
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOptimization of a CE-ICP-MS/MS method for the investigation of liposome–cisplatin nanosystems and their interactions with transferrin†
Anna Maria
Wróblewska
a,
Jan
Samsonowicz-Górski
a,
Ewelina
Kamińska
a,
Marcin
Drozd
bc and
Magdalena
Matczuk
 *a
*a
aChair of Analytical Chemistry, Faculty of Chemistry, Warsaw University of Technology, Noakowskiego St. 3, 00-664 Warsaw, Poland. E-mail: magdalena.matczuk@pw.edu.pl; Tel: +48 222347719
bChair of Medical Biotechnology, Faculty of Chemistry, Warsaw University of Technology, Noakowskiego St. 3, 00-664 Warsaw, Poland
cCentre for Advanced Materials and Technologies CEZAMAT, Warsaw University of Technology, Poleczki Str. 19, 02-822 Warsaw, Poland
First published on 1st June 2022
Abstract
As chemotherapy suffers from the limitation of non-selectivity towards cancer cells, resulting in severe side effects, the targeted delivery of anticancer drugs such as cisplatin using nontoxic nanomaterials has been under extensive examination. In such an approach, qualities such as biocompatibility, biodegradability, ease of synthesis (including the surface modifying possibility with targeting ligands), and the tunable encapsulation of chemotherapeutics make liposomes superior to other nanomaterials as drug nanocarriers. Despite ten liposome-drug formulations being approved for the market, none of them concern platinum-based anticancer drugs. This can be due to the problematic multistep synthesis of such systems and the use of ineffective analytical tools for their characterization. Consequently, the current study aims to propose a straightforward protocol for liposome–cisplatin system synthesis as well as a comprehensive method for their characterization based on the combination of capillary electrophoresis (CE) and inductively coupled plasma-tandem mass spectrometry (ICP-MS/MS). Although the application of CE-ICP-MS has been reported in previous studies on liposome–cisplatin systems, the advantages of the optimized method proposed in our study include not only the possibility of the direct quantitative monitoring of liposome–metallodrug systems (all analytes of interest) but also their interactions with proteins in close-to-physiological conditions of analysis due to the applied tandem mass mode of the spectrometer. The present approach thereby facilitates overcoming the limitations of the strategies previously described in the literature, especially in the case of sulfur monitoring.
1. Introduction
Currently, an increasing and significant health liability, especially in developed countries, is neoplastic disease. Despite the progress in medicine, the primary cancer treatment method remains chemotherapy, which suffers from many limitations. The real challenges facing novel approaches are the nonselective activity of the utilized cytotoxic chemotherapeutics towards cancer cells and the increasing mechanisms of resistance initiation. For that reason, increased localized efficacy of anticancer drug activity is essential, especially in the case of cisplatin—the first metal-based and one of the most frequently used anticancer drugs (ca. 50% of patients, mainly suffering from solid tumors, are treated with cisplatin).1 A solution can be found in augmenting existing therapies with nanomaterials, which have been under extensive examination. In such an approach, nanomaterials are used as drug nanocarriers that can deliver them selectively to cancer cells via passive or active transport mechanisms. The formed nanostructure–drug combinations are commonly called drug targeted delivery systems (DTDSs). The enhanced targeting capability of DTDSs can favor the reduction of the systemic toxicity of drugs and thereby improve treatment efficacy.2,3 Among the many available nanoscale drug carriers, liposomes are considered the nanomaterials with the greatest potential in clinical application. A majority of the FDA-approved nanomaterial–drug formulations employ liposomes.2 This type of sphere-shaped vesicle is composed of amphipathic phospholipids forming one or more bilayers surrounding an aqueous interior. The crucial feature of liposomes is biocompatibility due to their structure mimicking the cell membrane, which enables the incorporation of the liposome content into the cell interior. The additional advantage of using liposomes as nanocarriers is the possibility of enclosing various compounds (both lipophilic and hydrophilic) with various physicochemical properties.4 Despite the ten liposomal-based formulations already used, none of them are dedicated to delivering the well-known and frequently used platinum-based anticancer drugs. Therefore, the optimization of vehicle composition can be problematic, resulting in insufficient selectivity of drug transportation. In general, liposomes exploit the mechanism of passive drug delivery (via the enhanced permeability and retention effect) into the tumor. The efficiency of this targeting mode is different in different cancer types. Another substantial issue is the monitoring of the interactions of liposome-based DTDSs with plasma proteins, which can lead to the inactivation/decomposition of the DTDSs or the drug leaking during their trafficking into cell targets. Hence, great importance should be afforded to the development of improved liposomal formulations characterized by low immune recognition, enhanced stability, better targeting capability (based on tuned bilayer composition), and high cisplatin loading. Therefore, it is crucial to use advanced and comprehensive analytical tools that allow for the online quantitative monitoring of liposome-based cisplatin delivery system formation with the simultaneous analysis of each sample's component (product and substrates) at trace levels. As it is comprehensive but still not widespread in metallodrug targeted delivery system investigation, the application of the combination of separation modules and inductively coupled plasma-mass spectrometry (ICP-MS) has been considered.5–7 The ICP-MS technique, which features sensitivity and multi-elemental and isotopically selective analysis, is one of the few techniques that opens up the possibility of total phosphorus (liposome marker) quantification. Nevertheless, the direct ICP-MS analysis of phosphorus can be handicapped by the multiple spectral interferences8 resulting from its ionization under atmospheric pressure due to oxygen. As a solution, its diversification by introducing a collision or reaction cell module has been proposed, but until now, only a few protocols of DTDSs (composed of liposomes and platinum-based anticancer drugs) employing ICP-MS-based methods can be found in the literature. Gammelgaard and coworkers reviewed feasible methods based on ICP-MS equipped with a dynamic reaction cell (DRC), which enabled the simultaneous monitoring of phosphorus (31P+) and platinum (195Pt+, a cisplatin/oxaliplatin marker) isotopes and their quantification in analyzed samples. As collision gases in DRC, xenon7,9 or argon10–12 were used since they were found to be superior to oxygen in terms of interference removal. In initial studies, DRC-ICP-MS was coupled to size-exclusion chromatography9 (SEC-DRC-ICP-MS) to simultaneously monitor 31P+ and 195Pt+ species corresponding to liposomes and free and liposome-entrapped oxaliplatin forms. Although applicable in such DTDS characterization (including drug release monitoring), the method suffered from limitations related to liposomes and the column's stationary phase interactions. Without prior saturation, significant losses of the liposome's signal intensities were observed, and drug leakage from liposomes might occur.13 While chromatography-assisted methods feature good resolution (essential in identifying particular analytes), the conditions of the performed analyses are not appropriate to monitor the potential behavior of the formed DTDSs under physiological conditions after intravenous administration. With respect to maintaining a close-to-physiological environment during the analysis, standard eluents abundant in surfactants and organic solvents that may contribute to lipid bilayer disruption should not be used.Moreover, the presence of sulfur (protein marker in ICP-MS-based studies) in components used to prepare the mobile phase makes it impossible to examine protein influence on DTDSs directly. Additionally, the sample's and eluent's consumptions are substantial. Alternatively, capillary electrophoresis (CE) can be used as a separation module in DTDS examination. Besides its higher separation efficiency towards charged compounds and less consumption of reagents, the CE technique allows for adjusting the background electrolyte (BGE) to physiological fluids. Therefore, in further DTDS encapsulation, leakage, and stability (also in human plasma) studies by the authors, CE and DRC-ICP-MS coupling were used.10–12
Nonetheless, the performed studies on the human plasma (HS) protein influence on encapsulated vesicles are doubtful since the obtained results refer only to 31P+ and 195Pt+ analytes but not to sulfur isotopes (markers of proteins) directly. Moreover, the proposed BGE contained SDS and did not resemble human blood's natural conditions. Furthermore, SDS along with 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES) in BGE precludes the determination of protein impact on DTDSs, which could be assessed by sulfur monitoring5 and analyzing HS-containing samples. The DRC-ICP-MS system does not offer utility either due to the significant interference in the sulfur (32S+) case (mostly 16O2+ and 14N16OH2+).
In this study, we propose a solution to the problems mentioned above by utilizing a combination of CE and ICP-tandem mass spectrometry (ICP-MS/MS) equipped with a collision-reaction cell (CRC) with oxygen as a reaction gas. In ICP-MS/MS, the CRC is placed between two quadrupoles to obtain more selective ion filtering (the double filtering effect of ions).14 This results in greater sensitivity and significantly limits the interferences of sulfur and phosphorus, which can be quantified as 32S16O+ and 31P16O+ signals. Until now, the CE-ICP-MS/MS combination has been employed only once in a liposome-based DTDS survey by Ansar and Mudalige, who quantified the internal- and external-liposome sulfate in various (FDA-approved) liposome–doxorubicin formulations to control the doxorubicin encapsulation efficiency.14 However, the applied method did not provide information on the entrapped drug and proteinaceous sample analysis. In comparison, our research aims to optimize the CE-ICP-MS/MS method to simultaneously quantify proteins, liposomes, and Pt-based drug forms. The improved method presented herein offers an advantage of the direct quantification of protein-modified liposomes (as well as future targeting ligands) and DTDS–human serum protein interactions (by 32S16O+ signal monitoring), which is not affected by sulfur-related spectral interferences (compared to the DRC-ICP-MS-based method). However, the second aspect, not implemented until now, is performing the analyses under simulated physiological conditions during species separation.
2. Experimental
2.1. Reagents
Buffer solutions (5, 10, and 20 mM Trizma® base pH 7.4, 10 mM NH4HCO3 pH 7.4), incubation solution (1 mM calcium gluconate in 0.3% NaCl aqueous solution; IS), and 1 M NaOH solutions were prepared by dissolving analytical grade reagents (Sigma-Aldrich, USA) in ultrapure water (R ≥ 15 MΩ cm−1, Millipore Elix 3, Merck Millipore, France). The pH of the buffers was measured using an Elmetron pH-meter CP-411 (Poland) and adjusted to the desired value with acetic acid (99.9% trace metal basis, Honeywell, Poland). Stock solutions of monitored ions for ICP-MS/MS tuning (7Li, 59Co, 89Y, 140Ce, 205Tl; each at a concentration of 10 μg L−1) and calibration (115In and 195Pt at a concentration of 10 μg L−1, and 31P at 100 μg L−1) were obtained by the dilution of commercial standards (1 g L−1, Merck Millipore, Germany) in trace 2% HNO3 solution. A 2.5 μg L−1 indium solution of 10-times diluted BGE was used as a sheath liquid. All the solutions mentioned above were stored in a fridge in the dark. All used lipids of analytical grade were obtained from commercial sources: 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), and cholesterol (Chol) were purchased from Sigma-Aldrich (USA), hydrogenated soy phosphatidylcholine (HSPC), N-(carbonyl-methoxypolyethylene glycol 2000)-1,2-distearoyl-sn-glycero-3-phosphoethanolamine (DSPE-PEG2000), and 1,2-dioctadecanoyl-sn-glycero-3-phospho-(1-rac-glycerol) monosodium salt (DSPG-Na) were purchased from Avanti Polar Lipids Inc. (USA). Cis-diamminedichloroplatinum(II) (cisplatin, ≥99.9% trace metal basis, CDDP), oxaliplatin ([(1R,2R)-2-azanidylcyclohexyl]azanide, EP Reference Standard), and transferrin (human, ≥98%) were purchased from Sigma-Aldrich (USA). Anhydrous ethanol used for liposome preparation was of LC-MS analytical grade (Merck, USA). Methanol (LC-MS purity) was purchased from POCH (Poland).2.2. Synthesis of liposomes
Liposomes were synthesized using a sonication method15,16 and the ethanol injection method (EIM).7,17,18 In the sonication method, the defined portions of lipids (Table S1, ESI†) were dissolved in 1 mL IS and settled in an ultrasound bath (Bandelin Sonorex DT 52 H, Germany) at 60 °C for 20 min. Then, the samples were cooled at room temperature (RT) and filtered through polytetrafluoroethylene membrane (PTFE) filters (0.45 μm pore size; Roth, Germany). The filtrate was stored in a fridge and warmed to RT before CE-ICP-MS/MS analysis.In the second approach, first, IS was warmed up to 70 °C on a Multi-Therm incubator (Benchmark, USA). Subsequently, appropriate amounts of lipids were dissolved in 1 mL anhydrous ethanol at 50 °C, injected rapidly into 2 mL IS and stirred intensively (1000 rpm) at a temperature of 70 °C. Afterwards, the obtained mixture was set in an incubator (70 °C for 30 minutes at 1000 rpm, then 60 °C for ca. 2 h at 1000 rpm) to evaporate ethanol, resulting in liposome self-assembly (the concentration of phospholipids was ca. 10 mM). The obtained emulsions were cooled to RT and then filtered through PTFE 0.45 μm filters to remove non-liposomal agglomerates. The filtrate was stored in a fridge and warmed to RT before CE-ICP-MS/MS analysis. Liposome–cisplatin systems were prepared similarly to the non-encapsulated liposomes, but the lipids dissolved in ethanol were injected into IS containing cisplatin (0.2 mg mL−1). Cisplatin samples were prepared by dissolving appropriate amounts in IS (0.05 mg mL−1). During the investigation of drug encapsulation, the obtained emulsions were dialyzed (instead of filtration) using cellulose membranes (Sigma-Aldrich, Germany) for 1 h to remove the excess drug (up to 100 times more volume of the IS).
New samples of liposomes, cisplatin, and DTDSs were prepared each day. The pH values of the obtained liposomal suspensions were measured using an ERH-13-6 microelectrode (Hydromet, Poland).
2.3. DTDSs and transferrin interaction investigation
The current study investigated the impact of one of the HS proteins, namely transferrin, on the formed liposome–cisplatin systems. For this, transferrin was dissolved in 100 mM NaCl solution (3 mg mL−1) and mixed with liposome–cisplatin systems in a volume ratio of 1/1. The samples were then incubated at 37 °C under stirring (400 rpm) and measured after specified time intervals.2.4. DLS and ζ-potential measurements
Dynamic light scattering (DLS) and ζ-potential measurements were performed on a Zetasizer Nano ZS (Malvern Panalytical, UK) at 25 °C to obtain information on the unloaded liposomes, hydrodynamic size distribution, surface charge, and dispersion stability. Before DLS and ζ-potential analyses (conducted in 4 replications), each prepared liposomal suspension (not filtered) was diluted 200 times with IS to obtain optimal scattering intensity. The measurements were carried out in disposable polystyrene cuvettes using dip cells for ζ-potential equipped with palladium electrodes.2.5. CE-ICP-MS/MS instrumentation
During the investigation, a CE-ICP-MS/MS coupling was used, which employed a 7100 CE system (Agilent Technologies, Waldbronn, Germany) and an 8900 ICP-MS Triple Quad (equipped with a CRC, Agilent Technologies, USA). A CEI-100 nebulizer interface (Teledyne CETAC Technologies, USA) was used as a sample introduction system. It was composed of a low-volume spray chamber, a cross-piece merging the sheath liquid flow (containing indium, an internal standard), and a grounded platinum wire. The closing of the electrical circuit of the CE was provided by the constant flow of the sheath liquid (maintained by the self-aspiration mode of the nebulizer). Every day, CE-ICP-MS/MS analysis was initiated when the 115In+ signal was sufficiently high (counts per second > 5000, cps) and stable (relative standard deviation < 2%, RSD). The sheath liquid flow was monitored to control the instrument stability and nebulization efficiency. A thermostat maintained the temperatures of the sample tray and the capillary cassette (37 °C). An Agilent MassHunter Workstation and the Agilent Chemstation software were used for instrument control and data collection.A capillary (fused silica, 70 cm length, o.d. 375 μm, i.d. 75 μm) was conditioned with 1 M NaOH (15 min), H2O (10 min), and BGE (15 min) each day. Between analyses, the capillary was rinsed with 1 M NaOH, a washing mixture (1 M NaOH, methanol, and water; 1/2/1, v/v/v), H2O (0.5 min), and BGE (1 min).
3. Results and discussion
3.1. Liposome synthesis and characterization
During the investigation, liposomes with a simple composition of phospholipid bilayers were tested, the formation of which is described in the literature, to assess the applicability of the optimized CE-ICP-MS/MS analytical method. Although these vesicles (varying in composition) have been synthesized using various approaches, in this study, two simple and common methods were used: EIM and sonication. Initially, to confirm the liposome formation repeatability, and assess their stability and size, the DLS and ζ-potential measurements were conducted. In the case of liposomes formed by the sonication method (Samples S1 and S2; Table S1, ESI†), the vesicles featured distinct differences in hydrodynamic diameters and low stability; the outcomes were unrepeatable and the liposomes tended to aggregate. This was reflected in the significant variation in their hydrodynamic diameters and high polydispersity indexes. Using the EIM approach, three types of liposome compositions (Samples: E1, E4, E6; Table S1, ESI†) were tested and analyzed.According to the obtained results, the highest uniformity of the mean hydrodynamic diameter values and a low polydispersity index were observed in the case of E6 liposomes (d = 67.51 ± 1.96 nm, PDI = 0.189 ± 0.022; Fig. S1, ESI†), which were prepared with DSPE-PEG2000/Chol/DOPC in a molar ratio of 10/36/54. However, the highest stability during incubation at RT and the lowest polydispersity were observed in the case of E4 liposomes (d = 135.50 ± 2.21 nm, PDI = 0.095 ± 0.014; Fig. S2, ESI†), which were prepared with DSPE-PEG2000/HSPC/Chol in a molar ratio of 5.3/56.3/38.4. Due to the greater stability (up to 3 days) and size of the formed vesicles (which is related to enhanced drug encapsulation), the E4 liposomes were chosen for further analysis. All liposome types feature slightly anionic character and small standard deviation values of ζ-potential, indicating insubstantial differences in the liposome surface charge and thus in the bilayer composition. Therefore, the results verified that EIM is an efficient and straightforward method leading to the formation of stable liposomes. The presence of groups capable of protonation/deprotonation (phosphate and amine) shifts the pH of the liposome-containing samples towards lower values (E1 liposomes, 5.99; E4, 6.02; E6, 5.76; IS, 6.13).
3.2. Oxygen flow rate in CRC optimization
A tandem mass spectrometer (triple quadrupole) was used in the study to eliminate the sulfur and phosphorus spectral interferences. Indium was used as an internal standard since it is not a source of interference for any of the analyzed elements.11 Oxygen was used a reaction/collision gas in CRC, which improves the accuracy of sulfur measurements.19 The first quadrupole filters the selected ions with a mass-to-charge ratio (m/z) equal to 31, 32, and 195, corresponding to 31P+, 32S+, and 195Pt+. In the presence of oxygen in CRC, both sulfur20 and phosphorus undergo oxidation (Fig. S3 and S4, ESI†), which enables their measurements by the next quadrupole in a m/z shift-mode of +16, corresponding to 32S16O+ (m/z = 48) and 31P16O+ (m/z = 47). Platinum did not undergo oxidation and was indicated only as a 195Pt+ ion (on-mass mode) (Fig. S5†). In order to obtain the highest phosphorus conversion rate into an oxide form and its highest signals, along with minimizing the background level, the optimization of the oxygen flow rate was performed. In this regard, the signal-to-noise ratio (S/N) was calculated based on average counts of 31P+ and 31P16O+ ions for calibration and control (10 μg L−1 of 115In in 2% HNO3) (Fig. 1). According to our previous research,20 where the evaluation of the oxygen flow rate in the platinum and sulfur cases was performed in the same manner, the increase in O2 flow contributes to the slight growth of platinum S/N, while the sulfur S/N remains stable. An O2 flow of 0.39 mL min−1 (26%) was chosen as optimal since it ensured almost the highest S/N towards an m/z of 47 and a simultaneously high phosphorus oxidation rate. For this value, the 31P+ → 31P16O+ conversion rate yielded 72%. The chosen flow value did not affect the sensitivity towards platinum and sulfur.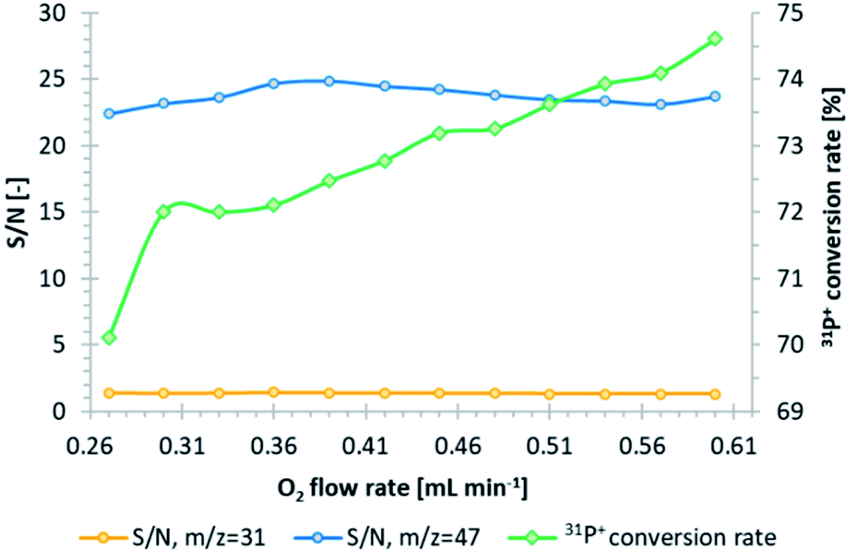 | ||
| Fig. 1 Evaluation of the oxygen flow rate in CRC, based on S/N in the case of m/z equal to 31 (orange line) and 47 (blue line), and the estimation of the 31P+ to 31P16O+ conversion rate (green line). | ||
3.3. CE separation condition optimization
Numerous examples of BGE used in liposome sample electrophoretic separation (e.g., phosphate buffer) can be found in the literature. However, BGE should not contain any phosphorus or sulfur ions that could coincide with the analytes of interest due to the aim of conducting optimization. Additionally, electrolytes should feature a pronounced buffering capacity close to pH 7.4 (physiological) to mimic the liposome environment after intravenous administration. Therefore, the crucial issue was to find appropriate chemicals that could be used at trace levels. Then, to obtain the best signal resolution (corresponding to free cisplatin, liposomes, and DTDSs), the highest peak intensity, and the best analyte recovery from the capillary, the optimization of the BGE and its concentration, the voltage applied during the analysis, and the sample volume should be performed.To fulfill the requirements concerning the BGE, two solutions were proposed: ammonium bicarbonate (NH4HCO3) and Trizma® Base (Tris). Both electrolytes (10 mM, pH 7.4) were compared under the criterion of their resolution (based on cisplatin and non-encapsulated liposome (substrates) migration times, MTs) and signal shape and intensity (based on the peak area, normalized by the average value of 115In signal intensity). Initially, electrophoretic separation was conducted at a voltage of +15 kV and a sample loading of 350 mbar × 5 s. The resolution (R) was calculated based on the cisplatin and liposome peak MTs and width at the base (eqn (1), ESI†). Additionally, the symmetry (S) of the phosphorus peaks was calculated (eqn (2), ESI†).
Both BGE types provided the initial separation of signals corresponding to the substrates. According to the calculations, better resolution and symmetry of the peak outcomes were observed in the case of the Tris electrolyte (1.54 and 1.02, respectively). The signals registered for NH4HCO3 as the BGE were broader, resulting in low resolution and poor symmetry (1.03 and 1.37, respectively). Moreover, greater peak areas (for both the 195Pt+ and 31P16O+ signals) were obtained in the case of Tris. As a result, for further analysis, Tris was chosen due to its superiority over the NH4HCO3 electrolyte.
Afterwards, the impact of BGE concentration on the liposome signals was examined (5, 10, and 20 mM), and the obtained electropherograms are presented in Fig. S6 (ESI).† The highest and most symmetric signals were registered when Tris solutions at 5 mM and 10 mM were used. As the optimal concentration of the BGE, 5 mM was chosen since compared to 10 mM, the current during the electrophoretic run was not affected, resulting in a stable signal baseline. In the case of the Tris concentration of 20 mM, the 31P16O+ signal was barely visible.
The next optimized operational parameter was the voltage applied during analysis. Due to the necessity of maintaining a persistent flow of the sheath liquid, the voltage and amperage during the electrophoretic separation should not exceed +25 kV and 50 μA, respectively. Therefore, to improve the separation (again, the cisplatin and liposomes samples were probed), the evaluated values ranged from +15 (∼4 μA) to +25 kV (∼8 μA). The value of +15 kV was chosen as optimal, as it ensured a stable amperage (not affecting the sheath liquid flow) and the best resolution.
Lastly, the sample volume was optimized to achieve the highest signal intensity obtained without impairing the recoveries of analytes from the capillary. For this purpose, various values of hydrodynamic injection pressure and time intervals were tested. Based on the best ratio of the registered signal's area to the applied sample volume, the injection with a pressure of 30 mbar for 5 s (3.7 nL of a sample loaded into the capillary) was chosen. Table 1 summarizes the parameters of the optimized CE-ICP-MS/MS method.
| CE system | |
|---|---|
| Capillary | Fused silica capillary, i.d. 75 μm, o.d. 375 μm, 70 cm length |
| BGE | Trizma® base (Tris), 5 mM, pH 7.4 |
| Temperature | 37 °C |
| Voltage | +15 kV |
| Current | 3–5 μA |
| Sample injection | Hydrodynamic, 30 mbar × 5 s (∼3.7 nL) |
| ICP-MS/MS system | |
|---|---|
| RF power | 1570 W |
| Sample depth | 8.4 mm |
| Plasma gas | 15.0 L min−1 |
| Nebulizer gas flow | 1.03 L min−1 |
| Sheath liquid | 2.5 ng mL−1 115In in 0.5 mM Tris |
| Sheath liquid flow | 10 μL min−1 |
| Cell gas (O2) flow | 0.39 μL min−1 |
| Monitored masses | 31P16O+, 32S16O+, 115In+, 195Pt+ |
3.4. The CE-ICP-MS/MS method's analytical parameters
The optimized CE-ICP-MS/MS method was evaluated based on the calculated figures of merit. In the case of the 31P16O+ analyte corresponding to liposomes, the obtained MTs were better for both intra- and inter-day measurements (RSD (n = 3) equal to 3.08% and 4.64%, respectively) than for the peak areas (4.85% and 5.34%). Until now, the quantification of phosphorus has not been performed in any approach using the CE-ICP-MS method. The described LODs relate only to the drug encapsulated or released from the liposome–oxaliplatin or cisplatin systems.7,11 Herein, the LOD and limit of quantification of the 31P16O+ signals equal 1.06 μM and 3.19 μM, respectively. The LOD of phosphorus was established, for instance, for the SEC-DRC-ICP-MS method (mentioned above),9 whose value is only a few times lower than that in the current method, i.e., 0.13 μM, despite usual chromatography yielding much lower LODs than electrophoresis. Detailed information on the method of LOD and LOQ calculation can be found in the ESI (eqn (3) and (4)).†Another parameter estimated to assess the method accuracy is the capillary recovery, which yielded 82.35%. The parameter was calculated as the ratio of the 31P16O+ average peak areas (n = 3) corresponding to the non-encapsulated liposomes, determined based on analyses executed in two ways (eqn (5), ESI†). In one, the samples were analyzed under optimized separation conditions, and in the other, the electrophoretic run was conducted with an applied internal pressure (20 mbar).
The calibration curves for E4 liposomes, transferrin, and cisplatin quantification (31P16O+, 195Pt+, and 32S16O+ signals) were drawn towards the optimized method (Fig. S7–S9, ESI†). The E4 liposomes were prepared with appropriate amounts of DSPE-PEG2000, HSPC, and Chol in a molar ratio of 5.3/56.3/38.4, which contained 0.666 μmol phosphorus. Its final concentration in the unfiltered and undiluted liposome samples was 0.351 mM. To obtain the curve, shown as the dependence of the 31P16O+ signal area (normalized) vs. the phosphorus concentration, the filtered stock sample and several dilutions of it (in IS) were measured. The probed samples of liposomes, transferrin, and cisplatin (several dilutions of the stock solution) were prepared in a medium composed of 100 mM NaCl and IS mixed in a v/v ratio of 1/1. The calculated correlation coefficients equal 0.9967, 0.9939, and 0.9971 for the phosphorus, cisplatin, and transferrin calibration curves, respectively. The obtained values prove the linearity of the method in the considered concentration range.
3.5. Liposome–drug formulations: initial investigations
Further investigations concerned the platinum-based anticancer drug encapsulation in the obtained E4 liposomes. According to the literature, the drug that can be used for encapsulation in these formulations is cisplatin.21 Therefore, CE-ICP-MS/MS examinations were conducted for the E4 liposome–cisplatin system samples. The E4 liposome–oxaliplatin systems were probed as well. Unfortunately, in both cases, the formation of DTDSs was not confirmed since the co-migration of the analytes of interest was not observed. Signals corresponding to oxaliplatin and cisplatin were registered at ca. 9.5 and 10 min, respectively, while the 31P16O+ signal was observed after 14 min of the analysis. Since the developed liposomes did not guarantee drug encapsulation, their composition was modified to enhance the DTDS formation. The tested compositions of liposomes are described in Table S1 (ESI†). In the first modification (E4_mod1), an increased concentration of Chol was applied, while in the second (E4_mod2), DSPC was used instead of HSPC. However, cisplatin and oxaliplatin encapsulation was not observed in both cases. It was decided to investigate the initially assessed liposomes in terms of the drug encapsulation in such circumstances. CE-ICP-MS/MS experiments confirmed cisplatin accumulation in the E6-type of liposomes (Fig. 2, signal no. 3). The composition of the prepared vesicles was almost the same as the E6 formulation, with a slight difference in the fatty acid in one of the components: instead of two oleoyl chains (DOPC), the phospholipid with palmitoyl and oleoyl chains (POPC) was used (E6_mod1, ESI†). According to the label on the Pfizer cisplatin concentrate (1 g L−1) used in chemotherapy, the drug should be administered at 20–140 mg of CDDP per 1 m2 of the human body surface with 1 L or 2 L of infusion fluid. Assuming an average dose of 100 mg and 2 m2 of the body surface, the final dose of administered cisplatin equals 0.2 mg mL−1. Based on this presumption, the objective of DTDS application in cancer treatment is the limitation of the dosage currently applied in classical chemotherapy1,22 by protecting the drug (entrapped in DTDSs) from deactivation processes caused by HS proteins23 and transporting this drug more selectively into cancer targets. Hence, the cytotoxic effect on cancer cells can be the same or even better, although the dose of the administered drug is much lower.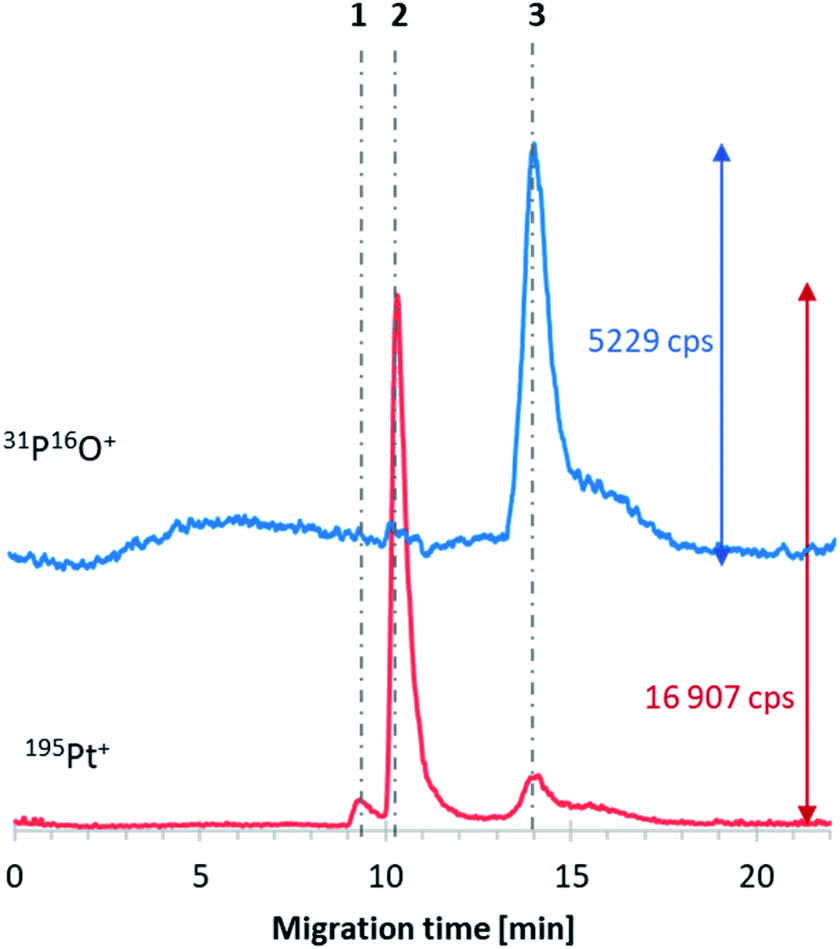 | ||
| Fig. 2 CE-ICP-MS/MS electropherogram of the E6 liposome–cisplatin–transferrin reaction mixture sample incubated for 20 h at 37 °C with stirring (400 rpm); the grey dot-dashed lines correspond to free cisplatin (2), its hydrolyzed form (1), the transferrin–cisplatin form (3), and encapsulated liposome–transferrin (4); voltage: +15 kV (current ∼3 μA), injection: 50 mbar × 7 s, capillary: fused silica, length: 70 cm (i.d. 75 μm). | ||
Based on the area under the 195Pt+ peak corresponding to DTDSs and the equation from the platinum calibration curve, the liposomes encapsulated 24 μg mL−1 of cisplatin. The value was not affected during 24 h of storage (in a fridge). The efficiency of cisplatin encapsulation can be further optimized by adjusting the liposome composition rather than changing the synthetic method.
3.6. Interactions of DTDS with transferrin
The next step was the investigation of the obtained DTDSs (E6_mod1) treated with transferrin. Transferrin was chosen as a model protein in this study. This iron transporting protein present in the blood can form protein corona on the surface of liposome-based DTDSs and play the role of a targeting ligand.24,25 The measurements performed after 2 h of incubation of the mixture at 37 °C (to mimic the physiological temperature and assumed average time of DTDS half-life in the bloodstream, dependent on their size and composition26,27) showed that the presence of transferrin does not affect the stability of DTDSs. The calculated efficiency of cisplatin encapsulation was almost the same as that in the sample without transferrin (23.7 μg mL1), which suggests that the protein may have a stabilizing effect on DTDSs. Moreover, no interaction between the protein and liposomes was noted (no co-migrated 31P16O+ and 32S16O+ signals). However, considering the analogical sample analyzed after 20 h of incubation, two new forms of analytes can be observed on the electropherogram presented in Fig. 3. The extended sample incubation time resulted in attaching ca. 7% (calculated as the percentage of both 32S16O+-containing signals) of the protein to the liposome–cisplatin system (line no. 4), which did not cause faster leakage of the drug from DTDSs compared to the natural process observed during its storage without protein. The following newly formed species (line no. 3) was hypothesized to be a transferrin adduct with free drug forms, mainly from non-encapsulated cisplatin (comparing signal 2 peak area). The described phenomenon confirms that for incubation times similar to those of conventional DTDSs in blood circulation, the affinity of transferrin to attach to the surface of liposomes and form the corona is low, which suggests that in the case of the developed liposome formulation, transferrin cannot be used as an active targeting agent. Simultaneously, transferrin presence in the media does not cause the decomposition of the DTDSs and drug release. The obtained results should be treated as a basis for further composition-adjustment experiments of E6-family liposomes using sample treatments that increase the value of the encapsulated drug and improve the long-term liposome stability.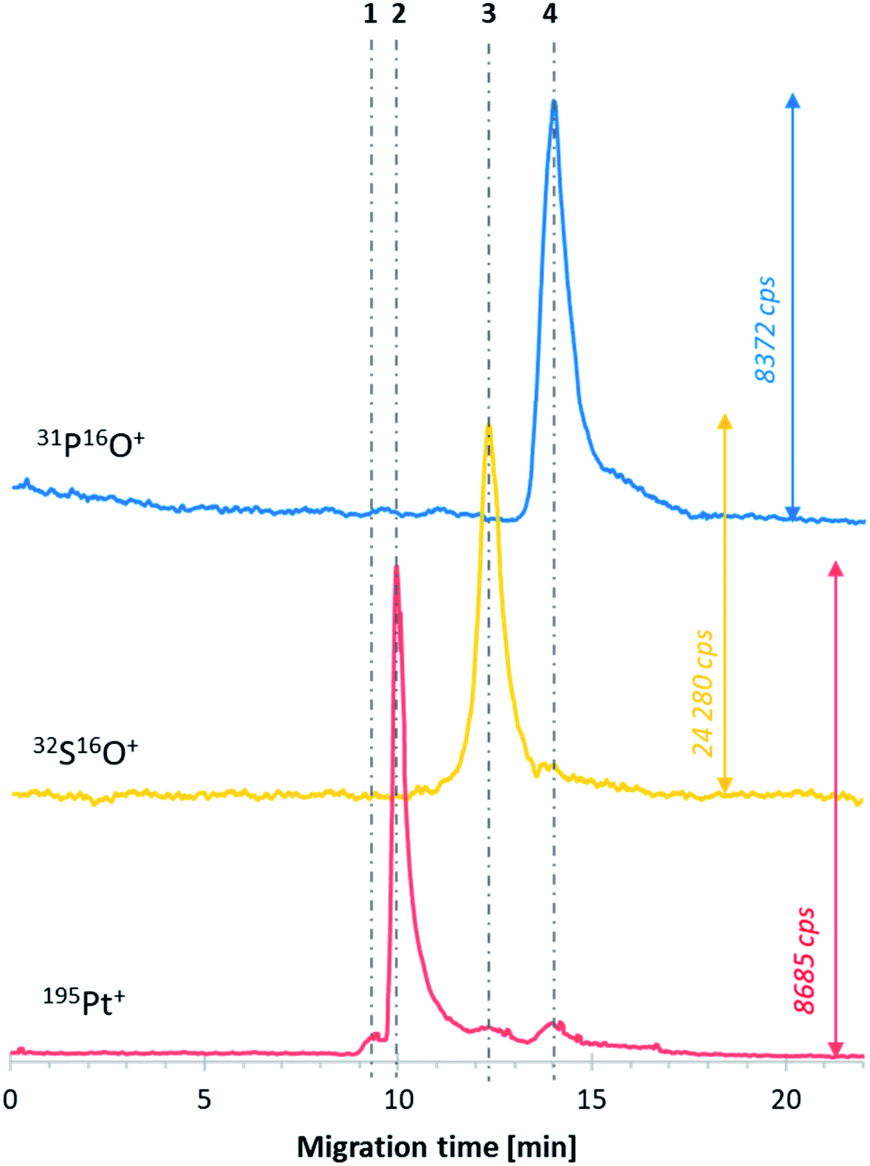 | ||
| Fig. 3 CE-ICP-MS/MS electropherogram of the E6 liposome–cisplatin systems; the grey dot-dashed lines correspond to free cisplatin (2), its hydrolyzed form (1), and the encapsulated liposome–cisplatin systems (3); voltage: +15 kV (current ∼3 μA), injection: 50 mbar × 7 s, capillary: fused silica, length: 70 cm (i.d. 75 μm). | ||
However, from an analytical point of view, attention should be paid to the resistance method in analyzing high-protein-concentration samples. The presence of transferrin in the mixture did not cause the shift of analyte migration times. Moreover, the choice of CE-ICP-MS/MS opens up the novel possibility of simultaneous determination of DTDSs (based on organic nanomaterials and metallodrugs) and proteins by sulfur monitoring. This was not possible till now using the HPLC-DRC-ICP-MS combination.9
4. Conclusions
In summary, the optimized CE-ICP-MS/MS method allows for the robust isotopic-selective quantitative monitoring of cisplatin–liposome system formation, stability, and its interactions with proteins, with satisfactory figures of merit. The significant advantages of this method, contributing to its superiority over those proposed until now, i.e., the CE- and HPLC-assisted methods, are the numerous interference limitations, the possibility of mimicking the physiological environment during separation, the lower reagent consumption, and the reasonable resolution. Moreover, the developed method overcomes the lack of approaches dedicated to the simultaneous and high-quality analysis of liposomes (phosphorus), drugs (platinum), and proteins (sulfur) using one measurement. This method can be a comprehensive tool for the detailed analyses of liposome-containing samples. Furthermore, the developed methodology paves a new way for effectively tracking the in vitro alterations of liposome-based DTDSs in the future.Additionally, instead of the sonication method, the ethanol injection method was found to enable the repeatable and straightforward formation of stable and homogenous liposomal suspensions, which opposes the hitherto mandatory application of the time-consuming and expensive lipid film method.28
Author contributions
Conceptualization: M. Matczuk; methodology: M. Matczuk, J. Samsonowicz-Górski, E. Kamińska, A. M. Wróblewska, M. Drozd; investigation: J. Samsonowicz-Górski, E. Kamińska, A. M. Wróblewska, M. Drozd; data curation: A. M. Wróblewska, J. Samsonowicz-Górski, M. Drozd; resources: M. Matczuk; writing – original draft: A. M. Wróblewska, J. Samsonowicz-Górski; writing – review & editing: M. Matczuk, M. Drozd; supervision: M. Matczuk; project administration: M. Matczuk; funding acquisition: M. Matczuk. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. M. Wróblewska acknowledges financial support from the IDUB project (Scholarship Plus program). The studies were financed by the Warsaw University of Technology.References
- S. Ghosh, Bioorg. Chem., 2019, 88, 102925 CrossRef CAS PubMed.
- J. A. Kemp and Y. J. Kwon, Nano Convergence, 2021, 8, 34 CrossRef CAS PubMed.
- K. T. Jin, Z. B. Lu, J. Y. Chen, Y. Y. Liu, H. R. Lan, H. Y. Dong, F. Yang, Y. Y. Zhao and X. Y. Chen, J. Nanomater., 2020, 2020, 9184284 Search PubMed.
- W. Yan, S. S. Y. Leung and K. K. W. To, Nanomedicine, 2019, 15, 303–318 CrossRef PubMed.
- J. Zajda, A. Wróblewska, L. Ruzik and M. Matczuk, J. Controlled Release, 2021, 335, 178–190 CrossRef CAS PubMed.
- A. Wróblewska and M. Matczuk, Electrophoresis, 2020, 41, 394–398 CrossRef PubMed.
- U. Franzen, T. T. T. N. Nguyen, C. Vermehren, B. Gammelgaard and J. Østergaard, J. Pharm. Biomed. Anal., 2011, 55, 16–22 CrossRef CAS PubMed.
- T. W. May and R. H. Wiedmeyer, At. Spectrosc., 1998, 19, 150–155 CAS.
- T. T. T. N. Nguyen, S. Stürup, J. Østergaard, U. Franzen and B. Gammelgaard, J. Anal. At. Spectrom., 2011, 26, 1466 RSC.
- T. T. T. N. Nguyen, J. Østergaard, S. Stürup and B. Gammelgaard, Anal. Bioanal. Chem., 2012, 402, 2131–2139 CrossRef CAS PubMed.
- T. T. T. N. Nguyen, J. Østergaard, S. Stürup and B. Gammelgaard, Int. J. Pharm., 2013, 449, 95–102 CrossRef CAS PubMed.
- T. T. T. N. Nguyen, J. Østergaard, S. Stürup and B. Gammelgaard, Anal. Bioanal. Chem., 2013, 405, 1845–1854 CrossRef CAS PubMed.
- U. Franzen and J. Østergaard, J. Chromatogr. A, 2012, 1267, 32–44 CrossRef CAS PubMed.
- S. M. Ansar and T. Mudalige, Int. J. Pharm., 2019, 561, 283–288 CrossRef CAS PubMed.
- S. Moghassemi and A. Hadjizadeh, J. Controlled Release, 2014, 185, 22–36 CrossRef CAS PubMed.
- A. Akbarzadeh, R. Rezaei-Sadabady, S. Davaran, S. W. Joo, N. Zarghami, Y. Hanifehpour, M. Samiei, M. Kouhi and K. Nejati-Koshki, Nanoscale Res. Lett., 2013, 8, 102 CrossRef PubMed.
- A. Jain, P. Hurkat and S. K. Jain, Chem. Phys. Lipids, 2019, 224, 104764 CrossRef CAS PubMed.
- D. Guimarães, J. Noro, A. Loureiro, A. Cavaco-Paulo and E. Nogueira, Colloids Surf., B, 2019, 179, 414–420 CrossRef PubMed.
- L. Balcaen, E. Bolea-Fernandez, M. Resano and F. Vanhaecke, Anal. Chim. Acta, 2015, 894, 7–19 CrossRef CAS PubMed.
- A. M. Wróblewska, A. Milewska, M. Drozd and M. Matczuk, Int. J. Mol. Sci., 2022, 23, 2324 CrossRef PubMed.
- S. H. Alavizadeh, J. Akhtari, A. Badiee, S. Golmohammadzadeh and M. R. Jaafari, Expert Opin. Drug Delivery, 2016, 13, 325–336 CrossRef CAS PubMed.
- X. Wang and Z. Guo, Chem. Soc. Rev., 2013, 42, 202–224 RSC.
- L. Messori and A. Merlino, Coord. Chem. Rev., 2016, 315, 67–89 CrossRef CAS.
- T. Huo, Y. Yang, M. Qian, H. Jiang, Y. Du, X. Zhang, Y. Xie and R. Huang, Biomaterials, 2020, 260, 120305 CrossRef CAS PubMed.
- X. M. Li, L. Y. Ding, Y. Xu, Y. Wang and Q. N. Ping, Int. J. Pharm., 2009, 373, 116–123 CrossRef CAS PubMed.
- H. Brochu, A. Polidori, B. Pucci and P. Vermette, Curr. Drug Delivery, 2004, 1, 299–312 CrossRef CAS PubMed.
- S. Fathi and A. K. Oyelere, Future Med. Chem., 2016, 8, 2091–2112 CrossRef CAS PubMed.
- A. F. Vikbjerg, S. A. Petersen, F. Melander, J. R. Henriksen, and K. Jørgensen, Liposomes for Drug Delivery and Methods for Preparation Thereof, WO2009EP56297 20090525[WO 2009/141450 A2], 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d1ja00459j |
| This journal is © The Royal Society of Chemistry 2022 |