sheets with a diameter of approximately 20 μm, which suggests a flexible architecture. To examine the flexural properties of individual particles, we used micropipettes with 10 and 25 μm diameter to perform aspiration with pressure ranging from 0 to −1000 mbar (Fig. 2f). Particle deformation or buckling could not be observed, suggesting the particles to be relatively stiff. However, the separation of sheets from hexagonal stacks was possible in some cases. Additional micropipette aspiration experiments on multiple particles with different geometries can be found in ESI Video 1.†
Microparticle assembly
Examination of the particles’ top layer at highest magnifications revealed each particle being composed of nanometer-thick stacked sheets with hexagonal geometry as shown by the sharp contour line (Fig. 3a). We also noted that each nanosheet is in a different stage of maturity, with bottom layers more expanded than the ones on top. A closer look at the center point suggests nucleation and growth is mediated through spiral growth by screw-dislocation with a minimum of two continuous paracrystal domains (Fig. 3a, higher resolution images, and an additional specimen can be found in Fig. S18†). Screw-dislocations have a dislocation line perpendicular to the growing lamellae and have been proven as the growth mechanism for a variety of facetted crystalline systems.48,49 Sharp domain boundaries remain present even as the lamellae grow large indicating that the growth process is energetically favorable. This observation is also consistent with the general understanding that 1D particles (here triple-helices) with attractive interaction would optimally arrange in a hexagonal 2D lattice which has the highest packing fraction. A steady growth of a 2D hexagonal lattice would hence yield hexagonal particles.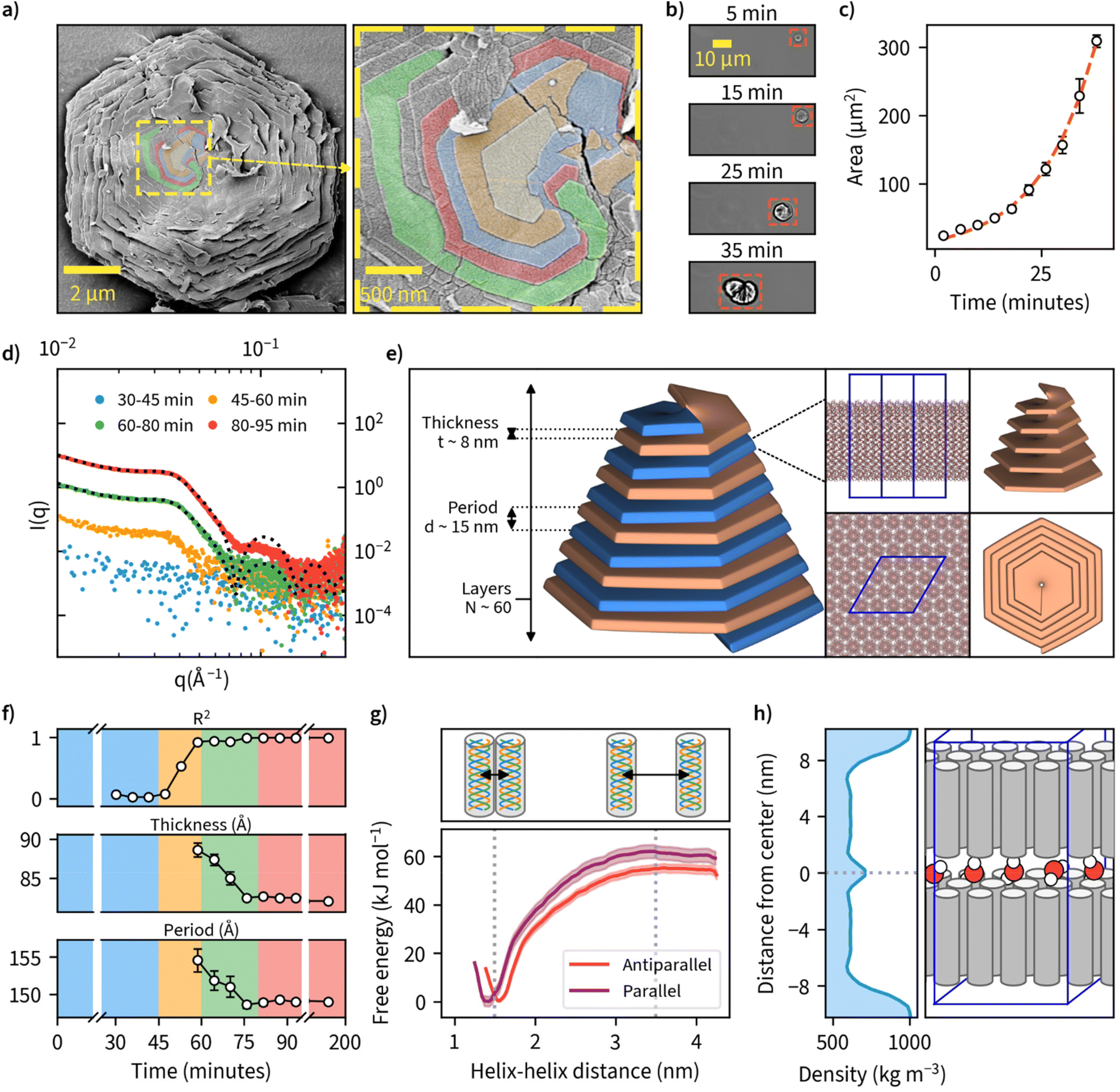 | ||
| Fig. 3 The detailed structure and mechanisms of β-1,3-D-glucan sheet and microparticle assembly. (a) SEM image of sheet-like lamellar structures observed on the surface of facetted hexagonal β-1,3-D-glucan particles when viewed under high magnification. Domain boundaries between the topmost layers at the center point of the crystal are colored in yellow, orange, blue, red, and green, indicating the spiral growth by screw-dislocation. (b) Timelapse of microparticle formation imaged using an optical microscope. Timescale corresponds to time since nucleation was first observed. (c) Particle area growth rate derived from microscopic observations in (b). A red dashed line indicates a fit to the exponential Gompertz function to illustrate exponential growth of particlea area. (d) Small-angle X-ray scattering (SAXS) data collected and averaged at different time points of enzymatic synthesis carried out at 50 °C. Exemplary model fittings for 60–80 min and 80–95 min data are shown with a black dotted line with R2 values of 0.986 and 0.996, respectively. (e) A schematic illustration of the key structural parameters of a single lamellae layer and their final values obtained by a fitting a theoretical lamellar model on the SAXS-data collected in (d).( f) Evolution of the key parameters obtained from the SAXS-model fitting as the reactions proceed over time. Background color corresponds to the color scheme in (d). (g) Molecular dynamics (MD) simulation result for free-energy profiles for parallel and antiparallel triple-helix orientations in helix–helix complexes forming lamellar sheets consisting of β-1,3-D-glucan triple-helices. (h) MD simulation result for water mass density in a β-1,3-glucan bilayer, and an illustration of the corresponding lamellar sheet structure with water molecules between the layers. | ||
To our knowledge, this is the first observation of β-1,3-glucans undergoing supramolecular complexation into microparticles. We investigated the mechanism of how these thin sheets assemble into well-ordered microparticles through a time-lapse image series of microparticle growth under the microscope at a reaction temperature of 50 °C in sealed capillary tubes (Fig. 3b). We observed multiple individual hexagonal particles forming after a reaction time of approximately 1 hour, but not during later stages of the reaction. This could be an indication that the growth of existing particles is preferred over the nucleation of new particles and is supported by the general notion that systems with screw-dislocation favor spiral growth over nucleation due to energetic reasons.50 From the individual frames of the timelapse, we quantified particle area over time and found this value to increase from an initial approximate value of 20 μm2 to over 300 μm2 over a time span of 40 minutes since nucleation was observed (Fig. 3c and ESI Video 2†).
To further examine the detailed particle structure, we carried out an in situ small-angle X-ray scattering (SAXS) measurement where data was collected during β-1,3-glucan synthesis at 50 °C (Fig. 3d). During the initial phase from 0 to 45 minutes, we couldn't detect any signal that could be discerned from the background. From 45 minutes onward, however, we could observe the formation of insoluble β-1,3-glucan inside the capillaries, and this corresponded with an increase in signal intensity detected using SAXS. After a reaction time of 80 minutes, no major differences in SAXS intensity could be observed anymore. To quantify the formed structures, we employed a lamellar paracrystal model51 to fit the averaged data obtained between 80 and 200 minutes. The obtained fit suggested that the aggregates consist of stacked sheets with a thickness of approximately 8 nm and average period of 15 nm between them (Fig. 3e). This is in good agreement with earlier reports of hexagonal β-1,3-glucan sheets25,52–54 where the sheet thickness has been reported to be approximately 8 nm despite these studies lacking the assembly of such sheets into larger microparticles. The acquired sheet thickness corresponds to the length of triple-helical β-1,3-glucan with a chain length of approximately 28 glucose units based on a pitch 0.2835 nm per glucose unit,18,19 which is in line with the MMDs we measured using MALDI-ToF-MS. The collected SAXS data for all measured time points is presented with and without background subtraction in Fig. S19.†
Since we had measured SAXS for the duration of the whole reaction with 1-minute intervals, we tried to fit this model for each individual point of measurement to quantify any changes in β-1,3-glucan particle structure over time (Fig. 3f). We could obtain good fits from 60-minute reaction time onwards, finding decreasing sheet thickness from 8.9 ± 0.1 to 8.2 ± 0.0 nm, and decreasing lamellar spacing from 15.5 ± 0.2 to 14.9 ± 0.0 nm between 60 and 80 minutes. For the number of layers, we could not obtain a reliable estimate because for large aggregates (number of lamellae larger than ∼100) the employed model is insensitive to variation in the number of lamellae per stack. Intriguingly, the obtained lamellar spacing suggested quite a large spacing between the layers. Our SAXS data does not cover more details or the ultimate cause of the spacing, but it could be related to topological defects. The corresponding data and fits are shown in Fig. S20,† and the evolution of the parameters of the lamellar paracrystal model during the time course of the reaction in Fig. S21.†
We were also interested in the enthalpic stabilization of hydrophilic and hydrophobic associations leading to β-1,3-glucan assemblies such as individual chains associating into helical entities, the demixing of solvent water and polysaccharide chains and the formation of stable inter-molecular associations between triple-helices. First, to examine the effect of triple-helix orientation in lamellar β-1,3-glucan sheets, we calculated the free-energy profiles for parallel and antiparallel helix–helix complexes using data obtained from molecular dynamics (MD) simulations (Fig. 3g). These results indicate a highly energetically favorable association of the two triple helices characterized by free energy change of a magnitude of approximately 55–60 kJ mol−1. The parallel helix–helix association is slightly more favorable than antiparallel (by approximately 5 kJ mol−1). The formation of helix–helix complexes is not correlated with crossing any significant free energy barriers and the only minimum of the free energy is the global one, corresponding to fully associated helices, maintaining contact with each other along their longest dimension. Furthermore, the free energy change associated with removing a single triple helix from a fully formed quasi-infinite layer composed of uniform triple helices is equal to 1490 ± 132 kJ mol−1. This indicates that free energy changes resulting from the formation of a layer are not additive with respect to those corresponding to the helix–helix association. However, such a large magnitude of free energy change suggests that the formation of a fully packed layer is a highly energetically favorable process resulting in a very stable final structure. The detailed information of several different modelled systems and their conformational characteristics are given in Fig. S22 and S23.† Two exemplary videos showing a single layer of 16 helices and the formation hexagonal-like clusters can be found in ESI Videos 3 and 4,† respectively. Calculation of water density between a β-1,3-glucan bilayer from one of these systems is shown in Fig. 3h, as a possible explanation for empty space between layers. Additionally, inherent irregularities in layer packing such as deviations from the ideal hexagonal structure and unaligned chains could also contribute towards the spacing between layers.
Finally, we carried out further characterization to evaluate structural integrity of the assemblies by direct mechanical agitation (Fig. S24†). For that, we performed tip sonication with amplitude ranging from 20–40% (selected based on the equipment's lowest and highest setting). Almost in all cases, we noted complete deconstruction. Only, tip sonicated samples with 20% amplitude exhibit the presence of small quantities of intact assemblies.
Application
Due to the large available surface area, well-defined structure, and spacing between the layers, we finally investigated the potential of β-1,3-glucan microparticles for drug encapsulation applications. For this purpose, we freeze-dried an amount corresponding to 80 mg of the dry weight of 1,3-glucan and compressed it into tablets with an approximate diameter of 13 mm and thickness of 0.55 mm (Fig. 4a). The compressed material has suitable properties for handling in tablet form as indicated by a flexural stress–strain measurement (Fig. 4b). Furthermore, the tablets remained fully intact in a hydrated environment. A cross-sectional view of the fractured tablet after the flexural measurement is shown in Fig. 4d.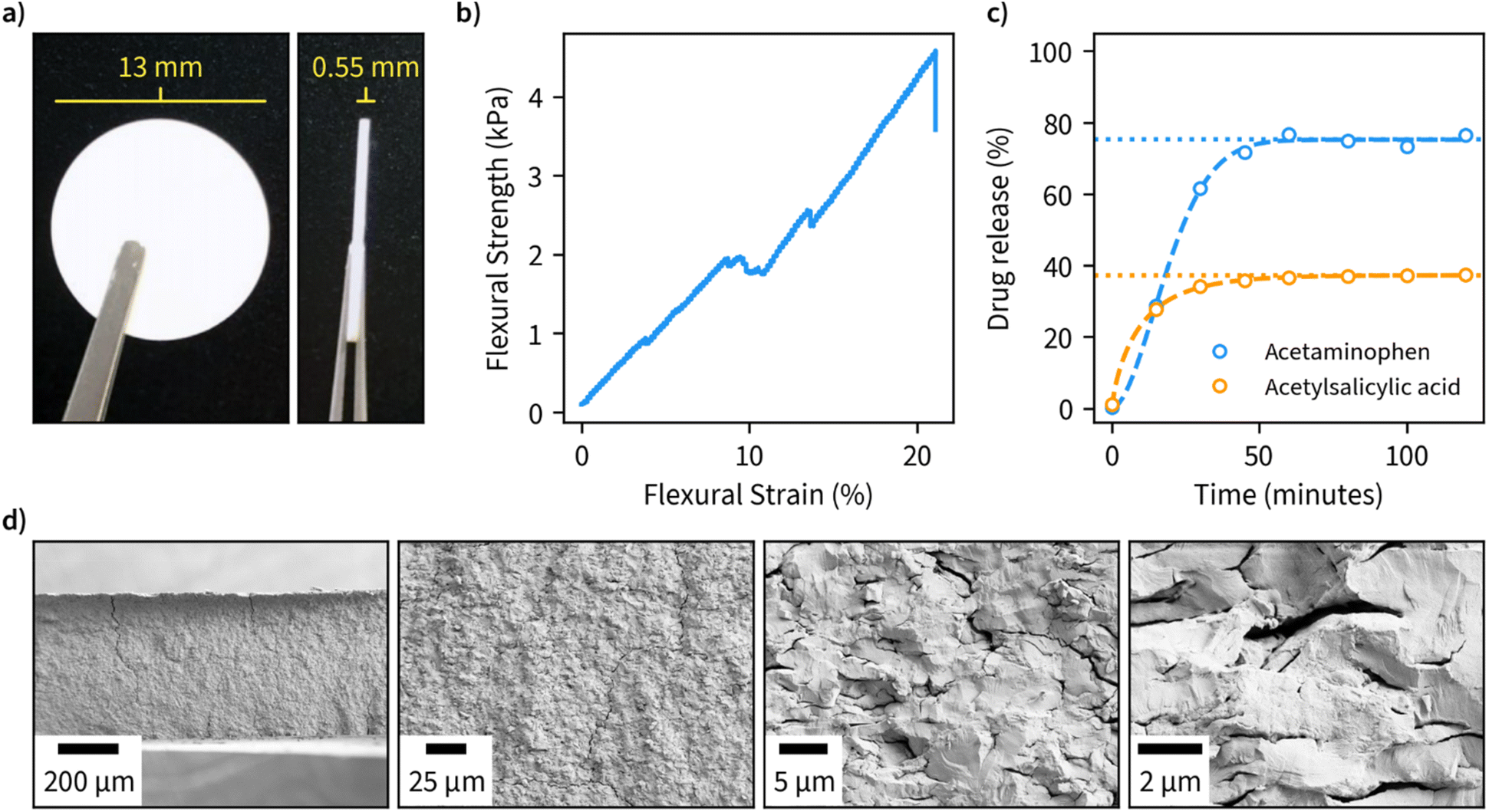 | ||
| Fig. 4 Application of hexagonal β-1,3-glucan microparticles synthesized at 50 °C as drug encapsulation tablets. (a) Tablets compressed from freeze-dried β-1,3-D-glucan (80 mg dry weight). (b) Flexural stress and strain properties of the compressed tablets. (c) Release profiles of tablets containing 5 mg acetaminophen or acetylsalicylic measured during incubation of tablets in phosphate buffer (pH 7) at 37 °C, with a dashed line indicating a fit for the drug release obtained using Weibull function and a dotted line indicating the cumulative amount of drug released at infinite time corresponding to the fit. (d) SEM images at different magnifications of the cross-sectional view of fractured specimens after bending test. | ||
As ligands for the drug release measurements, we opted for acetaminophen and acetylsalicylic acid due to both being well-characterized and having high water solubility. We performed encapsulation by incubating the drugs in an aqueous solution of β-1,3-glucan overnight, after which the samples were freeze-dried and compressed into tablets. Drug release measurements were performed in phosphate buffer, pH 7, at 37 °C and the obtained data was fitted using the Weibull function to discern the possible drug release mechanism (Fig. 4c). A maximum drug release of approximately 33 and 75% was obtained for acetylsalicylic acid and acetaminophen, respectively. In both cases, maximum drug release was obtained after a period of 50 minutes, which is in line with other fast-release matrices55 without using any other excipients than β-1,3-glucan. The value b of the fitted Weibull function, which is an indicator of the mechanism of drug transport through the polymer matrix, was found to be 1.8 for acetylsalicylic acid, suggesting a complex release mechanism and 0.8 for acetaminophen, suggesting a combined mechanism (Fickian diffusion and case II transport).56
Conclusions
In summary, we present a novel self-assembly mechanism for hexagonal β-1,3-glucan microparticles with defined lamellar nanostructure and potential application as a drug encapsulation matrix. The phosphorylase catalyzed enzymatic synthesis can be successfully carried out at temperatures ranging from 25 to 80 °C. At optimized temperatures (above 37 °C), β-1,3-glucans are assembled into well-ordered stacked hexagonal lamellar sheets with 8 nm thickness and triple-helical crystal structure. Optimally, the stacks form facetted paracrystalline particles with a particle diameter of approximately 10 μm. This type of assembly has not been previously reported, and it further highlights the untapped potential of using phosphorylases for glucan synthesis.We believe the produced microparticles to have many use cases in a wide range of different applications due to their large surface area, well-defined structure, and a large amount of space between the layers. Furthermore, the materials composed of these well-ordered β-1,3-glucan microparticles are chiral which could facilitate their use in separation of small molecules based on their chirality.
Author contributions
R. P., P. M., V. L., W. P., G. B., J. V. I. T., and M. P. have contributed to this research by designing the experiments and interpreting the results. All authors read and approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Jenny and Antti Wihuri Foundation (Centre for Young Synbio Scientists) and Academy of Finland project 348628. The computational part of the study was supported by the National Science Centre, Poland (contract financed in 2020–2024 under no. 2019/35/B/ST4/01149 OPUS 18). We thank Ernesto Scoppola, Chenghao Li and Wolfgang Wagermaier from the Max Planck Institute of Colloids and Interfaces in Potsdam, Germany for granting synchrotron beamtime and measurements at the μSpot beamline at BESSY at the Helmholtz-Zentrum Berlin für Materialien und Energie in Berlin, Germany. We acknowledge the provision of facilities and technical support by Aalto University at the OtaNano Nanomicroscopy Center (Aalto-NMC). We thank Atte Mikkelson for SEC measurements and Heidi Salminen for MALDI-ToF-MS experiments. We thank Jorg de Ruijter for critical comments on the manuscript.References
- M. J. Gidley and K. Nishinari, in Chemistry, Biochemistry, and Biology of 1-3 Beta Glucans and Related Polysaccharides, Elsevier, 2009, pp. 47–118 Search PubMed.
- A. L. Rae, P. J. Harris, A. Bacic and A. E. Clarke, Planta, 1985, 166, 128–133 CrossRef CAS PubMed.
- H. R. Huwyler, G. Franz and H. Meier, Plant Sci. Lett., 1978, 12, 55–62 CrossRef CAS.
- T. Harada, A. Misaki and H. Saito, Arch. Biochem. Biophys., 1968, 124, 292–298 CrossRef CAS PubMed.
- M. Portilho, G. Matioli, G. Zanin, F. F. de Moraes and A. R. P. Scamparini, Appl. Biochem. Biotechnol., 2006, 131, 864–869 CrossRef PubMed.
- P. S. Saudagar and R. S. Singhal, Appl. Biochem. Biotechnol., 2004, 118, 021–032 CrossRef CAS.
- S. Shivakumar and S. V. N. Vijayendra, Lett. Appl. Microbiol., 2006, 42, 477–482 CrossRef CAS PubMed.
- S. K. Ghai, M. Hisamatsu, A. Amemura and T. Harada, Microbiology, 1981, 122, 33–40 CAS.
- C. S. Buller and K. C. Voepel, J. Ind. Microbiol., 1990, 5, 139–145 CrossRef CAS.
- W. J. Kenyon and C. S. Buller, J. Ind. Microbiol. Biotechnol., 2002, 29, 200–203 CrossRef CAS PubMed.
- W. J. Kenyon, S. W. Esch and C. S. Buller, Antonie Van Leeuwenhoek, 2005, 87, 143–148 CrossRef CAS PubMed.
- S. N. Gummadi and K. Kumar, Biotechnol. Bioprocess Eng., 2005, 10, 546–551 CrossRef CAS.
- A. Archibald, W. Cunningham, D. Manners, J. Stark and J. Ryley, Biochem. J., 1963, 88, 444–451 CrossRef CAS PubMed.
- D. R. Kreger and J. van der Veer, Acta Bot. Neerl., 1970, 19, 401–402 CrossRef.
- J. Z. Kiss, A. C. Vasconcelos and R. E. Triemer, J. Phycol., 1988, 24, 152–157 CAS.
- J. Z. Kiss, A. C. Vasconcelos and R. E. Triemer, Am. J. Bot., 1987, 74, 877 CrossRef CAS.
- A. E. Clarke and B. A. Stone, Biochim. Biophys. Acta, 1960, 44, 161–163 CrossRef CAS.
- C. T. Chuah, A. Sarko, Y. Deslandes and R. H. Marchessault, Macromolecules, 1983, 16, 1375–1382 CrossRef CAS.
- Y. Deslandes, R. H. Marchessault and A. Sarko, Macromolecules, 1980, 13, 1466–1471 CrossRef CAS.
- K. Okuyama, A. Otsubo, Y. Fukuzawa, M. Ozawa, T. Harada and N. Kasai, J. Carbohydr. Chem., 1991, 10, 645–656 CrossRef CAS.
- M. Guberman and P. H. Seeberger, J. Am. Chem. Soc., 2019, 141, 5581–5592 CrossRef CAS PubMed.
- F. Jamois, V. Ferrières, J.-P. Guégan, J.-C. Yvin, D. Plusquellec and V. Vetvicka, Glycobiology, 2005, 15, 393–407 CrossRef CAS PubMed.
- H. He, F. Yang and Y. Du, Carbohydr. Res., 2002, 337, 1673–1678 CrossRef CAS PubMed.
- L. Pelosi, T. Imai, H. Chanzy, L. Heux, E. Buhler and V. Bulone, Biochemistry, 2003, 42, 6264–6274 CrossRef CAS PubMed.
- M. Hrmova, T. Imai, S. J. Rutten, J. K. Fairweather, L. Pelosi, V. Bulone, H. Driguez and G. B. Fincher, J. Biol. Chem., 2002, 277, 30102–30111 CrossRef CAS PubMed.
- G. S. Bulmer, P. de Andrade, R. A. Field and J. M. van Munster, Carbohydr. Res., 2021, 508, 108411 CrossRef CAS PubMed.
- H. Nakai, M. Kitaoka, B. Svensson and K. Ohtsubo, Curr. Opin. Chem. Biol., 2013, 17, 301–309 CrossRef CAS PubMed.
- R. Pylkkänen, P. Mohammadi, S. Arola, J. C. de Ruijter, N. Sunagawa, K. Igarashi and M. Penttilä, Biomacromolecules, 2020, 21, 4355–4364 CrossRef PubMed.
- C. Goedl, T. Sawangwan, M. Mueller, A. Schwarz and B. Nidetzky, Angew. Chem., Int. Ed., 2008, 47, 10086–10089 CrossRef CAS PubMed.
- M. Nishimoto and M. Kitaoka, Biosci. Biotechnol. Biochem., 2007, 71, 2101–2104 CrossRef CAS PubMed.
- S. Kuhaudomlarp, G. Pergolizzi, N. J. Patron, B. Henrissat and R. A. Field, J. Biol. Chem., 2019, 294, 6483–6493 CrossRef CAS PubMed.
- Y. Wu, G. Mao, H. Fan, A. Song, Y.-H. P. Zhang and H. Chen, Sci. Rep., 2017, 7, 4849 CrossRef PubMed.
- H. Zhang, K. Nishinari, M. A. K. Williams, T. J. Foster and I. T. Norton, Int. J. Biol. Macromol., 2002, 30, 7–16 CrossRef CAS PubMed.
- T. Funami and K. Nishinari, Food Hydrocolloids, 2007, 21, 59–65 CrossRef CAS.
- T. Funami, M. Funami, H. Yada and Y. Nakao, Food Hydrocolloids, 2000, 14, 509–518 CrossRef CAS.
- T. Funami, M. Funami, H. Yada and Y. Nakao, Food Hydrocolloids, 1999, 13, 317–324 CrossRef CAS.
- T. Funami, M. Unami, H. Yada and Y. Nakao, J. Food Sci., 1999, 64, 129–132 CrossRef CAS.
- T. Harada, A. Koreeda, S. Sato and N. Kasai, Microscopy, 1979, 28, 147–153 CrossRef CAS.
- Y. Kanzawa, T. Harada, A. Koreeda and A. Harada, Agric. Biol. Chem., 1987, 51, 1839–1843 CAS.
- Y. Kanzawa, T. Harada, A. Koreeda, A. Harada and K. Okuyama, Carbohydr. Polym., 1989, 10, 299–313 CrossRef CAS.
- M. Watase and K. Nishinari, in Food Hydrocolloids, Springer US, Boston, MA, 1994, pp. 125–129 Search PubMed.
- T. Harada, K. Okuyama, A. Konno, A. Koreeda and A. Harada, Carbohydr. Polym., 1994, 24, 101–106 CrossRef CAS.
- B. Nidetzky and C. Zhong, Biotechnol. Adv., 2021, 51, 107633 CrossRef CAS PubMed.
- Z. Ubiparip, M. de Doncker, K. Beerens, J. Franceus and T. Desmet, Appl. Microbiol. Biotechnol., 2021, 105, 4073–4087 CrossRef CAS PubMed.
- Y. Wu, G. Mao, H. Fan, A. Song, Y. H. P. Zhang and H. Chen, Sci. Rep., 2017, 7, 1–12 CrossRef PubMed.
- H. Saitô, E. Miyata and T. Sasaki, Macromolecules, 1978, 11, 1244–1251 CrossRef.
- B. Stone and A. E. Clarke, Chemistry, Biochemistry, and Biology of 1–3 Beta Glucans and Related Polysaccharides, Elsevier, 2009 Search PubMed.
- A. G. Shtukenberg, Z. Zhu, Z. An, M. Bhandari, P. Song, B. Kahr and M. D. Ward, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17195–17198 CrossRef CAS PubMed.
- S. A. Morin, A. Forticaux, M. J. Bierman and S. Jin, Nano Lett., 2011, 11, 4449–4455 CrossRef CAS PubMed.
- F. Meng, S. A. Morin, A. Forticaux and S. Jin, Acc. Chem. Res., 2013, 46, 1616–1626 CrossRef CAS PubMed.
- M. Bergström, J. S. Pedersen, P. Schurtenberger and S. U. Egelhaaf, J. Phys. Chem. B, 1999, 103, 9888–9897 CrossRef.
- T. Harada, A. Koreeda, S. Sato and N. Kasai, Microscopy, 1979, 28, 147–153 CrossRef CAS.
- H. Chanzy and R. Vuong, in Polysaccharides, Palgrave Macmillan UK, London, 1985, pp. 41–71 Search PubMed.
- Y. Ogawa, K. Noda, S. Kimura, M. Kitaoka and M. Wada, Int. J. Biol. Macromol., 2014, 64, 415–419 CrossRef CAS PubMed.
- S. Nyol and M. M. Gupta, J. Drug Delivery Ther., 2013, 3, 155–161 CAS.
- V. Papadopoulou, K. Kosmidis, M. Vlachou and P. Macheras, Int. J. Pharm., 2006, 309, 44–50 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr02731c |
| This journal is © The Royal Society of Chemistry 2022 |