
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Steps towards a nature inspired inorganic crystal engineering
Fabrizia
Grepioni
 *a,
Lucia
Casali
a,
Cecilia
Fiore
a,
Luca
Mazzei
b,
Renren
Sun
ac,
Oleksii
Shemchuk
d and
Dario
Braga
*a
*a,
Lucia
Casali
a,
Cecilia
Fiore
a,
Luca
Mazzei
b,
Renren
Sun
ac,
Oleksii
Shemchuk
d and
Dario
Braga
*a
aDipartimento di Chimica “Giacomo Ciamician”, Università di Bologna, Via Selmi 2, 40126 Bologna, Italy. E-mail: fabrizia.grepioni@unibo.it
bLaboratory of Bioinorganic Chemistry, Department of Pharmacy and Biotechnology, University of Bologna, Viale Giuseppe Fanin 40, 40127 Bologna, Italy
cSchool of Chemical Engineering, Zhengzhou University, 450001, Zhengzou, Henan Province, The People's Republic of China
dInstitute of Condensed Matter and Nanosciences, UCLouvain, 1 Place Louis Pasteur, B-1348, Belgium
First published on 20th April 2022
Abstract
This Perspective outlines the results obtained at the University of Bologna by applying crystal engineering strategies to develop nature inspired organic–inorganic materials to tackle challenges in the health and environment sectors. It is shown by means of a number of examples that co-crystallization of inorganic salts, such as alkali and transition metal halides, with organic compounds, such as amino acids, urea, thiourea and quaternary ammonium salts, can be successfully used for (i) chiral resolution and conglomerate formation from racemic compounds, (ii) inhibition of soil enzyme activity in order to reduce urea decomposition and environmental pollution, and (iii) preparation of novel agents to tackle antimicrobial resistance. All materials described in this Perspective have been obtained by mechanochemical solvent-free or slurry methods and characterized by solid state techniques. The fundamental idea is that a crystal engineering approach based on the choice of intermolecular interactions (coordination and hydrogen bonds) between organic and inorganic compounds allows obtaining materials with collective properties that are different, and often very much superior to those of the separate components. It is also demonstrated that the success of this strategy depends crucially on cross-disciplinary synergistic exchange with expert scientists in the areas of bioinorganics, microbiology, and chirality application-oriented developments of these novel materials.
 Dario Braga, Fabrizia Grepioni, Lucia Casali, Cecilia Fiore, Renren Sun, Oleksii Shemchuk and Luca Mazzei (from left to right). | Prof. Dario Braga and Prof. Fabrizia Grepioni lead the group of Molecular Crystal Engineering at the University of Bologna. The research of the group focuses on crystal engineering applied to multiple crystal forms (molecular and ionic co-crystals, polymorphs, solvates, inorganic/organic metal complexes) to tackle pharmaceutical, agrochemical and environmental issues related to solid-state chemistry. Dr Lucia Casali (PostDoc), Cecilia Fiore (PhD student) and Renren Sun (visiting PhD student) are all active members. Dr Oleksii Shemchuk has been part of the group for six years, and is currently an FNRS postdoctoral researcher at the Institute of Condensed Matter and Nanosciences, University of Louvain-la-Neuve, working on chiral systems and chiral resolution in the Molecular Chemistry, Materials and Catalysis group of Prof. Tom Leyssens. Dr Luca Mazzei is a Junior assistant professor at the Department of Pharmacy and Biotechnology, University of Bologna, working in the group of Prof. Stefano Ciurli in the Bioinorganic Chemistry field; his research involves characterization of metal-dependent proteins responsible for medical and agro-environmental issues, including the elucidation of their structure–function relationships. |
Introduction
Crystal engineering has been defined as the synthesis of functional solid-state structures from neutral or ionic building blocks, using intermolecular interactions in the design strategy.1 It is a dynamic discipline in continuous, rapid evolution, being a way to approach solid state issues, rather than an ensemble of techniques, a sequence of steps, a recipe or a computational model. Actually, modern crystal engineering is all of this together and allows tackling problems in many diverse fields of application/utilization of solid materials.2,3There is a vast literature available to the reader interested in revisiting the evolutionary steps of the discipline, an ad hoc entry point being the landmark 1989 book by Gautam Desiraju “Crystal Engineering: The Design of Molecular Solid”.4
Crystal engineering, though born in the organic chemistry field, rapidly expanded across all sub-fields of chemistry, as well as outside the domain of chemistry.5,6 Here we focus on some developments in the inorganic chemistry area. An interesting entry point in time is represented by the Dalton Discussion on “Inorganic Crystal Engineering” held at the University of Bologna in 2000.7 The meeting saw a great variety of contributions on coordination polymers, metal organic frameworks, networks, molecular complexes and polymorphs. The working definition of inorganic crystal engineering was “modelling, synthesis and evaluation of the properties of crystalline materials obtained from inorganic, organometallic and bioinorganic building blocks”.
In two decades the field has expanded beyond expectations. The success is certainly due to the enormous possibilities of innovation generated by the hybridization of the supramolecular approach (the chemistry beyond the molecule)8 with the utilitarian objectives of materials chemistry, in order to tackle high impact issues of our times, as environmental cleansing,9 gas storage,10 CO2 trapping,11 new drug delivery systems,12 and more efficient catalysts,13 among others.
The progress in experimental and computational techniques and the almost combinatorial possibilities offered by the convolution of transition metal properties (coordination geometry, charge, spin, etc.) with the library of organic ligands, each with its own specific supramolecular bonding capacities, were also responsible for the expansion of the area.
A fundamental evolutionary step took place in the area of coordination chemistry with the substitution of “convergent” ligand polydentation on a same metal, so typical of 0-D coordination complexes, by “divergent” polydentation on different metal atoms, which led to the development of coordination polymers, coordination networks and metal organic frameworks (see Fig. 1).14
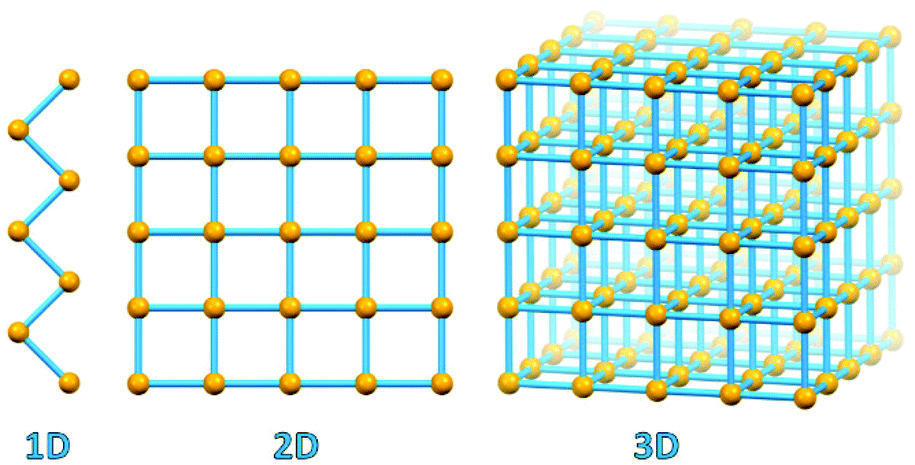 | ||
| Fig. 1 1D coordination polymer, 2D coordination network and 3D MOF geometries. Metal-based nodes in yellow, connectors/polydentate ligands in light blue. | ||
The quest for porous materials for gas uptake/trapping/release, etc. was and is one of the main drivers for the interest in MOFs.10,11,15,16
More recently, research in this direction was paralleled by the systematic preparation of analogous porous compounds obtained from organic building blocks capable of 1-, 2- and 3-strong hydrogen bonding interactions. Organic hydrogen bonded frameworks (HOFs), analogous to MOFs, are being investigated.17,18
In terms of relevance and impact in the field, the counterpart of porous materials, whether based on organic or metallo-organic building blocks, is certainly represented by co-crystals.19 Co-crystals engineering relies on the use of supramolecular interactions (hydrogen bonds,20 halogen and chalcogen bonds,21 weak π-interactions22etc.) to aggregate in the solid state two or more molecular components so as to attain collective properties that the separate components do not possess or to alter specific physico-chemical properties (solubility, dissolution rate, melting etc.) of an active ingredient by association with a coformer.23,24
Crystal engineering involving co-crystals owns its unquestionable success to the possibility of applying this approach to a great variety of crystalline aggregates, such as drugs,25 agrochemicals,26 fertilizers,27 food ingredients,28 organic semiconductors,29 and energetic materials,30 among others. In this respect, if coordination bonds are included (see Fig. 2) in the portfolio of supramolecular interactions available to devise crystalline materials, with collective properties resulting from the convolution of those of metal complexes and those of organic molecules, the distinction between coordination compounds and co-crystals becomes a matter of context.
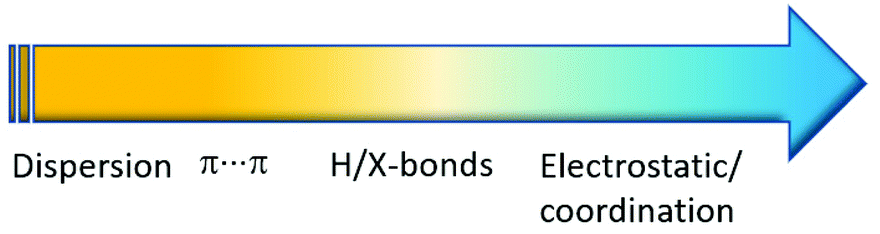 | ||
| Fig. 2 Interactions and corresponding energies (increasing from yellow to blue) at work between neutral molecules and ions in a supramolecular context. | ||
For this reason, in this Perspective, as in other related studies, the term co-crystallization has been used purposely to underline the fact that crystalline materials with specific application-oriented properties have been prepared by direct mixing of compounds that separately form stable crystalline phases at ambient conditions, which, incidentally, is an operative definition of co-crystals.19c It will also be seen that the preparative methods are mainly based on solvent-free mechanochemical approaches.31 Direct mixing of the solid reactants has been amply demonstrated to be well suited for the preparation of co-crystals whether these are molecular co-crystals (i.e. neutral molecules interacting in the solid state only via non-covalent bonds), ionic co-crystals (i.e. co-crystals formed by inorganic salts and neutral organic molecules) or complexes (formed via co-crystallization of organic molecules with transition metal salts).
Attention will be focused on the utilization of the principles outlined above towards three specific areas of nature inspired crystal engineering, namely chiral resolution of amino acids and drugs, soil enzyme inhibition and antimicrobial activity enhancement via co-crystallization. All results discussed herein originate from the collaborations between the molecular crystal engineering group of the University of Bologna and other groups of scientists in Italy and abroad and depend critically on synergistic interactions between different branches of science.
Co-crystallization with inorganic salts and chiral resolution
Chiral resolution of enantiomeric pairs is one of the “perpetual” challenges of applied chemistry. This is not only because chiral molecules are ubiquitous in nature but also, and very importantly, because enantiomers of a biologically active compound may exert very different pharmacological activity. Regulatory authorities, such as the European Medicines Agency (EMA) and the Food and Drug Administration (FDA), require independent pharmacological tests for each enantiomer, as well as an assessment of their combined effects, before authorizing administration of active principles in the form of racemic mixtures or compounds.32 Hence, methods to separate enantiomers are of great interest, and their applications are not limited to the pharmaceutical field.As mentioned above, co-crystallization of active pharmaceutical ingredients and/or molecules of pharmaceutical interest has proven useful to modify the physicochemical properties of a target molecule.25–28 One such application of the co-crystallization approach can indeed be in the direction of separating enantiomers in racemic compounds. To this goal, crystal engineering has introduced new approaches for chiral separation in addition to commonly used methods (e.g. chiral chromatography,33 kinetic resolution,34 diastereoisomeric salt formation,35 and preferential crystallization36,37). These approaches are based on (i) chiral resolution via enantiospecific co-crystal formation,38–41i.e. co-crystallization of a racemic compound with either an enantiopure coformer, and (ii) the use of an achiral co-crystallizing agent, whereby the role of co-crystallization is that of transforming a racemic compound into a conglomerate.42,43
Somewhat serendipitously we have found a third way to utilize achiral coformers to obtain chiral resolution and conglomerate formation starting from a racemic compound.44 In the course of the preparation of ionic co-crystals45 of the racemic amino acid histidine with lithium halides LiX (X = Cl, Br, I) we have observed that the Li+ cation selectively coordinates molecules of the same handedness, regardless of whether the crystalline product is racemic or a conglomerate.46 Depending on the type of halide, the crystalline materials may result in either a racemic crystal but composed of separate chains of the same chirality, or, in the case of X = I, separate out as a conglomerate (Fig. 3).
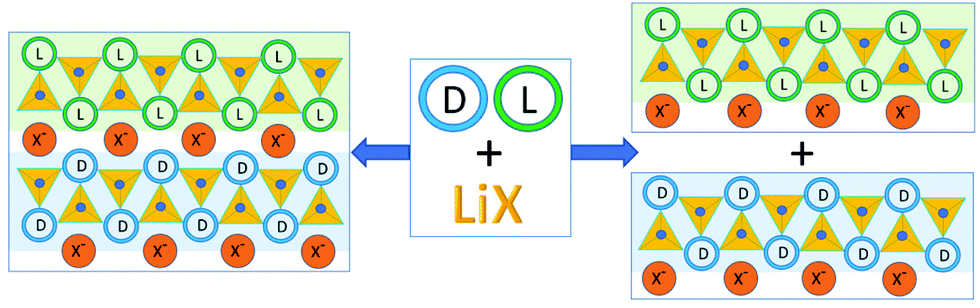 | ||
| Fig. 3 Co-crystallization of the investigated racemic amino acids with lithium halides. “L” and “D” stay for the L- and D-amino acid molecules respectively; small blue spheres represent water oxygens.46 | ||
In both the conglomerate and the racemic crystals the Li+ cations are coordinated to the amino acid of the same chirality (Fig. 4).
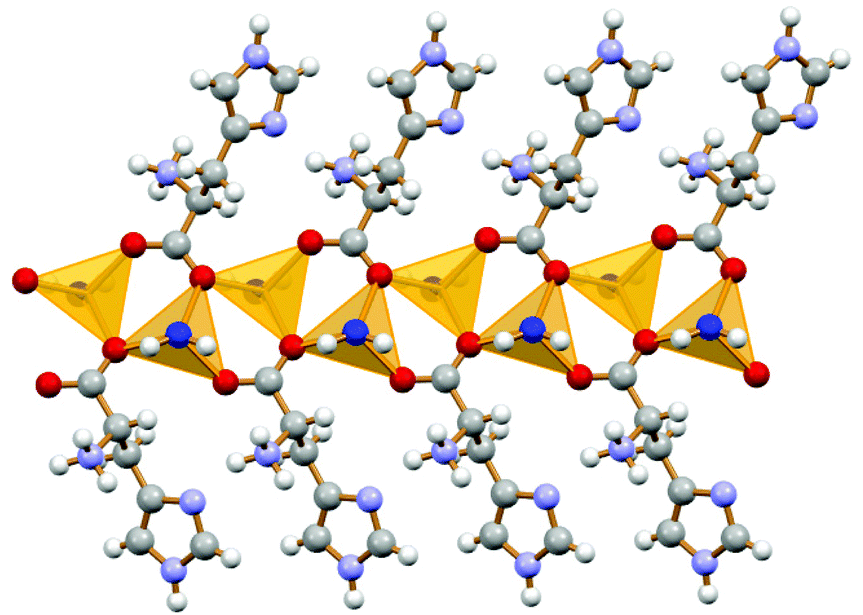 | ||
| Fig. 4 Homochiral chain observed in both conglomerate and racemic ICC of histidine with lithium chloride.46 | ||
It is useful to stress that, while chiral selection via co-crystallization of a racemic compound by means of an enantiopure coformer is predictable, the resolution of enantiomeric pairs via coordination to a small cation favouring tetrahedral coordination could not be anticipated.
Stimulated by this unexpected observation we decided to test the chiral selectivity and conglomerate formation34 for a number of amino acid racemic compounds44 (see Table 1). In the cases of the LiCl ionic co-crystals with valine, leucine, histidine and proline it was also possible to obtain both the racemic compound and the conglomerate, both types of crystals containing amino acids of the same chirality coordinating the Li+ cation. Coordination to Zn2+ does not lead to conglomerate formation. In most cases, however, the Zn2+ ions directly interact with amino acids of the same chirality, hence forming homochiral complexes within racemic crystals. It may be useful to focus on those cases where the enantiomeric selection by tetrahedral coordination is not observed, i.e. the family of Zn2+ complexes of the amino acids alanine, valine, proline, isoleucine, serine, asparagine, tyrosine, and threonine.49
| Amino acid | LiX46–48 | ZnCl249 | |
|---|---|---|---|
| Racemate | Conglomerate | Racemate | |
| DL-Alanine | DL-Ala·LiCl·H2O | — | DL-Ala2·ZnCl2 |
| DL-Valine | DL-Val·LiCl·H2O | D-/L-Val·LiCl·H2O | DL-Val2·ZnCl2 |
| DL-Leucine | DL-Leu·LiCl·1.5H2O | L-Leu·LiCl·H2O | — |
| DL-Isoleucine | — | D-/L-Ile·LiCl·H2O | DL-Ile2·ZnCl2 |
| DL-Histidine | DL-His·LiCl·H2O | ||
| DL-His·LiBr·1.5H2O | D-/L-His·LiI·1.5H2O | — | |
| DL-Proline | DL-Pro·LiCl·H2O | ||
| DL-Pro·LiI·H2O | |||
| DL-Pro·LiBr | |||
| DL-Pro·LiI | D-/L-Pro·LiCl·H2O | ||
| D-/L-Pro·LiBr·H2O | |||
| D-/L-Pro·LiCl | |||
| DL-Pro2·ZnCl2 | |||
| DL-Pro2·ZnCl2 | |||
| DL-Serine | — | — | DL-Ser2·ZnCl2 |
| DL-Threonine | — | — | DL-Thr2·ZnCl2·Thr |
| DL-Asparagine | — | — | DL-Asn2·ZnCl2 |
| DL-Tyrosine | — | — | DL-Tyr·ZnCl2 |
| DL-Glutamic acid | — | — | DL-Glu2·ZnCl2 |
The question then arose on whether the “tetrahedral selectivity” towards homochiral molecules could be observed also with other small metals favoring tetrahedral coordination. To this end, racemic amino acids were mechanochemically reacted with ZnCl2; the results are shown in Table 1.
With the exception of threonine, which crystallizes out as meso-threonine2ZnCl2, all other amino acids as their racemic compounds under different preparative conditions (manual grinding, liquid assisted grinding (LAG), ball milling etc.) generally leads to crystals of rac-(amino acid)2ZnCl2, formed by 0D homochiral complexes of formula L-(amino acid)2ZnCl2 and D-(amino acid)2ZnCl2, respectively (see Fig. 5).
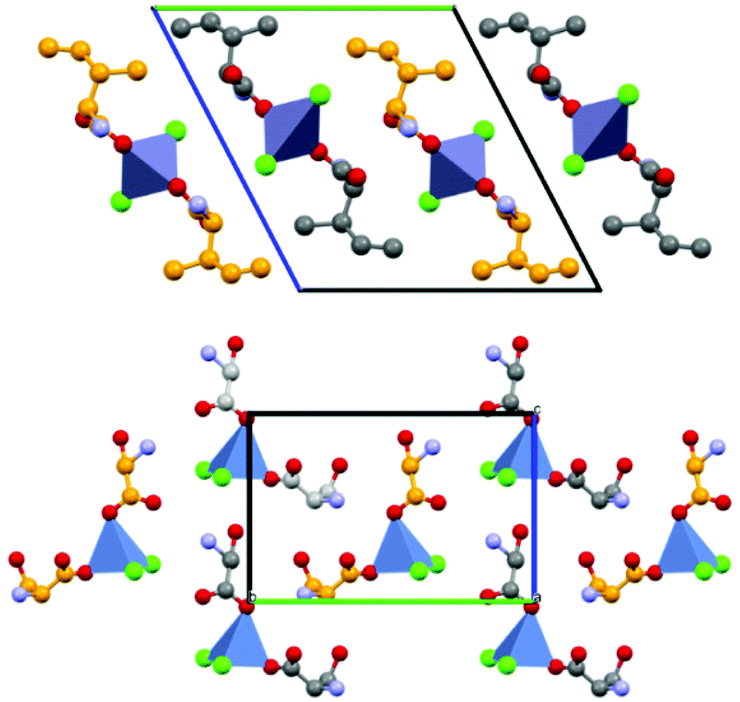 | ||
| Fig. 5 0D homochiral complexes of formula L-(amino acid)2ZnCl2 (top) and D-(amino acid)2ZnCl2 (bottom), respectively. Reproduced from ref. 49 with permission from the Royal Society of Chemistry. | ||
With DL-proline both the known racemic50 and the new meso-proline2ZnCl2 solids have been obtained (see Fig. 6).
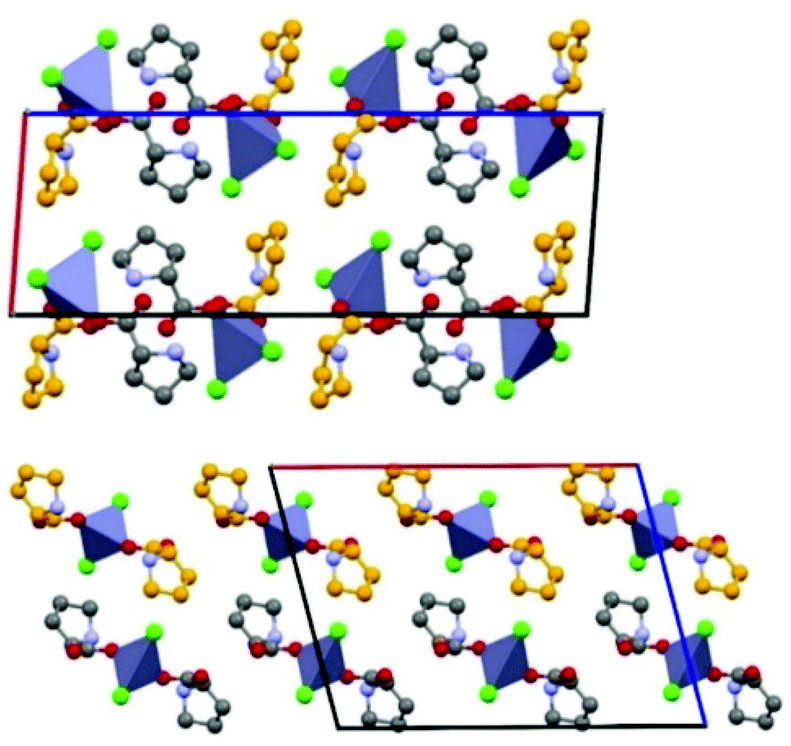 | ||
| Fig. 6 Comparison between crystal packings for homochiral (top) and heterochiral (bottom) DL-pro2·ZnCl2. Grey and orange spheres for carbons refer to proline molecules with opposite chirality; Zn2+ coordination polyhedra in blue-grey; chloride ions in lime green; H atoms omitted for clarity. Reproduced from ref. 49 with permission from the Royal Society of Chemistry. | ||
Formation of 1D coordination polymers has been observed in the cases of DL-asparagine and DL-tyrosine, with alternating D- and L-amino acids along the polymeric chain, as shown in Fig. 7.
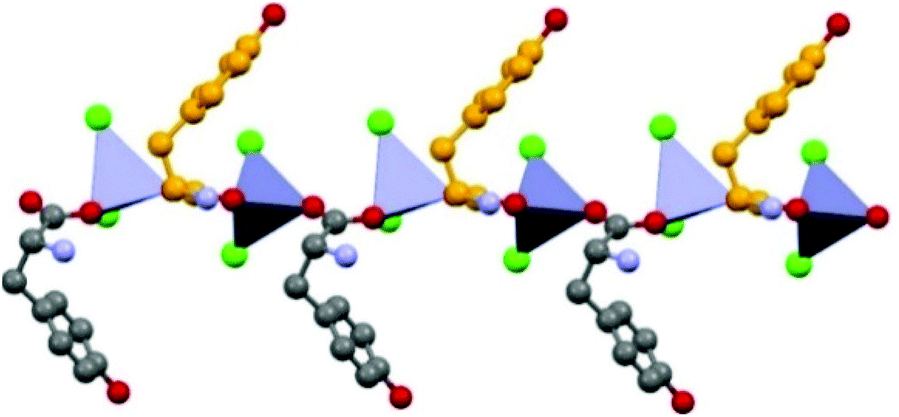 | ||
| Fig. 7 1-D coordination polymer observed in the case of crystalline catena-[(μ2-DL-tyrosine)ZnCl2]. Reproduced from ref. 49 with permission from the Royal Society of Chemistry. | ||
After discovering the potential of co-crystallization of lithium and zinc halides with the racemic amino acids in terms of chiral resolution, we decided to switch from model compounds to APIs. In close collaboration with the research group of Professor Tom Leyssens at the Catholic University of Louvain we have investigated the possibility to achieve chiral resolution of RS-etiracetam51 (ETI, Scheme 1, left) – the racemate of the antiepileptic drug levetiracetam (S-etiracetam, Scheme 1, right).
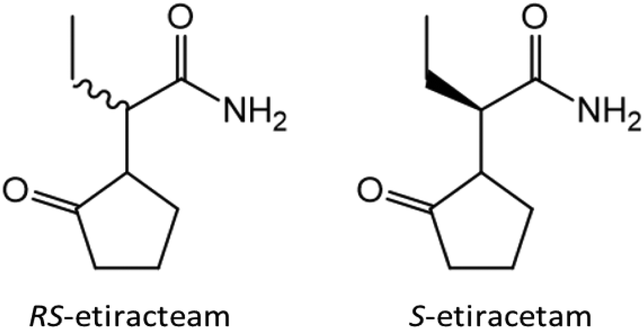 | ||
| Scheme 1 Chemical structures of RS-etiracetam (ETI) and levetiracetam (S-etiracetam). | ||
Based on the co-crystallization results obtained for amino acids, lithium halides seemed to be the most promising coformers in terms of potential conglomerate formation. However, all the co-crystallization experiments with etiracetam were unsuccessful. Consequently, we turned to different inorganic salts, and the most obvious choice was zinc chloride. Even though no conglomerate formation had been observed with amino acids,49 its homochiral preference increased the chances that complexes with etiracetam would crystallize as conglomerates. Unfortunately, the complexation of racemic etiracetam with ZnCl2 resulted in racemic RS-ETI2·ZnCl2: as expected, zinc cations were coordinated by etiracetam molecules of one chirality, forming distinct layers of R-ETI2·ZnCl2 and of S-ETI2·ZnCl2 in the overall racemic compound, but no chiral resolution was achieved (see Fig. 8).
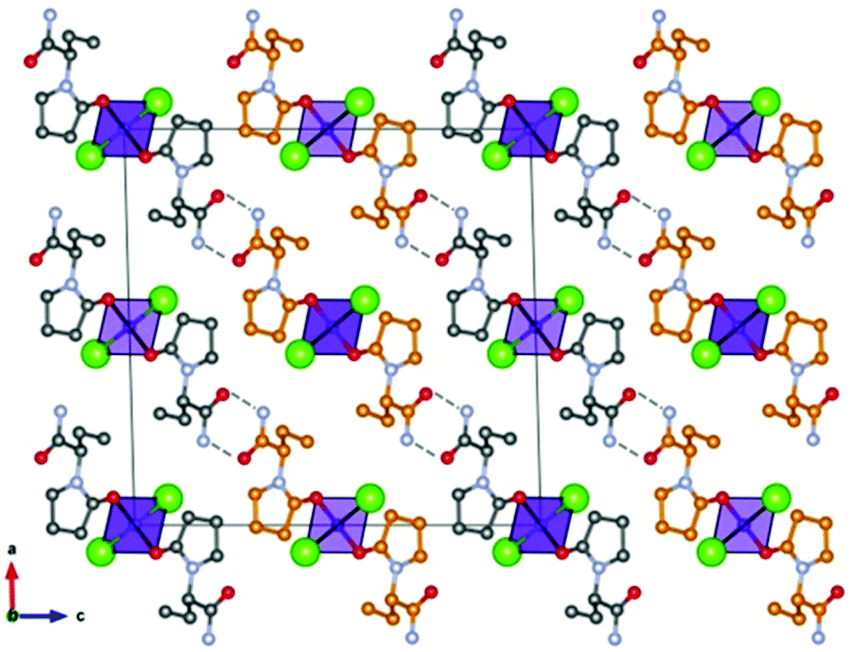 | ||
| Fig. 8 Crystalline RS-ETI2·ZnCl2: Projection in the ac-plane, showing the hydrogen bonds between the amido groups of etiracetam molecules of opposite chirality (coloured in grey and orange for clarity). “Orange” and “grey” layers of R-ETI2·ZnCl2 and S-ETI2·ZnCl2 can be seen in projection, parallel to the a-axis. Reproduced from ref. 51 with permission from the Royal Society of Chemistry. | ||
Interestingly, a subsequent increase of the amount of zinc chloride with respect to RS-etiracetam led to disruption of the racemic compound, with formation of the stable conglomerate R-ETI·ZnCl2 + S-ETI·ZnCl2 (Scheme 2).
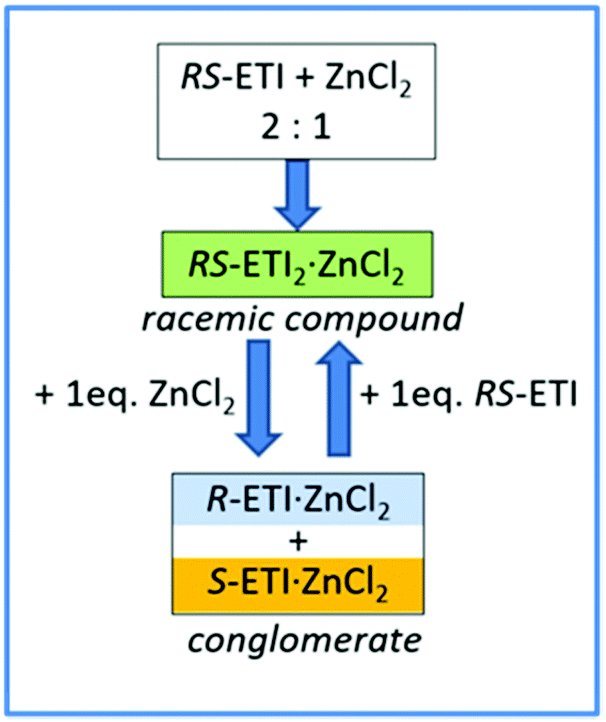 | ||
| Scheme 2 Graphic representation of the RS-ETI:ZnCl2 system in the solid-state, and the role of stoichiometry in the racemic compound/conglomerate switch mechanism. Reproduced from ref. 51 with permission from the Royal Society of Chemistry. | ||
Organic–inorganic adducts for soil enzyme inhibition
In this section we describe our inorganic crystal engineering approach to tackle an important agro-environmental and economic issue related to the global nitrogen (N) cycle (Fig. 9), namely the urgent need to ensure sustainable soil fertilization and crop productivity by minimizing N losses upon the use of N-containing plant fertilizers. Urea is the most widely used plant fertilizer, accounting for about 60% of the global nitrogen fertilizer used in the World.52 Upon its deposition in the soil, urea is degraded by urease (urea amidohydrolase, EC 3.5.1.5), a nickel-dependent enzyme widely spread in soils both inside living cells of plants and microorganisms (i.e. Sporosarcina pasteurii) and in an extracellular form adsorbed onto soil components.53a Urease catalyzes rapid hydrolysis of urea into hydrogen carbonate (HCO3−) and ammonium (NH4+), the latter serving as a nutrient to plants, at a rate 1015 times faster than in the non-catalyzed reaction53b and an overall pH increase is observed that leads to the formation of gaseous ammonia (NH3).53a,c Along with urease, the copper-dependent enzyme ammonia monooxygenase (AMO) present in ammonia-oxidizing microorganisms (i.e. Nitrosomonas europaea) catalyzes the oxidation of nascent ammonia into hydroxylamine (NH2OH),54 that is further oxidized to nitrite (NO2−) and nitrate (NO3−); NO3− is used as an available N form by plants, but it concomitantly undergoes leaching phenomena in soils. Moreover, both NO2− and NO3− act as precursors of gaseous species responsible for the greenhouse effect. As a consequence of this complex reactions network, a large amount (ca. 50%) of nitrogen fertilizer applied to soils as urea is lost, while the environment is polluted.55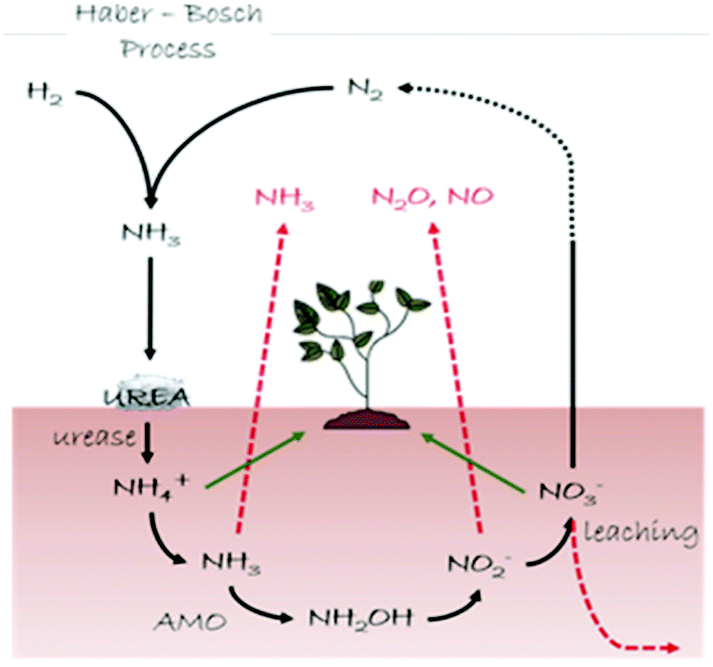 | ||
| Fig. 9 Schematic representation of the N cycle (black arrows). Loss pathways of the reactive N are shown with red arrows, while green arrows indicate the N species available for plants. | ||
To the present day, two main methods have been employed to tackle this problem: (i) coating/encapsulation of urea and (ii) amendment of urea-based fertilizers with inhibitors. The first approach is aimed to reduce the water solubility/dissolution rate of urea while the second directly modulates the reaction rates of urease and AMO, responsible for the ammonification and nitrification processes, thus enhancing N uptake by plant roots.
More recently, it has been shown that urea based ionic co-crystals with inorganic compounds can successfully be used to slow down urea decomposition, hence ammonia release. Good examples are provided by urea·MgSO4 co-crystals,56 as well as by co-crystals with Ca2+ salts obtained via mechanochemistry using the appropriate minerals.57 Beside co-crystals, inorganic salts such as Ca(NH4)2(HPO4)2·H2O and Mg(NH4)2(HPO4)2·4H2O as well as their struvite equivalents Ca(NH4)(PO4)·H2O and Mg(NH4)(PO4)·6H2O have been utilized.58
In our efforts to apply crystal engineering strategies to address this important nature inspired problem, we have joined strengths with the groups of Professor Baltrusaitis at the University of Leigh and of Professor Ciurli at the University of Bologna in the quest for hybrid organic–inorganic co-crystals capable to reconcile both the above-mentioned aspects, i.e. to improve the chemical–physical properties of urea by reducing its water solubility/dissolution rate and to exert an inhibition activity towards the enzymes urease and/or AMO, yet still providing nutrients and fertilizers to the soil.
We have found59 that when urea is co-crystallized with the inorganic metal salts KCl and ZnCl2, crystalline urea·ZnCl2·KCl (ZnKU) is obtained quantitatively in a simple, solvent free and scalable manner. It is worth mentioning that the compound is known in two polymorphic modifications depending on the preparation conditions (Fig. 10a).
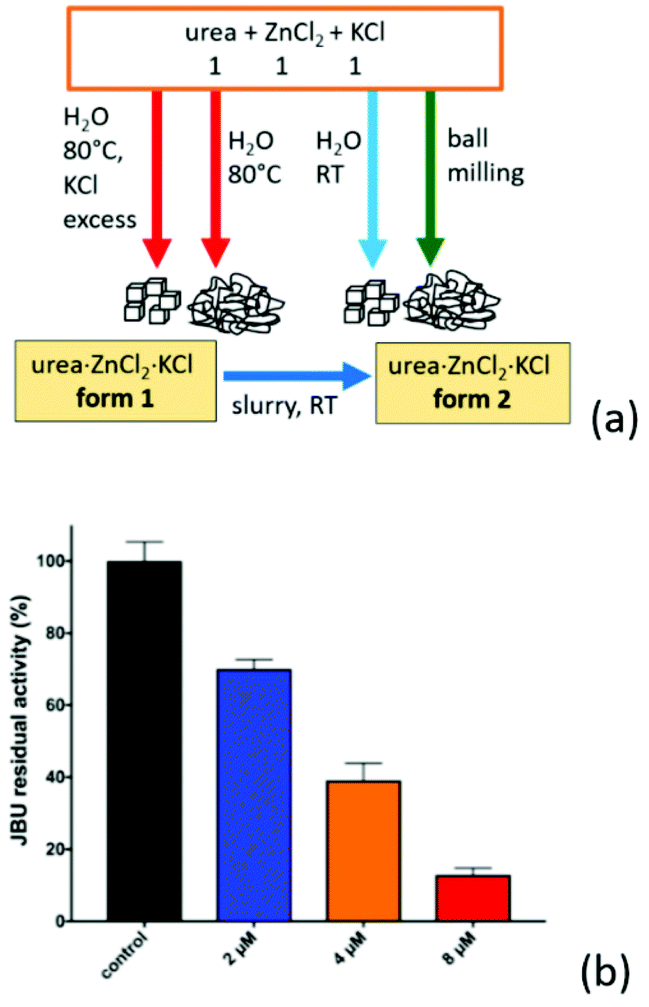 | ||
Fig. 10 (a) Urea·Zn·KCl forms 1 and 2 obtained by reacting urea, ZnCl2 and KCl in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 stoichiometric ratio. (b) Residual percentage activity of jack bean urease (JBU), referred to 100% (control, black bar) in the presence of increasing concentrations of ZnKU form 2, at pH 7.5. The blue, orange and red bars correspond to 2, 4 and 8 μM of inhibitor, respectively. Reproduced from ref. 10 with permission from the Royal Society of Chemistry. :1:1 stoichiometric ratio. (b) Residual percentage activity of jack bean urease (JBU), referred to 100% (control, black bar) in the presence of increasing concentrations of ZnKU form 2, at pH 7.5. The blue, orange and red bars correspond to 2, 4 and 8 μM of inhibitor, respectively. Reproduced from ref. 10 with permission from the Royal Society of Chemistry. | ||
We have found59 that when urea is co-crystallized with the inorganic metal salts KCl and ZnCl2, crystalline urea·ZnCl2·KCl is obtained quantitatively in a simple, solvent free and scalable manner. It is worth mentioning that the compound is known in two polymorphic modifications depending on the preparation conditions (Fig. 10a).
The compound obtained from aqueous solution at 80 °C is a metastable form, since it converts by slurry at RT into the stable form, which can be obtained by ball milling or crystallization at RT.27 The known ability of Zn(II) to act as a urease inhibitor60a prompted us to determine the inhibitory potency of increasing concentrations of ZnKU in its stable crystal form on enzyme activity (as shown in Fig. 10b), demonstrating a strong inhibition effect. Beside acting as an efficient modulator of urease activity, the co-crystal provides a nutrient component (KCl) together with the urea fertilizer.
We have also reported that co-crystallization of urea and thiourea (a known inhibitor of ammonia oxidation process by AMO60b with ZnCl2 produces the mixed system [ZnCl2(thiourea)(urea)] (ZnTU).60c,d This compound, besides providing the urea fertilizer, is able to act as a dual inhibitor. In particular, measurements carried out on bacterial cultures containing either S. pasteurii or N. europaea, as well as in tandem experiments in the presence of both the microorganisms, provide evidence that ZnTU is selectively effective in lowering the ammonification and oxygen consumption via the Zn2+ and thiourea components, respectively. By carrying the urea fertilizer and simultaneously inhibiting these two N cycle key enzymes, hence ZnTU is able to thus increasing N fertilization efficiency (see Fig. 11).
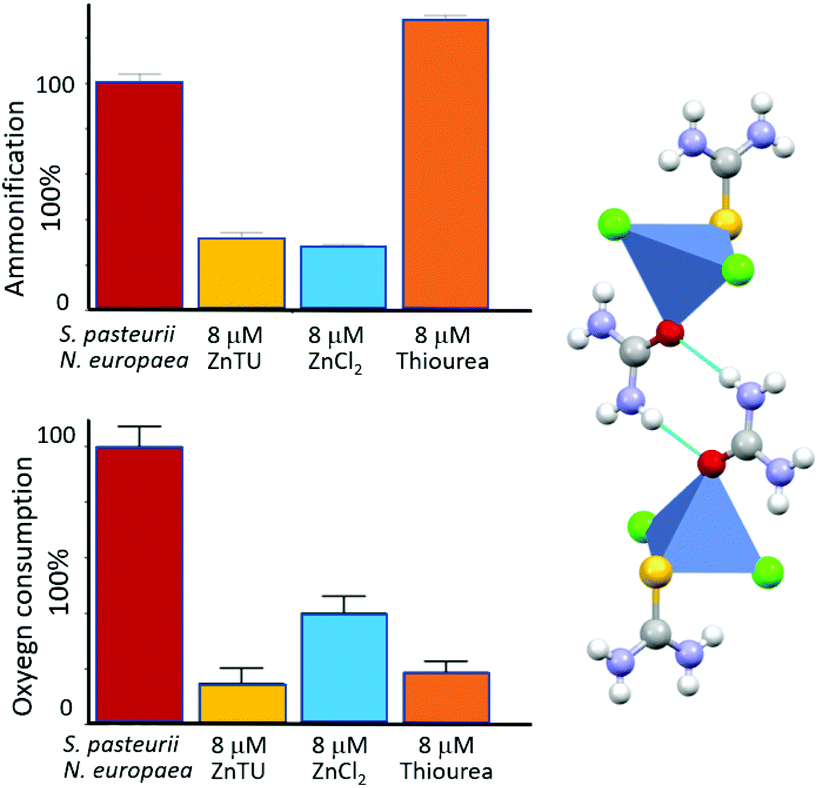 | ||
| Fig. 11 Inhibition activity of [Zn(thiourea)(urea)Cl2] (ZnTU) towards a mixture of S. pasteurii and N. europaea cells: residual ammonification (top left) and oxygen consumption (bottom left) in the absence (red bar) and in the presence of 8 μM ZnTU (yellow bars) are compared to the effect provided by stoichiometric amounts of ZnCl2 (light blue bars) and thiourea (orange bars), reported as controls. The structure of ZnTU is shown on the right.60 | ||
As a third example of application of crystal engineering and solid state strategies to investigate compounds with potentials in the agrochemical field is the recently reported in situ monitoring of the mechanochemical reaction between the AMO inhibitor dicyandiamide and inorganic salts of the urease inhibitor copper(II) [CuX2, where X = Cl−, NO3−].61 By getting a better understanding of the mechanochemical experimental conditions to obtain the new adducts it was possible to drive the reactions towards the desired compounds and to suggest a methodology to explore new compounds of agrochemical interest. A similar approach has been discussed by Friscic et al.62
Targeting antimicrobial resistance with organic–inorganic co-crystals and complexes
This section deals with a third nature inspired application of the crystal engineering. Herein we discuss our efforts in the quest for new materials to tackle another high-impact problem of our times, namely antimicrobial resistance.63 Crystal engineering approaches utilizing metal complexes and coordination networks with antimicrobial properties have been gaining popularity as a new means to deal with this challenge.64 As a matter of fact, metals have been used as antimicrobials65 long before the discovery of antibiotics and before their utilization to treat human, animal and plant diseases became such a widespread practice. However, it is the abuse of antibiotics that accelerates the development of antimicrobial resistance in microorganisms.In close collaboration with the group of Professor Ray Turner at the University of Calgary, we have investigated the co-crystallization of a series of metal complexes with active organic principles in the quest for new antimicrobial agents. While in the previous section we use co-crystallization techniques with the aim of obtaining materials able to inhibit enzyme activity, here the goal is quite the opposite: co-crystallization methods are used to prepare new compounds with old drugs to see if the antimicrobial activity can be enhanced.
More specifically, we have concentrated our efforts on a well-known disinfectant-bacteriostatic organic molecule belonging to the class of quaternary-cation compounds (QCC),66 namely proflavine,67e.g. (acridine-3,6-diamine). Proflavine (PF) and its protonated proflavinium cation (HPF+) shown in Scheme 3 have been used in the preparation and evaluation of a series of co-crystals obtained by reacting proflavine with salts and complexes of Cu, Zn, Ag and Ga.
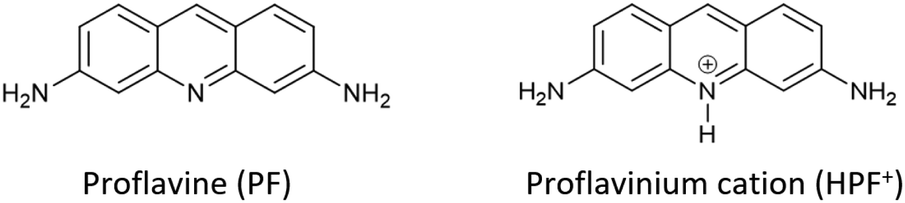 | ||
| Scheme 3 Neutral proflavine PF and the proflavinium cation HPF+. | ||
Compounds were obtained in most cases by mechanochemical mixing of the solid reactants or by slurry mediated reactions; this last method yielded higher purity target products, which were subsequently utilized for the investigation of the antimicrobial activity against three bacteria strains, namely Pseudomonas aeruginosa ATCC27853, Staphylococcus aureus ATCC25923, and Escherichia coli ATCC25922.
An example is shown in Fig. 12, where the structure of the copper(I) chloride co-crystal PF·CuCl is shown,68 formed by a 1-D polymer of CuCl monomers and by neutral proflavine molecules arranged in herring-bone fashion.
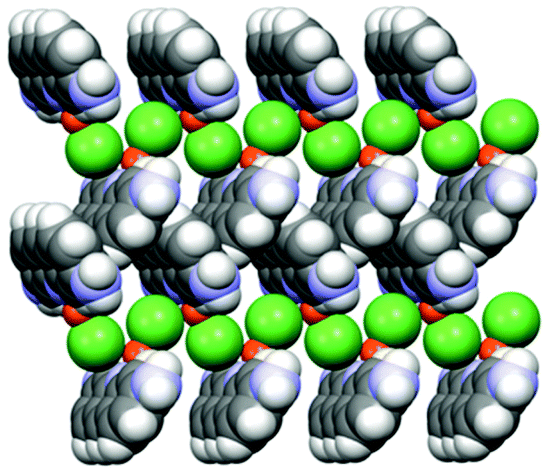 | ||
| Fig. 12 (CuCl⋯CuCl⋯)n chains and herring-bone arrangement of proflavine molecules in the proflavine·CuCl co-crystal. Reproduced from ref. 68 with permission from the Royal Society of Chemistry. | ||
The PF·AgNO3 and PF·CuCl compounds were subjected to microbiological assays against S. aureus, P. auriginosa and E. coli. Both compounds perform better than proflavine and the inorganic salts separately (see Fig. 13). We took this as a proof of concept for the possibility of using hybrid inorganic salts and active molecules to generate new materials for applications, for example, in surface coating.
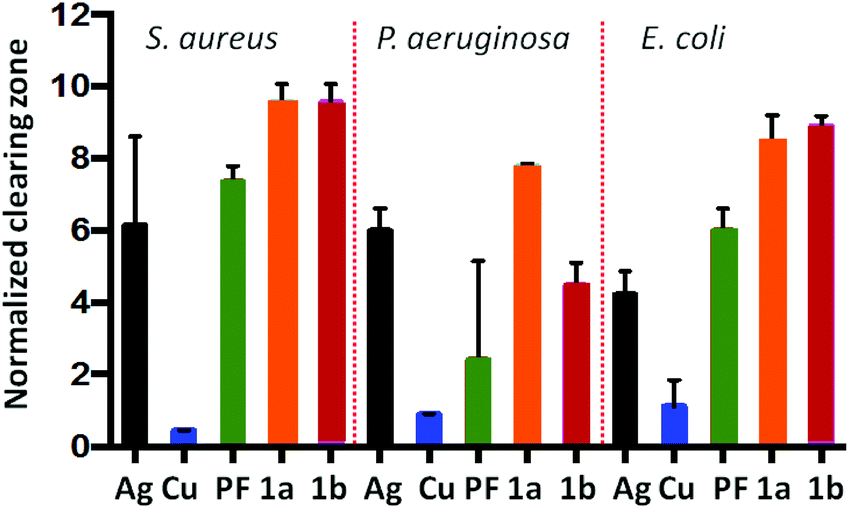 | ||
| Fig. 13 Efficacy of PF based compounds: Normalized zones of growth inhibition utilizing compound impregnated disks [Ag = AgNO3, PF = proflavine, Cu = CuCl, 1a = PF·CuCl, 1b = PF·AgNO3]. Reproduced from ref. 68 with permission from the Royal Society of Chemistry. | ||
With the same rationale, we then attempted preparation of analogous derivatives of ZnCl2.69 The nature of the products obtained by mechanochemical and solution methods varied depending on the Zn-proflavine stoichiometric ratio and on the experimental conditions. Two compounds were isolated and fully characterized as [HPF]ZnCl3 and as the monohydrate [HPF]2[ZnCl4]·H2O, respectively. Contrary to the copper and silver complexes both compounds contain proflavine as its proflavinium cation (HPF)+.
The structures of the two compounds are compared in Fig. 14. The most noteworthy feature of these two materials is the stacking of the proflavinium cations. The same interaction is predominant in the crystal structures of neutral proflavine (PF) and of the salt [HPF]Cl·2H2O.
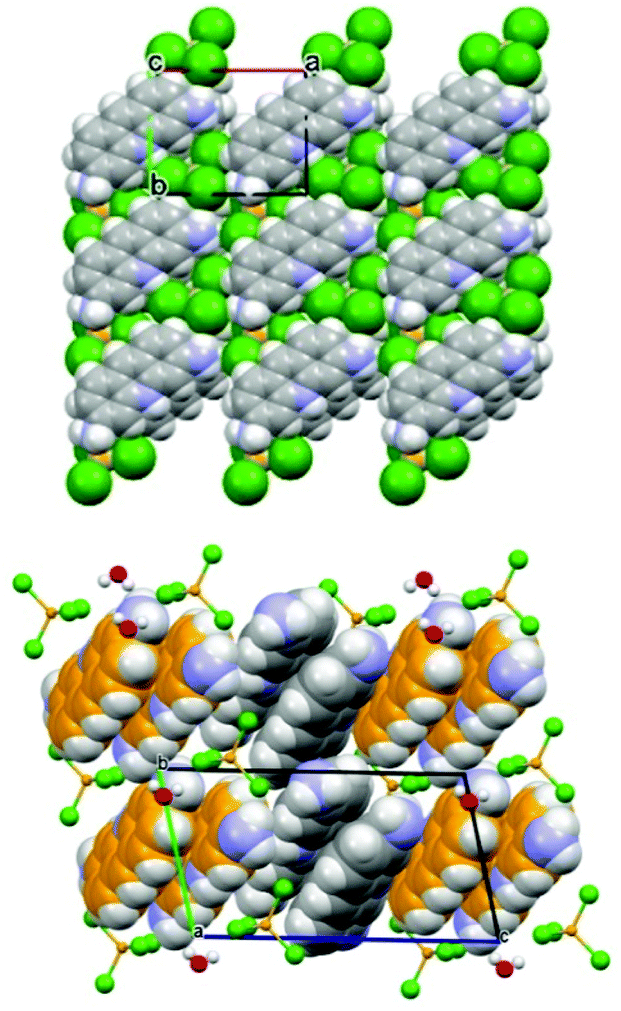 | ||
| Fig. 14 (Top) Projection in the ab-plane of crystalline ZnCl3(HPF) in which the molecules are arranged in piles along the c-axis direction. (Bottom) Projection down the crystallographic b-axis of crystalline [HPF]2[ZnCl4]·H2O, showing the herring-bone pattern of HPF+ cationic pairs. Reproduced from ref. 69 with permission from the Royal Society of Chemistry. | ||
When tested against the pathogen indicator strains the compounds showed a 50–125% enhancement of the antimicrobial activity with respect to AgNO3 used as a reference standard. The increase in antimicrobial activity is slightly less pronounced when compared to the physical mixture of the separate components, but it is still noteworthy, see Fig. 15. In summary, the association of proflavine, as its proflavinium cation, with Zn increases the antimicrobial efficacy of proflavine itself.
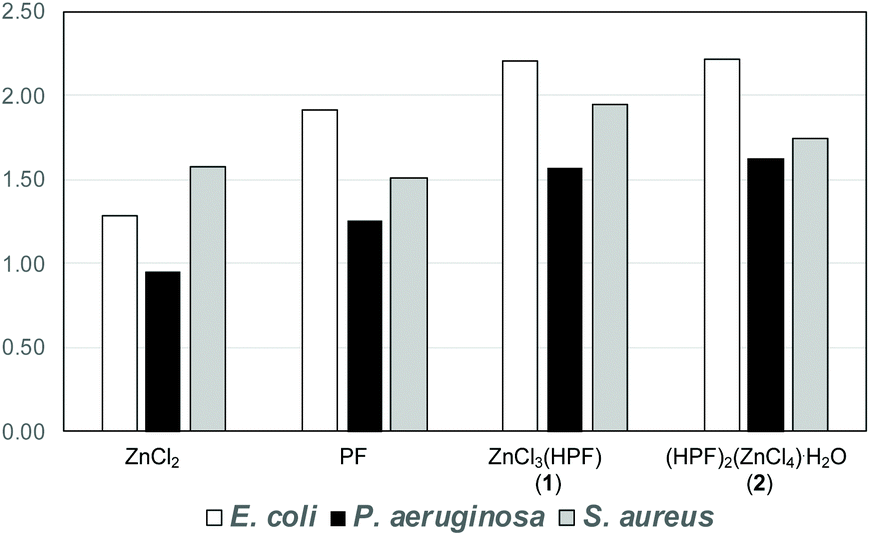 | ||
| Fig. 15 Fold antimicrobial activity of proflavine, ZnCl3(HPF) and (HPF)2(ZnCl4)·H2O compared to the activity of AgNO3 (the value of 1.00 is equal efficacy as silver nitrate). Reproduced from ref. 69 with permission from the Royal Society of Chemistry. | ||
More recently,70 we have extended the quest for novel proflavine-based co-crystals by using as a preformed building block, the gallium oxalate complex [Ga(ox)3]3−, in order to prepare the molecular salt [HPF]3[Ga(ox)3]·4H2O (see Fig. 16). The proflavinium cations envelope the [Ga(ox)3]3− anions forming the stackings of proflavine cations similar to those observed in the neutral co-crystals discussed above.
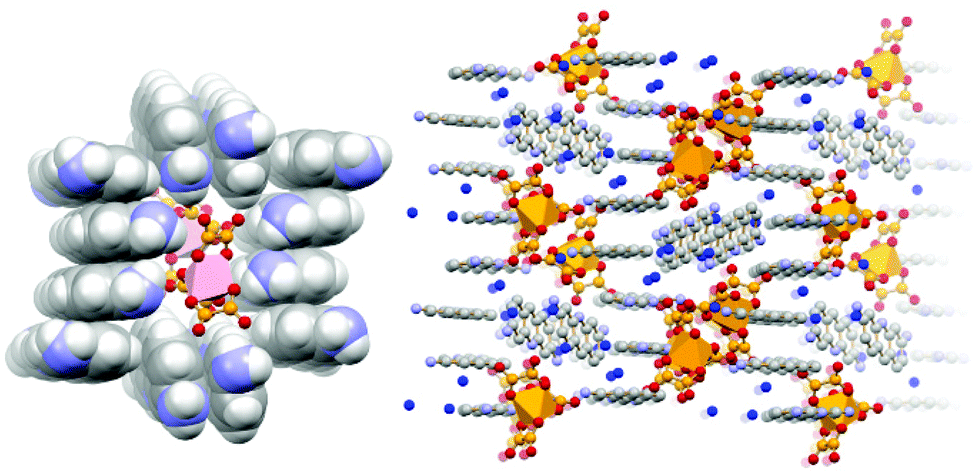 | ||
| Fig. 16 π-stacking of [HPF]+ cations in crystalline [HPF]3[Ga(ox)3]·4H2O (left); an envelope formed by proflavinium cations around two [Ga(ox)3]3− anions (right)70. | ||
As in the cases discussed above, the antimicrobial performance has been evaluated by disk diffusion assays, showing that also the gallium compound can be a valuable antimicrobial. It is worth mentioning that, while [HPF]3[Ga(ox)3]·4H2O is effective against all three strains, the gallium oxalate salt K3[Ga(ox)3] shows an impressive selectivity towards P. aeruginosa, with little to no antimicrobial activity against the other two organisms. This selectivity is per se remarkable and is being investigated further in its microbiological aspects.
In summary, these results show that co-crystallization of a known QCC compound, e.g. proflavine, with metal salts, such as CuCl, CuCl2 and AgNO3, but also with a preformed coordination compound such as [Ga(ox)3]3−, via mechanochemical or slurry methods, is a viable, eco-friendly, and inexpensive way to obtain new materials with enhanced antimicrobial properties. All crystalline materials investigated thus far, namely PF·CuCl, PF·AgNO3, ZnCl3(HPF), [HPF]2[ZnCl4]·H2O and [HPF]3[Ga(ox)3]·4H2O appear to perform better than PF and the metal salts separately. The antimicrobial performance of these compounds is noteworthy, even more so because the crystal mixtures by weight invariably contain less proflavine by moles.
These results ought to be looked at in the context of the work of other groups. The number of papers dealing with hybrid inorganic–organic materials to tackle antimicrobial resistance71 and /or to find new, metal based ways for drug delivery72 is rapidly increasing.
Conclusions and outlook
In closing the Dalton Trans. Perspective in 2000,7 one of us wrote “It is the challenge for the future to demonstrate that crystal engineering is providing a new way of thinking Chemistry…”. With hindsight, we might also add that crystal engineering was also providing a new way of thinking Crystallography, because many crystallographers realized that they could exploit their knowledge of supramolecular interactions in the solid state, combined with the experience in crystallization and solid-state characterization methods, to make their own solid materials, thanks also to a synergistic interaction with those expert in organic and inorganic synthesis. The transition from crystallography to crystal making has generated a bounty of results in all directions of solid-state chemistry, with important applications in very many diverse fields, causing an evolution from crystal making to crystal utility. Accordingly, we have chosen to apply the crystal engineering way of thinking to address some specific nature inspired societal challenges, such as antimicrobial resistance caused by the increasing use of antibiotics in humans, animals and agriculture, and the environmental and economic issues related to degradation of the most commonly used fertilizer, namely urea, by natural soil enzymes. To address these problems, as demonstrated by the papers presented in this Perspective, a multidisciplinary approach to problem solving is of paramount importance. Forces need to be joined with other scientists, in order to evaluate the performance of the materials produced by applying the crystal engineering paradigm, namely, going from molecules and complexes to supramolecular aggregates with collective solid state properties. The inorganic chemistry area offers plenty of opportunities and great diversity of physico-chemical and biological properties. During the two past decades, thanks to the possibility of providing new tools to tackle new and old problems, crystal engineering has overcome many disciplinary barriers, providing opportunities for new discoveries not only in terms of fundamental “blue sky” research, but also in terms of applied, industrially relevant, high impact research. A truly holistic science, as well pointed out by Desiraju.1Conflicts of interest
There are no conflicts to declare.Acknowledgements
As pointed out repeatedly in this Perspective, the work described here could only be done by virtue of a close synergistic interaction with other research groups. We gratefully acknowledge the fruitful collaboration with the groups of Professors Ray J. Turner (University of Calgary), Stefano Luciano Ciurli (University of Bologna), Jonas Baltrusaitis (University of Leigh), Tom Leyssens (University of Leuven), Franziska Emmerling (BAM, Berlin). The China Scholarship Council is acknowledged (Renren Sun) for a Visiting PhD Student State Scholarship. We also acknowledge financial support from MUR, project “Nature Inspired Crystal Engineering” (PRIN2020), and from the University of Bologna.References
- G. R. Desiraju, Angew. Chem., Int. Ed., 2007, 46, 8342–8356 CrossRef CAS PubMed.
- D. Braga and F. Grepioni, Making Crystals by Design, Wiley-VCH, 2007 Search PubMed.
- E. R. T. Tiekink and J. J. Vittal, Frontiers in Crystal Engineering, Wiley-VCH, 2006 Search PubMed.
- G. R. Desiraju, Crystal Engineering: the Design of Organic Solids, Elsevier, Amsterdam, 1989 Search PubMed.
- (a) E. R. T. Tiekink, J. J. Vittal and M. Zaworotko, Organic Crystal Engineering: Frontiers in Crystal Engineering, John Wiley & Sons, 2010 CrossRef; (b) J. J. Novoa, Intermolecular Interactions in Crystals: Fundamentals of Crystal Engineering, Royal Society of Chemistry, 2017 Search PubMed.
- (a) J. H. Williams, Crystal Engineering, Morgan and Claypool Publishers, 2017 Search PubMed; (b) G. R. Desiraju, J. J. Vittal and A. Ramanan, Crystal engineering: a textbook, World Scientific, 2011 CrossRef.
- Dalton Discussion ICE2000, The first meeting on inorganic crystal engineering, University of Bologna, D. Braga, J. Chem. Soc., Dalton Trans., 2000, 3705–3998 Search PubMed.
- J. M. Lehn, Angew. Chem., Int. Ed. Engl., 1990, 29, 1304–1319 CrossRef.
- Y. Yamini and M. Safari, Microchem. J., 2019, 146, 134–141 CrossRef CAS.
- (a) M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed; (b) M. O'Keeffe, M. A. Peskov, S. J. Ramsden and O. M. Yaghi, Acc. Chem. Res., 2008, 41, 1782–1789 CrossRef PubMed.
- (a) M. Ding, R. W. Flaig, H. L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC; (b) T. Ghanbari, A. Faisal and W. M. A. W. Daud, Sci. Total Environ., 2020, 707, 135090 CrossRef CAS PubMed.
- K. Suresh and A. J. Matzger, Angew. Chem., Int. Ed., 2019, 58, 16790–16794 CrossRef CAS PubMed.
- S. N. Zhao, X. Z. Song, S. Y. Song and H. J. Zhang, Coord. Chem. Rev., 2017, 337, 80–96 CrossRef CAS.
- D. Braga, Chem. Commun., 2003, 22, 2751–2754 RSC.
- (a) R. Lin, Z. Zhang and B. Chen, Acc. Chem. Res., 2021, 54, 3362–3376 CrossRef CAS PubMed; (b) G. Cai, P. Yan, L. Zhang, H. Zhou and H. Jiang, Chem. Rev., 2021, 121, 12278–12326 CrossRef CAS PubMed.
- (a) O. T. Qazvini, R. Babarao and S. G. Telfer, Nat. Commun., 2021, 12, 197–204 CrossRef CAS PubMed; (b) L. Yang, L. Yan, Y. Wang, Z. Liu, J. He, Q. Fu, D. Liu, X. Gu, P. Dai, L. Li and X. Zhao, Angew. Chem., Int. Ed., 2021, 60, 4570–4574 CrossRef CAS PubMed.
- P. Li, M. R. Ryder and J. F. Stoddard, Acc. Mater. Res., 2020, 1, 77–87 CrossRef CAS.
- R. B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Chem. Soc. Rev., 2019, 48, 1362–1389 RSC.
- (a) A. J. Cruz-Cabeza, S. M. Reutzel-Edens and J. Bernstein, Chem. Soc. Rev., 2015, 44, 8619 RSC; (b) S. Chaudhari, S. A. Nikam, N. Khatri and S. Wakde, J. Drug Delivery Ther., 2018, 8, 350–358 CrossRef; (c) C. B. Aakeröy and D. J. Salmon, CrystEngComm, 2005, 7, 439–448 RSC.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS.
- G. Cavallo, P. Metrangolo, T. Pilati, G. Resnati and G. Terraneo, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- (a) R. Thakuria, N. K. Nath and B. K. Saha, Cryst. Growth Des., 2019, 19, 523–528 CrossRef CAS; (b) Z. F. Yao, J. Y. Wang and J. Pei, Cryst. Growth Des., 2018, 18, 7–15 CrossRef CAS.
- (a) D. J. Good, N. Nair and R. Rodriguez-Hornedo, Cryst. Growth Des., 2010, 10, 1028–1032 CrossRef CAS; (b) A. Jayasankar, L. Sreenivas Reddy, S. J. Bethune and N. Rodríguez-Hornedo, Cryst. Growth Des., 2009, 9, 889–897 CrossRef CAS; (c) F. Keramatnia, A. Shayanfar and A. Jouyban, J. Pharm. Sci., 2015, 104, 2559–2565 CrossRef CAS PubMed.
- Y. Yan, J. M. Chen, N. Geng and T. B. Lu, CrystEngComm, 2015, 17, 612–620 RSC.
- J. Wouters and L. Quéré, Pharmaceutical Salts and Co-Crystals, RSC Drug Discovery Ser., 2012 Search PubMed.
- E. Nauha and M. Nissinen, J. Mol. Struct., 2011, 1006, 566–569 CrossRef CAS.
- B. Sandhu, A. S. Sinha, J. Desper and C. B. Aakeröy, Chem. Commun., 2018, 54, 4657–4660 RSC.
- (a) H. Oertling, CrystEngComm, 2016, 18, 1676–1692 RSC; (b) A. V. Karangutkar and L. Ananthanarayan, J. Food Eng., 2020, 27, 1–13 Search PubMed.
- A. A. Dar and S. Rashid, CrystEngComm, 2021, 23, 8007–8026 RSC.
- H. M. Titi, M. Arhangelskis, G. P. Rachiero, T. Friščić and R. D. Rogers, Angew. Chem., Int. Ed., 2019, 58, 18399–18404 CrossRef CAS PubMed.
- (a) D. Braga, L. Maini and F. Grepioni, Chem. Soc. Rev., 2013, 42, 7638–7648 RSC; (b) D. Braga, F. Grepioni and O. Shemchuk, CrystEngComm, 2018, 20, 2212–2220 RSC; (c) T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef PubMed.
- (a) US Food and Drug administration (2014) Development of Stereoisomeric Drugs, https://www.fda.gov/drugs/; (b) H. Leek, L. Thunberg, A. C. Jonson, K. Ohlen and M. Klarqvist, Drug Discovery Today, 2017, 22, 133–139 CrossRef CAS PubMed.
- T. E. Beesley and R. P. Scott, Chiral chromatography, John Wiley & Sons, 1999 Search PubMed.
- C. S. Chen, Y. Fujimoto, G. Girdaukas and C. J. Sih, J. Am. Chem. Soc., 1982, 104, 7294–7299 CrossRef CAS.
- D. Kozma, CRC handbook of optical resolutions via diastereomeric salt formation, Crc Press, 2001 Search PubMed.
- G. Coquerel, in Novel optical resolution technologies, Springer, 2006, 1–51 Search PubMed.
- G. Levilain and G. Coquerel, CrystEngComm, 2010, 12, 1983–1992 RSC.
- G. R. Springuel and T. Leyssens, Cryst. Growth Des., 2012, 12, 3374–3378 CrossRef CAS.
- O. Sánchez-Guadarrama, F. Mendoza-Navarro, A. Cedillo-Cruz, H. Jung-Cook, J. I. Arenas-García, A. Delgado-Díaz, D. Herrera-Ruiz, H. Morales-Rojas and H. Höpfl, Cryst. Growth Des., 2015, 16, 307–314 CrossRef.
- B. Harmsen and T. Leyssens, Cryst. Growth Des., 2017, 18, 441–448 CrossRef.
- L. He, Z. Liang, G. Yu, X. Li, X. Chen, Z. Zhou and Z. Ren, Cryst. Growth Des., 2018, 18, 5008–5020 CrossRef CAS.
- L. C. Harfouche, C. Brandel, Y. Cartigny, S. Petit and G. Coquerel, Chem. Eng. Technol., 2020, 43, 1093–1098 CrossRef CAS.
- X. Buol, C. Caro Garrido, K. Robeyns, N. Tumanov, L. Collard, J. Wouters and T. Leyssens, Cryst. Growth Des., 2020, 20, 7979–7988 CrossRef CAS.
- O. Shemchuk, F. Grepioni, T. Leyssens and D. Braga, Isr. J. Chem., 2021, 61, 563–572 CrossRef CAS.
- D. Braga, F. Grepioni, L. Maini, S. Prosperi, R. Gobetto and M. R. Chierotti, Chem. Commun., 2010, 46, 7715–7717 RSC.
- D. Braga, L. Degli Esposti, K. Rubini, O. Shemchuk and F. Grepioni, Cryst. Growth Des., 2016, 16, 7263–7270 CrossRef CAS.
- O. Shemchuk, E. Spoletti, D. Braga and F. Grepioni, Cryst. Growth Des., 2021, 21, 3438–3448 CrossRef CAS.
- O. Shemchuk, B. K. Tsenkova, D. Braga, M. T. Duarte, V. Andre and F. Grepioni, Chem. – Eur. J., 2018, 24, 12564–12573 CrossRef CAS PubMed.
- O. Shemchuk, F. Grepioni and D. Braga, CrystEngComm, 2020, 22, 5613–5619 RSC.
- M. Lutz and R. Bakker, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, 59, m18–m20 CrossRef PubMed.
- O. Shemchuk, L. Song, K. Robeyns, D. Braga, F. Grepioni and T. Leyssens, Chem. Commun., 2018, 54, 10890–10892 RSC.
- M. Prud'homme, Global fertilizer supply and trade 2016–2017, https://www.fertilizer.org/ItemDetail?iProductCode=10192Pdf&Category=ECO.
- (a) H. L. Mobley and R. P. Hausinger, Microbiol. Rev., 1989, 53, 85–108 Search PubMed; (b) B. P. Callahan, Y. Yuan and R. Wolfenden, J. Am. Chem. Soc., 2005, 127, 10828–10829 CrossRef CAS PubMed; (c) R. P. Hausinger, Microbiol. Rev., 1987, 51, 22–42 CrossRef CAS PubMed.
- F. Musiani, V. Broll, E. Evangelisti and S. Ciurli, J. Biol. Inorg. Chem., 2020, 25, 995–1007 CrossRef CAS PubMed.
- L. B. Fenn and L. R. Hossner, Adv. Soil Sci., 1985, 1, 123–169 CAS.
- (a) L. B. Fenn and J. Richards, Fert. Res., 1986, 9, 265–275 CrossRef CAS; (b) W. V. Rheinbaben, Fert. Res., 1987, 11, 149–159 CrossRef.
- (a) K. Honer, E. Kalfaoglu, C. Pico, J. McCann and J. Baltrusaitis, ACS Sustainable Chem. Eng., 2017, 5, 8546–8550 CrossRef CAS; (b) K. Honer, C. Pico and J. Baltrusaitis, ACS Sustainable Chem. Eng., 2018, 6, 4680–4687 CrossRef CAS.
- L. Sharma, D. Kiani, K. Honer and J. Baltrusaitis, ACS Sustainable Chem. Eng., 2019, 7, 6802–6812 CrossRef CAS.
- L. Casali, L. Mazzei, O. Shemchuk, K. Honer, F. Grepioni, S. Ciurli, D. Braga and J. Baltrusaitis, Chem. Commun., 2018, 54, 7637–7640 RSC.
- (a) B. Krajewska, J. Mol. Catal. B: Enzym., 2009, 59, 9–21 CrossRef CAS; (b) W. K. Keener, S. A. Russell and D. J. Arp, Biochim. Biophys. Acta, 1998, 1388, 373–385 CrossRef CAS PubMed; (c) L. Casali, L. Mazzei, O. Shemchuk, L. Sharma, K. Honer, F. Grepioni, S. Ciurli and D. Braga, ACS Sustainable Chem. Eng., 2019, 7, 2852–2859 CrossRef CAS; (d) L. Mazzei, V. Broll, L. Casali, M. Silva, D. Braga, F. Grepioni, J. Baltrusaitis and S. Ciurli, ACS Sustainable Chem. Eng., 2019, 7, 13369–13378 CrossRef CAS.
- L. Casali, T. Feiler, M. Heilmann, D. Braga, F. Emmerling and F. Grepioni, CrystEngComm, 2022, 24, 1292–1298 RSC.
- P. A. Julien, L. S. Germann, H. M. Titi, M. Etter, R. E. Dinnebier, L. Sharma, J. Baltrusaitis and T. Friščić, Chem. Sci., 2020, 11, 2350–2355 RSC.
- (a) S. B. Levy and M. Bonnie, Nat. Med., 2004, 10, S122–S129 CrossRef CAS PubMed; (b) E. D. Brown and G. D. Wright, Nature, 2016, 529, 336–343 CrossRef CAS PubMed; (c) L. M. Streicher, J. Global Antimicrob. Resist., 2021, 24, 285–295 CrossRef PubMed.
- (a) A. S. Abd-El-Aziz, C. Agatemor and N. Etkin, Biomaterials, 2017, 118, 27–50 CrossRef CAS PubMed; (b) S. Quaresma, V. André, A. Fernandes and M. T. Duarte, Inorg. Chim. Acta, 2017, 455, 309–318 Search PubMed; (c) I. Luz, I. E. Stewart, N. P. Mortensen and A. J. Hickey, Chem. Commun., 2020, 56, 13339–13342 RSC; (d) S. Quaresma, V. André, A. M. M. Antunes, S. M. F. Vilela, G. Amariei, A. Arenas-Vivo, R. Rosal, P. Horcajada and M. T. Duarte, Cryst. Growth Des., 2020, 20(1), 370–382 CrossRef CAS.
- (a) R. J. Turner, Microb. Biotechnol., 2017, 10, 1062–1065 CrossRef PubMed; (b) A. Pormohammad and R. J. Turner, Antibiotics, 2020, 9, 853 CrossRef CAS PubMed; (c) N. Gugala, D. Vu, M. D. Parkins and R. J. Turner, Antibiotics, 2019, 8, 51 CrossRef CAS PubMed.
- S. J. S. Qazi and R. J. Turner, Biochem. Biophys. Rep., 2018, 13, 129–140 Search PubMed.
- V. M. Hridya, J. T. Hynes and A. Mukherjee, J. Phys. Chem. B, 2019, 123, 10904–11914 CrossRef CAS PubMed.
- O. Shemchuk, D. Braga, F. Grepioni and R. J. Turner, RSC Adv., 2020, 10, 2146–2149 RSC.
- C. Fiore, O. Shemchuk, F. Grepioni, R. J. Turner and D. Braga, CrystEngComm, 2021, 23, 4494–4499 RSC.
- M. Guerrini, S. d'Agostino, F. Grepioni, D. Braga, A. Lekhan and R. J. Turner, Sci. Rep., 2022 DOI:10.1038/s41598-022-07813-0.
- (a) T. A. Fernandes, I. F. M. Costa, P. Jorge, A. C. Sousa, V. André, N. Cerca and A. M. Kirillov, ACS Appl. Mater. Interfaces, 2021, 13, 12836–12844 CrossRef CAS PubMed; (b) F. Wu, D. He, L. Chen, F. Liu, H. Huang, J. Dai, S. Zhang and J. You, RSC Adv., 2018, 8, 20829 RSC; (c) C. Pettinari, R. Pettinari, C. Di Nicola, A. Tombesi, S. Scuri and F. Marchetti, Coord. Chem. Rev., 2021, 446, 214121 CrossRef CAS.
- (a) Y. Sun, L. Zheng, Y. Yang, X. Qian, T. Fu, X. Li, Z. Yang, H. Yan, C. Cui and W. Tan, Nano-Micro Lett., 2020, 12, 103 CrossRef CAS PubMed; (b) M. N. Hasan, A. Bera, T. K. Maji and S. K. Pal, Inorg. Chim. Acta, 2021, 523, 120381–120395 CrossRef CAS; (c) S. He, L. Wu, X. Li, H. Sun, T. Xiong, J. Liu, C. Huang, H. Xu, H. Sun, W. Chen, R. Gref and J. Zhang, Acta Pharm. Sin. B, 2021, 11, 2362–2395 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |