
A sustainable way of recycling polyamides: dissolution and ammonolysis of polyamides to diamines and diamides using ammonia and biosourced glycerol†
Wouter
Stuyck
 a,
Kwinten
Janssens
a,
Mats
Denayer
c,
Free
De Schouwer
a,
Robin
Coeck
a,
Katrien V.
Bernaerts
b,
Jelle
Vekeman
d,
Frank
De Proft
c and
Dirk E.
De Vos
*a
a,
Kwinten
Janssens
a,
Mats
Denayer
c,
Free
De Schouwer
a,
Robin
Coeck
a,
Katrien V.
Bernaerts
b,
Jelle
Vekeman
d,
Frank
De Proft
c and
Dirk E.
De Vos
*a
aCentre for Membrane Separations, Adsorption, Catalysis and Spectroscopy for Sustainable Solutions (cMACS), KU Leuven, Celestijnenlaan 200F, Post box 2454, 3001 Leuven, Belgium. E-mail: dirk.devos@kuleuven.be
bSustainable Polymer Synthesis Group, Aachen-Maastricht Institute for Biobased Materials (AMIBM), Faculty of Science and Engineering, Maastricht University, Brightlands Chemelot Campus, Urmonderbaan 22, 6167 RD Geleen, The Netherlands
cEenheid Algemene Chemie (ALGC), Department of Chemistry, Vrije Universiteit Brussel (VUB), Pleinlaan 2, 1050 Brussel, Belgium
dCentre for Molecular Modeling (CMM), Ghent University, Technologiepark-Zwijnaarde 46, 9052 Zwijnaarde, Belgium
First published on 24th August 2022
Abstract
In order to make recycling a viable strategy for post-consumer plastics, economically feasible revalorization processes must be developed. The ammonolysis of polyamides can be such a cutting-edge recycling technology; however, due to the rigid structure of these polyamide plastics, operating conditions of current ammonolysis processes are harsh, including high temperatures (>300 °C) and high NH3 pressures. Here, we report a very green and elegant ammonolysis process of the widely abundant polyamide 66 by using a hard Lewis acid catalyst and 1 bar of NH3 in a simple glycol solvent at 200 °C. Computational studies revealed that especially the vicinal diol moiety of these glycol solvents plays a key role in activation of the ammonia nucleophile, with glycerol being the most effective solvent, reaching the depolymerization equilibrium after 20 h even without a catalyst. To our delight, a biosourced glycerol (obtained from the saponification of triglycerides) could also directly serve as a suitable solvent, even outperforming the ammonolysis process in highly purified glycerol.
Introduction
The great versatility, easy applicability and tunability of plastics have led to an enormous production of plastics1 and forecasted growth of the plastic market.2 However, current production goes hand in hand with the generation of large amounts of plastic waste, especially due to our embedded take-make-dispose approach regarding consumable materials.3,4 To achieve a transition from the current linear plastics economy to a desirable circular plastics economy, green and environmentally friendly recycling processes must be developed for plastics.5–7 Moreover, these recycling processes should be economically feasible in order to compete with the production of new plastics starting from virgin, petrochemical feedstocks.8 Polyamides could be used in this respect, because of the presence of secondary amide bonds in their polymer structure, as a potential source for the production of highly valuable, (bifunctionalized) nitrogen-rich molecules, thereby offering a definite economic incentive to recycle these polymers. Most of the polyamide plastics are used either in the automotive, electrical or textile industry and approximately 95% of these polymers consist of polyamide 6 (PA6) and polyamide 66 (PA66).9,10 Regarding recycling of polyamides, two main approaches can be followed: mechanical recycling or chemical recycling. The process of mechanical recycling is generally more simple and cheaper compared to chemical recycling. However, recurring drawbacks such as a loss of molecular weight due to chain scission result in inferior properties of the recycled polyamide compared to virgin polyamide.11 This downcycling can be circumvented by selective degradation of the polyamides to their respective monomers, which can then be reused in the synthesis of new polyamides. This approach is known as chemical recycling and can, in theory, result in the complete reusability of waste polyamides.12 Chemical recycling of PA6 has been investigated repeatedly, and different technologies provide efficient routes for recovering the monomer ε-caprolactam through pyrolysis,13 hydrolysis14 or ammonolysis,15 or by applying supercritical fluids16 or ionic liquids.17 However, high operational temperatures (>250 °C),13,16 high pressures16 and strong acidic or basic conditions14 often entail high energy input and operational difficulties that limit the applicability. Chemical recycling of PA66, on the other hand, is more challenging, because PA66 polymer chains interact more strongly with one another since each secondary amide bond participates in two strong hydrogen bonds. As a result, PA66 possesses a rigid semi-crystalline polymer structure, which translates into a higher tensile strength and higher melting point compared to PA6 on a macroscopic scale.18 Both the intrinsic strength of the secondary amide bond and the resulting semi-crystalline polymer matrix pose severe challenges for the chemical recycling of PA66, which is typically tackled by applying high temperatures (>275 °C),14a,15a,b,19,20 high pressures,21–23 supercritical fluids,24,25 and/or strong acidic26 or alkaline conditions.20,27 In this work, the successful chemocatalytic ammonolysis of PA66 is reported, which can be carried out under relatively mild conditions by applying a homogeneous Lewis acid La(OTf)3 catalyst and an environmentally friendly glycol solvent. In this way adipamide and hexamethylenediamine are generated, of which the latter is a monomer of PA66, while the former can be converted to the highly demanded adiponitrile or again to the monomer hexamethylene diamine.28,29 Compared to previous studies on the ammonolysis of PA66 (Fig. 1),15b,30,31 we have developed an ammonolysis process that operates at significantly lower temperatures and NH3 pressures. In addition, it is shown that especially the glycol solvent plays a key role in splitting of the polymer, which was supported with theoretical density functional (DFT) studies. Remarkably, the greenness of the recycling process could be further improved with the direct use of a biobased crude glycerol waste stream as a solvent leading to the successful ammonolysis of PA66. | ||
| Fig. 1 Relevant literature on the ammonolysis of secondary (poly)amides.15b,30–32 | ||
Results and discussion
The rigid structure and semi-crystalline nature of polyamide 66 (PA66) imply a low accessibility of the secondary amide bonds, which hampers the nucleophilic attack of ammonia during the ammonolysis. Therefore, McKinney applied high temperatures (300 °C–320 °C), which resulted in the melting of PA66 and thus in more accessible secondary amide bonds.15a,b Our first objective was to achieve the dissolution of PA66 at milder temperatures as the secondary amide bonds might be more accessible when PA66 is dissolved in a solvent compared to when PA66 is present as a melt. In addition, the use of a solvent might also facilitate the coordination of a potential catalyst to the secondary amide bond, eventually leading to a more efficient ammonolysis. With an emphasis on green and environmentally benign solvents, ethylene glycol was selected after a screening (details on the choice can be found in the ESI, Table S2†). This solvent, with its polar and protic properties, is able to interfere with the hydrogen bonds between the different PA66 chains, and it can successfully dissolve the polymer at 180 °C. The second objective of this work encompassed the chemical splitting of PA66 through a catalytic ammonolysis process. The ammonolysis of 0.1 M PA66 was conducted in ethylene glycol in presence of 1 bar NH3 at 200 °C, which resulted in 35% of broken polyamide bonds after 20 h, as measured by liquid 1H NMR (Fig. 2). All of the broken bonds are split to form primary amines and primary amides (or derivatives); no alcoholysis products of PA66 with ethylene glycol were observed, indicating that the large amount of ammonia in solution (18 M, ESI section 1.4†) successfully suppresses the alcoholysis side reaction.33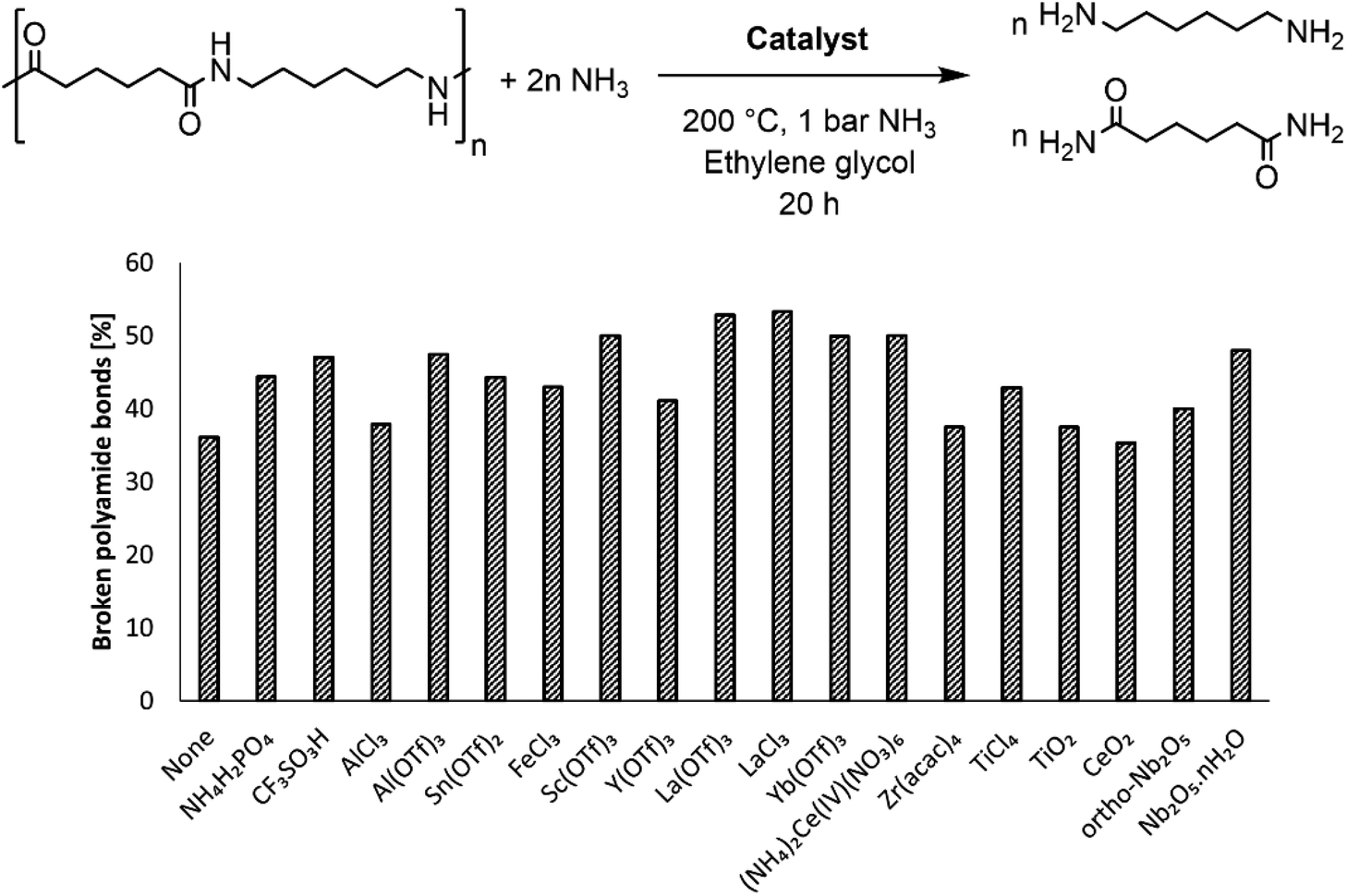 | ||
| Fig. 2 Effect of acid catalysts on the ammonolysis of PA66. Conditions: 0.1 M PA66 and 0.1 M benzyl alcohol (internal standard) in ethylene glycol (10 mL), 5 mol% homogeneous catalyst with respect to PA66, 200 °C, 1 bar NH3, 20 h. In the case of heterogeneous catalysts, 0.1 g was applied. | ||
To increase the number of broken polyamide bonds, a catalyst screening was performed, which included homogeneous Brønsted and Lewis acids and some heterogeneous acids (Fig. 2). While the reaction already proceeds without a catalyst, the degree of splitting is significantly increased in the presence of a catalyst (Fig. 2). Both homogeneous Brønsted and Lewis acids enhance the ammonolysis reaction. Brønsted acid catalysts will likely immediately be neutralized by the ammonia excess; the formed NH4+ might assist with the protonation of the amine leaving group.32 On the other hand, Lewis acid catalysts might facilitate the ammonolysis by coordination of the carbonyl oxygen atom. Cations like La3+ are in fact hard acids and do possess a strong oxophilic character,34 which results in a preferential coordination of the cation by the carbonyl oxygen atom instead of by NH3. This interaction withdraws electron density from the carbonyl oxygen atom, making the carbonyl carbon atom more prone to the nucleophilic attack of NH3 as depicted in the proposed catalytic cycle in Fig. 3. In presence of 5 mol% LaCl3 or La(OTf)3, 53% of the polyamide bonds were broken after 20 h (Fig. 2). Heterogeneous acids were generally found to be less efficient, presumably due to steric hindrance. Only in presence of Nb2O5·nH2O, a marked increase in the degree of splitting was achieved and 48% of the polyamide bonds were broken. Nb2O5·nH2O is a stable solid acid with a significant number of both Lewis and Brønsted acid sites and might thus assist in the ammonolysis through protonation of the amine leaving group, activation of the carbonyl group or a combination of both.31,35,36 Further optimizations of the ammonolysis of PA66 were conducted in presence of 5 mol% La(OTf)3.
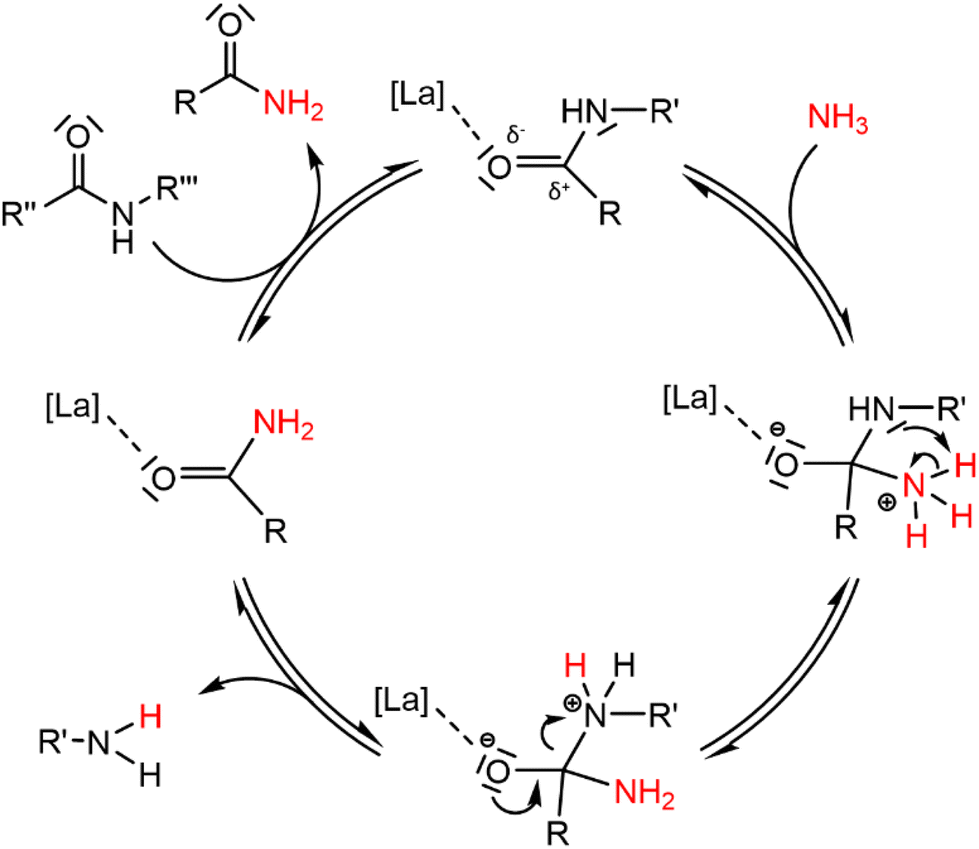 | ||
| Fig. 3 Proposed reaction mechanism for the ammonolysis of a secondary polyamide bond catalyzed by a hard Lewis acid La catalyst. | ||
Increasing the NH3 pressure, and thus the concentration of NH3 in the ethylene glycol solvent, did not significantly increase the number of broken polyamide bonds (ESI, Table S3†). Increasing the reaction temperature to 220 °C did result in an increase of the number of broken polyamide bonds to 62%; however more side products like nitriles or imidazoles were observed by 2D NMR analyses (ESI, section 4). Therefore, the reaction temperature was later kept at 200 °C. Changing the reaction solvent did have a clear effect on the ammonolysis of PA66 and a significant increase in the number of broken polyamide bonds is observed in the case of 2,2,2-trifluoroethanol (TFE) and glycerol (Fig. 4). Noteworthily, the formation of an imidazole function from the generated primary amides was not observed when glycerol or TFE were applied as solvent.
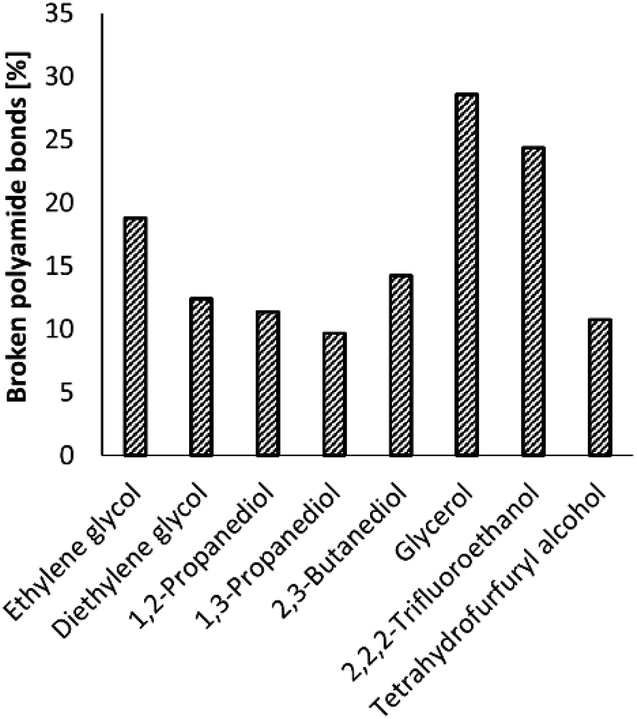 | ||
| Fig. 4 Effect of the reaction solvent on the ammonolysis of PA66. Conditions: 0.1 M PA66 and 0.1 M benzyl alcohol (internal standard) in solvent (10 mL), 5 mol% La(OTf)3, 200 °C, 1 bar NH3, 6 h. | ||
Although TFE and tetrahydrofurfuryl alcohol are both primary alcohols, large differences are observed in their performance as solvent for the ammonolysis of PA66, which can be attributed to their proton donating ability, which is weak for tetrahydrofurfuryl alcohol, but very strong for TFE. This strong proton donating ability not only assists in the dissolution of PA66, which can be achieved at room temperature in TFE,37 but might also assist in the ammonolysis of PA66 where the hydroxyl group of such an alcohol can serve as a proton shuttle catalyst.38 However, TFE is not preferred as a solvent in this process due to its toxicity39,40 and negative environmental impact.41
Regarding the polyfunctional alcohol solvents, ethylene glycol (19% broken PA66 bonds after 6 h) outperforms all the other diols, except for glycerol (29% broken PA66 bonds), while the concentration of NH3 is nearly equal in both solvents (±18 M, ESI 1.4†). Gordon et al. observed similar solvent effects of these polyfunctional alcohols on the ammonolysis of esters.42 They postulated that NH3 is activated by a vicinal diol through the formation of hydrogen-bonded ensembles. In addition, one of the hydroxyl groups can also serve as a proton shuttle (vide supra). Remarkably, small changes in solvent structure have strong effects on the reaction outcome: for instance, after 6 h, the reaction yield in glycerol is three times larger than in 1,3-propanediol, which lacks the central alcohol group.
Since all of the molecules involved, viz. the reactants ammonia and the amide group, as well as the diol can act both as a donor and as an acceptor of hydrogen bonds, computational studies were performed, scrutinizing the interactions in the solvent-polyamide-ammonia complex. In particular, the systems involving glycerol and 1,3-propanediol, the most and least favored of the investigated diol solvents (Fig. 4), were investigated. Note that instead of modeling the whole PA66 polymer, only a single amide center was considered, where methyl groups acted as R groups (Fig. 5). First, classical molecular dynamics simulations were performed, using the GROMACS package43 and OPLS-AA force field,44,45 and radial distribution functions (RDFs) between different atom pairs on the molecules of interest were calculated to obtain a reasonable estimate of the preferred orientations of the solvent around the reactants. The predominant geometry was subsequently optimized at the B3LYP/6-31++g(d,p) level of theory including Grimme's D3 dispersion correction,46 and a subsequent NCI calculation47 was conducted using the NCIplot4 program (ESI, section 8†).48Fig. 4 compares the non-covalent interactions in the amide-ammonia-glycerol complex (left) with those in the amide-ammonia-1,3-propanediol complex (right). A color gradient is used to distinguish strong non-covalent interactions with some covalent character, like hydrogen bonding (blue), from weaker van der Waals type interactions, such as dispersion (green), and destabilizing interactions (red), if any.
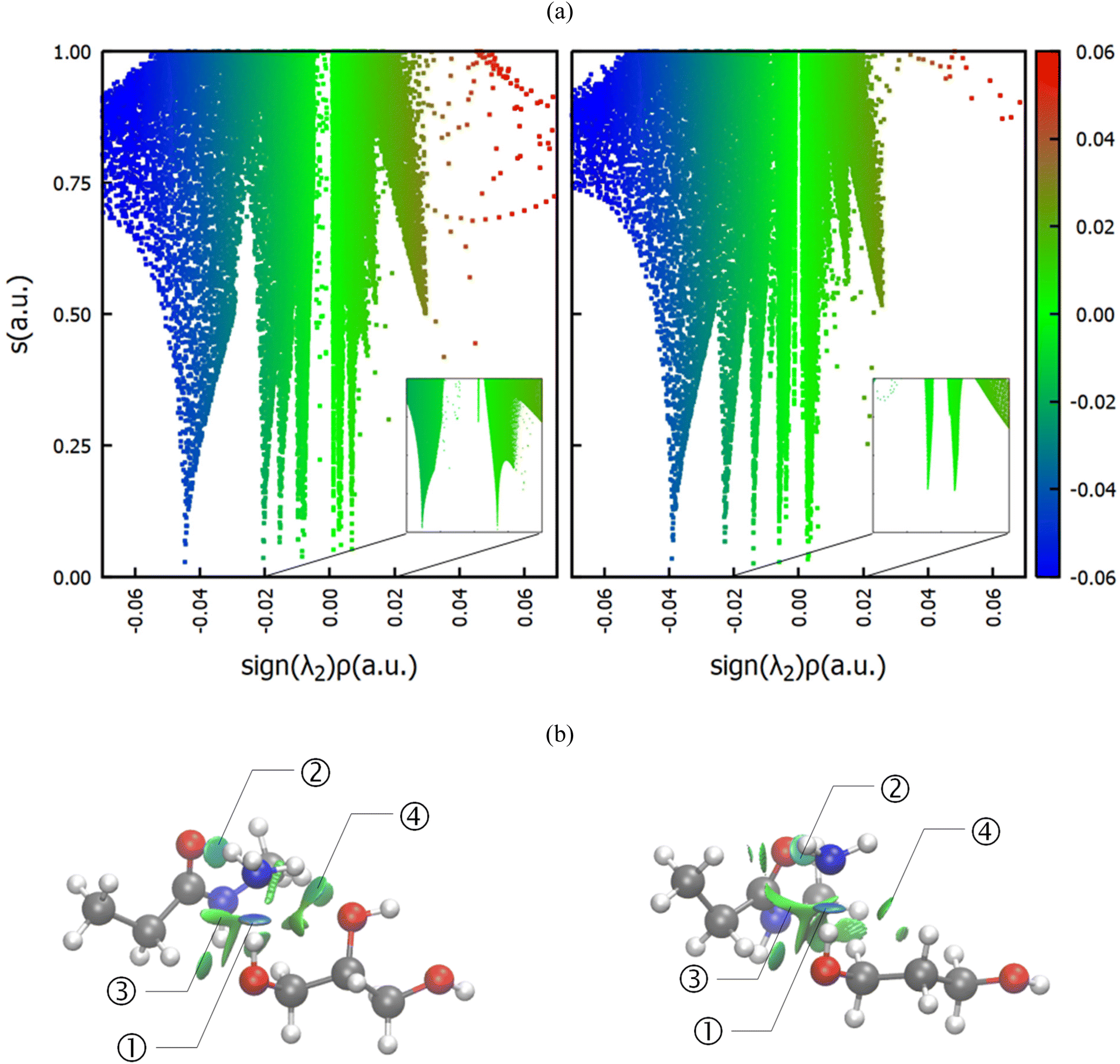 | ||
| Fig. 5 2D plot of s vs. sign(λ2)ρ (top) and 3D reduced gradient isosurfaces (bottom) for the amide-ammonia-glycerol complex (left) and the amide-ammonia-1,3-propanediol complex (right) with focus on the interaction between ammonia and the middle region of the solvent molecule pointed at by the black arrow. (isovalue = 0.5 a.u., b3lyp/6-31++g(d,p) GD3 level). Blue = nitrogen, red = oxygen, grey = carbon, white = hydrogen. | ||
It can be seen that in both cases a hydrogen bond is formed between the outer hydroxyl group (-CH2-OH) of the solvent molecule and ammonia's nitrogen atom as H-bond acceptor (Fig. 5b, ①). Additionally, ammonia interacts with the amide's oxygen atom through a hydrogen bond-like interaction with the carbonyl group as hydrogen bond acceptor and NH3 as hydrogen bond donor (②). Moreover, the relatively abundant green isosurfaces reveal that van der Waals interactions dominate the interaction between the compounds (③). An important difference between the glycerol complex and the 1,3-propanediol complex is highlighted when zooming in on the interaction between ammonia and the central section of glycerol/1,3-propanediol (④). In this region, the van der Waals interaction occurring in the glycerol complex is (much) more pronounced than in the complex with 1,3-propanediol moiety. This is evidenced by bigger peaks at a higher density in the 2D NCI plot (Fig. 5a), and a larger reduced gradient isosurface in the three-dimensional representation (Fig. 5b). These findings indicate that the central OH group in glycerol enables a stronger polarizing interaction with NH3; such an additional interaction is absent in the 1,3-propanediol complex. This is also supported by the radial distribution function of glycerol's inner oxygen atom with respect to ammonia's nitrogen atom, shown in Fig. 6. The peak at a distance of 0.30 nm indicates that there is a clear preferred distance between those atoms as a result of the van der Waals interaction between them, hindering free rotation around the C–C bond in glycerol, thus underlining the significance of this relatively weak non-covalent interaction. The vicinal hydroxyl structure thus appears crucial in enhancing the activation of the ammonia nucleophile through the formation of a true hydrogen bond on one end, and enhanced stabilization through polarizing van der Waals interactions on the other end. In addition, when looking at this stable ground state ensemble, it is clear that the nitrogen of the ammonia molecule is well-positioned to perform the nucleophilic attack on the carbonyl carbon atom (Fig. 7). It follows that glycerol, having two concatenated vicinal OH groups, is better at facilitating the nucleophile attack than ethylene glycol, a single vicinal diol. Glycerol is also superior to 1,2-propanediol, a similar scaffold but with an additional methyl group impeding the substitution reaction, and 1,3-propanediol which contains only isolated OH groups.
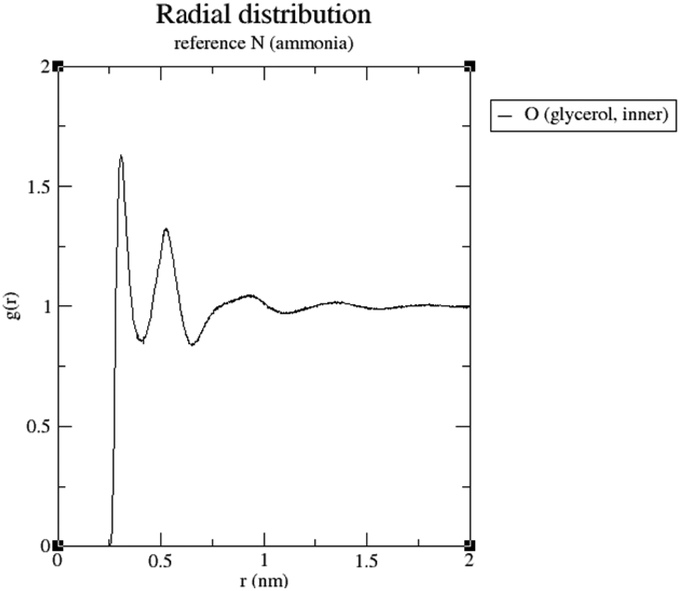 | ||
| Fig. 6 Radial distribution function of glycerol's inner oxygen with respect to ammonia's nitrogen atom. | ||
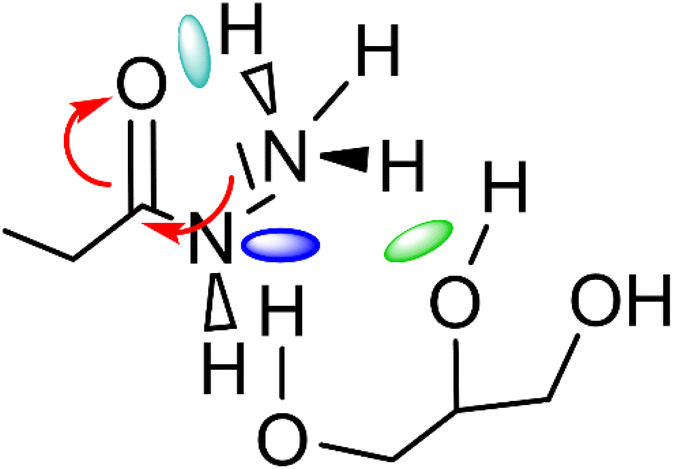 | ||
| Fig. 7 Main molecular interactions between ammonia-amide-glycerol during the nucleophilic attack of ammonia's nitrogen atom on the carbonyl group of the secondary amide. | ||
Gel permeation chromatography (GPC) was used to determine the polymer dispersity and molar mass distribution of the virgin PA polymer and two reaction mixtures after ammonolysis in ethylene glycol and glycerol, respectively (ESI, Fig. S7†). A clear shift is observed in the elution profile from a unimodal peak for PA66 to a significantly broader peak at higher elution volumes in the GPC chromatogram. For both glycerol and ethylene glycol a random chain scission is expected, explaining the random distribution at lower molecular weight (ESI, Fig. S8†).
Subsequently, the ammonolysis of PA66 was investigated over time in ethylene glycol and glycerol, with or without La(OTf)3 as a Lewis acid catalyst (Fig. 8). Whereas the addition of La(OTf)3 has a positive effect when ethylene glycol is applied, this enhancement becomes almost negligible with glycerol as the solvent: in presence of 5 mol% La(OTf)3 in glycerol, 69% of polyamide bonds were broken after 20 h, while without La(OTf)3 already 66% of the polyamide bonds were broken. Extending the reaction time for up to 72 h did not result in a significant increase of broken PA bonds, indicating that the equilibrium in the ammonolysis of PA66 has been reached under these conditions. To confirm this, the reverse reaction was studied starting from hexamethylene diamine and adipamide, resulting in 68% of primary amines after 20 h (ESI, Table S5†). Finally, since glycerol could on its own serve as a suitable solvent for the ammonolysis of PA66 in the absence of a Lewis acid catalyst, it can be argued to further improve the greenness of this reaction using a crude glycerol solvent obtained directly from a biorefinery process (e.g. the saponification of triglycerides). The use of lower grade glycerol would clearly be beneficial for the overall reduction of energy intensive purifications by finding a direct use for current biomass waste streams.49 In addition, the use of crude glycerol could result in better processability, since the presence of water (up to 20 wt%) results in a significant drop in viscosity (ηglycerol = 1410 mPa s vs. ηcrude glycerol = 56 mPa s at room temperature; ESI, section 1.8†). The use of industrial waste glycerol results in the same thermodynamic equilibrium, with 68% of the polyamide bonds broken after 20 hours (Fig. 8, green). Remarkably, the ammonolysis of PA66 in the crude glycerol solvent clearly surpasses the ammonolysis in pure glycerol at shorter reaction times, making it the best performing system hitherto. A possible explanation for the increased reaction rate, lies in the subtle differences in composition of the crude glycerol. The presence of a significant fraction of water not only lowers the viscosity of the solvent, but may also lead to a faster ammonolysis: water molecules can accelerate the dissolution of PA66, since these molecules might aid in interfering with the intermolecular hydrogen bonds between the different PA66 polymer chains. In addition, water molecules can also assist in the proton rearrangements that have to take place during the ammonolysis of a secondary amide bond. Evidently, this would only marginally change the overall thermodynamics of the reaction and therefore after 20h the expected equilibrium is achieved.
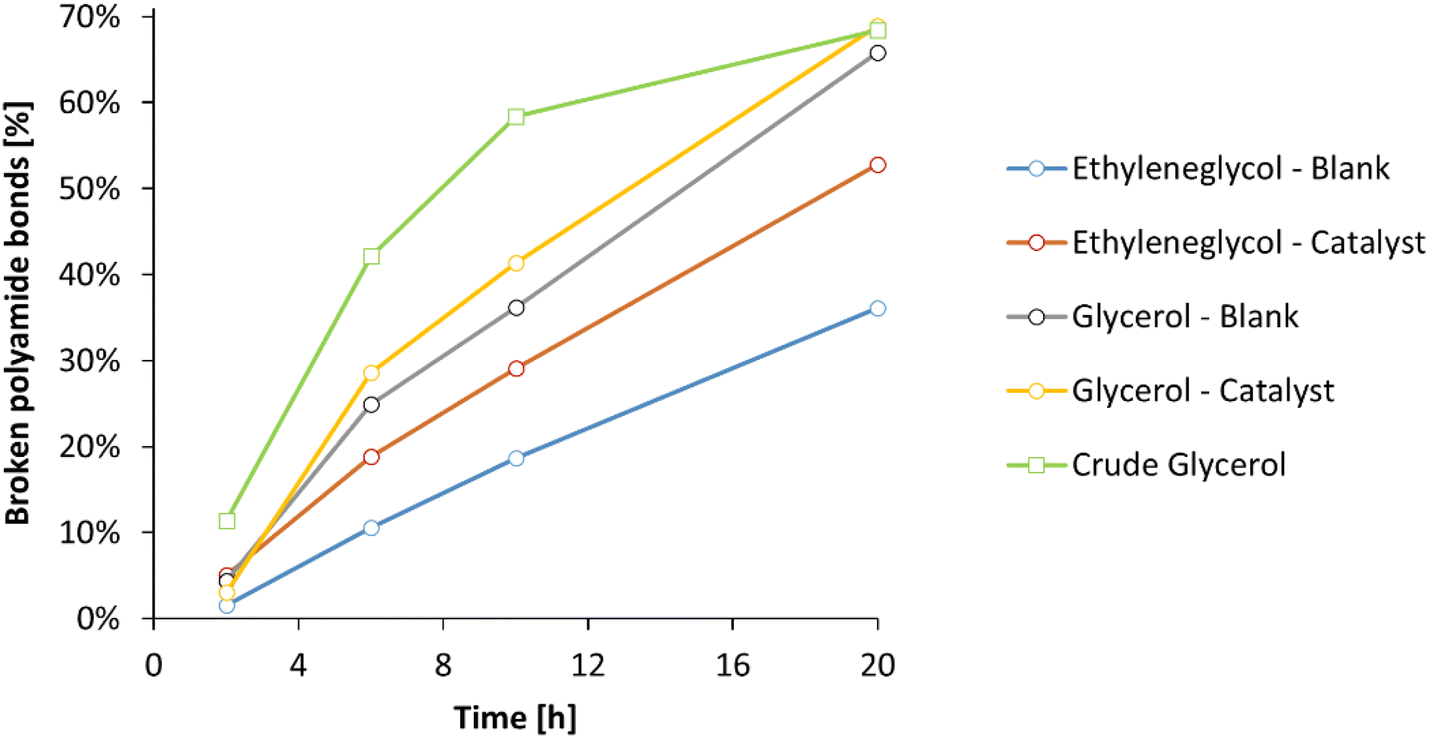 | ||
| Fig. 8 Number of broken polyamide bonds with time for different solvent systems. Conditions: 0.1 M PA66 and 0.1 M benzyl alcohol (internal standard) in glycol solvent (10 mL), 200 °C, 1 bar NH3. For the systems including a catalyst, 5 mol% La(OTf)3 was applied. | ||
The yield of hexamethylene diamine (HMD) was compared to the best results reported in literature on the ammonolysis of PA66 (ESI, Table S6†).15a,b,31 The best performing systems in literature reported a HMD yield around 40%,15b which was clearly met and surpassed since this work reached the overall thermodynamic equilibrium for the ammonolysis of PA66, by applying a lower reaction temperature, significantly less ammonia and crude biomass waste streams as the active solvent. This reduces several operating costs, energy consumption and improves the overall greenness of PA66 chemical recycling.
Conclusions
In short, we have developed a new approach for the chemical recycling of polyamide 66 via a sustainable and green ammonolysis process that can operate at mild reaction conditions in simple glycol solvents. Even in absence of a catalyst and with a crude glycerol solvent, directly obtained from a biorefinery process, 68% of the polyamide bonds were broken after 20 h of reaction time. To explain the high reactivity in these glycol solvents, computational studies were invoked which clearly illustrated that a vicinal diol moiety is essential for an efficient ammonolysis of the secondary amide bond. Due to equilibrium limitations, a higher number of broken polyamide bonds could not be achieved. Therefore, subsequent research should focus on circumventing these limitations by for instance evaporation of the hexamethylenediamine monomer once the equilibrium is reached, or by linking the ammonolysis of polyamide 66 to a subsequent (in situ) hydrogenation of the generated primary amides. As such, continuous depolymerization of polyamide 66 to hexamethylene diamine (and adipamide) should be feasible.Author contributions
W.S. performed the ammonolysis reactions. K.J., F.D.S. and R.C. helped with the experimental work. W.S. and K.J. wrote the manuscript. K.B. performed the GPC analyses. M.D., J.V. and F.D.P. conducted the computational studies on the intermolecular interactions in the solvent-polyamide-ammonia complex. W.S., F.D.S., F.D.P. and D.D.V. provided the funding, did the conceptualization and critical reading of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
W. S. and D. D. V. gratefully acknowledge the support of Catalisti-Vlaio through the Moonshot project CoRe2 (HBC2019.0116). W. S., K. J. and D. D. V. are grateful to OleonNV ‘A Natural Chemistry’ for a gift of ex-biodiesel waste glycerol. We like to thank Luca Passaro for the help with the viscosity measurements. W. S. is grateful to the FWO for his SB PhD fellowship (1SC1519N). The authors also thank Niels Van Velthoven for his contribution to the manuscript.References
- R. Geyer, R. J. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e170082 CrossRef PubMed.
- Ellen MacArthur Foundation, The New Plastics Economy: Rethinking the future of plastics, 2016, retrieved from: https://www.ellenmacarthurfoundation.org/ Search PubMed.
- E. Watkins, S. Gionfra, J.-P. Schweitzer, M. Pantzar, C. Janssens and P. ten Brink, EPR in the EU Plastics Strategy and the Circular Economy: a Focus on Plastic Packaging. Institute for European Environmental Policy, Brussels, Belgium, Study report 2017, 2017, 57pp.
- Institute European Environmental Policy, Single Use Plastics, 2016, retrieved from: https://ieep.eu.
- Plastic Recyclers Europe, 20 years later & the way forward – making more from plastics waste, 2016, retrieved from: https://plasticrecylers.eu Search PubMed.
- European Commission, A European strategy for plastics in a circular economy, 2018, retrieved from: https://ec.europa.eu/.
- F. Timmermans and J. Katainen, Plastic Waste: a European strategy to protect the planet, defend our citizens and empower our industries, European Commission – Press Release, 2018, retrieved from: https://ec.europa.eu/commission/presscorner/ Search PubMed.
- J. Hopewell, R. Dvorak and E. Kosior, Philos. Trans. R. Soc., B, 2009, 364, 2115–2126 CrossRef CAS PubMed.
- M. Bienmüller, J. Endtner, D. Joachimi, T. Linder, G. Margraf and H. Plaggenborg, Kunststoffe Int., 2017, 107(10), 20–26 Search PubMed.
- Y. Wang, Y. Zhang, M. Polk, S. Kumar and J. Muzzy, Recycling of Carpet and Textile Fibers in Plastics and the Environment: A Handbook, John Wiley & Sons, New York, United States of America, 2003, pp. 697–725 Search PubMed.
- P. M. Subramanian, US5430068, 1995.
- I. A. Ignatyev, W. Thielemans and B. Vander Beke, ChemSusChem, 2014, 7(6), 1579–1593 CrossRef CAS PubMed.
- (a) W. Nielinger, E. Ostlinning, K.-J. Idel, D. Freitag and H.-J. Buysch, US5233037, 1993; (b) S. Czernik, C. C. Elam, R. J. Evans, R. R. Meglen, L. Moens and K. Tatsumoto, J. Anal. Appl. Pyrolysis, 1998, 46(1), 54–64 CrossRef; (c) H. Bockhorn, A. Hornung, U. Hornung and J. Weichmann, Thermochim. Acta, 1999, 337, 97–110 CrossRef CAS; (d) H. Bockhorn, S. Donner, M. Gernsbeck, A. Hornung and U. Hornung, J. Anal. Appl. Pyrolysis, 2001, 58–59, 79–94 CrossRef CAS.
- (a) J. Weise, US2930790, 1960; (b) J. H. Bohnfield, R. C. Hecker, O. E. Snider and B. G. Apostle, US3182055, 1965; (c) O. Dicoi and E. Doerr, US4107160, 1978; (d) J. W. Mandoki, US4605762, 1986; (e) T. F. Corbin, E. A. Davis and J. A. Dellinger, US5169870, 1992; (f) R. Kotek, US5294707, 1994; (g) P. J. H. Thomissen, EP0875504A1, 1998; (h) S. Sifniades, A. B. Levy and J. A. J. Hendrix, US5932724, 1999; (i) U. Klun and A. Krzan, Polym. Adv. Technol., 2002, 13, 817–822 CrossRef CAS; (j) J. Chen, Z. Li, L. Jin, P. Ni, G. Liu, H. He, J. Zhang, J. Dong and R. Ruan, J. Mater. Cycles Waste Manage., 2010, 12, 321–325 CrossRef CAS.
- (a) R. J. McKinney, US5302756A, 1994; (b) R. J. McKinney, US5395974A, 1995; (c) J. A. J. Hendrix, M. Booij and Y. H. Frentzen, US5668277, 1997.
- (a) O. Sato and Y. Ikushima, Kobunshi Ronbunshu, 2001, 58(10), 533–540 CrossRef CAS; (b) M. Goto, M. Umdea, A. Kodama, T. Hirose and S. Nagaoka, Kobunshi Ronbunshu, 2001, 58(10), 548–551 CrossRef; (c) M. Goto, S. Sasaki and T. Hirose, J. Mater. Sci., 2006, 41, 1509–1515 CrossRef CAS; (d) A. Kamimura, Y. Oishi, K. Kaiso, T. Sugimoto and K. Kashiwagi, ChemSusChem, 2008, 1, 82–84 CrossRef CAS PubMed.
- (a) A. Kamimura and S. Yamamoto, Org. Lett., 2007, 9(13), 2532–2535 CrossRef PubMed; (b) A. Kamimura and S. Yamamoto, Polym. Adv. Technol., 2008, 19, 1391–1395 CrossRef CAS.
- M. I. Kohan, S. A. Mestemacher, R. U. Pagilagan and K. Redmond, Polyamides in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag, Germany, 2020, pp. 1–31 Search PubMed.
- M. L. Doerr, US4107160, 1978.
- E. F. J. Moran, EP0664787B1, 1995.
- W. Zhou, P. Neumann, M. Al Batal, F. Rominger, A. S. K. Hashmi and T. Schaub, ChemSusChem, 2020, 13, 1–6 CrossRef.
- A. Kumar, N. von Wolff, M. Rauch, Y.-Q. Zou, G. Shmul, Y. Ben-David, G. Leitus, L. Avram and D. Milstein, J. Am. Chem. Soc., 2020, 142, 14267–14275 CrossRef CAS PubMed.
- A. C. Fernandes, Green Chem., 2021, 23, 7330–7360 RSC.
- L. Meng, Y. Zhang, Y. Huang, M. Shibata and R. Yosomiya, Polym. Degrad. Stab., 2004, 83, 389–394 CrossRef CAS.
- H. Matsumoto, Y. Akinari, K. Kaiso and A. Kamimura, J. Mater. Cycles Waste Manage., 2017, 19, 326–331 CrossRef CAS.
- U. Cesarek, D. Pahovnik and E. Zagar, ACS Sustainable Chem. Eng., 2020, 8, 16274–16282 CrossRef CAS PubMed.
- X. Song, F. Liu, R. Zhao, S. Yu, S. Liu and M. Huang, CN107056624A, 2017.
- R. Coeck and D. E. De Vos, Green Chem., 2020, 22, 5105–5114 RSC.
- R. Coeck, S. Berden and D. E. De Vos, Green Chem., 2019, 21, 5326–5335 RSC.
- R. Simonnot and S. Simonnot, FR1532777, 1968.
- R. Coeck, A. De Bruyne, T. Borremans, W. Stuyck and D. E. De Vos, ACS Sustainable Chem. Eng., 2022, 10(9), 3048–3056 CrossRef CAS.
- N. A. Stephenson, S. H. Gellman and S. S. Stahl, RSC Adv., 2014, 4, 46840–46843 RSC.
- J. Datta, K. Blazek, M. Wloch and R. Bukowski, J. Polym. Environ., 2018, 26, 4415–4429 CrossRef CAS.
- R. G. Pearson, J. Chem. Educ., 1968, 45(9), 581–587 CrossRef CAS.
- K. Nakajima, Y. Baba, R. Noma, M. Kitano, J. N. Kondo, S. Hayashi and M. Hara, J. Am. Chem. Soc., 2011, 133, 4224–4227 CrossRef CAS PubMed.
- W. Stuyck, A. L. Bugaev, T. Nelis, R. de Oliveira-Silva, S. Smolders, O. A. Usoltsev, D. A. Esteban, S. Bals, D. Sakellariou and D. De Vos, J. Catal., 2022, 408, 88–97 CrossRef CAS.
- Tosoh F-Tech Inc., 2,2,2-Trifluoroethanol (TFEA) Its Production Process and Various Applications. Retrieved from: https://www.tosohusa.com.
- M. A. Rangelov, G. P. Petrova, V. M. Yomtova and G. N. Vayssilov, J. Org. Chem., 2010, 75, 6782–6792 CrossRef CAS PubMed.
- D. A. Blake, H. F. Cascorbi, R. S. Rozman and F. J. Meyer, Toxicol. Appl. Pharmacol., 1969, 15, 83–91 CrossRef CAS PubMed.
- M. J. Murphy, D. A. Dunbar and L. S. Kaminsky, Toxicol. Appl. Pharmacol., 1983, 71, 84–92 CrossRef CAS PubMed.
- S. R. Sellevåg, C. J. Nielsen, O. A. Søvde, G. Myhre, J. K. Sundet, F. Stordal and I. S. A. Isaksen, Atmos. Environ., 2004, 38, 6725–6735 CrossRef.
- M. Gordon, J. G. Miller and A. R. Day, J. Am. Chem. Soc., 1949, 71, 1245–1250 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Pall, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- W. L. Jorgensen and J. Tirado-Rives, J. Am. Chem. Soc., 1988, 110(6), 1657–1666 CrossRef CAS PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118(45), 11225–11236 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sanchez, J. Contreras-Garcia, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132(18), 6498–6506 CrossRef CAS PubMed.
- R. A. Boto, F. Peccati, R. Laplaza, C. Quan, A. Carbone, J.-P. Piquemal, Y. Maday and J. Contreras-Garcia, J. Chem. Theory Comput., 2020, 16(7), 4150–4158 CrossRef CAS PubMed.
- K. Janssens, M. Stalpaert, M. Henrion and D. E. De Vos, Chem. Commun., 2021, 57, 6324–6363 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, parameter optimization, dissolution experiments, ammonolysis equilibrium, details on computational studies. See DOI: https://doi.org/10.1039/d2gc02233h |
| This journal is © The Royal Society of Chemistry 2022 |