
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of 2,5-furandicarboxylic acid dimethyl ester from galactaric acid via dimethyl carbonate chemistry†
Giacomo
Trapasso
,
Mattia
Annatelli
,
Davide
Dalla Torre
and
Fabio
Aricò
 *
*
Department of Environmental Sciences, Informatics and Statistics, Ca’ Foscari University of Venice, 30172, Venice, Italy. E-mail: fabio.arico@unive.it
First published on 4th February 2022
Abstract
A convenient and simple procedure for the one-pot synthesis of 2,5-furandicarboxylic acid dimethyl ester (FDME) from galactaric (mucic) acid via dimethyl carbonate (DMC) chemistry is presented. Optimization of the reaction conditions showed that when galactaric acid was reacted with DMC in the presence of Amberlyst-36 for 2 hours at 200 °C, FDME formed as the main product. The compound was isolated as a pure crystalline powder in 70% yield using a simple custom-made purification protocol. The reaction intermediates of this one-pot procedure were identified and a possible reaction mechanism was proposed.
The global effort in reducing CO2 emissions and the need to convert the fossil-based economy into a bio-based circular economy have urged the development of innovative green technologies for the sustainable production of bioderived chemicals. Current global bio-based chemical and polymer production is estimated to be around 90 million tons, whereas 330 million tons are still produced starting from petrochemical feedstocks.1
Among the different bioderived compounds, a series of furan-based molecules easily synthesised from D-fructose and D-glucose have attracted scientists’ attention in consideration of their potential market applications. In this regard, 2,5-furandicarboxylic acid (FDCA) has been extensively investigated, especially as a monomer for polyester production2 such as polyethylene furanoate (PEF), a valuable alternative to petroleum-based polyethylene terephthalate (PET). Data collected showed that PEF has promising mechanical and thermal properties, a strong gas barrier, a low carbon footprint, and produces few greenhouse gases during its synthesis.3
The increasing interest in FDCA has also been recently boosted by a revolutionary catalytic process from Avantium that is able to produce this monomer from carbohydrates (i.e. cellulose, hemi-cellulose, starch and sucrose) at an industrial level.4 However, most of the synthetic approaches to FDCA are based on converting edible sugars such as glucose or fructose into 5-hydroxymethylfurfural (HMF) via an acid-catalysed dehydration reaction followed by an oxidation step (Scheme 1).5 One of the main issues of this procedure is that it relies on HMF as the key intermediate. In fact, 5-hydroxymethylfurfural is considered the quintessential bio-based platform chemical, but it has also been labelled “the sleeping giant”6 due to its many drawbacks including high cost (Scheme 1), difficult separation from reaction media, and purification and degradation issues due to the formation of humins that ultimately affect the product yield.7
 | ||
| Scheme 1 Synthetic process for the production of FDCA through the HMF route and galactaric acid route; prices available on Sigma-Merck website. | ||
In 1876, Fittig and Heinzelmann reported for the first time an alternative synthetic approach to FDCA that avoids the use of HMF as a substrate. In this procedure a water solution of galactaric acid (Gal) was heated in the presence of a strong acid (H2SO4 or HBr) to achieve FDCA via a cyclisation/aromatization reaction in approximately 50% yield (Scheme 1).8
Galactaric (mucic) acid belongs to the family of aldaric acids, which are dicarboxylic derivatives of sugars often referred to as “sugar acids”.9 This compound is produced in quantitative yield by galactose oxidation promoted by nitric acid (HNO3).10 Other greener synthetic routes involve an extraction process either from citrus peel,11 sugar beet,12 sound ripe peaches and pears13 or orange peel waste.14 An alternative synthesis employs genetically modified bacteria, yeast and fungi like Escherichia coli, Saccharomyces cerevisiae and Trichoderma reesei.15
Nowadays, galactaric acid is gaining increasing attention as a value-added chemical from biomass16 in consideration of its numerous applications as a biodegradable chelate, corrosion inhibitor, cosmetic ingredient, pharmaceutical conjugate and biopolymer precursor for nylon, polyanhydrides, polycations and coordination polymers.17
In this regard, additional studies conducted on the conversion of galactaric acid into FDCA demonstrated that benzene sulfonic acid and p-toluene sulfonic acid are viable homogenous catalysts for this dehydration/aromatization reaction, although FDCA was achieved in moderate yield (50%).17
A few investigations were also directed to the synthesis of FDCA esters (FDE) as these compounds are easier to isolate than FDCA due to their enhanced solubility in organic media. Diethyl 2,5-furandicarboxylate (FDEE) was prepared in moderate yield (30%) by a two-step procedure; first galactaric acid was converted into FDCA employing sulfonic acid, and then its esterification to FDEE was achieved by a reaction with ethanol.18 Heteropolyacids (H3PW12O40·nH2O) were shown to be slightly more efficient in the preparation of FDE allowing the direct synthesis of dibutyl 2,5-furandicarboxylate (FDBE) in approximately 40% yield.19
More recently, silica-supported sulfonic acids were also employed for converting Gal into a mixture of 2-furanmonocarboxylates and 2,5-furandicarboxylates.20 Under the best-found reaction conditions FDCA butyl esters were achieved in a two-step approach (Scheme 2a) encompassing galactaric acid esterification in the presence of butanol (24 h) followed by its cyclisation/aromatization reaction at 220 °C (4 h). FDCA esters were achieved in 80% yield, although this value includes the mono and dibutyl furandicarboxylates. The products selectivity was evaluated by GC, meanwhile there was no indication on the FDBE isolated yield although it was stated that the pure compound could be recovered by distillation.
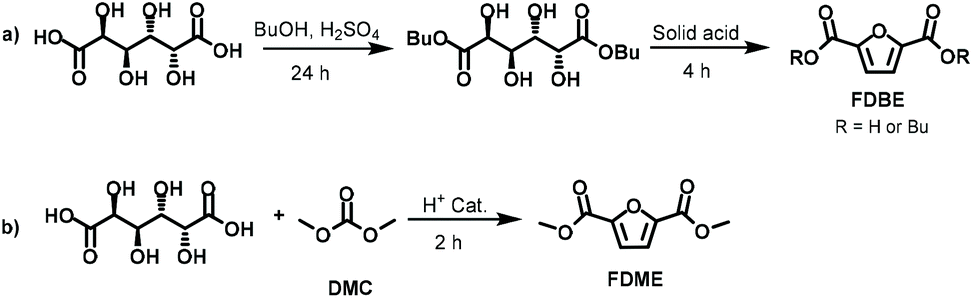 | ||
| Scheme 2 (a) Two-step approach to FDBE; (b) one-pot procedure of FDME via DMC chemistry. | ||
Following our interest in a simple synthetic approach to bio-based platform chemicals,21 in the present work we report a convenient one-pot procedure to convert galactaric acid into 2,5-furandicarboxylic acid dimethyl ester (FDME) via dimethyl carbonate (DMC) chemistry. In this synthesis, DMC was used as a green solvent and reagent in the presence of a commercially available and inexpensive acidic heterogeneous catalyst (Scheme 2b). Under the best-found reaction conditions, FDME was isolated as a pure compound in 70% yield.
The reaction pathway was also investigated, and data collected aided understanding DMC contribution in promoting FDME formation in such a short reaction time.
In a typical reaction, galactaric acid (1.0 g) was reacted with DMC (35 mL) in the presence of 0.5 mol eq. or 50 wt.% (0.5 g) of the selected homogeneous or heterogeneous catalyst, respectively (Table 1). The synthesis was carried out at 200 °C for three hours in a stainless-steel autoclave under autogenous pressure.
| # | Catalyst (0.5 mol eq./50 wt.%a) | P (bar) | t (h) | Conv. (%) | FDME yieldb (%) |
|---|---|---|---|---|---|
| Reaction conditions: 1.0 g of galactaric acid (0.13 M) and 35 mL of DMC in a stainless steel autoclave under pressure at 200 °C.a Isolated yield of FDME after the purification step with charcoal and diethyl ether.b For entries 4–9 the amount of catalyst used was 0.5 g (50 wt.% compared to Gal); the catalysts were dried in an oven at 100 °C overnight prior to their use. | |||||
| 1 | Formic acid | 20 | 3 | <5 | 0 |
| 2 | Oxalic acid | 15 | 3 | <5 | 0 |
| 3 | Acetic acid | 15 | 3 | <5 | 0 |
| 4 | CT275 | 55 | 3 | 99 | 57 |
| 5 | CT275DR | 95 | 3 | 99 | 46 |
| 6 | CT269 | 95 | 3 | 99 | 30 |
| 7 | CT151 | 100 | <2 | 99 | 51 |
| 8 | Amb-36 | 95 | 3 | 99 | 70 |
| 9 | Amb-15 | 80 | 3 | 99 | 60 |
None of the selected homogeneous acids (0.5 mol eq.) was capable of promoting the one-pot FDME formation (#1–3; Table 1). In all trials the conversion of galactaric acid was very poor.
In contrast, Purolite and Amberlyst catalysts, dried in an oven at 100 °C prior to use, led to a quantitative conversion of galactaric acid, although FDME yield varied depending on the catalyst employed (#4–9; Table 1).
Three different Purolite types were investigated (Table S1 in ESI†), all incorporating sulfonic groups:
• Purolite CT275, a macroporous catalyst showing excellent accessibility of active sites. CT275DR comprises the same chemical structure, the main difference being that the catalyst was dried to achieve a residual humidity ≤3%;
• Purolite CT269, a macroporous catalyst with very good mechanical resistance;
• Purolite CT151, a macroporous polystyrene crosslinked with divinylbenzene.
Of these, Purolite CT275 resulted in being the most efficient (#4; Table 1) possibly because of its high acidity in combination with an appropriate surface area.
Initially, FDME was isolated from the reaction mixture by gradient elution column chromatography (hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate 7:3 as the mobile phase). However, we were then able to develop a simpler and quicker purification protocol. In this procedure the brownish crude mixture, recovered after filtering off the catalyst and evaporating the DMC excess, was dissolved in diethyl ether (ca. 20 mL), a small amount of charcoal was added and the solution was heated at 40 °C for 30 minutes. The mixture was then filtered through a paper filter and the resulting transparent liquid dried under vacuum to recover FDME as a white crystalline powder.
:ethyl acetate 7:3 as the mobile phase). However, we were then able to develop a simpler and quicker purification protocol. In this procedure the brownish crude mixture, recovered after filtering off the catalyst and evaporating the DMC excess, was dissolved in diethyl ether (ca. 20 mL), a small amount of charcoal was added and the solution was heated at 40 °C for 30 minutes. The mixture was then filtered through a paper filter and the resulting transparent liquid dried under vacuum to recover FDME as a white crystalline powder.
Several column chromatography purification steps were performed to isolate the reaction by-products; however, in all cases FDME was the only compound present in good amount. Thin layer chromatography (TLC) showed the presence of numerous by-products that were either difficult to separate or present in very small amounts. Such complexity in the reaction mixture was elucidated when further studies on the reaction mechanism were conducted (Scheme 3; Table S3 in the ESI†).
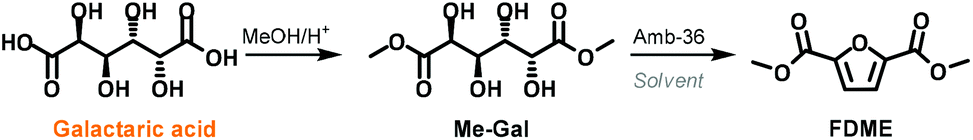 | ||
| Scheme 3 Synthesis of FDME starting from Me-Gal. | ||
According to the isolated yields reported in Table 1, although Amberlyst-15 was also able to promote the formation of FDME in good yield, Amberlyst-36 was the best catalyst for the one-pot conversion of galactaric acid into 2,5-furandicarboxylic acid dimethyl ester, i.e., 70% isolated yield. This result could be ascribed to the higher acidity of Amberlyst-36 in combination with a very defined surface area (Table S1; ESI†). As the pore volume and the average pore diameter greatly differed among the best performing catalysts, i.e., Ambelyst-36, Ambelyst-15 and CT275, we estimated that their influence on the reaction outcomes were not significant.
In all the trials conducted in the presence of a heterogeneous acid catalyst (#4–9; Table 1), the resulting autogenous pressure observed was relatively high (50–95 bar). This was ascribed to the numerous chemical transformations involved in the conversion of Gal into FDME (see also Scheme 4) that take place releasing a considerable amount of CO2, methanol and water. Furthermore, the residual liquid recovered at the end of the experiments was generally halved compared to the initial amount of DMC used. This suggested that part of the DMC was subjected to decarboxylation due to the combined effect of the acid catalyst and high temperature. In this regard, a blank experiment was conducted by heating the best performing catalyst, Amberlyst-36 (0.5 g), and DMC (35 mL) at 200 °C for 2 hours. In this case also the autogenous pressure was high (90 bar), and furthermore only 10 mL of DMC were recovered at the end of the reaction confirming its decarboxylation due to the activity of the acid catalyst (see the ESI†).
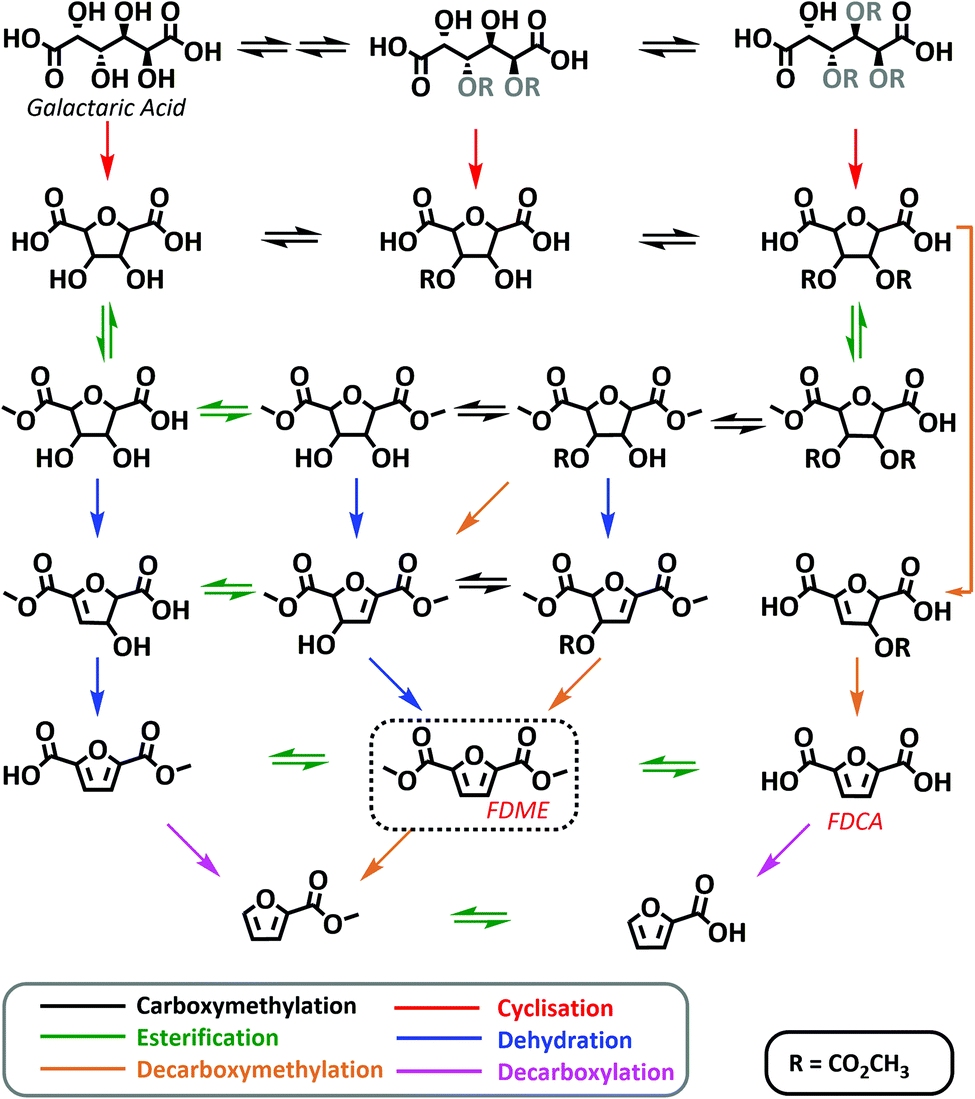 | ||
| Scheme 4 Possible reaction mechanism for the one-pot synthesis of FDME from galactaric acid via DMC chemistry. In the scheme the main species detected through HR-MS are reported. All species detected are shown in the ESI.† Chemical groups whose location and spatial orientation cannot be precisely determined are represented in grey. | ||
Further optimization trials of the one-pot conversion of Gal into FDME were carried out focusing on Amberlyst-36, by evaluating the effect of the reaction time, temperature, catalyst amount, and substrate concentration on product formation (Table 2).
| # | Gal (g) | Amb-36 (wt%) | T (°C) | P (bar) | t (h) | Conv. (%) | FDMEa yield (%) |
|---|---|---|---|---|---|---|---|
| Reaction conditions: Amberlyst-36 was dried in an oven at 100 °C and was in wt% compared to Gal; 35 mL of DMC, T = 200 °C in a stainless steel autoclave under pressure.a Isolated yield of FDME after the purification step with charcoal and diethyl ether.b Pressure >100 bar, nd = not detected.c This trial is the same as entry 8 in Table 1.d Isolated by column chromatography. | |||||||
| 1 | 1.0 | 50 | 200 | >100 | 4b | n.d. | n.d. |
| 2c | 1.0 | 50 | 200 | 95 | 3 | 99 | 70 |
| 3 | 1.0 | 50 | 200 | 95 | 2 | 99 | 70 |
| 5 | 1.0 | 100 | 200 | >100 | <1b | n.d. | n.d. |
| 6 | 1.0 | 25 | 200 | 80 | 2 | 80 | 24 |
| 7 | 1.0 | 50 | 160 | 45 | 2 | 99 | 14d |
| 8 | 1.0 | 50 | 180 | 80 | 4 | 99 | 71 |
| 9 | 1.0 | 50 | 190 | 85 | 2 | 99 | 71 |
| 10 | 2.0 | 50 | 200 | >100 | 0.1b | n.d. | n.d. |
| 11 | 2.0 | 50 | 180 | 95 | 4 | 99 | 63 |
| 12 | 3.0 | 50 | 180 | 95 | 4 | 99 | 61 |
An attempt was made to estimate the effect of a prolonged reaction time (#1, Table 2) on the reaction outcome; however, shortly after 3 hours the autogenous pressure generated during the reaction exceeded 100 bar, the functioning limit of our apparatus, thus the experiment was stopped.
On the other hand, when the reaction was performed for 2 hours, FDME was isolated in 70% yield (#7, Table 2) demonstrating that the reaction time can be further reduced without affecting the product yield.
The use of a 1.0:1.0 substrate:Ambelyst-36 weight ratio caused, once again, an autogenous pressure above 100 bar, therefore the reaction was stopped before the 2-hours reaction time (#5, Table 2). A catalyst load of 25% by weight led to an incomplete conversion of Gal and a scarce FDME yield, i.e., 24% (#6, Table 2).
Similarly, a test reaction carried out at 160 °C resulted in a poor yield; for this experiment our purification protocol was not effective and FDME has to be isolated by column chromatography.
The one-pot procedure can be efficiently conducted at 180 °C, but it required 4 hours to achieve an isolated yield comparable to the one obtained in 2 hours at 200 °C. On the other hand, performing the reaction at 190 °C also led to FDME in 71% yield after a 2-hour reaction time (#9, Table 2).
Finally, the effect of substrate concentration was also assessed to exploit the possibility of achieving a greater amount of FDME and reducing the excess of DMC. All the experiments so far reported employed a 0.13 M solution (1.0 g) of the substrate in DMC. When we performed a reaction at a 0.26 M concentration of galactaric acid (2.0 g) at 200 °C, the pressure rapidly increased above 100 bar and the experiment had to be stopped. On the other hand, reactions performed at 0.26 M and 0.39 M substrate concentrations were feasible at 180 °C prolonging the reaction time to 4 hours. In both cases the isolated yield of FDME was above 60% (#11–12, Table 2).
Preliminary experiments on catalyst recycling were also carried out (Table S3, ESI†). The reaction conducted at 180 °C for 4 hours was used as the case study (#8; Table 2). As the synthesis of FDME was performed under continuous stirring, at the end of the reaction the catalyst beads were reduced to powder and could not be properly recovered. To avoid this issue, the reaction was performed without mechanical stirring; interestingly the FDME yield remained almost unaltered (60% isolated). The Amberlyst-36 was recovered, washed with methanol and dried overnight at 100 °C. A second run was carried out under the same reaction conditions. In this case FDME was recovered by column chromatography due to the presence of numerous by-products and was finally isolated in 5% yield. Restoration of the catalyst acidic sites was then attempted by immersing the spheres in an acid solution (H2SO4) of water/methanol. Analysis of the reaction mixture obtained when employing the regenerated beads showed that the activity of the catalyst was not re-established as the presence of FDME was not detected either by NMR spectroscopy or TLC. This result was ascribed to the high temperature employed for the reaction as the Amberlyst-36 temperature limit is reported to be 150 °C (Table S1, ESI†). Most probably the high temperature required for the reaction impacts on the chemical structure and in particular on the sulfonic units of the beads rendering the catalyst not recyclable.
Despite the evident advantage of using Amberlyst-36 as a commercially available cheap catalyst for the preparation of FDME, further studies on developing new, recyclable catalytic systems are essential to improve this synthetic approach.
In order to have a better understanding of the role of DMC in this one-pot procedure, a series of tests were performed by substituting DMC with different solvents. Preliminary experiments showed that galactaric acid was mostly insoluble in all media if DMC was not employed. Thus we decided to use dimethyl galactarate (Me-Gal) as a substrate that we reckoned should have fewer solubility issues (Scheme 3).
Me-Gal was prepared on a multi-gram scale by adapting a procedure previously reported in the literature.22
According to our best-found reaction conditions, the cyclisation/aromatization reaction of Me-Gal was performed at 200 °C in the presence of 50 wt.% of Amberlyst-36. Acetonitrile, dioxane, tert-butanol, methyl lactate and water were tested as solvents in substitution of DMC (Table S2, ESI†). However, the formation of FDME was never observed in these experiments. On the other hand, when Me-Gal was reacted employing as a solvent, DMC or diethyl carbonate (DEC), FDME and FDEE were isolated in 60% and 38% yields, respectively.
These data confirmed that DMC (and in general DACs) played an important role not only in the dissolution of Gal or its derivative Me-Gal, but also in the subsequent cyclisation/aromatization steps.
As stated above, despite our best efforts we were unable to isolate other major reaction intermediates, thus we set up several experiments under the best-found reaction conditions, using high resolution mass spectrometry (HR-MS) to monitor the reaction outcome over time, i.e., at 0 min, 30 min, 1 h and 2 h (Table S3; ESI†). The reaction time zero was fixed when the autoclave reached a temperature of 200 °C. As the reaction required 50 minutes to reach 200 °C, three additional experiments were conducted where the heating was stopped 35 min (82 °C), 20 min (160 °C) and 10 min (184 °C) before the reaction time zero (Table S3, ESI†).
The crude mixture of the seven experiments was recovered and dried, the solid residue was dissolved in dimethyl sulfoxide (DMSO), filtered to remove the catalyst and analysed through HR-MS (Table S3, ESI†). Each sample was analysed by HR-MS both in positive and negative ion modes.
Data collected showed the presence of numerous intermediates formed by the reaction of DMC with the diverse hydroxyl and/or carboxylic moieties present in the galactaric acid backbone.
Compared to the two step procedure previously reported by Rautiainen et al.20 where esterification, cyclisation and dehydration (aromatization) were the main reactions involved, in our procedure, the presence of DMC rendered the reaction mechanism far more complicated.
Considering the compounds identified at different reaction times, Scheme 4 shows a tentative reaction mechanism. It appears that the one-pot reaction proceeds first through multiple carboxymethylation reactions involving galactaric acid hydroxyl groups, followed by a cyclisation step (Scheme 4). This observation is compatible with the numerous studies on DMC catalysed cyclisation reactions.23
Most probably, the carboxymethylation reaction(s) promoted by DMC facilitates the dissolution of Gal in the solvent, thus favouring the subsequent steps.
Once the temperature has reached 200 °C (samples at 0 min, 30 min and 1 hour), esterification of cyclic intermediate acidic groups takes place. In the last step (1 h and 2 h samples), the aromatization of the tetrahydrofuran ring leads to FDME either via dehydration or decarboxymethylation reactions. In the latter case methanol and CO2 are released as leaving groups instead of water.
Some decarboxylation reactions also seem to occur as demonstrated by the identification of 2-furoic acid and its ester in the crude mixture after 2 hours at 200 °C. This observation is consistent with the results previously reported in the acid catalysed two-step procedure leading to FDBE.20
It is finally interesting to note that methoxycarbonylation and cyclisation reactions via DMC chemistry are well-known and extensively studied;23 however, these preliminary data seem to suggest that DMC might also be capable of promoting other reactions such as aromatization via decarboxymethylation.
In conclusion, in this work the one-pot direct conversion of galactaric acid into FDCA methyl esters via DMC chemistry is reported. Amberlyst-36 was used as an inexpensive heterogeneous acidic catalyst and the reaction conditions were optimized taking into consideration numerous variables. A custom-made simplified purification protocol allowed the isolation of FDME as a pure crystalline powder from the reaction mixture in 70% yield. Investigation of the reaction pathway showed an intricate mechanism due to the versatile reactivity of DMC in combination with the numerous functional groups present in the substrate. Nonetheless, DMC chemistry was shown to be capable of efficiently driving the reaction outcome through all possible intermediates to achieve the wanted FDME in high yield. This procedure is yet another example of DMC demonstrating great versatility as a reagent/solvent for the sustainable valorisation of renewables.
Author contributions
Dr D. Dalla Torre and Dr M. Annatelli: investigation, data curation and methodology. Dr G. Trapasso: conceptualization, investigation, data curation, methodology and writing. Prof. F. Aricò: conceptualization, supervision, visualization and writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We want to acknowledge the Istituto Nazionale della Previdenza Sociale (INPS) for funding the PhD fellowship of Giacomo Trapasso. Prof. Aricò wants to acknowledge the Organization for the Prohibition of Chemical Weapons (OPCW); Project Number L/ICA/ICB/218789/19 for funding part of this research. This article is also within the framework of COST Action FUR4Sustain (CA18220-European network of FURan based chemicals and materials FOR a Sustainable development), supported by COST (European Cooperation in Science and Technology).Notes and references
- Data from IEA Bioenergy annual report 2020 https://task42.ieabioenergy.com.
- (a) G. R. Dick, A. D. Frankhouser, A. Banerjee and M. W. Kanan, Green Chem., 2017, 19, 2966 RSC; (b) B. M. Stadler, C. Wulf, T. Werner, S. Tin and J. G. de Vries, ACS Catal., 2019, 9, 8012–8067 CrossRef CAS; (c) M. Sajid, X. Zhao and D. Liu, Green Chem., 2018, 20, 5427 RSC; R. Sakuta and N. Nakamura, Int. J. Mol. Sci., 2019, 20, 3660 Search PubMed.
- (a) A. J. J. E. Eerhart, A. P. C. Faaij and M. K. Patel, Energy Environ. Sci., 2012, 5, 6407–6422 RSC; (b) S. K. Burgess, J. E. Leisen, B. E. Kraftschik, C. R. Mubarak, R. M. Kriegel and W. J. Koros, Macromolecules, 2014, 47, 1383–1391 CrossRef CAS; (c) S. K. Burgess, O. Karvan, J. Johnson, R. M. Kriegel and W. J. Koros, Polymer, 2014, 55, 4748–4756 CrossRef CAS; (d) S. K. Burgess, R. M. Kriegel and W. J. Koros, Macromolecules, 2015, 48, 2184–2193 CrossRef CAS; (e) G. R. Dick, A. D. Frankhouser, A. Banerjee and M. W. Kanan, Green Chem., 2017, 19, 2966 RSC; G. Chen, N. M. Van Straalen and D. Roelofs, Green Chem., 2016, 18, 4420–4431 Search PubMed; (f) A. S. Joshi, N. Alipourasiabi, Y. Kim, M. R. Coleman and J. G. Lawrence, React. Chem. Eng., 2018, 3, 447–453 RSC.
- (a) E. De Jong, M. A. Dam, L. Sipos and G. J. M. Gruter, ACS Symp. Ser., 2011, 1105, 1–13 Search PubMed; (b) Avantium YXY Technology, https://www.avantium.com/technologies/yxy/.
- (a) G. R. Dick, A. D. Frankhouser, A. Banerjee and M. W. Kanan, Green Chem., 2017, 19, 2966 RSC.
- (a) R.-J. van Putten, J. C. van der Waal, E. de Jong, C. B. Rasrendra, H. J. Heeres and J. G. de Vries, Chem. Rev., 2013, 113(3), 1499 CrossRef CAS PubMed; (b) K. I. Galkin and V. P. Ananikov, ChemSusChem, 2019, 12, 2976 CrossRef CAS PubMed.
- C. Thoma, J. Konnerth, W. Sailer-Kronlachner, P. Solt, T. Rosenau and H. W. G. van Herwijnen, ChemSusChem, 2020, 13, 3544 CrossRef CAS PubMed.
- R. Fittig and H. Heinzelmann, Chem. Ber., 1876, 9, 1198 Search PubMed.
- (a) M. J. Climent, A. Corma and S. Iborra, Green Chem., 2011, 13, 520–540 RSC; (b) F. H. Isikgor and C. R. Becer, Polym. Chem., 2015, 6, 4497–4559 RSC; (c) T. Mehtiö, M. Toivari, M. G. Wiebe, A. Harlin, M. Penttilä and A. Koivula, Crit. Rev. Biotechnol., 2016, 36, 904–916 CrossRef PubMed.
- (a) H. Chae-Hwan, J. Sung-Won, Y. Na-Kyong, K. Young-Gyu, S. Na-Ra and K. So-Hyun, Hyundai Motor Company, Seoul National University R&DB Foundation, KR2017/9521, 2017, A; (b) J. H. Jang, I. Ro, P. Christopher and M. M. Abu-Omar, ACS Catal., 2021, 11, 95–109 CrossRef CAS.
- D. Jeong, H. Park, B. K. Jang, Y. Ju, M. H. Shin, E. J. Oh, E. J. Lee and S. R. Kim, Bioresour. Technol., 2021, 323, 124603 CrossRef CAS PubMed.
- (a) J. N. Stark, A. E. Goodban and H. S. Owens, Proo. Amer. Soe. Sugar Beet Teoh., 1950, 6, 578 CAS; (b) H. Zhang, X. Li, X. Su, E. L. Ang, Y. Zhang and H. Zhao, ChemCatChem, 2016, 8, 1500–1506 CrossRef CAS.
- E. F. L. J. Anet and T. M. Reynolds, Division of Food Preservation and Transport, Commonwealth Scientific and Industrial Research Organization, Homebush, N.S.W., Nature, 1954, 174(4437), 930 CrossRef CAS.
- A. Ortiz-Sanchez, J. Solarte-Toro, J. González-Aguirre, K. E. Peltonen, P. Richard and C. A. C. Alzate, Pre-feasibility analysis of the production of mucic acid from orange peel waste under the biorefinery concept, Biochem. Eng. J., 2020, 161, 107680 CrossRef.
- (a) T. Paasikallio, A. Huuskonen and M. G. Wiebe, Microb. Cell Fact., 2017, 16, 119 CrossRef PubMed; (b) V. Vidgren, S. Halinen, A. Tamminen, S. Olenius and M. G. Wiebe, Microb. Cell Fact., 2020, 19, 156 CrossRef CAS PubMed; R. J. Protzko, L. N. Latimer, Z. Martinho, E. de Reus, T. Seibert, J. P. Benz and J. E. Dueber, Nat. Commun., 2018, 9, 5059 Search PubMed.
- T. Werpy and G. Peterson, Top Value Added Chemicals from Biomass Vol. 1-Results of Screening for Potential Candidates from Sugars and Synthesis Gas, National Renewable Energy Laboratory, 2004, https://www.nrel.gov/docs/fy04osti/35523.pdf Search PubMed.
- (a) D. E. Kiely, L. Chen and T. Lin, ACS Publications, 1994, pp. 149–158; (b) Y. Liu and T. M. Reineke, J. Am. Chem. Soc., 2005, 127, 3004–3015 CrossRef CAS PubMed; (c) R. Sakuta and N. Nakamura, Int. J. Mol. Sci., 2019, 20, 3660 CrossRef CAS PubMed.
- (a) G. Brătulescu, Rev. Roum. Chim., 2000, 45, 883 Search PubMed; (b) J. Lewkowski, Pol. J. Chem., 2001, 75, 1943 CAS; (c) D. Zhao, F. Delbecq and C. Len, Molecules, 2019, 24, 8 Search PubMed.
- Y. Taguchi, A. Oishi and H. Iida, Chem. Lett., 2008, 37, 50 CrossRef CAS.
- N. van Strien, S. Rautiainen, M. Asikainen, D. A. Thomas, J. Linnekoski, K. Niemelä and A. Harlin, Green Chem., 2020, 22, 8271 RSC.
- (a) M. Musolino, J. Andraos and F. Aricò, ChemistrySelect, 2018, 3, 2359 CrossRef CAS; (b) M. Annatelli, D. Dalla Torre, M. Musolino and F. Aricò, Catal. Sci. Technol., 2021, 11, 3411 RSC; (c) A. G. Sathicq, M. Annatelli, I. Abdullah and G. Romanelli, Sustainable Chem. Pharm., 2021, 19, 100352 CrossRef; (d) M. Musolino, M. J. Ginés-Molina, R. Moreno-Tost and F. Aricò, ACS Sustainable Chem. Eng., 2019, 7, 10221 CrossRef CAS.
- M. E. Davis and H. Han, A Location in patent: Paragraph 0230; 0231, California Institute of Technology, JP2019/108372, 2019 Search PubMed.
- (a) F. Aricò, P. Tundo, A. Maranzana and G. Tonachini, ChemSusChem, 2012, 5, 1578 CrossRef PubMed; (b) F. Aricò, U. Toniolo and P. Tundo, Green Chem., 2012, 14, 58 RSC; P. Tundo, M. Musolino and F. Aricò, Front. Chem., 2019, 7, 300 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1gc04408g |
| This journal is © The Royal Society of Chemistry 2022 |