
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAntioxidant silicone oils from natural antioxidants†
Michael A.
Brook
 *,
Akop
Yepremyan
,
Guanhua
Lu
,
Miguel
Melendez-Zamudio
,
Daniel J.
Hrabowyj
and
Cody B.
Gale
*,
Akop
Yepremyan
,
Guanhua
Lu
,
Miguel
Melendez-Zamudio
,
Daniel J.
Hrabowyj
and
Cody B.
Gale
Department of Chemistry and Chemical Biology, McMaster University, 1280 Main St. W., Hamilton, ON, Canada L8S 4M1. E-mail: mabrook@mcmaster.ca
First published on 25th October 2022
Abstract
Antioxidants are important mediators of radical processes. Nature exploits a variety of antioxidants, including vitamins, to moderate oxidation in living systems. Silicone oils and elastomers, widely used in personal care and biomedical applications, are rather unreactive under redox conditions and can therefore host such reactions. We report that phenolic antioxidants, including derivatives of vitamins A and E, and eugenol are readily tethered to silicone oils. These natural constituents dilute the energetic toll required to make silicones while delivering antioxidant properties to the product silicone oils.
Introduction
Oxidation is an important, but not always beneficial, process in normal biological and abiotic environmental processes; combustion is one relevant example. One of the most pernicious classes of oxidation involves singlet oxygen, peroxides, and/or hydroxy radicals – reactive oxygen species (ROS) – that aggressively attack materials, usually via radical processes. Antioxidants are frequently added to extend the lifetime of materials to impact, among other things, metal corrosion, biomaterials, and early aging of human skin.1–4Two classes of these type of natural antioxidants (AO) that function against ROS are particularly relevant for this work: highly conjugated, non-aromatic compounds that form conjugated radicals, and phenols that are converted to phenoxy radicals. For humans, vitamin A (retinol, retinal, retinoic acid, etc.),5 is exemplary of the former type of antioxidant, and vitamin E6 (α-tocopherol) or the amino acid tyrosine7 are representative of antioxidant phenols. Other relevant antioxidants to which humans may be exposed include a variety of essential oils like eugenol, a phenol that is found in cloves,8 and lignin, the network polymer that reinforces trees and other plants.9
There is an increasing desire to exploit natural materials in the marketplace. In part, this is due to public perception of the benefits that accrue from nature. From the perspective of Green Chemistry,10 this strategy is key because such materials are renewable and, when they finally enter the environment at end of life, should readily degrade to molecules that can be reused by natural processes (rules 7 and 10).
Silicone oils and elastomers are found in a myriad of technical areas because of their atypical properties when compared to organic polymers. Silicone oils show excellent degradability in the environment. Within a few weeks, depolymerization to low molecular weight oligomers and silanes occurs, depending on ambient temperature, humidity and the available of clay surfaces that catalyze the process.11,12 Best estimates for complete oxidation to water, CO2 and SiO2 depending on conditions, are several years.
All silicones are derived from silicon metal that, in turn, is prepared from SiO2 in a very energetically demanding process. By definition this synthetic route breaks rule 6 because very high temperatures ∼2000 °C are required for synthesis and, even if the thermal energy provided is from renewable sources, the reducing agent is carbon-based, such that at least one mole of the greenhouse gas CO2 is produced per mole of SiO2 reduced to silicon metal. The sustainability of silicones, with their wealth of interesting properties, would be improved by using less silicone for a given application by diluting the silicone with natural and renewable materials. Further benefit would accrue if the natural materials delivered functionality.
Silicones are highly resistant to oxidation/reduction and their lack of polarity is typically contraindicated for redox processes. Previously, silicones have been doped with active antioxidant compounds that are released to an external environment. For example, antibiotic surfaces arise from release of eugenol from silicone elastomers,13 and trans-retinoic acid undergoes release from silicone rubber directly,14,15 or after enzyme-induced ester hydrolysis. While there are many other examples, most of which are found in the patent literature, of silicones that carry and deliver antioxidants including vitamins, this work is focussed on compounds in which the antioxidant is grafted to the silicone polymer.
While antioxidant molecules have been previously grafted onto silicones, antioxidant efficacy was not measured. In most reports of eugenol silicones, for example, eugenol is typically first grafted via hydrosilylation of the allyl group, and then modified with further reactions that block the phenol group, for example, in polyurethanes,16 ether sulfones,17 or polycarbonates,18,19 or as crosslinkers for silicone20 or other networks.21 Eugenol-modified silicones in which the phenols are free can exhibit anticorrosion22 or antibiotic activities.23 The network polyphenol antioxidant lignin has been used in flame retardant silicone elastomers.24
Could vitamins and other natural antioxidants be used to dilute silicones and, once tethered to silicones, would they still convey antioxidant properties while maintaining the beneficial properties of silicone oils? We report the syntheses of grafted retinoate, tocopherol and eugenol-modified silicones and examine their ability to function as antioxidant films.
Results
One objective of this research was to increase, as much as practically possible, the quantity of natural materials in the antioxidant silicone product. The impact of very different functional group densities (from 6–25%) was examined using pendent starting materials of similar molar mass ∼1950 g mol−1, which facilitated comparison. These led to processible mixtures and, eventually, functional oils or elastomers. Very highly functional (50%) pendent and low functional density (4500 g mol−1) telechelic polymers were also used as starting materials.Retinol was judged to be too expensive, and retinal too chemically sensitive, to be considered for silicone composites. The allyl ester 1 of the more accessible vitamin A metabolite, retinoic acid, was readily prepared. The hydrosilylation reaction of the allyl group, a reactive handle that is widely used in the silicone industry (e.g., to make hydroxypropyl acrylate-modified coupling agents25), did not proceed as expected. The product polymers 1P, recovered as a yellow oil, contained the desired all trans hydrosilylated product, the silyl ester and a small fraction of propylated backbone; presumably all 3 constituents are present on the same silicone backbone. It is proposed that formation of the Pt/π-allyl complex26 was followed by reductive silylation to give the silyl ester, rather than the desired hydrosilylation (Fig. S2A, ESI†). To avoid this competing deallylation reaction, homoallyl retinoate 2 was instead prepared via an EDC coupling with 3-buten-1-ol mediated by DMAP. Reaction with this compound led only to hydrosilylation products 3T and 3P recovered in a 70/30 E/Z isomeric mixture, as shown by 1H NMR (nomenclature: T refers to a modified telechelic silicone, while P refers to a pendent polymer, Fig. 1, ESI†). The products were more viscous than the starting silicone oils. It is unclear why olefin scrambling was associated with the homoallyl but not the allyl educt. We note that this latter process fails many of the rules of Green Chemistry but pursued the route to determine the relative utility of the products. Simple Fisher esterification using 3-buten-1-ol was both inefficient and led to degradation of the polyene.
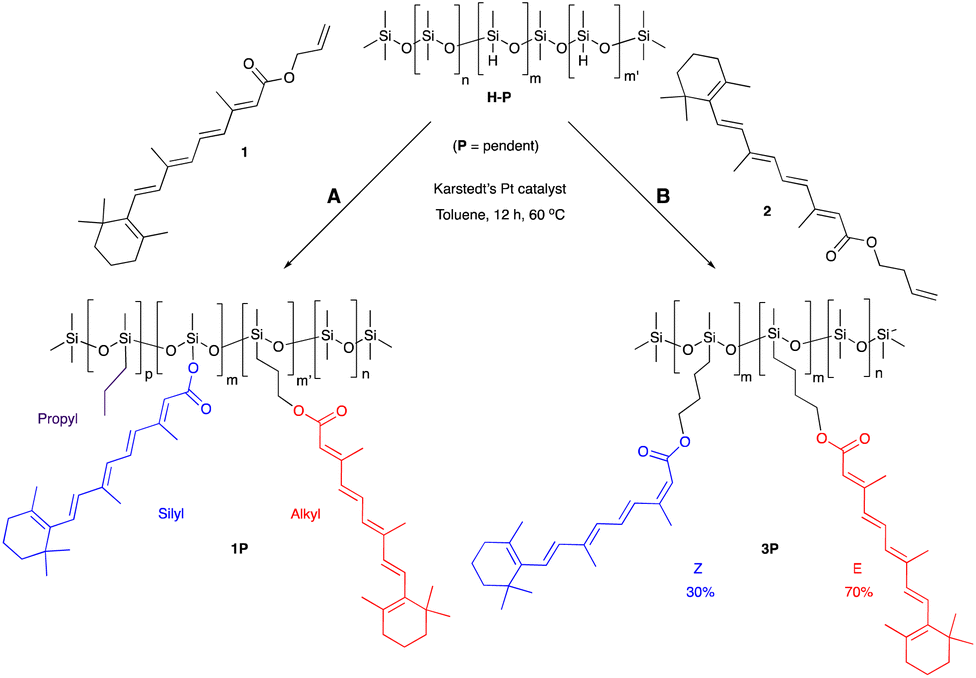 | ||
| Fig. 1 Hydrosilylation of (A) allyl retinoate to give 1P and (B) homoallyl retinoates to give 3P. | ||
Vitamin E was grafted to telechelic HSi-silicones by silylating/blocking the phenolic OH group using the Piers Rubinsztajn (PR) reaction in toluene to give, with use of methanol to remove any unreacted tocopherol, products 4T (for NMR data see ESI†), as a more viscous, clear polymer oil (Fig. 2A). The reaction is particularly useful to introduce phenols to silicones via formation of a hydrolytically stable Si–O–C bond.20 Analogous reactions with pendent SiH silicones gave 4P-17 (the number 17 represents the % monomer containing eugenol moieties in the product) or 4P-26, respectively. The two pendent compounds are comprised of 50% and 61% weight% tocopherol, respectively. That is, in 4P-26 the silicone components comprise just 39% of the final oil. Note that the molecular weights of the products are about twice that expected as SiH → Si-tocopherol. This is attributed to the presence of small amounts of water, which is known to dimerize SiH compounds → SiOSi + H2.27
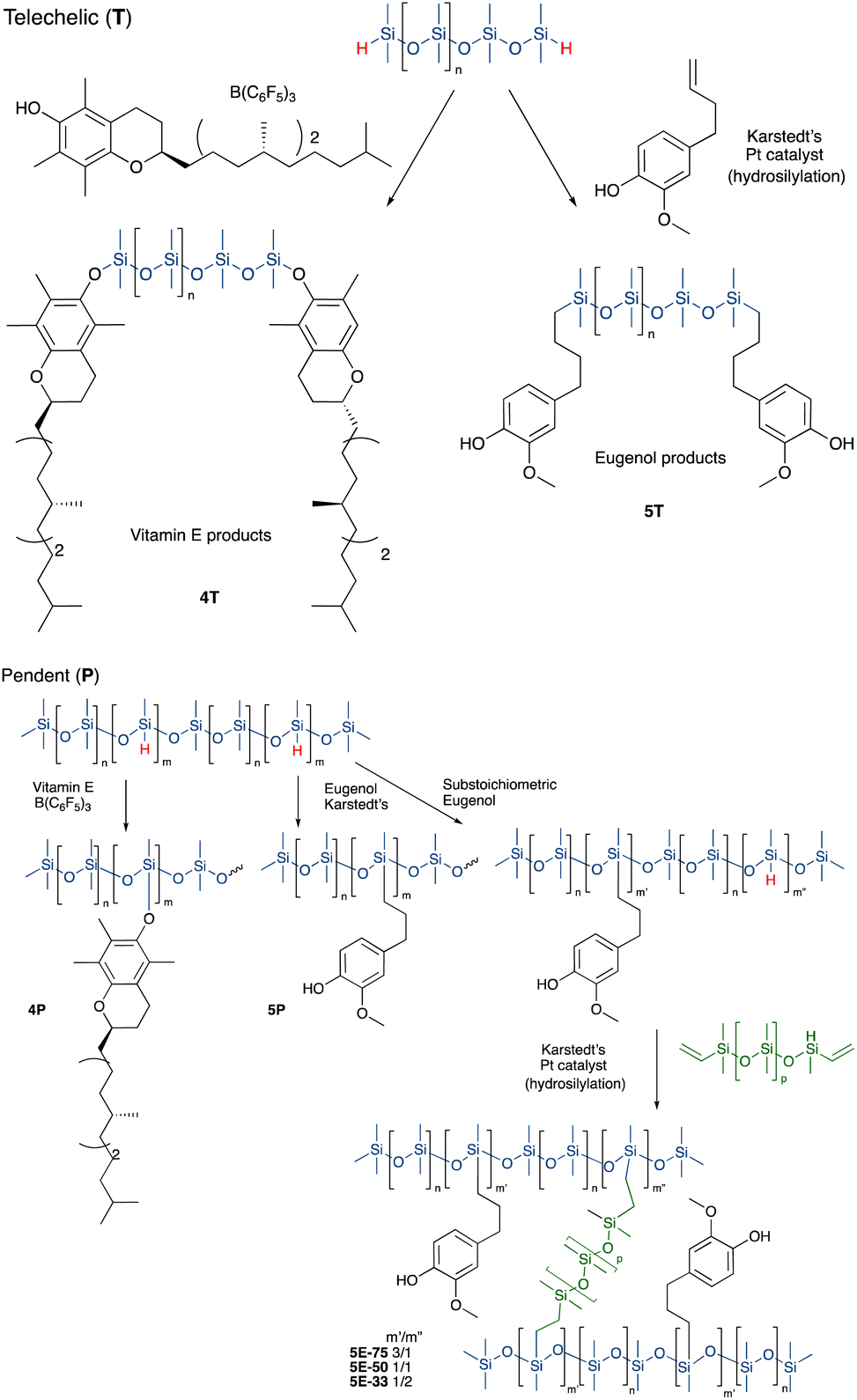 | ||
| Fig. 2 Preparation of antioxidant-modified silicones. | ||
It was anticipated that free phenols would be required to deliver antioxidant properties of AO-modified silicone oils or elastomers. Therefore, eugenol was introduced using platinum-catalyzed hydrosilylation of either telechelic or pendent HSi-containing silicones in toluene at 60 °C overnight to give, after washing with MeOH, eugenol tethered silicones with free phenols as pure pendant 5P-6, 5P-17 or telechelic 5T oils (Fig. 2B, for NMR data see ESI†). The viscosities of the products were much higher than the starting materials, which is attributed to both H-bonding and associative interactions between the aromatic units, and further depended on the ratio of eugenol-bearing/OSiMe2 monomers in the polymer.28
While, in the case of pendent silicones, it was possible to modify all the SiH groups with eugenol, an alternative process permitted simultaneous hydrosilylation of vinyl-terminated telechelic silicones to produce eugenol-modified silicone elastomers 5E-X (X = 33, 50, 75, the percent of SiH converted to eugenol moieties; the remaining served as crosslink sites, Fig. 2). Traditional strategies for controlling Mc allows one to choose both eugenol content in the elastomers and crosslink density; the latter property links directly to hardness/modulus and extension at break.
DPPH is a particularly useful molecule to quantify antioxidant activity (Fig. S2B, ESI†)29,30 by a readily observable colorimetric change from purple to yellow,31 along with other traditional spectroscopic means. The AO activities of PDMS modified with retinoate 3T, 3P, vitamin E (α-tocopherol) 4T (for 4P-17, 4P-26 see Fig. S10, ESI†) or eugenol 5T, 5P-6 were measured in 96-well plates at active antioxidant concentrations ranging from 0.1 mM to 100 mM (based on the respective antioxidant moiety); the pendent HSi silicone oil (P-4) and vitamin A, homoallyl retinoate, vitamin E, and eugenol were used as negative and positive controls, respectively. A stock solution of DPPH was added to each sample to give a final concentration of 0.1 mM DPPH. The absorbance at 520 nm was measured 15 minutes after addition. In all cases, the DPPH absorbance at 520 nm decreased over time (Fig. 3).
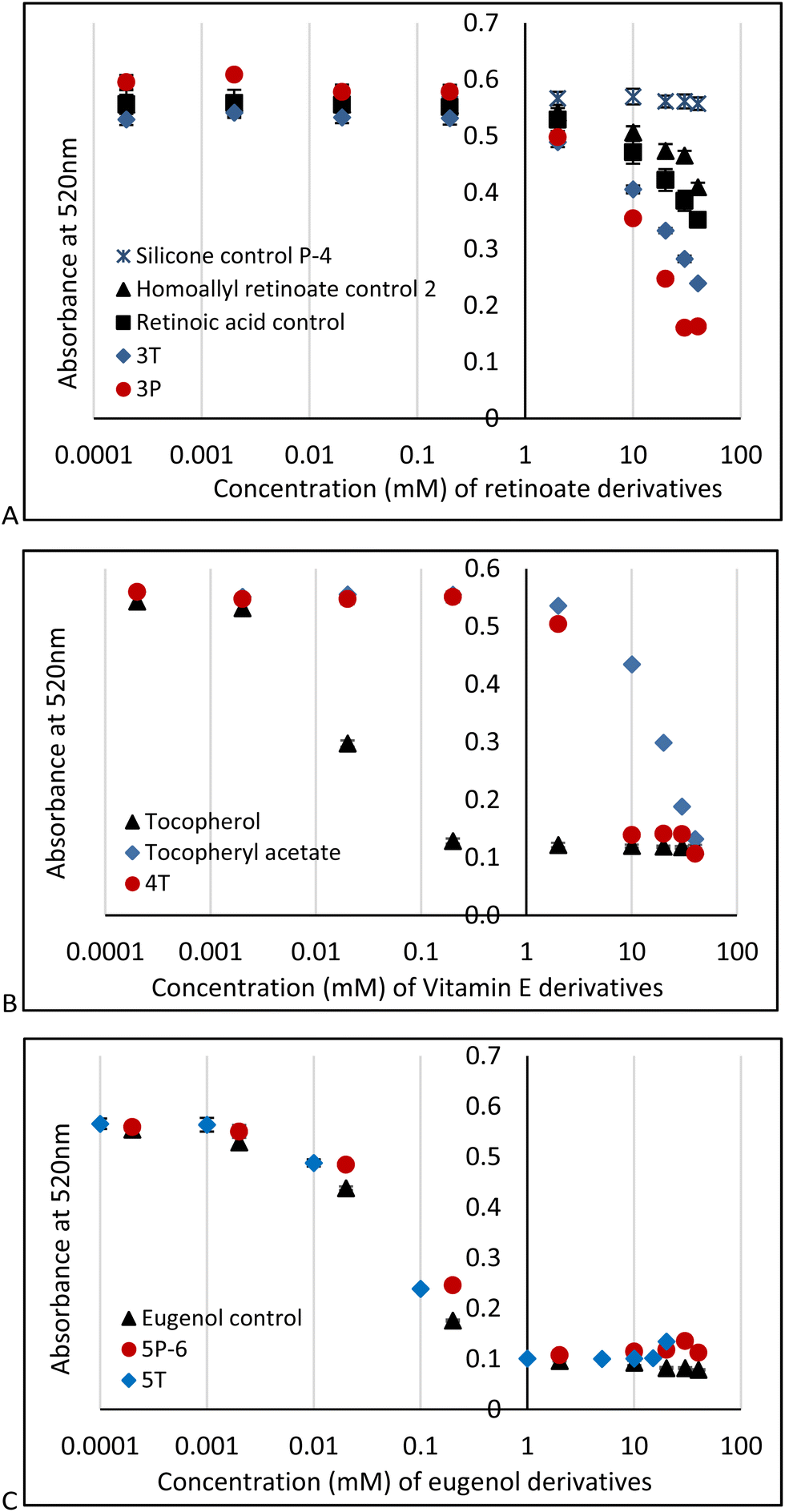 | ||
| Fig. 3 Log plots of DPPH UV assay at 520 nm wavelength, comparison of anti-oxidation activity of: (A) retinoic acid, homoallyl retinoate and silicone derivatives 3T and 3P-6; (B) tocopherol, tocopheryl acetate, and silicone adduct 4T; (C) eugenol and eugenol-modified silicones 5T and 5P-6. Error bars reflect the standard error of 3 UV measurement trials. | ||
These experiments showed that silicone polymers modified with retinoate 3 were weak but still viable antioxidants, as was the case with the starting homoallyl retinoate 2. Note that retinoates are less sensitive to DPPH assays than other methods.32 It is entirely expected that the AO activity of vitamin E silicone polymers 4T, 4P-17 and 4P-26 (for the latter two, see ESI†) were much less effective than the native vitamin with its free phenol;6,33 they performed slightly better than the phenoxy protected vitamin E acetate (Fig. 3B). The ability of tocopherols to undergo secondary reactions that involve ring opening has previously been noted, and may be responsible for the weak antioxidant activity here.33
Eugenol, a potent antioxidant,8 maintained essentially the identical ability to reduce DPPH after grafting to silicones. There was a very slight difference between the telechelic 5T and pendent silicones 5P-6, 5P-17 (Fig. 3C). Films of eugenol-based silicone pendent silicones 5P-17 oils were tested using a DPPH surface assay both by exposing DPPH solutions on cotton to drops of the eugenol-silicone oil, or vice versa. Elastomeric materials were similarly tested with drops of DPPH (Fig. 4, Fig. S9, ESI†). DPPH was spotted onto 5E-75 elastomeric films made by reaction of the pendent 15% SiH silicone oil (P-17) modified with 75% eugenol and 25% divinylsilicone (Fig. 4, ESI†). Over a few minutes, the DPPH solutions were effectively decolored, demonstrating that organic oxidants which penetrated the silicone are reduced. Note that the silicone is protective against aqueous oxidants that are unable to penetrate the hydrophobic silicone oil medium. Eugenol-derived oils exposed first to aqueous H2O2 solution and then to DPPH, underwent decolorization at a similar rate and degree as elastomers as those films not exposed to H2O2 (ESI†).
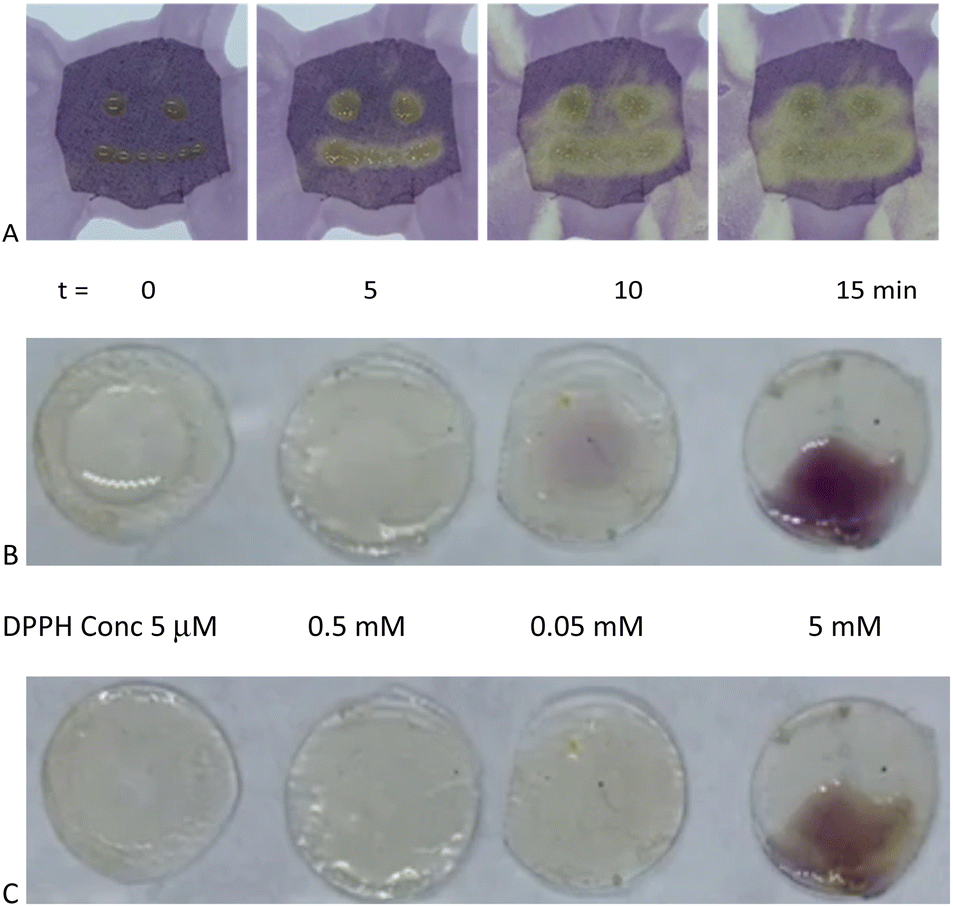 | ||
| Fig. 4 (A) 5P-17 spotted onto cotton fabric soaked with DPPH (purple, 5 mM) in IPA; time lapse over 5 minutes showing both antioxidant activity and diffusion of oil through the fabric. (B) From left to right, 5 μM, 0.05 mM, 0.5 mM, and, 5 mM DPPH in IPA solution spotted onto 5E-75t = 15 s, (C) t = 90 s. | ||
Discussion
It is important to confront, at the outset, the degree to which some of these syntheses fall within the rubric of green chemistry. Compounds 6T and 3P were made using very old school, effective methods with inappropriate solvents and active esters that generate stoichiometric waste. These syntheses fail the ‘Green Chemistry’ test. We include them as, in our hands, more appropriate syntheses failed because the polyene is simply too susceptible to undesired reactions. The data from 7T and 3P are presented simply to prove the point that: it is possible to tether vitamin A derivatives to silicones; doing so dilutes the silicone with natural products as desired; and the product maintains the desired, but relatively weak, antioxidant activity. Given the number of Green Chemistry rules that the syntheses break, the process is not easily justified.The reaction of tocopherol with hydrosilicones fares much better in this analysis, as the reaction is facile, and catalyzed by small quantities of B(C6F5)3. Compounds 4T, 4P-17 and 4P-26 prove the point that efficacious AO activity requires a free phenol in Vitamin E analogues; these compounds had only feeble AO activity. However, they achieve one of the other desired objectives: diluting silicones with natural products (rules 6, 7 and 10). The pendent compounds are comprised of 50 or 61 wt%, respectively, of tocopherol. That is, the silicone product, with silicone properties, has a much lower energy content by virtue of the contained vitamin E. These pale-yellow oils have a silicone ‘feel’, but are significantly more viscous than their precursors or more typical dimethyl- or phenylsilicone oils of comparable molecular weight.
The synthesis of the eugenol derivatives 5T, 5P-6, 5P-17 requires only very small quantities of platinum catalyst that are readily recovered during filtration (note: in the case of the elastomers 5E, the platinum is lost in the product). In all cases, the products possess free phenols and are potent antioxidants, equivalent to the parent eugenol. Thus, the eugenol-silicone polymers deliver AO function, and can be used in coatings or films (Fig. 3C, Fig. 4). In addition, in these products to the silicon is diluted with natural materials reducing, per gram of material, the quantity amount of silicone needed for a given purpose; 5P-17 is comprised of 28 wt% eugenol.
All three types of silicone antioxidants were as, or slightly more, potent AO than their starting material analogues. That is, the silicone has no significant impact on the AO activity even though the antioxidants were chemically tethered to the polymer. There was a small enhancement in reactivity of 5T over 5P-6 that can be ascribed to mobility. The AO terminus on telechelic 5T is not protected by the backbone as is the case with 5P and, moreover, it has more degrees of freedom to make contact with oxidizing radicals.
The eugenol oil 5P-17, as with all silicones, is exceptionally hydrophobic. Exposure to aqueous peroxide led to no noticeable change in DPPH activity. The antioxidants within the silicone were protected from aqueous oxidants by surface energy. This bodes well, in the sense that protective coatings based on silicone oils and elastomers that contain AO natural ingredients will repel aqueous oxidants and neutralize those that are partly or completely soluble in silicones.
A variety of polymers are noted for their antioxidant activity.34 Silicones such as those described above with AO properties could bring additional benefit to silicones that are already widely used in adhesives, sealants and coatings. For example, in the latter application, silicone oils are often used as flow control agents in paints because of their low surface tension and facility to rise to air interfaces. The materials described here can additionally provide surface functionality to different substrates with properties that both benefit from silicone per se and the AO functionality. To a degree, these materials exhibit the possibility of ‘having the cake and eating it too’. Many of the desirable properties of silicone are present to fulfil a specific objective, while reducing by up to 61% the actual amount of silicone needed and its attendant energy cost.
Experimental
Materials
Eugenol was purchased from Alfa Aesar. All trans-retinoic acid was purchased from Combi-Blocks. Telechelic HMe2SiO-terminated silicone DMS-H21 (∼4500 g mol−1), pendent HMeSiO (Me3SiO(SiMe2)n(OSiMeH)mOSiMe3, HMS-071 (∼1950 g mol−1, 6–7% SiH), HMS-151 (∼1950 g mol−1, 15–18% SiH); HMS-301 (∼1950 g mol−1, 25% SiH) and HMS-501 (∼1150 g mol−1, 50% SiH) were purchased from Gelest. α-Tocopherol and all other reagents, catalysts and solvents were purchased from Sigma Aldrich. 96-Well plates were obtained from Falcon.Methods
NMR analysis: 1H and 13C NMR were recorded on a Bruker Advance 600 MHz nuclear magnetic resonance spectrometer. High-resolution mass spectra (HR-MS) were recorded on an Agilent Technologies G1969 TOF spectrometer. Centrifugation was performed using a Thermo Fisher Durafuge 100 precision centrifuge. UV–vis spectra were obtained on a BioTek Syngery LX multimode reader.Synthesis of allyl retinoate
To a solution of retinoic acid (2 g, 6.67 mmol) in THF (25 mL) were added Cs2CO3 (5 g, 13.35 mmol) and allyl bromide (2.9 mL, 33.35 mmol); the reaction mixture was stirred at room temperature for 12 h, then filtered through Celite, which was rinsed with DCM. The solvents were removed by rotary evaporation under reduced pressure and extracted with H2O (100 mL) and DCM (100 mL × 2). The organic layer was collected, dried over Na2SO4, and filtered. Following concentration in vacuo, the crude product was purified by flash column chromatography (hexanes![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc 1:3 → 1:1) to afford allyl retinoate (1.55 g, 66%) as a light yellow oil (for NMR see Fig. S1†).
:EtOAc 1:3 → 1:1) to afford allyl retinoate (1.55 g, 66%) as a light yellow oil (for NMR see Fig. S1†).
1H NMR (600 MHz, chloroform-d, ppm) δ = 7.00 (dd, J = 15.0, 11.4 Hz, 1H), 6.29 (d, J = 15.1 Hz, 1H) 6.27 (d, J = 16.0 Hz, 1H), 6.14 (d, J = 10.8 Hz, 1H), 6.13 (d, J = 16.0 Hz, 1H), 5.95 (ddt, J = 17.1, 10.5, 5.7, 1H) 5.80 (s, 1H), 5.33 (dq, J = 17.2, 1.5 Hz, 1H), 5.22 (ddd, J = 10.5, 2.6, 1.3 Hz, 1H), 4.62 (dt, J = 5.7, 1.4 Hz, 2H), 2.36 (d, J = 1.0 Hz, 3H), 2.02 (t, J = 6.3 Hz, 2H), 2.00 (s, 3H), 1.71 (d, J = 0.6 Hz, 3H), 1.64–1.58 (m, 2H), 1.48–1.44 (m, 2H), 1.03 (s, 3H). 13C-NMR (151 MHz, chloroform-d, ppm) δ = 167.7, 153.3, 139.7, 137.7, 137.3, 135.1, 132.7, 131.1, 130.0, 129.5, 128.7, 118.1, 117.8, 64.4, 39.6, 34.3, 33.1 29.0, 21.7, 19.2, 13.9, 12.9 (Fig. S1†). HR-MS (ESI) m/z calcd for C23H32O2 [M + H]+ 341.2475; found 341.2481.
Synthesis of homoallyl retinoate 2
To a stirred solution of retinoic acid (5.0 g, 16.68 mmol) in anhydrous CH2Cl2 (237 mL) was added 3-buten-1-ol (1.72 ml, 20.02 mmol) followed by EDC·HCl (3.85 g, 20.02 mmol) and dimethylaminopyridine DMAP (4.89 g, 40.04 mmol). The reaction was stirred for 12 h at rt then extracted with H2O (100 mL) and DCM (100 mL × 2), dried over (Na2SO4), and filtered. Following concentration under reduced pressure, the crude product was purified by flash column chromatography (hexanes:EtOAc 4:1) to afford homoallyl retinoate 2 (4.1 g, 70%) as a light-yellow oil of a 70/30 E/Z mixture (for NMR see Fig. S3†).
2-E isomer: 1H-NMR (600 MHz, chloroform-d, ppm) δ = 6.99 (dd, J = 15.0, 11.3 Hz, 1H), 6.28 (d, J = 15.0 Hz, 1H) 6.27 (d, J = 16.5 Hz, 1H), 6.14 (d, J = 11.8 Hz, 1H), 6.13 (d, J = 15.8 Hz, 1H), 5.82 (ddt, J = 17.1, 10.3, 6.7, 1H), 5.77 (s, 1H), 5.14 (ddd, J = 17.2, 3.4, 1.6 Hz, 1H), 5.07 (ddd, J = 10.2, 2.7, 1.2 Hz, 1H), 4.17 (t, J = 6.7 Hz, 2H), 2.41 (q, J = 6.7 Hz, 2H), 2.35 (d, J = 1.0 Hz, 3H), 2.02 (t, J = 6.1 Hz, 2H), 2.00 (s, 3H), 1.71 (d, J = 0.5 Hz, 3H), 1.64–1.58 (m, 2H), 1.48–1.44 (m, 2H), 1.03 (s, 3H). 13C-NMR (151 MHz, chloroform-d, ppm) δ = 167.1, 152.8, 139.5, 137.7, 137.2, 135.1, 134.2, 131.0, 129.9, 129.5, 128.6, 118.4, 117.0, 62.8, 39.6, 34.2, 33.2, 33.1, 28.9, 21.7, 19.2, 13.8, 12.9.
2-Z isomer: 1H-NMR (600 MHz, chloroform-d, ppm) δ = 7.77 (d, J = 15.3 Hz, 1H), 6.97 (dd, J = 15.2, 10.1 Hz, 1H), 6.29–6.23 (m, 2H), 6.14 (d, J = 16.6 Hz, 1H), 5.82 (ddt, J = 17.1, 10.3, 6.7, 1H) 5.64 (s, 1H), 5.14 (ddd, J = 17.2, 3.4, 1.6 Hz, 1H), 5.07 (ddd, J = 10.2, 2.7, 1.2 Hz, 1H), 4.16 (t, J = 6.7 Hz, 2H), 2.41 (q, J = 6.7 Hz, 2H), 2.06 (d, J = 1.0 Hz, 3H), 2.02 (t, J = 6.1 Hz, 2H), 1.99 (s, 3H), 1.72–1.71 (m, 3H), 1.64–1.58 (m, 2H), 1.48–1.44 (m, 2H), 1.03 (s, 3H). 13C-NMR (151 MHz, chloroform-d, ppm) δ = 166.3, 151.2, 139.7, 137.7, 137.4, 134.3, 132.2, 130.3, 130.0, 129.3, 128.5, 117.0, 116.4, 62.8, 39.6, 34.2, 33.2, 33.1, 28.9, 20.9, 19.2, 13.8, 12.8. HR-MS (ESI) of the E/Z mixture. m/z calcd for C24H35O2 [M + H]+ 355.2632; found 355.2618.
Hydrosilylation of allyl retinoate
In a 100 mL round-bottomed flask equipped with a stir bar was added pendent SiH silicone (6–7% SiMeH, HMS-071, 2.0 g, 1.49 mmol) and allyl retinoate (0.65 g, 1.9 mmol) in toluene (10 ml). After stirring for 5 min, Pt in xylenes (0.05 mL of 2% solution, 5.0 μmol) was added, and the reaction was stirred for an additional 12 h at 60 °C. After the reaction was cooled to room temperature, charcoal was added and the reaction was stirred for 6 h. The crude mixture was then filtered through Celite and washed with DCM. After evaporating the solvents, methanol was added to the crude material, which was transferred to a Falcon tube and centrifuged. The methanol was then decanted from the oil, and the process was repeated 5 times (4000 rpm, 5 × 10 min) to obtain a 1:1 mixture of alkyl and silyl retinoates 1P (1.9 g, 76%) as a yellow oil. It is inferred that both structures will be found on the same polymer backbone. The structures are reported separately for clarity (Fig. 1A). The possible mode of reaction leading to these outcomes is shown in Fig. S2A;† for NMR spectra see Fig. S4.†
Alkyl retinoate (48%): 1H-NMR (600 MHz, chloroform-d, ppm) δ = 7.03–6.95 (m, 1H), 6.31–6.24 (m, 2H), 6.17–6.12 (m, 2H), 5.78 (s, 1H), 4.16 (t, J = 6.7 Hz, 2H), 2.35 (s, 3H), 2.03 (t, J = 6.1 Hz, 2H), 2.00 (s, 3H), 1.73–1.66 (m, 5H), 1.64–1.58 (m, 2H), 1.50–1.45 (m, 2H), 1.03 (s, 3H), 0.60–0.55 (m, 2H), 0.21 to −0.06 (m, 130H). 13C-NMR (151 MHz, chloroform-d, ppm) δ = 167.4, 152.7, 139.7, 137.9, 137.5, 135.4, 131.2, 130.1, 129.7, 128.8, 118.9, 66.3, 39.8, 34.4, 33.2, 29.1, 22.6, 21.9, 19.4, 14.0, 13.6, 13.0, 1.3.
Silyl retinoate (46%) 7.03–6.95 (m, 1H), 6.31–6.24 (m, 2H), 6.17–6.12 (m, 2H), 5.77 (s, 1H), 2.34 (s, 3H), 2.03 (t, J = 6.1 Hz, 2H), 2.00 (s, 3H), 1.73–1.66 (m, 5H), 1.71 (s, 3H), 1.64–1.58 (m, 2H), 1.50–1.45 (m, 2H), 1.03 (s, 3H), 0.32 (s, 6H), 0.21 to −0.06 (m, 130H). 13C-NMR (151 MHz, chloroform-d, ppm) δ = 165.7, 153.6, 139.6, 137.9, 137.5, 135.4, 131.2, 130.1, 129.7, 128.7, 120.3, 39.8, 34.4, 33.2, 22.6, 21.9, 19.4, 13.9, 13.0, 1.3.
Propyl modification (6%) 1.43–1.35 (m, 2H), 0.97–0.90 (m, 3H), 0.58–0.42 (m, 2H), 0.21 to −0.06 (m, 130H).
Hydrosilylation of homoallyl retinoate: telechelic silicones
In a 100 mL round-bottomed flask equipped with a stir bar was added telechelic DMS-121 (2.0 g, 0.89 mmol) and 2 (0.39 g, 1.1 mmol) in toluene (10 ml). After stirring for 5 min, Karstedt's platinum catalyst in xylenes was added (0.05 mL of 2% solution, 1.0 μmol), and stirred for an additional 12 h at 60 °C. After the reaction had cooled to room temperature, charcoal was added and stirred for 6 h. The crude mixture was then filtered through Celite and washed with DCM. After evaporating the solvents, methanol was added to the crude material, transferred to a Falcon tube and centrifuged. The methanol was then decanted from the oil, and the process was repeated 5 times (4000 rpm, 5 × 10 min) to obtain a pure 70/30 E/Z mixture of (3T) (2.1 g, 92%) as a yellow oil. Although both organic chains will present on the silicone backbone, we separate out the NMR data for clarity (Fig. S5†).Z-Isomer 3T (30%) δ = 7.77 (d, J = 15.3 Hz, 1H), 7.00–6.94 (m 1H), 6.29–6.23 (m, 2H), 6.17–6.12 (m, 1H), 5.64 (s, 1H), 4.12–4.09 (m, 2H), 2.06 (d, J = 1.0 Hz, 3H), 2.04–2.01 (m, 2H), 1.99 (s, 3H), 1.71 (s, 3H), 1.70–1.66 (m, 2H), 1.64–1.58 (m, 2H), 1.49–1.42 (m, 4H), 1.03 (s, 3H), 0.60–0.55 (m, 2H), 0.18 to −0.04 (m, 300H). 13C-NMR (151 MHz, chloroform-d, ppm) δ = 165.5, 149.9, 138.6, 136.7, 136.5, 131.1, 129.4, 128.5, 128.4, 115.6, 62.5, 38.6, 33.2, 32.1, 31.3, 27.9, 19.9, 18.8, 18.2, 12.8, 11.9, 0.0.
3T E-isomer (70%): 1H-NMR (600 MHz, chloroform-d, ppm) δ = E-isomer (70%) = 6.99 (dd, J = 15.3, 11.3 Hz, 1H), 6.28 (d, J = 15.0 Hz, 1H) 6.29–6.24 (m, 2H), 6.17–6.12 (m, 2H), 5.77 (s, 1H), 4.11 (t, J = 6.6 Hz, 2H), 2.35 (d, J = 1.0 Hz, 3H), 2.02 (t, J = 6.1 Hz, 2H), 2.00 (s, 3H), 1.71 (s, 3H), 1.70–1.66 (m, 2H), 1.64–1.58 (m, 2H), 1.49–1.42 (m, 4H), 1.03 (s, 3H), 0.60–0.55 (m, 2H), 0.18 to −0.04 (m, 300H). 13C-NMR (151 MHz, chloroform-d, ppm) δ = 166.3, 151.5, 138.5, 136.7, 136.3, 134.2, 129.9, 128.9, 128.5, 127.6, 117.7, 62.5, 38.6, 33.2, 32.1, 31.3, 27.9, 20.7, 18.8, 18.2, 16.8, 12.8, 11.9, 0.00 (Fig. S3A and B†).
An analogous process was run using the pendent HSi silicone HMS-071 (2.0 g, 1.49 mmol), 2 (0.67 g, 1.9 mmol) to give 3P (2.03 g, 81%) as a yellow oil.
Z isomer 3P (30%): 1H-NMR (600 MHz, chloroform-d, ppm) δ = 7.79 (d, J = 15.3 Hz, 1H), 7.00–6.94 (m 1H), 6.29–6.23 (m, 2H), 6.17–6.12 (m, 1H), 5.63 (s, 1H), 4.13–4.06 (m, 2H), 2.06 (s, 3H), 2.04–2.01 (m, 2H), 1.99 (s, 3H), 1.71 (s, 3H), 1.70–1.66 (m, 2H), 1.64–1.58 (m, 2H), 1.49–1.40 (m, 4H), 1.03 (s, 3H), 0.58–0.50 (m, 2H), 0.20 to −0.10 (m, 144H). 13C-NMR (151 MHz, chloroform-d, ppm) δ = 166.6, 151.1, 139.7, 137.9, 137.7, 132.2, 130.1, 129.6, 128.7, 116.8, 63.6, 39.8, 34.4, 33.3, 32.3, 29.1, 21.1, 19.7, 19.4, 17.2, 14.0, 13.0, 1.2.
E isomer 3P (70%): 1H-NMR (600 MHz, chloroform-d, ppm) δ = 6.98 (dd, J = 14.9, 11.4 Hz, 1H), 6.28 (d, J = 15.0 Hz, 1H) 6.29–6.24 (m, 2H), 6.17–6.12 (m, 2H), 5.76 (s, 1H), 4.13–4.06 (m, 2H), 2.35 (s, 3H), 2.02 (t, J = 6.1 Hz, 2H), 2.00 (s, 3H), 1.71 (s, 3H), 1.70–1.66 (m, 2H), 1.64–1.58 (m, 2H), 1.49–1.40 (m, 4H), 1.03 (s, 3H), 0.58–0.50 (m, 2H), 0.20 to −0.10 (m, 144H). 13C-NMR (151 MHz, chloroform-d, ppm) δ = 167.4, 152.7, 139.6, 137.9, 137.5, 135.4, 131.0, 130.1 129.7, 118.9, 63.7, 39.7, 34.4, 33.3, 32.3, 29.1, 21.9, 19.7, 19.4, 17.2, 14.0, 13.0, 1.2.
Silicone α-tocopherol derivatives 4T
To an oven-dried 100 ml round-bottom flask equipped with a stir bar under a N2 atmosphere, was added dry toluene (10 ml) and α-tocopherol (0.39 g, 0.90 mmol) and B(C6F5)3 as catalyst (2.3 μL of a 40 mg mL−1 solution, 0.05 mol%). Then telechelic DMS-121 (2.1 g, 0.93 mmol) was added and after 5 min bubbles began to form. After bubbling ceased, the reaction was stirred for an additional 4 h and quenched with neutral alumina. Following filtration and washing with DCM or toluene, the solvents were evaporated, then methanol was added to the crude material, transferred to a Falcon tube and centrifuged. The methanol was then decanted from the oil, and the process was repeated 5 times (4000 rpm, 5 × 10 minutes) to obtain 4T (1.5 g, 67%) as a clear oil.1H-NMR (600 MHz, chloroform-d, ppm) δ = 2.56 (t, J = 6.8 Hz, 2H), 2.12 (s, 3H), 2.07 (s, 6H), 1.84–1.72 (m, 2H), 1.61–1.54 (m, 1H), 1.52–1.46 (m, 2H), 1.44–1.34 (m, 4H), 1.33–1.23 (m, 8H), 1.22 (s, 3H), 1.16–1.11 (m, 2H), 1.10–1.03 (m, 4H), 0.87 (s, 3H), 0.86 (s, 3H), 0.85 (s, 3H), 0.84 (s, 3H), 0.17 (s, 6H), 0.07 (m, 228H). 13C-NMR (151 MHz, chloroform-d, ppm) δ = 145.1, 143.2, 124.7, 122.4, 121.3, 116.1, 73.4, 38.9, 38.4, 36.6, 36.5, 36.4, 36.3, 31.8, 31.7, 30.6, 27.0, 23.8, 23.4, 22.7, 21.7, 21.6, 20.0, 19.9, 18.7, 18.6, 12.9, 12.0, 10.9, 0.3, 0.0 (Fig. S6†).
A similar procedure was utilized to create pendent tocopherol derivatives 4P-17 and 4P-26
α-Tocopherol (1.11 g, 2.58 mmol); dry toluene (3 mL) in a dry, N2 purged 50 mL round-bottomed flask. B(C6F5)3 (99 μL, 3.86 μmol); HMS-151 (1 g, 2.15 mmol) was added dropwise using a syringe. The solution was allowed to stir at 60 °C for 12 h. Aluminum oxide was added to the solution and allowed to stir for 12 h. The resulting solution was filtered via vacuum filtration through a Celite pad, and the solvent removed under reduced pressure. The yellow viscous liquid was transferred to a 15 mL centrifuge tube and washed with acetone to remove excess of α-tocopherol via centrifugation at 4000 rpm, the washed was done 3 times. Polymer 4P-17 was obtained with a yield of 80% (1.6 g). 1H NMR (CDCl3, 600 MHz): δ 2.55 (m, 2H), 2.13–2.07 (m, 9H), 1.76 (m, 2H), 1.56–1.07 (m, 22H), 0.87–0.84 (m, 12H), 0.21–−0.04 (m, 39H). Molecular weight: HMS-151 (starting material) Mn (g mol−1) 2625 Mw (g mol−1) 3990, ĐM 1.51; 4P-17Mn (g mol−1) 7945, Mw (g mol−1) 11875, ĐM 1.49.
4P-26: The same procedure was employed to synthesize 301-VitE: HMS-301 (1.0 g, 3.977 mmol); tocopherol (2.05 g, 4.773 mmol); B(C6F5)3 (0.186 mL, 2.38 μmol); yield 76.67% 301-VitE (2.3 g). 1H NMR (CDCl3, 600 MHz): δ 2.54 (m, 2H), 2.13–2.06 (m, 9H), 1.75 (m, 2H), 1.55–1.07 (m, 22H), 0.88–0.84 (m, 12H), 0.20 to −0.03 (m, 25H). Molecular weight: HMS-301 (starting material) Mn (g mol−1) 2654, Mw (g mol−1) 4005, ĐM 1.50; 4P-26Mn (g mol−1) 8187, Mw (g mol−1) 14121, ĐM 1.72.
Eugenol-modified silicones: oils 5T and 5P-6 and 5P-17
5T: In a 100 mL round-bottomed flask equipped with a stir bar was added telechelic DMS-121 (2.0 g, 0.89 mmol) and eugenol (0.17 ml, 1.1 mmol, neat). After stirring for 5 min, Karstedt's platinum catalysts (0.05 mL of 2% solution, 1.0 μmol) was added. The reaction was stirred for 12 h at room temperature. Charcoal was added (100 mg) and the reaction further stirred for 6 h. The crude mixture was then filtered through Celite and washed with DCM. After evaporating the solvents, methanol was added to the crude material, transferred to a Falcon tube and centrifuged. The methanol was then decanted from the oil, and the process was repeated 5 times (4000 rpm, 5 × 10 minutes) to obtain pure 5T (2.0 g, 93%) as a clear oil.1H-NMR (600 MHz, chloroform-d, ppm) δ = 6.82 (d, J = 8.5 Hz, 1H), 6.69–6.61 (m, 2H) 5.43 (s, 1H), 3.87 (s, 3H), 2.55 (t, J = 7.6 Hz, 2H), 1.62 (dq, J = 16.1, 8.2 Hz, 2H), 0.61–0.54 (m, 2H), 0.18 to −0.04 (m, 320H). 13C-NMR (151 MHz, chloroform-d, ppm) δ = 145.2, 142.5, 133.7, 120.0, 113.0, 110.0, 54.8, 38.4, 24.6, 17.0, 0.0 (Fig. S7A and B†).
5P-6: The same protocol was utilized as above: 6–7% pendant HMS-071 (2.0 g, 1.49 mmol); eugenol (0.29 ml, 1.9 mmol). 5P-6 (1.9 g, 84%) as a clear oil.
1H-NMR (600 MHz, chloroform-d, ppm) δ = 6.82 (d, J = 8.5 Hz, 1H), 6.69–6.61 (m, 2H) 5.43 (s, 1H), 3.87 (s, 3H), 2.55 (t, J = 7.6 Hz, 2H), 1.68–1.60 (m, 2H), 0.61–0.54 (m, 2H), 0.18 to −0.04 (m, 320H). 13C-NMR (151 MHz, chloroform-d, ppm) δ = 145.2, 142.5, 133.6, 120.0, 113.0, 110.0, 54.8, 38.4, 24.3, 16.2, 0.0 (Fig. S7C and D†).
5P-17: Using the same protocol a starting material with a higher SiH density HMS-151 (15%) was also modified with eugenol (2.0 g, 4.3 mmol); eugenol (0.8 ml, 5.1 mmol) and Karstedt's platinum catalyst (0.1 mL of 2% solution, 10.0 μmol) to afford a light brown oil.
Synthesis of eugenol elastomers, shown for 5E-75
A similar protocol was used as for the synthesis of 5P-17. However, instead of consuming all of the backbone SiH groups with eugenol, 75% were eugenol modified and the other 25% were crosslinked with telechelic vinylsilicones.In a 40 mL polypropylene cup equipped with a stir bar was added pendent HMS-151 (2.0 g, 4.3 mmol SiH) in toluene (5 ml) and eugenol (0.5 ml, 3.2 mmol, 0.75 equiv.). After stirring for 5 min, Karstedt's platinum catalyst (0.1 ml of 2% Pt in xylenes, 10.0 μmol) was added. The reaction was stirred for 12 h at room temperature. Then telechelic DMS-V21 cross-linker (3.22 g, 1.08 mmol Vi, 0.25 equiv. vs. SiH) was added, the stir bar was removed and the mixture was placed in a 50 °C oven for 12 h to allow curing. After curing, the elastomer was extracted with DCM (5 × 10 ml) to remove, after evaporation, a light yellow elastomer resulted; <2 wt% of the oils were recovered during this process. This step is not necessary to produce the elastomer and was performed only to demonstrate that very low quantities of unreacted oils are found in the elastomer. This value is retained, which are not required.
5E-50 and 5E-33 were produced in a similar manner
5E-50: Pendent HMS-151 (2.0 g, 4.3 mmol) in toluene (5 ml), eugenol (0.33 ml, 2.1 mmol, 0.5 equiv.), Karstedt's platinum catalyst (0.1 ml of 2% Pt in xylenes, 10.0 μmol) and DMS-V21 (4.83 g, 1.62 mmol, 0.5 equiv.).
5E-33: Pendent HMS-151 (2.0 g, 4.3 mmol) in toluene (5 ml), eugenol (0.22 ml, 1.4 mmol, 0.33 equiv.), Karstedt's platinum catalyst (0.1 ml of 2% Pt in xylenes, 10.0 μmol) and DMS-V21 (7.30 g, 2.45 mmol).
AO activity using DPPH (Fig. 3)
First, 0.2 mM DPPH in IPA stock solution was prepared to be used with the analyte. Then antioxidant modified polymer solutions were prepared as 80 mM stock solution (with the exception of 5T, was prepared as 40 mM stock solution due to limited sample) and then serial diluted into 60, 40, 20, 4, 0.4 mM, 40, 4, 0.4 μM (with the exception of 5T, was diluted into 30, 20, 10, 2, 0.2 mM, 20, 2, 0.2 μM) to a total volume of 1 mL with IPA in 2 mL centrifuge tubes. Each solution (0.5 mL) was transferred to another 2 mL centrifuge, and to it was added DPPH (0.5 mL of 0.2 mM, i.e., total volume 1 mL). The solutions were vortexed and placed in the dark for 15 minutes to react. The resulting solution (200 μL) was added into a well of a 96 well plate in triplicate. DPPH solution was used as a control (0.1 mM), also in triplicate. Scans were taken for each well at 520 nm from the plate reader and the results were recorded. The DPPH reaction is shown in Fig. S2B (ESI†).DPPH film measurements
DPPH assay
A stock solution of DPPH (0.2 mM) was added to a 96 well plate containing AO silicones that had been serially diluted. Changes in visible spectra over time were recorded (Table S1,†Fig. 3, Fig. S8†).On fabric (oil deposited first)
To the cloth, 1 drop (25 mg) of 5P-17 was absorbed into the cloth for each ‘eye’, and 6 drops (150 mg) was absorbed and smeared to form the ‘smile’. Then DPPH solution (1 mL of 5 mM) was added to the cloth and a color change from purple to yellow was observed (Fig. S9†).On fabric (DPPH deposited first)
DPPH solution (2 mL of 5 mM) was absorbed onto the cloth, then 1 drop (25 mg) of 5P-17 was place on the cloth to give each ‘eye’, and 6 drops (150 mg) was absorbed to form the ‘smile’. The color change from purple to yellow was observed (Fig. 4A).DPPH analysis of elastomer 4P-17 and 4P-26
DPPH solution: 100 μM in toluene/IPA 1:1 mixture (1 mL), polymer solution: 0.5 g mL−1 in toluene/IPA mixture (1 mL). Very low antioxidant activity was observed with either 4P-17 and 4P-26 (Fig. S10†).
DPPH analysis of elastomer 5E-X
5E-75 pre-elastomer (HMS-151 75% eugenol and cross-linked with DMS V21 elastomeric films, see above) were cast as 200 μm films, and cured. Samples (circular diameter 0.85 cm) were cut from the film. Different concentrations of DPPH (0.1 mL, 5 mM, 0.5 mM, and 0.05 mM, and 0.005 mM DPPH in IPA) were spotted onto each sample and photographed at different time points (Fig. 4B and C).DPPH UV analysis of elastomers 5E
Samples of 5E-33, -50, -75, respectively (1 mg, 10 mg and 100 mg, respectively) were weighed into 1.5 mL centrifuge tubes as whole pieces. DPPH IPA solution (0.1 mM) was then added into the centrifuge tube. The tubes were shaken and elastomers allowed to swell/react for 15 min. The final eugenol concentration was calculated based on amount of eugenol present in the 1 mL IPA solution. The resulting solution (200 μL) was added into a well of a 96 well plate in triplicate. DPPH solution was used as a control (0.1 mM), also in triplicate. Scans were taken for each well at 520 nm from the plate reader and the results were recorded (Fig. S11, Table S2†).Attempted oxidation of eugenol-derived oils and elastomers with aqueous peroxide
To 5P-17 (100 mg) was added 30% H2O2 in water (3 ml); the sample was allowed to air dry for 12 h. The oil was then placed in a 24 well plate. 5E-X elastomers (100 mg) were similarly soaked in 30% H2O2 in water (3 ml) for 12 h. Then the solution was decanted from the elastomers and washed with water (5 ml × 5), and IPA (5 ml × 5). After drying in the oven at 70 °C for 3 h, the elastomers were placed in a 24 well plate. The time for decolorization of an IPA/DPPH solution (0.5 mM, 1 ml) by the oil/elastomer was essentially unaffected by exposure to the H2O2 solution showing that the water-based oxidant is not able to oxidize eugenol within the silicone material (Table S3†).Conclusions
The environmental impact of silicones can be mitigated while adding functionality by incorporating natural antioxidants. Chemical granting of polyenes and phenols to silicones via typical SiH chemistry led to products with approximately the same antioxidant performance as the starting natural material. The silicones could be effectively diluted, particularly with pendent oils, by up to 60 wt% by natural materials, yet exhibited typical silicone properties.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge with gratitude the financial support of the Natural Sciences and Engineering Research Council of Canada.References
- A. Yadav, R. Kumari, A. Yadav, J. P. Mishra, S. Srivatva and S. Prabha, Res. Environ. Life Sci., 2016, 9, 1328–1331 Search PubMed.
- M. Dexter, R. W. Thomas and R. E. King III, Encyclopedia of Polymer Science and Technology, 2002, DOI:10.1002/0471440264.pst020.
- A. C. Pedro, O. G. Paniz, I. d. A. A. Fernandes, D. G. Bortolini, F. T. V. Rubio, C. W. I. Haminiuk, G. M. Maciel and W. L. E. Magalhães, Antioxidants, 2022, 11, 1644 CrossRef CAS PubMed.
- I. Kusumawati and G. Indrayanto, in Studies in Natural Products Chemistry, ed. Atta-ur-Rahman, Elsevier, 2013, vol. 40, pp. 485–505 Search PubMed.
- W. S. Blaner, I. O. Shmarakov and M. G. Traber, Annu. Rev. Nutr., 2021, 41, 105–131 CrossRef PubMed.
- M. G. Traber and J. Atkinson, Free Radical Biol. Med., 2007, 43, 4–15 CrossRef CAS PubMed.
- F. W. P. C. van Overveld, G. R. M. M. Haenen, J. Rhemrev, J. P. W. Vermeiden and A. Bast, Chem.-Biol. Interact., 2000, 127, 151–161 CrossRef CAS PubMed.
- İ. Gülçin, J. Med. Food, 2011, 14, 975–985 CrossRef PubMed.
- A. S. Kabir, Z.-S. Yuan, T. Kuboki and C. Xu, in Production of Materials from Sustainable Biomass Resources, ed. Z. Fang, J. R. L. Smith and X.-F. Tian, Springer Singapore, Singapore, 2019, pp. 39–59, DOI:10.1007/978-981-13-3768-0_2.
- P. T. Anastas and J. C. Warner, Green Chemistry Theory and Practice, OUP, 2000 Search PubMed.
- R. G. Lehmann, S. Varaprath, R. B. Annelin and J. L. Arndt, Environ. Toxicol. Chem., 1995, 14, 1299–1305 CrossRef CAS.
- R. G. Lehmann, S. Varaprath and C. L. Frye, Environ. Toxicol. Chem., 1994, 13, 1061–1064 CrossRef CAS.
- J. A. Heredia-Guerrero, L. Ceseracciu, S. Guzman-Puyol, U. C. Paul, A. Alfaro-Pulido, C. Grande, L. Vezzulli, T. Bandiera, R. Bertorelli, D. Russo, A. Athanassiou and I. S. Bayer, Carbohydr. Polym., 2018, 192, 150–158 CrossRef CAS PubMed.
- S. Iida, A. Takushima, N. Ohura, S. Sato, M. Kurita and K. Harii, Dermatol. Surg., 2013, 39, 1237–1242 CrossRef CAS PubMed.
- R. van Lith, X. Wang and G. Ameer, ACS Biomater. Sci. Eng., 2016, 2, 268–277 CrossRef CAS PubMed.
- X. Zhang, Y. Cai, X. Zhang, T. Aziz, H. Fan and C. Bittencourt, J. Appl. Polym. Sci., 2021, 138, 50515 CrossRef CAS.
- J. Xu, W. Zhang, Q. Jiang, J. Mu and Z. Jiang, Polymer, 2015, 62, 77–85 CrossRef CAS.
- M. S. I. Mollah, Y.-D. Kwon, M. M. Islam, D.-W. Seo, H.-H. Jang, Y.-D. Lim, D.-K. Lee and W.-G. Kim, Polym. Bull., 2012, 68, 1551–1564 CrossRef CAS.
- X. Pang, X. Ge, J. Ji, W. Liang, R. Liu, X. Chen, G. Yin and J. Ge, Polymers, 2019, 11, 1302 CrossRef CAS PubMed.
- S. E. Laengert, A. F. Schneider, E. Lovinger, Y. Chen and M. A. Brook, Chem. – Asian J., 2017, 12, 1208–1212 CrossRef CAS PubMed.
- B. Kumar, D. O. Agumba, D. H. Pham, H. C. Kim and J. Kim, J. Appl. Polym. Sci., 2022, 139, 51532 CrossRef CAS.
- G. Chen, J. Feng, W. Qiu and Y. Zhao, RSC Adv., 2017, 7, 55967–55976 RSC.
- J. Ji, X. Ge, W. Liang, R. Liang, X. Pang, R. Liu, S. Wen, J. Sun, X. Chen and J. Ge, Polymers, 2019, 11, 1389 CrossRef PubMed.
- J. Zhang, E. Fleury, Y. Chen and M. A. Brook, RSC Adv., 2015, 5, 103907–103914 RSC.
- E. P. Plueddemann, Silane Coupling Agents, Plenum Press, New York, 2nd edn, 1991 Search PubMed.
- T. Suzuki and H. Fujimoto, Inorg. Chem., 1999, 38, 370–382 CrossRef CAS.
- M. Liao, A. F. Schneider, S. E. Laengert, C. B. Gale, Y. Chen and M. A. Brook, Eur. Polym. J., 2018, 107, 287–293 CrossRef CAS.
- A. S. Fawcett and M. A. Brook, Macromolecules, 2014, 47, 1656–1663 CrossRef CAS.
- S. B. Kedare and R. P. Singh, J. Food Sci. Technol., 2011, 48, 412–422 CrossRef CAS PubMed.
- M. C. Foti, J. Agric. Food Chem., 2015, 63, 8765–8776 CrossRef CAS PubMed.
- E. Ogliani, A. L. Skov and M. A. Brook, Adv. Mater. Technol., 2019, 4, 1900569 CrossRef CAS.
- M. Hiramatsu and L. Packer, Methods Enzymol., 1990, 190, 273–280 CAS.
- R. Yamauchi, Food Sci. Technol. Int., Tokyo, 1997, 3, 301–309 CAS.
- S. Nagarajan, R. Nagarajan, J. Kumar, A. Salemme, A. R. Togna, L. Saso and F. Bruno, Polymers, 2020, 12, 1646 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of products, DPPH assay protocol, additional AO charts and photos of DPPH assays. See DOI: https://doi.org/10.1039/d2gc03112d |
| This journal is © The Royal Society of Chemistry 2022 |