
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrobiogechemical interactions in the hyporheic zone of a sulfate-impacted, freshwater stream and riparian wetland ecosystem†‡
Joshua M.
Torgeson§¶
a,
Carla E.
Rosenfeld§
 b,
Aubrey J.
Dunshee
a,
Kelly
Duhn||
c,
Riley
Schmitter
a,
Patrick A.
O'Hara
a,
G. H. Crystal
Ng
ad and
Cara M.
Santelli
*ac
b,
Aubrey J.
Dunshee
a,
Kelly
Duhn||
c,
Riley
Schmitter
a,
Patrick A.
O'Hara
a,
G. H. Crystal
Ng
ad and
Cara M.
Santelli
*ac
aDepartment of Earth and Environmental Sciences, University of Minnesota, Minneapolis, MN 55455, USA. E-mail: santelli@umn.edu; torge158@umn.edu; duns0034@umn.edu; schm4386@umn.edu; ohara060@umn.edu; gcng@umn.edu; Fax: +1 612-625-5780; Tel: +1 612-624-9760
bSection of Minerals and Earth Sciences, Carnegie Museum of Natural History, USA. E-mail: rosenfeldc@carnegiemnh.org
cBioTechnology Institute, University of Minnesota, St. Paul, MN 55108, USA. E-mail: duhnx004@d.umn.edu
dSt. Anthony Falls Laboratory, University of Minnesota, Minneapolis, MN 55455, USA
First published on 25th May 2022
Abstract
Coupled abiotic and biotic processes in the hyporheic zone, where surface water and groundwater mix, play a critical role in the biogeochemical cycling of carbon, nutrients, and trace elements in streams and wetlands. Dynamic hydrologic conditions and anthropogenic pollution can impact redox gradients and biogeochemical response, although few studies examine the resulting hydrobiogeochemical interactions generated within the hyporheic zone. This study examines the effect of hyporheic flux dynamics and anthropogenic sulfate loading on the biogeochemistry of a riparian wetland and stream system. The hydrologic gradient as well as sediment, surface water, and porewater geochemistry chemistry was characterized at multiple points throughout the 2017 spring-summer-fall season at a sulfate-impacted stream flanked by wetlands in northern Minnesota. Results show that organic-rich sediments largely buffer the geochemical responses to brief or low magnitude changes in hydrologic gradient, but sustained or higher magnitude fluxes may variably alter the redox regime and, ultimately, the environmental geochemistry. This has implications for a changing climate that is expected to dramatically alter the hydrological cycle. Further, increased sulfate loading and dissolved or adsorbed ferric iron complexes in the hyporheic zone may induce a cryptic sulfur cycle linked to iron and carbon cycling, as indicated by the abundance of intermediate valence sulfur compounds (e.g., polysulfide, elemental sulfur, thiosulfate) throughout the anoxic wetland and stream-channel sediment column. The observed deviation from a classical redox tower coupled with potential changes in hydraulic gradient in these organic-rich wetland and stream hyporheic zones has implications for nutrient, trace element, and greenhouse gas fluxes into surface water and groundwater, ultimately influencing water quality and global climate.
Environmental significanceThe hyporheic zone below freshwater streams and wetlands, where mixing of surface water and groundwater occurs, plays a critical role in water quality and ecosystem health. Our understanding of the interactions between dynamic water fluxes and environmental geochemistry is limited. In this study, we examine fluctuating water flow through a sulfate-impacted riparian wetland and stream system to assess the geochemical impacts on the subsurface. We show that sediment and porewater composition can buffer the impacts of brief or small magnitude hydrologic changes, but more substantial flux changes can lead to measurable geochemistry outcomes. Further, we show that sulfur cycling is coupled to iron and methane cycling, which has implications for fluxes of carbon, nutrients, and contaminants in the environment. |
Introduction
The hyporheic zone, the layer of sediment where oxygen-rich surface water mixes with nutrient-rich shallow groundwater, plays a critical role in controlling water quality and global ecosystem health.1 In the hyporheic zones of streams and associated riparian wetlands, high concentrations of organic matter2,3 combined with saturated and typically flooded sediment beds promote the fast consumption of oxygen in these aquatic ecotones.4–6 These conditions set up steep oxidation-reduction (redox) gradients that promote biogeochemical activity and organic matter turnover – it is estimated that up to 96% of riparian-river ecosystem respiration occurs within the hyporheic zone.7 Water flow is also spatially and temporally heterogeneous in the hyporheic zone8–10 which influences solute transport and further drives redox zone fluctuations that alter the geochemistry as well as the microbial community composition, function and activity.3,8,11–18 These dynamic hydrobiogeochemical interactions drive the formation and fluxes of major nutrients,4,19,20 redox active metals,11,21 contaminants,22–24 and greenhouse gases,15,25 having implications that reach far beyond the ecotone boundaries.High organic matter turnover in the hyporheic zone is fueled by microbial metabolic processes that regulate the biogeochemical cycling of carbon and other nutrients.26 The “thermodynamic” redox ladder concept predicts the sequence of redox reactions that drive these metabolic processes where the more energetically favorable terminal electron acceptor (TEA) will be consumed before the next favorable electron acceptor.27 For example, dissimilatory nitrate reduction should not occur until all oxygen (the more favorable electron acceptor) is depleted. This can lead to vertical stratification of the redox zones in some environments (e.g., marine sediments), forming a redox gradient with more oxidized conditions at the surface due to transport of oxygen from the surface waters and more reduced conditions deeper in the subsurface.28
Typically in the anoxic portions of the hyporheic zone, microbial nitrate and ferric iron (Fe(III)) reduction as well as methanogenesis are dominant processes, resulting in elevated nitrous oxide, dinitrogen, ferrous iron (Fe(II)), and methane (CH4) concentrations in interstitial porewaters.15,21,29 Depending on the hydrologic conditions, these compounds can be transported to the surface water and atmosphere.30–32 With relatively low sulfate concentrations, dissimilatory sulfate reduction has traditionally not been expected to be an important process in freshwater environments based on the hierarchical redox ladder. This paradigm, however, has been changing as several studies33–35 identified fast microbial sulfate reduction rates sustained by cryptic sulfur (S) cycling coupled to Fe cycling in anoxic environments. These cryptic reactions involve oxidation of sulfide (produced by sulfate reduction) to intermediate-valence S forms (e.g., thiosulfate, S(0), polysulfide), and disproportionation of these S intermediates can yield more sulfate to sustain the cycle. Sulfate is abundant in many industrial and agricultural wastewaters,36–38 so cryptic S cycling may be increasingly recognized as an important driver in organic matter decomposition in many freshwater environments impacted by wastewaters. Further, cryptic S cycling has implications for methane fluxes to the atmosphere because methane can be anaerobically oxidized (consumed) coupled to sulfate reduction pathways in the subsurface.39–41 In natural environments that are physically and chemically heterogeneous, like hyporheic zones, the redox ladder concept is being further challenged. Studies show that several redox reactions may co-occur or deviate from the predicted hierarchical order due to factors such as geochemical microniches,42–44 hydrologic conditions,8,10 and redox phase metastability.45
Hydrobiogeochemical interactions in hyporheic zones are complex and dynamic, and our understanding of these interactions and their environmental impacts is rapidly evolving. It is expected that changes in hydrologic flux direction or magnitude in the hyporheic zone would have a measurable impact, perhaps reflected in microsites (“hot spots”) and/or for short times (“hot moments”), on the biogeochemistry of the sediments and porewaters.10,42,44,46–50 The change from a gaining stream (upward groundwater flux) to a losing stream (downward surface water flux), for example, could alter the redox gradient by recharging the system with thermodynamically favorable TEAs (e.g., oxygen, sulfate, iron oxides, etc.). This could lead to changes in biogeochemical reactions and rates that would be reflected in the sediment and porewater geochemistry.11–18 The oxidation of aqueous Fe(II) and Fe(II)-bearing sulfides to form Fe(III)-oxides was previously observed, for example, in mangrove sediments due to tidal fluctuations and fluxes of oxygenated water into highly reducing sediments.51 These changes, however, might be temporally and spatially variable for the different chemical species. Many questions remain about the impact of pollution and fluctuating hydraulic flux direction on the biogeochemistry of the system, such as the rates, spatial and temporal heterogeneity, and overall extent of geochemical change during or after changes in hydraulic regime.
To shed light on these poorly resolved processes, reactive transport modeling was conducted in previous studies by Ng et al.,52,53 of a sulfate-impacted freshwater stream and associated riparian wetlands. Calibrated simulation results showed that sulfate reduction rates exceeded thermodynamically favored Fe reduction. Cryptic S cycling was proposed as a driving mechanism for the high rates, but key intermediate-valence S forms were not yet confirmed and thus not included in the modeled reactions. Interestingly, with the incorporation of only complete reduction and aerobic reoxidation reactions between sulfate to sulfide, the model could not properly reproduce the measurable concentrations of sulfate observed at depth – modeled sulfate concentrations always decreased to 0. This suggested that, in reality, anoxic oxidation of sulfide (not represented in the model) could be regenerating sulfate at depth. Additionally, the model presumed Fe(III) presence throughout the hyporheic zone, despite the absence of geochemical measurements to confirm this. If Fe(III) is indeed present throughout the hyporheic zone, this would suggest that anaerobic sulfide oxidation could be coupled to abiotic Fe(III) reduction as part of cryptic S cycling in this Fe-rich ecotone. To identify the activity and impact of these potentially coupled biotic-abiotic geochemical reactions and their response to dynamic hydrologic conditions, further geochemical analyses (e.g., confirmation of S intermediates and Fe(III) where sulfate reduction was active) and field examination were needed.
Building on this previous research52,53 the present study provides a comprehensive suite of observations that demonstrates geochemical responses to changes in hydrologic gradients through time and space, including the occurrence of cryptic S biogeochemical cycling. The previous year's modeling study focused on conditions that were dominated by upwelling. For this study, we aimed to quantitatively and qualitatively characterize sediment and porewater geochemistry before, during, and after fluctuations in flux direction, with the expectation that downwelling conditions would supply more energetically favorable electron acceptors (e.g. oxygen, nitrate, Fe(III) oxides) and high sulfate concentrations (due to industrial pollution) from the stream water. We also sought to identify if conditions in the stream channel compared to the organic-rich, flanking riparian wetlands resulted in different geochemical responses to these fluctuating flux directions and sulfate-loading. To do this, hydrologic gradient as well as sediment, surface water, and porewater geochemistry was characterized at multiple points throughout the 2017 spring-summer-fall season at a sulfate-impacted stream flanked by wetlands in northern Minnesota (USA). X-ray absorption spectroscopy (XAS) was used to characterize the iron and sulfur speciation of sediments collected throughout the season to provide evidence for cryptic sulfur cycling, such as intermediate-valence sulfur compounds and Fe(III) phases co-existing in anoxic conditions. Our results provide a greater understanding of how the hyporheic zone geochemically responds to dynamic hydrologic conditions and the potential impact on water quality and atmospheric methane fluxes.
Materials and methods
Field site overview
Sediment cores, surface waters, porewaters, and hydrological data were collected from Second Creek, a SO42−-rich stream flanked by wetlands near Hoyt Lakes, Minnesota (USA), as described in Ng et al.,52,53 (Fig. 1). The high SO42− concentrations (reaching as high as 10 mM) in the surface waters are largely due to the discharge of waters from upstream iron mining rock waste pits.54 These rock-filled pits contain minerals associated with taconite mining in northern Minnesota (including hematite, siderite; goethite, and Fe sulfides),55 and weathering of this waste-rock produces waters with elevated concentrations of SO42− that influence the biogeochemistry of Second Creek. During the 2017 field season, relatively sparse macrophytes including wild rice (Zizania palustris) were growing in the sand-dominated stream channel where our field work was conducted. The riparian wetlands were much more densely vegetated with grasses and sedges, and sediments are rich in organic matter along with finer sediment particles.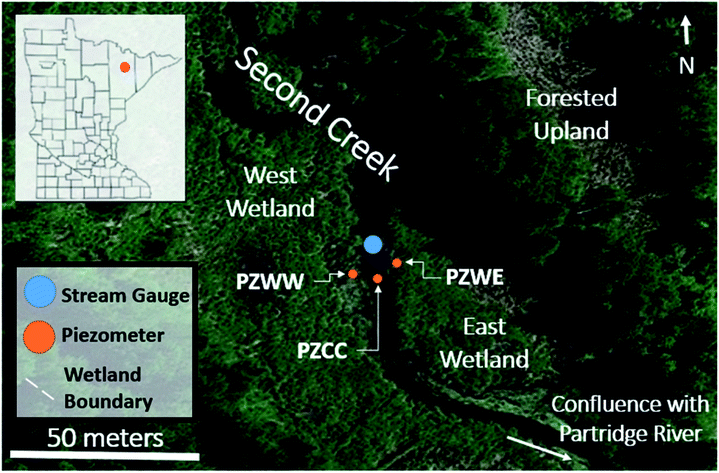 | ||
| Fig. 1 Second Creek study area showing the stream channel and riparian wetlands in northeastern Minnesota (inset) with the specific locations of groundwater piezometers (orange circles), stream gauge (blue circle) and wetland boundaries (white dashed lines). Samples for sediment and aqueous geochemistry were taken adjacent to the piezometers for the west wetland (PZWW), east wetland (PZEW), and stream channel center (PZCC). This figure is based on Fig. 1 in Ng et al. (2017). | ||
Hydrologic monitoring
Piezometers and stream gauges were installed from June 9 through October 21, 2017 to measure shallow groundwater head and surface water head, respectively. Three piezometers were manually installed at approximately 1 m depth: one in the center of the stream channel, one in the east wetland, and one in the west wetland (Fig. 1). The piezometers and the stream gauge contained Schlumberger Baro or Diver pressure transducers. The raw pressure data were compensated using air pressure records from Eveleth, MN (U.S. Local Climatological Data (LCD) from the National Centers for Environmental Information, https://www.ncei.noaa.gov/) and then calibrated to manual water level measurements and surveyed top-of-casing elevations taken at the start and end of the monitoring period. Resulting daily shallow groundwater and surface water head levels were used to calculate vertical hydraulic gradients and infer flux directions and magnitudes.Sample collection overview
Samples for aqueous geochemistry of porewaters and surface waters as well as solid-phase geochemistry of sediments were collected in a transect across the 2–3 m stream, including the east and west riparian wetlands flanking the main channel, in campaigns beginning May 24, June 21, August 1, August 29, and October 20, 2017. For the campaign on August 1, most of the samples collected are assumed to reflect sediment conditions during the end of July, and therefore this sampling period is hereafter referred to as ‘July’. Geochemistry samples were collected near the groundwater piezometers to understand the relationship of hydrologic flux to the hyporheic zone biogeochemistry. Sediment cores approximately 25–30 cm in length were retrieved using an HTH sediment gravity corer (Pylonex, Sweden) with 7 cm diameter polycarbonate tubes as described previously.52 Separate cores were retrieved from each location for porewater and sediment geochemistry analysis.Geochemistry analysis
Unfortunately, geochemical analysis showed that the “surface water” peeper samples from the sediment–water interface were likely impacted by anoxic groundwater intrusion into the peeper wells (potentially due to the hydrologic conditions or from changes in sediment suspension and vertical movement of the sediment–water interface after peeper emplacement). The actual surface water chemistry was assumed to be similar to that determined for samples previously collected using Nalgene bottles at this same field site52 and during June of the present study.
X-ray absorption spectroscopy (XAS)
All synchrotron-based XAS data collection was performed at the Advanced Photon Source, Argonne National Laboratory (Lemont, IL). Sediment samples were kept anoxic and shipped frozen to the facility inside N2-purged, sealed mylar bags and were thawed immediately before analysis. At the beamline, samples were prepared inside an anoxic chamber and transported to the beamline inside N2-purged mason jars to limit sample oxidation. Fe K-edge XAS spectra (X-ray absorption near edge structure; XANES, and extended X-ray absorption fine structure; EXAFS) and S K-edge XANES spectra were acquired from hydrated sediments collected at multiple depths and analyzed as described in the ESI.‡ Linear combination fitting (LCF) of the individual normalized spectra was conducted using a library of Fe and S reference compounds to identify and quantify structural components in sediment samples as further described in the ESI.‡Results
Hyporheic flux
The Second Creek stream and wetlands (including field sampling subsites) had inundated conditions throughout the summer of 2017. Vertical hyporheic flux directions were determined based on hydraulic head gradient signs calculated from surface water and subsurface water head measurements, and stronger fluxes were inferred for times with higher magnitude gradients. Hydraulic head gradients in Fig. 2 showed that the vertical flux between the surface water and groundwater was dynamic (in terms of direction and magnitude) with similar seasonal changes throughout all three subsites. Upward flux prevailed at all locations from June until mid-August, when a strong precipitation event caused a brief flux reversal. From mid-August to early October, flux directions and magnitudes were variable, with rainier conditions triggering multi-day to multi-week downward flux periods. Steady upward flux conditions then resumed for the rest of October. The central channel and west wetland hydraulic gradient time series show quite similar trends over time and magnitudes in 2017, consistent with similarities found between the two locations by Ng et al.52,53 in previous years, but the east wetland, which had not previously been monitored, showed some distinct differences. In particular, the east wetland showed two multi-day periods in late September to early October with very negative hydraulic gradient that suggest strong downward fluxes.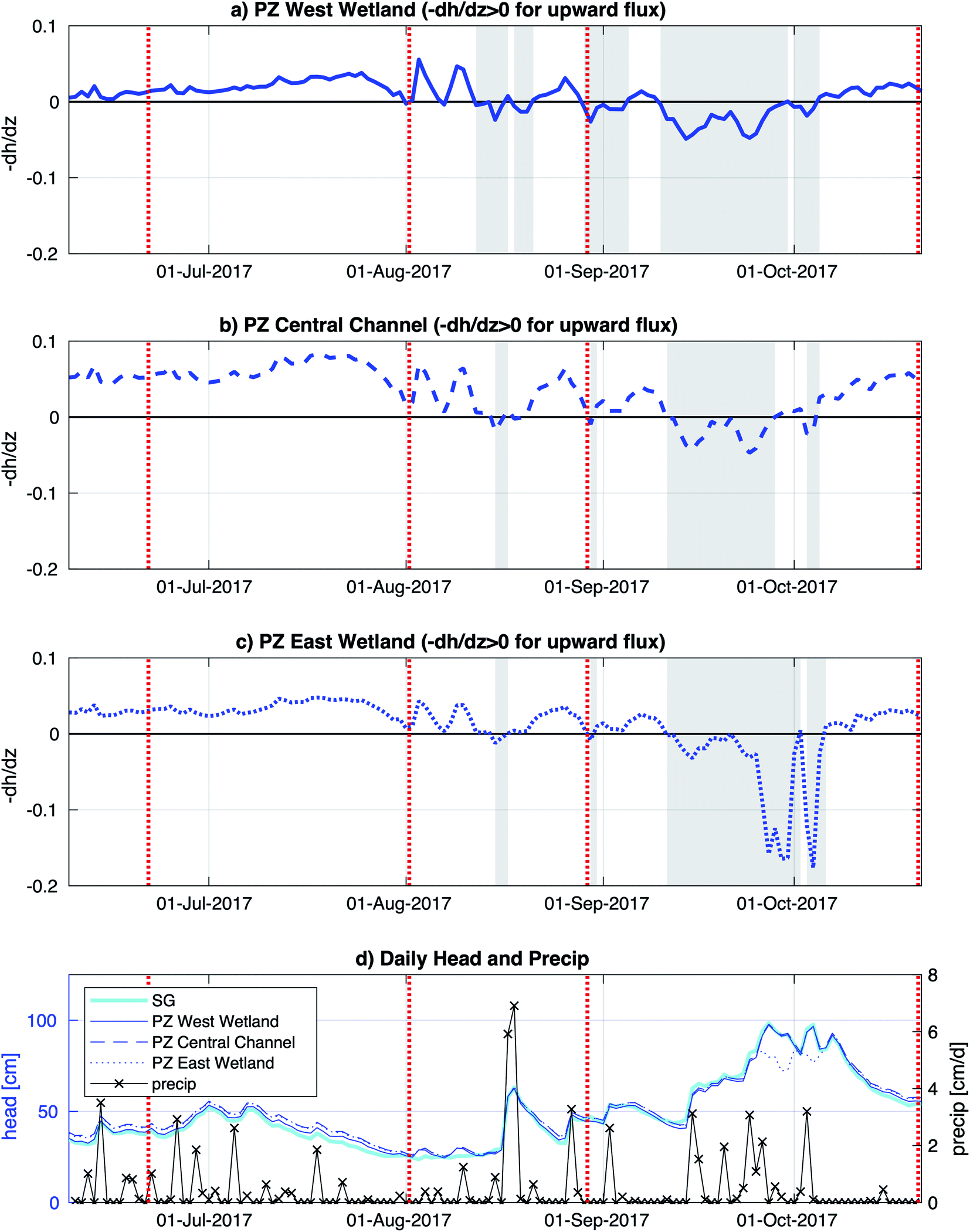 | ||
| Fig. 2 Vertical hydraulic head gradient for (a) the west wetland, (b) stream channel (center), and (c) east wetland, where the white (gray) shading demarcates times with upward (downward) flux. (d) Daily precipitation from Embarrass, MN (downloaded from the Midwestern Regional Climate Center, https://mrcc.purdue.edu/) and head levels, relative to the sediment–water interface in the channel, for surface water at the stream gauge (“SG”), the west wetland shallow groundwater (“PZ West Wetland”), the central channel shallow groundwater (“PZ Central Channel”), and the east wetland shallow groundwater (“PZ East Wetland”). Dashed red vertical lines indicate sampling time points for sediment and aqueous geochemistry analyses. | ||
Surface and porewater geochemistry
Compared to peeper-collected surface water phosphate (PO42−) (mean of 0.07 ppm), subsurface porewater PO42− levels were higher at all locations throughout the summer, with concentrations substantially higher (by 1–2 orders of magnitude in peeper samples) in the riparian wetlands than in the stream channel (Table S1 and Fig. S2‡). In contrast to PO42−, concentrations of NO3− + NO2− in the surface water and subsurface porewaters at all three locations were all consistently very low throughout the summer, with levels that were often too low to be accurately measured (Table S1 and Fig. S3‡). These trends are described in greater detail in the Results section (nutrient trends) of the ESI.‡
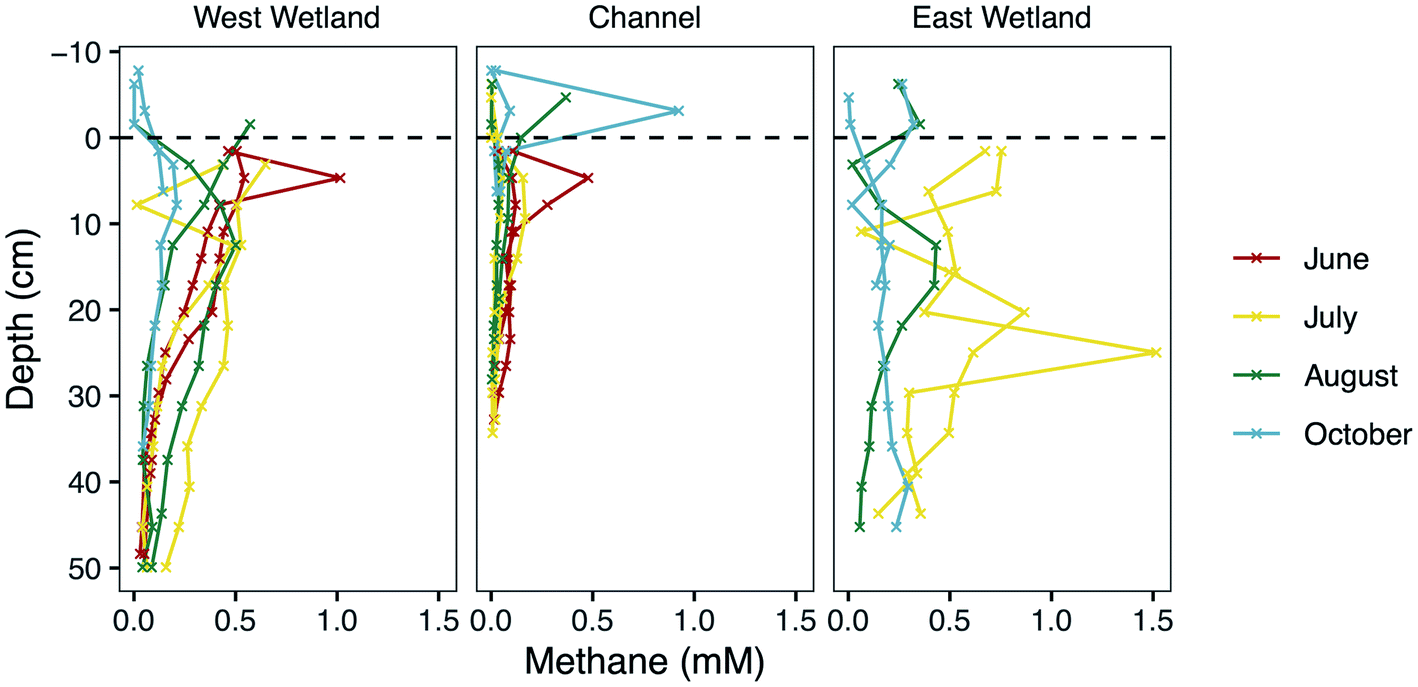 | ||
| Fig. 3 Methane concentrations (mM) of surface waters and porewaters with depth (cm) at Second Creek in the stream channel and both wetlands collected from replicate peepers during June, July, August, and October, 2017. Zero depth (indicated by the black dashed line) marks the sediment–water interface, increasingly positive depth values indicate deeper sediment depth, and negative values indicate surface water. Peepers were not installed in May or in the east wetland in June. | ||
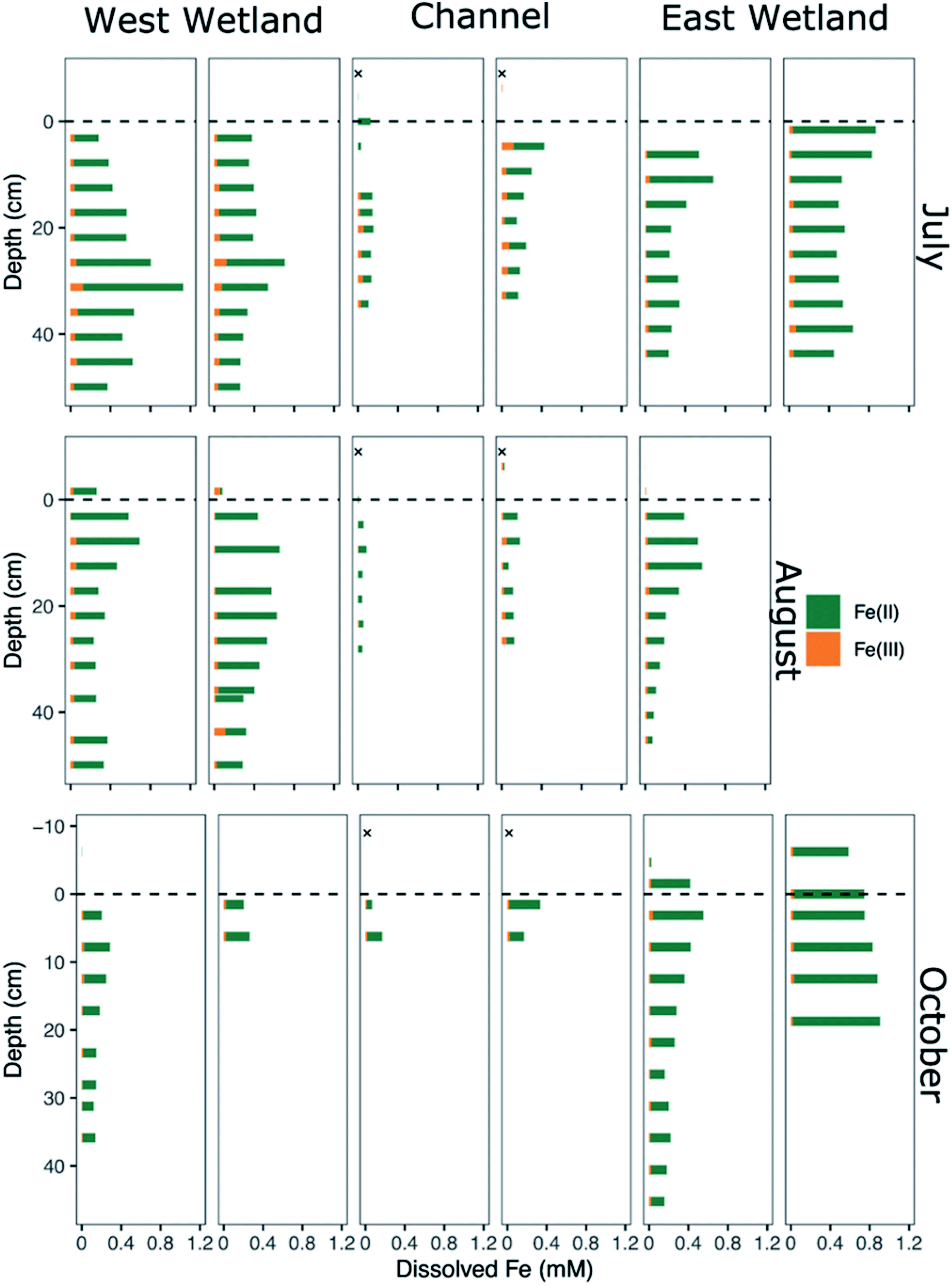 | ||
| Fig. 4 Dissolved iron speciation and concentration of porewaters collected from replicated peeper samplers as a function of sediment depth for July (top), August (middle), and October (bottom). Measured Fe(II) concentrations are denoted by green bars and calculated Fe(III) concentrations are denoted by orange bars. Channel surface water samples indicated by an “X” were collected in 2015 via Nalgene bottles46 and represent nearer surface conditions compared to peeper samples taken just above the sediment water-interface. | ||
Though Fe(II) was the dominant dissolved Fe species in all water samples including surface waters, dissolved Fe(III) was present in nearly all porewater samples, generally comprising between 10-20% of the total dissolved Fe, with several samples containing up to 25–40% (Fig. 4). In general, dissolved Fe(III) was a greater relative proportion of the total dissolved Fe concentrations in the channel porewaters than in either of the wetlands.
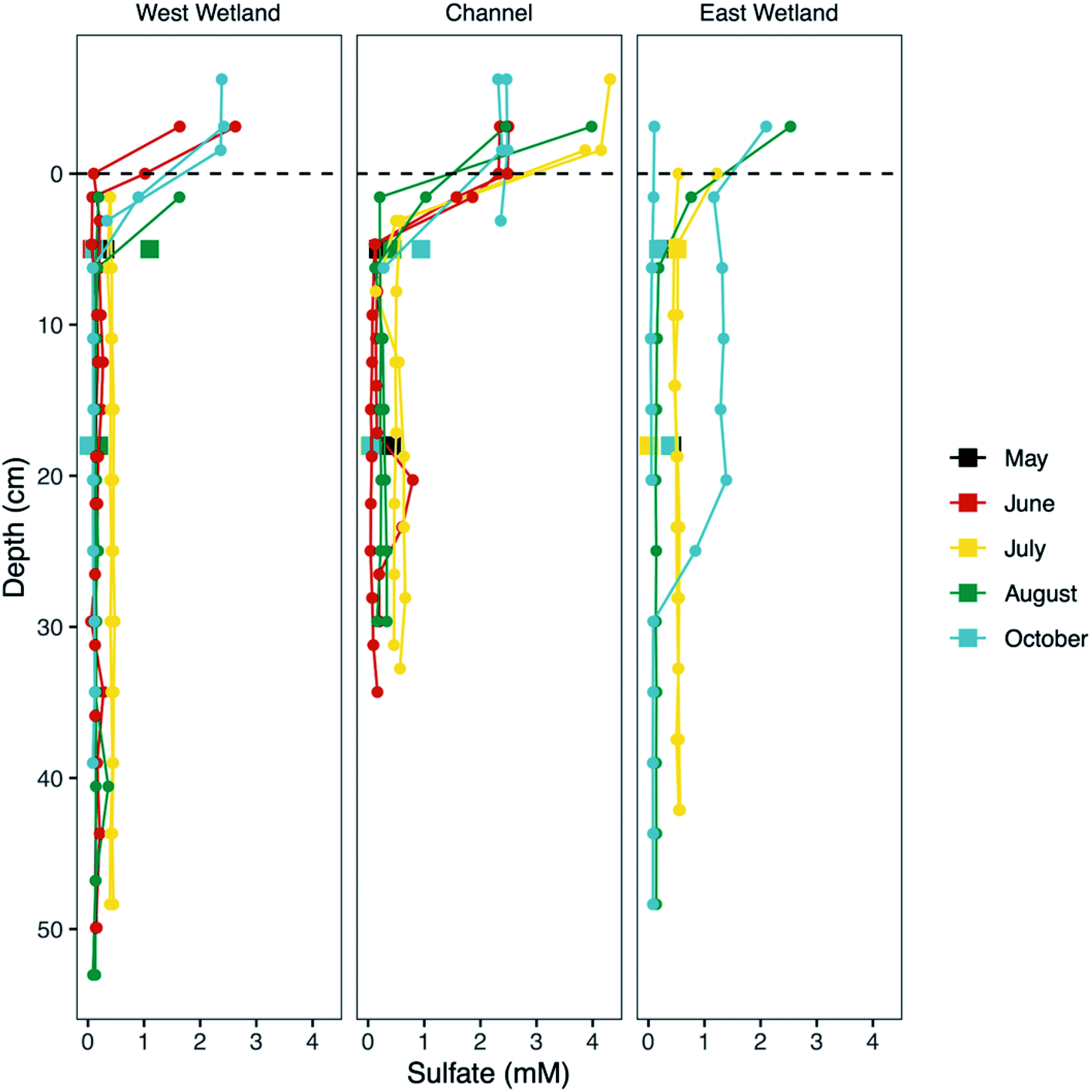 | ||
| Fig. 5 Dissolved sulfate concentration versus sediment depth at Second Creek in the stream channel and both wetlands during May, June, July, August, and October of 2017. Small solid circles represent ‘peeper’ porewater data whereas porewaters collected using Rhizon samplers are denoted by filled squares and represent averaged concentrations from 0–10 cm and 10–20 cm sediment depth. Zero depth (indicated by the black dashed line) marks the sediment–water interface, increasingly positive depth values indicate deeper sediment depth, and negative values indicate surface water. Peepers were not installed in May or in the east wetland in June. | ||
Concentrations of porewater SO42− rapidly decreased with increasing depth up to ∼5 cm in the channel and west wetland to 0.1–0.5 mM and 0.06–0.4 mM, respectively (Fig. 5). Below 5 cm, average SO42− concentrations were consistently low during the months of June and August in the channel (0.14 mM) and west wetland (0.21 mM), and slightly higher (0.50 mM and 0.43 mM, respectively) in July. East wetland porewater SO42− showed similar trends in July and August where concentrations decreased over the top ∼5 cm to 0.52 mM in July and 0.18 mM in August. In October, the replicate peepers in the east wetland indicated differing trends of porewater SO42−; one had low concentrations (≤0.05 mM) throughout the sediment profile, while the other showed concentrations in excess of 1.2 mM before decreasing to ∼0.08 mM below ∼30 cm. Rhizon samples generally showed slightly higher concentrations of SO42− relative to the peeper samples for the upper 10 cm, but concentrations at greater depths were consistent between the two sampling methods.
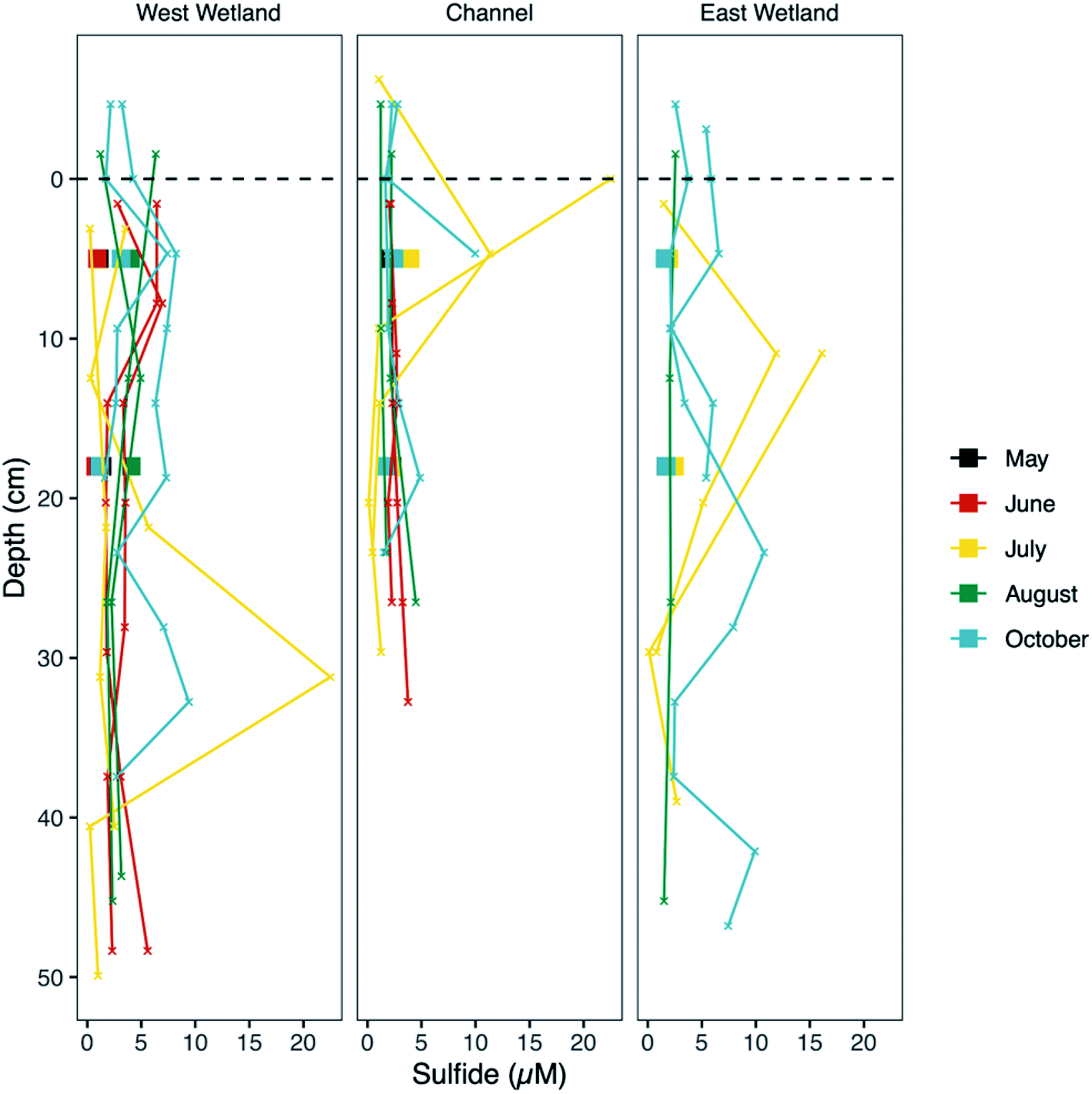 | ||
| Fig. 6 Dissolved sulfide concentration versus sediment depth at Second Creek in the stream channel and both wetlands during May, June, July, August, and October of 2017. Small “x's represent ‘peeper’ porewater data whereas porewaters collected using Rhizon samplers are denoted by filled squares and represent averaged concentrations from 0–10 cm and 10–20 cm sediment depth. Zero depth (indicated by the black dashed line) marks the sediment–water interface, increasingly positive depth values indicate deeper sediment depth, and negative values indicate surface water. Peepers were not installed in May or in the east wetland in June. | ||
Sediment AVS fraction
Measurements of unfiltered acid volatile sulfide, possible phases include reactive FeS and ZnS phases and free sulfide, represent the fraction of sulfide in the sediments readily influenced by biogeochemical cycling and fluctuations in redox gradients in the hyporheic zone. The average unfiltered AVS fraction (Fig. 7) was comparable in all three locations (30.0, 25.5, and 25.6 μmol S g−1 sediment, for west wetland, channel, and east wetland, respectively). There was a general decrease in AVS with increasing sediment depth with some exceptions (e.g., AVS in the west wetland was highest in the bottom interval in July and October).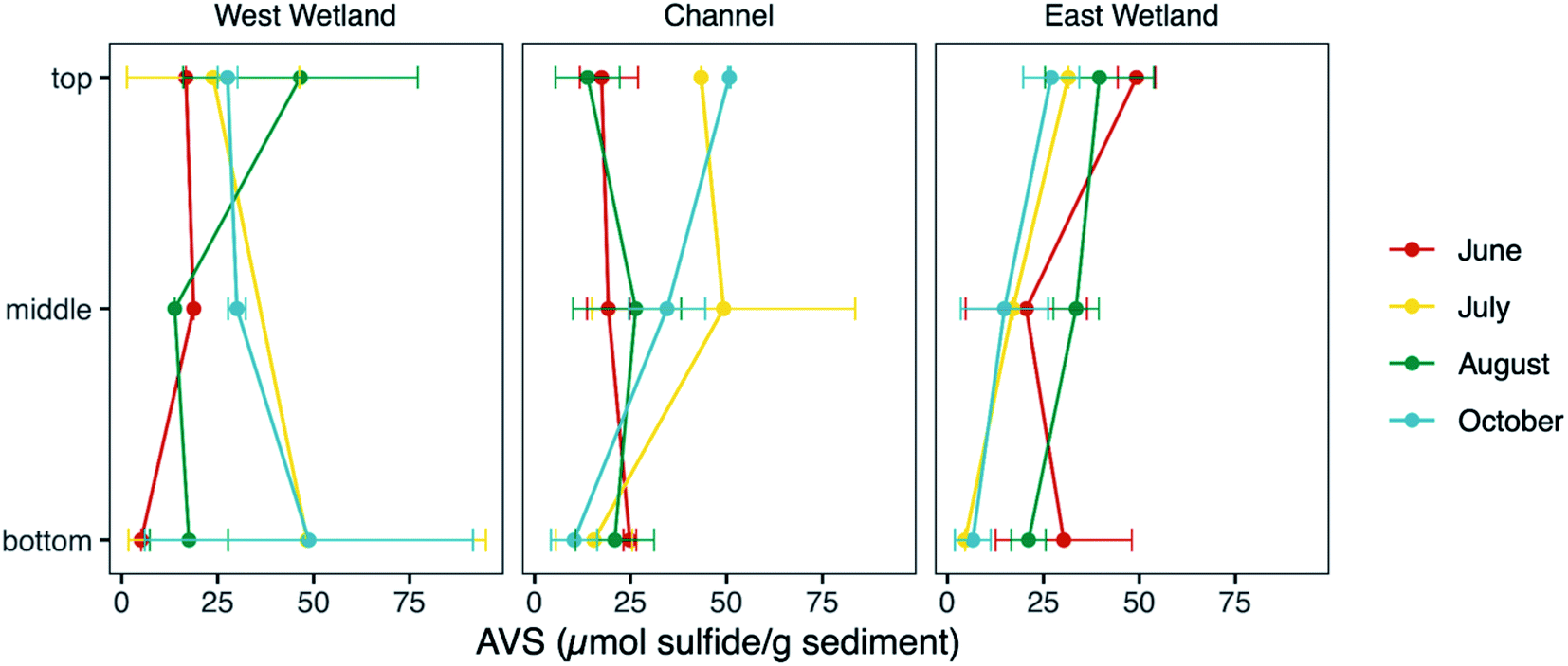 | ||
| Fig. 7 Measurement of unfiltered acid volatile sulfide (AVS) fraction in hyporheic zone sediments collected from the west wetland, channel, and east wetland (left to right, respectively) of Second Creek during the 2017 field season. The average AVS fraction is denoted by filled circles. Error bars represent the range of values measured for replicate samples. The ‘top’, ‘middle’, and ‘bottom’ correspond to sediment core intervals centered at approximately 4 cm, 10 cm, and 20 cm depths, respectively. | ||
Molecular-scale characterization of sediment iron
Fe K-edge X-ray absorption spectroscopy (XAS) was performed to characterize solid-phase Fe at different depths in the sediment profile. Linear combination fits of the Fe K-edge XANES spectra showed that all samples were mixed valence, with average Fe oxidation states ranging between 2.14 and 2.67 (Table S2 and Fig. S4‡). To determine individual contributions of Fe species, Fe K-edge EXAFS were also analyzed via LCF. Based on principal component analysis (PCA), most of the spectral variance (93.4–94.8%) in the 42-spectra dataset was explained by 4–5 components. Target transform analysis found that all reference compounds were potentially suitable components; therefore, all reference standards (Fig. S5‡) were used in the LCF fitting process. Optimal fits (Fig. S6‡) were selected using the minimum number of components, with additional components added only if the additional components improved the R-factor by 15% or more.Although there was substantial variability in the Fe-containing solid phases collected from the three locations across space, depth, and time, some trends were apparent (Fig. 8). Nearly all samples contained phyllosilicate-bound Fe (8–52%), as well as in sulfide phases including iron monosulfide (0–45%), iron disulfide (0–23%), and/or pyrrhotite (0–22%). Fe(III)-containing oxides such as hematite, goethite, and ferrihydrite were identified (between 0% and 31% maximum) in some locations and sampling times; no Fe(III)-containing oxides were detected in July. Dissolved or adsorbed Fe(II) and Fe(III) were also present in all samples (combined relative abundance 16–62%), with dissolved or adsorbed Fe(III) present at all depth intervals for most samples collected. The speciation of the dissolved and adsorbed Fe(II) and Fe(III) components, the Fe(III) phyllosilicate, and iron disulfide is described in greater detail in the ESI.‡
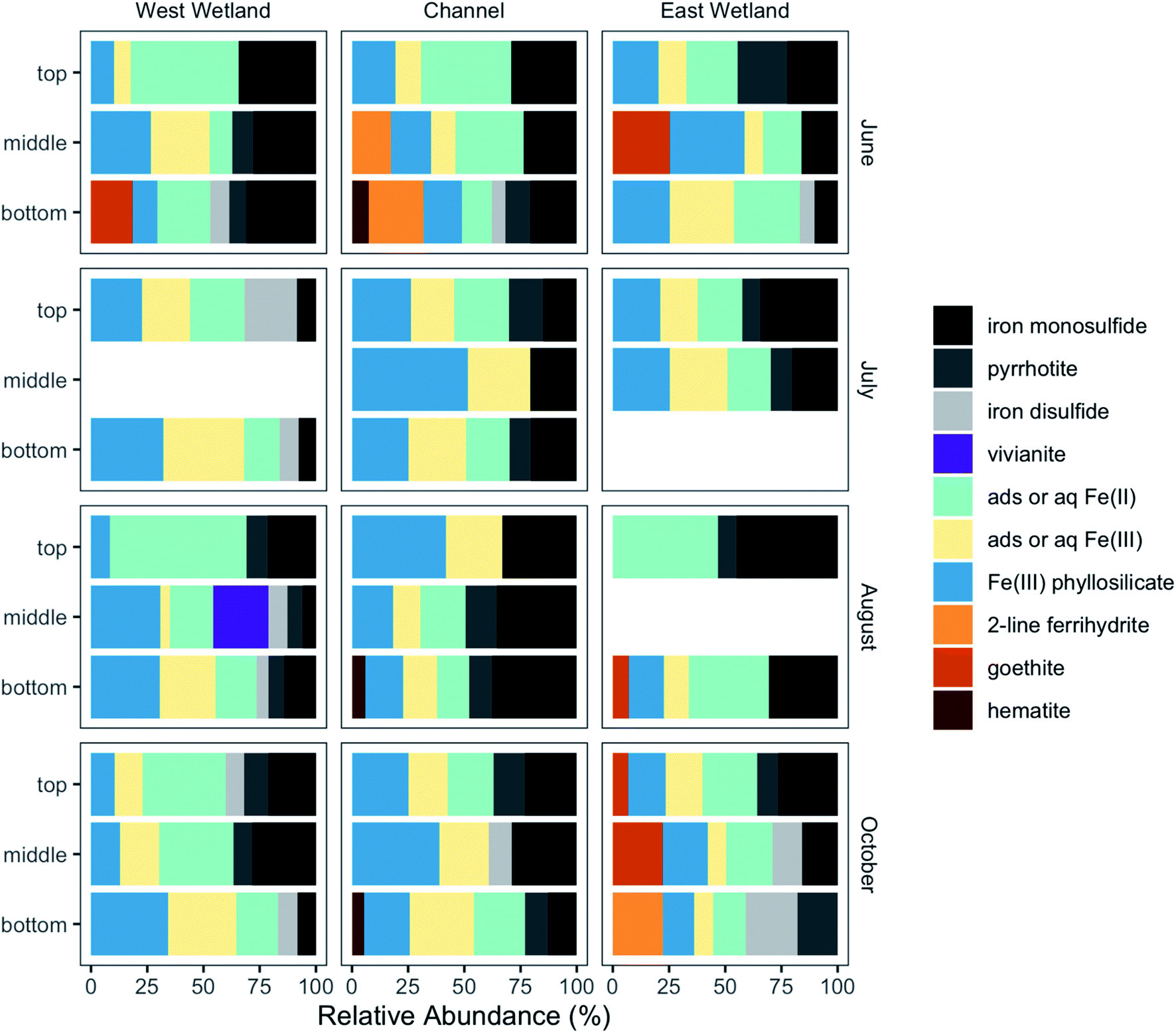 | ||
| Fig. 8 Linear Combination Fits (LCF)s of Fe K-edge EXAFS spectra from sediments collected from the west wetland, stream channel, and east wetland of Second Creek showing relative abundance for Fe-containing components in bulk sediment samples with depth through the 2017 sampling season. For LCFs, the sum of all Fe species was normalized to 100%. The ‘top’, ‘middle’, and ‘bottom’ correspond to sediment core intervals centered at approximately 4 cm, 10 cm, and 20 cm depths, respectively. | ||
One primary difference between the channel and wetlands was that while the vast majority of sulfide-containing Fe in the channel was identified as iron monosulfide and pyrrhotite, sulfide-containing Fe in the wetlands typically contained a mixture of iron monosulfide, pyrrhotite, and iron disulfide, especially deeper in the sediment profiles. As opposed to the other iron sulfides, pyrrhotite in this system is likely detrital due to its relatively common presence in geologic formations in the region.63 The range of total sulfide-containing Fe relative abundances in the three locations, however, were quite similar (west wetland = 16–47%, channel = 20–49%, east wetland = 16–53%). While iron disulfides in the wetlands increased with depth, the total relative abundance of iron sulfide phases in the wetlands tended to decrease or stay the same with increasing depth at most sampling times except June (west wetland). Conversely, the channel total sulfide abundance showed more variability but with no clear patterns across sampling times.
Molecular-scale analysis of sediment sulfur
Characterization of S-bearing solid phases was performed using S K-edge XANES. Principal component analysis indicated that 99.4% of the variance of the 22 S-XANES sample spectra could be fit using 4 components; adding the fifth component increased the cumulative variance to 99.8%. As with the Fe-EXAFS, target transform analysis indicated that all reference standards (Fig. S7‡) were potentially acceptable components; therefore, all reference standards were used in all LCF fits. Optimal fits were selected using the minimum number of components with additional components included only if they improved the R-factor by at least 15%.Nearly all sample spectra contained peaks in two primary regions (Fig. S8‡). Peaks in the 2470–2475 eV region result from more reduced S species including organic sulfide (e.g., methionine, cystine), inorganic sulfide (e.g. mackinawite (FeS), chalcopyrite), and elemental sulfur, while peaks in the 2480–2485 eV region result from more oxidized S components (e.g. sulfate, thiosulfate). S-XANES spectra were fit using reference standards selected to represent a range of S oxidation states ranging from −2 to +6, including a mix of inorganic and organic compounds (Table S3 and Fig. S7‡).
S XANES detected a wide range of S species and valence states in the Second Creek sediments, and nearly all sediment samples contained a mixture of S valence states. In the west wetland and channel, oxidized S forms (inorganic sulfate and organic oxidized S; oxidation indices = +5 − +6) were relatively consistent with depth, between ∼6–20% of the total S pool. The east wetland was more variable with respect to oxidized S content. For example in June, there was 0% oxidized S detected in the top interval and 65% oxidized S in the bottom interval. Both June and August saw increased oxidized S with increasing depth in the east wetland, while the greatest relative abundance of oxidized S in October occurred in the middle depth. In all locations, intermediate oxidation state S forms (organic with oxidation states +1 − +2, inorganic with mixed oxidation states of −1/+5 and −0.1/+3.3, S(0), and polysulfide with an oxidation state of −1.3/−0.1) were detected at nearly all depths, with relative abundances up to 46% organic forms and 28% inorganic mixed valence compounds. Note that while polysulfide also has a mixed valence state, it's chemistry in the hyporheic zone is expected to be different than the other inorganic mixed valence compounds (thiosulfate and tetrathionate) and was therefore not included in the inorganic mixed valence group. Elemental S (oxidation state = 0) and polysulfide (oxidation state = −1.3/−0.1) were also significant components of many of the sediment samples in all months and all locations, especially August (all locations, 35–66%), June in the west wetland (43–56%), and October in the east wetland (30–63%). Regarding the most reduced S phases, inorganic metal sulfides were also identified in most samples. In general, the metal sulfides were identified as iron monosulfide (mackinawite, oxidation state = −2), though iron disulfide (e.g., pyrite, oxidation state = −1) was identified in the bottom samples of the east and west wetlands (54 and 37%, respectively) in October and in the channel (31%) during August. Contrary to iron disulfide, iron monosulfide was typically most abundant in the shallower depths of the cores and the relative abundance decreased with increasing depth.
Discussion
Impact of hydrology on hyporheic zone geochemistry
A major goal of this study is to examine how major changes in vertical hyporheic flux impacts the subsurface geochemistry of riparian wetland and stream environments through time. Though we do encompass several small shifts in hyporheic flux within our sampling period, we notably do not observe major changes in porewater geochemistry after shifts from upward to downward fluxes, except for increased sulfate and Fe oxides in the east wetland in October (Fig. 5) at Second Creek. This suggests that the system can recover quickly to previous conditions and/or the system is buffered against major changes due to low magnitude or unsustained hyporheic flux changes or by geochemical controls related to metastable phase reactivity. Our data indicates that all these factors are contributing to the observed conditions. Flux magnitude variability across the wetlands and sediments is evident and can explain some of the geochemical excursions in sulfate and iron. For example, the ∼6 times greater magnitude hydraulic gradient in the east wetland in late September to early October compared to the channel and west wetland (Fig. 2) indicates that the east wetland experienced much stronger downward flux at that time which facilitated deeper penetration of elevated oxygen (resulting in Fe oxide formation) and sulfate (Fig. 5 and 8). Interestingly, previous inverse modeling at Second Creek suggests that the hydraulic conductivity of channel sediments can be up to 2–3 times greater than for wetland sediments,53 so elevated sulfate and Fe oxides at depth was expected in the stream rather than either wetland. The strong locally measured downward flux in the east wetland could potentially result from influences of the regional west to east groundwater flow regime, which would be more likely to support losing conditions on Second Creek's east bank compared to its west bank.52 Variable changes in geochemistry due to lateral transport has recently been demonstrated at topographic features such as stream meanders.64Because the Second Creek sediments have substantial quantities of organic matter52 that can generate highly reducing conditions, the system may be buffered against short-duration or small-scale hydrologic changes that allow only limited transport of surface water electron acceptors into the hyporheic zone relative to redox rates.65–67 In particular, previous model simulations52 showed that high sulfate concentrations from surface water moved only 5 cm or less into the sediment over a few days after a switch from upward to downward hyporheic flux, which is about the duration of most of the flow reversals observed in this 2017 study period. Unfortunately, the late August sampling coincided with the start of the flux reversal to downwelling conditions, which may not have allowed sufficient time to observe measurable geochemical changes, and the October sampling occurred two weeks after the strong downwelling conditions flipped to upwelling hyporheic flow. The higher porewater sulfate concentrations collected by peepers, which reflect a ∼2 weeks equilibration time, compared to Rhizons, which capture temporally instantaneous conditions, in the east wetland in October (2017) provide additional support that rates of SO42− reduction following the resumption of upwelling conditions were rapid enough to decrease porewater SO42− to previous values on short (<2 weeks) times scales.
Additional buffering against substantial geochemical responses during these brief hydrologic excursions could be due to the formation of redox active metastable phases (RAMPs) such as ferrihydrite and goethite, or mixed- and intermediate-valence sulfur compounds; which may only appear in “hot spots” and/or “hot moments”.45,68,69 These RAMPs have the potential to act as either/both electron donors and acceptors and their production is kinetically controlled, making their occurrence highly transient as they serve as powerful redox buffers.45 At Second Creek, the presence of Fe and S-bearing RAMPs at various times and depths is confirmed by bulk sediment Fe EXAFS, porewater Fe speciation, and S XANES analysis. Although bulk sediment Fe EXAFS analysis indicates that solid phase Fe(III) is mostly depleted except for in June and October, the analysis revealed a substantial portion of dissolved or adsorbed Fe(III) within the sediments (Fig. 8). Further, both dissolved Fe(II) and Fe(III) are present in porewaters at all depths in the hyporheic zone (Fig. 4), as has previously been observed in both anoxic and oxic environments.70–74 The stability of dissolved Fe, particularly Fe(III) in anoxic porewaters, is likely enhanced by complexation with various organic matter functional groups.75–78 These organic matter functional groups may further act as electron shuttles (i.e., as RAMPs), serving to oxidize or reduce Fe and other redox active analytes (e.g., sulfur) through chemical or microbial processes.45,75,79,80 The hydrologic regime changes (e.g., gaining to losing stream conditions) or general mixing of oxygenated surface waters and anoxic groundwaters in the hyporheic zone at Second Creek may facilitate the rapid recycling of these inorganic and organic RAMPs,45 which may be why metastable dissolved Fe(III) is consistently measured in the subsurface despite its likely reduction during cryptic S cycling.
Geochemical evidence for a cryptic sulfur cycle linked to Fe cycling
Cryptic S cycling has been invoked as an important process in previous modeling studies at Second Creek, though previous investigations have not shown substantiated geochemical evidence for this process. Porewater sulfate profiles measured in the current study provide one line of geochemical evidence that cryptic S cycling is occurring. Throughout the entire season, robust sulfate reduction is evident as porewater sulfate concentrations are substantially lower than surface waters (∼3–10 mM sulfate). Despite an abundance of electron donor, however, the sulfate concentrations never decrease to 0 mM in the porewater (Fig. 5) and remain moderately elevated (∼0.14–0.5 mM sulfate) at depth. These deep (to at least 50 cm depth) porewater sulfate concentrations are greater than what would result solely from downward dispersion and mixing of surface water (which occurs even during upwelling), as indicated by calibrated reactive transport model results for conservative solutes (see ESI‡ in Ng et al.52,53). This indicates that sulfate is being regenerated in the anoxic hyporheic zone at Second Creek, likely by sulfide oxidation processes81–83 as part of a cryptic S cycle.There are a variety of abiotic and biotic pathways that promote anoxic sulfide oxidation, ultimately leading to the production of sulfate that could explain this regeneration at Second Creek. In marine conditions (i.e., high sulfate concentrations, ∼3–10× that of Second Creek surface water), elemental S is the predominant product from sulfide oxidation coupled to Fe(III) oxide reduction, possibly formed via polysulfide intermediates.33,83–85 Polysulfide also forms from equilibrium with elemental sulfur and sulfide.86 Given the high relative amount of elemental S and polysulfide in all three locations (Fig. 9) and relatively low amounts of dissolved sulfide and solid iron sulfide phases (e.g., FeS or pyrite), sulfide oxidation seems to be a predominant pathway in the Second Creek hyporheic zone. Further reactions involving these S-intermediates, such as microbial S(0) and polysulfide oxidation87,88 and disproportionation reactions89,90 produce sulfate. High concentrations of aqueous sulfide have been shown to inhibit elemental S disproportionation,89 but the low sulfide concentrations detected throughout the hyporheic zone porewaters in both the wetlands and stream channel suggest that disproportionation is a viable pathway for sulfate regeneration at depth.
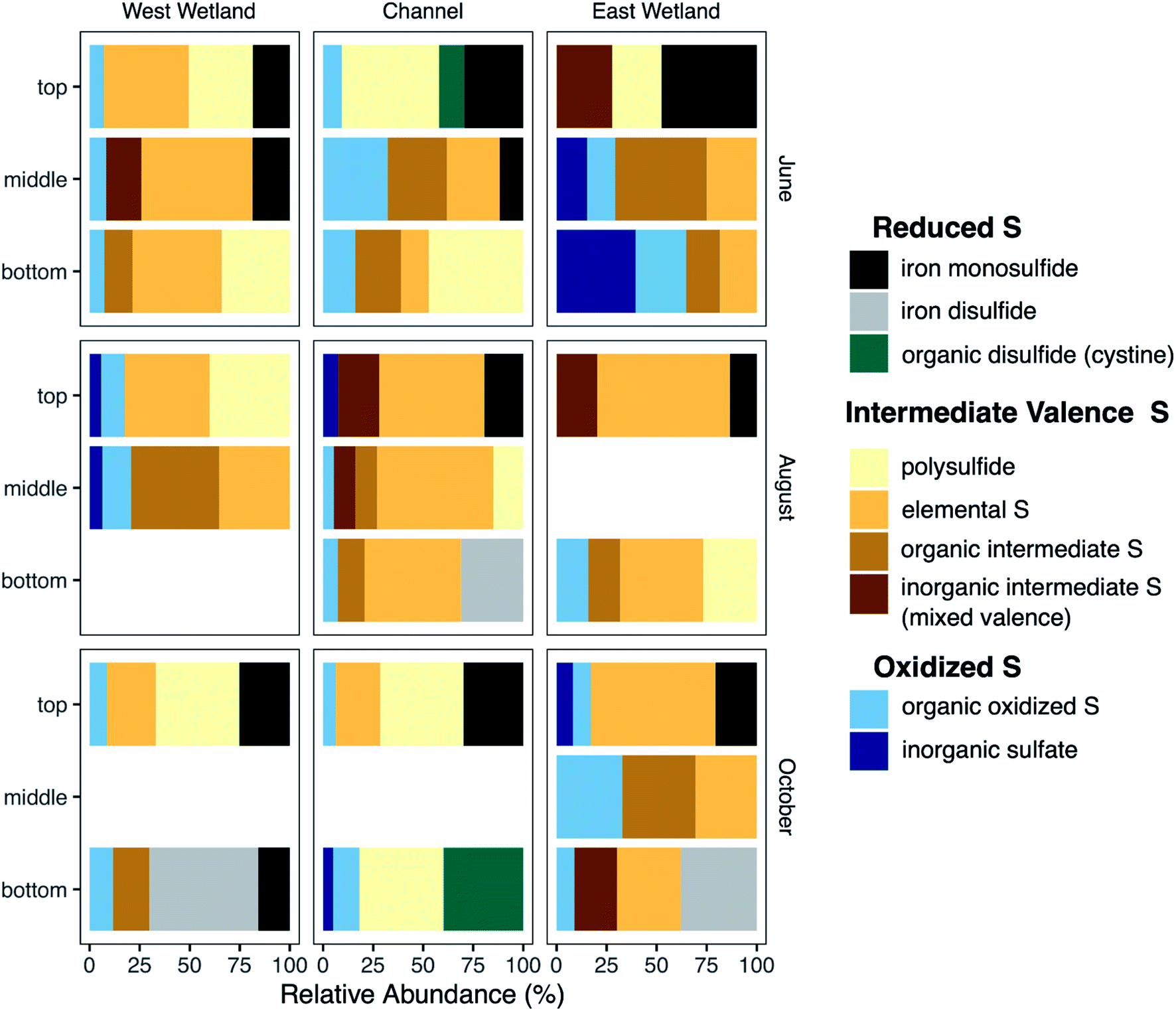 | ||
| Fig. 9 Linear Combination Fits (LCF)s of S K-edge XANES spectra from sediments collected from the west wetland, stream channel, and east wetland of Second Creek showing relative abundance for S-containing components in bulk sediment samples with depth through the 2017 sampling season. For LCFs, the sum of all S species was normalized to 100%. The ‘top’, ‘middle’, and ‘bottom’ correspond to sediment core intervals centered at approximately 4 cm, 10 cm, and 20 cm depths, respectively. The legend is approximately ordered with more reduced S forms at the top (iron monosulfide, iron disulfide, and organic disulfide), intermediate valence species in the middle (polysulfide, elemental S, organic intermediate S, and inorganic intermediate S (mixed valence)), and more oxidized species on the bottom (organic oxidized S and inorganic sulfate). In three cases, groups of references were combined into a single category based on oxidation index. “Organic oxidized S” (oxidation index = 5–6) includes sodium dodecyl sulfate (SDS), sulfanilamide, and sulfonate references. “Organic intermediate S” (oxidation index 1–2) includes sulfoxide and thiophene reference compounds. “Inorganic intermediate S (mixed valence)” includes thiosulfate and tetrathionate (oxidation indices −1/+5 and −0.1/−3.3, respectively). | ||
Both solid metastable Fe(III) oxyhydroxides and organic Fe(III) complexes are likely driving sulfide oxidation, and therefore cryptic S cycling, in the Second Creek sediments. Reactive, metastable Fe(III) oxyhydroxides, such as ferrihydrite identified in our Fe EXAFS analysis, can serve as the TEA driving aqueous sulfide oxidation in anoxic environments.33,91–93 Using an experimental laboratory approach, Taillafert et al.76 further showed that Fe(III) complexed by organic matter can be reduced by sulfide at circumneutral pH. This reaction is substantially faster (half-time of seconds) than the reduction of solid Fe(III) mineral phases (minutes to hours). A follow-up study showed that Fe(III)–organic complexes present in natural sediment porewaters reacts with aqueous sulfide, potentially through a S(0) intermediate, ultimately forming FeS in low sulfide environments and pyrite in high sulfide environments.94 Although organic–Fe(III) could not be specifically identified using the methods in this study, the measurable amounts of aqueous or adsorbed Fe(III) detected by Fe EXAFS sediment analysis (Fig. 8) and ferrozine-based valence state measurements of the porewaters (Fig. 4) suggest that Fe(III) was stabilized in the anoxic zone by organic complexes.
High quantities of DOC may also be playing a direct role in sulfide oxidation at Second Creek. Previous studies have shown that anoxic reactions between sulfide and aqueous humic acids are fast (minutes to hours) and produce S(0), thiosulfate, and a variety of different reduced organic S compounds.95,96 While DOC speciation was not conducted as part of this study, we suspect that humic acids were present, as indicated by the yellow-brown hues in the extracted porewaters, and may be playing a role in Fe(III) complexation. Mn(III/IV) oxides can also serve as a TEA and drive cryptic S cycling,97,98 and while these phases were not measured in the present study, elevated Mn2+ was detected previously in the porewaters.52,53 Further characterization of potential electron acceptors driving sulfide oxidation at Second Creek could yield valuable information about coupled redox cycles and turnover of organic matter in these dynamic environments.
Accumulation of intermediate valence S phases in hyporheic zone sediments
Several of the reactions that drive the cryptic S cycle at Second Creek likely produce various intermediate S species. In studies that have examined S speciation, elemental S and inorganic polysulfides are detected in sediments and porewaters,35,85,99,100 sometimes comprising a substantial portion of the total S pool depending on the environmental conditions of both terrestrial and marine ecosystems. In the present study, S XANES analysis shows that both S(0) and polysulfide are present in high relative abundance (Fig. 9), and are often the predominant S forms, throughout the ecosystem regardless of season, hydraulic flux, location, or depth (for further explanation of S XANES interpretation, see the ESI‡). The persistence of S(0) may be the result of organic matter S(0) stabilization, as has been observed recently.101–103 A portion of the elemental S analyzed in our study could also be organic polysulfides as some organic polysulfides have absorption energies and peak features that are overlapping with, and potentially indistinguishable from, that of elemental S.104 Organic polysulfides, though not targeted in most S speciation studies, have been found to be abundant in some anoxic environments such as salt marsh and marine sediments,28,100,104,105 supporting the hypothesis that these compounds are present in the organic rich sediments at Second Creek, particularly in the wetlands. Identification of these compounds would require additional S XANES database refinement and alternative analytical approaches.The relatively high contributions from intermediate valence S compounds compared to reduced iron monosulfide and disulfide forms (Fig. 9) were somewhat unexpected, given that AVS (presumably mostly FeS, as porewater sulfide measurements were low) was measured in all samples (Fig. 7), and reduced inorganic sulfides were consistently detected by Fe EXAFS analysis (Fig. 8). One possible explanation for this analytical discrepancy is that inorganic sulfides represent a relatively small (<5%) fraction of the total S pool (not quantified in this study) and were thus not detectable by S XANES. It is also plausible that the AVS results were inflated due to partial or complete extraction of non-target S phases such as organic polysulfides.106,107
Geochemical variability between the riparian wetland and stream hyporheic zone
Despite their close proximity, the riparian wetlands exhibit substantially different porewater geochemistry compared to the channel porewaters for some analytes, particularly P, Fe, and CH4 (Fig. 3, 4 and S2‡). These differences likely result from higher accumulations of total organic carbon in the wetland sediments compared to the stream sediments.52 Living and decaying aquatic plants are substantially more abundant in the wetlands than in the stream channel, although plants like wild rice (manoomin) grow sparsely throughout this section of the stream channel. This organic matter can serve as an electron donor, and even sometimes as an electron acceptor, for redox reactions that drive important biogeochemical cycles such as in N, Fe, S, and methane.45,95,108,109 Nitrate concentrations remained low at all sampling locations in both the surface waters and porewaters, which may suggest that biogeochemical nitrogen cycling is relatively limited in this system. It is also possible that the sampling regime in the current study was not well suited to capturing measurements indicative of nitrogen biogeochemistry, or that our analytical capabilities were not sensitive enough to resolve nitrate variations in our samples.110 The geochemistry data at Second Creek provides clear evidence, however, that Fe(III) and sulfate reduction are robust reactions in the hyporheic zone. The increased porewater phosphate concentrations could result from adsorbed phosphate released during reduction and dissolution of Fe(III) oxide and oxyhydroxide phases.111–113 Remineralization of this organic matter can also increase phosphate111,114 and methane,2,115 which aligns with our observations of high analyte concentrations in the wetlands relative to the channel. Previous modeling at Second Creek indicates more reducing conditions in the wetland hyporheic zones because of their higher concentrations of organic carbon compared to in the channel.52,53 Despite these concentration differences between wetland and stream channel sediments, the data indicate that cryptic S cycling linked to Fe and methane cycling is active in both environments.Methane cycling and fluxes in the hyporheic zone
In addition to Fe and sulfate reduction, the highly reducing conditions in the hyporheic zone support methanogenesis, particularly in the organic-rich wetlands as described above. Indeed, a diversity of methanogenic archaea were previously discovered in relatively high abundances throughout the wetlands and stream channel sediments.53 The rapid increase in CH4 concentrations (Fig. 3) just below the depth where dissolved Fe(II) concentrations increase and SO42− concentrations decrease shows that biogenic methanogenesis is spatially discrete from Fe(III) and sulfate reduction at most sampling times and locations. This is expected based on the lower redox potential of methanogenesis than for the other two redox reactions and which resulted in a deeper methanogenesis metabolic zone. Methane concentrations are generally lower in October than the other sampling months which also indicates that colder temperatures slightly suppressed methanogenesis, but there are still measurable amounts (near millimolar) in the wetlands (Fig. 3). Similar seasonal responses were previously observed in sediments and soils from a variety of environments resulting from decreased metabolic rates of methanogens due to lower temperatures and lower amounts of substrate availability, which also prompt shifts in the methanogen community composition and pathway (e.g., aceticlastic vs. hydrogenotrophic).116–119High CH4 concentrations measured near the sediment–water interface during the June and July sampling times, however, suggests that methanogenesis occurred in very shallow sediments during that time. This has major implications for methane fluxes to the atmosphere, especially because the wetlands had either very shallow surface water or periods without inundation. Indeed, wetlands are the largest natural source of atmospheric methane, contributing ∼20–39% of the global flux,120,121 and rivers contribute another ∼3% of the global emissions.122 Upward hydraulic flux through the hyporheic zone during these months could contribute to the elevated levels in shallow sediments. However, the previous modeling studies at the site did not include methanogenesis at shallow depths, and they under-simulated CH4 there.52,53 This suggests that hydraulic flux cannot entirely produce the measured methane concentrations, and thus, some methanogenesis is likely co-occurring with Fe or sulfate reduction, as previously observed in other studies.123–125 This is a particularly important consideration for implementing reactive transport models that prohibit co-occurring redox reactions that are contrary to thermodynamic ordering.
We suspect that the spuriously high CH4 concentrations at various depths and times (e.g., the channel sediment–water interface sample in October) resulted from trapping of methane bubbles in the porewater peepers. In dense, organic-rich sediments, CH4-rich bubbles break out of sediments and travel through bubble tubes, escaping sediments at discrete, point-source locations in a process known as ebullition.126–128 While sampling in the field, physical disruption of the wetland sediment from coring or from walking produced large amounts of bubbles, which could have contained CO2 and CH4. Upward hydrologic flux, diffusion, ebullition, and transport through arenchymous vascular plants (like those abundant in the wetlands) are all pathways of methane fluxes to the atmosphere,129 and it is likely that all these processes play a role at various times at Second Creek.
The net flux of methane to the atmosphere from wetland sediments is controlled not only by methane production and transport processes, but also the amount of methane oxidized in the subsurface. Reactive transport modeling in previous studies52,53 indicated that CH4 was largely consumed in shallow sediments during upwelling hyporheic fluxes at Second Creek, possibly coupled to sulfate, Fe, and nitrate reduction. With the exception of June and July in the present study, methane levels are substantially lower in the upper 5 cm than at deeper sediment depths during many of the sampling points. This is particularly true in the cooler months and in the stream channel, which may reflect not only lower production rates at those depths, but also oxidation of methane that is transported upward. Evaluation of methane consumption from a wide range of freshwater wetlands demonstrates that a substantial, but highly variable, fraction of CH4 generated in the subsurface, ranging from 1–90% of gross production, is consumed before reaching the atmosphere.31 A long-held assumption is that most terrestrial CH4 consumption occurs aerobically at the oxic–anoxic interface or near the rhizosphere, driven by methanotrophic bacteria that oxidize CH4 for energy and biomass.48,130,131 However, the anoxic conditions in the shallow reaches of the hyporheic zone at Second Creek during the warmer months with upwelling hydrologic conditions would have inhibited aerobic methane oxidation processes. Anaerobic methane oxidation (AOM) linked to abiotic Fe(III) reduction,132–135 sulfate reduction,39,40,136 or even humic acids,132,137 would have been favorable in this environment. A metabolically active community of anaerobic methanotrophic (ANME) archaea was discovered in terrestrial and freshwater subsurface environments that couple AOM to sulfate reduction138,139 and Fe reduction,134 and these biotic processes could be coupled with abiotic processes that drive carbon cycling in the hyporheic zone. In these dynamic and heterogeneous environments in which TEAs are periodically recharged through hyporheic fluxes, the co-existence of methane redox cycling with S and Fe cycling appears to be a dominant process. This deviation from classical redox theory is notable and important for our understanding and prediction of ecosystem functioning. Supporting this finding, previous studies have identified functional overlaps and co-occurring bacterial metabolisms within similarly complex and heterogeneous riparian systems140 and in coastal marine sediments.141 The overall impact of this heterogeneity, the dynamic hyporheic exchange, and the biogeochemical conditions on methane fluxes to the atmosphere from the hyporheic zone deserve greater investigation.
Conclusions
The substantial contribution of organic and inorganic intermediate S compounds (e.g., elemental sulfur, polysulfide, thiosulfate, sulfoxide, and thiophene) in the subsurface together with measurable Fe(III) oxyhydroxides and dissolved Fe(III)-complexes provide clear evidence of a cryptic S cycle coupled to Fe reduction in the Second Creek hyporheic zone. Importantly, the cryptic S cycle proceeds in stream and wetland sites and under both upwelling and downwelling conditions. Under dominantly upwelling conditions, including those with short-duration or low-magnitude shifts to downwelling, dissolved Fe(III)-complexes, as opposed to solid-phase Fe oxides, serve as TEAs for anaerobic sulfide oxidation that fuel the cryptic S cycle (Fig. 10, left panel). However, with high-magnitude or long-duration shifts to downwelling conditions, TEA recharge of O2 and sulfate from surface water pushes the reaction network to utilize the newly formed electron acceptors (e.g. Fe oxide minerals) to fuel the cryptic S cycle (Fig. 10, right panel). Incorporating TEA variability, co-occurring redox reactions, and cryptic S cycling into future modeling efforts will be important for accurately capturing these complex and dynamic systems and thus improving predicted reaction rates and mass balances. In the future, further identifying and quantifying the primary S intermediate forms and developing genomically informed models will also be critical to expand our predictive understanding for how coupled biogeochemical cycles respond to environmental perturbations.140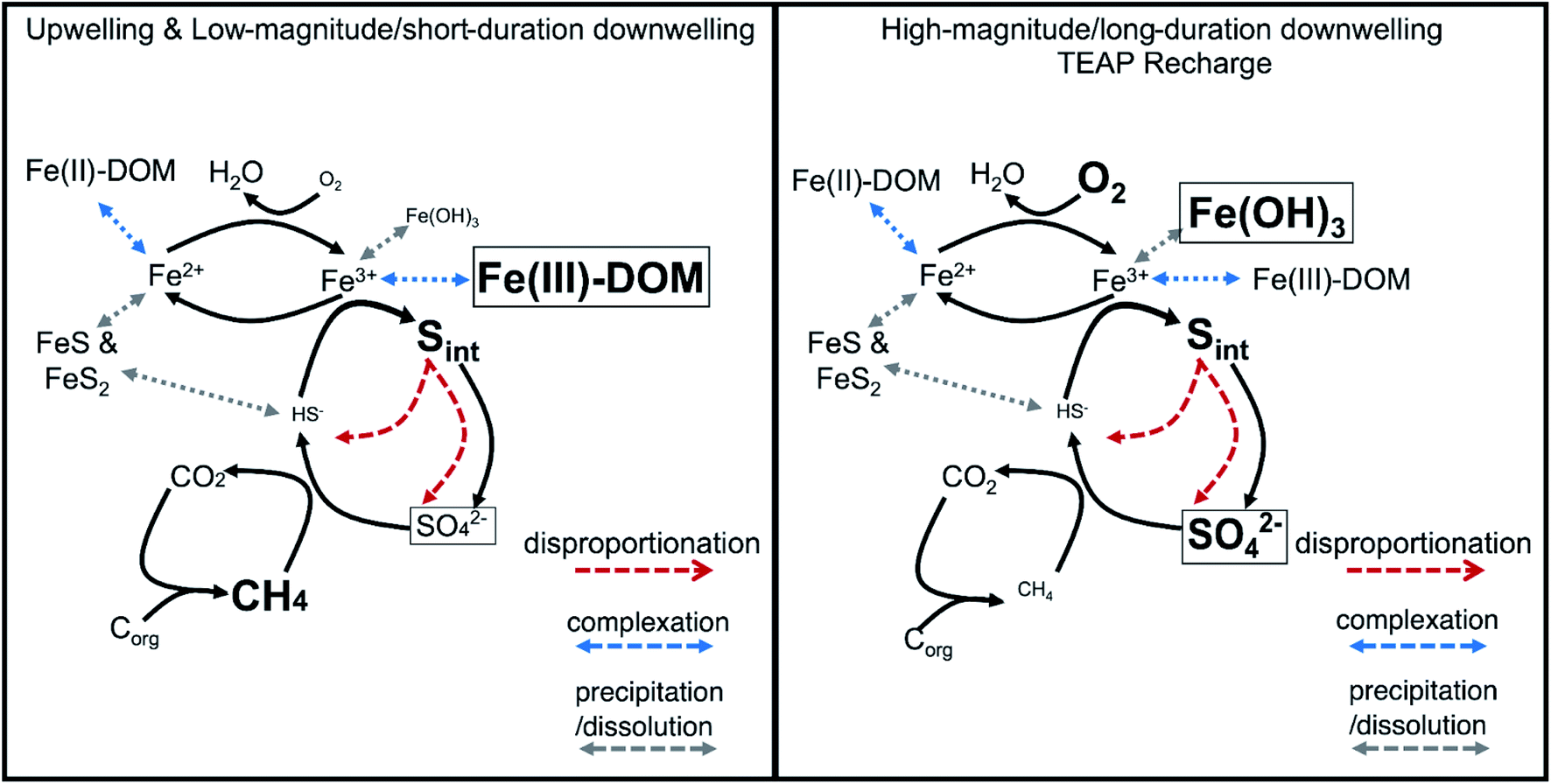 | ||
| Fig. 10 Conceptual model of a proposed reaction network that sustains a cryptic S cycle linked to Fe reduction and methane oxidation in the Second Creek hyporheic zone under two different hydrologic regimes: upwelling and low-magnitude/short-duration downwelling (left panel) and High magnitude/long-duration downwelling (right panel). Solid black arrows indicate generalized abiotic and biotic reactions (e.g., biological Fe(III) reduction could proceed via organic C oxidation instead of being coupled to sulfide oxidation to S intermediates). Species with boxes around them indicate important TEAs under the different conditions. Species with larger font size are expected to have increased concentrations in the system under the specific hydrologic conditions. | ||
At Second Creek, S recycling is likely fueled by high sulfate levels carried from the stream during downwelling hydrologic conditions and can have significant impacts on water quality. While some environmental sulfate loading has decreased with efforts to mitigate acid rain,142,143 high sulfate concentrations from industrial and agricultural waters38 remains problematic for many sensitive ecosystems, especially as sulfate is not federally regulated in all waste streams. Constructed wetlands have been used for the remediation of sulfate by promoting sulfate reduction to sulfide, and ultimately the precipitation of iron monosulfide (e.g., FeS) and iron disulfide phases (e.g., pyrite, chalcopyrite) that capture sulfide as well as heavy metals.144–146 As can be seen from this study and others, however, there could be significant intermediate S species forming in wetlands that can be recycled to replenish sulfate within the system rather than precipitated in sulfide minerals. If the formation of these intermediate phases is correlated with sulfate enrichment in surface waters, it will be important to determine whether they represent a net S sink or potential sulfate source, which could have ramifications for long-term water quality improvement in these wetlands. These intermediate S forms should be monitored more closely for ensuring the efficiency of these remediation approaches in controlling the fate and transport of contaminants in natural systems.
The fast recycling of S and Fe compounds in the hyporheic zone also has important implications for organic matter turnover, subsequently impacting CO2 and CH4 (greenhouse gas) emissions into the atmosphere. Some previous studies147–150 suggest that increased sulfate, either from surface sources or regenerated in the subsurface through sulfide oxidation, will suppress methanogenesis and stimulate anaerobic methane oxidation, thereby mitigating greenhouse gas fluxes. With rapid climate change, as warming temperatures stimulate methanogenesis, much uncertainty remains as to how further interactions with Fe and S cycling will mediate impacts on net CH4 fluxes, especially as hydrologic responses to intensified precipitation patterns will alter hyporheic zone mixing in unresolved ways. The complexity and heterogeneity of hyporheic zone hydrobiogeochemical interactions, such as those brought to light in this current study, necessitate further field-based examination. It is clear that advanced field methods, such as in situ geochemical sensors or real-time hydrologic flux monitoring for rapid-response sample collections, as well as higher spatial resolution geochemical analyses and isotopic analyses to capture reaction rates, would be beneficial to fully capture these transient and heterogeneous biogeochemical responses to dynamic hydrologic excursions. These additional methods, however, may still be unable to capture short-term hot moments/spots through ad hoc sampling approaches. Additionally, they can be expensive, not suitable for all environments, or require specialized analytical techniques not accessible to many (e.g., electrochemical sensors). Regardless, our data point to the importance of several enigmatic sediment processes that can be incorporated into current and future modeling efforts151–153 to accurately scale up and make predictions related to regional or global ecosystem, water quality, and climate impacts.
Data availability
Geochemistry data154 is publicly available at the Environmental Data Initiative (EDI) data portal: https://doi.org/10.6073/pasta/2a03c810518255ce147cccb10e92ace5. Hydrologic data155 is publicly available on Hydroshare at https://doi.org/10.4211/hs.cf9090408d1f4b8193e8ce2b7e8ed313.Author contributions
JMT and CER contributed equally to this work and share first authorship of this manuscript. JMT and CER collected field samples, conducted geochemical analyses, performed data analysis and interpretation, and wrote sections of the manuscript. AJD and RS contributed to field sampling and geochemical analysis. KD and PAO conducted field preparation and field research. GHCN and CMS contributed to all aspects of the research and manuscript preparation and devised and supported the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Research was supported by MnDRIVE Environment (CMS) and the College of Science and Engineering (GHCN) from the University of Minnesota-Twin Cities as well as by a United States Department of Energy grant (#DE-SC0019439) to CMS and GHCN. We are grateful for all the assistance with fieldwork, hydrogeology data analysis, and geochemistry analysis from many current and former researchers and students in the Santelli and Ng research groups: Ella Fadely, Jordan Loy, Jacqueline Mejia, Elizabeth Roepke, Mary Sabuda, Amanda Yourd, Alexander Waheed, Tingying Xu, and Christopher Schuler. This research used resources of the Advanced Photon Source, a U.S. Department of Energy Office of Science User Facility operated by Argonne National Laboratory under contract DE-AC02-06CH11357. We thank beamline scientists Carlo Segre, John Katsoudas, Yujia Ding, and Tianpin Wu for assistance with XAS data collection. We are grateful to Brandy Toner for guidance on S XANES analysis and for providing reference standards, as well as to Josh Feinberg for providing Fe EXAFS standards.References
- A. J. Boulton, S. Findlay, P. Marmonier, E. H. Stanley and H. M. Valett, The Functional Significance of the Hyporheic Zone in Streams and Rivers, Annu. Rev. Ecol. Syst., 1998, 29, 59–81 CrossRef.
- J. P. Megonigal and S. C. Neubauer, in Coastal Wetlands, 2019, pp. 641–683, DOI:10.1016/b978-0-444-63893-9.00019-8.
- J. D. Fudyma, R. K. Chu, N. Graf Grachet, J. C. Stegen and M. M. Tfaily, Coupled Biotic-Abiotic Processes Control Biogeochemical Cycling of Dissolved Organic Matter in the Columbia River Hyporheic Zone, Front. Water, 2021, 2, 574692 CrossRef.
- L. Bardini, F. Boano, M. B. Cardenas, R. Revelli and L. Ridolfi, Nutrient cycling in bedform induced hyporheic zones, Geochim. Cosmochim. Acta, 2012, 84, 47–61 CrossRef CAS.
- J. P. Zarnetske, R. Haggerty, S. M. Wondzell and M. A. Baker, Dynamics of nitrate production and removal as a function of residence time in the hyporheic zone, J. Geophys. Res., 2011, 116, G01025 Search PubMed.
- M. A. Baker, H. M. Valett and C. N. Dahm, Organic Carbon Supply and Metabolism in a Shallow Groundwater Ecosystem, Ecology, 2000, 81, 3133–3148 CrossRef.
- M. W. Naegeli and U. Uehlinger, Contribution of the Hyporheic Zone to Ecosystem Metabolism in a Prealpine Gravel-Bed-River, J. North Am. Benthol. Soc., 1997, 16, 794–804 CrossRef.
- A. R. Nelson, A. H. Sawyer, R. S. Gabor, C. M. Saup, S. R. Bryant, K. D. Harris, M. A. Briggs, K. H. Williams and M. J. Wilkins, Heterogeneity in Hyporheic Flow, Pore Water Chemistry, and Microbial Community Composition in an Alpine Streambed, J. Geophys. Res.: Biogeosci., 2019, 124, 3465–3478 CrossRef.
- E. T. Hester, M. B. Cardenas, R. Haggerty and S. V. Apte, The importance and challenge of hyporheic mixing, Water Resour. Res., 2017, 53, 3565–3575 CrossRef.
- F. Boano, J. W. Harvey, A. Marion, A. I. Packman, R. Revelli, L. Ridolfi and A. Worman, Hyporheic flow and transport processes: Mechanisms, models, and biogeochemical implications, Rev. Geophys., 2014, 52, 603–679 CrossRef.
- S. R. Bryant, A. H. Sawyer, M. A. Briggs, C. M. Saup, A. R. Nelson, M. J. Wilkins, J. N. Christensen and K. H. Williams, Seasonal manganese transport in the hyporheic zone of a snowmelt-dominated river (East River, Colorado, USA), Hydrogeol. J., 2020, 28, 1323–1341 CrossRef CAS.
- S. K. Wexler, K. M. Hiscock and P. F. Dennis, Catchment-scale quantification of hyporheic denitrification using an isotopic and solute flux approach, Environ. Sci. Technol., 2011, 45, 3967–3973 CrossRef CAS PubMed.
- J. C. Stegen, T. Johnson, J. K. Fredrickson, M. J. Wilkins, A. E. Konopka, W. C. Nelson, E. V. Arntzen, W. B. Chrisler, R. K. Chu, S. J. Fansler, E. B. Graham, D. W. Kennedy, C. T. Resch, M. Tfaily and J. Zachara, Influences of organic carbon speciation on hyporheic corridor biogeochemistry and microbial ecology, Nat. Commun., 2018, 9, 585 CrossRef PubMed.
- J. C. Stegen, J. K. Fredrickson, M. J. Wilkins, A. E. Konopka, W. C. Nelson, E. V. Arntzen, W. B. Chrisler, R. K. Chu, R. E. Danczak, S. J. Fansler, D. W. Kennedy, C. T. Resch and M. Tfaily, Groundwater-surface water mixing shifts ecological assembly processes and stimulates organic carbon turnover, Nat. Commun., 2016, 7, 11237 CrossRef CAS PubMed.
- J. A. Villa, G. J. Smith, Y. Ju, L. Renteria, J. C. Angle, E. Arntzen, S. F. Harding, H. Ren, X. Chen, A. H. Sawyer, E. B. Graham, J. C. Stegen, K. C. Wrighton and G. Bohrer, Methane and nitrous oxide porewater concentrations and surface fluxes of a regulated river, Sci. Total Environ., 2020, 715, 136920 CrossRef CAS PubMed.
- S. Zhao, B. Zhang, X. Sun and L. Yang, Hot spots and hot moments of nitrogen removal from hyporheic and riparian zones: A review, Sci. Total Environ., 2021, 762, 144168 CrossRef CAS PubMed.
- J. W. Harvey and C. C. Fuller, Effect of enhanced manganese oxidation in the hyporheic zone on basin-scale geochemical mass balance, Water Resour. Res., 1998, 34, 623–636 CrossRef CAS.
- M. H. Kaufman, M. B. Cardenas, J. Buttles, A. J. Kessler and P. L. M. Cook, Hyporheic hot moments: Dissolved oxygen dynamics in the hyporheic zone in response to surface flow perturbations, Water Resour. Res., 2017, 53, 6642–6662 CrossRef CAS.
- R. M. Burrows, H. Rutlidge, N. R. Bond, S. M. Eberhard, A. Auhl, M. S. Andersen, D. G. Valdez and M. J. Kennard, High rates of organic carbon processing in the hyporheic zone of intermittent streams, Sci. Rep., 2017, 7, 13198 CrossRef PubMed.
- F. J. Triska, J. H. Duff and R. J. Avanzino, in Nutrient Dynamics and Retention in Land/Water Ecotones of Lowland, Temperate Lakes and Rivers, ed. A. Hillbricht-Ilkowska and E. Pieczyńska, Springer Netherlands, Dordrecht, 1993, pp. 167–184, DOI:10.1007/978-94-011-1602-2_20.
- W. Dong, A. Bhattacharyya, P. M. Fox, M. Bill, D. Dwivedi, S. Carrero, M. Conrad and P. S. Nico, Geochemical Controls on Release and Speciation of Fe(II) and Mn(II) From Hyporheic Sediments of East River, Colorado, Front. Water, 2020, 2, 562298 CrossRef.
- L. L. Oram, D. G. Strawn, M. J. Morra and G. Moller, Selenium biogeochemical cycling and fluxes in the hyporheic zone of a mining-impacted stream, Environ. Sci. Technol., 2010, 44, 4176–4183 CrossRef CAS PubMed.
- A. Jaeger, M. Posselt, J. L. Schaper, A. Betterle, C. Rutere, C. Coll, J. Mechelke, M. Raza, K. Meinikmann, A. Portmann, P. J. Blaen, M. A. Horn, S. Krause and J. Lewandowski, Transformation of organic micropollutants along hyporheic flow in bedforms of river-simulating flumes, Sci. Rep., 2021, 11, 13034 CrossRef CAS PubMed.
- S. A. Nagorski and J. N. Moore, Arsenic mobilization in the hyporheic zone of a contaminated stream, Water Resour. Res., 1999, 35, 3441–3450 CrossRef CAS.
- G. J. Whiting and J. P. Chanton, Greenhouse carbon balance of wetlands: methane emission versus carbon sequestration, Tellus, 2001, 53, 521–528 Search PubMed.
- Y. Yamashita, B. D. Kloeppel, J. Knoepp, G. L. Zausen and R. Jaffé, Effects of Watershed History on Dissolved Organic Matter Characteristics in Headwater Streams, Ecosystems, 2011, 14, 1110–1122 CrossRef CAS.
- C. M. Bethke, R. a. Sanford, M. F. Kirk, Q. Jin and T. M. Flynn, The thermodynamic ladder in geomicrobiology, Am. J. Sci., 2011, 311, 183–210 CrossRef CAS.
- M. Yücel, Down the thermodynamic ladder: A comparative study of marine redox gradients across diverse sedimentary environments, Estuarine, Coastal Shelf Sci., 2013, 131, 83–92 CrossRef.
- J. T. Crawford, L. C. Loken, W. E. West, B. Crary, S. A. Spawn, N. Gubbins, S. E. Jones, R. G. Striegl and E. H. Stanley, Spatial heterogeneity of within-stream methane concentrations, J. Geophys. Res.: Biogeosci., 2017, 122, 1036–1048 CrossRef CAS.
- J. T. Crawford, N. R. Lottig, E. H. Stanley, J. F. Walker, P. C. Hanson, J. C. Finlay and R. G. Striegl, CO2and CH4emissions from streams in a lake-rich landscape: Patterns, controls, and regional significance, Global Biogeochem. Cycles, 2014, 28, 197–210 CrossRef CAS.
- R. Segers, Methane production and methane consumption: a review of processes underlying wetland methane fluxes, Biogeochemistry, 1998, 41, 23–51 CrossRef CAS.
- D. Dwivedi, C. I. Steefel, B. Arora, M. Newcomer, J. D. Moulton, B. Dafflon, B. Faybishenko, P. Fox, P. Nico, N. Spycher, R. Carroll and K. H. Williams, Geochemical Exports to River From the Intrameander Hyporheic Zone Under Transient Hydrologic Conditions: East River Mountainous Watershed, Colorado, Water Resour. Res., 2018, 54, 8456–8477 CrossRef.
- C. M. Hansel, C. J. Lentini, Y. Tang, D. T. Johnston, S. D. Wankel and P. M. Jardine, Dominance of sulfur-fueled iron oxide reduction in low-sulfate freshwater sediments, ISME J., 2015, 9, 2400–2412 CrossRef CAS PubMed.
- K.-H. Knorr, G. Lischeid and C. Blodau, Dynamics of redox processes in a minerotrophic fen exposed to a water table manipulation, Geoderma, 2009, 153, 379–392 CrossRef CAS.
- C. Blodau, B. Mayer, S. Peiffer and T. R. Moore, Support for an anaerobic sulfur cycle in two Canadian peatland soils, J. Geophys. Res., 2007, 112 Search PubMed.
- T. Hulsen, K. Hsieh and D. J. Batstone, Saline wastewater treatment with purple phototrophic bacteria, Water Res., 2019, 160, 259–267 CrossRef CAS PubMed.
- M. Mohammadi, D. Mowla, F. Esmaeilzadeh and Y. Ghasemi, Enhancement of sulfate removal from the power plant wastewater using cultivation of indigenous microalgae: Stage-wise operation, J. Environ. Chem. Eng., 2019, 7(1), 102870 CrossRef CAS.
- E.-L. S. Hinckley, J. T. Crawford, H. Fakhraei and C. T. Driscoll, A shift in sulfur-cycle manipulation from atmospheric emissions to agricultural additions, Nat. Geosci., 2020, 13, 597–604 CrossRef CAS.
- A. Boetius, K. Ravenschlag, C. J. Schubert, D. Rickert, F. Widdel, A. Gieseke, R. Amann, B. B. Jorgensen, U. Witte and O. Pfannkuche, A marine microbial consortium apparently mediating anaerobic oxidation of methane, Nature, 2000, 407, 623–626 CrossRef CAS PubMed.
- A. Hatzikioseyian, S. Bhattarai, C. Cassarini, G. Esposito and P. N. L. Lens, Dynamic modeling of anaerobic methane oxidation coupled to sulfate reduction: role of elemental sulfur as intermediate, Bioprocess Biosyst. Eng., 2021, 44, 855–874 CrossRef CAS PubMed.
- M. Pester, K. H. Knorr, M. W. Friedrich, M. Wagner and A. Loy, Sulfate-reducing microorganisms in wetlands - fameless actors in carbon cycling and climate change, Front. Microbiol., 2012, 3, 72 CAS.
- M. A. Briggs, F. D. Day-Lewis, J. P. Zarnetske and J. W. Harvey, A physical explanation for the development of redox microzones in hyporheic flow, Geophys. Res. Lett., 2015, 42, 4402–4410 CrossRef.
- A. H. Sawyer, L. A. Kaplan, O. Lazareva and H. A. Michael, Hydrologic dynamics and geochemical responses within a floodplain aquifer and hyporheic zone during Hurricane Sandy, Water Resour. Res., 2014, 50, 4877–4892 CrossRef CAS.
- M. E. McClain, E. W. Boyer, C. L. Dent, S. E. Gergel, N. B. Grimm, P. M. Groffman, S. C. Hart, J. W. Harvey, C. A. Johnston, E. Mayorga, W. H. McDowell and G. Pinay, Biogeochemical Hot Spots and Hot Moments at the Interface of Terrestrial and Aquatic Ecosystems, Ecosystems, 2003, 6, 301–312 CrossRef CAS.
- S. Peiffer, A. Kappler, S. B. Haderlein, C. Schmidt, J. M. Byrne, S. Kleindienst, C. Vogt, H. H. Richnow, M. Obst, L. T. Angenent, C. Bryce, C. McCammon and B. Planer-Friedrich, A biogeochemical–hydrological framework for the role of redox-active compounds in aquatic systems, Nat. Geosci., 2021, 14, 264–272 CrossRef CAS.
- R. E. Danczak, S. B. Yabusaki, K. H. Williams, Y. Fang, C. Hobson and M. J. Wilkins, Snowmelt Induced Hydrologic Perturbations Drive Dynamic Microbiological and Geochemical Behaviors across a Shallow Riparian Aquifer, Front. Earth Sci., 2016, 4, 57 Search PubMed.
- K. Feris, P. Ramsey, C. Frazar, J. Moore, J. Gannon and W. Holben, Differences in hyporheic-zone microbial community structure along a heavy-metal contamination gradient, Appl. Environ. Microbiol., 2003, 69, 5563 CrossRef CAS PubMed.
- S. Krause, M. Meima-Franke, M. M. Hefting and P. L. Bodelier, Spatial patterns of methanotrophic communities along a hydrological gradient in a riparian wetland, FEMS Microbiol. Ecol., 2013, 86, 59–70 CrossRef CAS PubMed.
- P. Vidon, C. Allan, D. Burns, T. P. Duval, N. Gurwick, S. Inamdar, R. Lowrance, J. Okay, D. Scott and S. Sebestyen, Hot Spots and Hot Moments in Riparian Zones: Potential for Improved Water Quality Management, J. Am. Water Resour. Assoc., 2010, 46, 278–298 CrossRef CAS.
- J. P. Zarnetske, R. Haggerty, S. M. Wondzell, V. a. Bokil and R. González-Pinzón, Coupled transport and reaction kinetics control the nitrate source-sink function of hyporheic zones, Water Resour. Res., 2012, 48, 1–15 CrossRef.
- V. Noël, C. Marchand, F. Juillot, G. Ona-Nguema, E. Viollier, G. Marakovic, L. Olivi, L. Delbes, F. Gelebart and G. Morin, EXAFS analysis of iron cycling in mangrove sediments downstream a lateralized ultramafic watershed (Vavouto Bay, New Caledonia), Geochim. Cosmochim. Acta, 2014, 136, 211–228 CrossRef.
- G. H. C. Ng, A. R. Yourd, N. W. Johnson and A. E. Myrbo, Modeling hydrologic controls on sulfur processes in sulfate-impacted wetland and stream sediments, J. Geophys. Res.: Biogeosci., 2017, 122, 2435–2457 CrossRef CAS.
- G. H. C. Ng, C. E. Rosenfeld, C. M. Santelli, A. R. Yourd, J. Lange, K. Duhn and N. W. Johnson, Microbial and Reactive Transport Modeling Evidence for Hyporheic Flux-Driven Cryptic Sulfur Cycling and Anaerobic Methane Oxidation in a Sulfate-Impacted Wetland-Stream System, J. Geophys. Res.: Biogeosci., 2020, 125 Search PubMed.
- N. W. Johnson, C. P. Mitchell, D. R. Engstrom, L. T. Bailey, J. K. Coleman Wasik and M. E. Berndt, Methylmercury production in a chronically sulfate-impacted sub-boreal wetland, Environ. Sci.: Processes Impacts, 2016, 18, 725–734 RSC.
- L. M. Zanko, H. B. Niles and J. A. Oreskovich, Mineralogical and microscopic evaluation of coarse taconite tailings from Minnesota taconite operations, Regul. Toxicol. Pharmacol., 2008, 52, S51–S65 CrossRef CAS PubMed.
- C. M. Koretsky, C. M. Moore, K. L. Lowe, C. Meile, T. J. DiChristina and P. Van Cappellen, Seasonal oscillation of microbial iron and sulfate reduction in saltmarsh sediments (Sapelo Island, GA, USA), Biogeochemistry, 2003, 64, 179–203 CrossRef CAS.
- H. P. Hansen and F. Koroleff, Methods Seawater Anal., 1999, 159–228, DOI:10.1002/9783527613984.ch10.
- J. D. Cline, Spectrophotometric determination of hydrogen sulfide in natural waters, Limnol. Oceanogr., 1969, 14, 454–458 CrossRef CAS.
- L. L. Stookey, Ferrozine-a New Spectrophotometric Reagent for Iron, 1970 Search PubMed.
- E. Viollier, P. W. Inglett, K. Hunter, A. N. Roychoudhury and P. Van Cappelen, The ferrozine method revisited: Fe(II)/Fe(III) determination in natural waters, Appl. Geochem., 2000, 15, 785–790 CrossRef CAS.
- H. E. Allen, G. Fu and B. Deng, Analysis of acid-volatile sulfide (AVS) and simultaneously extracted metals (SEM) for the estimation of potential toxicity in aquatic sediments, Environ. Toxicol. Chem., 1993, 12, 1441–1453 CrossRef CAS.
- A. Myrbo, E. B. Swain, D. R. Engstrom, J. Coleman Wasik, J. Brenner, M. Dykhuizen Shore, E. B. Peters and G. Blaha, Sulfide Generated by Sulfate Reduction is a Primary Controller of the Occurrence of Wild Rice (Zizania palustris) in Shallow Aquatic Ecosystems, J. Geophys. Res.: Biogeosci., 2017, 122, 2736–2753 CrossRef.
- R. D. Thériault and S. J. Barnes, Compositional variations in Cu-Ni-PGE sulfides of the Dunka Road deposit, Duluth Complex, Minnesota : the importance of combined assimilation and magmatic processes, Can. Mineral., 1998, 36, 869–886 Search PubMed.
- B. Arora, M. Burrus, M. Newcomer, C. I. Steefel, R. W. H. Carroll, D. Dwivedi, W. Dong, K. H. Williams and S. S. Hubbard, Differential C-Q Analysis: A New Approach to Inferring Lateral Transport and Hydrologic Transients Within Multiple Reaches of a Mountainous Headwater Catchment, Front. Water, 2020, 2, 24 CrossRef.
- C. J. Ocampo, C. E. Oldham and M. Sivapalan, Nitrate Attenuation in Agricultural Catchments: Shifting balances between transport and reaction, Water Resour. Res., 2006, 42, W01408 CrossRef.
- C. Gu, G. M. Hornberger, A. L. Mills, J. S. Herman and S. A. Flewelling, Nitrate reduction in streambed sediments: Effects of flow and biogeochemical kinetics, Water Resour. Res., 2007, 43, W12413 CrossRef.
- J. W. Harvey, J. K. Böhlke, M. A. Voytek, D. Scott and C. R. Tobias, Hyporheic zone denitrification: Controls on effective reaction depth and contribution to whole-stream mass balance, Water Resour. Res., 2013, 49, 6298–6316 CrossRef CAS.
- B. Arora, D. Dwivedi, S. S. Hubbard, C. I. Steefel and K. H. Williams, Identifying geochemical hot moments and their controls on a contaminated river floodplain system using wavelet and entropy approaches, Environ. Model. Software, 2016, 85, 27–41 CrossRef.
- A. Stockdale, W. Davison and H. Zhang, Micro-scale biogeochemical heterogeneity in sediments: A review of available technology and observed evidence, Earth-Sci. Rev., 2009, 92, 81–97 CrossRef.
- A. Bhattacharyya, M. P. Schmidt, E. Stavitski and C. E. Martínez, Iron speciation in peats: Chemical and spectroscopic evidence for the co-occurrence of ferric and ferrous iron in organic complexes and mineral precipitates, Org. Geochem., 2018, 115, 124–137 CrossRef CAS.
- B. M. Toner, S. C. Fakra, S. J. Manganini, C. M. Santelli, M. A. Marcus, J. Moffett, O. Rouxel, C. R. German and K. J. Edwards, Preservation of iron(II) by carbon-rich matrices in a hydrothermal plume, Nat. Geosci., 2009, 2, 197–201 CrossRef CAS.
- M. E. Jones, J. S. Beckler and M. Taillefert, The flux of soluble organic-iron(III) complexes from sediments represents a source of stable iron(III) to estuarine waters and to the continental shelf, Limnol. Oceanogr., 2011, 56, 1811–1823 CrossRef CAS.
- G. W. Luther, P. A. Shellenbarger and P. J. Brendel, Dissolved organic Fe(III) and Fe(II) complexes in salt marsh porewaters, Geochim. Cosmochim. Acta, 1996, 60, 951–960 CrossRef CAS.
- J. A. Hakala, R. L. Fimmen, Y.-P. Chin, S. G. Agrawal and C. P. Ward, Assessment of the geochemical reactivity of Fe-DOM complexes in wetland sediment pore waters using a nitroaromatic probe compound, Geochim. Cosmochim. Acta, 2009, 73, 1382–1393 CrossRef CAS.
- X. Wan, W. Xiang, N. Wan, S. Yan, Z. Bao and Y. Wang, Complexation and reduction of iron by phenolic substances: Implications for transport of dissolved Fe from peatlands to aquatic ecosystems and global iron cycling, Chem. Geol., 2018, 498, 128–138 CrossRef CAS.
- M. Taillefert, A. B. Bono and G. W. Luther, Reactivity of Freshly Formed Fe(III) in Synthetic Solutions and (Pore)Waters: Voltammetric Evidence of an Aging Process, Environ. Sci. Technol., 2000, 34, 2169–2177 CrossRef CAS.
- W. Stumm, in Aquatic Chemistry, American Chemical Society, 1995, vol. 244, ch. 1, pp. 1–32 Search PubMed.
- A. L. Rose and T. D. Waite, Kinetics of iron complexation by dissolved natural organic matter in coastal waters, Mar. Chem., 2003, 84, 85–103 CrossRef CAS.
- C. Peng, C. Bryce, A. Sundman and A. Kappler, Cryptic Cycling of Complexes Containing Fe(III) and Organic Matter by Phototrophic Fe(II)-Oxidizing Bacteria, Appl. Environ. Microbiol., 2019, 85, e02826-18 CrossRef PubMed.
- K. Küsel, M. Blöthe, D. Schulz, M. Reiche and H. L. Drake, Microbial reduction of iron and porewater biogeochemistry in acidic peatlands, Biogeosciences, 2008, 5, 1537–1549 CrossRef.
- A. Pellerin, G. Antler, H. Røy, A. Findlay, F. Beulig, C. Scholze, A. V. Turchyn and B. B. Jørgensen, The sulfur cycle below the sulfate-methane transition of marine sediments, Geochim. Cosmochim. Acta, 2018, 239, 74–89 CrossRef CAS.
- B. B. Jorgensen, A. J. Findlay and A. Pellerin, The Biogeochemical Sulfur Cycle of Marine Sediments, Front. Microbiol., 2019, 10, 849 CrossRef PubMed.
- L. Holmkvist, T. G. Ferdelman and B. B. Jorgensen, A cryptic sulfur cycle driven by iron in the methane zone of marine sediment (Aarhus Bay, Denmark), Geochim. Cosmochim. Acta, 2011, 75, 3581–3599 CrossRef CAS.
- M. Wan, A. Shchukarev, R. Lohmayer, B. Planer-Friedrich and S. Peiffer, Occurrence of surface polysulfides during the interaction between ferric (hydr)oxides and aqueous sulfide, Environ. Sci. Technol., 2014, 48, 5076–5084 CrossRef CAS PubMed.
- A. J. Findlay and A. Kamyshny, Turnover Rates of Intermediate Sulfur Species ([Formula: see text], S(0), S2[Formula: see text], S4[Formula: see text], [Formula: see text]) in Anoxic Freshwater and Sediments, Front. Microbiol., 2017, 8, 2551 CrossRef PubMed.
- A. J. Findlay, Microbial impact on polysulfide dynamics in the environment, FEMS Microbiol. Lett., 2016, 363(11), fnw103 CrossRef PubMed.
- C. G. Friedrich, F. Bardischewsky, D. Rother, A. Quentmeier and J. Fischer, Prokaryotic sulfur oxidation, Curr. Opin. Microbiol., 2005, 8, 253–259 CrossRef CAS PubMed.
- T. Zhang, T. S. Bain, M. A. Barlett, S. A. Dar, O. L. Snoeyenbos-West, K. P. Nevin and D. R. Lovley, Sulfur oxidation to sulfate coupled with electron transfer to electrodes by Desulfuromonas strain TZ1, Microbiology, 2014, 160, 123–129 CrossRef CAS PubMed.
- J. Milucka, T. G. Ferdelman, L. Polerecky, D. Franzke, G. Wegener, M. Schmid, I. Lieberwirth, M. Wagner, F. Widdel and M. M. Kuypers, Zero-valent sulphur is a key intermediate in marine methane oxidation, Nature, 2012, 491, 541–546 CrossRef CAS PubMed.
- K. Finster, W. Liesack and B. Thamdrup, Elemental sulfur and thiosulfate disproportionation by Desulfocapsa sulfoexigens sp. nov., a new anaerobic bacterium isolated from marine surface sediment, Appl. Environ. Microbiol., 1998, 64, 119–125 CrossRef CAS PubMed.
- S. W. Poulton, Sulfide oxidation and iron dissolution kinetics during the reaction of dissolved sulfide with ferrihydrite, Chem. Geol., 2003, 202, 79–94 CrossRef CAS.
- S. W. Poulton, M. D. Krom and R. Raiswell, A revised scheme for the reactivity of iron (oxyhydr)oxide minerals towards dissolved sulfide, Geochim. Cosmochim. Acta, 2004, 68, 3703–3715 CrossRef CAS.
- M. Dos Santos Afonso and W. Stumm, Reductive dissolution of iron(III) (hydr)oxides by hydrogen sulfide, Langmuir, 1992, 8, 1671–1675 CrossRef CAS.
- M. Taillefert, V. C. Hover, T. F. Rozan, S. M. Theberge and G. W. Luther, The influence of sulfides on soluble organic-Fe(III) in anoxic sediment porewaters, Estuaries, 2002, 25, 1088–1096 CrossRef CAS.
- T. Heitmann and C. Blodau, Oxidation and incorporation of hydrogen sulfide by dissolved organic matter, Chem. Geol., 2006, 235, 12–20 CrossRef CAS.
- Z. G. Yu, S. Peiffer, J. Gottlicher and K. H. Knorr, Electron transfer budgets and kinetics of abiotic oxidation and incorporation of aqueous sulfide by dissolved organic matter, Environ. Sci. Technol., 2015, 49, 5441–5449 CrossRef CAS PubMed.
- W. Yao and F. J. Millero, The rate of sulfide oxidation by δMnO2 in seawater, Geochim. Cosmochim. Acta, 1993, 57, 3359–3365 CrossRef CAS.
- M. E. Böttcher and B. Thamdrup, Anaerobic sulfide oxidation and stable isotope fractionation associated with bacterial sulfur disproportionation in the presence of MnO2, Geochim. Cosmochim. Acta, 2001, 65, 1573–1581 CrossRef.
- A. Kamyshny and T. G. Ferdelman, Dynamics of zero-valent sulfur species including polysulfides at seep sites on intertidal sand flats (Wadden Sea, North Sea), Mar. Chem., 2010, 121, 17–26 CrossRef CAS.
- E. Henneke, G. W. Luther, G. J. De Lange and J. Hoefs, Sulphur speciation in anoxic hypersaline sediments from the eastern Mediterranean Sea, Geochim. Cosmochim. Acta, 1997, 61, 307–321 CrossRef CAS.
- G. E. Lau, J. Cosmidis, S. E. Grasby, C. B. Trivedi, J. R. Spear and A. S. Templeton, Low-temperature formation and stabilization of rare allotropes of cyclooctasulfur (β-S8 and γ-S8) in the presence of organic carbon at a sulfur-rich glacial site in the Canadian High Arctic, Geochim. Cosmochim. Acta, 2017, 200, 218–231 CrossRef CAS.
- J. Cosmidis, C. W. Nims, D. Diercks and A. S. Templeton, Formation and stabilization of elemental sulfur through organomineralization, Geochim. Cosmochim. Acta, 2019, 247, 59–82 CrossRef CAS.
- B. Cron, J. L. Macalady and J. Cosmidis, Organic Stabilization of Extracellular Elemental Sulfur in a Sulfurovum-Rich Biofilm: A New Role for Extracellular Polymeric Substances?, Front. Microbiol., 2021, 12, 720101 CrossRef PubMed.
- M. A. Vairavamurthy, D. Maletic, S. Wang, B. Manowitz, T. Eglinton and T. Lyons, Characterization of Sulfur-Containing Functional Groups in Sedimentary Humic Substances by X-ray Absorption Near-Edge Structure Spectroscopy, Energy Fuels, 1997, 11, 546–553 CrossRef CAS.
- T. G. Ferdelman, T. M. Church and G. W. Luther, Sulfur enrichment of humic substances in a Delaware salt marsh sediment core, Geochim. Cosmochim. Acta, 1991, 55, 979–988 CrossRef CAS.
- D. Rickard and J. W. Morse, Acid volatile sulfide (AVS), Mar. Chem., 2005, 97, 141–197 CrossRef CAS.
- G. W. Luther, Acid volatile sulfide—a comment, Mar. Chem., 2005, 97, 198–205 CrossRef CAS.
- A. Kappler, M. Benz, B. Schink and A. Brune, Electron shuttling via humic acids in microbial iron(III) reduction in a freshwater sediment, FEMS Microbiol. Ecol., 2004, 47, 85–92 CrossRef CAS PubMed.
- D. R. Lovley, J. L. Fraga, J. D. Coates and E. L. Blunt-Harris, Humics as an electron donor for anaerobic respiration, Environ. Microbiol., 1999, 1, 89–98 CrossRef CAS PubMed.
- D. B. Rogers, M. E. Newcomer, J. H. Raberg, D. Dwivedi, C. Steefel, N. Bouskill, P. Nico, B. Faybishenko, P. Fox, M. Conrad, M. Bill, E. Brodie, B. Arora, B. Dafflon, K. H. Williams and S. S. Hubbard, Modeling the Impact of Riparian Hollows on River Corridor Nitrogen Exports, Front. Water, 2021, 3 Search PubMed.
- G. M. Filippelli and M. L. Delaney, Phosphorus geochemistry of equatorial Pacific sediments, Geochim. Cosmochim. Acta, 1996, 60, 1479–1495 CrossRef CAS.
- S. Ding, Y. Wang, D. Wang, Y. Y. Li, M. Gong and C. Zhang, In situ, high-resolution evidence for iron-coupled mobilization of phosphorus in sediments, Sci. Rep., 2016, 6, 24341 CrossRef CAS PubMed.
- E. Rydin, Potentially mobile phosphorus in Lake Erken sediment, Water Res., 2000, 34, 2037–2042 CrossRef CAS.
- S. R. Joshi, R. K. Kukkadapu, D. J. Burdige, M. E. Bowden, D. L. Sparks and D. P. Jaisi, Organic matter remineralization predominates phosphorus cycling in the mid-Bay sediments in the Chesapeake Bay, Environ. Sci. Technol., 2015, 49, 5887–5896 CrossRef CAS PubMed.
- J. E. Corbett, M. M. Tfaily, D. J. Burdige, P. H. Glaser and J. P. Chanton, The relative importance of methanogenesis in the decomposition of organic matter in northern peatlands, J. Geophys. Res.: Biogeosci., 2015, 120, 280–293 CrossRef CAS.
- M. Metje and P. Frenzel, Methanogenesis and methanogenic pathways in a peat from subarctic permafrost, Environ. Microbiol., 2007, 9, 954–964 CrossRef CAS PubMed.
- S. Das and T. K. Adhya, Dynamics of methanogenesis and methanotrophy in tropical paddy soils as influenced by elevated CO2 and temperature interaction, Soil Biol. Biochem., 2012, 47, 36–45 CrossRef CAS.
- L. I. Blake, A. Tveit, L. Ovreas, I. M. Head and N. D. Gray, Response of Methanogens in Arctic Sediments to Temperature and Methanogenic Substrate Availability, PLoS One, 2015, 10, e0129733 CrossRef PubMed.
- L. Fu, T. Song and Y. Lu, Snapshot of methanogen sensitivity to temperature in Zoige wetland from Tibetan plateau, Front. Microbiol., 2015, 6, 131 CrossRef PubMed.
- S. He, S. A. Malfatti, J. W. McFarland, F. E. Anderson, A. Pati, M. Huntemann, J. Tremblay, T. Glavina del Rio, M. P. Waldrop, L. Windham-Myers and S. G. Tringe, Patterns in wetland microbial community composition and functional gene repertoire associated with methane emissions, mBio, 2015, 6, e00066-15 CrossRef PubMed.
- K. L. Denman, G. Brasseur, A. Chidthaisong, P. Ciais, P. M. Cox, R. E. Dickinson, D. Hauglustaine, C. Heinze, E. Holland, D. Jacob, U. Lohmann, S. Ramachandran, P. L. da Silva Dias, S. C. Wofsy and X. Zhang, in Climate Change 2007: the Physical Science Basis. Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change, ed. S. Solomon, D. Qin, M. Manning, Z. Chen, M. Marquis, K. B. Averyt, M. Tignor and H. L. Miller, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2007 Search PubMed.
- S. Saarnio, W. Winiwarter and J. Leitão, Methane release from wetlands and watercourses in Europe, Atmos. Environ., 2009, 43, 1421–1429 CrossRef CAS.
- P. Dalcin Martins, D. W. Hoyt, S. Bansal, C. T. Mills, M. Tfaily, B. A. Tangen, R. G. Finocchiaro, M. D. Johnston, B. C. McAdams, M. J. Solensky, G. J. Smith, Y.-P. Chin and M. J. Wilkins, Abundant carbon substrates drive extremely high sulfate reduction rates and methane fluxes in Prairie Pothole Wetlands, Global Change Biol., 2017, 23, 3107–3120 CrossRef PubMed.
- M. Sela-Adler, Z. Ronen, B. Herut, G. Antler, H. Vigderovich, W. Eckert and O. Sivan, Co-existence of Methanogenesis and Sulfate Reduction with Common Substrates in Sulfate-Rich Estuarine Sediments, Front. Microbiol., 2017, 8, 766 CrossRef PubMed.
- R. S. Oremland and B. F. Taylor, Sulfate reduction and methanogenesis in marine sediments, Geochim. Cosmochim. Acta, 1978, 42, 209–214 CrossRef CAS.
- K. M. Walter Anthony and P. Anthony, Constraining spatial variability of methane ebullition seeps in thermokarst lakes using point process models, J. Geophys. Res.: Biogeosci., 2013, 118, 1015–1034 CrossRef.
- K. M. Walter, L. C. Smith and F. S. Chapin III, Methane bubbling from northern lakes: present and future contributions to the global methane budget, Philos. Trans. Royal Soc., 2007, 365, 1657–1676 CAS.
- S. Greene, K. M. Walter Anthony, D. Archer, A. Sepulveda-Jauregui and K. Martinez-Cruz, Modeling the impediment of methane ebullition bubbles by seasonal lake ice, Biogeosciences, 2014, 11, 6791–6811 CrossRef.
- S. C. Whalen, Biogeochemistry of Methane Exchange between Natural Wetlands and the Atmosphere, Environ. Eng. Sci., 2005, 22, 73–94 CrossRef CAS.
- T. R. Chowdhury and R. P. Dick, Ecology of aerobic methanotrophs in controlling methane fluxes from wetlands, Appl. Soil Ecol., 2013, 65, 8–22 CrossRef.
- Y. Cai, Y. Zheng, P. L. Bodelier, R. Conrad and Z. Jia, Conventional methanotrophs are responsible for atmospheric methane oxidation in paddy soils, Nat. Commun., 2016, 7, 11728 CrossRef CAS PubMed.
- K. E. Segarra, F. Schubotz, V. Samarkin, M. Y. Yoshinaga, K. U. Hinrichs and S. B. Joye, High rates of anaerobic methane oxidation in freshwater wetlands reduce potential atmospheric methane emissions, Nat. Commun., 2015, 6, 7477 CrossRef CAS PubMed.
- O. Sivan, G. Antler, A. V. Turchyn, J. J. Marlow and V. J. Orphan, Iron oxides stimulate sulfate-driven anaerobic methane oxidation in seeps, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E4139–E4147 CrossRef CAS PubMed.
- E. J. Beal, C. H. House and V. J. Orphan, Manganese- and iron-dependent marine methane oxidation, Science, 2009, 325, 184–187 CrossRef CAS PubMed.
- R. T. Amos, B. A. Bekins, I. M. Cozzarelli, M. A. Voytek, J. D. Kirshtein, E. J. Jones and D. W. Blowes, Evidence for iron-mediated anaerobic methane oxidation in a crude oil-contaminated aquifer, Geobiology, 2012, 10, 506–517 CrossRef CAS PubMed.
- D. E. Larowe, A. W. Dale and P. Regnier, A thermodynamic analysis of the anaerobic oxidation of methane in marine sediments, Geobiology, 2008, 6, 436–449 CrossRef CAS PubMed.
- Y. N. Bai, X. N. Wang, J. Wu, Y. Z. Lu, L. Fu, F. Zhang, T. C. Lau and R. J. Zeng, Humic substances as electron acceptors for anaerobic oxidation of methane driven by ANME-2d, Water Res., 2019, 164, 114935 CrossRef CAS PubMed.
- P. H. Timmers, D. A. Suarez-Zuluaga, M. van Rossem, M. Diender, A. J. Stams and C. M. Plugge, Anaerobic oxidation of methane associated with sulfate reduction in a natural freshwater gas source, ISME J., 2016, 10, 1400–1412 CrossRef CAS PubMed.
- H. S. Weber, K. S. Habicht and B. Thamdrup, Anaerobic Methanotrophic Archaea of the ANME-2d Cluster Are Active in a Low-sulfate, Iron-rich Freshwater Sediment, Front. Microbiol., 2017, 8, 619 Search PubMed.
- P. B. Matheus Carnevali, A. Lavy, A. D. Thomas, A. Crits-Christoph, S. Diamond, R. Meheust, M. R. Olm, A. Sharrar, S. Lei, W. Dong, N. Falco, N. Bouskill, M. E. Newcomer, P. Nico, H. Wainwright, D. Dwivedi, K. H. Williams, S. Hubbard and J. F. Banfield, Meanders as a scaling motif for understanding of floodplain soil microbiome and biogeochemical potential at the watershed scale, Microbiome, 2021, 9, 121 CrossRef CAS PubMed.
- J. Chen, A. Hanke, H. E. Tegetmeyer, I. Kattelmann, R. Sharma, E. Hamann, T. Hargesheimer, B. Kraft, S. Lenk, J. S. Geelhoed, R. L. Hettich and M. Strous, Impacts of chemical gradients on microbial community structure, ISME J., 2017, 11, 920–931 CrossRef CAS PubMed.
- J. A. Lynch, V. C. Bowersox and J. W. Grimm, Acid Rain Reduced in Eastern United States, Environ. Sci. Technol., 2000, 34, 940–949 CrossRef CAS.
- U. Dayan and D. Lamb, Influences of atmospheric circulation on the variability of wet sulfate deposition, Int. J. Climatol., 2008, 28, 1315–1324 CrossRef.
- Y. Chen, Y. Wen, Q. Zhou, J. Huang, J. Vymazal and P. Kuschk, Sulfate removal and sulfur transformation in constructed wetlands: The roles of filling material and plant biomass, Water Res., 2016, 102, 572–581 CrossRef CAS PubMed.
- W. Guo, A. R. Cecchetti, Y. Wen, Q. Zhou and D. L. Sedlak, Sulfur Cycle in a Wetland Microcosm: Extended (34)S-Stable Isotope Analysis and Mass Balance, Environ. Sci. Technol., 2020, 54, 5498–5508 CrossRef CAS PubMed.
- S. Wu, P. Kuschk, A. Wiessner, J. Müller, R. A. B. Saad and R. Dong, Sulphur transformations in constructed wetlands for wastewater treatment: A review, Ecol. Eng., 2013, 52, 278–289 CrossRef.
- V. Gauci, E. Matthews, N. Dise, B. Walter, D. Koch, G. Granberg and M. Vile, Sulfur pollution suppression of the wetland methane source in the 20th and 21st centuries, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 12583–12587 CrossRef CAS PubMed.
- V. Gauci, N. Dise and D. Fowler, Controls on suppression of methane flux from a peat bog subjected to simulated acid rain sulfate deposition, Global Biogeochem. Cycles, 2002, 16, 4-1–4-12 CrossRef.
- N. B. Dise and E. S. Verry, Suppression of Peatland Methane Emission by Cumulative Sulfate Deposition in Simulated Acid Rain, Biogeochemistry, 2001, 53, 143–160 CrossRef CAS.
- E. Rejmankova and R. A. Post, Methane in Sulfate-Rich and Sulfate-Poor Wetland Sediments, Biogeochemistry, 1996, 34, 57–70 CrossRef CAS.
- S. S. Hubbard, C. Varadharajan, Y. Wu, H. Wainwright and D. Dwivedi, Emerging technologies and radical collaboration to advance predictive understanding of watershed hydrobiogeochemistry, Hydrol. Process., 2020, 34, 3175–3182 CrossRef.
- L. Li, K. Maher, A. Navarre-Sitchler, J. Druhan, C. Meile, C. Lawrence, J. Moore, J. Perdrial, P. Sullivan, A. Thompson, L. Jin, E. W. Bolton, S. L. Brantley, W. E. Dietrich, K. U. Mayer, C. I. Steefel, A. Valocchi, J. Zachara, B. Kocar, J. McIntosh, B. M. Tutolo, M. Kumar, E. Sonnenthal, C. Bao and J. Beisman, Expanding the role of reactive transport models in critical zone processes, Earth-Sci. Rev., 2017, 165, 280–301 CrossRef CAS.
- X. Chen, R. M. Lee, D. Dwivedi, K. Son, Y. Fang, X. Zhang, E. Graham, J. Stegen, J. B. Fisher, D. Moulton and T. D. Scheibe, Integrating field observations and process-based modeling to predict watershed water quality under environmental perturbations, J. Hydrol., 2021, 602, 125762 CrossRef.
- C. E. Rosenfeld, J. M. Torgeson, A. J. Dunshee, K. D. Duhn, R. Schmitter, P. A. O'Hara, G. C. Ng and C. M. Santelli, Summer 2017 porewater and sediment geochemistry data at Second Creek, a sulfate-impacted riparian wetland in northeast Minnesota ver 1, Environmental Data Initiative, 2022, https://doi.org/10.6073/pasta/2a03c810518255ce147cccb10e92ace5 Search PubMed.
- G. C. Ng, P. O'Hara, J. Torgeson, C. E. Rosenfeld, A. Dunshee, K. Duhn, C. Santelli, E. Fadely and A. R. Yourd, Hydraulic Head Data for Second Creek, Minnesota, Summer 2017, Hydroshare, USA, 2022, DOI:10.4211/hs.cf9090408d1f4b8193e8ce2b7e8ed313.
Footnotes |
| † Special invited issue on “Biogeochemistry of the Trace Elements”. |
| ‡ Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2em00024e |
| § Co-first authorship: J. M. Torgeson and C. E. Rosenfeld contributed equally to this manuscript. |
| ¶ Present Address: Pacific Northwest National Laboratory, Richland, Washington 99352, USA. |
| || Present Address: Water Resources Science, University of Minnesota, Duluth, MN. |
| This journal is © The Royal Society of Chemistry 2022 |