
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of a multistep, electrochemical flow platform for automated catalyst screening†
Christiane
Schotten
a,
Jamie
Manson
 a,
Thomas W.
Chamberlain
a,
Richard A.
Bourne
*ab,
Bao N.
Nguyen
ab,
Nik
Kapur
*c and
Charlotte E.
Willans
*a
a,
Thomas W.
Chamberlain
a,
Richard A.
Bourne
*ab,
Bao N.
Nguyen
ab,
Nik
Kapur
*c and
Charlotte E.
Willans
*a
aSchool of Chemistry, University of Leeds, Leeds LS2 9JT, UK. E-mail: c.e.willans@leeds.ac.uk
bSchool of Chemical and Process Engineering, University of Leeds, Leeds LS2 9JT, UK
cSchool of Mechanical Engineering, University of Leeds, Leeds LS2 9JT, UK
First published on 25th May 2022
Abstract
The development of an integrated multistep flow platform that incorporates high-throughput electrochemical synthesis of metal catalysts and catalysis screening is described. Ligand libraries can be screened through the implementation of an autosampler, and online HPLC analysis facilitates continuous monitoring of the reaction. The equipment is controlled via a computer which enables the process to be automated, with the platform running ligand/catalysis screens autonomously. The platform has been validated using a ubiquitous Cu–NHC catalysed click reaction, with conditions chosen so that the reaction does not run at full conversion, which allows the effect of different ligand precursors to be observed. An efficient cleaning step is crucial to the reproducibility of reactions, and alternating polarity ensures the long-term stability of the electrochemical reactor. This technology will enable the profiling of catalysts in continuous systems and accelerate the process of developing more sustainable base-metal catalysts in manufacturing processes.
Introduction
The development of automated continuous flow systems with integrated analysis, that are capable of searching large regions of experimental space in comparatively quick periods of time, has opened up the potential to rapidly optimise processes.1 Recently, automated platforms for multistep reaction optimisation have been developed, further enhancing the speed of process development, and understanding of any synergistic effects in telescoped unit operations.2The use of electrons as reagents in synthetic chemistry has recently seen a renaissance.3–8 Electrochemistry offers a mild and atom efficient method for achieving selective transformations, by avoiding the use of harsh and often toxic chemical reducing and oxidising agents. Furthermore, complementary paths to traditional synthetic routes are often possible. The development of a range of user-friendly reactors, both batch and continuous, has allowed the more widespread adaption of this method, making electrochemistry a versatile tool in synthetic chemistry.9–21
Electrochemical screening is predominantly performed in batch conditions.22–24 There are very few continuous electrochemistry examples in synthetic reaction screening, and even fewer that make use of automation and inline or online analysis.17,25–28 Haider and co-workers integrated online mass spectrometry into the continuous methoxylation of methyl 2-furoate, thus enabling an effective reaction optimisation.28 Wirth and co-workers have used inline mass spectrometry and 2D-HPLC, together with DoE monitoring, to enable rapid screening of reaction conditions for asymmetric synthesis.17,26 The authors also developed a continuous protocol for the selenenylation of styrenes where the use of an autosampler enabled the automated screening of different substrates.27 Statistical methods for optimisation of electrochemical reactions have also been used by other groups. Waldvogel and co-workers developed batch and continuous protocols for the synthesis of periodates, for the C–H activation of (hetero)arenes, and for anodic dehydrogenative C–C coupling of phenols using DoE.29–31 Hilt and co-workers used DoE and multivariate linear regression for the rational optimisation of iodate(III)-mediated trifluoroethoxylactonisations.32 Jensen and co-workers developed a microscale electrochemical reactor, with a volume of only 15 μL, suitable for reaction condition screening and the use of cyclic voltammetry for measuring kinetics.20 Liquid handlers were used to inject reaction aliquots into a carrier gas and online LCMS for rapid analysis. This enabled optimisation of an α-amino C–H activation reaction in a 10 h runtime and obtained kinetic information on the TEMPO catalysed oxidation of alcohols and a mediated allylic C–H oxidation reaction using cyclic voltammetry.
Multistep electrochemistry is dominated by Yoshida's cation pool method, where a cation is generated electrochemically and subsequently reacts with a nucleophile.33,34 The continuous version was named “cation-flow”.35 The use of different cation precursors and nucleophiles allows for the quick build-up of product libraries and can be automated.36,37 The development of an anion pool method and subsequent reaction with an electrophile has been developed by Atobe and co-workers.38 Examples of the electrochemical synthesis of catalysts and the concurrent use in reactions in a multistep fashion remain underrepresented both in batch and under continuous reaction conditions.39–41 Due to electrons being used as reagents, electrochemistry enables the production of a clean catalyst solution, that can be used in a subsequent reaction without the need for purification. Separating the electrochemical formation of the catalyst and the catalytic steps allows the use of substrates that would decompose under the electrochemical conditions. However, the challenge lies in making both reaction steps compatible, particularly in terms of solvents and reactant concentrations, in addition to matching flow rates and maintaining a homogeneous regime. Feroci and co-workers demonstrated the electrochemical formation of NHCs (N-heterocyclic carbenes) for organocatalytic applications.41,42 A solution of the NHC precursor, which is often used neat as the ionic liquid, is electrolysed to produce the desired amount of NHC, and then either dispensed into vials with the substrate or the substrate is added to the NHC solution. We have previously demonstrated the continuous synthesis of Cu–NHCs and Cu(I) triflate for use in batch catalysis.14,40 The continuous setup for the electrochemical step allows for particularly mild reaction conditions, high-throughput and good reproducibility. Wirth and co-workers used electrogenerated hypervalent iodines for the oxidation of alcohols,39 in which the spacial separation was important, as some of the alcohols show lower oxidation potentials than the iodine precursor and would have undergone oxidation instead. As a result, performing the reaction continuously improved both selectivity and production.
Due to the need to wait for steady state in all steps of a multistep continuous process, reaction times are typically longer, and larger volumes are needed than screening steps individually. Therefore, most examples show an optimisation of each step individually and then combination of both for a synthetic scale application. However, performing and optimizing all steps continuously would offer the prospect of automating the full process and potentially capture parameters/reaction space and synergistic effects that may otherwise be missed when optimising each step individually. Recently, a screening platform involving the electrochemical generation of palladium species and examination in C–C coupling and C–H arylation reactions was described.43 Mechanistic studies were enabled using in situ MS to capture intermediates.
Herein, we describe the development of a flow platform that produces metal–NHC complexes electrochemically and in a high-throughput manner, with subsequent screening of the catalysts in a reaction of interest. The flow platform will enable the speed at which base-metal catalysts are discovered, optimised and implemented in industrially relevant transformations to be significantly accelerated.
Results and discussion
A miniaturised continuous electrochemical reactor was initially designed to allow for small reaction volumes, enabling small amounts of material to be used. A circular reactor with a 48 mm diameter, that is based on our previous continuous electrochemical reactors, was developed.14 The reactor consists of stacked electrodes and flow spacers held together by stainless steel plates (Fig. 1 and S1†).44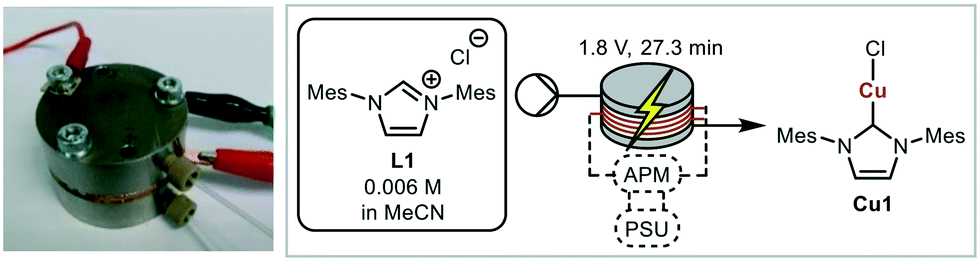 | ||
| Fig. 1 Miniaturised electrochemical flow reactor comprising circular stacked electrodes (48 mm diameter) separated by PTFE spacers (1 mm thick) with flow channels of 0.464 mL. Model electrochemical reaction; synthesis of Cu1 from L1 and copper electrodes. | ||
Suitable model reactions for both the electrochemical and catalytic reactions were identified in order to develop and validate the platform. NHCs have attracted considerable interest, particularly in the area of catalysis.45–52 Typically, NHCs are synthesised from the corresponding azolium salt via deprotonation with base, accessing a structurally diverse library of NHC analogues.53 However, base sensitive groups within the ligand architecture are generally not tolerated. Electrochemistry offers a complementary way to synthesise NHCs from azolium salts in a very selective and mild way (Fig. 2).42 The azolium is reduced at the cathode forming the carbene, with hydrogen as the only by-product.54 Therefore, the NHC can be produced in situ at a desired concentration before being added to the desired reaction. The counter reaction at the anode depends on the reaction system, with possible reactions being the decomposition of the solvent, oxidation of the counterion, or oxidation of a sacrificial anode to release metal ions for metal–NHC formation. As no further reactant or electrolyte is needed, the electrochemical synthesis of metal–NHCs from azolium salts results in a very clean reaction mixture, significantly simplifying downstream processes and purification.55–58 The direct use of these metal–NHC complexes in catalysis has been demonstrated to give comparable yields to the isolated and purified catalysts.14
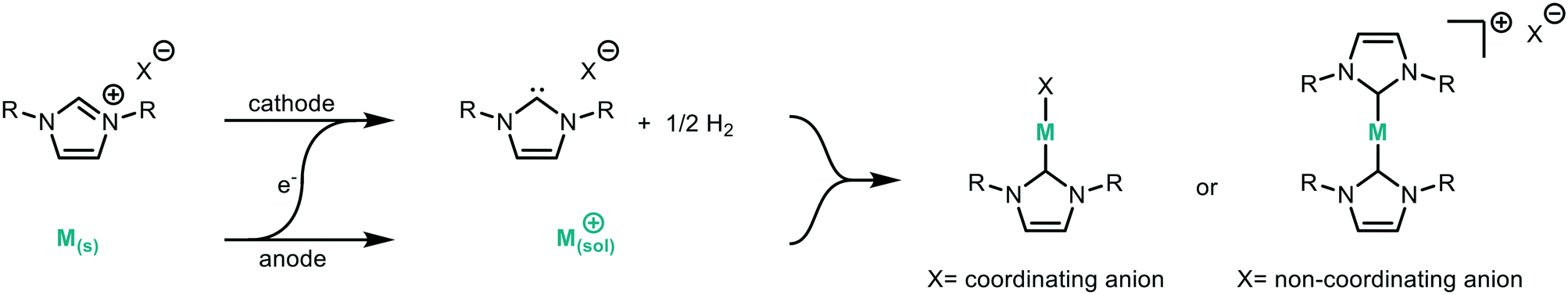 | ||
| Fig. 2 Electrochemical synthesis of metal–NHC complexes. An imidazolium salt is reduced at the cathode to form the NHC with concomitant oxidation of the sacrificial anode to produce metal ions to which the NHC coordinates. | ||
For initial optimisation of the electrochemical reaction the synthesis of the ubiquitous NHC complex Cu(IMes)Cl (Cu1) starting from IMes·HCl (L1) with copper electrodes was chosen (Fig. 2). Previous work has shown that single sweep voltammetry of this reaction at 1.8 V gave full conversion to Cu1 initially, though the reaction was unstable over a longer period of time which would be detrimental to a screening platform. To improve the long-term stability of the reactor, the use of alternating polarity was introduced (details in ESI† section 2.3), with a frequency range of 1/5 to 1/60 Hz being found optimal for the synthesis of Cu1.44 The reaction was run for 24 hours using HPLC monitoring with no decrease in conversion or yield.44 The click reaction was chosen as a model reaction for the catalysis step (Fig. 3). The copper-catalysed click reaction is well understood,59,60 and a continuous protocol addresses possible safety concerns related to azide starting materials.61 A continuous method was developed where the full reaction mixture (starting materials and catalyst in MeCN) was injected into and flowed through a hot tubular reactor. A residence time of 20 minutes at 120 °C gave 34% conversion from benzyl azide to the triazole when using catalyst Cu1. Two further catalysts, Cu2 and Cu4, were also examined, giving conversions of 89% and 57% respectively. The vast differences in catalytic activity when using different ligands provides an ideal model reaction for developing and validating a catalyst screening platform. With this in hand the electrochemical synthetic reaction and catalytic reaction were telescoped.
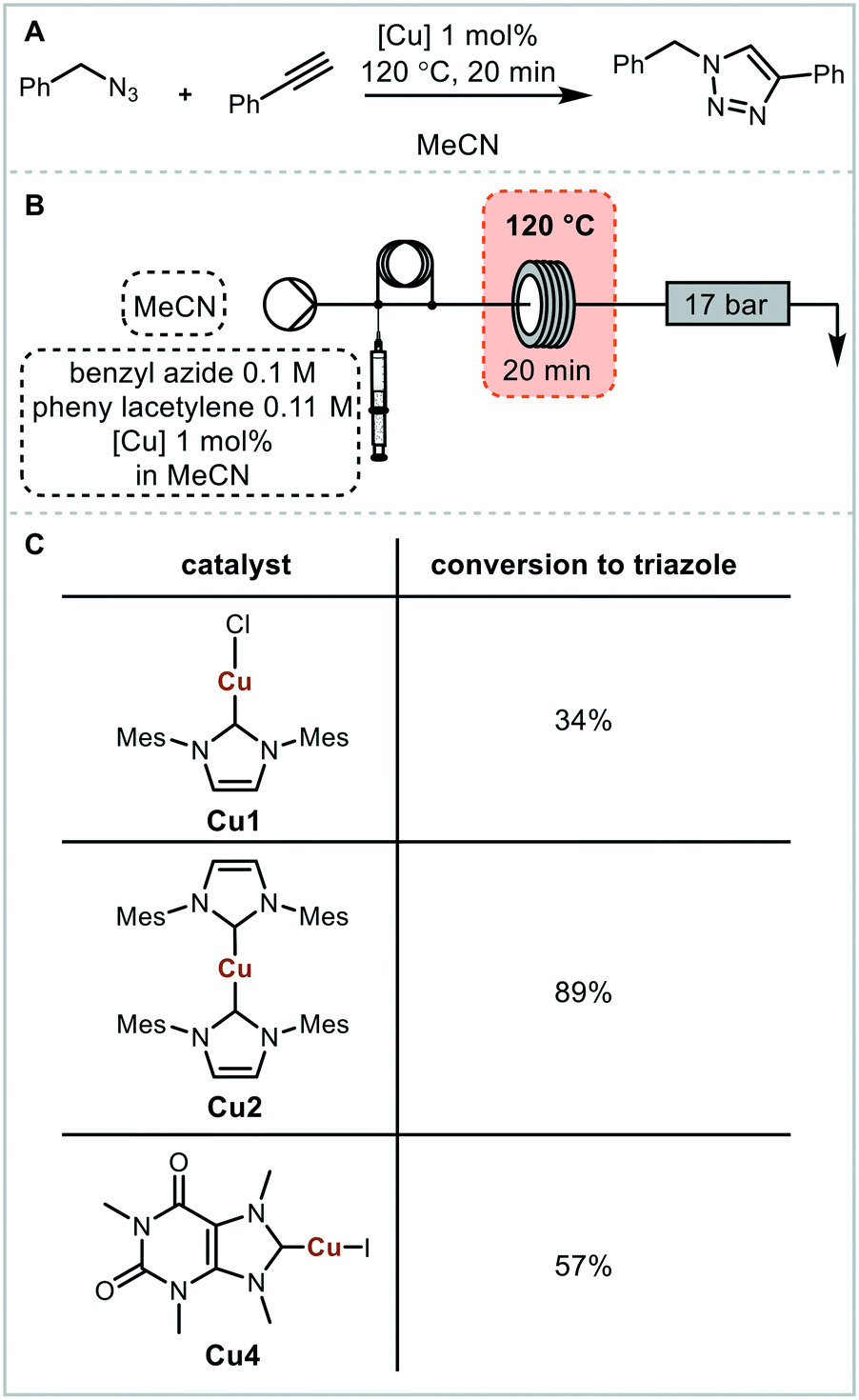 | ||
| Fig. 3 Click reaction using benzyl azide and phenyl acetylene as a model reaction. A: Reaction scheme; B: flow setup. Starting reagents and catalyst in MeCN are loaded into the sample loop and a flow rate of 1 mL min−1 is used. C: Catalyst screen, conversion from benzyl azide to triazole determined using 1H NMR spectroscopy. | ||
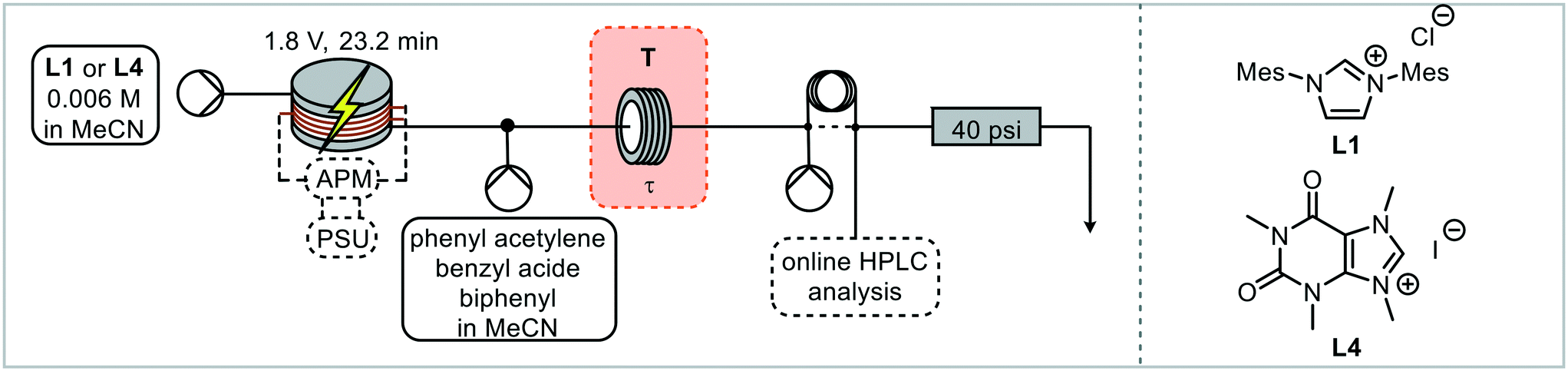
The ligand precursor (imidazolium salt) was pumped directly into the electrochemical reactor, with the output (catalyst) being merged with the reagents and standard solution before entering the catalysis reactor (Fig. 4). Subsequently the reaction solution passes through a valve for HPLC sampling. The reactors were filled with solution and the flow rates set before the power supply was turned on. HPLC samples were taken up until steady state was reached and beyond. The residence time in the catalysis step was reduced to 7.14 minutes, which enabled appropriate concentrations of both the catalyst and the reagents stream whilst allowing the catalyst loading to be adjusted.
 | ||
| Fig. 4 Telescoped continuous electrochemical synthesis of Cu–NHCs with catalytic click reaction and online HPLC monitoring. | ||
Continuous HPLC monitoring (every 6 minutes) allowed for understanding of the residence time distributions and ensured that steady state was reached prior to the yield being recorded (Fig. S17†). To enable a high-throughput approach to catalyst screening, an autosampler was introduced for the successive injection of different ligand precursors. In order to be able to handle air sensitive reaction mixtures, sealed microwave reaction vials were chosen so that the autosampler needle could pierce through the cap. The autosampler was incorporated into the system through a 2-way-6-port valve with a stainless-steel sample loop, which was connected to the electrochemical reactor via a stainless-steel tube (Fig. 5B). Stainless steel was necessary in this case to avoid the introduction of oxygen, with standard PFA tubing being slightly permeable to oxygen and was found to result in imidazolone formation following reduction of the imidazolium salt to an NHC and subsequent reaction with oxygen.62 A peristaltic pump was used to draw the solution through the autosampler needle, tube and sample loop which again avoids the introduction of oxygen whilst being able to handle gas slugs during sample withdrawing and needle cleaning. The remaining two ports were connected to the system pump and the system itself, with the pump ensuring a consistent flow through the reactors.
 | ||
| Fig. 5 A: Automated catalyst generation and activity screening. The platform generates in a stepwise fashion an array of catalysts through an electrochemical process followed by thermal reaction with online HPLC monitoring A: multistep continuous flow reactor platform setup; B: sample loop configuration. | ||
The setup was filled with solution, with MeCN only in the electrochemical reactor, and all pumps started. The autosampler sequence was started by switching the valve into the sample loop filling position. The autosampler arm moves to a defined vial position before the peristaltic pump withdraws a defined volume. The valve then switches for the aliquot to be injected into the reaction system. The autosampler needle rinses in a recirculating solution of methanol and then with fresh acetonitrile to be ready for the next precursor solution. The power supply was switched on after the autosampler cleaning, just before the precursor solution enters the reactor. HPLC monitoring started with the start of the autosampler sequence, with sampling every 6 minutes. Initially the reactor was taken apart and cleaned after every reaction. Ligand precursors L1 and L4 were injected into the electrochemical reactor, with the outcome of the catalytic step giving 2% and 9% triazole yield respectively. As the click reaction is very concentration dependent (batch reactions are usually performed neat),60 the triazole yields are lower than previously observed. HPLC is a very sensitive analytical method, hence even differences at this level (i.e. <10% yield) can be detected in a reproducible way. In this test, steady state was reached in 1.8 hours following the start of the autosampler sequence and was maintained until 2.6 hours, which indicates that the sample loop size could be reduced (Fig. S17†). The aliquot size and the time from reaching steady state at 1.8 h to the end of the aliquot were calculated to be 48 minutes, hence the minimum aliquot needed was 2.6 mL, so a 3 mL sample loop was used for all further reactions.
A cleaning step was implemented into the platform sequence which would enable several precursors to be run without having to take the reactor apart for offline cleaning in between each. The cleaning solution is required to dissolve deposits, mostly Cu deposited during the electrochemical reaction, off the electrode surface. A solution of AcOH in MeCN (2.5%, v/v) was tested with the power supply being switched off during the cleaning process. The same ligand precursor was run several times with the cleaning step in between, and triazole yield recorded. It was found that the cleaning step was required prior to every run, including prior to the first run, to ensure that the copper surface was consistent and the runs reproducible. The new sequence was tested with ligand precursors L1, L3 and L4 and achieved very reproducible results for two concurrent injections each (Fig. S19†). The cleaning cycle was 38 minutes (sample loop filling 6 minutes, 32 minutes for three total residence times), then the desired reaction was 145 minutes (sample loop filling 6 minutes, 139 minutes for 2 total residence times) giving a total reaction time of 3.1 hours. Product was observed after 1.5 hours, and steady state was reached at 2.5 hours for the first injection and 5.6 hours for the second injection corresponding to 2.5 residence times. After having established the reproducibility of the fully integrated platform, an initial ligand screen was undertaken (Fig. 6). 11 different azolium salts to make Cu–NHC complexes were examined in addition to acetic acid to make copper acetate.
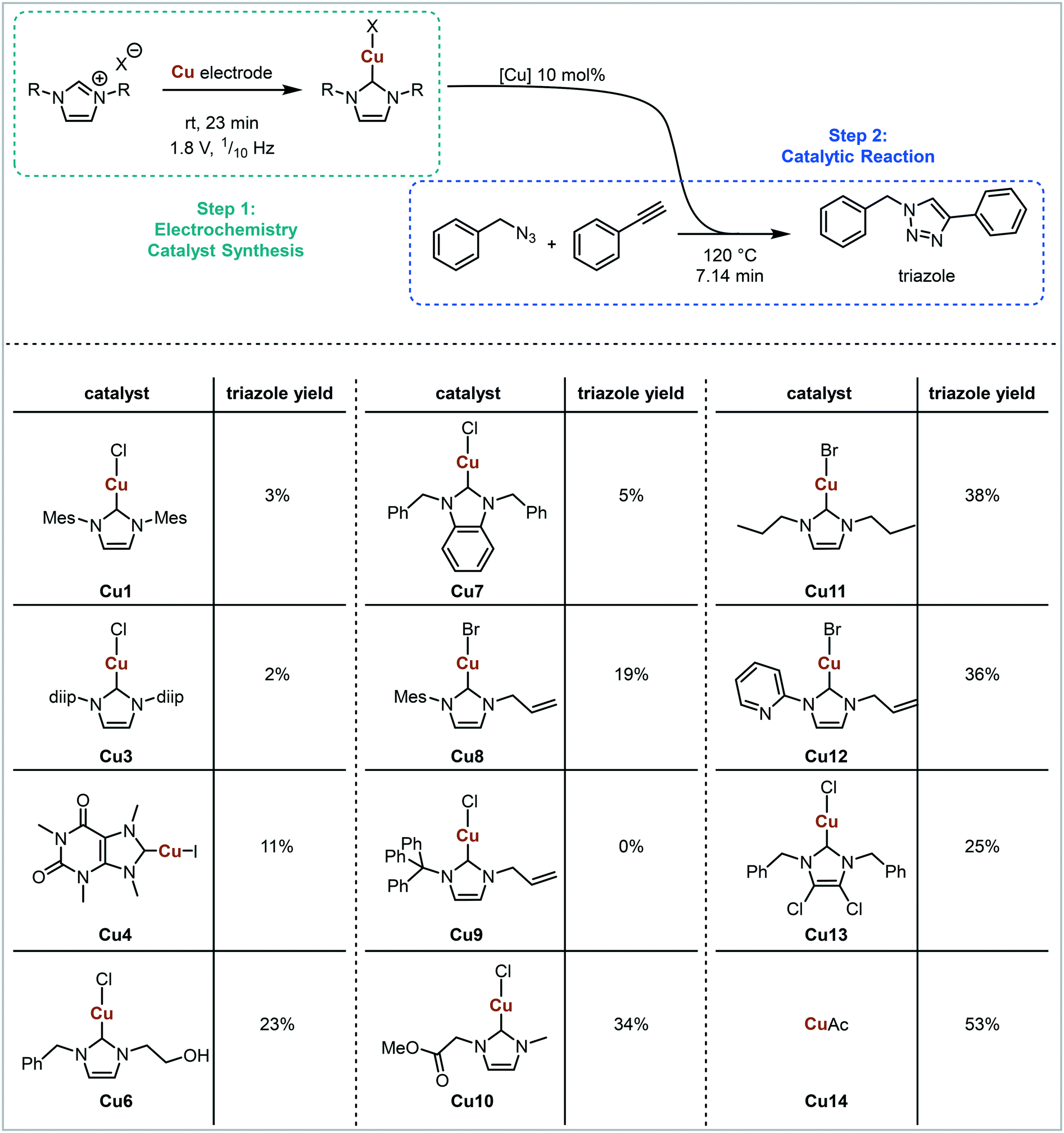 | ||
| Fig. 6 Ligand screen using the automated flow platform. Cu–NHCs are electrogenerated from the azolium starting materials (CuOAc from HOAC) and Cu electrodes, and telescoped into the catalytic reaction in flow, with online HPLC monitoring. | ||
Clear differences in triazole yields were observed, ranging from 2–3% for sterically demanding NHCs (Cu1 and Cu3) to 38% for less sterically demanding NHCs (Cu11). The electronic nature of the backbone appears to have less of an effect on triazole yield, with ligands in Cu4, Cu7 and Cu13 all having electron withdrawing backbones, but varying yields of 11%, 5% and 25% respectively. The injection of the imidazolium salt bearing a triphenyl N-substituent L9 resulted in no subsequent triazole formation. This is likely due to the imidazolium being unstable under electrochemical conditions forming a triphenylcarbenium. The least sterically demanding ligand bearing a protonated backbone (Cu11) provided the highest yield (38%) of all NHCs tested. However, in situ generated copper acetate (Cu14) resulted in an even higher triazole yield of 53%. The results presented in Fig. 6 demonstrate that in situ generated catalysts can be screened in a high-throughput manner, with conditions that allow differences between different catalysts to be observed. The screening platform can be used to assess various parameters and outcomes, with the possibility for automated optimisation. This would allow for optimal ligands/catalysts in conjunction with variables such as temperature, time, catalyst loading etc. to be rapidly identified.
Conclusions
An integrated and automated flow reactor platform has been developed that enables the electrochemical generation of metal catalysts and subsequent screening in a catalytic reaction. Traditional methods of catalyst screening often involve initial synthesis and purification of catalyst precursors. Electrochemical synthesis has a major advantage of minimal by-products; hence the catalyst can be dispensed directly into a reaction without the need for time-consuming work-up procedures, in addition to being amenable to continuous flow. Online analysis facilitates the rapid generation of data to understand reaction outcomes, which also has the potential to feed into evolutionary algorithms for automated optimisation. The platform will be relevant to both academia and industry for the rapid development of base-metal catalysed processes, enabling understanding through data generation and optimisation, thus reducing the growing and unsustainable demand for platinum group metals in manufacturing processes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from EPSRC (EP/R009406/1) and the University of Leeds. We would also like to acknowledge the industrial advisory board members, AstraZeneca, Johnson Matthey, Syngenta, CatSci, Advion, CTech Innovation and Innosyn, for valuable guidance and input into the project.Notes and references
- P. Sagmeister, R. Lebl, I. Castillo, J. Rehrl, J. Kruisz, M. Sipek, M. Horn, S. Sacher, D. Cantillo, J. D. Williams and C. O. Kappe, Angew. Chem., 2021, 60, 8139–8148 CrossRef CAS PubMed.
- A. D. Clayton, A. M. Schweidtmann, G. Clemens, J. A. Manson, C. J. Taylor, C. G. Niño, T. W. Chamberlain, N. Kapur, A. J. Blacker, A. A. Lapkin and R. A. Bourne, Chem. Eng. J., 2020, 384, 123340 CrossRef CAS.
- J. B. Sperry and D. L. Wright, Chem. Soc. Rev., 2006, 35, 605–621 RSC.
- K. D. Moeller, Chem. Rev., 2018, 118, 4817–4833 CrossRef CAS PubMed.
- R. D. Little and K. D. Moeller, Chem. Rev., 2018, 118, 4483–4484 CrossRef CAS PubMed.
- J.-I. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265–2299 CrossRef CAS PubMed.
- Q. Jing and K. D. Moeller, Acc. Chem. Res., 2020, 53, 135–143 CrossRef CAS PubMed.
- C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS PubMed.
- D. Pletcher, R. A. Green and R. C. D. Brown, Chem. Rev., 2018, 118, 4573–4591 CrossRef CAS PubMed.
- S. Maljuric, W. Jud, C. O. Kappe and D. Cantillo, J. Flow Chem., 2020, 10, 181–190 CrossRef CAS.
- H. R. Stephen, C. Schotten, T. P. Nicholls, M. Woodward, R. A. Bourne, N. Kapur and C. E. Willans, Org. Process Res. Dev., 2020, 24, 1084–1089 CrossRef CAS.
- R. A. Green, R. C. D. Brown, D. Pletcher and B. Harji, Org. Process Res. Dev., 2015, 19, 1424–1427 CrossRef CAS.
- C. Gütz, A. Stenglein and S. R. Waldvogel, Org. Process Res. Dev., 2017, 21, 771–778 CrossRef.
- M. R. Chapman, Y. M. Shafi, N. Kapur, B. N. Nguyen and C. E. Willans, Chem. Commun., 2015, 51, 1282–1284 RSC.
- A. A. Folgueiras-Amador, A. E. Teuten, D. Pletcher and R. C. D. Brown, React. Chem. Eng., 2020, 5, 712–718 RSC.
- A. Attour, P. Dirrenberger, S. Rode, A. Ziogas, M. Matlosz and F. Lapicque, Chem. Eng. Sci., 2011, 66, 480–489 CrossRef CAS.
- A. A. Folgueiras-Amador, K. Philipps, S. Guilbaud, J. Poelakker and T. Wirth, Angew. Chem., Int. Ed., 2017, 56, 15446–15450 CrossRef CAS PubMed.
- G. Laudadio, W. de Smet, L. Struik, Y. Cao and T. Noël, J. Flow Chem., 2018, 8, 157–165 CrossRef PubMed.
- M. Lehmann, C. C. Scarborough, E. Godineau and C. Battilocchio, Ind. Eng. Chem. Res., 2020, 59, 7321–7326 CrossRef CAS.
- Y. Mo, Z. Lu, G. Rughoobur, P. Patil, N. Gershenfeld, A. I. Akinwande, S. L. Buchwald and K. F. Jensen, Science, 2020, 368, 1352–1357 CrossRef CAS PubMed.
- J. Rein, J. R. Annand, M. K. Wismer, J. Fu, J. C. Siu, A. Klapars, N. A. Strotman, D. Kalyani, D. Lehnherr and S. Lin, ACS Cent. Sci., 2021, 7, 1347–1355 CrossRef CAS PubMed.
- C. Gütz, B. Klöckner and S. R. Waldvogel, Org. Process Res. Dev., 2016, 20, 26–32 CrossRef.
- M. Dörr, M. M. Hielscher, J. Proppe and S. R. Waldvogel, ChemElectroChem, 2021, 8, 2621–2629 CrossRef.
- T. Toshiki and N. Atsushi, Chem. Lett., 2009, 38, 160–161 CrossRef.
- Y. Mo, G. Rughoobur, A. M. K. Nambiar, K. Zhang and K. F. Jensen, Angew. Chem., Int. Ed., 2020, 59, 20890–20894 CrossRef CAS PubMed.
- M. Santi, J. Seitz, R. Cicala, T. Hardwick, N. Ahmed and T. Wirth, Chem. – Eur. J., 2019, 25, 16230–16235 CrossRef CAS PubMed.
- N. Amri and T. Wirth, Synthesis, 2020, 52, 1751–1761 CrossRef CAS.
- V. Mengeaud, O. Bagel, R. Ferrigno, H. H. Girault and A. Haider, Lab Chip, 2002, 2, 39–44 RSC.
- S. Arndt, D. Weis, K. Donsbach and S. R. Waldvogel, Angew. Chem., Int. Ed., 2020, 59, 8036–8041 CrossRef CAS PubMed.
- M. Dörr, J. L. Röckl, J. Rein, D. Schollmeyer and S. R. Waldvogel, Chem. – Eur. J., 2020, 26, 10195–10198 CrossRef PubMed.
- M. M. Hielscher, B. Gleede and S. R. Waldvogel, Electrochim. Acta, 2021, 368, 137420 CrossRef CAS.
- R. Möckel, E. Babaoglu and G. Hilt, Chem. – Eur. J., 2018, 24, 15781–15785 CrossRef PubMed.
- J.-I. Yoshida and S. Suga, Chem. – Eur. J., 2002, 8, 2650–2658 CrossRef CAS.
- J.-I. Yoshida, A. Shimizu and R. Hayashi, Chem. Rev., 2018, 118, 4702–4730 CrossRef CAS PubMed.
- J.-I. Yoshida, Chem. Commun., 2005, 4509–4516 RSC.
- K. Mitsudo, Y. Kurimoto, K. Yoshioka and S. Suga, Chem. Rev., 2018, 118, 5985–5999 CrossRef CAS PubMed.
- K. Mitsudo, Y. Kurimoto, K. Yoshioka and S. Suga, Curr. Opin. Electrochem., 2018, 8, 8–13 CrossRef CAS.
- Y. Matsumura, Y. Kakizaki, H. Tateno, T. Kashiwagi, Y. Yamaji and M. Atobe, RSC Adv., 2015, 5, 96851–96854 RSC.
- M. Elsherbini, B. Winterson, H. Alharbi, A. A. Folgueiras-Amador, C. Génot and T. Wirth, Angew. Chem., Int. Ed., 2019, 58, 9811–9815 CrossRef CAS.
- T. P. Nicholls, R. A. Bourne, B. N. Nguyen, N. Kapur and C. E. Willans, Inorg. Chem., 2021, 60, 6976–6980 CrossRef CAS PubMed.
- M. Feroci, M. Orsini, L. Rossi and A. Inesi, Curr. Org. Synth., 2012, 9, 40–52 CrossRef CAS.
- C. Schotten, R. A. Bourne, N. Kapur, B. N. Nguyen and C. E. Willans, Adv. Synth. Catal., 2021, 363, 3189–3200 CrossRef CAS.
- H. Cheng, T. Yang, M. Edwards, S. Tang, S. Xu and X. Yan, J. Am. Chem. Soc., 2022, 144, 1306–1312 CrossRef CAS PubMed.
- C. Schotten, C. J. Taylor, R. A. Bourne, T. W. Chamberlain, B. N. Nguyen, N. Kapur and C. E. Willans, React. Chem. Eng., 2021, 6, 147–151 RSC.
- S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed.
- D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed.
- D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307–9387 CrossRef CAS PubMed.
- F. Glorius, Topics in Organometallic Chemistry, Springer, 2007, pp. 1–20 Search PubMed.
- W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS PubMed.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- E. Peris, Chem. Rev., 2018, 118, 9988–10031 CrossRef CAS PubMed.
- A. A. Danopoulos, T. Simler and P. Braunstein, Chem. Rev., 2019, 119, 3730–3961 CrossRef CAS PubMed.
- L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz and V. César, Chem. Rev., 2011, 111, 2705–2733 CrossRef CAS PubMed.
- D. Rocco, I. Chiarotto, F. D'Anna, L. Mattiello, F. Pandolfi, C. Rizzo and M. Feroci, ChemElectroChem, 2019, 6, 4275–4283 CrossRef CAS.
- B. Liu, Y. Zhang, D. Xu and W. Chen, Chem. Commun., 2011, 47, 2883–2885 RSC.
- B. R. M. Lake, E. K. Bullough, T. J. Williams, A. C. Whitwood, M. A. Little and C. E. Willans, Chem. Commun., 2012, 48, 4887–4889 RSC.
- C. Galuppo, J. Alvarenga, A. C. Queiroz, I. Messias, R. Nagao and C. Abbehausen, Electrochem. Commun., 2020, 110, 106620 CrossRef CAS.
- A. Rodríguez and J. A. García-Vázquez, Coord. Chem. Rev., 2015, 303, 42–85 CrossRef.
- S. Díez-González, A. Correa, L. Cavallo and S. P. Nolan, Chem. – Eur. J., 2006, 12, 7558–7564 CrossRef PubMed.
- S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2008, 47, 8881–8884 CrossRef PubMed.
- M. Movsisyan, E. I. P. Delbeke, J. K. E. T. Berton, C. Battilocchio, S. V. Ley and C. V. Stevens, Chem. Soc. Rev., 2016, 45, 4892–4928 RSC.
- D. Li and T. Ollevier, Org. Lett., 2019, 21, 3572–3575 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cy00587e |
| This journal is © The Royal Society of Chemistry 2022 |