
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Overcoming mass transfer limitations in cross-linked polyethyleneimine-based adsorbents to enable selective CO2 capture at ambient temperature†
Louise B.
Hamdy‡
 *a,
Abel
Gougsa
a,
Wing Ying
Chow§
g,
James E.
Russell
b,
Enrique
García-Díez
c,
Viktoriia
Kulakova
c,
Susana
Garcia
c,
Andrew R.
Barron
adef,
Marco
Taddei¶
a and
Enrico
Andreoli
*a
*a,
Abel
Gougsa
a,
Wing Ying
Chow§
g,
James E.
Russell
b,
Enrique
García-Díez
c,
Viktoriia
Kulakova
c,
Susana
Garcia
c,
Andrew R.
Barron
adef,
Marco
Taddei¶
a and
Enrico
Andreoli
*a
aEnergy Safety Research Institute, Swansea University, Bay Campus, Swansea, SA1 8EN, UK. E-mail: e.andreoli@swansea.ac.uk
bAdvanced Imaging of Materials (AIM) Facility, College of Engineering, Swansea University, Bay Campus, Swansea, UK
cResearch Centre for Carbon Solutions (RCCS), School of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh EH14 4AS, UK
dArizona Institutes for Resilience (AIR), University of Arizona, Tucson, Arizona 85721, USA
eDepartment of Chemistry and Department of Materials Science and Nanoengineering, Rice University, Houston, Texas 77005, USA
fFaculty of Engineering, Universiti Teknologi Brunei, Brunei Darussalam
gLeibniz-Forschungsinstitut für Molekulare Pharmakologie im Forschungsverbund Berlin e.V. (FMP), Campus Berlin-Buch, Robert-Roessle-Str. 10, 13125 Berlin, Germany
First published on 16th February 2022
Abstract
New self-supported polyamine CO2 adsorbents are prepared by cross-linking branched polyethyleneimine (PEI) with 2,4,6-tris-(4-bromomethyl-3-fluoro-phenyl)-1,3,5-triazine (4BMFPT). Controlling the degree of cross-linking to ensure abundant free amine functionalities while maintaining a structure conducive to efficient mass transfer is key to accessing high CO2 adsorption and fast kinetics at ambient temperature. The polyamine-based adsorbent, PEI-4BMFPT, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (R), is composed of spherical particles up to 3 μm in diameter and demonstrates fast CO2 uptake of 2.31 mmol g−1 under 1 atm, 90% CO2/Ar at 30 °C. Its CO2/N2 selectivity, predicted by the ideal adsorbed solution theory is 575, equalling that of highly selective metal–organic frameworks. Based on humidified thermogravimetric analysis, it was observed that the presence of water promotes CO2 uptake capacity of 10:1 (R) to 3.27 mmol g−1 and results in strong chemisorption; likely by formation of ammonium carbonate and bicarbonate species. It is observed that CO2 uptake enhancement is highly subject to relative humidity and CO2 partial pressure conditions. When adsorption conditions combined low temperatures with low partial pressure CO2, 10:1 (R) showed reduced uptake. Tested under breakthrough conditions representative of post-combustion conditions, at 75% RH and 40 °C, CO2 uptake was reduced by 83% of the dry adsorption capacity. This body of work further advances the development of support-free CO2 adsorbents for ambient temperature applications and highlights the drastic effect that relative humidity and CO2 partial pressure have on uptake behaviour.
:1 (R), is composed of spherical particles up to 3 μm in diameter and demonstrates fast CO2 uptake of 2.31 mmol g−1 under 1 atm, 90% CO2/Ar at 30 °C. Its CO2/N2 selectivity, predicted by the ideal adsorbed solution theory is 575, equalling that of highly selective metal–organic frameworks. Based on humidified thermogravimetric analysis, it was observed that the presence of water promotes CO2 uptake capacity of 10:1 (R) to 3.27 mmol g−1 and results in strong chemisorption; likely by formation of ammonium carbonate and bicarbonate species. It is observed that CO2 uptake enhancement is highly subject to relative humidity and CO2 partial pressure conditions. When adsorption conditions combined low temperatures with low partial pressure CO2, 10:1 (R) showed reduced uptake. Tested under breakthrough conditions representative of post-combustion conditions, at 75% RH and 40 °C, CO2 uptake was reduced by 83% of the dry adsorption capacity. This body of work further advances the development of support-free CO2 adsorbents for ambient temperature applications and highlights the drastic effect that relative humidity and CO2 partial pressure have on uptake behaviour.
Introduction
It is scientifically acknowledged that atmospheric carbon dioxide (CO2) levels must be reduced in order to lessen the worst effects of climate change.1 Such is the quantity of this excess CO2, that large-scale industrial carbon capture technologies are an essential part of our toolkit;2 however, to meet this challenge, our current CO2 capture materials need to be significantly advanced beyond the liquid amine solutions that are currently used in the purification of natural gas.3,4 There is a wide consensus that CO2 capture technologies could be used to adsorb CO2 from flue gas, either from power stations or factories,5–8 but to reduce the absolute concentration of CO2 in our atmosphere, negative emission technologies (NETs) which result in an overall net reduction of CO2 emissions, will also likely become a necessity.9 NETs include changing land management to increase the CO2 taken up by soils, afforestation, reforestation, enhanced weathering, bioenergy with carbon capture and storage (BECCS) and direct air capture (DAC) of CO2.9,10 Of these, DAC has certain advantages in terms of its potential for developing to the gigatonne scale: it does not require arable land which could put pressure on food production and natural habitats, it has the smallest physical footprint and it is highly flexible in where it can be deployed.11 Therefore, there is much interest in DAC technologies and it is the focus of much research carried out today.12,13The separation of CO2 from the air is not facile, however, since the concentration of CO2 is around 400 ppm, compared with a concentration of up to 14 vol% for flue gas from a coal-fired power station.14 Therefore, a strong enthalpic drive and high CO2 selectivity over other gases (N2, O2, H2O) are fundamental for an effective adsorbent for DAC. Although aqueous amine absorbents have many drawbacks such as their regeneration costs15 and corrosive nature,16 one advantage is that due to the relatively strong and selective chemical bond formed on reaction with CO2, they can achieve their maximum uptake capacities at low CO2 partial pressures.17 Amine-based solid chemisorbents, which combine the advantages of amine chemistry with the easier regenerability of solid sorbents, have emerged among the most promising routes to designing an effective adsorbent for DAC. To this end, they are being employed commercially by several companies including Global Thermostat based in the US,18 Skytree based in the Netherlands,19 and Swiss-based Climeworks, which uses aminosilane-grafted nanofibrillated cellulose.20
Solid amine adsorbents are very often composed of long-chain amines or polyamines deposited onto high surface area materials such as silica or alumina.21,22 Both migration of the amine within the structure of the support and high loadings often lead to slower or reduced uptake,23,24 and reduced amine efficiency,25 (where amine efficiency is the number of moles of CO2 adsorbed per mole of amines). This is due to the saturation of pores, creating mass transfer limitations and reducing contact between CO2 and the adsorbent.25 Such diffusion resistance can often be overcome and higher adsorption may be achieved either under humid conditions,26,27 or at elevated temperatures above 50 °C,23,24 under which, the intermolecular interactions between polyamine chains may become weakened, chain mobility increases and CO2 diffusion is more efficient. Although viable under post-combustion adsorption systems, higher temperatures are not applicable under DAC conditions and high water adsorption cannot necessarily be relied upon due to its contribution to increased sensible heat required for desorption.28 Rather, features intrinsic to the adsorbent such as porosity, surface area and the nature of the interaction with the adsorbate must enable effective CO2 uptake at low temperatures.
In terms of maximising low temperature adsorption, porous solid sorbents, such as zeolites, activated carbons, and metal–organic frameworks (MOFs), have certain advantages.29 Unlike chemisorbents, they form weaker non-covalent interactions with the adsorbate, and have lower heats of adsorption (Qst) from 25 to around 50 kJ mol−1, while the Qst for chemisorption can be over 90 kJ mol−1.2,30 Adsorption is exothermic, therefore inversely proportional to temperature, however, where low adsorption temperatures may hinder CO2 uptake for amine-based chemisorbents due to poor diffusion, high surface area physisorbents may excel.31 The different environmental conditions under which chemisorption and physisorption may each be more efficiently deployed is depicted in Fig. 1. An advantage of a physisorbent with weakly adsorbed CO2 may be that it can be more easily regenerated than a chemisorbent.29 Accordingly, an ideal DAC CO2 adsorbent could combine the high selectivity of a chemisorbent, with the lower Qst of a physisorbent to enable efficient low temperature uptake of pure CO2.
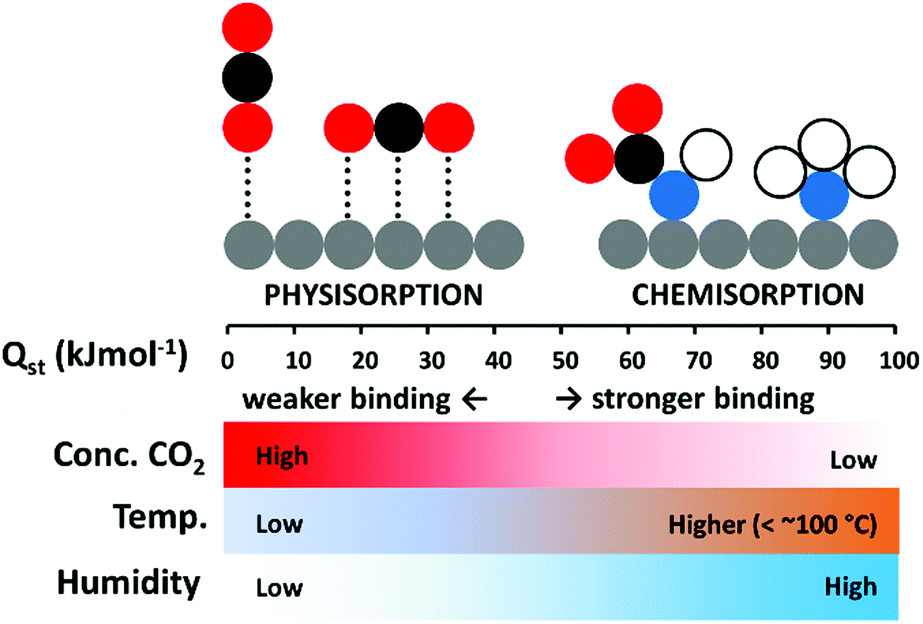 | ||
| Fig. 1 Depiction of physisorption and chemisorption with likely Qst values and physical conditions under which each may be preferred for CO2 capture. (Spheres represent atoms: grey, generic adsorbent; black, carbon; red, oxygen; blue, nitrogen; white, hydrogen.) | ||
Promising candidates to simultaneously access these adsorption phenomena are solid cross-linked polyamine-based materials. Supported cross-linked polyamine CO2 adsorbents have been reported. Polyethyleneimine (PEI) has been cross-linked with an epoxy resin and coated on a glass fibre matrix;32 or cross-linked with glyoxal, oxalic acid, 1,3-butadiene diepoxide and epichlorohydrin, and supported on a mesostructured cellular silica foam.33,34 A recent study demonstrated the formation of a new polyamine via the cross-linking of 1,3,5-tris(bromomethyl)benzene with ethylene diamine, which was then impregnated onto mesoporous silica SBA-15. Under air capture conditions, the material of 60 wt% organic loading adsorbed up to 1.9 mmol CO2 g−1 SiO2, equating to 0.75 mmol g−1 adsorbent.35
In their unsupported form, cross-linked polyamine adsorbents constitute a relatively new family of solid amine adsorbents, which has been significantly advanced in recent years by several groups, including ours.36,37 One particular advantage of these versatile materials over their supported polyamine counterparts is in their economy of mass: they are composed purely of amine-containing polymers that are cross-linked into solid structures. This negates the need for a support, enabling them to be lighter and potentially more easily regenerated and processed.37
Such adsorbents have been synthesised by Andreoli et al. using C60,38–41 and carbon nanotubes,42 to cross-link PEI or polypropylenimine. PEI (Mw 25000 Da) cross-linked with C60 showed excellent CO2 adsorption of 0.140 g g−1 (3.18 mmol) at 90 °C under 0.1 bar CO2, with high selectivity for CO2 over CH4 and N2.43 We recently swapped C60 for the epoxy resin bisphenol A diglycidyl ether as a more economical cross-linker to give an adsorbent with a CO2 uptake capacity of 0.101 g g−1 (2.30 mmol g−1) at 90 °C, 0.1 bar CO2.36 Hwang et al. have reacted PEI (Mw 25000 Da) with glutaraldehyde via an inverse emulsion technique.44 The adsorbent cPEI-GA36 had a surface area of 10 m2 g−1 and displayed the fastest adsorption and highest CO2 capacity of 2.18 mmol g−1 at 75 °C (1 atm CO2). Thompson et al. used polyaldehyde phosphorus dendrimers to cross-link PEI (Mw 600).45 At 65 °C, under 1 atm 30 mL min−1 CO2/60 mL min−1 He, PEI cross-linked with hexakis(4-formylphenoxy)cyclo(triphosphazene) (1-G0/600PEI) adsorbed 13.6 wt% CO2, (3.32 mmol g−1), reducing to 4.1 wt% (0.93 mmol g−1) at 25 °C. Hydrogel beads have been synthesised by Xu et al. by cross-linking PEI (Mw 25000 Da) with epichlorohydrin.46 Although CO2 uptake of the dry adsorbent was minimal, adsorption by hydrated PEI HB-4.0% EPC (under 15% CO2/N2 at ambient temperature) was 0.0602 g g−1 (1.37 mmol g−1).
As indicated in Table 1, these adsorbents are mostly prepared with a higher molar ratio of amine to cross-linking reactive site, therefore having plentiful free primary and secondary amines remaining within PEI for the chemisorption of CO2. They are most effective at elevated temperatures, or under hydrated conditions, due to the high flexibility and self-affinity of the cross-linked polymeric networks, resulting in insufficient gas diffusion at lower adsorption temperatures. This effect has been demonstrated by Yoo et al. who synthesised macroporous adsorbents from PEI (Mw 750000 Da) cross-linked with poly(ethylene glycol) diglycidyl ether via an ice-templating method.37 The optimum temperature of adsorption was dependent on the ratio of cross-linker used during preparation. Under 10% CO2/He, 1 atm, the adsorbent with the lowest cross-linker volume (E50-FZ) adsorbed the least CO2 at 25 °C, but the highest at 75 °C, at 3.00 mmol CO2 g−1. Meanwhile, the adsorbent with the highest cross-linker volume (E200-FZ) adsorbed 2.01 mmol CO2 g−1 at 25 °C, but less than 0.6 mmol g−1 at 75 °C. The latter adsorbent had a smaller average pore size of 13 μm and a larger surface area. In this case, more of the primary and secondary amines are involved in cross-linking, resulting in reduced basicity such that chemisorption is impaired, however this is over-compensated by the increased surface area and better diffusion enabling improved uptake at 25 °C.
| Adsorbent | (Poly)amine | CLRS | No. amines per CLRS | Ref. |
|---|---|---|---|---|
| PEI-C60 | PEI | C60 | 58 | 43 |
| BC-40 | PEI | Epoxy | 40 | 36 |
| cPEI-GA36 | PEI | Aldehyde | 7.6 | 44 |
| 1-G0/600PEI | PEI | Aldehyde | 4.6 | 45 |
| PEI HB–4.0% EPC | PEI | Alky Cl/epoxy | 27 | 46 |
| E50-FZ | PEI | Epoxy | 18.6 | 37 |
| E200-FZ | PEI | Epoxy | 4.1 | 37 |
| HCP-D | DETA | Acyl Cl | 6.0 | 48 |
| NUT-1 | ED | Alkyl Cl | 1 | 49 |
| NUT-11 | PEI | Alkyl Cl | 0.4 | 50 |
| 1-G0-TEPA | TEPA | Aldehyde | 4.2 | 51 |
Wang et al. developed porous polyamine particles synthesised via the precipitation polymerization of the monomer N-methyl-N-vinylformamide (MVF) and the cross-linker di[2-(N-vinylformamido)ethyl] (DVFE). In a similar trend as for E200-FZ, the resulting adsorbent – which had a surface area of 246 m2 g−1 – reached its optimum CO2 capacity of 2.30 mmol g−1 at 30 °C, in 1 atm CO2. Adsorption at 60 °C resulted in reduced uptake at all CO2 partial pressures to 1 atm.47 Huang et al. reported nanoporous crosslinked divinylbenzene-maleic anhydride copolymers functionalised with covalently bonded ‘bridging’ amines which exhibited an improved adsorption performance with lower temperature.48 The diethylenetriamine (DETA) grafted-copolymer, HCP-D, displayed both micro- and meso-porosity and a surface area of 343 m2 g−1. Its uptake under pure CO2 at 1 bar was 1.30 mmol g−1 at 25 °C, increasing to 1.53 mmol g−1 at 0 °C. Sun and co-workers also utilised small amine molecules, producing polyamine adsorbents from cross-linking diamines with 2,4,6-tris(chloromethyl)-mesitylene (TCM). NUT-1, synthesised from ethylene diamine (ED), adsorbed 1.43 mmol g−1 at 25 °C, raising to 1.87 mmol g−1 at 0 °C, under 1 bar CO2.49 From the same group, Mane et al. also observed CO2 adsorption increase inversely with temperature, in the performance of NUT-11, again synthesised using TCM, except this time to cross-link PEI (Mw 1800).50 This microporous adsorbent had a surface area of 598 m2 g−1. Its CO2 uptake at 25 °C was 2.23 mmol g−1, and 4.52 mmol g−1 at 0 °C. Its Qst value was between 46–49 kJ mol−1, more strongly indicative of physisorption rather than chemisorption – or it may be accessing both phenomena.
As shown in Table 1, E200-FZ, HCP-D, NUT-1 and NUT-11 were prepared using a more equal mol ratio of amine to cross-linker reactive site. Although more amine groups are therefore involved in cross-linking, reducing – or eliminating – the number of primary amines, the effect that the higher cross-linking density imparts on the structure, such as increased surface area, promotes higher CO2 uptake at lower temperatures. As in the case of HCP-D and NUT-1, a greater amount of cross-linker is indeed necessary when using small amine molecules to incorporate sufficient amine content. However, in addition to cross-linking density, the amine component can significantly affect adsorption behaviour. In a separate study, Thompson exchanged PEI for tetraethylenepentamine (TEPA) in their polyaldehyde phosphorus dendrimer cross-linked amines to give 1-G0-TEPA, using a similar amine:aldehyde ratio.51 Under the same conditions of adsorption tested for 1-G0/600PEI, at 25 °C, maximum CO2 uptake capacity increased to 10.7 wt% (2.43 mmol g−1).
The cross-linker structure itself is a crucial factor in imparting permanent porosity. The characteristic rigidity of conjugated π-systems is particularly attractive for this purpose.52 A series of CO2-sorbent, highly microporous organic polymers with surface areas of up to 809 m2 g−1 were reported by Liebl and Senker utilising the rigid and highly conjugated 2,4,6-tris(4-aminophenyl)-1,3,5-triazine and dianhydrides as building blocks.53
In our present work, we develop a new set of CO2 adsorbents, utilising the conjugated π-system of a triazine-based cross-linker with PEI (Mw 25000) to direct the formation of a rigid polymeric network and to access higher surface areas than achieved using an epoxy resin cross-linker. The bulky cross-linker acts as a spacer unit between polyamine chains, reducing intramolecular interactions and enabling good diffusion at ambient temperature. Through varying the ratios of amine to triazine units, the optimum cross-linking is achieved to enable low temperature adsorption and a highly CO2-selective adsorbent is produced, with an uptake capacity of 2.31 mmol g−1 CO2 at 30 °C, under 1 atm, 90% CO2/Ar. This demonstrates the potential of these materials to access low temperature adsorption applications. Finally, the material's behaviour under simulated post-combustion capture conditions is investigated.
Experimental
Chemicals
All starting material chemicals and solvents were used without further purification. THF (≥99.9%), acetone (≥99.5%), ethanol (≥99.8%), ammonium hydroxide solution (28.0–30.0% NH3 basis), and branched polyethyleneimine (PEI, average Mw = 25000 Da), were purchased from Sigma Aldrich. 4-Cyano-2-fluoro-benzyl bromide (98%, 214.037 g mol−1) and trifluoromethanesulphonic acid (99%, 150.07 g mol−1) were purchased from Fluorochem. Potassium hydroxide pellets (85%) were purchased from Alfa Aesar. Deionised water with 15 MΩ resistance from Merck Elix type 2 water purification system was used. Pureshield argon (99.9998%) and CO2 (99.8%) were supplied by BOC while 14.10% CO2/N2 (±2%) and 401.2 ppm CO2/N2 (±2%) were supplied by Air Liquide.
Materials synthesis
4BMFPT: 9.516 g of product were recovered (yield: 95.2%). Analysis found (calculated for C24H15N3F3Br3): C, 44.13 (44.89); H, 2.59 (2.36); N, 6.45 (6.54).
:alkyl bromide groups in the starting materials. Sorbents were either prepared in a 40 mL vial, indicated by (V), or a 250 mL round-bottom flask (R).
1:1 (V), 3:1 (V), 5:1 (V), 10:1 (V), 25:1 (V) were prepared in a 40 mL vial by adding a solution of PEI (31 mg, 0.72 mmol; 91 mg, 2.11 mmol; 140 mg, 3.25 mmol; 301 mg, 6.99 mmol; 731 mg, 16.97 mmol, respectively) dissolved in 6 mL THF at 82 °C, to a solution of 4BMFPT (150 mg, 0.234 mmol) dissolved in 29 mL THF at 82 °C. The PEI was washed from the vial with an additional volume of THF (3 mL; 3 mL; 3 mL; 6 mL; 6 mL, respectively) on combination of the reagents. The reaction mixtures were stirred at 82 °C for three nights, then the obtained solid products were separated from the solvent by centrifugation. Approx. 200 mL acetone was added to the products and the mixtures were stirred for 3 days then centrifuged to remove acetone. The products were stirred overnight in a solution of approximately 66 mg of KOH (1.18 mmol) dissolved in 106 mL of a 50:50 mixture of ethanol and water to remove the HBr formed during the reaction. The products were then washed in 106 mL of a 50:50 mixture of ethanol and water for 2.5 hours then this step was repeated. The products were stirred in approx. 112 mL ethanol for three nights, then centrifuged and dried in an oven overnight at 80 °C. The products were gently ground in a mortar prior to analysis. Yields: 1:1 (V), 8.0 mg; 3:1 (V), 109.9 mg; 5:1 (V), 123.1 mg; 10:1 (V), 326.7 mg; 25:1 (V), 544.8 mg.
10:1 (R) was prepared in a 250 mL round-bottom flask by adding a solution of PEI (317 mg, 7.36 mmol) dissolved in 5 mL THF at 60 °C to a solution of 4BMFPT (162 mg, 0.252 mmol) dissolved in 16 mL THF at 60 °C. The solution was stirred at 66 °C overnight, then the obtained solid product was separated from the solvent by centrifugation. Approx. 50 mL acetone was added to the product and the mixture stirred for approx. 1 hour, then centrifuged to remove the acetone. The product was stirred overnight in a solution of approximately 65 mg of KOH (1.18 mmol) dissolved in 100 mL of a 50:50 mixture of ethanol and water to remove the HBr formed during the reaction. The product was then washed in 100 mL of a 50:50 mixture of ethanol and water for 3 hours then this step was repeated. The product was stirred in approx. 75 mL ethanol overnight, centrifuged, and then dried in an oven for 3 nights at 70 °C. The product was gently ground in a mortar prior to analysis. 334.5 mg of product was obtained.
Materials characterisation
Elemental analysis data were collected on an Elementar vario MICRO cube equipped with a ceramic ash crucible in the combustion tube. 2–5 mg of sample was weighed into tin boats for analysis. A Thermo Scientific Nicolet iS10 FT-IR Spectrometer was used to collect the attenuated total reflectance infrared spectra of all samples. Spectra were recorded in the 650–4000 cm−1 region with 32 scans. N2 sorption isotherms at 77 K were measured with a Quantachrome Nova 2000e analyser. The samples (between 80–240 mg) were activated for a minimum of two hours under dynamic vacuum at 125 °C prior to analysis. BET surface areas were calculated in the 0.1–0.2 P/P0 range as detailed in Table S1 (ESI†). Thermal decomposition analysis was conducted on a TA Instruments SDT Q600 thermogravimetric analysis/differential scanning calorimeter (TGA/DSC). Between 5–15 mg of sample was placed in an open alumina crucible and the sample was heated until weight loss stabilised at a rate of 10 °C min−1 under a flow of 100 mL min−1 dry argon (Ar) which was filtered through a PerkinElmer Ultra Clean Moisture Filter. Scanning electron microscopy (SEM) images were collected using a ZEISS EVO LS25 SEM using an EHT of 10 or 15 kV with a probe current of 200 or 250 pA. Prior to SEM analysis samples were sputter coated with 10.3 nm of platinum to prevent charging. Solid-state NMR measurements were carried out at ambient temperature on a Bruker Avance III 400 MHz wide-bore spectrometer equipped with a 3.2 mm triple resonance probehead tuned to 1H, 13C, 15N. The magic angle spinning (MAS) rate was set at 12300 (PEI-4BMFPT) and 15000 Hz (4BMFPT) to avoid placing MAS sidebands over bona fide amine signals. Cross polarization (CP) experiments were used to improve sensitivity. For all samples, a recycle delay of 4 s was chosen based on 1H relaxation behaviour, a 1H excitation pulse of 80 kHz was used, followed by a 1H-13C CP contact time of 2.5 ms. The PEI-4BMFPT sample was signal-averaged for 17 hours, while the 4BMFPT sample was signal-averaged for 5.8 hours.
CO2 adsorption analyses
All gravimetric CO2 adsorption measurements were recorded using a TA Instruments SDT Q600 (TGA/DSC) at 1 atm throughout. Between 5–15 mg of sample was placed in an open alumina crucible. Prior to purging the sample, dry Ar was filtered through a PerkinElmer Ultra Clean Moisture Filter. Isothermal CO2 capture tests under 90% CO2 were carried out in the following sequence: (i) activation to remove the preadsorbed species at 125 °C under Ar flow of 100 mL min−1 for 3 hours; (ii) reduction of the temperature to 30 °C, (iii) once stabilised at temperature, reducing the Ar flow to 10 mL min−1 and introduction of dry CO2 at 80 mL min−1 and maintaining at temperature for 2 h, (iv) increase of Ar flow back to 100 mL min−1 and increase of temperature to 110 °C and remaining isothermal for 2 h, (v) reduction of temperature to 60 °C, (vi) once stabilised at temperature, reducing the Ar flow to 10 mL min−1 and introduction of dry CO2 at 80 mL min−1 and maintaining at temperature for 2 h, (vii) increase of Ar flow back to 100 mL min−1 and increase of temperature to 110 °C and remaining isothermal for 2 h, (viii) reduction of temperature to 90 °C, (ix) once stabilised at temperature, reducing the Ar flow to 10 mL min−1 and introduction of dry CO2 at 80 mL min−1 and maintaining at temperature for 2 hours.Cyclic CO2 adsorption–desorption experiments were carried out under pure, dry CO2 in the following sequence: (i) activation to remove the preadsorbed species at 120 °C under Ar flow of 100 mL min−1 for 6 hours; (ii) reduction of the temperature to 30 °C, (iii) stabilised at temperature for 1 hour; (iv) switch gas to dry CO2 flow at 95 mL min−1 and maintaining at temperature for 1 h, (v) switch gas to Ar flow at 100 mL min−1 and increase of temperature to 120 °C and remaining isothermal for 30 minutes. Then steps ii to v are repeated for 25 adsorption–desorption cycles.
Isothermal CO2 capture tests under dilute CO2 (either 10% CO2/N2 or 400 ppm CO2/N2) were carried out in the following sequence: (i) activation to remove the preadsorbed species at 125 °C under Ar flow of 100 mL min−1 for 3 h, (ii) reduction of the temperature to 30 °C, (iii) once stabilised at temperature, switching gas to dry CO2/N2 at a flow rate of 95 mL min−1 and maintaining at temperature for 3 h, (iv) switching gas to Ar flow of 100 mL min−1 and increase of temperature to 110 °C and remaining isothermal for 2 h, (v) reduction of temperature to 60 °C, (vi) once stabilised at temperature, switching gas to dry CO2/N2 at a flow rate of 95 mL min−1 and maintaining at temperature for 3 h, (vii) switching gas to Ar flow of 100 mL min−1 and increase of temperature to 110 °C and remaining isothermal for 2 h, (viii) reduction of temperature to 90 °C, (xi) once stabilised at temperature, switching gas to dry CO2/N2 at a flow rate of 95 mL min−1 and maintaining at temperature for 3 hours.
Prehydration experiments: water uptake and CO2 sorption experiments were carried out in the following sequence: (i) activation to remove the preadsorbed species at 120 °C under 100 mL min−1 Ar flow for 2 hours, (ii) the temperature was reduced and stabilised to 30 °C, (iii) the sorbent was equilibrated to a constant weight under a flow of 10 mL min−1 dry Ar with 80 mL min−1 Ar humidified using a water-filled bubbler (humidity calculated as 21.4% relative humidity (RH)),26 (iv) the humidified Ar was switched for humidified CO2 of flow rate 80 mL min−1 and flow maintained for 4 h; (v) the material was subsequently desorbed under a flow of dry Ar at 100 mL min−1 for 6 h, (vi) desorption continued as the temperature was ramped to 155 °C.
Volumetric CO2 and N2 adsorption isotherms were collected at 30 °C using a Quantachrome iSorb HP1 High Pressure Gas Sorption Analyser. For each low-pressure adsorption isotherm, up to 21 data points were collected from 0.01–1.05 bar. For each data point, 5 equilibrium points were measured with a 30-second interval. Prior to collection of the first isotherm, the sample (0.12–0.30 g) was activated under dynamic vacuum at ambient temperature for one hour, then degassed at 125 °C for four hours in an insulated thermal heat jacket, using a heating ramp rate of 10 °C min−1. Prior to collection of subsequent isotherms, the sample was degassed for 1 hour at 120 °C. Cell void volume was calibrated by introducing helium in two stages: first at 45 °C, then at the analysis temperature, controlled using an external temperature control system. The number of data points collected and equilibrium thresholds set for each isotherm are given in Table S2 (ESI†). After analysis, the sample was degassed at 120 °C for one to four hours.
Among all the synthesised material, the best performing at low temperature was selected for further H2O adsorption evaluation by dynamic vapour sorption and for CO2 uptake by dynamic breakthrough analysis.
H2O adsorption analysis
Water uptake was evaluated using a dynamic vapour sorption (DVS) analyser, Surface Measurement Systems (SMS), London, UK. The DVS analyser generates gas and/or vapour within a temperature range between 20 and 70 °C, and measures the sample's mass change over time as a function of RH or temperature. The temperature-controlled enclosure of the system allows a ±0.2 °C accuracy. The DVS system is equipped with rotary and turbomolecular pumps, to enable outgassing at high vacuum (maximum of 10−8 torr). The DVS experimental details are recorded in Table S3 (ESI†). The methodology first involved an outgassing step where 20 (±0.1) mg of sample was outgassed under high vacuum (10−6 Torr) and heated to 100 °C for at least 12 hours. Once the sample weight stabilised (indicating the dry mass), the sample was cooled down to the adsorption temperature of interest (30 or 40 °C). Then, the sample was exposed to different RH values ranging from 1 to 90%, following 5–10% increments. The mass equilibrium criteria of dm/dt = 0.001%/min and a maximum equilibrium time of 400 min was applied for each step of the relative humidity.Breakthrough CO2 experiments
Dynamic, i.e. breakthrough, adsorption experiments were carried out in a bespoke lab-scale rig (Fig. 2), specifically designed to mimic realistic typical coal-fired post-combustion conditions (∼1 bar, 40 °C and CO2 concentration of 14 vol%). The setup consists of a system of mass flow controllers from Bronkhorst (1) used to control the flow of gases fed into a fixed-bed reactor (3). The reactor is 20 cm long and has an internal diameter of 0.67 cm, and 250 mg of sample was used in the experiments. For wet conditions, N2 or He can be fed to a bubbler (2) that can be either heated or cooled at the desired temperature, hence creating the wet stream with the % RH of interest. The dry or wet mixture of gases is passed through the fixed bed column that is heated by a furnace specifically manufactured for this system. All lines from the bubbler to the analysers are heated to avoid water condensation. The composition at the outlet of the reactor is monitored online by a mass spectrometer (HPR20-HIDEN) (5) and the total flow is measured by a Coriolis flowmeter from Bronkhorst (4). | ||
| Fig. 2 Schematic of the dynamic setup used for conducting dry and wet CO2 breakthrough experiments. | ||
For dry CO2 capture experiments, 250 mg of sample, previously dried in a furnace for 12 hours at 150 °C, were placed in the reactor. A total flow of 40 ml N min−1 of He was fed to the fixed bed while heating to 160 °C. Once the temperature was reached it was kept constant for 30 minutes and then the bed was cooled to 40 °C under He flow. Once the sample was dried and cooled to 40 °C, the reactor was closed, and the lines were purged with a flow of 20 ml N min−1 of He. Next, the CO2/N2 gas mixture (1.4 ml N min−1 of CO2 and 8.6 ml N min−1 of N2) was fed to the reactor until sample saturation was reached.
For wet CO2 capture experiments, the sample was dried following the same procedure as in the dry experiments. Upon drying, two different protocols were followed: (1) in the first type of wet experiment, the sample was initially pre-saturated and the effect of H2O on CO2 adsorption was evaluated. A flow of He of 40 ml N min−1 was fed to the bubbler heated to 34.2 °C reaching 5.4 vol% of H2O in the stream. This wet He flow was continuously fed to the reactor until the observed signal of H2O in the mass spectrometer was constant, indicating saturation of the sample. Next, the reactor was closed and kept at a constant temperature of 40 °C, whilst the lines were purged with He (20 ml N min−1). A mixture of wet N2 and CO2 (CO2:H2O:N2 ratios equal to 1.4:0.54:8) was then fed to the reactor. (2) In the second type of wet experiment, the followed protocol was the same as previously described in (1) except for omitting pre-saturation of the sample. These experiments allowed for the evaluation of H2O–CO2 co-adsorption in the bed. Additionally, blank experiments were conducted to determine the dead volume and the gas response through the system.
Results and discussion
Elemental analysis and infrared spectroscopy characterisation
The cross-linker 4BMFPT was prepared following a procedure previously reported in the literature,54 except using 4-(bromomethyl)-3-fluorobenzonitrile in place of 4-(bromomethyl)-benzonitrile as the precursor for the preparation of 4BMFPT. The synthesis was confirmed successful by CHN analysis and FTIR-ATR spectroscopy (Fig. S1, ESI†). The absorption band at 2236 cm−1 displayed by 4-(bromomethyl)-3-fluorobenzonitrile, originating from the stretching vibration of the –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N group, is not present in the spectrum of 4BMFPT, showing the complete reaction of the nitrile group in the formation of the triazine ring. In the spectrum of 4BMFPT there are bands which are not in the spectrum of 4-(bromomethyl)-3-fluorobenzonitrile which originate from the triazine ring: at 1519 cm−1 and 1359 cm−1, relating to in-plane stretching vibrations, and at 816 cm−1 and 793 cm−1, relating to out-of-plane bending vibration bands.55
N group, is not present in the spectrum of 4BMFPT, showing the complete reaction of the nitrile group in the formation of the triazine ring. In the spectrum of 4BMFPT there are bands which are not in the spectrum of 4-(bromomethyl)-3-fluorobenzonitrile which originate from the triazine ring: at 1519 cm−1 and 1359 cm−1, relating to in-plane stretching vibrations, and at 816 cm−1 and 793 cm−1, relating to out-of-plane bending vibration bands.55
The formation of the cross-linked materials proceeded via an amine alkylation reaction between the alkyl bromide of the cross-linker and the amine groups of PEI, as shown in Fig. 3. The solids ranged in colour and texture from the pale yellow powder of 1:1 (V), becoming increasingly darker and coarser, to the deep orange slightly spongy material of 25:1 (V), shown in Fig. S2 (ESI†).
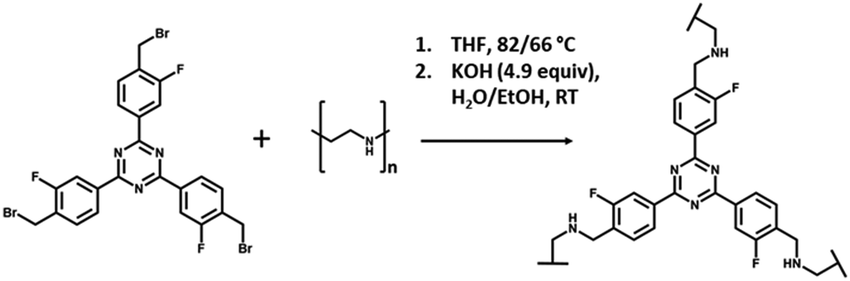 | ||
| Fig. 3 Reaction of 2,4,6-tris-[4-(bromomethyl-3-fluoro)-phenyl]-1,3,5,-triazine (4BMFPT) with PEI to produce cross-linked polymer PEI-4BMFPT. | ||
The samples were analysed for their elemental composition, as presented in Table 2 and Table S4 (ESI†). The C/N ratios of the products were used to calculate the amine:alkyl (featuring C–N bond of the reacted alkyl bromide, or 1/3 of the triazine ligand) ratios to indicate the extent of cross-linking which had taken place during the reaction, shown in Table 2. The calculation, using 10:1 (R) as an example, is presented in the ESI,† following Table S4 (ESI†). The lower the ratio of the amine to the alkyl cross-linker in the starting materials, the more cross-linked the product material, starting with 3:1 (V), for which there were almost two alkyl groups per amine. The ratio increased on increasing the amount of PEI starting material, and for both 10:1 (V) and 25:1 (V) there was excess amine to alkyl cross-linker in the product, with 1.5 amines per alkyl group in the latter.
:alkyl ratios of the product materials
| Sample | C/N | Amine:alkyl |
|---|---|---|
| 1:1 (V) |
3.40 | 0.68:1 |
| 3:1 (V) |
3.60 | 0.57:1 |
| 5:1 (V) |
3.37 | 0.71:1 |
| 10:1 (V) |
2.78 | 1.25:1 |
| 25:1 (V) |
2.65 | 1.52:1 |
| 10:1 (R) |
2.73 | 1.33:1 |
Branched chain PEI Mw 25000 has close to equal numbers of primary, secondary and tertiary amines in its structure, therefore, based on the expectation that the tertiary amines do not react with the alkyl bromide, and on the assumption that the secondary amines can, it can be assumed that a 1:1 ratio of amine:alkyl has no primary or secondary amines. It would be expected, however, that the steric hindrance of secondary amines would reduce the likelihood of reaction with the alkyl bromide. Thus, a 1.5:1 (or 3:2) ratio could have one secondary amine for every two tertiary amines, and a 0.5:1 (or 1:2) ratio would suggest that there are unreacted alkyl bromide groups present within the product. Given that the maximum amount of alkyl bromide cross-linker used in the starting materials was in a sufficient stoichiometric ratio for up to a 1:1 amine:alkyl ratio, that there is less than 1 equivalent of amine per alkyl for 1:1 (V), 3:1 (V) and 5:1 (V) suggests PEI is lost during the reaction, i.e., it does not get cross-linked and incorporated into the product material.
The FTIR-ATR spectra of the samples, from 2000 to 700 cm−1 are shown in Fig. 4, with the spectra of 4BMFPT and PEI above and below, respectively. Spectra from 4000 to 650 cm−1 are shown in Fig. S3 (ESI†). Going from 25:1 (V) to 1:1 (V), it is apparent that some bands associated with the PEI at about 3275 cm−1; 2930–2934 cm−1; 2818–2826 cm−1; 1456 cm−1, 1295 cm−1 and 1111 cm−1 decrease in intensity or disappear, this can be associated with the established lesser quantity of amine for the more intensely cross-linked materials. The band in the products’ spectra at 1653–1660 cm−1 is associated with the H–O–H bending vibration of adsorbed water,55 indicating that the lesser cross-linked materials, 10:1 (V) and 25:1 (V) contain more moisture. All products except 25:1 (V) exhibit an extremely weak band that can be associated with stretching of the C–F bond of the cross-linker at 1269–1270 cm−1. All of the products show bands associated with the triazine ring: out-of-plane bending vibration bands at 775–776 cm−1 and 816–817 cm−1, and in-plane stretching vibrations at 1519–1521 cm−1 and 1360–1362 cm−1. The band at 1420 cm−1 disappears in 10:1 (V) and 25:1 (V): this band, which could somewhat be masked by the band at 1456 cm−1, is present in the more cross-linked materials, and so may instead be associated with the CH2 deformation vibration of the CH2–Br group of unreacted cross-linker. Similarly, the weak band at 1184–1186 cm−1 in 1:1 (V), 3:1 (V) and 5:1 (V), very weak in 10:1 (V) and absent in 25:1 (V), may be associated with the CH2 twisting vibration of the CH2–Br group. This suggests that the more cross-linked materials have excess triazine linker such that some alkyl bromide groups are unreacted, as reflected in the low amine:alkyl ratio obtained from the elemental analysis data.
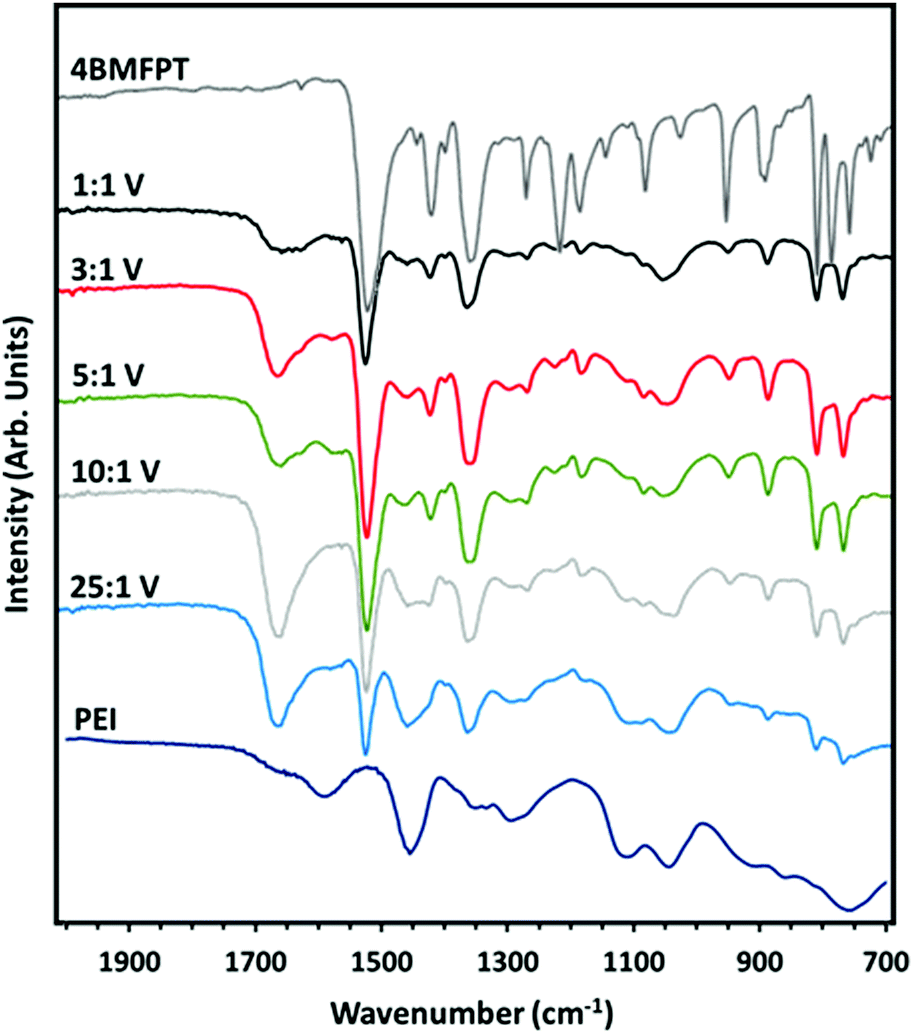 | ||
| Fig. 4 FTIR-ATR spectra of synthesised 4BMFPT and as-received PEI against spectra of PEI:4BMFPT cross-linked products. | ||
Morphology and textural properties
SEM images of the samples show differences in their morphologies which appear dependent on their amine:alkyl ratios, (Fig. 5). 1:1 (V) displays spherical particles of 0.1–0.7 μm in diameter, arranged singularly or in large dense clusters. There are also smoothened masses of material with a fairly flat morphology. 3:1 (V) and 5:1 (V) appear quite similar in that they are both composed of spherical particles clustered together in bunches ranging in size from around 0.2 – 1 μm. 10:1 (V) also features clustered spherical particles of about 0.3 – 1 μm in diameter. However, unlike 3:1 (V) and 5:1 (V), this sample has a very inhomogeneous morphology. Other locations studied by SEM showed an interconnected network of fused platelets forming a continuous solid composition, with the smallest globules of about 0.5 μm in the smallest dimensions. 25:1 (V) is very similar to the latter description of 10:1 (V). It too has an interconnected structure of an irregular globular morphology of a similar scale to that of 10:1 (V), with few discrete spherical structures. In general, for these materials, it appears that a lower amine content promotes the formation of smaller, discrete spherical particles, and a higher amine content results in a higher degree of connectivity between constituent particles.
 | ||
| Fig. 5 SEM images of: (a) 1:1 (V); (b) 3:1 (V); (c) 5:1 (V); (d) 10:1 (V); (e) 10:1 (V) and (f) 25:1 (V). | ||
The N2 adsorption–desorption isotherms at 77 K obtained for 3:1 (V), 10:1 (V) and 25:1 (V), shown in Fig. 6 (and separately in Fig. S4–S6, ESI†) further indicate these materials’ different structures. 3:1 (V) displays a type II adsorption–desorption isotherm with significant uptake between 0.9 and 1 P/P0, indicative of the presence of large mesopores and macropores. This isotherm shows hysteresis and some uptake at very low P/P0, (inset) suggesting 3:1 (V) may have a small share of micropores. 10:1 (V) and 25:1 (V) do not show this and can be considered non-porous to N2.
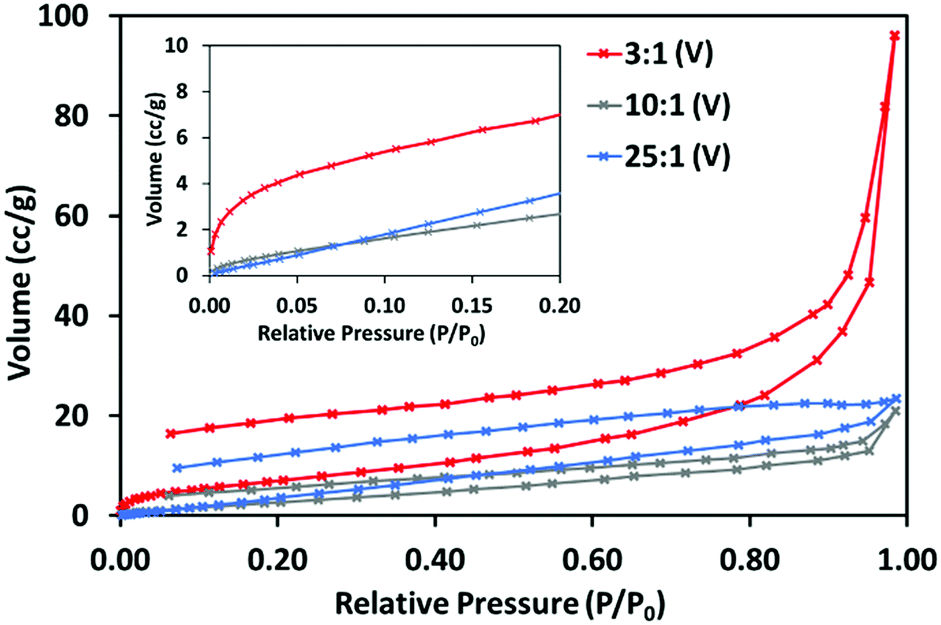 | ||
| Fig. 6 N2 adsorption–desorption isotherms for 3:1 (V), 10:1 (V) and 25:1 (V), at 77 K, with section of low pressure adsorption up to 0.20 P/P0 (inset). | ||
CO2 adsorption analysis
The CO2 adsorption behaviour of the PEI:4BMFPT materials was measured at 30 °C, 60 °C and 90 °C to assess their response to a range of temperatures relevant to both DAC and post-combustion CO2 capture. Analysis was conducted over a period of 2 hours under 90% CO2. Corresponding adsorption data are presented in Table S5 (ESI†). Fig. 7(a–c), shows that the maximum CO2 adsorption capacity is consistently higher for the lesser cross-linked materials and is consistently highest for 25:1 (V). This may be expected given that 25:1 (V) has the highest amount of primary and secondary amines relative to triazine cross-linker. 25:1 (V) and 10:1 (V) show maximum final CO2 uptake at 60 °C, reaching 2.45 mmol g−1 and 1.95 mmol g−1, respectively. Their maximum uptakes at 30 °C are 1.90 mmol g−1 and 1.73 mmol g−1, respectively, but saturation is not reached at this temperature within 2 hours, indicative of slow adsorption kinetics. Their adsorption rates increase with temperature, as after 10 minutes 10:1 (V) adsorbs 39.49% of its final capacity at 30 °C, 81.88% at 60 °C and 93.54% at 90 °C, and 25:1 (V) adsorbs 38.26%, 78.66% and 92.35%, of its final capacity at 30 °C, 60 °C and 90 °C, respectively. Their slower adsorption and lower uptakes at 30 °C suggest that 10:1 (V) and 25:1 (V) are limited in their adsorption potential by a diffusional barrier; however, increasing temperature results in decreased hydrogen bonding between polyamine chains and higher chain mobility, enabling CO2 to overcome this diffusion resistance, and uptake increases, as has been frequently observed for PEI-based CO2 adsorbents.23,24,27,56 From 30 °C to 60 °C, the maximum adsorption of 10:1 (V) increases by 12.47% (relative to adsorption at 30 °C), while 25:1 (V) improves by more than twice that at 29.00%. Although 25:1 (V) has better absolute adsorption capacity, 10:1 (V) may be experiencing relatively less diffusion resistance at 30 °C, which may suggest a structure that better enables gas transport. Both materials then show a reduction in their adsorption on moving from 60 °C to 90 °C, as previously observed on increasing temperature,23,24 due to the shifting of the thermodynamic adsorption–desorption equilibrium.57
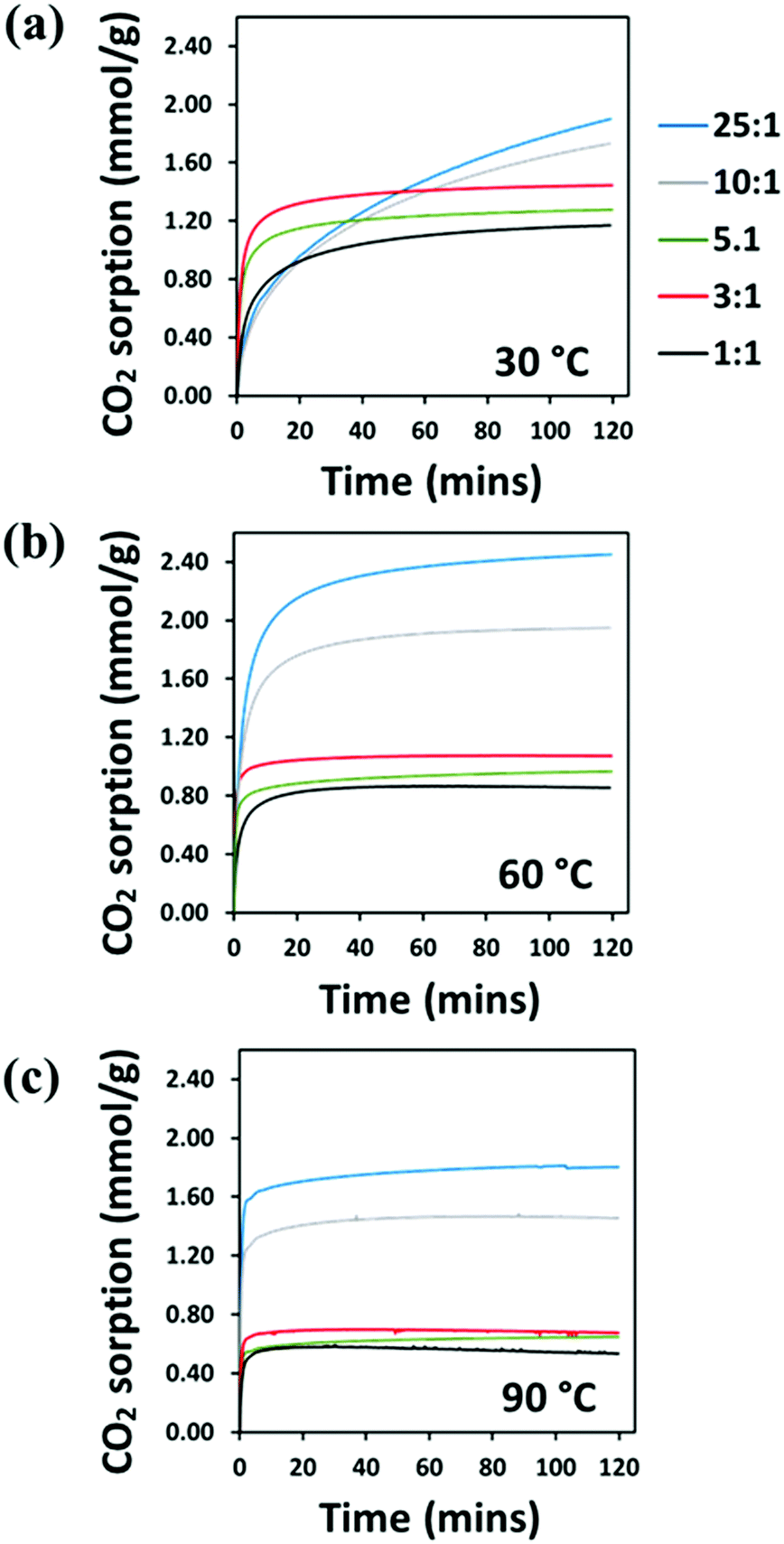 | ||
| Fig. 7 TGA–CO2 sorption (mmol g−1) of PEI-4BMFPT 1:1 (V), 3:1 (V), 5:1 (V), 10:1 (V), 25:1 (V) at (a) 30 °C; (b) 60 °C; (c) 90 °C, under 90% CO2. | ||
The more cross-linked materials, 1:1 (V), 3:1 (V) and 5:1 (V), have a lower final adsorption capacity compared with 10:1 (V) and 25:1 (V), and they generally show faster adsorption. After 10 minutes, at 30 °C, they show much higher uptake relative to their final capacities than 10:1 (V) and 25:1 (V), at 67.49%, 85.80% and 84.04% for 1:1 (V), 3:1 (V) and 5:1 (V), respectively, (Table S5, ESI†). The significant difference between the materials’ uptake kinetics at 30 °C can be rationalised by reference to their SEM images. The particles of 1:1 (V) are far more discrete than those of 25:1 (V), therefore offering greater access through the solid, i.e. they allow faster diffusion. The particles of 25:1 (V) appear fused together such that they form a dense, more impenetrable structure, thus less favourable for fast adsorption. This lack of clear passage into the bulk of the material is probably the source of the diffusion limitation observed for 25:1 (V) at 30 °C.
In common with the lesser cross-linked adsorbents, 1:1 (V), 3:1 (V) and 5:1 (V) also show faster CO2 uptake with increasing temperature, but conversely, increasing temperature is detrimental to their CO2 uptake. 1:1 (V), 3:1 (V) and 5:1 (V) reach their maximum adsorption of 1.17, 1.44, and 1.28 mmol CO2 g−1, respectively, at 30 °C. Given that these adsorbents have excess triazine linker to amine content, they may only contain tertiary amines which are ineffective at adsorbing CO2 under dry conditions;58 therefore, physisorption may be their primary mode of adsorption. These adsorbents lose weight at the higher temperatures; 1:1 (V) proceeds to lose weight after 75 minutes at 60 °C and after 30 minutes at 90 °C. Thermal decomposition data, (shown in Fig. S7, ESI†) confirms that these materials, and 4BMFPT, do not decompose below temperatures of about 200 °C, therefore this weight loss can be attributed to the desorption of CO2, possibly due to a shift in the adsorption–desorption equilibrium over time.
As 10:1 (V) and 25:1 (V) showed the highest uptake at 30 °C, they were further investigated to evaluate their CO2 uptake behaviours. The adsorbents were analysed for their adsorption at 30 °C from 0.01–1 bar. The isotherms are shown in Fig. 8. At 1 bar the interpolated CO2 uptake of 10:1 (V) is 1.87 mmol g−1 and of 25:1 (V) is 1.97 mmol g−1. These values are slightly higher than measured gravimetrically, possibly since here adsorption is measured under pure CO2 as opposed to the 90% CO2/Ar used in the TGA. Also, both materials may be closer to their equilibrium capacities. Significantly, 10:1 (V) shows higher CO2 adsorption in the low-pressure range of 0.01–0.5 bar. At 0.1 bar the interpolated CO2 uptake of 10:1 (V) is 1.15 mmol g−1, while the uptake of 25:1 (V) is lower at 0.90 mmol g−1. This demonstrates that, although the CHN data shows that 10:1 (V) has relatively fewer adsorption sites (amines) compared to 25:1 (V), it may present greater accessibility of these sites to CO2 such that, at lower pressures, there is a greater probability of CO2 coming into contact with them, improving uptake.
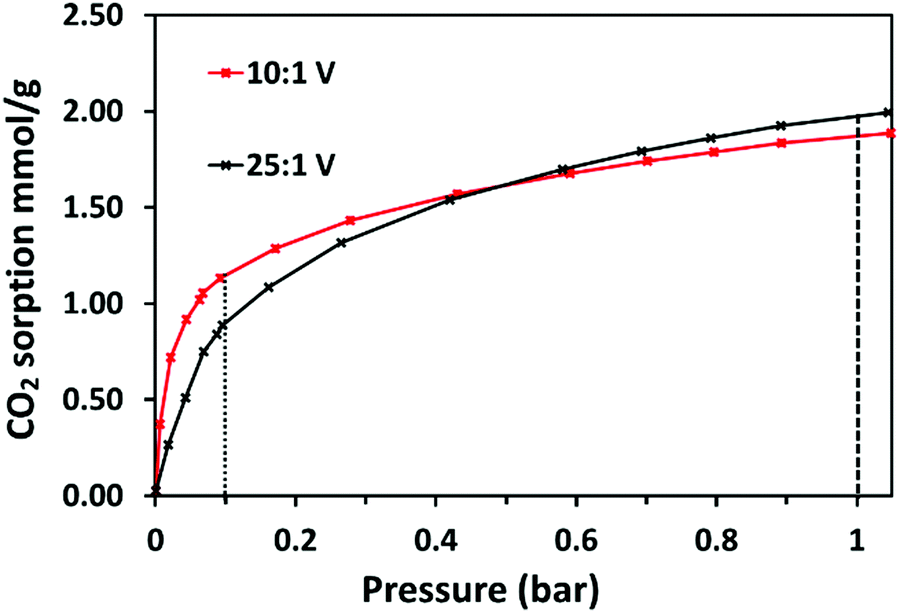 | ||
| Fig. 8 Single-component CO2 sorption isotherms of 10:1 (V) and 25:1 (V) at 30 °C from 0.1–1 bar. The dashed lines show where the uptakes have been interpolated at 0.1 bar and 1.0 bar CO2. | ||
In terms of developing a CO2 adsorbent that displays high capacity at low partial pressures, the former requirement is best fulfilled by 25:1 (V), and the latter by 10:1 (V). Due to the importance of efficient uptake under dilute conditions, further investigations were pursued based on the synthesis of 10:1 (V). During the initial screening stage, parallel syntheses were conducted in vials using a multi-well heating block to increase throughput. This synthesis procedure was adapted by synthesising the 10:1 material under reflux conditions at 66 °C in a round bottom flask. The RBF was employed to optimise the synthesis by more uniform heating. Also, with regards to eventual scaling up of the synthesis of the adsorbent, it is beneficial to use a larger vessel and the use of a round bottom flask is also a convenient move to build flexibility into the synthesis procedure, for adding further reactants etc. The product, 10:1 (R), is light peach-coloured and has a fine, sand-like consistency (Fig. S2, ESI†). Our group's previously reported epoxy cross-linked PEI adsorbents were often very dense spongey materials, unsuitable for industrial applications. Here we have advanced this work by utterly transforming these self-supported amine sorbents simply via switching the cross-linker to obtain a far more amenable texture in the powder-like 10:1 (R). The powder form can better enable the shaping of the sorbent into industrially practical forms, such as pellets or extrudates.59
The IR spectrum of 10:1 (R) is almost identical to that of 10:1 (V) (Fig. S8, ESI†). Interestingly, from the CHN data, 10:1 (R) also has a comparable amine:alkyl ratio to 10:1 (V) at 1.33:1, (Table 2) indicating that it has excess amine to alkyl cross-linker. Based on previous assumptions that secondary amines will react with the alkyl bromide, this would suggest that 10:1 (R) has one secondary amine for every three tertiary amines. In practice, it may be anticipated that steric hindrance would prevent the reaction of many secondary amines, so this ratio would indicate the presence of unreacted alkyl bromide groups. However, the signal associated with the carbon of the alkyl bromide (C8) is absent in the 13C NMR spectrum of 10:1 (R), suggesting that complete amine alkylation has taken place (Fig. S9, ESI†).
SEM imaging revealed that 10:1 (R) is entirely composed of discrete spherical particles ranging in diameter from about 0.4 to 1.9 μm, Fig. 9. There are also ‘doughnut’ shaped elements of about 2–3 μm across. The structure is more similar to that of the 3:1 (V) and 5:1 (V) than 25:1 (V), in that is has discrete individual particles, although those of 10:1 (R) have a broader size distribution. The difference in structures between 10:1 (R) and 10:1 (V) are likely to be due to the lower reaction temperature and shorter reaction time given to 10:1 (R), in that a coalescence process may have initiated in 10:1 (V) under the hotter and more prolonged heating conditions. Self-supported cross-linked amine adsorbents composed of spherical particles have previously been reported. Hwang et al. synthesised particles of 0.2–2 μm in diameter in cross-linking PEI with glutaraldehyde.44 Larger spherical particles of 10–300 μm in diameter were reported by Huang et al. who polymerised divinylbenzene and maleic anhydride before grafting diamines to the product.48
 | ||
| Fig. 9 SEM image of 10:1 (R). | ||
The CO2 adsorption behaviour of 10:1 (R) is superior to that of the other PEI-4BMFPT adsorbents at 30 °C, with a maximum capacity after 120 minutes of 2.31 mmol g−1, as shown in Fig. 10 (and against all PEI-4BMFPT adsorbents in Fig. S10, ESI†). This is 1 mmol higher than that reported by Huang et al. for their high surface area functionalised co-polymers,48 and 0.1 mmol higher than reported by Mane et al. for alkyl chloride cross-linked PEI,50 both analysed at 25 °C, and under 1 bar pure CO2. Compared to 10:1 (V) and 25:1 (V), CO2 uptake by 10:1 (R) is faster, adsorbing 59.51% of its final capacity after 10 minutes, reaching 1.38 mmol g−1 uptake. In common with all other adsorbents, its adsorption kinetics is improved by increasing temperature, and like the more cross-linked adsorbents, 1:1 (V), 3:1 (V) and 5:1 (V), 10:1 (R) shows its highest adsorption at the lowest temperature of 30 °C. Its final capacity reduces to 2.09 mmol g−1 and 1.45 mmol g−1 at 60 °C and 90 °C, respectively, therefore it is less effective than 25:1 (V) at higher temperatures, Fig. S10 (ESI†). The adsorption behaviour of 10:1 (R) may be linked with its morphology: the clear pathway through the bulk of the material formed between the distinct spherical particles likely allows good contact between the amine groups and CO2, enabling higher adsorption at 30 °C. Increasing temperature may aid diffusion, but any benefit is outweighed by the greater promotion of CO2 desorption, leading to reduced capacity.
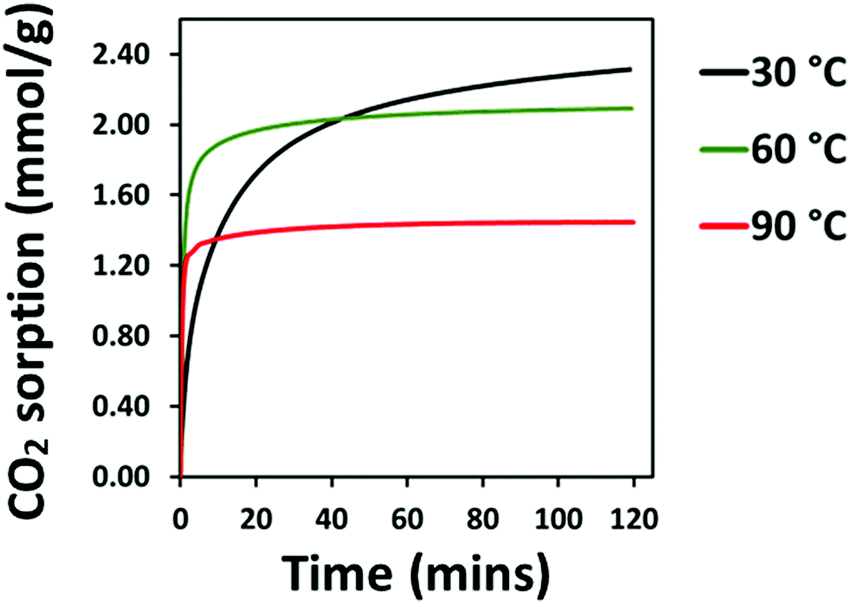 | ||
| Fig. 10 TGA–CO2 sorption (mmol g−1) of 10:1 (R) at: (a) 30 °C; (b) 60 °C; (c) 90 °C, under 90% CO2. | ||
The excellent low-temperature CO2 adsorption performance of 10:1 (R) prompted us to further explore the synthesis of the material. Our previous work has shown that hydrophobic functionality within cross-linked polyamine adsorbents promotes adsorption,36 therefore the fluorinated cross-linker 4BMFPT was selected for this study. However, unlike our previous work in which hydrocarbon and fluorocarbon chains were introduced to the adsorbent, 4BMFPT offers only three fluorine atoms per linker, situated in relative isolation from one another. To understand what extent this may have had on adsorption, we compared 10:1 (R) against its non-fluorinated analogue, 10:1 (RH), synthesised using a similar procedure (see ESI†). Although uptake is lower overall, 10:1 (RH), shows the same trend in CO2 adsorption as for 10:1 (R), (Fig. S11 and Table S5, ESI†). The maximum CO2 uptake of 10:1 (RH) at 30 °C is 2.09 mmol g−1, greater than both 10:1 (V) and 25:1 (V). Thus, it is seen that higher adsorption at low temperature can be achieved via this synthesis even using a non-fluorinated cross-linker which has potential economic and environmental advantages.
The gas sorption isotherm of 10:1 (R) from 0.01–1 bar at 30 °C was compared to those of 10:1 (V) and 25:1 (V), Fig. 11(a). The interpolated CO2 uptake of 10:1 (R) is 1.06 mmol g−1 at 0.1 bar and 2.01 mmol g−1 at 1 bar (Table S5, ESI†). Although slightly lower than the uptake at 0.1 bar for 10:1 (V), 10:1 (R) does also show higher low-pressure uptake than 25:1 (V). This is likely due to the greater access to the amine binding sites via the space between the spherical particles. To confirm this, the surface area of 10:1 (R) was measured by using the BET method. The N2 adsorption isotherm of 10:1 (R), shown in Fig. S12 (ESI†), is similar to that of 10:1 (V), indicating a non-porous material. The surface area, calculated as 11.827 m2 g−1 was even lower than for the other adsorbents, which showed no clear trend (Table S1, ESI†). The surface areas do not corroborate the differences observed in the materials’ microstructures, in that those composed of smaller, more discrete spherical particles would have a significantly higher surface area to those whose morphologies are of a more dense and continuous network. Gaining accurate surface area measurements for these materials is challenging. Firstly, they have very low surface areas. Secondly, the N2 adsorption data are not necessary equilibrium isotherms; the affinity for these materials to N2 is very low, and further, at 77 K, the polymers are likely in a glassy state, lacking flexibility. It is probable that it is this flexibility that, at ambient temperatures, affords the formation of small voids receptive to the inclusion of the CO2 molecule, with a smaller kinetic diameter than N2, enabling access to amines with which CO2 chemisorbs. Such a phenomenon has been suggested in previous work in which positron annihilation spectroscopy was carried out on a cross-linked CO2 sorbent.60
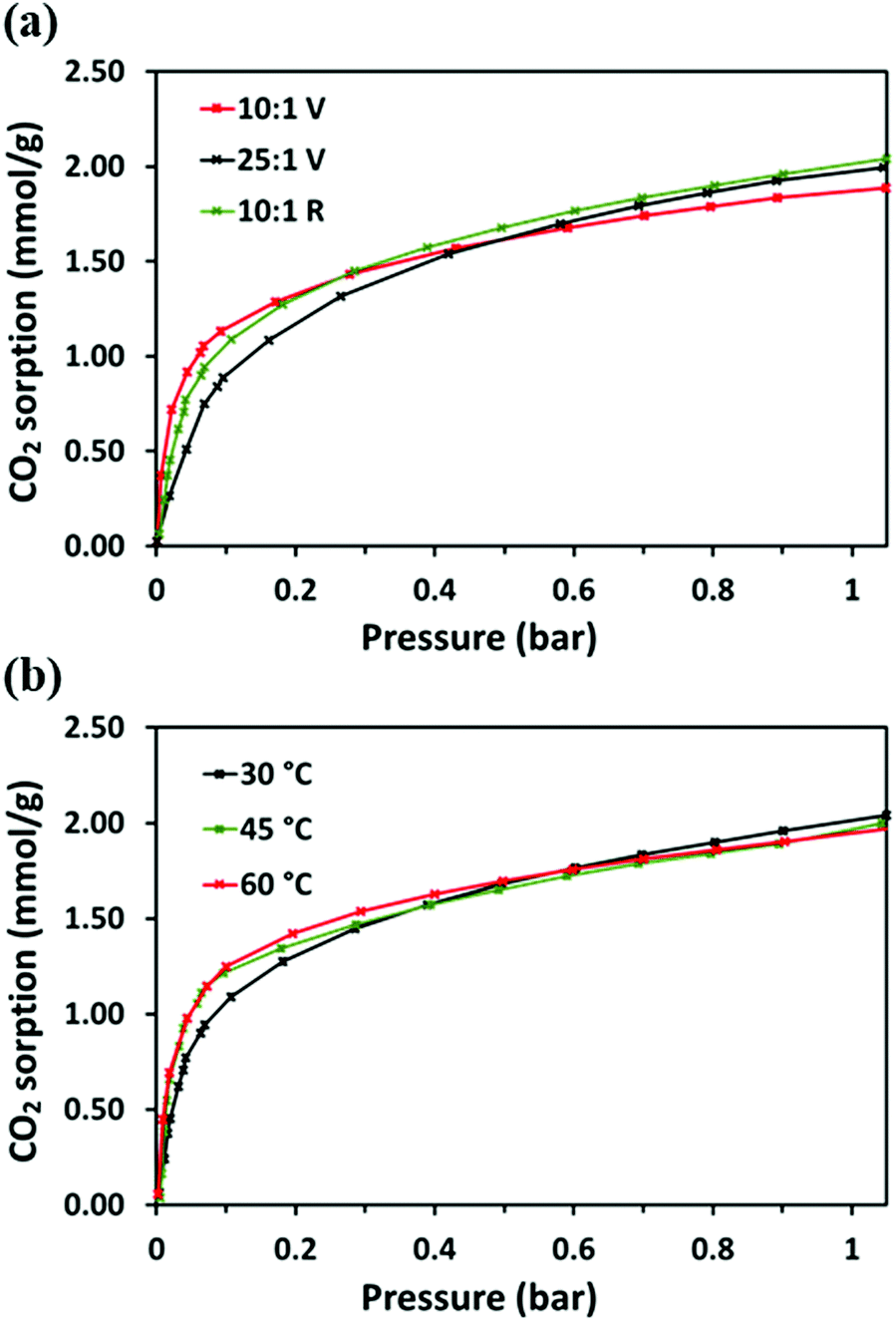 | ||
| Fig. 11 (a) Single-component CO2 sorption isotherms of 10:1 (V), 25:1 (V) and 10:1 (R) (in green) at 30 °C from 0.1–1 bar. (b) Single-component CO2 sorption isotherms of 10:1 (R) at 30 °C, 45 °C and 60 °C from 0.1–1 bar. | ||
CO2 adsorption isotherms of 10:1 (R) from 0.01–1 bar were compared at 45 °C and 60 °C, Fig. 11(b). The isotherms become steeper with temperature, showing higher adsorption at lower pressures. At 0.1 bar the interpolated CO2 uptakes are 1.21 mmol g−1 and 1.25 mmol g−1, at 45 °C and 60 °C, respectively, higher than adsorption at 30 °C. However, temperature also influences the kinetics of uptake, particularly at lower pressure: at 60 °C, the adsorption points reach equilibrium faster than at 30 °C, at which temperature, equilibrium is not consistently obtained (Fig. S13, ESI†). Therefore, it appears that at low pressure, temperature-enhanced diffusion enables more amine–CO2 reactions to take place faster.
The selectivity towards CO2 is a crucial factor for the success of an industrial adsorbent in order that it may release the purest CO2 product. To predict the adsorption equilibrium of components within a gas mixture from pure component adsorption isotherms, the ideal adsorbed solution theory (IAST) model,61 may be applied. Single-component CO2 and N2 sorption isotherms of 10:1 (R) were collected at 30 °C from 0.1–1 bar, Fig. S14 (ESI†). Taking the partial pressures of CO2 and N2 as 0.1 bar and 0.9 bar, respectively, 10:1 (R) adsorbs 1.06 mmol CO2 and 0.017 mmol g−1 N2, giving 10:1 (R) a predicted selectivity of 575 for CO2. This is higher than reported for comparable cross-linked polyamine adsorbents at similar temperatures,50 and is on a par with the most selective MOFs.62
For its economical industrial application, it is essential that an adsorbent be regenerable and capable of multiple adsorption–desorption cycles.2 To test its repetitive CO2 uptake, 10:1 (R) was subjected to multiple adsorption events at 30 °C, with each adsorption event lasting for one hour. The uptake of each cycle is presented in Fig. 12, with full data shown in Fig. S15 (ESI†). Over the initial cycle, the adsorption is 1.899 mmol g−1 CO2, and this drops by 10% to 1.706 mmol g−1 for the second cycle. Uptake then becomes stable, with the 25th cycle adsorbing 1.594 mmol g−1. Between the 2nd and 25th cycles, uptake deviates by a maximum of 0.154 mmol g−1. A slight drop in adsorption between the initial and subsequent adsorption cycles is also apparent in the cyclic adsorption of diethylenetriamine (DETA) modified porous organic polymers synthesised by Yang et al. for which adsorption was carried out under breakthrough conditions using a CO2/N2 mixture (20:80 v/v) at 25 °C, with desorption at 100 °C.63 It may be that for 10:1 (R), on the first cycle, the most strongly CO2-adsorbing amine sites, likely the primary amines on the most exposed, external surface, are saturated and the condition of desorption (120 °C for 30 minutes) is insufficient to regenerate them thereafter.
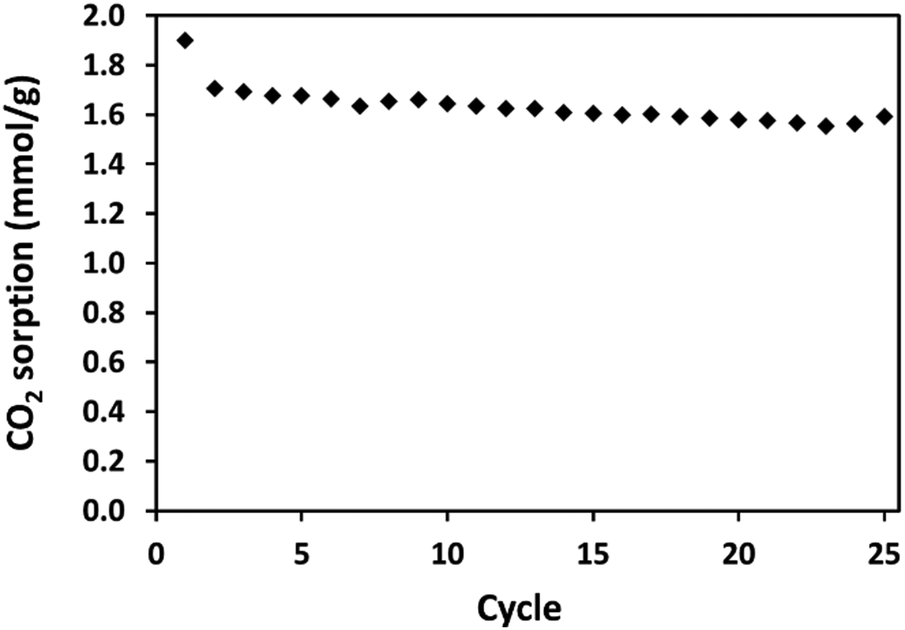 | ||
| Fig. 12 CO2 uptake of 10:1 (R) under temperature swing adsorption–desorption cycles. Uptake and desorption were carried out at 30 °C and 120 °C, respectively, in 1 atm pure, dry CO2. | ||
It can be seen from the data presented in Fig. S15 (ESI†), that CO2 adsorption (weight increase) remains more constant than does the weight of adsorbent. From the beginning of the first sample to the end of the last sample, the adsorbent weight reduces by 3.4%. A slight reduction in adsorbent weight is common in amine-based adsorbents over the duration of multiple cycles, as observed in our previous work,36 and that of others in which PEI is supported on a polyamide–carbon nanotube composite membrane,64 or graphene oxide.65 Goeppert et al. reported a reduction in adsorbent weight over the course of 50 adsorption–desorption cycles on silica-supported, epoxide-modified TEPA, with adsorption under 95% CO2/N2 at 85 °C – this was attributed to amine leaching.66 Despite the reduction in adsorbent mass, adsorption remains consistent to within 7% from the second to the last cycle.
Gravimetric experiments were carried out using dilute CO2 mixtures of 10% CO2/N2 and 400 ppm CO2/N2, simulating the concentration of CO2 in flue gas and air, respectively. Due to the high selectivity for CO2, the weight increase from the gravimetric experiment is interpreted as pure CO2 uptake. As generally expected for an amine-based adsorbent, performance is lower under a more dilute CO2 source.27 Under 10% CO2/N2, 10:1 (R) adsorbed 1.50 mmol g−1 CO2 at 30 °C, 1.42 mmol g−1 at 60 °C, and 0.55 mmol g−1 at 90 °C, after 180 minutes dynamic adsorption, as shown in Fig. 13(a) and Table 3 (with normalised adsorption data shown in Fig. S16, ESI†). Here, 10:1 (R) follows a similar trend of adsorption seen under 90% CO2: maximum adsorption is highest at 30 °C. After 10 minutes at 60 °C, 10:1 (R) takes up 1.01 mmol g−1 CO2, while at 30 °C, it adsorbs less than half of this at 0.47 mmol g−1. It is only after 115 minutes that adsorption at 30 °C starts to surpass adsorption at 60 °C; this is much later than under 90% CO2, for which adsorption at 30 °C overtakes that at 60 °C after just 43 minutes. The slow kinetic uptake at 30 °C suggests that under the more dilute CO2 environment of 10% CO2, a higher temperature to overcome diffusion limitation is more beneficial for uptake than it is detrimental, over a longer duration. Under dilute CO2, the probability of each amine coming into contact with CO2 is reduced (therefore maximising the number of accessible amines is imperative). Once the surface amines are saturated, the CO2 reacts with the internalised amines, and due to temperature assisted diffusion enhancement, this occurs more readily at 60 °C than at 30 °C. Given sufficient time at 30 °C, in terms of total uptake, the thermodynamic benefit outweighs the benefit of enhanced diffusion.
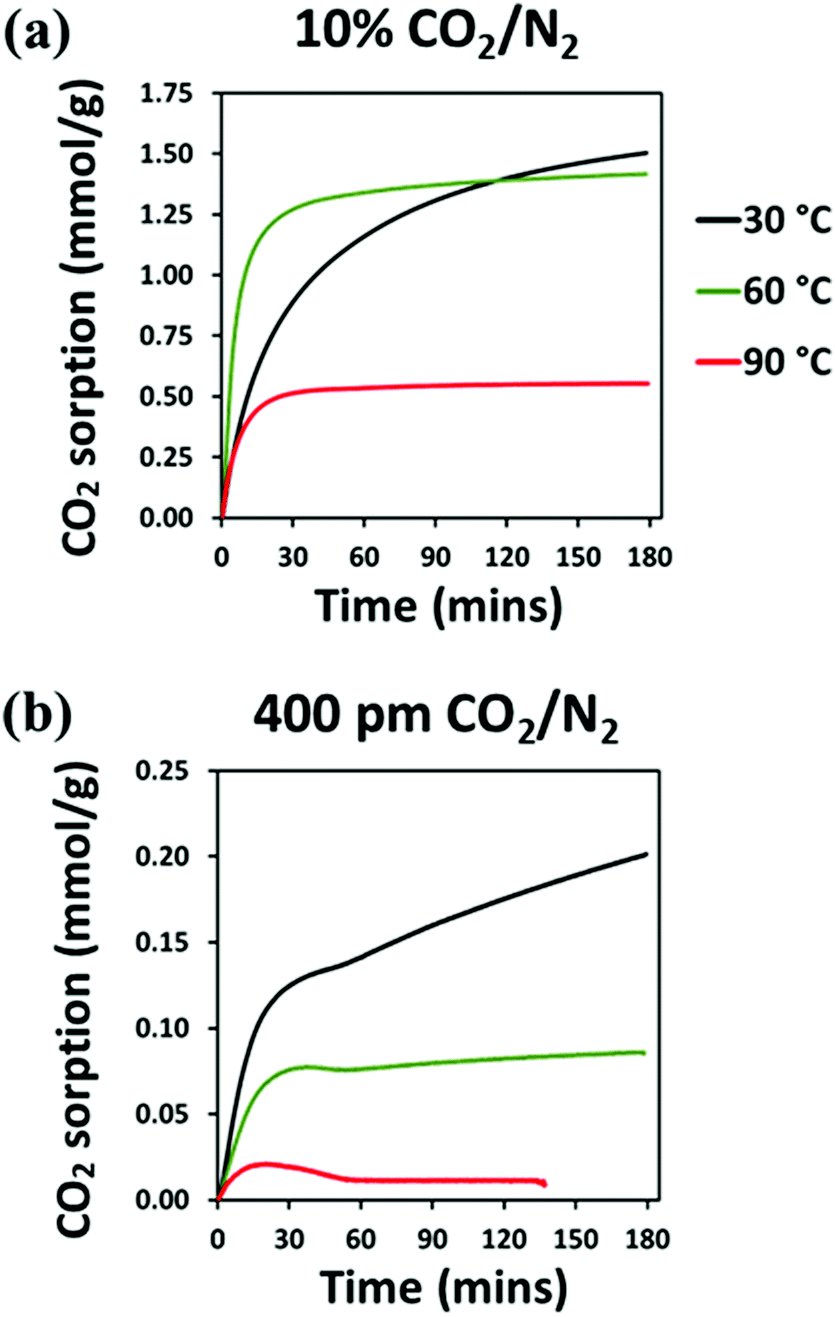 | ||
| Fig. 13 TGA–CO2 sorption (mmol g−1) of 10:1 (R), in (a) 1 atm 10% CO2/N2 (b) 1 atm 400 ppm CO2/N2. | ||
:1 (R) after 180, 120 and after 10 minutes in 10% CO2/N2 and 400 ppm CO2/N2
| Time (min) | Uptake at 1 bar, 10% CO2, (mmol g−1) | Uptake at 1 bar, 400 ppm CO2, (mmol g−1) | |||||
|---|---|---|---|---|---|---|---|
| 30 °C | 60 °C | 90 °C | 30 °C | 60 °C | 90 °C | ||
| 180 | 1.50 | 1.42 | 0.55 | 0.20 | 0.09 | 0.01 | |
| 120 | 1.40 | 1.39 | 0.55 | 0.18 | 0.08 | 0.01 | |
| 10 | 0.47 | 1.01 | 0.38 | 0.07 | 0.04 | 0.02 | |
Under 400 ppm CO2 10:1 (R) behaves entirely differently and uptake is significantly lower, Fig. 13(b). Adsorption is both fastest, and highest at 30 °C, with a final capacity of 0.20 mmol g−1 CO2, decreasing to 0.09 mmol g−1 and 0.01 mmol g−1 at 60 °C and 90 °C, respectively. Uptake is fastest until about 30 minutes, then the rate of adsorption decreases sharply at 30 °C and more so at 60 °C, whereas for 90 °C, desorption occurs after about 20 minutes. Under lower CO2 partial pressures, the heats of adsorption for amine-based adsorbents are higher due to the dominance of the most strongly interacting amine sites.67 It is likely that within the first half-hour, the most reactive surface amines become saturated. The evolved heat may not be fully dissipated, resulting in some desorption after around 40 and 20 minutes at 60 and 90 °C, respectively. After saturation of the surface amines, adsorption is via the less reactive and the more internalised amines, therefore it may occur at a lower rate partly due to slow diffusion of CO2. Rather than promoting diffusion and increasing adsorption, higher temperatures have a severely detrimental effect. This trend has also been reported by Goeppert et al. for silica-supported PEI adsorbents operating under air capture conditions.68 The lower driving force under the lower partial pressure of 400 ppm may overcome the benefit of enhanced diffusion at higher temperatures, as a greater decrease in entropy is required for adsorption.
Comparing the final adsorption capacities at 30 °C under 400 ppm and 10% CO2, adsorption is reduced by a factor of 7.5 at the lower partial pressure. This is in line with what was observed in lower amine-loaded hyperbranched aminosilica adsorbents reported by Choi et al.69 Therefore, it can be inferred that with greater amine functionality, the CO2 capture performance of 10:1 (R) under 400 ppm CO2 may be improved.
H2O Adsorption analysis
Amine-based chemisorbents generally increase their CO2 adsorption capacity in the presence of water due to better diffusion,26,27 and improved amine efficiency from the formation of ammonium bicarbonate, theoretically enabling each amine to adsorb one molecule of CO2.58 The influence of water on the CO2 adsorption capacity of 10:1 (R) was measured using different techniques. Initially, an investigation using a TGA experiment was conducted in which the adsorbent was pre-humidified for almost four hours using a flow of wet argon at 30 °C, prior to contact with CO2, Fig. 14. The adsorbent took up a maximum of 0.207 g g−1 water after 94 minutes, then lost some of this over the next hour, but regained weight and began to stabilise until reaching hydration of 0.198 g H2O g−1 (11.00 mmol g−1). This was taken to be the maximum hydration or saturation adsorption of the material. On subsequent exposure to wet CO2, sorption was extremely rapid, reaching 0.342 g g−1 within 28 minutes, therefore CO2 accounting for 0.144 g g−1 (3.27 mmol g−1), exceeding the maximum adsorption under dry conditions (2.31 mmol g−1) by 0.96 mmol CO2 g−1. During four hours under wet CO2, the adsorption equilibrium appears to shift resulting in slight desorption followed by re-adsorption until the final total adsorption was 0.347 g g−1. Exposure to dry argon at 30 °C for eight hours reduced the adsorbed loading to 0.088 g g−1via mass displacement. On heating, complete desorption was achieved at 125 °C. Contrary to what we have previously observed in wet CO2 uptake experiments on crosslinked PEI materials,26,36 more adsorbate is removed (0.259 g g−1) under the low temperature desorption step than water is adsorbed during initial hydration. Therefore, it can be inferred that at least some CO2 is desorbed at low temperature, which may have been weakly chemisorbed or possibly physisorbed, while the remaining CO2 is strongly chemisorbed, most probably in the form of carbonate and bicarbonate species.
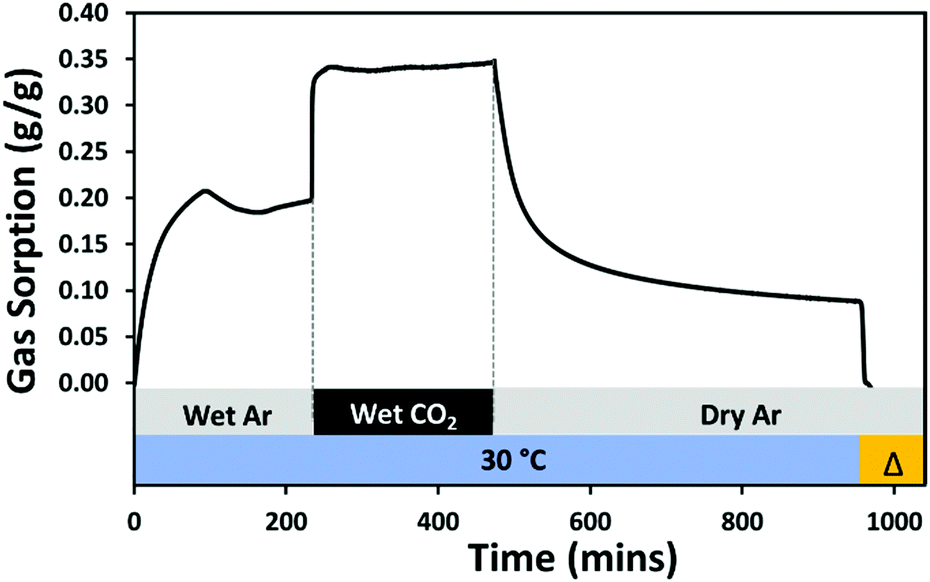 | ||
| Fig. 14 TGA–CO2 and H2O sorption (g g−1) of 10:1 (R) in humid environment at 30 °C, under 80 ml min−1 CO2, 10 ml min−1 Ar and 21.4% RH. | ||
An enhancement in CO2 uptake and adsorption kinetics was observed under humid conditions. Therefore, it was decided to further investigate the sorbent behaviour when exposed to water vapour using a DVS setup. Fig. 15 shows H2O uptake by 10:1 (R) at 30 and 40 °C at RH values between 0 and 95%. At both temperatures, an H2O isotherm type III was measured which is characteristic of monolayer–multilayer adsorption onto favourable sites,70 common in water adsorption by non-porous or macroporous amine adsorbents. The water capacity increases exponentially with RH, and largely independently of temperature. At 21% RH, the capture capacity measured by DVS was 1.84 mmol H2O vapour per g, higher than the typical water uptake reported for amine functionalised sorbents (1.10 mmol g−1).71 It can be seen that the amount of adsorbed H2O vapour obtained by DVS significantly differs from the water capacity measured by TGA at the same RH. However, comparisons between these experiments are not entirely direct. Firstly, the activation conditions are different which may significantly affect capture capacities. Secondly, the TGA analyser was operated at 1 atm, with the material exposed to both argon and water. For DVS the material was exposed only to water and the pressure inside the DVS chamber was kept at 0.015 bar to reach the desired 21% RH at 40 °C. Such high variation in the gas pressure may result in the reduction of the surface energy, leading to adsorption-induced strains, a deformation of solid surface in non-porous or macroporous materials, which can impact the amount of H2O adsorbed.72
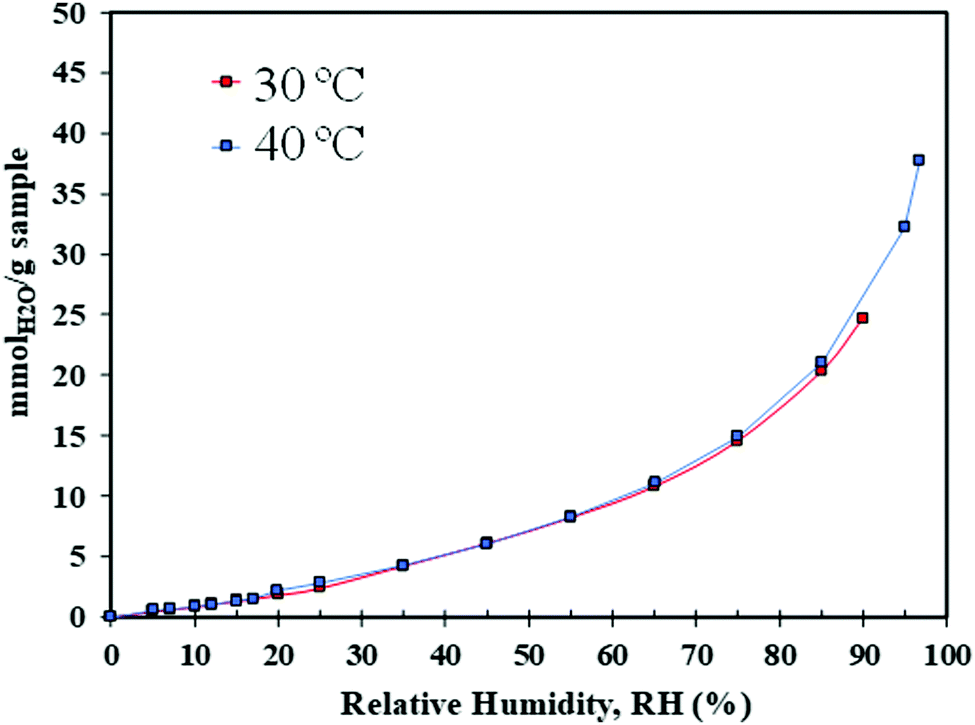 | ||
| Fig. 15 H2O isotherms of 10:1 (R) at 30 and 40 °C. | ||
As for 75% RH, (representing typical water content in post-combustion capture processes), 10:1 (R) adsorbs a high amount of water vapour at 14.55 mmol g−1, compared to the H2O capacity of the commercial amine-based adsorbent Lewatit® VP OC 1065.73 For practical CO2 capture applications it is vital to evaluate the effect of water on the CO2 capture performance of 10:1 (R).
Breakthrough experiments
The dynamic CO2 adsorption capacity of 10:1 (R) was measured at 40 °C, atmospheric pressure and 14 vol% CO2, under dry and wet conditions. The breakthrough curves of N2 and CO2 obtained for the dry experiment are presented in Fig. 16, where the outlet-inlet flow ratio of each gas versus time is shown. It can be observed that CO2 is the favoured adsorbed component whereas N2 is the more weakly adsorbed, as indicated by the difference in breakthrough times, i.e. time that it takes for the gas to be detected at the outlet of the reactor (3 minutes for N2versus 6 minutes for CO2). The breakthrough curve of N2 also shows that there is competitive adsorption between the two gas components, as indicated by the presence of a roll-up effect in the curve.74
The breakthrough curves of N2, H2O and CO2 obtained from wet experiments at 75% RH are presented in Fig. 17. Two different scenarios were tested: one in which the pristine sorbent is exposed to a stream of N2, H2O and CO2 (co-adsorption case), the other in which a pre-water saturated sorbent is exposed to the same stream (pre-saturated case). In the co-adsorption case (Fig. 17(a)), it can be observed that H2O is strongly and preferentially adsorbed, with a much longer breakthrough time (12 min) compared to those measured for CO2 and N2 (3 and 2 minutes respectively). The roll up effect in the N2 and CO2 curves also indicate competitive adsorption with H2O. The H2O breakthrough curve is far more extended than those of CO2 and N2, which are rather steep. In an ideal situation, when there are no hydrodynamic (axial dispersion) or kinetic (resistance to mass transfer) effects in the bed, the shape of the breakthrough curve or concentration front would be that of a perfect step change in concentration. In this case, the breakthrough curve would appear vertical, from a value of 0 to 1. When this is not the case, the steepness of the breakthrough curve gives an indication of the length of the mass transfer zone relative to the length of the bed.74 In the case of CO2 and N2, both curves are rather steep, indicating a shorter mass transfer zone. In the case of H2O, the mass-transfer and axial dispersion effects clearly influence the dynamics of adsorption, which is far slower than that of the other two gases. On the other hand, when the material is pre-saturated with H2O (Fig. 17(b)), the profiles for both H2O and N2 break simultaneously at about 1.5 minutes and no competitive adsorption is observed, with the CO2 breaking after about 2.5 minutes. In this case, there is not much difference between the slopes of the curves, although the H2O front is still slower than those of CO2 and N2. The CO2 capture capacities under dry and wet conditions were obtained from the breakthrough curves and are presented in Table 4.
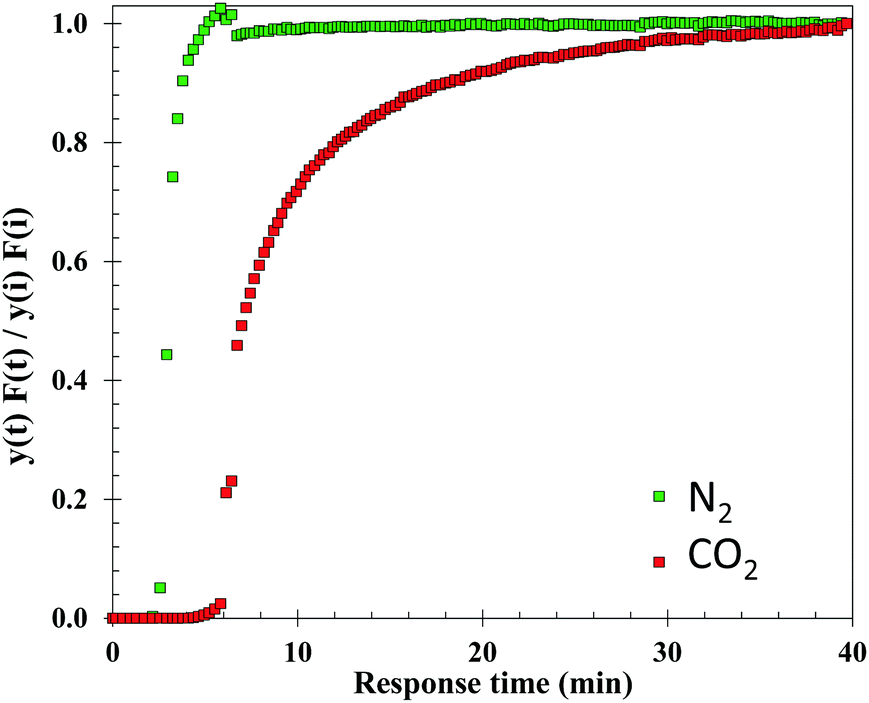 | ||
| Fig. 16 N2 and CO2 breakthrough curves of 10:1 (R) under dry conditions, atmospheric pressure, 40 °C and 14% vol. CO2/N2. | ||
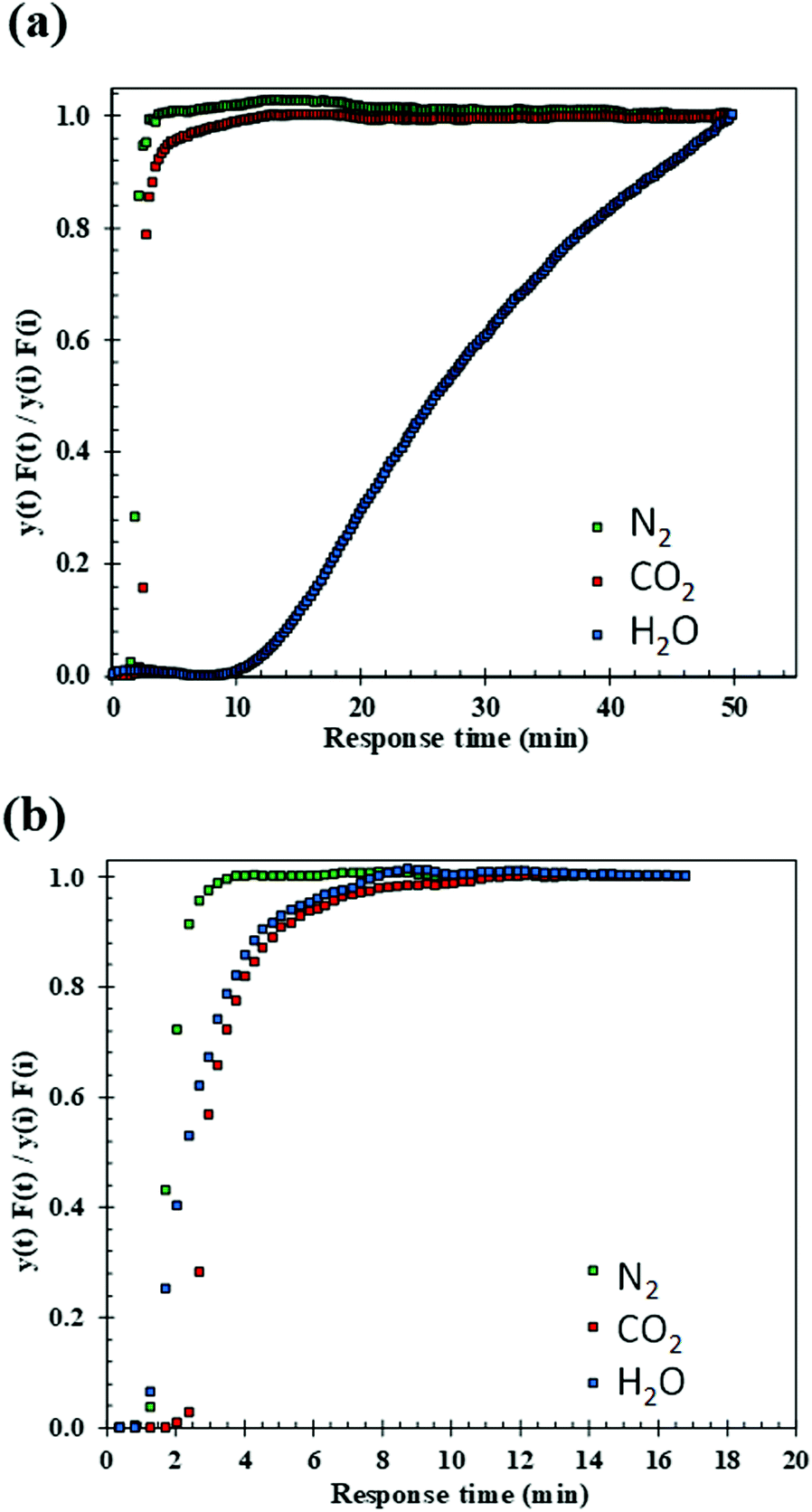 | ||
| Fig. 17 N2, H2O and CO2 breakthrough curves of 10:1 (R) under atmospheric pressure, 40 °C, 14 vol% CO2/N2, under: (a) wet co-adsorption conditions; (b) wet pre-saturation conditions, (75% RH in each case). | ||
:1 (R) under dry conditions and wet conditions (75% RH), with and without H2O pre-saturation
| CO2 capacity (mmol g−1) | |
|---|---|
| Dry conditions | 1.15 |
| Wet conditions co-adsorption | 0.2 |
| Wet conditions pre-saturation | 0.3 |
Under the dynamic breakthrough conditions of 40 °C, 14 vol% CO2/N2, 75% RH, 1 atm, the presence of water in the gas stream had a deleterious effect on the material's CO2 capture performance. In the breakthrough experiment, uptake reduced by up to 83% under wet conditions, with little difference in uptake observed between co-adsorption and pre-saturation conditions. Although studies on amine-based adsorbents generally report improved uptake under humid conditions, there are instances where there appears to be a threshold at which higher RH values do not translate into significant increases in CO2 uptake or can result in reduced adsorption. This has been reported for several adsorbents: a PEI-impregnated MCM-41 at 75 °C under simulated flue gas where CO2 uptake did not improve noticeably when RH exceeded 34%;75 a 50 wt% TEPA-modified silica adsorbent, when CO2 uptake under 10% CO2/N2 at 60 °C increased to a maximum at 37% RH;76 and for a 50 wt% PEI-loaded resin, where CO2 uptake under 5000 ppm reached a maximum at 40% RH and declined at 60% RH.77 At elevated humidity levels, such as 75% RH, reduced CO2 uptake has been attributed to competitive adsorption between water and CO2. It is established that CO2 capture by amine functionalised adsorbents in the presence of water takes place through the formation of different adsorbed species and can follow different mechanisms.58 One is through the formation of ammonium bicarbonate, and another is through the formation of hydronium carbonate in which carbamic acid is stabilised by a water molecule.78,79 However, hydronium formation may lead to the growth of a complex cluster of hydrogen-bonded solvating water molecules, which under high RH leads to the blocking of active sites to CO2, decreasing capture capacity. This was observed in a recent study on an amine functionalised adsorbent where the CO2 capture under wet conditions was not favoured, specifically as the RH and CO2 partial pressure increased.73
The observations in the dynamic breakthrough experiments are counter to what was observed by TGA, where humidity drastically improved CO2 uptake. The different observations between experiments may be attributed not only to the differences in RH, where TGA used a lower humidity, but by their different CO2 partial pressures. (Based on the DVS experiment, it is not believed that the differences in temperature, 30 °C for TGA, and 40 °C for the breakthrough, had a significant impact on the results.) The TGA experiment used a significantly higher concentration of CO2 in the feed gas, at approx. 90% CO2, versus 14% CO2 in the breakthrough experiment. The lower partial pressure of CO2 in the breakthrough experiment, along with the higher RH, may have resulted in the competition for adsorption sites being too high for efficient CO2 uptake, especially given that TGA results show drastically different maximum CO2 uptakes for 10:1 (R) at 30 °C under 90 and 10% CO2/N2, at 2.31 and 1.40 mmol g−1, respectively, under dry conditions. In the TGA experiment, it may be that the water content of the system was sufficient to benefit CO2 uptake, without out-competing CO2 for adsorption sites given the significantly higher partial pressure of CO2, at 90%.
Lastly, in addition to the differences in adsorption conditions in terms of the feed gas composition, high RH values may affect the hydrodynamics inside the breakthrough reactor, which could negatively affect the gas–solid interaction and lead to decreased capture capacity. This is an area worth further investigation.
Conclusions
A set of new unsupported solid polyamine-based CO2 adsorbents have been synthesised by cross-linking PEI with 2,4,6-tris-[4-(bromomethyl-3-fluoro)-phenyl]-1,3,5,-triazine. The ratio of amine to cross-linker in the products is consistently lower than the relative proportions used in the starting materials.The most densely cross-linked adsorbents (with less than one amine per alkyl group of the reacted alkyl bromide) show evidence of microporosity and have faster uptake of CO2, but lower equilibrium capacity compared with the less cross-linked adsorbents, with uptake under 90% CO2/Ar decreasing on increasing temperature from 30 °C–90 °C. The lesser cross-linked adsorbents (with more than one amine per alkyl group of the reacted alkyl bromide), 10:1 (V) and 25:1 (V), consistently show higher CO2 capacities due to their higher reactive amine content. Both perform most effectively at 60 °C, with 10:1 (V) obtaining CO2 uptake of 1.95 mmol g−1, and 25:1 (V) reaching 2.45 mmol g−1. The particles of 25:1 (V) appear highly fused forming a less penetrable material, giving rise to the diffusion barrier limiting its CO2 uptake at 30 °C.
Adjustment of the synthesis conditions of these adsorbents influences their structure and gas-diffusion properties and the number of accessible CO2-reactive amines. 10:1 (R) has a similar cross-linking density to 10:1 (V), yet it shows its highest adsorption of 2.31 mmol g−1 at 30 °C – the highest of all the adsorbents at this temperature, and with faster uptake than 10:1 (V) and 25:1 (V). The superior performance of 10:1 (R) is due to the accessibility of reactive amine: the adsorbent is composed of spherical particles up to 3 μm in size, the incomplete packing of which enables greater access to CO2 through the material. The non-fluorinated analogue of 10:1 (R) also shows its highest adsorption at 30 °C, with uptake of 2.09 mmol g−1, suggesting that the fluorination is less important compared to the overall structure and amine content for effective low temperature adsorption.
10:1 (R) exhibits good selectivity of CO2 over N2 of 575. In TGA experiments, under dilute CO2 multicomponent gas mixtures, 10:1 (R) reaches 1.50 mmol g−1 CO2 capacity under 10% CO2/N2, and up 0.20 mmol g−1 under 400 ppm CO2/N2. In 10% CO2/N2, uptake is faster at 60 °C, leading to higher adsorption at 60 °C than at 30 °C over a longer period than compared to adsorption under 90% CO2/Ar. The TGA experiment under humid conditions at 30 °C showed that CO2 adsorption of 10:1 (R) increased to 3.27 mmol g−1 with faster kinetics than under dry conditions. However, when the sample was further tested using a dynamic rig set-up at a high RH (75%), low CO2 partial pressure (14%), and slightly higher temperature (40 °C), the CO2 capacity drastically reduced, by up to 83% of the capacity under dry conditions. These findings suggest that the different water content in the gas streams, along with the differences in CO2 partial pressure, play a significant role in the material's performance. The results from the TGA do not allow us to accurately infer the performance of the adsorbent under the more closely replicated post-combustions provided by the dynamic rig set-up and comprehensive comparisons between the two techniques is worth further investigation.
From the thorough analysis of a set of unsupported cross-linked polyamine adsorbents, it is clear that with further development, these materials are becoming ever more credible candidates for application in industrial carbon capture technologies. Key to the success of this family of adsorbents is the selection of an effective cross-linker and tuning the cross-linking degree to optimise the structure for efficient CO2 diffusion. The novel cross-linker 2,4,6-tris-(4-bromomethyl-3-fluoro-phenyl)-1,3,5,-triazine, as a rigid, conjugated spacer unit, has proven successful in developing and optimising this unsupported polyamine-based material for high CO2 capture at ambient temperature.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is part of the Flexible Integrated Energy Systems (FLEXIS) and Reducing Industrial Carbon Emissions (RICE) research operations funded by the Welsh European Funding Office (WEFO) through the Welsh Government. Support was provided by the Engineering and Physical Sciences Research Council through the SUSTAIN Manufacturing Hub EP/S018107/1. Financial support was also provided by the Sêr Cymru Chair Programme and the Robert A. Welch Foundation (C-0002). We also acknowledge funding from European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no 663830. The authors would like to acknowledge Stephen Shearan for assistance with material synthesis and Dr Matthew J. McPherson for assistance in performing BET measurements. We would like to acknowledge the assistance provided by Swansea University College of Engineering AIM Facility, which was funded in part by the EPSRC (EP/M028267/1), the European Regional Development Fund through the Welsh Government (80708) and the Ser Solar project via the Welsh Government. We acknowledge postdoctoral fellowship funding from the German Academic Exchange Service (DAAD) and Leibniz Association for WYC. This work benefited from access to the FMP Berlin NMR facility. This work was also supported in part by the PrISMa Project (299659), funded through the ACT Programme (Accelerating CCS Technologies, Horizon 2020 Project 294766).References
- IPCC, 2018: Global Warming of 1.5 °C. An IPCC Special Report on the impacts of global warming of 1.5 °C above pre-industrial levels and related global greenhouse gas emission pathways, in the context of strengthening the global response to the threat of climate change, sustainable development, and efforts to eradicate poverty [Masson-Delmotte, V., P. Zhai, H.-O. Pörtner, D. Roberts, J. Skea, P.R. Shukla, A. Pirani, W. Moufouma-Okia, C. Péan, R. Pidcock, S. Connors, J.B.R. Matthews, Y. Chen, X. Zhou, M.I. Gomis, E. Lonnoy, T. Maycock, M. Tignor, and T. Waterfield (eds.)]. In Press.
- H. A. Patel, J. Byun and C. T. Yavuz, ChemSusChem, 2017, 10, 1303–1317 CrossRef CAS PubMed.
- P. Luis, Desalination, 2016, 380, 93–99 CrossRef CAS.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef CAS PubMed.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- A. Samanta, A. Zhao, G. K. H. Shimizu, P. Sarkar and R. Gupta, Ind. Eng. Chem. Res., 2012, 51, 1438–1463 CrossRef CAS.
- S. D. Kenarsari, D. Yang, G. Jiang, S. Zhang, J. Wang, A. G. Russell, Q. Wei and M. Fan, RSC Adv., 2013, 3, 22739–22773 RSC.
- B. Dutcher, M. Fan and A. G. Russell, ACS Appl. Mater. Interfaces, 2015, 7, 2137–2148 CrossRef CAS PubMed.
- European Academies’ Science Advisory Council, Negative emissions technologies: what role in meeting paris agreement targets?, EASAC policy report, 35 Search PubMed.
- R. Socolow, M. Desmond, R. Aines, J. Blackstock, O. Bolland, T. Kaarsberg, N. Lewis, M. Mazzotti, A. Pfeffer and K. Sawyer, Direct air capture of CO2 with chemicals: a technology assessment for the APS Panel on Public Affairs, American Physical Society, 2011 Search PubMed.
- C. Beuttler, L. Charles and J. Wurzbacher, Front. Clim., 2019, 1, 00010 CrossRef.
- M. Samari, F. Ridha, V. Manovic, A. Macchi and E. J. Anthony, Mitigation and Adaptation Strategies for Global Change, 2019, pp. 1–17 Search PubMed.
- A. R. Sujan, S. H. Pang, G. Zhu, C. W. Jones and R. P. Lively, ACS Sustainable Chem. Eng., 2019, 7, 5264–5273 CrossRef CAS.
- S. E. Park, J. S. Chang and K. W. Lee, Carbon Dioxide Utilization for Global Sustainability: Proceedings of the 7th International Conference on Carbon Dioxide Utilization, Seoul, Korea, October 12–16, 2003, Elsevier Science, 2004 Search PubMed.
- A. Chakma, Energy Convers. Manage., 1997, 38, S51–S56 CrossRef CAS.
- S. A. Mazari, L. Ghalib, A. Sattar, M. M. Bozdar, A. Qayoom, I. Ahmed, A. Muhammad, R. Abro, A. Abdulkareem, S. Nizamuddin, H. Baloch and N. M. Mubarak, Int. J. Greenhouse Gas Control, 2020, 96, 103010 CrossRef CAS.
- J. C. Meerman, E. S. Hamborg, T. van Keulen, A. Ramírez, W. C. Turkenburg and A. P. C. Faaij, Int. J. Greenhouse Gas Control, 2012, 9, 160–171 CrossRef CAS.
- A. R. Sujan, D. R. Kumar, M. Sakwa-Novak, E. W. Ping, B. Hu, S. J. Park and C. W. Jones, ACS Appl. Polym. Mater., 2019, 1, 3137–3147 CrossRef CAS.
- C. J. E. Bajamundi, J. Koponen, V. Ruuskanen, J. Elfving, A. Kosonen, J. Kauppinen and J. Ahola, J. CO2 Util., 2019, 30, 232–239 CrossRef.
- C. Gebald, J. A. Wurzbacher, P. Tingaut, T. Zimmermann and A. Steinfeld, Environ. Sci. Technol., 2011, 45, 9101–9108 CrossRef CAS PubMed.
- A. Goeppert, M. Czaun, R. B. May, G. K. S. Prakash, G. A. Olah and S. R. Narayanan, J. Am. Chem. Soc., 2011, 133, 20164–20167 CrossRef CAS PubMed.
- W. Chaikittisilp, R. Khunsupat, T. T. Chen and C. W. Jones, Ind. Eng. Chem. Res., 2011, 50, 14203–14210 CrossRef CAS.
- A. Heydari-Gorji, Y. Belmabkhout and A. Sayari, Langmuir, 2011, 27, 12411–12416 CrossRef CAS PubMed.
- M. Niu, H. Yang, X. Zhang, Y. Wang and A. Tang, ACS Appl. Mater. Interfaces, 2016, 8, 17312–17320 CrossRef CAS PubMed.
- W. Chaikittisilp, H.-J. Kim and C. W. Jones, Energy Fuels, 2011, 25, 5528–5537 CrossRef CAS.
- A. Koutsianos, A. R. Barron and E. Andreoli, J. Phys. Chem. C, 2017, 121, 21772–21781 CrossRef CAS.
- H. T. Kwon, M. A. Sakwa-Novak, S. H. Pang, A. R. Sujan, E. W. Ping and C. W. Jones, Chem. Mater., 2019, 31, 5229–5237 CrossRef CAS.
- D. V. Quang, A. Dindi, A. V. Rayer, N. E. Hadri, A. Abdulkadir and M. R. M. Abu-Zahra, Greenhouse Gases: Sci. Technol., 2015, 5, 91–101 CrossRef CAS.
- M. Khraisheh, S. Mukherjee, A. Kumar, F. Al Momani, G. Walker and M. J. Zaworotko, J. Environ. Manage., 2020, 255, 109874 CrossRef CAS PubMed.
- M. E. Potter, S. H. Pang and C. W. Jones, Langmuir, 2017, 33, 117–124 CrossRef CAS PubMed.
- C. A. Hibbitts and J. Szanyi, Icarus, 2007, 191, 371–380 CrossRef CAS.
- P. Li, S. Zhang, S. Chen, Q. Zhang, J. Pan and B. Ge, J. Appl. Polym. Sci., 2008, 108, 3851–3858 CrossRef CAS.
- S. Jeon, H. Jung, D. H. Jo and S. H. Kim, Energy Procedia, 2017, 114, 2287–2293 CrossRef CAS.
- H. Jung, S. Jeon, D. H. Jo, J. Huh and S. H. Kim, Chem. Eng. J., 2017, 307, 836–844 CrossRef CAS.
- D. R. Kumar, C. Rosu, A. R. Sujan, M. A. Sakwa-Novak, E. W. Ping and C. W. Jones, ACS Sustainable Chem. Eng., 2020, 8, 10971–10982 CAS.
- L. B. Hamdy, R. J. Wakeham, M. Taddei, A. R. Barron and E. Andreoli, Chem. Mater., 2019, 31(13), 4673–4684 CrossRef CAS.
- C.-J. Yoo, P. Narayanan and C. W. Jones, J. Mater. Chem. A, 2019, 7, 19513–19521 RSC.
- E. Andreoli and A. R. Barron, J. Mater. Chem. A, 2015, 3, 4323–4329 RSC.
- E. Andreoli and A. R. Barron, Energy Fuels, 2015, 29, 4479–4487 CrossRef CAS.
- E. Andreoli, L. Cullum and A. R. Barron, Ind. Eng. Chem. Res., 2015, 54, 878–889 CrossRef CAS.
- E. Andreoli and A. R. Barron, ChemSusChem, 2015, 8, 2635–2644 CrossRef CAS PubMed.
- E. P. Dillon, E. Andreoli, L. Cullum and A. R. Barron, J. Exp. Nanosci., 2015, 10, 746–768 CrossRef CAS.
- E. Andreoli, E. P. Dillon, L. Cullum, L. B. Alemany and A. R. Barron, Sci. Rep., 2014, 4, 7304 CrossRef CAS PubMed.
- K.-S. Hwang, H.-Y. Park, J.-H. Kim and J.-Y. Lee, Korean J. Chem. Eng., 2018, 35, 798–804 CrossRef CAS.
- S. J. Thompson, M. Soukri and M. Lail, Chem. Eng. J., 2018, 350, 1056–1065 CrossRef CAS.
- X. Xu, B. Pejcic, C. Heath and C. D. Wood, J. Mater. Chem. A, 2018, 6, 21468–21474 RSC.
- H.-B. Wang, P. G. Jessop and G. Liu, ACS Macro Lett., 2012, 1, 944–948 CrossRef CAS.
- J. Huang, J. Zhu, S. A. Snyder, A. J. Morris and S. R. Turner, Polymer, 2018, 154, 55–61 CrossRef CAS.
- L.-B. Sun, Y.-H. Kang, Y.-Q. Shi, Y. Jiang and X.-Q. Liu, ACS Sustainable Chem. Eng., 2015, 3, 3077–3085 CrossRef CAS.
- S. Mane, Y.-X. Li, D.-M. Xue, X.-Q. Liu and L.-B. Sun, Ind. Eng. Chem. Res., 2018, 57, 12926–12934 CrossRef CAS.
- S. J. Thompson, M. Soukri and M. Lail, Energy Fuels, 2018, 32, 8658–8667 CrossRef CAS.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291–1295 CrossRef CAS.
- M. R. Liebl and J. Senker, Chem. Mater., 2013, 25, 970–980 CrossRef CAS.
- M. Taddei, F. Costantino, F. Marmottini, A. Comotti, P. Sozzani and R. Vivani, Chem. Commun., 2014, 50, 14831–14834 RSC.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies: Tables and Charts, Wiley, 2001 Search PubMed.
- A. Saha, Int. J. Greenhouse Gas Control, 2018, 78, 198–209 CrossRef CAS.
- W. Xie, M. Yu and R. Wang, Aerosol Air Qual. Res., 2017, 17, 2715–2725 CrossRef CAS.
- P. Bollini, S. A. Didas and C. W. Jones, J. Mater. Chem., 2011, 21, 15100–15120 RSC.
- F. Rezaei, M. A. Sakwa-Novak, S. Bali, D. M. Duncanson and C. W. Jones, Microporous Mesoporous Mater., 2015, 204, 34–42 CrossRef CAS.
- A. Koutsianos, L. B. Hamdy, C.-J. Yoo, J. J. Lee, M. Taddei, J. M. Urban-Klaehn, J. Dryzek, C. W. Jones, A. R. Barron and E. Andreoli, J. Mater. Chem. A, 2021, 9, 10827–10837 RSC.
- K. S. Walton and D. S. Sholl, AIChE J., 2015, 61, 2757–2762 CrossRef CAS.
- H.-M. Wen, C. Liao, L. Li, A. Alsalme, Z. Alothman, R. Krishna, H. Wu, W. Zhou, J. Hu and B. Chen, J. Mater. Chem. A, 2019, 7, 3128–3134 RSC.
- Y. Yang, C. Y. Chuah and T.-H. Bae, Chem. Eng. J., 2019, 358, 1227–1234 CrossRef CAS.
- G. Zainab, N. Iqbal, A. A. Babar, C. Huang, X. Wang, J. Yu and B. Ding, Compos. Commun., 2017, 6, 41–47 CrossRef.
- Y. He, Y. Xia, J. Zhao, Y. Song, L. Yi and L. Zhao, Appl. Phys. A: Mater. Sci. Process., 2019, 125, 160 CrossRef CAS.
- A. Goeppert, H. Zhang, R. Sen, H. Dang and G. K. S. Prakash, ChemSusChem, 2019, 12, 1712–1723 CrossRef CAS PubMed.
- C. Knöfel, J. Descarpentries, A. Benzaouia, V. Zeleňák, S. Mornet, P. L. Llewellyn and V. Hornebecq, Microporous Mesoporous Mater., 2007, 99, 79–85 CrossRef.
- A. Goeppert, H. Zhang, M. Czaun, R. B. May, G. K. Prakash, G. A. Olah and S. R. Narayanan, ChemSusChem, 2014, 7, 1386–1397 CrossRef CAS PubMed.
- S. Choi, J. H. Drese, P. M. Eisenberger and C. W. Jones, Environ. Sci. Technol., 2011, 45, 2420–2427 CrossRef CAS PubMed.
- M. Thommes, K. Kaneko, A. V. Neimark, J. P. Olivier, F. Rodriguez-Reinoso, J. Rouquerol and K. S. W. Sing, Pure Appl. Chem., 2015, 87, 1051–1069 CAS.
- W. Jung and K. S. Lee, J. Nat. Gas Sci. Eng., 2020, 84, 103489 CrossRef CAS.
- G. Y. Gor, P. Huber and N. Bernstein, Appl. Phys. Rev., 2017, 4, 011303 Search PubMed.
- J. Young, E. García-Díez, S. Garcia and M. van der Spek, Energy Environ. Sci., 2021, 14, 5377–5394 RSC.
- N. S. Wilkins, A. Rajendran and S. Farooq, Adsorption, 2021, 27, 397–422 CrossRef CAS.
- X. Xu, C. Song, B. G. Miller and A. W. Scaroni, Ind. Eng. Chem. Res., 2005, 44, 8113–8119 CrossRef CAS.
- Y. Liu, Q. Ye, M. Shen, J. Shi, J. Chen, H. Pan and Y. Shi, Environ. Sci. Technol., 2011, 45, 5710–5716 CrossRef CAS PubMed.
- J. Wang, M. Wang, W. Li, W. Qiao, D. Long and L. Ling, AIChE J., 2015, 61, 972–980 CrossRef CAS.
- S. A. Didas, M. A. Sakwa-Novak, G. S. Foo, C. Sievers and C. W. Jones, J. Phys. Chem. Lett., 2014, 5, 4194–4200 CrossRef CAS PubMed.
- J. Yu and S. S. C. Chuang, Energy Fuels, 2016, 30, 7579–7587 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma01072g |
| ‡ Present address (LBH): CGG, Tyn-y-Coed, Pentywyn Road, Llandudno, LL30 1SA, UK. |
| § Present address (WYC): Department of Physics, University of Warwick, Coventry CV4 7AL, UK. |
| ¶ Present address (MT): Department of Chemistry and Industrial Chemistry, University of Pisa, Via Giuseppe Moruzzi, 13, Pisa, 56124, Italy. |
| This journal is © The Royal Society of Chemistry 2022 |