
Design strategies for improving the crystallinity of covalent organic frameworks and conjugated polymers: a review
Jie
Yang†
a,
Fangyuan
Kang†
a,
Xiang
Wang
a and
Qichun
Zhang
 *ab
*ab
aDepartment of Materials Science and Engineering, City University of Hong Kong, Hong Kong, SAR 999077, P. R. China. E-mail: qiczhang@cityu.edu.hk
bCenter of Super-Diamond and Advanced Films (COSDAF), City University of Hong Kong, Hong Kong, SAR 999077, P. R. China
First published on 19th July 2021
Abstract
Highly crystalline covalent organic frameworks (COFs) or conjugated polymers (CPs) are very important and highly desirable because these materials would display better performance in diverse devices and provide more structure–property related information. However, how to achieve highly crystalline or single-crystal COFs and CPs is very challenging. Recently, many research studies have demonstrated the possibility of enhancing the crystallinity of COFs and CPs. Thus, it is timely to offer an overview of the important progress in improving the crystallinity of COFs and CPs from the viewpoint of design strategies. These strategies include polycondensation reaction optimization, improving the planarity, fluorine substitution, side chain engineering, and so on. Furthermore, the challenges and perspectives are also discussed to promote the realization of highly crystalline or single-crystal COFs and CPs.
 Jie Yang | Jie Yang received his BS and PhD degrees from the School of Chemistry and Chemical Engineering, Huazhong University of Science and Technology in 2014 and 2019, respectively. He is now working as a postdoctoral fellow in Prof. Qichun Zhang's group, City University of Hong Kong. His research interests include the synthesis and applications of novel covalent organic frameworks and conjugated polymers. |
 Fangyuan Kang | Fangyuan Kang received his Master's degree from Chengdu Institute of Organic Chemistry, Chinese Academy of Sciences in 2015. He is a PhD candidate in Prof. Qichun Zhang's group, City University of Hong Kong. His research interests include the design, synthesis, and applications of carbon-rich nanomaterials and crystalline covalent organic networks. |
 Xiang Wang | Xiang Wang received his Master's degree from the School of Chemistry and Material Science, Nanjing Normal University in 2019. He is a PhD candidate in Prof. Qichun Zhang's group, City University of Hong Kong. His research focuses on developing novel methods for the fabrication of two-dimensional covalent organic frameworks and their single crystals. |
 Qichun Zhang | Qichun Zhang is a full professor at the Department of Materials Science and Engineering at City University of Hong Kong, China. His research focuses on carbon-rich conjugated materials and applications. |
1. Introduction
Covalent organic frameworks (COFs) and linear conjugated polymers (CPs) have attracted extensive attention in the fields of science and technology since their discovery.1–7 Both COFs and CPs are made from light elements such as C, H, O, N, and S through covalent bonds. COFs are a class of porous polymers with two-dimensional (2D) or three-dimensional (3D) networks, while CPs are one family of one-dimensional (1D) semiconducting polymers whose repeated units are linked by sp2 hybridized atoms.8–11 Both COFs and CPs have several important advantages including light weight, high structure stability, and good chemical tunability.12–24 To date, hundreds of COFs and CPs have been explored based on different organic building blocks such as benzene, pyridine, thiophene, acene, and natural dye molecules.25–36 The good chemical tunability of COFs and CPs endows them with wide-ranging applications in energy storage and optoelectronic devices such as rechargeable batteries,37–48 sensors,49–52 organic field-effect transistors (OFETs),53–57 organic light-emitting diodes (OLEDs),58–61 and organic photovoltaics (OPVs).49,62,63Crystallinity of COFs and CPs indicates the degree of long-range ordered periodic structure of atom arrangement in these materials, and thus it is one of the crucial features to determine the properties and performance of COFs and CPs in diverse devices.64–66 For example, high crystallinity (even single crystals) of COFs not only is favorable for their structural determination with lots of solid structural parameters including the pore size, atomic positions, and bond lengths and angles, but also significantly improves their performance in photocatalysis, proton conduction, energy storage, electronics, and other fields.64 As to CPs, high crystallinity would offer rapid charge carrier (hole or electron) transport pathways through π–π stacking, which is very important to realize high-performance electronic applications such as OFETs, sensors, and logic circuits.66 In particular, single-crystal CPs can offer a perfect platform to study the structure–property relationship and investigate the charge transport mechanisms in polymer systems. However, it still remains a big challenge to obtain highly crystalline or single-crystal COFs and CPs owing to the less reversibility or irreversibility of their covalent bond formation compared with the coordination bond and hydrogen bond formation in metal–organic frameworks (MOFs) and hydrogen-bonded organic frameworks (HOFs).67–72
Recently, lots of efforts have been made to achieve highly crystalline COFs and CPs. Strategies for achieving highly-crystalline or single-crystal COFs and CPs can be divided into two main categories: the optimization of polymerization reactions and the enhancement of the intramolecular or intermolecular interactions. For instance, Ma et al. developed a method to grow micrometer scale single-crystal COFs by adding a modulator.64 The added modulator slowed down the rate of imine exchange and enhanced the reversibility of imine bond formation, offering enough time for the growth of COF single crystals. In this review, we will first provide a general introduction of COF and CP materials. Next, we will describe the strategies for improving the crystallinity of COFs and CPs. For COFs, the strategies include polycondensation reaction optimization, improving the planarity, hydrogen bond and fluorine substitution, and so on. For CPs, the strategies include thermal annealing, molecular mass modulation, improving the planarity, molecular geometry modulation, side chain engineering, fluorine substitution, and so on. This review will summarize the mechanisms of the crystallinity enhancement for each design strategy. The effect of crystallinity enhancement on practical applications will also be discussed. Finally, the conclusion and outlook of these strategies are provided.
2. Synthesis and comparison of COFs and CPs
COFs and CPs have several similar and different features. The similar features include the following: (1) both COFs and CPs are light-weight materials because they only contain light elements such as C, H, O, N, and S; (2) compared with MOFs and HOFs, COFs and CPs display high chemical stability since COFs and CPs are constructed through strong covalent bonds; (3) the properties of COFs and CPs can be tuned through the selection or modification of building blocks (Fig. 1 and 2), where these units can be benzene, pyridine, thiophene, acene, triazine, diketopyrrolopyrrole (DPP),73–76 isoindigo,77–80 naphthalene diimide (NDI),81–83 and so on.84,85 However, the different features between COFs and CPs are also obvious: (1) generally, the polymerization reactions of COFs are reversible, while those of CPs are not; (2) COFs have 2D or 3D porous structures, while CPs are 1D materials with negligible surface areas; (3) in most cases, CPs are solution-processable due to their bulky side chains, which can be processed through spin coating or printing technologies, while most of the COFs are insoluble in organic solvents.56,86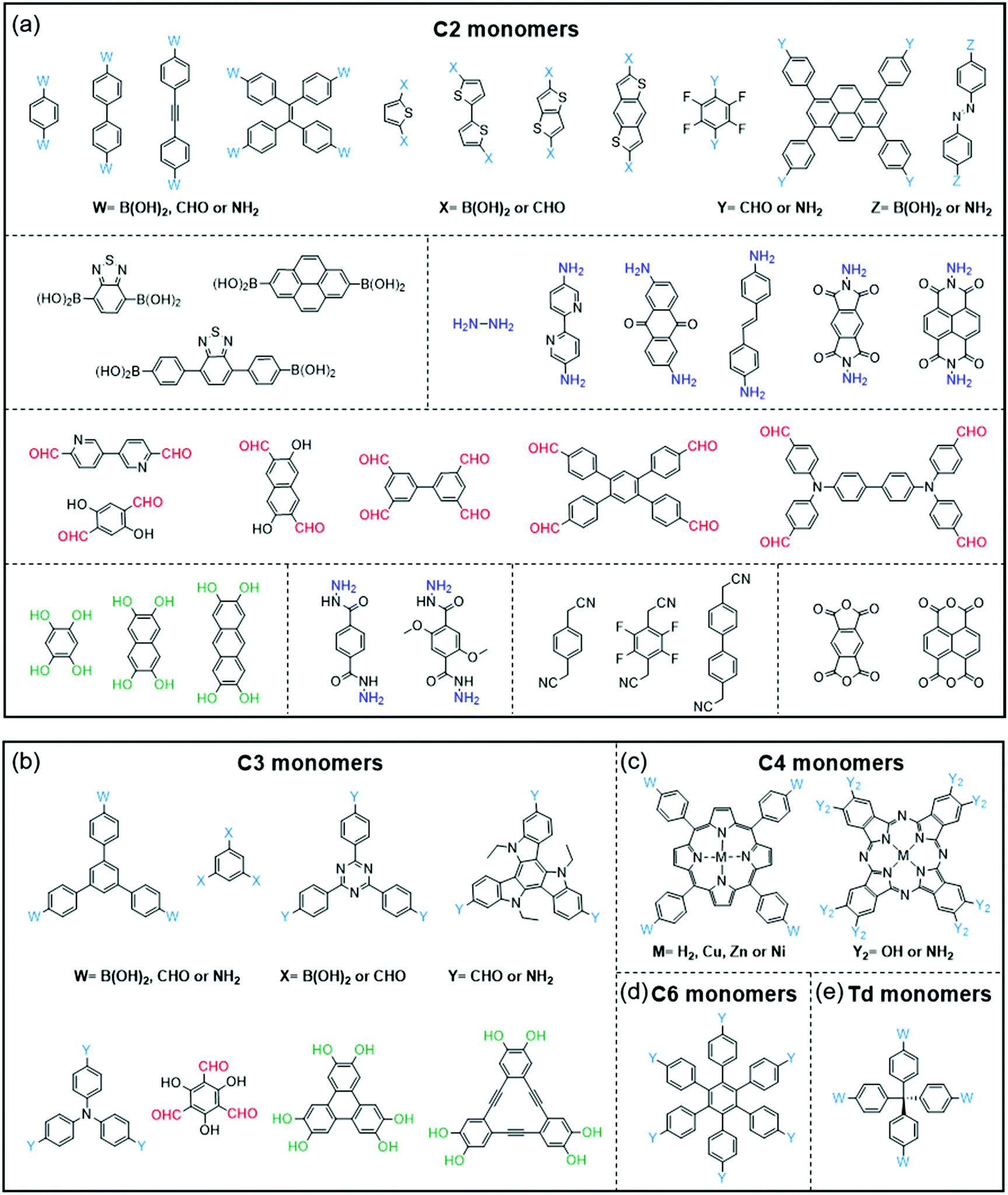 | ||
| Fig. 1 Monomers with different geometries and reactive groups for the synthesis of COFs. The monomers can be divided into (a) C2, (b) C3, (c) C4, (d) C6 or (e) Td geometry. | ||
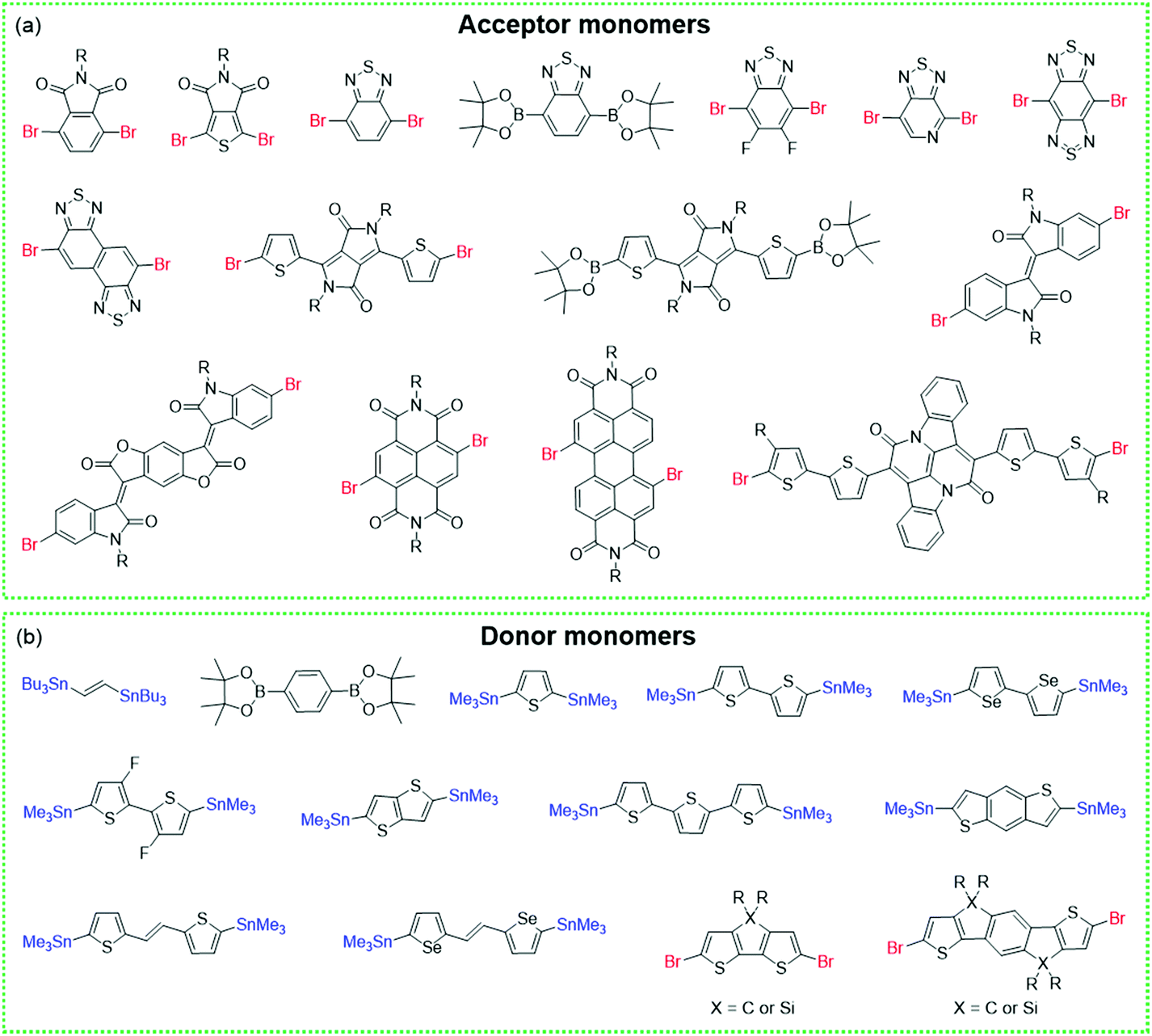 | ||
| Fig. 2 (a) Selected acceptor or (b) donor monomers for the synthesis of CPs. | ||
Since both COFs and CPs have wide applications in energy storage and optoelectronic devices such as LIBs, sensors, OFETs, OLEDs, and OPVs, developing different methods for their preparation is very important and highly desirable. Fig. 3 shows various synthetic methods to synthesize COFs and CPs. Most of the COFs are synthesized via reversible condensation reactions such as boronated ester formation, boronic acid trimerization, and Schiff base reactions (Fig. 3a).87–92 The reversible nature of condensation reactions offers COF materials with a self-correction ability that can repair the structural defects, which permits the formation of crystalline COFs. In contrast, most CPs are obtained via irreversible polymerization reactions such as Stille, Suzuki, and direct arylation reactions (Fig. 3b).93–99 Since these synthetic methods have been summarized in several reviews,10,12–14 here, we only focus on the strategies to improve the crystallinity of COFs and CPs during synthesis or processing.
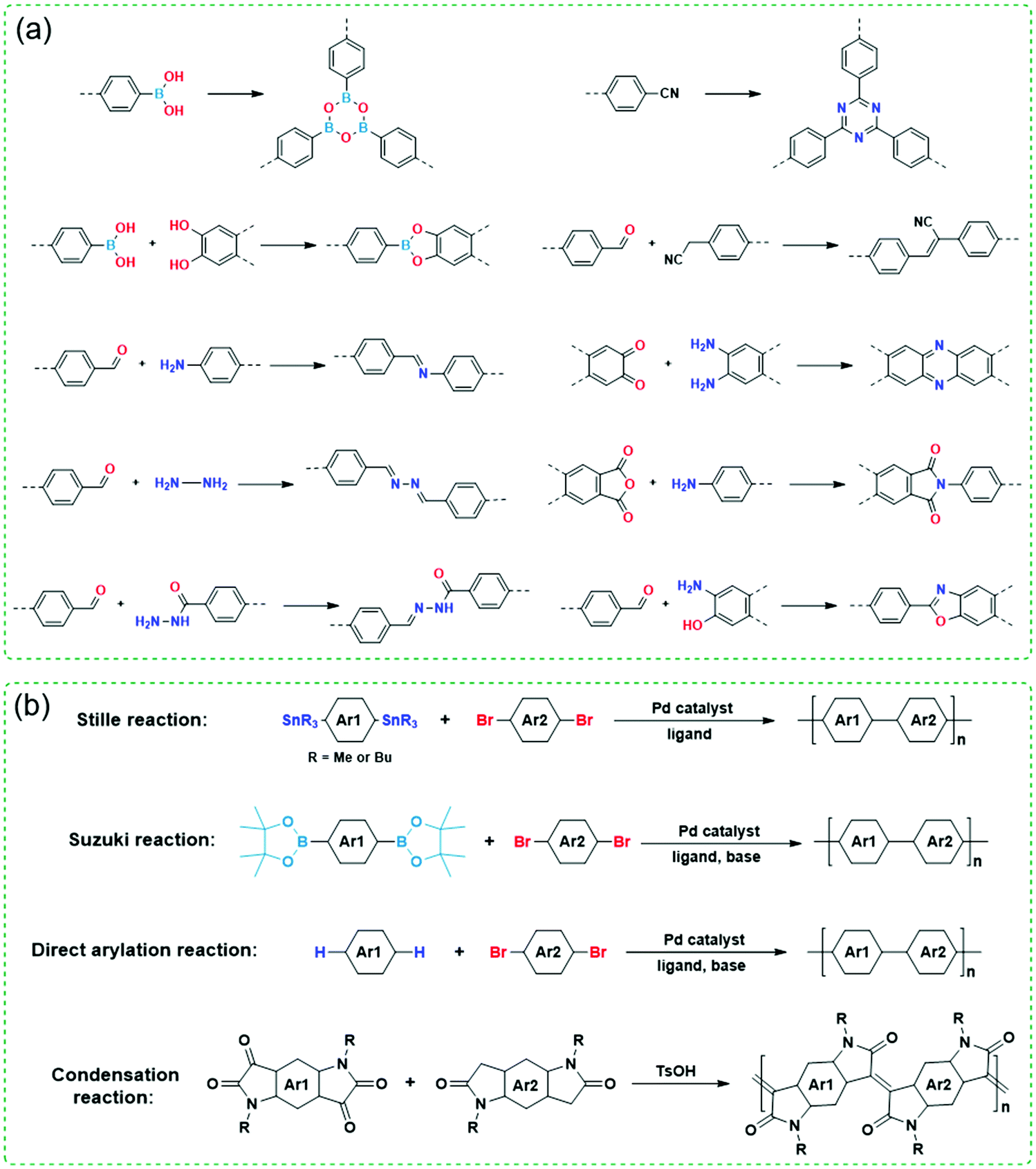 | ||
| Fig. 3 Polymerization reactions for the synthesis of (a) COFs and (b) CPs. Ar represents aryl units and R represents side chains. | ||
3. Strategies for improving the crystallinity of COFs
Strategies for improving the crystallinity of COFs can be divided into several categories: polycondensation reaction optimization, improving the planarity, hydrogen bond and fluorine substitution, and so on. The advances of single-crystal COFs are also described.3.1. Polycondensation reaction optimization
Polycondensation conditions of COFs are one of the common factors that can affect the crystallinity. The reaction conditions include catalyzed acids or bases, adding a modulator, the feeding rate of monomers, and so on. For example, highly crystalline COFs can be obtained by slowly adding the monomers because this operation can slow down the nucleation and crystal growth rates.100,101 In fact, lots of studies have reported the effect of polycondensation conditions on the crystallinity of COFs. Here, we described several representative studies on polycondensation reaction optimization. Fig. 4 shows the representative examples of polycondensation reaction optimization to improve the crystallinity.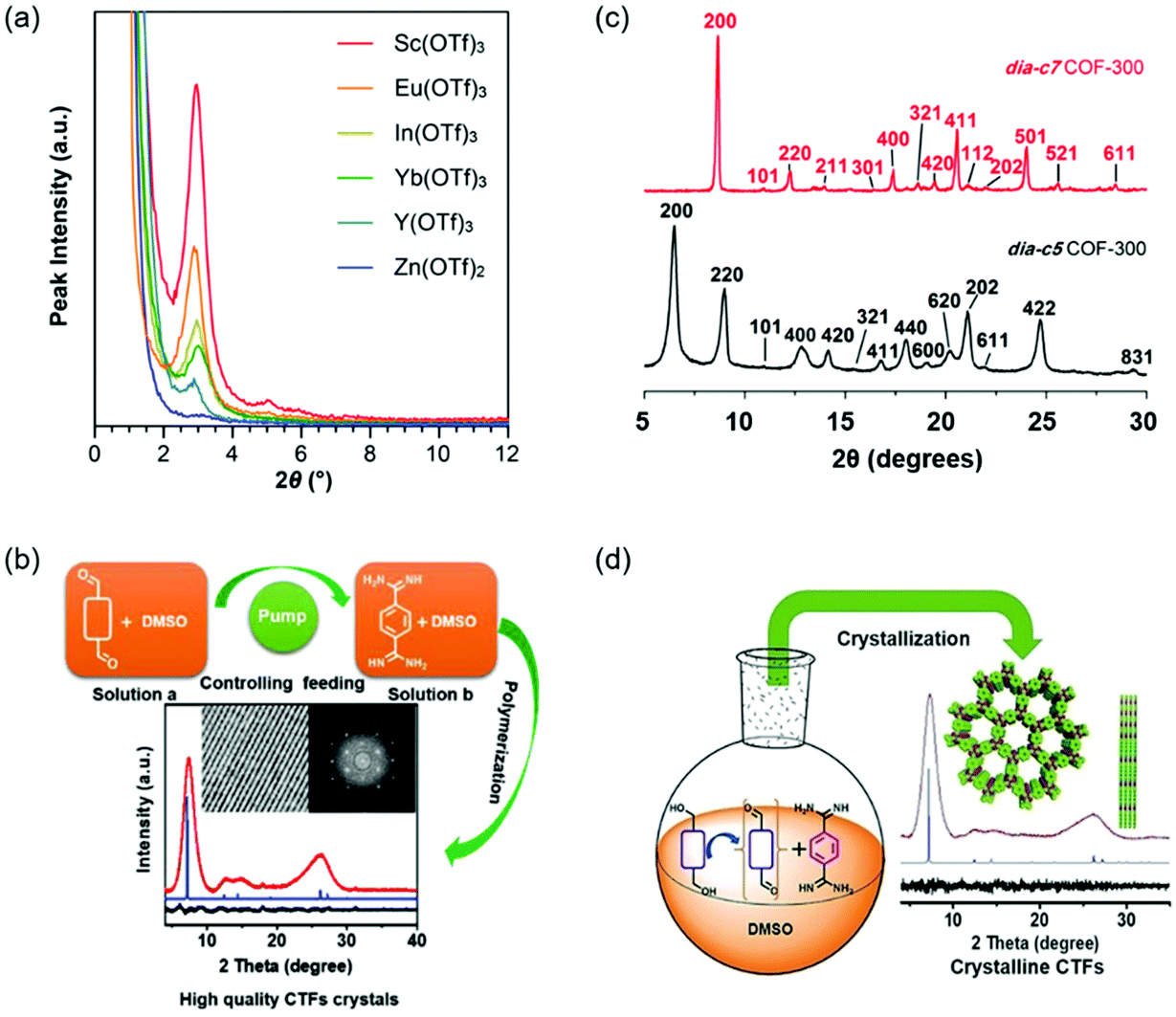 | ||
| Fig. 4 Polycondensation reaction optimization of COFs. (a) PXRD patterns of the TAPB–PDA COF prepared using different metal triflate catalysts. Reproduced with permission.77 Copyright 2017, American Chemical Society. (b) The method of controlling the nucleation rate and crystal growth by decreasing feeding rates to obtain highly crystalline CTFs. Reproduced with permission.101 Copyright 2019, Wiley-VCH. (c) Comparison of the PXRD patterns of dia-c5 and dia-c7 COF-300 interpenetration isomers. Reproduced with permission.107 Copyright 2018, American Chemical Society. (d) The method of improving the crystallinity of CTFs through in situ formation of aldehydes by controlled oxidation of alcohols. Reproduced with permission.108 Copyright 2018, Wiley-VCH. | ||
The crystallinity of COFs can be influenced by many reaction conditions, such as catalyzed acids or bases, adding a modulator, the feeding rate of monomers, and adding an aging process. Among these reaction conditions, adding a modulator and slowly adding the monomers are effective methods to improve the crystallinity by slowing down the formation rates of COFs.101,104 The replacement of the traditional acetic acid catalyst with Lewis acidic metal triflates offers an important approach to synthesize crystalline COFs at room temperature.77,109 In general, the relationship between the polycondensation reaction conditions and the crystallinity is complicated. It is highly desirable to explore other general and facile polycondensation methods for highly crystalline COFs.
3.2. Improving the planarity
Backbone planarity of 2D COFs is an important factor that can influence the crystallinity. Good backbone planarity of 2D COFs can promote interlayer π–π stacking of COF units, thus improving the crystallinity. Some monomers containing twisted groups may disrupt the interlayer π-stacking interactions and finally form amorphous porous polymers. The approaches to improve the planarity include introducing nitrogen atoms, removing twisted groups, and so on. For example, the replacement of C–H units in phenyl rings with nitrogen atoms can decrease the steric hindrance, resulting in an enhanced planarity.110Fig. 5 shows the crystallinity enhancement by planarity modulation.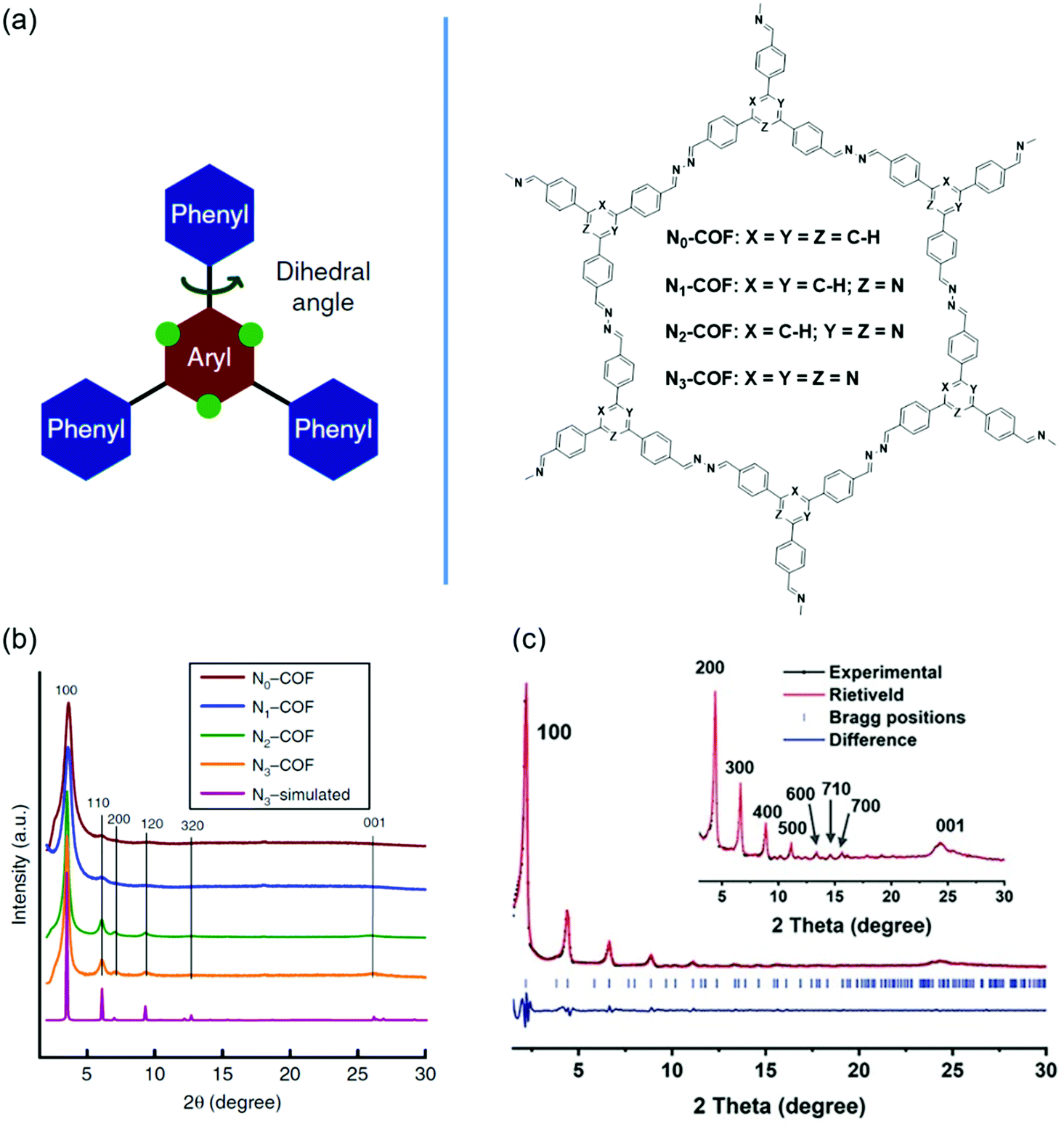 | ||
| Fig. 5 Improving the crystallinity of COFs by planarity modulation. (a) Design and molecular structures of Nx-COFs with tunable N atoms. Reproduced with permission.110 Copyright 2015, Springer Nature. (b) Experimental PXRD patterns of Nx-COFs. Reproduced with permission.110 Copyright 2015, Springer Nature. (c) Experimental PXRD patterns (black dots) of DBC-2P. Reproduced with permission.112 Copyright 2019, Wiley-VCH. | ||
Vyas et al. synthesized a series of 2D azine-linked COFs denoted as Nx-COF (x = 0, 1, 2, 3) based on the triphenylarene units with the central aryl moiety containing 0–3 nitrogen atoms (Fig. 5a).110 As the number of nitrogen atoms was increased from zero to three, the planarity of Nx-COF gradually became better. For example, the dihedral angles between the central aryl and peripheral phenyl rings were 38.7° for N0-COF and 0° for N3-COF. The improvement of planarity was beneficial for higher crystallinity. PXRD indicated that the diffraction peak intensity of Nx-COF gradually increased with the increase of the content of nitrogen atoms (Fig. 5b). In addition, some crystalline domains with sizes of 50–100 nm could be observed in N3-COF by TEM analysis. However, N0-COF and N1-COF did not display any crystalline domains. The Nx-COFs were used as photocatalysts in photocatalytic water splitting. The hydrogen evolution gradually increased from N0-COF to N3-COF, which was consistent with the enhanced crystallinity. Thompson et al. synthesized one COF (FLT-COF-1) and two covalent organic polymers (FLT-COP-2 and FLT-COP-3) based on fluoranthene units with different substitutions around the periphery.111 Molecular dynamics (MD) simulations showed that the torsion angles around fluoranthene cores were increased as the attachment of more substituted phenyl groups. The extra steric hindrance of the attached phenyl rings led to poor planarity. As a result, the BET surface area of FLT-COF-1 was 1180 m2 g−1, which was about two times that of FLT-COP-2 or FLT-COP-3. Moreover, FLT-COF-1 was a mesoporous material, while FLT-COP-2 and FLT-COP-3 were microporous materials. PXRD indicated that FLT-COF-1 showed intense diffraction peaks with an eclipsed conformation. However, FLT-COP-2 and FLT-COP-3 were amorphous without obvious diffraction peaks. Xie et al. developed a hetero-porous COF (DBC-2P) based on dibenzo[g,p]chrysene units (Fig. 5c).112 For comparison, a similar COF (4PE-2P) with tetraphenylethene (4PE) units as the knots was also prepared.113 DBC units had a better planar backbone than 4PE units because the surrounding four phenyl rings were strictly fixed on the core by additional C–C single bonds. Compared with 4PE-2P, DBC-2P achieved enhanced crystallinity with much higher order and stronger diffraction peaks. DBC-2P showed stronger interlayer interactions than 4PE-2P due to the larger conjugated structure and better backbone planarity. The interlayer distance of DBC-2P (0.36 nm) was much shorter than that of 4PE-2P (0.45 nm). As a result, the crystallinity of DBC-2P was maintained after treatment with HCl, NaOH or boiling water. In contrast, the crystallinity of 4PE-2P disappeared after treatment with the above reagents. Based on the ordered 1D dual-pore nanochannels with high stability and crystallinity of DBC-2P, they prepared a hybrid material by incorporating linear polyethylene glycol (PEG) and PEG-LiBF4 salt into the nanochannels and achieved a high ionic conductivity of 2.31 × 10−3 S cm−1.
Improving the planarity of 2D COFs is an effective strategy to obtain crystalline COFs by promoting the π–π stacking of COF layers. The planarity can be improved by introducing sp2-N atoms, avoiding twisted groups, or synthesizing large π-conjugated monomers. In particular, the introduction of sp2-N atoms has been demonstrated as a general method to improve the planarity of aromatic molecules.20 Except for the above approaches, more effective strategies to improve the planarity should be developed in the future.
3.3. Hydrogen bond and fluorine substitution
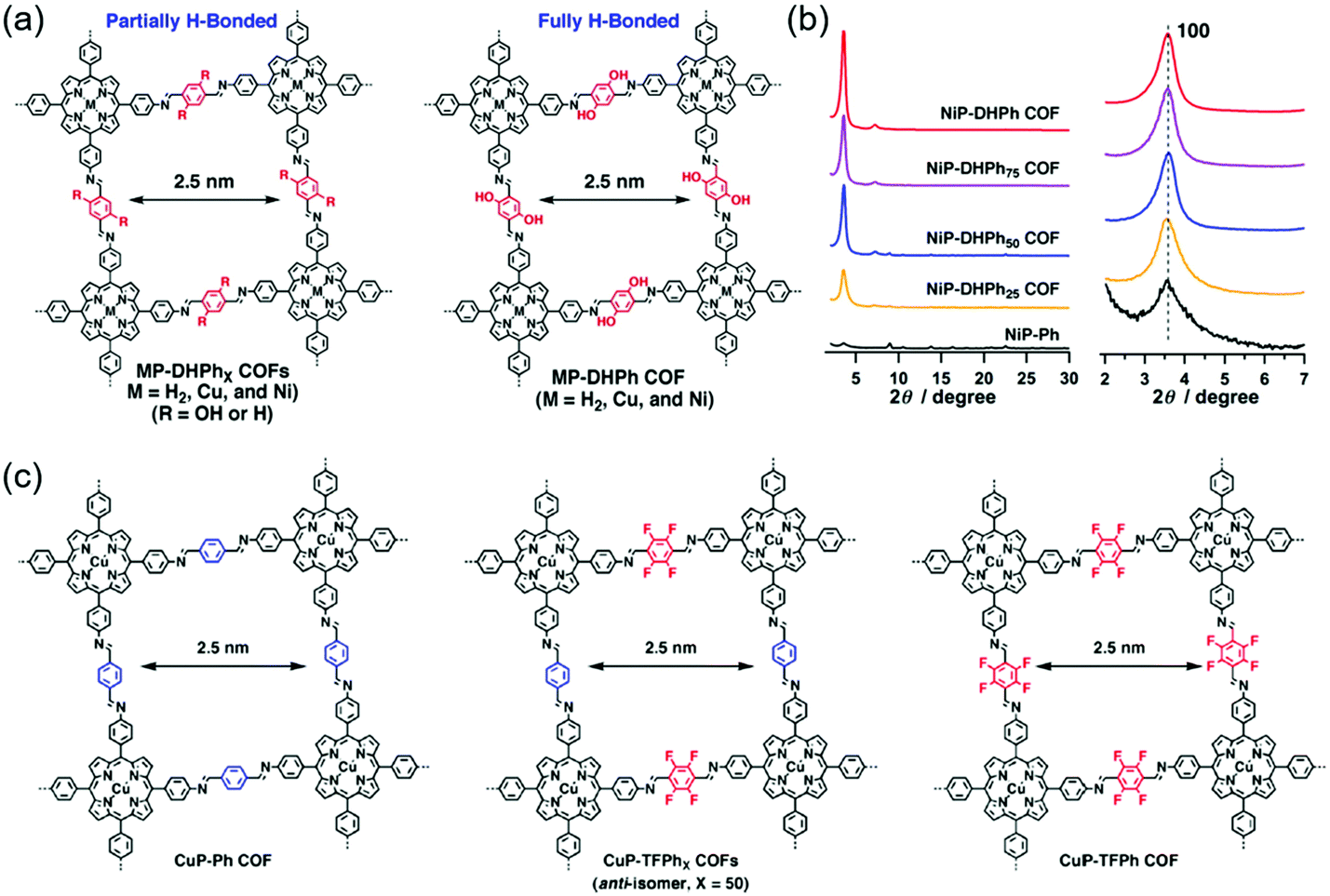
The introduction of hydrogen bonds or fluorine atoms into 2D COFs has been demonstrated as an effective approach to improve their crystallinity.114 The intralayer hydrogen bonds in 2D COFs can lock the molecular structure, improve the planarity of the 2D layers and thus enhance the crystallinity. Besides, the introduction of fluorine atoms can enhance interlayer π–π interactions based on self-complementary π-electronic forces, which will also improve the crystallinity.115Hydrogen bonds can be divided into intralayer hydrogen bonds and interlayer hydrogen bonds. Fig. 6a and b show the examples of the intralayer hydrogen bond strategy to improve the crystallinity. Kandambeth et al. synthesized a 2D porphyrin COF (DhaTph) containing –OH units, which could form intramolecular O–H⋯N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C hydrogen bonds with adjacent CN groups.116 For comparison, a methoxy-substituted COF (DmaTph) without hydrogen bonds was also prepared. XRD data indicated that DhaTph showed a much more enhanced crystallinity compared with DmaTph. The intramolecular O–H⋯NC hydrogen bonds in DhaTph could hold the phenyl units in one plane and enhance the structural rigidity, thus improving the crystallinity. However, the planarity between the phenyl units in DmaTph was lost, which was unfavorable for the 2D layer stackings and thus reduced the crystallinity. The surface area of DhaTph was much larger than that of DmaTph, which resulted from the improved crystallinity. In addition, DhaTph retained its crystallinity in HCl and boiling water, while DmaTph was unstable in these conditions. Chen et al. elucidated the effect of intramolecular H-bonding interactions on the crystallinity, porosity, and photochemical activity of 2D COFs by three-component condensation reactions to synthesize a series of 2D porphyrin COFs (named MP-DHPhX COFs when X = 25, 50, 75 and where M = H2, Cu, and Ni; and MP-DHPh COFs when X = 100 and where M = H2, Cu, and Ni), in which intramolecular H-bonding sites were tunable (Fig. 6a).117 For comparison, amorphous porphyrin polymers (named MP-Ph polymers when X = 0) were also prepared. XRD patterns showed that the crystallinity of MP-DHPhX COFs was much higher than that of the amorphous MP-Ph polymer without H-bonding interactions. The diffraction peak intensity of MP-DHPhX COFs gradually increased with the increase of the content of dihydroxyterephthalaldehyde (DHTA) (Fig. 6b). Theoretical calculations indicated that the dihedral angles between the imine linkage and meso-phenyl units of porphyrin in MP-DHPhX were quite smaller than those of MP-Ph polymers, where the total crystal stacking energy in these materials was opposite. Therefore, the amorphous MP-Ph polymer displayed inferior planarity and needed more energy to form crystalline frameworks, indicating that the H-bonding interactions improved the π-stacking interactions and enhanced the crystallinity by promoting the planar conformation. These COFs were applied in photocatalytic conversion reactions of molecular oxygen into singlet oxygen. The photocatalytic activity of MP-DHPhX gradually increased with the increase of the content of H-bonding sites, which was consistent with the crystallinity tendency. Guo et al. synthesized a series of 2D COFs, namely TPT–BD COF, TPT–DHBDX COFs (X = 25, 50, and 75 mol%), and TPT–DHBD COF.118 These COFs were composed of benzidine (BD) and 3,3′-dihydroxybenzidine (DHBD) units with molar ratios of 0/100, 25/75, 50/50, 75/25, and 100/0. PXRD patterns indicated that all the COFs showed (100), (110), (200), (210), and (220) diffraction peaks. The diffraction peak intensities of the TPT–BD COF, TPT-DHBDX COFs, and TPT–DHBD COF gradually increased with the increase of the content of DHBD. The enhanced crystallinity of the COFs was attributed to the formation of intramolecular hydrogen bonds between the phenolic hydroxyl group and the imine–nitrogen atom through a five-membered ring. The additional H-bonding interactions locked the partial structure of the COFs, which improved the planarity of the 2D layers and thus enhanced the crystallinity.
C hydrogen bonds with adjacent CN groups.116 For comparison, a methoxy-substituted COF (DmaTph) without hydrogen bonds was also prepared. XRD data indicated that DhaTph showed a much more enhanced crystallinity compared with DmaTph. The intramolecular O–H⋯NC hydrogen bonds in DhaTph could hold the phenyl units in one plane and enhance the structural rigidity, thus improving the crystallinity. However, the planarity between the phenyl units in DmaTph was lost, which was unfavorable for the 2D layer stackings and thus reduced the crystallinity. The surface area of DhaTph was much larger than that of DmaTph, which resulted from the improved crystallinity. In addition, DhaTph retained its crystallinity in HCl and boiling water, while DmaTph was unstable in these conditions. Chen et al. elucidated the effect of intramolecular H-bonding interactions on the crystallinity, porosity, and photochemical activity of 2D COFs by three-component condensation reactions to synthesize a series of 2D porphyrin COFs (named MP-DHPhX COFs when X = 25, 50, 75 and where M = H2, Cu, and Ni; and MP-DHPh COFs when X = 100 and where M = H2, Cu, and Ni), in which intramolecular H-bonding sites were tunable (Fig. 6a).117 For comparison, amorphous porphyrin polymers (named MP-Ph polymers when X = 0) were also prepared. XRD patterns showed that the crystallinity of MP-DHPhX COFs was much higher than that of the amorphous MP-Ph polymer without H-bonding interactions. The diffraction peak intensity of MP-DHPhX COFs gradually increased with the increase of the content of dihydroxyterephthalaldehyde (DHTA) (Fig. 6b). Theoretical calculations indicated that the dihedral angles between the imine linkage and meso-phenyl units of porphyrin in MP-DHPhX were quite smaller than those of MP-Ph polymers, where the total crystal stacking energy in these materials was opposite. Therefore, the amorphous MP-Ph polymer displayed inferior planarity and needed more energy to form crystalline frameworks, indicating that the H-bonding interactions improved the π-stacking interactions and enhanced the crystallinity by promoting the planar conformation. These COFs were applied in photocatalytic conversion reactions of molecular oxygen into singlet oxygen. The photocatalytic activity of MP-DHPhX gradually increased with the increase of the content of H-bonding sites, which was consistent with the crystallinity tendency. Guo et al. synthesized a series of 2D COFs, namely TPT–BD COF, TPT–DHBDX COFs (X = 25, 50, and 75 mol%), and TPT–DHBD COF.118 These COFs were composed of benzidine (BD) and 3,3′-dihydroxybenzidine (DHBD) units with molar ratios of 0/100, 25/75, 50/50, 75/25, and 100/0. PXRD patterns indicated that all the COFs showed (100), (110), (200), (210), and (220) diffraction peaks. The diffraction peak intensities of the TPT–BD COF, TPT-DHBDX COFs, and TPT–DHBD COF gradually increased with the increase of the content of DHBD. The enhanced crystallinity of the COFs was attributed to the formation of intramolecular hydrogen bonds between the phenolic hydroxyl group and the imine–nitrogen atom through a five-membered ring. The additional H-bonding interactions locked the partial structure of the COFs, which improved the planarity of the 2D layers and thus enhanced the crystallinity.
 | ||
| Fig. 6 Improving the crystallinity of COFs by hydrogen bond and fluorine substitution. (a) The molecular structures of 2D COFs with tunable content of hydrogen bonds. Reproduced with permission.117 Copyright 2015, American Chemical Society. (b) XRD patterns of the NiP-DHPhX COFs. Reproduced with permission.117 Copyright 2015, American Chemical Society. (c) The molecular structures of 2D COFs with tunable content of F atoms. Reproduced with permission.115 Copyright 2013, American Chemical Society. | ||
The introduction of interlayer hydrogen bonds can also improve the crystallinity of COFs. Halder et al. developed a COF (TpOMe-Pa1) based on 2,4,6-trimethoxy-1,3,5-benzenetricarbaldehyde (TpOMe) via a p-toluenesulfonic acid (PTSA) mediated solid-state mixing approach.119 For comparison, a similar polymer (Tf-Pa1) based on 1,3,5-benzenetricarbaldehyde (Tf) was also prepared. PXRD indicated that TpOMe-Pa1 showed high crystallinity with clear (100), (110), (210), and (001) diffraction peaks. In contrast, Tf-Pa1 was an amorphous polymer. The BET surface area of TpOMe-Pa1 was 1164 m2 g−1, which was much higher than that of Tf-Pa1. Moreover, TpOMe-Pa1 displayed high chemical stability in extreme conditions such as strong acids and bases. Theoretical calculations indicated that a number of interlayer C–H⋯N hydrogen bonds between the methoxy C–H units of a particular layer and the imine N atoms of the adjacent layers formed in TpOMe-Pa1. The presence of interlayer hydrogen bonds in TpOMe-Pa1 improved the crystallinity and chemical stability.
Fig. 6c shows the examples of fluorine substitution to improve the crystallinity. Chen et al. proposed a strategy to improve the crystallinity of COFs by controlling the interlayer interactions based on self-complementary π-electronic forces.115 To demonstrate the strategy, they synthesized a series of COFs, namely CuP–Ph COF, CuP–TFPhX COFs (X = 25, 50, and 75 mol%), and CuP–TFPh COF (Fig. 6c). These COFs were composed of tetrafluorophenyl (TFPh) and phenyl (Ph) units with molar ratios of 0/100, 25/75, 50/50, 75/25, and 100/0. XRD patterns showed clear (100), (200), and (001) diffraction peaks, indicating that all the COFs displayed good crystallinity. The XRD intensity gradually increased from the CuP–Ph COF to the CuP–TFPh50 COF. Further increase in the content of TFPh led to the decrease in the XRD intensity. Among all the COFs, the CuP–TFPh50 COF exhibited the strongest XRD intensity. The BET surface areas of the COFs also displayed a similar tendency with CuP–TFPh50 COF showing the largest value of 1389 m2 g−1. Theoretical calculations revealed that the interlayer π–π interactions between fluoro-substituted and non-substituted arenes increased the total crystal stacking energy of the COFs, thus resulting in high crystallinity. All the results elucidated that the self-complementary π-electronic interactions could improve the crystallinity of COFs. Alahakoon et al. synthesized two COFs (TF-COF 1 and TF-COF 2) containing fluorine atoms.120 For comparison, a non-fluorinated COF (NF-COF) was also prepared. PXRD patterns showed that the (100), (110), (120), (130), and (001) diffraction peaks could be clearly observed in TF-COF 1 and TF-COF 2, indicating their good crystallinity. However, only two diffraction peaks at 3.67° and 6.09° were observed for the NF-COF, suggesting a lower degree of crystalline order. The BET surface areas of TF-COF 1 and TF-COF 2 were 1820 and 2044 m2 g−1, respectively, which were much higher than that of the NF-COF. Moreover, TF-COF 1 and TF-COF 2 showed more well-defined pore structures compared with the NF-COF. All the results demonstrated that the rational introduction of fluorine atoms into the COF structures could significantly improve the crystallinity.
The crystallinity of COFs can be improved by introducing hydrogen bonds or fluorine atoms. Both hydrogen bond and fluorine substitution can enhance the π-stacking interactions of COFs, which will promote the COF crystallization. Besides, hydrogen bonds can enhance the planar conformations of COFs, which will further improve the crystallinity. However, the studies of hydrogen bond and fluorine substitution mainly focus on 2D COFs.115,116 The effect of hydrogen bond and fluorine substitution on the crystallinity of 3D COFs needs further studies. In addition, other functional groups such as Cl, CN, and CF3 can also be introduced into COFs to improve the crystallinity.
3.4. Other strategies
Other strategies to improve the crystallinity of COFs include a two-in-one strategy, a group protection approach, molecular symmetry or side chain modulation, and so on.121–125Fig. 7 lists some of them.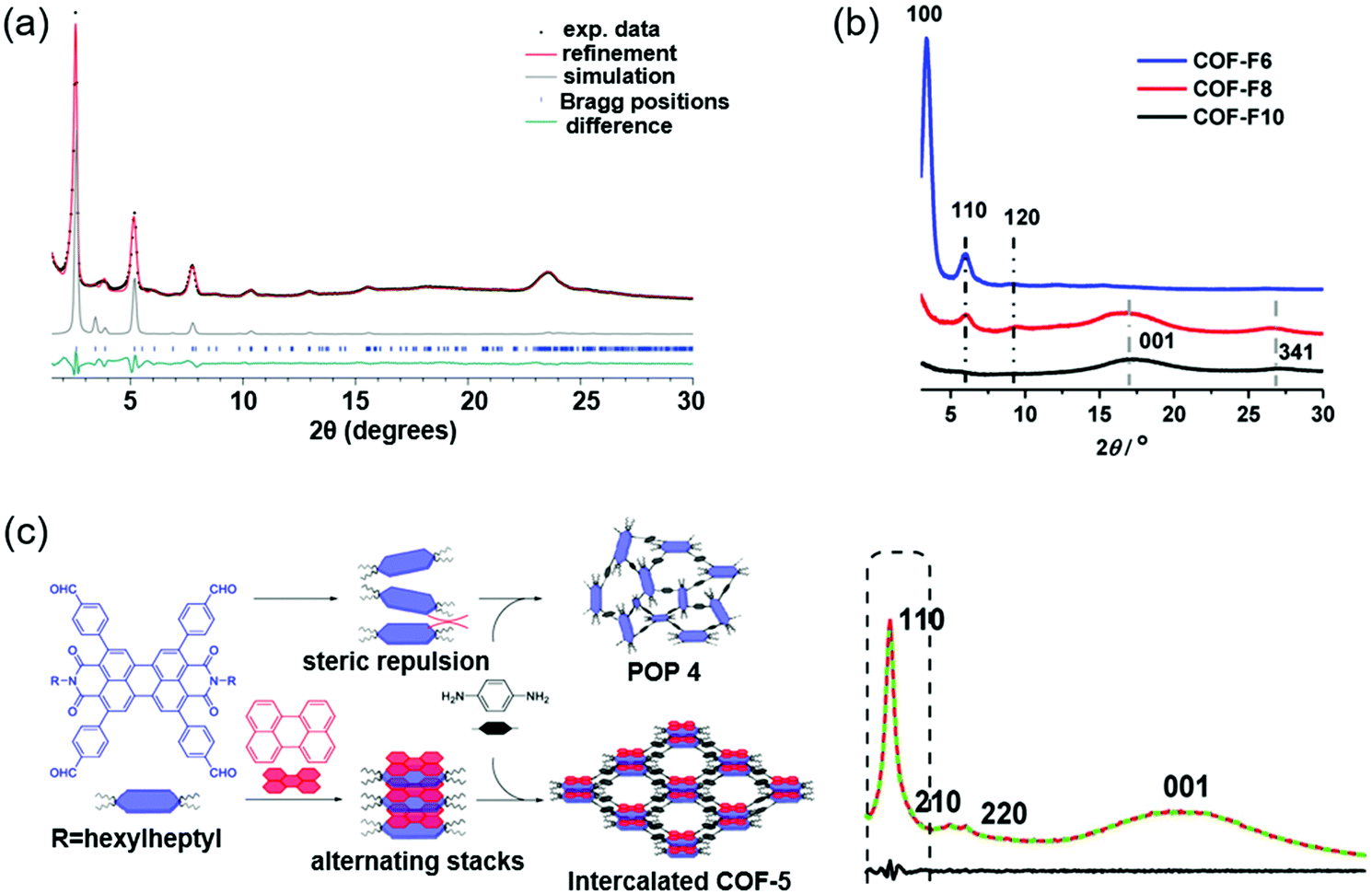 | ||
| Fig. 7 Improving the crystallinity of COFs by other strategies. (a) Experimental PXRD data (black dots) of Py–a4T COF. Reproduced with permission.123 Copyright 2017, American Chemical Society. (b) PXRD data of COF-FX. Reproduced with permission.124 Copyright 2020, American Chemical Society. (c) Synthetic schematic of POP 4 in the absence and intercalated COF-5 in the presence of D–A interactions (left). PXRD data of intercalated COF-5 (right). Reproduced with permission.130 Copyright 2020, American Chemical Society. | ||
Recently, a two-in-one strategy and a group protection approach have been developed to improve the crystallinity of COFs.121 Li et al. reported a two-in-one design strategy to synthesize 2D imine COFs in facile conditions. A bifunctional monomer with two aldehyde and amino groups in one pyrene unit was used to synthesize a 2D pyrene-based COF (Py-COF) through self-condensation.121 The stoichiometry of the aldehyde and amino groups in the monomer was strictly guaranteed (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), which was favorable for the formation of crystalline COFs. A series of highly crystalline Py-COFs were achieved not only in mixed organic solvents but also in single solvents such as methanol, acetonitrile, dichloromethane, and tetrahydrofuran. For comparison, the reported Py–Py COF, which had a similar molecular structure to the Py-COF, was prepared by the traditional co-condensation reaction.126 The crystallinity of the Py-COF was better than that of the Py–Py COF, as revealed by the PXRD results. Moreover, the Py–Py COFs obtained through a co-condensation method in a single solvent showed poor crystallinity. The two-in-one strategy was also used to grow crystalline Py-COF films on substrates such as glass and silicon wafer. Such COF films could be applied as charge-transport layers in perovskite solar cells. To demonstrate the generality of this method, another kind of dual-pore COFs (BFBAEB-COF and BFBAEPy-COF) were successfully prepared.121,127 These COFs could not be synthesized by the traditional co-condensation approach. Chen et al. developed a “two-in-one” strategy to synthesize a bicarbazole-based COF (BCzP-COF) by a self-polycondensation reaction of the A2B2 monomer with two amine and two neopentyl acetal units.128 The neopentyl acetal unit was an aldehyde protecting group. For comparison, another COF (BCz-COF) was also prepared by a reaction of the A2B2 monomer with free amines and aldehydes. PXRD indicated that both COFs showed (110), (220), (330), and (001) diffraction peaks. However, the diffraction peak intensity of the BCzP-COF was stronger than that of the BCz-COF. Scanning electron microscopy (SEM) patterns revealed that both COFs displayed rod-like microcrystals. The crystal length of the BCzP-COF was much longer (about 20 μm) than that of the BCz-COF. The BET surface area of the BCzP-COF was 2200 m2 g−1, which was much higher than that of the BCz-COF. All these results demonstrated that the BCzP-COF achieved better crystallinity than the BCz-COF. Bai et al. synthesized a COF (PI-2-COF) through a condensation reaction between an aryl amine and an aldehyde.129 The as-prepared COF showed moderate crystallinity with a surface area of 1286 m2 g−1. Later, Vitaku et al. synthesized a BND–TFB COF through a modulated condensation reaction between a benzophenone–imine protected monomer and an aldehyde.122 PXRD diffraction peaks indicated that the BND–TFB COF showed an improved crystallinity compared with the PI-2-COF. The higher crystallinity was achieved by the controlled release of free amine groups via slow removal of the benzophenone imine protecting groups. As a result, the BND–TFB COF displayed a high surface area of 2618 m2 g−1, which was much higher than that of the PI-2-COF. Moreover, the generality of this method was further demonstrated by the successful synthesis of other COFs including TAPB–PDA COF, DAB–TFP COF, and BND–TFP COF.
:1), which was favorable for the formation of crystalline COFs. A series of highly crystalline Py-COFs were achieved not only in mixed organic solvents but also in single solvents such as methanol, acetonitrile, dichloromethane, and tetrahydrofuran. For comparison, the reported Py–Py COF, which had a similar molecular structure to the Py-COF, was prepared by the traditional co-condensation reaction.126 The crystallinity of the Py-COF was better than that of the Py–Py COF, as revealed by the PXRD results. Moreover, the Py–Py COFs obtained through a co-condensation method in a single solvent showed poor crystallinity. The two-in-one strategy was also used to grow crystalline Py-COF films on substrates such as glass and silicon wafer. Such COF films could be applied as charge-transport layers in perovskite solar cells. To demonstrate the generality of this method, another kind of dual-pore COFs (BFBAEB-COF and BFBAEPy-COF) were successfully prepared.121,127 These COFs could not be synthesized by the traditional co-condensation approach. Chen et al. developed a “two-in-one” strategy to synthesize a bicarbazole-based COF (BCzP-COF) by a self-polycondensation reaction of the A2B2 monomer with two amine and two neopentyl acetal units.128 The neopentyl acetal unit was an aldehyde protecting group. For comparison, another COF (BCz-COF) was also prepared by a reaction of the A2B2 monomer with free amines and aldehydes. PXRD indicated that both COFs showed (110), (220), (330), and (001) diffraction peaks. However, the diffraction peak intensity of the BCzP-COF was stronger than that of the BCz-COF. Scanning electron microscopy (SEM) patterns revealed that both COFs displayed rod-like microcrystals. The crystal length of the BCzP-COF was much longer (about 20 μm) than that of the BCz-COF. The BET surface area of the BCzP-COF was 2200 m2 g−1, which was much higher than that of the BCz-COF. All these results demonstrated that the BCzP-COF achieved better crystallinity than the BCz-COF. Bai et al. synthesized a COF (PI-2-COF) through a condensation reaction between an aryl amine and an aldehyde.129 The as-prepared COF showed moderate crystallinity with a surface area of 1286 m2 g−1. Later, Vitaku et al. synthesized a BND–TFB COF through a modulated condensation reaction between a benzophenone–imine protected monomer and an aldehyde.122 PXRD diffraction peaks indicated that the BND–TFB COF showed an improved crystallinity compared with the PI-2-COF. The higher crystallinity was achieved by the controlled release of free amine groups via slow removal of the benzophenone imine protecting groups. As a result, the BND–TFB COF displayed a high surface area of 2618 m2 g−1, which was much higher than that of the PI-2-COF. Moreover, the generality of this method was further demonstrated by the successful synthesis of other COFs including TAPB–PDA COF, DAB–TFP COF, and BND–TFP COF.
Molecular symmetry and side chains can have a remarkable effect on the crystallinity of COFs. Keller et al. prepared a symmetric unit and an asymmetric unit of the dibutyl-quaterthiophene (4T).123 No crystalline COF could be formed based on the symmetric 4T and 1,3,6,8-tetrakis(4-aminophenyl)pyrene (Py). Cofacial stacking of the symmetric 4T unit could bring the alkyl chains of adjacent layers very close, causing obvious steric repulsion and impeding the formation of a crystalline COF. In contrast, a crystalline COF (Py–a4T COF) was formed based on the asymmetric 4T and Py because the steric repulsion could be avoided in the asymmetric 4T unit. PXRD indicated that the Py–a4T COF showed multiple higher-order diffraction peaks, which suggested the high degree of long-range order formed in the COF (Fig. 7a). The asymmetric functionalization strategy could also be used in other building blocks. For example, crystalline COFs could be obtained based on asymmetric 4H-thieno[3,4-c]pyrrole-4,6(5H)-dione (TPD) or thieno[3,4-b]thiophene (TT) units. Charge transfer states between the subunits of the COFs were observed, as revealed by the time-correlated single photon counting (TCSPC) traces. In another study, Wu et al. synthesized a series of perfluoroalkyl-functionalized 2D COFs (COF-F6, COF-F8, and COF-F10) and investigated the influence of different lengths of perfluoroalkyl chains on the crystallinity and proton conductivity.124 PXRD indicated that COF-F6 showed multiple high-order diffraction peaks, suggesting the high crystallinity of COF-F6. The diffraction peak intensity gradually decreased from COF-F6 to COF-F10 because the longer perfluoroalkyl chains might cause steric repulsion and could be unfavorable for the crystallinity (Fig. 7b). The COFs displayed good chemical stability and hydrophobic properties due to the presence of perfluoroalkyl chains. These COFs could be used as hosts to load H3PO4 for proton conduction. The highest proton conductivity of COF-F6 was 4.2 × 10−2 S cm−1 after H3PO4 doping. In comparison, COF-F8 and COF-F10 displayed lower proton conductivities, which was due to the facts that (i) the longer perfluoroalkyl chains could block the proton transport pathway in the COFs, and (ii) the lower crystallinity further decreased the proton transport efficiency. Li et al. developed a donor–acceptor (D–A)-type intercalated COF by a supramolecular assembly strategy.130 A solvothermal reaction between 2,5,8,11-tetra(p-formylphenyl)-perylene diimide (PDI) and p-phenylenediamine afforded an amorphous porous polymer. This was because the long alkyl chains of PDI could cause obvious steric repulsion, which hindered the interlayer stacking of COFs. However, if perylene was added before the solvothermal reaction, an intercalated COF was obtained. An associating D–A complex could form between the electron-deficient PDI and electron-rich perylene, which decreased the steric repulsion of the alkyl chains. As a result, an intercalated COF based on PDI and p-phenylenediamine could be synthesized (Fig. 7c). XRD indicated that the intercalated COF showed good crystallinity with AA-stacking.
Recently, other strategies including a two-in-one strategy, a group protection approach, and molecular symmetry or side chain modulation have been developed to improve the crystallinity of COFs. The two-in-one strategy can guarantee the precise stoichiometry of the monomers in the polycondensation reaction, which is beneficial for the formation of crystalline COFs.121 In the group protection approach, a free amine or aldehyde monomer will be slowly released via the removal of the protecting groups, thus decreasing the reaction rate of the reversible imine formation and improving the crystallinity.122 Molecular symmetry or side chain modulation can improve the crystallinity of COFs by decreasing the steric repulsion of side chains.123,124 Despite the advances of the above strategies, their studies are still limited. These strategies can be applied in new COFs to improve the crystallinity.
3.5. Single-crystal COFs
The realization of single-crystal COFs is challenging but very important for several reasons: (1) the determination of COF structures from PXRD is extremely difficult, especially for the COFs with complicated topology structures; (2) single-crystal data can enable precise structural characterization of COFs such as the porous structure, atomic positions, and bond lengths and angles; (3) compared with polycrystalline COFs, single-crystal COFs generally display better performance in gas absorption, photocatalysis, electronic devices, and so on.64 The first single-crystal COF was achieved by Beaudoin et al. in 2013.131 Recently, Evans et al. reported a single-crystal COF by a two-step seeded growth strategy.100 Ma et al. developed a general method to grow micrometer scale single-crystal COFs by adding a modulator.64 The effect of single-crystal size on the crystallinity, structural flexibility, and gas absorption properties was further investigated.132 The key point to achieve single-crystal COFs is to modulate the reversible process during the linkage bond formation, offering enough time for self-correction of defects. Fig. 8 shows the reported single-crystal COFs. | ||
| Fig. 8 Single crystals of COFs. (a) 3D reciprocal lattice of mCOF-Ag obtained from the SCED data and SEM image of mCOF-Ag. Scale bar: 1 μm. Reproduced with permission.135 Copyright 2020, Springer Nature. (b) A single-crystal COF with non-interpenetrated pts topology. Reproduced with permission.138 Copyright 2020, Wiley-VCH. (c) SEM images of different-sized LZU-111 crystals. Reproduced with permission.132 Copyright 2020, Springer Nature. | ||
Beaudoin et al. successfully synthesized high quality 3D COF crystals by reversible self-addition polymerizations, allowing the structures of these materials to be characterized by single-crystal XRD.131 A series of single-crystal COFs, NPN-1, NPN-2, and NPN-3, were synthesized via oxidation polymerization of the nitroso compounds of tetrakis(4-nitrosophenyl)methane, tetrakis(4-nitrosophenyl)silane, and 1,3,5,7-tetrakis(4-nitrosophenyl)adamantane, respectively. The single crystals were large and uniform in morphology, with their size reaching 0.5 mm in the case of NPN-3. NPN-1 and NPN-2 were both observed to crystallize in the tetragonal space group P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) and give diamondoid networks with four-fold interpenetration. NPN-3 was found to crystallize in the tetragonal space group P42/n and showed a diamondoid network with six-fold interpenetration. Zhang et al. reported another single-crystal 3D COF (COF-320).133 SEM revealed that COF-320 showed a uniform rice-like morphology with a crystal dimension of about 200 nm. The single-crystal structure of COF-320 was investigated by 3D RED for the first time. COF-320 had a highly porous diamond net with 9-fold interpenetration. The RED data indicated that COF-320 formed a body-centered orthorhombic crystal structure. COF-320 showed a large Langmuir surface area of 2400 m2 g−1 due to the good crystallinity. As a result, COF-320 achieved a high methane uptake capacity of 15.0 wt% at 298 °C and 80 bar. Gao et al. designed three isostructural 3D COFs with –H, –Me, or –F substituents (3D-TPB-COF-H, 3D-TPB-COF-Me, and 3D-TPBCOF-F).134 SEM indicated that these 3D-TPB-COFs had uniform morphology with a crystal dimension of about 1 μm. All 3D-TPB-COFs showed sharp and intense diffraction peaks, indicating a highly crystalline nature. Their crystal structures were investigated by RED with the resolution reaching up to 0.9–1.0 Å, elucidating the positions of all non-hydrogen atoms in COFs. All 3D-TPB-COFs showed the same five-fold interpenetrated topologies with the different substituents being visible. All 3D-TPB-COFs displayed different gas absorption properties, such as selectivity for CO2 over N2. 3D-TPBCOF-F with C–F bonds on the porous structure achieved the highest selectivity. In another study, Xu et al. reported a single-crystalline 1D metallo-COF (mCOF-Ag) by combining metal–ligand coordination and dynamic imine condensation (Fig. 8a).135 mCOF-Ag was synthesized by a one-pot reaction between 4,4′-(1,10-phenanthroline-2,9-diyl)dianiline (I) and 2,9-bis(4-(dimethoxymethyl)phenyl)-1,10-phenanthroline (II) in the presence of AgBF4. Monomer II with acetal groups showed good solubility and was favorable for the self-assembly process. Compared with amine groups, the acetal groups displayed lower reactivity, which could decrease the crystal nucleation rate to form good crystals. The coordination bonds between silver ions and phenanthroline units of I and II were beneficial for the crystal formation. SEM revealed that mCOF-Ag possessed a uniform rod-like morphology with crystal lengths exceeding 2 μm. The single-crystal nature of mCOF-Ag was confirmed by high-resolution TEM and selected-area electron diffraction (SAED). Single-crystal electron diffraction (SCED) indicated that mCOF-Ag showed a monoclinic crystal structure. Since mCOF-Ag had amine groups, it could react with glyoxal to give wCOF-Ag through crystalline-state polymerization. The crystal structure of wCOF-Ag was maintained, as established by PXRD.
and give diamondoid networks with four-fold interpenetration. NPN-3 was found to crystallize in the tetragonal space group P42/n and showed a diamondoid network with six-fold interpenetration. Zhang et al. reported another single-crystal 3D COF (COF-320).133 SEM revealed that COF-320 showed a uniform rice-like morphology with a crystal dimension of about 200 nm. The single-crystal structure of COF-320 was investigated by 3D RED for the first time. COF-320 had a highly porous diamond net with 9-fold interpenetration. The RED data indicated that COF-320 formed a body-centered orthorhombic crystal structure. COF-320 showed a large Langmuir surface area of 2400 m2 g−1 due to the good crystallinity. As a result, COF-320 achieved a high methane uptake capacity of 15.0 wt% at 298 °C and 80 bar. Gao et al. designed three isostructural 3D COFs with –H, –Me, or –F substituents (3D-TPB-COF-H, 3D-TPB-COF-Me, and 3D-TPBCOF-F).134 SEM indicated that these 3D-TPB-COFs had uniform morphology with a crystal dimension of about 1 μm. All 3D-TPB-COFs showed sharp and intense diffraction peaks, indicating a highly crystalline nature. Their crystal structures were investigated by RED with the resolution reaching up to 0.9–1.0 Å, elucidating the positions of all non-hydrogen atoms in COFs. All 3D-TPB-COFs showed the same five-fold interpenetrated topologies with the different substituents being visible. All 3D-TPB-COFs displayed different gas absorption properties, such as selectivity for CO2 over N2. 3D-TPBCOF-F with C–F bonds on the porous structure achieved the highest selectivity. In another study, Xu et al. reported a single-crystalline 1D metallo-COF (mCOF-Ag) by combining metal–ligand coordination and dynamic imine condensation (Fig. 8a).135 mCOF-Ag was synthesized by a one-pot reaction between 4,4′-(1,10-phenanthroline-2,9-diyl)dianiline (I) and 2,9-bis(4-(dimethoxymethyl)phenyl)-1,10-phenanthroline (II) in the presence of AgBF4. Monomer II with acetal groups showed good solubility and was favorable for the self-assembly process. Compared with amine groups, the acetal groups displayed lower reactivity, which could decrease the crystal nucleation rate to form good crystals. The coordination bonds between silver ions and phenanthroline units of I and II were beneficial for the crystal formation. SEM revealed that mCOF-Ag possessed a uniform rod-like morphology with crystal lengths exceeding 2 μm. The single-crystal nature of mCOF-Ag was confirmed by high-resolution TEM and selected-area electron diffraction (SAED). Single-crystal electron diffraction (SCED) indicated that mCOF-Ag showed a monoclinic crystal structure. Since mCOF-Ag had amine groups, it could react with glyoxal to give wCOF-Ag through crystalline-state polymerization. The crystal structure of wCOF-Ag was maintained, as established by PXRD.
Evans et al. developed a two-step seeded growth strategy to prepare a single-crystalline 2D COF (COF-5).100 A stable colloidal COF-5 suspension was prepared in the first step.136 Then single-crystalline COF-5 was formed in the second growth step, in which the monomers were introduced slowly into the solution. The monomer addition rates in the second step were crucial for the formation of COF single crystals. Dynamic light scattering (DLS) suggested that the COF-5 particle size increased from 400 nm to 1000 nm after 4.0 equiv. of 2,3,6,7,10,11-hexahydroxytriphenylene (HHTP) was added at a slow rate of 0.1 equiv. h−1. Wide-angle X-ray scattering (WAXS) indicated that the diffraction peak intensity gradually increased with slow addition of the monomers. In contrast, when HHTP was added rapidly (1.0 equiv. h−1), the average particle size gradually decreased. To demonstrate the generality of the seeded growth strategy, two other 2D COFs (COF-10 and TP-COF) were synthesized using this strategy. Single-crystalline COF-10 and TP-COF were also obtained when the monomers were added slowly. TEM characterization indicated that the maximum sizes of COF-5 and COF-10 could reach 1 and 5 μm, respectively. Later, Evans et al. developed a general strategy to synthesize a series of single-crystalline colloidal COFs (Ph-COF, BPh-COF, DBD-COF, Py-COF, and TMPh-COF) by introducing nitrile-containing cosolvents.137 DLS measurements suggested that the Ph-COF particle size increased from 100 nm to 400 nm with the increase of monomer concentration from 5 mM to 15 mM. The crystallinity of the COFs was confirmed by synchrotron XRD. TEM suggested that the COFs showed crystal sizes of several hundred nanometers. Fast Fourier transform (FFT) indicated that all COF particles displayed crystalline spot patterns, suggesting the single-grain crystallinity. Solution fluorescence spectroscopy indicated that the COF suspensions were highly emissive compared with their boronic acid monomer solutions, which was attributed to the through-space electronic communication of chromophores between COF sheets. Ma et al. developed a general approach to grow large single crystals of 3D imine-linked COFs by using aniline as a modulator.64 In the synthesis of a typical imine-linked COF, an amorphous solid firstly formed and then transformed into a crystalline phase via imine exchange conversion. It is a feasible approach to improve the crystallinity of the COF by decreasing the rate of imine exchange. Monofunctional aniline can act as a nucleation inhibitor to modulate the crystallization process because its reactivity is similar to that of the amino monomer. After the addition of aniline, the imine bond formation remained fast. The rate of imine exchange was slowed down, which provided enough time for the growth of COF single crystals. By using this method, a single crystal of COF-300 up to 100 μm was obtained. In addition, the crystal size of COF-300 was tunable by controlling the amount of aniline. To demonstrate the generality of this method, several single crystals of other 3D imine-linked COFs including COF-303, LZU-79, and LZU-111 were obtained. Based on the single-crystal XRD data of COF-300, its degree of interpenetration was confirmed, and the arrangement of water guests in hydrated COF-300 was determined. Liang et al. reported the first non-interpenetrated single-crystal COF (LZU-306) by employing the aniline-based modulation method (Fig. 8b).138 The crystals with a size of 200 nm were synthesized under the traditional reaction conditions without aniline. In the presence of aniline, single crystals of LZU-306 were obtained. The single crystal size became larger with the increase of the amount of aniline. Single-crystal XRD indicated that LZU-306 showed a non-interpenetrated pts structure. PXRD of LZU-306 displayed sharp peaks with a very small FWHM due to the high quality of the crystals. LZU-306 exhibited a large BET surface area of 2059 m2 g−1 with an average pore width of 10.9 Å. With a highly open structure, LZU-306 was used to explore the intrinsic dynamics of the tetraphenylethylene (TPE) as the individual aggregation induced-emission (AIE) unit. Later, Ma et al. systematically studied the effect of the crystal size of COFs on their respective properties by synthesizing a series of LZU-111 and COF-300 with different crystal sizes (Fig. 8c).132 Single-crystal LZU-111 and COF-300 with various crystal sizes (200 nm, 1 μm, and 30 μm for LZU-111; 500 nm, 1 μm, and 30 μm for COF-300) were prepared by adjusting the amount of modulator. For LZU-111, the FWHM of the diffraction peaks in the PXRD patterns of variable-sized crystals decreased with the increase of the crystal size from 200 nm to 30 μm, which indicated that the crystallinity of LZU-111 was enhanced from nanocrystal to micrometer crystals. For COF-300, the FWHM of the diffraction peaks of variable-sized crystals only slightly decreased with the increase of crystal size. The effect of crystal sizes on the gas sorption behaviors of the COFs was explored. The N2 and Ar uptakes of LZU-111 increased with the increase of crystal size. However, the N2 and Ar uptakes of COF-300 decreased with the increase of crystal size. This was because COF-300 had a flexible structure. Small crystals of COF-300 were more flexible and could be more easily stretched or compressed, which was favorable for N2 and Ar uptakes. Large single crystals of COF-300 were more rigid and unfavorable for gas uptake.
Lots of efforts have been made to synthesize single-crystal COFs. Until now, only several single-crystal COFs have been reported based on several methods such as reversible self-addition polymerizations, adding a modulator, and a two-step seeded growth strategy.64,100,131 In particular, a general method has been demonstrated to grow micrometer scale single-crystal COFs by adding a modulator.64 The largest crystal size by this method can reach 100 μm. However, the reported single-crystal COFs are mainly based on the boronate ester formation and Schiff base reactions. Single-crystal COFs synthesized by other reactions remain to be explored. Moreover, it is challenging to synthesize 2D single-crystal COFs, which offer potential applications in organic electronics such as transistors and luminescent devices.
4. Strategies for improving the crystallinity of CPs
In general, CPs are prepared using irreversible coupling reactions. The main strategies for improving the crystallinity of CPs are based on the mechanism of enhancing the intramolecular or intermolecular interactions. The strategies can be divided into several categories: thermal annealing, molecular mass modulation, improving the planarity, molecular geometry modulation, side chain engineering, fluorine substitution, and so on. The advances of conjugated polymer crystals are also discussed. Although crystalline CPs have wide applications in various optoelectronic devices, in this part, we focus only on their applications in OFETs.4.1. Thermal annealing
Thermal annealing is a widely used approach to improve the crystallinity of CPs. Since CP films can achieve the best crystallinity at the optimal annealing temperature, the effect of thermal annealing on polymer crystallinity has been extensively studied. Here, we introduced several representative studies in this part. Fig. 9 shows some examples of thermal annealing to improve the crystallinity.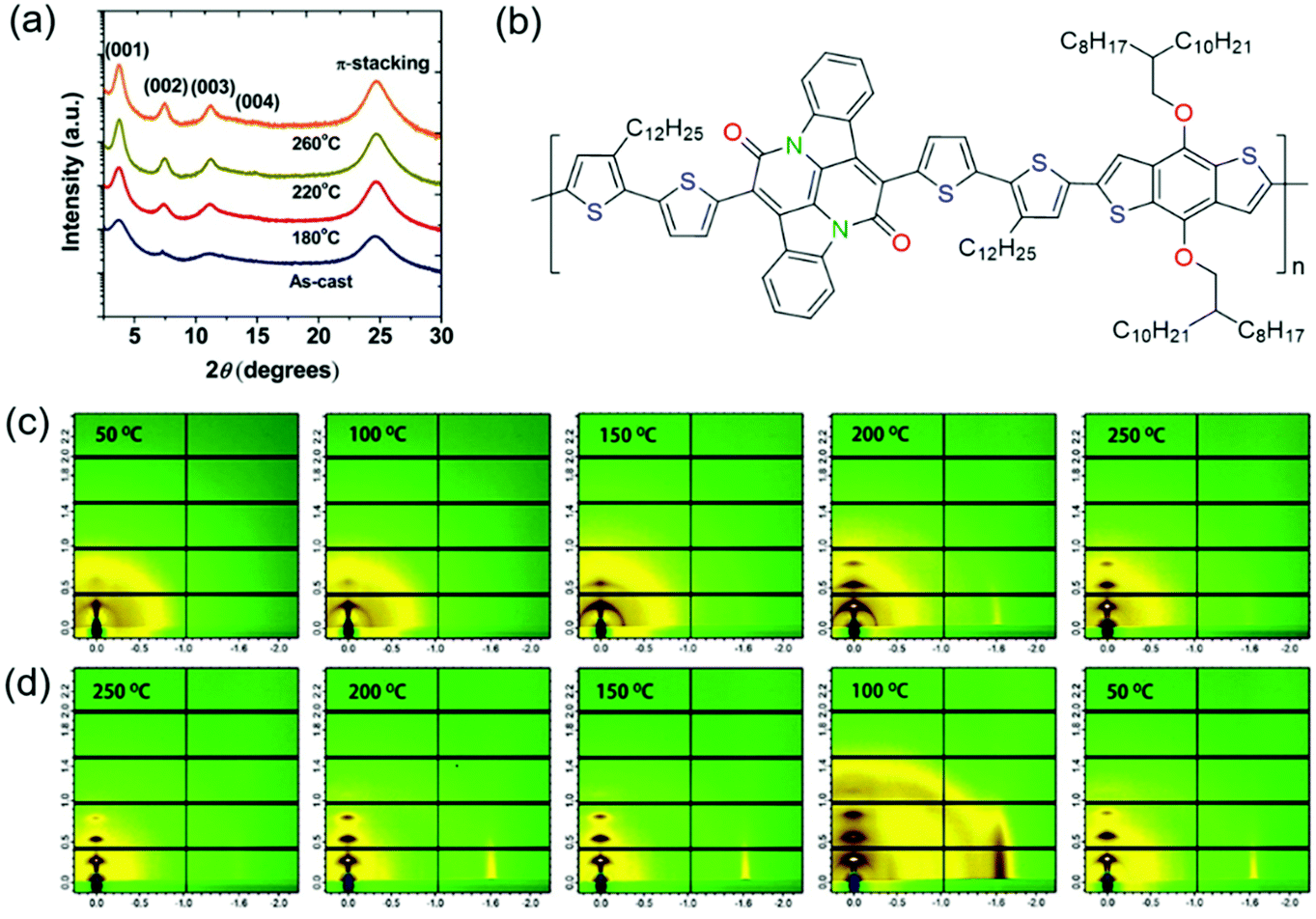 | ||
| Fig. 9 Improving the crystallinity of CPs by thermal annealing. (a) XRD patterns of PTDPPSe–Si films depending on the annealing temperature. Reproduced with permission.141 Copyright 2012, American Chemical Society. (b) The molecular structure of a BAI-based polymer (P1). GIWAXS patterns of P1 films during in situ (c) heating and (d) cooling cycles. Reproduced with permission.142 Copyright 2014, American Chemical Society. | ||
Sonar et al. developed a DPP-based polymer (PDPP-TBT).139 The influence of thermal annealing on polymer crystallinity was systematically investigated. XRD characterization suggested that the as-spun polymer film was almost amorphous without diffraction peaks. When the films were subjected to thermal annealing, the diffraction peaks appeared and became sharper. The diffraction peak intensity became stronger with increasing annealing temperature. After annealing at 200 °C, the film displayed the strongest (100) and (010) diffraction peaks. Atomic force microscopy (AFM) phase images indicated that the annealed films became more ordered and formed interconnected networks between the nanofiber domains with increasing annealing temperature. As a result, OFETs based on PDPP-TBT showed higher mobilities as the annealing temperature was increased. The devices annealed at 200 °C achieved the highest hole and electron mobilities of 0.35 and 0.4 cm2 V−1 s−1, respectively. Ha et al. reported a DPP-based polymer (P(DPP-alt-DTBSe)).140 XRD characterization indicated that the as-spun and annealed polymer films showed (h00) peaks up to fourth order. Annealing the films at 150 or 180 °C increased the intensity of the (h00) peaks, indicating the enhancement of the crystallinity. The film displayed the strongest (h00) peaks after annealing at 180 °C. OFETs based on P(DPP-alt-DTBSe) exhibited higher mobilities with increasing annealing temperature. The devices that were annealed at 180 °C achieved the highest hole mobility of 1.5 cm2 V−1 s−1. Lee et al. synthesized a DPP-based polymer (PTDPPSe–Si) containing hybrid siloxane-solubilizing side chains.141 The polymer films were annealed at different temperatures of 180, 220, and 260 °C. The as-spun film showed relatively broad (100) and (200) diffraction peaks. After annealing at 180, 220 or 260 °C, the (100) and (200) peaks became sharper along with the presence of a (300) peak. The film displayed the strongest (h00) peaks after annealing at 220 °C, which was determined as the optimal annealing temperature (Fig. 9a). As a result, OFETs based on the film annealed at 220 °C achieved the highest hole and electron mobilities of 1.69 and 0.2 cm2 V−1 s−1, respectively. In contrast, the as-spun film showed inferior hole and electron mobilities of 0.59 and 4.58 × 10−2 cm2 V−1 s−1, respectively. He et al. developed a novel bay-annulated indigo (BAI) based polymer (P1) from a natural dye (indigo) (Fig. 9b).142 The polymer films were annealed at different temperatures of 50, 100, 150, 200, and 250 °C. 2D grazing incidence wide-angle X-ray scattering (2D-GIWAXS) tests at different temperatures were conducted under in situ annealing to study the crystallinity evolution (Fig. 9c and d). During the heating process, the diffraction peaks became stronger with increasing annealing temperature. The films annealed below 150 °C showed weak (100) and (200) diffraction peaks. After annealing at 200 or 250 °C, the (100) and (200) peaks became stronger along with the presence of (300) and (010) peaks. During the cooling process, the diffraction peaks were retained. OFETs based on the annealed films achieved excellent ambipolar transport with hole and electron mobilities of 1.5 and 0.41 cm2 V−1 s−1, respectively.
In general, the as-spun polymer films are amorphous or display inferior crystallinity. Then thermal annealing is adopted to improve the crystallinity of polymer films. In an annealing process, the polymer chains can rearrange into long-range ordered packing modes controlled by the intermolecular interactions. There is an optimal annealing temperature for each polymer, which is confirmed by experiments. In general, the optimal annealing temperature is in the range from 100 to 250 °C. Note that the annealing temperature should be below the decomposition temperature of polymers.
4.2. Molecular mass modulation
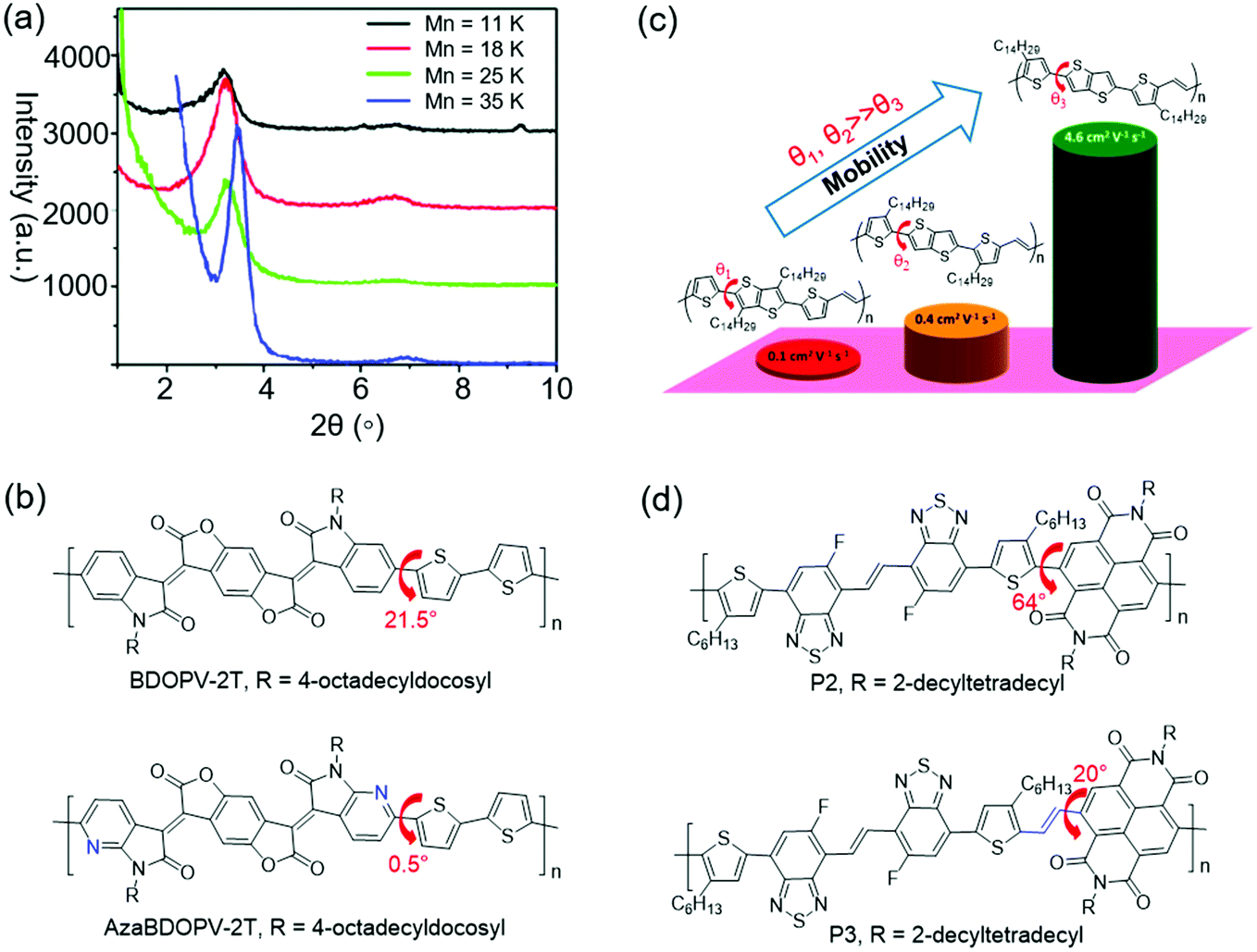
Molecular mass of CPs is one of the important factors that can influence the crystallinity. For D–A CPs, increasing the number-average molecular mass (Mn) can improve the crystallinity. One possible explanation might be that the polymers with higher Mn have stronger D–A interactions, which could promote long-range ordered packing of the polymer molecules.143Zhang et al. presented a D–A copolymer (CDT–BTZ) containing cyclopentadithiophene (CDT) as the donor and benzothiadiazole (BTZ) as the acceptor.144 The polymer showed a low Mn of 10.2 kDa. WAXS characterization suggested that CDT–BTZ showed poor crystallinity with only one (010) diffraction peak. OFETs based on CDT–BTZ displayed a hole mobility of 0.11 cm2 V−1 s−1. In another study, Tsao et al. synthesized CDT–BTZ with a higher Mn of 50 kDa by improving the purity of the monomers.145 This polymer displayed enhanced crystallinity with (100) and (010) diffraction peaks. As a result, the OFETs achieved better charge transport with a mobility of 0.67 cm2 V−1 s−1. Later, Tsao et al. synthesized several CDT–BTZs with various Mns of 11, 18, 25, and 35 kDa (Fig. 10a).143 XRD characterization suggested that the polymer crystallinity increased with the growing molecular weight. The polymer with the highest Mn showed the most intense (h00) diffraction peaks. One possible explanation might be that the polymer with a higher Mn had a stronger D–A interaction, which could promote higher order long-range packing of the polymer molecules. OFETs based on these polymers showed increasing hole mobilities from 0.28 cm2 V−1 s−1 for the lowest molecular weight sample (11 kDa) to 3.3 cm2 V−1 s−1 for the highest molecular weight sample (35 kDa). Li et al. synthesized DPP-based polymers (PDPP-TT) with various molecular masses by optimizing the polymerization reaction conditions.146 The molecular masses of these polymers were 91, 344, and 501 kDa. XRD characterization indicated that all these polymers displayed (h00) diffraction peaks up to third order. However, the diffraction peak intensity significantly increased with increasing molecular weight. As a result, OFETs based on these polymers showed increasing hole mobilities from about 1 cm2 V−1 s−1 for the low molecular weight sample (91 kDa) to 9 cm2 V−1 s−1 for the high molecular weight sample (501 kDa). These results clearly demonstrated that the as-synthesized polymers with high molecular masses were favorable for the achievement of high crystallinity and good semiconducting performance.
 | ||
| Fig. 10 Improving the crystallinity of CPs by molecular mass modulation or improving the planarity. (a) XRD patterns of CDT–BTZ depending on molecular mass. Reproduced with permission.143 Copyright 2011, American Chemical Society. (b) The planarity variations by N substitution. (c) The planarity variations by adjusting the positions of side chains. Reproduced with permission.151 Copyright 2014, American Chemical Society. (d) The planarity variations by adding a vinylene unit. | ||
The crystallinity of CPs can be improved by optimizing the molecular mass. The optimal molecular mass can be confirmed by tuning the catalyst amount or polymerization time. Although larger molecular mass may improve crystallinity, it also decreases the solubility of CPs in chlorinated solvents, which will increase the difficulty of polymer processing. Therefore, the molecular mass should be controlled within an appropriate range.
4.3. Improving the planarity
Good backbone planarity of CPs is beneficial for intermolecular π–π stacking of polymer chains, thus improving the crystallinity.143 The effective strategies include the introduction of sp2-nitrogen atoms and vinylene units into polymers and so on.147,148 For example, the replacement of C–H in aromatic rings with nitrogen atoms can decrease the steric hindrance, thus improving the coplanarity. Moreover, N atoms may induce intramolecular or intermolecular N⋯H and N⋯S interactions, which further improve the coplanarity. Fig. 10b–d show the crystallinity enhancement by improving the planarity.Guo et al. reported the synthesis of 4,4′-dialkoxy-5,5′-bithiazole (BTzOR) and 3,3′-dialkoxy-2,2′-bithiophene (BTOR).149 Incorporation of these building blocks into polymers afforded P1b and P4b, respectively. The replacement of (thiophene) C–H with (thiazole)N reduced the steric hindrance in –BTOR–Ar– units by eliminating C–H⋯H–C repulsions with adjacent arene units, thus enhancing the backbone coplanarity. The dihedral angle was about 23° between the alkoxythiophene and phthalimide planes in P4b, while it was 0° between the alkoxythiazole and phthalimide planes in P1b. The greater planarity in P1b was due to the elimination of C–H⋯H–C repulsions and the presence of S⋯O interactions in BTzOR units. XRD characterization indicated that the film of P4b was almost amorphous without diffraction peaks. However, the film of P1b showed a much more enhanced crystallinity with (h00) diffraction peaks up to third order. OFET devices based on P4b and P1b displayed hole mobilities of 0.04 and 0.13 cm2 V−1 s−1, respectively. The higher mobility of P1b could be attributed to the enhanced crystallinity endowed by the higher degree of backbone planarity. Dai et al. synthesized two polymers (BDOPV-2T and AzaBDOPV-2T) based on benzodifurandione-based oligo(p-phenylene vinylene) (BDOPV) and nitrogen-substituted BDOPV.147,150 Incorporation of nitrogen atoms decreased the dihedral angle between the BDOPV and neighboring thiophene from 21.5° (BDOPV-2T) to 0.5° (AzaBDOPV-2T) (Fig. 10b). The enhanced planarity of AzaBDOPV-2T was attributed to the intramolecular N⋯S interactions and less steric hindrance of sp2-N atoms than the CH units. 2D grazing-incidence X-ray diffraction (2D-GIXRD) characterization indicated that AzaBDOPV-2T achieved stronger diffraction peaks compared with BDOPV-2T. The π–π stacking distance of AzaBDOPV-2T (3.44 Å) was shorter than that of BDOPV-2T (3.55 Å). As a result, OFETs based on AzaBDOPV-2T demonstrated a remarkable electron mobility of 3.22 cm2 V−1 s−1, which was approximately two times that of the devices based on BDOPV-2T. Fei et al. designed three isomeric CPs (P1, P2 and P3), where the positions of alkyl chains on the polymer backbone were altered.151 Density functional theory (DFT) calculations indicated that both P1 and P2 showed obvious torsional angles of 30.5° and 31.3° between the thiophene and thieno[3,2-b]thiophene moieties, while P3 adopted a good coplanar backbone with a dihedral angle of 3.7° between the two aromatics (Fig. 10c). The absorption maxima of P3 in solution and in film red-shifted compared to those of P1 and P2, suggesting that P3 had the best backbone planarity and the largest effective conjugation in these three polymers. The 2D-GIWAXS of P1 and P2 only displayed (100) diffraction peaks. In contrast, P3 showed (h00) diffraction peaks up to fourth order and a strong π–π stacking peak. The carrier mobilities of P1, P2, and P3 were 0.1, 0.4, and 4.6 cm2 V−1 s−1, respectively. The significant improvement in the transistor performance of P3 was consistent with its enhanced crystallinity. Wang et al. reported the synthesis of two polymers (P2 and P3) based on benzothiadiazole.148 Intramolecular O⋯H hydrogen bonds formed in P3. DFT calculations indicated that P2 adopted a highly twisted backbone with a dihedral angle of 64° between the alkylated thiophene and neighboring NDI. P3 had additional vinylene bridges between NDI and thiophene units, resulting in a better backbone planarity due to the lower steric hindrance. The dihedral angle between the vinylene and NDI in P3 was 20° (Fig. 10d). The enhanced backbone planarity was beneficial for molecular packing. 2D-GIWAXS characterization indicated that both P2 and P3 showed second-order (h00) and (010) diffraction peaks. However, the (h00) diffraction peak intensity of P3 was stronger than that of P2. Moreover, the π–π stacking distance of P3 was much shorter than that of P2. OFETs based on P3 displayed an electron mobility of 3.95 cm2 V−1 s−1, which was one order of magnitude higher than P2.
Generally, functional groups (CN, CH3, Br, etc.) with large steric hindrance in CPs can hinder the molecular packing, which is harmful for polymer crystallization. Therefore, the crystallinity of CPs can be improved by introducing functional groups with small steric hindrance into the polymer backbone. These groups include sp2-N atoms and vinylene units. The introduction of sp2-N atoms not only decrease the steric hindrance, but also induce intermolecular N⋯H and N⋯S interactions, both of which can improve the crystallinity. Despite the aforementioned advantages, it is difficult to synthesize N-substituted units, especially for N-substituted acceptors.147 More effective synthetic approaches for N-substituted polymers should be explored in the future.
4.4. Molecular geometry modulation
Molecular geometric structures of the polymer backbone can have a remarkable effect on the crystallinity. Molecular geometric structures include molecular symmetry, regularity, and linearity. Recent studies indicate that polymers with centrosymmetric or regioregular monomers generally showed improved crystallinity than their counterparts with axisymmetric or regiorandom ones.152–154 A linear polymer backbone instead of a curved one is beneficial for polymer crystallization.155 The reason might be that regioregular and linear polymers can form better intermolecular π–π stacking in the solid state, thus improving the crystallinity. Fig. 11 shows the examples of molecular geometry modulation to improve the crystallinity.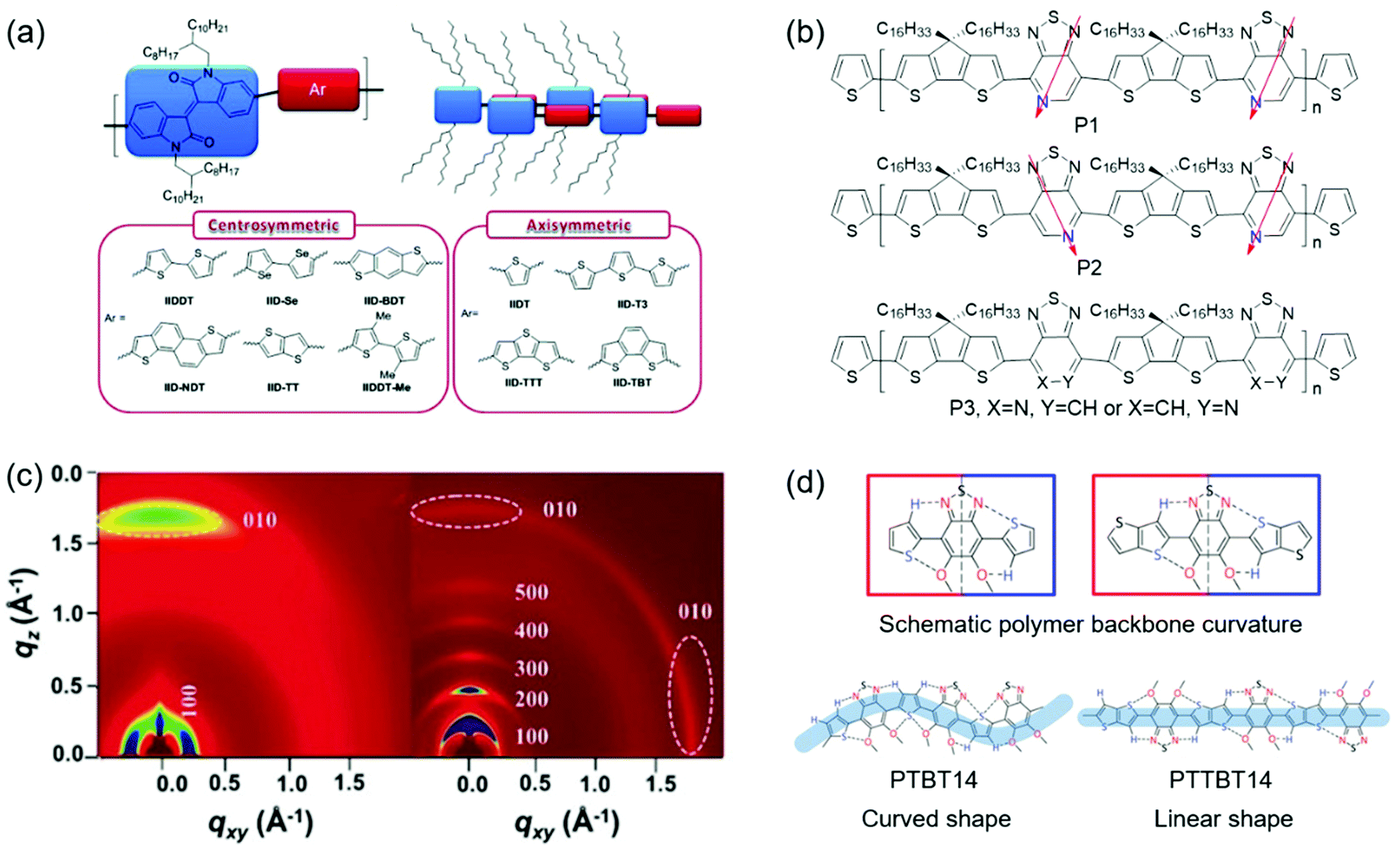 | ||
| Fig. 11 Improving the crystallinity of CPs by molecular geometry modulation. (a) The molecular structures of isoindigo-based polymers containing centrosymmetric and axisymmetric units. Reproduced with permission.152 Copyright 2012, American Chemical Society. (b) The molecular structures of regioregular polymers (P1 and P2) and a regiorandom polymer (P3). (c) 2D-GIWAXS of s-BTI2-FT (left) and f-BTI2-FT (right). Reproduced with permission.158 Copyright 2017, Wiley-VCH. (d) The molecular structures of two polymers (PTBT14 and PTTBT14) with different backbone curvatures. Reproduced with permission.159 Copyright 2014, American Chemical Society. | ||
The effect of molecular symmetry and regularity on crystallinity has been investigated. Lei et al. developed two D–A CPs (IIDDT and IIDT), which contained centrosymmetric acceptors (isoindigo).156 However, the centrosymmetric donor (bithiophene) was found for IIDDT, while the axisymmetric one (thiophene) was found for IIDT. Since the symmetry of donors had a remarkable effect on the polymer crystallinity, the film based on IIDT displayed a relatively amorphous structure with one weak diffraction peak, while the film based on IIDDT showed a much more enhanced crystallinity with (h00) diffraction peaks up to fourth order. Consequently, IIDDT-based FETs provided a hole mobility of 0.79 cm2 V−1 s−1, which was one order of magnitude higher than IIDT-based devices. Later, Lei et al. synthesized ten isoindigo-based polymers containing centrosymmetric or axisymmetric donors to systematically study the influence of symmetry and backbone curvature on crystallinity and semiconductor properties (Fig. 11a).152 Different from the polymers containing axisymmetric donors, the polymers containing centrosymmetric ones showed a linear backbone for good interchain π–π stacking interactions. The 2D-GIXRD of polymers containing centrosymmetric donors displayed fourth-order (h00) diffraction peaks. However, all polymers containing axisymmetric donors showed poor crystallinity with one or no (h00) diffraction peak. As a result, OFETs based on these six polymers containing centrosymmetric donors demonstrated hole-transporting behaviors with mobilities exceeding 0.1 cm2 V−1 s−1, which were one or three orders of magnitude higher than those of the devices based on the polymers with axisymmetric donors. These results demonstrated that designing polymers containing both centrosymmetric acceptors and donors was favorable for the achievement of high crystallinity and good semiconducting performance. Ying et al. developed two regioregular copolymers (P1 and P2) and one regiorandom copolymer (P3) based on pyridal[2,1,3]thiadiazole (PT) units (Fig. 11b).153 The optical band gaps of P1 and P2 were smaller than that of P3, indicating that molecular regioregularity could decrease the band gap of the polymers. Moreover, the backbone regioregularity had a direct impact on the molecular packing of the polymer films. 2D-GIWAXS characterization indicated that P1 and P2 showed stronger (h00) diffraction peaks compared with P3. Consequently, the regioregular polymers (P1 and P2) demonstrated remarkable hole mobilities of 0.4 and 0.6 cm2 V−1 s−1, respectively, which were two orders of magnitude higher than that of the regiorandom polymer (P3). Wang et al. reported the synthesis of a regioregular polymer (P2F) and a regiorandom polymer (PRF) based on 5-fluorobenzo[c][1,2,5]thiadiazole units.157 As shown in 2D-GIWAXS, films of both P2F and PRF displayed third-order (h00) diffraction peaks and π–π stacking peaks. However, the diffraction peak intensity of P2F was stronger than that of PRF. The regiorandom polymer PRF showed a hole mobility of 0.4 cm2 V−1 s−1, while its regioregular counterpart P2F achieved a mobility of 1.2 cm2 V−1 s−1. The significant improvement in the transistor performance of P2F was attributed to the enhanced crystallinity.
Molecular linearity of the polymer backbone can have a remarkable effect on the crystallinity. Rieger et al. designed five polymers (P1–P5) containing different benzodithiophene isomers with varying degrees of curvature.155 The backbones of P1–P3 were linear, while those of P4 and P5 were curved. The influence of the curvature on optical gaps and crystallinity was systematically investigated. UV-vis spectroscopy indicated that the optical gaps of the polymers became larger with increasing curvature due to decreased effective conjugation lengths. Wide-angle XRD experiments suggested that the linear polymers displayed enhanced crystallinity compared with the curved ones. For example, P3 showed strong third-order (h00) and (010) diffraction peaks, while P4 and P5 only displayed weak (100) and (010) diffraction peaks. As a result, the carrier mobilities of P1, P2, and P3 were one order of magnitude higher than those of P4 and P5. These results demonstrated that designing polymers with a linear backbone was favorable for the achievement of high crystallinity and good semiconducting performance. Wang et al. designed two bithiophene imide (BTI)-based n-type copolymers (s-BTI2-FT and f-BTI2-FT).158 s-BTI2-FT possessed a BTI dimer connected through a single bond, while f-BTI2-FT contained a fused BTI dimer. DFT calculations revealed that the molecular shapes of s-BTI2-FT and f-BTI2-FT were sinewave and pseudo-linear, respectively. f-BTI2-FT possessed a more linear backbone than s-BTI2-FT. The 2D-GIXRD of s-BTI2-FT showed (100) and (010) diffraction peaks. However, f-BTI2-FT displayed enhanced crystallinity with pronounced fifth-order (h00) diffraction peaks and a (010) peak (Fig. 11c). OFETs based on f-BTI2-FT showed an electron mobility of 1.13 cm2 V−1 s−1, which was higher than that of OFETs based on s-BTI2-FT. Lee et al. reported thiophene and thienothiophene based copolymers (PTBT14 and PTTBT14) with different backbone curvatures.159 DFT calculations indicated that both polymers possessed a similar planar structure via intramolecular noncovalent C–H⋯N and S⋯O attractive interactions. However, PTTBT14 was a linear polymer, while PTBT14 was a curved one (Fig. 11d). The linearity of the polymer backbone significantly influenced the crystallinity of polymer films. The film of PTBT14 annealed at 200 °C only showed weak (100) and (200) diffraction peaks. In contrast, the annealed film of PTTBT14 displayed pronounced third-order (h00) diffraction peaks and a π–π stacking peak. The carrier mobilities of PTBT14 and PTTBT14 were 0.3 and 1.2 cm2 V−1 s−1, respectively. The improvement in the mobility of PTTBT14 was consistent with its enhanced crystallinity.
Molecular geometric structures of CPs can influence the molecular packing in film, thus affecting the crystallinity. It has been demonstrated that the crystallinity can be improved by designing polymers with centrosymmetric, regioregular, and linear monomers.152–155 Different from other factors that can influence the crystallinity, molecular geometry, especially for molecular linearity, is generally ignored. Therefore, molecular geometry of CPs should be paid more attention when designing new polymers with high crystallinity.
4.5. Side chain engineering
CPs generally contain side chains, which not only endow the polymers with solubility in organic solvents, but also affect the intermolecular packing and thus the crystallinity. The side chain engineering strategy includes the optimization of the branched point position of side chains and the introduction of semifluorinated or hydrogen bond-containing alkyl chains and so on.160–164 This strategy can improve the polymer crystallinity by enhancing the intermolecular interactions. For instance, the strong self-organization behaviors of the semifluorinated alkyl chains can promote the polymer molecules to form a long-range crystalline structure.163 Yang et al. provided a good review of the side-chain modulation strategy for OFETs.165Fig. 12a and b show some examples of side chain engineering to improve the crystallinity.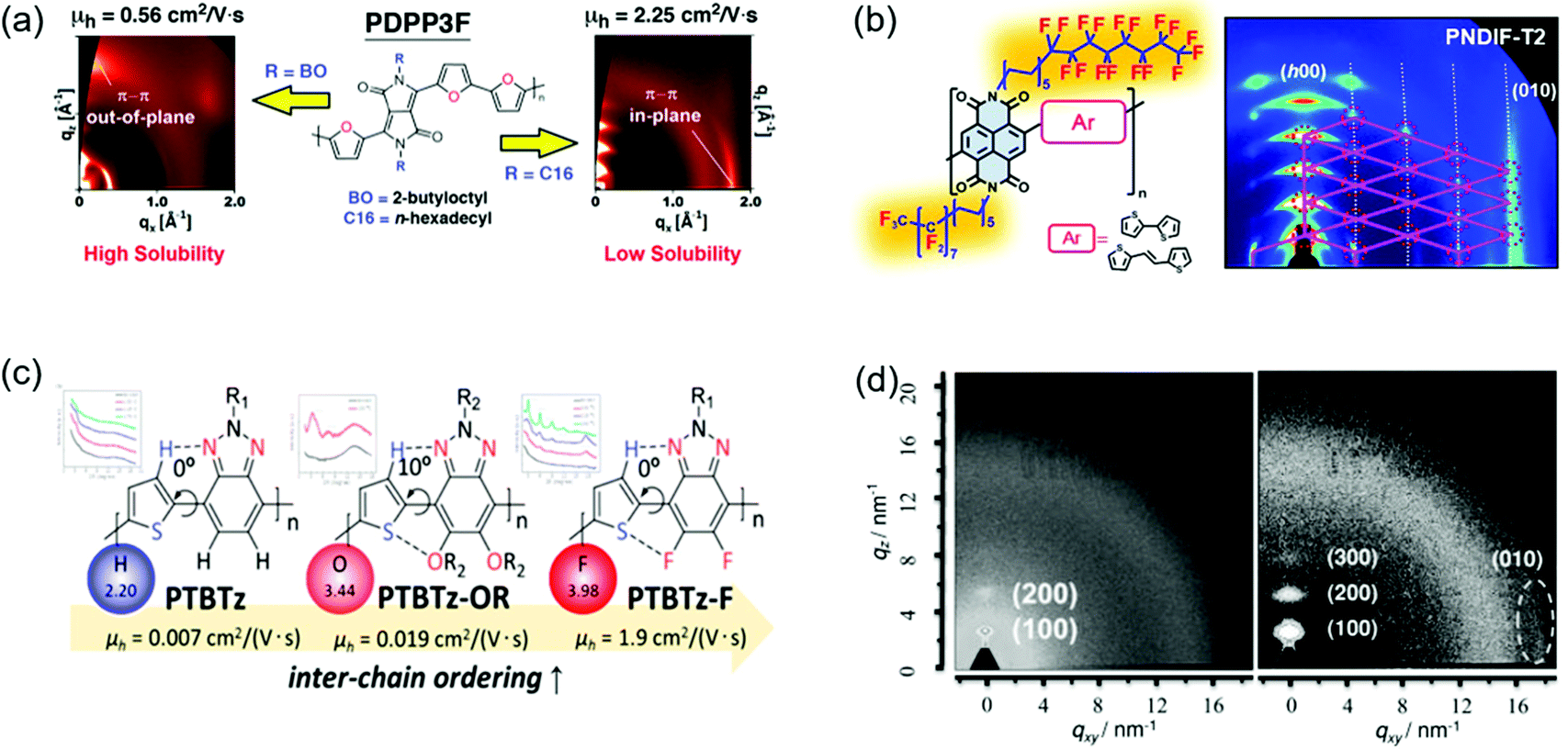 | ||
| Fig. 12 Improving the crystallinity of CPs by side chain engineering and fluorine substitution. (a) GIXRD patterns of PDPP3F containing different side chains. Reproduced with permission.167 Copyright 2013, American Chemical Society. (b) The molecular structures and GIXRD patterns of NDI-based polymers containing semifluorinated side chains. Reproduced with permission.163 Copyright 2016, American Chemical Society. (c) The molecular structures of benzotriazole-based polymers containing different substituted atoms. Reproduced with permission.171 Copyright 2014, American Chemical Society. (d) 2D GIWAXS of the annealed films of PDPP-TVT (left) and PDPP-4FTVT (right). Reproduced with permission.172 Copyright 2015, Wiley-VCH. | ||
Kang et al. developed four DPP-based polymers (P-24DPPDBTE, P-24-DPPDTSE, P-29DPPDBTE, and P-29-DPPDTSE).166 Compared with P-24DPPDBTE and P-24-DPPDTSE (P-24s), P-29DPPDBTE and P-29-DPPDTSE (P-29s) possessed extended branching-position-adjusted alkyl chains, which were favorable for molecular packing. XRD characterization suggested that the crystallinity of P-29s was much more improved than P-24s. For example, P-24DPPDBTE showed weak (100) and (200) diffraction peaks, whereas P-29DPPDBTE displayed pronounced (h00) diffraction peaks up to fourth order. The π–π stacking distance of P-29DPPDBTE was much shorter than that of P-24DPPDBTE. As a result, P-29s showed better charge transport compared with P-24s. Particularly, P-29-DPPDTSE achieved a remarkable hole mobility of 12.04 cm2 V−1 s−1, which was about three times that of P-24-DPPDTSE. Chen et al. reported two furan-containing DPP-based polymers (PDPP3F-BO and PDPP3F-C16).167 PDPP3F-BO and PDPP3F-C16 contained branched 2-butyloctyl and linear n-hexadecyl side chains, respectively (Fig. 12a). The linear side chains were expected to enhance the polymer crystallinity due to the less steric hindrance for molecular packing. 2D-GIXRD characterization indicated that PDPP3F-BO showed poor crystallinity with (100) and (200) diffraction peaks. In contrast, PDPP3F-C16 displayed strong fourth-order (h00) diffraction peaks. Compared with PDPP3F-BO, PDPP3F-C16 achieved tighter π–π stacking with a shorter distance. OFETs based on PDPP3F-C16 demonstrated a high hole mobility of 2.25 cm2 V−1 s−1, which was about four times that of the device based on PDPP3F-BO. Wang et al. developed two DPP-based polymers (PDPPSe-10 and PDPPSe-12) containing both branched and linear alkyl chains.168 A polymer (PDPPSe) only containing branched alkyl chains was synthesized for comparison. The replacement of bulky branched alkyl chains with linear ones could reduce the steric hindrance, thus improving the backbone planarity. 2D-GIWAXS characterization indicated that PDPPSe showed poor crystallinity with (100) and (200) diffraction peaks. In contrast, PDPPSe-10 and PDPPSe-12 displayed clear fourth-order (h00) diffraction peaks. OFETs based on PDPPSe-10 and PDPPSe-12 achieved hole mobilities of 8.1 and 9.4 cm2 V−1 s−1, respectively, which were about six times that of the devices based on PDPPSe.
The introduction of semifluorinated or hydrogen bond-containing alkyl chains can also improve the crystallinity of CPs. Kang et al. developed two novel NDI-based polymers (PNDIF-T2 and PNDIF-TVT) containing linear semifluorinated side chains (Fig. 12b).163 Two polymers (PNDI2OD-T2 and PNDI2DT-TVT) containing branched alkyl chains were synthesized for comparison. The strong self-organization behaviors of the semifluorinated chains induced a high degree of long-range order in the polymer backbone by forming a superstructure composed of “backbone crystals” and “side-chain crystals”. The annealed films of PNDIF-T2 and PNDIF-TVT displayed much more enhanced crystallinity compared with those of PNDI2OD-T2 and PNDI2DT-TVT. For example, PNDI2OD-T2 only showed (100), (200), and (010) diffraction peaks, while PNDIF-T2 displayed clear (h00) peaks up to fifth order and an intense (010) peak. Moreover, (00l), (30l), and (501) peaks were found for PNDIF-T2. All these peaks suggested the superior crystallinity of PNDIF-T2. As a result, OFETs based on PNDIF-T2 and PNDIF-TVT achieved outstanding electron mobilities of 3.95 and 3.75 cm2 V−1 s−1, respectively, which were much higher than those of PNDI2OD-T2 and PNDI2DT-TVT. Heo et al. reported two thienylenevinylene (TV) based polymers (PC12TVC5F7T and PC12TV12T) containing semi-fluorinated alkyl and hydrocarbon alkyl side chains.169 Highly crystalline structures formed in PC12TVC5F7T films through strong intermolecular interactions between semi-fluorinated alkyl chains. 2D-GIWAXS characterization indicated that PC12TVC5F7T exhibited intense (h00) diffraction peaks up to fifth order and a (010) peak. Several (00l) peaks were also observed for PC12TVC5F7T. Moreover, AFM images revealed that PC12TVC5F7T films displayed well-defined plate-like morphology with a domain size of about 40 nm. In contrast, PC12TV12T only showed (h00) peaks up to third order. OFET devices based on PC12TVC5F7T and PC12TV12T displayed hole mobilities of 1.91 and 1.05 cm2 V−1 s−1, respectively. The higher mobility of PC12TVC5F7T could be attributed to the improved crystallinity endowed by semi-fluorinated alkyl chains. Yao et al. designed three DPP-based polymers (pDPP4T-1, pDPP4T-2, and pDPP4T-3), in which the proportions of the branched alkyl chains and urea-containing alkyl chains were 30:1, 20:1, and 10:1, respectively.164 Fourier transform infrared and 1H NMR spectra demonstrated that the urea-containing alkyl chains could induce intermolecular hydrogen bonds. The number of hydrogen bonds increased from pDPP4T-1 to pDPP4T-3. GIXRD indicated that the diffraction peak intensity of the out-of-plane (h00) and in-plane (010) peaks increased from pDPP4T-1 to pDPP4T-3. As a result, pDPP4T-3 achieved the highest hole mobility of 13.1 cm2 V−1 s−1 among the polymers.
The side chains of CPs can have a remarkable effect on the crystallinity. The crystallinity of CPs can be improved by optimizing the branched point position of side chains or introducing semifluorinated chains and hydrogen bond-containing chains. The introduction of semifluorinated chains or hydrogen bond-containing chains can enhance the intermolecular interactions of CPs by strong self-organization or hydrogen bond interactions, which will promote the polymer crystallization.163,164 However, the optimal branched point positions of side chains vary for different polymers, which needs to be confirmed by experiments. Therefore, the side chains of CPs should be carefully selected to achieve good crystallinity.
4.6. Fluorine substitution
Fluorine substitution in the polymer backbone is an effective approach to improve the crystallinity. Fluorine atom is a strong electron-withdrawing unit with a small van der Waals radius (1.35 Å). The introduction of fluorine atoms can induce intramolecular or intermolecular F⋯S and F⋯H noncovalent interactions, which may improve the backbone planarity. Both noncovalent interactions and better planarity can improve the polymer crystallinity. Fig. 12c and d show some examples of fluorine substitution strategies to improve the crystallinity.Lei et al. presented a fluorinated (FBDPPV-1) and a non-fluorinated polymer (BDPPV) based on BDOPV units.170 Theoretical calculations indicated that FBDPPV-1 showed an almost planar conjugated backbone. Both polymer films exhibited strong out-of-plane (h00) diffraction peaks with edge-on lamellar packings. FBDPPV-1 displayed pronounced fifth-order (h00) diffraction peaks, whereas BDPPV displayed only fourth-order (h00) peaks. FBDPPV-1 showed a stronger (010) diffraction peak with a shorter π–π stacking distance of 3.42 Å compared with BDPPV. In addition, the root-mean-square (RMS) analysis of the AFM height figures indicated that the film of FBDPPV-1 showed a larger surface roughness compared with BDPPV. Both 2D-GIXRD and AFM data demonstrated that fluorination in polymers could improve the crystallinity. The electron mobilities of BDPPV and FBDPPV-1 were 1.1 and 1.7 cm2 V−1 s−1, respectively. The improvement in the mobility of FBDPPV-1 was consistent with its enhanced crystallinity. In another study, Yum et al. developed three functionalized benzotriazole-based polymers (PTBTz, PTBTz-OR, and PTBTz-F) (Fig. 12c).171 The dihedral angles between the thiophene and the benzotriazole units in PTBTz, PTBTz-OR, and PTBTz-F were 0°, 10°, and 0°, respectively. In PTBTz, only N⋯H noncovalent interactions were observed. In contrast, N⋯H and S⋯O or S⋯F noncovalent interactions were observed for PTBTz-OR or PTBTz-F. These noncovalent interactions were likely to induce strong intermolecular packing. XRD characterization indicated that PTBTz showed only a weak (100) diffraction peak, while PTBTz-OR or PTBTz-F displayed second- or fourth-order (h00) diffraction peaks. Moreover, both PTBTz-OR and PTBTz-F showed π–π stacking peaks. The hole mobilities of PTBTz, PTBTz-OR, and PTBTz-F were 0.007, 0.019, and 1.9 cm2 V−1 s−1, respectively. The highest mobility of PTBTz-F was consistent with its best crystallinity. Gao et al. developed a polymer (PDPP-4FTVT) based on tetrafluorinated (E)-2-(2-(thiophen-2-yl)vinyl)thiophene (TVT) via direct arylation polycondensation.172 A non-fluorinated polymer (PDPP-TVT) was also synthesized for comparison. DFT calculations revealed that PDPP-4FTVT showed a completely planar backbone due to the presence of intramolecular F⋯S noncovalent interactions. The film of PDPP-4FTVT annealed at 200 °C displayed obvious third-order (h00) diffraction peaks and a (010) peak. In contrast, the annealed film of PDPP-TVT exhibited only relatively weak (100) and (200) diffraction peaks without a (010) peak (Fig. 12d). OFETs based on PDPP-4FTVT showed an ambipolar transport with hole and electron mobilities of 3.40 and 5.86 cm2 V−1 s−1, respectively, which were higher than those of PDPP-TVT. Wang et al. designed three polymers (PhF0, PhF1, and PhF2,5) containing fluorinated phenyl units with increasing number of F atoms (0, 1, 2F).173 Theoretical calculations indicated that the backbone planarity of PhF2,5 was enhanced compared with PhF0 and PhF1. 2D-GIWAXS characterization indicated that the films of PhF0 and PhF1 were almost amorphous. In contrast, the film of PhF2,5 displayed (h00) diffraction peaks up to third order. A strong π–π stacking peak was also observed in the in-plane direction. The carrier mobility values of the polymers increased with substitution of more fluorine atoms. PhF2,5 displayed a hole mobility of 0.92 cm2 V−1 s−1, which was three orders of magnitude higher than that of PhF0. Sun et al. reported a general synthetic approach to obtain methoxylated quinoidal bithiophene (MQBT) units with a single regioisomer.174 A fluorinated (P2FMQBT-DFBT) and a non-fluorinated (PMQBT-BT) polymer based on MQBT units were prepared. DFT calculations suggested that P2FMQBT-DFBT possessed a better planar backbone compared with PMQBT-BT. 2D-GIXRD characterization indicated that PMQBT-BT showed a weak (100) diffraction peak, while P2FMQBT-DFBT displayed a much more enhanced crystallinity with (h00) diffraction peaks up to third order. Moreover, P2FMQBT-DFBT had a stronger (010) diffraction peak with a much shorter π–π stacking distance of 3.59 Å compared with PMQBT-BT. As a result, P2FMQBT-DFBT showed a hole mobility of 2.7 cm2 V−1 s−1, which was one order of magnitude higher than that of PMQBT-BT.
Fluorine substitution is a widely used method for improving the crystallinity of CPs. The introduction of fluorine atoms can enhance the intermolecular interactions of CPs by F⋯S and F⋯H noncovalent interactions, which will promote the polymer crystallization. Various fluorinated polymers or even multifluorinated polymers have been developed and achieved good crystallinity.175 However, it is very challenging to synthesize fluorinated polymers.172 Facile and effective synthetic methods for fluorinated polymers are highly desired. Except for fluorination, other halogenation methods like chlorination can be adopted to improve the crystallinity, which remains to be explored.
4.7. Single crystals of conjugated polymers
CPs generally contain branched alkyl chains, which are unfavorable for polymer crystallization. Also, the Suzuki or Stille polymerization reactions are irreversible. As a result, the as-synthesized polymers normally are polycrystalline rather than single-crystalline. Since single crystals of CPs offer a perfect platform to study the structure–property relationship and investigate the charge transport mechanisms in polymer systems, it is highly desirable to grow such single crystals. Moreover, polymer crystals usually exhibit better semiconducting performance than films due to the long-range ordered molecular packing. However, conjugated polymer crystals amenable to single-crystal XRD are still in the early stage. Most of the reported polymer crystals are obtained by a self-assembly method as nanowires or microwires.65,66Fig. 13 shows the reported polymer crystals. | ||
| Fig. 13 Conjugated polymer crystals. (a) Optical micrographs of the single crystals of P3HT-26. Reproduced with permission.176 Copyright 2012, Wiley-VCH. (b) Polymer nanowires of DPPBTSPE and its applications in electronic devices. Reproduced with permission.65 Copyright 2015, American Chemical Society. (c) GIXRD patterns of poly-PCDA crystals. Reproduced with permission.178 Copyright 2017, Wiley-VCH. (d) 3D schematic of the polymer crystals by a self-seeding approach. Reproduced with permission.66 Copyright 2021, Wiley-VCH. | ||
Rahimi et al. obtained large single crystals of a regioregular poly(3-hexylthiophene) polymer (P3HT-26) by a self-seeding method (Fig. 13a).176 This method contained three steps: dissolution, seed forming at a seeding temperature (TSS), and crystal growth at a lower temperature (TC). Large single crystals with a length of 20 μm were obtained by precisely controlling TSS and TC. SAED indicated that single crystals had a monoclinic crystal structure. The π–π stacking distance was 4.3 Å. Single crystals of P3HT samples with other molecular weights (P3HT-4 and P3HT-5) were also obtained by this method. Kim et al. reported single crystal nanowires based on a copolymer (PDTTDPP) of DPP and dithieno[3,2-b:2′,3′-d]thiophene (DTT) by a self-assembly method.177 The molecular rigidity and planarity of DTT could enhance intermolecular interactions to promote the self-assembly of the polymer backbone. The polymer nanowires (PNWs) were obtained by storing the dissolved polymer solution at room temperature for several days. The PNWs displayed a small curvature and surface uniformity with an average length of 15.9 μm and a width of 65 nm. SAED patterns indicated that the PNWs showed highly crystalline nature with an orthorhombic crystal structure. OFETs based on PNWs achieved a remarkable hole mobility of 7.0 cm2 V−1 s−1, which was one order of magnitude higher than that of the devices based on spin-coated films. Um et al. synthesized two fractions of DPPBTSPE with high and low number-average molecular weights (MW) of 8 and 68 kDa, respectively (Fig. 13b).65 Both high- and low-MW polymers could form single-crystalline nanowires by a self-assembly method. However, only the low-MW polymer formed well-isolated nanowires with a higher aspect ratio, which could be applied to fabricate single nanowire-based devices. The isolated nanowires had lengths of 50–100 μm and widths of 170 nm. The nanowires showed highly crystalline nature with an orthorhombic crystal structure. 2D-GIXRD characterization indicated that the single-crystalline nanowires displayed enhanced crystallinity compared with spin-coated films. The (010) diffraction peak of nanowires was stronger and sharper than that of spin-coated films. As a result, OFETs based on nanowires achieved a superior hole mobility of 24 cm2 V−1 s−1, which was one order of magnitude higher than that of the devices based on spin-coated films. Yao et al. achieved polymer nanocrystals by an in situ topochemical polymerization method.178 Firstly, single crystals of 10,12-pentacosadiynoic acid (PCDA) were obtained in a physical vapor transport (PVT) system. Then, single crystals of PCDA were directly transformed into poly-PCDA crystals by white light irradiation. The length and width of poly-PCDA crystals were tens of micrometers and hundreds of nanometers, respectively. The poly-PCDA crystals showed highly crystalline nature and long-range order with an orthorhombic crystal structure. 2D-GIXRD characterization indicated that poly-PCDA crystals displayed intense (h00), (020), and (001) diffraction peaks (Fig. 13c). SAED of poly-PCDA crystals showed very sharp diffraction spots, suggesting the high degree of crystallinity of this polymer. As a result, OFETs based on poly-PCDA crystals achieved outstanding charge transport with a maximum mobility of 50 cm2 V−1 s−1. Yao et al. reported a facile approach to obtain microwire crystals of a benzodifurandione-based oligo(p-phenylene vinylene) polymer (F4BDOPV-2T) by a controlled self-seeding growth process.66 The self-seeding approach contained three steps: dissolution at 140 °C, self-seeding and growth through slow cooling, and further crystal growth at room temperature (Fig. 13d). The obtained polymer crystals displayed a microwire shape with a length of 40 μm and a width of 0.5–7 μm. PXRD patterns of the polymer crystals showed intense diffraction peaks, which was consistent with Pawley-simulated XRD data. However, some diffraction peaks (0.5–1.5 Å) disappeared in the GIWAXS patterns of spin-coated polymer films. These results indicated the improved crystallinity in microwire crystals than in thin films. As a result, OFETs based on microwire crystals demonstrated a remarkable electron mobility of 3.46 cm2 V−1 s−1, which was about four times that of the devices based on spin-coated films. To demonstrate the generality of this method, another n-type polymer (P(NDI2OD-T2)) was selected for crystal growth. P(NDI2OD-T2) microwire crystals displayed an electron mobility of 2.56 cm2 V−1 s−1, which was much higher than that of P(NDI2OD-T2) films.
To date, conjugated polymer crystals have been achieved by a self-assembly method, which mainly contains three steps: polymer dissolution, seed forming, and crystal growth.66,176 The obtained polymer crystals by this method are nanowires or microwires, which have been demonstrated to exhibit a much better semiconducting performance than films in OFET applications.66 However, large-sized polymer crystals amenable to single-crystal XRD have not been achieved, which need further studies. It may be a feasible way to synthesize polymer single crystals by adopting reversible polymerization reactions.
5. Conclusion and outlook
Crystallinity of COFs and CPs is of great importance for structural determination and practical applications. The enhancement of crystallinity can significantly improve the performance of COFs and CPs in photocatalysis, electronics, and other fields. In recent years, diverse design strategies have been developed to improve the crystallinity of COFs and CPs. The key point for improving the crystallinity and even achieving single crystals is to modulate the reversible process during the bond formation, offering enough time for self-correction of defects. For COFs, the strategies include polycondensation reaction optimization, improving the planarity, hydrogen bond and fluorine substitution, and so on. For CPs, the strategies include thermal annealing, molecular mass modulation, improving the planarity, molecular geometry modulation, side chain engineering, fluorine substitution, and so on. To date, CP crystals and COF single crystals have been achieved based on the above effective strategies. However, there are still several fundamental issues that need further studies.It is well known that the polycondensation reaction conditions can have significant effects on the crystallinity of COFs. The relationship between the polycondensation reaction conditions and the crystallinity is complicated and requires further studies. Although single-crystal COFs have been achieved, more universal synthetic methods to obtain single crystals are still desired to offer precise structural information of COFs. Moreover, the reported single-crystal COFs are mainly based on boronate ester formation and Schiff base reactions. Single-crystal COFs based on other reactions remain to be explored.4 Although the effect of single-crystal size on the gas absorption properties was recently investigated,132 it is still very interesting to explore the applications of single-crystal COFs in other fields such as transistors and luminescent devices. For CPs, nitrogen or fluorine substitution can improve the crystallinity. However, it is difficult to synthesize the monomers containing nitrogen or fluorine atoms.172,179 Despite the fact that various strategies have been developed to improve the crystallinity of CPs, the polymer crystals amenable to single-crystal XRD are still in the early stage. Clearly, it is very difficult to obtain single crystals of the reported polymers synthesized based on irreversible coupling reactions. Thus, adopting reversible reactions may promote the realization of polymer single crystals.
Despite the challenges remaining for the crystallinity of COFs and CPs, various strategies have been demonstrated to be effective to achieve highly crystalline COFs and CPs. These strategies can also be applied in other new polymers, especially for those that are difficult to crystallize. The highly crystalline COFs and CPs will be attractive for practical applications, which can provide feasible solutions to address energy and environment issues.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
QZ acknowledges the support of starting funds from City University of Hong Kong.References
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS.
- C. S. Diercks and O. M. Yaghi, Science, 2017, 355, 6328 CrossRef.
- E. Jin, M. Asada, Q. Xu, S. Dalapati, M. A. Addicoat, M. A. Brady, H. Xu, T. Nakamura, T. Heine, Q. Chen and D. Jiang, Science, 2017, 357, 673–676 CrossRef CAS.
- C. K. Chiang, C. R. Fincher, Y. W. Park, A. J. Heeger, H. Shirakawa, E. J. Louis, S. C. Gau and A. G. MacDiarmid, Phys. Rev. Lett., 1977, 39, 1098–1101 CrossRef CAS.
- (a) A. Tsumura, H. Koezuka and T. Ando, Appl. Phys. Lett., 1986, 49, 1210–1212 CrossRef CAS; (b) Y. Zhi, Z. Wang, H.-L. Zhang and Q. Zhang, Small, 2020, 16, 2001070 CrossRef CAS.
- H. Yan, Z. Chen, Y. Zheng, C. Newman, J. R. Quinn, F. Dotz, M. Kastler and A. Facchetti, Nature, 2009, 457, 679–686 CrossRef CAS.
- J. W. Colson and W. R. Dichtel, Nat. Chem., 2013, 5, 453–465 CrossRef CAS.
- R.-R. Liang, S.-Y. Jiang, A. Ru-Han and X. Zhao, Chem. Soc. Rev., 2020, 49, 3920–3951 RSC.
- X. Guan, F. Chen, Q. Fang and S. Qiu, Chem. Soc. Rev., 2020, 49, 1357–1384 RSC.
- J. D. Yuen and F. Wudl, Energy Environ. Sci., 2013, 6, 392–406 RSC.
- Y. Jin, Y. Hu and W. Zhang, Nat. Rev. Chem., 2017, 1, 0056 CrossRef.
- J.-S. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS.
- D. Rodríguez-San-Miguel and F. Zamora, Chem. Soc. Rev., 2019, 48, 4375–4386 RSC.
- X.-H. Liu, C.-Z. Guan, D. Wang and L.-J. Wan, Adv. Mater., 2014, 26, 6912–6920 CrossRef CAS.
- S. Kandambeth, K. Dey and R. Banerjee, J. Am. Chem. Soc., 2019, 141, 1807–1822 CrossRef CAS.
- S. Seth and S. Jhulki, Mater. Horiz., 2021, 8, 700–727 RSC.
- X. Huang, C. Sun and X. Feng, Sci. China: Chem., 2020, 63, 1367–1390 CrossRef CAS.
- S. Z. Bisri, C. Piliego, J. Gao and M. A. Loi, Adv. Mater., 2014, 26, 1176–1199 CrossRef CAS.
- J. Yang, Z. Zhao, S. Wang, Y. Guo and Y. Liu, Chem, 2018, 4, 2748–2785 CAS.
- H. Huang, L. Yang, A. Facchetti and T. J. Marks, Chem. Rev., 2017, 117, 10291–10318 CrossRef CAS.
- A. J. Heeger, Chem. Soc. Rev., 2010, 39, 2354–2371 RSC.
- Z. B. Henson, K. Müllen and G. C. Bazan, Nat. Chem., 2012, 4, 699–704 CrossRef CAS.
- X. Guo and A. Facchetti, Nat. Mater., 2020, 19, 922–928 CrossRef CAS.
- N. Huang, P. Wang and D. Jiang, Nat. Rev. Mater., 2016, 1, 16068 CrossRef CAS.
- R. Liu, K. T. Tan, Y. Gong, Y. Chen, Z. Li, S. Xie, T. He, Z. Lu, H. Yang and D. Jiang, Chem. Soc. Rev., 2021, 50, 120–242 RSC.
- S.-Y. Ding and W. Wang, Chem. Soc. Rev., 2013, 42, 548–568 RSC.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- D. Jiang, Chem, 2020, 6, 2461–2483 CAS.
- F. Yu, W. Liu, B. Li, D. Tian, J.-L. Zuo and Q. Zhang, Angew. Chem., Int. Ed., 2019, 58, 16101–16104 CrossRef CAS.
- F. Yu, W. Liu, S.-W. Ke, M. Kurmoo, J.-L. Zuo and Q. Zhang, Nat. Commun., 2020, 11, 5534 CrossRef CAS.
- A. F. Paterson, S. Singh, K. J. Fallon, T. Hodsden, Y. Han, B. C. Schroeder, H. Bronstein, M. Heeney, I. McCulloch and T. D. Anthopoulos, Adv. Mater., 2018, 30, 1801079 CrossRef.
- H. Sun, X. Guo and A. Facchetti, Chem, 2020, 6, 1310–1326 CAS.
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS.
- Y. Zhao, Y. Guo and Y. Liu, Adv. Mater., 2013, 25, 5372–5391 CrossRef CAS.
- J. Zhou and B. Wang, Chem. Soc. Rev., 2017, 46, 6927–6945 RSC.
- X. Zhao, P. Pachfule and A. Thomas, Chem. Soc. Rev., 2021, 50, 6871–6913 RSC.
- J. Li, X. Jing, Q. Li, S. Li, X. Gao, X. Feng and B. Wang, Chem. Soc. Rev., 2020, 49, 3565–3604 RSC.
- S. Muench, A. Wild, C. Friebe, B. Häupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS.
- Y. Yusran, Q. Fang and V. Valtchev, Adv. Mater., 2020, 32, 2002038 CrossRef CAS.
- T. Sun, J. Xie, W. Guo, D.-S. Li and Q. Zhang, Adv. Energy Mater., 2020, 10, 1904199 CrossRef CAS.
- J. Wu, X. Rui, C. Wang, W.-B. Pei, R. Lau, Q. Yan and Q. Zhang, Adv. Energy Mater., 2015, 5, 1402189 CrossRef.
- J. Wu, X. Rui, G. Long, W. Chen, Q. Yan and Q. Zhang, Angew. Chem., Int. Ed., 2015, 54, 7354–7358 CrossRef CAS.
- W. Yang, X. Du, J. Zhao, Z. Chen, J. Li, J. Xie, Y. Zhang, Z. Cui, Q. Kong, Z. Zhao, C. Wang, Q. Zhang and G. Cui, Joule, 2020, 4, 1557–1574 CrossRef CAS.
- Z.-Q. Lin, J. Xie, B.-W. Zhang, J.-W. Li, J. Weng, R.-B. Song, X. Huang, H. Zhang, H. Li, Y. Liu, Z. J. Xu, W. Huang and Q. Zhang, Nano Energy, 2017, 41, 117–127 CrossRef CAS.
- J. Xie, F. Yu, J. Zhao, W. Guo, H.-L. Zhang, G. Cui and Q. Zhang, Energy Storage Mater., 2020, 33, 283–289 CrossRef.
- Y. Song, Q. Sun, B. Aguila and S. Ma, Adv. Sci., 2019, 6, 1801410 CrossRef.
- N. Keller and T. Bein, Chem. Soc. Rev., 2021, 50, 1813–1845 RSC.
- X. Liu, D. Huang, C. Lai, G. Zeng, L. Qin, H. Wang, H. Yi, B. Li, S. Liu, M. Zhang, R. Deng, Y. Fu, L. Li, W. Xue and S. Chen, Chem. Soc. Rev., 2019, 48, 5266–5302 RSC.
- H. Wang, Z. Zeng, P. Xu, L. Li, G. Zeng, R. Xiao, Z. Tang, D. Huang, L. Tang, C. Lai, D. Jiang, Y. Liu, H. Yi, L. Qin, S. Ye, X. Ren and W. Tang, Chem. Soc. Rev., 2019, 48, 488–516 RSC.
- (a) Y. H. Lee, M. Jang, M. Y. Lee, O. Y. Kweon and J. H. Oh, Chem, 2017, 3, 724–763 CrossRef CAS; (b) G. Sonmez, H. Meng, Q. Zhang and F. Wudl, Adv. Funct. Mater., 2003, 13, 726–731 CrossRef CAS.
- M. D. Allendorf, R. Dong, X. Feng, S. Kaskel, D. Matoga and V. Stavila, Chem. Rev., 2020, 120, 8581–8640 CrossRef CAS.
- S. Thomas, H. Li, R. R. Dasari, A. M. Evans, I. Castano, T. G. Allen, O. G. Reid, G. Rumbles, W. R. Dichtel, N. C. Gianneschi, S. R. Marder, V. Coropceanu and J.-L. Brédas, Mater. Horiz., 2019, 6, 1868–1876 RSC.
- A. M. Evans, A. Giri, V. K. Sangwan, S. Xun, M. Bartnof, C. G. Torres-Castanedo, H. B. Balch, M. S. Rahn, N. P. Bradshaw, E. Vitaku, D. W. Burke, H. Li, M. J. Bedzyk, F. Wang, J.-L. Brédas, J. A. Malen, A. J. H. McGaughey, M. C. Hersam, W. R. Dichtel and P. E. Hopkins, Nat. Mater., 2021, 20, 1142–1148 CrossRef CAS.
- A. C. Arias, J. D. MacKenzie, I. McCulloch, J. Rivnay and A. Salleo, Chem. Rev., 2010, 110, 3–24 CrossRef CAS.
- H. Sirringhaus, Adv. Mater., 2014, 26, 1319–1335 CrossRef CAS.
- H. Ding, J. Li, G. Xie, G. Lin, R. Chen, Z. Peng, C. Yang, B. Wang, J. Sun and C. Wang, Nat. Commun., 2018, 9, 5234 CrossRef CAS.
- W. K. Haug, E. M. Moscarello, E. R. Wolfson and P. L. McGrier, Chem. Soc. Rev., 2020, 49, 839–864 RSC.
- S. Xu and Q. Zhang, Mater. Today Energy, 2021, 20, 100635 CrossRef CAS.
- D.-W. Zhang, M. Li and C.-F. Chen, Chem. Soc. Rev., 2020, 49, 1331–1343 RSC.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K. Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3, 18003 CrossRef CAS.
- L. Lu, T. Zheng, Q. Wu, A. M. Schneider, D. Zhao and L. Yu, Chem. Rev., 2015, 115, 12666–12731 CrossRef CAS.
- T. Ma, E. A. Kapustin, S. X. Yin, L. Liang, Z. Zhou, J. Niu, L.-H. Li, Y. Wang, J. Su, J. Li, X. Wang, W. D. Wang, W. Wang, J. Sun and O. M. Yaghi, Science, 2018, 361, 48–52 CrossRef CAS.
- H. A. Um, D. H. Lee, D. U. Heo, D. S. Yang, J. Shin, H. Baik, M. J. Cho and D. H. Choi, ACS Nano, 2015, 9, 5264–5274 CrossRef CAS.
- Z.-F. Yao, Y.-Q. Zheng, J.-H. Dou, Y. Lu, Y.-F. Ding, L. Ding, J.-Y. Wang and J. Pei, Adv. Mater., 2021, 33, 2006794 CrossRef CAS.
- R. Freund, S. Canossa, S. M. Cohen, W. Yan, H. Deng, V. Guillerm, M. Eddaoudi, D. G. Madden, D. Fairen-Jimenez, H. Lyu, L. K. Macreadie, Z. Ji, Y. Zhang, B. Wang, F. Haase, C. Wöll, O. Zaremba, J. Andreo, S. Wuttke and C. S. Diercks, Angew. Chem., Int. Ed., 2021, 60, 2–31 CrossRef.
- I. Hisaki, C. Xin, K. Takahashi and T. Nakamura, Angew. Chem., Int. Ed., 2019, 58, 11160–11170 CrossRef CAS.
- Q. Huang, W. Li, Z. Mao, H. Zhang, Y. Li, D. Ma, H. Wu, J. Zhao, Z. Yang, Y. Zhang, L. Gong, M. P. Aldred and Z. Chi, Chem, 2021, 7, 1321–1332 CAS.
- G. Chakraborty, I.-H. Park, R. Medishetty and J. J. Vittal, Chem. Rev., 2021, 121, 3751–3891 CrossRef CAS.
- L. Feng, K.-Y. Wang, G. S. Day, M. R. Ryder and H.-C. Zhou, Chem. Rev., 2020, 120, 13087–13133 CrossRef CAS.
- Y. Liu, Y. Wei, M. Liu, Y. Bai, X. Wang, S. Shang, J. Chen and Y. Liu, Angew. Chem., Int. Ed., 2021, 60, 2887–2891 CrossRef CAS.
- B. Gole, V. Stepanenko, S. Rager, M. Grüne, D. D. Medina, T. Bein, F. Würthner and F. Beuerle, Angew. Chem., Int. Ed., 2018, 57, 846–850 CrossRef CAS.
- C. B. Nielsen, M. Turbiez and I. McCulloch, Adv. Mater., 2013, 25, 1859–1880 CrossRef CAS.
- Z. Yi, S. Wang and Y. Liu, Adv. Mater., 2015, 27, 3589–3606 CrossRef CAS.
- J. Yang, H. Wang, J. Chen, J. Huang, Y. Jiang, J. Zhang, L. Shi, Y. Sun, Z. Wei, G. Yu, Y. Guo, S. Wang and Y. Liu, Adv. Mater., 2017, 29, 1606162 CrossRef.
- M. Matsumoto, R. R. Dasari, W. Ji, C. H. Feriante, T. C. Parker, S. R. Marder and W. R. Dichtel, J. Am. Chem. Soc., 2017, 139, 4999–5002 CrossRef CAS.
- D. Bessinger, L. Ascherl, F. Auras and T. Bein, J. Am. Chem. Soc., 2017, 139, 12035–12042 CrossRef CAS.
- T. Lei, J.-Y. Wang and J. Pei, Acc. Chem. Res., 2014, 47, 1117–1126 CrossRef CAS.
- E. Wang, W. Mammo and M. R. Andersson, Adv. Mater., 2014, 26, 1801–1826 CrossRef CAS.
- L. Jiang, Y. Tian, T. Sun, Y. Zhu, H. Ren, X. Zou, Y. Ma, K. R. Meihaus, J. R. Long and G. Zhu, J. Am. Chem. Soc., 2018, 140, 15724–15730 CrossRef CAS.
- N. Sakai, J. Mareda, E. Vauthey and S. Matile, Chem. Commun., 2010, 46, 4225–4237 RSC.
- S.-L. Suraru and F. Würthner, Angew. Chem., Int. Ed., 2014, 53, 7428–7448 CrossRef CAS.
- I. Osaka and K. Takimiya, Adv. Mater., 2017, 29, 1605218 CrossRef.
- J. Yang, Y. Jiang, Z. Tu, Z. Zhao, J. Chen, Z. Yi, Y. Li, S. Wang, Y. Yi, Y. Guo and Y. Liu, Adv. Funct. Mater., 2019, 29, 1804839 CrossRef.
- K.-J. Baeg, M. Caironi and Y.-Y. Noh, Adv. Mater., 2013, 25, 4210–4244 CrossRef CAS.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS.
- J. L. Segura, M. J. Mancheño and F. Zamora, Chem. Soc. Rev., 2016, 45, 5635–5671 RSC.
- Y. Li, W. Chen, G. Xing, D. Jiang and L. Chen, Chem. Soc. Rev., 2020, 49, 2852–2868 RSC.
- J. L. Segura, S. Royuela and M. Mar Ramos, Chem. Soc. Rev., 2019, 48, 3903–3945 RSC.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- R. K. Sharma, P. Yadav, M. Yadav, R. Gupta, P. Rana, A. Srivastava, R. Zbořil, R. S. Varma, M. Antonietti and M. B. Gawande, Mater. Horiz., 2020, 7, 411–454 RSC.
- B. Carsten, F. He, H. J. Son, T. Xu and L. Yu, Chem. Rev., 2011, 111, 1493–1528 CrossRef CAS.
- J.-R. Pouliot, F. Grenier, J. T. Blaskovits, S. Beaupré and M. Leclerc, Chem. Rev., 2016, 116, 14225–14274 CrossRef CAS.
- Z. Ni, H. Wang, H. Dong, Y. Dang, Q. Zhao, X. Zhang and W. Hu, Nat. Chem., 2019, 11, 271–277 CrossRef CAS.
- N. S. Gobalasingham and B. C. Thompson, Prog. Polym. Sci., 2018, 83, 135–201 CrossRef CAS.
- Y. Ran, Y. Guo and Y. Liu, Mater. Horiz., 2020, 7, 1955–1970 RSC.
- G. Zhang, Y. Dai, Y. Liu, J. Liu, H. Lu, L. Qiu and K. Cho, Polym. Chem., 2017, 8, 3448–3456 RSC.
- A. Onwubiko, W. Yue, C. Jellett, M. Xiao, H.-Y. Chen, M. K. Ravva, D. A. Hanifi, A.-C. Knall, B. Purushothaman, M. Nikolka, J.-C. Flores, A. Salleo, J.-L. Bredas, H. Sirringhaus, P. Hayoz and I. McCulloch, Nat. Commun., 2018, 9, 416 CrossRef.
- A. M. Evans, L. R. Parent, N. C. Flanders, R. P. Bisbey, E. Vitaku, M. S. Kirschner, R. D. Schaller, L. X. Chen, N. C. Gianneschi and W. R. Dichtel, Science, 2018, 361, 52–57 CrossRef CAS.
- M. Liu, K. Jiang, X. Ding, S. Wang, C. Zhang, J. Liu, Z. Zhan, G. Cheng, B. Li, H. Chen, S. Jin and B. Tan, Adv. Mater., 2019, 31, 1807865 CrossRef.
- S. Karak, S. Kumar, P. Pachfule and R. Banerjee, J. Am. Chem. Soc., 2018, 140, 5138–5145 CrossRef CAS.
- S. Zhang, G. Cheng, L. Guo, N. Wang, B. Tan and S. Jin, Angew. Chem., Int. Ed., 2020, 59, 6007–6014 CrossRef CAS.
- M. Calik, T. Sick, M. Dogru, M. Döblinger, S. Datz, H. Budde, A. Hartschuh, F. Auras and T. Bein, J. Am. Chem. Soc., 2016, 138, 1234–1239 CrossRef CAS.
- D. Zhu, L. B. Alemany, W. Guo and R. Verduzco, Polym. Chem., 2020, 11, 4464–4468 RSC.
- F. J. Uribe-Romo, J. R. Hunt, H. Furukawa, C. Klöck, M. O’Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 4570–4571 CrossRef CAS.
- T. Ma, J. Li, J. Niu, L. Zhang, A. S. Etman, C. Lin, D. Shi, P. Chen, L.-H. Li, X. Du, J. Sun and W. Wang, J. Am. Chem. Soc., 2018, 140, 6763–6766 CrossRef CAS.
- M. Liu, Q. Huang, S. Wang, Z. Li, B. Li, S. Jin and B. Tan, Angew. Chem., Int. Ed., 2018, 57, 11968–11972 CrossRef CAS.
- S. Wang, Z. Zhang, H. Zhang, A. G. Rajan, N. Xu, Y. Yang, Y. Zeng, P. Liu, X. Zhang, Q. Mao, Y. He, J. Zhao, B.-G. Li, M. S. Strano and W.-J. Wang, Matter, 2019, 1, 1592–1605 CrossRef.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef CAS.
- C. M. Thompson, G. Occhialini, G. T. McCandless, S. B. Alahakoon, V. Cameron, S. O. Nielsen and R. A. Smaldone, J. Am. Chem. Soc., 2017, 139, 10506–10513 CrossRef CAS.
- Z. Xie, B. Wang, Z. Yang, X. Yang, X. Yu, G. Xing, Y. Zhang and L. Chen, Angew. Chem., Int. Ed., 2019, 58, 15742–15746 CrossRef CAS.
- Z.-F. Pang, S.-Q. Xu, T.-Y. Zhou, R.-R. Liang, T.-G. Zhan and X. Zhao, J. Am. Chem. Soc., 2016, 138, 4710–4713 CrossRef CAS.
- S. B. Alahakoon, S. D. Diwakara, C. M. Thompson and R. A. Smaldone, Chem. Soc. Rev., 2020, 49, 1344–1356 RSC.
- X. Chen, M. Addicoat, S. Irle, A. Nagai and D. Jiang, J. Am. Chem. Soc., 2013, 135, 546–549 CrossRef CAS.
- S. Kandambeth, D. B. Shinde, M. K. Panda, B. Lukose, T. Heine and R. Banerjee, Angew. Chem., Int. Ed., 2013, 52, 13052–13056 CrossRef CAS.
- X. Chen, M. Addicoat, E. Jin, L. Zhai, H. Xu, N. Huang, Z. Guo, L. Liu, S. Irle and D. Jiang, J. Am. Chem. Soc., 2015, 137, 3241–3247 CrossRef CAS.
- X. Guo, Y. Tian, M. Zhang, Y. Li, R. Wen, X. Li, X. Li, Y. Xue, L. Ma, C. Xia and S. Li, Chem. Mater., 2018, 30, 2299–2308 CrossRef CAS.
- A. Halder, S. Karak, M. Addicoat, S. Bera, A. Chakraborty, S. H. Kunjattu, P. Pachfule, T. Heine and R. Banerjee, Angew. Chem., Int. Ed., 2018, 57, 5797–5802 CrossRef CAS.
- S. B. Alahakoon, G. T. McCandless, A. A. K. Karunathilake, C. M. Thompson and R. A. Smaldone, Chem. – Eur. J., 2017, 23, 4255–4259 CrossRef CAS.
- Y. Li, Q. Chen, T. Xu, Z. Xie, J. Liu, X. Yu, S. Ma, T. Qin and L. Chen, J. Am. Chem. Soc., 2019, 141, 13822–13828 CrossRef CAS.
- E. Vitaku and W. R. Dichtel, J. Am. Chem. Soc., 2017, 139, 12911–12914 CrossRef CAS.
- N. Keller, D. Bessinger, S. Reuter, M. Calik, L. Ascherl, F. C. Hanusch, F. Auras and T. Bein, J. Am. Chem. Soc., 2017, 139, 8194–8199 CrossRef CAS.
- X. Wu, Y.-l. Hong, B. Xu, Y. Nishiyama, W. Jiang, J. Zhu, G. Zhang, S. Kitagawa and S. Horike, J. Am. Chem. Soc., 2020, 142, 14357–14364 CrossRef CAS.
- J.-K. Sun, Y. I. Sobolev, W. Zhang, Q. Zhuang and B. A. Grzybowski, Nature, 2020, 579, 73–79 CrossRef CAS.
- L. Ascherl, E. W. Evans, M. Hennemann, D. Di Nuzzo, A. G. Hufnagel, M. Beetz, R. H. Friend, T. Clark, T. Bein and F. Auras, Nat. Commun., 2018, 9, 3802 CrossRef.
- Y. Lv, Y. Li, G. Zhang, Z. Peng, L. Ye, Y. Chen, T. Zhang, G. Xing and L. Chen, CCS Chem., 2021, 3, 1773–1779 CrossRef.
- D. Chen, W. Chen, G. Xing, T. Zhang and L. Chen, Chem. – Eur. J., 2020, 26, 8377–8381 CrossRef CAS.
- L. Bai, S. Z. F. Phua, W. Q. Lim, A. Jana, Z. Luo, H. P. Tham, L. Zhao, Q. Gao and Y. Zhao, Chem. Commun., 2016, 52, 4128–4131 RSC.
- H. Li, P. Shao, S. Chen, G. Li, X. Feng, X. Chen, H.-J. Zhang, J. Lin and Y.-B. Jiang, J. Am. Chem. Soc., 2020, 142, 3712–3717 CrossRef CAS.
- D. Beaudoin, T. Maris and J. D. Wuest, Nat. Chem., 2013, 5, 830–834 CrossRef CAS.
- T. Ma, L. Wei, L. Liang, S. Yin, L. Xu, J. Niu, H. Xue, X. Wang, J. Sun, Y.-B. Zhang and W. Wang, Nat. Commun., 2020, 11, 6128 CrossRef CAS.
- Y.-B. Zhang, J. Su, H. Furukawa, Y. Yun, F. Gándara, A. Duong, X. Zou and O. M. Yaghi, J. Am. Chem. Soc., 2013, 135, 16336–16339 CrossRef CAS.
- C. Gao, J. Li, S. Yin, G. Lin, T. Ma, Y. Meng, J. Sun and C. Wang, Angew. Chem., Int. Ed., 2019, 58, 9770–9775 CrossRef CAS.
- H.-S. Xu, Y. Luo, X. Li, P. Z. See, Z. Chen, T. Ma, L. Liang, K. Leng, I. Abdelwahab, L. Wang, R. Li, X. Shi, Y. Zhou, X. F. Lu, X. Zhao, C. Liu, J. Sun and K. P. Loh, Nat. Commun., 2020, 11, 1434 CrossRef CAS.
- B. J. Smith, L. R. Parent, A. C. Overholts, P. A. Beaucage, R. P. Bisbey, A. D. Chavez, N. Hwang, C. Park, A. M. Evans, N. C. Gianneschi and W. R. Dichtel, ACS Cent. Sci., 2017, 3, 58–65 CrossRef CAS.
- A. M. Evans, I. Castano, A. Brumberg, L. R. Parent, A. R. Corcos, R. L. Li, N. C. Flanders, D. J. Gosztola, N. C. Gianneschi, R. D. Schaller and W. R. Dichtel, J. Am. Chem. Soc., 2019, 141, 19728–19735 CrossRef CAS.
- L. Liang, Y. Qiu, W. D. Wang, J. Han, Y. Luo, W. Yu, G.-L. Yin, Z.-P. Wang, L. Zhang, J. Ni, J. Niu, J. Sun, T. Ma and W. Wang, Angew. Chem., Int. Ed., 2020, 59, 17991–17995 CrossRef CAS.
- P. Sonar, S. P. Singh, Y. Li, M. S. Soh and A. Dodabalapur, Adv. Mater., 2010, 22, 5409–5413 CrossRef CAS.
- J. S. Ha, K. H. Kim and D. H. Choi, J. Am. Chem. Soc., 2011, 133, 10364–10367 CrossRef CAS.
- J. Lee, A. R. Han, J. Kim, Y. Kim, J. H. Oh and C. Yang, J. Am. Chem. Soc., 2012, 134, 20713–20721 CrossRef CAS.
- B. He, A. B. Pun, D. Zherebetskyy, Y. Liu, F. Liu, L. M. Klivansky, A. M. McGough, B. A. Zhang, K. Lo, T. P. Russell, L. Wang and Y. Liu, J. Am. Chem. Soc., 2014, 136, 15093–15101 CrossRef CAS.
- H. N. Tsao, D. M. Cho, I. Park, M. R. Hansen, A. Mavrinskiy, D. Y. Yoon, R. Graf, W. Pisula, H. W. Spiess and K. Müllen, J. Am. Chem. Soc., 2011, 133, 2605–2612 CrossRef CAS.
- M. Zhang, H. N. Tsao, W. Pisula, C. Yang, A. K. Mishra and K. Müllen, J. Am. Chem. Soc., 2007, 129, 3472–3473 CrossRef CAS.
- H. N. Tsao, D. Cho, J. W. Andreasen, A. Rouhanipour, D. W. Breiby, W. Pisula and K. Müllen, Adv. Mater., 2009, 21, 209–212 CrossRef CAS.
- J. Li, Y. Zhao, H. S. Tan, Y. Guo, C.-A. Di, G. Yu, Y. Liu, M. Lin, S. H. Lim, Y. Zhou, H. Su and B. S. Ong, Sci. Rep., 2012, 2, 754 CrossRef.
- Y.-Z. Dai, N. Ai, Y. Lu, Y.-Q. Zheng, J.-H. Dou, K. Shi, T. Lei, J.-Y. Wang and J. Pei, Chem. Sci., 2016, 7, 5753–5757 RSC.
- Y. Wang, T. Hasegawa, H. Matsumoto and T. Michinobu, J. Am. Chem. Soc., 2019, 141, 3566–3575 CrossRef CAS.
- X. Guo, J. Quinn, Z. Chen, H. Usta, Y. Zheng, Y. Xia, J. W. Hennek, R. P. Ortiz, T. J. Marks and A. Facchetti, J. Am. Chem. Soc., 2013, 135, 1986–1996 CrossRef CAS.
- T. Lei, J.-H. Dou, X.-Y. Cao, J.-Y. Wang and J. Pei, Adv. Mater., 2013, 25, 6589–6593 CrossRef CAS.
- Z. Fei, P. Pattanasattayavong, Y. Han, B. C. Schroeder, F. Yan, R. J. Kline, T. D. Anthopoulos and M. Heeney, J. Am. Chem. Soc., 2014, 136, 15154–15157 CrossRef CAS.
- T. Lei, Y. Cao, X. Zhou, Y. Peng, J. Bian and J. Pei, Chem. Mater., 2012, 24, 1762–1770 CrossRef CAS.
- L. Ying, B. B. Y. Hsu, H. Zhan, G. C. Welch, P. Zalar, L. A. Perez, E. J. Kramer, T.-Q. Nguyen, A. J. Heeger, W.-Y. Wong and G. C. Bazan, J. Am. Chem. Soc., 2011, 133, 18538–18541 CrossRef CAS.
- L. Ying, F. Huang and G. C. Bazan, Nat. Commun., 2017, 8, 14047 CrossRef CAS.
- R. Rieger, D. Beckmann, A. Mavrinskiy, M. Kastler and K. Müllen, Chem. Mater., 2010, 22, 5314–5318 CrossRef CAS.
- T. Lei, Y. Cao, Y. Fan, C.-J. Liu, S.-C. Yuan and J. Pei, J. Am. Chem. Soc., 2011, 133, 6099–6101 CrossRef CAS.
- M. Wang, M. Ford, H. Phan, J. Coughlin, T.-Q. Nguyen and G. C. Bazan, Chem. Commun., 2016, 52, 3207–3210 RSC.
- Y. Wang, Z. Yan, H. Guo, M. A. Uddin, S. Ling, X. Zhou, H. Su, J. Dai, H. Y. Woo and X. Guo, Angew. Chem., Int. Ed., 2017, 56, 15304–15308 CrossRef CAS.
- W. Lee, G.-H. Kim, S.-J. Ko, S. Yum, S. Hwang, S. Cho, Y.-H. Shin, J. Y. Kim and H. Y. Woo, Macromolecules, 2014, 47, 1604–1612 CrossRef CAS.
- T. Lei, J.-H. Dou and J. Pei, Adv. Mater., 2012, 24, 6457–6461 CrossRef CAS.
- J. Mei and Z. Bao, Chem. Mater., 2014, 26, 604–615 CrossRef CAS.
- J. Lee, A. R. Han, H. Yu, T. J. Shin, C. Yang and J. H. Oh, J. Am. Chem. Soc., 2013, 135, 9540–9547 CrossRef CAS.
- B. Kang, R. Kim, S. B. Lee, S.-K. Kwon, Y.-H. Kim and K. Cho, J. Am. Chem. Soc., 2016, 138, 3679–3686 CrossRef CAS.
- J. Yao, C. Yu, Z. Liu, H. Luo, Y. Yang, G. Zhang and D. Zhang, J. Am. Chem. Soc., 2016, 138, 173–185 CrossRef CAS.
- Y. Yang, Z. Liu, G. Zhang, X. Zhang and D. Zhang, Adv. Mater., 2019, 31, 1903104 CrossRef CAS.
- I. Kang, H.-J. Yun, D. S. Chung, S.-K. Kwon and Y.-H. Kim, J. Am. Chem. Soc., 2013, 135, 14896–14899 CrossRef CAS.
- M. S. Chen, O. P. Lee, J. R. Niskala, A. T. Yiu, C. J. Tassone, K. Schmidt, P. M. Beaujuge, S. S. Onishi, M. F. Toney, A. Zettl and J. M. J. Fréchet, J. Am. Chem. Soc., 2013, 135, 19229–19236 CrossRef CAS.
- Z. Wang, Z. Liu, L. Ning, M. Xiao, Y. Yi, Z. Cai, A. Sadhanala, G. Zhang, W. Chen, H. Sirringhaus and D. Zhang, Chem. Mater., 2018, 30, 3090–3100 CrossRef CAS.
- Y.-J. Heo, H.-G. Jeong, J. Kim, B. Lim, J. Kim, Y. Kim, B. Kang, J.-M. Yun, K. Cho and D.-Y. Kim, ACS Appl. Mater. Interfaces, 2020, 12, 49886–49894 CrossRef CAS.
- T. Lei, X. Xia, J.-Y. Wang, C.-J. Liu and J. Pei, J. Am. Chem. Soc., 2014, 136, 2135–2141 CrossRef CAS.
- S. Yum, T. K. An, X. Wang, W. Lee, M. A. Uddin, Y. J. Kim, T. L. Nguyen, S. Xu, S. Hwang, C. E. Park and H. Y. Woo, Chem. Mater., 2014, 26, 2147–2154 CrossRef CAS.
- Y. Gao, X. Zhang, H. Tian, J. Zhang, D. Yan, Y. Geng and F. Wang, Adv. Mater., 2015, 27, 6753–6759 CrossRef CAS.
- M. Wang, M. J. Ford, A. T. Lill, H. Phan, T.-Q. Nguyen and G. C. Bazan, Adv. Mater., 2017, 29, 1603830 CrossRef.
- Y. Sun, Y. Zhang, Y. Ran, L. Shi, Q. Zhang, J. Chen, Q. Li, Y. Guo and Y. Liu, J. Mater. Chem. C, 2020, 8, 15168–15174 RSC.
- Y. Gao, Y. Deng, H. Tian, J. Zhang, D. Yan, Y. Geng and F. Wang, Adv. Mater., 2017, 29, 1606217 CrossRef.
- K. Rahimi, I. Botiz, N. Stingelin, N. Kayunkid, M. Sommer, F. P. V. Koch, H. Nguyen, O. Coulembier, P. Dubois, M. Brinkmann and G. Reiter, Angew. Chem., Int. Ed., 2012, 51, 11131–11135 CrossRef CAS.
- J. H. Kim, D. H. Lee, D. S. Yang, D. U. Heo, K. H. Kim, J. Shin, H.-J. Kim, K.-Y. Baek, K. Lee, H. Baik, M. J. Cho and D. H. Choi, Adv. Mater., 2013, 25, 4102–4106 CrossRef CAS.
- Y. Yao, H. Dong, F. Liu, T. P. Russell and W. Hu, Adv. Mater., 2017, 29, 1701251 CrossRef.
- Z. Ni, H. Dong, H. Wang, S. Ding, Y. Zou, Q. Zhao, Y. Zhen, F. Liu, L. Jiang and W. Hu, Adv. Mater., 2018, 30, 1704843 CrossRef.
Footnote |
| † These authors contribute equally. |
| This journal is © The Royal Society of Chemistry 2022 |