
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A computational and experimental investigation of deep-blue light-emitting tetraaryl-benzobis[1,2-d:4,5-d′]oxazoles†
D.
Wheeler
*a,
S.
Tannir
 a,
E.
Smith
b,
A.
Tomlinson
b and
M.
Jeffries-EL
*ac
a,
E.
Smith
b,
A.
Tomlinson
b and
M.
Jeffries-EL
*ac
aDepartment of Chemistry, Boston University, Boston, MA 02215, USA. E-mail: malikaj@bu.edu
bDepartment of Chemistry and Biochemistry, University of North Georgia, Dahlonega, GA 30041, USA
cDivision of Materials Science and Engineering, Boston University, Boston, MA 02215, USA
First published on 10th February 2022
Abstract
In an effort to design deep-blue light emitting materials for use in OLEDs, the optical and electronic properties of a series of tetraarylbenzobis[1,2-d:4,5-d′]oxazole (BBO) cruciforms were evaluated using density functional theory (DFT) and time-dependent DFT (TD-DFT). Of the nine possible combinations of phenyl-, furan-2-yl-, and thiophen-2-yl-substituted BBO cruciforms, five were predicted to have ideal optical and electronic properties for use in blue-light emitting diodes. These five cruciforms were synthesized and then characterized electrochemically and spectroscopically. Additionally, they were solution-processed into functional organic light-emitting diodes (OLED). Several of the OLEDs exhibited deep-blue EL (λEL < 452 nm; CIEy ≤ 0.12) with maximum luminance efficacies reaching 0.39 lm W−1 and maximum current efficiencies of 0.59 cd A−1. A comparison of identical device architectures showed that heterocycles such as furan and thiophene helped improve device efficiencies with only a minor red-shift of the electroluminescence (EL). Although these BBO cruciforms produced the desired deep-blue emission their modest performance in host–guest OLEDs demonstrates the incorporation of heterocycles onto the BBO cruciform motif is detrimental to the fluroescence quantum yield. These results add to the knowledge base on structure–property relationships that will inform the design of better blue emitting materials.
Introduction
In the past four decades, the escalation of organic semiconductor (OSC) research has led to the discovery of numerous materials with properties suitable for use in applications such as photovoltaics,1–4 electrochromics,5–8 and organic light-emitting diodes (OLED)s.9–11 As a result, these OSCs can be used to replace the expensive, sometimes toxic, inorganic materials currently used in modern technology. The semiconducting properties of OSC is a result of the extensive delocalization of the π-electrons within these materials. One positive aspect of OSC is that the opto-electronic properties can be fine-tuned via structural modification for specific applications. As a result, understanding the relationship between and OSCs structure and it's properties is imperative for developing a useful material.Recently, there has been renewed interest in OLEDs due to the advent of new display and lighting applications. OLEDs have several advantages over conventional inorganic LEDs. For example, OLED displays can be fabricated via solution-processing techniques like inkjet printing, eliminating the need for thermal evaporation of various layers. This enables the facile production of large area devices with lower fabrication costs.12 OLEDs can convert up to 100% of injected charges into photons, maximizing light output while minimizing the amount of energy wasted as thermal radiation.13 Furthermore, unlike inorganic LEDs, OLEDs do not require the use of backlighting and thus can achieve the high contrast ratio that is considered in the industry as “true-black”.14 As a result of these advantages, OLEDs are currently being used in cell phones and displays.13
While OLEDs have a bright future for use in consumer applications, their operational lifetimes impede the wide-spread commercial-utility of this technology. It is common for blue-emissive materials to degrade an order of magnitude faster than both the red and green emissive materials.15 This is due to several factors including the greater amount of heat generated by blue-fluorescent OLEDs and the high S1 energy level of the blue fluorescent materials (∼3.0 eV). This is higher than the C–P bond energy and equal to the energy of the C–N bond favoring bond homolysis.15,16 As most display and lighting technologies rely on a combination of different colors to produce a gamut of detectable chromaticities, the degradation of one component renders the device unusable. While there have been some recent breakthroughs, such as the development of hyperfluorescent OLEDs,17,18 the development of electrochemically stable deep-blue OLEDs remains challenge.
Our group has focused on the development of novel organic semiconductors based on benzobis[1,2-d;4,5-d′]oxazole (BBOs) cruciforms and their application in organic electronics.19–22 These materials are comprised of four aryl-substituents along the central benzene ring (4,8-(2,6-position/axis)), Fig. 1.23,24 This arrangement creates spatially segregated frontier molecular orbitals (FMOs), facilitating the nearly independent tuning of the HOMO and LUMO levels, respectively using aromatic substitution.24–26 Using steric hindrance, the HOMO and LUMO energies can be further altered for better alignment between various charge-transport layers without altering the optical band gap (Eoptg).21 These substituents can be further functionalized with alkyl-chains to increase their solubility for solution-processing capabilities. Materials based on this heterocycle are also known to have high thermal decomposition temperatures and excellent oxidative stability.27–30 While the tunable nature of cruciforms makes them exciting candidates for use in a myriad of technologies,24–26 the BBO moiety shows great promise for use in OLED application.31–36 Previously, we reported a series of BBO cruciforms bearing phenyl- and fluoren-2-yl-substituents, which displayed deep-blue emission with maximum luminance efficiencies as high as ∼2 cd A−1 and Commission Internationale de L’Eclairage (CIE) coordinates of (0.15 ≤ x ≤ 0.17, 0.05 ≤ y ≤ 0.11).32
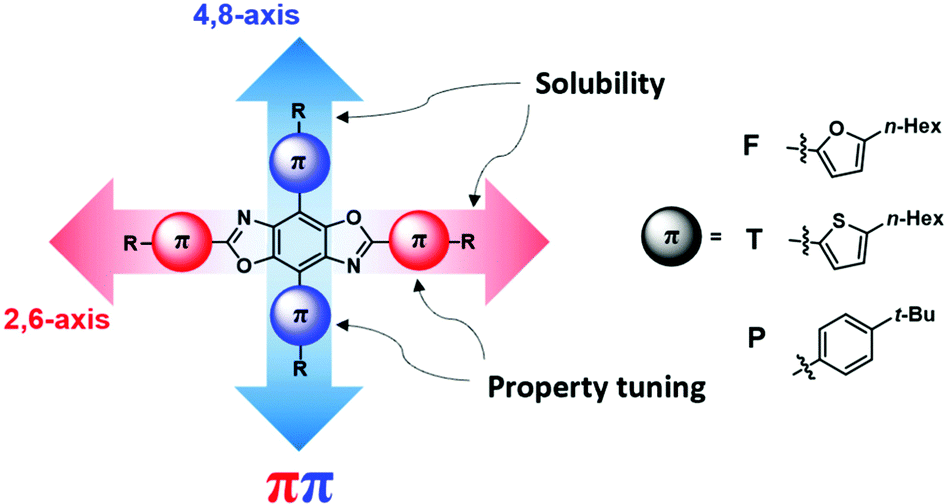 | ||
| Fig. 1 General structure of benzobis[1,2-d:4,5-d′]oxazole cruciforms with aryl substituents used in the study. | ||
To improve upon the cruciform design motif, we have designed a series of nine BBO cruciforms bearing all possible pairings using phenyl-, furan-2-yl-, or thiophene-2-yl-substituents at the two axes. Benzenoid compounds have been shown to produce wide-band gap materials. OSCs incorporating the heterocycle furan have been shown to be highly fluorescent materials.37–39 Whereas, thiophene-containing materials are known to exhibit high electrochemical stabilities and good charge-transport properties for a variety of electronic technologies.40,41 While benzene and thiophene-containing BBOs have been synthesized, these materials have not been investigated in active OLED devices. Herein, we report the theoretical and experimental findings of a series of BBO cruciforms and their performance in solution-processed OLEDs.
Results and discussion
Computational predictions
To identify the most promising candidates (those with Eoptg greater than 2.80 eV), density functional theory (DFT) and time-dependent DFT (TD-DFT) computations were performed at the mPW3PBE/SV level.21 All simulated spectra can be found in the supplemental information section along with detailed excited state analyses. The calculated electronic energy levels, FMOs assigned with percentage of electron location, and ground state dihedral angles are collated in Table 1. A discussion of calculated LUMO energies is not included due to the variability in theoretical modelling of electron affinity.21| Energy Levels (eV) | Localization (%) | Dihedral Angles (°) | ||||||
|---|---|---|---|---|---|---|---|---|
| HOMO | LUMO | |||||||
| BBO | HOMO | E optg | 4,8 | 2,6 | 4,8 | 2,6 | 4,8 | 2,6 |
| PP | −5.94 | 3.17 | 84.4 | 15.6 | 28.8 | 71.2 | 153.8 | 176.9 |
| PF | −5.57 | 2.83 | 95.8 | 4.2 | 39.1 | 60.9 | 180.0 | 180.0 |
| PT | −5.64 | 2.79 | 95.8 | 4.2 | 48.8 | 51.2 | 180.0 | 180.0 |
| FP | −5.82 | 2.96 | 81.4 | 18.6 | 28.4 | 71.6 | 180.0 | 180.0 |
| FF | −5.53 | 2.78 | 92.0 | 8.0 | 29.7 | 70.3 | 180.0 | 180.0 |
| FT | −5.61 | 2.74 | 92.9 | 7.1 | 44.8 | 55.2 | 180.0 | 180.0 |
| TP | −5.91 | 2.97 | 73.8 | 26.2 | 18.1 | 81.9 | 157.0 | 178.2 |
| TF | −5.56 | 2.66 | 91.6 | 8.4 | 20.5 | 79.5 | 180.0 | 180.0 |
| TT | −5.64 | 2.63 | 92.5 | 7.5 | 32.1 | 67.9 | 180.0 | 180.0 |
The structure–property study began with an examination of the HOMO level. The two lowest HOMO levels were PP and TP both of which were the only two which possessed non-planar structures with an out-of-plane contortion along the 4,8-axis of 26.2° and 23.0°, respectively. The lack of planarity in these two cruciforms produced levels that were ≥ 0.09 eV lower than the third lowest lying HOMO exhibited by FP. Overall, placement of the phenyl ring along the 4,8-axis produced the lowest levels likely due to the strong aromaticity of the benzene. Additionally, substitutions along the 4,8-axis produced HOMO levels differences of 0.37 eV (PP, PF, PT), 0.28 eV (FP, FF, FT) and 0.35 eV (TP, TF, TT) thereby suggesting that the HOMO level may be tuned utilizing this axis. Irrespective of the aryl group identity at the 4,8-axis, the HOMO level was most stabilized by a phenyl ring followed by thiophenyl and finally furanyl with differences ranging from 0.03 to 0.12 eV which indicates that changes along the 2,6-axis do not significantly impact the HOMO level. Finally, in all cases the electron density for the HOMO was localized along the 4,8-axis with PF and PP demonstrating the largest percentage of localization at 95.8% (Fig. 2).
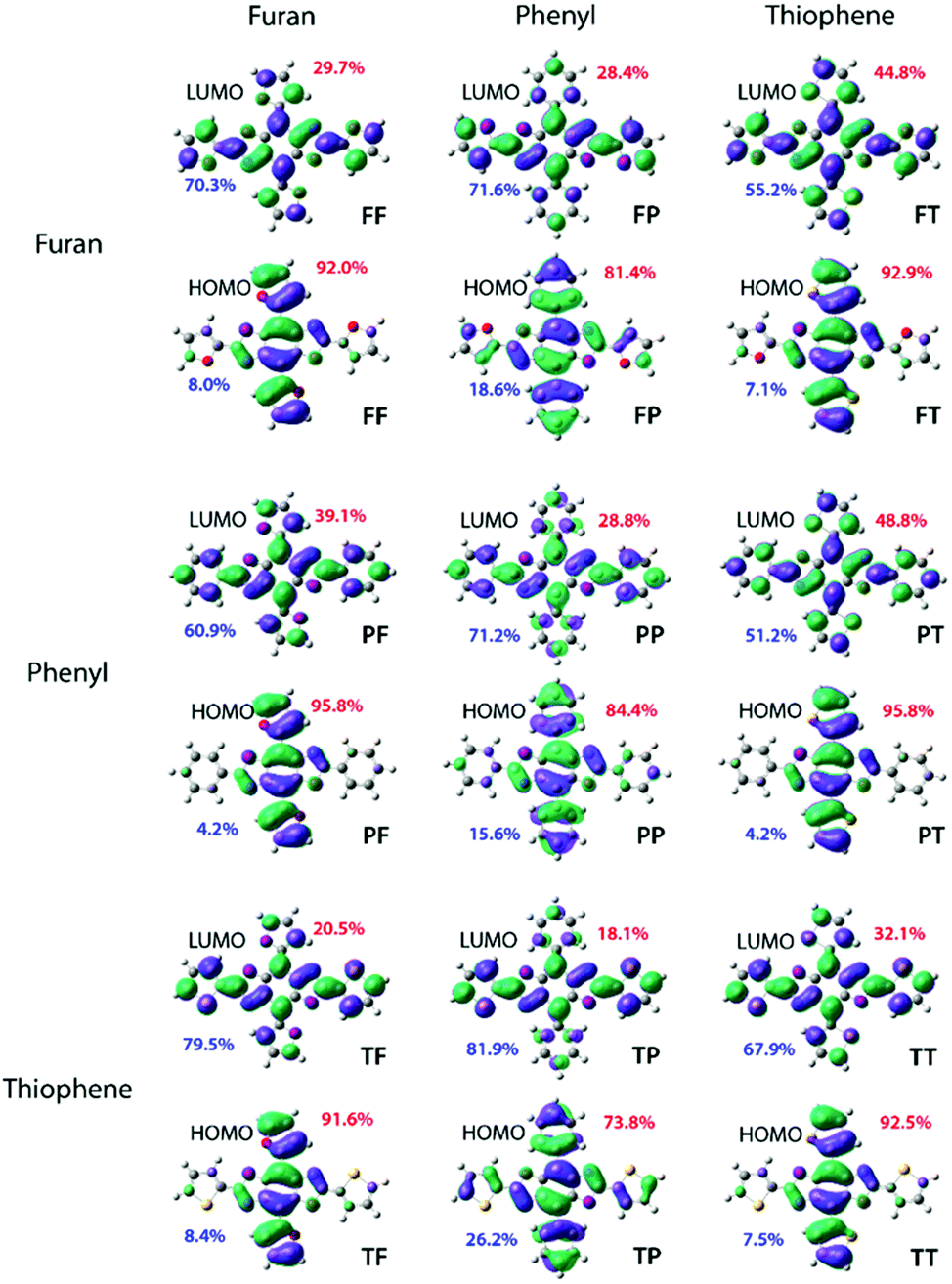 | ||
| Fig. 2 FMOs of the nine computationally studied systems. Compound names are listed bottom right of each structure. Percentages in red correspond to the total electron density that lies along the 4,8-axis while that in blue quantifies the 2,6-axis. | ||
Compounds bearing thiophene at the 4,8-positions have an average HOMO of −5.63 eV while those with furan are slightly increased to approximately −5.55 eV. Since all cruciforms without benzene rings at the 4,8-axis are predicted to be completely planar, this indicates the energy differences are solely due to aryl-substituent identity. Thus, the slight increase in energy for cruciforms with furan at the 4,8-axis is most likely due to the decreased aromaticity of the furan unit.
Based on the calculated band gap (Eoptg) there were several promising deep-blue emissive candidates. Of the nine cruciforms, those bearing benzene rings produced the largest Eoptg. Three cruciforms (PP, TP, and FP) had band gaps ranging from 2.96–3.17 eV. Moreover, the simulated UV-Vis spectra for these compounds all show very little tailing in the visible spectrum, further supporting their classification as potential deep-blue emitters. Two other compounds, PT and PF, were found to exhibit Eoptg values close to the lower limit of blue-emitters (2.79–2.83 eV) with minor absorption in the visible spectrum. Although their structural isomers show more promise as deep-blue dopants, these BBOs were also included in the study to investigate structure–property relationships. The other cruciforms had smaller Eoptg (2.78–2.63 eV), thereby red-shifting the absorbance profiles. Thus, TT, TF, TF, and FF were omitted as synthetic targets.
Synthesis
The synthetic pathways for the functional arenes are shown in the ESI.† All five BBOs were synthesized using a tandem approach: a condensation reaction to introduce aryl substituents along the 2,6-axis followed by a cross-coupling reaction to attach substituents at the 4,8-axis. The formation of the final BBOs is outlined in Scheme 1. First, 2,5-diamino-3,6-dibromo-p-hydroquinone was condensed with either the benzoic acid to form 26P48Br or orthoesters to form 26F48Br, and 26T48Br. The benzene substituent was coupled along 4,8-axis using traditional Suzuki–Miyaura cross-coupling conditions, providing compounds FP, TP, and PP in good yields. The furan and thiophene units were attached to the 4,8-positions using Stille cross-coupling techniques, yielding PF and PT in good yields. Upon purification, all compounds were found to be moderately soluble in chloroform and characterized by 1H and 13C NMR spectroscopy, HR-MS (ESI), thermogravimetric analysis (TGA), and melting point determination.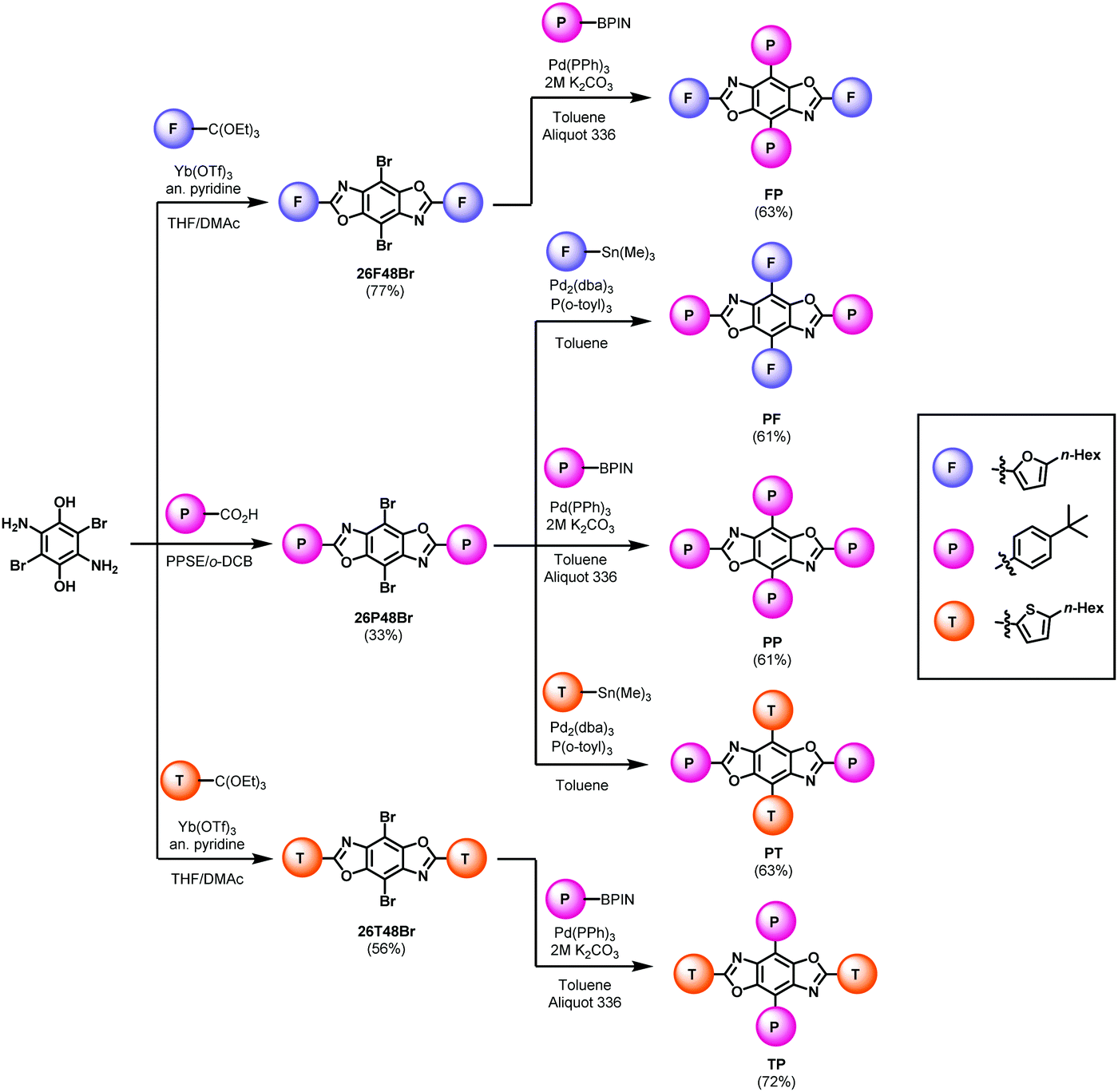 | ||
| Scheme 1 Synthesis of the final BBO cruciform. | ||
Electronic and optical properties
The experimental electronic and optical properties of all synthesized compounds were investigated using cyclic voltammetry, UV-Vis spectroscopy, and photoluminescence spectroscopy in solution and thin film. All pertinent data can be found in Table 2. The electrochemical oxidation potentials (Fig. S25–S29, ESI†) were used to calculate the HOMO energy while the LUMO level was found by adding the onset of absorbance to the HOMO level. The HOMO levels of each material range from −5.74 to −5.25 eV while the LUMO energies range from −2.68 to −2.52 eV. It was observed that when the phenyl substituents are placed through the 4,8-axis, the HOMO energy was lowered when compared to the placement of electron-rich substituents, such as furan and thiophene. Moreover, the solution-state voltammograms indicate good stability of most BBOs as seen by the reproducibility of three consecutive redox cycles. However, PF a exhibits slight change with each sequential redox cycle, indicating this material exhibits the highest level of instability of the set.| Electronic | Optical | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Solution State | Solid State | ||||||||||
| BBO | HOMOa (eV) | LUMOb (eV) |
E
optg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) c (eV) c (eV) |
εd (104) (M−1 cm−1) |
λ
absmaxe [CF] (nm) |
λ
emmaxe [CF] (nm) |
λ
emmaxe [Tol] (nm) |
Lifetimef (ns) | Φ (%) |
λ
absmaxe (nm) |
λ
emmaxe (nm)e |
| a HOMO values were determined from onset of oxidation using cyclic voltammetry and compared to a ferrocene standard. b LUMO = HOMO + Eoptg. c Optical band gaps determined from onset of absorbance in solution. d Experimental absorption coefficients were calculated from peak maxima at concentrations of 0.01 g L−1, 0.005 g L−1, and 0.0017 g L−1. e Bold values indicate peak of maximum absorbance/emission. Values with * following indicate spectral shoulders. f Acquired in chloroform. g Found in chloroform. | |||||||||||
| PP | −5.74 | −2.60 | 3.14 | 7.0 | 291, 354 | 422, 406 | 427, 408 | 1.96 | 44 | 299, 362 | 445 |
| PF | −5.25 | −2.52 | 2.73 | 5.1 | 371, 355, 321, 303 | 478*, 453 | 494, 472 | 1.94 | 22 | 388, 300 | 546 |
| PT | −5.28 | −2.55 | 2.73 | 5.4 | 407, 377, 359, 330, 318 | 479, 451 | 472, 445 | 2.20 | 26 | 418, 380, 365, 334, 277 | 569*, 544 |
| FP | −5.65 | −2.59 | 3.06 | 6.4 | 390, 371, 356 | 440, 413 | 459*, 430, 410 | 1.38 | 60 | 402, 380, 364, 301 | 489, 461* |
| TP | −5.65 | −2.68 | 2.97 | 6.2 | 402, 382, 365, 306 | 456, 432 | 471*, 444, 421 | NA | 45 | 414, 390, 372, 309, 266 | 458 |
The experimental UV-Vis results show very good correlation to the theoretical model, see ESI.† Due to the limited solubility of these compounds, all five BBOs were able to be dissolved in either toluene or chloroform as 0.01 gL−1 equimolar solutions. The peak maxima for each BBO show very little difference (<6 nm) between the two solvent media (see ESI†), suggesting that all 5 BBOs exhibit more locally excited (LE) character between the ground and excited states. In chloroform, there is a distinct pattern between aryl-substitution and absorbance profiles, Fig. 3. The spectrum for PP exhibits a peak maximum centred at 354 nm with small spectral features contributing to the peak breadth (∼345 nm and 376 nm) When the 2,6-axis substituent is changed to either furan or thiophene, the absorbance profile undergoes a bathochromic shift of 17 nm and 28 nm for FP and TP, respectively. For both compounds, the low energy transitions occur between ∼325 to ∼415 nm which is ideal for deep-blue OLED dopants. As thin films, the peak maximum for FP is red-shifted by 9 nm while TP is shifted by 8 nm due to π–π stacking interactions. When the aryl substituents along the 4,8-axis of PP are switched to furan or thiophene (PF and PT), the absorbance profiles experience a bathochromic shift. Cruciform PF is shifted by 23 nm while PT shifts by 28 nm due to the chalcogens. Both BBOs also exhibit a broad series of low-energy transitions at ∼385 to ∼460 nm demonstrating that even dispersed in solution, these compounds start to absorb in the visible spectrum. Moreover, when compounds PF and PT are cast as thin films, the absorbance profile becomes quite broad, thus losing the resolution between observable transitions.
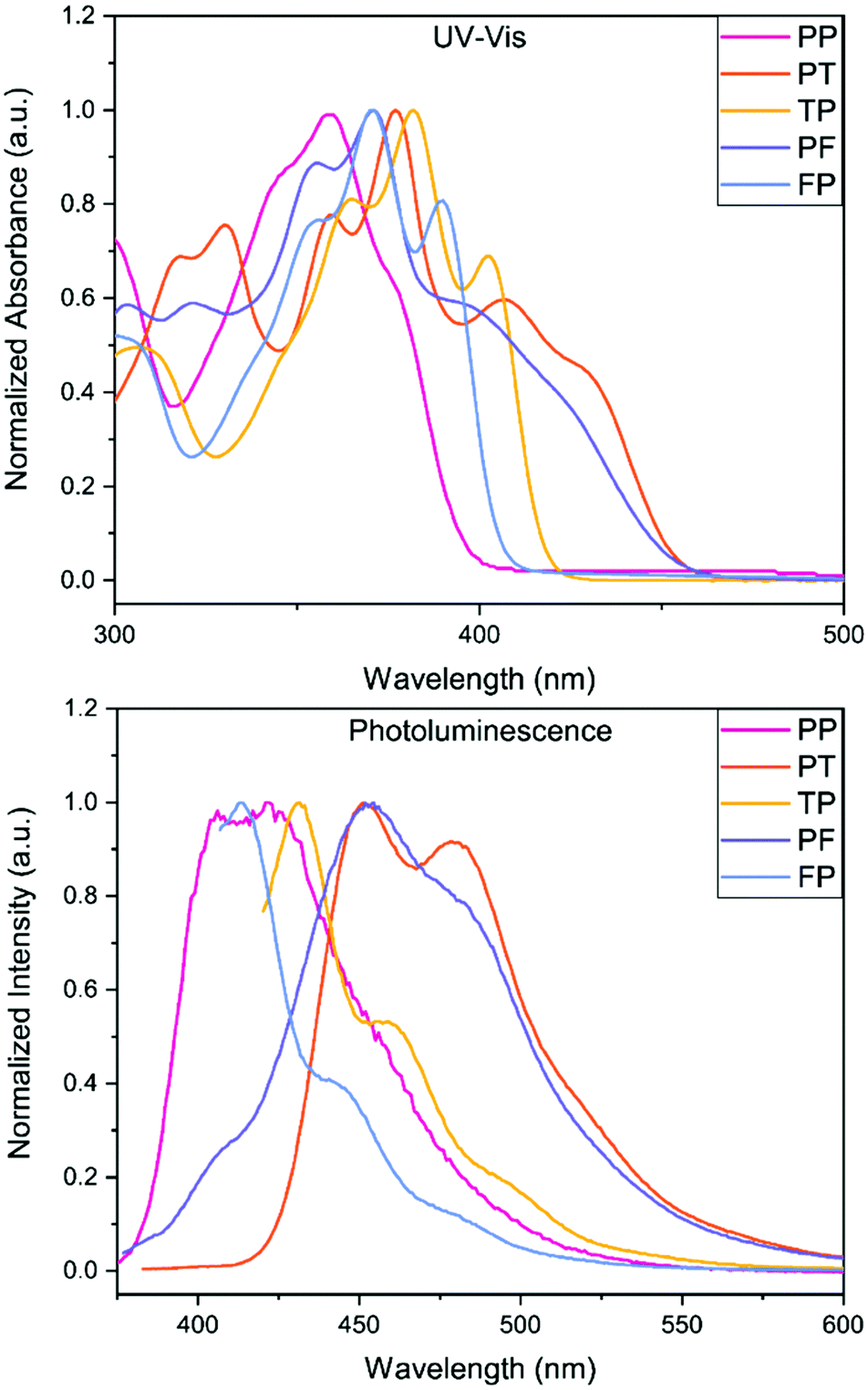 | ||
| Fig. 3 UV-Vis spectra (top) and photoluminescence (bottom) of the five BBOs in chloroform (0.01 g L−1). | ||
The photoluminescence (PL) spectra were measured in CHCl3 to determine emission profiles and quantum yields for each BBO (Fig. 3; Table 2). Since a host–guest active layer is used in the OLED devices for this study, the neat film data, available in the ESI,† will not be discussed. In solution, PP exhibited two emission peaks at 406 and 422 nm with a quantum yield of 44%. Switching the 2,6-substituent to furan (FP) provides a slight hypsochromic shift in emission by 8 nm whereas the thiophene substituent (TP) bathochromically shifts the PL spectra by 10 nm. Interestingly, the substitution of furan increases the quantum yield of the compound to 60% while the use of thiophene kept the quantum yield relatively constant (45%) when compared to PP (44%). FP, TP and PP are in a suitable range of emission for deep-blue OLEDs. As hypothesized, changes to the 4,8-aryl substituent (PF and PT) produced more cyan emission where the furan variant exhibits a PL maximum red-shift of 31 nm while the thiophene counterpart shows a comparable shift of 29 nm. Moreover, this conjugation pattern considerably lowers the PL quantum yields (22–26%) compared to that of PP.
We next investigated the radiative lifetimes of the five BBOs to assess their photoluminescent behaviour. Excluding TP which was unable to be measured, the BBOs emit light under 2.30 ns, further supporting the hypothesized LE character of the materials and suggesting traditional fluorophore behaviour as OLED emitters. While PF and PT are believed to be less than ideal as deep-blue dopants, these materials were used in OLED devices to elucidate the structure–property trends when compared to their counterparts FP and TP.
OLED device properties
The device characteristics of each BBO were evaluated as dopants in a mixed-host matrix. The host–guest architecture has proven to be a beneficial motif for small molecule-based OLEDs. The mixed host strategy was employed to reduce the energetic barrier from PEDOT:PSS to the active layer by integrating in common hole transport layer (HTL) material, 4,4′,4′′-tris(N-carbazolyl)triphenylamine (TCTA). By blending this material into the active layer, better positive charge injection into the A electrode occurs and is met with better recombination.42,43 The device configuration for all studies was as follows: ITO (100 nm)/PEDOT:PSS (35 nm)/5% BBO, 1:1 26DCzPPY:TCTA/TmPyPB (35 nm)/LiF (1 nm)/Al (100 nm). The electroluminescence (EL) spectra, external quantum efficiencies (EQE), and energy level diagram for all compounds used are illustrated in Fig. 4. Photoluminescence spectra of the active layers are displayed in the ESI† (Fig. S26, ESI†)
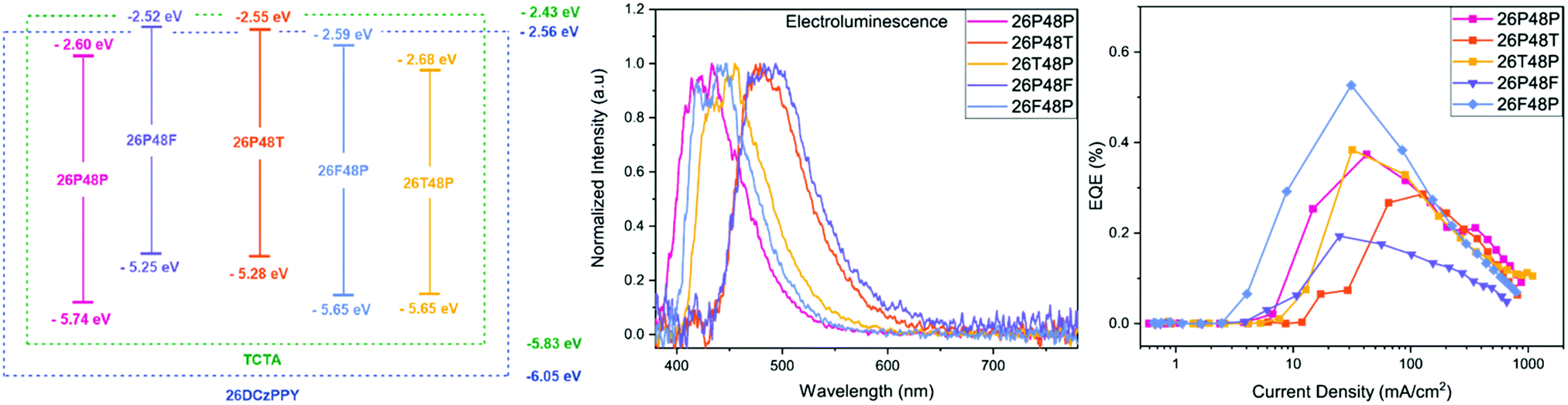 | ||
| Fig. 4 (Left) Band diagram of five BBO dopants in mixed-host matrix. (Middle) Electroluminescence of BBO-based devices. (Right) EQE vs. Current Density map of the five devices. | ||
Each cruciform exhibited EL that closely matched their solid-state PL spectra corresponding to the same active layer composition. Compounds PF and PT exhibited the most red-shifted EL with peak maxima at 487 nm and 476 nm, respectively, producing sea-green emission. The EL for the for isomers hypsochromically shifted to 442 and 451 for FP and TP, respectively. Resulting from the large Eoptg, cruciform PP had the bluest EL with a peak maxima of 434 nm. Of the five BBOs, compounds FP, TP and PP all exhibited CIEx components around 0.15 and CIEy components < 0.12, making them ideal candidates for deep-blue dopants.44–50
We next examined the device parameters to determine the performance of each BBO. The device efficiencies/outputs are shown in Table 3. All compounds exhibited low turn-on voltages (measured at 1 cd m−2), ranging between 3.3–4.0 V, while driving voltages (100 cd m−2) were found between 4.4–5.1 V. We attribute the low driving voltages to the mixed-host system and donor/acceptor units of each BBO, allowing good injection of positive and negative charges into the active layer. The maximum achievable luminance (Lmax) for PP and FP were comparable at ∼530 cd m−2 whereas TP had more than twice the luminance (1288 cd m−2). While blue emission was not obtained for PF and PT, compounds with thiophene substituents did significantly enhance each OLED's Lmax. The placement of aryl substituents also provides a moderate luminance enhancement. For example, compared to TP, the isomer PT had a Lmax value of 1572 cd m−2. The same effect is noted between the isomers FP and PF. All five compounds produce luminance levels suitable for display technology but do exhibit complications with regards to stability. Compounds PP, PT and PF all show irreversible device degradation after surpassing the potential corresponding to Lmax. However, by exchanging the location of aryl substituents, compounds TP and FP show an overall enhanced stability. These results are interesting as they could indicate an important structure–property trend: the 4,8-axis controls the brightness of the device while the 2,6-axis extends the stability.
| BBO | λ EL (nm) | CIE (x, y) | V on (V) | V 100 (V) | L max (cd m−2) | CE100 (cd A−1) | CEMax (cd A−1) | PE100 (lm W−1) | PEMax (lm W−1) | EQEMax (%) | RMS (nm) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PP | 434, 419 | 0.16, 0.06 | 3.3 | 4.5 | 552.6 | 0.23 | 0.23 | 0.16 | 0.16 | 0.37 | 0.333 |
| PT | 476 | 0.17, 0.33 | 3.7 | 4.5 | 1572 | 0.32 | 0.59 | 0.22 | 0.35 | 0.29 | 0.322 |
| TP | 451, 433 | 0.15, 0.12 | 3.6 | 4.4 | 1288 | 0.33 | 0.40 | 0.24 | 0.28 | 0.38 | 0.394 |
| PF | 487 | 0.19, 0.36 | 4.0 | 5.1 | 784.4 | 0.32 | 0.36 | 0.20 | 0.21 | 0.21 | 0.339 |
| FP | 442, 420 | 0.15, 0.08 | 3.5 | 4.4 | 532.2 | 0.37 | 0.42 | 0.26 | 0.39 | 0.53 | 0.353 |
The device efficiencies for all BBO dopants showed results comparable to other small molecule, BBO-based OLEDs.32 Compound PP was found to exhibit the lowest current efficiency (CE) and luminous efficacy (LE) of all emissive BBOs with an overall EQE of 0.37%. Our previous hypothesis stated this compound exhibited poor device performance in part due to the planarity of the cruciform, causing luminescent quenching during device operation.5 However, comparing this data to observations for TP refutes this claim. Specifically, TP experiences nearly equivalent torsion about the 2,6- and 4,8-dihedral angles as PP and has higher CE and LE. Furthermore, compound FP, whose ground-state geometry is calculated to be completely planar experiences one of the highest CE/LE outputs with the highest EQE of 0.53%. Even reversing the axis, PF and PT all exhibit higher CE and LE values when compared to PP with EQEs of 0.21% and 0.29%, respectively. Therefore, we believe the reason PP can undergo EL in this study is due to the mixed-host system, while the poor efficiencies are due to the lack of aryl substituents which allow for more effective hole-transport to the dopant, such as thiophene and furan. However, the efficiencies of each device are overall poor and thus, it is evident that improvements must be made by altering fabrication conditions or utilizing new hosts.
To determine if the device performance was inhibited due to morphological constraints, each active layer was measured using atomic force microscopy (AFM). All AFM images are shown in Fig. S23 (ESI†) and roughness values are collated in Table 3. It is evident that the host–guest system employed using the mixed-host matrix provides excellent thin-films. The roughness for all active layers is <4 Å, suggesting that the morphological contributions towards poor device performance are negligible and that the issues could be due to factors at the molecular level. One possible explanation for the low CE/LE outputs could be due to the triplet state (T1) of each BBO cruciform. If a drastic energetic difference between the singlet (S1) and T1 levels exists, this could result in the accumulation of charge-traps in the active layer. To assess this claim, preliminary DFT calculations show that the energy difference between the S1 and T1 states are between 0.94–1.09 eV (ESI†), supporting the hypothesis. Therefore, a more detailed degradation study of these materials is required to see what chemical or fabrication parameters can be altered to improve device performance and efficiencies.
Conclusions
Guided by DFT predictions, we successfully identified a series of BBO cruciforms with deep-blue emissive character for OLEDs. The five candidates were synthesized in a few steps and solution-processed into functional devices for evaluation. The experimental opto-electronic properties closely matched DFT outputs and the results will be used to help improve the computational model for future predictions. Of the five-candidates, compounds PP and FP all exhibited deep-blue emission (CIEy ≤ 0.08) and luminance output useful for display technology. However, the lifetimes and efficiencies for the OLED devices were too low for practical usage. These experimental results indicate that we have the potential to control device stability and brightness through aryl-substitution at a specific BBO axis. The solution-processed active layer formed excellent thin-films, suggesting improvements should be made at the molecular level. Thus, our results affirms that BBO cruciforms are useful building blocks for the development of blue light-emitting materials, whose efficiencies can be enhanced through the inclusion of electron-rich heterocycles. Further studies into the photophysical properties and degradation analysis will improve the design of our BBOs, increasing the device efficiencies and lifetimes. With this information, potential of incorporating the structure of the deep-blue materials into polymeric backbones could produce emissive polymers with mechanical properties suited for foldable technology.Experimental
Computational methodology
All computations were performed using Gaussian 09 using the Comet supercomputer cluster provided by the San Diego Supercomputing Center through the Extreme Science and Engineering Discovery Environment (XSEDE). Compounds were then analysed through GaussView 6 GUI interface program package. Electronic ground state geometries were optimized using density functional theory (DFT) employing a mPW3PBE functional and an SV basis set verified through a frequency calculation. The first 15 excited states were generated through time dependent density functional theory (TD-DFT) applied to the optimized ground state for each oligomer. The HOMO, LUMO, band gap, FMOs, and UV-Vis simulations were generated from these excited computations.Materials and methods
4,8-Dibromo-2,6-diethylbenzobisoxazole,19 and 2,5-diaminohyroquinone bishydrochloride51 were synthesized according to literature procedures. THF was dried using an Innovative Technologies solvent purification system. All other chemical reagents were purchased from commercial sources and used without further purification unless otherwise noted. All column chromatography described was performed using a Teledyne CombiFlash® Rf+ system with solvents as specified. Nuclear magnetic resonance (NMR) experiments were carried out in CDCl3 at 500 MHz (1H) AND 125 MHz (13C). In all spectra, chemical shifts are given in δ relative to residual protonated solvent peak, CHCl3 (7.26 ppm, 1H; 77.16 ppm, 13C). Coupling constants are reported in hertz (Hz). High-resolution mass spectra were recorded on a double-focusing magnetic sector mass spectrometer using ESI. Melting Points were determined using a MelTemp II apparatus equipped with thermometer. The electrochemical properties of the BBOs were investigated by cyclic voltammetry (CV) using a platinum working electrode in dichloromethane (solution) and acetonitrile (film), with 0.1 M Bu4NPF6 as the electrolyte and an Ag/Ag+ reference electrode. The onsets were referenced to Fc/Fc+. All UV-Vis and fluorescence spectra were obtained using quartz cuvettes with a 10 mm path length in CHCl3 or as spin-casted thin-films on a quartz slide (filtered 5 mg mL−1 solution in CHCl3 spun at 1500 RPM). Active layer fluorescence spectra were obtained using quartz slides filtered from 10 mg mL−1 solution in chlorobenzene spun at 2000 RPM. UV-vis spectra were collected on a Shimadzu UV-1800 UV spectrophotometer. Photoluminescence spectra and absolute solution fluorescence quantum yields were obtained using a Varian Cary Eclipse Fluorimeter. Quantum yields were obtained with a HORIBA spectrophotometer Nanolog FL3-2iHR equipped with a Quanta-phi integrating sphere, respectively. The chloroform solution time-resolved fluorescence data for all the samples were collected at 298 K on a time-correlated single-photon counting Edinburgh LifeSpec II and analyzed with the F980 software. A NanoLED of 405nm was used as excitation source.
Synthesis of precursors
General Suzuki–Miyaura cross-coupling conditions
All equivalence/mole percentage values are in reference to the starting BBO. To a condenser-fitted flask, equipped with stir bar, was added the starting BBO, 3 mol eq of 9, and 5 mol% of PEPPSI–iPr. The flask was purged with argon three times before the addition of 2 M K2CO3 (30 mol eq.) and toluene. The solution was degassed with argon for 15 minutes and allowed to reflux overnight at 120 °C. Upon cooling, the solution was diluted with chloroform, washed with DI water, and separated from the aqueous layer. The organic layer was dried over Na2SO4 and filtered into a flask with silica. Removal of solvent facilitated the formation of solid-load product necessary for column chromatography (hexanes:CHCl3 gradient) to purifying the products. Below are the physical characteristics, percent yields, and characterization data for all compounds using this method::689.4107; found: 689.4094.
:725.4318; found: 725.4323.
:757.3861; found: 757.3862.
General Stille cross-coupling conditions
All equivalence/mole percentage values are in reference to the starting BBO. To a condenser-fitted flask equipped with stir bar was added the starting BBO, 2.5 mol% of Pd2(dba)3, and 10 mol% of tri(o-tolyl)phosphine. The flask was purged with argon three times before the addition of corresponding organotin reagent (3 mol eq) and toluene. The solution was degassed with argon for 15 minutes and allowed to reflux overnight at 120 °C. Upon cooling, the solution was diluted with chloroform and stirred in a 1:1 mixture of silica gel:K2CO3 for at least 30 minutes. The solids were removed via vacuum filtration and washed with excess chloroform. To the solution was added fresh silica gel. Removal of solvent facilitated the formation of solid-load product necessary for column chromatography (hexanes:CHCl3 gradient) to purifying the products. Below are the physical characteristics, percent yields, and characterization data for all compounds using this method:
Device fabrication and characterization
OLED devices were fabricated on pre-patterned ITO (100 nm)/glass substrates with 20 Ω sq−1 resistance (Ossila, Ltd.). Substrates were cleaned by sonicating in Mucasol, water (2×), acetone, isopropanol and finally dried in an oven for at least one hour. Substrates were then subject to 15 minutes of UV-ozone curing to aide in hole-injection. Next, PEDOT:PSS (Heraeus Clevios P VP AI 4083) was spin-coated onto each substrate (3500 RPM, 2 minutes) followed by annealing at 120 °C for 20 minutes under a nitrogen atmosphere, producing an appx. 35 nm layer. The active layer solution (conc.: 9 mg mL−1) composed of a mixed-host system (with 5% w/w dopant) dissolved in chlorobenzene. The host matrix was a 1:1 ratio of 26DCzPPY:TCTA. Solutions were allowed to stir at 50 °C for at least one hour and filtered prior to solution processing. The active layer was spin-coated on top of the PEDOT:PSS at 4000 RPM for 15 seconds and then annealed at 120 °C for 10 minutes. Substrates were then transferred to a thermal evaporator where TmPyPB (30 nm; 0.5 Å s−1), LiF (1 nm; 0.1 Å s−1), and Al (100 nm; 2 Å s−1) were sequentially evaporated at 10−6 Torr. Finally, substrates were coated with a UV-curing epoxy and glass coverslip and cured at 365 nm for 25 minutes to perform measurements in ambient conditions. OLED electrical properties were characterized using an Ossila Ltd. Lifetime System. Electroluminescence was recorded using a Konica Minolta LS-160 luminance meter and electroluminescent spectra were recorded using an Ocean Insight HR2000+ ES spectrometer, calibrated using a HL-3-plus light source. Tapping-mode AFM measurements were made using a Bruker Dimension 3000 microscope.
Author contributions
David Wheeler: conceptualization, methodology, investigation, writing – original draft, writing – review & editing, and visualization. Shambhavi Tannir: investigation, writing – revision writing – review & editing. Evan Smith: investigation and data curation. Aimeé Tomlinson: conceptualization, methodology, investigation, resources, writing – original draft, writing – review & editing, visualization and funding acquisition. Malika Jeffries-EL: conceptualization, methodology, investigation, writing – original draft, writing – review & editing, visualization supervision, project administration and funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Science Foundation (CHE-1808402, and CHE-1808414) as well as supercomputer allocation from XSEDE (DMR-160146) for financial support of this work. We also would like to thank Dr. Paul Ralifo and Dr. Norman Lee of the Boston University Chemistry Instrumentation Center for NMR and HR-MS access and measurements (respectively), Professor Allison Dennis for fluorimeter/integrating sphere access for quantum yield measurements, Dr. Paul Mak for profilometry access, and Dr. Anlee Krupp for AFM access. We also thanks Professors Joseph Shinar, Dr. Ruth Shinar, Mr. Michael Fralaide and Mr. Erik Dykstra from Iowa State University for help with EQE calculations.Notes and references
- C. Lee, S. Lee, G.-U. Kim, W. Lee and B. J. Kim, Chem. Rev., 2019, 119, 8028–8086 CrossRef CAS PubMed.
- L. Ying, F. Huang and G. C. Bazan, Nat. Commun., 2017, 8, 14047 CrossRef CAS PubMed.
- Y.-W. Su, Y.-C. Lin and K.-H. Wei, J. Mater. Chem. A, 2017, 5, 24051–24075 RSC.
- D. T. Christiansen, S. Ohtani, Y. Chujo, A. L. Tomlinson and J. R. Reynolds, Chem. Mater., 2019, 31, 6841–6849 CrossRef CAS.
- P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268–320 CrossRef CAS PubMed.
- G. Yang, Y.-M. Zhang, Y. Cai, B. Yang, C. Gu and S. X.-A. Zhang, Chem. Soc. Rev., 2020, 49, 8687–8720 RSC.
- A. M. Österholm, D. E. Shen and J. R. Reynolds, in Conjugated Polymers: Properties, Processing, and Applications, ed. J. R. Reynolds, B. C. Thompson and T. A. Skotheim, CRC Press, 4th edn, 2019, ch. 6 DOI:10.1201/9780429190520-chapter-6.
- D. T. Christiansen, A. L. Tomlinson and J. R. Reynolds, J. Am. Chem. Soc., 2019, 141, 3859–3862 CrossRef CAS PubMed.
- M. Zhu and C. Yang, Chem. Soc. Rev., 2013, 42, 4963–4976 RSC.
- X. Yang, X. Xu and G. Zhou, J. Mater. Chem. C, 2015, 3, 913–944 RSC.
- J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539–541 CrossRef CAS.
- H. Gorter, M. J. J. Coenen, M. W. L. Slaats, M. Ren, W. Lu, C. J. Kuijpers and W. A. Groen, Thin Solid Films, 2013, 532, 11–15 CrossRef CAS.
- S.-J. Zou, Y. Shen, F.-M. Xie, J.-D. Chen, Y.-Q. Li and J.-X. Tang, Mater. Chem. Front., 2020, 4, 788–820 RSC.
- J. Han and H.-J. Suk, J. Infect. Dis., 2019, 20, 31–38 CAS.
- D. Wang, C. Cheng, T. Tsuboi and Q. Zhang, CCS Chem., 2020, 2, 1278–1296 CrossRef CAS.
- D. Y. Kondakov, W. C. Lenhart and W. F. Nichols, J. Appl. Phys., 2007, 101, 024512 CrossRef.
- C.-Y. Chan, M. Tanaka, Y.-T. Lee, Y.-W. Wong, H. Nakanotani, T. Hatakeyama and C. Adachi, Nat. Photonics, 2021, 15, 203–207 CrossRef CAS.
- S. O. Jeon, K. H. Lee, J. S. Kim, S.-G. Ihn, Y. S. Chung, J. W. Kim, H. Lee, S. Kim, H. Choi and J. Y. Lee, Nat. Photonics, 2021, 15, 208–215 CrossRef CAS.
- B. C. Tlach, A. L. Tomlinson, A. G. Ryno, D. D. Knoble, D. L. Drochner, K. J. Krager and M. Jeffries-EL, J. Org. Chem., 2013, 78, 6570–6581 CrossRef CAS PubMed.
- A. J. Zucchero, P. L. McGrier and U. H. F. Bunz, Acc. Chem. Res., 2010, 43, 397–408 CrossRef CAS PubMed.
- D. L. Wheeler, A. V. Diodati, A. L. Tomlinson and M. Jeffries-EL, ACS Omega, 2020, 5, 12374–12384 CrossRef CAS PubMed.
- J. Lim, T. A. Albright, B. R. Martin and O. Š. Miljanić, J. Org. Chem., 2011, 76, 10207–10219 CrossRef CAS PubMed.
- J. E. Klare, G. S. Tulevski and C. Nuckolls, Langmuir, 2004, 20, 10068–10072 CrossRef CAS PubMed.
- J. E. Klare, G. S. Tulevski, K. Sugo, A. de Picciotto, K. A. White and C. Nuckolls, J. Am. Chem. Soc., 2003, 125, 6030–6031 CrossRef CAS PubMed.
- M. A. Saeed, H. T. M. Le and O. Š. Miljanić, Acc. Chem. Res., 2014, 47, 2074–2083 CrossRef CAS PubMed.
- A. A. Burney-Allen, J. Shaw, D. L. Wheeler, L. Diodati, V. Duzhko, A. L. Tomlinson and M. Jeffries-EL, Asian J. Org. Chem., 2021, 10, 215–223 CrossRef CAS.
- R. C. Evers, F. E. Arnold and T. E. Helmeniak, Macromolecules, 1981, 14, 925–930 CrossRef CAS.
- Q. Fu, H. Zhang, B. Song, X. Liu, Q. Zhuang and Z. Han, J. Appl. Polym. Sci., 2011, 121, 1734–1739 CrossRef CAS.
- S. J. Krause, T. B. Haddock, D. L. Vezie, P. G. Lenhert, W. F. Hwang, G. E. Price, T. E. Helminiak, J. F. O'Brien and W. W. Adams, Polymer, 1988, 29, 1354–1364 CrossRef CAS.
- D. B. Roitman, R. A. Wessling and J. McAlister, Macromolecules, 1993, 26, 5174–5184 CrossRef CAS.
- M. M. Alam and S. A. Jenekhe, Chem. Mater., 2002, 14, 4775–4780 CrossRef CAS.
- R. Chavez III, M. Cai, B. Tlach, D. L. Wheeler, R. Kaudal, A. Tsyrenova, A. L. Tomlinson, R. Shinar, J. Shinar and M. Jeffries-EL, J. Mater. Chem. C, 2016, 4, 3765–3773 RSC.
- X. Yin, T. Zhang, Q. Peng, T. Zhou, W. Zeng, Z. Zhu, G. Xie, F. Li, D. Ma and C. Yang, J. Mater. Chem. C, 2015, 3, 7589–7596 RSC.
- J. J. Intemann, E. S. Hellerich, M. D. Ewan, B. C. Tlach, E. D. Speetzen, R. Shinar, J. Shinar and M. Jeffries-EL, J. Mater. Chem. C, 2017, 5, 12839–12847 RSC.
- J. J. Intemann, E. S. Hellerich, B. C. Tlach, M. D. Ewan, C. A. Barnes, A. Bhuwalka, M. Cai, J. Shinar, R. Shinar and M. Jeffries-EL, Macromolecules, 2012, 45, 6888–6897 CrossRef CAS.
- E. S. Hellerich, J. J. Intemann, M. Cai, R. Liu, M. D. Ewan, B. C. Tlach, M. Jeffries-EL, R. Shinar and J. Shinar, J. Mater. Chem. C, 2013, 1, 5191–5199 RSC.
- O. Gidron, A. Dadvand, Y. Sheynin, M. Bendikov and D. F. Perepichka, Chem. Commun., 2011, 47, 1976–1978 RSC.
- O. Gidron and M. Bendikov, Angew. Chem., Int. Ed., 2014, 53, 2546–2555 CrossRef CAS PubMed.
- S. S. Zade, N. Zamoshchik and M. Bendikov, Acc. Chem. Res., 2010, 44, 14–24 CrossRef PubMed.
- R. D. McCullough, Adv. Mater., 1998, 10, 93–116 CrossRef CAS.
- I. Osaka and R. D. McCullough, Acc. Chem. Res., 2008, 41, 1202–1214 CrossRef CAS PubMed.
- B. S. Kim and J. Y. Lee, Adv. Funct. Mater., 2014, 24, 3970–3977 CrossRef CAS.
- Y. J. Doh, J. S. Park, W. S. Jeon, R. Pode and J. H. Kwon, Org. Electron., 2012, 13, 586–592 CrossRef CAS.
- S.-K. Kim, B. Yang, Y. Ma, J.-H. Lee and J.-W. Park, J. Mater. Chem., 2008, 18, 3376–3384 RSC.
- Y. Yu, Z. Wu, Z. Li, B. Jiao, L. Li, L. Ma, D. Wang, G. Zhou and X. Hou, J. Mater. Chem. C, 2013, 1, 8117–8127 RSC.
- C.-C. Huang, M.-M. Xue, F.-P. Wu, Y. Yuan, L.-S. Liao and M.-K. Fung, Molecules, 2019, 24, 353–362 CrossRef PubMed.
- A. Pachariyangkun, P. Wongkaew, T. Sudyoadsuk, S. Namuangruk and V. Promarak, Dyes Pigm., 2021, 186, 109065 CrossRef CAS.
- Y. Tan, Z. Wang, C. Wei, Z. Liu, Z. Bian and C. Huang, Org. Electron., 2019, 69, 77–84 CrossRef CAS.
- Z. Li, G. Gan, Z. Ling, K. Guo, C. Si, X. Lv, H. Wang, B. Wei and Y. Hao, Org. Electron., 2019, 66, 24–31 CrossRef CAS.
- J. Santos, J. H. Cook, H. A. Al-Attar, A. P. Monkman and M. R. Bryce, J. Mater. Chem. C, 2015, 3, 2479–2483 RSC.
- J. F. Mike, A. J. Makowski and M. Jeffries-EL, Org. Lett., 2008, 10, 4915–4918 CrossRef CAS PubMed.
- Y. Qiu, A. Fortney, C.-H. Tsai, M. A. Baker, R. R. Gil, T. Kowalewski and K. J. T. Noonan, ACS Macro Lett., 2016, 5, 332–336 CrossRef CAS.
- B. Shaik, J. H. Park, T. K. An, Y. R. Noh, S. B. Yoon, C. E. Park, Y. J. Yoon, Y.-H. Kim and S.-G. Lee, Tetrahedron, 2013, 69, 8191–8198 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra, Photoluminescent spectra, AFM images, cyclic voltammograms. See DOI: 10.1039/d1ma00990g |
| This journal is © The Royal Society of Chemistry 2022 |