
Red-emissive nanocrystals of Cs4MnxCd1−xSb2Cl12 layered perovskites†
Emanuela
Sartori
 ab,
Marta
Campolucci
ab,
Dmitry
Baranov
b,
Min
Zeng
cd,
Stefano
Toso
be,
Giorgio
Divitini
f,
Maurizio
Ferretti
a,
Zeger
Hens
d,
Liberato
Manna
b and
Federico
Locardi
*abd
ab,
Marta
Campolucci
ab,
Dmitry
Baranov
b,
Min
Zeng
cd,
Stefano
Toso
be,
Giorgio
Divitini
f,
Maurizio
Ferretti
a,
Zeger
Hens
d,
Liberato
Manna
b and
Federico
Locardi
*abd
aDepartment of Chemistry and Industrial Chemistry, Università degli Studi di Genova, Via Dodecaneso 31, 16146 Genova, Italy. E-mail: federico.locardi@unige.it
bNanochemistry Department, Istituto Italiano di Tecnologia, Via Morego 30, 16163, Italy
cHubei Key Laboratory of Ferro- & Piezoelectric Materials and Devices, Faculty of Physics and Electronic Science, Hubei University, Wuhan, 430062, P. R. China
dPhysics and Chemistry of Nanostructures group (PCN), Ghent University, Krijgslaan 281, Gent 9000, Belgium
eInternational Doctoral Program in Science, Università Cattolica del Sacro Cuore, 25121 Brescia, Italy
fElectron Spectroscopy and Nanoscopy, Istituto Italiano di Tecnologia, Via Morego 30, 16163, Italy
First published on 30th November 2021
Abstract
Layered double perovskites are currently being investigated as emerging halide-based materials for optoelectronic applications. Herein, we present the synthesis of Cs4MnxCd1−xSb2Cl12 (0 ≤ x ≤ 1) nanocrystals (NCs). X-ray powder diffraction evidences the retention of the same crystal structure for all the inspected compositions; transmission electron microscopy revealed monodisperse particles with a mean size between 10.7 nm and 12.7 nm. The absorption spectra are mostly determined by transitions related to Sb3+, whereas Mn2+ induced a red emission in the 625–650 nm range. The photoluminescence emission intensity and position vary with the Mn2+ content and reach the maximum for the composition with x = 0.12. Finally, we demonstrate that the photoluminescence quantum yield of the latter NCs was increased from 0.3% to 3.9% through a post-synthesis treatment with ammonium thiocyanate. The present work expands the knowledge of colloidal layered double perovskite nanocrystals, stimulating future investigations of this emerging class of materials.
 Federico Locardi | Dr Federico Locardi received his PhD in Chemical Science from the University of Genoa (Italy), in 2015. He completed his first postdoctoral studies at the University of Genoa and Nanochemistry Department of IIT (Istituto Italiano di Tecnologia), focusing on the synthesis of halide perovskite nanocrystals. In 2019, he moved to the Physics and Chemistry of Nanostructures group at Ghent University (Belgium); after one year, he returned to the University of Genoa, where he is an assistant professor at the Department of Chemistry and Industrial Chemistry. His research focuses on the development of new inorganic (nano)materials for photocatalysis and optoelectronics. |
Introduction
In recent years, extensive research on lead halide perovkistes1 has led to increasing interest in new metal halides with a wide variety of structures and compositions based on different transition metals.2 In particular, the concerns about the toxicity and limited stability of lead-based compositions have inspired researchers to explore alternatives, with comparable or, ideally, even better optoelectronic properties.3,4 Double perovskites (DPs) represent one of the first attempts towards lead substitution. In DPs, the original 3D structure of lead halides, i.e. a framework composed of corner-sharing [PbX6] (X = Cl−, Br−, I−) octahedra, is retained by the alternation of octahedra formed by monovalent and trivalent cations.5 In this way, charge neutrality is maintained, as expressed through the general chemical formula A2M+M3+X6 (M = metal, X = Cl−, Br−, I−). Several combinations of M+ and M3+ cations have been tested, both in bulk and in nanocrystals (NCs), for example M+ = Ag+, Na+, K+ and M3+ = In3+, Bi3+, Sb3+.6–12 In this kind of structure, the need to maintain the charge balance and the constraints imposed by the ionic radii make the introduction of bivalent metal cations (M2+) generally possible only at low concentrations.12–14Complete substitution of the M+ cations with M2+ cations is possible with the concomitant introduction of a vacancy (Vac), resulting in a general A4M2+M3+2X12 stoichiometry; these materials, due to their peculiar structure, are commonly known as 〈111〉 oriented layered double perovskites (LDPs).2 Indeed, the structure can be described as a cubic substructure of Cs+ cations15 filled alternatively by layers of corner-sharing [M2+X6]–[M3+X6]–[M2+X6]–[VacX6] octahedra along the 〈111〉 direction. The first LDP, Cs4CuSb2Cl12, was synthesized as a bulk material by Solis-Ibarra's group in 2017 and had an optical bandgap of 1 eV, which is promising for photovoltaics;16 Cai et al. prepared the same composition as NCs, with optical properties in line with those of the corresponding powders.17 The partial substitution of Cu2+ with Mn2+ tunes the band gap to higher energy, and a low-intensity red emission was measured from Cs4MnSb2Cl12.18 A more efficient emission has been observed with the partial introduction of Cd2+,19 leading to a photoluminescence quantum yield (PLQY) of 37.5% for Cs4Cd0.75Mn0.25Sb2Cl12.20 The replacement of Sb3+ with Bi3+ resulted in an increase of the PLQY21 up to 79.5% for the Cs4Cd1−xMnxBi2Cl12 (x = 0.1) composition.22 Yang et al. successfully synthesized the same series in the form of nanocrystals; however, the resulting materials exhibited a 20-fold drop of the PLQY (4.6%) compared to bulk crystals.23
LDPs have been proposed as optoelectronic materials for LEDs2,19 and high-speed photodetection20,24 due to the strong anisotropy in their crystal structure. Moreover, preliminary studies have been conducted to investigate their magnetic behavior for potential applications in ferroelectrics and spintronics.2,25
Motivated by the aforementioned studies and by the need to expand the range of the available stoichiometries of LDPs at the nanoscale, we devised a synthesis of Cs4Cd1−xMnxSb2Cl12 in the form of NCs. Using a hot injection approach, we found that alloying Cd2+ and Mn2+ is possible (that is, x in the above formula can range from 0 to 1) with the formation of phase-pure cuboidal nanoparticles. The optical characterization has revealed a red emission centered at 625–650 nm for all the samples, except for x = 0, with the emission position and intensity depending on the Mn2+ content. The sample with x = 0.12 yielded the most intense emission of the series, with a PLQY of 0.3% that was further enhanced ten-fold to 3.9% by means of surface treatment with ammonium thiocyanate. Overall, our work expands the knowledge of the layered double perovskite at the nanoscale, highlighting the potential of these compounds as optoelectronic materials.
Results and discussion
We prepared Cs4MnxCd1−xSb2Cl12 NCs by a hot-injection method. Briefly, the synthesis consisted of degassing the precursors (metal acetates and carbonates) under vacuum in a mixture of dioctyl ether, oleylamine, and oleic acid for 20 minutes at 115 °C, followed by an injection of benzoyl chloride at 160 °C under a N2 atmosphere (Scheme 1; see the ESI† for details). The resulting nanoparticles had a cuboidal shape for all the inspected samples, with a Mn2+ content tunable by systematically changing the Mn2+/(Cd2+ + Mn2+) precursor ratio from 0 to 1. The ICP-OES analysis revealed that, in general, an amount of Mn2+ lower than the one loaded actually enters the structure, especially when the Mn2+/(Cd2+ + Mn2+) ratio of the precursors is lower than 0.5 (Table S1 and Fig. S1†); a similar phenomenon was observed in Cs4MnxCd1−xBi2Cl12.21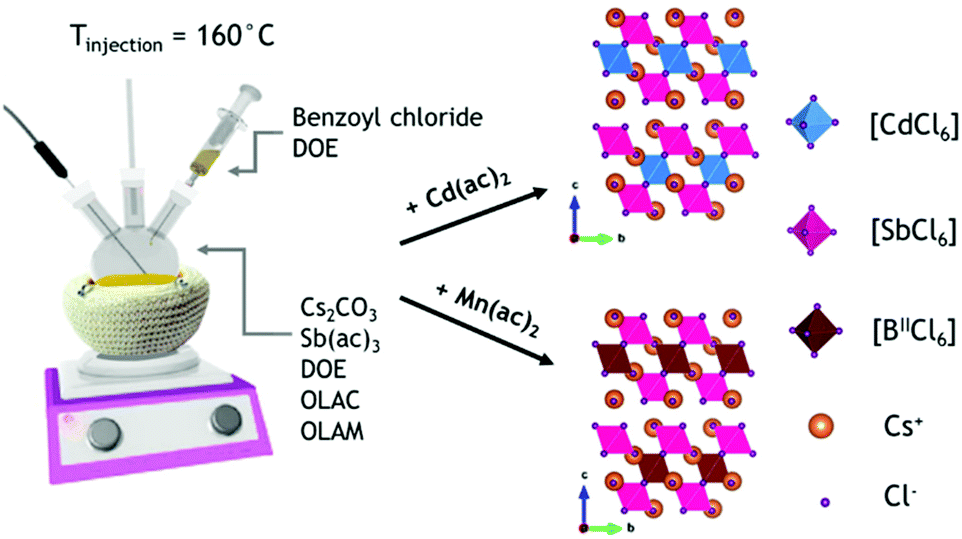 | ||
| Scheme 1 Colloidal synthesis of Cs2MnxCd1−xSb2Cl12 (0 ≤ x ≤ 1) NCs. | ||
The powder X-ray diffraction (XRD) data indicated that all the synthesized compositions crystallize always in the trigonal R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m structure (Fig. 1a and S2†) and the samples with intermediate x were phase-pure and isostructural to the terminal compositions (x = 0 and 1, Fig. 1b and S2†). The shape of nanocrystals reflected the influence of the cuboidal Cs+ substructure, as commonly observed for other metal–halide perovskite nanocrystals.15 The slight shift of the diffraction peaks toward higher 2θ angles (Fig. 1b right) originates from the contraction of the unit cell due to the substitution of the Cd2+ cations with the smaller Mn2+ cations. Indeed, the cell parameters shifted from a = 7.586 Å, c = 37.165 Å to a = 7.549 Å, c = 36.994 Å for 0% Mn2+ and 100% Mn2+, respectively. The unit cell contracted by 1.44% in volume, and the contraction was found to be isotropic (a = −0.49%; c = −0.46%), suggesting a random Mn2+/Cd2+ replacement in the structure (Fig. S3†). These results agree with the data reported on the corresponding powders.19 Transmission electron microscopy (TEM) micrographs of Cs4MnxCd1−xSb2Cl12 NCs evidenced the formation of NCs with a cuboidal shape (Fig. 1c–f), and a mean size of 10.7 ± 3.5 nm and 12.4 ± 3.8 nm for 0% and 100% Mn2+, respectively; for the larger Mn2+ fractions (x = 0.9, 1) smoothing of the NC edges occurred (Fig. 1f and S4†). High-angle annular dark-field (HAADF) scanning TEM imaging confirmed the crystallinity of the NCs (Fig. 1g and S5†), most often composed of a single crystal domain; occasionally, multiple domains were observed in particles larger than 20 nm (Fig. S5†). Elemental analysis confirmed that all the expected elements are included in the NCs (Fig. S6†). Upon changing the injection temperature (i.e. 140–150–170 °C), we observed no changes in the crystal structure (Fig. S7a†), yet higher temperatures led to NC samples with a more polydisperse distribution of size and morphology (Fig. S7b–e†).
m structure (Fig. 1a and S2†) and the samples with intermediate x were phase-pure and isostructural to the terminal compositions (x = 0 and 1, Fig. 1b and S2†). The shape of nanocrystals reflected the influence of the cuboidal Cs+ substructure, as commonly observed for other metal–halide perovskite nanocrystals.15 The slight shift of the diffraction peaks toward higher 2θ angles (Fig. 1b right) originates from the contraction of the unit cell due to the substitution of the Cd2+ cations with the smaller Mn2+ cations. Indeed, the cell parameters shifted from a = 7.586 Å, c = 37.165 Å to a = 7.549 Å, c = 36.994 Å for 0% Mn2+ and 100% Mn2+, respectively. The unit cell contracted by 1.44% in volume, and the contraction was found to be isotropic (a = −0.49%; c = −0.46%), suggesting a random Mn2+/Cd2+ replacement in the structure (Fig. S3†). These results agree with the data reported on the corresponding powders.19 Transmission electron microscopy (TEM) micrographs of Cs4MnxCd1−xSb2Cl12 NCs evidenced the formation of NCs with a cuboidal shape (Fig. 1c–f), and a mean size of 10.7 ± 3.5 nm and 12.4 ± 3.8 nm for 0% and 100% Mn2+, respectively; for the larger Mn2+ fractions (x = 0.9, 1) smoothing of the NC edges occurred (Fig. 1f and S4†). High-angle annular dark-field (HAADF) scanning TEM imaging confirmed the crystallinity of the NCs (Fig. 1g and S5†), most often composed of a single crystal domain; occasionally, multiple domains were observed in particles larger than 20 nm (Fig. S5†). Elemental analysis confirmed that all the expected elements are included in the NCs (Fig. S6†). Upon changing the injection temperature (i.e. 140–150–170 °C), we observed no changes in the crystal structure (Fig. S7a†), yet higher temperatures led to NC samples with a more polydisperse distribution of size and morphology (Fig. S7b–e†).
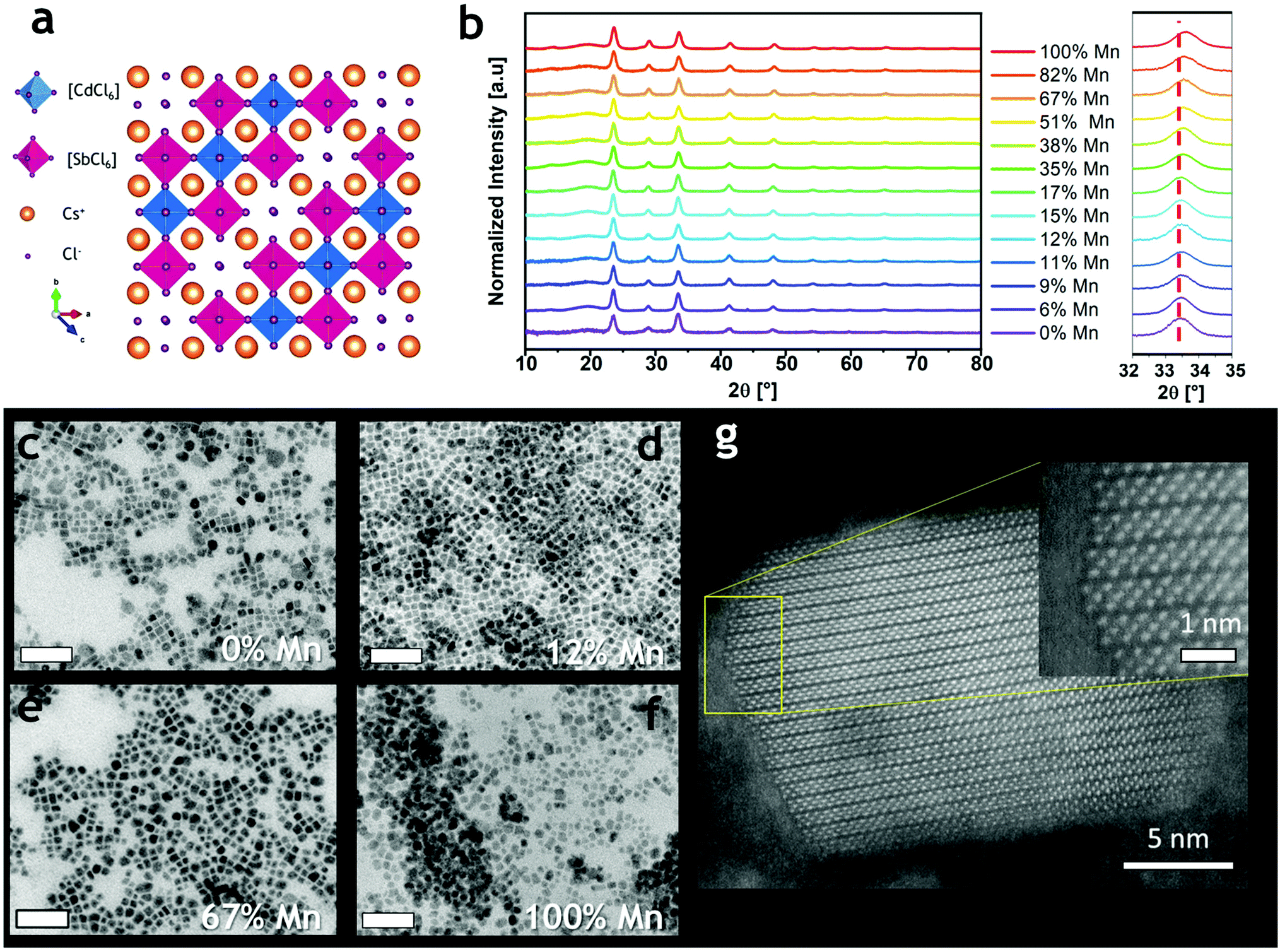 | ||
| Fig. 1 (a) Crystal structure of Cs4CdSb2Cl12; (b) XRD patterns of Cs4MnxCd1−xSb2Cl12 NCs; a shift to higher angles on increasing the Mn2+ amount was observed (right); (c–f) bright-field TEM micrographs of Cs4MnxCd1−xSb2Cl12 NCs. Scale bars are 100 nm; (g) HAADF-STEM image of a single nanocrystal of the Cs4Mn0.12Cd0.88Sb2Cl12 sample. | ||
All samples displayed an absorption spectrum characterized by three different features: two broad peaks between 300 nm and 400 nm (Fig. 2a and S8a†) that were independent of the Mn2+/(Cd2+ + Mn2+) ratio, and a well-defined peak at 267 nm (for x = 0) whose intensity decreased with a concomitant blue shift with the increasing Mn2+ amount (Fig. S8a†). The observed behavior is consistent with the corresponding bulk material except for the intensity variation and shift of the last spectral feature (250–300 nm), which was previously not observed by Solis-Ibarra's group.19 Overall, the absorption spectrum of Cs4MnxCd1−xSb2Cl12 seems to be mostly determined by transitions related to Sb3+, not unlike previous observations on Cs3Sb2Cl9.26 Sb3+ is an ns2 ion characterized by the 1S0 ground state and four higher-energy levels denoted as 3P0, 3P1, 3P2 and 1P1.27 The absorption in the 300–400 nm range can be ascribed to the partially allowed 1S0 → 3P1 transition, commonly named the A band, split into a doublet; similarly, the allowed 1S0 → 1P1 transition (C band) is responsible for the absorption found at ∼270 nm.11,19,27,28 Instead, the d → d Mn2+ transition, generally observed for Mn-based materials between 400 nm and 550 nm, was not detected. All the Cs4MnxCd1−xSb2Cl12 compositions, except for x = 0, showed a red emission centered at around 625–650 nm under UV excitation (Fig. S8b†). The photoluminescence excitation (PLE) spectra exhibited an intense peak centered at ∼325 nm with a shoulder at ∼285 nm (Fig. 2b). The PLE behavior resembled that of absorption, indicating that the excitation occurs through the Sb3+ bands. Interestingly, even if Sb3+ is known for exhibiting optical emission in metal halides,27,29 we did not detect any PL signals in Cs4CdSb2Cl12, whereas the 4T1 → 6A1 transition of Mn2+ induced the red emission in all the other samples. We explain this behavior by an energy transfer between antimony(III) and manganese(II) ions, Sb3+ → Mn2+.30 Indeed, the PLE profiles were the same for all the samples, with no considerable shift (Fig. S8c†), while significant differences were detected both in the PL intensity and positions (Fig. 2c, d and S8b†). No differences in the PL were observed when exciting the samples at a different wavelength (Fig. 2b). The intensity increased with the Mn2+ ratio (Fig. 2c) up to a maximum at the experimental composition of 11.5% Mn2+. Similar results were reported for the corresponding bulk system.19 By increasing the Mn2+ concentration the emission gradually shifted from 624 nm (6% Mn2+), a value higher than that in bulk Cs4MnxCd1−xSb2Cl12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 19,20 but close to reports for the Bi-based counterpart Cs2MnBi2Cl12,21,23 to a roughly constant spectral position of ∼648 nm for x > 0.5 (Fig. 2d). The Mn2+ emission shift was already reported in Cs4Mn(Bi1−xInx)2Cl1231 and CsPbCl3,32,33 and ascribed to a contraction of the unit cell; according to the Tanabe–Sugano diagram for the d5 ion (e.g. Mn2+), an increase of the crystal field induces a red shift in the 4T1 → 6A1 transition. Indeed, the progressive substitution of Cd2+ with the smaller Mn2+ decreased the cell volume by shrinking the [MnCl6] octahedra,19 thus inducing the observed red shift. The variation of the PL intensity can be instead explained considering that the maximum intensity is reached, in principle, when the MnCl6 octahedra are surrounded by [SbCl6] and [CdCl6] octahedra, thus, avoiding any Mn–Mn self-quenching.23
19,20 but close to reports for the Bi-based counterpart Cs2MnBi2Cl12,21,23 to a roughly constant spectral position of ∼648 nm for x > 0.5 (Fig. 2d). The Mn2+ emission shift was already reported in Cs4Mn(Bi1−xInx)2Cl1231 and CsPbCl3,32,33 and ascribed to a contraction of the unit cell; according to the Tanabe–Sugano diagram for the d5 ion (e.g. Mn2+), an increase of the crystal field induces a red shift in the 4T1 → 6A1 transition. Indeed, the progressive substitution of Cd2+ with the smaller Mn2+ decreased the cell volume by shrinking the [MnCl6] octahedra,19 thus inducing the observed red shift. The variation of the PL intensity can be instead explained considering that the maximum intensity is reached, in principle, when the MnCl6 octahedra are surrounded by [SbCl6] and [CdCl6] octahedra, thus, avoiding any Mn–Mn self-quenching.23
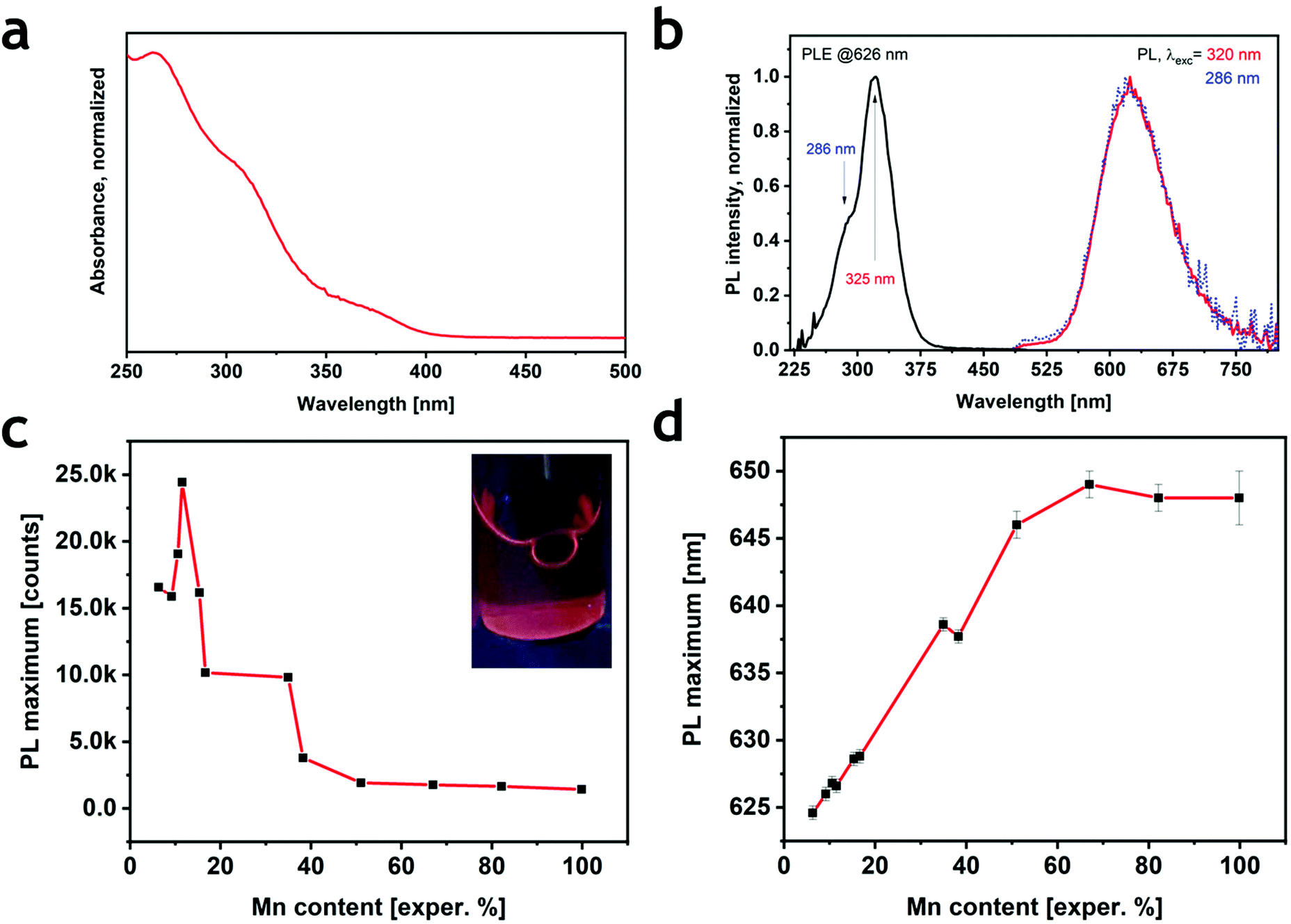 | ||
| Fig. 2 (a) Optical absorption at 250 nm and (b) PLE and PL spectra measured at two different excitation wavelengths (320 nm and 286 nm) of the Cs4Mn0.12Cd0.88Sb2Cl12 sample chosen as a representative. (c) PL intensity (normalized at the absorption at 320 nm) behavior at increasing Mn2+ content; the inset shows a photograph of the most emissive sample under UV light. (d) Position of the PL peak with increasing Mn2+ content. | ||
The PLQY of the most emissive colloidal sample was found to be 0.3%, a value that is much lower than that reported for the corresponding bulk material (28.5%).19 The decrease of PLQY with reduced particle size has been previously observed in Cs4MnxCd1−xBi2Cl12: when lowering the dimensions of the particles from micrometers to nanometers, the PLQY collapsed from 79.5%22 to 4.6%.23 This unfavorable effect, already documented in several metal–halide perovskites, can be ascribed to the formation of non-radiative relaxation paths induced by surface traps. To test this hypothesis, we attempted three different post-synthetic treatments to increase the PLQY of the nanocrystals: a small fraction of the nanocrystal suspension was added to (i) a mixture of CdCl2, OA and OlAm in hexane;32,34 (ii) a solution of didodecyldimethylammonium chloride (DDAC) in toluene;35 and (iii) a mixture of ammonium thiocyanate (NH4SCN) in hexane36,37 (see the ESI and Table S2†). The first two treatments slightly increased the photoluminescence, while a significant enhancement was observed when using NH4SCN (Fig. S9†); consequently, only the sample treated with NH4SCN was investigated. During the treatment, the sample emission kept increasing for one hour (Fig. S9†), after which it started decreasing due to the progressive decomposition of the sample, which completely evolved in other phases after three hours (Fig. S10†). The XRD and TEM analyses on the brightest sample (Fig. 3a) showed that the crystalline phase was preserved, even if a small amount of a secondary phase, identified as CsCl, was present. As determined by TEM, the morphology of the NCs was largely preserved, but cuboidal shapes appeared more rounded compared to the pristine sample (Fig. 3b and c). The absorption spectrum revealed a small decrease in the 350–400 nm range and an enhancement of the C band (λ < 300 nm) (Fig. 3d). Furthermore, the PLE spectrum narrowed without a significant shift (Fig. 3e). More importantly, an enhanced PL emission was observed, which resembled the same broadness of the pristine material with a small shift of ∼2 nm (Fig. 3f and S11†). The conservation of the PLE and PL spectral shapes and positions suggests that the post-treatment does not influence the environment around Mn2+ (i.e., the crystal structure); as previously demonstrated in other halide perovskites,36,38,39 the thiocyanate group passivates the surface sites removing the shallow trap and reducing the number of non-radiative relaxation channels. Indeed, the PLQY of the material after the post-synthesis treatment reached a value of 3.9% (Fig. S12†), which is 10 times higher than that in the as-synthesized particles, and comparable to that of the Bi-based counterpart.
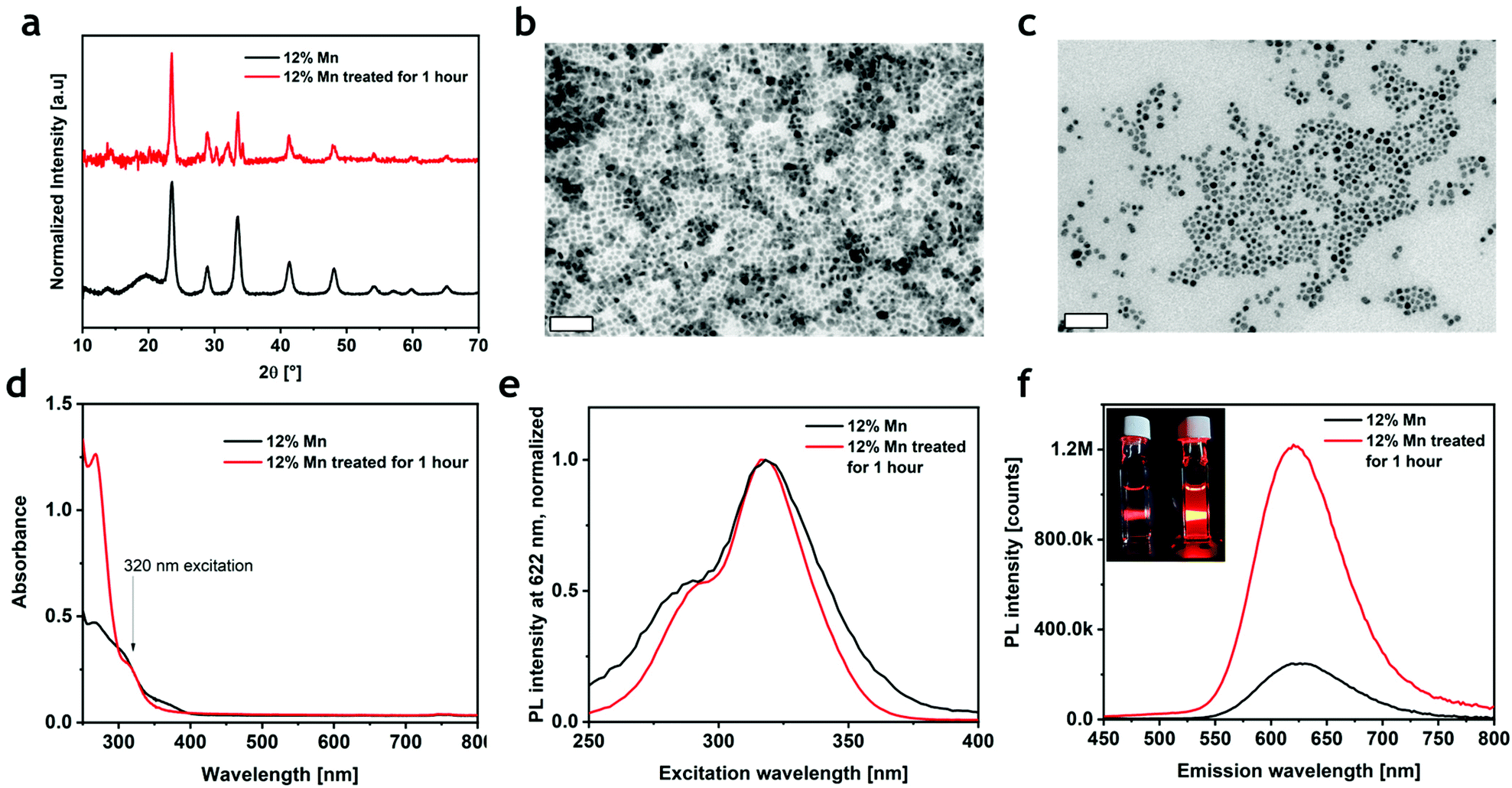 | ||
| Fig. 3 (a) XRD pattern of Cs4Mn0.12Cd0.88Sb2Cl12 before (black line) and after the thiocyanate treatment (red line). (b) and (c) bright-field TEM images before and after the thiocyanate treatment, respectively. (d) ABS, (e) PLE, and (f) PL of Cs4Mn0.12Cd0.88Sb2Cl12 before (black line) and after thiocyanate treatment (red line). Inset in (f) shows a photograph under a UV lamp of the sample with (bright orange) and without the treatment (dark orange). | ||
Conclusions
In summary, we successfully synthesized Cs4MnxCd1−xSb2Cl12 nanocrystals with tunable composition in the x = 0–1 range. All the samples showed a UV absorption and a red emission stemming from Sb3+ and Mn2+ ions (except for the Mn-free x = 0), respectively. The PL intensity changed with the Mn2+ amount, reaching a maximum for x = 0.12. The pristine sample featured a low PLQY (<1%) that was increased to 3.9% by a post-synthesis treatment with ammonium thiocyanate. The intriguing optical properties of Cs4MnxCd1−xSb2Cl12 nanocrystals should stimulate additional studies on this emerging family of layered double perovskites and their application as optoelectronic materials.Author contributions
E.S. – conceptualization, data curation, formal analysis, investigation (synthesis, XRD, TEM, ICP-OES, and optical analysis), validation, visualization (figures) and writing (original draft); M.C. – data curation, formal analysis, investigation (synthesis, XRD, TEM, and optical analysis), and validation; D.B. – data curation, formal analysis, investigation (optical analysis), and writing (editing); M.Z. – data curation, formal analysis, and investigation (XRD, optical analysis); S.T. – formal analysis and investigation (XRD); G.D. – investigation (HAADF – STEM); M.F. – conceptualization, supervision, and writing (review and editing); Z.H. – conceptualization, supervision, and writing (review and editing); L.M. – conceptualization, supervision, and writing (review and editing); and F.L. – conceptualization, formal analysis, visualization (figures), supervision, and writing (original draft, review and editing). Contributions are assigned using CRediT taxonomy (https://casrai.org/credit/).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Materials Characterization Facility and Electron Microscopy Facility at the Fondazione Istituto Italiano di Tecnologia for use of XRD and TEM equipment and technical support and Filippo Drago of Nanochemistry Facility for elemental analysis using ICP-OES. Z. H. acknowledges Ghent University (GOA 01G01019) and FWO-Vlaanderen (SBO Proceed) for research funding. E. S., M. C. and F. L. thank Matilde Cirignano for the fruitful suggestions.Notes and references
- Q. A. Akkerman, G. Rainò, M. V. Kovalenko and L. Manna, Nat. Mater., 2018, 17, 1–12 CrossRef PubMed.
- B. Vargas, G. Rodrĺguez-López and D. Solis-Ibarra, ACS Energy Lett., 2020, 5, 3591–3608 CrossRef CAS.
- W. Ke and M. G. Kanatzidis, Nat. Commun., 2019, 10, 1–4 CrossRef CAS PubMed.
- X. Li, X. Gao, X. Zhang, X. Shen, M. Lu, J. Wu, Z. Shi, V. L. Colvin, J. Hu, X. Bai, W. W. Yu and Y. Zhang, Adv. Sci., 2021, 8, 1–33 Search PubMed.
- F. Giustino and H. J. Snaith, ACS Energy Lett., 2016, 1, 1233–1240 CrossRef CAS.
- J. Luo, X. Wang, S. Li, J. Liu, Y. Guo, G. Niu, L. Yao, Y. Fu, L. Gao, Q. Dong, C. Zhao, M. Leng, F. Ma, W. Liang, L. Wang, S. Jin, J. Han, L. Zhang, J. Etheridge, J. Wang, Y. Yan, E. H. Sargent and J. Tang, Nature, 2018, 563, 541–545 CrossRef CAS PubMed.
- F. Locardi, E. Sartori, J. Buha, J. Zito, M. Prato, V. Pinchetti, M. L. Zaffalon, M. Ferretti, S. Brovelli, I. Infante, L. De Trizio and L. Manna, ACS Energy Lett., 2019, 4, 1976–1982 CrossRef CAS.
- N. R. Wolf, B. A. Connor, A. H. Slavney and H. I. Karunadasa, Angew. Chem., Int. Ed., 2021, 60, 16264–16278 CrossRef CAS PubMed.
- G. Volonakis, A. A. Haghighirad, R. L. Milot, W. H. Sio, M. R. Filip, B. Wenger, M. B. Johnston, L. M. Herz, H. J. Snaith and F. Giustino, J. Phys. Chem. Lett., 2017, 8, 772–778 CrossRef CAS PubMed.
- S. E. Creutz, E. N. Crites, M. C. De Siena and D. R. Gamelin, Nano Lett., 2018, 18, 1118–1123 CrossRef CAS PubMed.
- A. Noculak, A. Noculak, V. Morad, V. Morad, K. M. McCall, S. Yakunin, Y. Shynkarenko, S. Yakunin, Y. Shynkarenko, M. Wörle and M. V. Kovalenko, Chem. Mater., 2020, 32, 5118–5124 CrossRef CAS PubMed.
- F. Locardi, M. Cirignano, D. Baranov, Z. Dang, M. Prato, F. Drago, M. Ferretti, V. Pinchetti, M. Fanciulli, S. Brovelli, L. De Trizio and L. Manna, J. Am. Chem. Soc., 2018, 140, 12989–12995 CrossRef CAS PubMed.
- Q. Liao, J. Chen, L. Zhou, T. Wei, L. Zhang, D. Chen, F. Huang, Q. Pang and J. Z. Zhang, J. Phys. Chem. Lett., 2020, 11, 8392–8398 CrossRef CAS PubMed.
- N. K. Nila and N. Angshuman, Chem. Commun., 2018, 54, 5205–5208 RSC.
- S. Toso, D. Baranov and L. Manna, ACS Energy Lett., 2020, 5, 3409–3414 CrossRef CAS.
- B. Vargas, E. Ramos, E. Pérez-Gutiérrez, J. C. Alonso and D. Solis-Ibarra, J. Am. Chem. Soc., 2017, 139, 9116–9119 CrossRef CAS PubMed.
- T. Cai, W. Shi, S. Hwang, K. Kobbekaduwa, Y. Nagaoka, H. Yang, K. Hills-Kimball, H. Zhu, J. Wang, Z. Wang, Y. Liu, D. Su, J. Gao and O. Chen, J. Am. Chem. Soc., 2020, 142, 11927–11936 CrossRef CAS PubMed.
- B. Vargas, R. Torres-Cadena, J. Rodríguez-Hernández, M. Gembicky, H. Xie, J. Jiménez-Mier, Y. S. Liu, E. Menéndez-Proupin, K. R. Dunbar, N. Lopez, P. Olalde-Velasco and D. Solis-Ibarra, Chem. Mater., 2018, 30, 5315–5321 CrossRef CAS.
- B. Vargas, E. Coutiño-Gonzalez, O. Ovalle-Encinia, C. Sánchez-Aké and D. Solis-Ibarra, J. Phys. Chem. Lett., 2020, 11, 10362–10367 CrossRef CAS PubMed.
- C. Peng, Q. Wei, L. Chen, R. Zeng, Q. Zhang, Q. Hu and B. Zou, J. Mater. Chem. C, 2021, 9, 15522 RSC.
- N. P. Holzapfel, J. D. Majher, T. A. Strom, C. E. Moore and P. M. Woodward, Chem. Mater., 2020, 32, 3510–3516 CrossRef CAS.
- B. Vargas, D. T. Reyes-Castillo, E. Coutino-Gonzalez, C. Sánchez-Aké, C. Ramos, C. Falcony and D. Solis-Ibarra, Chem. Mater., 2020, 32, 9307–9315 CrossRef CAS.
- H. Yang, W. Shi, T. Cai, K. Hills-Kimball, Z. Liu, L. Dube and O. Chen, Nanoscale, 2020, 12, 23191–23199 RSC.
- T. Cai, W. Shi, D. J. Gosztol, K. Kobbekaduwa, H. Yang, N. Jin, Y. Nagaoka, L. Dube, J. Schneider, S. Hwang, J. Gao, X. Ma and O. Chen, Matter, 2021, 4, 2936–2295 CrossRef.
- T. T. Tran, T. T. Tran, C. A. Pocs, Y. Zhang, Y. Zhang, M. J. Winiarski, M. J. Winiarski, J. Sun, M. Lee, T. M. McQueen and T. M. McQueen, Phys. Rev. B, 2020, 101, 235107 CrossRef CAS.
- B. Pradhan, G. S. Kumar, S. Sain, A. Dalui, U. K. Ghorai, S. K. Pradhan and S. Acharya, Chem. Mater., 2018, 30, 2135–2142 CrossRef CAS.
- Y. Jing, Y. Liu, M. Li and Z. Xia, Adv. Opt. Mater., 2021, 9, 1–15 Search PubMed.
- F. Locardi, M. Samoli, A. Martinelli, O. Erdem, D. V. Magalhaes, S. Bals and Z. Hens, ACS Nano, 2021, 15, 17729–17737 CrossRef CAS PubMed.
- H. Arfin, A. S. Kshirsagar, J. Kaur, B. Mondal, Z. Xia, S. Chakraborty and A. Nag, Chem. Mater., 2020, 32, 10255–10267 CrossRef CAS.
- X. Liu, X. Xu, B. Li, L. Yang, Q. Li, H. Jiang and D. Xu, Small, 2020, 16, 1–7 Search PubMed.
- S. He, S. Fang, T. Han, T. Lang, M. Cai, H. You, L. Peng, S. Cao, B. Liu, Q. Qiang, J. Chen and B. Lei, J. Phys. Chem. C, 2021, 125(31), 16938–16945 CrossRef CAS.
- S. Ji, X. Yuan, S. Cao, W. Ji, H. Zhang, Y. Wang, H. Li, J. Zhao and B. Zou, J. Phys. Chem. Lett., 2020, 11, 2142–2149 CrossRef CAS PubMed.
- H. Liu, Z. Wu, J. Shao, D. Yao, H. Gao, Y. Liu, W. Yu, H. Zhang and B. Yang, ACS Nano, 2017, 11, 2239–2247 CrossRef CAS PubMed.
- N. Mondal, A. De and A. Samanta, ACS Energy Lett., 2019, 4, 32–39 CrossRef CAS.
- M. Imran, J. Ramade, F. Di Stasio, M. De Franco, J. Buha, S. Van Aert, L. Goldoni, S. Lauciello, M. Prato, I. Infante, S. Bals and L. Manna, Chem. Mater., 2020, 32, 10641–10652 CrossRef CAS PubMed.
- M. Lu, J. Guo, P. Lu, L. Zhang, Y. Zhang, Q. Dai, Y. Hu, V. L. Colvin and W. W. Yu, J. Phys. Chem. C, 2019, 123, 22787–22792 CrossRef CAS.
- V. G. V. Dutt, S. Akhil and N. Mishra, ChemNanoMat, 2020, 6, 1730–1742 CrossRef CAS.
- B. A. Koscher, J. K. Swabeck, N. D. Bronstein and A. P. Alivisatos, J. Am. Chem. Soc., 2017, 139, 6566–6569 CrossRef CAS PubMed.
- J. T. Lin, Y. K. Hu, C. H. Hou, C. C. Liao, W. T. Chuang, C. W. Chiu, M. K. Tsai, J. J. Shyue and P. T. Chou, Small, 2020, 16, 2–9 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nr06200j |
| This journal is © The Royal Society of Chemistry 2022 |