
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium-nitrosyl complexes as NO-releasing molecules, potential anticancer drugs, and photoswitches based on linkage isomerism
Iryna
Stepanenko
*a,
Michal
Zalibera
 *b,
Dominik
Schaniel
*c,
Joshua
Telser
*d and
Vladimir B.
Arion
*a
*b,
Dominik
Schaniel
*c,
Joshua
Telser
*d and
Vladimir B.
Arion
*a
aUniversity of Vienna, Institute of Inorganic Chemistry, Währinger Strasse 42, A-1090 Vienna, Austria. E-mail: vladimir.arion@univie.ac.at
bInstitute of Physical Chemistry and Chemical Physics, Faculty of Chemical and Food Technology, Slovak University of Technology in Bratislava, Radlinského 9, SK-81237 Bratislava, Slovak Republic. E-mail: michal.zalibera@stuba.sk
cUniversité de Lorraine, CNRS, CRM2, 54500 Nancy, France. E-mail: dominik.schaniel@univ-lorraine.fr
dDepartment of Biological, Physical and Health Sciences, Roosevelt University, 430 S. Michigan Avenue, Chicago, Illinois 60605, USA. E-mail: jtelser@roosevelt.edu
First published on 28th February 2022
Abstract
The synthesis of new types of mono- and polynuclear ruthenium nitrosyl complexes is driving progress in the field of NO generation for a variety of applications. Light-induced Ru-NO bond dissociation in solution may involve transient linkage isomers MS1 (Ru-ON) and MS2 (Ru-η2-NO), which can be detected spectroscopically and analyzed computationally. The investigation of photoisomerization processes in the solid state may be useful for potential application of such complexes for data storage, photochromic or photomagnetic materials or even non-linear optics. Herein we describe the major developments in the synthesis of ruthenium nitrosyl complexes, their photoinduced linkage isomerization (PLI) processes, their NO release both in the solid state and in solution, and their application as potential anticancer drugs. Illustrative examples of such innovations made mainly in the last decade are provided.
1. Introduction
Nitric oxide (NO) was for a long time considered a poisonous gas, a carcinogen, and a pollutant. This is not surprising since NO is a highly reactive free radical. However, the negative reputation completely changed with the discoveries of Ignarro, Furchgott, and Murad, who established NO as an important neurotransmitter in mammalian biology.1 Today, we understand that the effects of NO depend on its cellular concentration.2 At low levels (<200 nM) NO is beneficial: it induces smooth muscle relaxation resulting in a decrease in blood pressure; macrophages generate NO against pathogens and microorganisms;3 in neuronal cells, NO controls the release of neurotransmitters and is involved in synaptogenesis, synaptic plasticity, memory function, and neuroendocrine secretion.4 Similarly, NO has been implicated in carcinogenesis and inhibition of tumor growth. At low levels, NO induces cell proliferation by activating oncogenes, stimulates angiogenesis, and inhibits apoptosis by S-nitrosylation and inactivation of caspases. At high concentrations (>200 nM), NO becomes toxic and causes cell death by apoptosis.5The promise of NO in cancer therapeutics has been realized by the introduction of several classes of NO donors and the recent development of novel hybrid drugs.6 However, considering the variable half-life, complex pharmacokinetics, and multiple reactions of NO with bystander molecules, it remains a challenge to fine-tune the molecular properties of drug candidates containing NO. The coordination chemistry of nitric oxide has been a long time solely a subject of academic interest, aimed at the study of the structure and bonding in metal nitrosyl complexes.7 Nitric oxide is a small diatomic molecule with an unpaired number of electrons, which, in addition, is redox-active and can be involved in both one electron oxidation to NO+, and one electron reduction to NO−, both being electron-paired species. The protonated form of the NO− HNO is of particular interest in biological systems.8 The non-innocence of NO in coordination compounds was recognized many years ago,9 and these three Lewis forms, NO+,0,−, as ligands have been encountered in numerous metal complexes. Recently, a fourth form, NO2−, was discovered in {[(Me3Si)2N]2(THF)Y}2(μ–η2:η2-NO),10 and confirmed by EPR spectroscopy and additional chemical transformations of this dimeric complex. All four Lewis forms of nitric oxide which can act as ligands in metal complexes are shown in Scheme 1.11
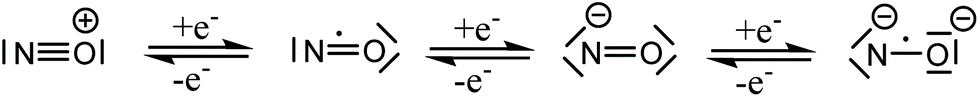 | ||
| Scheme 1 Different redox states of “nitrosyl” in metal complexes. | ||
The ambiguity in defining the formal charge of the central metal and the NO ligand in the transition metal nitrosyls led to the introduction of the Enemark–Feltham notation {M-NO}x,12 where M defines the corresponding central metal and x represents the sum of electrons in the d orbitals of the metal and π* orbital of the NO moiety.
Nowadays, the coordination chemistry of nitric oxide has regained momentum with attempts to use metal complexes with a high NO affinity for scavenging free NO, while metal-nitrosyl complexes have been used for the delivery of NO in biological environments.2,7,13
In vivo release of NO from metal-nitrosyl complexes may occur through biological redox processes. However, this route is usually slow, nonspecific, and produces low amounts of NO. This is useful in the case of a vasodilator but is ineffective in providing high doses of NO to induce apoptosis in cancer cells. In this context, the photorelease of NO from metal-nitrosyl complexes seems to be an easier way to deliver the required NO amounts to specific targets.
Only in the last two decades have researchers turned to metal-nitrosyl complexes that release NO by photoactivation for therapeutic applications.14 Photoexcitation of metal-nitrosyl complexes leads to NO release or isomerism of the NO linkage, and its photosensitivity depends mainly on the location of the dpπ(M) → π*(NO) metal to ligand charge transfer (MLCT) transition in its electronic absorption spectrum.15 Most of these clinical trials focused on ruthenium-nitrosyl complexes because of the photolability of the RuNO bond and because of the fewer toxicity problems of Ru compared to other metals, as documented from clinical experience with ruthenium anticancer drugs. Unfortunately, most of the ruthenium-nitrosyls known today require excitation by biologically harmful UV radiation. Pushing the limit of MLCT excitation to the Vis range represents one of the major challenges in the design of NO-releasing anticancer metallodrugs. Once achieved, the complexes can be combined with upconverting nanoparticle antennas that utilize multiphoton NIR excitation and take advantage of the deeper penetration of NIR radiation into living tissue.16
It has been recently suggested that light-induced Ru-NO bond dissociation in solution17 may involve transient linkage isomers MS1 (Ru-ON) and MS2 (Ru-η2-NO).18,19 Therefore, the investigation of these intermediates is of great interest. Moreover, these photoisomerization processes in the solid state have potential to be used for data storage,20,21 and in the design of photochromic materials22 or hologram gratings because of the difference in refractive index of ground and metastable states.23 Another interest is the use of ruthenium nitrosyl complexes as building blocks for assembly of systems combining a photo-switchable unit with a paramagnetic moiety in the same crystal or compound.24,25 Such systems may allow for light induced linkage isomerization, which will trigger changes in magnetic behavior leading potentially to novel functional materials.26
Herein we will briefly discuss the most common synthetic routes towards ruthenium nitrosyl complexes with a variety of ligands and the compounds often used as starting materials (section 2). Several examples of preparation of related chalcogenonitrosyl complexes will be also presented. Rare mono- and heteropolynuclear complexes, as well as double complex salts, bearing a ruthenium nitrosyl moiety will be covered in a separate section along with their potential application (section 3). Other important topics addressed herein will be linkage isomerism and its possible application along with NO release in the solid state (section 4), as well as NO release in solution triggered by 1e reduction and photoexcitation (section 5). Recent developments in the field of ruthenium nitrosyl complexes as potential anticancer drugs will be reported in section 6. Section 7 will be dealing with antibacterial activity and other applications of ruthenium nitrosyl complexes. Conclusions and outlook will be presented in section 8.
2. Synthesis of ruthenium complexes with chalcogenonitrosyl (NE, E = O, S, Se, Te) ligands
The most common synthetic routes to ruthenium nitrosyl complexes include: (a) the use of specific starting materials, which already contain the ruthenium nitrosyl moiety, via a variety of substitution reactions (NO2 by Cl,27 NH3,28,29 pyridines,29–33 pyrazine;34 NO3 by H2O, F;35 OH by F;22,36,37 Cl by pyridines,38–40 tetradentate Schiff bases,41,42 tetradentate 2-hydroxybenzamidobenzene derivatives,43 bis-phosphine monoxide ligands;44 H2O by Cl,45 SO4;46 NH3 by Cl46) or metathesis reactions of Na+ to Ba2+,29 Ba2+ to NH4+,29 Cl− to ClO4−,47 PF6−,48 in solution and in the solid state;45,46 (b) conversion of the coordinated nitro ligand into nitrosyl in acidic media (HCl, TFA, HFP6, HNO3) reported for ruthenium triammine complex,45 as well as for compounds with pyridine, bipyridine, terpyridine, phenanthroline, triazine, and indazole ligands (see references in Table 1); (c) direct reaction of ruthenium pyridine, bipyridine, terpyridine, porphyrin, corrole species (see Table 1) or ruthenium azole complexes49 with NO via substitution reactions of labile monodentate ligands, e.g., Cl,50–54 H2O,55,56 DMSO,57etc. As a source of nitric oxide, NO gas was used.52–54,57 This gas can also be produced by reactions of sodium nitrite with HBF4 or H2SO4,51,55 and copper with HNO3.50 NOBF4 was used to introduce the second NO ligand into the coordination sphere of ruthenium.58 Other sources of NO such as oximes59 and alkyl nitrites60 were also successfully used for the generation of the RuNO unit. The most ubiquitous precursors recently exploited are summarized in Table 1.| Ref. | |
|---|---|
| Ru(NO) precursors | |
| cis-[RuNO(NH3)2(NO2)2OH] | 27 |
| fac-[RuNO(NH3)2(NO3)3] | 35 |
| trans-(NO,NO3)cis-(NH3,NH3)[RuNO(NH3)2(L)2(NO3)](NO3)2, where L = pyridine | 35 |
| Na2[RuNO(NO2)4OH] | 28 and 30–34 |
| (NH4)2[RuNO(NO2)4OH] | 29 |
| trans-[RuNO(NH3)4OH](NO3)2 | 22, 36 and 37 |
| trans-[RuNO(NH3)4OH]Cl2 | 46 and 61 |
| K2[RuNOCl5] | 38–40 |
| Hydrated Ru(NO)Cl3 | 41–44 and 62–64 |
| [Ru(NO)Cl3(PPh3)2] | 44 and 65 |
| mer-[RuNO(NH3)3(H2O)Cl]Cl2 | 45 |
| trans-[Ru(NO)(NH3)4(H2O)](SO4)HSO4 | 46 |
| Ru(NO 2 ) precursors | |
| Na[fac-Ru(NH3)3(NO2)3] | 45 |
| trans-[Ru(NO2)2(L)4], where L = 4-methyl pyridine, 4-chloropyridine, 1H-indazole | 17 and 66, 67 |
| cis-[Ru(NO2)(L1)2L2]m, where L1 = 2,2′-bipyridine; L2 = 4-benzoylpyridine, 4-methylpyridine, a modified lysine residue (L−) | 68–70 |
| [Ru(NO2)(L1)(L2)]+, where L1 = substituted on the 4′-position of terpyridine ligands, N,N-dimethyl-4,6-di(pyridin-2-yl)-1,3,5-triazin-2-amine; L2 = un- or substituted 2,2′-bipyridines, 1,10-phenanthroline | 19 and 71–78 |
| cis-[Ru(NO2)2(L)2], where L = 5,5′-dimethyl-2,2′-bipyridine | 79 |
| Ru precursors used for direct reaction with NO | |
| trans-[Ru(NH3)4(L)H2O]2+, where L = isonicotinamide | 55 |
| cis-[RuCl2(L)2], where L = 2,2′-bipyridine-4,4′-dicarboxylic acid | 50 |
| [RuCl(L1)(L2)2]PF6, where L1 = thiourea, thiobenzamide, L2 = phenanthroline | 51 |
| trans-[RuCl2(dmso-S)4] | 57 and 59 |
| cis-[RuCl2(dmso-S)3(dmso-O)] | 57 and 59 |
| [(dmso)2H][trans-RuCl4(dmso-S)2] | 57 and 59 |
| mer-[RuCl3(dmso)3] | 57 and 59 |
| mer-[RuIIICl3(L)], where L = substituted on the 4′-position of terpyridine ligands | 52–54 |
| [Ru(NO)(PR3)2X3] (X = Cl, Br, I) | 58 and 80 |
| [Ru(CO)(L)], where H2L = tetra(p-methoxyphenyl)porphyrin, tetra(p-tolyl)porphyrin | 60 |
| [Ru(L)]2, where H3L = meso-tris(p-X-phenyl)corroles, X = CF3, H, Me, and OMe | 81 |
| [Ru(COD)Cl2]x (x ≥ 2; COD = 1,5-cyclooctadiene) | 81 |
A textbook example of using different synthetic routes to ruthenium nitrosyls is the synthesis of ruthenium nitrosyl tetraazamacrocycles reviewed recently82 and involves three approaches: the use of precursors that already contains a nitrosyl group, i.e., K2[Ru(NO)Cl5] or “RuNOCl3·nH2O”, exposure of ruthenium(III) complexes with coordinated tetraazamacrocycles to NO or conversion of a bound nitrite to coordinated nitrosyl.
The possibility of chemical conversion of small-molecule bioregulator such as NO into other signaling molecules in biological organisms, and the interaction between redox signaling agents must be also considered.83 Therefore, it seems appropriate to cover some ruthenium chalcogenonitrosyl (i.e., thionitrosyl, selenonitrosyl, and telluronitrosyl) complexes which appear to be relevant from this aspect.
Ruthenium thionitrosyl complexes can be synthesized by: (a) sulfur atom transfer to ruthenium(VI) nitrido species,84,85 (b) reaction of ruthenium(III) complexes with trithiazyl chloride (N3S3Cl3),86 (c) halide abstraction from thiazyl complexes, and (d) reaction of tetrasulfur tetranitride (S4N4) with metal halides or nitrides.87 Selenonitrosyl complexes can be prepared by treatment of ruthenium nitride species with elemental selenium.84
In contrast to NO, the chemistry of heavier congeners NS (nitric sulfide), NSe (nitric selenide) and NTe (nitric telluride), as well as of HNO, has gained much less attention. This is because these are elusive molecules; the NE species can be detected only at low temperatures.88–90 Coordination chemistry offers an opportunity to stabilize these elusive molecules by binding them to metals to gain insight into the chemistry of these labile and/or unstable molecules. Even though metal complexes containing NE (E = S, Se) have been prepared and comprehensively characterized for different metals, e.g. iridium,91,92 and osmium,93 herein we will briefly compare the ruthenium complexes containing the set of NE molecules with E = O, S and Se. Two series of such complexes are well-documented so far, namely charge neutral metal complexes mer,trans-[RuCl3(NE)(AsPh3)2] (see Scheme 2) and anionic complexes [Ph4P][Ru(NE)(hybebR1,R2)(DMAP)], where hybebR1,R2 = tetradentate 2-hydroxybenzamidobenzene derivatives, DMAP = dimethylaminopyridine (see Chart 1). Other ruthenium complexes with NE molecules characterized by SC-XRD and IR spectroscopy are shown in Table 2.
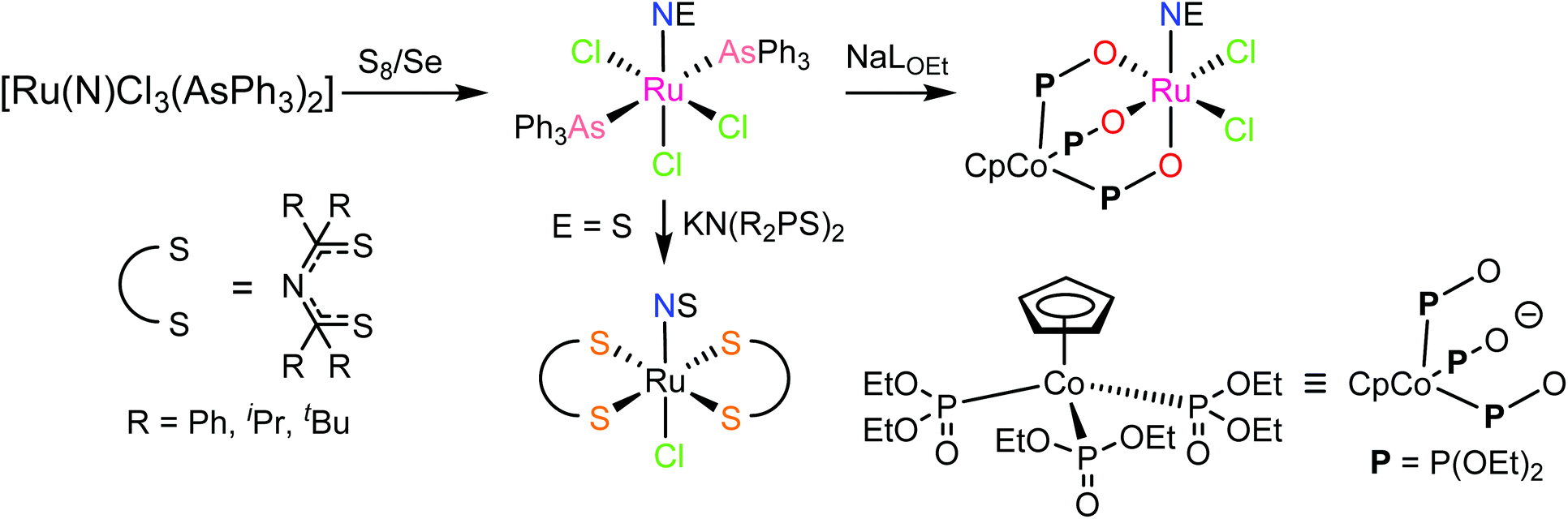 | ||
| Scheme 2 Synthesis of mer,trans-[RuCl3(NE)(AsPh3)2] (E = S, Se) and their conversion into other species. | ||
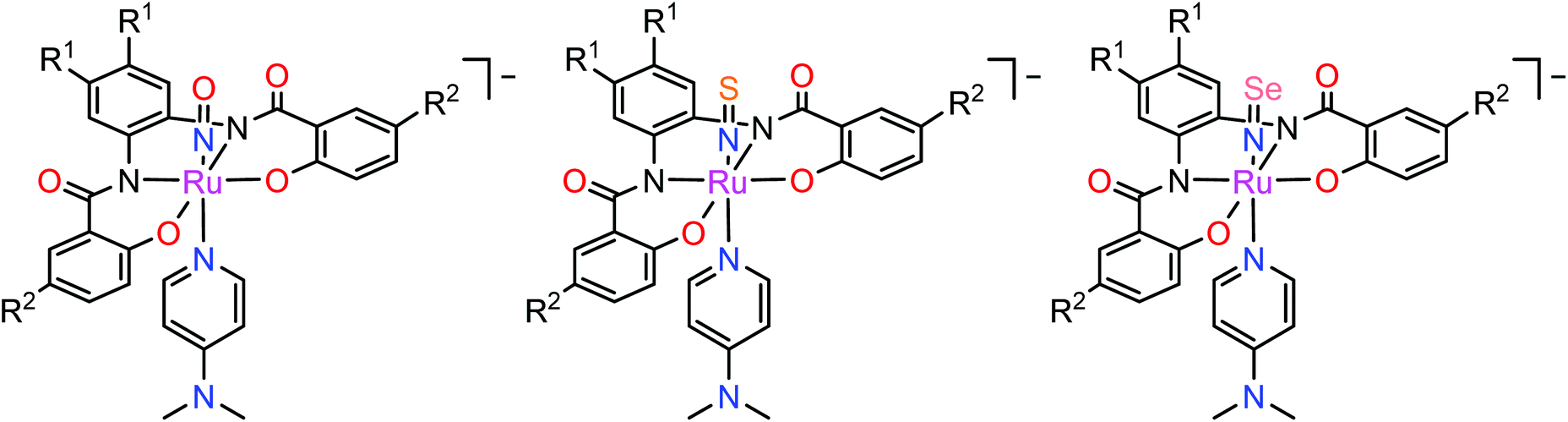 | ||
| Chart 1 The line drawings of the complex anions [Ph4P][Ru(NE)(hybebR1,R2)(DMAP)] (R1 = R2 = Cl, R1 = H, R2 = CF3, R1 = Cl, R2 = CF3; E = O, S, Se) isolated as tetraphenylphosphonium salts. | ||
| E | Ru-NE, Å | RuN-E, Å | Ru–N–E, ° | ν NE, cm−1 | Ref. |
|---|---|---|---|---|---|
| a These complexes contain two crystallographically independent molecules in the asymmetric unit. | |||||
| [RuCl 3 (NE)(AsPh 3 ) 2 ] | |||||
| O | 1.729(7) | 1.151(9) | 180.0 | 1869 | 95 |
| S | 1.753(4) | 1.502(4) | 180.0 | 1310 | 84 |
| Se | 1.756(3) | 1.650(3) | 171.2(2) | 1137 | 84 |
| [RuCl 2 (NE)(CpCo{PO(OEt) 2 } 3 ] | |||||
| O | 1.729(3) | 1.127(3) | 177.6(3) | 1865 | 97 |
| S | 1307 | 97 | |||
| Se | 1.731(4) | 1.651(4) | 178.4(3) | 84 | |
| [Ph 4 P][Ru(NE)(hybeb H,CF 3 )(DMAP)] | |||||
| O | 1.752(2) | 1.144(3) | 174.9(2) | 1810 | 43 |
| 1.744(2) | 1.149(3) | 177.27(19) | |||
| S | 1.746(4) | 1.511(4) | 171.0(3) | 43 | |
| 1.746(4) | 1.516(4) | 172.2(3) | |||
| Se | 1.734(4) | 1.670(4) | 171.2(3) | 43 | |
| 1.732(4) | 1.671(4) | 169.9(3) | |||
| [Ph 4 P] 2 [RuCl 4 (NE)] 2 ·4CH 2 Cl 2 | |||||
| S | 1.752(6) | 1.466(7) | 177.3(5) | 1280 | 99 |
| [Ph 4 P][trans-RuCl 4 (NE)(H 2 O)] | |||||
| S | 1.729(4) | 1.504(4) | 170.9(3) | 1318 | 100 |
| [RuCl(NE)TTP] (TTP = meso-tetratoluylporphyrin) | |||||
| O | 1845 | 86 | |||
| S | 1.768(4) | 1.489(5) | 169.1(3) | 1271 | 86 |
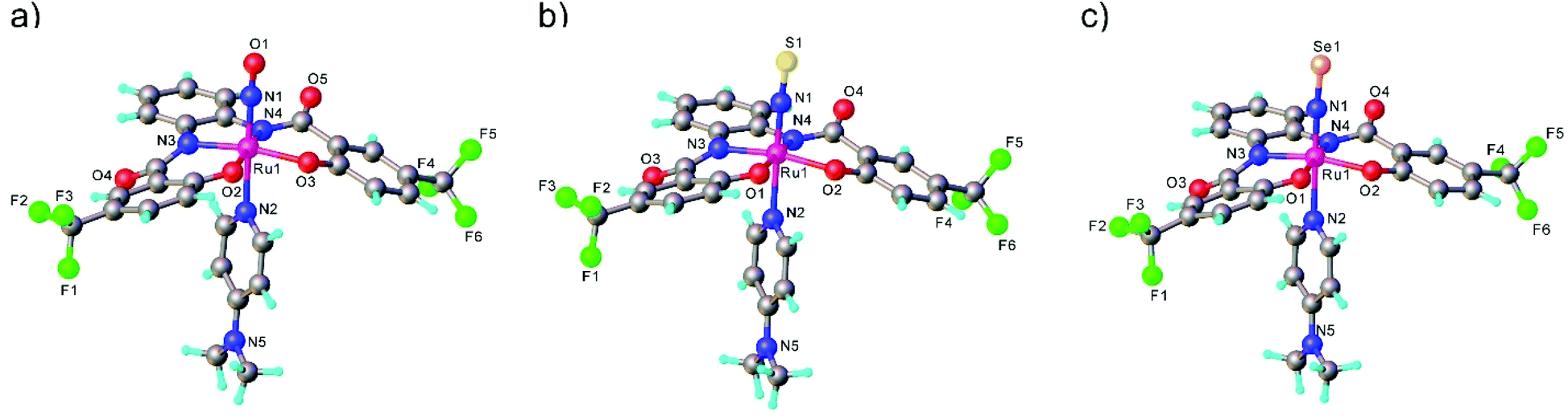
mer,trans-[RuCl3(NO)(AsPh3)2] was synthesized from nitrosylruthenium trichloride94 and AsPh3 in anoxic boiling methanol.95 The structure was confirmed by SC-XRD (sp. gr. C2/c; Ru–N 1.729(7) Å; N–O 1.151(9) Å; Ru–N–O 180.0(3)°, which is crystallographically imposed). The reaction of the nitrido complex mer-[RuCl3(N)(AsPh3)2] with elemental sulfur and selenium in boiling THF produced the chalcogenonitrosyl complexes mer,trans-[RuCl3(NE)(AsPh3)2] (E = S, Se), which are air stable both in the solid state and in solution (Scheme 2).84 The synthesis of thionitrosyl complex from [RuCl3(AsPh3)2L] (L = DMSO, DMF or THF) and N3S3Cl3 was also reported.96 Further reaction of mer-[RuCl3(NS)(AsPh3)2] with KN(R2PS)2 afforded trans-[RuCl(NS){N(R2PS)2}2] with R = Ph, iPr, tBu (Scheme 2). The thionitrosyl complex trans-[RuCl(NS){N(tBu2PS)2}2] was also obtained by treatment of [nBu4N][RuCl4(N)] with KN(tBu2PS)2 in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio, presumably via sulfur atom transfer from [N(tBu2PS)2]− to the nitride. Treatment of mer,trans-[RuCl3(NE)(AsPh3)2] (E = S, Se) with NaLOEt (LOEt− = [Co(η5-C5H5){P(O)(LOEt)2}3]−) gave [RuCl2(NE)(LOEt)], where E = S, Se (Scheme 2).84 The complex [RuCl2(NS)(LOEt)] was also prepared in 70% yield by reaction of electrophilic ruthenium(VI) nitride complex [RuCl2(N)(LOEt)] with electrophilic species S2O32−.97 A similar ruthenium nitrosyl complex with Kläui's tripodal ligand [CpCo{P(O)(OEt)2}3]− was obtained from the same starting material when treated with Me3NO in 75% yield.97 A series of {Ru-NO}6 species with Kläui's tripodal ligand was recently extended by introducing several substituted catecholates as a second potential redox non-innocent ligand in addition to NO.98 These complexes will be discussed in more detail in section 3.
:2 molar ratio, presumably via sulfur atom transfer from [N(tBu2PS)2]− to the nitride. Treatment of mer,trans-[RuCl3(NE)(AsPh3)2] (E = S, Se) with NaLOEt (LOEt− = [Co(η5-C5H5){P(O)(LOEt)2}3]−) gave [RuCl2(NE)(LOEt)], where E = S, Se (Scheme 2).84 The complex [RuCl2(NS)(LOEt)] was also prepared in 70% yield by reaction of electrophilic ruthenium(VI) nitride complex [RuCl2(N)(LOEt)] with electrophilic species S2O32−.97 A similar ruthenium nitrosyl complex with Kläui's tripodal ligand [CpCo{P(O)(OEt)2}3]− was obtained from the same starting material when treated with Me3NO in 75% yield.97 A series of {Ru-NO}6 species with Kläui's tripodal ligand was recently extended by introducing several substituted catecholates as a second potential redox non-innocent ligand in addition to NO.98 These complexes will be discussed in more detail in section 3.
All three compounds mer,trans-[RuCl3(NE)(AsPh3)2] (E = O, S, Se) were characterized by SC-XRD and the results are shown in Fig. 1.
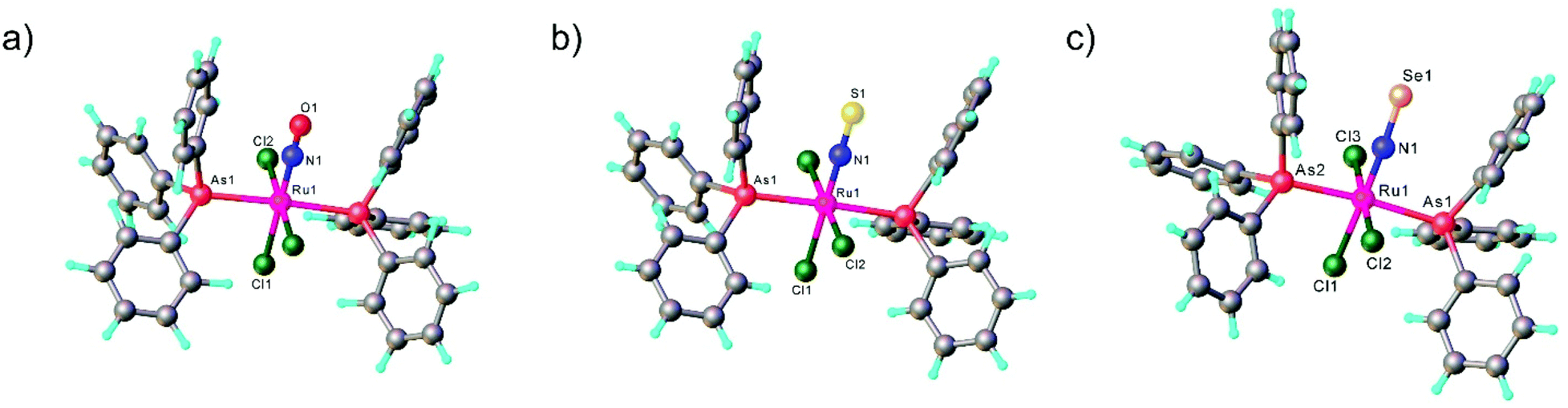 | ||
| Fig. 1 X-ray diffraction structures of the series of chalcogenonitrosyl complexes mer,trans-[RuCl3(NE)(AsPh3)2], where E = O (a), S (b) and Se (c). The structures are taken from those reported on the CSD; codes: (a) YUCFEH, (b) LUVPEZ, (c) LUVSEC. | ||
In the case of the second series, the complex with NO was prepared by reaction of RuNOCl3 with tetradentate ligands treated first with sodium hydride, followed by addition of DMAP to give Na[Ru(NO)(hybebR1,R2)(DMAP)]. Metathesis reaction of the latter complex with [Ph4P]Br afforded the desired product [Ph4P][Ru(NO)(hybebR1,R2)(DMAP)] (Chart 1). The other two types of complexes [Ph4P][Ru(NE)(hybebR1,R2)(DMAP)] with E = S, Se were synthesized by reaction of the nitrido complexes [Ph4P][Ru(N)(hybebR1,R2)] with elemental sulfur and elemental selenium, respectively, in the presence of DMAP.
It should be stressed that reaction with elemental tellurium in the presence of DMAP failed presumably due to low solubility of Te in common organic solvents. A full set of complexes [Ph4P][Ru(NE)(hybebR1,R2)(DMAP)] with R1 = H, R2 = CF3 was characterized by SC-XRD (Fig. 2).
 | ||
| Fig. 2 X-ray diffraction structures of chalcogenonitrosyl anionic complexes [Ru(NE)(hybebH,CF3) L(DMAP)]−, where E = O (a), S (b) and Se (c). The structures are taken from those reported on the CSD; codes: (a) ROWDAL, (b) ROWMOI, (c) ROWCUE. | ||
Comparison of Ru-NE and (DMAP)N-RuNE bond lengths for [Ph4P][Ru(NE)(hybebR1,R2)(DMAP)] with R1 = H, R2 = CF3 (see Table 2) indicates that the NSe ligand is a better π-acceptor and stronger σ-donor than the lighter chalcogen counterparts. The Ru-NSe bond is slightly shorter than Ru-NO and Ru-NS. Increased π-back donation from Ru to NSe ligand was also in accord with natural population analysis (NPA) charge distribution on RuNSe unit (more positive charge on Ru and more negative on the nitrogen atom when compared to that in the other two complexes). The stronger σ-donor ability of the NSe ligand and its resulting greater trans influence relative to that of NO and NS are corroborated by the increase of the (DMAP)N-RuNE bond lengths for E = O to S to Se. The calculated Wiberg bond indices indicate double bond character of both Ru-NE and N-E bonds.43
Discovered as a gas with a bad reputation NO nowadays is a well-recognized signaling molecule in important biological processes (i.e. blood pressure regulation, wound healing, memory formation).101 The signaling step in mammals is believed to involve coordination of NO to the iron(II) in the heme protein soluble guanylyl cyclase (sGC).102–105 NO binding activates the enzyme by labilization of the trans-coordinated (proximal) histidine ligand. Even though NO and NO−/HNO are assumed to bind to this protein in a similar way, it has been suggested that only NO and NO− may activate the enzyme because of their strong trans effect. HNO in contrast is supposed to have a weaker σ-donor capability, and as consequence, lower trans effect power. Therefore, the acidity of this coordinated group is of interest, as this can help to understand the biological role of the HNO molecule, if it can deprotonate with formation of nitroxyl under physiological conditions.
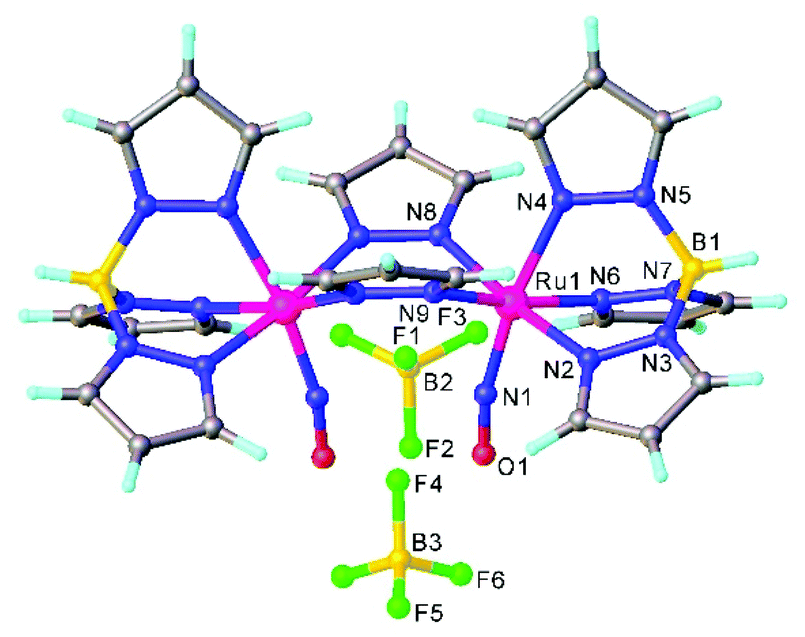
The first examples of {Ru(NO)}7 complexes characterized by SC-XRD were reported only recently.106,107 The complexes [Ru(Me3[9]aneN3)(L)(NO](BF4)2, where L = bpy, MeObpy or bpym) were obtained by one-electron (1e) reduction of [Ru(Me3[9]aneN3)(L)(NO](BF4)3 with iodide in acetone (Scheme 3). The SC-XRD structures of one of the starting materials [Ru(Me3[9]aneN3)(bpy)(NO](BF4)3 and its 1e reduced counterpart are shown in Fig. 3. Reduction resulted in lengthening of the N–O bond from 1.135(5) to 1.177(3) Å and the trans Ru–N bond from 2.128(4) to 2.169(2) Å. Another feature is the bending of the Ru–N–O angle from almost linear [172.5(4)°] to severely bent [141.6(2)°]. This is accompanied by stretching vibration shift from 1898 cm−1 to 1616 cm−1 (KBr). The X-band EPR spectrum of [Ru(Me3[9]aneN3)(L)(NO](BF4)2 in frozen acetonitrile (MeCN) at 85 K showed signals consistent with an S = 1/2 spin state, better described as RuIINO˙ (g = [2.030, 1.993, 1.880] and A(14N) = [11.0, 30.4, 3.9] × 10−4 cm−1). All these features, structural and spectroscopic, are well predicted by DFT. They revealed the simultaneous weakening of the N–O, Ru–NO, and Ru–N(trans) bonds upon reduction. The singly occupied molecular orbital (SOMO) is mostly located in one of the split π*NO σ orbitals, partially delocalized over the metal-centered dz2/dxz orbitals and to a lower degree over the trans N atom. The SOMO is Ru-NO σ-bonding in character and at the same time Ru–N(trans) σ-antibonding, thus weakening the Ru–L bond trans to the NO.
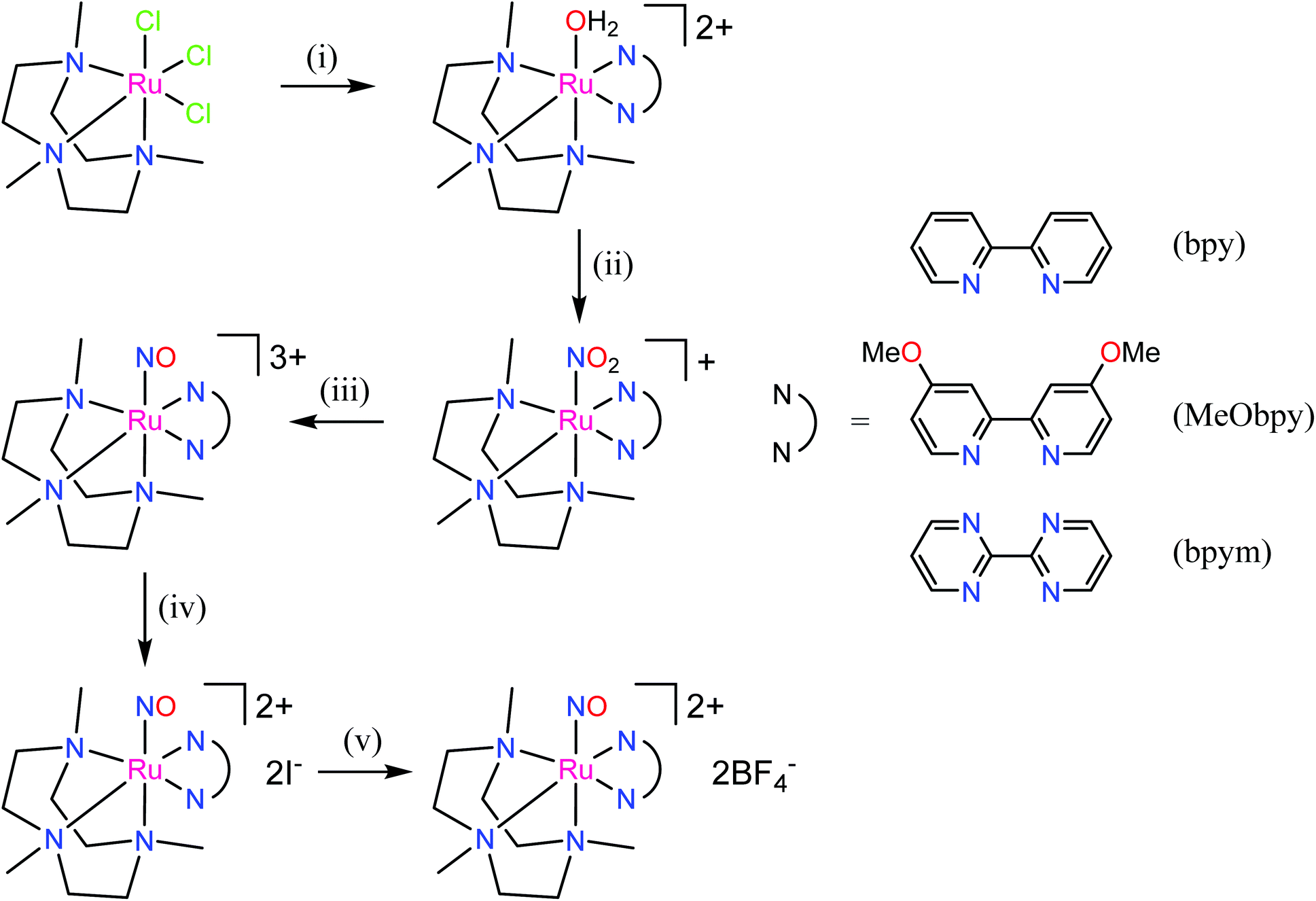 | ||
| Scheme 3 Synthetic route to {Ru(NO)}7 complexes. Reagents and conditions: (i) bpy, MeObpy or bpym, Zn powder, aqueous solution; (ii) NO2−; (iii) HOTf; (iv) acetone, I−; (v) Dowex 22 (BF4−), then lyophilization. | ||
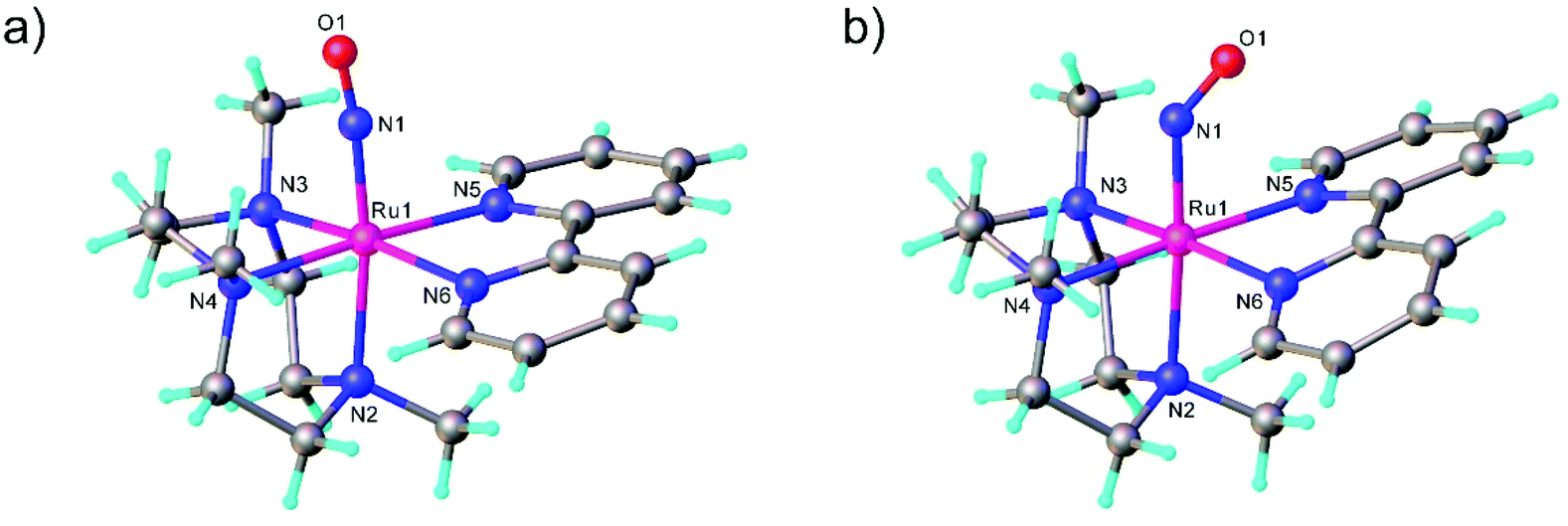 | ||
| Fig. 3 Linear Ru–N–O unit in (a) trication [Ru(Me3[9]aneN3)(bpy)(NO]3+ ({Ru(NO)}6) and bent Ru–N–O unit in (b) dication [Ru(Me3[9]aneN3)(bpy)(NO]2+ ({Ru(NO)}7). The SC-XRD structures are taken from those reported on the CSD; codes: (a) GIGPAQ, (b) ATAKAJ. | ||
The electron donor/acceptor properties of the three co-ligands used (bpy, MeObpy and bpym) did not affect the metric parameters of the complexes studied by SC-XRD, but exerted a strong effect on both the redox potentials and on the acidity of coordinated HNO (pKa variation from 7.7(1) to 10.5(2)).107 The oxidation potentials for the three different {RuNO}6/7 couples were found to be pH independent, while the reduction potentials of the {RuNO}7/8 couples were strongly dependent on pH. In the alkaline pH region the reduction is pH independent, while in acidic region the redox potentials decreased by ∼60 mV per pH unit in accord with eqn (1) and (2):
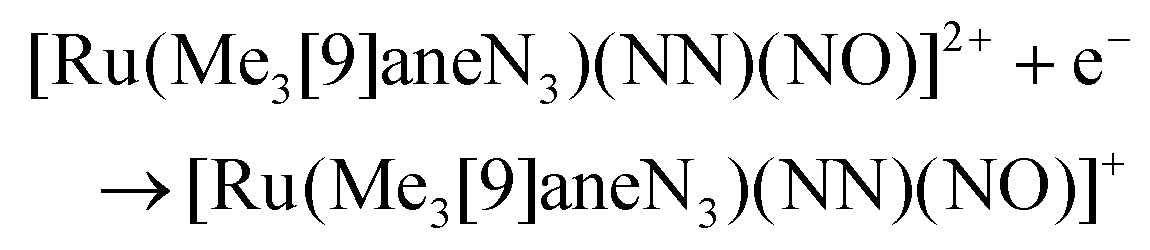 | (1) |
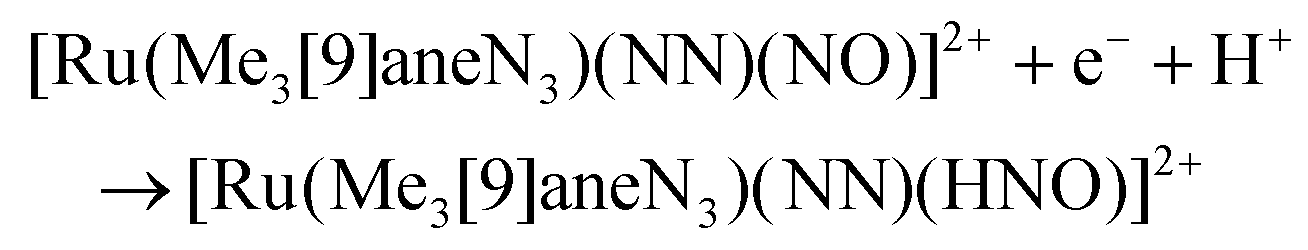 | (2) |
These transformations are of particular relevance for better understanding of the role of these particles in biological signaling system.
Another exciting family of compounds which deserves to be presented in this review are the ruthenium complexes featuring two nitrosyl groups bound to ruthenium. In contrast to the chemistry of the homologous dinitrosyl iron compounds (DNICs)7 ruthenium-dinitrosyl chemistry appears to be much less developed, but has become more intensively investigated in recent years. A series of dinitrosyl complexes of general formula [Ru(NO)2(L)2X](BF4) (L = monodentate phosphane, X = Cl, Br, I) was reported.58,80 These five-coordinate dinitrosyl complexes [Ru(NO)2(L)2X](BF4) were synthesized by treatment of the phosphine-containing mononitrosyl precursor with a zinc/copper alloy to reduce it to the {RuNO}8 species followed by insertion of the second nitrosyl ligand by the oxidative addition of NOBF4. Interestingly, these compounds adopt two coordination geometries: trigonal-bipyramidal with two nearly linear NO ligands and/or vacant octahedral with one linear and one bent RuNO moiety. Dinitrosyl complexes offer attractive possibilities for investigation of photo-induced isomerization of RuNO moiety in a selective manner and these data will be more specifically discussed in section 4.
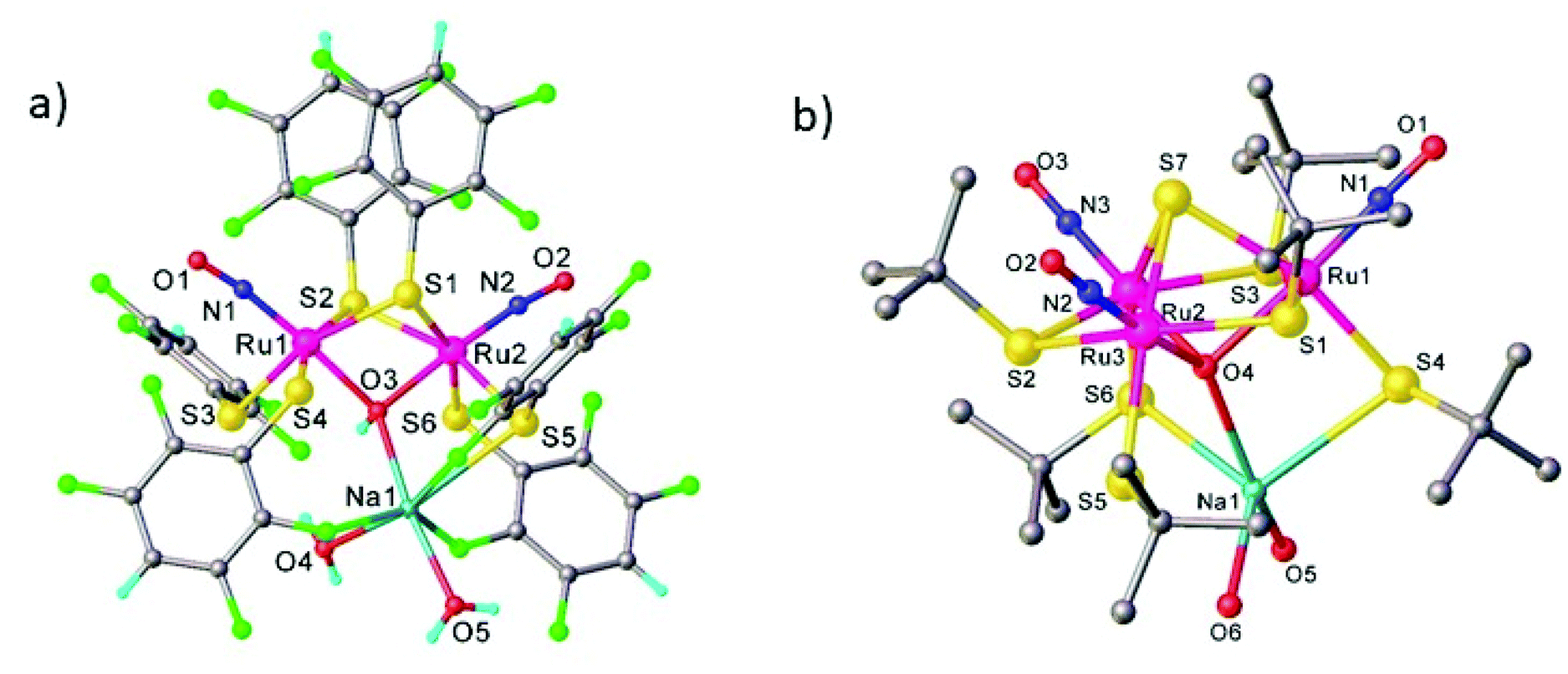
Among other recent achievements in the field of synthesis of ruthenium nitrosyl complexes, the assembly of double complex salts and, especially, mono- and heteropolynuclear complexes and clusters bearing one or several coordinated NO units deserve to be mentioned as well.
3. Ruthenium nitrosyl moiety in double complex salts, mono- and heteronuclear metal complexes and clusters
Double complex salts (DCS), i.e., complexes in which both the anion and cation are, in this case, transition metal coordination complexes, have been shown to be convenient single-source heterometallic precursors for the preparation of nanoalloys under reductive thermal decomposition. The heterometallic complexes [Pd(L)4][RuNO(NO2)4OH], (L = NH3, pyridine), represent one example. These complexes were prepared by reaction of Na2[RuNO(NO2)4OH] with [Pd(L)4](NO3)2.108,109 In the case of [Pd(NH3)4][RuNO(NO2)4OH], the palladium and ruthenium subunits are bound by hydrogen bonds between NH3 and NO2− ligands (Fig. 4). Some restrictions on the choice of ligands were suggested109 and a metastable solid solution Pd0.5Ru0.5 was prepared only in the case of ammine analogue.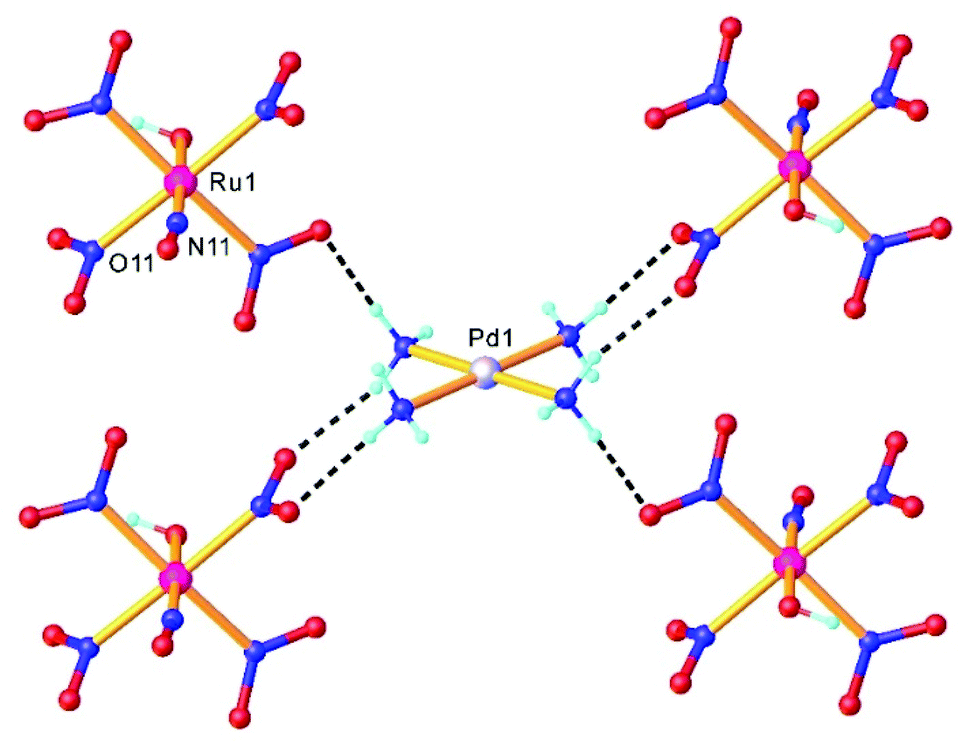 | ||
| Fig. 4 A fragment of the crystal structure of [Pd(NH3)4][RuNO(NO2)4OH] showing intermolecular H-bonding interactions between a complex cation [Pd(NH3)4]2+ and complex anions [Ru(NO)(NO2)4(OH)]2−. The structure is taken from those reported on the CSD; CCDC number: 1871869. | ||
Three other DCS derived from the cation trans-[RuNO(NH3)4F]2+ and the noble-metal anions [PtCl6]2−, [PdCl4]2−, and [PtCl4]2−, crystallized in a non-centrosymmetric structure, determined by N–H⋯halogen intermolecular interactions. The complexes were prepared so as to understand how the counterion can affect the metastable state properties and how these properties can be improved by rational design.36
The design of new catalysts based on solid solutions of ruthenium with non-platinum metals (for hydrogen production from ammonia–borane110 or Fischer–Tropsch catalysts for conversion of synthesis gas into higher hydrocarbons111) can reduce the cost of the catalyst, and in some cases lead to a synergistic increase in catalytic activity. The synthesis of heterometallic complexes [RuNO(NO2)2(μ-NO2)2(μ-OH)M(H2O)3] with two bridging μ(N,O)-NO2 and one μ-OH ligands was realized via an exchange reaction between Ba[RuNO(NO2)4OH] and cobalt(II) or nickel(II) sulfate.112 These complexes proved to be convenient precursors for catalytic systems. Their low decomposition temperatures in reductive (H2), inert (He), and oxidative (O2) atmosphere and the absence of carbon-containing ligands, along with high water solubility allows for direct application of these compounds for the impregnation of carriers without side-salt contamination.112
The synthesis of multifunctional molecular compounds by combination of different building blocks in one molecule allows for control of specific features (magnetic, spectral, etc.) of one fragment influencing another sensor fragment (through irradiation, temperature, pH) was extensively developed over the last decades. In particular, a photoswitchable Ru-NO fragment (as potential trigger of the change in the second building block) was combined with heavier lanthanides (paramagnetic units, exhibiting single molecule magnet (SMM) properties) through short bridges, starting from Na2[RuNO(NO2)4OH] and the corresponding lanthanide nitrate. A series of tetranuclear butterfly-like Ru2Ln2 compounds with a {Ln[RuNO(μ-NO2)4(μ3-OH)]2Ln} (Ln = Gd, Tb, Dy, Ho, Lu) core, in which the coordination unit includes two lanthanide ions and two ruthenium based anionic moieties bound through eight bridging nitro ligands and two μ3-OH groups were reported and structurally characterized (Fig. 5).113
 | ||
| Fig. 5 X-ray diffraction structure of a tetranuclear butterfly-like Ru2Lu2 complex. The structure is taken from those reported on the CSD; code: XELTOA. | ||
Heteropolynuclear complexes are also of interest for the design of new potential theranostics for cancer diseases by rationalized combination of the luminescence emission of the lanthanide complexes and therapeutic anticancer potential of ruthenium nitrosyl complexes. A series of pentanuclear lanthanide-labeled ruthenium-nitrosyl (4d–4f) complexes of the general formula (nBu4N)5[Ln{RuCl3(μ-C2O4)(NO)}4], where Ln = Y, Gd, Tb, Dy were prepared by treatment of (nBu4N)2[RuCl3(μ-C2O4)(NO)] with the respective lanthanide salt and their pentanuclear structure was confirmed by SC-XRD (Fig. 6).114
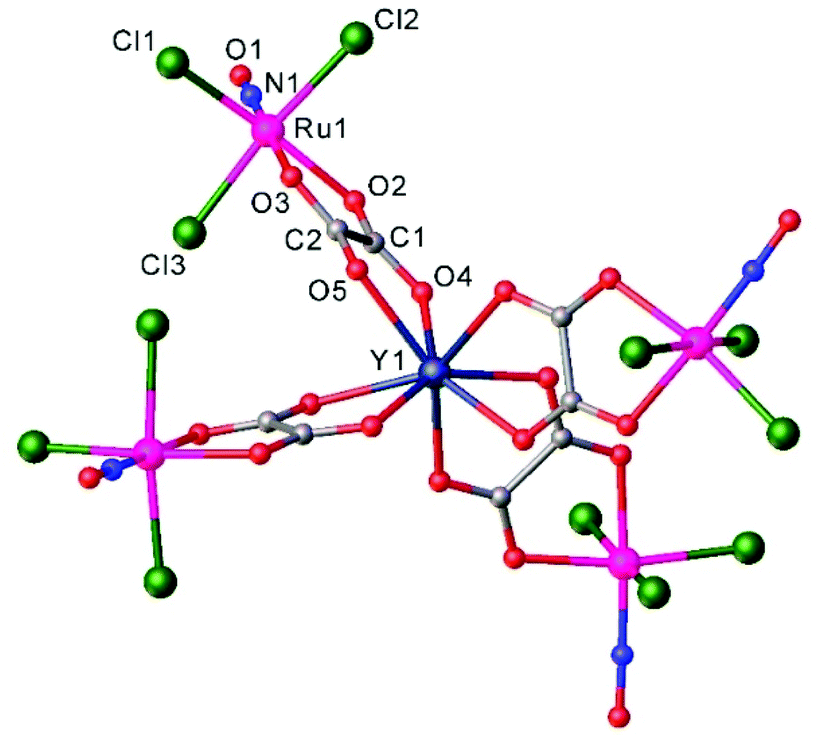 | ||
| Fig. 6 X-ray diffraction structure of the complex anion [Ln{RuCl3(μ-C2O4)(NO)}4]5−. The structure is taken from those reported on the CSD; code: YUWLEJ. | ||
Another example of a compound with photo-triggered properties is the heterometallic complex, featuring both 3d-V and 4d-Ru, [Ru(NO)(NO2)2(μ-NO2)(μ-CH3COO)(μ-O)VO(L)], where L = 4,4′-di-tert-butyl-2,2′-bipyridyl.115 Since both metals, ruthenium and vanadium, are potential catalytic centers, the combination of both in one complex might lead to synergism. The complex was obtained from a pre-organized starting material, namely Na2[RuNO(NO2)4OH] and an oxidovanadium(IV) species [VO(L)(H2O)Cl2] in acetonitrile. During the reaction vanadium(IV) was oxidized by air oxygen to diamagnetic vanadium(V). The unexpected presence of the coordinated bridging acetate is presumably due to the hydrolysis of acetonitrile upon the reaction, in which the vanadium complex is likely involved. The V2–O1 bond length equals to 1.70(1) Å indicating the coordination of the μ-oxido rather than μ-hydroxido ligand (Fig. 7). It is also of note that the presence of vanadium strongly influences the photochemical properties of ruthenium nitrosyl both in the solid phase and in solution. Unoccupied orbitals of an almost linear ON-Ru–O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) V chain were suggested to be responsible for the efficiency of nitric oxide release. A change in the orientation of the vanadium center with respect to the Ru–N–O linkage is expected to lead to strong modification of the photochemical properties.
V chain were suggested to be responsible for the efficiency of nitric oxide release. A change in the orientation of the vanadium center with respect to the Ru–N–O linkage is expected to lead to strong modification of the photochemical properties.
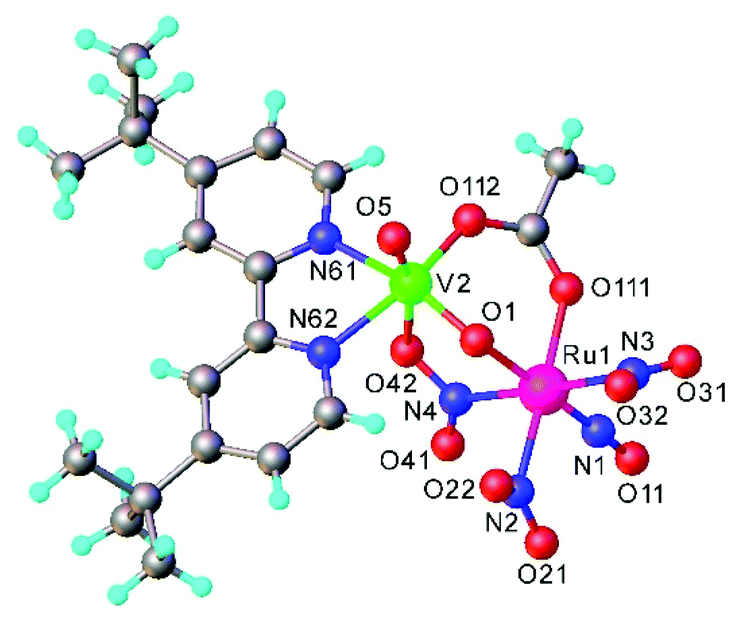 | ||
| Fig. 7 X-ray diffraction structure of [Ru(NO)(NO2)2(μ-NO2)(μ-CH3COO)(μ-O)VO(L)], where L = 4,4′-di-tert-butyl-2,2′-bipyridyl. The structure is taken from those reported on the CSD; code: FAFMEI. | ||
The [Ru(LOMe)(NO)(cat)] complexes with the Kläui's tripodal oxygen ligand, [CpCo{P(O)(OMe)2}3]− (LOMe), and substituted catecholates (cat = dianion of catechol, 3,5-di-tert-butylcatechol, tetrabromocatechol, or 2,3-dihydroxynaphthalene), prepared by reactions of [Ru(LOMe)(NO)Cl2] with catecholates, represent rare examples of ruthenium nitrosyl complexes with oxygen based ligands. They are regarded as molecular models for heterogeneous catalysts comprising noble metal atoms dispersed on metal oxide supports.98 Moreover the combination of catecholates with Kläui's tripodal oxygen ligand gave rise to compounds that exhibited reversible redox events centered on the catecholate framework.
An interesting class of ruthenium nitrosyl thiolate complexes as photoactive NO releasing agents, as well as models of the active sites of metalloenzymes and potential heterogeneous catalysts, was also reported.116 It was found that the outcome of the reaction between Ru(NO)Cl3 and NaSR is dependent on substituent R. Thus, in the case of 2,3,5,6-tetrafluorothiophenol the dinuclear hydroxido-bridged dimer Na(H2O)2[Ru(NO)(L)2]2(μ-L)2(μ-OH) was produced (Fig. 9a), in which two ruthenium centers are bridged by two thiolates and one hydroxido ligand and in addition, each ruthenium binds to two terminal thiolates and one nitrosyl ligand. The {Na(H2O)2}+ moiety binds to the diruthenium core via the μ-hydroxido ligand and three ortho fluorine atoms of the thiolate ligands. When the sodium salt of tert-butylthiol was used, a C–S cleavage of the thiolate ligand also occurred and a trinuclear oxido-sulfido cluster, Na(H2O)2[Ru(NO)(L)(μ-L)]3(μ3-S)(μ3-O), was obtained that contains a trinuclear {Ru3(SBut)3} core capped by a μ3-oxido and a μ3-sulfido ligand. Each ruthenium is also coordinated by a linear nitrosyl and a terminal thiolate ligand. The {Na(H2O)2}+ moiety binds to the triruthenium core via the μ3-oxido and two terminal thiolate ligands as shown in Fig. 9b.
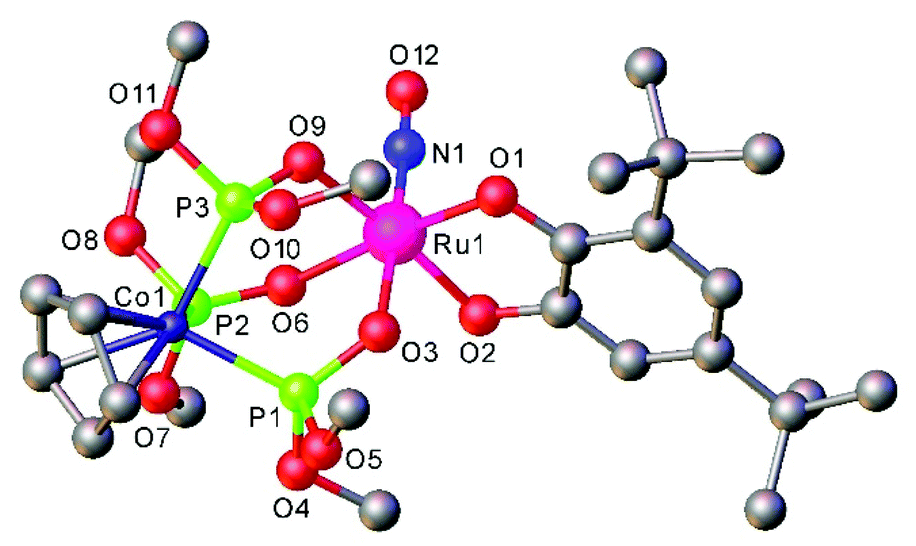 | ||
| Fig. 8 X-ray diffraction structure of [Ru(LOMe)(NO)(cat)] complex, where cat = dianion of 3,5-di-tert-butylcatechol, LOMe = [CpCo{P(O)(OMe)2}3]−. The structure is taken from those reported on the CSD; code: CALXAR. | ||
 | ||
| Fig. 9 Ball-and-stick plot of SC-XRD structure of (a) Na(H2O)2[Ru(NO)(L)2]2(μ-L)2(μ-OH) (L = 2,3,5,6-tetrafluorothiophenolate) and (b) Na(H2O)2[Ru(NO)(L)(μ-L)]3(μ3-S)(μ3-O) (L = tert-buthylthiolate) with H atoms omitted for clarity. The structures are taken from those reported on the CSD; codes: (a) IQIDOD, (b) MAPWAF. | ||
μ-Oxido trinuclear ruthenium mononitrosyl clusters [Ru3(μ3-O)(μ-OOCCH3)6(NO)L2]PF6 (L = pyridine, 4–acetylpyridine or 4-tert-butylpyridine) were obtained by passing a continuous NO gas flow through a solution of the aqua precursor [Ru3(μ3-O)(μ-OOCCH3)6(H2O)L2]PF6 in dichloromethane.117 These compounds have proven to be useful vasodilation agents which relaxed pre-contract rat aorta up to 100%.
trans-[Ru(OH)(NO)(Hpz)4]Cl2, where Hpz = 1H-pyrazole, was disclosed recently to show a high reactivity as a metalloligand towards oxophile metal ions (i.e., Mg2+, Fe3+, Ga3+) and form heteronuclear species via μ-hydroxido, μ3- and/or μ4-oxido bridging of the oxophilic metal centers. Several heterometallic complexes have been synthesized and characterized by spectroscopic methods (1H NMR, UV–vis, IR in the solid state in the absence and under light irradiation), magnetochemistry and Mössbauer spectroscopy.26 The results of SC-XRD studies of these complexes are shown in Fig. 10. These complexes can be also regarded in the paradigm of “inverse coordination” with oxygen as coordination center.118,119
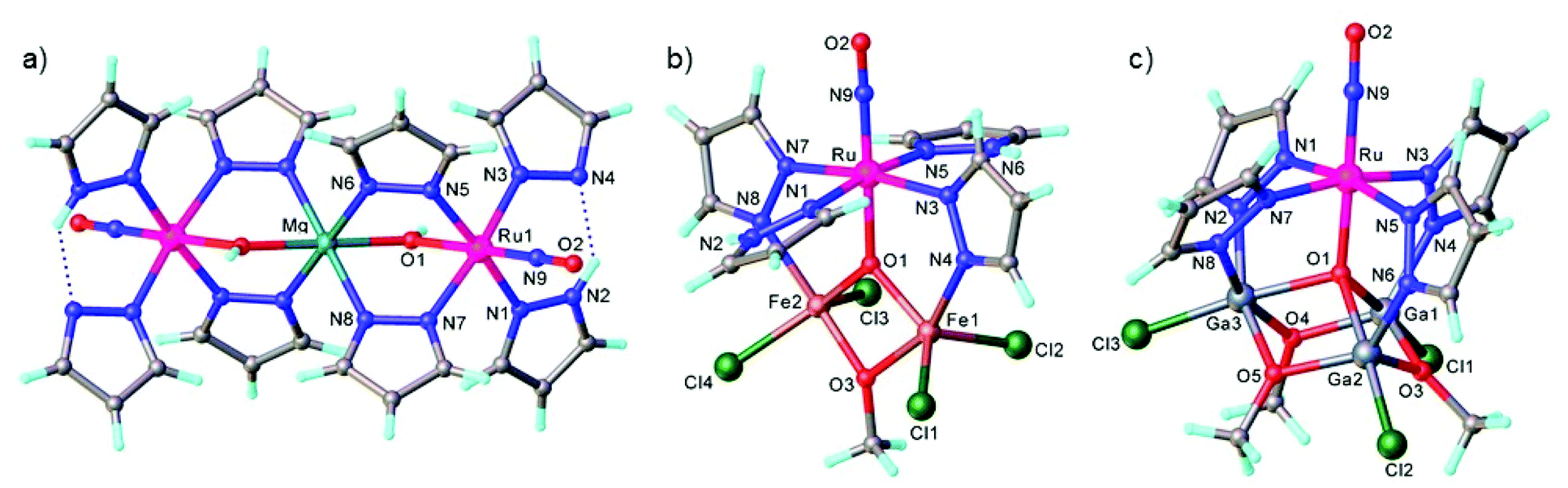 | ||
| Fig. 10 The structures of (a) {[Ru(μ-OH)(μ-pz)2(pz)(NO)(Hpz)]2Mg}, (b) [Fe2RuCl4(μ3-O)(μ-OMe)(μ-pz)2(NO)(Hpz)2] and (c) [Ga3RuCl3(μ4-O)(μ-OMe)3(μ-pz)4(NO)]. The structures are taken from those reported on the CSD; codes: (a) LAWGUP, (b) LAWHOK, (c) LAWHIE. | ||
A synthetic cycle proposed in 2007 for NO reduction (2NO + 2H+ + 2e− → N2O + H2O) on a dinuclear platform has recently confirmed that the production/elimination of N2O via the N–N coupling of two NO molecules on metals, occurs by light irradiation and is not proton-assisted.120 The dinitrosyl complex [{TpRu(NO)}2(μ-pz)2](BF4)2 (where Tp = HB(pyrazol-1-yl)3) with linear-type NO ligands was generated by bubbling NO gas through a solution of bis(μ-pyrazolato)diruthenium complex [(TpRu)2(μ-O)(μ-pz)2] in acetone in the presence of HBF4. The SC-XRD structure of this complex is shown in Fig. 11. The unusual neutral (ON–NO) moiety in [(TpRu)2{μ-N(O)–N(O)}(μ-pz)2] was formed by the two-electron reduction of [{TpRu(NO)}2(μ-pz)2](BF4)2 with [Cp*2Fe] and is only the second example of an N–N coupling on a dinuclear ruthenium complex (Scheme 4). Photo-induced conversion of the N–N coupled complex then resulted in the oxido-bridged starting complex [(TpRu)2(μ-O)(μ-pz)2]. It was supposed that such an NO reduction cycle could occur during the human immune response, controlled by NO reductase (NOR) and flavodiiron NO reductase (FDP), when the reduction of increased levels of NO takes place in order to protect the organism from nitrosative stress.
 | ||
| Fig. 11 The X-ray diffraction structure of [{TpRu(NO)}2(μ-pz)2](BF4)2. The structure is taken from those reported on the CSD; code: LIGWOQ. | ||
 | ||
| Scheme 4 Synthesis of [{TpRu(NO)}2(μ-pz)2](BF4)2 and photo-induced conversion of the N–N coupled complex into oxido-bridged starting complex [(TpRu)2(μ-O)(μ-pz)2]. | ||
These examples will foster the synthesis of unprecedented mono- and heteropolynuclear complexes bearing NO moieties in the future. In addition, some of them showed light-induced NO linkage isomerism and even NO release both in the solid state and in solution.
4. Ruthenium nitrosyl complexes: linkage isomerism and possible application
Ruthenium nitrosyl complexes may exhibit photoresponsive behavior, either by forming nitrosyl linkage isomers upon light irradiation121 or photoinduced nitric oxide release.14 While linkage isomers are mostly observed in the solid-state, photorelease of NO is in general studied in solutions, even though NO release was also reported in the solid-state highlighting the reactivity and particularity of the NO ligand in this respect.17,122,123 Furthermore, there is an ongoing discussion as to whether linkage isomers might be involved in the NO photorelease mechanism.124 With respect to applications, nitrosyl linkage isomers have been considered for information storage due to the absorptive and photorefractive changes occurring upon their photogeneration,23,125 but also for fast optical correlation techniques126 or switchable nonlinear optics.127Photoinduced linkage isomerism (PLI) is the generation of linkage isomers by irradiation with light. In the case of ruthenium mononitrosyl complexes two linkage isomers can be induced by light. Starting from the ground state (GS) with its linear Ru–N–O configuration, an iso-nitrosyl configuration with a linear Ru–O–N configuration can be induced, in general by irradiation in the blue spectral range corresponding to the HOMO–LUMO transition. A second linkage isomer can be generated by irradiating the complex in its iso-nitrosyl configuration using infrared light, the complex then adopting a so-called side-on configuration, where the Ru–N–O angle is of the order of 80–90 degrees (see Fig. 12).20,128 These linkage isomers correspond to minima on the potential energy surface of the complex, separated by activation barriers from the GS.129 Therefore, they are stable at low temperatures (lifetime of several years), which has yielded them also the term “metastable state” (MS), MS1 corresponding to the iso-nitrosyl configuration and MS2 corresponding to the side-on configuration of the nitrosyl ligand. The long lifetime at low temperature allows for a thorough investigation of their structure and properties.130 For information storage applications a long lifetime at room temperature would be beneficial, while short lifetimes might be exploited in other applications, such as optical correlators. In all cases a significant conversion efficiency from the GS to the MS is desired in order to enhance the optical response of the material.
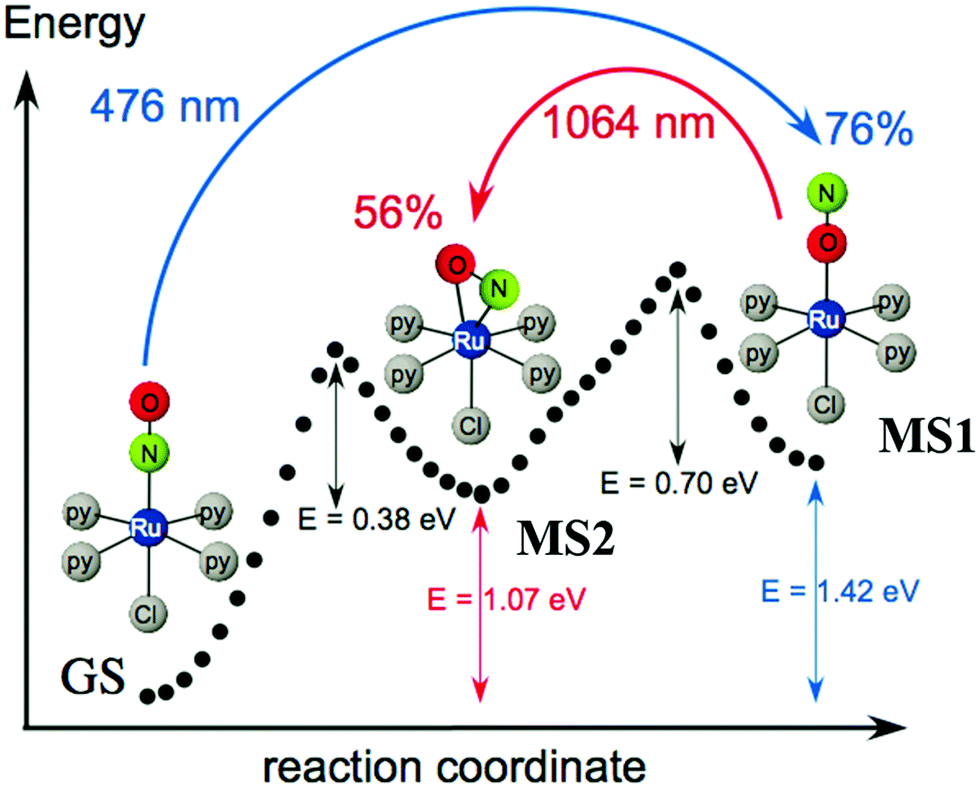 | ||
| Fig. 12 Scheme illustrating the photogeneration of nitrosyl linkage isomers in [RuCl(py)4NO]2+ according to results obtained from investigations by calorimetry, infrared spectroscopy and DFT.20 Indicated values correspond to experimentally determined energies (1.0 eV = 96.49 kJ mol−1 = 23.06 kcal mol−1), while the dotted curve corresponds to the ground state potential energy surface according to DFT. | ||
Concerning photoinduced linkage isomerism (PLI), there has been important progress in the last few years in two aspects. On the one hand the mechanism for the generation of PLI in octahedral mono-nitrosyl complexes has been studied both by theory and experiment. Based on earlier established knowledge in the complex [Ru(NH3)5NO]2+/3+, where a two-step mechanism for the generation of MS1 has been proposed131 and the generation of MS2 by infrared transfer out of MS1 has been shown,132 several studies have been performed on [RuL(py)4NO]2+ (L = F, Cl, Br, I) complexes, corroborating this mechanism. [RuCl(py)4NO]2+ is a particularly suitable system for such studies since it offers an almost complete conversion of GS to MS120,128 and thus recent theoretical studies have focused on this system in order to elucidate the photoisomerization mechanism.133–135 Using DFT and TDDFT a model was proposed for the photogeneration of MS1, showing that a sequential two-photon absorption mechanism involving the MS2 state as an intermediate state, GS → MS2 → MS1, can explain the observations.133 This mechanism implies that the MS2 state absorption needs to overlap with the GS absorption in order to allow an efficient conversion towards MS1 using a single wavelength of irradiation. In this study it was also pointed out that along the isomerization pathway, the singlet and triplet potential energy surfaces are highly entangled, suggesting that triplet states are involved in the isomerization.134 The authors used multiconfigurational second order perturbation theory (CASPT2) to corroborate the earlier finding concerning the photoisomerization process. In particular, their calculations indicated efficient intersystem crossings along the reaction pathway, which supports the involvement of triplet states in the photoisomerization. Talotta et al.135 then studied in more detail the early relaxation dynamics, still in the [RuCl(py)4NO]2+ system, using non-adiabatic dynamics for assessing internal conversions and intersystem crossings. Their results indicate three possible mechanisms for photoisomerization in the first 200 fs, two pathways including both internal conversion and intersystem crossings, while the third involves only internal conversions. The main conclusion of all these theoretical studies is that a two-step mechanism, GS → MS2 → MS1, is the origin of MS1. Using visible absorption spectroscopy at low temperatures, Khadeeva et al.136 have pointed to the MS2 state as an intermediate transient state during the generation of MS1 in [RuCl(py)4NO]2+, even though no structural study could be performed due to the low population of this transient state. Finally, Mikhailov et al.137 have demonstrated experimentally this two-step mechanism on a [RuF(py)4NO]2+ complex and pointed out the importance of the stability of the MS2 state for the generation of MS1 at room temperature (see Fig. 13). Given the involvement of MS2 as an intermediate state in the GS → MS2 → MS1 sequential two-step excitation, it seems natural that MS2 has to exhibit a certain minimum lifetime, in order to guarantee a sufficiently high efficiency of this population pathway. These authors thus performed a temperature dependent study of the achievable population of MS1, and found that when the lifetime of MS2 for a given temperature drops below a few seconds, then the population of MS1 drops to zero (when using standard modest power light sources). In the case of [RuF(py)4NO]2+ this occurs at approximately 230 K, a temperature at which the lifetime of MS1 is of the order of 107 seconds (>100 days). Using a high-power pulsed laser to increase the photon density and thereby increasing the probability of the two-step process, it was then shown that MS1 could be populated even at room temperature, where MS1 exhibits a lifetime of only 150 seconds. All these studies thus clearly confirm the GS → MS2 → MS1 sequential two-step photoisomerization mechanism in these ruthenium nitrosyl complexes. An important open question that remains concerning the PLI mechanism is the involvement of triplet states that are strongly suggested by theory, but for which up to now experimental evidence is missing. For this purpose, time-resolved ultrafast spectroscopic measurements are required. Another important issue concerns the competition between photoisomerization and photoinduced NO release. As pointed out by Talotta et al.,138 based on CASPT calculations, the photodissociation of NO is a two-step sequential process coupled to a partial population of the MS2 side-on isomer. The above described recent progress in understanding of the photoisomerization mechanism as well as the detection of NO release in the solid state17,122,123 opens the way to investigate the competition between these two processes.
 | ||
| Fig. 13 Scheme illustrating the two-step mechanism GS → MS2 → MS1 in the photogeneration of nitrosyl linkage isomers in [RuF(py)4NO]2+ according to ref. 137. In this compound a record stability for MS1 at room temperature with a lifetime of 151 s was also reported. | ||
On the other hand, significant progress has been made concerning the stability of the PLI, important for potential applications. In particular, a record high stability has been achieved by introducing F as a trans-to-NO ligand, and it could be shown that MS1 can be generated at room temperature with a lifetime of 120–150 s.48,137 The increase in stability for F-based complexes was predicted by theory139 and has been further underlined by additional studies on a series of [RuF(NH3)4NO]2+ complexes with different counteranions22,36,37 as well as on RuNOF complexes mixing pyridine and ammine equatorial ligands.35 These high stabilities open the path to combining PLI with other properties, such as nonlinear optical (NLO) response. For this purpose, a non-centrosymmetric crystal of a [RuF(NH3)4NO]2+ complex, [RuF(NH3)4NO]SiF6, has been studied for PLI and NLO response.37 While this compound shows both a high stability of MS1 and a significant second harmonic emission, a modification of the second harmonic emission by the PLI could not be unambiguously confirmed. However, the increased stability of the PLI and the possibility to design non-centrosymmetric structures opens the door to a new family of compounds with photoswitchable non-linear optical properties. Furthermore, several nitro-nitrosyl ruthenium complexes have been investigated with respect to their ability to form PLI, by introducing pyridine ligands in view of increasing the conversion efficiency towards the linkage isomers33 and study the influence on the biological properties.31 Such nitro-nitrosyl ruthenium complexes were also used as anions with tetranuclear ruthenium lanthanide complexes in order to combine the PLI with single molecular magnets,113 even though the potential interplay between the magnetic and photoswitchable properties was not reported. Nitro-nitrosyl complexes offer in principle the possibility to observe both linkage isomers of the NO and the NO2 ligand,140,141 and thus a multitude of PLI combinations that might be useful for applications. A non-centrosymmetric structure of such a nitro-nitrosyl ruthenium complex with 3-cyanopyridine ligand was reported, but only nitrosyl isomers could be observed and the effect of this PLI generation on the nonlinear optical properties was not reported.30 The interplay between NO2 ligands and NO ligands was studied in a binuclear heterometallic ruthenium–vanadium complex, where PLI in the solid state and NO release in solution were both observed.115
The search for multiple PLI motivated the synthesis and physico-chemical characterization of ruthenium-dinitrosyl complexes. While dinitrosyl iron complexes (DNICs) are widely investigated,142 being also a subject of a recent “Forum on renaissance in NO chemistry”,13 there are far fewer studies on dinitrosyl ruthenium complexes and only a few with respect to the PLI phenomenon. Gallien et al. have established in 2014 a whole family of bis-phosphane salts of the formula [Ru(NO)2(PR3)2X]BF4 (X = Cl, Br, I), for which PLI was detected.58 In the last few years several photocrystallographic studies have revealed the nature of the different PLI in this family. The complexes in this family in the GS adopt a molecular structure either in the form of a vacant octahedron (vOC-5) with one (strongly) bent and one (almost) linear NO ligand, or a trigonal bipyramid (TBPY-5) with two almost linear NO ligands.58 In the vOC-5 type compounds, up to three independent PLI were observed,143 while in the TBPY-5 type compounds only one PLI was found.144 For the latter, it was demonstrated that only one of the NO ligands significantly changes its conformation during the PLI, the Ru–N–O angle decreases from 166° in GS to 114° in the PLI.144 The analysis of PLI in the vOC-5 type compounds allowed for the distinction of three independent PLI, which depending on the used light wavelength for photogeneration may occur in different ratios, but always exhibiting a dominating structural configuration for PLI-1 with about 30%, while PLI-2 and PLI-3 remain below 10% of population. Again, each PLI involves the structural reconfiguration of only one of the NO ligands, the second NO ligand only slightly adapts to the new situation. For PLI-1, the NO ligand that is already bent in the GS rotates from a configuration with ∠Ru–N–O ≈ 130–140° oriented toward the second (linear) NO ligand to a configuration with ∠Ru–N–O ≈ 108–118° oriented toward the halogen ligand.80,143 The second NO ligand remains linear in the PLI and both NO ligands are N-bound to ruthenium. By choosing an appropriate irradiation wavelength and temperature, the PLI-2 in one of the complexes could be isolated and its structural configuration was investigated by photocrystallography.145 The results point in this case to an iso-nitrosyl configuration, i.e. Ru–O–N, exhibiting ∠Ru–O–N = 125°, close to ∠Ru–N–O = 136° found in the GS. Again the second NO ligand remains linear and is nearly unaffected by the reorganization of the first one (see Fig. 14). These results show that one of the NO ligands can be manipulated quite independently from the other one and that selective switching is possible to some extent. If one succeeds in optimizing the selectivity and increasing the conversion efficiency for the different isomers, e.g. by designing appropriate ligands, then this opens the way for all-optical switching within a multi-state system.
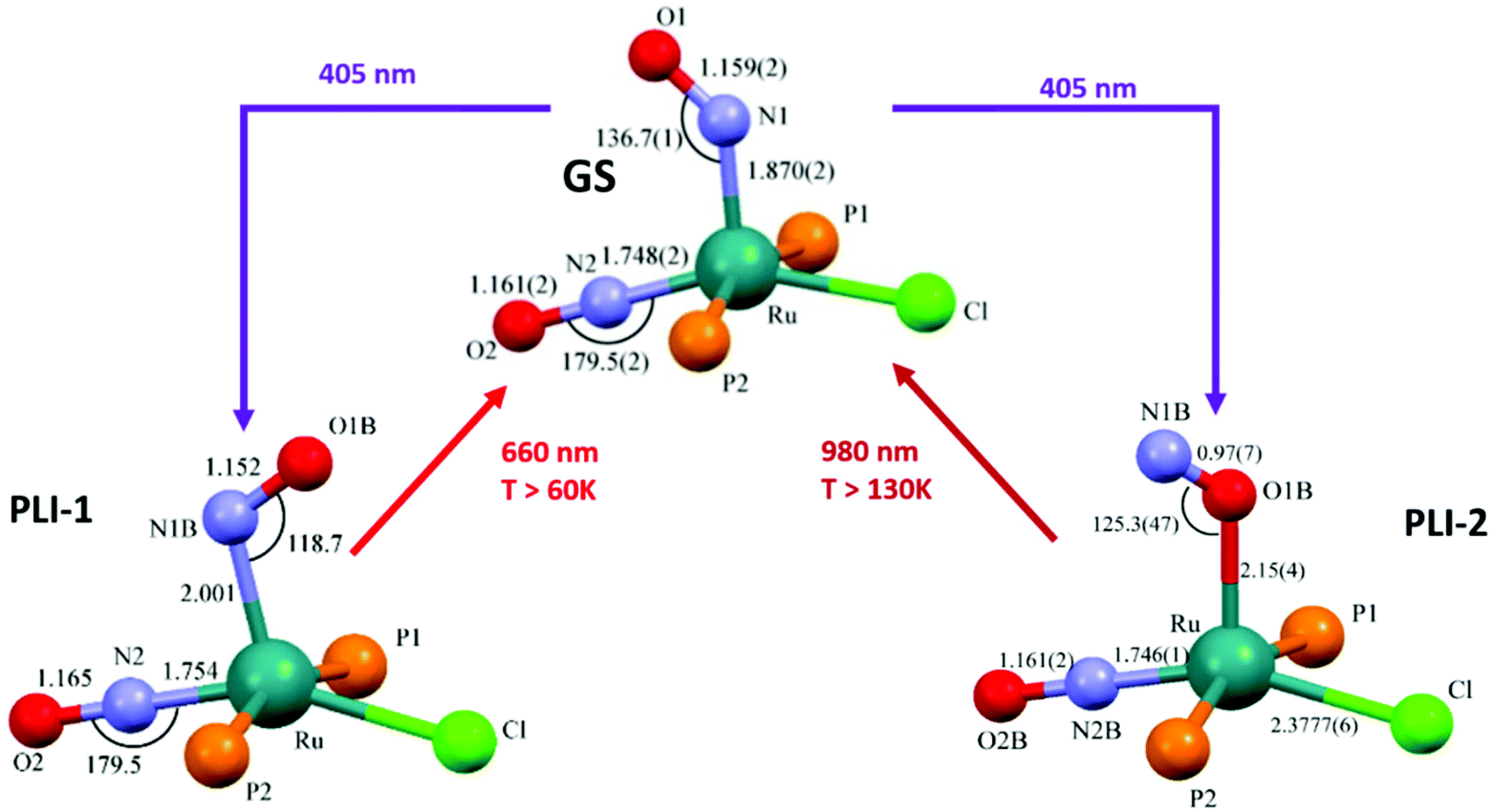 | ||
| Fig. 14 Scheme illustrating the photogeneration of nitrosyl linkage isomers in RuCl(NO)2(PCy3)2BF4 according to results reported recently.80,145 By using different irradiation wavelengths and adjusting the temperature the complex can selectively undergo either PLI-1 or PLI-2, e.g. irradiation with 405 nm in the temperature range 70–130 K results in PLI-2 (right branch) while irradiation with 405 nm below 60 K followed by irradiation with 980 nm results in PLI-1 (left branch, through depopulation of PLI-2 by 980 nm irradiation). | ||
5. NO release via one-electron reduction and photorelease: theoretical background and experimental evidence, near-IR light-induced photorelease for bioapplications
The majority of Ru-NO complexes are isolated in their {Ru-NO}6 state, where spectroscopic evidence indicates the formal RuII-NO+ charge distribution. The relatively short Ru–N bond and the linear NO coordination with the Ru–N–O bond angle close to 180° are the typical structural features of the {Ru-NO}6 state originating from the electron backbonding interaction between the Ru d orbitals and the empty π* of the NO+.146,147 Redistribution of electrons, either via a single electron reduction and/or by the photo-excitation of the complex, leads to the occupation of the π* orbital with antibonding Ru–N character, resulting in the weakening of the Ru–N bond and in the bending of the NO group coordination defined by an Ru–N–O angle of about 140°. Depending on the electronic properties of the ancillary ligands and especially the group in the trans position to NO, the weakening of the Ru–N bond can result in NO dissociation.At the beginning of the 21st century the NO-releasing properties of a series of tetrammine Ru-NO complexes of the general formula trans-[Ru(NH3)4(NO)X]3+/2+ induced by a single electron reduction have been thoroughly reviewed.146,148 The reduction proceeds at the NO+ ligand forming the {Ru-NO}7 (RuII-NO˙) state which undergoes an aquation reaction in an aqueous environment, accompanied by the release of NO˙, according to eqn (3) and (4).
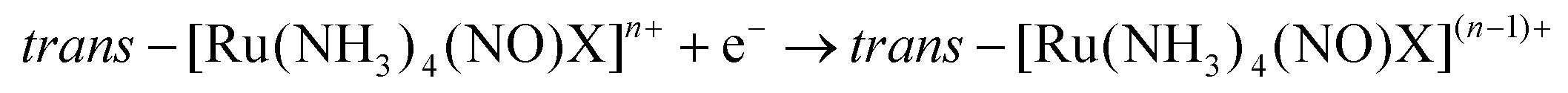 | (3) |
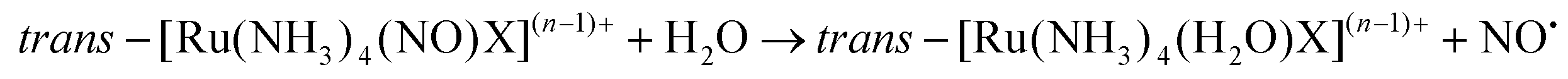 | (4) |
For X = Cl−, the halide was aquated first followed by a second aquation-NO dissociation step. The reviewed complexes had a reduction potential (E1(NO+/NO0)) in the range accessible to biologically relevant reductants (−0.4 to +0.8 V vs. NHE). Furthermore, the effect of the trans ligand X on the increase in the reduction potential, the increase in the vibrational frequencies (νNO), and the decrease in the NO release rate (k−NO) with the rise of the π-withdrawing power was observed in this series. Such clear trends were proposed as a guide for the preparation of NO donors suitable for bioapplications.
In aqueous solution, {Ru-NO}6 (RuII-NO+) nitrosyls are prone to electrophilic attack of the hydroxide anion followed by conversion of the nitrosyl group to nitrite, as illustrated with example of tetrammine in eqn (5). In fact, the inverse reaction, induced by the acidification of the nitrite complexes, is often the synthetic route to M-NO compounds, as reviewed in section 2. The Ru-NO/NO2 ratio will therefore depend on pH and the susceptibility of the NO+ for OH− attack should increase with higher electron deficiency in the nitrosyl group, as a consequence of the [Ru(NH3)4X]2+ core π-acidity.146,148 Within the tetrammine complexes, the situation proved to be more complicated due to the formation of H-bonds149 and Keq did not match the expected trends induced by the trans-ligand effect.
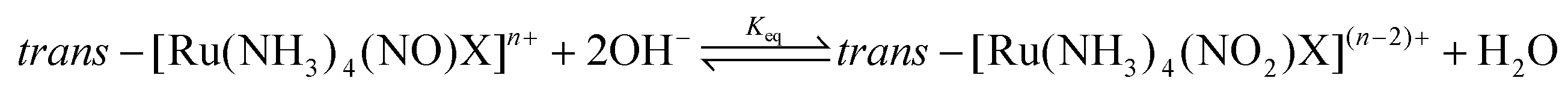 | (5) |
However, the reported Keq values confirm the domination of nitrosyl complexes at pH 7 and lower, making them suitable for evaluation as NO donors under physiological conditions.146,148 The vasorelaxant effect in the aorta ring without endothelium, induced by NO liberated from the trans-[Ru(NH3)4(NO)X]3+ complex series involving P(OEt)3, 4-picoline (4-pic), pyridine (py), pyrazine (pz), imidazole (imN), nicotineamide (nic), SO3, and NH3 as the trans X ligand was observed. All ruthenium tetrammines induced a vasorelaxation effect, but the intensity and time course were different. Complete relaxation was achieved only with complexes carrying X = P(OEt)3, 4-pic, and py and closely resembled the trend in the specific rate constant of NO release (k−NO).150,151 Rodrigues et al.55 recently used the trans-[Ru(NH3)4(NO)(isn)]3+ (isn = isonicotinamide) tetrammine nitrosyl as the NO donor activated by NADH reduction and exhibiting NO release directly inside the A549 cancer cell. The {Ru-NO}6/7 reduction was monitored in situ in a single cancer cell utilizing a novel approach of multiplex Fourier-transform infrared microscopy (μFT-IR).
The properties of related tetraazamacrocyclic ruthenium nitrosyls [Ru(mac)(NO)X]n+ (mac = macrocycle – cyclic ligand with several typically N-coordinating sites) have been reviewed by Doro et al.82 Most explored compounds involve the [14]aneN4 (cyclam)152,153 mac and its derivatives,154–157 but complexes have been reported that include smaller (imcyclen)158 and larger ([15]aneN4)159 mac rings. The trans-[Ru(mac)(NO)X]n+ scaffold with the mac ligand occupying the equatorial plane has been observed in most complexes, but cis- (cis-syn–syn/anti-[Ru(imcyclen)(NO)Cl]2+,158 [Ru(N-(2-mepy)cyclam)(NO)]3+)155,160 or fac- (fac-[Ru(κ3N4,N8,N11(1-carboxypropyl)cyclam)(NO)Cl2]+)161 geometries were also established. Although not explored in such detail as in the tetrammine complexes, the trans-[Ru(mac)(NO)X]n+ configuration offers the opportunity to tune the redox and NO releasing properties with judicious choice of the X ligand. The reactivity of [Ru(mac)(NO)X]n+ upon one-electron reduction (E1(NO+/NO0)) is similar to tetrammines (eqn (3) and (4), Scheme 5), involving NO dissociation at the expense of H2O exchange, which is preceded by rapid release of halides when X = Cl−. The NO release rate (aquation rate) from the {Ru-NO}7trans-[Ru(cyclam)(NO)(OH2)]2+ (ref. 152) is considerably slower compared to analogous tetrammines and was related to the strong interaction of NO with the [RuII(mac)]2+ fragment. Hydrogen bonding between coordinated NO+ (or NO) and HN cyclam function could also contribute to slow release.82 The enhanced persistence of the {Ru-NO}7trans-[Ru(cyclam)(NO)(OH2)]2+ makes it accessible for a second electron reduction (E3(NO0/NO−)) at more negative potentials generating the {Ru-NO}8trans-[Ru(cyclam)(NO)(OH2)]+, and is followed by a rapid release of NO− (or HNO depending on pH). The diaqua complex formed by NO or NO− liberation could act as a nitrite (NO2−) scavenger, forming the nitro complex trans-[Ru(cyclam)(NO2)(OH2)]+, which in turn, converts back to the ruthenium nitrosyl complex trans-[Ru(cyclam)(NO)(OH2)]3+ (or trans-[Ru(cyclam)(NO)(OH)]2+ at physiological pH).82,151,162,163 As nitrite is one of the major NOx pools in blood,164 these reactions would constitute a catalytic conversion of nitrite to NO, increasing the efficiency of the mac and tertrammine ruthenium nitrosyls as NO donors (Scheme 5).
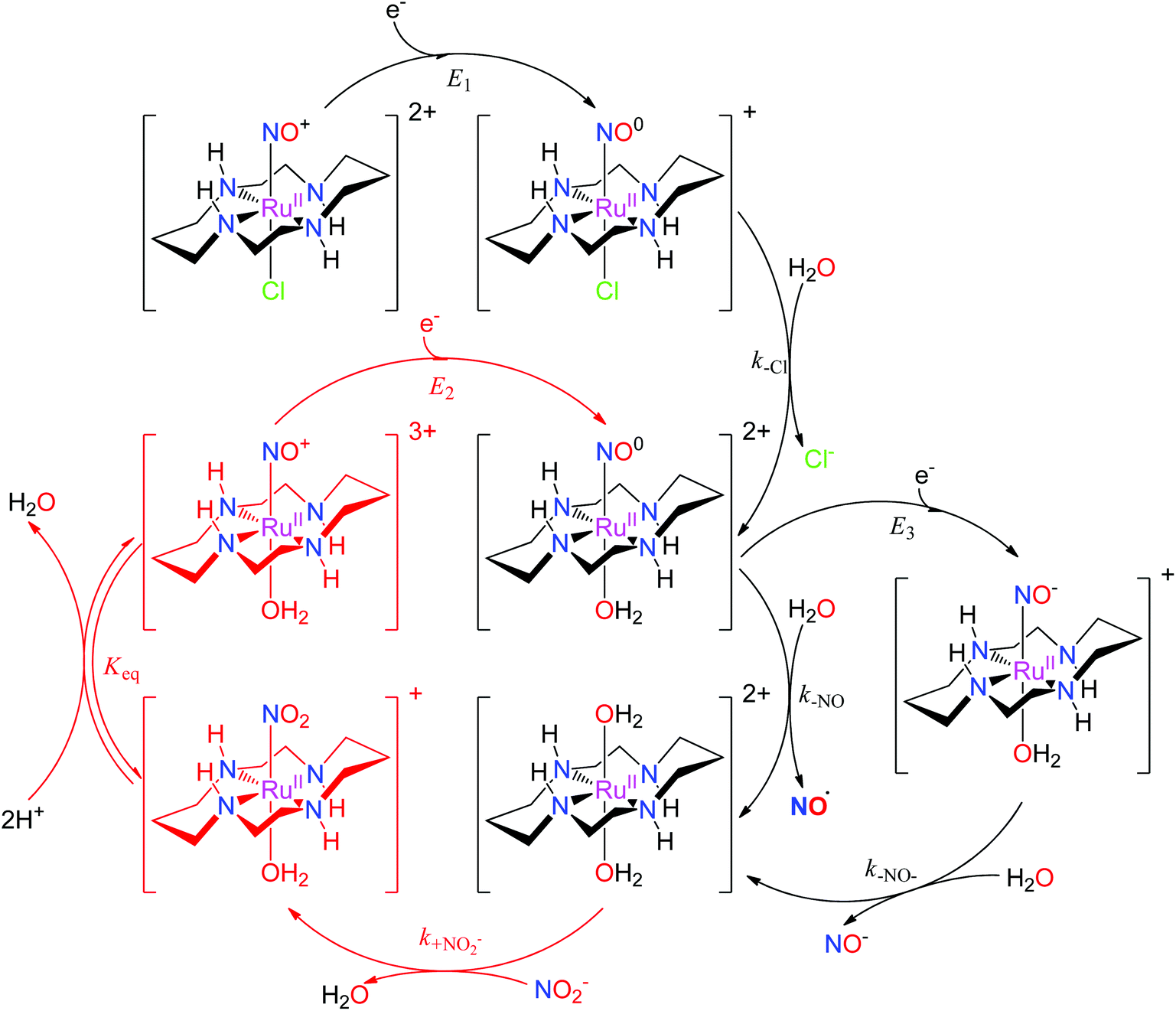 | ||
| Scheme 5 The reaction mechanism of trans-[Ru(cyclam)(NO)Cl]2+ involving reduction, NO release, and the catalytic cycle for NO2−/NO conversion proposed in the literature.82,151,162,163 | ||
The complex trans-[Ru(cyclam)(NO)Cl](PF6)2 induced a reduction in blood pressure in normotensive and hypertensive Wistar rats, and the effect lasted 20 times longer than with the same dose of sodium nitroprusside (SNP) used as the reference standard.151,164 The hypotensive effect was completely blocked by 2-(4-carboxyphenyl)-4,5-dihydro-4,4,5,5-tetramethyl-1H-imidazol-1-yloxy-3-oxide potassium salt (cPTIO), a specific NO scavenger, indicating its origin in NO liberated in vivo by chemical reduction of the cyclam Ru nitrosyl. The complex trans-[Ru([15]aneN4)(NO)Cl]2+ caused the vascular relaxation of rat aortas after in vitro reduction with norepinephrine.151,165 Efficiency was similar to that observed with SNP, and relaxation was completely abolished by oxyhemoglobin, a known NO scavenger. Recently, control of vascular smooth muscle cell growth was mediated by trans-[Ru(cyclam)(NO)Cl](PF6)2 and the observed long-lasting effect connected with NO release after reduction could protect against the development of intimal hyperplasia (IH – vessel wall thickening), a common complication in vascular remodeling surgery.163
The ability of mac to modulate the stability of the three NO redox states (NO+ ⇌ NO˙ ⇌ NO−) in ruthenium nitrosyls was recently documented with the first example of a {Ru-NO}7 complex characterised by SC-XRD, [Ru(Me3[9]aneN3)(bpy)(NO)](BF4)2 (see Scheme 3 in section 2).106 Complete spectroscopic, redox, and acid–base characterization of the three redox states of the complex cation ({Ru-NO}6,7,8) was performed in water.166 Furthermore, a systematic exploration of the acidity of the most labile {Ru-NO}8 (HNO/NO−) state was achieved by modulating the electron donating properties of the bipyridine coligand. Remarkably, a variation of the pKa HNO over almost three units (7.7–10.5) was found. More recently, such comprehensive investigations were extended to the complex featuring a robust five-coordinate tetraazamacrocyclic complex cis-[Ru(N-(2-mepy)cyclam)(NO)](PF6)3.160 The established correlation between the acidity of coordinated HNO and the redox potentials of the {RuNO}n fragment matched the trend observed in the Me3[9]aneN3-family and the pKa HNO of 13.0 expanded the range of HNO acidity in [RuII(mac)]2+ complexes over five pH units. The results confirm the sensitivity of the pKa to the nature of the coordination sphere and provide synthetic hints to design species that allow the coexistence of HNO and NO− under physiological conditions that might be of bioinorganic relevance for the elucidation of differential biological roles of HNO and NO−.
The family of ruthenium nitrosyl complexes featuring pyridyl and polypyridyl ligands comprises another set of potential redox-activated NO donors. Roberto Santana da Silva reviewed the extensive contribution of his group to this field recently.167 In the series of cis-[Ru(bpy)2(NO)X]n+ complexes, the backbonding in {Ru-NO}6 (RuII-NO+), and the electron density on the nitrosyl ligand augments with increasing σ- and π-donor character of the X ligand under the metal ion, and results in a decrease of the E1(NO+/NO0) and νNO similar to that in trans-tetrammine complexes (vide supra). In aqueous solution, chemical reduction of the cis/trans-[Ru(bpy)2(NO)X]n+ (ref. 168–173) as well as related complexes [Ru(tpy)(NO)L]n+ (ref. 174) (tpy = terpyridine) released NO, as attested by measurement with an NO sensor. The k−NO rate constants were similar for the whole group but generally lower than those for the tetrammines. cis-[Ru(bpy)2(NO)X]n+ (ref. 51) also showed a higher susceptibility to OH− attack and conversion to nitrite form, an thus requiring a lower pH to stabilize the nitrosyl complex. The complex [Ru(NO)(tpy)(NH·NHq)]3+ was found to be a vasodilator in in vitro experiments with rat aorta rings precontracted by norepinephrine and phenylephrine that could act as reducing agents.174 More recently, vasodilation activity through NO generation was confirmed for cis-[Ru(phen)2(NO)X]3+ (phen = phenanthroline, X = thiourea or thiobenzamide), and along with the stabilization of nitrosyl- over the nitro-form of the complex at physiological pH, such compounds show potential for pharmacological applications that deserves further biological studies.51
Ruthenium nitrosyls with other than nitrogen donor coordination sphere are much less abundant, but series of [Ru(P–N)(PR3)(NO)]PF6, (P–N = o-diphenylphosphino-N,N-dimethylaniline, and R = Ph and p-X-C6H4 (X = OMe, Me, F))175 or [Ru(LOMe)(NO)(cat)] (LOMe = [CpCo{P(O)(OMe)2}3]−, cat = catecholate derivative)98 (see Fig. 8 in section 3) have recently been reported. However, since their water solubility seems limited, the utility for NO delivery in biosystems would require further synthetic modification.
The light-activated release of NO represents another attractive approach for NO delivery in biological systems. In particular, the spaciotemporal control over the NO dose that can be achieved by the light pulse targeted at the required spot, as part of the photodynamic therapy (PDT) of e.g., tumors, motivates the extensive research in Ru-NO photochemistry. Progress in the field throughout the twentieth century and the beginning of the twenty-first century has been compiled in several excellent reviews.14,176–178 In the simplest case, photoexcitation of {Ru-NO}6 (RuII-NO+) in solution results in the dissociation of NO˙ followed by its exchange with a solvent molecule forming an RuIII complex according to eqn (6).
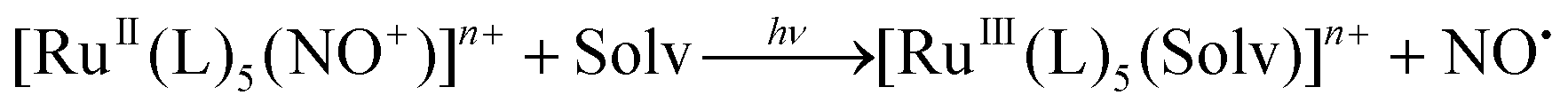 | (6) |
Excitation represents a charge transfer reaction between the molecular orbitals dominated by Ru d orbitals (often containing an admixed contribution from ancillary ligands) into the NO centred π* orbital of antibonding character with respect to Ru–N (MLCT transition(s)). The exact theoretical descriptions of the photoexcitation and successive transformation of Ru-NOs within the NO-release pathway are still a matter of debate. However, most recent studies suggest that the energetically most preferred dissociation proceeds from the triplet state of the MS2 photoisomer (3MS2).124,138 This mechanism requires the absorption of two photons, and the 3MS2 state represents a branching point that binds the NO release with the photoisomerization reactions described above. The corresponding MLCT transitions for many known Ru-NO species fall into the UV or near-Vis range of the optical spectrum.14 This is true for all the complexes with tetrammine (NO release quantum yields (ϕNO) of 8 × 10−5–0.23 mol per einstein at 313 or 330 nm)146 and tetraazamacrocylic (ϕNO of 0.008–0.61 mol per einstein at 313, 330 or 355 nm)82 ligands discussed above, where the highest ϕNO of 0.61 mol per einstein at 355 nm was reported for trans-[Ru([15]aneN4)(NO)Cl]2+.159 UV light was also required to cleave the NO in the series of [Ru(tpa)(Y)(NO)]n+ complexes (tpa = tris(2-pyridylmethyl)amine; Y = 2Cl, ONO, urea) (ϕNO of 0.002–0.06 mol per einstein), where a considerable decrease in efficiency was observed by moving from organic solvents to water.146,147 However, this high-energy UV radiation is not compatible with medical applications due to its harmful effects on living tissues and low penetration depth. The “therapeutic window” preferred for photodynamic therapy treatment covers light from the far-Vis to the NIR region (600–1100 nm) and significant effort has been devoted in the last two decades to the preparation of NO donors photosensitive in this range.
The Mascharak group systematically developed a library of ruthenium nitrosyls carrying polypyridyl ligands with carboxyamide moiety(ies).176,178 The first set of complexes with pentadentate polypyridines was derived from [Ru(PaPy3)(NO)](BF4)2 (ϕNO of 0.05 mol per einstein at 410 nm; PaPy3H = N,N-bis(2-pyridylmethyl)amine-N-ethyl-2-pyridine-2-carboxamide), which successfully delivered NO to biologically relevant proteins such as cytochrome c oxidase (CcO)18 and soluble guanylate cyclase (sGC).179 An extension in the conjugation of the ligand frame in [Ru(PaPy2Q)(NO)](BF4)2 (ϕNO of 0.17 mol per einstein at 410 nm; PaPy2QH = N,N-bis(2-pyridylmethyl)amine-N-ethyl-2-quinaldine-2-carboxamide)180 or the incorporation of a second carboxyamide in [Ru(Py3P)(NO)](BF4)2 (ϕNO of 0.05 mol per einstein at 532 nm; Py3PH2 = N,N-bis(2-(2-pyridylethyl)amine-N-ethyl-2,6-dicarboxamide))181 led to an increase in NO release efficiency as well as the desired shift of the photoband to longer wavelengths.
The second family of complexes was designed with tetradentate ligands involving two carboxamide groups that coordinate the Ru center in the equatorial plane (Chart 2). Systematic tuning of the donor–acceptor properties of the equatorial binding groups and substituents at the periphery of the tetradentate ligands, resulted in the bathochromic shift of the photoband from 320 nm in [Ru(hybeb)(NO)(OEt)]2−,182 through 380, 385, and 395 nm in [Ru(bpb)(NO)Cl], [Ru(bpb)(NO)(OEt)], and [Ru(Me2bpb)(NO)Cl],183 up to 420 nm in [Ru((OMe)2bpb)(NO)(Cl]183 and [Ru(hypyb)(NO)(OEt)]−,182 respectively. When the pyridine moieties of the bpb frame were replaced with more conjugated quinoline and isoquinoline units (bQb and IQ1, Chart 2), the resulting complexes [Ru((OMe)2bQb)(NO)Cl]183 and [Ru((OMe)2IQ1)(NO)Cl]184 exhibited photobands at 490 and 475 nm, respectively. The steric interaction of the quinoline rings in the bQb ligand led to a twisted equatorial plane in [Ru((OMe)2bQb)(NO)Cl].183 Removal of this steric strain in [Ru((OMe)2IQ1)(NO)Cl] increased both the extinction coefficient (ε = 8700 M−1 cm−1) and NO release quantum yield (ϕNO of 0.035 mol per einstein at 500 nm) making this complex a leader among those shown in Chart 2.
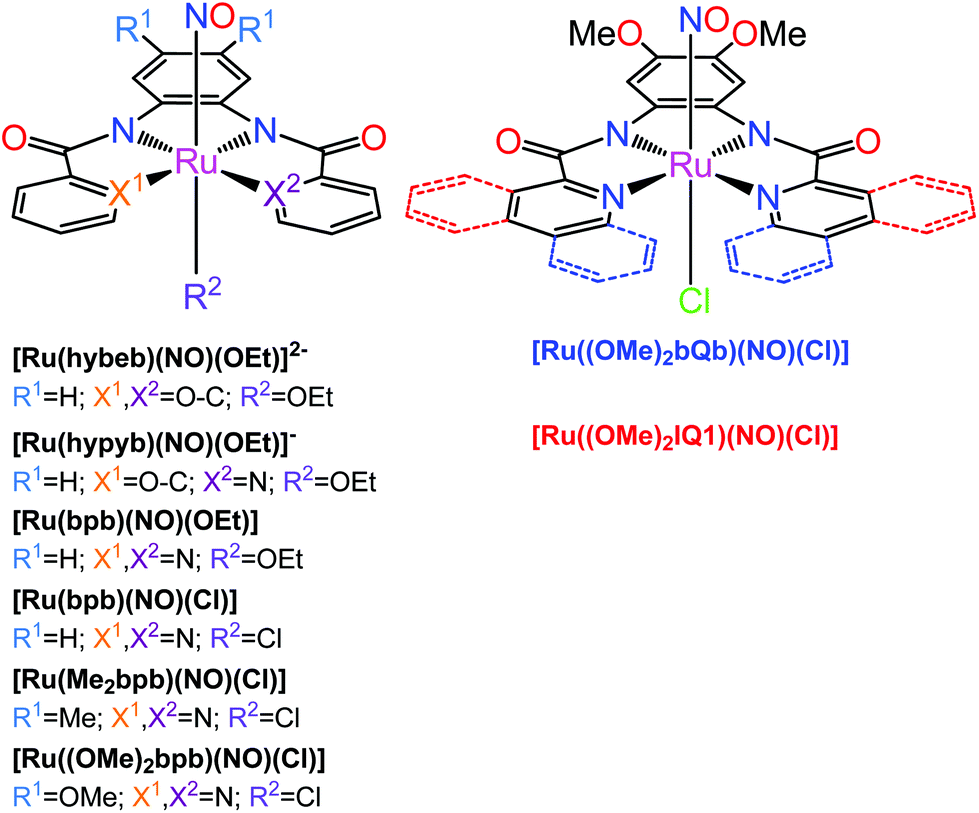 | ||
| Chart 2 {Ru-NO}6 nitrosyl complexes with tetradentate dicarboxamide ligands investigated by the Mascharak group.176,178 The axial Cl in e.g., [Ru(Me2bpb)(NO)Cl] (left structure, R2 = Cl) and [Ru((OMe)2bQb)(NO)Cl] (right structure) can be replaced by a dye molecule which greatly increases the quantum yield of NO release (see text). | ||
Further development of this scaffold involved the photosensitization of Ru-NO complexes via incorporation of intensely absorbing chromophores into the trans-axial position of the octahedral complex. The derivatives of [Ru(Me2bpb)(NO)Cl] with Cl exchanged for resorufin (Resf−) and fluorescein ethyl ester (FlEt−) dyes,15,185 showed ϕNO of 0.052 and 0.306 mol per einstein at 500 nm, respectively. Moreover, the study of [Ru((OMe)2bQb)(NO)Cl] derivatives with Resf− and its analogues containing heavier chalcogen atoms S (Thienol, Thnl) and Se (Selenophore, Seln) instead of oxygen originally in the phenoxazine ring of Resf−, revealed an increasing NO release efficiency with the larger overlap of the dye and Ru-NO MLCT absorptions (ϕNO at 500 nm [Ru((OMe)2bQb)(NO)(Resf)] 0.124 < [Ru((OMe)2bQb)(NO)(Thnl)] 0.155 < [Ru((OMe)2bQb)(NO)(Seln)] 0.189 mol per einstein).186 The Se-derivative [Ru((OMe)2bQb)(NO)(Seln)] exhibited NO photolability even under 600 nm light. Collectively, these results clearly indicated that the molecular orbitals of the dye and Ru-NO moiety in the dye-nitrosyl conjugates are intrinsically linked because of the direct coordination of the dye units to the metal center and such mixing results in efficient photosensitization toward visible light.176 As a positive side effect, the use of fluorescent dye antennas allows one to visualize the complex inside cancer cells. Moreover, the dye-nitrosyl conjugate [Ru((OMe)2bQb)(NO)(Resf)] behaved as an NO donor with a fluorometric on/off switch and quenching of the dye fluorescence due to interaction with paramagnetic RuIII in the photoproduct has been used to track the NO delivery employed to kill MDA-MB-231 cells.185 Similar results were also reported for the [Ru(Me2bpb)(NO)(Ds-im)]BF4 complex carrying the green fluorescent dansyl-imidazole (Ds-im) tag.187
In the last five years Malfant and co-workers extensively examined a series of ruthenium nitrosyls featuring substituted terpyridine ligands. The complexes cis- and trans-(Cl,Cl)-[Ru(R-Phtpy)(NO)Cl2]+ (R-Phtpy = 4′-phenyl-2,2′:6′,2′′-terpyridine; R = NO2, H, Br, OMe)54 isomers showed a progressive bathochromic shift of the MLCT transitions with increasing electron donating strength of the R-Ph substituent (λmax for R = NO2 → OMe, trans – 320 → 387 nm, cis – 290 → 366 nm). DFT calculations on the trans complex series revealed the dominant HOMO → LUMO character of these low energy transitions. The unoccupied orbitals (LUMOs) were strongly localized on the Ru-NO fragment, which led to similar energy levels in the whole complex series. On the other hand, the occupied orbitals were localized on the organic R-Phtpy ligands, and their energies increased with the strength of the electron-donating groups (MeO > H ∼ Br > NO2), as reflected in the drop of the HOMO–LUMO gaps and red shift of the corresponding transition. Efficient NO release was observed after excitation with 365 nm light, following eqn (6) and the RuIII-Solv photoproducts were formed exclusively in the trans-Cl,Cl configuration as confirmed by SC-XRD.
The ϕNO values were larger in the cis complexes (0.24–0.39 vs. 0.05–0.12 mol per einstein in the trans series) but the expected correlations with donating capabilities of the substituents were not found. Additionally, an NO release after two photon absorption (TPA) was observed upon excitation with the 810 nm light, thus involving radiation well within the PDT therapeutic window.
The TPA approach utilizes two low energy NIR photons to deliver the energy equivalent of a single photon UV excitation (Fig. 15), and offers appealing perspectives in PDT connected to: (i) the high levels of spatial resolution arising from the quadratic dependence of TPA on the pulsed light intensity; (ii) deeper penetration of NIR radiation into the tissue; (iii) decrease of harmful side-effects due to the absence of UV radiation and the use of ultrashort infrared laser pulses. The NO release induced by a TPA is a function of the product of ϕNO and TPA molecular cross-section (σTPA), a parameter describing the efficiency of TPA. A fluorenyl chromophore was reported to increase the TPA efficiency and several Ru-NO complexes featuring the 4′-(2-fluorenyl)-2,2′:6′,2′′-terpyridine (FT) ligand were thus prepared and tested. The complex cis-[Ru(FT)(NO)Cl2]+ (ref. 73, 192 and 193) showed the photoband at 389 nm and the trans-[Ru(FT)(NO)Cl2]+ (ref. 73, 192 and 193) featured a shifted absorption at 414 nm. They possessed moderate single photon ϕNO at 405 nm of (cis-) 0.31 and (trans-) 0.10 mol per einstein as well as moderate σTPA at 800 nm of (cis-) 102 and (trans-) 87 GM (Göppert–Mayer units, 1 GM = 10−50 cm4 s per photon per molecule), respectively. The dichloride tpy complexes in water environment undergo an exchange of one Cl ligand, exclusively forming the trans-(NO,OH) isomer, and the transformation is connected with a decrease in NO release quantum yield.52 Substitution of the chlorido ligands for bipyridine in [Ru(FT)(bpy)(NO)]3+ (ref. 73) led to a minor increase of σTPA to 108 GM but was connected with a drop of the ϕNO at 405 nm to 0.06 mol per einstein (0.03 at 436 nm).74 Interestingly, during the photolysis of the bpy complex a rapid reduction of the RuIII-Solv photoproduct to a RuII-Solv complex was observed and the released NO was speculated to act as the reducing agent.76 Exchange of the fluorenyl substituent on tpy for carbazole, as in [Ru(CzT)(bpy)(NO)]3+ (ref. 74) (CzT = 4′-(N-ethylcarbazol-3-yl)-2,2′:6′,2′′-terpyridine) further improved the σTPA at 800 nm to 159 GM, but decreased the ϕNO (436 nm) to 0.01. Finally, a significant jump in the TPA efficiency was achieved in a trinuclear Ru-NO complex utilizing the three branch architecture of a truxene-based ligand TX.74 The strong electronic coupling between the peripheral metal centers in RuTX, in conjunction with the symmetry of the complex, yielded σTPA of 1600 GM at 800 nm, making it an appealing candidate for PDT treatments (Table 3).
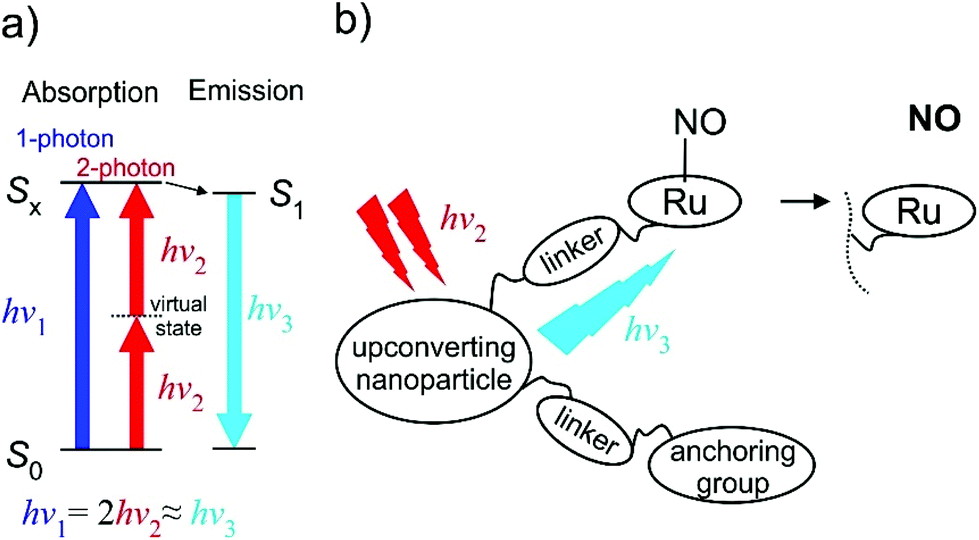 | ||
| Fig. 15 (a) The two-photon absorption (TPA) principle in energy diagram sketch. (b) Schematic drawing of the “nanoplatform puzzle” approach for NO photo-delivery utilized by Liu et al.16,188–191 | ||
| Complex | pH (in H2O), or solventa |
λ
irrb/nm |
ϕ NO/mol per einstein |
λ
TPAirrc/nm |
σ TPA/GM | Ref. |
|---|---|---|---|---|---|---|
| a pH in H2O, or the organic solvent where ϕNO determination was performed (MeCN – acetonitrile, DMF – N,N-dimethylformamide). b Irradiation wavelength in single-photon photolysis. c Irradiation wavelength in two-photon photolysis. | ||||||
| trans-[Ru(NH3)4(NO)(py)]3+ | 3.0 | 330 | 0.11 ± 0.01 | — | — | 146 |
| 4.4 | 330 | 0.13 ± 0.01 | — | — | 146 | |
| trans-[Ru(NH3)4(NO)(4-pic)]3+ | 2.0 | 330 | 0.00008 | — | — | 146 |
| 3.45 | 330 | 0.00012 | — | — | 146 | |
| 5.0 | 330 | 0.00026 | — | — | 146 | |
| trans-[Ru(NH3)4(NO)(pz)]3+ | 3.45 | 313 | 0.17 ± 0.03 | — | — | 146 |
| 4.32 | 313 | 0.23 ± 0.03 | — | — | 146 | |
| trans-[Ru(NH3)4(NO)(4-acpy)]3+ | 3.45 | 313 | 0.13 ± 0.03 | — | — | 146 |
| 4.32 | 313 | 0.23 ± 0.03 | — | — | 146 | |
| trans-[Ru(NH3)4(NO)(L-hist)]3+ | 3.45 | 313 | 0.067 ± 0.004 | — | — | 146 |
| 4.32 | 313 | 0.086 ± 0.004 | — | — | 146 | |
| trans-[Ru(1-pramcyclam)(NO)Cl]3+ | 1.0 | 313 | 0.009 ± 0.001 | — | — | 82 |
| 334 | 0.008 ± 0.001 | — | — | 82 | ||
| 370 | <0.001 | — | — | 82 | ||
| 4.9 | 313 | 0.030 ± 0.002 | — | — | 82 | |
| 334 | 0.011 ± 0.001 | — | — | 82 | ||
| 370 | <0.002 | — | — | 82 | ||
| 7.4 | 313 | 0.100 ± 0.002 | — | — | 82 | |
| 334 | 0.090 ± 0.010 | — | — | 82 | ||
| 370 | <0.003 | — | — | 82 | ||
| trans-[Ru(cyclam)(NO)Cl]2+ | 1.0 | 334 | 0.008 ± 0.001 | — | — | 82 |
| 4.9 | 313 | 0.010 ± 0.002 | — | — | 82 | |
| 334 | 0.009 ± 0.001 | — | — | 82 | ||
| 355 | <0.002 | — | — | 82 | ||
| 7.4 | 334 | 0.100 ± 0.010 | — | — | 82 | |
| 355 | 0.160 ± 0.050 | — | — | 82 | ||
| trans-[Ru([15]aneN4)(NO)Cl]2+ | 7.4 | 355 | 0.61 ± 0.05 | — | — | 82 and 159 |
| [Ru(Hind)4(NO)Cl]2+ | MeCN | 405 | 0.045 ± 0.005 | — | — | 17 |
| [Ru(Hind)4(NO)OH]2+ | MeCN | 405 | 0.040 ± 0.005 | — | — | 17 |
| [Ru(Hind)2(ind)2(NO)Cl] | MeCN | 405 | 0.038 ± 0.003 | — | — | 17 |
| [Ru(Hind)2(ind)2(NO)OH] | MeCN | 405 | 0.062 ± 0.005 | — | — | 17 |
| [Ru(tpa)Cl2(NO)]+ | MeCN | 365 | 0.06 ± 0.03 | — | — | 147 |
| DMF | 365 | 0.03 ± 0.02 | — | — | 147 | |
| [Ru(tpa)(ONO)(NO)]2+ | DMF | 365 | 0.01 ± 0.002 | — | — | 147 |
| [Ru(tpa)(Urea)(NO)]3+ | DMF | 365 | 0.007 ± 0.001 | — | — | 147 |
| 7 | 365 | 0.002 ± 0.001 | — | — | 147 | |
| [Ru(PaPy3)(NO)]2+ | 7 | 355 | 0.12 | — | — | 180 |
| MeCN | 300 | 0.06 | — | — | 180 | |
| 410 | 0.05 | — | — | 180 | ||
| [Ru(PaPy2Q)(NO)]2+ | 7 | 355 | 0.20 | — | — | 180 |
| MeCN | 300 | 0.14 | — | — | 180 | |
| 410 | 0.17 | — | — | 180 | ||
| [Ru(Py3P)(NO)]+ | 7 | 302 | 0.15 ± 0.01 | — | — | 181 |
| MeCN | 532 | 0.050 ± 0.004 | — | — | 181 | |
| [Ru(hybeb)(NO)(OEt)]2− | MeCN | 300 | 0.025 | — | — | 182 |
| [Ru(hypyb)(NO)(OEt)]− | MeCN | 300 | 0.067 | — | — | 182 |
| 400 | 0.008 | — | — | 182 | ||
| [Ru(Me2bpb)(NO)Cl] | DMF | 500 | 0.0008 ± 0.0002 | — | — | 184 |
| [Ru(Me2bQb)(NO)Cl] | DMF | 500 | 0.010 ± 0.003 | — | — | 184 |
| [Ru((OMe)2bQb)(NO)Cl] | DMF | 500 | 0.025 ± 0.003 | — | — | 184 |
| MeCN | 500 | 0.025 ± 0.010 | — | — | 186 | |
| [Ru((OMe)2bpb)(NO)Cl] | DMF | 500 | 0.010 ± 0.005 | — | — | 184 |
| [Ru((OMe)2IQ1)(NO)Cl] | DMF | 500 | 0.035 ± 0.005 | — | — | 184 |
| [Ru(Me2bpb)(NO)(Resf)] | DMF | 500 | 0.052 ± 0.008 | — | — | 184 |
| [Ru(Me2bQb)(NO)(Resf)] | DMF | 500 | 0.102 ± 0.009 | — | — | 184 |
| [Ru((OMe)2bQb)(NO)(Resf)] | DMF | 500 | 0.206 ± 0.008 | — | — | 184 |
| MeCN | 500 | 0.124 ± 0.010 | — | — | 186 | |
| 525 | 0.078 ± 0.010 | — | — | 186 | ||
| [Ru((OMe)2bpb)(NO)(Resf)] | DMF | 500 | 0.120 ± 0.008 | — | — | 184 |
| [Ru((OMe)2IQ1)(NO)(Resf)] | DMF | 500 | 0.271 ± 0.008 | — | — | 184 |
| [Ru(Me2bpb)(NO)(FlEt)] | DMF | 500 | 0.306 | — | — | 176 |
| [Ru((OMe)2bQb)(NO)(Thnl)] | MeCN | 500 | 0.155 ± 0.010 | — | — | 186 |
| 525 | 0.190 ± 0.010 | — | — | 186 | ||
| 550 | 0.131 ± 0.010 | — | — | 186 | ||
| [Ru((OMe)2bQb)(NO)(Seln)] | MeCN | 500 | 0.189 ± 0.010 | — | — | 186 |
| 525 | 0.214 ± 0.010 | — | — | 186 | ||
| 550 | 0.176 ± 0.010 | — | — | 186 | ||
| 575 | 0.152 ± 0.010 | — | — | 186 | ||
| 600 | 0.040 ± 0.010 | — | — | 186 | ||
| trans-(Cl,Cl)-[Ru(NO2-Phtpy)(NO)Cl2]+ | MeCN | 365 | 0.05 | — | — | 54 |
| trans-(Cl,Cl)-[Ru(H-Phtpy)(NO)Cl2]+ | MeCN | 365 | 0.12 | — | — | 54 |
| trans-(Cl,Cl)-[Ru(Br-Phtpy)(NO)Cl2]+ | MeCN | 365 | 0.11 | — | — | 54 |
| trans-(Cl,Cl)-[Ru(MeO-Phtpy)(NO)Cl2]+ | MeCN | 365 | 0.07 | — | — | 54 |
| cis-(Cl,Cl)-[Ru(NO2-Phtpy)(NO)Cl2]+ | MeCN | 365 | 0.24 | — | — | 54 |
| cis-(Cl,Cl)-[Ru(H-Phtpy)(NO)Cl2]+ | MeCN | 365 | 0.39 | — | — | 54 |
| cis-(Cl,Cl)-[Ru(Br-Phtpy)(NO)Cl2]+ | MeCN | 365 | 0.32 | — | — | 54 |
| cis-(Cl,Cl)-[Ru(MeO-Phtpy)(NO)Cl2]+ | MeCN | 365 | 0.28 | — | — | 54 |
| cis-[Ru(FT)(NO)Cl2]+ | MeCN | 405 | 0.31 | 800 | 102 | 73, 192 and 193 |
| trans-[Ru(FT)(NO)Cl2]+ | MeCN | 405 | 0.10 | 800 | 87 | 73, 192 and 193 |
| [Ru(FT)(bpy)(NO)]3+ | MeCN | 405 | 0.06 | 800 | 108 | 74 |
| 436 | 0.03 | |||||
| [Ru(CzT)(bpy)(NO)]3+ | MeCN | 436 | 0.01 | 800 | 159 | 74 |
| RuTX | MeCN | — | — | 800 | 1600 | 194 |
An alternative strategy for the application of low energy excitation in NO release PDT involves the incorporation of Ru-NO complexes in supramolecular systems with upconverting nanoparticles (UCNPs). The nanoparticles emit light in the UV-Vis range upon absorption of two NIR photons (Fig. 15), and this light is used to cleave the Ru-NO bond of the attached complex directly at the spot of the drug system location within the cell.195 Liu et al.188 developed a multifunctional nanoplatform {Lyso-Ru-NO@FA@C-TiO2} containing a ruthenium nitrosyl donor, [Ru(tpyCOOH)(Lyso-NINO)(NO)](PF6)3, (Lyso-Ru-NO), a cancer cell directing group of folic acid (FA), and a carrier of biocompatible carbon-doped titanium dioxide nanoparticles (C-TiO2 NPs). The Lyso-NINO ligand featured a morpholine moiety that served as a lysosomal targeting group. The FA group was used as a tumor-targeting component on the basis of its high affinity to folate receptor (FR), which is often overexpressed on the surface of a wide range of human cancerous cells (e.g., HeLa and KB cell lines). The nanoplatform was used for cellular and subcellular delivery of NO and ROS to HeLa cells under mild 808 nm NIR light irradiation. Moreover, the inherent blue fluorescence of the nanoplatform enabled it to be tracked during its cellular internalization and subcellular distribution. The cytotoxicity assay manifested the highest anticancer efficacy of the dual-targeted nanoplatform, when compared with non-targeted counterparts. This multifunctional platform thus garnered cancer cell selectivity, subcellular lysosome-targetable, mild NIR light-triggered NO and ROS release, and cell imaging functionalities all into one system. The same research group later described several luminescent nanoplatforms consisting of N-doped graphene quantum dots (N-GQDs, diameters 7–10 nm)16 functionalized with a [Ru(tpyCOOH)(OPDA)(NO)](PF6)3 (@Ru-NO; OPDA = o-phenylenediamine).189 This nanoplatform {N-GQDs@Ru-NO@TPP} was also decorated with a triphenylphosphonium moiety that, when administered to HeLa cancer cells, localizes the system in the mitochondria.16 The in vitro generation of NO in these cells upon 808 nm irradiation was demonstrated using the intracellular NO sensitive fluorescent probe, DAF-FM DA (4-amino-5-methylamino-20,70-difluorofluorescein diacetate). Furthermore, this treatment significantly decreased the viability of these HeLa cells by triggering apoptosis. Similar effects were noted after intratumoral injection of this platform into HeLa tumor-bearing mice followed by 808 nm irradiation. Notably, continuous 808 nm irradiation of a suspension of this nanoplatform in aqueous buffer at a relatively low power density led to a substantial temperature change in the bulk solvent. Such a photothermal effect needs to be considered whenever such absorbers are subjected to continuous NIR excitation. The total energy deposited at a site is likely to be much larger under such conditions than when using a pulsed laser, which has much higher peak power, but much lower average power. Nevertheless, photothermal therapy, induced in this fashion, can boost the overall effectiveness of PDT and contribute to the cytotoxicity effect of the nanoplatform drug. The same ruthenium nitrosyl NO donor was also recently reported in another platform based on N-doped graphene QDs decorated with a galactose derivative {N-GQDs@Ru-NO@Gal} that selectively targets liver cancer cells over normal liver cells.190 In a follow-up study,196 an approach combining targeted chemotherapy and NO-mediated multimodal phototherapy was tested with several cancer cell lines. The Ru-NO donor was covalently linked with the anti-cancer PtIV prodrug (cis-[PtIV(NH3)2(Cl)2(OH)2]) and combined with N-GQDs. The system was further equipped with FA for improved cancer cell recognition forming a new {N-GQDs@Ru-NO-Pt@FA} system. With the properties of inherent fluorescence-tracing and FR-targeting, the nanoplatform selectively attacked folate sensitive cancer cell lines over normal cells, and co-delivered NO and PtII into the tumor site upon 808 nm NIR light irradiation resulting in a high cytotoxicity toward cancer cells. The anticancer efficacy was more pronounced when compared with both the NO-release-only nanoplatform {N-GQDs@Ru-NO@FA} and cis-platin alone. More recently, N-GQDs have been exchanged with magnetic polydopamine (PDA)-coated iron oxide (Fe3O4@PDA) in {Fe3O4@PDA@Ru-NO@FA} nanoplatform, aiming at a better control over the spatial drug distribution in mice tissue via magnetic field guiding.191 The Fe3O4 magnetic core and the incorporated FA-directing groups enabled the nanoplatform to accumulate selectively in tumors via magnetic field-guidance and thus to target the FR-positive cancer cell lines. Moreover, the strong magnetic response allowed a precise tracking of the drug location through the dark contrast in T2-weighted magnetic resonance imaging (MRI). Both the in vitro and in vivo experiments showed noteworthy antitumor efficacy of the nanoplatform by the synergistic effect of the spatiotemporal-controlled release of NO and photothermal therapy under 808 nm NIR light irradiation.
6. Ruthenium nitrosyl complexes as potential anticancer drugs
Ruthenium nitrosyl complexes as NO donors as well as the wide application of NO in the treatment of different diseases (cancer, cardiovascular, mental, parasitic, etc.) were reviewed recently.151,197 Nitric oxide is a key regulator of cellular processes. NO can produce different effects in the body in addition to activation of NO/sGC/cGMP signaling pathway. These imply (i) nitrosylation of metalloenzymes; (ii) the regulatory effects associated with post-translational modification (e.g., S-nitrosothiols, 3-nitrotyrosine, 8-nitroguanine, etc.); (iii) free radical scavenging (e.g., detoxification of ROS); and (iv) compensatory changes in NOS expression and related stress response mechanisms.198 As mentioned previously, NO is able either to stimulate or inhibit tumor growth depending on the local concentration of delivered NO and other factors, e.g., rate of NO release, cell type, etc.199 Low levels of NO (20–100 nM) promote angiogenesis,200 while higher levels of NO (200–600 nM) inhibit cell proliferation and can induce apoptosis.201 Among cellular changes,202,203 which finally can end in apoptosis, nitrosylation of metalloenzymes plays an important role. This process is reversible at low NO levels, but irreversible at higher concentrations of NO leading to irreversible inhibition of the mitochondrial respiratory chain mediated by inhibition of cytochrome oxidase or even to leakage of cytochrome c through the mitochondrial membrane, followed by activation of several caspases in a signaling cascade. In addition, modification of cellular DNA or its fragmentation can occur and disturb cellular protein homeostasis.201Here we would like to highlight briefly the most significant recent achievements in the therapeutic application of ruthenium nitrosyl complexes as potential anticancer drugs. Several research groups around the world are engaged in the development of compounds that can release NO at target sites after site-specific delivery to cells by exposure to light, cell pH or to specific enzymes.2,178,195 The therapeutic effect of NO is highly dependent on its concentration at the site where it is delivered. Therefore, precise control of NO delivery is a crucial point for successful application of lead drug candidates.
The evaluation of photocytotoxic potential of the ruthenium nitrosyl complexes [RuCl3(L1)(NO)]−, where HL1 = 8-hydroxyquinoline and its derivatives, confirmed the photo-enhanced cytotoxicity on the HepG2 cells in vitro.62 For example, [RuCl3(L1)(NO)]− with HL1 = 2-chloro-8-hydroxyquinoline, showed superior activity with an IC50 value of 21.5 μM under irradiation with 420 nm light compared to 100 μM in the absence of photoirradiation. In comparison, the photocytotoxicity of complexes [RuCl(L1)(L2)(NO)]+, where HL1 = 8-hydroxyquinoline, L2 = 2,2′-bipyridine derivatives, evaluated against HeLa cells, increased by a factor of ∼1.5.204
The cytotoxicity of mer-[Ru(NO)(L)2Cl3], where L = different nicotinates, on Hep2 and HepG2 cells, carried out using dual staining with Hoechst 33342 and propidium iodide (PI), was found to be in the concentration range of 1–50 μM. The complexes with isonicotinic acid esters are the most cytotoxic, while the antiproliferative activity of the compounds with nicotinic acid derivatives is lower and well comparable with that of the pyridine complex.39
The complexes trans-[Ru(NO)(L)4X]Cl2 (where X = Cl, OH; L = 1H-indazole) showed very good antiproliferative activity in human cancer cell lines HCT116 and A2780 with IC50 values in the low micromolar or submicromolar concentration range.17 The effects of NO release on the cytotoxicity of these complexes with or without light irradiation (by blue LED light (max emission at around 470 nm)) was examined and found to increase upon irradiation by ∼30%, even though the cytotoxicity was not reduced in the presence of NO scavenger carboxy-PTIO.
The complexes (indazolium)[cis/trans-RuCl4(NO)(1H-indazole)] revealed remarkable antiproliferative activity in cancer cell lines (A549, CH1/PA-1 and SW480) and yielded IC50 values in the low micromolar range.205,206 In addition, the trans-configured ruthenium complex is up to 2.4-fold more potent than its cis analog. It should be pointed out that the exchange of one indazole in KP1019 for NO increased the cytotoxic potency of trans-ruthenium nitrosyl analogue by a factor of 72 and 34 in SW480 and CH1/PA-1 cells, respectively. The studied ruthenium nitrosyl complexes induced apoptosis by the mitochondrial pathway, which is at least in part associated with ROS generation.
To investigate the reactivity of potential anticancer ruthenium-nitrosyl-based drug candidates towards amino acids as important biological ligands in the blood serum and in the cytosol, a series of ruthenium nitrosyl complexes of the general formula (nBu4N)[RuCl3(AA-H)(NO)], where AA = glycine (Gly), L-alanine (L-Ala), L-valine (L-Val), L-proline (L-Pro), D-proline (D-Pro), L-serine (L-Ser), L-threonine (L-Thr), and L-tyrosine (L-Tyr), were prepared.207 SC-XRD studies revealed the same isomer type, namely mer(Cl),trans(NO,O)-[RuCl3(AA-H)(NO)], for all complexes. The compounds were more cytotoxic in the chemosensitive CH1 cancer cells (IC50 = 7.5–27 μM) than in SW480 cells (IC50 = 20–71 μM) and even more chemoresistant A549 cells (IC50 > 100 μM). Variation of the amino acid as ligand had generally a small impact on the antiproliferative activity within the range of amino acids employed.
Human serum albumin (HSA) as a delivery vehicle for controlled nitric oxide release was investigated on nitrosyl ruthenium complexes [RuCl3(L1)(NO)]−, where HL1 = 8-hydroxyquinoline and its derivatives62,63,208,209 and [RuCl(L1)(L2)(NO)]+, where HL1 = 8-hydroxyquinoline, L2 = 2,2′-bipyridine derivative.204 The binding constants of the ruthenium complexes with HSA (as well as DNA) were determined by fluorescence spectroscopy.62,204 The binding mode of the HSA with [RuCl3(L)(NO)]− complex, where HL = 5-chloro-8-quinoline, was established by protein X-ray crystallography (Fig. 16, PDB ID: 7DL4).63,208,209 It should be noted that two other structures of HSA adducts with ruthenium complexes were also reported, namely HSA-[RuCl5(indazole)]2−,208 and HSA-[RuCl4(indazole)2]−.209
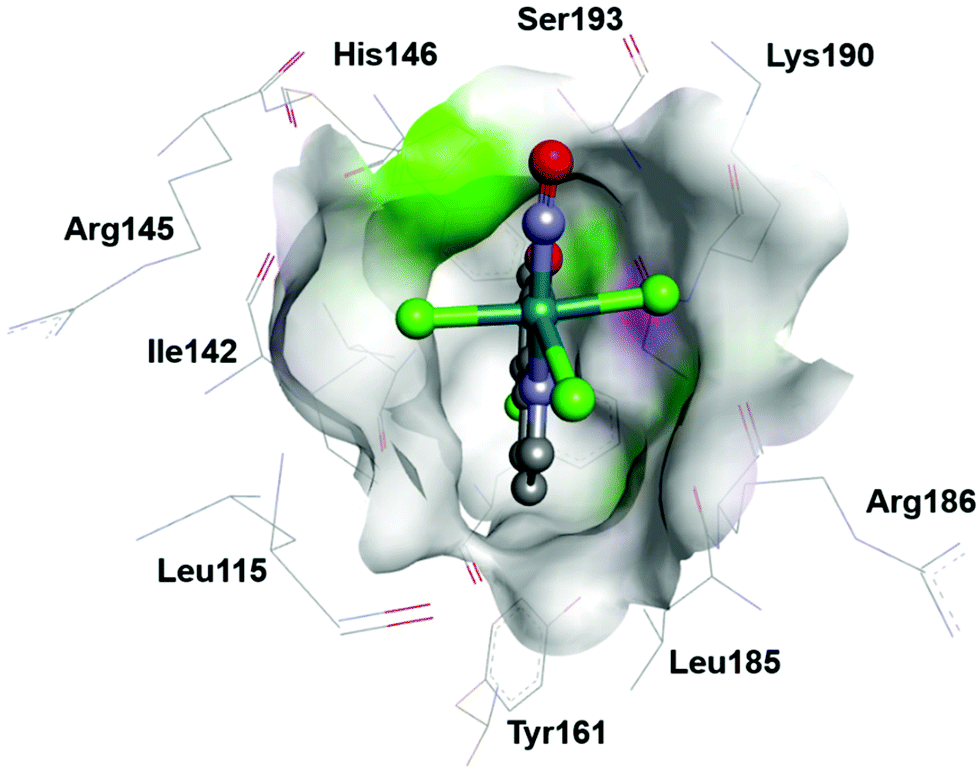 | ||
| Fig. 16 The [RuCl3(L)(NO)]− complex (HL = 5-chloro-8-quinoline) with occupancy higher than 0.70 located in the HSA subdomain IB in a hydrophobic cavity surrounded by Leu115, Ile142, Tyr161, Ser193, Arg145, His146, Arg186, and Lys190. Taken from PDB 7DL4. | ||
The investigation of the photoinduced inactivation of DNA repair enzymes (DNA glycosylases) in vitro by [RuNO(L)2(NO2)2OH], where L = 3-methylpyridine,210 revealed that the activity of DNA glycosylases decreased by more than 90% after irradiation.
The voltage-dependent anion channel (VDAC) is a protein in the mitochondrial outer membrane that plays a central role in mitochondria-mediated apoptosis and is thus a potential target for NO donor agents based on ruthenium complexes. The complex cis-[RuCl(NO)(L)2]2−, where H2L is 2,2′-bipyridine-4,4′-dicarboxylic acid, was coupled to a polyclonal antibody immunoglobulin IgG (anti-VDAC) via amide bond formation, and found to recognize a cell surface marker. The reported cytotoxicity of (RuNO)-IgG conjugate against the liver hepatocellular carcinoma cell line HepG2 was up to 80% greater than that of the IgG-free complex. It was suggested that this increase of cytotoxicity was due to site-specific interaction of the complex followed by NO release.50
Several research groups studied the photochemistry of Ru-NO complexes to evaluate their NO releasing properties under light irradiation. Generally, most ruthenium nitrosyls exhibit photoactivation by UV light (<400 nm) with relatively low quantum yields for NO release. Therefore, the attention was focused on sensitising Ru-NOs to visible light.195
The development of different ruthenium nitrosyl nanoplatforms for photocontrolled targeted NO delivery to cancer cells followed by NO release in a widen window of therapeutic wavelengths has been recently reported, and is reviewed in detail previously (see the end of section 5).16,189–191,195,196
The use of liposome delivery systems was reported for other ruthenium nitrosyl species, namely for cis-[Ru(NO)(L1)2(L2)](PF6)3 (where L1 = bipyridine, L2 = 4-picoline). This conjugate was able to release NO inside cancer cells. The NO release was monitored by fluorescent microscopy using a non-fluorescent probe, 4-amino-5-methylamino-2′,7′-difluorofluorescein (DAF-FM), which reacts with NO to give a fluorescent benzotriazole. The death-induction mechanism in hepatocarcinoma cell line (HepG2) was assessed via cytochrome c release from mitochondria and by activating the caspases 3 and 9.69
7. Antibacterial activity and other applications of ruthenium nitrosyl complexes
A magnetic multifunctional nanoplatform, {Fe3O4@PDA@Ru-NO}, in which the ruthenium nitrosyl donor is [Ru(NO)(L1)(L2)]3+ where L1 is 2,2′:6′,2′′-terpyridine functionalized by a carboxylic group at the 4′-position and L2 is a modified 1,2-benzoquinone-diimine, while Fe3O4@PDA is polydopamine (PDA)-coated mixed iron oxide, was imbedded into the matrix of a chitosan (CS)-poly(vinyl alcohol) (PVA) to give a hybrid thermosensitive CS-PVA/NO hydrogel. The CS-PVA/NO hydrogel demonstrated mild (<150 mW cm−2) NIR light-controlled NO release and, in addition, produced an efficient antibacterial effect evaluated by the inhibition zone test for both Gram-negative Escherichia coli and Gram-positive Staphylococcus aureus.191A series of complexes, namely fac-[Ru(NO)Cl3(P,P-L)], where P,P-L = bis[(2-diphenylphosphino)phenyl] ether, and trans-(NO,OMe)-[RuCl(OMe)(NO)(P,P-L)(L)]PF6 (L = pyridine, 4-methylpyridine, 1-methylimidazole and 1H-benzimidazole) were tested against strains of E. coli, P. aeruginosa, S. aureus, S. epidermidis, and E. faecalis by minimum inhibitory concentration (MIC) assay.64 The tests revealed that after 24 and 48 h, all complexes exhibited MIC values higher than 500 μg mL−1 for both the Gram-negative bacteria strains E.coli and P. aeruginosa, and for the Gram-positive E. faecalis. On the other hand, the complexes were active against the Gram-positive bacteria strains S. aureus and S. epidermidis with MIC values in the range 4.2–80.5 μmol L−1 (S. aureus) and 8.0–322.2 μmol L−1 (S. epidermidis). Interestingly, the cationic complexes were 4.8 times more active than the neutral complex against S. aureus and 20-fold against S. epidermidis.
The effect of trans-(NO, OH)-[Ru(NO)(OH)Cl(L)](PF6), where L is 4′-(2-fluorenyl)-2,2′:6′,2′′-terpyridine, on both methicillin antibiotic sensitive and methicillin antibiotic resistant to S. epidermidis has been studied.211 At concentration >1 μM, this complex induced a 40% decrease in viability even in the absence of irradiation. Under irradiation, toxicity could be already observed at 0.1 μM, with a 2-fold decrease in cell viability, as determined by colony counting. Interestingly, in combination with the antibiotic methicillin, this low dose of NO dramatically decreased bacterial resistance and made methicillin-resistant S. epidermidis bacteria 100-fold more sensitive.
Antimicrobial activity studies against Gram-positive and Gram-negative bacteria were also carried out with [Ru(NO)Cl(P,P-L1)(L2)](PF6)2 (L1 = 1,4-bis(diphenylphosphino)butane, L2 = 4,4′-dimethyl-2,2′-bipyridine), and its precursor cis-[RuCl2(P,P-L1)(L2)].212 Both complexes showed promising antibacterial activity against Gram-positive bacterial strains of S. aureus and S. epidermidis with MIC values ranging from 125.0 to 7.8 mg mL−1, while were devoid of activity against Gram-negative bacteria P. aeruginosa and E. coli even at 500 mg mL−1, the highest concentration applied. It is notable that the nitrosyl complex showed a greater antibiotic effect than its precursor, supporting the important role of NO in this assay.
Antileishmanial213,214 and anti-inflammatory (as caused by gout arthritis)215 activities of cis-[Ru(L)2(SO3)(NO)](PF6), where L = bipyridine, were also reported recently. Anti-asthmatic complexes, cis-[Ru(L1)2(L2)(NO)](PF6)3, where L1 = 2,2′-bipyridine, L2 = 2-methyl-1H-imidazole,216 and [Ru(NO)(L1)(L2)]3+, where L1 = terpyridine, L2 = 1,2-benzoquinone-diimine,217 were recently reported as well. Some attention was also payed to ruthenium nitrosyl compounds as nitric oxide donors for the treatment of cardiovascular diseases.41,51,68,163,218–220
8. Conclusions and outlook
Remarkable progress in the design and synthesis of mono- and heteropolynuclear complexes and clusters bearing RuNE moiety(ies) (E = O, S, Se) has been achieved over the last decade. The synthesis of RuNTe complexes still remains a challenge. A series of ruthenium(II) nitrosyl complexes supported by Me3[9]aneN3 and bpy ligands proved to be suitable for the synthesis of one-electron reduced {Ru(NO)}7 species, their isolation and characterization by SC-XRD. These works allowed for further insight into the role of NO−/HNO as endogenous species and their bioinorganic relevance. Use of complexes such as trans-[Ru(OH)NO(Hpz)4] as a source of μ-hydroxido, μ3-oxido and μ4-oxido groups appears to be a reliable and prospective general way to new heteropolynuclear complexes and clusters. Of potential interest is the further exploitation of common coordination features of carboxylates and pyrazolates as effective bridging ligands. The new compounds might possess the required robustness in solution and preserve the essential properties searched for in nitrosyl compounds, such as photoinduced linkage isomerism and/or NO release. In this respect the significant progress of recent years opens some interesting perspectives. The establishment of a large family of ruthenium nitrosyl complexes exhibiting high conversion efficiencies and high stabilities of PLI enables the design of functional materials for optical applications at room temperature, either by exploiting the intrinsic photochromic and photorefractive properties due to PLI generation, or by combining them in assemblies for non-linear optics or photomagnetic devices. From ultrafast studies in SNP in solution221 it is known that an intermediate state is involved in the photorelease of NO and the recent studies on ruthenium nitrosyls suggest that this might be the MS2 state for one of the possible reaction paths. The gained insight in the photoexcitation mechanism, mainly pushed by theory, needs to be pursued further by employing time-resolved experimental methods in order to confirm the models and to gain more insight into the competition between photoinduced NO release and PLI formation, especially at the branching points. Dinitrosyl complexes bear the potential for multi-state photoswitching materials and as such may even be of interest in the framework of generation of entangled states.222 The direct photoactivation of NO release triggered by low energy light within the PDT therapeutic window (600–1100 nm) still remains a tedious challenge for Ru-NO complexes. Scarce examples where conjugation in the ligand framework successfully shifted the photoband close to the edge of the required range might indicate the limits of such a synthetic strategy. Fortunately, workaround approaches involving two photon absorption, and/or combination of blue absorbing Ru-NO chromophores with upconverting nanoparticles show promising results, including efficient cancer phototherapy in animal models. More progress might be also expected from polynuclear complexes bearing several NO groups. These complexes are expected to act as a double-edged sword, releasing in the tumor upon reduction or photoactivation high concentrations of nitric oxide along with toxic metal complexes. Moreover, they might be relevant as platforms to combine magnetism and photoinduced changes to produce new functional materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
MZ acknowledges the support of the Science and Technology Assistance Agency (contract no., APVV-19-0024, DS-FR-19-0035) and Scientific Grant Agency of the Slovak Republic VEGA (contracts no. 1/0078/21, 1/0504/20). VBA is thankful to the Austrian Agency for International Cooperation in Education and Research OEAD for financial support via grant no. MULT 08/2020. We thank Dr Jóhannes Reynisson for preparation of Fig. 16.Notes and references
- https://www.nobelprize.org/prizes/medicine/1998/summary/ .
- M. J. Rose and P. K. Mascharak, Curr. Opin. Chem. Biol., 2008, 12, 238–244 CrossRef CAS PubMed.
- P. L. Feldman, O. W. Griffith, H. Hong and D. J. Stuehr, J. Med. Chem., 1993, 36, 491–496 CrossRef CAS PubMed.
- R. B. Silverman, Acc. Chem. Res., 2009, 42, 439–451 CrossRef CAS PubMed.
- L. A. Ridnour, D. D. Thomas, C. Switzer, W. Flores-Santana, J. S. Isenberg, S. Ambs, D. D. Roberts and D. A. Wink, Nitric Oxide, 2008, 19, 73–76 CrossRef CAS PubMed.
- S. Huerta, Future Sci. OA, 2015, 1 CAS , fso.15.44.
- N. Lehnert, E. Kim, H. T. Dong, J. B. Harland, A. P. Hunt, E. C. Manickas, K. M. Oakley, J. Pham, G. C. Reed and V. S. Alfaro, Chem. Rev., 2021, 121, 14682–14905 CrossRef CAS PubMed.
- P. Farmer and F. Sulc, J. Inorg. Biochem., 2005, 99, 166–184 CrossRef CAS PubMed.
- C. K. Jørgensen, Coord. Chem. Rev., 1966, 1, 164–178 CrossRef.
- W. J. Evans, M. Fang, J. E. Bates, F. Furche, J. W. Ziller, M. D. Kiesz and J. I. Zink, Nat. Chem., 2010, 2, 644–647 CrossRef CAS PubMed.
- W. Kaim, A. Das, J. Fiedler, S. Záliš and B. Sarkar, Coord. Chem. Rev., 2020, 404, 213114 CrossRef CAS.
- J. H. Enemark and R. D. Feltham, Coord. Chem. Rev., 1974, 13, 339–406 CrossRef CAS.
- P. C. Ford, Y. Shiro and R. van Eldik, Inorg. Chem., 2021, 60, 15831–15834 CrossRef CAS PubMed.
- M. J. Rose and P. K. Mascharak, Coord. Chem. Rev., 2008, 252, 2093–2114 CrossRef CAS PubMed.
- M. J. Rose, M. M. Olmstead and P. K. Mascharak, J. Am. Chem. Soc., 2007, 129, 5342–5343 CrossRef CAS PubMed.
- M. Guo, H.-J. Xiang, Y. Wang, Q.-L. Zhang, L. An, S.-P. Yang, Y. Ma, Y. Wang and J.-G. Liu, Chem. Commun., 2017, 53, 3253–3256 RSC.
- E. Orlowska, M. V. Babak, O. Dömötör, E. A. Enyedy, P. Rapta, M. Zalibera, L. Bučinský, M. Malček, C. Govind, V. Karunakaran, Y. C. S. Farid, T. E. McDonnell, D. Luneau, D. Schaniel, W. H. Ang and V. B. Arion, Inorg. Chem., 2018, 57, 10702–10717 CrossRef CAS PubMed.
- I. Szundi, M. J. Rose, I. Sen, A. A. Eroy-Reveles, P. K. Mascharak and Ó. Einarsdóttir, Photochem. Photobiol., 2006, 82, 1377 CrossRef CAS PubMed.
- M. Roose, M. Tassé, P. G. Lacroix and I. Malfant, New J. Chem., 2019, 43, 755–767 RSC.
- D. Schaniel, B. Cormary, I. Malfant, L. Valade, T. Woike, B. Delley, K. W. Krämer and H.-U. Güdel, Phys. Chem. Chem. Phys., 2007, 9, 3717–3724 RSC.
- B. Cormary, S. Ladeira, K. Jacob, P. G. Lacroix, T. Woike, D. Schaniel and I. Malfant, Inorg. Chem., 2012, 51, 7492–7501 CrossRef CAS PubMed.
- A. A. Mikhailov, T. S. Sukhikh, N. V. Kuratieva, D. P. Pishchur and G. A. Kostin, Dalton Trans., 2021, 50, 2864–2871 RSC.
- D. Schaniel, M. Imlau, T. Weisemoeller, T. Woike, K. W. Krämer and H.-U. Güdel, Adv. Mater., 2007, 19, 723–726 CrossRef CAS.
- L. A. Kushch, L. S. Kurochkina, E. B. Yagubskii, G. V. Shilov, S. M. Aldoshin, V. A. Emel'yanov, Y. N. Shvachko, V. S. Mironov, D. Schaniel, T. Woike, C. Carbonera and C. Mathonière, Eur. J. Inorg. Chem., 2006, 4074–4085 CrossRef CAS.
- D. Schaniel, Th. Woike, L. Kushch and E. Yagubskii, Chem. Phys., 2007, 340, 211–216 CrossRef CAS.
- I. Stepanenko, P. Mizetskyi, E. Orlowska, L. Bučinský, M. Zalibera, B. Vénosová, M. Clémancey, G. Blondin, P. Rapta, G. Novitchi, W. Schrader, D. Schaniel, Y.-S. Chen, M. Lutz, J. Kožíšek, J. Telser and V. B. Arion, Inorg. Chem., 2022, 61, 950–967 CrossRef CAS PubMed.
- V. Vorobyev, V. A. Emelyanov, I. A. Valuev and I. A. Baidina, Inorg. Chem. Commun., 2017, 76, 40–43 CrossRef CAS.
- V. Vorobyev, G. A. Kostin, N. V. Kuratieva and V. A. Emelyanov, Inorg. Chem., 2016, 55, 9158–9161 CrossRef CAS PubMed.
- G. A. Kostin, A. O. Borodin, N. V. Kuratieva, A. A. Mikhailov and P. E. Plusnin, J. Mol. Struct., 2019, 1176, 402–407 CrossRef CAS.
- A. A. Mikhailov, D. P. Pishchur, N. V. Kuratieva and G. A. Kostin, Mendeleev Commun., 2020, 30, 719–721 CrossRef CAS.
- G. A. Kostin, A. A. Mikhailov, N. V. Kuratieva, D. P. Pischur, D. O. Zharkov and I. R. Grin, New J. Chem., 2017, 41, 7758–7765 RSC.
- I. A. Yakovlev, A. A. Mikhailov, J. A. Eremina, L. S. Klyushova, V. A. Nadolinny and G. A. Kostin, Dalton Trans., 2021, 50, 13516–13527 RSC.
- G. A. Kostin, A. A. Mikhailov, N. V. Kuratieva, S. V. Tkachev, D. Schaniel and T. Woike, Eur. J. Inorg. Chem., 2016, 4045–4053 CrossRef CAS.
- G. A. Kostin, V. Vorobyev, A. A. Mikhailov and N. V. Kuratieva, J. Mol. Struct., 2019, 1193, 334–341 CrossRef CAS.
- A. A. Mikhailov, V. Yu. Komarov, D. P. Pishchur, D. Schaniel and G. A. Kostin, New J. Chem., 2021, 45, 8192–8202 RSC.
- A. A. Mikhailov, V. Yu. Komarov, A. S. Sukhikh, D. P. Pishchur, D. Schaniel and G. A. Kostin, New J. Chem., 2020, 44, 18014–18024 RSC.
- A. Mikhailov, V. Vuković, C. Kijatkin, E. Wenger, M. Imlau, T. Woike, G. Kostin and D. Schaniel, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2019, 75, 1152–1163 CrossRef CAS PubMed.
- E. D. Rechitskaya, V. A. Vorobiev, N. V. Kuratieva and G. A. Kostin, J. Struct. Chem., 2021, 62, 256–264 CrossRef CAS.
- E. D. Rechitskaya, N. V. Kuratieva, E. V. Lider, J. A. Eremina, L. S. Klyushova, I. V. Eltsov and G. A. Kostin, J. Mol. Struct., 2020, 1219, 128565 CrossRef CAS.
- A. N. Makhinya, M. A. Il'in, R. D. Yamaletdinov, I. A. Baidina, S. V. Tkachev, A. P. Zubareva, I. V. Korol'kov and D. A. Piryazev, Russ. J. Coord. Chem., 2016, 42, 768–774 CrossRef CAS.
- M. A. Crisalli, L. P. Franco, B. R. Silva, A. K. M. Holanda, L. M. Bendhack, R. S. Da Silva and P. C. Ford, J. Coord. Chem., 2018, 71, 1690–1703 CrossRef CAS.
- F. Wu, C.-J. Wang, H. Lin, A.-Q. Jia and Q.-F. Zhang, Inorg. Chim. Acta, 2018, 471, 718–723 CrossRef CAS.
- C.-W. Fung, G. Fukada, Y. Mutoh, N. Tsuchida and S. Saito, Dalton Trans., 2020, 49, 613–624 RSC.
- L. R. Walton, S. E. Knight, S. K. Herold, K. J. Olsonowski, D. S. Amenta, J. W. Gilje and G. P. A. Yap, Polyhedron, 2018, 144, 44–54 CrossRef CAS.
- V. Vorobyev, V. A. Emelyanov, O. A. Plusnina, E. M. Makarov, I. A. Baidina, A. I. Smolentsev, S. V. Tkachev and T. I. Asanova, Eur. J. Inorg. Chem., 2017, 971–978 CrossRef CAS.
- A. N. Makhinya, M. A. Il'in, I. A. Baidina and I. V. Korol'kov, J. Struct. Chem., 2016, 57, 1182–1187 CrossRef CAS.
- V. Vorobyev, A. A. Mikhailov, V. Yu. Komarov, A. N. Makhinya and G. A. Kostin, New J. Chem., 2020, 44, 4762–4771 RSC.
- G. A. Kostin, A. A. Mikhailov, N. V. Kuratieva, D. P. Pishchur and A. N. Makhinya, New J. Chem., 2018, 42, 18928–18934 RSC.
- B. Cebrián-Losantos, E. Reisner, C. R. Kowol, A. Roller, S. Shova, V. B. Arion and B. K. Keppler, Inorg. Chem., 2008, 47, 6513–6523 CrossRef PubMed.
- L. C. B. Ramos, F. P. Rodrigues, J. C. Biazzotto, S. de Paula Machado, L. D. Slep, M. R. Hamblin and R. S. da Silva, J. Biol. Inorg. Chem., 2018, 23, 903–916 CrossRef CAS PubMed.
- C. D. S. Silva, I. A. Paz, F. D. Abreu, A. P. de Sousa, C. P. Veríssimo, N. R. F. Nascimento, T. F. Paulo, D. Zampieri, M. N. Eberlin, A. C. S. Gondim, L. C. Andrade, I. M. M. Carvalho, E. H. S. Sousa and L. G. F. Lopes, J. Inorg. Biochem., 2018, 182, 83–91 CrossRef CAS PubMed.
- P. Labra-Vázquez, M. Bocé, M. Tassé, S. Mallet-Ladeira, P. G. Lacroix, N. Farfán and I. Malfant, Dalton Trans., 2020, 49, 3138–3154 RSC.
- I. Sasaki, S. Amabilino, S. Mallet-Ladeira, M. Tassé, A. Sournia-Saquet, P. G. Lacroix and I. Malfant, New J. Chem., 2019, 43, 11241–11250 RSC.
- S. Amabilino, M. Tasse, P. G. Lacroix, S. Mallet-Ladeira, V. Pimienta, J. Akl, I. Sasaki and I. Malfant, New J. Chem., 2017, 41, 7371–7383 RSC.
- F. P. Rodrigues, L. J. A. Macedo, L. N. C. Máximo, F. C. P. F. Sales, R. S. da Silva and F. N. Crespilho, Nitric Oxide, 2020, 96, 29–34 CrossRef CAS PubMed.
- L. Bučinský, G. E. Büchel, R. Ponec, P. Rapta, M. Breza, J. Kožíšek, M. Gall, S. Biskupič, M. Fronc, K. Schiessl, O. Cuzan, D. Prodius, C. Turta, S. Shova, D. A. Zając and V. B. Arion, Eur. J. Inorg. Chem., 2013, 2505–2519 CrossRef.
- I. Bratsos and E. Alessio, Eur. J. Inorg. Chem., 2018, 2996–3013 CrossRef CAS.
- A. K. E. Gallien, D. Schaniel, T. Woike and P. Klüfers, Dalton Trans., 2014, 43, 13278–13292 RSC.
- R. Y. Ahmed Alshwafy, A. A. Dahy, I. Warad and R. M. Mahfouz, J. Coord. Chem., 2019, 72, 2200–2214 CrossRef.
- D. Awasabisah, N. Xu, K. P. S. Gautam, D. R. Powell, M. J. Shaw and G. B. Richter-Addo, Eur. J. Inorg. Chem., 2016, 509–518 CrossRef CAS.
- C. Lefebvre, T. Dumas, C. Tamain, T. Ducres, P. L. Solari and M.-C. Charbonnel, Ind. Eng. Chem. Res., 2017, 56, 11292–11301 CrossRef CAS.
- L. Xu, Z. Ma, W. Wang, L. Xie, L. Liu, J. Liu, X. Zhao and H. Wang, Polyhedron, 2017, 137, 157–164 CrossRef CAS.
- L. Xie, H. Bai, L. Song, C. Liu, W. Gong, W. Wang, X. Zhao, C. Takemoto and H. Wang, Inorg. Chem., 2021, 60, 8826–8837 CrossRef CAS PubMed.
- G. A. Barbosa, J. P. da Silva, P. Appelt, O. Fuganti, F. S. Murakami and M. P. de Araujo, Inorg. Chem. Commun., 2018, 90, 108–111 CrossRef CAS.
- C. Chen, J. Ji, C.-J. Wang, A.-Q. Jia and Q.-F. Zhang, J. Coord. Chem., 2020, 73, 1306–1313 CrossRef CAS.
- H. Shamran Mohammed, S. Mallet-Ladeira, B. Cormary, M. Tassé and I. Malfant, IUCrData, 2017, 2 CrossRef CAS , x171761.
- M. Tassé, H. S. Mohammed, C. Sabourdy, S. Mallet-Ladeira, P. G. Lacroix and I. Malfant, Polyhedron, 2016, 119, 350–358 CrossRef.
- A. de Sousa, A. Fernandes, I. Paz, N. Nascimento, J. Ellena, E. Sousa, L. Lopes and A. Holanda, J. Braz. Chem. Soc., 2017, 28, 2117–2129 CAS.
- F. P. Rodrigues, Z. A. Carneiro, P. Mascharak, C. Curti and R. S. da Silva, Coord. Chem. Rev., 2016, 306, 701–707 CrossRef CAS.
- A. P. S. Gaspari, R. S. da Silva, Z. A. Carneiro, M. R. de Carvalho, I. Carvalho, L. Pernomian, L. P. Ferreira, L. C. B. Ramos, G. A. de Souza and A. L. B. Formiga, Anti-Cancer Agents Med. Chem, 2021, 21, 1602–1611 CrossRef CAS PubMed.
- V. Bukhanko, P. G. Lacroix, I. Sasaki, M. Tassé, S. Mallet-Ladeira, Z. Voitenko and I. Malfant, Inorg. Chim. Acta, 2018, 482, 195–205 CrossRef CAS.
- B. Giri, T. Saini, S. Kumbhakar, K. Selvan K, A. Muley, A. Misra and S. Maji, Dalton Trans., 2020, 49, 10772–10785 RSC.
- A. Enriquez-Cabrera, I. Sasaki, V. Bukhanko, M. Tassé, S. Mallet-Ladeira, P. G. Lacroix, R. M. Barba-Barba, G. Ramos-Ortiz, N. Farfán, Z. Voitenko and I. Malfant, Eur. J. Inorg. Chem., 2017, 1446–1456 CrossRef CAS.
- A. Enriquez-Cabrera, P. G. Lacroix, I. Sasaki, S. Mallet-Ladeira, N. Farfán, R. M. Barba-Barba, G. Ramos-Ortiz and I. Malfant, Eur. J. Inorg. Chem., 2018, 531–543 CrossRef CAS.
- V. Bukhanko, A. F. León-Rojas, P. G. Lacroix, M. Tassé, G. Ramos-Ortiz, R. M. Barba-Barba, N. Farfán, R. Santillan and I. Malfant, Eur. J. Inorg. Chem., 2021, 1670–1684 CrossRef CAS.
- M. Roose, I. Sasaki, V. Bukhanko, S. Mallet-Ladeira, R. M. Barba-Barba, G. Ramos-Ortiz, A. Enriquez-Cabrera, N. Farfán, P. G. Lacroix and I. Malfant, Polyhedron, 2018, 151, 100–111 CrossRef CAS.
- B. Giri, S. Kumbhakar, K. Selvan K, A. Muley and S. Maji, New J. Chem., 2020, 44, 18732–18744 RSC.
- B. Giri, S. Kumbhakar, K. Kalai Selvan, A. Muley and S. Maji, Inorg. Chim. Acta, 2020, 502, 119360 CrossRef CAS.
- M. I. F. Barbosa, G. G. Parra, R. S. Correa, R. N. Sampaio, L. N. Magno, R. C. Silva, A. C. Doriguetto, J. Ellena, N. M. B. Neto, A. A. Batista and P. J. Gonçalves, J. Photochem. Photobiol., A, 2017, 338, 152–160 CrossRef CAS.
- N. Casaretto, B. Fournier, S. Pillet, E. E. Bendeif, D. Schaniel, A. K. E. Gallien, P. Klüfers and T. Woike, CrystEngComm, 2016, 18, 7260–7268 RSC.
- A. B. Alemayehu, H. Vazquez-Lima, K. J. Gagnon and A. Ghosh, Inorg. Chem., 2017, 56, 5285–5294 CrossRef CAS PubMed.
- F. G. Doro, K. Q. Ferreira, Z. N. da Rocha, G. F. Caramori, A. J. Gomes and E. Tfouni, Coord. Chem. Rev., 2016, 306, 652–677 CrossRef CAS.
- K. M. Miranda, L. A. Ridnour, C. L. McGinity, D. Bhattacharyya and D. A. Wink, Inorg. Chem., 2021, 60, 15941–15947 CrossRef CAS PubMed.
- H.-Y. Ng, W.-M. Cheung, E. Kwan Huang, K.-L. Wong, H. H.-Y. Sung, I. D. Williams and W.-H. Leung, Dalton Trans., 2015, 44, 18459–18468 RSC.
- H.-Y. Ng, N.-M. Lam, M. Yang, X.-Y. Yi, I. D. Williams and W.-H. Leung, Inorg. Chim. Acta, 2013, 394, 171–175 CrossRef CAS.
- D. S. Bohle, C.-H. Hung, A. K. Powell, B. D. Smith and S. Wocadlo, Inorg. Chem., 1997, 36, 1992–1993 CrossRef CAS PubMed.
- K. K. Pandey, Prog. Inorg. Chem, Wiley, New York, S. Lippard, 1992, vol. 40 Search PubMed.
- S. B. Yaghlane and M. Hochlaf, J. Phys. B: At., Mol. Opt. Phys., 2008, 42, 015101 CrossRef.
- D. Shi, P. Li, J. Sun and Z. Zhu, Spectrochim. Acta, Part A, 2014, 117, 109–119 CrossRef CAS PubMed.
- R. T. Boeré and T. L. Roemmele, in Comprehensive Inorganic Chemistry II, Elsevier, 2013, pp. 375–411 Search PubMed.
- M. G. Scheibel, I. Klopsch, H. Wolf, P. Stollberg, D. Stalke and S. Schneider, Eur. J. Inorg. Chem., 2013, 3836–3839 CrossRef CAS.
- M. G. Scheibel, B. Askevold, F. W. Heinemann, E. J. Reijerse, B. de Bruin and S. Schneider, Nat. Chem., 2012, 4, 552–558 CrossRef CAS PubMed.
- T. J. Crevier, S. Lovell, J. M. Mayer, A. L. Rheingold and I. A. Guzei, J. Am. Chem. Soc., 1998, 120, 6607–6608 CrossRef CAS.
- J. M. Fletcher, I. L. Jenkins, F. M. Lever, F. S. Martin, A. R. Powell and R. Todd, J. Inorg. Nucl. Chem., 1955, 1, 378–401 CrossRef CAS.
- D. H. F. Souza, G. Oliva, A. Teixeira and A. A. Batista, Polyhedron, 1995, 14, 1031–1034 CrossRef CAS.
- M. Gupta, J. Seth and U. C. Agarwala, Bull. Chem. Soc. Jpn., 1989, 62, 3397–3399 CrossRef CAS.
- Yi, T. C. H. Lam, Y.-K. Sau, Q.-F. Zhang, I. D. Williams and W.-H. Leung, Inorg. Chem., 2007, 46, 7193–7198 CrossRef CAS PubMed.
- V. M. Krishnan, H. D. Arman and Z. J. Tonzetich, Dalton Trans., 2017, 46, 1186–1193 RSC.
- W. Willing, U. Müller, U. Demant and K. Dehnicke, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1986, 41, 560–566 CrossRef.
- J. W. Bats, K. K. Pandey and H. W. Roesky, J. Chem. Soc., Dalton Trans., 1984, 2081–2083 RSC.
- W. R. Montfort, J. A. Wales and A. Weichsel, Antioxid. Redox Signal., 2017, 26, 107–121 CrossRef CAS PubMed.
- N. Lehnert, T. C. Berto, M. G. I. Galinato and L. E. Goodrich, in Handbook of Porphyrin Science (Vol. 14), World Scientific Publishing Company, 2011, vol. 15, pp. 1–247 Search PubMed.
- L. E. Goodrich and N. Lehnert, J. Inorg. Biochem., 2013, 118, 179–186 CrossRef CAS PubMed.
- T. Shimizu, D. Huang, F. Yan, M. Stranava, M. Bartosova, V. Fojtíková and M. Martínková, Chem. Rev., 2015, 115, 6491–6533 CrossRef CAS PubMed.
- Y. Guo, D. L. M. Suess, M. A. Herzik, A. T. Iavarone, R. D. Britt and M. A. Marletta, Nat. Chem. Biol., 2017, 13, 1216–1221 CrossRef CAS PubMed.
- N. Levin, N. O. Codesido, E. Bill, T. Weyhermüller, A. P. Segantin Gaspari, R. S. da Silva, J. A. Olabe and L. D. Slep, Inorg. Chem., 2016, 55, 7808–7810 CrossRef CAS PubMed.
- N. Levin, N. O. Codesido, J. P. Marcolongo, P. Alborés, T. Weyhermüller, J. A. Olabe and L. D. Slep, Inorg. Chem., 2018, 57, 12270–12281 CrossRef CAS PubMed.
- G. A. Kostin, E. Yu. Filatov, D. P. Pischur, N. V. Kuratieva and S. V. Korenev, CrystEngComm, 2020, 22, 3692–3700 RSC.
- G. A. Kostin, P. E. Plyusnin, E. Yu. Filatov, N. V. Kuratieva, A. A. Vedyagin and D. B. Kal'nyi, Polyhedron, 2019, 159, 217–225 CrossRef CAS.
- K. Mori, K. Miyawaki and H. Yamashita, ACS Catal., 2016, 6, 3128–3135 CrossRef CAS.
- T. O. Eschemann, J. Oenema and K. P. de Jong, Catal. Today, 2016, 261, 60–66 CrossRef CAS.
- G. A. Kostin, A. O. Borodin, N. V. Kuratieva, E. Yu. Filatov and P. E. Plyusnin, Inorg. Chim. Acta, 2017, 457, 145–149 CrossRef CAS.
- G. A. Kostin, A. O. Borodin, N. V. Kuratieva, A. S. Bogomyakov and A. A. Mikhailov, Inorg. Chim. Acta, 2018, 479, 135–140 CrossRef CAS.
- P.-S. Kuhn, L. Cremer, A. Gavriluta, K. K. Jovanović, L. Filipović, A. A. Hummer, G. E. Büchel, B. P. Dojčinović, S. M. Meier, A. Rompel, S. Radulović, J. B. Tommasino, D. Luneau and V. B. Arion, Chem. – Eur. J., 2015, 21, 13703–13713 CrossRef CAS PubMed.
- I. S. Fomenko, A. A. Mikhailov, V. Vorobyev, N. V. Kuratieva, G. A. Kostin, D. Schaniel, V. A. Nadolinny and A. L. Gushchin, J. Photochem. Photobiol., A, 2021, 407, 113044 CrossRef CAS.
- K.-W. Chan, W.-M. Ng, W.-M. Cheung, C.-S. Lai, H. H. Y. Sung, I. D. Williams and W.-H. Leung, J. Organomet. Chem., 2016, 812, 151–157 CrossRef CAS.
- C. F. N. da Silva, B. Possato, L. P. Franco, L. C. B. Ramos and S. Nikolaou, J. Inorg. Biochem., 2018, 186, 197–205 CrossRef CAS PubMed.
- I. Haiduc, Ann. Chem. Sci. Res., 2021, 2, 1–2 Search PubMed.
- I. Haiduc, in Comprehensive Coordination Chemistry III, Elsevier, 2021, pp. 66–120 Search PubMed.
- Y. Arikawa, J. Hiura, C. Tsuchii, M. Kodama, N. Matsumoto and K. Umakoshi, Dalton Trans., 2018, 47, 7399–7401 RSC.
- P. Coppens, I. Novozhilova and A. Kovalevsky, Chem. Rev., 2002, 102, 861–884 CrossRef CAS PubMed.
- H. Giglmeier, T. Kerscher, P. Klüfers, D. Schaniel and T. Woike, Dalton Trans., 2009, 9113–9116 RSC.
- A. A. Mikhailov, T. Woike, A. Gansmüller, D. Schaniel and G. A. Kostin, Spectrochim. Acta, Part A, 2021, 263, 120217 CrossRef CAS PubMed.
- J. S. García, F. Alary, M. Boggio-Pasqua, I. M. Dixon and J.-L. Heully, J. Mol. Model., 2016, 22, 284 CrossRef PubMed.
- Th. Woike, W. Kirchner, G. Schetter, Th. Barthel, K. Hyung-sang and S. Haussühl, Opt. Commun., 1994, 106, 6–10 CrossRef CAS.
- M. Goulkov, D. Schaniel and T. Woike, J. Opt. Soc. Am. B, 2010, 27, 927–932 CrossRef CAS.
- J. Akl, C. Billot, P. G. Lacroix, I. Sasaki, S. Mallet-Ladeira, I. Malfant, R. Arcos-Ramos, M. Romero and N. Farfán, New J. Chem., 2013, 37, 3518 RSC.
- B. Cormary, I. Malfant, M. Buron-Le Cointe, L. Toupet, B. Delley, D. Schaniel, N. Mockus, T. Woike, K. Fejfarová, V. Petříček and M. Dušek, Acta Crystallogr., Sect. B: Struct. Sci., 2009, 65, 612–623 CrossRef CAS PubMed.
- D. Schaniel and T. Woike, Phys. Chem. Chem. Phys., 2009, 11, 4391–4395 RSC.
- P. Gütlich, Y. Garcia and T. Woike, Coord. Chem. Rev., 2001, 219–221, 839–879 CrossRef.
- S. I. Gorelsky and A. B. P. Lever, Int. J. Quantum Chem., 2000, 80, 636–645 CrossRef CAS.
- D. Schaniel, T. Woike, C. Boskovic and H.-U. Güdel, Chem. Phys. Lett., 2004, 390, 347–351 CrossRef CAS.
- J. Sanz García, F. Alary, M. Boggio-Pasqua, I. M. Dixon, I. Malfant and J.-L. Heully, Inorg. Chem., 2015, 54, 8310–8318 CrossRef PubMed.
- F. Talotta, J.-L. Heully, F. Alary, I. M. Dixon, L. González and M. Boggio-Pasqua, J. Chem. Theory Comput., 2017, 13, 6120–6130 CrossRef CAS PubMed.
- F. Talotta, M. Boggio-Pasqua and L. González, Chem. – Eur. J., 2020, 26, 11522–11528 CrossRef CAS PubMed.
- L. Khadeeva, W. Kaszub, M. Lorenc, I. Malfant and M. Buron-Le Cointe, Inorg. Chem., 2016, 55, 4117–4123 CrossRef CAS PubMed.
- A. A. Mikhailov, E. Wenger, G. A. Kostin and D. Schaniel, Chem. – Eur. J., 2019, 25, 7569–7574 CrossRef CAS PubMed.
- F. Talotta, L. González and M. Boggio-Pasqua, Molecules, 2020, 25, 2613 CrossRef CAS PubMed.
- R. D. Yamaletdinov and I. L. Zilberberg, Eur. J. Inorg. Chem., 2017, 2951–2954 CrossRef CAS.
- A. Y. Kovalevsky, G. King, K. A. Bagley and P. Coppens, Chem. – Eur. J., 2005, 11, 7254–7264 CrossRef CAS PubMed.
- D. Schaniel, E.-E. Bendeif, T. Woike, H.-C. Böttcher and S. Pillet, CrystEngComm, 2018, 20, 7100–7108 RSC.
- D. R. Truzzi, N. M. Medeiros, O. Augusto and P. C. Ford, Inorg. Chem., 2021, 60, 15835–15845 CrossRef CAS PubMed.
- N. Casaretto, S. Pillet, E. E. Bendeif, D. Schaniel, A. K. E. Gallien, P. Klüfers and T. Woike, IUCrJ, 2015, 2, 35–44 CrossRef CAS PubMed.
- N. Casaretto, S. Pillet, E.-E. Bendeif, D. Schaniel, A. K. E. Gallien, P. Klüfers and T. Woike, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2015, 71, 788–797 CrossRef CAS PubMed.
- D. Schaniel, N. Casaretto, E.-E. Bendeif, T. Woike, A. K. E. Gallien, P. Klüfers, S. E. Kutniewska, R. Kamiński, G. Bouchez, K. Boukheddaden and S. Pillet, CrystEngComm, 2019, 21, 5804–5810 RSC.
- E. Tfouni, M. Krieger, B. R. McGarvey and D. W. Franco, Coord. Chem. Rev., 2003, 236, 57–69 CrossRef CAS.
- A. C. Merkle, A. B. McQuarters and N. Lehnert, Dalton Trans., 2012, 41, 8047–8059 RSC.
- J. C. Toledo, H. A. S. Silva, M. Scarpellini, V. Mori, A. J. Camargo, M. Bertotti and D. W. Franco, Eur. J. Inorg. Chem., 2004, 1879–1885 CrossRef CAS.
- Y. Chen, F.-T. Lin and R. E. Shepherd, Inorg. Chem., 1999, 38, 973–983 CrossRef CAS PubMed.
- P. G. Zanichelli, H. F. G. Estrela, R. C. Spadari-Bratfisch, D. M. Grassi-Kassisse and D. W. Franco, Nitric Oxide, 2007, 16, 189–196 CrossRef CAS PubMed.
- E. Tfouni, D. R. Truzzi, A. Tavares, A. J. Gomes, L. E. Figueiredo and D. W. Franco, Nitric Oxide, 2012, 26, 38–53 CrossRef CAS PubMed.
- D. R. Lang, J. A. Davis, L. G. F. Lopes, A. A. Ferro, L. C. G. Vasconcellos, D. W. Franco, E. Tfouni, A. Wieraszko and M. J. Clarke, Inorg. Chem., 2000, 39, 2294–2300 CrossRef CAS PubMed.
- R. da S. Vidal, F. G. Doro, K. Q. Ferreira, Z. N. da Rocha, E. E. Castellano, S. Nikolaou and E. Tfouni, Inorg. Chem. Commun., 2012, 15, 93–96 CrossRef CAS.
- K. Q. Ferreira and E. Tfouni, J. Braz. Chem. Soc., 2010, 21, 1349–1358 CrossRef CAS.
- A. G. De Candia, J. P. Marcolongo and L. D. Slep, Polyhedron, 2007, 26, 4719–4730 CrossRef CAS.
- F. G. Doro, I. M. Pepe, S. E. Galembeck, R. M. Carlos, Z. N. da Rocha, M. Bertotti and E. Tfouni, Dalton Trans., 2011, 40, 6420–6432 RSC.
- K.-Y. Wong, C.-M. Che, W.-H. Yip, R.-J. Wang and T. C. W. Mak, J. Chem. Soc., Dalton Trans., 1992, 1417–1421 RSC.
- K. Q. Ferreira, F. G. Santos, Z. N. Da Rocha, T. Guaratini, R. S. Da Silva and E. Tfouni, Inorg. Chem. Commun., 2004, 7, 204–208 CrossRef CAS.
- F. de S. Oliveira, V. Togniolo, T. T. Pupo, A. C. Tedesco and R. S. da Silva, Inorg. Chem. Commun., 2004, 7, 160–164 CrossRef CAS.
- J. Perdoménico, M. M. Ruiz, N. Osa Codesido, A. G. De Candia, J. P. Marcolongo and L. D. Slep, Dalton Trans., 2021, 50, 1641–1650 RSC.
- F. G. Doro, E. E. Castellano, L. A. B. Moraes, M. N. Eberlin and E. Tfouni, Inorg. Chem., 2008, 47, 4118–4125 CrossRef CAS PubMed.
- E. Tfouni, F. G. Doro, L. E. Figueiredo, J. C. M. Pereira, G. Metzke and D. W. Franco, Curr. Med. Chem., 2010, 17, 3643–3657 CrossRef CAS PubMed.
- M. G. de Oliveira, F. G. Doro, E. Tfouni and M. H. Krieger, J. Pharm. Pharmacol., 2017, 69, 1155–1165 CrossRef CAS PubMed.
- F. G. Marcondes, A. A. Ferro, A. Souza-Torsoni, M. Sumitani, M. J. Clarke, D. W. Franco, E. Tfouni and M. H. Krieger, Life Sci., 2002, 70, 2735–2752 CrossRef CAS PubMed.
- D. Bonaventura, F. de S. Oliveira, V. Togniolo, A. C. Tedesco, R. S. da Silva and L. M. Bendhack, Nitric Oxide, 2004, 10, 83–91 CrossRef CAS PubMed.
- N. O. Codesido, T. Weyhermüller, J. A. Olabe and L. D. Slep, Inorg. Chem., 2014, 53, 981–997 CrossRef CAS PubMed.
- R. S. da Silva, R. G. de Lima and S. de Paula Machado, in Adv. Inorg. Chem, Elsevier, 2015, vol. 67, pp. 265–294 Search PubMed.
- V. Togniolo, R. Santana da Silva and A. C. Tedesco, Inorg. Chim. Acta, 2001, 316, 7–12 CrossRef CAS.
- M. G. Sauaia and R. S. da Silva, Transition Met. Chem., 2003, 28, 254–259 CrossRef.
- M. G. Sauaia, R. G. de Lima, A. C. Tedesco and R. S. da Silva, J. Am. Chem. Soc., 2003, 125, 14718–14719 CrossRef CAS PubMed.
- M. G. Sauaia, F. de Souza Oliveira, R. G. de Lima, A. De Lima Cacciari, E. Tfouni and R. S. Da Silva, Inorg. Chem. Commun., 2005, 8, 347–349 CrossRef CAS.
- F. O. N. Silva, S. X. B. Araújo, A. K. M. Holanda, E. Meyer, F. A. M. Sales, I. C. N. Diógenes, I. M. M. Carvalho, Í. S. Moreira and L. G. F. Lopes, Eur. J. Inorg. Chem., 2006, 2020–2026 CrossRef CAS.
- A. N. de Carvalho, E. C. Fornari, W. R. Gomes, D. M. S. Araújo, A. E. H. Machado and S. Nikolaou, Inorg. Chim. Acta, 2011, 370, 444–448 CrossRef CAS.
- R. G. de Lima, M. G. Sauaia, D. Bonaventura, A. C. Tedesco, L. M. Bendhack and R. S. da Silva, Inorg. Chim. Acta, 2006, 359, 2543–2549 CrossRef.
- J. P. da Silva, F. D. Fagundes, D. F. Back, J. Ellena, B. R. James and M. P. de Araujo, Inorg. Chim. Acta, 2017, 454, 40–45 CrossRef CAS.
- T. R. deBoer and P. K. Mascharak, in Adv. Inorg. Chem, Elsevier, 2015, vol. 67, pp. 145–170 Search PubMed.
- A. A. Mikhailov, E. D. Stolyarova and G. A. Kostin, J. Struct. Chem., 2021, 62, 497–516 CrossRef CAS.
- N. L. Fry and P. K. Mascharak, Acc. Chem. Res., 2011, 44, 289–298 CrossRef CAS PubMed.
- M. Madhani, A. K. Patra, T. W. Miller, A. A. Eroy-Reveles, A. J. Hobbs, J. M. Fukuto and P. K. Mascharak, J. Med. Chem., 2006, 49, 7325–7330 CrossRef CAS PubMed.
- M. J. Rose, M. M. Olmstead and P. K. Mascharak, Polyhedron, 2007, 26, 4713–4718 CrossRef CAS.
- M. J. Rose, A. K. Patra, E. A. Alcid, M. M. Olmstead and P. K. Mascharak, Inorg. Chem., 2007, 46, 2328–2338 CrossRef CAS PubMed.
- N. L. Fry, M. J. Rose, D. L. Rogow, C. Nyitray, M. Kaur and P. K. Mascharak, Inorg. Chem., 2010, 49, 1487–1495 CrossRef CAS PubMed.
- A. K. Patra, M. J. Rose, K. A. Murphy, M. M. Olmstead and P. K. Mascharak, Inorg. Chem., 2004, 43, 4487–4495 CrossRef CAS PubMed.
- N. L. Fry, B. J. Heilman and P. K. Mascharak, Inorg. Chem., 2011, 50, 317–324 CrossRef CAS PubMed.
- M. J. Rose, N. L. Fry, R. Marlow, L. Hinck and P. K. Mascharak, J. Am. Chem. Soc., 2008, 130, 8834–8846 CrossRef CAS PubMed.
- M. J. Rose and P. K. Mascharak, Inorg. Chem., 2009, 48, 6904–6917 CrossRef CAS PubMed.
- M. J. Rose and P. K. Mascharak, Chem. Commun., 2008, 33, 3933–3935 RSC.
- H.-J. Xiang, Q. Deng, L. An, M. Guo, S.-P. Yang and J.-G. Liu, Chem. Commun., 2016, 52, 148–151 RSC.
- Q. Deng, H.-J. Xiang, W.-W. Tang, L. An, S.-P. Yang, Q.-L. Zhang and J.-G. Liu, J. Inorg. Biochem., 2016, 165, 152–158 CrossRef CAS PubMed.
- Y.-H. Li, M. Guo, S.-W. Shi, Q.-L. Zhang, S.-P. Yang and J.-G. Liu, J. Mater. Chem. B, 2017, 5, 7831–7838 RSC.
- Y.-T. Yu, S.-W. Shi, Y. Wang, Q.-L. Zhang, S.-H. Gao, S.-P. Yang and J.-G. Liu, ACS Appl. Mater. Interfaces, 2020, 12, 312–321 CrossRef CAS PubMed.
- J. Akl, I. Sasaki, P. G. Lacroix, I. Malfant, S. Mallet-Ladeira, P. Vicendo, N. Farfán and R. Santillan, Dalton Trans., 2014, 43, 12721–12733 RSC.
- J. Akl, I. Sasaki, P. G. Lacroix, V. Hugues, P. Vicendo, M. Bocé, S. Mallet-Ladeira, M. Blanchard-Desce and I. Malfant, Photochem. Photobiol. Sci., 2016, 15, 1484–1491 CrossRef CAS PubMed.
- M. R. Ávila, A. F. León-Rojas, P. G. Lacroix, I. Malfant, N. Farfán, R. Mhanna, R. Santillan, G. Ramos-Ortiz and J. P. Malval, J. Phys. Chem. Lett., 2020, 11, 6487–6491 CrossRef PubMed.
- H. Xiang, M. Guo and J. Liu, Eur. J. Inorg. Chem., 2017, 1586–1595 CrossRef CAS.
- S.-W. Shi, Y.-H. Li, Q.-L. Zhang, S.-P. Yang and J.-G. Liu, J. Mater. Chem. B, 2019, 7, 1867–1874 RSC.
- L. B. Negri, T. J. Martins, L. C. B. Ramos and R. S. da Silva, in Nitric Oxide Donors, Elsevier, 2017, pp. 1–24 Search PubMed.
- Editorial for special issue: Pharmacology and Medicinal Chemistry of Nitric Oxide. Nitric Oxide, 2019, 85, 53.
- R. J. Jones, Y. T. Gao, T. M. Simone, J. C. Salerno and S. M. E. Smith, Nitric Oxide, 2006, 14, 228–237 CrossRef CAS PubMed.
- L. J. Ignarro, Nitric oxide biology and pathobiology, Elsevier, Amsterdam, 2010 Search PubMed.
- G. C. Brown, Biochim. Biophys. Acta, 2001, 1504, 46–57 CrossRef CAS.
- B. Brüne, Cell Death Differ., 2003, 10, 864–869 CrossRef PubMed.
- D. Hirst and T. Robson, J. Pharm. Pharmacol., 2010, 59, 3–13 CrossRef PubMed.
- L. Song, H. Bai, C. Liu, W. Gong, A. Wang, L. Wang, Y. Zhao, X. Zhao and H. Wang, Molecules, 2021, 26, 2545 CrossRef CAS PubMed.
- G. E. Büchel, A. Gavriluta, M. Novak, S. M. Meier, M. A. Jakupec, O. Cuzan, C. Turta, J.-B. Tommasino, E. Jeanneau, G. Novitchi, D. Luneau and V. B. Arion, Inorg. Chem., 2013, 52, 6273–6285 CrossRef PubMed.
- M. S. Novak, G. E. Büchel, B. K. Keppler and M. A. Jakupec, J. Biol. Inorg. Chem., 2016, 21, 347–356 CrossRef CAS PubMed.
- A. Rathgeb, A. Böhm, M. S. Novak, A. Gavriluta, O. Dömötör, J. B. Tommasino, É. A. Enyedy, S. Shova, S. Meier, M. A. Jakupec, D. Luneau and V. B. Arion, Inorg. Chem., 2014, 53, 2718–2729 CrossRef CAS PubMed.
- Y. Zhang, A. Ho, J. Yue, L. Kong, Z. Zhou, X. Wu, F. Yang and H. Liang, Eur. J. Med. Chem., 2014, 86, 449–455 CrossRef CAS PubMed.
- A. Bijelic, S. Theiner, B. K. Keppler and A. Rompel, J. Med. Chem., 2016, 59, 5894–5903 CrossRef CAS PubMed.
- A. A. Mikhailov, D. V. Khantakova, V. A. Nichiporenko, E. M. Glebov, V. P. Grivin, V. F. Plyusnin, V. V. Yanshole, D. V. Petrova, G. A. Kostin and I. R. Grin, Metallomics, 2019, 11, 1999–2009 CrossRef CAS PubMed.
- M. Bocé, M. Tassé, S. Mallet-Ladeira, F. Pillet, C. Da Silva, P. Vicendo, P. G. Lacroix, I. Malfant and M.-P. Rols, Sci. Rep., 2019, 9, 4867 CrossRef PubMed.
- A. P. de Sousa, A. C. S. Gondim, E. H. S. Sousa, L. G. de França Lopes, E. H. Teixeira, M. A. Vasconcelos, P. H. R. Martins, E. J. T. Medeiros, A. A. Batista and A. K. M. Holanda, New J. Chem., 2020, 44, 21318–21325 RSC.
- N. R. F. do Nascimento, F. L. N. de Aguiar, C. F. Santos, A. M. L. Costa, D. de J. Hardoim, K. da S. Calabrese, F. Almeida-Souza, E. H. S. de Sousa, L. G. de F. Lopes, M. J. Teixeira, V. S. Pereira, R. S. N. Brilhante and M. F. G. Rocha, Acta Trop., 2019, 192, 61–65 CrossRef PubMed.
- T. M. Orsini, N. Y. Kawakami, C. Panis, A. P. F. S. Thomazelli, F. Tomiotto-Pellissier, A. H. D. Cataneo, D. Kian, L. M. Yamauchi, F. S. Gouveia Júnior, L. G. de França Lopes, R. Cecchini, I. N. Costa, J. J. N. da Silva, I. Conchon-Costa and W. R. Pavanelli, Mediat. Inflammation, 2016, 2631625 Search PubMed.
- A. C. Rossaneis, D. T. Longhi-Balbinot, M. M. Bertozzi, V. Fattori, C. Z. Segato-Vendrameto, S. Badaro-Garcia, T. H. Zaninelli, L. Staurengo-Ferrari, S. M. Borghi, T. T. Carvalho, A. J. C. Bussmann, F. S. Gouveia, L. G. F. Lopes, R. Casagrande and W. A. Verri, Front. Pharmacol., 2019, 10, 229 CrossRef CAS PubMed.
- P. P. C. Costa, S. B. Waller, G. R. dos Santos, F. de L. Gondim, D. S. Serra, F. S. Á. Cavalcante, F. S. Gouveia Júnior, V. F. de Paula Júnior, E. H. S. Sousa, L. G. de F. Lopes, W. L. C. Ribeiro and H. S. A. Monteiro, PLoS One, 2021, 16, e0248394 CrossRef CAS PubMed.
- P. F. da S. Castro, D. L. de Andrade, C. de F. Reis, S. H. N. Costa, A. C. Batista, R. S. da Silva and M. L. Rocha, Clin. Exp. Pharmacol. Physiol., 2016, 43, 221–229 CrossRef CAS PubMed.
- P. P. C. Costa, R. Campos, P. H. B. Cabral, V. M. Gomes, C. F. Santos, S. B. Waller, E. H. S. de Sousa, L. G. de F. Lopes, M. C. Fonteles and N. R. F. do Nascimento, Res. Vet. Sci., 2020, 130, 153–160 CrossRef CAS PubMed.
- S. R. Potje, J. A. Troiano, M. D. Grando, M. E. Graton, R. S. da Silva, L. M. Bendhack and C. Antoniali, Life Sci., 2018, 201, 130–140 CrossRef CAS PubMed.
- B. Pauwels, C. Boydens, L. Vanden Daele and J. Van de Voorde, J. Pharm. Pharmacol., 2016, 68, 293–304 CrossRef CAS PubMed.
- M. S. Lynch, M. Cheng, B. E. Van Kuiken and M. Khalil, J. Am. Chem. Soc., 2011, 133, 5255–5262 CrossRef CAS PubMed.
- S. M. Aldoshin, A. I. Zenchuk, E. B. Fel'dman and M. A. Yurishchev, Russ. Chem. Rev., 2012, 81, 91–104 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2022 |