
Heterostructured Bi–Cu2S nanocrystals for efficient CO2 electroreduction to formate†
Xue
Han‡
 a,
Tianyou
Mou‡
a,
Shikai
Liu
b,
Mengxia
Ji
ac,
Qiang
Gao
a,
Qian
He
b,
Hongliang
Xin
a and
Huiyuan
Zhu
*a
a,
Tianyou
Mou‡
a,
Shikai
Liu
b,
Mengxia
Ji
ac,
Qiang
Gao
a,
Qian
He
b,
Hongliang
Xin
a and
Huiyuan
Zhu
*a
aDepartment of Chemical Engineering, Virginia Polytechnic Institute and State University, Blacksburg, Virginia 24061, USA. E-mail: huiyuanz@vt.edu
bDepartment of Materials Science and Engineering, National University of Singapore, 117575, Singapore
cInstitute of Energy Research, Jiangsu University, Zhenjiang, 212013, P. R. China
First published on 15th February 2022
Abstract
The electrochemical CO2 reduction reaction (ECO2RR) driven by renewable electricity holds promise to store intermittent energy in chemical bonds, while producing value-added chemicals and fuels sustainably. Unfortunately, it remains a grand challenge to simultaneously achieve a high faradaic efficiency (FE), a low overpotential, and a high current density of the ECO2RR. Herein, we report the synthesis of heterostructured Bi–Cu2S nanocrystals via a one-pot solution-phase method. The epitaxial growth of Cu2S on Bi leads to abundant interfacial sites and the resultant heterostructured Bi–Cu2S nanocrystals enable highly efficient ECO2RR with a largely reduced overpotential (240 mV lower than that of Bi), a near-unity FE (>98%) for formate production, and a high partial current density (2.4- and 5.2-fold higher JHCOO− than Cu2S and Bi at −1.0 V vs. reversible hydrogen electrode, RHE). Density functional theory (DFT) calculations show that the electron transfer from Bi to Cu2S at the interface leads to the preferential stabilization of the formate-evolution intermediate (*OCHO).
New conceptsIn this work, we report a colloidal route to synthesize a new class of heterostructures consisting of a p-block metal (Bi) and a transition metal chalcogenide (Cu2S). During the synthesis, Bi nucleated first and served as seeds for the shape-controlled epitaxial growth of Cu2S nanorods. A new concept of the interfacial synergy between p-block metals and transition metal chalcogenides is also demonstrated. These heterostructured Bi–Cu2S nanocrystals with abundant interfacial sites enable highly efficient electrochemical CO2 reduction. The electrochemical investigation shows that Bi–Cu2S demonstrated the highest faradaic efficiency toward formate production with a simultaneously largely reduced overpotential and a high partial current density compared with the control samples (Bi NPs, Cu2S NRs, and a physical mixture of Bi NPs and Cu2S NRs), providing convincing evidence of the important role played by the interfacial synergy in promoting electrocatalysis. Density functional theory calculations show that electron transfer from Bi to Cu2S leads to the preferential stabilization of formate-evolution intermediates compared with CO formation and hydrogen evolution, which is consistent with the experimental results. This work highlights a unique interfacial design and synthesis of p-block metal and transition metal chalcogenide heterostructures to refine active sites for advanced electrocatalysis. |
Introduction
The electrochemical CO2 reduction reaction (ECO2RR) is considered a promising strategy to sustainably recycle CO2 into the anthropogenic carbon cycle, paving the road to a clean-energy future.1–10 Despite its promise, the ECO2RR usually suffers from: (i) a significant overpotential, leading to low energy efficiency; (ii) a broad product distribution in aqueous electrolytes resulting from the multi-proton/electron transfer steps, and (iii) competition with the hydrogen evolution reaction (HER) and, thus, limited selectivity.1,11,12Metal and metal-based catalysts have been commonly used for the ECO2RR to CO, formate, and C2+ products.1,3,7,13,14 Among those catalysts, nanostructured p-block metals such as Pb, In, and Bi demonstrate high selectivity toward formate production due to their passivation of the HER.11,12,15,16 Bi has become especially attractive because of its low cost and non-toxicity.17 However, monometallic Bi usually suffers from a high overpotential (>800 mV) and a low current density (<10 mA cm−2).18,19 This can be attributed to the relatively weak binding of ECO2RR intermediates (e.g., *OCHO, *COOH) on Bi surfaces. Thus, a more reductive potential is required to stabilize these intermediates to enable electron and proton transfer steps. Alloying Bi with other transition metals is a common approach to tune the stability of the reaction intermediate, as shown in the Bi–Sn,11,20–22 Bi–Cu,23–25 and Bi–Mo26 systems. Nevertheless, a trade-off among overpotentials, faradaic efficiencies (FEs), and current densities is commonly observed due to adsorption-energy scaling relations of alloy surfaces that impose constraints on the attainable catalytic performance.27–29 It is therefore imperative to develop design strategies of ECO2RR catalysts that demonstrate simultaneous high current densities, low overpotentials, and high FEs. To achieve this goal, constructing interfacial sites that can selectively stabilize relevant ECO2RR intermediates on Bi surfaces could be a viable solution to improve electrokinetics, while maintaining the high FE toward formate. To design an efficient interfacial site with Bi for the ECO2RR, transition metal chalcogenides (TMCs), especially Cu2S, are promising as they demonstrate stronger binding of ECO2RR intermediates and thus a lower overpotential and higher current density than Bi.30,31 However, the HER is inevitable in Cu2S systems.
Herein, we report a proof-of-concept experimental and theoretical design of interfacial sites with a selectivity-governing and HER-suppressing domain (Bi) in concomitance with an electrokinetics-promoting domain (Cu2S) for efficient ECO2RR to formate. We developed a one-pot solution-phase synthesis method for preparing a new class of heterostructured Bi–Cu2S nanocrystals with a microphone-like morphology. In 0.1 M KHCO3, the Bi–Cu2S catalyst manifests a much higher current density, lower overpotential, and higher FE toward formate production than its single component (Bi, Cu2S) counterparts for the ECO2RR. Compared with Bi nanoparticles (NPs), the ECO2RR onset potential on Bi–Cu2S is positively shifted by 240 mV. By controlling the synthesis conditions, e.g., the reaction temperature, the amount of reducing agent, and degassing temperature, the optimal Bi–Cu2S catalyst can achieve near-unity formate selectivity at −1.2 V (vs. reversible hydrogen electrode, RHE) with a high current density of 18.2 mA cm−2, outperforming many previously reported catalysts.32–34 Density functional theory (DFT) calculations showed that the interfacial site preferentially stabilizes the formate-evolution intermediate *OCHO compared with the *COOH intermediate toward CO formation and the *H intermediate toward H2 generation. The stabilization of key intermediates was attributed to the electron transfer from Bi to Cu2S moieties at the interface.
Results and discussion
Heterostructured Bi–Cu2S nanocrystals were synthesized via a one-pot wet chemical approach (see ESI†). The TEM images of the as-synthesized Bi–Cu2S (Fig. 1a b and Fig. S1, ESI†) show that the Bi ‘head’ with a size of 76 nm and the 103 nm Cu2S ‘body’ are well-connected, forming a heterostructure interface. A clear interfacial boundary was observed in a bright-field (BF) STEM image (Fig. 1d), suggesting the epitaxial growth between Cu2S and Bi. From the STEM image, we can observe an interplanar distance of 3.3 Å in the Bi domain, corresponding to the (012) crystalline plane of rhombohedral Bi. The lattice spacing of Cu2S is 3.4 Å, which can be attributed to the (002) plane of hexagonal Cu2S. High-angle annular dark-field STEM (HAADF-STEM) (Fig. 1c) X-ray energy dispersive spectrometry (X-EDS) mapping (Fig. 1e) suggests the enrichment of Bi at the head, while the signals of Cu and S are distributed across the entire structure and enriched in the rod, confirming the formation of abundant Bi–Cu2S interfacial sites. Fig. 2a shows a schematic illustration of the nucleation and growth of heterostructured Bi–Cu2S nanocrystals. The transmission electron microscopy (TEM) images of Bi–Cu2S along the time sequence after the injection of a sulfur-containing surfactant suggest that the Bi nucleates first and serves as a seed for the deposition, nucleation, and growth of Cu2S domains (Fig. 2b). For comparison, Bi NPs and Cu2S nanorods (NRs) were also synthesized according to reported methods (Fig. S2, ESI†).12,35 A typical rhombohedral crystal structure of Bi–Cu2S and Bi and a hexagonal structure of Cu2S in the X-ray diffraction (XRD) patterns were observed (Fig. 1f), consistent with the lattice spacing obtained in Fig. 1d. No obvious Cu2S peak is present in Bi–Cu2S due to the overlapping of the diffraction peaks between Bi and Cu2S. The X-ray photoelectron spectroscopy (XPS) of the Bi 4f spectrum for Bi–Cu2S (Fig. 1g) shows peaks at 156.93 and 158.91 eV, which are attributed to Bi0 and Bi3+, respectively. Compared to the XPS result of pure Bi NPs (Fig. S8b, ESI†), Bi–Cu2S has no peak shift, suggesting that the chemical composition and valence state of Bi–Cu2S in the bulk remain unchanged. The Cu 2p XPS spectrum of Bi–Cu2S (Fig. 1h) shows a single Cu 2p3/2 feature at 932.3 eV, which can be assigned to Cu1+ or Cu0. The deconvolution of the Cu LMM spectra (Fig. S7, ESI†) further distinguishes the existence of Cu1+ from Cu0 in Bi–Cu2S.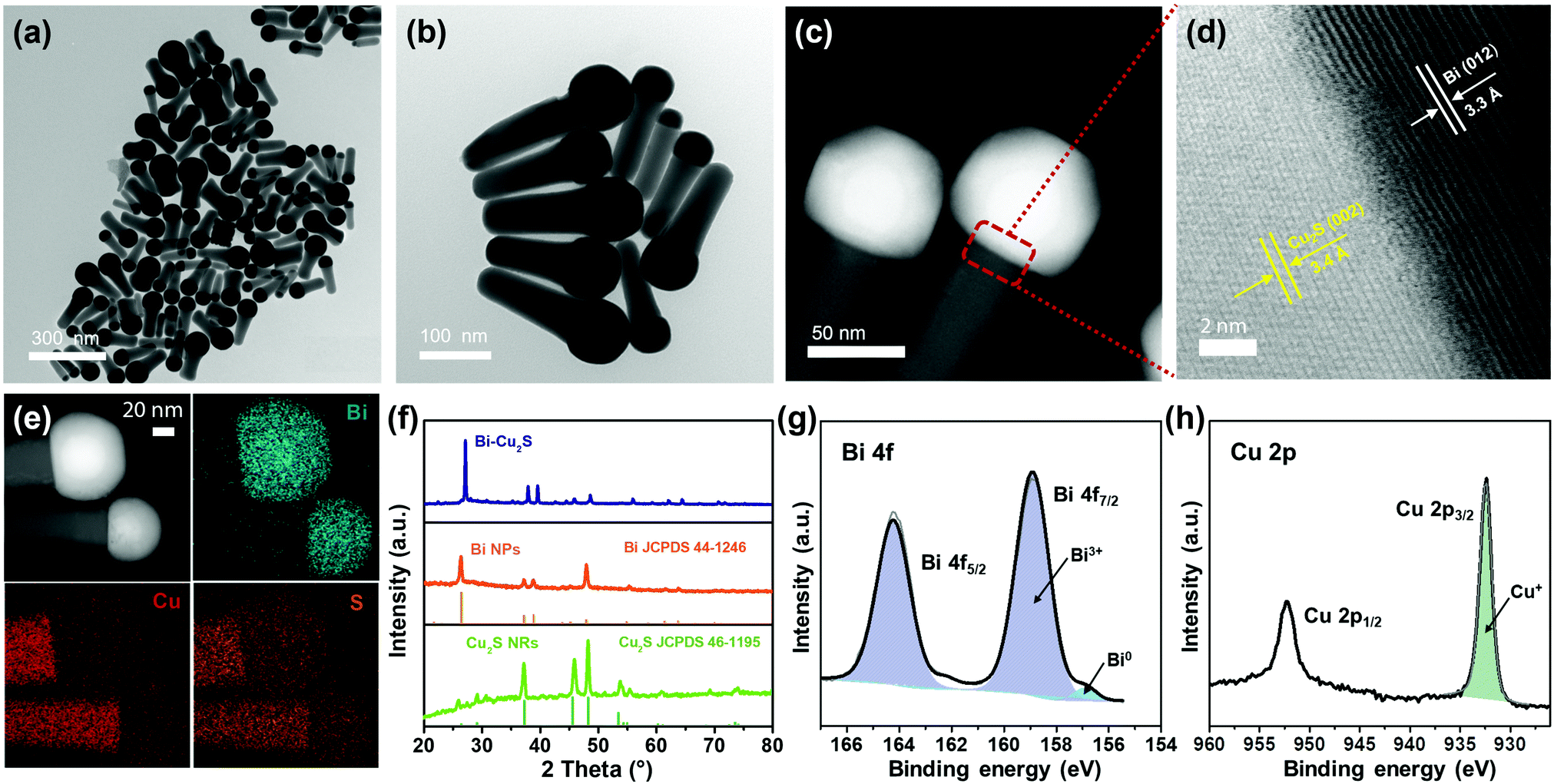 | ||
| Fig. 1 Structural characterizations of heterostructured Bi–Cu2S nanocrystals. (a and b) TEM images; (c) HAADF-STEM image and (d) BF-STEM image of Bi–Cu2S. (e) The HAADF-STEM image and the corresponding X-EDS mappings of Bi–Cu2S. (f) XRD patterns of Bi NPs, Cu2S NRs and Bi–Cu2S. (g) Bi 4f XPS spectrum and (h) Cu 2p XPS spectrum of Bi–Cu2S. | ||
 | ||
| Fig. 2 (a) Schematic illustration of the nucleation and growth of the heterostructured Bi–Cu2S nanocrystals. (b) TEM images of Bi–Cu2S at different time intervals after introducing 1-dodecanethiol at 220 °C. | ||
The as-synthesized Bi–Cu2S and control samples of Bi NPs and Cu2S NRs were deposited on activated carbon and surface-activated through a ligand-stripping method (Fig. S5, ESI†). The catalyst ink was prepared and airbrushed onto carbon paper (ESI†). The ECO2RR testing was conducted in CO2-saturated 0.1 M KHCO3 in a gas-tight H-cell. Before the measurement, all the samples were further activated via potential cycling between −1.2 V and 0 V vs. RHE in an Ar-saturated solution for 10 cycles at a scan rate of 20 mV s−1. Cyclic voltammetry (CV) curves were then recorded first in Ar- and then in CO2-saturated solutions (Fig. S10–S12, ESI†). To further evaluate the catalytic performance of Bi–Cu2S, linear sweep voltammetry (LSV) curves were obtained. In Fig. 3a, Bi–Cu2S not only shows a higher current density than the two control samples (Bi NPs and Cu2S NRs) in a wide range of potentials from −0.8 V to −1.2 V vs. RHE, but also demonstrates a 240 mV more positive onset potential than Bi. At −1.0 V vs. RHE, Bi–Cu2S demonstrates a 4.9-fold increase of total current density compared with Bi. To probe the product distribution, the electrolysis measurements in a wide range of applied potentials (−0.8 to −1.2 V) were carried out. Gas chromatography (GC) was used to detect gas products, while 1H nuclear magnetic resonance (NMR) spectrometry was employed to analyze liquid-phase products. The Bi–Cu2S demonstrated the highest partial current density for formate production (JHCOO−) at all applied potentials compared with the Bi NPs and Cu2S NRs (Fig. 3b). Meanwhile, the JHCOO− of Bi–Cu2S rapidly increased from −0.9 V, exhibiting a striking difference from the control samples. The FE of formate production (FEHCOO−) on Bi–Cu2S is shown in Fig. S10c (ESI†). Over 90% of FEHCOO− was achieved at the applied potential of −1.0 V. In particular, near-unity HCOO− selectivity was obtained at the potential of −1.2 V, exceeding most of the state-of-art catalysts for formate production.7,19Fig. 3e shows the FEs over Bi–Cu2S, Bi, and Cu2S. The FEHCOO− of the designed Bi–Cu2S was higher than that of the two control samples at all applied potentials. In particular, the FEHCOO− of Bi–Cu2S was 92.4% at −1.0 V, which is ∼1.4 times higher than that of the Cu2S NRs (65% FEHCOO−). The production rate of formate on Bi–Cu2S reached 131 μmol cm−2 h−1 at −1.0 V, which is 3.5 times higher than that of the Bi NPs (Fig. S16, ESI†). Furthermore, Bi–Cu2S showed a much lower FE of H2 (FEH2), indicating that Bi–Cu2S preserved the HER-inhibiting property. To verify that the interfacial sites of Bi–Cu2S play a key role in the ECO2RR, Bi NPs and Cu2S NRs were physically mixed (Bi NPs+Cu2S NRs) as a control sample (Fig. S3, ESI†). Compared with the physical mixture of Bi NPs and Cu2S NRs (Fig. S13 and S14, ESI†), Bi–Cu2S showed a larger current density and higher FEHCOO−, indicating the importance of a well-connected interface on Bi–Cu2S in optimizing the ECO2RR kinetics. Moreover, we tested the ECO2RR performance of Bi–Cu2S heterostructures with 5-, 15-, 30-, and 120 minutes of reaction time after injecting the sulfur-containing surfactant, denoted as Bi–Cu2S/5, Bi–Cu2S/15, Bi–Cu2S/30 (which is the Bi–Cu2S catalyst in this work), and Bi–Cu2S/120. The TEM images (Fig. S4, ESI†) show that there are many impurities and unreacted precursors in the Bi–Cu2S/5 and Bi–Cu2S/15. The reaction is complete and there are no more impurities in the sample when the reaction time is more than 30 min. In Fig. S15, (ESI†) Bi–Cu2S under complete reaction conditions (Bi–Cu2S/30 and Bi–Cu2S/120) demonstrates more positive onset potentials than the incomplete ones (Bi–Cu2S/5 and Bi–Cu2S/15). Bi–Cu2S/30 shows the highest partial current density (JHCOO−) and faradaic efficiency of formate (FEHCOO−) production at potentials from −0.9 to −1.2 V vs. RHE. In particular, Bi–Cu2S/30 demonstrates a much higher JHCOO− and FEHCOO− than the Bi–Cu2S/5 and Bi–Cu2S/15, indicating the negative impact of the incomplete reactants in the CO2 reduction reaction. Furthermore, both JHCOO− and FEHCOO− on B–Cu2S/120 catalysts slightly decrease compared to that of Bi–Cu2S/30. This can be attributed to the larger size and fewer active sites of Bi–Cu2S/120. This result provides evidence that the interfacial synergy in Bi–Cu2S plays an important role in promoting electrocatalysis. Taking cumulatively, Bi–Cu2S/30 demonstrates an extremely high HCOO− selectivity, improved current density, and much lower overpotential due to the synergistic effect at the interfacial sites of Bi–Cu2S. The durability test of Bi–Cu2S (Fig. S10d, ESI†) showed that the current density and FE stay stable after 10 h electrolysis.
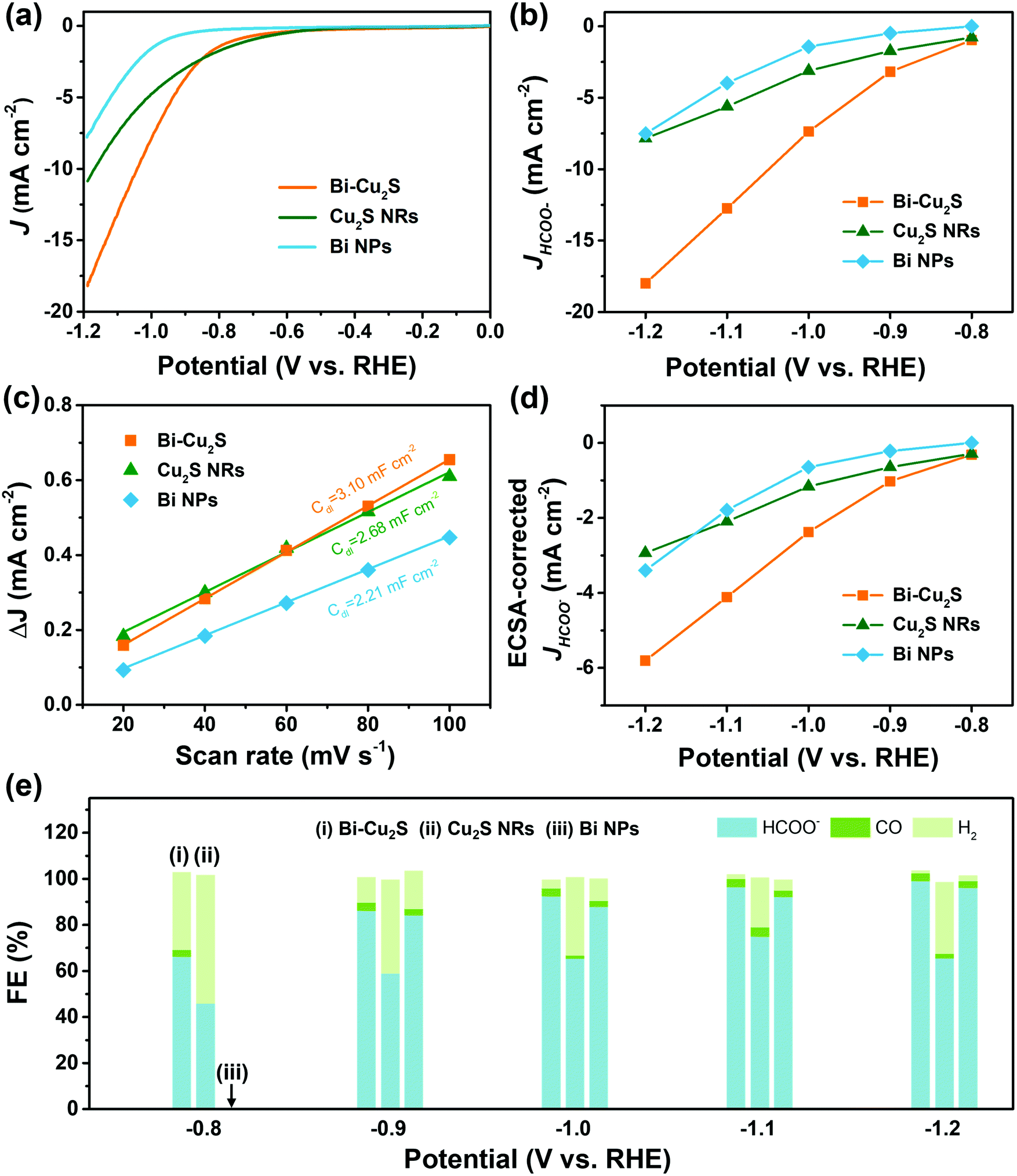 | ||
| Fig. 3 ECO2RR on Bi–Cu2S, Cu2S NRs, and Bi NPs in the CO2-saturated 0.1 M KHCO3 electrolyte: (a) LSV curves. (b) Partial current density of formate at different potentials. (c) ECSAs of Bi–Cu2S, Cu2S NRs and Bi NPs. (d) ECSA-corrected current densities for formate. (e) Comparison of FEs for the ECO2RR on Bi–Cu2S, Bi and Cu2S electrodes at various applied potentials. | ||
To gain insight into the origin of the ECO2RR enhancement on Bi–Cu2S, electrochemical surface area (ECSA) was measured by calculating double-layer capacitance (Cdl) to evaluate their ECSA-corrected current density (Fig. S17 and S18, ESI†). The calculated values of Cdl (Fig. 3c) for Bi–Cu2S, Bi, and Cu2S are 3.10, 2.21, and 2.68 mF cm−2, respectively, indicating that Bi–Cu2S has more active sites than the controls. Meanwhile, the normalized ECSA current of formate (Fig. 3d) shows that Bi–Cu2S has higher intrinsic activity than Bi NPs and Cu2S NRs at all applied potentials. The XPS was performed before and after the ECO2RR to gain a deeper insight into the valence states and surface compositions of the Bi–Cu2S catalyst (ESI†). In the Bi 4f XPS spectrum of pure Bi NPs (Fig. S8, ESI†), only a Bi3+ peak is observed, indicating that Bi is surface-oxidized when re-exposed to air after the CO2 reduction. On the contrary, the XPS results of Bi–Cu2S (Fig. S6, ESI†) reveal that Bi partially keeps the metallic state and the Bi0/Bi3+ ratio increases from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :26 to 1:3 before and after the ECO2RR. This observation suggests that the existence of Cu2S stabilizes metallic Bi and suppresses its surface oxidation upon air exposure to promote the ECO2RR.12
:26 to 1:3 before and after the ECO2RR. This observation suggests that the existence of Cu2S stabilizes metallic Bi and suppresses its surface oxidation upon air exposure to promote the ECO2RR.12
To understand the interfacial synergy, DFT calculations were performed to investigate the energetics of key surface intermediates governing the kinetics of the ECO2RR and the competing HER. Motivated by the X-EDS mappings of the synthesized Bi–Cu2S showing the dispersed Cu2S moieties on the Bi domain (Fig. 1e), we built an interface model using a rhombohedral Bi(001) surface decorated with hexagonal Cu2S NRs. Cu2S NRs were constructed with the sulfur-terminated (001) orientation on all sides (Fig. S19, ESI†), which allows the formation of a stable interface between the two low-index surfaces. Under relevant ECO2RR conditions, Cu2S is partially reduced according to previous reports.36–38 The free energy of the sulfur vacancy formation of the stoichiometric Cu2S(001) surface to generate H2S(g) is −1.23 eV at potential −1.0 V vs. RHE (Fig. S20 and Fig. 4b, see ESI† for computational details). In Fig. 4b, the free energy of the sulfur vacancy formation is plotted as a function of the operating potential U and pH. The red line is the potential U at which the vacancy starts forming. The free energy of the vacancy formation is negative below the red line indicating the feasibility of losing sulfur atoms on the surface. The reaction operating conditions are plotted as the black line, starting from −1.21 V to −1.61 V vs. SHE at pH = 7, and under these conditions, vacancies are formed on the surface. The generation of a second sulfur vacancy is energetically unfavorable, resulting in surface reconstruction. Thus, the interface model of Bi–Cu2S contains one S vacancy in our model systems. This is consistent with our EDS spectra (Fig. S9, ESI†) where the ratio of S/Cu decreases after the electrolysis. Since ECO2RR on Bi surfaces mainly produces formate and CO with two-electron transfers, we consider the first proton-coupled electron transfer with the protonation of the C atom to form the *OCHO intermediate or O atom to form the *COOH intermediate.39 The *OCHO and *COOH intermediates can be further reduced to HCOO− and CO, respectively (Fig. 4a). The HER via the *H intermediate is also considered at the interfacial sites. The reaction pathway and the optimized geometries for each intermediate are shown in Fig. 4c–e, for both Bi and Bi–Cu2S systems (see Fig. S21, ESI† for detailed geometries). The formation of the *COOH intermediate is significantly uphill in free energy compared to the *OCHO intermediate on both surfaces, leading to a favorable production of HCOO−, consistent with the experimental results of a much higher FEHCOO− than FECO. The Volmer step of the HER is highly unfavorable since the *H intermediates bind very weakly on Bi sites, resulting in a suppressed HER under CO2 reduction conditions on Bi.18,40
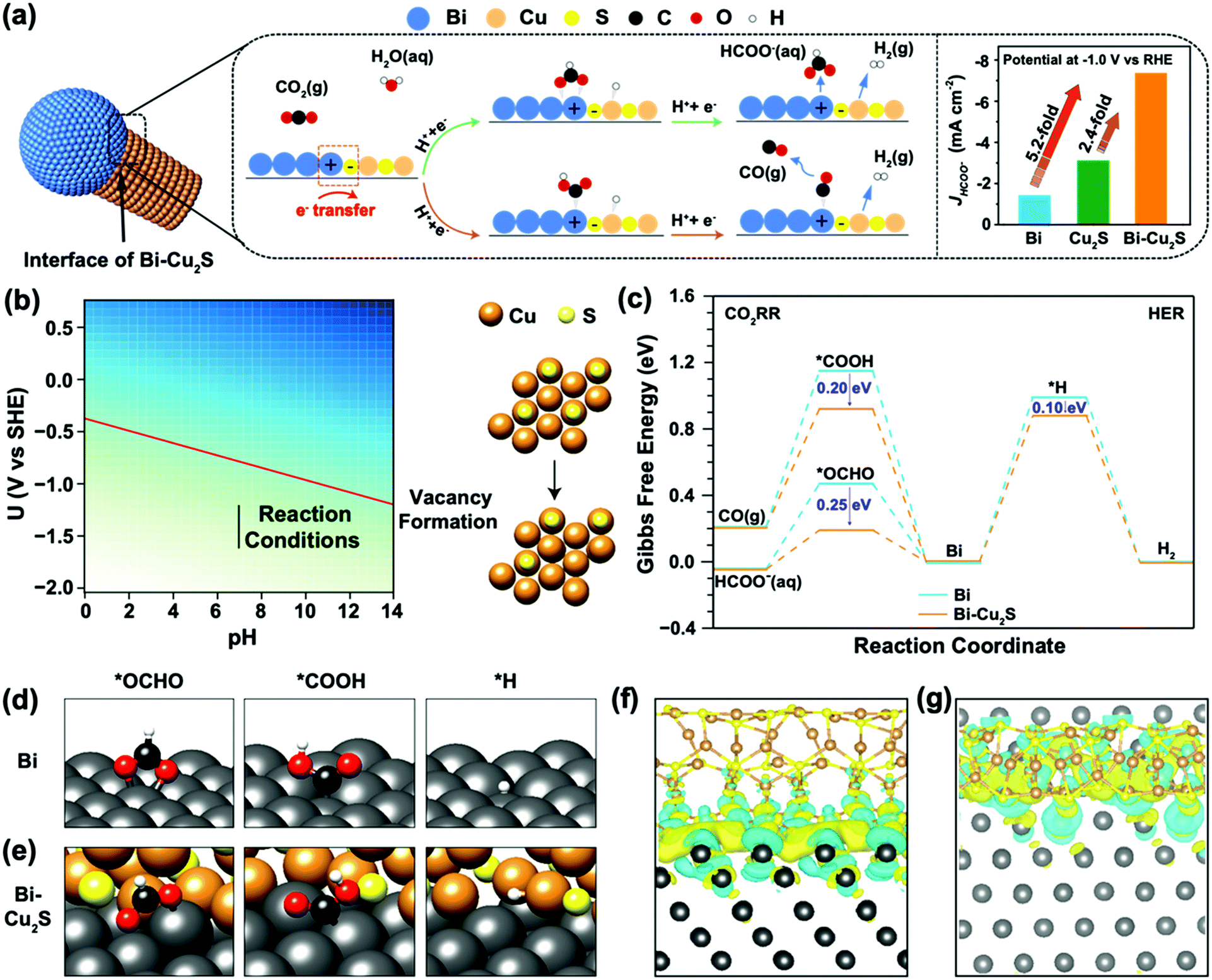 | ||
| Fig. 4 (a) Proposed CO2 reduction mechanism on the Bi–Cu2S interfacial system. (b) The sulfur vacancy formation Pourbaix diagram. (c) Free energy diagrams of the ECO2RR and HER on Bi (001) and Bi–Cu2S model systems. Optimized geometry structures of key intermediates (*OCHO, *COOH, and *H) on Bi (001) and Bi–Cu2S systems are shown in (d and e), respectively. (Dark grey, brown, yellow, black, red, and white spheres denote Bi, Cu, S, C, O, and H atoms, respectively.) (f and g) are the top and front views of the charge density difference for the Bi–Cu2S interfacial surface, respectively. Cyan corresponds to an isosurface of −0.001 e bohr−3 and yellow to +0.001 e bohr−3. | ||
While the ECO2RR is facilitated in both Bi and Bi–Cu2S systems, the extent of stabilization of the CO- and HCOO−-evolution intermediates varies. Attributed to the interactions between Bi and Cu2S, *OCHO and *COOH are stabilized by 0.25 eV and 0.20 eV on the Bi–Cu2S interfacial site, respectively, leading to a reduced overpotential and improved selectivity toward formate production compared with that on pure Bi. The *H intermediate is merely stabilized by 0.10 eV due to its adsorption on the Cu site of Bi–Cu2S. Therefore, the HER is still inhibited in Bi–Cu2S, consistent with our experimental results in Fig. 3e. We further analyzed the local electronic structures of the interface and the adsorbates to gain insights into the improved performance. Fig. S22 and S23 (ESI†) show the isosurfaces of the Cu2S-induced charge density difference, and Fig. S24 (ESI†) shows the Bader charge analysis,41 both of which show that Bi is electron-deficit, which is consistent with the fact that a Schottky interface between Bi and Cu2S could lead to electron flow from Bi to Cu2S.42,43 For a bare Bi–Cu2S surface (Fig. 4f–g), the Bi substrate is electron-deficit, especially for Bi atoms forming bonds with S atoms, which plays an important role in electrostatically stabilizing the *OCHO and *COOH intermediates, since both are negatively charged as shown by the adsorbate-induced charge density difference on Bi (001) shown in Fig. S23 (ESI†).
Conclusions
In summary, we have reported a one-pot synthesis of heterostructured Bi–Cu2S nanocrystals for the ECO2RR toward formate production. Detailed structural characterizations proved the successful construction of a well-connected, epitaxial interface between Bi and Cu2S. Systematic electrochemical investigations demonstrated much enhanced ECO2RR activity without the sacrifice of high selectivity at the interface between Bi and Cu2S. Compared with Bi NPs, Cu2S NRs, and the Bi+Cu2S physical mixture, Bi–Cu2S presented the highest FE at all applied potentials toward formate production, with partial current densities increasing by 5.2-fold, 2.4-fold, and 5.3-fold at −1.0 V vs. RHE, respectively. The onset potential of Bi–Cu2S is 240 mV more positive than that of Bi NPs. These results provide clear evidence that the interfacial synergy in Bi–Cu2S plays a pivotal role in promoting electrocatalysis. DFT calculations suggest that the electron transfer from Bi to Cu2S at the interface stabilized the ECO2RR intermediates, particularly for *OCHO toward HCOO− evolution compared with *COOH to CO and *H to H2. This work highlights a unique design strategy of p-block metal and TMC heterostructures to fine-tailor active sites for advanced electrocatalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the US National Science Foundation (CHE-2102363). Q. H. would like to acknowledge the support by National Research Foundation (NRF) Singapore, under its NRF Fellowship (NRF-NRFF11-2019-0002) and the National University of Singapore Flagship Green Energy Program (#R-284-000-185-733). M. X. J. would like to acknowledge financial support from China Scholarship Council. H. X. acknowledges the NSF CAREER program (CBET-1845531). The computational resource used in this work is provided by Advanced Research Computing at Virginia Polytechnic Institute and State University.Notes and references
- J. Zhao, S. Xue, J. Barber, Y. W. Zhou, J. Meng and X. B. Ke, J. Mater. Chem. A, 2020, 8, 4700–4734 RSC.
- J. Wang, T. Cheng, A. Q. Fenwick, T. N. Baroud, A. Rosas-Hernández, J. H. Ko, Q. Gan, W. A. Goddard Iii and R. H. Grubbs, J. Am. Chem. Soc., 2021, 143, 2857–2865 CrossRef CAS PubMed.
- A. M. Liu, M. F. Gao, X. F. Ren, F. N. Meng, Y. N. Yang, L. G. Gao, Q. Y. Yang and T. L. Ma, J. Mater. Chem. A, 2020, 8, 3541–3562 RSC.
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen and J. Yang, Adv. Mater., 2020, 32, e2001848 CrossRef PubMed.
- S. Garg, M. R. Li, A. Z. Weber, L. Ge, L. Y. Li, V. Rudolph, G. X. Wang and T. E. Rufford, J. Mater. Chem. A, 2020, 8, 1511–1544 RSC.
- L. Fan, C. Xia, F. Q. Yang, J. Wang, H. T. Wang and Y. Y. Lu, Sci. Adv., 2020, 6 Search PubMed.
- W. J. Zhang, Y. Hu, L. B. Ma, G. Y. Zhu, Y. R. Wang, X. L. Xue, R. P. Chen, S. Y. Yang and Z. Jin, Adv. Sci., 2018, 5 CAS.
- Y. Quan, J. Zhu and G. Zheng, Small Sci., 2021, 2100043, DOI:10.1002/smsc.202100043.
- X. Han, Q. Gao, Z. H. Yan, M. X. Ji, C. Long and H. Y. Zhu, Nanoscale, 2021, 13, 1515–1528 RSC.
- Q. Wang, C. Cai, M. Dai, J. Fu, X. Zhang, H. Li, H. Zhang, K. Chen, Y. Lin, H. Li, J. Hu, M. Miyauchi and M. Liu, Small Sci., 2021, 1, 2000028 CrossRef.
- Z. Wu, H. Wu, W. Cai, Z. Wen, B. Jia, L. Wang, W. Jin and T. Ma, Angew. Chem., Int. Ed., 2021, 60, 12554–12559 CrossRef CAS PubMed.
- Z. Y. Zhang, M. F. Chi, G. M. Veith, P. F. Zhang, D. A. Lutterman, J. Rosenthal, S. H. Overbury, S. Dai and H. Y. Zhu, ACS Catal., 2016, 6, 6255–6264 CrossRef CAS.
- F. Franco, C. Rettenmaier, H. S. Jeon and B. Roldan Cuenya, Chem. Soc. Rev., 2020, 49, 6884–6946 RSC.
- J. L. Yu, J. Wang, Y. B. Ma, J. W. Zhou, Y. H. Wang, P. Y. Lu, J. W. Yin, R. Q. Ye, Z. L. Zhu and Z. X. Fan, Adv. Funct. Mater., 2021, 31, 2102151 CrossRef CAS.
- Q. F. Gong, P. Ding, M. Q. Xu, X. R. Zhu, M. Y. Wang, J. Deng, Q. Ma, N. Han, Y. Zhu, J. Lu, Z. X. Feng, Y. F. Li, W. Zhou and Y. G. Li, Nat. Commun., 2019, 10 Search PubMed.
- Q. Gao, T. Mou, S. Liu, G. Johnson, X. Han, Z. Yan, M. Ji, Q. He, S. Zhang, H. Xin and H. Zhu, J. Mater. Chem. A, 2020, 8, 20931–20938 RSC.
- E. H. Zhang, T. Wang, K. Yu, J. Liu, W. X. Chen, A. Li, H. P. Rong, R. Lin, S. F. Ji, X. S. Zhene, Y. Wang, L. R. Zheng, C. Chen, D. S. Wang, J. T. Zhang and Y. D. Li, J. Am. Chem. Soc., 2019, 141, 16569–16573 CrossRef CAS PubMed.
- F. Yang, A. O. Elnabawy, R. Schimmenti, P. Song, J. W. Wang, Z. Q. Peng, S. Yao, R. P. Deng, S. Y. Song, Y. Lin, M. Mavrikakis and W. L. Xu, Nat. Commun., 2020, 11 Search PubMed.
- Y. Y. Guan, M. M. Liu, X. F. Rao, Y. Y. Liu and J. J. Zhang, J. Mater. Chem. A, 2021, 9, 13770–13803 RSC.
- Z. Li, Y. Feng, Y. Li, X. Chen, N. Li, W. He and J. Liu, Chem. Eng. J., 2022, 428, 130901 CrossRef CAS.
- G. B. Wen, D. U. Lee, B. H. Ren, F. M. Hassan, G. P. Jiang, Z. P. Cano, J. Gostick, E. Croiset, Z. Y. Bai, L. Yang and Z. W. Chen, Adv. Energy Mater., 2018, 8, 1802427 CrossRef.
- Y. Xing, X. Kong, X. Guo, Y. Liu, Q. Li, Y. Zhang, Y. Sheng, X. Yang, Z. Geng and J. Zeng, Adv. Sci., 2020, 7, 1902989 CrossRef CAS PubMed.
- W. X. Lv, J. Zhou, J. J. Bei, R. Zhang, L. Wang, Q. Xu and W. Wang, Appl. Surf. Sci., 2017, 393, 191–196 CrossRef CAS.
- Z. J. Wang, Q. Yuan, J. J. Shan, Z. H. Jiang, P. Xu, Y. F. Hu, J. G. Zhou, L. N. Wu, Z. Z. Niu, J. M. Sun, T. Cheng and W. A. Goddard, J. Phys. Chem. Lett., 2020, 11, 7261–7266 CrossRef CAS PubMed.
- Z. Yang, H. Wang, X. Fei, W. Wang, Y. Zhao, X. Wang, X. Tan, Q. Zhao, H. Wang, J. Zhu, L. Zhou, H. Ning and M. Wu, Appl. Catal., B, 2021, 298, 120571 CrossRef CAS.
- X. F. Sun, Q. G. Zhu, X. C. Kang, H. Z. Liu, Q. L. Qian, Z. F. Zhang and B. X. Han, Angew. Chem., Int. Ed., 2016, 55, 6771–6775 CrossRef CAS PubMed.
- M. M. Montemore and J. W. Medlin, Catal. Sci. Technol., 2014, 4, 3748–3761 RSC.
- Y. W. Li and Q. Sun, Adv. Energy Mater., 2016, 6 Search PubMed.
- Z. Yan, M. Ji, J. Xia and H. Zhu, Adv. Energy Mater., 2019, 10 Search PubMed.
- Y. Chen, K. Chen, J. Fu, A. Yamaguchi, H. Li, H. Pan, J. Hu, M. Miyauchi and M. Liu, Nano Mater. Sci., 2020, 2, 235–247 CrossRef.
- Y. L. Deng, Y. Huang, D. Ren, A. D. Handoko, Z. W. Seh, P. Hirunsit and B. S. Yeo, ACS Appl. Mater. Interfaces, 2018, 10, 28572–28581 CrossRef CAS PubMed.
- S. B. Liu, X. F. Lu, J. Xiao, X. Wang and X. W. Lou, Angew. Chem., Int. Ed., 2019, 58, 13828–13833 CrossRef CAS PubMed.
- P. L. Deng, H. M. Wang, R. J. Qi, J. X. Zhu, S. H. Chen, F. Yang, L. Zhou, K. Qi, H. F. Liu and B. Y. Xia, ACS Catal., 2020, 10, 743–750 CrossRef CAS.
- Y. Zhang, F. W. Li, X. L. Zhang, T. Williams, C. D. Easton, A. M. Bond and J. Zhang, J. Mater. Chem. A, 2018, 6, 4714–4720 RSC.
- Y. Zhai and M. Shim, ChemPhysChem., 2016, 17, 741–751 CrossRef CAS PubMed.
- N. T. P. Thao, S. Tsuji, S. Jeon, I. Park, C. B. Tabelin, M. Ito and N. Hiroyoshi, Hydrometallurgy, 2020, 194 Search PubMed.
- Y. Zhang, D. Yao, B. Xia, H. Xu, Y. Tang, K. Davey, J. Ran and S.-Z. Qiao, Small Sci., 2021, 1, 2000052 CrossRef.
- K. A. Persson, B. Waldwick, P. Lazic and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85 Search PubMed.
- H. S. Pillai and H. L. Xin, Ind. Eng. Chem. Res., 2019, 58, 10819–10828 CrossRef CAS.
- N. Han, Y. Wang, H. Yang, J. Deng, J. H. Wu, Y. F. Li and Y. G. Li, Nat. Commun., 2018, 9 Search PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- Y. Zhang, Y. Qiu, Z. Ma, Y. Wang, Y. Zhang, Y. Ying, Y. Jiang, Y. Zhu and S. Liu, J. Mater. Chem. A, 2021, 9, 10893–10908 RSC.
- X. Ji, Y. Zhang, Z. Ma and Y. Qiu, ChemSusChem, 2020, 13, 5004–5014 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1nh00661d |
| ‡ X. H. and T. M. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |