
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHydrogen-bonded cobalt(II)-organic framework: normal and reverse spin-crossover behaviours†
Takuya
Kanetomo
 *,
Zhen
Ni
and
Masaya
Enomoto
*,
Zhen
Ni
and
Masaya
Enomoto
Department of Chemistry, Faculty of Science Division 1, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8603, Japan. E-mail: kanetomo@rs.tus.ac.jp
First published on 8th March 2022
Abstract
A novel hydrogen-bonded metal-organic framework (H-MOF) [Co(HL)2](DMF)1.2(H2O)2.4 (1·solv), in which L = 2,2′:6′,2′′-terpyridine-5,5"-diyl biscarboxylate, was prepared. An intermolecular single H-bond between carboxy and carboxylate sites was present in this compound. The crystal structure of 1·solv showed a 4-fold interpenetrating H-bonded diamond framework. After the desorption of the crystal solvents, 1·desolv exhibited normal and reverse spin-crossover (SCO) behaviours, providing an asymmetric thermal hysteresis loop.
Introduction
Hydrogen-bonded metal-organic frameworks (H-MOFs) have a metal complex as a building unit, in which they are connected through H-bonding.1–11 They have been applied in molecular porous materials like the typical MOFs12–14 and covalent organic frameworks (COFs).15,16 The magnitude of H-bonding determines the flexibility between units and can be controlled via the electrostatic interactions between the donor and acceptor moieties. Recently, organic/inorganic compounds undergoing self-assembly through H-bonds have been intensively investigated in terms of their porosity, where these materials are referred to as hydrogen-bonded organic/inorganic frameworks (HOFs/HIFs).17–22 However, the strategic molecular construction of these systems depending on intermolecular interactions remains challenging. In particular, H-MOFs require the coexistence of coordination and hydrogen bonds.2,2′:6′,2′′-Terpyridine (tpy) is a three N-donor ligand that can be chemically modified,23,24 and it, therefore, has been applied to the formation of supermolecules25 and coordination polymers.26,27 In tpy-metal complexes, those involving a Co2+ ion often exhibit unique magnetic properties such as the spin crossover (SCO) phenomenon,28–33 which is a reversible spin transition based on changes in the electron configuration due to external stimuli.34 The Co2+ centre exhibits high-spin (HS; S = 3/2) and low-spin (LS; S = 1/2) states, and the difference in the spin entropy is small. Cobalt(II)-SCO compounds can exhibit a sensitive response toward the absorption/desorption of guest molecules.35–40 SCO-active Co-tpy complexes may act as switching components that are compatible with porous architectures such that functional materials may be developed.
A novel tpy-based ligand 2,2′:6′,2′′-terpyridine-5,5′′-diyl biscarboxylic acid (H2L) as shown in Scheme 1 not only possesses the tpy moiety and contributes to the SCO behaviour, but it also contains two carboxy groups for intermolecular H-bonding. Carboxy groups have often been employed in H-bonded architectures.17,18 Here, two H2L ligands are coordinated to one Co2+ centre, and the molecular structure exhibits a cruciform-like structure, providing two- or three-dimensional frameworks through the H-bonds at the four carboxy sites. In this study, we prepared a novel cobalt(II) complex [Co(HL)2](DMF)1.2(H2O)2.4 (1·solv), in which HL has carboxy and carboxyl sites as shown in Scheme 1. Crystallographic studies showed that an intermolecular H-bond was present between the carboxy and carboxylate sites, thereby forming the H-bonded 3-D framework. There were crystal solvents in the void space, and they can be removed above 300 K. The 1·desolv exhibited normal and reverse SCO behaviours with an asymmetric thermal hysteresis loop.
 | ||
| Scheme 1 H2L and its derivatives. | ||
Results and discussion
Synthesis and characterization
We prepared H2L using the 5,5′′-dimethyl-2,2′:6′,2′′-terpyridine41,42via oxidation with KMnO4. After complexation between H2L and CoCl2 in N,N-dimethylformamide (DMF) and H2O, the product was purified by recrystallization through the vapour diffusion approach using the above mixture and 1,4-dioxane, affording black block crystals in the form of [Co(HL)2](DMF)1.2(H2O)2.4 (1·solv). The HL anion could be prepared during the above complexation process. For elemental analysis, the experimental values agreed with the calculated values, including 1.2 DMF and 2.4 H2O molecules after collection and immediate measurement. The presence of these solvents was also confirmed by thermal and X-ray crystallographic analysis (for details, see below).Single-crystal X-ray crystallography
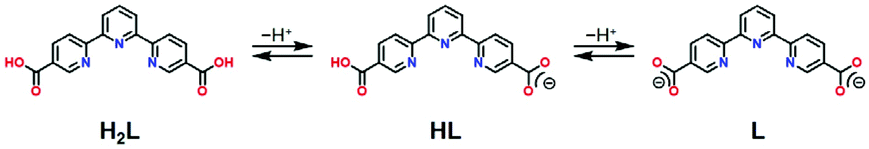
The crystal structure of 1·solv was evaluated at 93 K, and it crystallized in a tetragonal P42/n space group, as shown in Table 1. In Fig. 1, the Co2+ centre is shown to be 6-coordinated with two tpy moieties, providing the N6 environment. The Co1–N1, Co1–N2, and Co1–N3 bond lengths were 2.1326(11), 2.0327(10), and 2.1313(11) Å, respectively. The mean of the Co–N lengths (dmean) was 2.099 Å. In addition, the octahedral distortion parameter (Σ)43 was estimated to be 119.53°. These geometric parameters result in a suppressed octahedron environment along the N2, Co1, and N2* atoms. The dmean and Σ parameters have been used to predict the spin state of the Co2+ centre. The relationship between these parameters was described in our previous work,40 and the relationship observed in this study indicates the HS state of the Co2+ centre.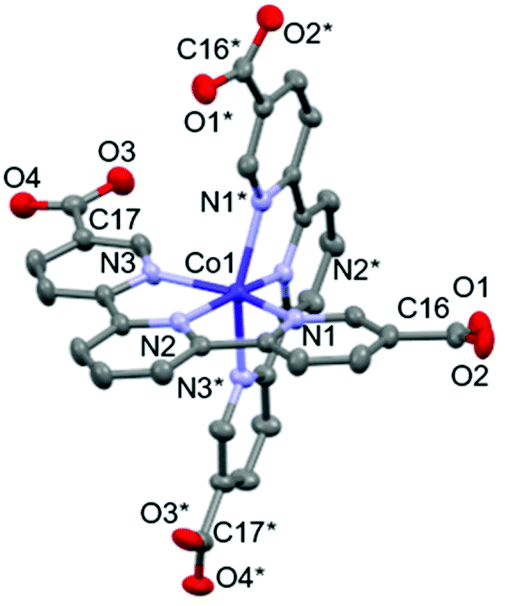 | ||
| Fig. 1 Molecular structure of 1·solv. The H atoms are omitted for clarity. The thermal ellipsoids for non-hydrogen atoms are drawn at a 50% probability. Symmetric code: * = 3/2 − x, 3/2 − y, z. | ||
| a R = ∑||Fo| − |Fc||/∑|Fo|. b R w = [∑w(|Fo| − |Fc|)2/∑w|Fo|2]1/2. | |
|---|---|
| Formula | C34H20CoN6O8 |
| Fw | 699.49 |
| T/K | 93 |
| Crystal system | Tetragonal |
| Space group | P42/n |
| a/Å | 14.3814(2) |
| c/Å | 18.3531(4) |
| V/Å3 | 3795.87(13) |
| Z | 4 |
| d calcd/g cm−3 | 1.224 |
| μ(Mo Kα)/mm−1 | 0.505 |
| R(F)a (I > 2σ(I)) | 0.0311 |
| R w(F2)b (all data) | 0.0807 |
| Goodness of fit | 1.050 |
| No. unique reflns | 4512 |
The structural investigations of this system indicated the production of the HL anion. We next identified the four C–O bond lengths in 1·solv. The C16–O1, C16–O2, C17–O3, and C17–O4 bond lengths were 1.2127(19), 1.292(2), 1.2661(19), and 1.2311(19) Å, respectively. The typical C–O and C–OH bond lengths of the carboxy group are 1.226(20) and 1.305(20) Å, whereas the typical C–O bond length of the carboxylate group is 1.255(10) Å.44 These findings allow the C16, O1, and O2 and the C17, O3, and O4 moieties to be assigned to the carboxy and the carboxylate groups, respectively.
As shown in Fig. 2a, the nearest O2⋯O3 distance of 2.4782(13) Å was smaller than the sum of the van der Waals radii (O/O: 3.04 Å),45 indicating a strong single {O–H⋯O−}-type H-bond. Considering the symmetry of this structure, the [Co(HL)2] unit extends in four directions, exhibiting a diamond framework (Fig. 2b). In addition, a 4-fold interpenetrating structure was present, as shown in Fig. 2c and d. The packing exhibited two species of 1-D pores along the [110] and [1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0] directions (Fig. 3). The space was occupied by the disordered crystal solvents, which were accounted for in the SQUEEZE/PLATON program. From the calculations, the solvent-accessible void volume was estimated to be 27.9% per unit cell. An electron count of 78 electrons per formula unit was obtained, which was close to the sum of the 1.2 DMF (48 electrons) and 2.4 H2O (24 electrons) solvents. This finding is consistent with the elemental analysis results.
0] directions (Fig. 3). The space was occupied by the disordered crystal solvents, which were accounted for in the SQUEEZE/PLATON program. From the calculations, the solvent-accessible void volume was estimated to be 27.9% per unit cell. An electron count of 78 electrons per formula unit was obtained, which was close to the sum of the 1.2 DMF (48 electrons) and 2.4 H2O (24 electrons) solvents. This finding is consistent with the elemental analysis results.
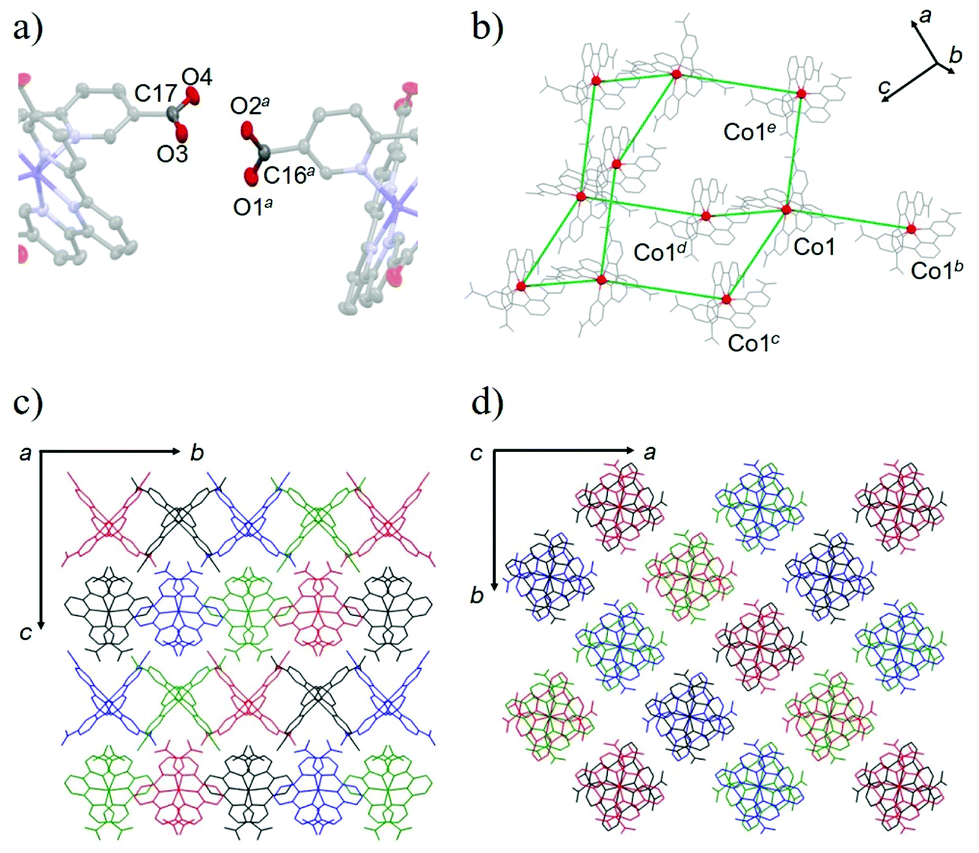 | ||
| Fig. 2 (a) Intermolecular H-bonding between carboxylate and carboxy sites. Symmetric code: a = −1/2 + y, 2 −x, −1/2 + z. (b) H-bonding network. The red and grey colours represent Co and non-metal atoms, respectively. The green lines represent the connection of the Co centres via H-bonding. Symmetric code: b = 1 − y, 1/2 + x, −1/2 + z. c = 1 − y, −1/2 + x, 1/2 + z. d = 2 − y, 1/2 + x, 1/2 + z. e = 2 − y, −1/2 + x, −1/2 + z. The H atoms are omitted for clarity. Packing structures of 1·solv along the (c) a- and (d) c-axes. The red, green, blue and black colours represent the independent H-bonding networks. | ||
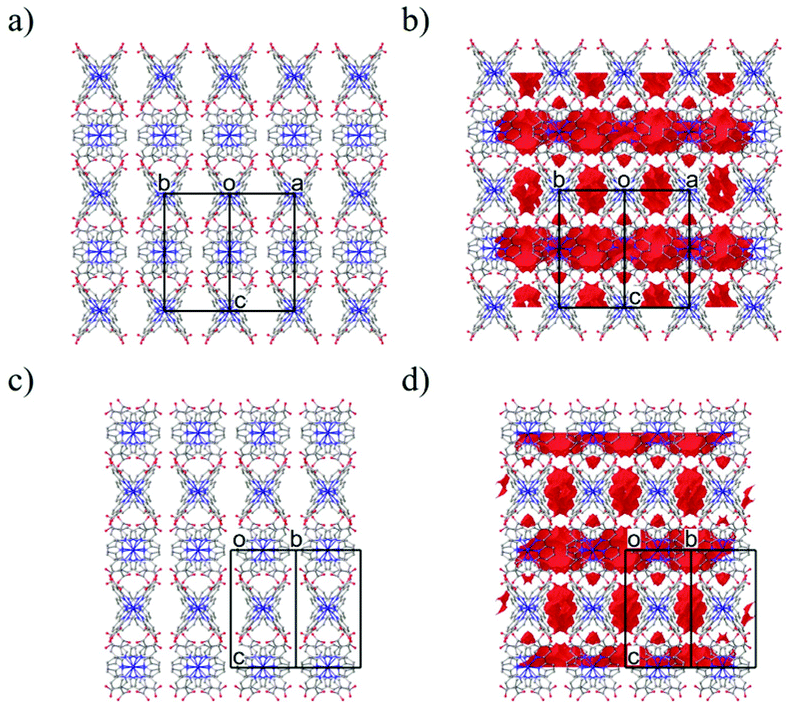 | ||
| Fig. 3 Packing structure of 1·solv along the [110] (a and b) and [10] (c and d) directions. (a) The only packing structure. (b) The packing structure with the solvent accessible void space (resolution, 0.1 Å−1; a red surface). The H atoms are omitted for clarity. | ||
Thermal analyses
Compound 1·solv was evaluated via thermogravimetry (TG) and differential thermal analysis (DTA) techniques, as shown in Fig. 4. The TG curve exhibited three-step decreasing processes at 320, 380, and 420 K. The weight loss (Δw) for these processes were approximately −3.3%, −7.0%, and −4.8%, respectively. These findings indicate the desorption of the crystal solvents. Upon further heating, the TG curve exhibited a plateau at 440 to 560 K. This plateau implies that 1·solv was fully desolvated (1·desolv, fw: 699.49). The Δw of approximately 15.1% at 300 to 440 K is equivalent to fw 124.4, corresponding to the sum of the 1.2 DMF (fw 87.7) and 2.4 H2O (fw 43.2) solvents observed in the results of the elemental and crystallographic analyses.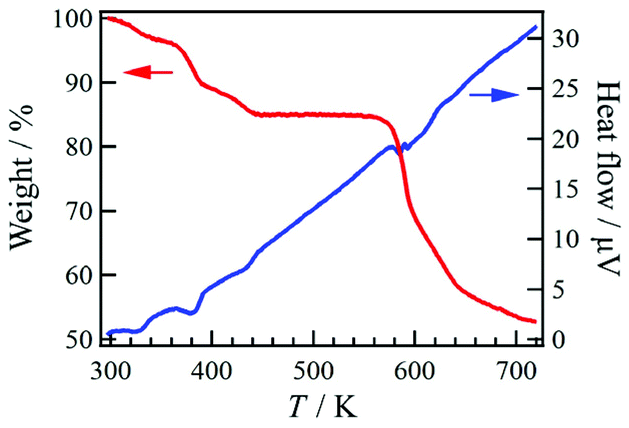 | ||
| Fig. 4 TG (red) and DTA (blue) results for 1·solv in the temperature range 300–720 K. | ||
Powder X-ray crystallography
Variable-temperature powder X-ray diffraction (VT-PXRD) was applied to 1·solv (Fig. 5). The simulation pattern from the above single-crystal XRD analysis was superposed onto Fig. 5 (indicated by the black line). The PXRD pattern at 298 K agrees well with the simulation results. Upon heating, the PXRD patterns changed three times at 333, 383, and 423 K. These temperatures agreed with those associated with the desorption of the crystal solvents, and the spectra above 423 K represent the PXRD pattern for 1·desolv. Interestingly, the structural change that occurred around 383 K showed a significant difference in the PXRD patterns, and the change of spectra indicates the reduced symmetry of crystal structure compared with that of 1·solv.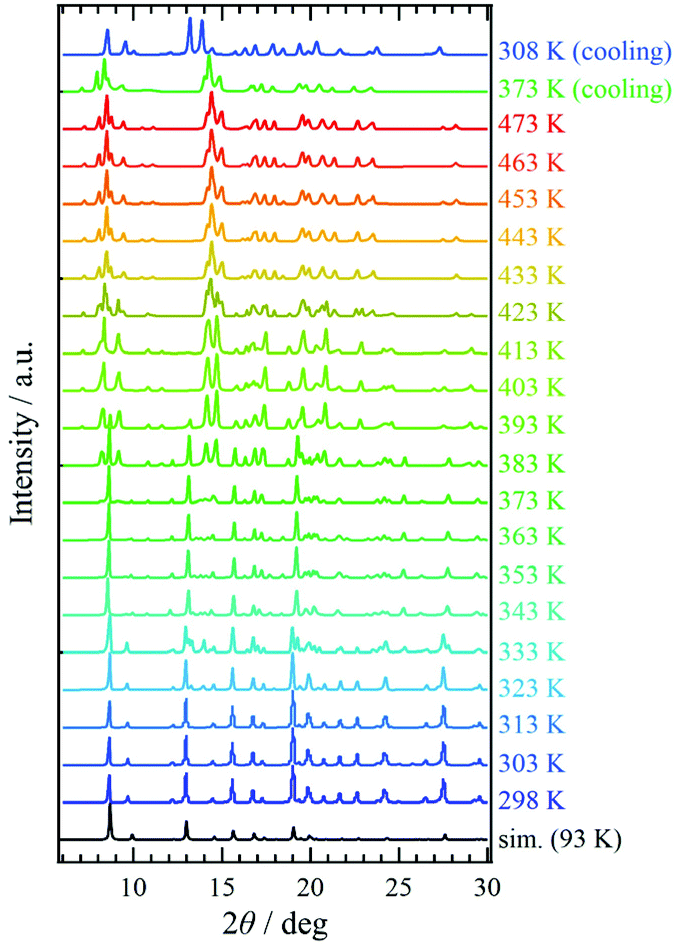 | ||
| Fig. 5 Variable-temperature PXRD measurements of 1·solv from 298 to 473 K and from 473 to 308 K. The simulation results (black line) were derived from the single-crystal X-ray crystallographic results obtained at 93 K. | ||
On cooling from 473 K, the PXRD pattern showed no change until 373 K. The spectrum at 308 K, however, changed and was close to that at 403 K in the heating process with the shift of peaks, implying the irreversible structural change owing to the desorption of crystal solvents. In addition, the finding indicates that 1·desolv captured the guest molecules, in which the water molecule is a good candidate, from the ambient environment in the cooling process from 373 K.
Magnetic properties
The temperature dependence of χmT for 1·solv was measured at 0.5 T in the range of 10 to 350 K, as shown in Fig. 6. The χmT value of 1·solv was 2.37 cm3 K mol−1 at 300 K, which is close to the expected value of 2.37 cm3 K mol−1 derived from the HS-cobalt(II) centre with g = 2.25. Upon cooling, the χmT value gradually decreased. This behaviour implies an intermolecular antiferromagnetic (AF) interaction or zero-field splitting (zfs) for the Co2+ centre. Conversely, when this system was heated from 320 K, χmT exhibited a sharply decreasing behaviour, indicating a structural change triggered by the desorption of crystal solvents.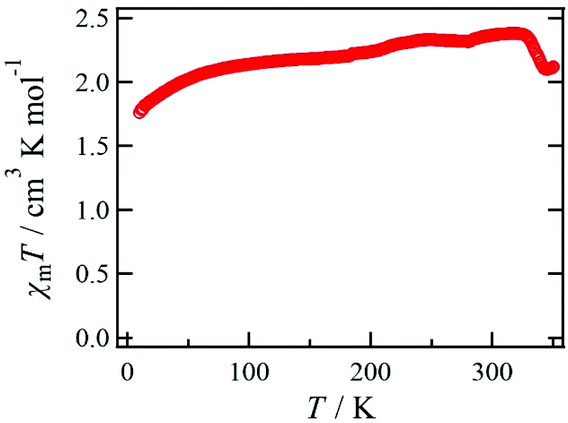 | ||
| Fig. 6 Temperature dependence of the product χmT measured at 0.5 T for 1·solv. | ||
The χmT vs. T plots for 1·desolv upon heating, cooling, and 2nd heating processes from 10 to 400 K are shown in Fig. 7. The sample 1·desolv used for these measurements was used after undergoing the above PXRD measurements. The χmT values above 350 K and below 250 K showed almost the same behaviour in all processes. The χmT values obtained at 400 and 200 K during the 1st heating process were 2.43 and 1.40 cm3 K mol−1, respectively. The former is close to 2.42 cm3 K mol−1 derived from the HS-cobalt(II) centre with g = 2.27, whereas the latter agrees well with 1.45 cm3 K mol−1 derived from the HS/LS state of 1/1, in which the expected value of the LS-cobalt(II) centre with g = 2.27 is 0.483 cm3 K mol−1. These χmT values indicate that 1·desolv exhibited an incomplete SCO behaviour in the range of 200 to 400 K. Conversely, the χmT values obtained during the cooling process traced the outside of those obtained during heating; therefore, a thermal hysteresis loop occurred. Below 250 K, χmT gradually decreased and reached 0.92 cm3 K mol−1 at 10 K. This finding may have resulted from the intermolecular AF interaction or the zfs of the Co centre.
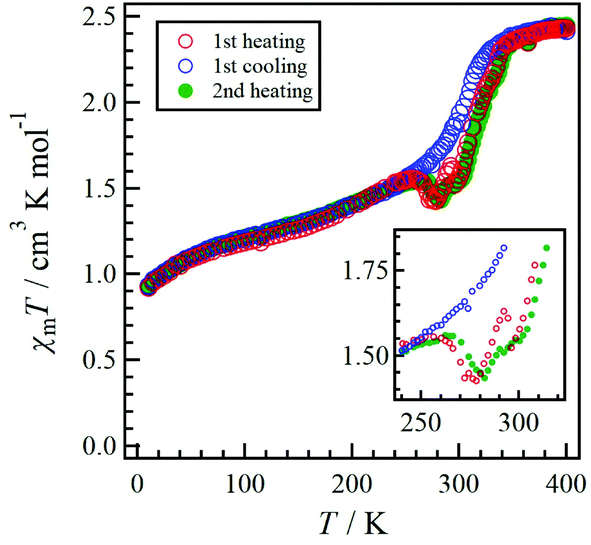 | ||
| Fig. 7 Temperature dependence of the product χmT measured at 0.5 T for 1·desolv. The red, blue, and green colours represent the 1st heating, 1st cooling, and 2nd heating processes, respectively. The inset shows an enlarged view of the 240 to 320 K region. | ||
Although 1·desolv exhibited typical SCO behaviour during the cooling process, a decrease of χmT was observed from 250 to 270 K during the 1st and 2nd heating processes, as shown in the inset of Fig. 7. Note that a peak can be observed around 290 K in the 1st heating process, which nearly vanished in the 2nd heating process, implying the desorption of the reabsorbed guest molecules. To understand the atypical SCO behaviour that was observed during the heating process, we propose the mechanism shown in Scheme 2. The χmT behaviour from 400 to 200 K exhibitions a spin transition between the HS and 1/1 HS/LS states. The spin state at 200 K indicates the presence of two non-equivalent Co2+ ions, although 1·solv showed only one crystallographic independent Co2+ ion. To illustrate this spin state, it may be reasonable to assume that at least two crystallographic independent systems arise from the 4-fold interpenetrating diamond frameworks owing to the structural change resulting from the desorption of solvents. Therefore, the SCO behaviour in half of the frameworks upon cooling is as shown in Scheme 2. The spin alignment at 200 K should be alternating for the HS and LS states to achieve an even chemical pressure from the HS frameworks.
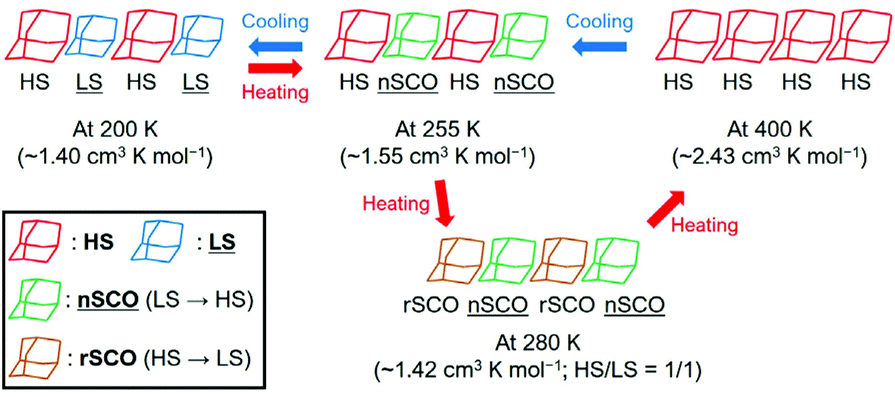 | ||
| Scheme 2 Proposed mechanism of the asymmetric SCO behaviour for 1·desolv in the temperature range 200–400 K. The normal and reverse SCO behaviours represent nSCO and rSCO, respectively. | ||
Upon heating from 200 K, the χmT value increased owing to the SCO behaviour, and it then decreased from 255 K and returned to the 1/1 HS/LS state at 280 K. These findings indicate a reverse SCO (rSCO) phenomenon, which is the spin transition from the HS to LS states. Owing to the reverse and normal SCO behaviour, 1 exhibits the steady-state conditions between the HS and LS groups, as shown in Scheme 2. These conditions were broken upon further heating, and all Co2+ centres showed the HS state.
The rSCO behaviour has been reported in the iron(II)46–49 and cobalt(II) complexes,33,37,50–53 and it is mainly explained as originating in the structural phase transition such as an order–disorder transition. For the rSCO behaviour of 1·desolv, we have focused on the intermolecular H-bond between carboxy and carboxylate sites. From the crystallographic studies of 1·solv, there was the intermolecular single {O–H⋯O−}-typed H-bond, which has double-well energy potential. When the distance between carboxy and carboxylate sites is shortened, the H-bond can exhibit the {O⋯H⋯O}− form having the single-well potential.54 It is known to demonstrate the crossover between the above two-typed H-bonds depending on the temperature, and the crossover gives the structural phase transition.55–58 Therefore, we have concluded the rSCO observed in 1·desolv may be triggered by the structural phase transition corresponding to the above crossover of H-bonds.
Conclusions
In summary, we prepared a novel H-MOF 1·solv formed by cobalt(II)-SCO units. Compound 1·solv exhibited a H-bonded diamond framework and a 4-fold interpenetrating structure with 1-D pores. The crystal solvents were occupied in the void spaces, and the desorption of those solvents caused three structural changes. Compound 1·desolv exhibited normal and reverse SCO behaviours with asymmetric thermal hysteresis. H-MOF 1·desolv exhibited a porous architecture and magnetic switching characteristics. For improvement of the reversibility and selectivity of guest molecules in this system, a multi-component H-MOF system is a good candidate; for example, a two-component H-MOF, which is a cocrystal of the [Co(HL)2] unit and an H-bonding formable linker. The multi-component H-MOF system presented in this study may be used to form a variety of porous structures.Experimental
Materials and methods
5,5′′-Dimethyl-2,2′:6′,2′′-terpyridine was prepared by the reported procedure.41,42 Anhydrous N,N-dimethylformamide (DMF) was purchased from KANTO Chemical Co., Inc. and used without further purification. 1H and 13C NMR experiments (400 and 100 MHz, respectively) were performed on JNM-ECZ400S (JEOL). The chemical shift (given in ppm) was measured versus a reference peak of tetramethylsilane (TMS). The splitting patterns are designated as follows: s (singlet), d (doublet), t (triplet), and dd (doublet of doublets). Elemental analyses were carried out with a PerkinElmer Series II CHNS/O 2400 analyzer. Melting point measurement was performed on ATM-02 (AS ONE). High-resolution mass spectra (HRMS) were recorded in electrospray ionization (ESI) mode using an AccuTOF-JMS-T100LP (JEOL) spectrometer. The specimen was dissolved in a small amount of DMF, and then the solution was diluted with methanol. Infrared (IR) spectra were obtained on an FT/IR-4600 (JASCO) spectrometer using a diamond attenuated total reflectance (ATR) method. The spectral data are obtained as major peaks in wavenumbers (cm−1) are recorded in a spectral window of 4000–400 cm−1. Electronic spectra (ultraviolet–visible, UV–Vis) were obtained on a JASCO V-650 spectrometer. Thermogravimetry (TG) and differential thermal analysis (DTA) of 1·solv were carried out on a Bruker AXS TG2010SA instrument. The temperature scan rate was 5 K min−1 in the range of 300–720 K. Powder X-ray diffraction (PXRD) spectra were recorded using a Rigaku MiniFlex600 diffractometer (Cu Kα radiation: λ = 1.541862 Å) in the range of 298–473 K.Synthesis of 2,2′:6′,2′′-terpyridine-5,5′′-diyl biscarboxylic acid (H2L)
5,5′′-Dimethyl-2,2′:6′,2′′-terpyridine (720.0 mg, 2.76 mmol) was dissolved in 4/3 pyridine/H2O (70 mL), and thus KMnO4 (4.06 g, 25.7 mmol) was added to the solution. After the mixture was refluxed for 4 h, KMnO4 (2.02 g, 12.8 mmol) was added, again. The mixture was refluxed for 12 h and quenched by ethanol. The filtrate was concentrated under reduced pressure. The crude was dissolved in a small amount of H2O, and the solution was made pH 3 by a dilute HCl aqueous, affording colorless precipitants. The obtained colorless powder H2L was a yield of 719.8 mg (2.24 mol, 81%). Mp. >200 °C. 1H NMR (400 MHz, DMSO-d6, Fig. S1†): δ 13.57 (br s, 2H), 9.21 (s, 2H), 8.77 (d, J = 8.2 Hz, 2H), 8.57 (d, J = 7.7 Hz, 2H), 8.48 (dd, J = 8.2, J′ = 1.6 Hz, 2H), and 8.20 (t, J = 7.7 Hz, 1H). 13C NMR (100 MHz, DMSO-d6, Fig. S2†): δ 166.13, 158.00, 154.02, 150.20, 138.98, 138.35, 126.78, 122.28, and 120.54. HRMS (ESI+, Fig. S3†): m/z calcd for C17H12N3O4 [M + H]+: 322.08278, found 322.08251. IR (ATR): 2841, 1682, 1591, 1557, 1421, 1380, 1298, 1138, 1020, 921, 824, 722, 649, and 541 cm−1. UV-vis (DMF) λmax/nm (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε) 294 (1.06).
ε) 294 (1.06).
Synthesis of [Co(HL)2](DMF)1.2(H2O)2.4 (1·solv)
The H2L (81.73 mg, 0.262 mmol) and CoCl2 (14.12 mg, 0.109 mmol) were dissolved in DMF (10 mL) and H2O (3 mL), respectively. The above two solutions were mixed and stirred for 15 minutes at 0 °C. The red mixture was crystallized under 1,4-dioxane vapor diffusion for 5 days. The black block crystals were collected, and the yield was 53.05 mg (0.0674 mmol, 62%). Mp. 272 °C (decomp.). Anal. Calc. for C37.6H33.2CoN7.2O11.6 (1·(DMF)1.2(H2O)2.4): C, 54.38; H, 4.03; N, 12.14%. Found: C, 54.38; H, 3.84; N, 12.00%. IR (ATR): 3069, 1600, 1449, 1354, 1249, 1132, 1025, 770, and 718 cm−1.Single-crystal X-ray diffraction
The X-ray diffraction data of 1·solv at 93 K were collected on a Rigaku VariMax Dual (Mo Kα radiation: λ = 0.71073 Å). The selected crystallographic data are given in Table 1. X-ray data analyses were carried out using the SHELXT59 and SHELXL60 programs operated with the Olex2 interface.61 All the hydrogen atoms were refined as “riding”. The thermal displacement parameters of the non-hydrogen atoms were refined anisotropically. The contribution of the disordered solvent was removed using the SQUEEZE option from PLATON operated with the Olex2 interface.61 The estimated total solvent-accessible void space (1.2 Å probe) was 1060.9 Å3 per the unit cell (27.9%) and 78 electrons per unit cell. The squeeze electron count correlates with the electron density of 1.2 DMF and 2.4 H2O molecules of unit cell (72 electrons). Fig. 3 represents the packing structure along with the [110] and [10] directions, respectively. Each figure also shows the solvent-accessible void space (a red surface). The CCDC number is 2143196.†
Magnetic measurements
The direct current magnetic susceptibilities of 1·solv and 1·desolv were measured on a Quantum Design MPMS-XL7AC SQUID magnetometer equipped with a 7 T coil under the static field of 0.5 T. The sample 1·desolv was used after the PXRD measurement, in which the sample was heated to 473 K. The 1·solv was measured on heating in the 10–350 K range shown in Fig. 6, whereas 1·desolv was measured on heating, cooling, and then 2nd heating processes in the 10–400 K range shown in Fig. 7. The magnetic data were corrected using diamagnetic blank data of sample holder measured separately. The diamagnetic contribution of the sample itself was estimated from Pascal's constant.62Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was financially supported by KAKENHI (JSPS. 21K14604). The crystallography work for 1·solv was conducted at the Advanced Characterization Nanotechnology Platform of the University of Tokyo, supported by the “Nanotechnology Platform” of MEXT, Japan.Notes and references
- D. S. Reddy, S. Duncan and G. K. H. Shimizu, Angew. Chem., Int. Ed., 2003, 42, 1360 CrossRef CAS PubMed.
- C.-L. Chen and A. M. Beatty, J. Am. Chem. Soc., 2008, 130, 17222 CrossRef CAS PubMed.
- S. S. Mondal, A. Bhunia, A. Kelling, U. Schilde, C. Janiak and H.-J. Holdt, Chem. Commun., 2014, 50, 5441 RSC.
- J. Wu, M. Guo, X.-L. Li, L. Zhao, Q.-F. Sun, R. A. Layfield and J. Tang, Chem. Commun., 2018, 54, 12097 RSC.
- Z. Bao, D. Xie, G. Chang, H. Wu, L. Li, W. Zhou, H. Wang, Z. Zhang, H. Xing, Q. Yang, M. J. Zaworotko, Q. Ren and B. Chen, J. Am. Chem. Soc., 2018, 140, 4596 CrossRef CAS PubMed.
- G. Gerer, F. Melin, P. Hellwig, M. W. Hosseini and S. Ferlay, Inorg. Chem., 2019, 58, 1541 CrossRef CAS PubMed.
- H.-L. Wang, X.-F. ma, Z.-H. Zhu, Y.-Q. Zhang, H.-H. Zou and F.-P. Liang, Inorg. Chem. Front., 2019, 6, 2906 RSC.
- Z.-Q. Shi, N.-N. Ji, K.-M. Guo and G. Li, Appl. Surf. Sci., 2020, 504, 144484 CrossRef CAS.
- Z.-H. Zhu, H.-L. Wang, H.-H. Zou and F.-P. Liang, Dalton Trans., 2020, 49, 10708 RSC.
- B. Yuan, Y. Tang, G. Gou, W. Dan and L. Li, CrystEngComm, 2020, 22, 3921 RSC.
- D. I. Konovalov, A. A. Ivanov, Y. A. Vorotnikov, N. V. Kuratieva, I. V. Eltsov, K. A. Kovalenko and M. A. Shestopalov, Inorg. Chem., 2021, 60, 14687 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- Z.-F. Wu, B. Tan, W. P. Lustig, E. Velasco, H. Wang, X.-Y. Huang and J. Li, Coord. Chem. Rev., 2019, 399, 213025 CrossRef CAS.
- L. S. Xie, G. Skorupskii and M. Dincă, Chem. Rev., 2020, 120, 8536 CrossRef CAS PubMed.
- X. Chen, K. Geng, R. Liu, K. T. Tan, Y. Gong, Z. Li, S. Tao, Q. Jiang and D. Jiang, Angew. Chem., Int. Ed., 2020, 59, 5050 CrossRef CAS PubMed.
- H. L. Nguyen, Chem. Sci., 2021, 12, 8632 RSC.
- I. Hisaki, C. Xin, K. Takahashi and T. Nakamura, Angew. Chem., Int. Ed., 2019, 58, 11160 CrossRef CAS PubMed.
- I. Hisaki, J. Inclusion Phenom. Macrocyclic Chem., 2020, 96, 215 CrossRef CAS.
- R. Liang, J. Samanta, B. Shao, M. Zhang, R. J. Staples, A. D. Chen, M. Tang, Y. Wu, I. Aprahamian and C. Ke, Angew. Chem., Int. Ed., 2021, 60, 23176 CrossRef CAS PubMed.
- J. Zhang, Y. Feng, R. J. Staples, J. Zhang and J. M. Shreeve, Nat. Commun., 2021, 12, 2146 CrossRef CAS PubMed.
- Y.-H. Luo, L. Zhang, W.-X. Fang, S.-H. Ma, H. Dong, S. Su, Z.-Y. Zheng, D.-N. Li and L.-H. Zhai, Chem. Commun., 2021, 57, 5901 RSC.
- M. Igarashi, T. Nozawa, T. Matsumoto, F. Yagihashi, T. Kikuchi and K. Sato, Nat. Commun., 2021, 12, 7025 CrossRef CAS PubMed.
- A. M. W. C. Thompson, Coord. Chem. Rev., 1997, 160, 1 CrossRef CAS.
- A. Wild, A. Winter, F. Schlütter and U. S. Schubert, Chem. Soc. Rev., 2011, 40, 1459 RSC.
- E. C. Constable, Chem. Soc. Rev., 2007, 36, 246 RSC.
- P. R. Andres and U. S. Schubert, Adv. Mater., 2004, 16, 1043 CrossRef CAS.
- J. Veliks, J.-C. Tseng, K. I. Arias, F. Weissher, A. Linden and J. S. Siegel, Chem. Sci., 2014, 5, 4317 RSC.
- S. Hayami, Y. Komatsu, T. Shimizu, H. Kamihata and Y. H. Lee, Coord. Chem. Rev., 2011, 255, 1981 CrossRef CAS.
- X. Zhang, H. Xie, M. Ballesteros-Rivas, Z.-X. Wang and K. R. Dunbar, J. Mater. Chem. C, 2015, 3, 9292 RSC.
- Y. Guo, X.-L. Yang, R.-J. Wei, L.-S. Zheng and J. Tao, Inorg. Chem., 2015, 54, 7670 CrossRef CAS PubMed.
- A. Ondo and T. Ishida, Crystals, 2018, 8, 155 CrossRef.
- Y. Pankratova, D. Aleshin, I. Nikovskiy, V. Novikov and Y. Nelyubina, Inorg. Chem., 2020, 59, 7700 CrossRef CAS PubMed.
- R. Akiyoshi, R. Ohtani, L. F. Lindoy and S. Hayami, Dalton Trans., 2021, 50, 5065 RSC.
- Spin Crossover in Transition Metal Compounds I, II and III, ed. P. Gütlich and H. A. Goodwin, Springer, Berlin, Germany, 2004 Search PubMed.
- D. Shao, L. Shi, F.-X. Shen, X.-Q. Wei, O. Sato and X.-Y. Wang, Inorg. Chem., 2019, 58, 11589 CrossRef CAS PubMed.
- M. Nakaya, W. Kosaka, H. Miyasaka, Y. Komatsumaru, S. Kawaguchi, K. Sugimoto, Y. Zhang, M. Nakamura, L. F. Lindoy and S. Hayami, Angew. Chem., Int. Ed., 2020, 59, 10658 CrossRef CAS PubMed.
- F. Kobayashi, Y. Komatsumaru, R. Akiyoshi, M. Nakamura, Y. Zhang, L. F. Lindoy and S. Hayami, Inorg. Chem., 2020, 59, 16843 CrossRef CAS PubMed.
- H. Zenno, F. Kobayashi, M. Nakamura, Y. Sekine, L. F. Lindoy and S. Hayami, Dalton Trans., 2021, 50, 7843 RSC.
- Y.-F. Deng, Y.-N. Wang, X.-H. Zhao and Y.-Z. Zhang, CCS Chem., 2021, 3, 3277 CrossRef.
- T. Kanetomo, K. Inokuma, Y. Naoi and M. Enomoto, Dalton Trans., 2021, 50, 11243 RSC.
- T. Seckin, I. Özdemir, S. Köytepe and N. Gürbüz, J. Inorg. Organomet. Polym., 2009, 19, 143 CrossRef CAS.
- B. Golesorkhi, L. Guénée, H. Nozary, A. Fürstenberg, Y. Suffren, S. V. Eliseeva, S. Petoud, A. Hauser and C. Piguet, Chem. – Eur. J., 2018, 24, 13158 CrossRef CAS PubMed.
- C. A. Kilner and M. A. Halcrow, Dalton Trans., 2010, 39, 9008 RSC.
- F. H. Allen, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, in International Tables for Crystallography, ed E. Prince, Springer, Berlin, 2006, vol. C, pp. 790 Search PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441 CrossRef CAS.
- M. Seredyuk, M. C. Muñoz, M. Castro, T. Romero-Morcillo, A. B. Gaspar and J. A. Real, Chem. – Eur. J., 2013, 19, 6591 CrossRef CAS PubMed.
- M. Yamashita and T. Ishida, J. Mater. Chem. C, 2015, 3, 7784 RSC.
- M. Weselski, M. Książek, P. Mess, J. Kusz and R. Bronisz, Chem. Commun., 2019, 55, 7033 RSC.
- R. Kulmaczewski, F. Bamiduro, N. Shahid, O. Cespedes and M. A. Halcrow, Chem. – Eur. J., 2021, 27, 2082 CrossRef CAS PubMed.
- G. Agustí, C. Bartual, V. Martínez, F. J. Muñoz-Lara, A. B. Gaspar, M. C. Muñoz and J. A. Real, New J. Chem., 2009, 33, 1262 RSC.
- S. Hayami, M. Nakaya, H. Ohmagari, A. S. Alao, M. Nakamura, R. Ohtani, R. Yamaguchi, T. Kuroda-Sowa and J. K. Clegg, Dalton Trans., 2015, 44, 9345 RSC.
- K. Kuroiwa, T. Arie, S. Sakurai, S. Hayami and T. J. Deming, J. Mater. Chem. C, 2015, 3, 7779 RSC.
- Z.-S. Yao, S.-Q. Wu, Y. Kitagawa, S.-Q. Su, Y.-G. Huang, G.-L. Li, Z.-H. Ni, H. Nojiri, Y. Shiota, K. Yoshizawa, S. Kang, S. Kanegawa and O. Sato, Angew. Chem., Int. Ed., 2017, 56, 717 CrossRef CAS PubMed.
- S. J. Grabowski, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2006, 102, 131 RSC.
- E. J. Samuelsen and D. Semmingsen, J. Phys. Chem. Solids, 1977, 38, 1275 CrossRef CAS.
- T. Mitani, G. Saito and H. Urayama, Phys. Rev. Lett., 1988, 60, 2299 CrossRef CAS PubMed.
- A. Ueda, Bull. Chem. Soc. Jpn., 2017, 90, 1181 CrossRef CAS.
- C. Shi, X. Zhang, C.-H. Yu, Y.-F. Yao and W. Zhang, Nat. Commun., 2018, 9, 481 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- L. J. Bourhis, O. V. Dolomanov, R. J. Gildea, J. A. K. Howard and H. Puschmann, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 59 CrossRef CAS PubMed.
- O. Kahn, Molecular Magnetism, VCH-Verlag, Weinheim, New York, 1993 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2143196. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2dt00453d |
| This journal is © The Royal Society of Chemistry 2022 |