
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolymer-templated mesoporous lithium titanate microspheres for high-performance lithium batteries†
Minh Tri
Nguyen
 a,
Preston
Sutton‡
ab,
Andrea
Palumbo
a,
Michael G.
Fischer§
a,
Xiao
Hua
c,
Ilja
Gunkel
*a and
Ullrich
Steiner
*a
a,
Preston
Sutton‡
ab,
Andrea
Palumbo
a,
Michael G.
Fischer§
a,
Xiao
Hua
c,
Ilja
Gunkel
*a and
Ullrich
Steiner
*a
aAdolphe Merkle Institute, University of Fribourg, Chemin des Verdiers 4, 1700 Fribourg, Switzerland. E-mail: ilja.gunkel@unifr.ch; ullrich.steiner@unifr.ch
bInstitute for Frontier Materials, Deakin University, Burwood, VIC 3125, Australia
cDepartment of Chemical and Biological Engineering, University of Sheffield, UK
First published on 2nd November 2021
Abstract
The spinel Li4Ti5O12 (LTO) is a promising lithium ion battery anode material with the potential to supplement graphite as an industry standard, but its low electrical conductivity and Li–ion diffusivity need to be overcome. Here, mesoporous LTO microspheres with carbon-coatings were formed by phase separation of a homopolymer from microphase-separated block copolymers of varying molar masses containing sol–gel precursors. Upon heating the composite underwent a sol–gel condensation reaction followed by the eventual pyrolysis of the polymer templates. The optimised mesoporous LTO microspheres demonstrated an excellent electrochemical performance with an excellent specific discharge capacity of 164 mA h g−1, 95% of which was retained after 1000 cycles at a C-rate of 10.
1 Introduction
The continued growth of lithium–ion batteries (LIBs) for transportation and power applications requires cell-level performance improvements.1 While these improvements can be realised by optimising all battery components,2 the anode is the principal focus of this paper. Graphite is currently the most widely used commercial anode material, although its low rate performance, safety concerns related to lithium dendrite growth, and material degradation are slowing battery development for vehicles and high power systems.3,4 A promising alternative to graphite and its limitations is lithium titanate, Li4Ti5O12 (LTO).5–7LTO has proven to be a safe, low-cost, and electrochemically stable anode material with excellent thermal stability and increased cyclability compared to graphite.8,9 An important advantage of LTO is its flat (de)lithiation potential well above the voltage of lithium plating (0 V vs. Li+/Li), preventing the fire and explosion risks caused by dendrite formation in graphite cells. In addition, the stable LTO spinel structure (Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group) exhibits negligible volume change during (de)lithiation, which enables fast (dis)charging.4,9 This is in contrast to graphite, which expands up to 13% by volume during lithiation,10 causing a host of degradation issues. While the high redox potential of LTO with respect to lithium reduces the voltage of any cell, and thus its energy density (175 mA h g−1 discharged to 1.0 V vs. Li+/Li, compared to 372 mA h g−1 discharged to almost 0 V vs. Li+/Li, for graphite),8,11 the high potential inhibits the decomposition of contemporary carbonate-based electrolytes, extending the useful cycle life well beyond that of graphite-based cells.2
m space group) exhibits negligible volume change during (de)lithiation, which enables fast (dis)charging.4,9 This is in contrast to graphite, which expands up to 13% by volume during lithiation,10 causing a host of degradation issues. While the high redox potential of LTO with respect to lithium reduces the voltage of any cell, and thus its energy density (175 mA h g−1 discharged to 1.0 V vs. Li+/Li, compared to 372 mA h g−1 discharged to almost 0 V vs. Li+/Li, for graphite),8,11 the high potential inhibits the decomposition of contemporary carbonate-based electrolytes, extending the useful cycle life well beyond that of graphite-based cells.2
However, to fully realise the benefits of LTO over graphite, its intrinsically low electrical conductivity (ca. 10−8 to 10−13 S cm−1) and its low lithium–ion diffusion coefficient (10−8 to 10−13 cm2 s−1),12 must be addressed. These underlying rate-limiting properties of LTO can be improved through several strategies, including surface coatings, doping, or control of particle size and morphology, which determine the effective reaction area and Li–ion diffusion lengths.11–14 Nanostructuring, for example, generally reduces diffusion paths and increases surface area, which allows for higher (dis)charging rates by increasing the number of reaction sites. A mere size reduction of the traditionally micrometer-sized LTO particles to the nanoscale causes however also a low volumetric energy density and poor particle–particle interconnections.11 These drawbacks of LTO nanoparticles can be circumvented by introducing nanometer-sized pores into micrometer-sized particles. Such hierarchical structures, which are referred to as mesoporous microspheres combine short lithium–ion diffusion paths and high surface areas with a high volumetric energy density and structural stability.11,12
Mesoporous LTO microspheres can be prepared using various synthetic approaches, including hydrothermal15,16 and solvothermal methods,17,18 which both improve the rate performance. For example, Tang et al. used a hydrothermal process to prepare mesoporous LTO spheres that showed excellent high-rate capabilities with a specific capacity of 114 mA h g−1 at 30 C as well as good cycle performance with a 94.5% capacity retention after 200 cycles at 4 C.15 To further improve the rate performance of these hierarchically structured materials, fine control over particle and pore size is desirable. In this regard, the use of polymer templates provides a powerful tool. In an earlier study, we have shown the efficacy of combining block copolymer (BCP) self-assembly and polymer phase separation with a sol–gel chemistry for precise structure templating in TiO2 microspheres, i.e. in a similar material to the LTO studied here.19 BCP self-assembly was also previously used to introduce mesoporosity into LTO.20
Here, we extend the facile one-pot synthesis approach using polymer templates to the fabrication of mesoporous LTO microspheres with tunable mesopore sizes. The desired micron-sized spherical particles were produced via polymer phase separation, while co-assembly of LTO sol–gel precursors with amphiphilic BCPs created a mesoporous structure upon sol–gel condensation during high-temperature annealing in argon. This annealing process forms a very thin graphitic layer on the mesoporous surface of the LTO microspheres, substantially increasing the intrinsically low electric conductivity of LTO.21 The mesoporosity of the microspheres was adjusted by varying the BCP molar mass, enabling the optimization of the LTO rate performance. The resulting polymer-templated anode material showed excellent properties, achieving 113 mA h g−1 at 30 C with a capacity retention of 95% after 1000 cycles at 10 C, demonstrating superior cyclability compared to earlier studies.15,17,18,22,23
2 Results and discussion
2.1 Fabrication of mesoporous Li4Ti5O12 (LTO) microspheres
Mesoporous LTO microspheres were prepared by confining their sol–gel synthesis in hierarchical polymer templates, in a two-step fabrication process consisting of the coassembly of a sol precursor with a suitable BCP followed by a temperature-induced condensation reaction and calcination to carbon-coated LTO, Fig. 1. The amphiphilic poly(styrene-b-ethylene oxide) (PS-b-PEO) BCPs enable the selective complexation of LTO precursor alkoxides with the hydrophilic PEO block.20,24 The self-assembly of this complex (i.e. the microphase separation of the hydrophilic PEO-complex and the hydrophobic PS block) causes the formation of a nanostructure in solution.25 The addition of a PS homopolymer to this blend causes the mixture to further phase separate at a different length scale, giving rise to micrometer-sized spheres of the LTO-BCP coassembly. Nanostructured microspheres are thus formed in a one-pot synthesis by mixing the polymers and the LTO precursors in a common solvent (tetrahydrofuran (THF)), plus oxalic acid to stabilise the sol–gel process,19 followed by slow solvent evaporation. The resulting mesoporous LTO microspheres are calcined at 600 °C or 700 °C in argon, causing the confined crystallisation of LTO inside the polymer template. The organic matrix is gradually pyrolysed and partially carbonised at these temperatures, resulting in LTO microparticles with carbon-coated nanopores.21 This approach allows tuning of the pores sizes, as previously demonstrated for mesoporous TiO2 microspheres.19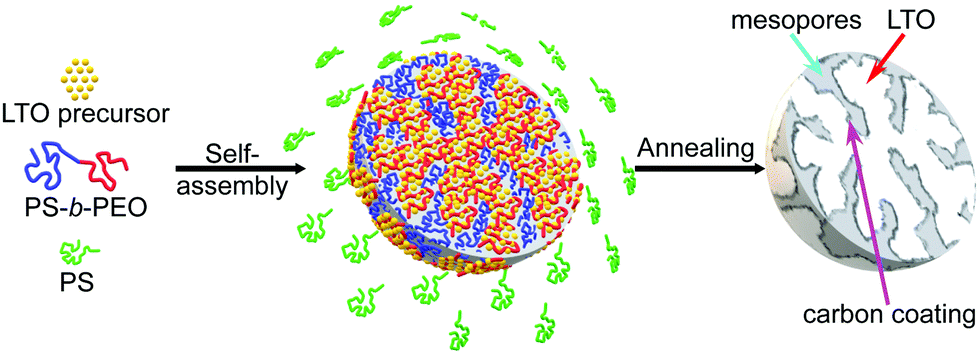 | ||
| Fig. 1 Schematic of the polymer-templated fabrication of carbon-coated mesoporous LTO microspheres. Initially, LTO precursors, PS-b-PEO block copolymers (BCPs) and PS homopolymer are mixed in a common solvent. Upon solvent evaporation, the BCP and the PS homopolymer phase separate into BCP spheres, in which the BCP blocks coassemble into a nanostructured morphology with the LTO precursor molecules preferentially residing in the PEO domains. Annealing this blend in an argon atmosphere causes confined crystallisation of the LTO while the polymer template is burnt away and partially carbonised, thereby creating LTO microspheres with carbon-coated mesopores. | ||
To adjust the size of the mesopores,19 PS-b-PEO BCPs with different molar masses but similar block volume fractions were used. The molar mass of the added PS homopolymer was either higher or similar to that of the PS blocks in the PS-b-PEO BCPs to ensure phase separation and microparticle formation. The different mesoporous LTO microsphere samples synthesised in this work were named based on the employed BCPs and the calcination temperatures, Table 1.
| LTO name | BCP name | BCP | M n (kg mol−1) | w PS | M n (kg mol−1) | Calcination | |
|---|---|---|---|---|---|---|---|
| T (°C) | Time (h) | ||||||
| LTO-A-600 | BCPA | PS-b-PEO | 10-b-3.5 | ∼0.74 | 35 | 600 | 2.5 |
| LTO-A-700 | BCPA | PS-b-PEO | 10-b-3.5 | ∼0.74 | 35 | 700 | 2.5 |
| LTO-B-600 | BCPB | PS-b-PEO | 18-b-7.5 | ∼0.71 | 35 | 600 | 2.5 |
| LTO-B-700 | BCPB | PS-b-PEO | 18-b-7.5 | ∼0.71 | 35 | 700 | 2.5 |
| LTO-C-700 | BCPC | PS-b-PEO | 38-b-15 | ∼0.72 | 35 | 700 | 2.5 |
2.2 Phase structure and morphology of mesoporous LTO microspheres
m) for all Li4Ti5O12 samples, as well as the absence of any discernible impurity phases such as anatase or rutile. The XRD patterns show a decreasing peak width, and thus an increasing crystallite size with increasing calcination temperature. The average crystallite sizes in Table 2 were calculated using the Scherrer equation (K = 0.9) by averaging the values obtained for the (111), (131), (040), (151), and (404) planes.26
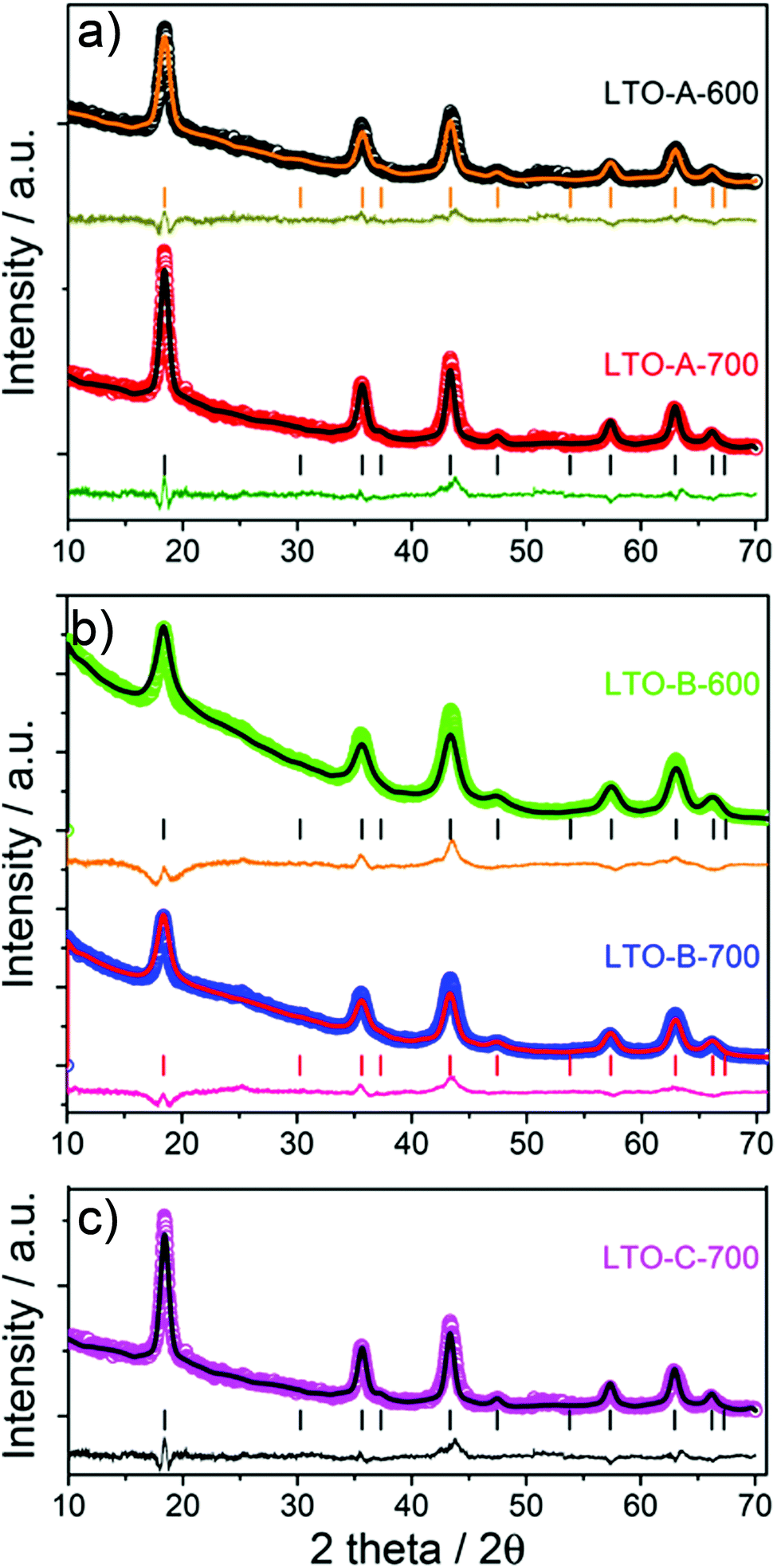 | ||
| Fig. 2 XRD patterns of the mesoporous LTO microspheres listed in Tables 1 and 2, (a) LTO-A, (b) LTO-B and (c) LTO-C. The symbols show the experimental data and the lines are fitted Rietveld refinements. The vertical bars indicate the tabulated peak positions for spinel LTO, below which the differences between experimental data and fits are plotted. In (c) the peaks expected for a spinel structure (space group: Fdm) are indexed with the (hkl) values of the corresponding lattice planes. | ||
| Sample | Crystallite-size (nm) | S BET (m2 g−1) | V pore (cm3 g−1) | Rate test (mA h g−1) | CE (%) | Cycle test (mA h g−1) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.5 C | 10 C | 30 C | 0.5 C | 10 C | ΔE (V) | Total resistances (Ω) | |||||||||
| BJH | 1st | 10th | 10th | 10th | 1st | 5th | 1000th | a | b | c | a | c | |||
| LTO-A-600 | 10.02 | 75.6 | 0.133 | 156 | 143 | 113 | 93 | 94.7 | 108 | 96 | 0.26 | 0.09 | 0.09 | 210 | 58 |
| LTO-A-700 | 12.17 | 54.4 | 0.122 | 156 | 139 | 107 | 85 | 92.3 | 99 | 86 | 0.28 | 0.10 | 0.11 | 144 | 51 |
| LTO-B-600 | 8.42 | 123.3 | 0.168 | 161 | 143 | 104 | 54 | 92.2 | 101 | 97 | 0.16 | 0.09 | 0.07 | 263 | 100 |
| LTO-B-700 | 9.78 | 110.6 | 0.162 | 164 | 147 | 127 | 113 | 91.9 | 128 | 122 | 0.22 | 0.09 | 0.09 | 159 | 48 |
| LTO-C-700 | 10.90 | 68.3 | 0.078 | 88 | 72 | 28 | 16 | 91.9 | 26 | 24 | 0.15 | 0.09 | 0.11 | 140 | 133 |
This careful analysis of crystallite sizes shows two interesting trends. First, the crystallite size increases with increasing calcination temperatures, as expected. Note that this increase is more pronounced in LTO-A compared to LTO-B. A moderate increase in crystallite size with calcination temperature reflects temperature-dependent crystallisation kinetics, which explain the small crystallite-size increase in LTO-B-700 compared to LTO-B-600. Second, the crystallite size is larger in LTO-A than in LTO-B, despite the fact that the lower molar mass of BCPA should yield a tighter LTO confinement than LTO-B. Indeed, LTO-A crystallite sizes are comparable to or larger than those of LTO-C, despite the 4-fold larger molar mass of the confining BCPC template. These observations are indicative of a less effective confinement of LTO crystallisation provided by the low-molar mass BCPA template compared to the other two BCPs, which is likely due to a weaker segregation in BCPs with shorter blocks. This hypothesis is further substantiated by the nitrogen physisorption experiments described below.
The spinel structure of all mesoporous LTO microspheres was further confirmed by Raman spectroscopy, Fig. 3. The spinel LTO has five first-order Raman modes, namely, 1 × A1g, 1 × Eg, 3 × F2g, according to group theory.27,28 These bands were observed at around 230 cm−1 (F2g), 404 cm−1 (Eg) and 675 cm−1 (A1g) along with a shoulder at about 750 cm−1, for all LTO samples, which is in good agreement with spinel LTO,29–31 and corroborates the XRD results. The band at 230 cm−1 is assigned to the bending vibration of the O–Ti–O bonds.30 The band at 404 cm−1 is attributed to the stretching vibration of the Li–O bonds in tetrahedral LiO4 and polyhedral LiO6.27,30 The bands at 675 and 750 cm−1 (A1g) correspond to the vibration of the Ti–O bonds in octahedral TiO6.28,31 Furthermore, two weak bands at around 1340 cm−1 and 1600 cm−1 corresponding to the D band and the G band of carbon, respectively, confirm the presence of carbon resulting from pyrolysis of the polymers upon the annealing at high temperature.32,33 While the D and G bands are clearly seen in the spectra of the LTO-A-600 and LTO-B-600 samples, their intensity is lower in the LTO-A-700 and LTO-B-700 spectra indicating a lower carbon content in samples calcined at the higher temperature. The peak area ratios AD/AG of about 1.33 for LTO-A-600, 1.23 for LTO-A-700, 1.28 for LTO-B-600, 1.25 for LTO-B-700, and 1.53 for LTO-C-700, imply predominantly disordered (amorphous) carbon layers in all samples.34
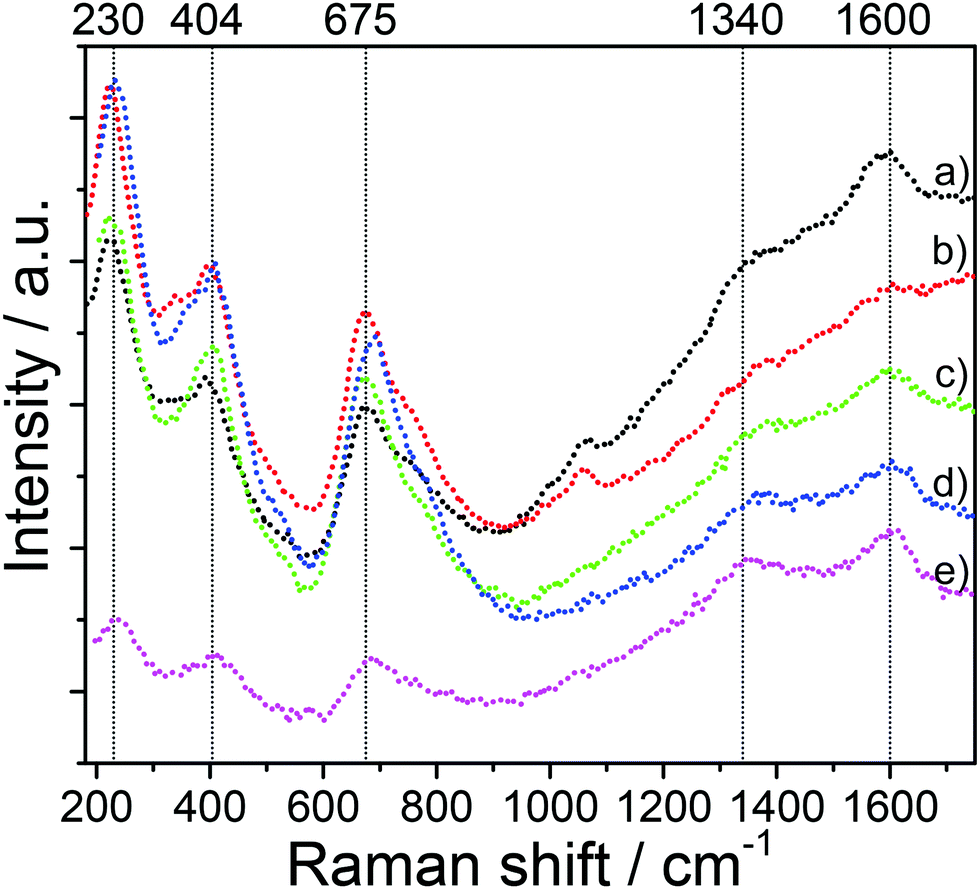 | ||
| Fig. 3 Raman spectra of the mesoporous LTO microspheres. (a) LTO-A-600, (b) LTO-A-700, (c) LTO-B-600, (d) LTO-B-700, and (e) LTO-C-700. | ||
While XRD and Raman measurements reveal no impurities any LTO samples, lithium carbonate and hydroxy groups are seen in the FTIR spectra, Fig. S1 (ESI†). The presence of Li2CO3 in the synthesised LTO samples probably arises from the reaction of lithium ions with CO2 at the sample surface, while the presence of OH groups is associated with adsorbed H2O from ambient air.35 Small amounts of (disordered) carbon (≤6 wt%) and water (≤3 wt%) in the synthesised LTO were found also in thermogravimetric analysis (TGA), Fig. S1 (ESI†). Similar amounts of carbon were detected in mesoporous TiO2 microspheres that were coated with a 1.0 to 1.5 nm thick carbon layer as seen in high-resolution TEM imaging (Fig. S11 (ESI†), M.G. Fischer et al.).19
 | ||
| Fig. 4 Low to high magnification SEM images (left to right columns) of mesoporous LTO-A-600 (a), LTO-A-700 (b), LTO-B-600 (c), LTO-B-700 (d), and LTO-C-700 (e) showing the predominance of LTO spheres exhibiting mesoporosity. Left column scale bar: 20 μm, centre column scale bar: 2 μm, right column scale bar: 100 nm. | ||
The pore size, volume, and surface area of the five LTO sample types were quantified by measuring nitrogen physisorption isotherms, which were analysed by the Brunauer–Emmett–Teller (BET) formalism, Fig. 5a. All LTO samples show type-IV isotherms, which are typical for mesoporous materials.36 Their different hysteresis loops imply differences in their pore structures. The type-H1 hysteresis loop observed for the LTO-A-600, LTO-A-700, LTO-B-600 and LTO-B-700 samples is indicative of highly uniform pore sizes, high pore connectivities, and cylindrical pore geometries. In contrast, the type-H4 hysteresis loop of the LTO-C-700 sample suggests the presence of some large mesopores in addition to a large fraction of much smaller pores.36,37 Note the decrease in porosity with increasing annealing temperatures, which correlates with the increased crystallite sizes.19
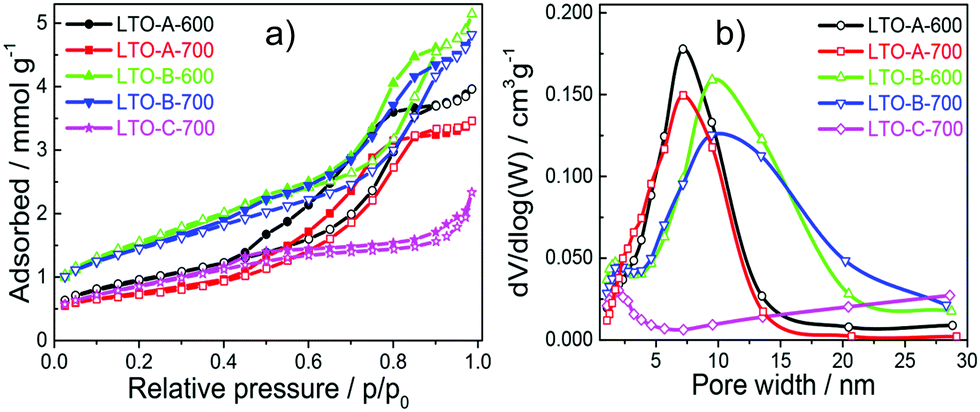 | ||
Fig. 5 (a) Nitrogen physisorption isotherms of the mesoporous LTO microspheres at 77 K. (b) Pore size distribution as determined from the adsorption branch using the BJH method, where the derivative pore volume normalised to the natural logarithm of pore-width interval, dV/d![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) log(W), is shown as a function of the pore width. log(W), is shown as a function of the pore width. | ||
The Barrett–Joyner–Halenda (BJH) analysis in Fig. 5b and in Fig. S7 (ESI†) was used to determine the pore-size distribution of the samples, revealing bimodal pore size distributions. The larger-sized pores are assigned to the polymer templating, while the smaller sized pores are intrinsic to the sol–gel chemistry, which is known to give rise to nanopores even in the absence of any macromolecular additives.38 The smaller, ca. 2 nm pores are similar in all samples, while the larger pores vary from ∼5 nm to ∼20 nm as a function of the chosen BCP and calcination temperature, giving rise to two trends. First, an increase in pore size with increasing BCP molar mass is observed, as expected.19 Second, an interesting aspect arises from the comparison of BET and BJH isotherms of the two LTO-A and the two LTO-B samples. Note that the ∼7 nm LTO-B pore size is invariant with the calcination temperature, while the 4.6 nm pores size of LTO-A-600 increases to 5.7 nm in LTO-A-700. The BET pore volumes of the LTO-A samples (see Table 2) are much lower compared to the LTO-B samples, despite the expectation that the lower-molar mass BCP should give rise to a higher porosity. Furthermore, the pore volume of LTO-A-700 is reduced compared to LTO-A-600, while the two LTO-B samples have identical pore volumes.
Combining these observations with the Scherrer analysis of the XRD data in Fig. 2 leads to conclusions concerning the structure formation in LTO-A and LTO-B. The invariance of porosity in LTO-B with the calcination temperature indicates that the LTO morphology is robustly templated by the BCP. The pore volume is stable at the two calcination temperatures and the crystallite and pore diameters are comparable. In LTO-A, however, the pore volume is comparably lower and decreases further with the calcination temperature, indicating structural degradation and the formation of fewer larger pores. The crystallite sizes are not only larger in LTO-A, they are substantially larger than the LTO-B crystallite sizes, and both increase with increasing calcination temperature. These observations are indicative of break-out crystallisation, where the crystallisation process of LTO partially destroys the confining polymeric template.
Finally, the LTO-C sample has a low porosity caused by the high molar mass of BCP-C, resulting in an inferior material in terms of mesoporosity compared to LTO-B, as qualitatively expected.
2.3 Electrochemical performance of mesoporous LTO microspheres
In order to optimise electrochemical performance, mesoporous LTO microsphere composite electrodes with different porosities, surface areas, and crystallite sizes were tested. The lithium-ion storage properties of these samples were analysed under galvanostatic conditions in Swagelok cells at various C-rates (a C-rate of 1 equals a current of 175 mA g−1) with a voltage range of 1.0 to 2.5 V vs. Li+/Li, using Li metal as counter electrode.To determine the initial specific capacities and the Coulombic efficiencies of the samples, the galvanostatic discharge and charge profiles of the first four cycles were measured at a C-rate of 0.5, Fig. 6. The initial specific discharge capacities for LTO-A-600, LTO-A-700, LTO-B-600, LTO-B-700, and LTO-C-700 were found to be 156, 156, 161, 164, and 88 mA h g−1, Table 2. LTO-B samples showed the highest specific capacities, correlating with the highest specific surface area of the samples. The slightly higher specific capacity of LTO-B-700 compared to LTO-B-600 might arise from the larger LTO crystallite size and potentially an overall higher crystallinity.39–41
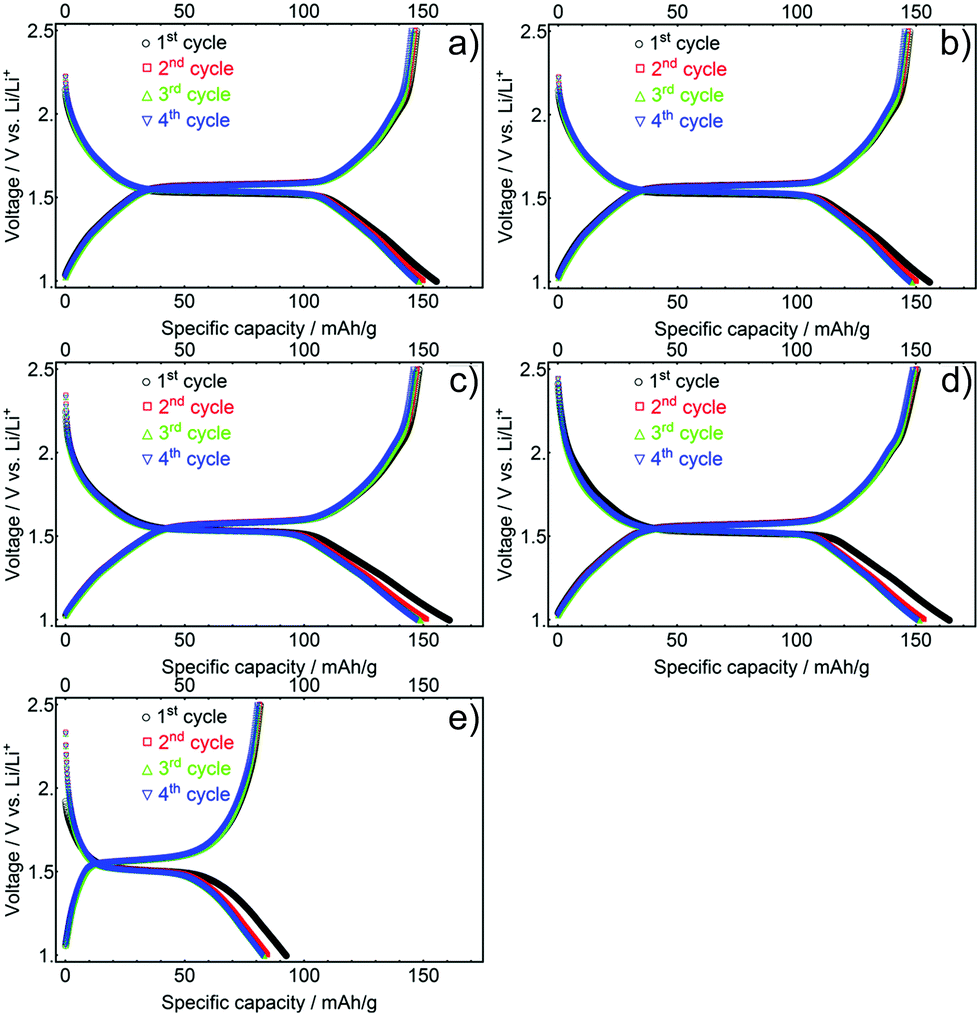 | ||
| Fig. 6 First four galvanostatic discharge and charge profiles of mesoporous LTO-A-600 (a), LTO-A-700 (b), LTO-B-600 (c), LTO-B-700 (d) and LTO-C-700 (e), C-rate of 0.5. | ||
The relatively low capacity of LTO-C-700 probably arises from the low porosity of this material, i.e. its low pore volume and low specific surface area, (Fig. 5b and Table S1, ESI†). While the initial capacities of the LTO-B and LTO-A samples are lower than the theoretical capacity value of 175 mA h g−1, they are similar if not better than the best-performing state-of-the-art mesoporous LTO materials.15,17,20,42 Differences between experimental and theoretical capacities during the initial discharge cycles are commonly justified by surface defects, irreversible lithium insertion, and contaminants like residual trace water common to high-surface area materials.20,41,43,44 It is also possible that the differing pore structures have different electrolyte wettability and interconnectivity, limiting access to electrochemically active material, particularly in LTO-C.
The aforementioned capacity losses during the initial charge–discharge cycle were observed for all samples with Coulombic efficiencies of the initial cycle for the LTO-A-600, LTO-A-700, LTO-B-600, LTO-B-700, and LTO-C-700 being 94.7, 92.3, 92.2, 91.9, and 91.9%, respectively. The efficiency increased upon further cycling, with a value above 97% in the second cycle for all samples. Furthermore, charge/discharge plateau potentials in between 1.5 and 1.6 V were observed for all LTO samples. These plateaus correspond to the topotactic transformation of spinel Li4Ti5O12 into rock salt type Li7Ti5O12, with the exact voltage also affected by the crystallite size of the sample.8,27 The discharge potentials decreased with increasing crystallite size, resulting from a higher calcination temperature, i.e. 1.54 V compared to 1.52 V for calcination at 600 °C (e.g. LTO-A-600 and LTO-B-600) vs. 700 °C (e.g. LTO-A-700, LTO-B-700, and LTO-C-700) Fig. S3 (ESI†).40,45,46
The influence of the current density on the electrochemical performance of the mesoporous LTO microsphere samples was analysed by rate testing. The goal was to quantify the effects of morphology factors including different crystallite sizes, specific surface areas and porosity on the rate performance. The initial charge–discharge profiles for all LTO samples cycled at various C-rates show a decrease in capacity, and an increase in polarization between discharge–charge plateaus as current density is increased, a typical result for rate testing,12 see Fig. 7. LTO-B-700 exhibited the highest specific charge–discharge capacity at every C-rate, followed by the LTO-A-600, LTO-A-700, LTO-B-600 and LTO-C-700. These results are attributed to an interplay between the pore structure, surface area, and crystallite sizes.47–49 For example, the crystallite size is known to influence the specific capacity.40,41,45 This is reflected in the LTO-B-700 and LTO-B-600, samples with relatively similar pore structures (volume and surface area) but different crystallite sizes (LTO-B-700 = 9.78 nm, LTO-B-600 = 8.42 nm). At low C-rates i.e., from 0.5 to 2 C their capacity performance is similar, while it differs greatly at higher C-rates i.e., from 5 to 30 C. At 30 C, the capacity for LTO-B-700 was 113 mA h g−1 while that of LTO-B-600 was only 54 mA h g−1, Table S1 (ESI†). This suggests that the Li host sites in the LTO are somehow restricted by the crystallite boundaries.41,50 However, the capacity does not directly depend on the crystallite size, as is evident from the comparison of LTO-C, with the two LTO-A samples, which have similar crystallite sizes (which are larger than those of the best performing LTO-B-700 sample), but vastly differing specific capacities. This comparison indicates that the smaller surface area and pore volume of the LTO-C sample is also significant (Table S1, ESI†).
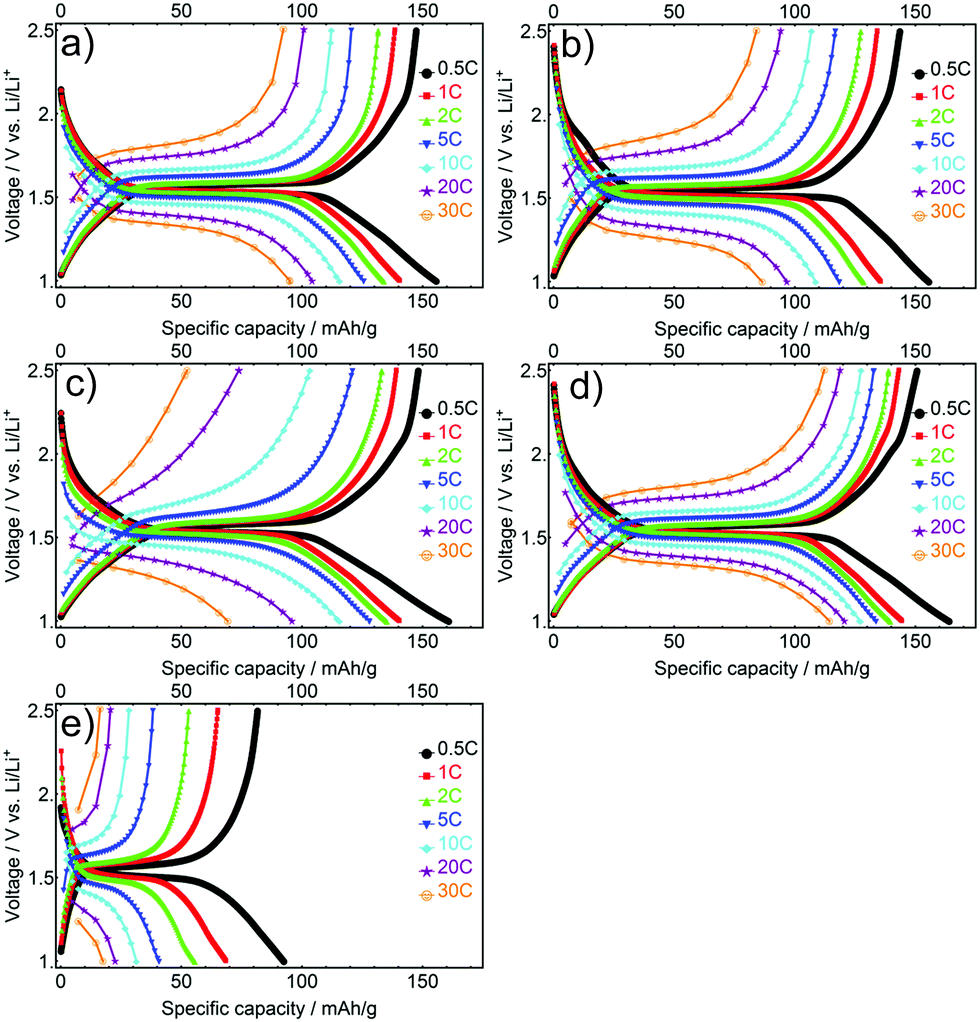 | ||
| Fig. 7 Initial galvanostatic discharge/charge profiles at different C-rates for the mesoporous LTO-A-600 (a), LTO-A-700 (b), LTO-B-600 (c), LTO-B-700 (d) and LTO-C-700 (e). | ||
The full performance of LTO samples was investigated by rate tests, 10 cycles at increasing C-rates, followed by a cycle test of 1000 cycles at a C-rate of 10, Fig. 8.
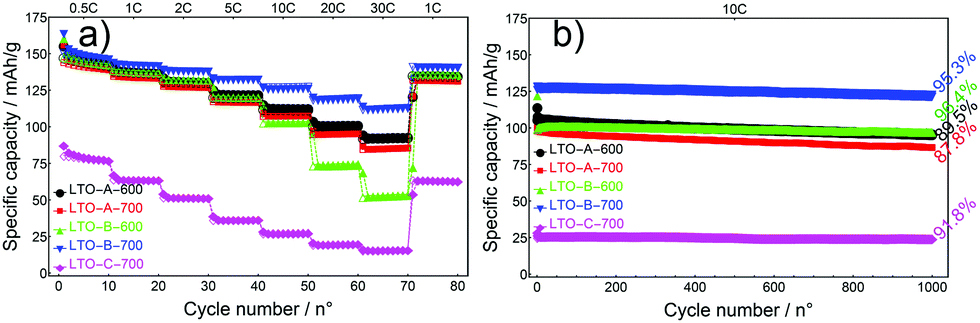 | ||
| Fig. 8 Rate test (a) and cycle test (b) of mesoporous LTO microsphere composite electodes. Cycle testing at a C-rate of 10. Percentages indicate capacity retention during cycle testing. The measurements of each sample in (a) and (b) were carried out with the same cells. | ||
At any given rate, the capacities of all devices were relatively stable except for the initial series at 0.5 C, which reflects the conditioning period of the cells. As the current rates increased from a C-rate of 0.5 to 30, the LTO-A-600, LTO-A-700 and LTO-B-700 samples showed very good rate capabilities i.e., their specific capacities decayed from approx. 143, 139, and 147 mA h g−1 (at a C-rate of 0.5 after 10 cycles) to 93, 85, and 113 mA h g−1 (at a C-rate of 30 after 10 cycles), respectively, Table 2. Also, their specific capacities mostly recovered upon returning to a C-rate of 1 (Fig. 8a, capacity retention after 75 cycles for LTO-A-600, LTO-A-700, LTO-B-600, LTO-B-700, and LTO-C-700 were 97.9%, 98.2%, 98.4%, 98.8%, and 99.5%, respectively). Note that the rate-dependent capacity of LTO-B-600 in Fig. 8a lies below that which could be expected from most key parameters in Table 2. The much larger-sized particles in Fig. 4c1 and c2 (compared to d1, d2), and their eventual packing in a finished composite electrode might, however, account for the low specific capacities of this material at high C-rates.
The long-term cycling at 10 C for 1000 cycles (post EIS, CV and rate capability tests) showed good stability, Fig. 8b. These results show capacity retention of 89.5% for LTO-A-600, 87.8% for LTO-A-700, 96.4% for LTO-B-600, 95.3% for LTO-B-700, and 91.8% for LTO-C-700 after 1000 cycles with the 5th cycle selected as the reference, Table 2. This performance is comparable to previous mesoporous LTO microspheres systems but at a significantly higher cycle number (Table 3), e.g., 94.5% capacity retention after 200 cycles at 4 C by Tang et al.,15 97.4% capacity retention after 100 cycles at 1 C by Shen et al.,17 82% capacity retention after 200 cycles at 1 C by Nugroho et al.,22 86% capacity retention after 100 cycles at 10 C by Lin et al.18
| Sample | Crystallite size (nm) | S BET (m2 g−1) | d pore (nm) | V pore (cm3 g−1) | Specific capacity (mA h g−1) | Capacity retention %/number of cycles/C-rate | |||
|---|---|---|---|---|---|---|---|---|---|
| BJH | BJH | 1 C | 10 C | 20 C | 30 C | ||||
| LTO-B-700* | 9.78 | 110 | 7.17 | 0.167 | 144 | 126 | 119 | 113 | 95%/1000/10 C |
| Tang et al.,15 | n.a. | 165.9 | 9.5 | 0.46 | 150 | 136 | n.a. | 114 | 94.5%/200/4 C |
| Shen et al.,17 | 11 | 159.4 | 4.3 | 0.2 | 157 | 140 | 125 | n.a. | 97.4%/100/1 C |
| Nugroho et al.,22 | 32.2 | 60.2 | n.a. | n.a. | 159 | 117 | 94 | 61 | 82%/200/1 C |
| Lin et al.18 | n.a. | 40.2 | 4.67 | 0.077 | 185 | 115 | n.a. | n.a. | 86%/100/10 C |
These results highlight that the detailed LTO morphology including the crystallinity and sample porosity play a pivotal role in the rate performance and cycling stability. The ability to control these parameters, by employing different annealing temperatures and BCP molar masses, therefore enables the optimisation of the rate capability and cycle stability of LTO material. Specifically, this research suggests that the LTO-B-700 is the most promising candidate for use in LIB electrodes.
Cyclic voltammetry (CV) measurements were taken on each of the LTO samples, using the same cells at 3 different stages, post-assembly, post-rate test, and post-cycle test. The CV results show increasing peak currents ip with increasing scan rate ν, following the Randles–Ševčík equation, which assumes a diffusion limitation of the active species in the electrode solids,
| ip = 0.4463nFAC(nFνD/RT)1/2, | (1) |
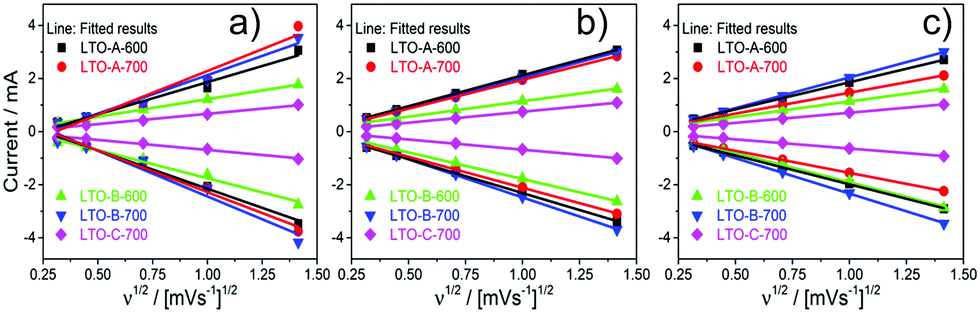 | ||
| Fig. 9 Peak current ipvs. scan rate ν1/2 of the five mesoporous LTO microsphere samples, (a) post-assembly, (b) post-rate test, and (c) post-cycle test. The slopes in the positive peak current region correspond to the anodic processes, the slopes in the negative peak current region correspond to the cathodic processes. | ||
Electrochemical impedance spectroscopy (EIS) was employed to measure the ohmic effects of the mesoporous LTO microsphere morphology. For each sample, EIS measurements were recorded before the rate test and after the cycle test. The EIS spectra of all samples consist of one depressed semicircle in the high-frequency region and a linear tail in the low-frequency region, Fig. 10. The apparent impedance differences after cycling are attributed to the varying LTO morphologies since the samples are otherwise identical, including the Li–metal counter electrode. Note that all samples show higher total impedance before cycling, Fig. 10, suggesting that there is a conditioning period of the cell in addition to the morphological effects. While the lowering impedance as cycling progresses is consistent with an active material that requires a conditioning period before optimal performance, the Li–metal electrode should also be considered. Even though a solid–electrolyte interface (SEI) does not form on LTO under these conditions, Schweikert et al. have identified SEI formation on the Li metal/electrolyte interface in Li/LTO cells as a significant source of initial cell resistance.56,57 This may also be the source of the impedance variation shown in Fig. 10, where a high initial impedance is lowered through the stable forming of a SEI upon conditioning. Therefore, the EIS spectra of Fig. 10b after conditioning were selected as the accurate reflection of the morphological differences of the LTO samples.
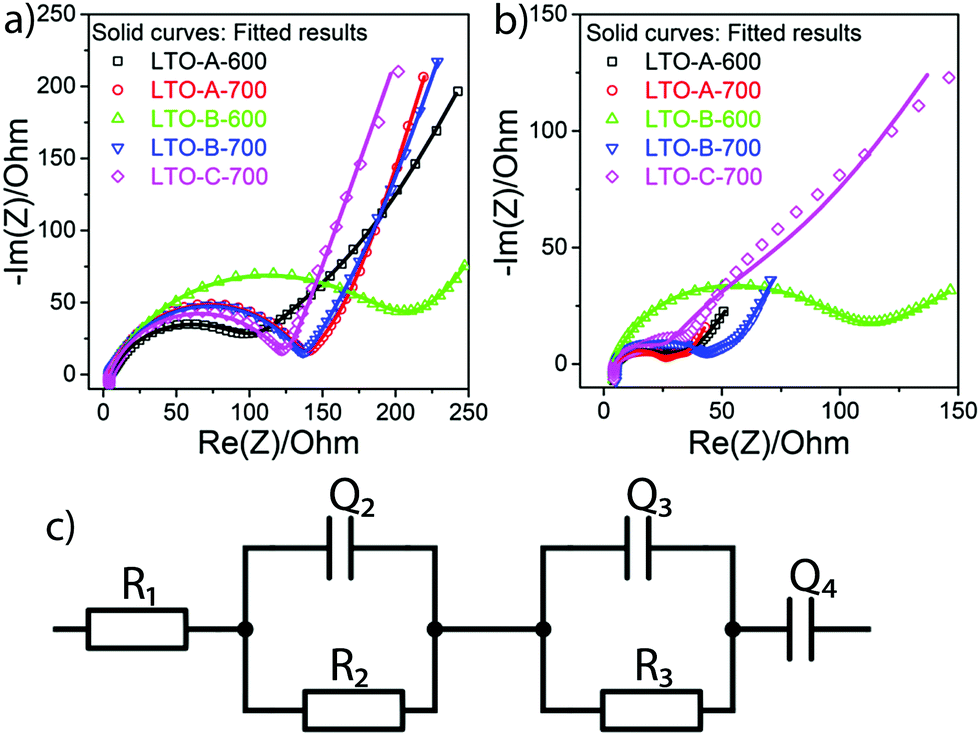 | ||
| Fig. 10 Nyquist plots of Li-metal/LTO half-cells, before the rate test (a), and after the cycle test (b). The equivalent circuit (c) with R1 the apparatus resistance and two R–Q elements, the Li- metal/electrolyte and electrolyte/LTO interfaces. Q4 accounts for Li–ion diffusion at low frequencies. | ||
The total resistance values of all LTO samples were calculated based on the equivalent circuit in Fig. 10c, and are summarized in Table 2. R1, assigned to the test apparatus including the connections and the Swagelok cell, was approximately 4.5 Ω in all measurements, remaining nearly unchanged throughout testing. R2 and R3 constitute two distinct R–Q elements that are related to the charge transfer kinetics at the two electrolyte interfaces of the electrodes. They vary from 48 to 263 Ω across samples and tests. Q4 models the low-frequency Li–ion diffusion in the samples. Post-cycling tests revealed a much lower ohmic resistance of the Li metal/electrolyte interface compared to the other two resistances, so that its R–Q element can be eliminated in the analysis of the EIS measurements.
It is important to reiterate that an equivalent circuit model is a vast simplification of a complex process. The modelling of the two electrode/electrolyte interfaces in particular are unlikely to perfectly capture the entire scope of all electrochemical and transport processes taking place. However, as the cells are similarly conditioned, post-cycle test EIS should primarily reflect impedance changes caused only by the LTO morphological differences and can therefore be used to effectively compare the different morphological effects on the electrochemical performance.
The total resistances before the rate test 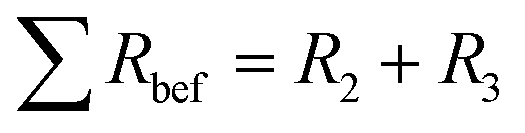 were 210 Ω and 144 Ω for LTO-A-600 and LTO-A-700, respectively, 263 Ω and 159 Ω for LTO-B-600 and LTO-B-700, respectively, and 140 Ω for LTO-C-700. The total resistances after the cycle test
were 210 Ω and 144 Ω for LTO-A-600 and LTO-A-700, respectively, 263 Ω and 159 Ω for LTO-B-600 and LTO-B-700, respectively, and 140 Ω for LTO-C-700. The total resistances after the cycle test 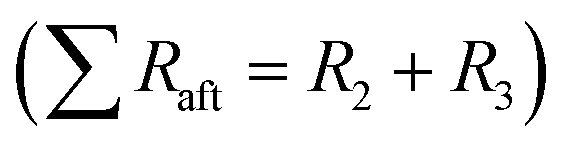 were 58 Ω and 51 Ω for LTO-A-600 and LTO-A-700, respectively, 100 Ω and 48 Ω for LTO-B-600 and LTO-B-700, respectively and 133 Ω for LTO-C-700. Again, the reduction of the total resistance after the cycling test is associated with a combination of a material conditioning and the formation of a stable SEI layer on the Li–metal electrode.20,56,57
were 58 Ω and 51 Ω for LTO-A-600 and LTO-A-700, respectively, 100 Ω and 48 Ω for LTO-B-600 and LTO-B-700, respectively and 133 Ω for LTO-C-700. Again, the reduction of the total resistance after the cycling test is associated with a combination of a material conditioning and the formation of a stable SEI layer on the Li–metal electrode.20,56,57
While Schweikert et al. suggest that different mass loadings of active material may contribute to differences in impedance,56 we conclude here that the LTO morphology rather than material loading lies at the origin of the performance variations, since loadings were relatively similar. Impedance performance alone though, does not guarantee high performing material. The two LTO-A samples exhibit reduced resistance upon cycling, but the relatively low average pore size and low specific surface area (Table 2) result in a loss of specific capacity at high C-rates compared to the LTO-B-700, highlighting the intricate interplay of design parameters that must be controlled to optimize LTO performance.
3 Conclusions
The goal of this study was the fabrication of mesoporous LTO microspheres as an important next step in LIB materials development. By using block copolymer and homopolymer blends to create templates for sol–gel LTO synthesis, we show that both the molar mass of the BCP, the overall BCP/homopolymer composition, and the annealing temperature yields control over particle size, pore size, crystallite size, and specific surface area of electrode materials. This enables the tunability of key material parameters, allowing the improvement of the electrochemical performance of LTO.Specifically, a rise in annealing temperature increased the crystallite size and decreased the specific surface area of the LTO material. For instance, the average crystallite size of LTO annealed at 600 °C was smaller than that of samples annealed at 700 °C (approximately 9 nm vs. 11 nm, respectively). This comes, however, at the cost of specific surface area, which was larger for samples annealed at 600 °C compared to those annealed at 700 °C. The mesoporosity of the LTO spheres was controlled through the molar mass of PS in the BCP, maintaining a constant volume fraction ratio of the blocks, yielding pore sizes spanning 5 nm to 20 nm. An anaerobic calcination step caused the carbonisation of the polymer templates, leading to a nanometer-thin carbon layer which provides good electrical conductivity of the resulting LTO material. The optimised balance of these parameters yielded a material with an excellent electrochemical performance, employing BCP-B annealed at 700 °C for 2.5 h, exhibiting a relatively small particle size, and a large specific surface area combined with a large pore size. Apart from high discharge capacities up to C-rates of 30, electrodes made from mesoporous LTO spheres yielded a capacity retention of 95% after 1000 cycles at a C-rate of 10.
The control over detailed morphology demonstrated by this polymer templating method and its resulting effect on LTO electrode performance suggests that even further increases in rate capability and cycle stability may be possible, opening the door to increased LTO utilisation in commercial lithium batteries.
4 Experimental
4.1 Materials
Polystyrene-b-poly(ethylene oxide) (PS-b-PEO) block copolymers (BCPs) with a total molar mass of Mn = 10-b-3.5 kg mol−1, Mn = 18-b-7.5 kg mol−1, and Mn = 38-b-15 kg mol−1 were purchased from Polymer Source, Inc. Polystyrene (PS) homopolymer with a total molar mass of 35 kg mol−1 was purchased from Sigma-Aldrich. Anhydrous tetrahydrofuran (THF, containing 250 ppm butylated hydroxytoluene (BHT) as inhibitor, ≥99.9%), titanium(IV) isopropoxide (Ti[OCH(CH3)2]4, 97%), 1.0 M lithium ethoxide (CH3CH2OLi) solution in THF, oxalic acid (C2H2O4, puriss. p.a, anhydrous, ≥99.0%), and N-methyl-2-pyrrolidone (NMP) (anhydrous, 99.5%) were purchased from Sigma-Aldrich. Conductive carbon black (Super C65) was kindly provided by Imerys Graphite & Carbon, Switzerland Ltd. Polyvinylidene fluoride (PVDF, Kynar) was provided by ARKEMA Innovative Chemistry. Lithium chips were purchased from Gelon LIB Group and GF/B glass microfiber from Healthcare Life Sciences. 1 M lithium hexafluorophosphate (LiPF6) in 1:1 (v/v) ethylene carbonate (EC):dimethyl carbonate (DMC) was purchased from Solvionic. All chemicals were used as received.
4.2 LTO Synthesis
The preparation of mesoporous LTO microspheres was carried out using a three-neck round-bottom flask, which was vacuum-dried overnight at 100 °C before use. During the synthesis, a constant flow of 2.0 mL min−1 of N2 was maintained while the flask was sealed with a rubber stopper and parafilm. Mixing of all precursor solutions was achieved by magnetic stirring. Prior to the synthesis, a 5.6% (w/w) stock solutions of the PS-b-PEO BCPs, PS HP, and oxalic acid in anhydrous THF were prepared. The quantities of all employed chemicals are listed in Table 4.| Chemical compounds | Amount (mL) |
|---|---|
| THF | 62 |
| CH3CH2OLi | 1.81 |
| Ti[OCH(CH3)2]4 | 0.65 |
| C2H2O4 | 3.956 |
| PS-b-PEO (BCP) | 2.019 |
| PS (HP) | 10.1 |
The LTO synthesis consisted of six steps.
(i) Under constant magnetic stirring, 62 mL THF were first injected into the flask, followed by adding 1.81 mL lithium ethoxide, before slowly adding 0.65 mL titanium(IV) isopropoxide. This initial solution was stirred for two hours, during which its color changed to bright gold before further chemicals were added
(ii) 3.956 mL of oxalic acid were slowly added to the solution, changing its color first to yellow and then back to bright gold. Note that oxalic acid swells the hydrophilic PEO domains24
(iiii) 2.019 mL of a 5.6% (w/w) solution of PS-b-PEO BCP in THF were slowly added to the precursor solution before adding 10.1 mL of a 5.6% (w/w) solution of PS HP in THF, followed by stirring of the precursor solution for two hours
(iv) to evaporate the solvent, the flask was submerged into an oil bath while the temperature was ramped from 40 to 120 °C over the course of two days
(v) the precipitate was vacuum-dried at 100 °C overnight to inhibit water uptake
(vi) to form the spinel Li4Ti5O12 structure, the dried precipitate was calcined at 600 °C or 700 °C (Table 1) in a tube furnace under a constant argon flow of 5 L min−1. This calcination also burns off the organic compounds and partially converts the polymers into a carbon coating.21
4.3 Materials characterisation
X-Ray diffraction (XRD) was performed on a Rigaku Ultima IV equipped with a copper target. Fourier-transform infrared spectroscopy (FTIR) spectra were recorded on a PerkinElmer Spectrum 65 spectrometer between 4000 and 450 cm−1 with a resolution of 8 cm−1, averaging 5 scans per sample. Raman spectroscopy was carried out at room temperature on a custom-built setup using an excitation wavelength of 633 nm at a power of 60 mW (LuxX633, Omicron), and acquisition times of 0.5 s. Scanning electron microscope (SEM) images were acquired on a Tescan Mira 3 LMH scanning electron microscope at accelerating voltages of 10 to 20 kV. Thermogravimetric analysis (TGA) was performed on a Mettler Toledo TGA/DSC 1 instrument in a temperature range of 25 to 600 °C with a heating rate of 10 °C min−1 under N2 flow of 30 mL min−1. The specific surface area and the pore size distribution of the samples were determined with a Micromeritics Gemini V surface area and pore size analyzer.4.4 Electrochemical properties of mesoporous LTO microspheres
To characterise the electrochemical performance of the synthesized mesoporous LTO microspheres, composite electrodes were prepared by mixing the LTO particles with carbon black, and poly(vinylidene fluoride) (PVDF) at a ratio of 8:1:1 using aluminum foil as current collector. A homogeneous slurry was created by mixing all powders using pestle and mortar before adding NMP as a solvent. The slurry was cast onto aluminum foil and subsequently doctor-bladed into a 100 μm thick electrode film, and then dried under a fume hood for two days. The dried electrode film was cut into 7/16 inch diameter discs, vacuum-dried overnight at 100 °C, and then transferred into an argon-filled glovebox for assembly into Swagelok cells. A 1/2 inch diameter lithium metal chip was used as the counter electrode and a Grade GF/B Glass microfiber filter was used as separator. Galvanostatic charge–discharge tests were conducted using an Arbin BT 2043 multiple channel cell test system in a voltage range of 1.0 to 2.5 V (vs. Li+/Li). Cyclic voltammetry (CV) and electrochemical impedance spectroscopy (EIS) were performed with a BioLogic VMP 300 test system. CV was recorded in a voltage range of 1.0 to 2.5 V (vs. Li+/Li), at scan rates of 0.1, 0.2, 0.5, 1.0, and 2.0 mV s−1. EIS was measured in a frequency range of 1 MHz to 100 mHz with a voltage amplitude of 20 mV. CV was performed at different stages: post-assembly, post-rate test (in order of the following C-rates: 0.5, 1, 2, 5, 10, 20, 30, back to 1; 10 cycles at each C-rate) and post-cycle test (C-rate of 10 for 1000 cycles). While, EIS was conducted post-assembly and post-cycle test.
Author contributions
MTN carried out the experiments, PS supervised EIS and CV analysis and discussion, AP discussed material characterization and electrochemical analysis, MGF provided preliminary data, XH, MGF, IG, and US designed the experimental study, IG and US supervised the project. MTN, PS, IG, and US wrote and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Dr Dimitri Vanhecke and Dr Bodo D. Wilts for help interpreting the SEM and Raman data. Access to XRD was kindly provided by the Department of Geosciences, University of Fribourg. The authors gratefully acknowledge the financial support from the Swiss National Science Foundation (SNSF). P. S. has been funded by the SNSF under project number P2FRP2-191846. U. S. has been funded by the SNSF through the NRP70 program, Grant 153764. U. S., I. G., and M. T. N. have been funded by the National Center of Competence in Research (NCCR) Bio-Inspired Materials Grant 51NF40-182881. We acknowledge the support of the Adolphe Merkle Foundation.Notes and references
- G. Zubi, R. Dufo-Lopez, M. Carvalho and G. Pasaoglu, Renewable Sustainable Energy Rev., 2018, 89, 292–308 CrossRef.
- M. Li, J. Lu, Z. Chen and K. Amine, Adv. Mater., 2018, 30, 1800561 CrossRef.
- M. V. Reddy, G. V. Subba Rao and B. V. R. Chowdari, Chem. Rev., 2013, 113, 5364–5457 CrossRef CAS.
- Y. Ding, Z. P. Cano, A. Yu, J. Lu and Z. Chen, Electrochem. Energy Rev., 2019, 2, 1–28 CrossRef CAS.
- G.-N. Zhu, Y.-G. Wang and Y.-Y. Xia, Energy Environ. Sci., 2012, 5, 6652–6667 RSC.
- Z. Chen, H. Li, L. Wu, X. Lu and X. Zhang, Chem. Rec., 2018, 18, 350–380 CrossRef CAS PubMed.
- X. Zeng, M. Li, D. A. El-Hady, W. Alshitari, A. S. Al-Bogami, J. Lu and K. Amine, Adv. Energy Mater., 2019, 9, 1900161 CrossRef.
- X. Sun, P. V. Radovanovic and B. Cui, New J. Chem., 2014, 39, 38–63 RSC.
- T.-F. Yi, S.-Y. Yang and Y. Xie, J. Mater. Chem. A, 2015, 3, 5750–5777 RSC.
- S. Schweidler, L. de Biasi, A. Schiele, P. Hartmann, T. Brezesinski and J. Janek, J. Phys. Chem. C, 2018, 122, 8829–8835 CrossRef CAS.
- T. Yuan, Z. Tan, C. Ma, J. Yang, Z.-F. Ma and S. Zheng, Adv. Energy Mater., 2017, 7, 1601625 CrossRef.
- B. Zhao, R. Ran, M. Liu and Z. Shao, Mater. Sci. Eng., R, 2015, 98, 1–71 CrossRef.
- G. Yang and S.-J. Park, J. Mater. Chem. A, 2020, 8, 2627–2636 RSC.
- J. Liu, A. Wei, G. Pan, S. Shen, Z. Xiao, Y. Zhao and X. Xia, J. Energy Chem., 2021, 54, 754–760 CrossRef.
- Y. Tang, L. Yang, Z. Qiu and J. Huang, J. Mater. Chem., 2009, 19, 5980–5984 RSC.
- L. Yu, H. B. Wu and X. W. D. Lou, Adv. Mater., 2013, 25, 2296–2300 CrossRef CAS PubMed.
- L. Shen, C. Yuan, H. Luo, X. Zhang, L. Chen and H. Li, J. Mater. Chem., 2011, 21, 14414–14416 RSC.
- C. Lin, X. Fan, Y. Xin, F. Cheng, M. On Lai, H. Zhou and L. Lu, Nanoscale, 2014, 6, 6651–6660 RSC.
- M. G. Fischer, X. Hua, B. D. Wilts, I. Gunkel, T. M. Bennett and U. Steiner, ACS Appl. Mater. Interfaces, 2017, 9, 22388–22397 CrossRef CAS PubMed.
- E. Kang, Y. S. Jung, G.-H. Kim, J. Chun, U. Wiesner, A. C. Dillon, J. K. Kim and J. Lee, Adv. Funct. Mater., 2011, 21, 4349–4357 CrossRef CAS.
- M. Christopher Orilall and U. Wiesner, Chem. Soc. Rev., 2011, 40, 520–535 RSC.
- A. Nugroho, K. Y. Chung and J. Kim, J. Phys. Chem. C, 2014, 118, 183–193 CrossRef CAS.
- J. Ma, Y. Wei, L. Gan, C. Wang, H. Xia, W. Lv, J. Li, B. Li, Q.-H. Yang, F. Kang and Y.-B. He, J. Mater. Chem. A, 2019, 7, 1168–1176 RSC.
- M. Stefik, J. Song, H. Sai, S. Guldin, P. Boldrighini, M. Christopher Orilall, U. Steiner, S. M. Gruner and U. Wiesner, J. Mater. Chem. A, 2015, 3, 11478–11492 RSC.
- G. J. d. A. A. Soler-Illia, A. Louis and C. Sanchez, Chem. Mater., 2002, 14, 750–759 CrossRef.
- A. L. Patterson, Phys. Rev., 1939, 56, 978–982 CrossRef CAS.
- L. Aldon, P. Kubiak, M. Womes, J. C. Jumas, J. Olivier-Fourcade, J. L. Tirado, J. I. Corredor and C. Perez, Vicente, Chem. Mater., 2004, 16, 5721–5725 CrossRef CAS.
- C. M. Julien, M. Massot and K. Zaghib, J. Power Sources, 2004, 136, 72–79 CrossRef CAS.
- I. A. Leonidov, O. N. Leonidova, L. A. Perelyaeva, R. F. Samigullina, S. A. Kovyazina and M. V. Patrakeev, Phys. Solid State, 2003, 45, 2183–2188 CrossRef CAS.
- X. Li, H.-C. Lin, W.-J. Cui, Q. Xiao and J.-B. Zhao, ACS Appl. Mater. Interfaces, 2014, 6, 7895–7901 CrossRef CAS.
- K. Mukai, Y. Kato and H. Nakano, J. Phys. Chem. C, 2014, 118, 2992–2999 CrossRef CAS.
- T. Yuan, X. Yu, R. Cai, Y. Zhou and Z. Shao, J. Power Sources, 2010, 195, 4997–5004 CrossRef CAS.
- A. Dillon, M. Yudasaka and M. Dresselhaus, J. Nanosci. Nanotechnol., 2004, 4, 691–703 CrossRef CAS.
- Y. Xu, X. Chen, L. Wang, K. Bei, J. Wang, I.-M. Chou and Z. Pan, J. Raman Spectrosc., 2020, 51, 1874–1884 CrossRef CAS.
- Y. Gao, Z. Wang and L. Chen, J. Power Sources, 2014, 245, 684–690 CrossRef CAS.
- M. Kruk and M. Jaroniec, Chem. Mater., 2001, 13, 3169–3183 CrossRef CAS.
- P. A. Monson, Microporous Mesoporous Mater., 2012, 160, 47–66 CrossRef CAS.
- Q. Wang, H. Wang, Y. Wu, L. Cheng, L. Zhu, J. Zhu, Z. Li and Y. Ke, J. Sol-Gel Sci. Technol., 2020, 94, 186–194 CrossRef CAS.
- E. Pohjalainen, T. Rauhala, M. Valkeapaa, J. Kallioinen and T. Kallio, J. Phys. Chem. C, 2015, 119, 2277–2283 CrossRef CAS.
- M. Wagemaker and F. M. Mulder, Acc. Chem. Res., 2013, 46, 1206–1215 CrossRef CAS PubMed.
- W. J. H. Borghols, M. Wagemaker, U. Lafont, E. M. Kelder and F. M. Mulder, J. Am. Chem. Soc., 2009, 131, 17786–17792 CrossRef CAS PubMed.
- X. Zhang, C. Lu, H. Peng, X. Wang, Y. Zhang, Z. Wang, Y. Zhong and G. Wang, Electrochim. Acta, 2017, 246, 1237–1247 CrossRef CAS.
- B. Kurc, Ionics, 2018, 24, 121–131 CrossRef CAS.
- J. Yue, C. Suchomski, T. Brezesinski and B. M. Smarsly, ChemNanoMat, 2015, 1, 415–421 CrossRef CAS.
- S. Ganapathy and M. Wagemaker, ACS Nano, 2012, 6, 8702–8712 CrossRef CAS PubMed.
- A. Van der Ven and M. Wagemaker, Electrochem. Commun., 2009, 11, 881–884 CrossRef CAS.
- Y. Zhang, Y. Luo, Y. Chen, T. Lu, L. Yan, X. Cui and J. Xie, ACS Appl. Mater. Interfaces, 2017, 9, 17145–17154 CrossRef CAS PubMed.
- H. Utsunomiya, T. Nakajima, Y. Ohzawa, Z. Mazej, B. Zemva and M. Endo, J. Power Sources, 2010, 195, 6805–6810 CrossRef CAS.
- J. M. Feckl, K. Fominykh, M. Doblinger, D. Fattakhova-Rohlfing and T. Bein, Angew. Chem., Int. Ed., 2012, 51, 7459–7463 CrossRef CAS PubMed.
- L. Kavan, J. Prochazka, T. M. Spitler, M. Kalbac, M. Zukalova, T. Drezen and M. Gratzel, J. Electrochem. Soc., 2003, 150, A1000 CrossRef CAS.
- J. Rikarte, B. Acebedo, A. Vilalta-Clemente, F. Bonilla, A. J. Wilkinson, M. Galceran, A. Lousa, J. Rubio-Zuazo and M. A. Munoz-Marquez, Adv. Mater. Interfaces, 2020, 7, 1902164 CrossRef CAS.
- M. Kitta, T. Akita, Y. Maeda and M. Kohyama, Langmuir, 2012, 28, 12384–12392 CrossRef CAS.
- D. Shao, J. He, Y. Luo, W. Liu, X. Yu and Y. Fang, J. Solid State Electrochem., 2012, 16, 2047–2053 CrossRef CAS.
- S. Hao, X. Xiao, Z. Hu, L. Sun, S. Han, D. Chen and X. Liu, J. Phys. Chem. C, 2013, 117, 26889–26895 CrossRef CAS.
- N. Elgrishi, K. J. Rountree, B. D. McCarthy, E. S. Rountree, T. T. Eisenhart and J. L. Dempsey, J. Chem. Educ., 2018, 95, 197–206 CrossRef CAS.
- N. Schweikert, H. Hahn and S. Indris, Phys. Chem. Chem. Phys., 2011, 13, 6234–6240 RSC.
- N. Schweikert, R. Heinzmann, A. Eichhofer, H. Hahn and S. Indris, Solid State Ionics, 2012, 226, 15–23 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ma00708d |
| ‡ Present address: BeDimensional S. p. A., Via Lungo Torrente Secca 30R, 16163 Genova, Italia |
| § Present address: Sensirion AG, Laubisruetistrasse 50, 8712 Staefa ZH, Switzerland. |
| This journal is © The Royal Society of Chemistry 2022 |