
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bimolecular reactions of CH2CN2+ with Ar, N2 and CO: reactivity and dynamics†
Sam
Armenta Butt
* and
Stephen D.
Price
 *
*
Department of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. E-mail: sam.butt.16@ucl.ac.uk; s.d.price@ucl.ac.uk; Fax: +44 (0)20 7679 7463; Tel: +44 (0)20 7679 4600 Tel: +44 (0)20 7679 4606
First published on 15th June 2022
Abstract
The reactivity, energetics and dynamics of bimolecular reactions between CH2CN2+ and three neutral species (Ar, N2 and CO) have been studied using a position sensitive coincidence methodology at centre-of-mass collision energies of 4.3–5.0 eV. This is the first study of bimolecular reactions involving CH2CN2+, a species relevant to the ionospheres of planets and satellites, including Titan. All of the collision systems investigated display two collision-induced dissociation (CID) channels, resulting in the formation of C+ + CH2N+ and H+ + HC2N+. Evidence for channels involving further dissociation of the CID product HC2N+, forming H + CCN+, were detected in the N2 and CO systems. Proton-transfer from the dication to the neutral species occurs in all three of the systems via a direct mechanism. Additionally, there are product channels resulting from single electron transfer following collisions of CH2CN2+ with both N2 and CO, but interestingly no electron transfer following collisions with Ar. Electronic structure calculations of the lowest energy electronic states of CH2CN2+ reveal six local geometric minima: both doublet and quartet spin states for cyclic, linear (CH2CN), and linear isocyanide (CH2NC) molecular geometries. The lowest energy electronic state was determined to be the doublet state of the cyclic dication. The ready generation of C+ ions by collision-induced dissociation suggests that the cyclic or linear isocyanide dication geometries are present in the [CH2CN]2+ beam.
1. Introduction
Doubly-charged positive ions (dications) can be found in a variety of energised environments including the ionospheres of planets and their satellites.1–5 A number of atomic dications have been detected in planetary ionospheres,6 and, recently, the molecular dication CO22+ has been detected in the ionosphere of Mars; the first detection of a molecular dication in such an environment.7 The presence of molecular dications in planetary ionospheres is predicted by modelling,8–10 and the CO22+ dication has also been detected in a cometary gas envelope.11 Despite, in most cases, their inherent thermodynamic instability towards charge separation, the metastable electronic states of small molecular dications have been shown to possess lifetimes sufficient to allow collisions with other species in ionospheric and other environments.12 Indeed, the lifetimes of atomic dications in planetary ionospheres are expected to be primarily determined by collisional processes.13 Significant bimolecular reactivity has been observed following collisions of both atomic and molecular dications with neutral species.12,14–18 This bimolecular reactivity suggests that dication chemistry can play a role in ionospheric processes;13,19 for example, dications could be involved in the chemistry of complex molecule formation through carbon chain-growth reactions.13,20–23 In order to identify dication reactions of ionospheric interest, experiments to probe dicationic reactivity are vital.24 For example, recent experimental work has identified the role molecular dications can play in atmospheric erosion processes, such as that involving CO22+ dissociation in the atmosphere of Mars.25–28 This paper presents an investigation of the reactions between the molecular dication CH2CN2+ and Ar, N2 and CO. This work provides the first probe of the reactivity, reaction mechanisms and energetics of this dication, enhancing our understanding of the role of such species in energised environments such as planetary atmospheres.The atmosphere of Titan is primarily comprised of molecular nitrogen (N2, ∼95%) and methane (CH4, ∼5%).29,30 It follows that nitriles, organic compounds containing a –CN moiety, are also relevant to the atmospheric chemistry of this satellite. Indeed, a number of nitrile compounds, including acetonitrile, have been detected in the atmosphere of Titan.31,32 Acetonitrile, CH3CN, is the simplest nitrile, and has also been detected in the atmosphere of the Earth,33–35 as well as in the interstellar medium (ISM).36,37 A recent investigation of the electron ionization and photoionization of CH3CN detected the formation of long-lived molecular dications including, predominantly, CH2CN2+.38 In ionospheres, dications are readily formed from their precursors by solar radiation and by collisions with high-energy particles.13 Therefore, the CH2CN2+ dication is likely to be produced from CH3CN in the ionosphere of Titan, as well as in the atmospheres of other planets and satellites. In addition to its relevance in planetary ionospheres, the neutral radical CH2CN has been detected in the interstellar medium, where nitriles are considered to play an important role in mechanisms for formation of complex organic molecules.36,39–43
Our choice of neutral collision targets is stimulated by their simplicity and relevance to planetary atmospheres. Argon is often present in the atmospheres of planets and satellites, for example in the atmospheres of Earth, Mars and Titan.30,44–49 Molecular nitrogen is the dominant species in the atmosphere of the Earth, but also in the atmosphere of Titan.13,19,45–48,50–52 While CO has been detected in comets,53–55 is widely distributed across the ISM;56,57 and is also present in the atmospheres of various planets and satellites including the Earth,58 Venus,59 and Titan.60–62
To the authors’ knowledge, no bimolecular reactions involving CH2CN2+ have previously been reported. However, comparisons can be made with the reactivity of similar molecular dications when they encounter the neutral collision partners used in this study. The reactions resulting from dication collisions with Ar have been well studied.63–76 Along with other channels, proton-transfer (PT) forming ArH+ was detected in collisions between Ar and the hydrogen-containing dications CHCl2+ and C2H22+.77,78 Additionally, Ar + CHCl2+ collisions generated HCCAr2+.78 The reactions resulting from dication collisions with N2 have been the subject of several previous studies, including recent work by the current authors.63,69–71,79–86 PT reactions have been observed following the interactions of N2 with C4H32+, resulting in the formation of N–H bonds.87 CO has been used as a collision partner in several studies of dication reactivity.70,84,88–91 Collisions between HCl2+ and CO resulted in PT forming COH+.89 In the light of these previous studies, we may hypothesise that PT is a likely consequence of CH2CN2+ collisions with neutrals.
Single electron transfer (SET), from the neutral to the dication, is a common outcome of dication-neutral collisions. Such dication-neutral electron transfer reactions are generally well represented by the Landau–Zener (LZ) ‘reaction window’ model.12 In this framework, an electron moves from the neutral to the dication at the point where the reactant (dication + neutral) and product (monocation + monocation) potential energy surfaces intersect. If this crossing lies within the ‘reaction window’, typically 2–6 Å, efficient electron-transfer can occur. At larger interspecies separations, there is weak coupling between the reactant and product potential energy surfaces and electron-transfer is disfavoured. For curve crossings at interspecies separations smaller than the ‘reaction window’, the coupling between the reactant and product potential energy surfaces is strong. In this situation it is likely that an electron will transfer twice between the neutral and dication, once on the neutral's approach and once upon departure, resulting in no net electron transfer. Thus, when crossings lie within the reaction window, where the reactant and product potentials are not too strongly or too weakly coupled, efficient SET can occur. Typically, electron transfer exothermicities from 2–6 eV result in curve crossings that lie in the reaction window.92,93
Roithová et al.94 reported a comprehensive experimental study of the competition between PT and SET in the reactions of the hydrogen-containing dications CHX2+ (X = F, Cl, Br, I) with several atomic, non-polar and polar neutral species including the neutral collision partners used in this study: Ar, N2, and CO. From their observations, Roithová et al.94 proposed that if SET falls within the ‘reaction window’ (i.e. has an exothermicity of 2–6 eV) then PT is supressed, despite the latter pathway often nominally involving energetically favoured products. Roithová et al. argued that SET usually occurs at longer distances than PT, rendering the geometry of the collision less important, whereas PT requires at least a minimal degree of orientation of the reactants. Therefore, they deduced that ET is kinetically favoured. Roithová et al. also observed that when the neutral collision partner was polar, the relative intensity of PT increased. The authors accounted for this observation by invoking the increased attractive forces between the dication and polar neutral. These increased forces would result in an increased likelihood of a collision complex forming, thus extending the lifetime of the interaction and allowing more efficient access to the thermodynamic PT products. Despite the small dipole moment of CO, Roithová et al.94 surmised this dipole was reason for the increased relative intensity of PT with respect to ET in collisions with this molecule. Based on this study by Roithová et al.94 we might expect that, of the neutral collision partners used in our experiments (Ar, N2, and CO), CO might exhibit the most intense PT channel.
As noted above, this work presents results from the experimental investigation of the bimolecular reactivity of CH2CN2+, a dication derived from the simplest nitrile, with the neutral species Ar, N2 and CO. PT channels were observed in all the collision systems and these reactions are shown to proceed via direct, long-range, mechanism. The competition between the PT and SET reactions of CH2CN2+ does not follow the trends observed by Roithová et al.94 Specifically, SET does not occur following collisions between CH2CN2+ and Ar despite the presence of accessible pathways. Additionally, the relative intensity of the PT channel in the CH2CN2+ + CO system is lower than in the systems with non-dipolar collision partners. We also observe the presence of two CID channels, producing CH2N+ + C+ and CHCN+ + H+. In order to explain our results, we carried out electronic structure calculations of the CH2CN2+ dication, which reveal a complex energy landscape. Given the nature of the CID reactions observed, and the lowest energy structures revealed by our calculations, it appears that the CH2CN2+ dication involved in our experiments explores a number of conformations, including cyclic and linear isocyanide structures.
2. Experimental
Coincidence techniques involve the simultaneous detection of two or more products from a single reactive event. Dication interactions with neutrals often generate pairs of monocations, and these pairs of ions are detected in coincidence in our position-sensitive coincidence (PSCO) mass spectrometry (MS) experiment. The apparatus used in this study has been described in detail in the literature.65,95,96 In brief, a pulsed beam of dications is directed into the field-free source region of a time-of-flight mass spectrometer (TOF-MS). In this region the dications interact with an effusive jet of the neutral reactant. Subsequent application of an extraction voltage to the source region allows the TOF-MS to detect the cation pairs generated from the dication-neutral interactions. The detection of these ions involves recording their arrival times, and arrival positions, at a large microchannel-plate detector. From this raw data, a list of flight times and arrival positions of the ions detected in pairs, a two-dimensional mass spectrum, can be generated, revealing the different reactive channels. The positional data accompanying the ionic arrivals yields the relative motion of the products of each detected event, providing a detailed insight into the mechanisms of each reactive channel.96The CH2CN2+ ions used in the experiments described in this work were generated via electron ionization of CH3CN (BDH, >99.9%, purified by repeated freeze–pump–thaw cycles) by 100 eV electrons in a custom-built ion source. Previous studies have shown that whilst several molecular dications are generated following the bombardment of CH3CN with electrons, CH2CN2+ (m/z = 20) is the most abundant.38 Following electron ionization the positively charged ions are extracted from the ion source and pass through a hemispherical energy analyser to restrict the translational energy spread of the final CH2CN2+ beam to ∼0.3 eV. The continuous beam of ions exiting the hemispherical analyser is then pulsed, using a set of electrostatic deflectors, before being accelerated and focussed into a commercial velocity filter. The velocity filter is set to transmit just the CH2CN2+ (m/z = 20) ions. The resulting pulsed beam of energy-constrained CH2CN2+ ions is then decelerated to less than 10 eV in the laboratory frame before entering the source region of the TOF-MS. In the TOF-MS source region the beam of dications is crossed with an effusive jet of the appropriate neutral species: Ar (BOC, 99.998%), N2 (BOC, >99.998%), or CO (Aldrich, >99.0%). Single-collision conditions97 are achieved by employing an appropriately low pressure of the neutral collision partner. Under these conditions most dications do not undergo a collision and only a small percentage experience one collision. Such a pressure regime ensures no secondary reactions, due to successive collisions with two neutral species, influences the CH2CN2+ reactivity we observe. As noted above, an electric field is applied across the TOF-MS source when the dication pulse reaches the centre of this region. This electric field accelerates positively charged species into the second electric field (acceleration region) of the TOF-MS and then on into the flight tube. At the end of the flight tube, the cations are detected by a position-sensitive detector comprising a chevron-pair of microchannel plates (diameter = 12.7 cm) located in front of a dual delay-line anode.95 The voltage pulse applied to the source region also starts the ion timing circuitry, to which the signals from the detector provide stop pulses. The experiments in this work employed a TOF-MS source field of 183 V cm−1.
Signals from the detector are amplified and discriminated before being passed to a PC-based time-to-digital converter. If two ions are detected in the same TOF cycle, a coincidence event is recorded and each ion's arrival time and impact position on the detector are stored for off-line analysis. The use of single-collision conditions ensures ‘false’ coincidences are kept to a minimum. The ion pairs data can be plotted as a 2D histogram, a ‘pairs spectrum’, where the time of flights (t1, t2) of each ion in the pair are used as the (x, y) co-ordinates. Peaks in the pairs spectrum readily identify bimolecular reaction channels that result in a pair of positively-charged product ions. Each such peak, the group of events corresponding to an individual reaction channel, can then be selected for further off-line analysis.
As shown in previous work, the positional and time of flight information for each ion of a pair can be used to generate their x, y and z velocity vectors in the laboratory frame; here the z-axis is defined by the principal axis of the TOF-MS.95 The x and y velocity vectors of an ion are determined from the associated positional information and the ion's flight time; the z vector is determined from the deviation of the observed TOF from the expected TOF of the same ion with zero initial kinetic energy. The laboratory frame velocities are then converted into the centre-of-mass (CM) frame using the initial dication velocity.95 Often, the pair of monocations resulting from the reaction between a dication and a neutral are accompanied by a neutral species: a three-body reaction. A powerful feature of the PSCO-MS experiment is that the CM velocity of such a neutral product can be determined from the CM velocities of the detected ionic products via conservation of momentum.95
To reveal the dynamics of a given reaction channel, a CM scattering diagram can be generated from the velocities of the product ions. Such CM scattering diagrams are radial histograms that, for each event collected in a given reaction channel, plot the magnitude of the products’ CM velocity |wi| as the radial co-ordinate and the scattering angle θ between wi and the CM velocity of the incident dication as the angular coordinate. In the kinematics that apply in our experiment, where the dication has significantly more momentum than the neutrals, the velocity of the incident dication is closely oriented with the velocity of the centre of mass. In our CM scattering diagrams, since 0° ≤ θ ≤ 180°, the data for one product can be shown in the upper semi-circle of the figure and the data for another product in the lower semi-circle, as the scattering of each ion is azimuthally symmetric. The typical angular resolution of the scattering data achieved by the PSCO apparatus operating with a high source field is 4°.72 It should be noted that in our data treatment, to enable presentation in a two-dimensional figure on the page, we are integrating over the azimuthal angle: that is, we are binning events according to their value of θ irrespective of the azimuthal scattering angle. This integration over the azimuthal angle results in an isotropic scattering distribution yielding a sin(θ) distribution in our scattering diagrams, giving our experimental arrangement a low relative detection efficiency at values of θ very close to 0° or 180°. This phenomenon is clearly visible in the scattering diagrams where even strongly forward scattered reactions exhibit a peak in θ away from θ = 0. A more detailed discussion of the construction and form of these scattering diagrams can be found in the literature.96
In all the scattering diagrams presented in this work, a logarithmic scale is used for the scattering intensity. Such scales allow the diagrams to reveal subtleties of the scattering in the areas with low intensities. However, one should be aware that such logarithmic scales can, at first glance, overemphasise the importance of areas in the scattering diagrams with low intensities. In the ESI† (Fig. SI1) we contrast scattering diagrams with linear and logarithmic intensity scales.
For three-body reactions, internal-frame scattering diagrams can be a powerful aid in interpreting the reaction dynamics. In this class of scattering diagram |wi| is again the radial coordinate, but the angular coordinate is now the CM scattering angle with respect to CM velocity of one of the other product species.
From the CM velocities of the product species the total kinetic energy release (KER) T for a given reactive event can also be determined using the individual CM velocities of the products.95 The exoergicity of the reaction ΔE can then be determined from T and the CM collision energy, Ecom:
| ΔE = T − Ecom = −(Eproducts − Ereactants) | (1) |
2.1. Electronic structure calculations
Electronic structure calculations using Gaussian16 (Rev.A03)98 have been used to support the experimental findings. Specifically, the equilibrium geometries, and corresponding adiabatic and vertical ionisation energies, were determined for various species and structures of interest. These quantities were calculated for the lowest energy states of each of the relevant multiplicities. Stationary points were located using the MP2 algorithm with a cc-pVTZ basis set and the vibrational frequencies were analysed to confirm minima had been located. The energetics of these minima were then determined using single-point CCSD(T) calculations with the cc-pVTZ basis set. Zero-point energies from the MP2 geometry optimisations were used to correct the CCSD(T) single point energies. For the quartet isocyanate dication (4ID), where one MP2 vibrational frequency appeared unreliable, a B3LYP determination of the vibrational frequencies was used for the zero-point energy correction, the B3LYP algorithm converging to essentially the same geometry as the MP2 algorithm. The above methodology has been successfully used in previous work to determine the geometries and energetics of dications.88,99 Readers are referred to the SI for the full geometrical data for each dication structure.3. Results and discussion
The collisions of CH2CN2+ with Ar, N2 and CO were investigated, at CM collision energies of 5.0 eV, 4.5 eV, and 4.3 eV respectively, and the reactions observed are shown in Tables 1–3. In all the systems, a collision-induced dissociation (CID) channel was detected that involves the formation of CH2N+ + C+ (Rxns. I, IV, and X). Another CID channel, generating CHCN+ + H+, was also detected following collisions of CH2CN2+ with all three of the neutrals (Rxns. II, V, and XI). The collisions of CH2CN2+ with N2 and CO also result in C2N+ + H+ due to the further dissociation of the primary CID product HC2N+ (Rxns. VI and XII). All of the collision systems exhibit a PT channel forming HC2N+ and the protonated collision partner, XH+ (X = Ar, N2, CO, Rxns. III, VII, and XIII). Following collisions of CH2CN2+ with N2 and CO, non-dissociative single electron-transfer (NDSET) channels are observed, forming CH2CN+ + Y+ (Y = N2, CO, Rxns. VIII and XIV). The collisions of CH2CN2+ with CO and N2 also result in products from dissociative single electron-transfer (DSET) pathways, involving the fragmentation of the CH2CN+* ion initially generated by SET (Rxns. IX, XV, and XVI). Finally, following the collisions of CH2CN2+ and CO, a DSET channel was detected, involving the fragmentation of CO+, generating CH2CN+ + C+ (Rxn. XVII).| Reaction | Products | Relative intensity/% |
|---|---|---|
| I | CH2N+ + C+ | 59.8 ± 3.3 |
| II | HC2N+ + H+ | 22.0 ± 1.8 |
| III | HC2N+ + ArH+ | 18.2 ± 1.6 |
| Reaction | Products | Relative intensity/% |
|---|---|---|
| IV | CH2N+ + C+ | 41.0 ± 0.6 |
| V | HC2N+ + H+ | 15.6 ± 0.3 |
| VI | C2N+ + H+ | 1.0 ± 0.1 |
| VII | HC2N+ + N2H+ | 38.6 ± 0.5 |
| VIII | CH2CN+ + N2+ | 2.8 ± 0.1 |
| IX | HC2N+ + N2+ | 1.1 ± 0.1 |
| Reaction | Products | Relative intensity/% |
|---|---|---|
| X | CH2N+ + C+ | 21.3 ± 0.4 |
| XI | HC2N+ + H+ | 3.7 ± 0.1 |
| XII | C2N+ + H+ | 0.4 ± 0.1 |
| XIII | HC2N+ + COH+ | 14.7 ± 0.3 |
| XIV | CH2CN+ + CO+ | 52.4 ± 0.7 |
| XV | CO+ + CH+ | 2.3 ± 0.1 |
| XVI | HC2N+ + CO+ | 0.8 ± 0.1 |
| XVIII | CH2CN+ + C+ | 4.3 ± 0.2 |
To determine the accessible electronic states of CH2CN2+ in the reactant beam the lowest energy structures of the CH2CN2+ dication were investigated using the computational methodology discussed above. Three local geometric minima of CH2CN2+ were discovered for both the doublet and quartet multiplicities (see Table 4). The lowest energy conformation is a doublet, with a cyclic C–C–N arrangement (2CD) shown in Fig. 1a, and an adiabatic double ionization energy of 28.9 eV from the ground state neutral. The ground state neutral structure determined from our calculations, a doublet with a linear geometry, agrees well with the literature.100 A linear doublet dication structure (2LD), shown in Fig. 1b, and a linear doublet isocyanide dication structure (2ID), shown in Fig. 1c, were also located. Each dication conformation also has an associated higher energy quartet state (Table 4). Whilst 2CD is the lowest energy conformation revealed by our calculations, the linear and linear isocyanide structures do not lie far above the 2CD state energetically. Therefore, it is quite possible that the CH2CN2+ ions in our beam are fluxional, sampling different minima on the dicationic potential energy surface. Here we can directly analogise with the benzene dication which has a number of metastable minima on the dicationic potential energy surface which are sampled by C6H62+ ions.101–103
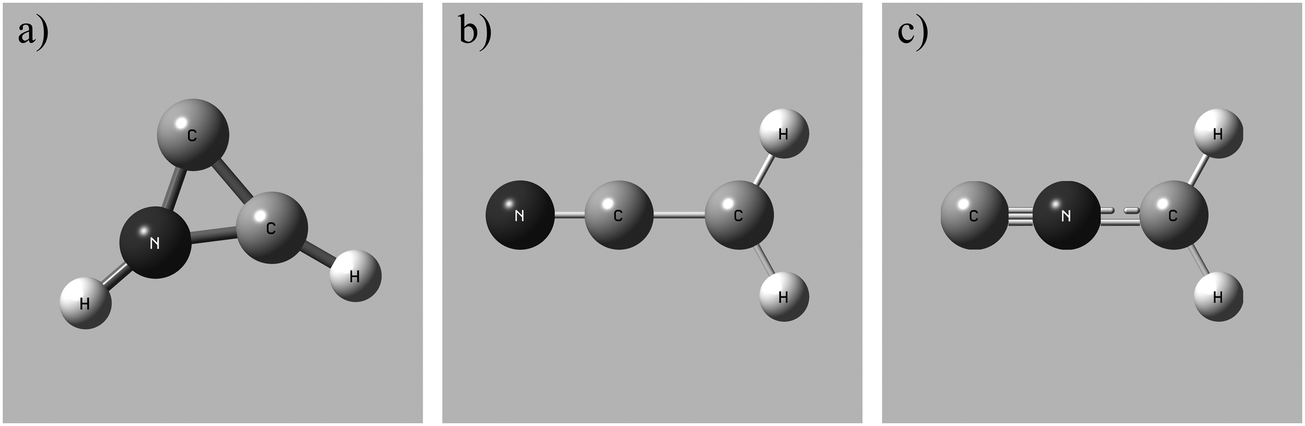 | ||
| Fig. 1 Geometries of the local minima of the reactant CH2CN2+ dication (doublet states). (a) Cyclic conformation, 2CD, (b) linear conformation, 2LD, (c) linear isocyanide conformation, 2ID. See text for details. | ||
The six metastable dication states revealed by the calculations (Table 4) have adiabatic double ionization potentials, from CH2CN, of between 28.9 eV and 32.9 eV. These double ionization potentials are significantly lower than the double ionization potentials of Ar (43.39 eV),104 N2 (∼43 eV),105–107 and CO (∼41.5 eV).108–111 Therefore, as expected, there are no product peaks in any of the spectra from the CH2CN2+ collision systems that result from double electron-transfer (DET), as such processes are significantly endoergic.
Similar computational investigations were also undertaken to determine likely structures of the CH2CN+ monocation, the ion which results from the dication acquiring an electron in a SET reaction. These calculations reveal a singlet and triplet state for each of the cyclic, linear, and linear isocyanide geometries, shown in Table 5. The vertical ionization energies determined for the linear and linear isocyanide geometries are a good match (∼0.1 eV difference) with experimental data from photoelectron spectroscopy.112–114 In addition the energetics and geometries we derive are in good agreement with recent computational studies.115
Calculations were also employed to determine the energy of the lowest energy structure of the HC2N+ monocation formed when the dication loses a proton. The lowest energy state of HC2N+ determined from these calculations is a doublet that has a linear N–C–C geometry, with the H bonded to the terminal C slightly off the N–C–C axis. This state has a calculated adiabatic ionization energy of 10.4 eV. The structure of the corresponding neutral species, HCCN, was also determined to corroborate our methodology. This investigation revealed a staggered conformation, in accord with the geometry calculated by Nimlos et al.116
In the following sections the different reaction mechanisms revealed by our scattering data for the collisions of CH2CN2+ + X (X = Ar, N2, and CO) are discussed.
3.1. The formation of C+ + CH2N+ (CID)
Fig. 2 shows the CM scattering diagrams of the C+ and CH2N+ products resulting from the collisions of CH2CN2+ with Ar, N2 and CO. It is clear, from the dependence of the number of counts in this channel on the neutral target gas pressure, that these products result from bimolecular reactions. The scattering diagrams (Fig. 2) for each of the reactions are remarkably similar, with the C+ and CH2N+ ionic products isotropically scattered about the CM velocity of the reactant dication. Therefore, the mechanism responsible for this channel is clearly CID: the CH2CN2+ dication dissociates as a result of a collision with the neutral species, forming CH2N+ + C+. CID involves energy transfer between the dication and neutral species promoting the dication to a state which subsequently dissociates. We are unable to directly resolve the energy transfer between the dication and neutral species in this CID channel because we cannot directly determine the change in kinetic energy of the neutral product. However, determination of the CM velocity associated with the ionic products reveals, within our experimental uncertainty, no resolvable difference to the velocity of the reactant dication. This observation clearly indicates there only a small (markedly less than 1 eV) average energy exchange between the dication and neutral species. Interestingly, modelling shows that CH2N+, the product of this CID reaction, is one of the most abundant ionic species in the atmosphere of Titan.117–119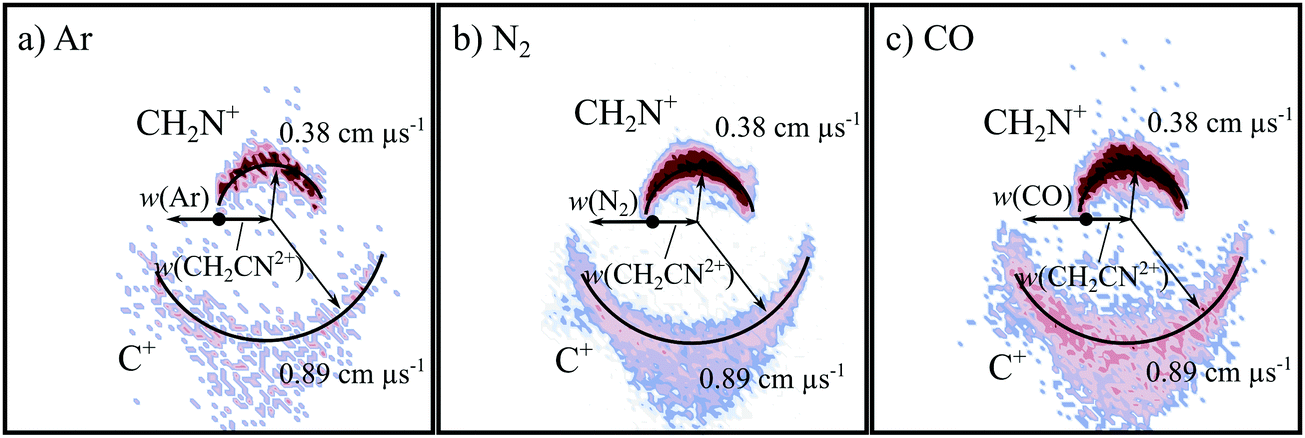 | ||
| Fig. 2 CM scattering diagrams for the CID channel producing CH2N+ + C+ resulting from the reactions of (a) CH2CN2+ + Ar at a CM collision energy of 5.0 eV, (b) CH2CN2+ + N2 at a CM collision energy of 4.5 eV, and (c) CH2CN2+ + CO at a CM collision energy of 4.3 eV. | ||
The precursor used to produce CH2CN2+via electron ionization is acetonitrile, CH3CN. CH3CN has a well-defined structure (C3v) involving a –CH3 moiety bound to the nitrile group (–CN). Given this connectivity, the presence of a CID channel generating C+ (CH2N+ + C+) points to the involvement of a dication structure significantly different from the structure of the CH3CN precursor, which has no terminal carbon atoms. As discussed above, the minimum energy CH2CN2+ structure, 2CD, revealed by the calculations has a cyclic C–C–N arrangement (Fig. 1a), whilst slightly higher in energy is the linear isocyanide structure, 2ID (Fig. 1c). It is easy to see how either the 2CD or 2ID CH2CN2+ structures could readily dissociate to form CH2N+ + C+. The presence of this CID channel therefore hints that our dication beam includes dications in the cyclic or linear isocyanide geometries. Of course, an alternative explanation could be that upon collision with the neutral, the linear dication rearranges to the cyclic or isocyanide state before dissociation. Our data cannot reveal which of these detailed mechanisms is operating. However, the similarity in scattering across the three CH2CN2+ + neutral systems strongly suggests the cyclic or linear isocyanide dication structures are involved.
Extracting the ionic velocity distributions from our data for this channel, we see that velocity distributions of the CH2N+ and C+ products are independent of the neutral species involved in the collision, as shown qualitatively in Fig. 2. As well as the dominant isotropic angular distribution, the scattering diagrams (Fig. 2) have a slight ‘bump’ at angles near 90 degrees, where the product ions are scattered with higher velocities. This bump is also reflected in the experimental exoergicity spectrum (Fig. 3, discussed below), giving rise to the structure from ∼8–11 eV.
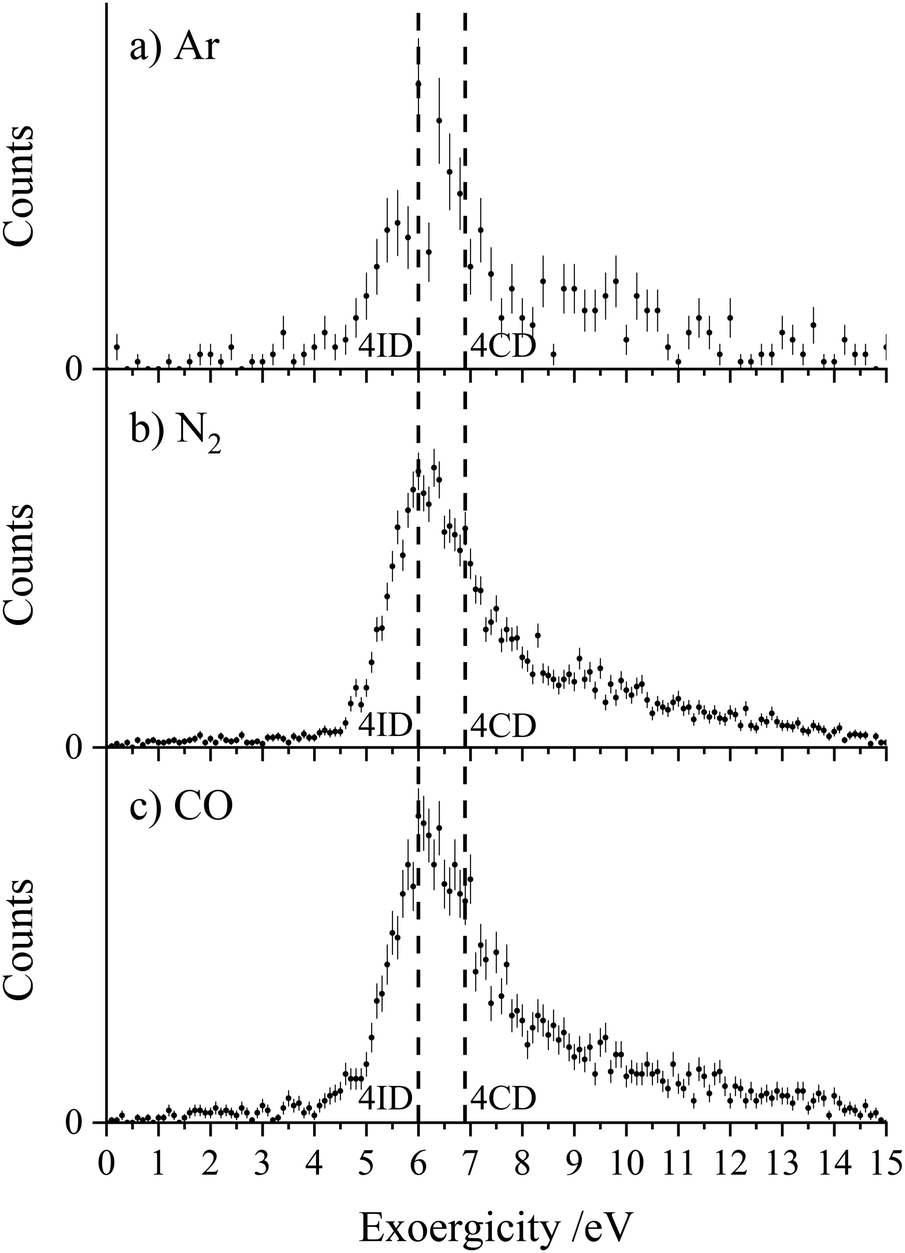 | ||
| Fig. 3 Experimental exoergicity distributions resulting from the CID channel CH2CN2+ + X → CH2N+ + C+ + X, where X = (a) Ar, (b) N2, and (c) CO. Dashed lines are marked at 6.0 eV and 6.9 eV in each of the distributions to show the predicted literature exoergicities associated with the involvement of the quartet linear isocyanide dication 4ID and the cyclic quartet dication 4CD respectively. The error bars represent two standard deviations of the counts. | ||
Fig. 3 shows the experimental exoergicity distributions for the dissociation of CH2CN2+ in these CID channels forming CH2N+ + C+. The exoergicity distributions for the different collision systems are again very similar, rising sharply from ∼5 eV to a maximum at ∼6 eV before slowly falling. The experimental exoergicity distributions all have FWHMs of ∼5.5–7.5 eV, with a shoulder to higher exoergicities. In order to rationalise the experimental exoergicity distribution we must consider the relative energies of the reactant and product species that could be involved in this reaction. For this system, energetic data for C+ is readily available and the relevant energy levels for CH2N+ can be estimated using the proton affinity and heat of formation of HCN, which are also in the literature.120,121 By combining the energy of a proton, the heat of formation of HCN and the proton affinity of HCN, we determine an estimate of the heat of formation of CH2N+ as 9.87 eV and will assume that no other electronic states of CH2N+ are involved in the reaction. For simplicity, and given our experimental observations above, we will assume that, given the level of accuracy of these energetic estimates, there is negligible energy transfer between the dication and neutral species. With the above energetics, and using the double ionization energies from our calculations of CH2CN2+ and the heat of formation of CH2CN (there is some uncertainty in this latter value, we will use +2.5 eV),122–124 we are then in a position to estimate the literature exoergicities expected for this reaction. These energetics indicate that, in an exothermic reaction, C+ could be formed in either its ground, 2P, or first excited state, 4P.
Using the above energetics, we find three pathways with literature exoergicities that fall within the FWHMs observed experimentally for these CID channels. The energy releases from 4CD and 4ID dissociating to produce C+ + CH2N+ in their electronic ground states are 6.9 eV and 6.0 eV respectively, fitting nicely with the experimental exoergicity distributions in Fig. 3. The corresponding energy release from the linear quartet CH2CN2+ dication is 6.7 eV, also falling within the observed experimental exoergicity distribution. However, due to the fragments formed we think that the dication reactant is likely not in the regular linear conformation (4LD). If either of the doublet 2CD (the ground state) or 2ID states were involved, the expected exoergicity released from this CID channel would be 2.8 eV and 3.3 eV respectively, clearly outside of the observed experimental exoergicity range (Fig. 3). Of course, both the reactant CH2CN2+ and product CH2N+ ions could be vibrationally excited, which would act to broaden the experimentally observed exoergicities. If the reactant dication was in the 2CD state and significantly vibrationally excited (on the order of ∼3 eV) the resulting exoergicities would be a match with the observed distribution. However, this level of vibrational excitation is very unlikely as such dications would almost certainly dissociate before reaching the interaction region. Further detailed computational work, beyond the scope of this study, on the nature of the CH2CN2+ dication states, including their lifetimes and ability to support vibrational excitation, would be valuable to further interpret the exoergicity data.
To summarise, one of the dominant channels following the collisions of CH2CN2+ with Ar, N2, and CO results in the generation of CH2N+ + C+, revealed by the dynamics to be due to CID. This CID channel is the most intense reaction channel following the collisions of CH2CN2+ with Ar and N2, whilst with CO, it is the second most intense channel. The presence of this CID channel, resulting in the ejection of a C+ from the reactant dication, suggests that the dications participating in this CID channel are likely not in the linear H2CCN conformation derived directly from the structure of the CH3CN precursor. The experimental exoergicity distributions of these three channels are in excellent agreement with each other and point to the involvement of either the quartet cyclic state, 4CD, or the quartet linear isocyanide state, 4ID, of the CH2CN2+ dication, and result in the formation of C+ + CH2N+ in their electronic ground states. The CH2N+ product is likely formed with some vibrational excitation.
3.2. The formation of H+ + HC2N+ (CID)
Each of the coincidence spectra resulting from the CH2CN2+ + X (X = Ar, N2, CO) collision systems displays a peak corresponding to the formation of a pair of ions with m/z = 1 and m/z = 39, corresponding to H+ + HC2N+, clearly another CID channel. The form of the pairs spectra and the associated scattering diagrams from the channels generating HC2N+ + H+ in each of the three CH2CN2+ + X (X = Ar, N2, CO) systems look close to identical, pointing to the involvement of the same mechanism in each system (see Fig. SI2 and SI3 in the ESI†). It is difficult to say too much about the mechanism of this channel from the dynamics because the velocity of the H+ product ion is dominant, leading to markedly increased uncertainty in the scattering of the molecular ion; however, the form of the scattering (with product monocation distributions centred on the dication velocity) clearly indicates that CID is the mechanism involved.The experimental exoergicity distributions recorded following the CH2CN2+ + X → HC2N+ + H+ + X (X = Ar, N2, CO) reactions are shown in Fig. 4. All three exoergicity distributions have a similar structure, with a peak centred at ∼2.5 eV and a FHWM from approximately 0.5 eV to 6 eV. In order to rationalise these experimental exoergicities, we must again consider the product and reactant states involved. As before, we will consider the six metastable states of CH2CN2+ found from our calculations: 2CD, 2LD, 2ID, 4CD, 4LD, and 4ID. The neutral reactants will, of course, be in their ground vibronic states. As before, given our experimental observations, we will neglect any energy transferred to the dication in the collision. The heat of formation of H is +2.26 eV, and its ionization energy is 13.60 eV.104 The lowest energy structure of the HC2N+ monocation revealed by our calculations (a doublet with a linear N–C–C geometry) has an adiabatic ionization energy of 10.4 eV relative to the neutral, and the neutral has a heat of formation of +4.8 eV.116 The literature exoergicities of the reaction CH2CN2+ + X → HC2N+ + H+ + X (X = Ar, N2, CO) using the above constraints are marked on the experimental exoergicity distributions in Fig. 4 for each of the six possible CH2CN2+ dication states. These exoergicities are 2CD = 0.4 eV, 2LD = 1.9 eV, 2ID = 0.9 eV, 4CD = 4.5 eV, 4LD = 4.2 eV, and 4ID = 3.6 eV. The literature exoergicities resulting from the involvement of the 2LD structure and all of the quartet structures (4CD, 4ID, 4LD) are good matches to the bulk of the experimental data (Fig. 4). It is tempting to attribute the linear dication structures 2LD and 4LD to this CID reaction, given the good match with the experimental exoergicity distribution and the fact that we think that these structures are not involved in the CID channel forming C+ + CH2N+. However, the data does not rule out the involvement of the 4ID and 4CD dication states in this CID channel. The signals at lower exoergicities in Fig. 4 could involve reactions of the 2CD and 2ID geometries or could result from vibrational excitation of the HCCN+ product.
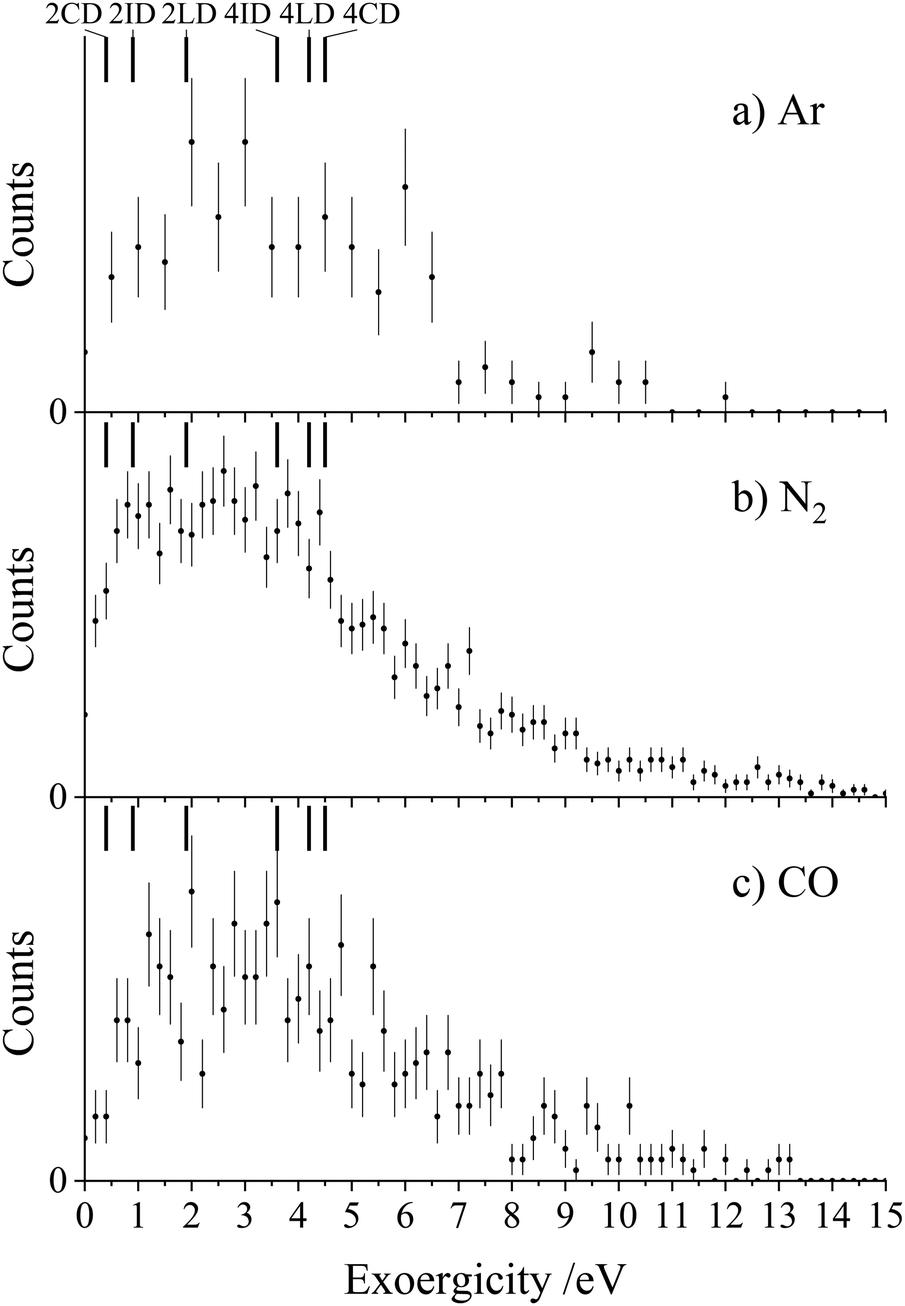 | ||
| Fig. 4 Experimental exoergicity distributions resulting from the channel CH2CN2+ + X → HC2N+ + H+ + X, where X = (a) Ar, (b) N2, and (c) CO. Lines are marked in each of the distributions to show the literature exoergicities for reactions involving each of the six dication states. The error bars represent two standard deviations of the counts. | ||
To summarise, the peak observed in the coincidence spectrum corresponding to the formation of HC2N+ + H+ following the collisions of CH2CN2+ + X (X = Ar, N2, CO) results from CID. The experimental exoergicity distributions suggest the involvement of, predominantly, the linear 2LD state and the three quartet states (4LD, 4CD and 4ID) of the dication resulting in the formation of the products in their ground electronic states. It is interesting to note that the bulk of the experimental exoergicity distributions in both CID channels can be primarily explained with the higher energy quartet states of the CH2CN2+ dication. The lower energy doublet CH2CN2+ states appear to be more stable to collisional excitation at the energies we are employing.
3.3. Proton-transfer (PT) channels
PT is observed in all three CH2CN2+ + neutral collision systems, resulting in the formation of HC2N+ and the protonated neutral. PT is often observed following the collisions of hydrogen-containing dications with neutrals, and has been detected before in different reactive systems involving each of the neutral species used in this investigation: N2,87 CO,89 and Ar.74,77,78,94 Considering the products of PT, the argonium ion, ArH+, is an example of a molecular species involving a rare gas and has been detected in the ISM,125,126 including in other galaxies.127 N2H+ has been detected in the interstellar medium,128 protoplanetary disks,129 and in the atmosphere of Titan (where modelling also predicts the presence of N2H2+). In such environments N2H+ can be formed via a number of processes.130,131 OCH+ has also been detected in the ISM.132,133Fig. 5 shows the CM scattering diagrams of the ionic products resulting from the PT reactions. In each of the scattering diagrams, strong forward scattering is observed. Such a scattering pattern is indicative of a long-range, direct, mechanism. If a proton transfers from the dication to the neutral at a large interspecies separation, the deflection of the reactant species is minimal and therefore the product velocities are strongly oriented with the velocities of their corresponding reactants. Evidence shows that dication-neutral reactions involving the formation of new bonds more commonly occur via the formation of a collision complex, where the reactants temporarily associate in order for new bonds to be made.96,134–136 However, previous experiments have also shown that dication-neutral PT reactions can occur via direct mechanisms.87,137 For example, PT occurs via a direct mechanism following collisions of C2H22+ with Ar.78 The direct nature of these PT reactions is not surprising given that just a proton is being transferred between the reactants, and thus the operation of a direct mechanism, analogous to LZ style electron transfer, seems dynamically reasonable.
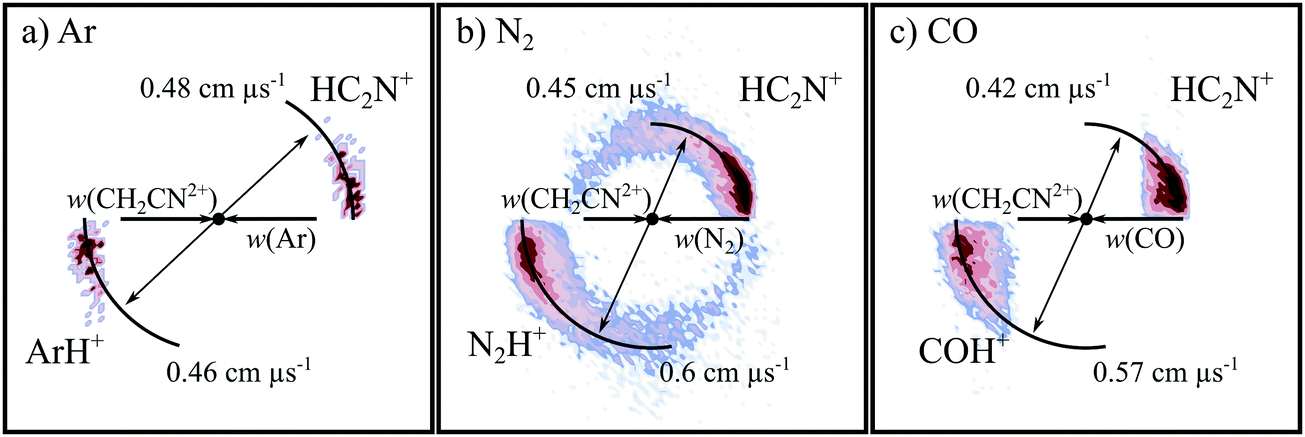 | ||
| Fig. 5 CM scattering diagrams for the PT channel CH2CN2+ + X → HC2N+ + XH+. (a) X = Ar at a CM collision energy of 5.0 eV, (b) X = N2 at a CM collision energy of 4.5 eV, and (c) X = CO at a CM collision energy of 4.3 eV. | ||
The experimental exoergicity distributions recorded for the PT channels are shown in Fig. 6. The distributions have similar shapes for all three collision systems, with maxima at ∼5 eV. In order to rationalise the experimental exoergicity spectra we again must consider the reactant and product electronic states that could be involved. We will again consider the six CH2CN2+ dication states obtained from our calculations (Table 4), and use the adiabatic ionization energy from our calculations of lowest energy structure of the HC2N+ monocation, together with the heat of formation from Nimlos et al.116 The energetics of the protonated species ArH+ and N2H+ are easily determined from proton affinities that are readily available, giving heats of formation of 12.0 eV and 10.7 eV respectively.104 CO can be protonated at either the carbon, forming OCH+, or the oxygen, forming COH+, giving heats of formation of 8.6 eV and 10.3 eV respectively.104
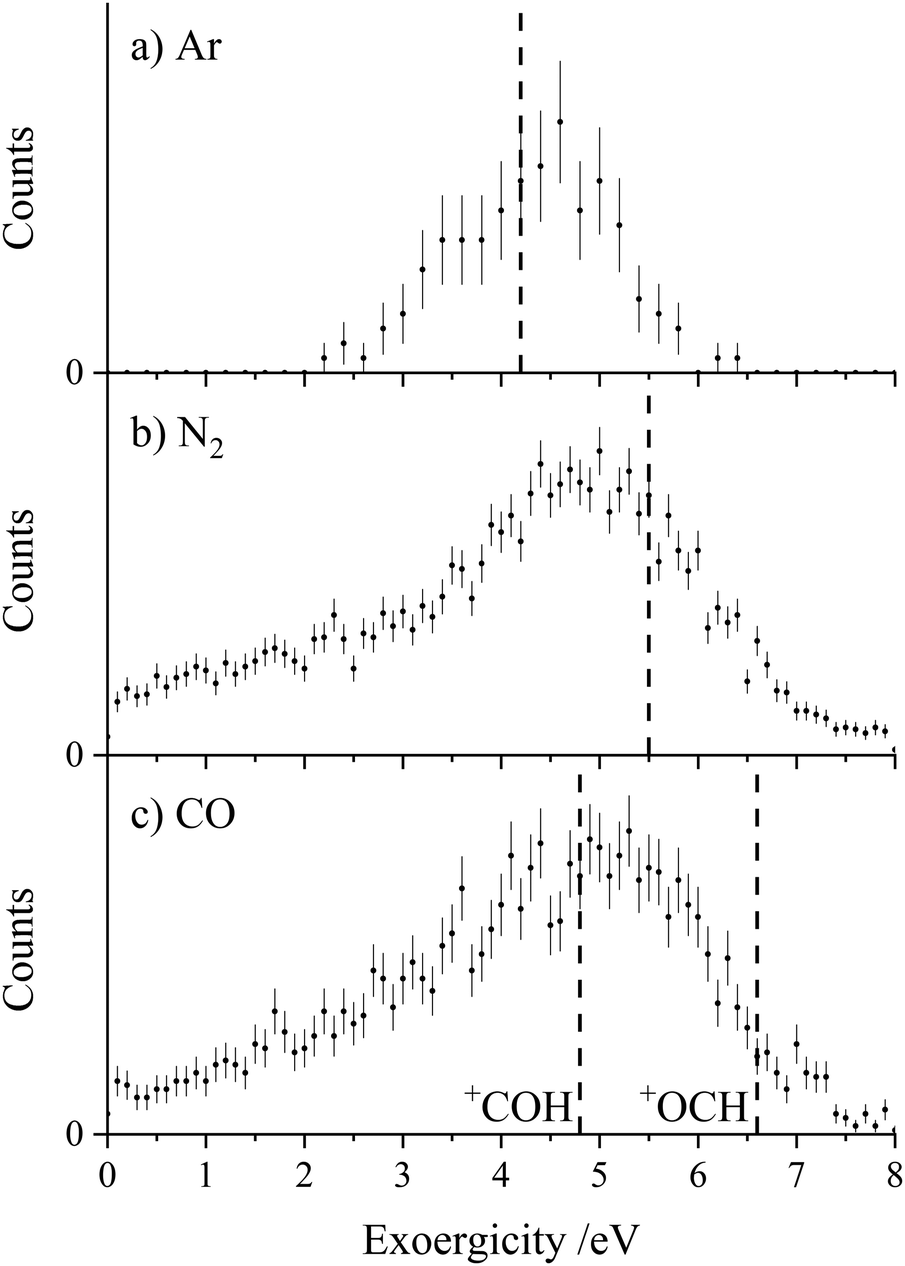 | ||
| Fig. 6 Experimental exoergicity distributions resulting from the PT reactions CH2CN2+ + X → HC2N+ + XH+. (a) X = Ar, (b) X = N2, (c) X = CO. Lines are marked on each distribution corresponding to the expected literature exoergicity from forming XH+ + HC2N+ in their ground states from the ground state dication, 2CD. For CO, there are two possible XH+ products, COH+ and OCH+. The error bars represent two standard deviations of the counts. | ||
Using these energetics, the literature exoergicities for the PT reactions involving the cyclic doublet dication 2CD, resulting in the formation of the XH+ and HC2N+ products in their ground states are 4.2 eV, 5.5 eV, 4.8 eV and 6.6 eV for the reactions with Ar, N2, CO (forming COH+) and CO (forming OCH+) respectively. These literature exoergicities are marked on the corresponding distributions in Fig. 6 and fit nicely with the experimental data. Of course, if the linear isocyanide dication structure was involved (2ID), similar exoergicities (∼0.4 eV higher) would result. Therefore, for simplicity, only 2CD is discussed below. Predicted literature exoergicities involving the quartet dication states are ∼3 eV higher than the literature exoergicities shown in Fig. 5, clearly outside of the range of the experimental exoergicity distributions. Therefore, the involvement of the quartet states in this PT channel is, at most, minor. As seen in Fig. 6c, the literature exoergicity predicted for the formation of COH+ is a better fit with the experimental exoergicity distribution than the formation of the lower energy OCH+ isomer.
If the reactant dication was vibrationally excited, this would act to increase the observed experimental exoergicity. Conversely, if the product HC2N+ or XH+ ions were formed with vibrational excitation, the observed experimental exoergicity would be lower. So given the likely broadening of the experimental distributions due to such vibrational excitation, the experimental data (Fig. 6) can clearly be accounted for with the involvement of the 2CD (and 2ID) dication geometries. The experimental exoergicity distributions (Fig. 6) are clearly broader for the molecular targets, N2 and CO. Such broadening, particularly to low exoergicities is likely due to the increased number of vibrational modes in the triatomic (N2H+, COH+) product ion facilitating vibrational excitation of the XH+ product in comparison with the diatomic ArH+.
To summarise, PT reactions occur in each of the CH2CN2+ + X (X = Ar, N2 and CO) systems, resulting in the formation of HC2N+ and XH+. The dynamics show that PT occurs via a direct, long-range, mechanism. The experimental exoergicity distributions reveal that the 2CD and 2ID dication states are the principal reactants, generating the monocationic products in their ground electronic states.
3.4. Non-dissociative single electron-transfer (NDSET) channels
Fig. 7 shows the CM scattering diagrams for the NDSET reactions CH2CN2+ + X → CH2CN+ + X+, for X = N2 and CO. Strong forward scattering is observed for both reactions. That is, the velocity of the CH2CN+ product ion is oriented with the velocity of the reactant dication, CH2CN2+. Such a scattering pattern is typical for NDSET reactions in dication-neutral systems and is indicative of a direct LZ electron-transfer process occurring at a significant interspecies separations.65,72,92,97,138 For the NDSET reaction with N2, in the coincidence spectrum only the forward scattered counts could be isolated from counts from the PT channel, hence the absence of signal at higher angles in the scattering diagram (Fig. 7a).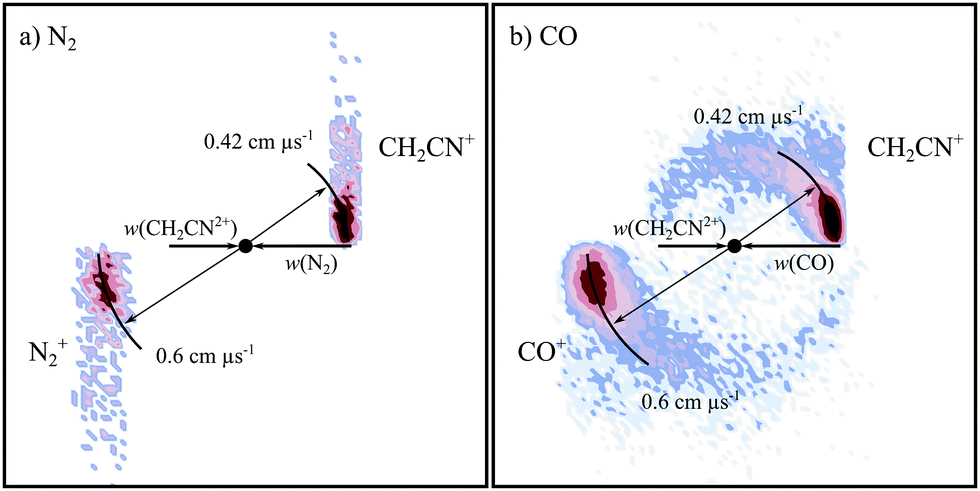 | ||
| Fig. 7 CM scattering diagrams for the NDSET channel CH2CN2+ + X → CH2CN+ + X+. (a) X = N2 at a CM collision energy of 4.5 eV (b) X = CO at a CM collision energy of 4.3 eV. | ||
Fig. 8 and 9 show the experimental exoergicity distributions recorded following the NDSET reactions of CH2CN2+ with N2 and CO respectively. As above, in order to rationalise the experimental exoergicity spectra we must first consider the reactant and product states that could be involved. Again, we consider the six electronic states of CH2CN2+ (Table 4). Calculations probing the structure of CH2CN+reveal a singlet and triplet state for the each of cyclic, linear, and linear isocyanide geometries (See Table 5): 1CM, 1LM, 1IM, 3CM, 3LM, and 3IM. The lowest energy CH2CN+ state is the singlet linear conformation (1LM). The ground state of N2+ (X2Σg+) is 15.58 eV higher in energy than the ground state of N2.106,139 The lowest energy dissociation asymptote of N2+ (N+(3P) + N(4S)) lies at ∼24.3 eV above ground state N2. The energy of this dissociation asymptote corresponds to the energy of N2+(C2Σu+v = 3).104,140 Photoionization studies of N2 show that N2+ states generated at an energy higher than 24.3 eV have dissociation lifetimes significantly less than the timescale of our experiment and therefore will not contribute to the observed N2+ counts in this channel.140–143 The ground state of CO+ (X2Σ+) lies 14.0 eV above CO(X1Σ+).144,145 The lowest energy dissociation asymptote of CO+, corresponding to C+(2P) + O(3P), lies at 22.4 eV above CO(X1Σ+).104 Photoionization investigations of CO also show that if CO+ is formed at an energy of greater than 22.4 eV, it will dissociate within the lifetime of the PSCO experiment and therefore will not contribute to the counts observed in this reaction channel.145–147
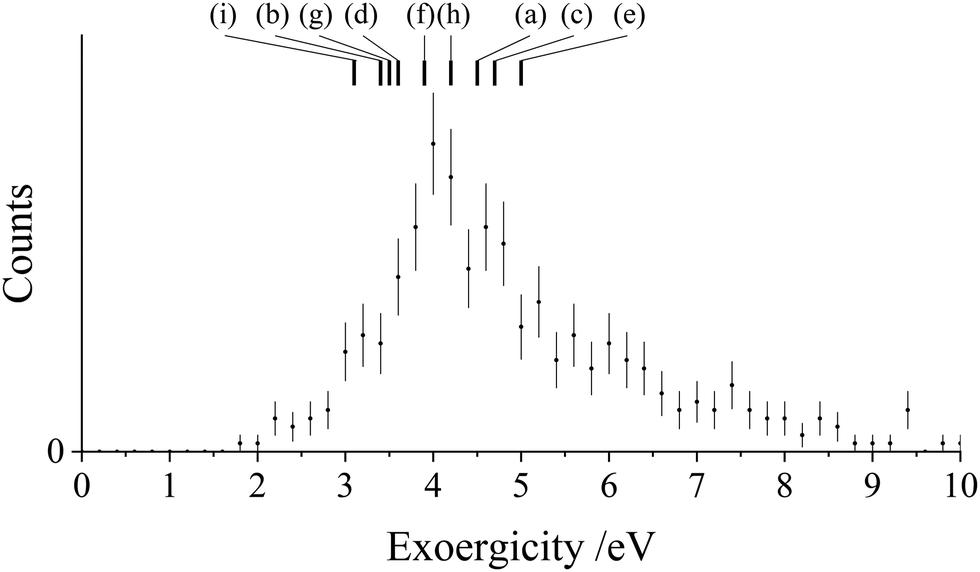 | ||
| Fig. 8 Experimental exoergicity distributions resulting from the NDSET reaction between CH2CN2+ + N2, resulting in the formation of CH2CN+ + N2+. The literature exoergicities resulting from pathways (a)–(i) are marked, see Table 6 for details. The error bars represent two standard deviations of the counts. | ||
To use the above energetic information to analyse the experimental exoergicity distributions and determine the electronic states involved in the NDSET channels we will also assume that the transitions will be spin allowed, and that the geometry of the dication will not change upon accepting an electron; for example, a cyclic dication must result in the formation of a cyclic monocation when it accepts an electron. Indeed, experimental work shows “vertical” transitions dominate dicationic electron transfer.
The experimental exoergicity distribution for the NDSET reaction with N2 (Fig. 8) has a maximum at 4.0 eV, with a FWHM from 3.6–5.2 eV. Using the energetics and assumptions presented above, we find that there are 9 pathways that have literature exoergicities that match the experimental data, which we list in Table 6. The exoergicities of these channels are also marked on Fig. 8. For completeness, we report all possible pathways which meet the above criteria (vertical and spin-allowed) in the ESI† (Table SI1). As shown in Table 6, the pathways which match the experimental exoergicity distribution involve all but the lowest energy dication state (2CD). Involvement of the 2CD dicationic ground state would result in an exoergicity below the bulk of the experimental distribution so therefore this dication state does not appear to contribute significantly to this channel.
| Pathway | Reactant CH2CN2+ state | Product CH2CN+ state populated | Product N2+ state populated | Literature exoergicity (eV) |
|---|---|---|---|---|
| (a) | 4CD | 3CM | X2Σg+ | 4.5 |
| (b) | 4CD | 3CM | A2Πu | 3.4 |
| (c) | 2LD | 1LM | X2Σg+ | 4.7 |
| (d) | 2LD | 1LM | A2Πu | 3.6 |
| (e) | 4LD | 3LM | X2Σg+ | 5.0 |
| (f) | 4LD | 3LM | A2Πu | 3.9 |
| (g) | 2ID | 1IM | X2Σg+ | 3.5 |
| (h) | 4ID | 3IM | X2Σg+ | 4.2 |
| (i) | 4ID | 3IM | A2Πu | 3.1 |
The experimental exoergicity distribution for the NDSET reaction with CO, shown in Fig. 9, has a maximum at 4.4 eV, with a FWHM from 3.4–5.5 eV. Using the energetic information above, and constrained by the same assumptions, we find that there are 12 pathways for the NDSET reaction between CH2CN2+ and CO that give literature exoergicities within the experimentally observed range (Table 7). These 12 pathways include all of the six reactant dication states.
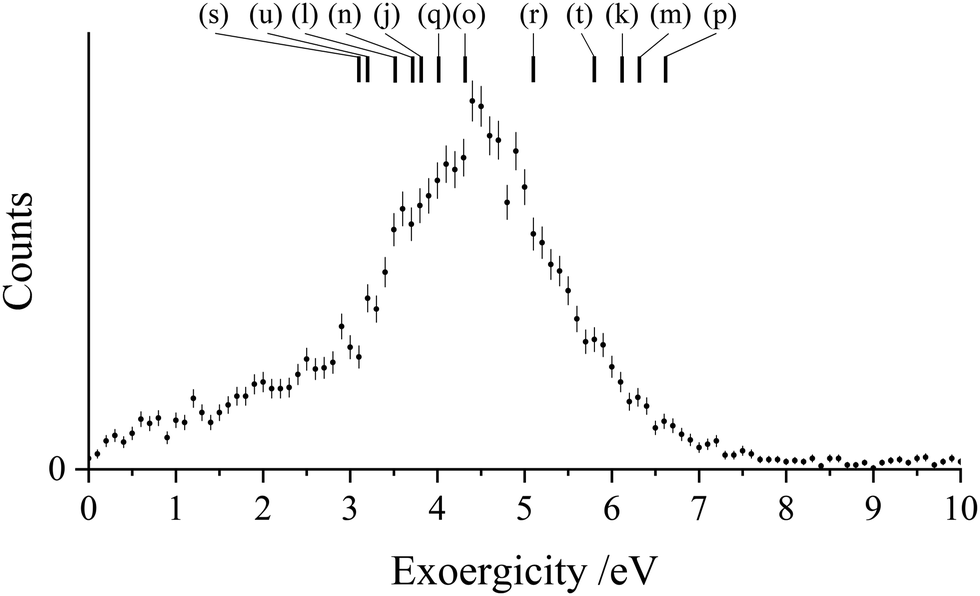 | ||
| Fig. 9 Experimental exoergicity distributions resulting from the NDSET reaction between CH2CN2+ + CO, resulting in the formation of CH2CN+ + CO+. The literature exoergicities resulting from pathways (j)–(u) are marked, see Table 7 for details. The error bars represent two standard deviations of the counts. | ||
| Pathway | Reactant CH2CN2+ state | Product CH2CN+ state populated | Product CO+ state populated | Literature exoergicity (eV) |
|---|---|---|---|---|
| (j) | 2CD | 1CM | X2Σ+ | 3.8 |
| (k) | 4CD | 3CM | X2Σ+ | 6.1 |
| (l) | 4CD | 3CM | A2Π | 3.5 |
| (m) | 2LD | 1LM | X2Σ+ | 6.3 |
| (n) | 2LD | 1LM | A2Π | 3.7 |
| (o) | 2LD | 3LM | X2Σ+ | 4.3 |
| (p) | 4LD | 3LM | X2Σ+ | 6.6 |
| (q) | 4LD | 3LM | A2Π | 4.0 |
| (r) | 2ID | 1IM | X2Σ+ | 5.1 |
| (s) | 2ID | 3IM | X2Σ+ | 3.1 |
| (t) | 4ID | 3IM | X2Σ+ | 5.8 |
| (u) | 4ID | 3IM | A2Π | 3.2 |
With N2 the NDSET channel has a relative intensity of 2.8 ± 0.1%, whilst with CO, NDSET is the most intense channel (52.4 ± 0.7%). However, for the CH2CN2+ + Ar system, there are no experimental signals associated with SET reactions occurring in the interaction region. This absence of a SET reaction with Ar is surprising as there are several possible pathways that would result in literature exoergicities that fall within the LZ reaction window. These available pathways involve all of the six metastable dication states revealed by our calculations. Hence there are no energetic grounds for the absence of NDSET reactivity following collisions of CH2CN2+ with Ar. The absence of any SET channels in the CH2CN2+ + Ar collision system, despite energetically accessible product channels, clearly points to a kinetic barrier in this pathway. Indeed, ‘Coulomb barriers’ on dication/neutral interaction potentials have been proposed to account for the stability of both molecular dications and dicationic collision complexes, preventing charge separation by the routes of CID and SET.148 In a dication-neutral collision system such a Coulomb barrier could impede access to the SET product asymptote. For the CH2CN2+ + Ar collision system, we see that SET is supressed, but PT is efficient. Thus, any barrier is clearly only significant in the SET exit channel. There is further experimental evidence supporting the existence of a kinetic barrier to the SET products. Specifically, we do observe signals, in the coincidence spectra, for SET between CH2CN2+ with Ar in collisions beyond the interaction region of our TOF-MS; when the dications are moving much faster and the collision energy is correspondingly higher.149 These higher collision energies clearly allow the barrier to SET in the collision system with Ar to be overcome. Indeed, despite the observation by Roithová et al.94 that the involvement of polar neutral collision partners increases the chance of PT reactions occurring, in the current study the CH2CN2+ + CO system exhibits the highest ratio of SET to PT reactivity. Clearly there are many subtleties in individual dication-neutral interaction potentials that can undermine global generalizations regarding dicationic reactivity.
In order to dissociate, accessing the C+ + O asymptote, CO+ must be formed with an energy of at least 22.4 eV relative to CO. Using the energies of the CH2CN2+ dication states and CH2CN+ monocation states we determined computationally, we see that forming CO+ at such an energy is possible but requires the formation of a singlet CH2CN+ state from a quartet CH2CN2+ state, which is nominally spin forbidden if forming a doublet state of CO+. This spin forbidden nature likely accounts for the very low intensity of this DSET channel. In order for CO+ to fragment to produce O+ + C, which we do not observe experimentally, CO+ must be formed with an energy at least 24.6 eV above that of neutral CO. Our calculated energetics show that it is not possible to form CO at such an energy from the CH2CN2+ and CH2CN+ states we believe to be involved in this collision system. Such agreement between the conclusions from our energetics and the experimental observations strongly supports the robustness of our computational conclusions concerning the accessible CH2CN2+ and CH2CN+ states.
4. Conclusions
In this work we have explored the reactivity of the molecular dication CH2CN2+ with Ar, N2, and CO. To the author's knowledge, this is the first study of the bimolecular reactivity of CH2CN2+, a species relevant to the ionospheres of planets and satellites including Titan. Calculations of the lowest energy electronic states of CH2CN2+ reveal six low-lying (and presumably metastable) bound electronic states. Specifically, we locate both doublet and quartet states for cyclic (2CD & 4CD), linear (2LD & 4LD), and linear isocyanide (2ID & 4ID) geometries. The lowest energy electronic state was determined to be the doublet cyclic dication, 2CD with an adiabatic double ionization energy of 28.9 eV.All of the collision systems we investigated display an intense collision induced dissociation (CID) channel resulting in the formation of C+ + CH2N+. This channel exhibits similar dynamics and experimental exoergicity distributions for all three collision systems. The formation of a C+ fragment suggests the involvement of the cyclic or linear isocyanide dication states, and the experimental exoergicity distributions confirm the involvement of the quartet cyclic or isocyanide (4CD, 4ID) dication states.
Each CH2CN2+ + neutral collision system also results in a CID reaction generating H+ + HC2N+. Again, the products exhibit similar dynamics and experimental exoergicity distributions in all three collision systems. The experimental exoergicity distributions primarily point to the involvement of the 2CD and 2ID dication states in this CID reaction, with 2LD also involved. Channels involving further dissociation of the HC2N+ product, forming H+ + CCN+, were also detected in the CH2CN2+ + N2/CO systems.
Proton transfer (PT) occurs in all three of the collision systems via a direct mechanism, giving further evidence that hydrogen-containing dications are effective proton donors. The experimental exoergicity distributions resulting from the PT reactions firmly point to the involvement of the 2CD or 2ID dication states as the reactants.
Finally, we observe reaction channels involving single-electron transfer (SET) following the collisions of CH2CN2+ with both N2 and CO. The experimental exoergicity distributions show that the non-dissociative SET channels could involve any of the dication states in the beam. There is no SET reactivity with Ar which we attribute, with experimental evidence, to an energy barrier restricting access to the appropriate product asymptote. In this work, SET channels are only more intense than PT processes in collisions with the CO molecule. These observations differ from the trends observed by Roithová et al.94 for reactions of halogenated dications.
Our experimental data indicates the reactivity we observe involves all three dication conformations (cyclic, linear, isocyanide) identified by our computational investigation of the accessible structures of CH2CN2+. This observation points towards considerable fluxionality in the structure of the low-lying metastable electronic states of CH2CN2+.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support of the EPSRC (EP/J010839/1), the Leverhulme Trust (RPG-2017-309), the Royal Society of Chemistry (E21-4679591105) and UCL. We also acknowledge valuable discussions with Dr Mike Parkes and Dr Lilian Ellis-Gibbings.References
- H. S. Bridge, J. W. Belcher, A. J. Lazarus, J. D. Sullivan, R. L. McNutt, F. Bagenal, J. D. Scudder, E. C. Sittler, G. L. Siscoe, V. M. Vasyliunas, C. K. Goertz and C. M. Yeates, Science, 1979, 204, 987–991 CrossRef CAS PubMed.
- S. Ghosh, K. K. Mahajan, J. M. Grebowsky and N. Nath, J. Geophys. Res., 1995, 100, 23983–23991 CrossRef CAS.
- J. H. Hoffman, C. Y. Johnson, J. C. Holmes and J. M. Young, J. Geophys. Res., 1969, 74, 6281–6290 CrossRef CAS.
- E. Dubinin, R. Modolo, M. Fraenz, J. Woch, G. Chanteur, F. Duru, F. Akalin, D. Gurnett, R. Lundin, S. Barabash, J. D. Winningham, R. Frahm, J. J. Plaut and G. Picardi, J. Geophys. Res.: Space Phys., 2008, 113, A10217 CrossRef.
- J. Lilensten, C. Simon, O. Witasse, O. Dutuit, R. Thissen and C. Alcaraz, Icarus, 2005, 174, 285–288 CrossRef CAS.
- L. A. Frank, W. R. Paterson, K. L. Ackerson, V. M. Vasyliunas, F. V. Coroniti and S. J. Bolton, Science, 1996, 274, 394–395 CrossRef CAS PubMed.
- H. Gu, J. Cui, D. Niu, L. Dai, J. Huang, X. Wu, Y. Hao and Y. Wei, Earth Planet. Phys., 2020, 4, 396–402 Search PubMed.
- C. Simon, J. Lilensten, O. Dutuit, R. Thissen, O. Witasse, C. Alcaraz and H. Soldi-Lose, Ann. Geophys., 2005, 23, 781–797 CrossRef CAS.
- O. Witasse, O. Dutuit, J. Lilensten, R. Thissen, J. Zabka, C. Alcaraz, P.-L. Blelly, S. W. Bougher, S. Engel, L. H. Andersen and K. Seiersen, Geophys. Res. Lett., 2002, 29, 104 CrossRef.
- J. Lilensten, O. Witasse, C. Simon, H. Soldi-Lose, O. Dutuit, R. Thissen and C. Alcaraz, Geophys. Res. Lett., 2005, 32, L03203 CrossRef.
- A. Beth, K. Altwegg, H. Balsiger, J.-J. Berthelier, M. R. Combi, J. De Keyser, B. Fiethe, S. A. Fuselier, M. Galand, T. I. Gombosi, M. Rubin and T. Sémon, Astron. Astrophys., 2020, 642, A27 CrossRef CAS.
- S. D. Price, J. D. Fletcher, F. E. Gossan and M. A. Parkes, Int. Rev. Phys. Chem., 2017, 36, 145–183 Search PubMed.
- R. Thissen, O. Witasse, O. Dutuit, C. S. Wedlund, G. Gronoff and J. Lilensten, Phys. Chem. Chem. Phys., 2011, 13, 18264–18287 RSC.
- D. Ascenzi, J. Aysina, E. L. Zins, D. Schröder, J. Žabka, C. Alcaraz, S. D. Price and J. Roithová, Phys. Chem. Chem. Phys., 2011, 13, 18330–18338 RSC.
- J. Roithová, H. Schwarz and D. Schröder, Chem. – Eur. J., 2009, 15, 9995–9999 CrossRef PubMed.
- W. Wolff, H. Luna, R. Schuch, N. D. Cariatore, S. Otranto, F. Turco, D. Fregenal, G. Bernardi and S. Suárez, Phys. Rev. A, 2016, 94, 022712 CrossRef.
- P. Bhatt, T. Sairam, A. Kumar, H. Kumar and C. P. Safvan, Phys. Rev. A, 2017, 96, 022710 CrossRef.
- R. Plašil, S. Rednyk, A. Kovalenko, T. D. Tran, Š. Roučka, P. Dohnal, O. Novotný and J. Glosík, Astrophys. J., 2021, 910, 155 CrossRef.
- O. Dutuit, N. Carrasco, R. Thissen, V. Vuitton, C. Alcaraz, P. Pernot, N. Balucani, P. Casavecchia, A. Canosa, S. Le Picard, J.-C. Loison, Z. Herman, J. Zabka, D. Ascenzi, P. Tosi, P. Franceschi, S. D. Price and P. Lavvas, Astrophys. J., Suppl. Ser., 2013, 204, 20 CrossRef.
- C. L. Ricketts, D. Schröder, C. Alcaraz and J. Roithová, Chem. – Eur. J., 2008, 14, 4779–4783 CrossRef CAS PubMed.
- E.-L. Zins and D. Schröder, J. Phys. Chem. A, 2010, 114, 5989–5996 CrossRef CAS PubMed.
- J. Roithová and D. Schröder, Chem. – Eur. J., 2007, 13, 2893–2902 CrossRef PubMed.
- J. Roithová and D. Schröder, Phys. Chem. Chem. Phys., 2007, 9, 731–738 RSC.
- H. Sabzyan, E. Keshavarz and Z. Noorisafa, J. Iran. Chem. Soc., 2014, 11, 871–945 CrossRef CAS.
- S. Falcinelli, F. Pirani, M. Alagia, L. Schio, R. Richter, S. Stranges, N. Balucani and F. Vecchiocattivi, Atmosphere, 2016, 7, 112 CrossRef.
- S. Falcinelli, M. Rosi, P. Candori, F. Vecchiocattivi, J. M. Farrar, F. Pirani, N. Balucani, M. Alagia, R. Richter and S. Stranges, Planet. Space Sci., 2014, 99, 149–157 CrossRef CAS.
- J. Lilensten, C. Simon Wedlund, M. Barthélémy, R. Thissen, D. Ehrenreich, G. Gronoff and O. Witasse, Icarus, 2013, 222, 169–187 CrossRef CAS.
- M. Alagia, N. Balucani, P. Candori, S. Falcinelli, F. Pirani, R. Richter, M. Rosi, S. Stranges and F. Vecchiocattivi, Rend. Lincei, 2013, 24, 53–65 CrossRef.
- R. L. Hudson and M. H. Moore, Icarus, 2004, 172, 466–478 CrossRef CAS.
- H. B. Niemann, S. K. Atreya, S. J. Bauer, G. R. Carignan, J. E. Demick, R. L. Frost, D. Gautier, J. A. Haberman, D. N. Harpold, D. M. Hunten, G. Israel, J. I. Lunine, W. T. Kasprzak, T. C. Owen, M. Paulkovich, F. Raulin, E. Raaen and S. H. Way, Nature, 2005, 438, 779–784 CrossRef CAS PubMed.
- M. A. Cordiner, M. Y. Palmer, C. A. Nixon, P. G. J. Irwin, N. A. Teanby, S. B. Charnley, M. J. Mumma, Z. Kisiel, J. Serigano, Y.-J. Kuan, Y.-L. Chuang and K.-S. Wang, Astrophys. J., 2015, 800, L14 CrossRef.
- A. Coustenis, B. Schmitt, R. K. Khanna and F. Trotta, Planet. Space Sci., 1999, 47, 1305–1329 CrossRef CAS.
- S. Hamm, G. Helas and P. Warneck, Geophys. Res. Lett., 1989, 16, 483–486 CrossRef CAS.
- H. Böhringer and F. Arnold, Nature, 1981, 290, 321–322 CrossRef.
- E. Murad, W. Swider, R. A. Moss and S. Toby, Geophys. Res. Lett., 1984, 11, 147–150 CrossRef CAS.
- P. M. Solomon, K. B. Jefeerts, A. A. Penzias and R. W. Wilson, Astrophys. J., 1971, 168, L107–L110 CrossRef CAS.
- B. E. Turner, P. Friberg, W. M. Irvine, S. Saito and S. Yamamoto, Astrophys. J., 1990, 355, 546 CrossRef CAS PubMed.
- M. A. Parkes, K. M. Douglas and S. D. Price, Int. J. Mass Spectrom., 2019, 438, 97–106 CrossRef CAS.
- C. Vastel, S. Yamamoto, B. Lefloch and R. Bachiller, Astron. Astrophys., 2015, 582, L3 CrossRef.
- M. Agúndez, J. P. Fonfría, J. Cernicharo, J. R. Pardo and M. Guélin, Astron. Astrophys., 2008, 479, 493–501 CrossRef.
- E. Herbst and C. M. Leung, Astron. Astrophys., 1990, 233, 177–180 CAS.
- A. D. Volosatova, M. A. Lukianova, P. V. Zasimov and V. I. Feldman, Phys. Chem. Chem. Phys., 2021, 23, 18449–18460 RSC.
- M. A. Zdanovskaia, P. M. Dorman, V. L. Orr, A. N. Owen, S. M. Kougias, B. J. Esselman, R. C. Woods and R. J. McMahon, J. Am. Chem. Soc., 2021, 143, 9551–9564 CrossRef CAS PubMed.
- D. F. Mark, F. M. Stuart and M. de Podesta, Geochim. Cosmochim. Acta, 2011, 75, 7494–7501 CrossRef CAS.
- A. G. W. Cameron, Space Sci. Rev., 1973, 15, 121–146 CrossRef CAS.
- S. A. Stern, Rev. Geophys., 1999, 37, 453–491 CrossRef CAS.
- A. O. Nier, W. B. Hanson, A. Seiff, M. B. McElroy, N. W. Spencer, R. J. Duckett, T. C. Knight and W. S. Cook, Science, 1976, 193, 786–788 CrossRef CAS PubMed.
- D. D. Bogard, R. N. Clayton, K. Marti, T. Owen and G. Turner, in Space Science Reviews, ed. R. Kallenbach, J. Geiss and W. K. Hartmann, Springer, Dordrecht, 2001, vol. 12, pp. 425–458 Search PubMed.
- D. F. Strobel, D. T. Hall, X. Zhu and M. E. Summers, Icarus, 1993, 103, 333–336 CrossRef.
- H. B. Niemann, S. K. Atreya, J. E. Demick, D. Gautier, J. A. Haberman, D. N. Harpold, W. T. Kasprzak, J. I. Lunine, T. C. Owen and F. Raulin, J. Geophys. Res., 2010, 115, E12006 CrossRef.
- O. Badr and S. D. Probert, Appl. Energy, 1993, 46, 1–67 CrossRef CAS.
- E. W. Schwieterman, T. D. Robinson, V. S. Meadows, A. Misra and S. Domagal-Goldman, Astrophys. J., 2015, 810, 57 CrossRef.
- D. Bockelée-Morvan, V. Debout, S. Erard, C. Leyrat, F. Capaccioni, G. Filacchione, N. Fougere, P. Drossart, G. Arnold, M. Combi, B. Schmitt, J. Crovisier, M.-C. de Sanctis, T. Encrenaz, E. Kührt, E. Palomba, F. W. Taylor, F. Tosi, G. Piccioni, U. Fink, G. Tozzi, A. Barucci, N. Biver, M.-T. Capria, M. Combes, W. Ip, M. Blecka, F. Henry, S. Jacquinod, J.-M. Reess, A. Semery and D. Tiphene, Astron. Astrophys., 2015, 583, A6 CrossRef.
- M. A. DiSanti, M. J. Mumma, N. Dello Russo and K. Magee-Sauer, Icarus, 2001, 153, 361–390 CrossRef CAS.
- M. J. Mumma, M. A. DiSanti, N. Dello Russo, M. Fomenkova, K. Magee-Sauer, C. D. Kaminski and D. X. Xie, Science, 1996, 272, 1310–1314 CrossRef CAS PubMed.
- E. B. Burgh, K. France and S. R. McCandliss, Astrophys. J., 2007, 658, 446–454 CrossRef CAS.
- D. C. B. Whittet and W. W. Duley, Astron. Astrophys. Rev., 1991, 2, 167–189 CrossRef.
- P. C. Novelli, L. P. Steele and P. P. Tans, J. Geophys. Res., 1992, 97, 20731–20750 CrossRef.
- A. C. Vandaele, A. Mahieux, S. Chamberlain, B. Ristic, S. Robert, I. R. Thomas, L. Trompet, V. Wilquet and J. L. Bertaux, Icarus, 2016, 272, 48–59 CrossRef CAS.
- D. O. Muhleman, G. L. Berge and R. T. Clancy, Science, 1984, 223, 393–396 CrossRef CAS PubMed.
- B. L. Lutz, C. De Bergh and T. Owen, Science, 1983, 220, 1374–1375 CrossRef CAS PubMed.
- B. Fleury, N. Carrasco, T. Gautier, A. Mahjoub, J. He, C. Szopa, E. Hadamcik, A. Buch and G. Cernogora, Icarus, 2014, 238, 221–229 CrossRef CAS.
- D. Smith, D. Grief and N. G. Adams, Int. J. Mass Spectrom. Ion Phys., 1979, 30, 271–283 CrossRef CAS.
- D. Smith, N. G. Adams, E. Alge, H. Villinger and W. Lindinger, J. Phys. B: At. Mol. Phys., 1980, 13, 2787–2799 CrossRef CAS.
- W.-P. Hu, S. M. Harper and S. D. Price, Mol. Phys., 2005, 103, 1809–1819 CrossRef CAS.
- M. Manning, S. D. Price and S. R. A. Leone, J. Chem. Phys., 1993, 99, 8695–8704 CrossRef CAS.
- S. D. Price, M. Manning and S. R. Leone, Chem. Phys. Lett., 1993, 214, 553–558 CrossRef CAS.
- J. F. Lockyear, K. Douglas, S. D. Price, M. Karwowska, K. J. Fijalkowski, W. Grochala, M. Remeš, J. Roithová and D. Schröder, J. Phys. Chem. Lett., 2010, 1, 358–362 CrossRef CAS.
- G. Dupeyrat, J. B. Marquette, B. R. Rowe and C. Rebrion, Int. J. Mass Spectrom. Ion Processes, 1991, 103, 149–156 CrossRef CAS.
- G. C. Shields and T. F. Moran, J. Phys. B: At. Mol. Phys., 1983, 16, 3591–3607 CrossRef CAS.
- E. Y. Kamber, P. Jonathan, A. G. Brenton and J. H. Beynon, J. Phys. B: At. Mol. Phys., 1987, 20, 4129–4142 CrossRef CAS.
- S. M. Harper, W.-P. Hu and S. D. Price, J. Phys. B: At., Mol. Opt. Phys., 2002, 35, 4409–4423 CrossRef CAS.
- H. R. Koslowski, H. Lebius, V. Staemmler, R. Fink, K. Wiesemann and B. A. Huber, J. Phys. B: At., Mol. Opt. Phys., 1991, 24, 5023–5034 CrossRef CAS.
- E. L. Zins and D. Schröder, Int. J. Mass Spectrom., 2011, 299, 53–58 CrossRef CAS.
- N. Tafadar, N. Kaltsoyannis and S. D. Price, Int. J. Mass Spectrom., 1999, 192, 205–214 CrossRef CAS.
- P. W. Burnside and S. D. Price, Int. J. Mass Spectrom., 2006, 249–250, 279–288 CrossRef.
- J. Roithová, R. Thissen, J. Žabka, P. Franceschi, O. Dutuit and Z. Herman, Int. J. Mass Spectrom., 2003, 228, 487–495 CrossRef.
- D. Ascenzi, P. Tosi, J. Roithová, C. L. Ricketts, D. Schröder, J. F. Lockyear, M. A. Parkes and S. D. Price, Phys. Chem. Chem. Phys., 2008, 10, 7121–7128 RSC.
- B. K. Chatterjee and R. Johnsen, J. Chem. Phys., 1989, 91, 1378–1379 CrossRef CAS.
- S. Armenta Butt and S. D. Price, Phys. Chem. Chem. Phys., 2021, 23, 11287–11299 RSC.
- H. Störi, E. Alge, H. Villinger, F. Egger and W. Lindinger, Int. J. Mass Spectrom. Ion Phys., 1979, 30, 263–270 CrossRef.
- H. M. Holzscheiter and D. A. Church, J. Chem. Phys., 1981, 74, 2313–2318 CrossRef CAS.
- P. Tosi, R. Correale, W. Lu, S. Falcinelli and D. Bassi, Phys. Rev. Lett., 1999, 82, 450–452 CrossRef CAS.
- A. Ehbrecht, N. Mustafa, C. Ottinger and Z. Herman, J. Chem. Phys., 1996, 105, 9833–9846 CrossRef CAS.
- J. H. Agee, J. B. Wilcox, L. E. Abbey and T. F. Moran, Chem. Phys., 1981, 61, 171–179 CrossRef CAS.
- S. M. Harper, S. W.-P. Hu and S. D. Price, J. Chem. Phys., 2004, 120, 7245–7248 CrossRef CAS PubMed.
- J. Jašik, J. Roithová, J. Žabka, R. Thissen, I. Ipolyi and Z. Herman, Int. J. Mass Spectrom., 2006, 255–256, 150–163 CrossRef.
- J. D. Fletcher, M. A. Parkes and S. D. Price, Mol. Phys., 2015, 113, 2125–2137 CrossRef CAS.
- P. W. Burnside and S. D. Price, Phys. Chem. Chem. Phys., 2007, 9, 3902–3913 RSC.
- P. Tosi, W. Lu, R. Correale and D. Bassi, Chem. Phys. Lett., 1999, 310, 180–182 CrossRef CAS.
- M. W. Wong and L. Radom, J. Phys. Chem., 1989, 93, 6303–6308 CrossRef CAS.
- Z. Herman, Int. Rev. Phys. Chem., 1996, 15, 299–324 Search PubMed.
- S. D. Price, J. Chem. Soc., Faraday Trans., 1997, 93, 2451–2460 RSC.
- J. Roithová, Z. Herman, D. Schröder and H. Schwarz, Chem. – Eur. J., 2006, 12, 2465–2471 CrossRef PubMed.
- W.-P. Hu, S. M. Harper and S. D. Price, Meas. Sci. Technol., 2002, 13, 1512–1522 CrossRef CAS.
- S. D. Price, Int. J. Mass Spectrom., 2007, 260, 1–19 CrossRef CAS.
- K. Yamasaki and S. R. Leone, J. Chem. Phys., 1989, 90, 964–976 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- L. K. Ellis-Gibbings, W. G. Fortune, B. Cooper, J. Tennyson and S. D. Price, Phys. Chem. Chem. Phys., 2021, 23, 11424–11437 RSC.
- H. G. Cho and L. Andrews, J. Phys. Chem. A, 2011, 115, 8638–8642 CrossRef CAS PubMed.
- W. Wolff, A. Perlin, R. R. Oliveira, F. Fantuzzi, L. H. Coutinho, F. D. A. Ribeiro and G. Hilgers, J. Phys. Chem. A, 2020, 124, 9261–9271 CrossRef CAS PubMed.
- S. Anand and H. B. Schlegel, J. Phys. Chem. A, 2005, 109, 11551–11559 CrossRef CAS PubMed.
- J. Jašík, D. Gerlich and J. Roithová, J. Am. Chem. Soc., 2014, 136, 2960–2962 CrossRef PubMed.
- NIST Atomic Spectra Database (version 5.6.1), ed. A. Kramida, Y. Ralchenko, J. Reader and NIST ASD Team, National Institute of Standards and Technology, Gaithersburg, MD, 2020 Search PubMed.
- M. Lundqvist, D. Edvardsson, P. Baltzer and B. Wannberg, J. Phys. B: At., Mol. Opt. Phys., 1996, 29, 1489–1499 CrossRef CAS.
- A. J. Yencha, K. Ellis and G. C. King, J. Electron Spectrosc. Relat. Phenom., 2014, 195, 160–173 CrossRef CAS.
- M. Hochlaf, R. I. Hall, F. Penent, H. Kjeldsen, P. Lablanquie, M. Lavollée and J. H. D. Eland, Chem. Phys., 1996, 207, 159–165 CrossRef CAS.
- L. H. Andersen, J. H. Posthumus, O. Vahtras, H. Agren, N. Elander, A. Nunez, A. Scrinzi, M. Natiello and M. Larsson, Phys. Rev. Lett., 1993, 71, 1812–1815 CrossRef CAS PubMed.
- F. Penent, R. I. Hall, R. Panajotović, J. H. D. Eland, G. Chaplier and P. Lablanquie, Phys. Rev. Lett., 1998, 81, 3619–3622 CrossRef CAS.
- J. H. D. Eland, M. Hochlaf, G. C. King, P. S. Kreynin, R. J. LeRoy, I. R. McNab and J.-M. Robbe, J. Phys. B: At., Mol. Opt. Phys., 2004, 37, 3197–3214 CrossRef CAS.
- M. Lundqvist, P. Baltzer, D. Edvardsson, L. Karlsson and B. Wannberg, Phys. Rev. Lett., 1995, 75, 1058–1061 CrossRef CAS PubMed.
- B. Gans, S. Hartweg, G. A. Garcia, S. Boyé-Péronne, O. J. Harper, J. C. Guillemin and J. C. Loison, Phys. Chem. Chem. Phys., 2020, 22, 12496–12501 RSC.
- R. P. Thorn, P. S. Monks, L. J. Steif, S. C. Kuo, Z. Zhang, S. K. Ross and R. B. Klemm, J. Phys. Chem. A, 1998, 102, 846–851 CrossRef CAS.
- G. A. Garcia, J. Krüger, B. Gans, C. Falvo, L. H. Coudert and J. C. Loison, J. Chem. Phys., 2017, 147, 13908 CrossRef PubMed.
- H. B. A. Cerqueira, J. C. Santos, F. Fantuzzi, F. D. A. Ribeiro, M. L. M. Rocco, R. R. Oliveira and A. B. Rocha, J. Phys. Chem. A, 2020, 124, 6845–6855 CrossRef CAS PubMed.
- M. R. Nimlos, G. Davico, C. M. Geise, P. G. Wenthold, W. C. Lineberger, S. J. Blanksby, C. M. Hadad, G. A. Petersson and G. B. Ellison, J. Chem. Phys., 2002, 117, 4323–4339 CrossRef CAS.
- D. B. Milligan, C. G. Freeman, R. G. A. R. Maclagan, M. J. McEwan, P. F. Wilson and V. G. Anicich, J. Am. Soc. Mass Spectrom., 2001, 12, 557–564 CrossRef CAS PubMed.
- V. Vuitton, R. V. Yelle and M. J. McEwan, Icarus, 2007, 191, 722–742 CrossRef.
- C. N. Keller, V. G. Anicich and T. E. Cravens, Planet. Space Sci., 1998, 46, 1157–1174 CrossRef CAS.
- M. W. Chase Jr., NIST-JANAF Themochemical Tables, J. Phys. C, 4th edn, 1998, pp. 1–1951 Search PubMed.
- E. P. L. Hunter and S. G. Lias, J. Phys. Chem. Ref. Data, 2009, 27, 413–656 CrossRef.
- J. Berkowitz, G. B. Ellison and D. Gutman, J. Phys. Chem., 1994, 98, 2744–2765 CrossRef CAS.
- D. F. McMillen and D. M. Golden, Annu. Rev. Phys. Chem., 1982, 33, 493–532 CrossRef CAS.
- J. L. Holmes and P. M. Mayer, J. Phys. Chem., 1995, 99, 1366–1370 CrossRef CAS.
- P. Schilke, D. A. Neufeld, H. S. P. Müller, C. Comito, E. A. Bergin, D. C. Lis, M. Gerin, J. H. Black, M. Wolfire, N. Indriolo, J. C. Pearson, K. M. Menten, B. Winkel, Á. Sánchez-Monge, T. Möller, B. Godard and E. Falgarone, Astron. Astrophys., 2014, 566, 29 CrossRef.
- M. J. Barlow, B. M. Swinyard, P. J. Owen, J. Cernicharo, H. L. Gomez, R. J. Ivison, O. Krause, T. L. Lim, M. Matsuura, S. Miller, G. Olofsson and E. T. Polehampton, Science, 2013, 342, 1343–1345 CrossRef CAS PubMed.
- H. S. P. Müller, S. Muller, P. Schilke, E. A. Bergin, J. H. Black, M. Gerin, D. C. Lis, D. A. Neufeld and S. Suri, Astron. Astrophys., 2015, 582, 4 CrossRef.
- F. Daniel, M. L. Dubernet, M. Meuwly, J. Cernicharo and L. Pagani, Mon. Not. R. Astron. Soc., 2005, 363, 1083–1091 CrossRef CAS.
- V. N. Salinas, M. R. Hogerheijde, E. A. Bergin, L. Ilsedore Cleeves, C. Brinch, G. A. Blake, D. C. Lis, G. J. Melnick, O. Panic, J. C. Pearson, L. Kristensen, U. A. Ylldlz and E. F. Van, Dishoeck, Astron. Astrophys., 2016, 591, 122 CrossRef.
- V. Brites and M. Hochlaf, J. Phys. Chem. A, 2009, 113, 11107–11111 CrossRef CAS PubMed.
- J. L. Fox and R. V. Yelle, NASA Contractor Report, 1996, NASA-CR-200657.
- E. Herbst, J. M. Norbeck, P. R. Certain and W. Klemperer, Astrophys. J., 1976, 207, 110–112 CrossRef CAS.
- J. R. Najita, J. S. Carr, S. E. Strom, D. M. Watson, I. Pascucci, D. Hollenbach, U. Gorti and L. Keller, Astrophys. J., 2010, 712, 274–286 CrossRef CAS.
- Z. Herman, Int. J. Mass Spectrom., 2015, 378, 113–126 CrossRef CAS.
- M. A. Parkes, J. F. Lockyear and S. D. Price, Int. J. Mass Spectrom., 2014, 365–366, 68–74 CrossRef CAS.
- C. L. Ricketts, S. M. Harper, S. W. P. Hu and S. D. Price, J. Chem. Phys., 2005, 123, 134322 CrossRef PubMed.
- J. Roithová, J. Žabka, J. Hrušák, R. Thissen and Z. Herman, J. Phys. Chem. A, 2003, 107, 7347–7354 CrossRef.
- S. Armenta Butt and S. D. Price, Phys. Chem. Chem. Phys., 2020, 22, 8391–8400 RSC.
- P. Baltzer, M. Larsson, L. Karlsson, B. Wannberg and M. Carlsson Göthe, Phys. Rev. A: At., Mol., Opt. Phys., 1992, 46, 5545–5553 CrossRef CAS PubMed.
- H. R. Hrodmarsson, R. Thissen, D. Dowek, G. A. Garcia, L. Nahon and T. R. Govers, Front. Chem., 2019, 7, 222 CrossRef CAS PubMed.
- E. M. Bahati, J. J. Jureta, D. S. Belic, H. Cherkani-Hassani, M. O. Abdellahi and P. Defrance, J. Phys. B: At., Mol. Opt. Phys., 2001, 34, 2963–2973 CrossRef CAS.
- X. Tang, Y. Hou, C. Y. Ng and B. Ruscic, J. Chem. Phys., 2005, 123, 074330 CrossRef PubMed.
- L. Åsbrink and C. Fridh, Phys. Scr., 1974, 9, 338–340 CrossRef.
- B. Wannberg, D. Nordfors, K. L. Tan, L. Karlsson and L. Mattsson, J. Electron Spectrosc. Relat. Phenom., 1988, 47, 147–166 CrossRef CAS.
- Y. Zhao, M. Cao, Y. Li, X. Shan, F. Liu, L. Sheng, L. Li and W. Liu, J. Electron Spectrosc. Relat. Phenom, 2014, 196, 181–186 CrossRef CAS.
- P. Baltzer, M. Lundqvist, B. Wannberg, L. Karlsson, M. Larsson, M. A. Hayes, J. B. West, M. R. F. Siggel, A. C. Parr and J. L. Dehmer, J. Phys. B: At., Mol. Opt. Phys., 1994, 27, 4915–4932 CrossRef CAS.
- R. C. Shiell, M. Evans, S. Stimson, C.-W. Hsu, C. Y. Ng and J. W. Hepburn, Chem. Phys. Lett., 1999, 315, 390–396 CrossRef CAS.
- J. Roithová and D. Schröder, Angew. Chem., Int. Ed., 2009, 48, 8788–8790 CrossRef PubMed.
- J. F. Lockyear, PhD thesis, UCL, 2011.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cp01523d |
| This journal is © the Owner Societies 2022 |