
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural insights into Lewis acid- and F4TCNQ-doped conjugated polymers by solid-state magnetic resonance spectroscopy†
Alana L.
Dixon
a,
Hervé
Vezin
b,
Thuc-Quyen
Nguyen
 *a and
G. N. Manjunatha
Reddy
*c
*a and
G. N. Manjunatha
Reddy
*c
aCenter for Polymers and Organic Solids, University of California Santa Barbara (UCSB), Santa Barbara, California 93106, USA. E-mail: quyen@chem.ucsb.edu
bUniversity of Lille, CNRS UMR8516, LASIRE, Lille, F-59000, France
cUniversity of Lille, CNRS, Centrale Lille Institut, Univ. Artois, UMR 8181–UCCS–Unité de Catalyse et Chimie du Solide, Lille, F-59000, France. E-mail: gnm.reddy@univ-lille.fr
First published on 20th December 2021
Abstract
Molecular doping strategies facilitate orders of magnitude enhancement in the charge carrier mobility of organic semiconductors (OSCs). Understanding the different doping mechanisms and molecular-level constraints on doping efficiency related to the material energy levels is crucial to develop versatile dopants for OSCs. Given the compositional and structural heterogeneities associated with OSC thin films, insight into dopant–polymer interactions by long-range techniques such as X-ray scattering and electron microscopy is exceedingly challenging to obtain. This study employs short-range probes, solid-state (ss)NMR and EPR spectroscopy, to resolve local structures and intermolecular interactions between dopants such as F4TCNQ (2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane), Lewis acid BCF (tris[pentafluorophenyl] borane) and Lewis base conjugated polymer, PCPDTBT (P4) (poly[2,6-(4,4-bis(2-hexadecyl)-4H-cyclopenta[2,1-b;3,4-b′]dithiophene)-alt-4,7(2,1,3-benzothiadiazole)]). Analysis of 1H and 13C ssNMR spectra of P4, P4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :F4TCNQ and P4:BCF blends indicates that the addition of dopants induces local structural changes in the P4 polymer, and causes paramagnetism-induced signal broadening and intensity losses. The hyperfine interactions in P4:BCF and P4:F4TCNQ are characterized by two-dimensional pulsed EPR spectroscopy. For P4:F4TCNQ, 19F ssNMR analysis indicates that the F4TCNQ molecules are distributed and aggregated into different local chemical environments. By comparison, BCF molecules are intermixed with the P4 polymer and interact with traces of water molecules to form BCF–water complexes that serve as Brønsted acid sites, as revealed by 11B ssNMR spectroscopy. These results indicate that the P4–dopant blends exhibit complex morphology with different distributions of dopants, whereby the combined use of ssNMR and EPR provides essential insights into how higher doping efficiency is observed with BCF and a mediocre efficiency is associated with F4TCNQ molecules.
:F4TCNQ and P4:BCF blends indicates that the addition of dopants induces local structural changes in the P4 polymer, and causes paramagnetism-induced signal broadening and intensity losses. The hyperfine interactions in P4:BCF and P4:F4TCNQ are characterized by two-dimensional pulsed EPR spectroscopy. For P4:F4TCNQ, 19F ssNMR analysis indicates that the F4TCNQ molecules are distributed and aggregated into different local chemical environments. By comparison, BCF molecules are intermixed with the P4 polymer and interact with traces of water molecules to form BCF–water complexes that serve as Brønsted acid sites, as revealed by 11B ssNMR spectroscopy. These results indicate that the P4–dopant blends exhibit complex morphology with different distributions of dopants, whereby the combined use of ssNMR and EPR provides essential insights into how higher doping efficiency is observed with BCF and a mediocre efficiency is associated with F4TCNQ molecules.
New conceptsPolymeric organic semiconductors (OSCs) are of high fundamental and technological interest for a range of optoelectronics, though they exhibit a lower number of charge carriers than their inorganic counterparts. While molecular doping of OCSs by Lewis acids or by F4TCNQ is an effective strategy to augment charge carrier mobilities, the complex doping mechanisms and intermolecular interactions are difficult to define at the atomic-level. The lack of knowledge on dopant–polymer intermolecular interactions hinders further development of efficient dopants. It is unclear why the doping efficiency of most conjugated polymers is only a few percent, and in this study for the same conjugated polymer the BCF-water complex is a much better dopant than F4TCNQ. Thus, there is an urgent need for further investigation to gain insight into the molecular origins of different doping efficiencies. We propose a combined two-dimensional (2D) solid-state NMR and EPR approach to investigate local structures and intermolecular interactions in a Lewis base PCPDTBT (P4) polymer doped with F4TCNQ and a Lewis acid BCF–water complex that serves as a Brønsted acid. These findings offer a greater molecular-level understanding of polymer–dopant interactions and are expected to provide guidance for the development of simple and efficient molecular dopants for OSC-based optoelectronics. |
Introduction
Organic semiconductors (OSCs) are attractive due to their flexibility and light weight, which make them suitable for optoelectronic applications such as organic light emitting diodes (OLEDs) for large area displays, organic photovoltaic cells (OPVs), organic photodiodes, organic field-effect transistors (OFETs), thermoelectrics, and emerging applications in bioelectronics. A key parameter for such applications is the energy gap between electronic states of π-conjugated building blocks, and charge transport within and in between π-conjugated chains. For OSCs, the conductivity is limited by the relatively low number of charge carriers compared to their inorganic counterparts and hybrid compositions.1,2 This limitation is due in part to the relatively large energy band gaps of OSCs, meaning a sufficiently high energy (above the thermal energy provided at room temperature) must be supplied to excite an electron from the ground state into the excited state. Additionally, poor electric field screening in organic semiconductors leads to the attractive Coulombic force between the excited electron and its positively charged ‘hole’ in the lower energy level to provide an additional energy barrier to be overcome.3 One way to enhance the number of charge carriers is through doping, the addition of dopants to the OSC, which either serve as electron donors (n-type doping) or electron acceptors (p-type doping).Early OSC doping strategies relied on the use of metals and volatile halides4,5 that unfortunately tended to diffuse through the organic layers, resulting in irreproducible doping and device instability.6,7 The advent of molecular dopants has allowed for stable doping to be achieved in OSCs, improving the performance of devices such as OFETs, OLEDs, and OPVs.8–13,36 Specifically, acid–base complexation and reduction–oxidation (redox) reactions between small molecule dopants and conjugated polymers are among the widely explored chemical doping strategies to adjust the optical and electronic properties in OSCs. In redox doping, an integer charge transfer occurs from the LUMO (lowest unoccupied molecular orbital) of the dopant to the HOMO (highest occupied molecular orbital) of the OSC, which depends strongly on their energy level alignment.14 F4TCNQ is one of the most common p-dopants because its LUMO matches with the HOMO of many OSCs and the material is commercially available. However, F4TCNQ has a very low solubility in common organic solvents and tends to aggregate in thin films.15–19 In the case of acid–base reactions, doping occurs by proton transfer to the basic polymer backbones upon addition of small molecule acids. Several Lewis and Brønsted acids have been proposed as molecular dopants to OSCs, whereby Lewis and Brønsted acids lead to weak and strong doping, respectively.20–27 Recently, OSC doping driven by ion exchange has been suggested.28 In order to take full advantage of the opportunity presented by molecular doping, a detailed understanding of the polymer–dopant intermolecular interactions upon doping with specific small molecules is needed.
The impact of dopants on the optoelectronic properties of OSCs has been rationalized based on morphological studies and dopant–host interactions. For example, in the poly(3-hexylthiophene) (P3HT):F4TCNQ system, it was observed that doping by vapor diffusion resulted in minimal disturbance to the ordering of the conjugated backbone along which charges are transported in OSC materials, yielding significantly higher conductivity values when compared to solution-processed doping that more drastically altered the OSC morphology.29,30 In the case of the poly[2,5-bis(3-tetradecylthiophen-2-yl)thieno[3,2-b]thiophene] (PBTTT-C14):F4TCNQ blend, F4TCNQ molecules are ordered in a cofacial arrangement with PBTTT-C14, leading to an efficient charge transfer.31,32 In other studies, varied processing conditions were used to tune the ratio of doping by F4TCNQ, which occurred through the integer charge transfer (ICT) or partial charge transfer mechanisms as explained by the location of the dopant molecules in the polymer structure.33–36 As partial charge transfer involves orbital hybridization between the dopant and the host material,37,38 it was shown to be favored when the dopant intercalates between the conjugated backbones of the OSC.31,39 In contrast to the multitude of doping studies involving F4TCNQ molecules, the doping mechanism employed by the relatively nascent Lewis acid dopant BCF is still unclear and ripe for further investigation.40–42 The emerging Lewis acid dopants such as BCF are of great current interest to the OSC community due their high doping efficiency, defined as the ratio of the free charge carriers to the dopant molecules, and good solubility in common organic solvents when compared to other dopants such as F4TCNQ.25,40,43–45 When BCF molecules interact with traces of water molecules, BCF–water complexes are formed, which perform as strong Brønsted acids that are shown to dope conjugated polymers.40 In a recent study by Yurash et al., the proposed BCF doping mechanism is based on proton transfer from a BCF·H2O adduct to the polymer backbone, followed by electron transfer from a nearby neutral polymer chain. It is then proposed to result in a neutral radical on the polymer backbone chain containing the extra proton, a positive radical on the adjacent polymer backbone chain that already donated an electron, and a coexisting BCF-OH− anion.40 In a later study by Arvind et al., it is the neutral protonated radical that was not observed in electron paramagnetic resonance (EPR) measurements of P3HT:BCF blends.41 Therefore, it has been suggested that the overall P3HT–BCF reaction resulted in the formation of the H2 product, with one possible route for H2 formation being the reaction of two neutral radical species in the neighboring polymer chains. In addition, density-functional theory (DFT) calculations of various scenarios from both mechanisms show highly endergonic processes (positive free energy) unless the formation of a larger anion, [BCF(OH)(OH2)BCF]− in which [BCF(OH)]− is hydrogen bonded to another BCF(OH2) complex, occurs.42 The formation of a large [BCF(OH)(OH2)BCF]− anion has been previously observed by Doerrer and Green in their study on the oxidation of metallocenes by BCF(OH2) in non-aqueous solvents, in which H2 was also proposed to be the side product.46 The formation of H2 proposed in these BCF oxidation reactions of both P3HT and metallocenes has not, however, been observed experimentally. Thus, molecular-level understanding of the doping mechanism is expected to help the rational development of design rules to produce Lewis acids for doping OSCs. However, such knowledge is rarely fully established, due to compositional and structural heterogeneity associated with molecularly doped conjugated polymers.
Studies on dopant–host morphology have mostly been based on techniques such as X-ray scattering that is limited to detecting the crystalline and well-ordered regions of a material, and atomic force or optical microscopy which are limited by their nature as surface techniques. Very few OSC doping studies have utilized complementary analytical techniques such as solid-state NMR (ssNMR) spectroscopy.32,47 Information on intermolecular interactions at the atomic-scale can be obtained through ssNMR techniques that cover the entirety of the material by probing both crystalline and amorphous regions in conjugated polymers and their blends.48–54 Specifically, 2D EPR techniques enable the electron–nuclear hyperfine interactions in polymer–dopant blends to be identified and distinguished at complementary length scales to ssNMR, as the latter technique probes predominantly the undoped regions of the polymer.
In this study, 1D and 2D ssNMR and EPR techniques were employed to study the intermolecular interactions between the dopants BCF–water complex and F4TCNQ with the OSC polymer referred to as P4 (Fig. 1). In P4:F4TCNQ, fully doped F4TCNQ molecules lead to strong hyperfine interactions with P4 backbones as revealed by 2D EPR analysis (e.g., charge transfer complexes that may disrupt π–π stacking), and the undoped/aggregated F4TCNQ molecules (i.e., diamagnetic regions) can be detected by 19F ssNMR spectroscopy. In the P4:BCF blend, the local boron environments of BCF dopants are characterized by 11B NMR and the hyperfine structure is characterized by 2D EPR techniques. Furthermore, the results discussed herein elucidate that the BCF doping mechanism does not rely on BCF association (or intercalation) with the conjugated backbone, as it does not require the formation of a charge-transfer complex. The results provide essential insights into polymer–dopant interactions, and partly explain the lower doping efficiency observed in F4TCNQ doped P4 (<4%) as compared to that of BCF doped P4 (>14%).55 The addition of these solid-state NMR and EPR results is expected to help experimental characterization and modelling studies of different BCF–polymer interactions and doping mechanisms.
 | ||
| Fig. 1 Structures of the host polymer P4 along with dopants BCF and F4TCNQ. | ||
Results and discussion
We separately examined the local structures and interactions in pristine P4 polymer and P4:dopant blends at different molar ratios. Based on our previous morphological studies of BCF-doped P4 using grazing incidence wide angle X-ray scattering (GIWAXS) and atomic force microscopy (AFM), we did not expect BCF to significantly alter the degree of molecular order in the P4 polymer.40 Nevertheless, the addition of a dopant to a polymer is expected to result in a variety of local chemical environments such as diamagnetic and paramagnetic species, which can be investigated by ssNMR and EPR techniques. For example, a molecular-level assessment of local structures and interactions in the neat P4, P4:BCF and P4:F4TCNQ samples can be made by analyzing the 1H and 13C magic-angle spinning (MAS) NMR spectra. The compositional and structural heterogeneity of the P4–dopant blends is indicated by broad signals covering a wider range of 1H and 13C chemical shift (δ) values than those of the pristine P4 polymer (ESI,† Fig. S1 and S2).56 Of particular note, the origin of the NMR signal broadening in the doped P4 polymers can be related to several short and long range interactions, anisotropic bulk magnetic susceptibility (ABMS) effects, hyperfine interactions, locally (dis)ordered structures and relaxation-induced broadening, which are difficult to deconvolute. However, 1H → 13C cross polarization (CP)-MAS NMR57 spectra of the P4–dopant blends exhibit higher signal broadening and intensity losses in their aromatic regions than the alkyl peaks, indicating that the noteworthy changes occur in the vicinity of the P4 backbone moieties (ESI,† Fig. S2). Notably, broad distributions of 13C peaks in the range of 105–175 ppm (∼70 ppm wide for P4:BCF and P4–F4TCNQ blends compared to ∼50 ppm for pristine P4) indicate the different local chemical environments in doped polymers and the presence of free radicals, which could also lead to signal intensity loss due to paramagnetism induced relaxation.58,59 Consequently, a large number of transients were coadded to acquire the 13C CP-MAS spectra of the P4:dopants with good signal-to-noise ratios (20480 and 16384 coadded transients, respectively, for P4:F4TCNQ and P4:BCF), compared to the coaddition of 4098 transients for the P4 polymer. Although 1H → 13C CP-MAS experiments of these materials do not provide quantitative assessment of 13C{1H} peak enhancements, a comparison of alkyl/aromatic 13C signal intensities indicates that the major intensity losses occur in the P4–dopant blends (Fig. S2, ESI†) compared to the neat P4 polymer.
To gain further insights into the signal intensity losses induced by the addition of a dopant to the P4 polymers, 1H MAS NMR spectra of undoped and doped P4 polymers (∼2.5 mg each) were acquired at identical experimental conditions and compared (ESI,† Fig. S3 and Table S1). For the pristine P4 polymer, the aliphatic signal (0–4 ppm) intensity is calibrated to 66, which leads to the estimation of the aromatic signal (5–9 ppm) intensity of 4.7, consistent with the composition of the CPDT-BT unit with 4 aromatic protons and 66 alkyl sidechain protons. By comparison, the peak integral of the alkyl sidechains in the P4:F4TCNQ (1:0.2 molar ratio) blend is reduced to 36.3 (calibrated with respect to 66 in the pristine P4), and the aromatic signal is reduced to 1.7, which suggests that the intensity losses occurred for both alkyl and aromatic peaks, though greater intensity losses were observed for aromatic peaks than the aliphatic signals. A similar trend is observed for the P4:BCF blend, whereby the peak integrals of alkyl (49.5) and aromatic (1.4) groups are decreased (Table S1, ESI†). It can be reasoned that the presence of both paramagnetic and diamagnetic entities in the vicinity of the P4 polymer backbones, as characterized by the EPR and NMR techniques discussed below, contribute to such peak broadening and intensity losses. For example, previous studies have shown that the F4TCNQ molecules intercalate between the π–π stacks and/or lamellar stacks of PBTTT-C14 and P3HT polymers, leading to charge transfer complexes.29,32,35,36,60–63 In addition, the contributions from the undoped polymer regions with self-aggregated dopant molecules to the NMR peak broadening cannot be ruled out.
To examine the backbone and sidechain moieties in the (un)doped P4 polymer more closely, we employed 2D 1H–13C heteronuclear correlation and 1H–1H double quantum-single quantum (DQ–SQ) correlation NMR spectroscopy (ESI,† Fig. S4 and S5). In measurements of this type, the 2D signals are created for through-space and dipolar coupled 1H–13C and 1H–1H spin pairs, which correspond to inter- and intramolecular interactions. Specifically, 2D 1H DQ–SQ combines several key advantages; 1H signals are detected in two different frequency dimensions, whereby the DQ signals originating from dipolar coupled 1H–1H pairs are manifested as the sum of 1H SQ chemical shifts (see Fig. 2, the vertical DQ axis has a chemical shift range twice as large as the horizontal SQ axis). The DQ intensities are characteristic of the strength of dipolar interactions between specific 1H–1H pairs, which manifest as on- and off-diagonal correlation peaks corresponding to chemically equivalent and distinct sites, respectively. This is illustrated with greater detail in the previous studies.64–66 A comparison of 2D 1H–1H DQ–SQ correlation spectra of P4, P4:BCF and P4:F4TCNQ blends acquired with 20 microseconds of recoupling time is shown in Fig. 2. For the neat P4 polymer (Fig. 2b), the broad distribution of 1H DQ peaks centered at ∼3 ppm corresponds to dipolar coupled 1H–1H pairs in the alkyl sidechains. The off-diagonal DQ signals at 7.5 and 9.3 ppm are due to the dipolar interactions between sidechain and aromatic 1H–1H pairs. The distribution of on-diagonal 1H DQ peaks in the range of 11–14 ppm (Fig. 2b, solid oval) is attributable to the close 1H–1H proximities in the BT units of the P4 polymer. Although the on-diagonal DQ peak associated with through-space H–H proximities in CPDT moieties is expected to resonate in the range of 14–16 ppm, this peak is too weak to be detected due to weak inter- and intramolecular dipolar interactions between the thiophene protons. Nonetheless, a 1H DQ–SQ spectrum of the neat P4 acquired with a large recoupling time of 40 microseconds (ESI,† Fig. S7a) shows a weak signal intensity at ∼15 ppm. In addition, the off-diagonal peak associated with the aromatic groups of the P4 appearing at 5.4 and 5.9 ppm on the SQ axis (DQ, 11.3 ppm) is attributable to the different local chemical environments of 1H sites with different aromatic ring current effects in the π–π stacked PDT units. In contrast to the pristine P4 material, the doped polymers display different DQ–SQ features whereby the aliphatic signals are detected with appreciable intensities but not the aromatic regions (Fig. 2c, d, dashed ovals) indicating that the free radicals are closely associated with the aromatic moieties of P4 polymers. In addition, the 1H DQ coherences relax much faster than the SQ coherences, which may contribute to further signal intensity losses, particularly in the localities of the free radicals. This is further evaluated by examining the horizontal line-cut spectra of 2D 1H DQ spectra of doped systems acquired with different recoupling times as shown in the ESI† (Fig. S6 and S7b, c). The weak intensity signals depicted in dashed ovals are either artifacts or any residual aromatic 1H DQ peaks originating from the undoped diamagnetic regions. This result is in line with the analysis of 1D 1H and 13C CP-MAS spectra, although 1H DQ–SQ NMR notably allows observation of the specific moieties of the polymer impacted by the doping process. It can be reasoned that the different doping mechanisms and the presence of different radical species contribute to the intensity losses, as will be investigated by EPR techniques discussed below.
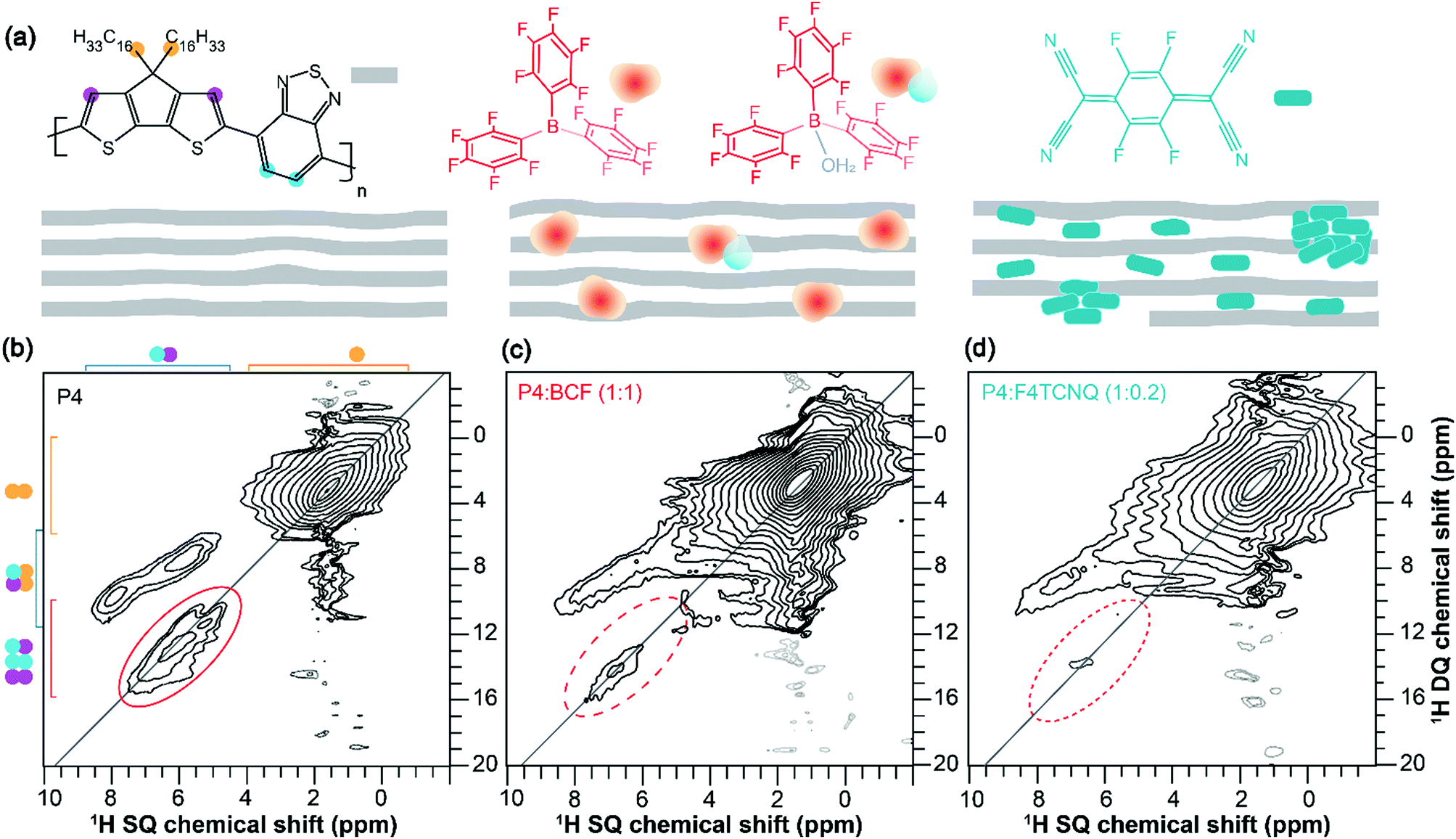 | ||
| Fig. 2 (a) Schematic diagram of the different distributions of dopant molecules in the host polymer. Solid-state 2D 1H–1H DQ–SQ correlation NMR spectra of (b) neat P4, (c) P4:BCF (1:1 molar equiv.) and (d) P4:F4TCNQ (1:0.2 molar equiv.) indicating the signal intensity losses in the aromatic regions of doped P4 polymers. Weak intensity signals in the dashed ovals are likely to be artefacts or residual DQ signals originating from the undoped diamagnetic aromatic regions of the P4–dopant blends. | ||
Information on the hyperfine structure and local chemical environments of dopants in organic semiconductors can be obtained by EPR spectroscopy,67–69 which has a single-site resolution to investigate paramagnetic species. Fig. 3 compares the X-band continuous-wave length (CW) EPR and pulsed EPR hyperfine sublevel correlation (HYSCORE) spectra of the P4, P4:BCF and P4:F4TCNQ blends. The CW spectrum of the P4 polymer (Fig. 3a) shows a very weak EPR signal with a 5 Gauss (G) linewidth centered at a g-factor of 2.0026 indicating the mobility of the delocalized electrons at minuscule concentrations. The addition of dopants to the P4 induces a spike in the signal intensity and a dramatic change in the EPR linewidth that decreases to 1.7 G with a g-factor that resembles the free electron g-value. However, the P4:BCF blend leads to a relatively stronger EPR signal than the P4:F4TCNQ, consistent with the increased doping efficiency associated with the BCF molecules. More elaboratively, the localization of dopant molecules in the P4 polymer network is analyzed by the 2D-HYSCORE technique that provides information on the hyperfine interactions between electrons and nuclear spins. The physical origins that contribute to hyperfine couplings are (i) anisotropic dipole–dipole interaction that depends on the relative orientation of magnetic moments, and (ii) isotropic Fermi-contact interaction that arises when there is a finite spin density of the electron spin at the nucleus. For the neat P4 polymer, a weak Fermi-contact hyperfine coupling associated with 1H (14.5 MHz) with a maximum anti-diagonal ridge extent of 8.2 MHz is detected in the (+, +) quadrant (Fig. 3b). In addition, weak hyperfine couplings associated with 13C and with 14N sites are observed. In the (−, + and +, +) quadrant of the same spectrum, the single-quantum and double-quantum signals associated with 14N are observed as indicated by red arrows. By comparison, the 2D HYSOCRE spectrum of P4:BCF (Fig. 3c) exhibits weak hyperfine couplings with the two isotopes of boron 10B (ν = 1.6 MHz) and 11B (ν = 4.8 MHz) on the diagonal (red arrows), and does not show anti-diagonal signals, indicating that these hyperfine interactions arise purely from dipolar interaction. A well-resolved 19F (ν = 13.7 MHz) signal with a maximum hyperfine coupling of 2.1 MHz indicates that the fluorine atoms are spin polarized by the electrons, whereas the 1H signal (ν = 14.9 MHz) with a hyperfine coupling of 5 MHz (with an electron-1H distance greater than 0.4 nm) suggests that the bulky aromatic BCF molecules are located in the vicinity of the P4 backbone moieties rather than intercalated between the π–π stacked P4 backbones. In the case of P4:F4TCNQ (Fig. 3d), a 1H signal at ν = 14.9 MHz originates from a pure dipolar interaction with a vertical shift of 1.4 MHz with a distance of less than 0.38 nm between the unpaired electron and 1H, as estimated based on the dipole single point approximation. This hyperfine coupling is within the regime of typical π–π stacking interactions in conjugated polymers, suggesting that the F4TCNQ molecules are intercalated, at least in part, between the π–π stacked P4 polymers. Combined, the 2D ssNMR and EPR results corroborate that the BCF and F4TCNQ molecules exhibit different intermolecular interactions with the P4 polymer, leading to different doping mechanisms and efficiencies.
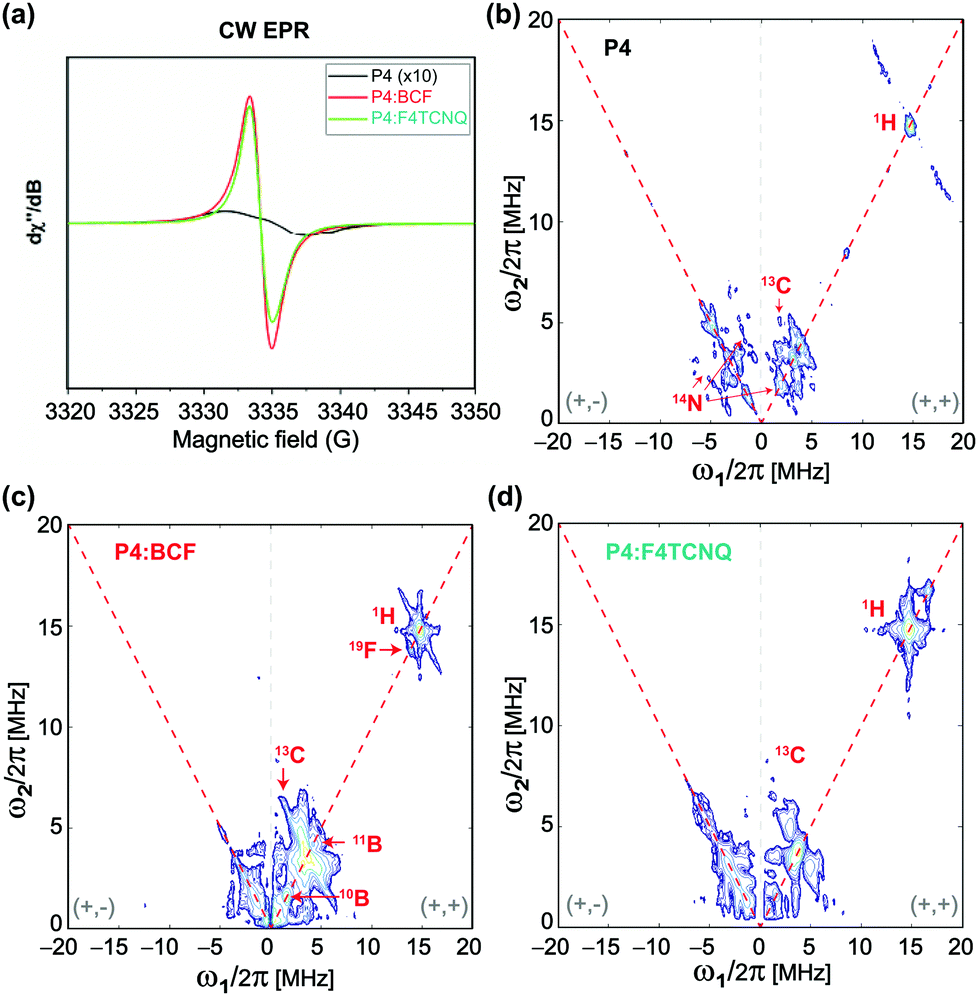 | ||
| Fig. 3 Solid-state (a) continuous-wave EPR spectra of the P4 and P4:dopant blends and 2D HYSCORE spectra of the (b) P4, (c) P4:BCF (1:1 molar equiv.) and (d) P4:F4TCNQ (1:0.2 molar equiv.) indicating the different hyperfine interactions between the P4 and dopant molecules. | ||
Further insights into the different doping effects in the P4:BCF and P4:F4TCNQ blends can be obtained by analyzing NMR longitudinal relaxation times (T1) of 1H sites in the P4 polymer. The presence of paramagnetic species in doped polymers influences the associated longitudinal relaxation (T1) times of nuclear spins.59 For the neat P4 and P4:dopant blends, the T1 relaxation times are given in Table S2 (ESI†). Aromatic 1H sites in P4:BCF show relatively longer T1 values (∼0.89 s) than the same sites in the P4:F4TCNQ blend (0.03 s), indicating different nuclear spin relaxation behaviors in these blends. However, the P4:BCF–water complexes exhibit much shorter T1 values for both alkyl and aromatic 1H sites. The different T1 values are hypothesized to originate from different distributions of dopant molecules in the polymers and the different hyperfine interactions in the P4:F4TCNQ, P4:BCF and P4:BCF–water blends. Although the 1H T1 relaxation measurements and analysis corroborate that the loss of 1H DQ signal intensities in the P4:dopant blends (Fig. 2c) is due to the presence of paramagnetic species in much closer proximity to the P4 backbone moieties (as confirmed by EPR analysis, Fig. 3), further investigation of the factors that contribute to site-specific T1 relaxation times is required to gain insight into the different relaxation behaviors in the doped polymers.
By analyzing the 11B (spin magnetic moment I = 3/2 and exhibits quadrupolar interaction) MAS NMR spectra of the BCF and BCF:P4 blends, we provide further insight into the changes in the local chemical environments of BCF molecules upon addition to the P4 polymer. As seen in Fig. 4a, the 1D 11B spectrum of BCF shows a broad quadrupolar lineshape between 45 and 60 ppm produced by the tri-coordinated boron atoms in BCF, and a relatively narrow lineshape near 0 ppm attributed to tetracoordinated boron nuclei in the BCF·H2O complex. As a result of the increased coordination number of boron, and reduced quadrupolar interactions, the tetracoordinated BCF·H2O complex exhibits a relatively narrower signal.57,70 Although the neat BCF material was stored under a nitrogen atmosphere in a glovebox, trace amounts of water present (or exposure to ambient atmosphere during the sample preparation for ssNMR studies) were enough to result in the BCF·H2O complex,40 and different types of BCF–water complexes.42 Upon addition to P4, there is further narrowing of the 11B peak appearing at −4 ppm, which could be attributed to the BCF molecules embedded into the polymer chains and the hyperfine interactions between BCF–water/BCF·OH− and the P4 polymer radicals. When a higher concentration of BCF is incorporated into the P4 polymer (1:1 molar ratio), an additional 11B signal emerges at ∼18 ppm, indicating the formation of additional BCF·water, BCF·OH− like or bridged complexes involving two BCF molecules.40,42 It is noteworthy that the conjugated polymers with extended sidechains are lipophilic, and thus enhance the moisture stability of the BCF molecules embedded into the P4 polymer. Addition of water molecules promotes the formation of BCF·water and bridged complexes, which influence the doping efficiency of the P4:BCF blend. To test this, we carried out 11B and 1H MAS NMR experiments as a function of the amount of water added to the P4:BCF blend, whereby the 11B signal at 15–19 ppm is detected with much higher intensity (Fig. 4a). The 1H NMR spectra also show the emergence of a water 1H signal resonating at ∼4.5 ppm (Fig. 4b), and the 1H signal integral values associated with aliphatic and aromatic protons (13.2 and 0.5) indicate further signal intensity loss upon addition of water to the P4:BCF blend (Table S1, ESI†). In addition, a comparison of the 1D X-band CW-EPR spectra of the P4, P4:F4TCNQ, P4:BCF and P4:BCF + water complexes (ESI,† Fig. S8) suggests that the P4:BCF–water complex has a relatively higher concentration of radical species as indicated by the stronger signal intensity than the P4:BCF blend. The distinct EPR features of the P4:BCF + water complex are due to the different local chemical environments of dopant molecules and hyperfine interactions between P4 and BCF–water complexes. Overall, this analysis suggests that the increased concentrations of the paramagnetic species are formed when a drop of water is added to the P4:BCF complex. This result is in line with our previous observation, whereby the addition of a drop of water to P4:BCF resulted in enhanced charge carrier mobility.40 Conversely, when deuterated water (D2O) is added to the P4:BCF blend, there is no such trend in the emergence of the 11B signal intensity (15–20 ppm) or the additional 1H signal intensity loss in the aromatic regions of the 1H MAS NMR spectra. It can be reasoned that the deuteration of water molecules can affect the formation of bridged BCF–water complexes (for example, large anions), due to the geometric isotopic effects on the hydrogen bonding interactions, which may ultimately influence the proton transfer process to the P4 polymer chains.
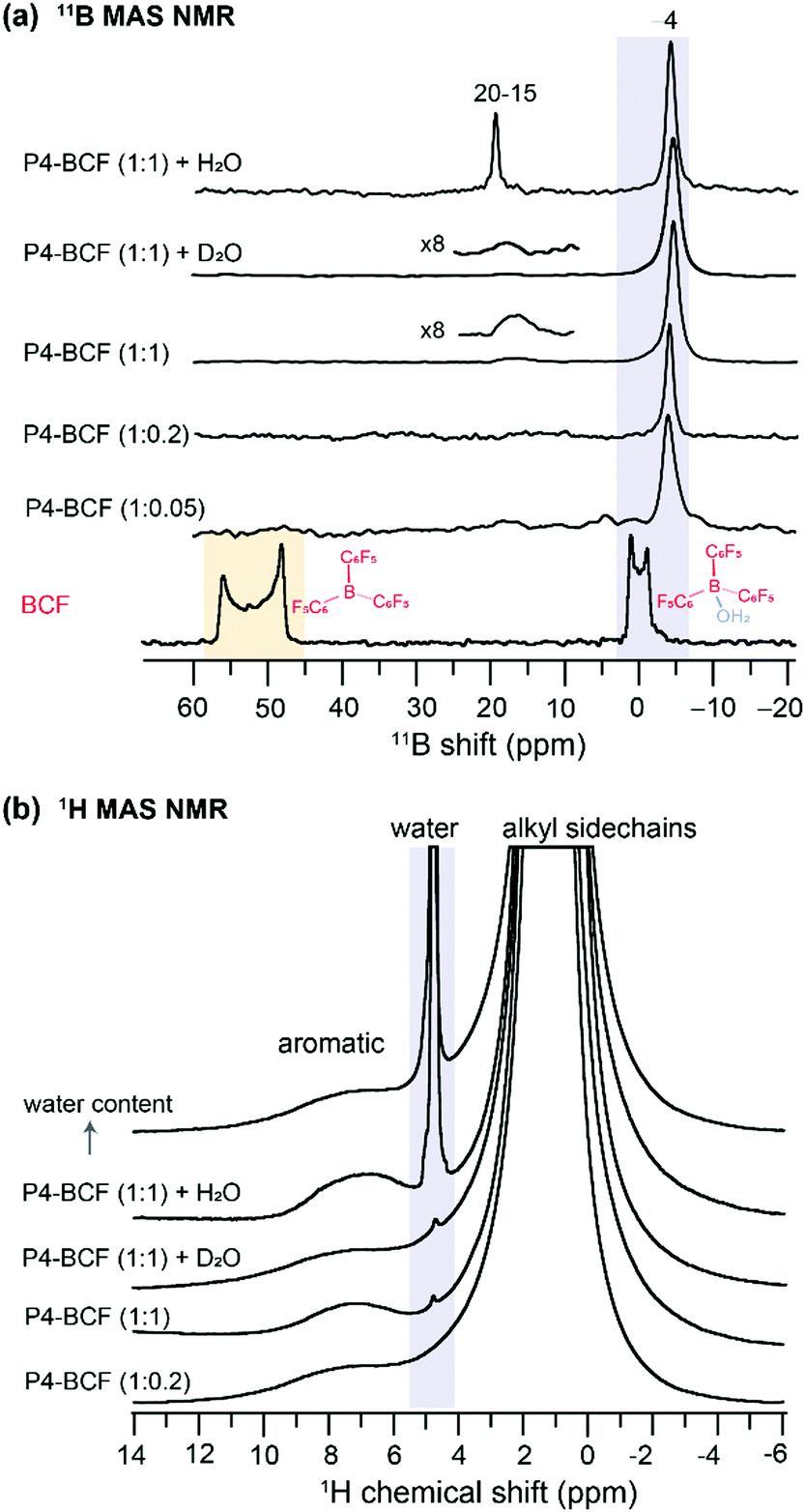 | ||
| Fig. 4 1D 11B MAS NMR spectra of (a) BCF, P4:BCF at different molar ratios and P4:BCF with a drop of added water and water-d. (b) 1H MAS NMR spectra of P4:BCF with and without added water or water-d. All spectra were acquired at 18.8 T (1H = 800.1 MHz, 11B = 256.7 MHz) at room temperature. | ||
From the 1D 19F NMR of the neat F4TCNQ (Fig. 5a), we observe two distinct peaks at −129 and −133 ppm corresponding to the unique local environments for fluorine atoms in the crystal structure of F4TCNQ.71,72 For the P4:F4TCNQ blends (1:0.02 and 1:0.2 molar ratios), the detection of 19F signals is challenging due to minuscule concentrations of F4TCNQ molecules and longer 19F longitudinal relaxation delays of up to several minutes.32 This is further exacerbated by the presence of delocalized electrons in the vicinity of intercalated F4TCNQ molecules between the polymer backbones, as revealed by the EPR analysis and the intensity loss in the 1H NMR spectrum of P4:F4TCNQ as shown in Fig. 2. Here we used a spin-echo filter to suppress background signals from the NMR probe, and a large number of transients were coadded (1600 scans) to acquire the 19F spectrum of P4:F4TCNQ (1:0.02) in order to identify two distinct 19F signals at −128 and −150 ppm. Further addition of F4TCNQ (0.2 molar equivalents) produces two additional local environments for fluorine nuclei, resulting in partially resolved peaks at −134, −135, −138 and −143 ppm. Although 19F signals corresponding to the fully doped P4:F4TCNQ regions (i.e., intercalated F4TCNQ molecules) may not be observed in ssNMR experiments,32 the NMR-detected 19F signals are expected to originate from the nonconducting regions and/or locally aggregated F4TCNQ molecules in the proximity of lamellar regions.35 The combined ssNMR and EPR results suggest that the F4TCNQ molecules are distributed into different local chemical environments in the P4 polymer, some of which, but not all, are involved in the doping process. However, from the 1D 19F ssNMR data alone we cannot discern which peaks would correspond to backbone intercalated, crystalline and disordered aggregates of F4TCNQ molecules in the P4:F4TCNQ blends. To further resolve these signals, we used a spin-echo filter73 of 10 μs, which suppresses the signals from the probe background and a series of 1D 19F NMR spectra (Fig. 5b) were acquired by varying the longitudinal relaxation delays in the range from 0.5 to 50 s. This experiment allows fast and slow relaxing 19F sites in the aggregated and ordered F4TCNQ molecules to be distinguished and identified. Distribution of F4TCNQ molecules closely associated with P4 backbone moieties produces broad peaks at −143 and −134 ppm, visible after a short relaxation delay (0.5 s), whereas the aggregated F4TCNQ results in narrow signals at −138 and −135 ppm which were detected at higher intensities when a relaxation delay of greater than 20 s is used. It is noteworthy that these relaxation delays are much smaller than the T1 relaxation delays of the neat F4TCNQ (up to 600s), suggesting that the added F4TCNQ molecules to the P4 polymer exhibit different local structures and dynamics than the pristine F4TCNQ. For example, the literature reports detail F4TCNQ aggregate formation at high doping concentrations, which can be avoided at low dopant concentrations.44,74 This trend is observed in the P4:F4TCNQ system as revealed by the 1D 19F NMR experiment carried out for a lower P4:F4TCNQ molar ratio of 1.0:0.02 (Fig. 5a), which showed only the peaks near −128 and −150 ppm. These 19F chemical shifts are identical to those reported earlier, which correspond to dopant molecules located in the vicinity of the P4 backbone moieties, further supporting the assignments made by the spin echo experiments.
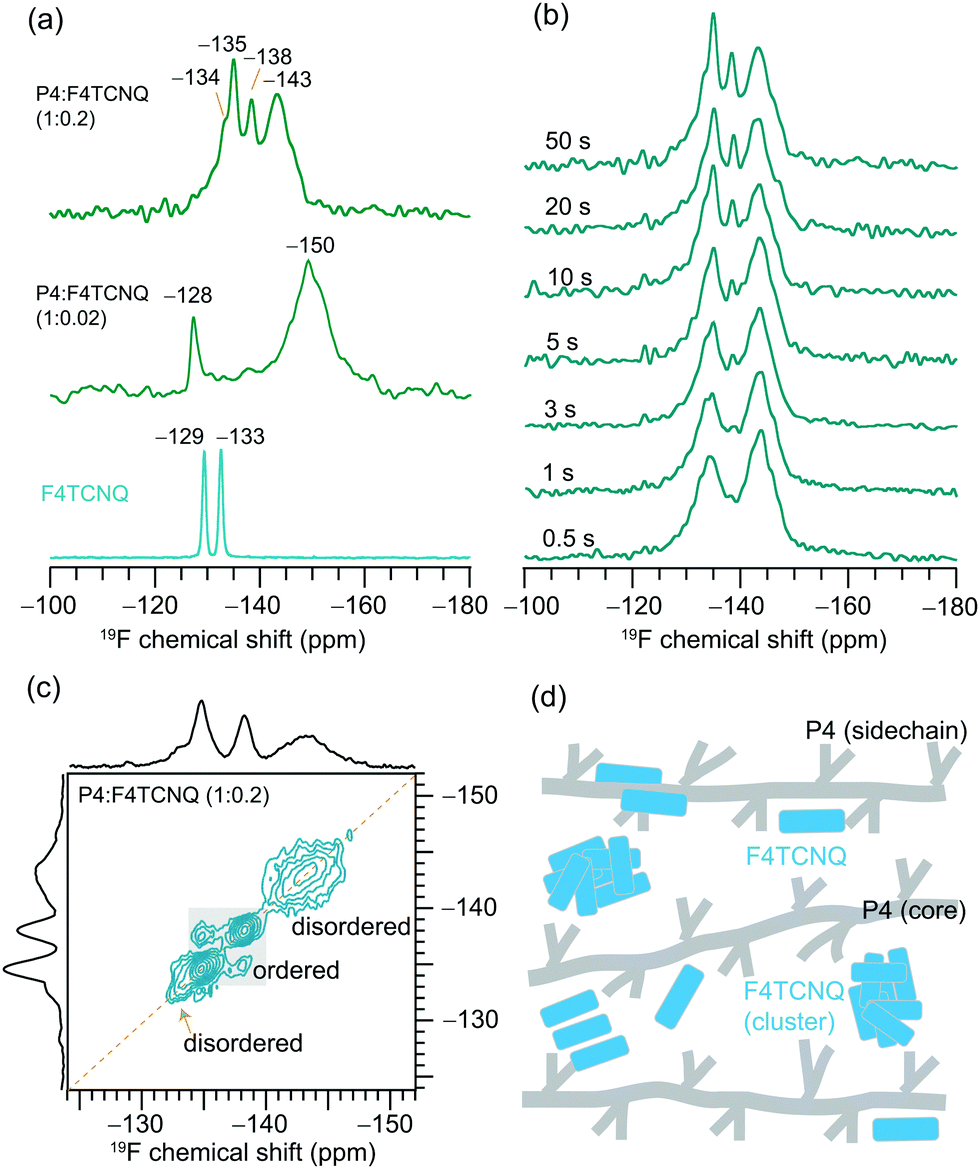 | ||
| Fig. 5 Solid-state (a) 1D 19F MAS NMR spectra of the neat F4TCNQ and P4:F4TCNQ blends with different F4TCNQ molar ratios, and (b) T2-filtered 19F MAS NMR spectra of P4:F4TCNQ (1:0.2) as a function of spin-echo time, and (c) 2D 19F–19F spin-diffusion NMR spectrum of P4:F4TCNQ (1:0.2) acquired with 500 ms mixing time. (d) Schematic representation of different distributions of F4TCNQ molecules in the P4 polymer chains. | ||
Correspondingly, 2D 19F–19F spin diffusion measurements also provided resolution of the aggregated and distributed F4TCNQ molecules in the doped P4 polymer (Fig. 5c and d). These experiments use a spin diffusion delay (also referred to as mixing time) to allow magnetization exchange between dipolar coupled 19F spins. A small mixing time of 10 ms is not enough for the exchange of the magnetization between 19F sites to occur (Fig. S9, ESI†), resulting in only self-correlation peaks that appear along the diagonal. The longer spin diffusion time of 500 ms allows a markedly different spectrum to be obtained, showing cross-correlation peaks between the signals at −138 and −135 ppm on both axes, which indicates spin magnetization exchange between chemically distinct 19F nuclei in the F4TCNQ aggregates. In contrast, the lack of cross (off-diagonal) peaks between intercalated (or closely associated with the P4 backbone) 19F nuclei producing peaks at −143 ppm and those in the aggregates producing peaks at −135 ppm suggests that these two 19F sites are too distant or locally disordered to allow for spin magnetization exchange to occur between them. The F4TCNQ aggregation in the blend and the partial disruption of the P4 backbone by F4TCNQ explain the lower doping efficiency observed in P4:F4TCNQ films (<4%). Further analysis of cooperative interactions that drive molecular self-assembly in OSCs are expected to help to understand the aggregation behaviors of small molecular dopants in polymers.75
Owing to the inherently heterogeneous chemical nature, the π-conjugated polymers and their blends represent a characterization challenge. The addition of dopant molecules causes changes to the molecular self-assembly and local structures at sub-nanometer distances, which are often difficult to analyze by X-ray scattering and microscopy techniques, and insights offered by infrared spectroscopy are also limited. On the other hand, electroanalytical and UV-visible spectroscopy techniques, though they provide information on the macroscopic changes such as conductivity and optical absorption, do not resolve atomic-level structures and interactions. Therefore, it is less straightforward to reach a consensus about structure–property relationships. To this end, the ssNMR and EPR analysis presented here enabled the specific moieties of doped polymers impacted such as backbone and sidechain structures, and hyperfine interactions to be identified and distinguished. Our results indicate that the addition of F4TCNQ and BCF to the P4 polymer leads to different intermolecular interactions and structural changes due to different doping mechanisms. Some of the F4TCNQ molecules, but not all, dope the P4 polymer by forming charge transfer complexes (free radicals that can be detected by EPR and confirmed by the signal intensity loss in the aromatic 1H NMR spectra of the P4 polymer), and the self-aggregated F4TCNQ molecules distributed within the polymer network can be analyzed by 19F MAS NMR and relaxation measurements. In addition, ssNMR and EPR analyses provide essential insights into the BCF-doped P4 polymers that exhibit an entirely different doping mechanism, whereby BCF molecules interact with traces of water molecules to serve as Brønsted acid sites to transfer a proton to the P4 polymer leading to BCF·OH− and other BCF–water Brønsted acid complexes that are detected by 11B MAS NMR. These magnetic resonance techniques present several opportunities and obvious challenges for the study of doped polymers. For example, it is less straightforward to distinguish between the doped and undoped polymer regions and polymer–dopant interfaces precisely and quantitively. It is also difficult to determine the domain sizes and shapes of the doped and undoped polymers due to the short–range nature of both EPR and NMR techniques. More in general, polymer blends consisting of dia- and paramagnetic species lead to signal broadening due to several short and long-range interactions such as Fermi contacts, pseudocontacts, and anisotropic bulk magnetic susceptibility (ABMS)-induced shifts, as well as the dipolar contributions and locally disordered regions, which are additive, and deconvolution of these factors to rationalize the NMR peak broadening requires the ability to quantify them separately.
Conclusions
The use of high resolution ssNMR and EPR techniques in this study has provided valuable insights into the intermolecular interactions at sub-nanometer distances, which underlie the previously observed differences in doping efficiency between the different molecular dopants. In the case of the P4:BCF blend, the BCF molecules are closely associated with the P4 backbones, though they cause minimal disruption to the conjugated backbone stacking, which is critical for charge transport in organic semiconductors. In addition, BCF can interact with traces of water molecules to form Brønsted acid BCF–water complexes. The 11B NMR signals corresponding to tri- and tetracoordinated BCF complexes are distinguished and identified, and EPR analysis further corroborates the close polymer–dopant interactions, suggesting that the BCF–water complexes serve as Brønsted acid sites to enhance the doping efficiency (>14%). By comparison, the combined EPR and NMR results indicate that the fully doped F4TCNQ molecules are closely associated with the P4 backbones, and the undoped F4TCNQ molecules are distributed into different aggregates, specifically by illustrating the 2D 1H-1H and 19F-19F NMR measurements and analysis. This observation together with F4TCNQ aggregation helps to explain the much lower doping efficiency (<4%) of P4:F4TCNQ.40 Insight into the hyperfine interactions in the P4, P4:BCF and P4:F4TCNQ blends is obtained by analyzing the pulse 2D HYSCORE spectra. Furthermore, these results are complemented by 1H longitudinal relaxation (T1) measurements and analysis that indicate that the addition of dopants has an impact on the relaxation behaviors of 1H sites in the P4 polymers. Therefore, a high-resolution ssNMR and EPR approach can be applied to obtain essential structural insights and have much wider relevance to characterize intermolecular interactions in small molecule doped π-conjugated polymers.
Experimental
Materials, synthesis and deposition
The polymer poly[2,6-(4,4-bis(2-hexadecyl)-4H-cyclopenta[2,1-b;3,4-b′]dithiophene)-alt-4,7(2,1,3-benzothiadiazole)] (P4), BCF and F4TCNQ were purchased from commercial suppliers, and P4 and P4:dopants at the indicated molar ratios were prepared according to the procedure described in the ESI.†
Solid-state NMR and EPR spectroscopy
All solid-state NMR and EPR experiments were carried out at the high field NMR/EPR facility at the University of Lille, France. Sample preparation for ssNMR and EPR experiments, 1D and 2D MAS NMR data acquisition and spectral analysis details are given in the ESI.†Author contributions
This manuscript was written through the contributions of all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. L. D. and T.-Q. N. acknowledge the support from the US Department of Energy under Award no. DE-SC0017659. G. N. M. R. acknowledges financial support from the University of Lille, UCCS laboratory and IR-RMN-THC FR-3050 CNRS France for conducting solid-state NMR measurements. H. V. acknowledges the support from IR RENARD (FR384).References
- B. Lüssem, M. Riede and K. Leo, Doping of organic semiconductors, John Wiley & Sons, Ltd, 2013, vol. 210 Search PubMed.
- J. Euvrard, Y. Yan and D. B. Mitzi, Nat. Rev. Mater., 2021, 6, 531–549 CrossRef CAS.
- A. Köhler and H. Bässler, Electronic processes in organic semiconductors: An introduction, 2015 Search PubMed.
- H. Shirakawa, E. J. Louis, A. G. MacDiarmid, C. K. Chiang and A. J. Heeger, J. Chem. Soc., Chem. Commun., 1977, 36, 578–580 RSC.
- Y. Yamamoto, K. Yoshino and Y. Inuishi, J. Phys. Soc. Jpn., 1979, 47, 1887–1891 CrossRef CAS.
- G. Parthasarathy, C. Shen, A. Kahn and S. R. Forrest, J. Appl. Phys., 2001, 89, 4986–4992 CrossRef CAS.
- M. Pfeiffer, K. Leo, X. Zhou, J. S. Huang, M. Hofmann, A. Werner and J. Blochwitz-Nimoth, Org. Electron., 2003, 4, 89–103 CrossRef CAS.
- K. Walzer, B. Männig, M. Pfeiffer and K. Leo, Chem. Rev., 2007, 107, 1233–1271 CrossRef CAS PubMed.
- Y. Han, G. Barnes, Y. H. Lin, J. Martin, M. Al-Hashimi, S. Y. Alqaradawi, T. D. Anthopoulos and M. Heeney, Chem. Mater., 2016, 28, 8016–8024 CrossRef CAS.
- A. F. Paterson, L. Tsetseris, R. Li, A. Basu, H. Faber, A. H. Emwas, J. Panidi, Z. Fei, M. R. Niazi, D. H. Anjum, M. Heeney and T. D. Anthopoulos, Adv. Mater., 2019, 31, 1–8 Search PubMed.
- A. Basu, M. R. Niazi, A. D. Scaccabarozzi, H. Faber, Z. Fei, D. H. Anjum, A. F. Paterson, O. Boltalina, M. Heeney and T. D. Anthopoulos, J. Mater. Chem. C, 2020, 8, 15368–15376 RSC.
- M. H. An, R. Ding, G. Da Ye, Q. C. Zhu, Y. N. Wang, B. Xu, M. L. Xu, X. P. Wang, W. Wang, J. Feng and H. B. Sun, Org. Electron., 2021, 91, 106089 CrossRef CAS.
- Y. Lin, Y. Firdaus, M. I. Nugraha, F. Liu, S. Karuthedath, A. H. Emwas, W. Zhang, A. Seitkhan, M. Neophytou, H. Faber, E. Yengel, I. McCulloch, L. Tsetseris, F. Laquai and T. D. Anthopoulos, Adv. Sci., 2020, 7, 1–9 Search PubMed.
- I. Salzmann, G. Heimel, M. Oehzelt, S. Winkler and N. Koch, Acc. Chem. Res., 2016, 49, 370–378 CrossRef CAS PubMed.
- F. Deschler, D. Riedel, A. Deák, B. Ecker, E. von Hauff and E. Da Como, Synth. Met., 2015, 199, 381–387 CrossRef CAS.
- J. Gao, J. D. Roehling, Y. Li, H. Guo, A. J. Moulé and J. K. Grey, J. Mater. Chem. C, 2013, 5638–5646 RSC.
- D. Tyler Scholes, S. A. Hawks, P. Y. Yee, H. Wu, J. R. Lindemuth, S. H. Tolbert and B. J. Schwartz, J. Phys. Chem. Lett., 2015, 6, 4786–4793 CrossRef PubMed.
- J. Li, G. Zhang, D. M. Holm, I. E. Jacobs, B. Yin, P. Stroeve, M. Mascal and A. J. Moulé, Chem. Mater., 2015, 27, 5765–5774 CrossRef CAS.
- D. T. Duong, H. Phan, D. Hanifi, P. S. Jo, T. Q. Nguyen and A. Salleo, Adv. Mater., 2014, 26, 6069–6073 CrossRef CAS PubMed.
- D. Kiefer, R. Kroon, A. I. Hofmann, H. Sun, X. Liu, A. Giovannitti, D. Stegerer, A. Cano, J. Hynynen, L. Yu, Y. Zhang, D. Nai, T. F. Harrelson, M. Sommer, A. J. Moulé, M. Kemerink, S. R. Marder, I. McCulloch, M. Fahlman, S. Fabiano and C. Müller, Nat. Mater., 2019, 18, 149–155 CrossRef CAS PubMed.
- Y. Yamashita, J. Tsurumi, T. Kurosawa, K. Ueji, Y. Tsuneda, S. Kohno, H. Kempe, S. Kumagai, T. Okamoto, J. Takeya and S. Watanabe, Commun. Mater., 2021, 2, 1–9 CrossRef.
- B. Lüssem, C. M. Keum, D. Kasemann, B. Naab, Z. Bao and K. Leo, Chem. Rev., 2016, 116, 13714–13751 CrossRef PubMed.
- R. Fujimoto, Y. Yamashita, S. Kumagai, J. Tsurumi, A. Hinderhofer, K. Broch, F. Schreiber, S. Watanabe and J. Takeya, J. Mater. Chem. C, 2017, 5, 12023–12030 RSC.
- Z. Zhao, Z. Liu, Q. Zhong, Y. Qin, A. Xu, W. Li and J. Shi, ACS Appl. Energy Mater., 2020, 3, 8774–8785 CrossRef CAS.
- C. R. Bridges and T. Baumgartner, J. Phys. Org. Chem., 2020, 33, 1–17 Search PubMed.
- C. Bergquist, B. M. Bridgewater, C. J. Harlan, J. R. Norton, R. A. Friesner and G. Parkin, J. Am. Chem. Soc., 2000, 122, 10581–10590 CrossRef CAS.
- E. H. Suh, J. G. Oh, J. Jung, S. H. Noh, T. S. Lee and J. Jang, Adv. Energy Mater., 2020, 10, 2002521 CrossRef CAS.
- Y. Yamashita, J. Tsurumi, M. Ohno, R. Fujimoto, S. Kumagai, T. Kurosawa, T. Okamoto, J. Takeya and S. Watanabe, Nature, 2019, 572, 634–638 CrossRef CAS PubMed.
- E. Lim, K. A. Peterson, G. M. Su and M. L. Chabinyc, Chem. Mater., 2018, 30, 998–1010 CrossRef CAS.
- M. T. Fontana, D. A. Stanfield, D. T. Scholes, K. J. Winchell, S. H. Tolbert and B. J. Schwartz, J. Phys. Chem. C, 2019, 123, 22711–22724 CrossRef CAS.
- H. Hase, K. O’Neill, J. Frisch, A. Opitz, N. Koch and I. Salzmann, J. Phys. Chem. C, 2018, 122, 25893–25899 CrossRef CAS.
- J. E. Cochran, M. J. N. Junk, A. M. Glaudell, P. L. Miller, J. S. Cowart, M. F. Toney, C. J. Hawker, B. F. Chmelka and M. L. Chabinyc, Macromolecules, 2014, 47, 6836–6846 CrossRef CAS.
- K. E. Watts, B. Neelamraju, E. L. Ratcliff and J. E. Pemberton, Chem. Mater., 2019, 31, 6986–6994 CrossRef CAS.
- S. Nagamatsu and S. S. Pandey, Sci. Rep., 2020, 10, 1–10 CrossRef PubMed.
- V. Untilova, T. Biskup, L. Biniek, V. Vijayakumar and M. Brinkmann, Macromolecules, 2020, 53, 2441–2453 CrossRef CAS.
- I. E. Jacobs, C. Cendra, T. F. Harrelson, Z. I. Bedolla Valdez, R. Faller, A. Salleo and A. J. Moulé, Mater. Horiz., 2018, 5, 655–660 RSC.
- I. E. Jacobs and A. J. Moulé, Adv. Mater., 2017, 29, 1–39 Search PubMed.
- C. D. Dong and S. Schumacher, J. Phys. Chem. C, 2019, 123, 30863–30870 CrossRef CAS.
- O. Zapata-Arteaga, B. Dörling, A. Perevedentsev, J. Martín, J. S. Reparaz and M. Campoy-Quiles, Macromolecules, 2020, 53, 609–620 CrossRef CAS PubMed.
- B. Yurash, D. X. Cao, V. V. Brus, D. Leifert, M. Wang, A. Dixon, M. Seifrid, A. E. Mansour, D. Lungwitz, T. Liu, P. J. Santiago, K. R. Graham, N. Koch, G. C. Bazan and T. Q. Nguyen, Nat. Mater., 2019, 18, 1327–1334 CrossRef CAS PubMed.
- M. Arvind, C. E. Tait, M. Guerrini, J. Krumland, A. M. Valencia, C. Cocchi, A. E. Mansour, N. Koch, S. Barlow, S. R. Marder, J. Behrends and D. Neher, J. Phys. Chem. B, 2020, 124, 7694–7708 CrossRef CAS PubMed.
- P. S. Marqués, G. Londi, B. Yurash, T. Q. Nguyen, S. Barlow, S. R. Marder and D. Beljonne, Chem. Sci., 2021, 12, 7012–7022 RSC.
- P. Pingel, M. Arvind, L. Kölln, R. Steyrleuthner, F. Kraffert, J. Behrends, S. Janietz and D. Neher, Adv. Electron. Mater., 2016, 2, 1600204 CrossRef.
- D. T. Duong, C. Wang, E. Antono, M. F. Toney and A. Salleo, Org. Electron., 2013, 14, 1330–1336 CrossRef CAS.
- E. F. Aziz, A. Vollmer, S. Eisebitt, W. Eberhardt, P. Pingel, D. Neher and N. Koch, Adv. Mater., 2007, 19, 3257–3260 CrossRef CAS.
- L. H. Doerrer and M. H. Green, Dalton Trans., 1999, 4325–4329 RSC.
- Z. Zujovic, P. A. Kilmartin and J. Travas-Sejdic, Molecules, 2020, 25, 444 CrossRef CAS PubMed.
- M. T. Seifrid, G. N. M. Reddy, C. Zhou, B. F. Chmelka and G. C. Bazan, J. Am. Chem. Soc., 2019, 141, 5078–5082 CrossRef CAS PubMed.
- M. Seifrid, G. N. M. Reddy, B. F. Chmelka and G. C. Bazan, Nat. Rev. Mater., 2020, 5, 910–930 CrossRef CAS.
- A. Melnyk, M. J. N. Junk, M. D. McGehee, B. F. Chmelka, M. R. Hansen and D. Andrienko, J. Phys. Chem. Lett., 2017, 8, 4155–4160 CrossRef CAS PubMed.
- A. Karki, J. Vollbrecht, A. J. Gillett, S. S. Xiao, Y. Yang, Z. Peng, N. Schopp, A. L. Dixon, S. Yoon, M. Schrock, H. Ade, G. N. M. Reddy, R. H. Friend and T. Q. Nguyen, Energy Environ. Sci., 2020, 13, 3679–3692 RSC.
- A. Karki, J. Vollbrecht, A. L. Dixon, N. Schopp, M. Schrock, G. N. M. Reddy and T. Nguyen, Adv. Mater., 2019, 31, 1903868 CrossRef CAS PubMed.
- B. Yurash, D. Leifert, G. N. M. Reddy, D. X. Cao, S. Biberger, V. V. Brus, M. Seifrid, P. J. Santiago, A. Köhler, B. F. Chmelka, G. C. Bazan and T. Q. Nguyen, Chem. Mater., 2019, 31, 6715–6725 CrossRef CAS.
- A. Karki, G. J. A. H. Wetzelaer, G. N. M. Reddy, V. Nádaždy, M. Seifrid, F. Schauer, G. C. Bazan, B. F. Chmelka, P. W. M. Blom and T. Q. Nguyen, Adv. Funct. Mater., 2019, 29, 1901109 CrossRef.
- B. Yurash, D. X. Cao, V. V. Brus, D. Leifert, M. Wang, A. Dixon, M. Seifrid, A. E. Mansour, D. Lungwitz, T. Liu, P. J. Santiago, K. R. Graham, N. Koch, G. C. Bazan and T. Q. Nguyen, Nat. Mater., 2019, 18, 1327–1334 CrossRef CAS PubMed.
- I. Farnan, P. J. Grandinetti, J. H. Baltisberger, J. F. Stebbins, U. Werner, M. A. Eastman and A. Pines, Nature, 1992, 358, 31–35 CrossRef CAS PubMed.
- M. J. Duer, Solid-State NMR Spectroscopy Principles and Applications, Blackwell Science Ltd, Oxford, UK, 2001 Search PubMed.
- F. Devreux, G. Bidan, A. A. Syed and C. Tsintavis, J. Phys., 1985, 46, 1595–1601 CrossRef CAS.
- A. C. Kolbert, S. Caldarelli, K. F. Thier, N. S. Sacriciftci, Y. Cao and A. J. Heeger, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 1541–1545 CrossRef CAS PubMed.
- H. Méndez, G. Heimel, S. Winkler, J. Frisch, A. Opitz, K. Sauer, B. Wegner, M. Oehzelt, C. Röthel, S. Duhm, D. Többens, N. Koch and I. Salzmann, Nat. Commun., 2015, 6, 8560 CrossRef PubMed.
- D. T. Scholes, P. Y. Yee, J. R. Lindemuth, H. Kang, J. Onorato, R. Ghosh, C. K. Luscombe, F. C. Spano, S. H. Tolbert and B. J. Schwartz, Adv. Funct. Mater., 2017, 27, 1–13 CrossRef.
- J. Hynynen, D. Kiefer, L. Yu, R. Kroon, R. Munir, A. Amassian, M. Kemerink and C. Müller, Macromolecules, 2017, 50, 8140–8148 CrossRef CAS PubMed.
- C. E. Tait, A. Reckwitz, M. Arvind, D. Neher, R. Bittl and J. Behrends, Phys. Chem. Chem. Phys., 2021, 23, 13827–13841 RSC.
- S. P. Brown, Prog. Nucl. Magn. Reson. Spectrosc., 2007, 50, 199–251 CrossRef CAS.
- S. P. Brown and H. W. Spiess, Chem. Rev., 2001, 101, 4125–4155 CrossRef CAS PubMed.
- S. P. Brown, Solid State Nucl. Magn. Reson., 2012, 41, 1–27 CrossRef CAS PubMed.
- A. Privitera, R. Warren, G. Londi, P. Kaienburg, J. Liu, A. Sperlich, A. E. Lauritzen, O. Thimm, A. Ardavan, D. Beljonne and M. Riede, J. Mater. Chem. C, 2021, 9, 2944–2954 RSC.
- T. Biskup, Front. Chem., 2019, 7, 10 CrossRef CAS PubMed.
- C. E. Tait, A. Reckwitz, M. Arvind, D. Neher, R. Bittl and J. Behrends, Phys. Chem. Chem. Phys., 2021, 23, 13827 RSC.
- M. Hušák, A. Jegorov, J. Rohlíček, A. Fitch, J. Czernek, L. Kobera and J. Brus, Cryst. Growth Des., 2018, 18, 3616–3625 CrossRef.
- L. Misseeuw, A. Krajewska, I. Pasternak, T. Ciuk, W. Strupinski, G. Reekmans, P. Adriaensens, D. Geldof, F. Blockhuys, S. Van Vlierberghe, H. Thienpont, P. Dubruel and N. Vermeulen, RSC Adv., 2016, 6, 104491 RSC.
- Y. Krupskaya, M. Gibertini, N. Marzari and A. F. Morpurgo, Adv. Mater., 2015, 27, 2453–2458 CrossRef CAS PubMed.
- M. H. Levitt, Spin Dynamics: Basics of Nuclear Magnetic Resonance, John Wiley & Sons, Ltd, Chichester, 2nd edn, 2015 Search PubMed.
- A. Fediai, A. Emering, F. Symalla and W. Wenzel, Phys. Chem. Chem. Phys., 2020, 22, 10256–10264 RSC.
- S. Samanta, P. Raval, G. N. M. Reddy and D. Chaudhuri, ACS Cent. Sci., 2021, 7, 1391–1399 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Sample preparation, experimental details, ssNMR and EPR spectra and analysis. See DOI: 10.1039/d1mh01574e |
| This journal is © The Royal Society of Chemistry 2022 |