
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-nanocluster science and technology: my personal history and outlook
Yuichi
Negishi
 ab
ab
aDepartment of Applied Chemistry, Faculty of Science, Tokyo University of Science, Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan
bResearch Institute for Science and Technology, Tokyo University of Science, Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: negishi@rs.tus.ac.jp
First published on 27th January 2022
Abstract
Metal nanoclusters (NCs) are one of the leading targets in research of nanoscale materials, and elucidation of their properties (science) and development of control techniques (technology) have been continuously studied for the past 60 years or so. I have been continuously studying metal NCs since 1995 while changing my own interests, and thereby research topics, according to development of the field and the global situation. Specifically, I started my research on gas-phase metal NCs, and then investigated systematic isolation of glutathionate-protected gold NCs. Thereafter, I worked on development of flexible and precise techniques for controlling ligand-protected metal NCs and creation of highly functional energy and environmental catalysts using ligand-protected metal NCs. This personal account summarizes my previous studies on metal NCs. Through this personal history, I would like to describe my motivation for each previous subject, what type of research I want to perform for each subject in the future, and how I consider the outlook of the field.
 Yuichi Negishi | Yuichi Negishi is a Professor in the Department of Applied Chemistry at Tokyo University of Science. He received his PhD degree in Chemistry in 2001 under the supervision of Prof. Atsushi Nakajima at Keio University. Before joining Tokyo University of Science in 2008, he was employed as an Assistant Professor at Keio University (with Associate Prof. Atsushi Nakajima) and at the Institute for Molecular Science (with Associate Prof. Tatsuya Tsukuda). His current research interests include the precise synthesis of stable and functionalized metal nanoclusters and their applications in energy and environmental materials. |
1. Introduction
Research on nanoscale materials is generally believed to have begun with a talk by Feynman in 1959 entitled “There's Plenty of Room at the Bottom”.1 In this talk, he stated that in the nanoscale region of a few atoms, properties that are completely different from the macroscopic world appear. He also claimed that it is very important for humanity to conduct research in this area, and he predicted that in the future we would be able to arrange atoms one by one as we wish to create desired materials. Later, high-resolution analysis devices, such as scanning tunneling microscopes2 and atomic force microscopes,3 were developed, which made it possible to directly observe atoms on a surface. In addition, instruments such as lasers and molecular beams4,5 have been successively developed, and much knowledge has been gained about the stability and electronic/geometric structure of aggregates consisting of metal atoms in a vacuum, namely, gas-phase metal nanoclusters (NCs). Since 2000, many countries have adopted nanotechnology as a national policy,6 which has led to dramatic advancement in the technology and knowledge related to these materials, as well as a dramatic increase in the number of researchers involved in research of nanoscale materials. Through this knowledge, it is now possible to synthesize metal NCs consisting of certain metal elements with atomic precision.7–27 In addition, through these studies, it has been experimentally demonstrated that in the nanoscale region, novel electronic/geometric structures and physicochemical properties different from those of ordinary metals actually emerge. In recent years, a number of studies on creation of new materials that take advantage of the unique properties of such nanoscale materials have been reported. Thus, with regard to nanoscale materials, both elucidation of their properties (science) and development of control techniques (technology) have made great progress in the past 60 years or so.28–38I have been continuously studying metal NCs since 1995 (Fig. 1). In this time, I have witnessed dramatic progress of the “science” and “technology” in related fields. In response to development of these fields and the state of the world, I have changed my own interests and research themes many times. Specifically, I started my research on gas-phase metal NCs (Fig. 1(a)),39–41 and then investigated systematic isolation of thiolate (SR)-protected gold (Au) NCs (Aun(SR)m; Fig. 1(b)).42–47 Since 2008, I have been working on development of flexible and precise control techniques for ligand-protected metal NCs (Fig. 1(c)) and their use in creation of highly functional energy and environment catalysts (Fig. 1(d)).48 I have already written several accounts49–57 and reviews58–66 on each research topic. Therefore, in this personal account, I will describe my personal history in metal NCs. Through this personal history, I will explain what has led me to perform each type of research, what type of research I would like to perform in the future, and what I consider to be the future prospects in the field.
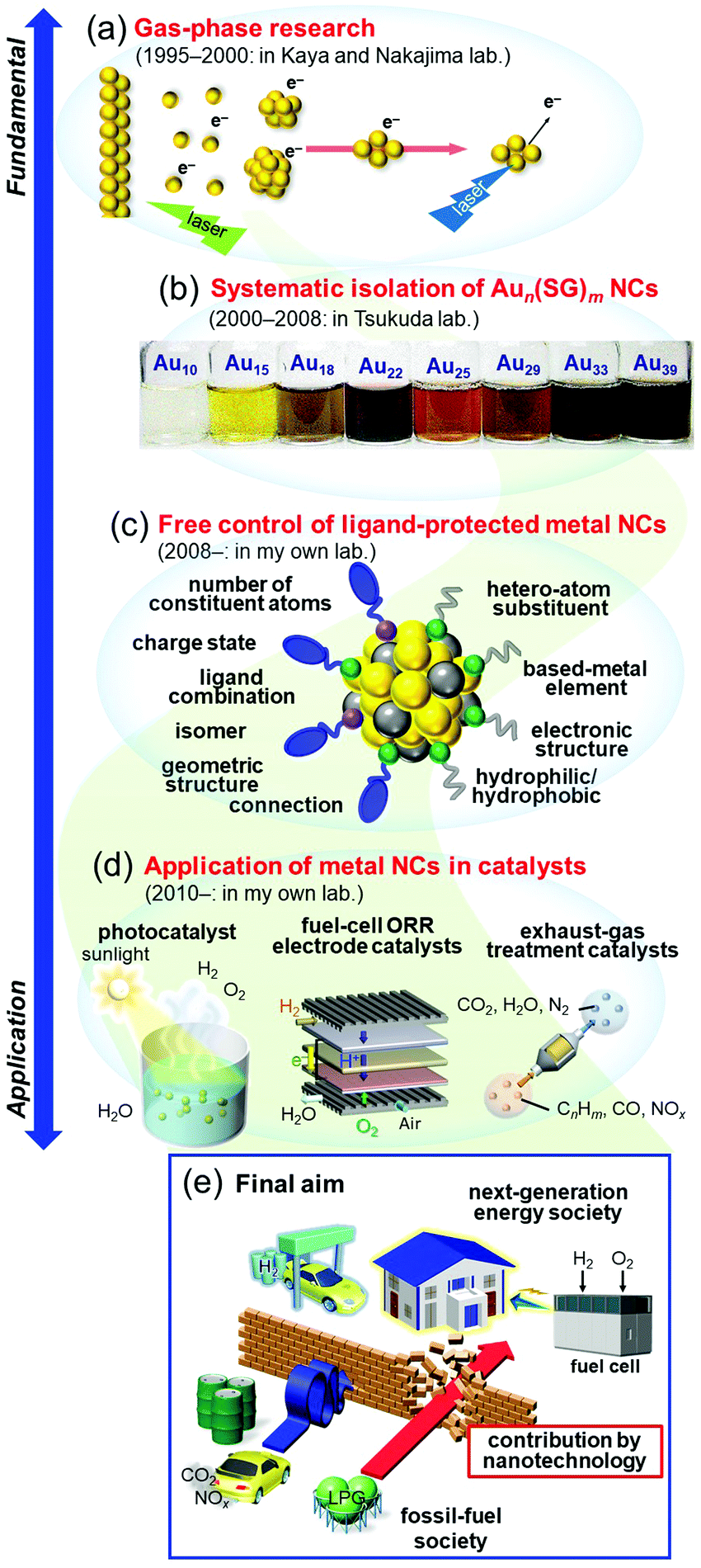 | ||
| Fig. 1 (a–d) History of my research on NCs: (a) gas-phase research, (b) systematic isolation of glutathionate-protected Au NCs, (c) development of flexible and precise control techniques for ligand-protected metal NCs, and (d) application of metal NCs to energy and environmental catalysts. (e) Final aim of my research and what I would like to contribute through my studies. In this figure, LPG indicates the liquefied petroleum gas. | ||
2. Gas-phase research
Isolation of metal NCs in a vacuum where there is no interaction with the outside world and investigation of their formation distribution, structure, and physical properties can provide an essential and fundamental understanding of the origin of the stability and mechanism of the size-specific structures and properties of metal NCs. Investigation of isolated metal NCs and supported metal NCs in the gas phase has a great advantage in this respect, and it has therefore been carried out for many years, with various improvements in the equipment.67–80I conducted research on gas-phase NCs from 1995 to 2000 (Fig. 1(a)) under the guidance of Professor Kaya and Associate Professor Nakajima. In this research, NCs were produced by laser vaporization4,5 and their electronic structures were investigated by negative-ion photoelectron spectroscopy (Fig. 2(a)).81 This research revealed that in the bulk, group 14 elements are classified as semiconductors for silicon (Si) and germanium (Ge), semi-metals for tin (Sn), and metals for lead (Pb), whereas in the fine size range all NCs have highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) gaps and overall similar electronic structures (Fig. 2(b)).40,82,83 In a study of Au–silver (Ag) alloy NCs, we experimentally demonstrated that not only controlling the number of constituent atoms, but also mixing the different elements is very effective for creating a new electronic structure (Fig. 2(c)).84 Through this research, I confirmed that new materials can be created by controlling the number of constituent atoms and chemical composition of NCs with atomic precision.
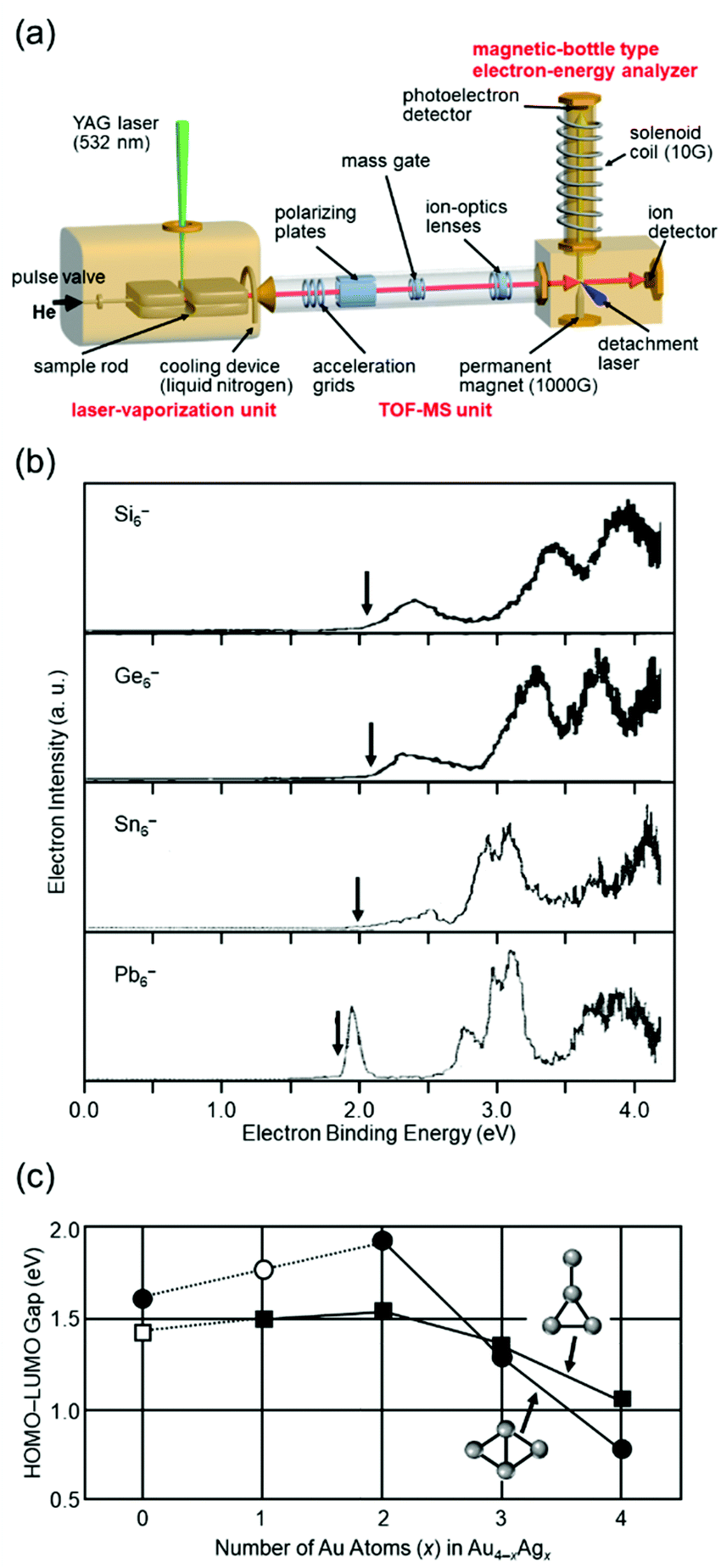 | ||
| Fig. 2 (a) Schematic illustration of the apparatus used in my research on gas-phase NCs. (b) Photoelectron spectra of Si6−, Ge6−, Sn6−, and Pb6− obtained by irradiating a 266 nm laser as a detachment laser. The vertical arrows indicate the thresholds energies.82,83 (c) Dependence of the HOMO–LUMO gaps of the rhombic and “T-form” isomers on the number of the Ag atoms (x) for Au4−xAgx (x = 0–4). The solid circles are the rhombic isomers and the solid squares are the T-form isomers.84 Reproduced with permission from ref. 82–84. Copyright 1997 Elsevier Science B.V., 2000 Elsevier Science B. V., and 2001 American Institute of Physics. | ||
However, through this research, I also strongly felt that it is essential to develop a large-scale synthesis method for NCs to create new materials that take advantage of their size-specific properties while understanding their fundamental properties. In addition, at this time, I got the chance to deposit germanium oxide (GenOm) NCs generated in a vacuum on a support and investigate their luminescence properties in air.85 Because the GenOm NCs deposited at that time were originally oxides, their electronic structure did not significantly change even when exposed to air, and they exhibited blue luminescence in air. Through this experiment, I was impressed that it was essential to develop NCs that are stable in air to create NCs that can be used as materials.
3. Isolation of glutathionate-protected Au NCs
While my interest was shifting from gas-phase experiments to materials chemistry research, in 2000, I got the chance to work as an Assistant Professor in the laboratory of Associate Professor Tsukuda, who had just established his own laboratory at the Institute for Molecular Science. Tsukuda also originally studied gas-phase NCs, but he started to work on liquid-phase synthesis of metal NCs at the end of his previous position.86 After joining his laboratory, we worked together to develop a precise synthesis method for metal NC in solution (Fig. 1(b)). These efforts were not immediately successful, and it took several years to achieve the first isolation. After a series of selections87 and abandonments of various material systems, we finally arrived at Aun(SG)m NCs in 2003.42,88Au and SR form strong bonds, and thereby SR forms self-assembled monolayers on Au surfaces.89–91 For these reasons, many technologies that take advantage of the combination of Au and SR have been developed in nanotechnology, such as dip-pen nanolithography92,93 and microcontact printing.94 In 1994, Brust et al.95 reported that combination of Au and SR is also effective for formation of metal NCs. They found that Aun(SC12)m NCs (SC12 = dodecanethiolate) can be formed by reduction of Au salts with sodium borohydride in the presence of dodecanethiol in solution. The Aun(SC12)m NCs reported at this time had a particle-size distribution of 1–3 nm. Regarding metal NCs, synthesis of metal NCs using phosphine (PR3), carbon monoxide (CO), and halogens as ligands, such as Aun(PR3)m(Cl)l, Ptn(PR3)m(CO)l (Pt = platinum) and Pdn(PR3)m(CO)l (Pd = palladium), had been reported before the report of Aun(SC12)m NCs. Those had been isolated with atomic precision, and their geometric structures had been determined by single-crystal X-ray diffraction (SC-XRD).23,96 However, the number of constituent atoms of Aun(SC12)m NCs could not be controlled at atomic precision at this stage. Because Aun(SC12)m NCs are easier to synthesize and more stable in air than Aun(PR3)m(Cl)l, Ptn(PR3)m(CO)l, and Pdn(PR3)m(CO)l NCs, Aun(SR)m NCs were easy for researchers who had not previously worked with NCs to investigate. Although the number of researchers working on metal NCs dramatically increased after 2000 when many countries had adopted nanotechnology as a national policy, this was probably the reason why the number of researchers working on Aun(SR)m NCs particularly increased.
Whetten and co-workers97 and Murray and co-workers98 had worked on high-resolution separation of Aun(SR)m NCs since the 1990s. Among their studies, the work on Aun(SG)m NCs (SG = glutathionate) reported by Whetten and co-workers in 199899 and 2000100 deserves special mention. They successfully separated a series of Aun(SG)m NCs at high resolution by polyacrylamide gel electrophoresis (PAGE, Fig. 3(a)) and reported that the chemical composition of the main product was Au28(SG)16.99
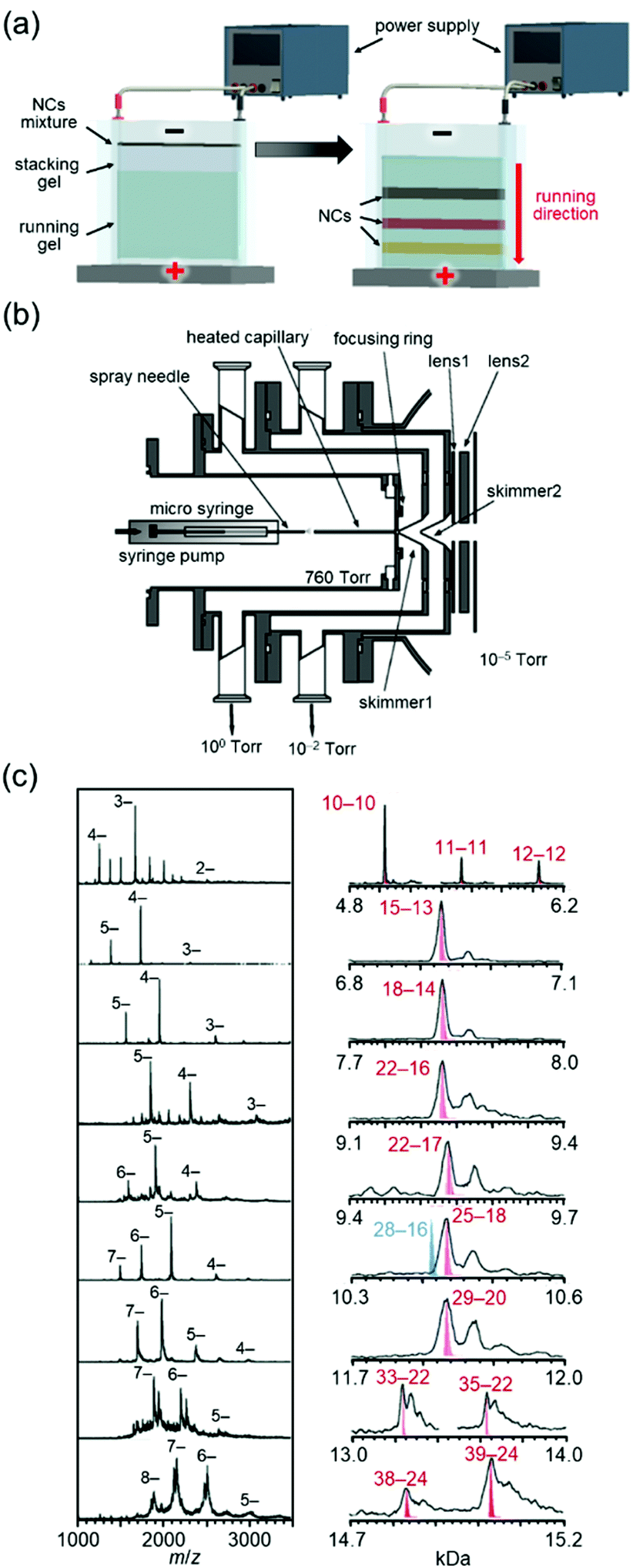 | ||
| Fig. 3 (a) Schematic illustration of PAGE separation techniques.61 (b) Details of the ESI source. (c) Negative-ion ESI mass spectrum of each fraction separated by PAGE.102 In right figure, Aun(SG)m is described as n–m. Reproduced with permission from ref. 61 and 102. Copyright 2020 The Royal Society of Chemistry and 2005 American Chemical Society. | ||
After these reports, Tsukuda and I started to work on Aun(SG)m NCs, where our first aim was to determine the chemical compositions of Aun(SG)m NCs other than the main product by mass spectrometry (MS). We had experienced development of the vacuum apparatus at our former poisitions.101 Therefore, we developed an electrospray ionization (ESI) unit that could deliver Aun(SG)m NCs synthesized in solution to a mass spectrometer without dissociation (Fig. 3(b)).102 Using a mass spectrometer equipped with the original ESI unit, we succeeded in obtaining the ESI-MS spectra of all of the Aun(SG)m NCs separated by PAGE. In a paper published in 2004, we assigned the Aun(SG)m NCs to Au18(SG)11, Au21(SG)12, Au25±1(SG)14±1, Au28(SG)16, Au32(SG)18, and Au39(SG)23.103 However, further studies with modification of sample purification and the MS sections revealed that the chemical compositions reported in this paper were incorrect. Therefore, the chemical compositions of the series of Aun(SG)m NCs were corrected to Au10–12(SG)10–12, Au15(SG)13, Au18(SG)14, Au22(SG)16, Au22(SG)17, Au25(SG)18, Au29(SG)20, Au33(SG)22, and Au39(SG)24 in a paper published in 2005 (Fig. 3(c)).102 Note that the chemical composition of the stable Au25(SR)18 NC46,105 was determined for the first time in this work. Determination of the chemical compositions of a series of Au NCs was also achieved for the Au NCs protected by mercaptosulfuric acid (S(SA)) or N-(2-mercaptopropionyl)glycine (S(SG)).104 Through these studies, we succeeded in demonstrating that Aun(SR)m NCs can be treated as compounds with defined chemical compositions, similar to organic and complex compounds. Research of Aun(SR)m NCs has provided a deep understanding of the correlation between the chemical composition, stability, electronic structure, and luminescence properties of Aun(SR)m NCs.43,45,103–106
4. Toward development of the ultimate nanotechnology and a deep understanding of ligand-protected metal NCs
After reporting Aun(SG)m NCs, I got the opportunity to run my own laboratory at Tokyo University of Science in 2008. Therefore, after 2008, I began new projects. I had the feeling that atomic control of ligand-protected metal NCs would be the ultimate nanotechnology for metal–molecule complex systems. However, the NCs that we could control at my previous institute were limited to small Aun(SR)m NCs (n = 10–39) protected by certain SRs (SG, S(SA), and S(SG)). To develop the ultimate nanotechnology for metal–molecule complex systems and thereby gain a better understanding of the correlation between the chemical composition, structure, and physical properties for ligand-protected metal NCs, it was necessary to develop new precise synthesis methods for (1) Au NCs with a large number of constituent atoms, (2) alloy NCs composed of several metal elements, (3) metal NCs composed of other elements, and (4) metal NCs protected by other ligands. According to the report by Teo et al.,107 certain metal NCs could become the constituent units of “clusters of clusters”. To create new materials using such connections in the future, it was necessary to also make further progress in (5) the connection techniques of ligand-protected metal NCs.4.1. Our strategy for flexible and precise control of ligand-protected metal NCs
Since 2008, we have been working in parallel to develop the following six techniques: (1) isolation of Au NCs with a large number of constituent atoms108,109 (Section 4.2 and Fig. 4(a)), (2) isolation of alloy NCs110–117 (Section 4.3 and Fig. 4(b)), (3) isolation of metal NCs consisting of Ag or Pt118–121 (Section 4.4 and Fig. 4(c)), (4) isolation of metal NCs protected by other ligands122–128 (Section 4.5 and Fig. 4(d)), (5) connection of metal NCs129–131 (Section 4.6 and Fig. 4(e)), and (6) high-resolution separation of ligand-protected metal NCs by high-performance liquid chromatography (HPLC)53,55,61,108–110,132–138 (Section 4.6 and Fig. 4(f)). Through these six techniques, we have been attempting to develop flexible and precise control techniques for ligand-protected metal NCs, that is, the ultimate nanotechnology for metal–molecule complex systems (Fig. 4(g)), and thereby gaining a deeper understanding of the correlation between the chemical composition, structure, and physicochemical properties of ligand-protected metal NCs.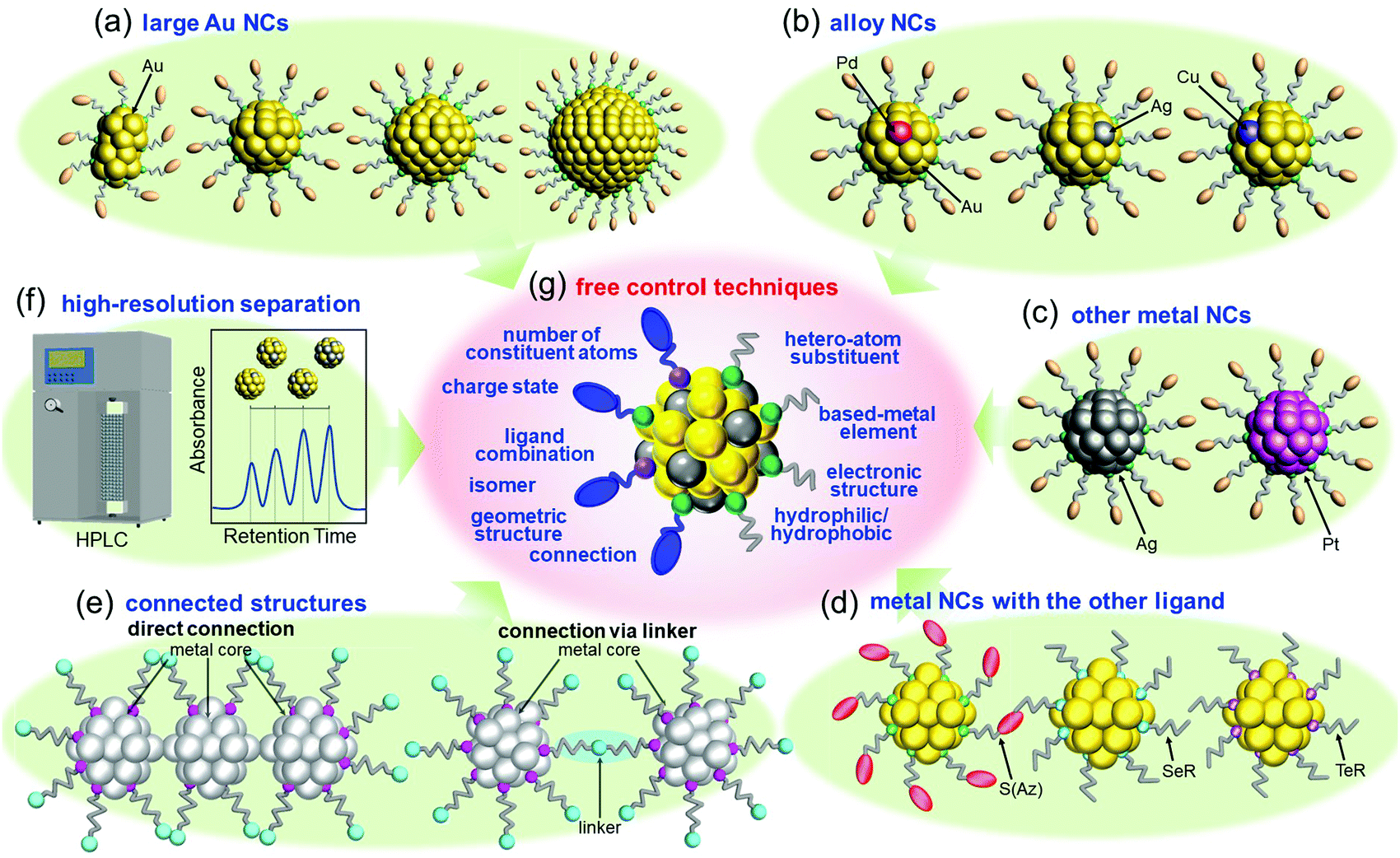 | ||
| Fig. 4 Our strategy for developing ultimate control methods for ligand-protected metal NCs. Development of isolation methods for (a) large Au NCs, (b) Au-based alloy NCs, (c) Ag and Pt NCs, (d) metal NCs protected by S(Az), SeR, and TeR, (e) connected structures. (f) Development of high-resolution separation methods using HPLC. (g) Controllable parameters achieved by (a–f). | ||
4.2. Isolation of relatively large Au NCs
In recent years, it has become possible to size-selectively synthesize relatively large Aun(SR)m NCs.139,140 However, as of 2008, the largest Aun(SR)m NCs that could be isolated with atomic precision were Au144 NCs,47,141,142 for which I was also involved in determining the chemical composition. Therefore, since 2008, we have been working on isolating Aun(SR)m NCs larger than Au144.When the hydrophilic SRs (SG or S(SA)) described in Section 3 are used as ligands, relatively large Aun(SR)m NCs are difficult to synthesize.143 However, Whetten and co-workers97 and Wolfe and Murray144 reported that Aun(SR)m NCs larger than Au144 NCs can be synthesized when hydrophobic SRs, such as alkanethiolate and 2-phenylethanethiolate (PET), are used as ligands. Therefore, we attempted to separate each Aun(SC12)m NC from a mixture at high resolution by reversed-phase (RP) HPLC and then determine their chemical compositions by ESI-MS (Fig. 5(a)).108,109 Using these methods, we succeeded in systematically isolating Aun(SC12)m NCs up to Au∼520(SC12)∼130 in 2014 (the paper was published in 2015).109
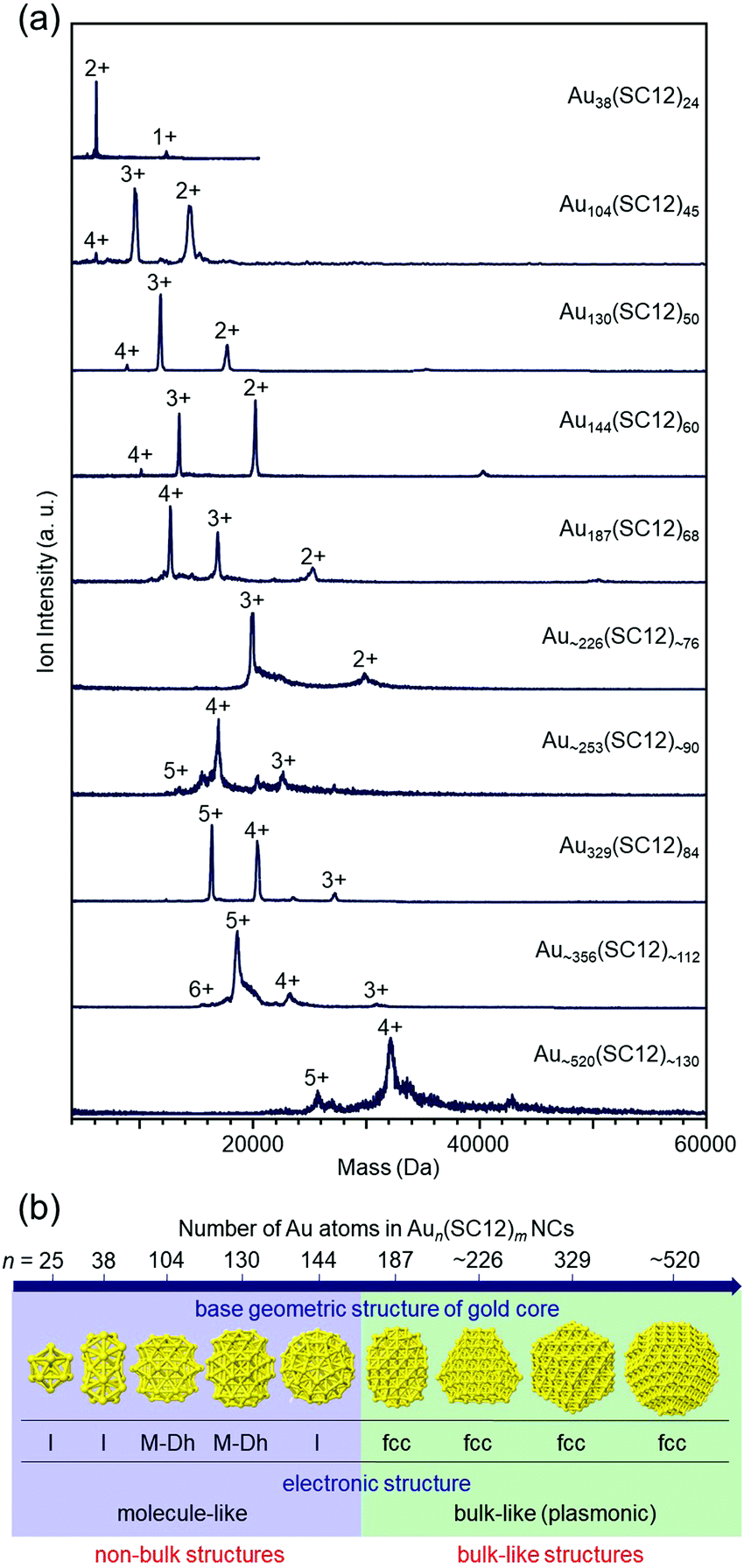 | ||
| Fig. 5 (a) Positive-ion ESI mass spectra of Aun(SC12)m NCs. (b) Non-bulk to bulk transition between Au144(SC12)60 and Au187(SC12)68 in the Aun(SC12)m–NC system.109 Reproduced with permission from ref. 109. Copyright 2015 American Chemical Society. | ||
In parallel with us, Jin and co-workers and Dass and co-workers were also working on isolation of relatively large Aun(SR)m NCs, and as of 2014, they had reported isolation of Au329(PET)84 (or Au333(PET)79), Au∼500(PET)∼120, and Au∼940(PET)∼160.145–148 Through optical absorption spectroscopy of those NCs145–148 and density functional theory (DFT) calculations by Häkkinen and co-workers,149 it had been predicted that non-bulk to bulk transition occurs at around Au144(SR)60 in the Aun(SR)m–NC system. We worked with Tsukuda and Häkkinen to clarify the electronic/geometric structures of a series of isolated Aun(SC12)m NCs (n = 38–∼520) to gain a better understanding of the details of the size at which non-bulk to bulk transition occurs. The results showed that non-bulk to bulk transition occurs between Au144(SC12)60 and Au187(SC12)68 in the Aun(SC12)m–NC system (Fig. 5(b)).109 In subsequent papers by Jin and co-workers and Dass and co-workers,139,150–152 these electronic/geometric transitions were reported to occur in different size regions when the bulkier SR is used as the ligand.
4.3. Isolation of Au-based alloy NCs
Even if only one metal element is used as a constituent element, various electronic structures and physicochemical properties can be obtained by controlling the number of constituent atoms in the metal NCs. However, as mentioned in Section 2, if the size effect is combined with the synergy effect caused by mixing different elements, it is expected that a variety of different electronic structures and physicochemical properties can be increased. Therefore, since 2008, we have been working on isolation of alloy NCs and elucidation of their geometric/electronic structures and physicochemical properties.110–117When I started this research in 2008, it was not easy to control the number of constituent atoms, even for metal NCs consisting of a single metal element. Furthermore, when I started my laboratory at Tokyo University of Science, I only had undergraduate students in my laboratory. Therefore, I thought that it was necessary to investigate metal-NC systems in which I had some experience to create new alloy NCs. Accordingly, Aun(SR)m NCs, which I had experience in handling, were selected as the base metal NCs for alloying.46,47 Specifically, we attempted to isolate alloy NCs in which some of the Au atoms in [Au25(SR)18]− and [Au38(SR)24]0 (SR = SC12 or PET)153–157 were replaced by Pd, Ag, or Cu atoms.
Fortunately, we isolated [Au24Pd(SC12)18]0 (Fig. 6(a)),110 [Au25−xAgx(SR)18]− (SR = SC12 or PET, x = 1–11, Fig. 6(b)),111 [Au25−xCux(PET)18]− (x = 1–5, Fig. 6(c)),112 and [Au36Pd2(PET)24]0 (Fig. 6(d))114 at an early stage. Studies on the obtained alloy NCs showed that (1) these three different elements have different preferential substitution sites (Fig. 6(e)),110–112,114,158 (2) Pd substitution is effective for creating more stable metal NCs than Aun(SR)m NCs,110,114 (3) Ag substitution is effective for creating metal NCs with larger HOMO–LUMO gaps than Aun(SR)m NCs (Fig. 6(f)),111 (4) Cu substitution is effective for creating metal NCs with smaller HOMO–LUMO gaps than Aun(SR)m NCs (Fig. 6(g)),112,158 (5) heteroatom substitution with Pd, Ag, or Cu enhances the speed of ligand-exchange reaction of NCs,52,159,160 and (6) Pd substitution improves the hydrogen evolution reaction (HER), oxygen evolution reaction, and oxygen reduction reaction (ORR) activities of the NCs.48 In addition, studies on tri-metal115 and tetra-metal116 alloy NCs have revealed that simultaneous substitution with different elements at different substitution sites causes superimposed addition of each substitution effect on the base metal NCs.116 In these early studies, the substitution sites were predicted based on DFT calculations by Nobusada and/or X-ray absorption fine structure (XAFS) analysis by Tsukuda and Yamazoe. However, since 2016,128,161 we have also been able to determine the geometric structures of metal NCs by SC-XRD, and such studies have enabled us to (1) create [Au∼20MAg∼4(PET)18]− (M = Pd or Pt), where both the number and position of Ag substitution are controlled (Fig. 6(h)),117 and (2) change the Cu substitution site by combining Cu and Pt substitution.162
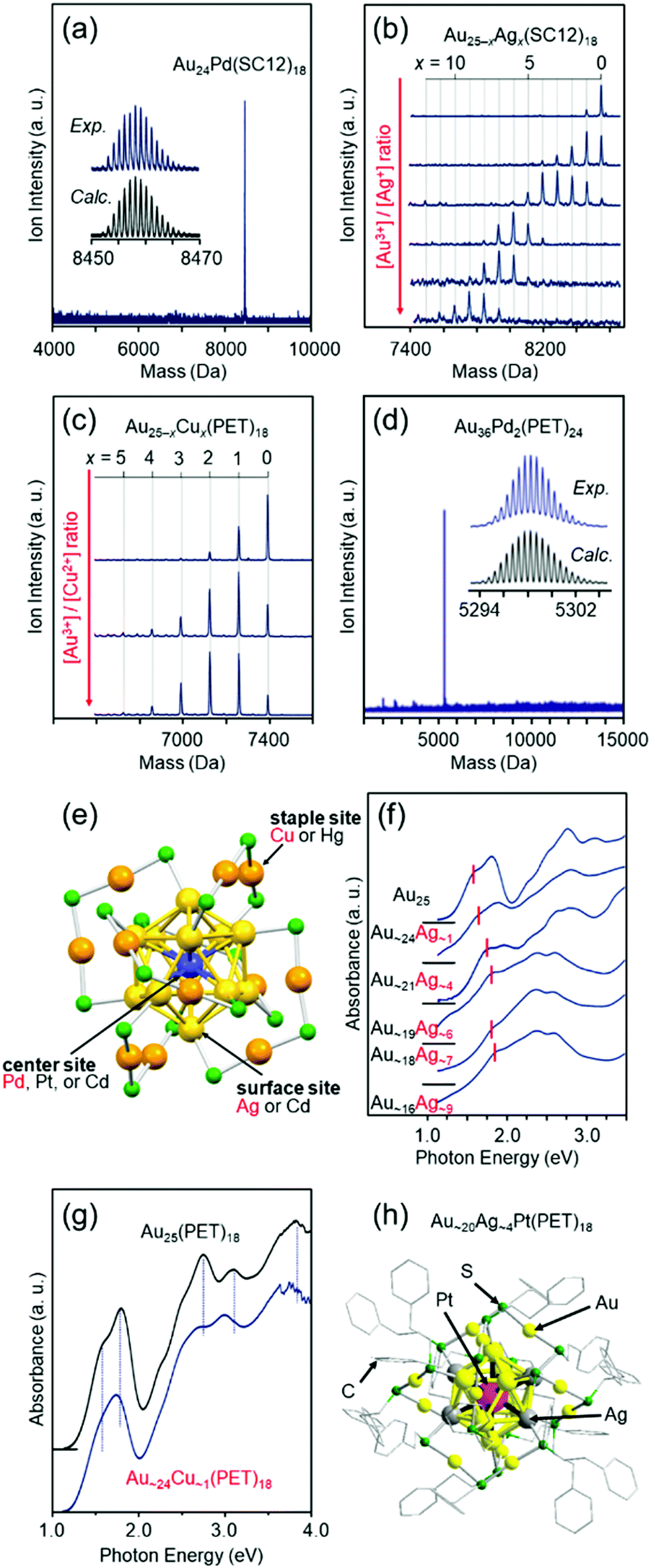 | ||
| Fig. 6 Negative-ion MALDI mass spectra of (a) Au24Pd(SC12)18,110 (b) Au25−xAgx(SC12)18,111 and (c) Au25−xCux(SC12)18.112 (d) Negative-ion ESI mass spectrum of Au36Pd2(PET)24.114 (e) Preferential substitution site in Au25(SR)18 for each element.116 Blue, yellow, and orange spheres represent Au atoms, and green spheres represent S atoms. The R groups are omitted for clarity. Optical absorption spectra of (f) [Au25−xAgx(SC12)18]− and (g) [Au∼24Cu∼1(PET)18]−.112,158 (h) Geometric structure of [Au∼20Ag∼4Pt(PET)18]0.117 Reproduced with permission from ref. 110–112, 114, 116 and 117. Copyright 2010 The Royal Society of Chemistry, 2012 American Chemical Society, 2012 The Royal Society of Chemistry, 2016 The Royal Society of Chemistry, and 2018 American Chemical Society. | ||
4.4. Isolation of Ag and Pt NCs
When I started my own laboratory, it was not easy to control the number of constituent atoms, even for Aun(SR)m NCs. Therefore, until 2010, the examples of isolated Agn(SR)m NCs were limited to Ag7(DMSA)4 (DMSA = meso-2,3-dimercaptosuccinic) reported by Jin and co-workers163 and Ag9(S(SA))8 reported by Pradeep and co-workers.164 Thus, we attempted to isolate larger Agn(SR)m NCs.Based on the report of Murray and co-workers,165 it was expected that using 4-(tert-butyl)benzylmercaptan (SBB) as a ligand would result in synthesis of stable Agn(SR)m NCs. Therefore, we first synthesized a mixture of Agn(SR)m NCs and then applied the reaction with thiols, which had been frequently used in isolation of Aun(SR)m NCs,105,166 to the mixture to convert the unstable species to stable species. As a result, Agn(SBB)m NCs with an average particle size of 2.1 nm were successfully obtained in a size-selective manner.118 The obtained stable Agn(SBB)m NCs were assigned to Ag∼280(SBB)∼120 based on the results of ESI-MS. However, in this experiment, which was conducted in 2010 (the paper was published in 2011), the chemical composition was determined by a home-made MS apparatus without high resolution. In recent years, many ESI-MS apparatuses with high sensitivity and resolution have been developed. It is expected that the chemical composition of the stable Agn(SBB)m NCs obtained in that study will be determined more accurately using such ESI mass spectrometers in the future.
Agn NCs are attractive because of their high quantum yield of luminescence167 and high selectivity for carbon dioxide reduction.168 Since we have also been working on catalytic application of metal NCs, we have continued to work on development of isolation techniques for Agn NCs also after the report of Agn(SBB)m NCs. As a result, for example, in 2013, we successfully isolated Ag44(SePh)30 (SePh = phenylselenolate) in collaboration with Pradeep and co-workers (Fig. 7(a)).119 DFT calculations by Häkkinen and co-workers showed that Ag44(SePh)30 has a geometric structure similar to that of [Ag44(SR)30]4− (SR = SPhF2 or 4-mercaptobenzoic acid (p-MBA)), whose geometric structure was determined just before our reports by Zheng and co-workers169 and Bigioni and co-workers.170 In addition to this NC, we recently succeeded in isolating several new Ag NCs171 using anions as templates (the paper is being prepared for submission).
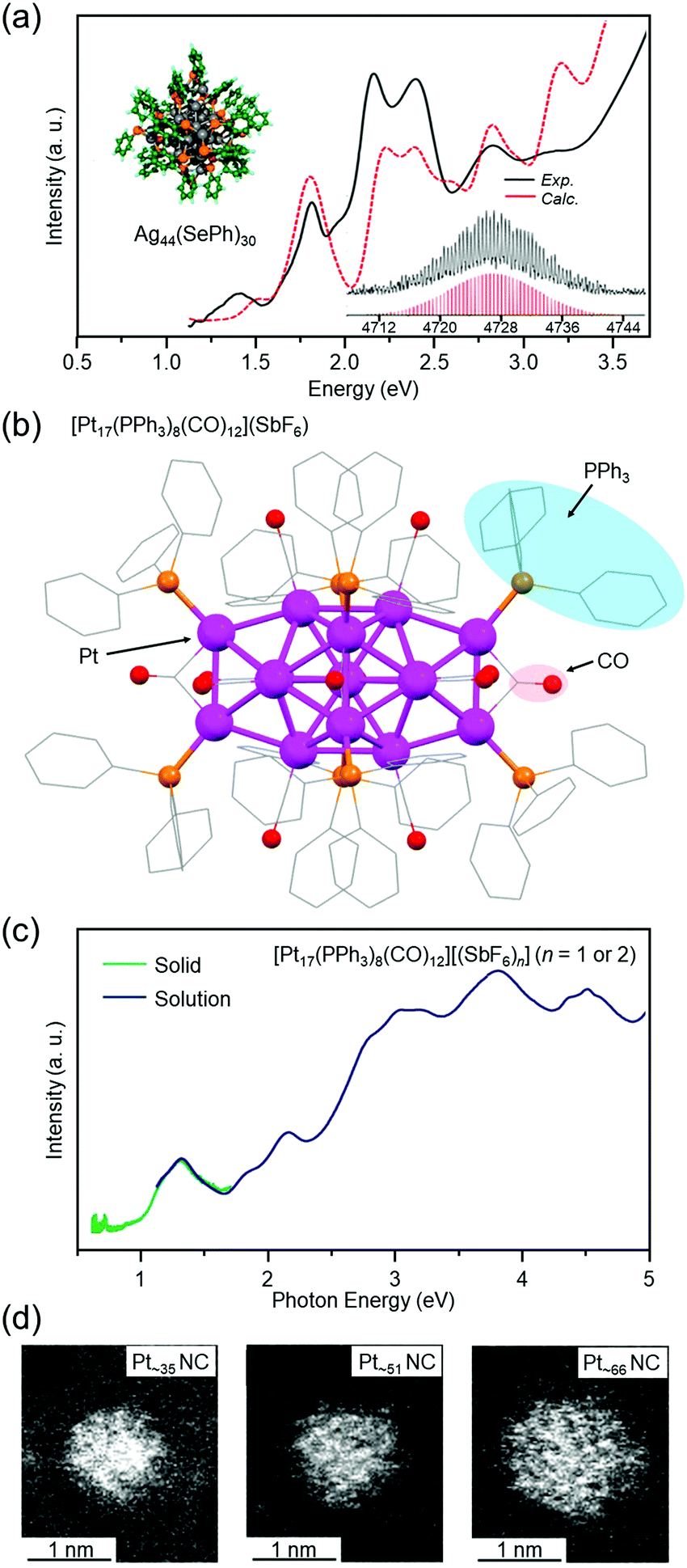 | ||
| Fig. 7 (a) Optical absorption spectra, computationally relaxed structure, and expanded ESI mass spectrum of Ag44(SePh)30.119 (b) Geometric structure of [Pt17(PPh3)8(CO)12](SbF6) (SbF6− = hexafluoroantimonate) determined by SC-XRD.120 (c) Optical absorption spectra of [Pt17(PPh3)8(CO)12][(SbF6)n] (n = 1 or 2) in dichloromethane solution (blue) and the solid state [Pt17(PPh3)8(CO)12][(SbF6)n] (n = 1 or 2) (green).120 (d) High-angle scattering dark-field scanning transmission electron microscopy (HAADF-STEM) images of Ptn(PET)m(CO)l NCs with n = ∼35 (left), ∼51 (center), and ∼66 (right).121 Reproduced with permission from ref. 119–121. Copyright 2013 American Chemical Society, 2017 American Chemical Society, and 2021 The Royal Society of Chemistry. | ||
Isolation of Ptn NCs has been reported by Chini and co-workers and Longoni and co-workers.22,27 In their studies, CO was used as the main ligand, but in later studies on Ptn and its alloy NCs by Dahl, Slovokhotov, Zacchini, etc., PR3 was also used as a ligand.23,172,173 In these studies, the geometric structures of the metal NCs were determined by SC-XRD. For Aun(SR)m NCs, in 2007, Kornberg, Ackerson and co-workers reported the geometric structure of [Au102(p-MBA)44]0, which was the first report of the geometric structure of Aun(SR)m NCs determined by SC-XRD.174 The advancement of research of Ptn and its alloy NCs at that time was astonishing. However, synthesis and the subsequent handling of the reported Ptn NCs had to be carried out under CO or an inert-gas atmosphere,23,172,173 which was not easy for those who were not used to using CO gas or Schlenk techniques. Therefore, we searched for Ptn NCs that could be synthesized by mixing reagents in air, similar to Aun(SR)m NCs.
When a Pt salt is dissolved in polyol and the resulting solution is heated, the Pt salt is reduced to generate Pt atoms (polyol reduction).175–179 The polyol generates CO, and the generated CO coordinates to the Pt surface. Addition of PR3 to the solution of the resulting product is expected to give Ptn(PR3)m(CO)l NCs without the need for a CO atmosphere. Indeed, we successfully isolated [Pt17(PPh3)8(CO)12]z+ (PPh3 = triphenylphosphine, z = 1 or 2) with atomic precision by this method (Fig. 7(b)).120 The obtained NC had a geometric structure similar to previously reported [Pt17(PEt3)8(CO)12]0 (PEt3 = triethylphosphine)23 and [Pt13Au4(PPh3)8(CO)12]0.172 Optical absorption spectroscopy revealed that [Pt17(PPh3)8(CO)12]z+ (z = 1 or 2) has a discrete electronic structure similar to a small Aun(SR)m NC (Fig. 7(c)).102
For Ptn NCs, we have also succeeded in obtaining a series of Ptn(PET)m(CO)l NCs (n = ∼35, ∼51, and ∼66) in high yields (∼80% yield for Pt∼66) by changing the ligand from PPh3 to PET (Fig. 7(d)).121 Unfortunately, the chemical compositions of these Ptn(PET)m(CO)l NCs cannot be controlled with atomic precision at present. Therefore, the chemical compositions and geometric structures of these NCs have not been accurately determined. Thus, we are now attempting to apply various size-focusing methods166,180 to Ptn(PET)m(CO)l NCs to isolate the Ptn(PET)m(CO)l NCs with a single chemical composition. If these efforts are successful and the chemical compositions and geometric structures of the isolated Ptn(PET)m(CO)l NCs are accurately determined, it is expected that a deeper understanding of the chemical compositions and geometric structures of the Ptn(PET)m(CO)l NCs that can be isolated will be obtained.
4.5. Isolation of metal NCs protected by functional ligands
For ligand-protected metal NCs, it is possible to impart new functions by controlling not only the metal core, but also the functional groups of the ligands. The solvent solubility, redox potential,181–184 luminescence properties,185 optical activity,186 and catalytic activity of Aun(SR)m NCs are remarkably dependent on the functional group structure of SR. If Aun(SR)m NCs with these characteristics are protected by SR, which changes its molecular structure in response to light irradiation, it is expected that the physicochemical properties of Aun(SR)m NCs can be reversibly changed by light irradiation.Azobenzene is a typical photoresponsive molecule.187,188 It changes from trans to cis by ultraviolet (UV)-light irradiation and from cis to trans by visible-light irradiation. When SR containing azobenzene is used as a protective ligand, Aun(SR)m NCs, whose physicochemical properties repeatedly change by light irradiation, could be created. Accordingly, we have synthesized Au25(S(Az))18 protected by an azobenzene derivative thiolate (S(Az)) (Fig. 8(a)) to create such photoresponsive Aun(SR)m NCs.122 When the obtained Au25(S(Az))18 was irradiated with light, S(Az) repeatedly underwent photoisomerization (Fig. 8(b)). In addition, the redox potential of Au25(S(Az))18 reversibly changed with such structural changes (Fig. 8(c)). These results demonstrated that the combination of azobenzene and Au25 NCs can be used to create Aun(SR)m NCs with photoresponsive redox potentials.
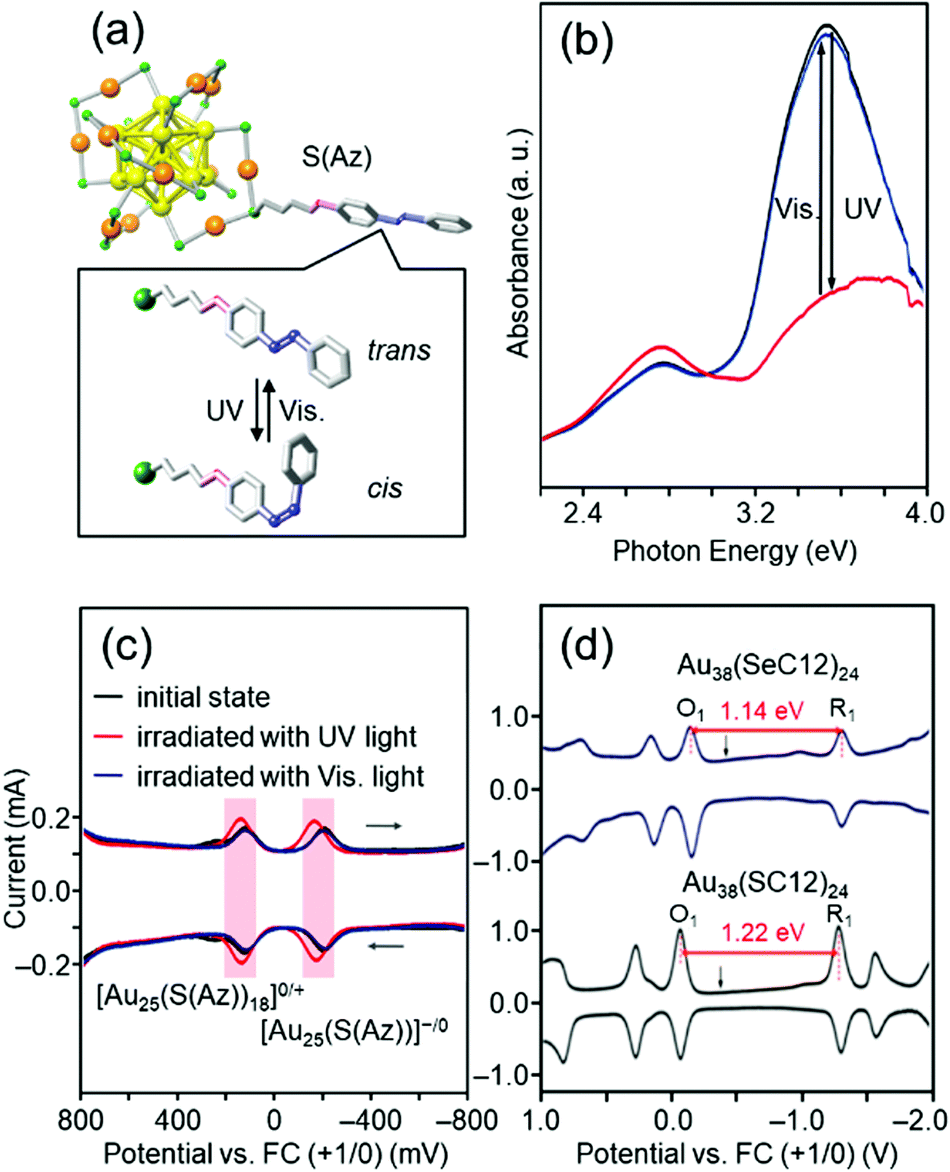 | ||
| Fig. 8 (a) Proposed geometric structure of [Au25(S(Az))18]− and (b) the photoresponsive behavior in the optical absorption spectra of [Au25(S(Az))18]− in toluene solution in the energy range 2.2–4.0 eV.122 The visible photostationary state (blue line) overlaps with the initial state (black line). (c) Photoresponse of the differential pulse voltammetry (DPV) curve of [Au25(S(Az))18]−.122 (d) DPV curves of [Au38(SeC12)24]0 and [Au38(SC12)24]0 in CH2Cl2 containing 0.1 M (C4H9)4NClO4.126 O1 and R1 indicate the first oxidation and reduction peaks of the NCs, respectively. The black arrows indicate the solution open-circuit potentials. The red arrows indicate the electrochemical HOMO–LUMO gaps of [Au38(SeC12)24]0 and [Au38(SC12)24]0. Reproduced with permission from ref. 122 and 126. Copyright 2012 The Royal Society of Chemistry and 2013 American Chemical Society. | ||
Au25(SR)18 has a compact and symmetric metal core (Fig. 8(a)).153,154 Therefore, the metal-core structure of Au25(S(Az))18 remains almost unchanged even after isomerization of the ligand by photoirradiation. The change in the redox potential is considered to be because of the change in the dipole moment of the S(Az) molecule caused by isomerization of azobenzene, which in turn changes the orbital energy of Au25(S(Az))18.189,190 In 2016, Jin and co-workers reported that [Au28(SR)20]0 can change the metal-core structure depending on the functional group structure of the ligand.191 Therefore, in the future, we will combine S(Az) with a Aun(SR)m NC whose metal core was easily deformable,192 and thereby create “intelligent” Aun(SR)m NCs in which the metal-core structure changes by light irradiation, leading to significant changes in the redox potential, luminescence properties, optical activity, and catalytic activity.
In the above study of Au25(S(Az))18, a new function was provided to the Aun NCs by including a function in a functional group that was not directly bound to Au. However, changing the functional group directly bound to Au (i.e., S) to Se is expected to change the binding mode between Au and the ligand,193 thereby providing Aun NCs with new physical and chemical properties. With this expectation, we precisely synthesized [Au25(SeR)18]− (SeR = octaneselenolate (SeC8) or dodecaneselenolate (SeC12)),123,124 [Au25(PET)18−y(SePh)y]− (y = 1–18),128 [Au25−xCux(SeC8)18]− (x = 1–9),125 and [Au38(SeC12)24]−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 126 with selenolate (SeR) ligands. Studies on the obtained Au NCs revealed that changing the ligand from SR to SeR (1) changes the binding mode between Au and the ligand, thereby making the Au NCs more stable,124 and (2) has the effect of narrowing the HOMO–LUMO gap of the Au NCs (Fig. 8(d)).124,126,128 We have also systematically synthesized [Au25(SC8)18−y(TePh)y]− (TePh = phenyltellurolate, y = 1–18) containing a tellurolate (TeR) in the ligand layer.127,128 We found that changing the ligand from SR to TeR further narrows the HOMO–LUMO gap of the Au NCs.127,128
126 with selenolate (SeR) ligands. Studies on the obtained Au NCs revealed that changing the ligand from SR to SeR (1) changes the binding mode between Au and the ligand, thereby making the Au NCs more stable,124 and (2) has the effect of narrowing the HOMO–LUMO gap of the Au NCs (Fig. 8(d)).124,126,128 We have also systematically synthesized [Au25(SC8)18−y(TePh)y]− (TePh = phenyltellurolate, y = 1–18) containing a tellurolate (TeR) in the ligand layer.127,128 We found that changing the ligand from SR to TeR further narrows the HOMO–LUMO gap of the Au NCs.127,128
Based on a study of two-dimensional self-assembled monolayers,194 changing the ligand from SR to SeR or TeR is expected to induce an increase in the conductivity between the Au core and the ligand. High conductivity is a desired feature in creation of single-molecule devices based on Au NCs,195 and we hope to clarify this point in future studies by comparing the conductivities of Aun(SR)m, Aun(SeR)m, and Aun(TeR)m NCs.
4.6. Connection of ligand-protected metal NCs
Among ligand-protected metal NCs, icosahedral NCs have been reported to become constituent units of “clusters of clusters”.196 For example, in the 1990s, Teo and co-workers reported synthesis of [(p-tol3P)10Au13Ag12Cl7]2+ (p-tol3P = tris(4-methylphenyl)phosphine) consisting of two Au13−xAgx197 and [(p-tol3P)12Au18Ag20Cl14]0 consisting of three Au13−xAgx.198 In 2007, I was also involved in isolation of [Au25(PPh3)10(SCn)5Cl2]2+ (SCn = alkanethiolate, n = 2–18), which consists of two Au13 NCs.199 Further development of techniques for connecting metal NCs could lead to fabrication of ultra-thin metal nanowires. Therefore, we have also been working on development of techniques to connect metal NCs.The first connected structure that was successfully isolated was [Au24Pd(PPh3)10(PET)5Cl2]+ (Fig. 9(a)).129 This connected structure was size-selectively synthesized under appropriate conditions. In this connected structure, only one central Au atom is substituted by a different atom, unlike most of the connected structures reported in previous papers.200–203 DFT calculations by Jiang and co-workers showed that [Au24Pd(PPh3)10(PET)5Cl2]+ NCs with these hetero bi-icosahedral cores have dipole moments in the NC.129 Very recently, we also succeeded in synthesizing [Ag23Pd2(PPh3)10Cl7]0 consisting of two Ag12Pd cores (Fig. 9(b)).131 Stability experiments and DFT calculations by Iwasa and co-workers indicated that it is very important to strengthen the framework of individual icosahedral cores and increase their stability to form the connected structures described by the chemical formula [Ag23M2(PPh3)10Cl7]z (M = Ag, Pd, or Pt, z = 2+ or 0).131
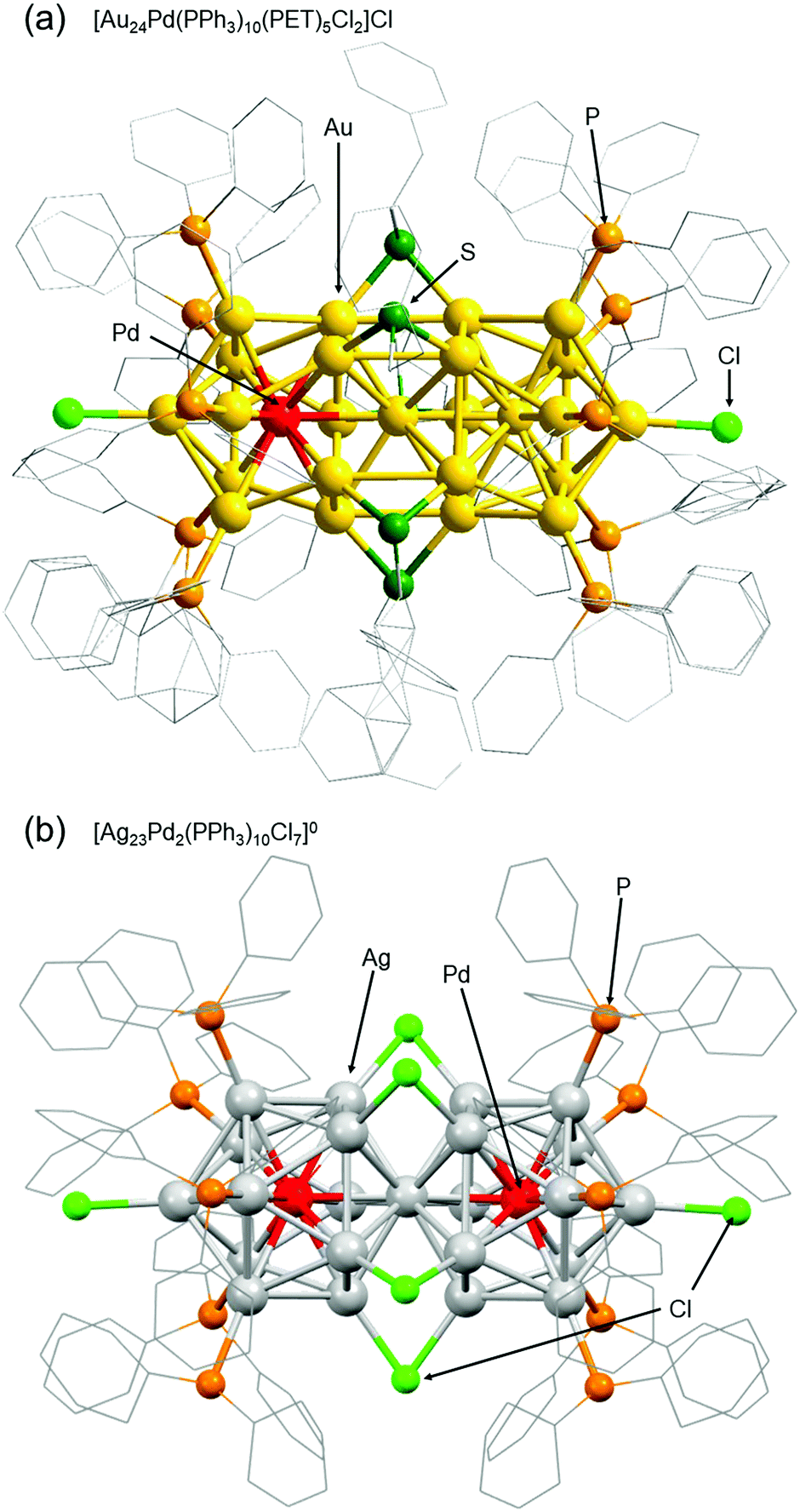 | ||
| Fig. 9 Geometric structures of (a) [Au24Pd(PPh3)10(PET)5Cl2]Cl and (b) [Ag23Pd2(PPh3)10Cl7]0.129,131 Reproduced with permission from ref. 129 and 131. Copyright 2018 The Royal Society of Chemistry and 2021 American Institute of Physics. | ||
In the above study, two metal NCs were connected. On the other hand, in the study on [Au4Pt2(SR)8]0 (SR = PET (Fig. 10(a)) or 2-chlorobenzylmercaptane (SCH2PhCl)), we successfully connected them continuously in one dimension.130 This was the second report of formation of a one-dimensional connected structure, following that reported by Maran and co-workers for [Au24M(SR)18]0 (M = Au, Hg, or Cd, SR = butanethiolate or pentanethiolate)204–206 (note that although it was not mentioned in the paper,207 [Au4Pd2(PET)8]0 reported by Wu and co-workers also seemed to form a similar connected structure). A series of studies with SR ligands revealed that it is important to design the ligand interaction within [Au4Pt2(SR)8]0 to form one-dimensional connected structures and control their geometry (Fig. 10(b)).130 The obtained one-dimensional connected structures have a semiconductor band gap, and the band gap size can be modulated by the ligand structure. These findings are expected to provide clear design guidelines for creation of metal-NC connected structures with desired geometric structures and physicochemical properties. We are now also working on connection of these metal NCs via linker molecules.62,208–213
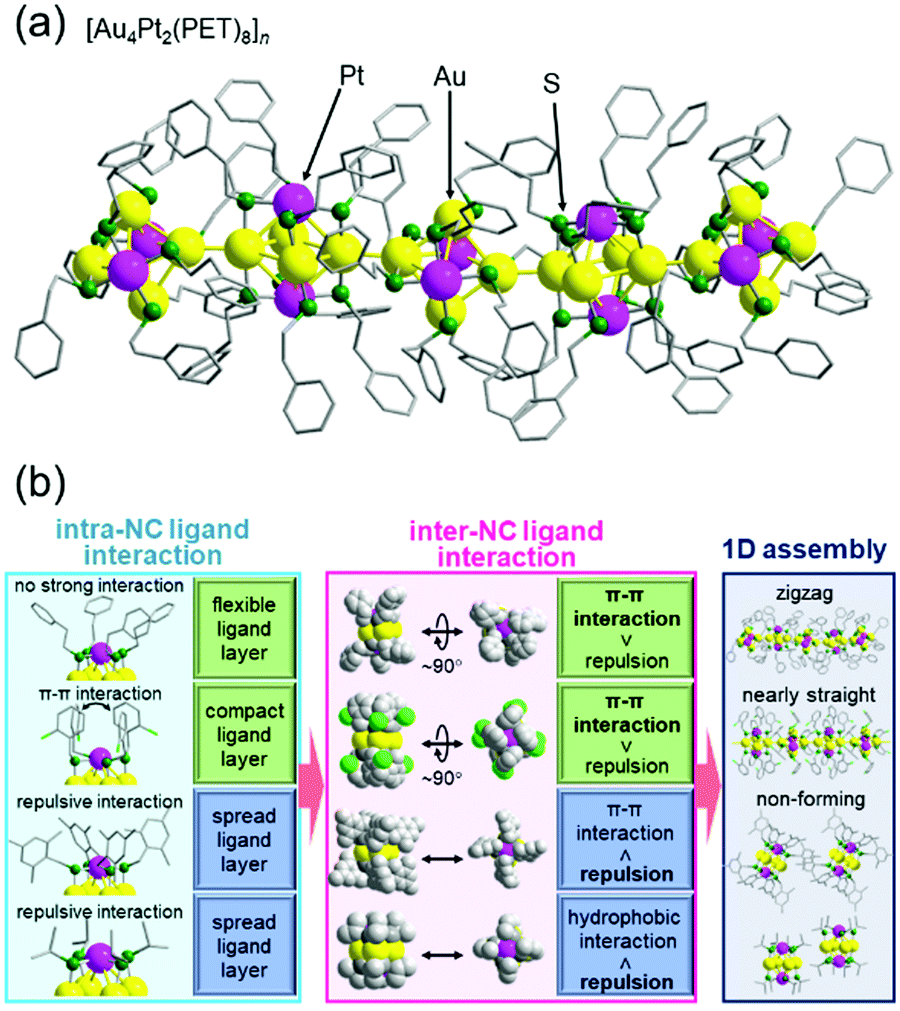 | ||
| Fig. 10 (a) Crystal structure of [Au4Pt2(PET)8]n. (b) Relationships between intra-NC ligand interactions, which are related to the distribution of the ligands within each NC, inter-NC ligand interactions, and one-dimensional assembly. Reproduced with permission from ref. 130. Copyright 2020 The Royal Society of Chemistry. | ||
4.7. Development of high-resolution separation methods
Most of the ligand-protected metal NCs and their connected structures discussed in Sections 4.2–4.6 can be isolated by only mixing the reagents and washing the by-products in the ambient atmosphere. However, all NCs cannot be necessary isolated by such a simple process in the case of Aun(SC12)m NCs109 described in Section 4.2. Therefore, it was necessary to develop new high-resolution separation techniques for ligand-protected metal NCs to isolate the metal NCs precisely and systematically, and thereby to understand the correlation between the chemical composition and the structure/properties.PAGE (Fig. 3(a)), which is described in Section 3, is suitable for high-resolution separation of hydrophilic compounds, but it is not so suitable for high-resolution separation of hydrophobic compounds. On the other hand, HPLC (Fig. 11(a)) is suitable for separation of both hydrophilic and hydrophobic compounds. Indeed, in 2003, Murray and co-workers succeeded in isolating Au NCs, such as Au38, Au140, and Au225 (the chemical composition is reported one in the papers), from a mixture of Aun(SC6)m NCs (SC6 = hexanethiolate) by RP-HPLC.144,214,215
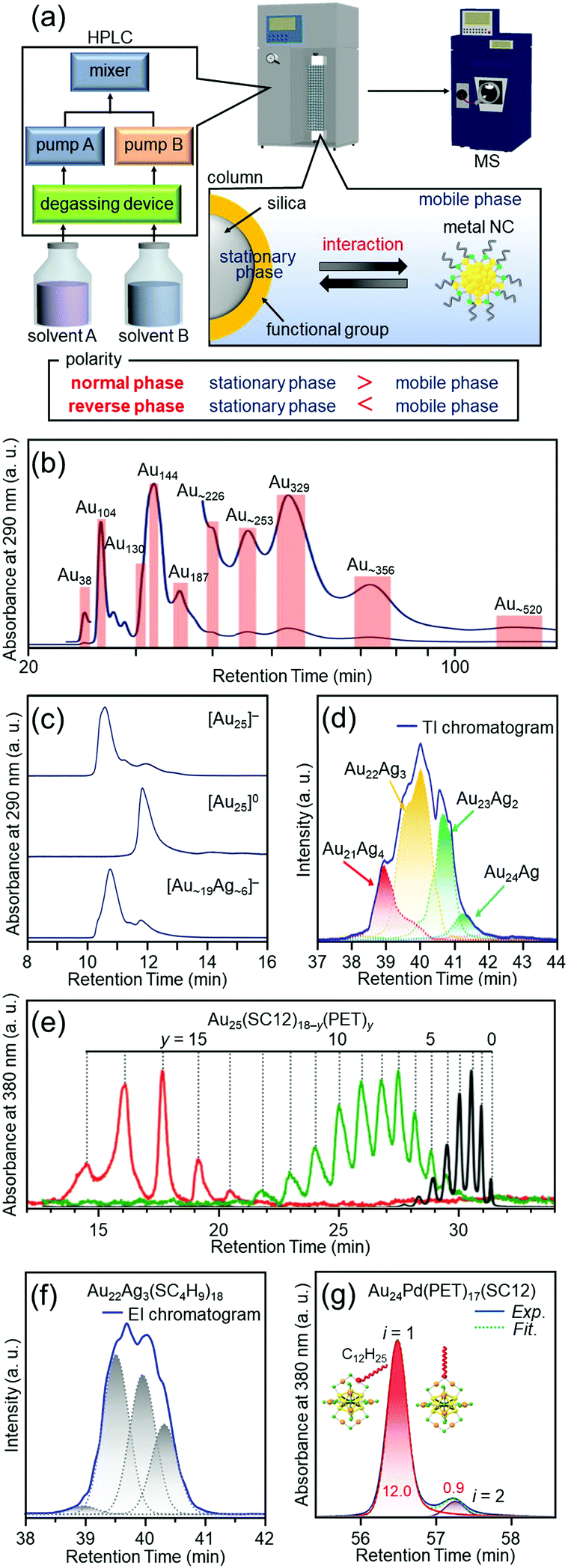 | ||
| Fig. 11 (a) Schematic illustration of HPLC separation techniques.61 Chromatograms of (b) the crude sample of Aun(SC12)m NCs109 and (c) [Au25(SC12)18]−, [Au25(SC12)18]0, and [Au∼19Ag∼6(SC12)18]−.111 (d) Extracted ion (EI) chromatogram of [Au25−xAgx(SC4H9)18]0.132 TI means total ion. (e) Chromatogram of Au25(SC12)18−y(PET)y.134 (f) EI chromatogram of Au22Ag3, which represents separation of several types of isomers.132 (g) Chromatogram of Au24Pd(PET)17(SC12), which represents separation of coordination isomers.137 Reproduced with permission form ref. 61, 109, 111, 132, 134 and 137. Copyright 2020 The Royal Society of Chemistry, 2015 American Chemical Society, 2010 The Royal Society of Chemistry, 2018 American Chemical Society, and 2013 American Chemical Society. | ||
Inspired by the work of Murray and co-workers, we attempted to separate metal NCs, which were difficult to isolate by only washing the by-products, at the atomic and molecular levels by advanced use of RP-HPLC.53 As a result, in addition to separation depending on the number of constituent atoms (Fig. 11(b)),108,109 ligand-protected metal NCs were also successfully separated depending on the charge state (Fig. 11(c)),110 alloy composition (Fig. 11(d)),132,138 ligand combination (Fig. 11(e)),133,134 and structural isomer (Fig. 11(f and g)).132,137 The separation techniques described in Fig. 11(f and g) led to not only control of the structural isomers, but also elucidation of the reaction mechanisms of the metal-exchange138 and ligand-exchange137 reactions. Although these high-resolution separations were achieved for hydrophobic metal NCs, in later studies, we also succeeded in separating hydrophilic metal NCs depending on their constituent atoms with high resolution by ion-pair chromatography135 or hydrophilic-interaction chromatography.136
In the future, we would like to use two-dimensional HPLC216 for such high-resolution separations. It allows the use of two different separation modes. We expect that this will make it possible, for example, to first separate the products generated by ligand exchange depending on the number of exchanged ligands,134 and then to separate the products of the same chemical composition depending on the optical isomers217 generated by the position of the exchanged ligands.
4.8. Summary of control of ligand-protected metal NCs
We have succeeded in developing several new techniques for (1) isolation of large Au NCs, (2) isolation of alloy NCs, (3) isolation of metal NCs consisting of Ag or Pt, (4) isolation of metal NCs protected by functional ligands, (5) connection of metal NCs, and (6) high-resolution separation of metal NCs. Other groups have also been simultaneously working in these areas, and they have developed several other isolation methods.7,218 Through these studies, the control techniques for ligand-protected metal NCs, that is, “bottom-up nanotechnology for complexes composed of metals and molecules”, has been dramatically enhanced in recent years. In the future, it is expected that the “science of ligand-protected metal NCs” will be further deepened by further development of these control techniques.5. Toward creation of high-performance energy and environmental catalysts
When I started investigating gas-phase NCs in 1995, I was not concerned about the applications of metal NCs as materials. However, after I had deposited GenOm NCs on a support and observed their luminescence in air with my own eyes, I thought to contribute to society by applying metal NCs as materials in future studies. Because energy and environmental issues have become major global challenges, I decided to contribute to the field of energy and environmental materials.5.1. Social problems to be solved by chemistry
In the 21st century, there are serious issues regarding energy and the environment, such as depletion of fossil fuels and destruction of the environment on a global scale. Therefore, chemistry in the 21st century is expected to address these issues, and thereby build a clean and sustainable society. The energy conversion system shown in Fig. 12 is one of the systems necessary to build such a society. In this system, hydrogen (H2) is produced by photocatalysts or electrocatalysts, and the obtained H2 is converted to electricity by fuel cells. Using such a system, it is possible to obtain energy from only water and sunlight, which are almost unlimited on earth. In addition, this system allows the energy medium (H2) to be circulated, thus preventing the issues of energy depletion. Furthermore, this system does not generate carbon dioxide, which leads to global warming. In this system, the properties of the photocatalyst,219 electrocatalyst,63,220–232 and fuel cell60,224,233–238 are highly dependent on the chemistry. Therefore, modern chemistry is expected to improve the functions of these components. Considering the fact that the shift to H2 society cannot be realized immediately, it is necessary to improve the current system at the same time. In other words, automotive exhaust-gas purification catalysts,239 which inhibit emission of harmful gases, need to be further improved. Therefore, we have been working on development of highly functionalized water-splitting photocatalysts, fuel-cell electrocatalysts, and automotive exhaust-gas purification catalysts (Fig. 1(d and e)) using precise metal NCs, in addition to developing flexible and precise control techniques for metal NCs.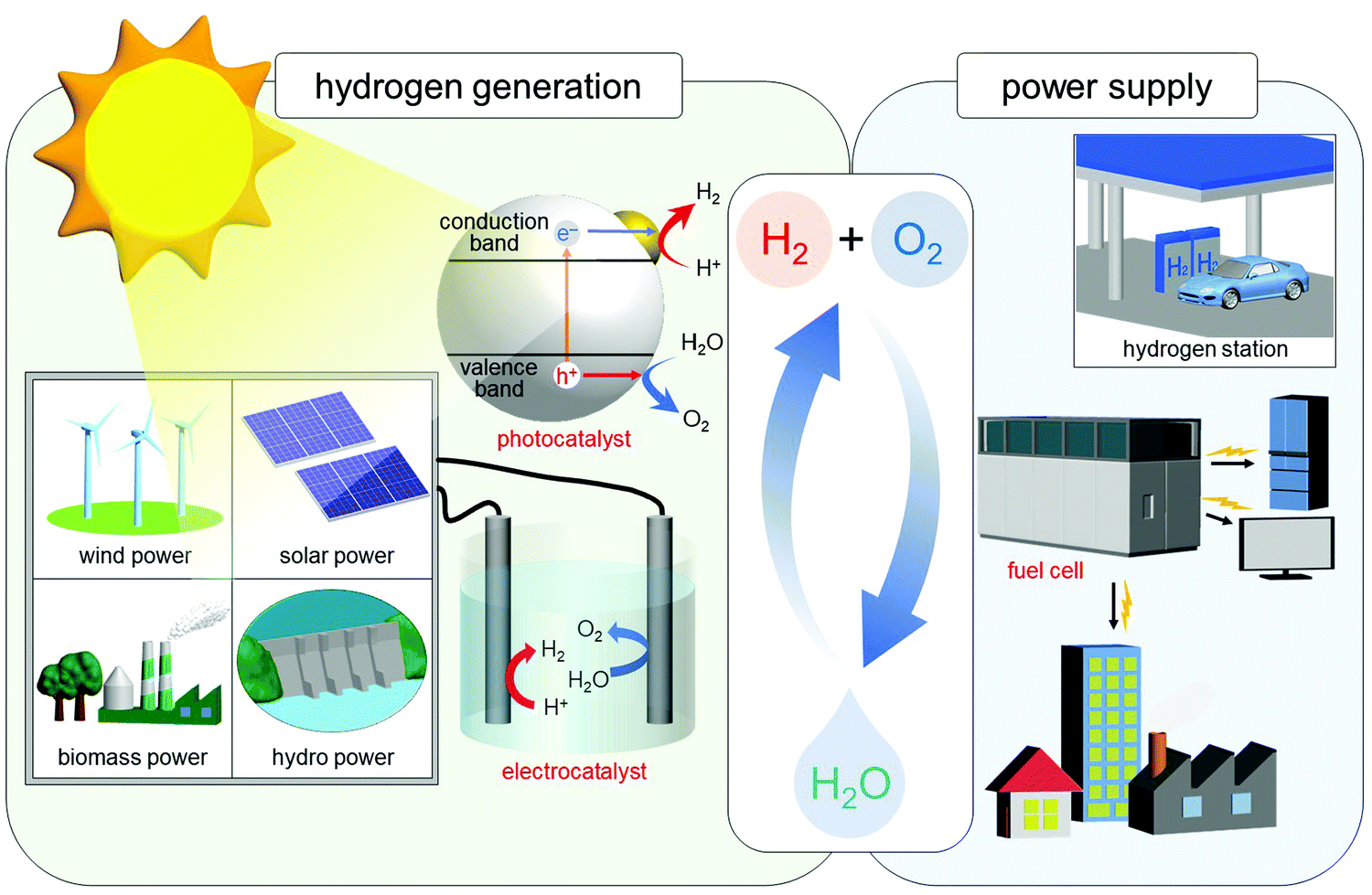 | ||
| Fig. 12 Schematic of the energy-conversion system for constructing a sustainable society. Note that in addition to solar power, sunlight also produces wind, biomass, and hydro power. Reproduced with permission from ref. 56. Copyright 2021 The Royal Society of Chemistry. | ||
5.2. Our strategy for activation of energy and environmental catalysts
For preparation of water-splitting photocatalysts, fuel-cell electrocatalysts, and automotive exhaust-gas purification catalysts, metal nanoparticles (NPs) are generally loaded on the support by impregnation (Fig. 13(a))240 or photodeposition method (Fig. 13(b)).241 In the impregnation method, metal NPs are immobilized on the support by stirring the support and metal salt in solution followed by calcination (Fig. 13(a)). In the photodeposition method, the support is dispersed in a solution containing the precursor metal salts of the metal NPs, which is then irradiated with light. The electrons and holes generated by photoexcitation are used to reduce or oxidize the precursor metal salts of the metal NPs, thereby immobilizing the metal NPs on the surface of the support (Fig. 13(b)). These loading methods are widely used because of their simplicity. However, the size distributions of the metal NPs obtained by these methods are wide because the metal atoms are aggregated on the support. Therefore, it is extremely difficult to load precisely controlled metal NPs on the support by these methods.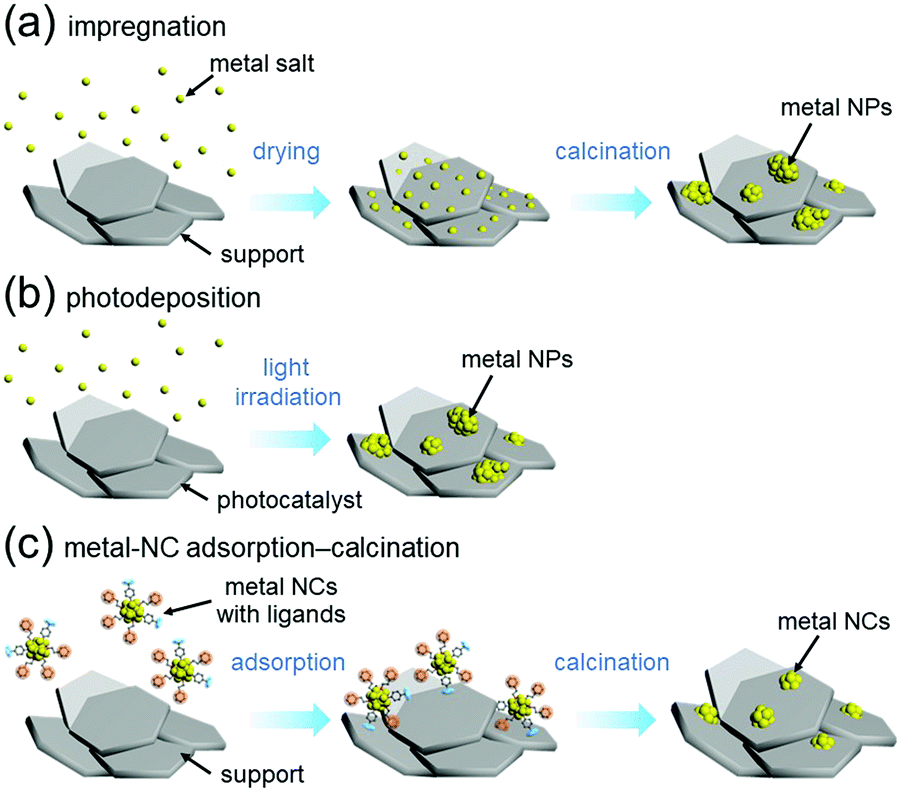 | ||
| Fig. 13 Comparison of (a) impregnation, (b) photodeposition, and (c) metal-NC adsorption–calcination methods. | ||
On the other hand it is possible to load metal NCs with a controlled particle size on the support by adsorbing ligand-protected metal NCs synthesized in the liquid phase onto the support and then removing the ligand by calcination (Fig. 13(c)).242–244 If the chemical composition of the loaded metal NCs can be controlled with atomic precision, it could be possible to create high-performance heterogeneous catalysts based on selective loading of highly active metal NCs.66 For atomically controlled loaded metal NCs, a deep understanding of the electronic/geometric structure of the loaded metal NCs and the electronic/geometric interaction between the loaded-metal NCs and the catalyst surface can be obtained by various high-resolution analyses and theoretical calculations (Fig. 14(a)).11,245–255 It is expected that this will provide a deep understanding of the important factors for high activation and clear design guidelines for further activation (Fig. 14(b)). Design and creation of appropriate metal NCs based on the obtained information are expected to lead to further functionalization of water-splitting photocatalysts, fuel-cell electrocatalysts, and automotive exhaust-gas purifying catalysts (Fig. 14). However, when we started this work, there were no examples of atomically precise control of loaded metal NCs on these catalysts using ligand-protected metal NCs as precursors.
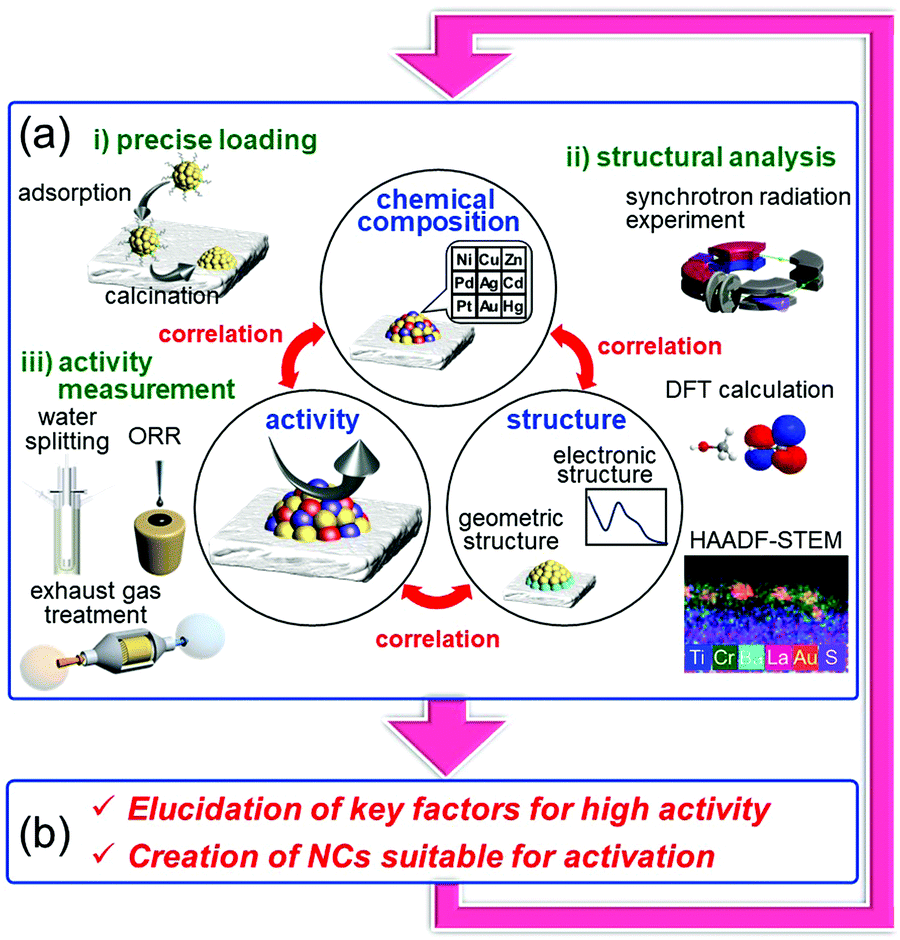 | ||
| Fig. 14 Our strategy for activation of energy and environmental catalysts: elucidation of (a) the correlation between the chemical composition, structure, and activity and (b) the key factors for the activity and therefore for creation of NCs suitable for activation. This cycle is repeated to create high-performance energy and environmental catalysts. | ||
5.3. Activation of water-splitting photocatalysts
Using photocatalytic reactions (Fig. 12),256–267 it is possible to produce H2 directly from water and sunlight. Therefore, water-splitting photocatalysis is considered to be one of the cleanest energy production reactions. However, at present, the energy conversion efficiency from sunlight to H2 (STH) is only about 1.1%.268 To realize practical use of water-splitting photocatalysts, it is essential to improve the STH to about 5–10% in the future.264In many cases, metal/metal-oxide NPs/NCs (cocatalysts) should be loaded on water-splitting photocatalysts as the active sites (Fig. 12). Controlling their particle sizes is very effective for improving the water-splitting activity.64,258,269 Therefore, we have focused on (1) precisely controlling the loaded metal NCs on the photocatalyst using precise ligand-protected metal NCs as precursors, (2) elucidating the details of how control of the cocatalyst affects the water-splitting activity, and (3) actually improving the functionality of the water-splitting photocatalyst.
The first step was to precisely load Aun NCs onto BaLa4Ti4O15 (an UV-light-driven water-splitting photocatalyst),270 which is one of the most advanced photocatalysts developed by our collaborator, Kudo and co-workers.256 We successfully determined the appropriate precursor NCs for precise loading (Fig. 15(a)), the appropriate adsorption conditions, and the appropriate calcination conditions.271,272 Activity measurement (Fig. 15(b)) of the obtained Au25 NC-loaded BaLa4Ti4O15 (Au25/BaLa4Ti4O15) revealed that the small Au cocatalyst accelerates not only the HER, but also the oxygen (O2)-photoreduction reaction, which is one of the reverse reactions.273 Therefore, we next attempted to form a layer to prevent the reverse reaction on the Au25 cocatalyst. Maeda, Domen and co-workers reported that formation of a chromium oxide (Cr2O3) layer is extremely effective for preventing the reverse reaction.244,274–283 However, using their formation method, it was difficult to form the Cr2O3 layer on the Au25 cocatalyst while maintaining the number of constituent atoms of the cocatalyst. Therefore, we developed a new method for formation of the Cr2O3 layer.273 Specifically, the Cr2O3 layer was first loaded on BaLa4Ti4O15 (Cr2O3/BaLa4Ti4O15), and then Au25(SG)18 was adsorbed on Cr2O3/BaLa4Ti4O15. The obtained catalyst (Au25(SG)18/Cr2O3/BaLa4Ti4O15) was calcined in an electric furnace under low vacuum to remove the ligands of Au25(SG)18 and embed Au25 into the Cr2O3 layer (Fig. 15(c)). The obtained Cr2O3/Au25/BaLa4Ti4O15 photocatalyst showed high water-splitting activity. Thus, we succeeded in creating a highly active water-splitting photocatalyst with the advantage of high HER activity of fine Au NCs by combining miniaturization of the Au cocatalyst with formation of the reverse-reaction prevention layer (Fig. 15(c)). In a later study using Au25 as the cocatalyst, it was found that even higher activity can be obtained by substituting one Pt atom for one Au atom in the Au25 cocatalyst.284 On the basis of the various experimental results, we concluded that Au24Pt/BaLa4Ti4O15 showed higher water-splitting activity than Au25/BaLa4Ti4O15, mainly because (1) photoexcited electrons can more easily transfer from BaLa4Ti4O15 to the metal NCs in Au24Pt/BaLa4Ti4O15 than in Au25/BaLa4Ti4O15 and (2) Au in Au24Pt is more negatively charged than Au in Au25 (Fig. 15(d)).
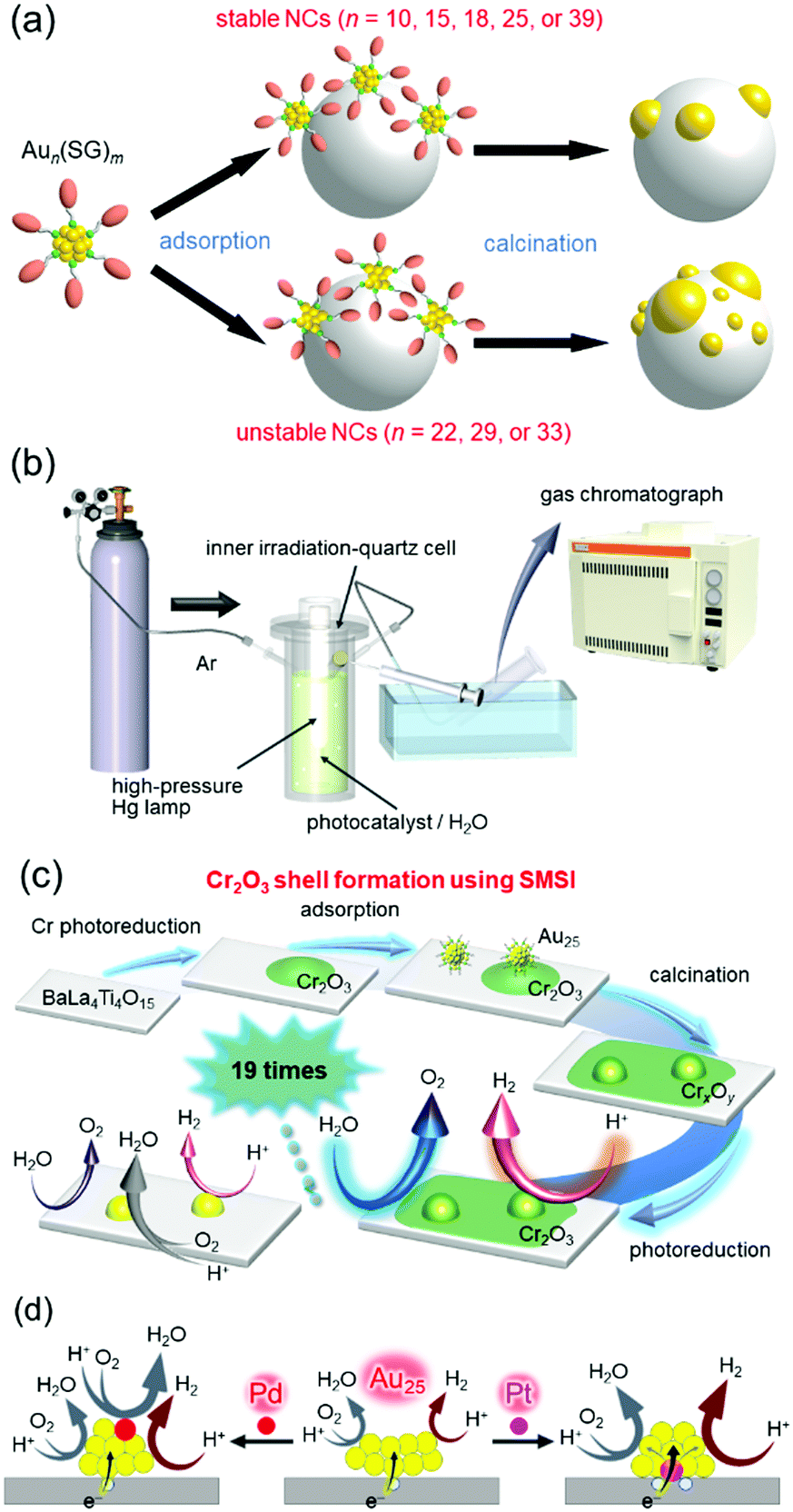 | ||
| Fig. 15 (a) Aun(SG)m suitable for loading Aun NCs on the photocatalyst while retaining the number of constituent atoms.272 (b) Schematic of the system used to estimate the photocatalytic activity.273 (c) Schematic of preparation of Cr2O3/Au25/BaLa4Ti4O15 and its water-splitting activity.273 (d) Proposed structures of Au24M/BaLa4Ti4O15 for M = Au, Pd, and Pt and the effect of the geometric/electronic structure on each reaction over metal-NC cocatalysts.284 Reproduced with permission from ref. 272, 273 and 284. Copyright 2015 American Chemical Society, 2018 American Chemical Society, and 2019 American Chemical Society. | ||
In the above study, Au was used as the metal element for the cocatalyst. However, based on the volcano plot285 for H2 adsorption and desorption, rhodium (Rh) is predicted to have higher catalytic activity than Au for the HER. Therefore, it is expected that highly active water-splitting photocatalysts can be created by loading a fine cocatalyst consisting of rhodium and chromium oxides (Rh2−xCrxO3)286–289 on the photocatalysts. Therefore, we also attempted to load fine Rh2−xCrxO3 NCs on BaLa4Ti4O15. Unfortunately, there have been no reports of precise synthesis of Rh2−xCrxO3 NCs. Therefore, in this experiment, we used Rh2(SG)2 complexes (Rh–SG) as precursors, which were aggregated on BaLa4Ti4O15 and then calcined. As a result, we succeeded in loading monodispersed Rh2−xCrxO3 NCs with particle sizes of 1.3 ± 0.3 nm on BaLa4Ti4O15 (Fig. 16(a and b)).290 The obtained photocatalyst showed the highest apparent quantum yield of 16% for BaLa4Ti4O15 (excitation wavelength = 270 nm, Fig. 16(c)). The loading method of Rh2−xCrxO3 NCs developed in this study can in principle be applied to other photocatalysts. In addition, Rh2−xCrxO3 NCs are useful cocatalysts for many water-splitting photocatalysts.286–289,291,292 In the future, it is expected that high quantum yields will be obtained for many other water-splitting photocatalysts using this technique.
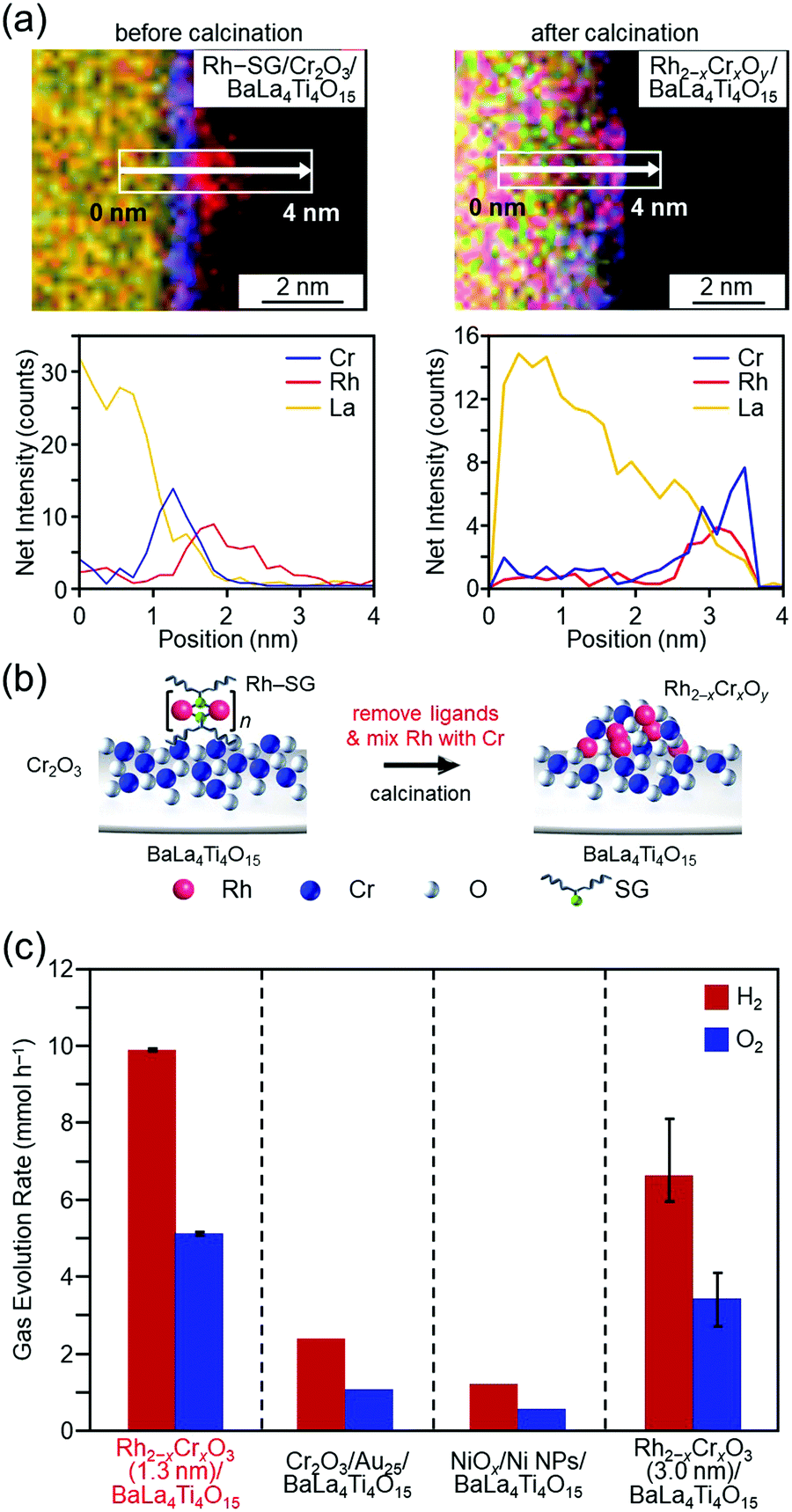 | ||
| Fig. 16 HAADF-STEM images of Rh–SG complexes adsorbed on the Cr2O3/BaLa4Ti4O15 photocatalyst before and after calcination (Rh–SG/Cr2O3/BaLa4Ti4O15 and Rh2−xCrxOy/BaLa4Ti4O15, respectively). (a) Line analysis of elemental mapping of Rh–SG/Cr2O3/BaLa4Ti4O15 (left) and Rh2−xCrxOy/BaLa4Ti4O15 (right). (b) Schematic of the phenomenon that occurred during the calcination process. (c) Comparison of the gas-evolution rates over different photocatalysts: Rh2−xCrxO3 (1.3 nm)/BaLa4Ti4O15 (0.09 wt% Rh and 0.10 wt% Cr), Cr2O3/Au25/BaLa4Ti4O15 (0.10 wt% Au and 0.50 wt% Cr), Ni NiOx/NPs/BaLa4Ti4O15 (0.50 wt% Ni), and Rh2−xCrxO3 (3.0 nm)/BaLa4Ti4O15 (0.10 wt% Rh and 0.15 wt% Cr). Reproduced with permission from ref. 290. Copyright 2020 Willey-VCH. | ||
For practical use of a water-splitting photocatalyst, it is essential that the photocatalyst causes water splitting by visible light (visible-light-driven water-splitting photocatalyst), which accounts for about 40% of solar energy. Currently, there are only a few semiconductor photocatalysts that enable complete water splitting in one step using visible light.264 However, overall water splitting by visible light can also be achieved by photocatalytic systems using a two-step reaction called the Z-scheme, which mimics plant photosynthesis.257,260 We are currently working on improving the functionality of both of these visible-light-driven water-splitting photocatalysts based on the knowledge obtained in research of BaLa4Ti4O15.
5.4. Activation of ORR electrode catalysts
The polymer electrolyte fuel cell (PEFC, Fig. 12) is already in practical use. However, a significant amount of Pt is used in current PEFCs, which makes the current PEFCs extremely expensive. This has impeded widespread use of PEFCs, and thereby transition to a society using H2 as an energy source (Fig. 12) has stagnated. Even if the cost of PEFCs can be lowered by improving the parts other than the Pt catalyst, we will inevitably encounter a shortage of Pt if the current Pt catalyst is used. Therefore, it is essential to reduce the amount of Pt used in PEFC electrocatalysts to build a clean and sustainable society.In PEFCs, the ORR at the cathode is the rate-limiting reaction. Pt NPs with particle sizes of 2–3 nm loaded on carbon black (CB) (Pt NPs/CB) are widely used ORR cathodes. Recently, the following facts were reported by Yamamoto and co-workers,293,294 Nakajima and co-workers,295 and Fischer and co-workers296 for smaller ∼1 nm particle size Ptn NCs: (1) the Ptn NCs have higher mass activity than the currently used Pt NPs and (2) the mass activity of the Ptn NCs dramatically varies depending on the number of constituent atoms. Therefore, it is expected that if highly active ∼1 nm Ptn NCs can be size-selectively synthesized and then loaded on CB, highly active ORR catalysts can be created, which will lead to cost reduction of PEFCs.
As described in Section 4.4, we have already developed a method to isolate [Pt17(PPh3)8(CO)12]z+ (z = 1 or 2) with atomic precision by simple manipulation in air.120 In addition, by further modification of this method, we successfully synthesized a series of Ptn(PET)m(CO)l NCs (n = ∼35, ∼51, and ∼66) in a size-selective manner with a narrow particle-size distribution.121 Ptn/CB catalysts were then prepared by adsorbing these Ptn NCs on CB and partially removing the ligands at appropriate temperatures (Fig. 17(a and b)). We found that the mass activities of Ptn/CB (n = ∼35, ∼51, and ∼66) were higher than that of commercial Pt NPs/CB (Fig. 17(c)).121 Among the Ptn/CB catalysts, Pt∼51/CB showed 2.1 times higher mass activity than the commercial Pt NPs/CB catalyst. On the basis of estimation of the electrochemical activity area and specific activity, we concluded that Ptn/CB shows higher ORR mass activity than commercial Pt NPs/CB because both the percentage of surface Pt atoms and specific activity of each surface Pt atom are higher for Ptn/CB than for commercial Pt NPs/CB.
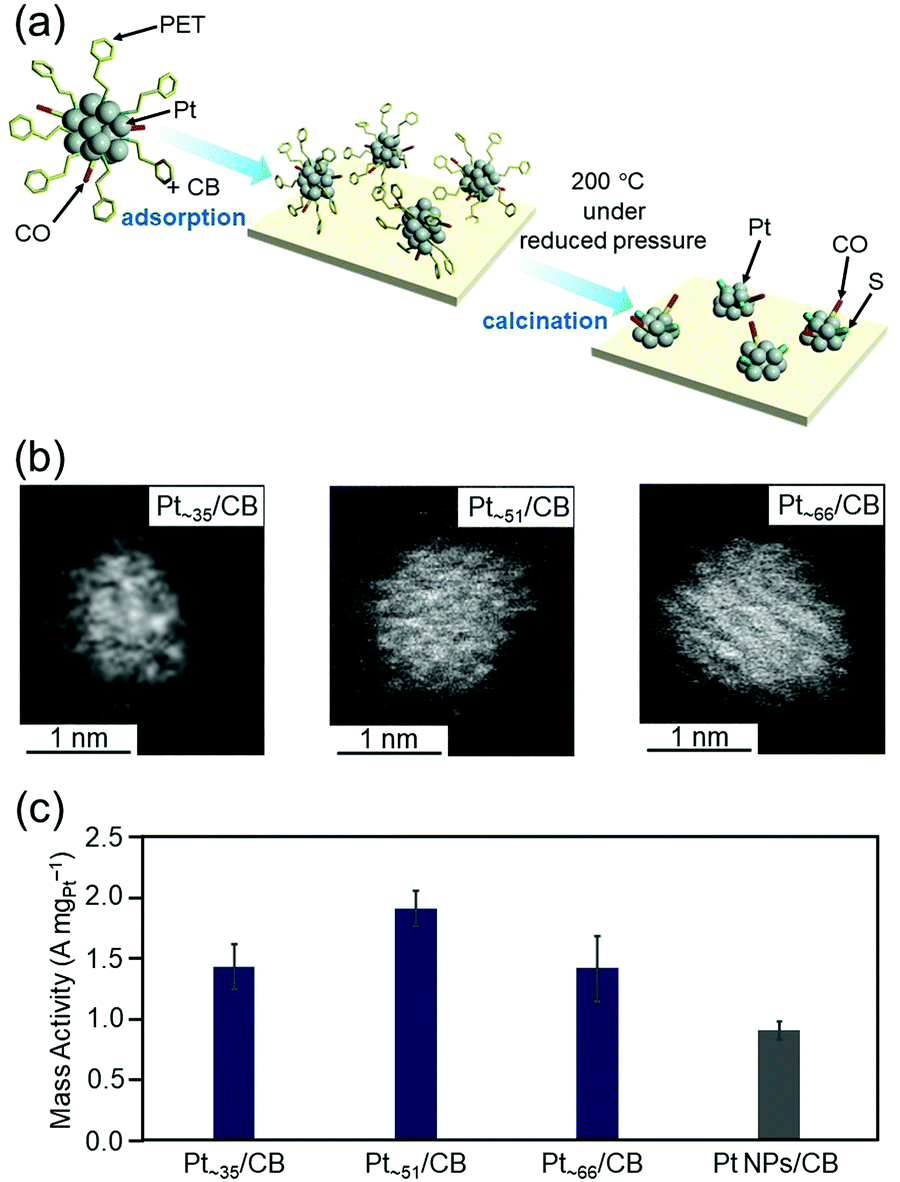 | ||
| Fig. 17 (a) Schematic illustration of controlled loading of ∼1 nm Pt NCs on CB. (b) HAADF-STEM images of Ptn/CB with n = ∼35 (left), ∼51 (center), and ∼66 (right). (c) ORR mass activity at 0.6 V vs. the reversible hydrogen electrode for Pt∼35/CB, Pt∼51/CB, Pt∼66/CB, and commercial Pt NPs/CB. In this figure, the average values obtained by performing the experiments three times are reported. Reproduced from ref. 121. Copyright 2021 The Royal Society of Chemistry. | ||
Regarding the ORR activity of Pt NCs, it has been reported that the presence of Pt atoms in the generalized coordination number (GCN) region of 7.7–8.3 on the surface of the Pt NCs induces high activity.297,298 The GCN is a coordination number that takes into account the second nearest neighbor atoms in addition to the nearest neighbor atoms. When surface atoms are removed and concave cavities are created on the Pt-NC surface, Pt atoms with such GCNs are generated on the surface of the Pt NC. Unfortunately, in our previous studies, we did not obtain detailed information about the geometric structures of Ptn/CB (n = ∼35, ∼51, and ∼66). Therefore, we are currently working on elucidating the details of their geometric structures by obtaining high-resolution high-angle scattering dark-field scanning transmission electron microscopy (HAADF-STEM) images and analyzing the GCNs in the obtained geometric structures by DFT calculations. This is expected to provide a deep understanding of the factors that lead to high activity and clear design guidelines for further enhancement of the activity.
5.5. Activation of automobile exhaust-gas purification catalysts
It is expected that electric vehicles and fuel-cell vehicles will be widely used in the future. However, conventional vehicles (gasoline and diesel vehicles) are also estimated to account for 30–50% of the total market until 2030. Pt is widely used as an exhaust-gas purification catalyst, along with Pd and Rh, because of its catalytic activity to purify the harmful substances contained in exhaust gases.239 However, reduction of Pt use is a critical issue at present because Pt is a rare and expensive precious metal. In addition, with the widespread use of automobiles, such as hybrid vehicles, where the engine is repeatedly stopped and restarted, high activation at low temperatures is also an issue that needs to be overcome.239Studies on Pt-free catalysts are now being performed to overcome the above issues. However, the activity and durability of Pt are overwhelmingly superior to those of non-metallic materials. Reducing the particle size of the Pt catalyst is an extremely effective way to both reduce the amount of Pt used and improve the functionality of the Pt catalyst. Indeed, Anderson and co-workers demonstrated that Ptn NCs (n = 14–18) loaded on alumina (Ptn/Al2O3) have high catalytic activity for CO oxidation, one of the automotive exhaust-gas purification reactions, using a vacuum apparatus.70
We have prepared Pt17/γ-Al2O3 catalysts by adsorbing [Pt17(PPh3)8(CO)12]z+ (z = 1 or 2)120 on γ-Al2O3 and then partially removing the ligands at appropriate temperatures.299 HAADF-STEM images and XAFS measurements showed that Pt does not become oxidized300 in Pt17/γ-Al2O3, and it maintains the skeletal structure of Pt17 NC (Fig. 18(a–c)). To evaluate the catalytic performance of the obtained catalysts under conditions as close as possible to those of actual vehicles, Pt17/γ-Al2O3 was coated on a honeycomb substrate and the catalytic performance for CO or propylene (C3H6) oxidation was evaluated under these conditions (Fig. 18(d)). The measurements of the HAADF-STEM images and catalytic activity were performed in collaboration with Nagaoka of Johnson Matthey Japan.299 We found that Pt17/γ-Al2O3 has higher CO oxidation activity at each temperature than Pt NPs/γ-Al2O3 loaded with Pt NPs with average particle size of 3.1 ± 3.1 nm by the impregnation method, and, therefore, it is capable of purifying CO at lower temperature than Pt NPs/γ-Al2O3 (Fig. 18(e)). Previous studies have suggested that in oxidation of CO, CO and O2 are activated at Pt on the terrace and Pt in the step, respectively.72,301,302 In the structures shown in Fig. 18(b and c), most of the terrace Pt atoms are located near the Pt step. This seems to be one of the factors that leads to effective reaction of CO and O2, that is, oxidation of CO, over Pt17/γ-Al2O3. High oxidation capacity of Pt17/γ-Al2O3 at low temperatures was also observed for oxidation of C3H6. These results indicate that Pt17/γ-Al2O3 is a promising catalyst for reducing the amount of Pt used and improving the catalytic performance at low temperatures.
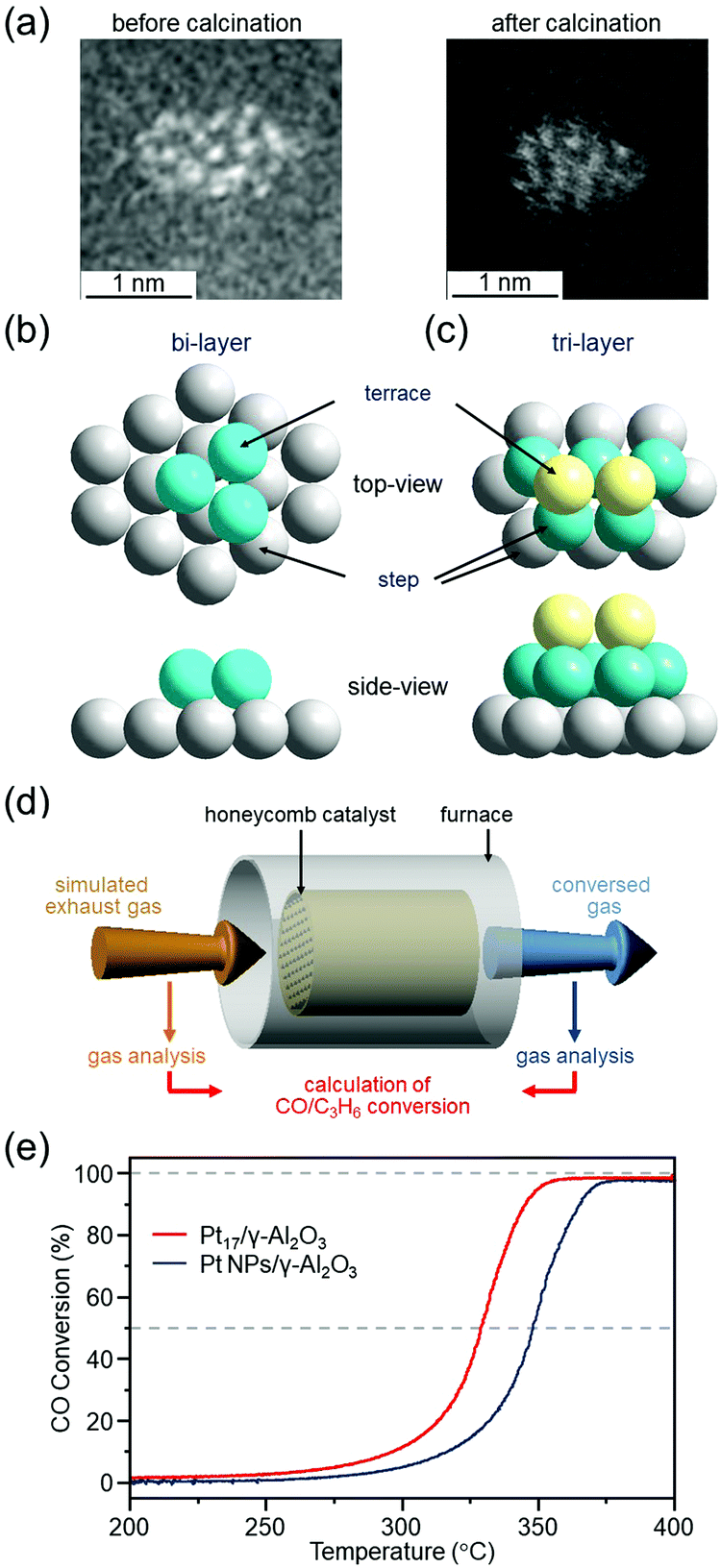 | ||
| Fig. 18 (a) HAADF-STEM images of Pt17(PPh3)8(CO)12/γ-Al2O3 (left) and Pt17/γ-Al2O3 (right). Two proposed structures for Pt17 on γ-Al2O3 based on the HAADF-STEM images of Pt17/γ-Al2O3: (b) bi-layer and (c) tri-layer. In (b) and (c), both top and side views are shown. (d) Schematic illustration of estimation of CO and C3H6 conversion over Pt17/γ-Al2O3 or Pt NPs/γ-Al2O3 coated on a cordierite honeycomb substrate. (e) CO conversion over Pt17/γ-Al2O3 (red curve) and Pt NPs/γ-Al2O3 (gray curve). Reproduced from ref. 299. Copyright 2020 The Royal Society of Chemistry. | ||
To use Pt17/γ-Al2O3 as a practical catalyst, it is necessary to investigate the catalytic activity and durability of the catalyst with the loading amount in the actual vehicle and at the exhaust-gas mixture ratio302–305 during actual operation. We are currently performing measurements under such conditions in collaboration with industry.
5.6. Elucidation of the ligand-removal mechanism
In our catalyst preparation process (Fig. 13(c)), (1) metal NCs are precisely synthesized using ligands and then (2) the resulting NCs are adsorbed on a support. The presence of the ligand on the surface of the metal NCs generally leads to a decrease in the activity306,307 because it prevents the approach of the reactants to the metal-NC surface and induces modulation of the electronic structure of the metal NCs. Therefore, (3) some or all of the ligands are removed by calcination to produce high-activity metal NCs. However, removal of the ligands simultaneously induces aggregation of the metal NCs and a resulting decrease in the catalytic activity. Thus, in calcination, it is extremely important to remove only the ligands while maintaining the number of constituent atoms of the metal NCs (Fig. 19(a)).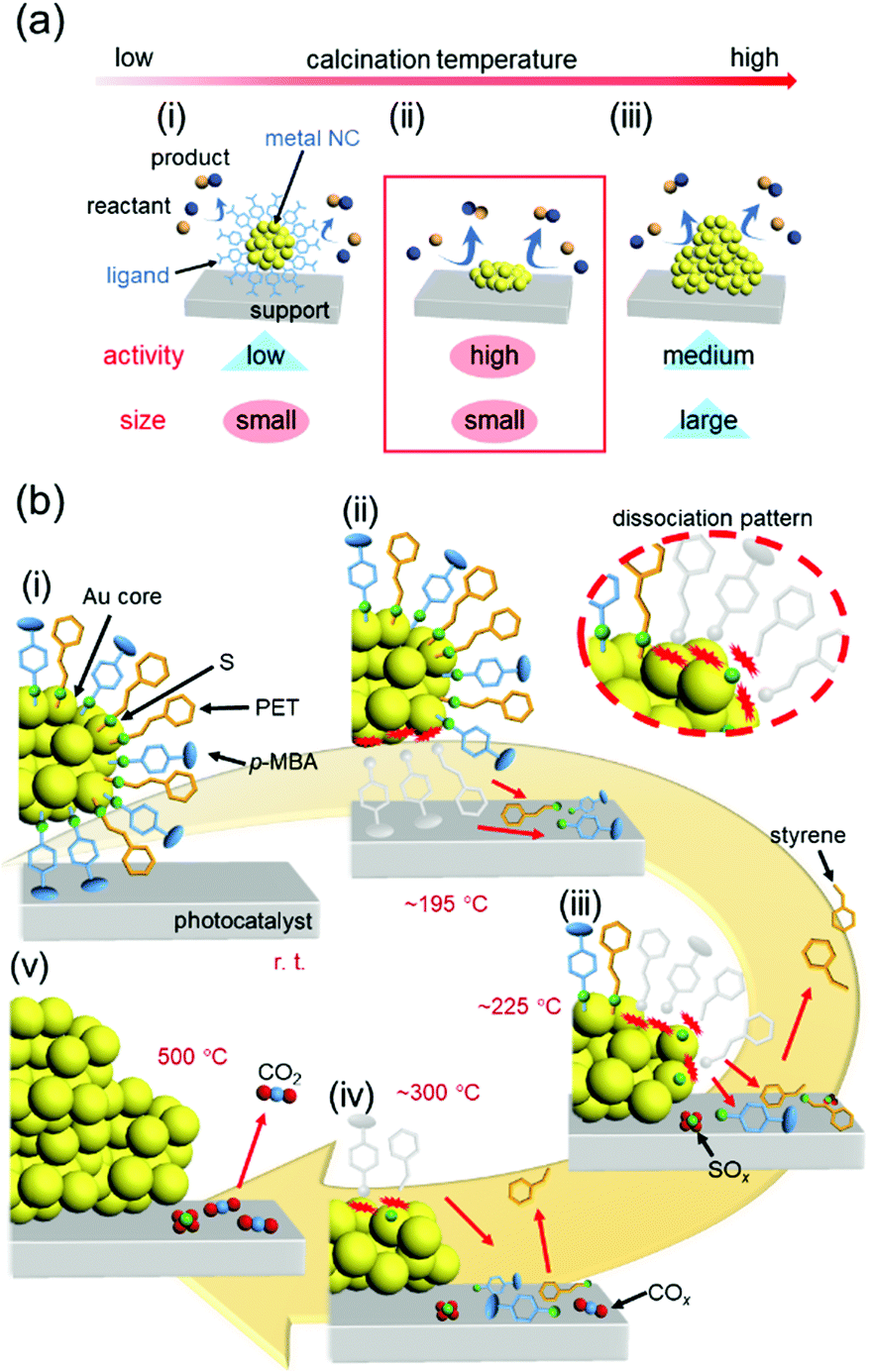 | ||
| Fig. 19 (a) Schematic illustrations of the typical phenomena caused by increasing the calcination temperature in metal-oxide-supported ligand-protected metal NCs: (i) a small size is maintained but the activity is low, (ii) high activity is achieved while maintaining a small size, and (iii) the activity decreases owing to aggregation. (b) Proposed phenomenon occurring at each temperature during calcination of Au25(PET)∼9(p-MBA)∼9/BaLa4Ti4O15 at (i) room temperature, (ii) ca. 195 °C, (iii) ca. 225 °C, (iv) ca. 300 °C, and (v) 500 °C. Reproduced from ref. 308. Copyright 2021 Willey-VCH. | ||
Regarding removal of the ligands, in the studies reported in Section 5.3–5.5, the calcination conditions were determined by confirming the presence or absence of ligands in the catalysts after calcination by X-ray photoelectron spectroscopy (XPS) and XAFS. However, through these studies, we felt that it was essential to understand the reaction mechanisms occurring during calcination to perform calcination under appropriate conditions and thereby create high-performance heterogeneous catalysts. Therefore, we recently worked on elucidation of the reaction mechanism during ligand calcination.308 Specifically, the ligand desorption process in Au25(PET)∼9(p-MBA)∼9/BaLa4Ti4O15 during calcination was followed by five techniques: direct insertion probe-mass spectrometry, XAFS, Fourier-transform infrared spectroscopy, XPS, and TEM observation. As a result, we succeeded in elucidating that the ligand desorption process consists of dissociation of the ligand on the metal-NC surface, adsorption of the generated compound on the support, and desorption of the compound from the support, and at what temperatures these processes occur (Fig. 19(b)). Based on the obtained knowledge, we also succeeded in developing a method to form the Cr2O3 shell on the surface of Au NCs while preventing their aggregation, and thereby obtained a highly active and stable water-splitting photocatalyst.308
The desorption temperature of SR from the Au-NC surface is strongly related to the interactions between the ligands on the Au-NC surface, as well as the strengths of the Au–S and S–C bonds.308 In addition, the temperature for desorption from the support is related to the strength of the compound–support interaction.309 Furthermore, the easiness of dissociation/desorption of the ligands and the resulting aggregation of the Au NCs slightly varies depending on the calcination atmosphere.309,310 However, previous calcinations with different SR functional groups, supports, and atmospheres have often suggested occurrence of the phenomena in Fig. 19(b).243,271,309,311–314 Therefore, it is assumed that a phenomenon similar to Fig. 19(b) also occurs in calcination of the other Aun(SR)m NCs/metal oxides, although the temperature at which it occurs is different. There had not been a unified view on the behavior in Aun(SR)m NCs/metal oxides during calcination before this study.312,315 We succeeded in elucidating the details of the phenomena occurring during calcination of Au25(PET)∼9(p-MBA)∼9/metal-oxide catalysts by combining five measurements. The knowledge gained in this study is expected to provide clear design guidelines for creation of high-performance heterogeneous catalysts.
5.7. Summary of activation of energy and environmental catalysts using controlled metal NCs
We succeeded in precisely controlling the metal NCs on the support with atomic precision using precise ligand-protected metal NCs as precursors. Studies of the obtained energy and environmental catalysts revealed the correlation between the number of constituent atoms/chemical composition of the loaded metal NCs and the material function of each catalyst. Through these studies, each catalyst has been successfully highly functionalized. Specifically, we have achieved producing the highest water-splitting activity for BaLa4Ti4O15,290 one of the most advanced water-splitting photocatalyst, creating electrode catalysts with higher ORR activity than the Pt catalysts currently used in fuel cells121 and improving the functionality of automotive exhaust-gas purification catalysts.299 These results strongly suggest that the bottom-up nanotechnology established in the metal-NC field can contribute to construction of a clean and sustainable society, that is, provide solutions for the issues faced by modern society.For these catalytic applications, in the future, we will work on alloying316 with various metal elements for the metal NC contained in each catalyst. In addition, the geometric and electronic structures of the loaded metal NCs during the reaction317 will be revealed to gain a better understanding of the structure–property relationship of the loaded metal NCs and provide design guidelines for further enhancement of their activity. We believe that these catalysts could be further enhanced by repeating the cycle shown in Fig. 14 while incorporating the obtained knowledge. Regarding these applied studies, we would also like to continue our study while deepening both the science and technology of metal NCs.
6. Conclusions
Nanoscale materials have the following interesting and important features: (1) they possess great potential and value that cannot be found in bulk materials and (2) the nanoscale is the level of emergence of material functions, and thus if materials can be controlled at the nanoscale, we can control the properties of the larger materials. In this paper, I have described the contents of and motivation for my own studies in this field. The knowledge and skills required to study metal NCs are likely to change in the future, depending on the stage of the study and the situation in the world. However, the importance of understanding and controlling metal NCs will not change because they are deeply related to understanding and controlling various materials in the fields of energy, environment, and materials. I hope that the science and technology in the metal-NC field will be further advanced in the future through collaboration among researchers in many fields across physics, chemistry, and biology.7. Outlook
The studies that we will perform in the future or are currently being performed have already been described at the end of each section. Therefore, in this section, I will describe the studies to be performed in the future for further development of the field of metal NCs (Fig. 20).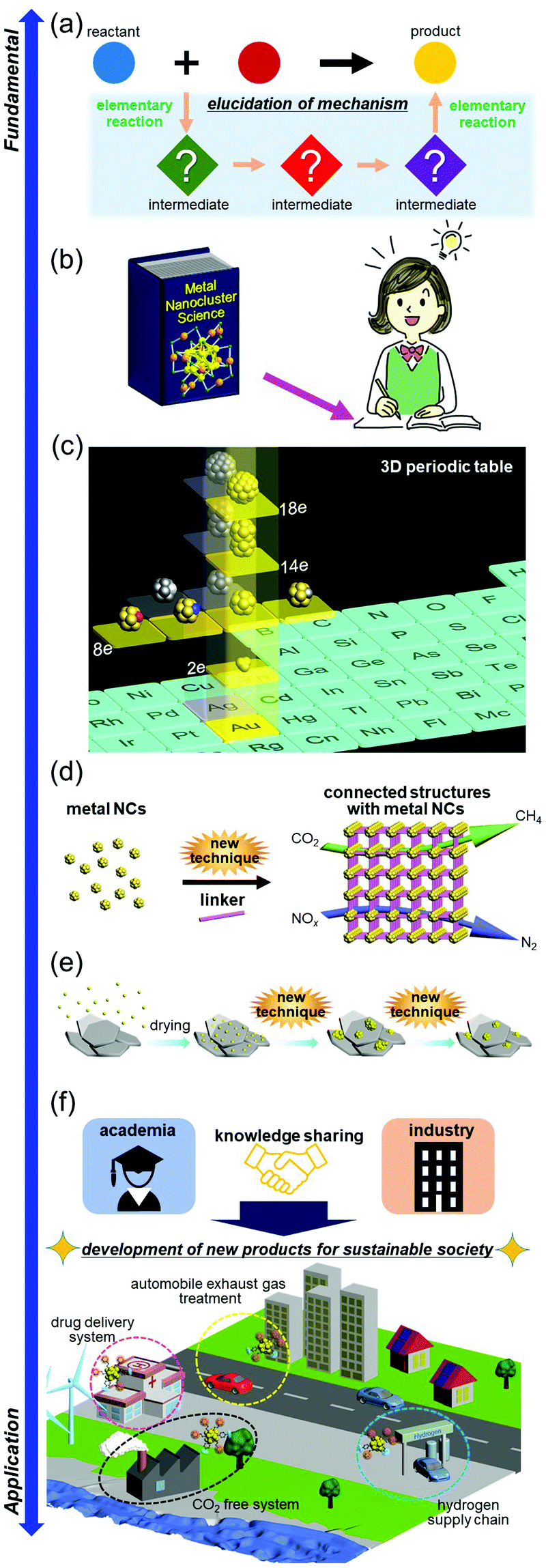 | ||
| Fig. 20 Outlook in the metal-NC field. (a) Elucidation of the synthesis mechanism. (b) Preparation of a textbook for students. (c) Development of super-atomic chemistry. This figure was reproduced with permission from ref. 330. (d) Control of the connected structures. (e) Control of the supported metal NCs without calcination at high temperature. (f) Industry–academia collaboration to realize industrial development and a more comfortable society. Copyright 2021 Willey-VCH. | ||
7.1. Elucidation of the synthesis mechanism
Recent work by Xie and co-workers has revealed the details of the reaction mechanism for synthesis of hydrophilic SR-protected metal NCs.318,319 In addition, recent studies by Pradeep and co-workers,320–322 Bürgi and co-workers,323 and Pei and co-workers324 have advanced our understanding of the reaction mechanism for synthesis of ligand-protected alloy NCs through metal exchange. However, the mechanism for synthesis of hydrophobic ligand-protected metal NCs in a two-phase system, which was first reported by Brust and co-workers,95 has yet to be elucidated at the atomic and molecular levels. Similarly, ligand-exchange-induced structure transformation325 is not yet understood at the atomic or molecular level with respect to the chemical composition that induces the structural transformation. In the future, it is expected that high-resolution separation, mass spectrometry, ion-mobility experiments, and theoretical calculations will be used to clarify the details of reactions whose mechanisms are currently unknown (Fig. 20(a)), which will make it possible to further control synthesis of metal NCs.7.2. Preparation of a textbook for students
For ligand-protected metal NCs, there was not a good textbook until 2014.10 However, taking into account the number of syntheses, reactions, and applications reported so far, it seems that metal-NC chemistry has reached a stage where many textbooks for undergraduate and graduate students should be published, like for organic chemistry326 and coordination chemistry.327 Publication of such a textbook would dramatically increase the number of students and researchers working on metal-NC research, thereby further deepening and expanding the field of metal NCs. Publication of a textbook that includes the latest information on metal NCs is an urgent issue for creation of new materials with size-specific properties and thereby solving many issues in the world (Fig. 20(b)).7.3. Development of super-atomic chemistry
Metal NCs, which consist of metal atoms such as Au and Ag, become stable when the total number of valence electrons fills a closed-shell electronic structure. Because this phenomenon is similar to that for atoms, metal NCs can be considered to be super-atoms.328 Super-atoms are expected to be the constituent units of a variety of functional materials, like conventional atoms. The use of super-atoms as constituent units is expected to lead to creation of materials that are difficult to create with conventional atoms. Several researchers have been actively working on super-atomic metal NCs, in the fields of both gas-phase metal NCs67,329 and ligand-protected metal NCs.330,331 However, more researchers need to be involved in this field to enrich the library of super-atomic metal NCs, just like not all conventional atoms were discovered by only a few researchers. It is expected that the number of researchers working on super-atomic metal NCs will increase in the future, which will lead to completion of a three-dimensional periodic table (Fig. 20(c)).3307.4. Control of connected structures
Although studies of applications of ligand-protected metals NCs have been performed, at present, their main applications are limited to catalysts and chemical sensors in the dispersed state. If ligand-protected metal NCs could be connected in a regular pattern, they could be applied to designable electronic and luminescent devices and reaction-selective highly active catalysts. Connection techniques of ligand-protected metal NCs would also be indispensable for creation of materials with super-atomic metal NCs (Section 7.3) as a constituent unit. However, at present, there are few examples of direct connection of ligand-protected metal NCs through metal–metal bonds. Since 2017, connection of metal NCs through linker molecules has also been vigorously studied by several groups, including Zang and co-workers.208 However, there have been few reports on connected structures, except for Ag NCs. In the future, it is expected that techniques to connect various ligand-protected metal NCs (synthesis of connected structures) will be developed, which will lead to creation of more diverse functions and expansion of the application fields (Fig. 20(d)).7.5. Control of supported metal NCs without calcination at high temperature
As described in Section 5, it has recently become possible to control the number of constituent atoms and chemical composition of the loaded metal NCs on the support using ligand-protected metal NCs as a precursor.332 However, the method involves removal of the ligands by calcination at high temperature, which wastes energy. This waste of energy might become a hindrance to industrial application of metal NCs. Therefore, it is expected that a new method to control the number of constituent atoms and chemical composition of the loaded metal NCs on the support will be developed in the future (Fig. 20(e)). I expect that such control can be realized if a size-convergence method can be found for loaded metal NCs, similar to the case of ligand-protected metal NCs dispersed in solution. In other words, it is expected that the number of constituent atoms and chemical composition of the loaded metal NCs can be controlled by combining the conventional loading method and size-convergence method. If such a method can be developed, it might also be possible to control the geometric structure of the loaded metal NCs. Although control of ligand-protected metal NCs dispersed in solution was once thought to be extremely difficult to achieve with atomic precision, many control methods have been developed.7 I hope that control of the number of constituent atoms and chemical composition of the loaded metal NCs on the support by an easier method will also be achieved through the enthusiasm of the readers of this paper.7.6. Collaboration between academia and industry
The researchers in the field of metal NCs have discovered potential applications of metal NCs in various fields, as well as new facts hidden in nanoscale materials. For example, the applications described in Section 5 are such examples. Collaboration between industry and academia seems to be essential for successfully connecting the scientific discoveries found in these academic fields to development of industry. It is not necessary for all researchers in the academic field to collaborate with industry because the challenges that need to be overcome in the academic field and in industry are probably different. Thus, I would like to encourage young researchers to continue to make great effort to explore the new science hidden in the metal-NC field. However, the field of metal NCs is important in terms of both science and technology, and the techniques established in the field of metal NCs also have the potential to contribute to construction of the next-generation society (Section 5). For this reason, I urge researchers who already have the seeds of technology to also engage in industry–university collaborations so that the knowledge and techniques discovered in the field of metal NCs can be successfully linked to development of industry and society (Fig. 20(f)).Conflicts of interest
There are no conflicts to declare.Acknowledgements
I thank the collaborators of the work summarized in this review, especially Prof. Koji Kaya, Prof. Atsushi Nakajima (Keio University), and Prof. Tatsuya Tsukuda (The University of Tokyo), for their guidance in the early stage of my career. I also thank Mr Masanobu Kawachi, Mr Shun Kato, and Mr Yusuke Horita for their assistance with making the figures. This work was supported by the Japan Society for the Promotion of Science (JSPS) KAKENHI (grant numbers 20H02698 and 20H02552), Scientific Research on Innovative Areas “Coordination Asymmetry” (grant numbers 17H05385 and 19H04595), Scientific Research on Innovative Areas “Innovations for Light-Energy Conversion” (grant numbers 18H05178 and 20H05115), and the JST Adaptable and Seamless Technology transfer Program through Target-driven R&D (A-STEP, grant number JPMJTM20MS). Funding from Nissanken, the Yashima Environment Technology Foundation, and the Yazaki Memorial Foundation for Science and Technology is gratefully acknowledged.References
- R. P. Feynman, There's Plenty of Room at the Bottom An Invitation to Enter a New Field of Physics, 1960 Search PubMed.
- G. Binnig, H. Rohrer, C. Gerber and E. Weibel, Phys. Rev. Lett., 1982, 49, 57–61 CrossRef.
- G. Binnig, C. F. Quate and C. Gerber, Phys. Rev. Lett., 1986, 56, 930–933 CrossRef PubMed.
- V. E. Bondybey and J. H. English, J. Chem. Phys., 1982, 76, 2165–2170 CrossRef CAS.
- R. E. Smalley, Laser Chem., 1983, 2, 167–184 CrossRef CAS.
- N. Lane, National Nanotechnology Initiative: The Initiative and Its Implementation Plan, Washington, D. C., 2000 Search PubMed.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- X. Kang, Y. Li, M. Zhu and R. Jin, Chem. Soc. Rev., 2020, 49, 6443–6514 RSC.
- T. Tsukuda and H. Häkkinen, Protected Metal Clusters: From Fundamentals to Applications, Elsevier, Amsterdam, 2015 Search PubMed.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526–622 CrossRef CAS PubMed.
- H. Hirai, S. Takano, T. Nakamura and T. Tsukuda, Inorg. Chem., 2020, 59, 17889–17895 CrossRef CAS PubMed.
- K. Konishi, M. Iwasaki and Y. Shichibu, Acc. Chem. Res., 2018, 51, 3125–3133 CrossRef CAS PubMed.
- C. E. Briant, B. R. C. Theobald, J. W. White, L. K. Bell, D. M. P. Mingos and A. J. Welch, J. Chem. Soc., Chem. Commun., 1981, 201–202 RSC.
- M. McPartlin, R. Mason and L. Malatesta, J. Chem. Soc. D, 1969, 334 RSC.
- E. G. Mednikov and L. F. Dahl, Philos. Trans. R. Soc., A, 2010, 368, 1301–1332 CrossRef CAS PubMed.
- G. Schmid, Chem. Rev., 1992, 92, 1709–1727 CrossRef CAS.
- M. Schulz-Dobrick and M. Jansen, Z. Anorg. Allg. Chem., 2007, 633, 2326–2331 CrossRef CAS.
- B. K. Teo, X. Shi and H. Zhang, J. Am. Chem. Soc., 1992, 114, 2743–2745 CrossRef CAS.
- J. D. Roth, G. J. Lewis, L. K. Safford, X. Jiang, L. F. Dahl and M. J. Weaver, J. Am. Chem. Soc., 1992, 114, 6159–6169 CrossRef CAS.
- A. Ceriotti, N. Masciocchi, P. Macchi and G. Longoni, Angew. Chem., Int. Ed., 1999, 38, 3724–3727 CrossRef CAS PubMed.
- I. Ciabatti, C. Femoni, M. C. Iapalucci, G. Longoni and S. Zacchini, J. Cluster Sci., 2014, 25, 115–146 CrossRef CAS.
- S. S. Kurasov, N. K. Eremenko, Y. L. Slovokhotov and Y. T. Struchkov, J. Organomet. Chem., 1989, 361, 405–408 CrossRef CAS.
- L. Hao, G. J. Spivak, J. Xiao, J. J. Vittal and R. J. Puddephatt, J. Am. Chem. Soc., 1995, 117, 7011–7012 CrossRef CAS.
- V. G. Albano, P. L. Bellon, M. Manassero and M. Sansoni, J. Chem. Soc. D, 1970, 1210–1211 RSC.
- P. Chini, J. Organomet. Chem., 1980, 200, 37–61 CrossRef CAS.
- M. Paolieri, I. Ciabatti and M. Fontani, J. Cluster Sci., 2019, 30, 1623–1631 CrossRef CAS.
- H. Yu, B. Rao, W. Jiang, S. Yang and M. Zhu, Coord. Chem. Rev., 2019, 378, 595–617 CrossRef CAS.
- M. Agrachev, M. Ruzzi, A. Venzo and F. Maran, Acc. Chem. Res., 2019, 52, 44–52 CrossRef CAS PubMed.
- K. Kwak and D. Lee, Acc. Chem. Res., 2019, 52, 12–22 CrossRef CAS PubMed.
- B. Nieto-Ortega and T. Bürgi, Acc. Chem. Res., 2018, 51, 2811–2819 CrossRef CAS PubMed.
- N. A. Sakthivel and A. Dass, Acc. Chem. Res., 2018, 51, 1774–1783 CrossRef CAS PubMed.
- R. L. Whetten, H.-C. Weissker, J. J. Pelayo, S. M. Mullins, X. López-Lozano and I. L. Garzón, Acc. Chem. Res., 2019, 52, 34–43 CrossRef CAS PubMed.
- B. Bhattarai, Y. Zaker, A. Atnagulov, B. Yoon, U. Landman and T. P. Bigioni, Acc. Chem. Res., 2018, 51, 3104–3113 CrossRef CAS PubMed.
- Z. Gan, N. Xia and Z. Wu, Acc. Chem. Res., 2018, 51, 2774–2783 CrossRef CAS PubMed.
- A. Ghosh, O. F. Mohammed and O. M. Bakr, Acc. Chem. Res., 2018, 51, 3094–3103 CrossRef CAS PubMed.
- J. Yan, B. K. Teo and N. Zheng, Acc. Chem. Res., 2018, 51, 3084–3093 CrossRef CAS PubMed.
- S. Lee, M. S. Bootharaju, G. Deng, S. Malola, W. Baek, H. Häkkinen, N. Zheng and T. Hyeon, J. Am. Chem. Soc., 2020, 142, 13974–13981 CrossRef CAS PubMed.
- H. Kawamata, Y. Negishi, R. Kishi, S. Iwata, A. Nakajima and K. Kaya, J. Chem. Phys., 1996, 105, 5369–5376 CrossRef CAS.
- Y. Negishi, H. Kawamata, F. Hayakawa, A. Nakajima and K. Kaya, Chem. Phys. Lett., 1998, 294, 370–376 CrossRef CAS.
- Y. Negishi, T. Yasuike, F. Hayakawa, M. Kizawa, S. Yabushita, A. Nakajima and K. Kaya, J. Chem. Phys., 2000, 113, 1725–1731 CrossRef CAS.
- Y. Negishi and T. Tsukuda, J. Am. Chem. Soc., 2003, 125, 4046–4047 CrossRef CAS PubMed.
- Y. Shichibu, Y. Negishi, T. Tsukuda and T. Teranishi, J. Am. Chem. Soc., 2005, 127, 13464–13465 CrossRef CAS PubMed.
- Y. Negishi, H. Tsunoyama, Y. Yanagimoto and T. Tsukuda, Chem. Lett., 2005, 34, 1638–1639 CrossRef CAS.
- Y. Negishi, H. Tsunoyama, M. Suzuki, N. Kawamura, M. M. Matsushita, K. Maruyama, T. Sugawara, T. Yokoyama and T. Tsukuda, J. Am. Chem. Soc., 2006, 128, 12034–12035 CrossRef CAS PubMed.
- Y. Negishi, N. K. Chaki, Y. Shichibu, R. L. Whetten and T. Tsukuda, J. Am. Chem. Soc., 2007, 129, 11322–11323 CrossRef CAS PubMed.
- N. K. Chaki, Y. Negishi, H. Tsunoyama, Y. Shichibu and T. Tsukuda, J. Am. Chem. Soc., 2008, 130, 8608–8610 CrossRef CAS PubMed.
- T. Kawawaki, A. Ebina, Y. Hosokawa, S. Ozaki, D. Suzuki, S. Hossain and Y. Negishi, Small, 2021, 17, 2005328 CrossRef CAS PubMed.
- Y. Negishi, W. Kurashige, Y. Niihori and K. Nobusada, Phys. Chem. Chem. Phys., 2013, 15, 18736–18751 RSC.
- Y. Negishi, Bull. Chem. Soc. Jpn., 2014, 87, 375–389 CrossRef CAS.
- W. Kurashige, Y. Niihori, S. Sharma and Y. Negishi, Coord. Chem. Rev., 2016, 320–321, 238–250 CrossRef CAS.
- Y. Niihori, S. Hossain, S. Sharma, B. Kumar, W. Kurashige and Y. Negishi, Chem. Rec., 2017, 17, 473–484 CrossRef CAS PubMed.
- Y. Niihori, C. Uchida, W. Kurashige and Y. Negishi, Phys. Chem. Chem. Phys., 2016, 18, 4251–4265 RSC.
- S. Hossain, Y. Niihori, L. V. Nair, B. Kumar, W. Kurashige and Y. Negishi, Acc. Chem. Res., 2018, 51, 3114–3124 CrossRef CAS PubMed.
- Y. Niihori, K. Yoshida, S. Hossain, W. Kurashige and Y. Negishi, Bull. Chem. Soc. Jpn., 2019, 92, 664–695 CrossRef CAS.
- T. Kawawaki, Y. Kataoka, S. Ozaki, M. Kawachi, M. Hirata and Y. Negishi, Chem. Commun., 2021, 57, 417–440 RSC.
- T. Kawawaki, N. Shimizu, Y. Mitomi, D. Yazaki, S. Hossain and Y. Negishi, Bull. Chem. Soc. Jpn., 2021, 94, 2853–2870 CrossRef CAS.
- W. Kurashige, Y. Niihori, S. Sharma and Y. Negishi, J. Phys. Chem. Lett., 2014, 5, 4134–4142 CrossRef CAS PubMed.
- Y. Niihori, S. Hossain, B. Kumar, L. V. Nair, W. Kurashige and Y. Negishi, APL Mater., 2017, 5, 053201 CrossRef.
- T. Kawawaki, Y. Negishi and H. Kawasaki, Nanoscale Adv., 2020, 2, 17–36 RSC.
- Y. Negishi, S. Hashimoto, A. Ebina, K. Hamada, S. Hossain and T. Kawawaki, Nanoscale, 2020, 12, 8017–8039 RSC.
- A. Ebina, S. Hossain, H. Horihata, S. Ozaki, S. Kato, T. Kawawaki and Y. Negishi, Nanomaterials, 2020, 10, 1105 CrossRef CAS PubMed.
- T. Kawawaki and Y. Negishi, Nanomaterials, 2020, 10, 238 CrossRef CAS PubMed.
- T. Kawawaki, Y. Mori, K. Wakamatsu, S. Ozaki, M. Kawachi, S. Hossain and Y. Negishi, J. Mater. Chem. A, 2020, 8, 16081–16113 RSC.
- T. Kawawaki, Y. Imai, D. Suzuki, S. Kato, I. Kobayashi, T. Suzuki, R. Kaneko, S. Hossain and Y. Negishi, Chem. – Eur. J., 2020, 26, 16150–16193 CrossRef CAS PubMed.
- T. Kawawaki, Y. Kataoka, M. Hirata, Y. Iwamatsu, S. Hossain and Y. Negishi, Nanoscale Horiz., 2021, 6, 409–448 RSC.
- M. Akutsu, K. Koyasu, J. Atobe, N. Hosoya, K. Miyajima, M. Mitsui and A. Nakajima, J. Phys. Chem. A, 2006, 110, 12073–12076 CrossRef CAS PubMed.
- A. Sanchez, S. Abbet, U. Heiz, W.-D. Schneider, H. Häkkinen, R. N. Barnett and U. Landman, J. Phys. Chem. A, 1999, 103, 9573–9578 CrossRef CAS.
- S. Nagaoka, T. Matsumoto, K. Ikemoto, M. Mitsui and A. Nakajima, J. Am. Chem. Soc., 2007, 129, 1528–1529 CrossRef CAS PubMed.
- F. S. Roberts, M. D. Kane, E. T. Baxter and S. L. Anderson, Phys. Chem. Chem. Phys., 2014, 16, 26443–26457 RSC.
- J. Li, X. Li, H.-J. Zhai and L.-S. Wang, Science, 2003, 299, 864–867 CrossRef CAS PubMed.
- Y. Watanabe, X. Wu, H. Hirata and N. Isomura, Catal. Sci. Technol., 2011, 1, 1490–1495 RSC.
- A. Bettac, L. Köller, V. Rank and K. H. Meiwes-Broer, Surf. Sci., 1998, 402–404, 475–479 CrossRef CAS.
- T. P. Martin, T. Bergmann, H. Göhlich and T. Lange, J. Phys. Chem., 1991, 95, 6421–6429 CrossRef CAS.
- S. C. Richtsmeier, E. K. Parks, K. Liu, L. G. Pobo and S. J. Riley, J. Chem. Phys., 1985, 82, 3659–3665 CrossRef CAS.
- P. Fayet, F. Granzer, G. Hegenbart, E. Moisar, B. Pischel and L. Wöste, Z. Phys. D: At., Mol. Clusters, 1986, 3, 299–302 CrossRef CAS.
- K. Tono, A. Terasaki, T. Ohta and T. Kondow, Chem. Phys. Lett., 2007, 449, 276–281 CrossRef CAS.
- K. Sattler, J. Mühlbach and E. Recknagel, Phys. Rev. Lett., 1980, 45, 821–824 CrossRef CAS.
- S. Gilb, K. Jacobsen, D. Schooss, F. Furche, R. Ahlrichs and M. M. Kappes, J. Chem. Phys., 2004, 121, 4619–4627 CrossRef CAS PubMed.
- W. E. Kaden, T. Wu, W. A. Kunkel and S. L. Anderson, Science, 2009, 326, 826–829 CrossRef CAS PubMed.
- P. Kruit and F. H. Read, J. Phys. E: Sci. Instrum., 1983, 16, 313–324 CrossRef CAS.
- Y. Negishi, H. Kawamata, T. Hayase, M. Gomei, R. Kishi, F. Hayakawa, A. Nakajima and K. Kaya, Chem. Phys. Lett., 1997, 269, 199–207 CrossRef CAS.
- Y. Negishi, H. Kawamata, A. Nakajima and K. Kaya, J. Electron Spectrosc. Relat. Phenom., 2000, 106, 117–125 CrossRef CAS.
- Y. Negishi, Y. Nakamura, A. Nakajima and K. Kaya, J. Chem. Phys., 2001, 115, 3657–3663 CrossRef CAS.
- Y. Negishi, S. Nagao, Y. Nakamura, A. Nakajima, S. Kamei and K. Kaya, J. Appl. Phys., 2000, 88, 6037–6043 CrossRef CAS.
- T. Tsukuda, N. Kimura, T. Sasaki and T. Nagata, Trans. MRS-J., 2000, 25, 929–932 CAS.
- Y. Negishi, H. Murayama and T. Tsukuda, Chem. Phys. Lett., 2002, 366, 561–566 CrossRef CAS.
- Y. Negishi and T. Tsukuda, Chem. Phys. Lett., 2004, 383, 161–165 CrossRef CAS.
- R. G. Nuzzo and D. L. Allara, J. Am. Chem. Soc., 1983, 105, 4481–4483 CrossRef CAS.
- C. A. Widrig, C. A. Alves and M. D. Porter, J. Am. Chem. Soc., 1991, 113, 2805–2810 CrossRef CAS.
- L. Strong and G. M. Whitesides, Langmuir, 1988, 4, 546–558 CrossRef CAS.
- R. D. Piner, J. Zhu, F. Xu, S. Hong and C. A. Mirkin, Science, 1999, 283, 661–663 CrossRef CAS PubMed.
- D. S. Ginger, H. Zhang and C. A. Mirkin, Angew. Chem., Int. Ed., 2004, 43, 30–45 CrossRef PubMed.
- A. Kumar and G. M. Whitesides, Appl. Phys. Lett., 1993, 63, 2002–2004 CrossRef CAS.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- E. G. Mednikov, N. K. Eremenko, Y. L. Slovokhotov and Y. T. Struchkov, J. Organomet. Chem., 1986, 301, C35–C37 CrossRef CAS.
- T. G. Schaaff, M. N. Shafigullin, J. T. Khoury, I. Vezmar, R. L. Whetten, W. G. Cullen, P. N. First, C. Gutiérrez-Wing, J. Ascensio and M. J. Jose-Yacamán, J. Phys. Chem. B, 1997, 101, 7885–7891 CrossRef CAS.
- V. L. Jimenez, D. G. Georganopoulou, R. J. White, A. S. Harper, A. J. Mills, D. Lee and R. W. Murray, Langmuir, 2004, 20, 6864–6870 CrossRef CAS PubMed.
- T. G. Schaaff, G. Knight, M. N. Shafigullin, R. F. Borkman and R. L. Whetten, J. Phys. Chem. B, 1998, 102, 10643–10646 CrossRef CAS.
- T. G. Schaaff and R. L. Whetten, J. Phys. Chem. B, 2000, 104, 2630–2641 CrossRef CAS.
- B. Palpant, Y. Negishi, M. Sanekata, K. Miyajima, S. Nagao, K. Judai, D. M. Rayner, B. Simard, P. A. Hackett, A. Nakajima and K. Kaya, J. Chem. Phys., 2001, 114, 8459–8466 CrossRef CAS.
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261–5270 CrossRef CAS PubMed.
- Y. Negishi, Y. Takasugi, S. Sato, H. Yao, K. Kimura and T. Tsukuda, J. Am. Chem. Soc., 2004, 126, 6518–6519 CrossRef CAS PubMed.
- Y. Negishi, Y. Takasugi, S. Sato, H. Yao, K. Kimura and T. Tsukuda, J. Phys. Chem. B, 2006, 110, 12218–12221 CrossRef CAS PubMed.
- Y. Shichibu, Y. Negishi, H. Tsunoyama, M. Kanehara, T. Teranishi and T. Tsukuda, Small, 2007, 3, 835–839 CrossRef CAS PubMed.
- K. Ikeda, Y. Kobayashi, Y. Negishi, M. Seto, T. Iwasa, K. Nobusada, T. Tsukuda and N. Kojima, J. Am. Chem. Soc., 2007, 129, 7230–7231 CrossRef CAS PubMed.
- B. K. Teo and H. Zhang, Coord. Chem. Rev., 1995, 143, 611–636 CrossRef CAS.
- Y. Negishi, C. Sakamoto, T. Ohyama and T. Tsukuda, J. Phys. Chem. Lett., 2012, 3, 1624–1628 CrossRef CAS PubMed.
- Y. Negishi, T. Nakazaki, S. Malola, S. Takano, Y. Niihori, W. Kurashige, S. Yamazoe, T. Tsukuda and H. Häkkinen, J. Am. Chem. Soc., 2015, 137, 1206–1212 CrossRef CAS PubMed.
- Y. Negishi, W. Kurashige, Y. Niihori, T. Iwasa and K. Nobusada, Phys. Chem. Chem. Phys., 2010, 12, 6219–6225 RSC.
- Y. Negishi, T. Iwai and M. Ide, Chem. Commun., 2010, 46, 4713–4715 RSC.
- Y. Negishi, K. Munakata, W. Ohgake and K. Nobusada, J. Phys. Chem. Lett., 2012, 3, 2209–2214 CrossRef CAS PubMed.
- W. Kurashige and Y. Negishi, J. Cluster Sci., 2012, 23, 365–374 CrossRef CAS.
- Y. Negishi, K. Igarashi, K. Munakata, W. Ohgake and K. Nobusada, Chem. Commun., 2012, 48, 660–662 RSC.
- S. Sharma, W. Kurashige, K. Nobusada and Y. Negishi, Nanoscale, 2015, 7, 10606–10612 RSC.
- S. Sharma, S. Yamazoe, T. Ono, W. Kurashige, Y. Niihori, K. Nobusada, T. Tsukuda and Y. Negishi, Dalton Trans., 2016, 45, 18064–18068 RSC.
- S. Hossain, T. Ono, M. Yoshioka, G. Hu, M. Hosoi, Z. Chen, L. V. Nair, Y. Niihori, W. Kurashige, D.-e. Jiang and Y. Negishi, J. Phys. Chem. Lett., 2018, 9, 2590–2594 CrossRef CAS PubMed.
- Y. Negishi, R. Arai, Y. Niihori and T. Tsukuda, Chem. Commun., 2011, 47, 5693–5695 RSC.
- I. Chakraborty, W. Kurashige, K. Kanehira, L. Gell, H. Häkkinen, Y. Negishi and T. Pradeep, J. Phys. Chem. Lett., 2013, 4, 3351–3355 CrossRef CAS PubMed.
- L. V. Nair, S. Hossain, S. Wakayama, S. Takagi, M. Yoshioka, J. Maekawa, A. Harasawa, B. Kumar, Y. Niihori, W. Kurashige and Y. Negishi, J. Phys. Chem. C, 2017, 121, 11002–11009 CrossRef CAS.
- T. Kawawaki, N. Shimizu, K. Funai, Y. Mitomi, S. Hossain, S. Kikkawa, D. J. Osborn, S. Yamazoe, G. F. Metha and Y. Negishi, Nanoscale, 2021, 13, 14679–14687 RSC.
- Y. Negishi, U. Kamimura, M. Ide and M. Hirayama, Nanoscale, 2012, 4, 4263–4268 RSC.
- Y. Negishi, W. Kurashige and U. Kamimura, Langmuir, 2011, 27, 12289–12292 CrossRef CAS PubMed.
- W. Kurashige, M. Yamaguchi, K. Nobusada and Y. Negishi, J. Phys. Chem. Lett., 2012, 3, 2649–2652 CrossRef CAS PubMed.
- W. Kurashige, K. Munakata, K. Nobusada and Y. Negishi, Chem. Commun., 2013, 49, 5447–5449 RSC.
- W. Kurashige, S. Yamazoe, K. Kanehira, T. Tsukuda and Y. Negishi, J. Phys. Chem. Lett., 2013, 4, 3181–3185 CrossRef CAS.
- W. Kurashige, S. Yamazoe, M. Yamaguchi, K. Nishido, K. Nobusada, T. Tsukuda and Y. Negishi, J. Phys. Chem. Lett., 2014, 5, 2072–2076 CrossRef CAS PubMed.
- S. Hossain, W. Kurashige, S. Wakayama, B. Kumar, L. V. Nair, Y. Niihori and Y. Negishi, J. Phys. Chem. C, 2016, 120, 25861–25869 CrossRef CAS.
- L. V. Nair, S. Hossain, S. Takagi, Y. Imai, G. Hu, S. Wakayama, B. Kumar, W. Kurashige, D.-E. Jiang and Y. Negishi, Nanoscale, 2018, 10, 18969–18979 RSC.
- S. Hossain, Y. Imai, Y. Motohashi, Z. Chen, D. Suzuki, T. Suzuki, Y. Kataoka, M. Hirata, T. Ono, W. Kurashige, T. Kawawaki, T. Yamamoto and Y. Negishi, Mater. Horiz., 2020, 7, 796–803 RSC.
- S. Hossain, S. Miyajima, T. Iwasa, R. Kaneko, T. Sekine, A. Ikeda, T. Kawawaki, T. Taketsugu and Y. Negishi, J. Chem. Phys., 2021, 155, 024302 CrossRef CAS PubMed.
- Y. Niihori, Y. Koyama, S. Watanabe, S. Hashimoto, S. Hossain, L. V. Nair, B. Kumar, W. Kurashige and Y. Negishi, J. Phys. Chem. Lett., 2018, 9, 4930–4934 CrossRef CAS PubMed.
- Y. Niihori, M. Matsuzaki, C. Uchida and Y. Negishi, Nanoscale, 2014, 6, 7889–7896 RSC.
- Y. Niihori, M. Matsuzaki, T. Pradeep and Y. Negishi, J. Am. Chem. Soc., 2013, 135, 4946–4949 CrossRef CAS PubMed.
- Y. Niihori, Y. Kikuchi, D. Shima, C. Uchida, S. Sharma, S. Hossain, W. Kurashige and Y. Negishi, Ind. Eng. Chem. Res., 2017, 56, 1029–1035 CrossRef CAS.
- Y. Niihori, D. Shima, K. Yoshida, K. Hamada, L. V. Nair, S. Hossain, W. Kurashige and Y. Negishi, Nanoscale, 2018, 10, 1641–1649 RSC.
- Y. Niihori, Y. Kikuchi, A. Kato, M. Matsuzaki and Y. Negishi, ACS Nano, 2015, 9, 9347–9356 CrossRef CAS PubMed.
- Y. Niihori, S. Hashimoto, Y. Koyama, S. Hossain, W. Kurashige and Y. Negishi, J. Phys. Chem. C, 2019, 123, 13324–13329 CrossRef CAS.
- N. A. Sakthivel, S. Theivendran, V. Ganeshraj, A. G. Oliver and A. Dass, J. Am. Chem. Soc., 2017, 139, 15450–15459 CrossRef CAS PubMed.
- C. Zeng, Y. Chen, K. Kirschbaum, K. J. Lambright and R. Jin, Science, 2016, 354, 1580–1584 CrossRef CAS PubMed.
- C. A. Fields-Zinna, R. Sardar, C. A. Beasley and R. W. Murray, J. Am. Chem. Soc., 2009, 131, 16266–16271 CrossRef CAS PubMed.
- H. Qian and R. Jin, Nano Lett., 2009, 9, 4083–4087 CrossRef CAS PubMed.
- K. Kimura, N. Sugimoto, S. Sato, H. Yao, Y. Negishi and T. Tsukuda, J. Phys. Chem. C, 2009, 113, 14076–14082 CrossRef CAS.
- R. L. Wolfe and R. W. Murray, Anal. Chem., 2006, 78, 1167–1173 CrossRef CAS PubMed.
- C. Kumara and A. Dass, Anal. Chem., 2014, 86, 4227–4232 CrossRef CAS PubMed.
- H. Qian, Y. Zhu and R. Jin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 696–700 CrossRef CAS PubMed.
- C. Kumara, X. Zuo, J. Ilavsky, K. W. Chapman, D. A. Cullen and A. Dass, J. Am. Chem. Soc., 2014, 136, 7410–7417 CrossRef CAS PubMed.
- C. Kumara, X. Zuo, D. A. Cullen and A. Dass, ACS Nano, 2014, 8, 6431–6439 CrossRef CAS PubMed.
- S. Malola, L. Lehtovaara, J. Enkovaara and H. Häkkinen, ACS Nano, 2013, 7, 10263–10270 CrossRef CAS PubMed.
- M. Zhou, C. Zeng, Y. Song, J. W. Padelford, G. Wang, M. Y. Sfeir, T. Higaki and R. Jin, Angew. Chem., Int. Ed., 2017, 56, 16257–16261 CrossRef CAS PubMed.
- T. Higaki, M. Zhou, K. J. Lambright, K. Kirschbaum, M. Y. Sfeir and R. Jin, J. Am. Chem. Soc., 2018, 140, 5691–5695 CrossRef CAS PubMed.
- N. A. Sakthivel, M. Stener, L. Sementa, A. Fortunelli, G. Ramakrishna and A. Dass, J. Phys. Chem. Lett., 2018, 9, 1295–1300 CrossRef CAS PubMed.
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754–3755 CrossRef CAS PubMed.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS PubMed.
- J. Akola, M. Walter, R. L. Whetten, H. Häkkinen and H. Grönbeck, J. Am. Chem. Soc., 2008, 130, 3756–3757 CrossRef CAS PubMed.
- H. Qian, W. T. Eckenhoff, Y. Zhu, T. Pintauer and R. Jin, J. Am. Chem. Soc., 2010, 132, 8280–8281 CrossRef CAS PubMed.
- O. Lopez-Acevedo, H. Tsunoyama, T. Tsukuda, H. Häkkinen and C. M. Aikens, J. Am. Chem. Soc., 2010, 132, 8210–8218 CrossRef CAS PubMed.
- S. Yamazoe, W. Kurashige, K. Nobusada, Y. Negishi and T. Tsukuda, J. Phys. Chem. C, 2014, 118, 25284–25290 CrossRef CAS.
- Y. Niihori, W. Kurashige, M. Matsuzaki and Y. Negishi, Nanoscale, 2013, 5, 508–512 RSC.
- Y. Niihori, M. Eguro, A. Kato, S. Sharma, B. Kumar, W. Kurashige, K. Nobusada and Y. Negishi, J. Phys. Chem. C, 2016, 120, 14301–14309 CrossRef CAS.
- S. Hossain, Y. Imai, D. Suzuki, W. Choi, Z. Chen, T. Suzuki, M. Yoshioka, T. Kawawaki, D. Lee and Y. Negishi, Nanoscale, 2019, 11, 22089–22098 RSC.
- S. Hossain, D. Suzuki, T. Iwasa, R. Kaneko, T. Suzuki, S. Miyajima, Y. Iwamatsu, S. Pollitt, T. Kawawaki, N. Barrabés, G. Rupprechter and Y. Negishi, J. Phys. Chem. C, 2020, 124, 22304–22313 CrossRef CAS.
- Z. Wu, E. Lanni, W. Chen, M. E. Bier, D. Ly and R. Jin, J. Am. Chem. Soc., 2009, 131, 16672–16674 CrossRef CAS PubMed.
- T. U. B. Rao, B. Nataraju and T. Pradeep, J. Am. Chem. Soc., 2010, 132, 16304–16307 CrossRef CAS PubMed.
- M. R. Branham, A. D. Douglas, A. J. Mills, J. B. Tracy, P. S. White and R. W. Murray, Langmuir, 2006, 22, 11376–11383 CrossRef CAS PubMed.
- H. Qian, Y. Zhu and R. Jin, ACS Nano, 2009, 3, 3795–3803 CrossRef CAS PubMed.
- Z. Han, X.-Y. Dong, P. Luo, S. Li, Z.-Y. Wang, S.-Q. Zang and T. C. W. Mak, Sci. Adv., 2020, 6, eaay0107 CrossRef CAS PubMed.
- M. Yamamoto, T. Yoshida, N. Yamamoto, T. Nomoto, Y. Yamamoto, S. Yagi and H. Yoshida, J. Mater. Chem. A, 2015, 3, 16810–16816 RSC.
- H. Yang, Y. Wang, H. Huang, L. Gell, L. Lehtovaara, S. Malola, H. Häkkinen and N. Zheng, Nat. Commun., 2013, 4, 2422 CrossRef PubMed.
- A. Desireddy, B. E. Conn, J. Guo, B. Yoon, R. N. Barnett, B. M. Monahan, K. Kirschbaum, W. P. Griffith, R. L. Whetten, U. Landman and T. P. Bigioni, Nature, 2013, 501, 399–402 CrossRef CAS PubMed.
- X.-Y. Li, Z. Wang, H.-F. Su, S. Feng, M. Kurmoo, C.-H. Tung, D. Sun and L.-S. Zheng, Nanoscale, 2017, 9, 3601–3608 RSC.
- N. de Silva and L. F. Dahl, Inorg. Chem., 2005, 44, 9604–9606 CrossRef CAS PubMed.
- I. Ciabatti, C. Femoni, M. C. Iapalucci, G. Longoni, T. Lovato and S. Zacchini, Inorg. Chem., 2013, 52, 4384–4395 CrossRef CAS PubMed.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS PubMed.
- H. Dong, Y.-C. Chen and C. Feldmann, Green Chem., 2015, 17, 4107–4132 RSC.
- C. Bock, C. Paquet, M. Couillard, G. A. Botton and B. R. MacDougall, J. Am. Chem. Soc., 2004, 126, 8028–8037 CrossRef CAS PubMed.
- Y. Wang, J. Ren, K. Deng, L. Gui and Y. Tang, Chem. Mater., 2000, 12, 1622–1627 CrossRef CAS.
- B.-J. Hwang, L. S. Sarma, C.-H. Chen, C. Bock, F.-J. Lai, S.-H. Chang, S.-C. Yen, D.-G. Liu, H.-S. Sheu and J.-F. Lee, J. Phys. Chem. C, 2008, 112, 19922–19929 CrossRef CAS.
- I. Schrader, J. Warneke, S. Neumann, S. Grotheer, A. A. Swane, J. J. K. Kirkensgaard, M. Arenz and S. Kunz, J. Phys. Chem. C, 2015, 119, 17655–17661 CrossRef CAS.
- R. Jin, H. Qian, Z. Wu, Y. Zhu, M. Zhu, A. Mohanty and N. Garg, J. Phys. Chem. Lett., 2010, 1, 2903–2910 CrossRef CAS.
- D. Lee, R. L. Donkers, G. Wang, A. S. Harper and R. W. Murray, J. Am. Chem. Soc., 2004, 126, 6193–6199 CrossRef CAS PubMed.
- R. Guo and R. W. Murray, J. Am. Chem. Soc., 2005, 127, 12140–12143 CrossRef CAS PubMed.
- A. H. Holm, M. Ceccato, R. L. Donkers, L. Fabris, G. Pace and F. Maran, Langmuir, 2006, 22, 10584–10589 CrossRef CAS PubMed.
- M. S. Devadas, K. Kwak, J.-W. Park, J.-H. Choi, C.-H. Jun, E. Sinn, G. Ramakrishna and D. Lee, J. Phys. Chem. Lett., 2010, 1, 1497–1503 CrossRef CAS.
- Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568–2573 CrossRef CAS PubMed.
- S. Kumar and R. Jin, Nanoscale, 2012, 4, 4222–4227 RSC.
- M. Irie, Chem. Rev., 2000, 100, 1683–1684 CrossRef CAS PubMed.
- N. Tamai and H. Miyasaka, Chem. Rev., 2000, 100, 1875–1890 CrossRef CAS PubMed.
- M. Suda, N. Kameyama, M. Suzuki, N. Kawamura and Y. Einaga, Angew. Chem., Int. Ed., 2008, 47, 160–163 CrossRef CAS PubMed.
- M. Suda, N. Kameyama, A. Ikegami and Y. Einaga, J. Am. Chem. Soc., 2009, 131, 865–870 CrossRef CAS PubMed.
- Y. Chen, C. Liu, Q. Tang, C. Zeng, T. Higaki, A. Das, D.-E. Jiang, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2016, 138, 1482–1485 CrossRef CAS PubMed.
- T. Kawai and M. Hashizume, Stimuli-Responsive Interfaces Fabrication and Application, Springer, Singapore, 2017 Search PubMed.
- C. K. Yee, A. Ulman, J. D. Ruiz, A. Parikh, H. White and M. Rafailovich, Langmuir, 2003, 19, 9450–9458 CrossRef CAS.
- L. V. Romashov and V. P. Ananikov, Chem. – Eur. J., 2013, 19, 17640–17660 CrossRef CAS PubMed.
- S. Kano, Y. Azuma, M. Kanehara, T. Teranishi and Y. Majima, Appl. Phys. Express, 2010, 3, 105003 CrossRef.
- J.-i. Nishigaki, K. Koyasu and T. Tsukuda, Chem. Rec., 2014, 14, 897–909 CrossRef CAS PubMed.
- B. K. Teo and H. Zhang, Inorg. Chem., 1991, 30, 3115–3116 CrossRef CAS.
- B. K. Teo, H. Zhang and X. Shi, J. Am. Chem. Soc., 1990, 112, 8552–8562 CrossRef CAS.
- Y. Shichibu, Y. Negishi, T. Watanabe, N. K. Chaki, H. Kawaguchi and T. Tsukuda, J. Phys. Chem. C, 2007, 111, 7845–7847 CrossRef CAS.
- R. Jin, C. Liu, S. Zhao, A. Das, H. Xing, C. Gayathri, Y. Xing, N. L. Rosi, R. R. Gil and R. Jin, ACS Nano, 2015, 9, 8530–8536 CrossRef CAS PubMed.
- Y. Song, F. Fu, J. Zhang, J. Chai, X. Kang, P. Li, S. Li, H. Zhou and M. Zhu, Angew. Chem., Int. Ed., 2015, 54, 8430–8434 CrossRef CAS PubMed.
- M. S. Bootharaju, S. M. Kozlov, Z. Cao, M. Harb, N. Maity, A. Shkurenko, M. R. Parida, M. N. Hedhili, M. Eddaoudi, O. F. Mohammed, O. M. Bakr, L. Cavallo and J.-M. Basset, J. Am. Chem. Soc., 2017, 139, 1053–1056 CrossRef CAS PubMed.
- S. Yang, J. Chai, Y. Lv, T. Chen, S. Wang, H. Yu and M. Zhu, Chem. Commun., 2018, 54, 12077–12080 RSC.
- M. De Nardi, S. Antonello, D.-E. Jiang, F. Pan, K. Rissanen, M. Ruzzi, A. Venzo, A. Zoleo and F. Maran, ACS Nano, 2014, 8, 8505–8512 CrossRef CAS PubMed.
- S. Antonello, T. Dainese, F. Pan, K. Rissanen and F. Maran, J. Am. Chem. Soc., 2017, 139, 4168–4174 CrossRef CAS PubMed.
- W. Fei, S. Antonello, T. Dainese, A. Dolmella, M. Lahtinen, K. Rissanen, A. Venzo and F. Maran, J. Am. Chem. Soc., 2019, 141, 16033–16045 CrossRef CAS PubMed.
- J. Chen, L. Liu, X. Liu, L. Liao, S. Zhuang, S. Zhou, J. Yang and Z. Wu, Chem. – Eur. J., 2017, 23, 18187–18192 CrossRef CAS PubMed.
- R.-W. Huang, Y.-S. Wei, X.-Y. Dong, X.-H. Wu, C.-X. Du, S.-Q. Zang and T. C. W. Mak, Nat. Chem., 2017, 9, 689–697 CrossRef CAS PubMed.
- M. J. Alhilaly, R.-W. Huang, R. Naphade, B. Alamer, M. N. Hedhili, A.-H. Emwas, P. Maity, J. Yin, A. Shkurenko, O. F. Mohammed, M. Eddaoudi and O. M. Bakr, J. Am. Chem. Soc., 2019, 141, 9585–9592 CrossRef CAS PubMed.
- S.-H. Lu, Y. Li, S.-X. Yang, R.-D. Zhao, Z.-X. Lu, X.-L. Liu, Y. Qin, L.-Y. Zheng and Q.-E. Cao, Inorg. Chem., 2019, 58, 11793–11800 CrossRef CAS PubMed.
- Z.-K. Wang, M.-M. Sheng, S.-S. Qin, H.-T. Shi, M. Strømme, Q.-F. Zhang and C. Xu, Inorg. Chem., 2020, 59, 2121–2126 CrossRef CAS PubMed.
- X.-H. Wu, P. Luo, Z. Wei, Y.-Y. Li, R.-W. Huang, X.-Y. Dong, K. Li, S.-Q. Zang and B. Z. Tang, Adv. Sci., 2019, 6, 1801304 CrossRef PubMed.
- Z. Lei, X.-L. Pei, Z.-G. Jiang and Q.-M. Wang, Angew. Chem., Int. Ed., 2014, 53, 12771–12775 CrossRef CAS PubMed.
- Y. Song, M. LAV Heien, V. Jimenez, R. M. Wightman and R. W. Murray, Anal. Chem., 2004, 76, 4911–4919 CrossRef CAS PubMed.
- Y. Song, V. Jimenez, C. McKinney, R. Donkers and R. W. Murray, Anal. Chem., 2003, 75, 5088–5096 CrossRef CAS.
- D. R. Stoll and P. W. Carr, J. Am. Chem. Soc., 2005, 127, 5034–5035 CrossRef CAS PubMed.
- I. Dolamic, S. Knoppe, A. Dass and T. Bürgi, Nat. Commun., 2012, 3, 798 CrossRef PubMed.
- S. Takano, S. Ito and T. Tsukuda, J. Am. Chem. Soc., 2019, 141, 15994–16002 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- K. Kwak, W. Choi, Q. Tang, M. Kim, Y. Lee, D.-E. Jiang and D. Lee, Nat. Commun., 2017, 8, 14723 CrossRef PubMed.
- S. Zhao, R. Jin, Y. Song, H. Zhang, S. D. House, J. C. Yang and R. Jin, Small, 2017, 13, 1701519 CrossRef PubMed.
- W. Choi, G. Hu, K. Kwak, M. Kim, D.-E. Jiang, J.-P. Choi and D. Lee, ACS Appl. Mater. Interfaces, 2018, 10, 44645–44653 CrossRef CAS PubMed.
- D. Eguchi, M. Sakamoto and T. Teranishi, Chem. Sci., 2018, 9, 261–265 RSC.
- B. Kumar, T. Kawawaki, N. Shimizu, Y. Imai, D. Suzuki, S. Hossain, L. V. Nair and Y. Negishi, Nanoscale, 2020, 12, 9969–9979 RSC.
- K. Kwak, W. Choi, Q. Tang, D.-E. Jiang and D. Lee, J. Mater. Chem. A, 2018, 6, 19495–19501 RSC.
- E. J. Popczun, J. R. McKone, C. G. Read, A. J. Biacchi, A. M. Wiltrout, N. S. Lewis and R. E. Schaak, J. Am. Chem. Soc., 2013, 135, 9267–9270 CrossRef CAS PubMed.
- J. Xie, J. Zhang, S. Li, F. Grote, X. Zhang, H. Zhang, R. Wang, Y. Lei, B. Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 17881–17888 CrossRef CAS PubMed.
- P. Xiao, M. A. Sk, L. Thia, X. Ge, R. J. Lim, J.-Y. Wang, K. H. Lim and X. Wang, Energy Environ. Sci., 2014, 7, 2624–2629 RSC.
- H. Jin, J. Wang, D. Su, Z. Wei, Z. Pang and Y. Wang, J. Am. Chem. Soc., 2015, 137, 2688–2694 CrossRef CAS PubMed.
- Y. Du, J. Xiang, K. Ni, Y. Yun, G. Sun, X. Yuan, H. Sheng, Y. Zhu and M. Zhu, Inorg. Chem. Front., 2018, 5, 2948–2954 RSC.
- C. Jiang, S. J. A. Moniz, A. Wang, T. Zhang and J. Tang, Chem. Soc. Rev., 2017, 46, 4645–4660 RSC.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. A. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- W. Chen and S. Chen, Angew. Chem., Int. Ed., 2009, 48, 4386–4389 CrossRef CAS PubMed.
- Y. Lu, Y. Jiang, X. Gao and W. Chen, Chem. Commun., 2014, 50, 8464–8467 RSC.
- L. Wang, Z. Tang, W. Yan, H. Yang, Q. Wang and S. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 20635–20641 CrossRef CAS PubMed.
- L. Sumner, N. A. Sakthivel, H. Schrock, K. Artyushkova, A. Dass and S. Chakraborty, J. Phys. Chem. C, 2018, 122, 24809–24817 CrossRef CAS.
- T. C. Jones, L. Sumner, G. Ramakrishna, M. B. Hatshan, A. Abuhagr, S. Chakraborty and A. Dass, J. Phys. Chem. C, 2018, 122, 17726–17737 CrossRef CAS.
- M. Liu, Z. Zhao, X. Duan and Y. Huang, Adv. Mater., 2019, 31, 1802234 CrossRef PubMed.
- R. M. Heck, R. J. Farrauto and S. T. Gulati, Catalytic Air Pollution Control: Commercial Technology: Third edition, Wiley-VCH, Weinheim, 2009 Search PubMed.
- K. Domen, S. Naito, M. Soma, T. Onishi and K. Tamaru, J. Chem. Soc., Chem. Commun., 1980, 543–544 RSC.
- B. Kraeutler and A. J. Bard, J. Am. Chem. Soc., 1978, 100, 4317–4318 CrossRef CAS.
- Y. Liu, H. Tsunoyama, T. Akita, S. Xie and T. Tsukuda, ACS Catal., 2011, 1, 2–6 CrossRef CAS.
- S. Xie, H. Tsunoyama, W. Kurashige, Y. Negishi and T. Tsukuda, ACS Catal., 2012, 2, 1519–1523 CrossRef CAS.
- N. Sakamoto, H. Ohtsuka, T. Ikeda, K. Maeda, D. Lu, M. Kanehara, K. Teramura, T. Teranishi and K. Domen, Nanoscale, 2009, 1, 106–109 RSC.
- D. Bahnemann, A. Henglein, J. Lilie and L. Spanhel, J. Phys. Chem., 1984, 88, 709–711 CrossRef CAS.
- H. N. Ghosh, J. B. Asbury and T. Lian, J. Phys. Chem. B, 1998, 102, 6482–6486 CrossRef CAS.
- B. Ohtani, R. M. Bowman, D. P. Colombo Jr., H. Kominami, H. Noguchi and K. Uosaki, Chem. Lett., 1998, 579–580 CrossRef CAS.
- A. Yamakata, T.-a. Ishibashi and H. Onishi, Chem. Phys. Lett., 2001, 333, 271–277 CrossRef CAS.
- T. Yoshihara, R. Katoh, A. Furube, Y. Tamaki, M. Murai, K. Hara, S. Murata, H. Arakawa and M. Tachiya, J. Phys. Chem. B, 2004, 108, 3817–3823 CrossRef CAS.
- J. Lee, H. S. Shim, M. Lee, J. K. Song and D. Lee, J. Phys. Chem. Lett., 2011, 2, 2840–2845 CrossRef CAS.
- X.-Q. Gong, A. Selloni, O. Dulub, P. Jacobson and U. Diebold, J. Am. Chem. Soc., 2008, 130, 370–381 CrossRef CAS PubMed.
- M. R. Nellist, F. A. L. Laskowski, J. Qiu, H. Hajibabaei, K. Sivula, T. W. Hamann and S. W. Boettcher, Nat. Energy, 2018, 3, 46–52 CrossRef CAS.
- M. R. Nellist, J. Qiu, F. A. L. Laskowski, F. M. Toma and S. W. Boettcher, ACS Energy Lett., 2018, 3, 2286–2291 CrossRef CAS.
- G. N. Vayssilov, Y. Lykhach, A. Migani, T. Staudt, G. P. Petrova, N. Tsud, T. Skála, A. Bruix, F. Illas, K. C. Prince, V. V. Matolin, K. M. Neyman and J. Libuda, Nat. Mater., 2011, 10, 310–315 CrossRef CAS PubMed.
- K. He, J. Xie, Z.-Q. Liu, N. Li, X. Chen, J. Hu and X. Li, J. Mater. Chem. A, 2018, 6, 13110–13122 RSC.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- K. Maeda, ACS Catal., 2013, 3, 1486–1503 CrossRef CAS.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900–1909 CrossRef CAS PubMed.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- Y. Wang, H. Suzuki, J. Xie, O. Tomita, D. J. Martin, M. Higashi, D. Kong, R. Abe and J. Tang, Chem. Rev., 2018, 118, 5201–5241 CrossRef CAS PubMed.
- G. Zhang, Z.-A. Lan, L. Lin, S. Lin and X. Wang, Chem. Sci., 2016, 7, 3062–3066 RSC.
- X. Li, J. Yu, J. Low, Y. Fang, J. Xiao and X. Chen, J. Mater. Chem. A, 2015, 3, 2485–2534 RSC.
- T. Hisatomi, J. Kubota and K. Domen, Chem. Soc. Rev., 2014, 43, 7520–7535 RSC.
- Z. Wang, C. Li and K. Domen, Chem. Soc. Rev., 2019, 48, 2109–2125 RSC.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- S. Sato and J. M. White, Chem. Phys. Lett., 1980, 72, 83–86 CrossRef CAS.
- T. Takata, J. Jiang, Y. Sakata, M. Nakabayashi, N. Shibata, V. Nandal, K. Seki, T. Hisatomi and K. Domen, Nature, 2020, 581, 411–414 CrossRef CAS PubMed.
- Q. Wang, T. Hisatomi, Q. Jia, H. Tokudome, M. Zhong, C. Wang, Z. Pan, T. Takata, M. Nakabayashi, N. Shibata, Y. Li, I. D. Sharp, A. Kudo, T. Yamada and K. Domen, Nat. Mater., 2016, 15, 611–615 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- Y. Miseki, H. Kato and A. Kudo, Energy Environ. Sci., 2009, 2, 306–314 RSC.
- Y. Negishi, M. Mizuno, M. Hirayama, M. Omatoi, T. Takayama, A. Iwase and A. Kudo, Nanoscale, 2013, 5, 7188–7192 RSC.
- Y. Negishi, Y. Matsuura, R. Tomizawa, W. Kurashige, Y. Niihori, T. Takayama, A. Iwase and A. Kudo, J. Phys. Chem. C, 2015, 119, 11224–11232 CrossRef CAS.
- W. Kurashige, R. Kumazawa, D. Ishii, R. Hayashi, Y. Niihori, S. Hossain, L. V. Nair, T. Takayama, A. Iwase, S. Yamazoe, T. Tsukuda, A. Kudo and Y. Negishi, J. Phys. Chem. C, 2018, 122, 13669–13681 CrossRef CAS.
- T. Ikeda, A. Xiong, T. Yoshinaga, K. Maeda, K. Domen and T. Teranishi, J. Phys. Chem. C, 2013, 117, 2467–2473 CrossRef CAS.
- K. Maeda, A. Xiong, T. Yoshinaga, T. Ikeda, N. Sakamoto, T. Hisatomi, M. Takashima, D. Lu, M. Kanehara, T. Setoyama, T. Teranishi and K. Domen, Angew. Chem., Int. Ed., 2010, 49, 4096–4099 CrossRef CAS PubMed.
- A. Xiong, T. Yoshinaga, T. Ikeda, M. Takashima, T. Hisatomi, K. Maeda, T. Setoyama, T. Teranishi and K. Domen, Eur. J. Inorg. Chem., 2014, 767–772 CrossRef CAS.
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851–7861 CrossRef CAS.
- K. Maeda, J. Photochem. Photobiol., C, 2011, 12, 237–268 CrossRef CAS.
- K. Maeda, D. Lu and K. Domen, Chem. – Eur. J., 2013, 19, 4986–4991 CrossRef CAS PubMed.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, Angew. Chem., Int. Ed., 2006, 45, 7806–7809 CrossRef CAS PubMed.
- M. Yoshida, K. Takanabe, K. Maeda, A. Ishikawa, J. Kubota, Y. Sakata, Y. Ikezawa and K. Domen, J. Phys. Chem. C, 2009, 113, 10151–10157 CrossRef CAS.
- K. Maeda, K. Teramura, D. Lu, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. C, 2007, 111, 7554–7560 CrossRef CAS.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- W. Kurashige, R. Hayashi, K. Wakamatsu, Y. Kataoka, S. Hossain, A. Iwase, A. Kudo, S. Yamazoe and Y. Negishi, ACS Appl. Energy Mater., 2019, 2, 4175–4187 CrossRef CAS.
- S. Trasatti, J. Electroanal. Chem. Interfacial Electrochem., 1972, 39, 163–184 CrossRef CAS.
- T. Hisatomi, K. Maeda, K. Takanabe, J. Kubota and K. Domen, J. Phys. Chem. C, 2009, 113, 21458–21466 CrossRef CAS.
- K. Maeda, K. Teramura, H. Masuda, T. Takata, N. Saito, Y. Inoue and K. Domen, J. Phys. Chem. B, 2006, 110, 13107–13112 CrossRef CAS PubMed.
- T. Ohno, L. Bai, T. Hisatomi, K. Maeda and K. Domen, J. Am. Chem. Soc., 2012, 134, 8254–8259 CrossRef CAS PubMed.
- K. Maeda, D. Lu, K. Teramura and K. Domen, Energy Environ. Sci., 2010, 3, 471–478 RSC.
- W. Kurashige, Y. Mori, S. Ozaki, M. Kawachi, S. Hossain, T. Kawawaki, C. J. Shearer, A. Iwase, G. F. Metha, S. Yamazoe, A. Kudo and Y. Negishi, Angew. Chem., Int. Ed., 2020, 59, 7076–7082 CrossRef CAS PubMed.
- Y. Sakata, T. Hayashi, R. Yasunaga, N. Yanaga and H. Imamura, Chem. Commun., 2015, 51, 12935–12938 RSC.
- T. H. Chiang, H. Lyu, T. Hisatomi, Y. Goto, T. Takata, M. Katayama, T. Minegishi and K. Domen, ACS Catal., 2018, 8, 2782–2788 CrossRef CAS.
- T. Imaoka, H. Kitazawa, W.-J. Chun, S. Omura, K. Albrecht and K. Yamamoto, J. Am. Chem. Soc., 2013, 135, 13089–13095 CrossRef CAS PubMed.
- T. Imaoka, H. Kitazawa, W.-J. Chun and K. Yamamoto, Angew. Chem., Int. Ed., 2015, 54, 9810–9815 CrossRef CAS PubMed.
- H. Tsunoyama, A. Ohnuma, K. Takahashi, A. Velloth, M. Ehara, N. Ichikuni, M. Tabuchi and A. Nakajima, Chem. Commun., 2019, 55, 12603–12606 RSC.
- B. Garlyyev, K. Kratzl, M. Rück, J. Michalička, J. Fichtner, J. M. Macak, T. Kratky, S. Günther, M. Cokoja, A. S. Bandarenka, A. Gagliardi and R. A. Fischer, Angew. Chem., Int. Ed., 2019, 58, 9596–9600 CrossRef CAS PubMed.
- F. Calle-Vallejo, J. Tymoczko, V. Colic, Q. H. Vu, M. D. Pohl, K. Morgenstern, D. Loffreda, P. Sautet, W. Schuhmann and A. S. Bandarenka, Science, 2015, 350, 185–189 CrossRef CAS PubMed.
- B. Garlyyev, J. Fichtner, O. Piqué, O. Schneider, A. S. Bandarenka and F. Calle-Vallejo, Chem. Sci., 2019, 10, 8060–8075 RSC.
- Y. Negishi, N. Shimizu, K. Funai, R. Kaneko, K. Wakamatsu, A. Harasawa, S. Hossain, M. E. Schuster, D. Ozkaya, W. Kurashige, T. Kawawaki, S. Yamazoe and S. Nagaoka, Nanoscale Adv., 2020, 2, 669–678 RSC.
- J. H. Kwak, J. Hu, D. Mei, C.-W. Yi, D. H. Kim, C. H. F. Peden, L. F. Allard and J. Szanyi, Science, 2009, 325, 1670–1673 CrossRef CAS PubMed.
- C. Yin, F. R. Negreiros, G. Barcaro, A. Beniya, L. Sementa, E. C. Tyo, S. Bartling, K.-H. Meiwes-Broer, S. Seifert, H. Hirata, N. Isomura, S. Nigam, C. Majumder, Y. Watanabe, A. Fortunelli and S. Vajda, J. Mater. Chem. A, 2017, 5, 4923–4931 RSC.
- U. Heiz, A. Sanchez, S. Abbet and W.-D. Schneider, J. Am. Chem. Soc., 1999, 121, 3214–3217 CrossRef CAS.
- V. Matsouka, M. Konsolakis, R. M. Lambert and I. V. Yentekakis, Appl. Catal., B, 2008, 84, 715–722 CrossRef CAS.
- J. Březina, P. Boutikos, A. B. Arvajová, R. Pečinka and P. Kočí, Top. Catal., 2019, 62, 252–258 CrossRef.
- S. B. Kang, C. Kalamaras, V. Balakotaiah and W. Epling, Ind. Eng. Chem. Res., 2017, 56, 13628–13633 CrossRef CAS.
- D. R. Kauffman, D. R. Alfonso, D. N. Tafen, C. Wang, Y. Zhou, Y. Yu, J. W. Lekse, X. Deng, V. Espinoza, J. Trindell, O. K. Ranasingha, A. Roy, J.-S. Lee and H. L. Xin, J. Phys. Chem. C, 2018, 122, 27991–28000 CrossRef CAS.
- N. Austin, S. Zhao, J. R. McKone, R. Jin and G. Mpourmpakis, Catal. Sci. Technol., 2018, 8, 3795–3805 RSC.
- T. Kawawaki, Y. Kataoka, M. Hirata, Y. Akinaga, R. Takahata, K. Wakamatsu, Y. Fujiki, M. Kataoka, S. Kikkawa, A. S. Alotabi, S. Hossain, D. J. Osborn, T. Teranishi, G. G. Andersson, G. F. Metha, S. Yamazoe and Y. Negishi, Angew. Chem., Int. Ed., 2021, 60, 21340–21350 CrossRef CAS PubMed.
- B. Zhang, S. Kaziz, H. Li, M. G. Hevia, D. Wodka, C. Mazet, T. Bürgi and N. Barrabés, J. Phys. Chem. C, 2015, 119, 11193–11199 CrossRef CAS.
- J. Liu, K. S. Krishna, Y. B. Losovyj, S. Chattopadhyay, N. Lozova, J. T. Miller, J. J. Spivey and C. S. S. R. Kumar, Chem. – Eur. J., 2013, 19, 10201–10208 CrossRef CAS PubMed.
- C. García, S. Pollitt, M. van der Linden, V. Truttmann, C. Rameshan, R. Rameshan, E. Pittenauer, G. Allmaier, P. Kregsamer, M. Stöger-Pollach, N. Barrabés and G. Rupprechter, Catal. Today, 2019, 336, 174–185 CrossRef.
- B. Zhang, A. Sels, G. Salassa, S. Pollitt, V. Truttmann, C. Rameshan, J. Llorca, W. Olszewski, G. Rupprechter, T. Bürgi and N. Barrabés, ChemCatChem, 2018, 10, 5372–5376 CrossRef CAS PubMed.
- Z. Wu, G. Hu, D.-E. Jiang, D. R. Mullins, Q.-F. Zhang, L. F. Allard, Jr., L.-S. Wang and S. H. Overbury, Nano Lett., 2016, 16, 6560–6567 CrossRef CAS PubMed.
- H. A. Almukhlifi and R. C. Burns, Appl. Catal., A, 2015, 502, 174–187 CrossRef CAS.
- L. M. Rossi, J. L. Fiorio, M. A. S. Garcia and C. P. Ferraz, Dalton Trans., 2018, 47, 5889–5915 RSC.
- H. Yano, M. Kataoka, H. Yamashita, H. Uchida and M. Watanabe, Langmuir, 2007, 23, 6438–6445 CrossRef CAS PubMed.
- H. Asakura, S. Hosokawa, T. Ina, K. Kato, K. Nitta, K. Uera, T. Uruga, H. Miura, T. Shishido, J. Ohyama, A. Satsuma, K. Sato, A. Yamamoto, S. Hinokuma, H. Yoshida, M. Machida, S. Yamazoe, T. Tsukuda, K. Teramura and T. Tanaka, J. Am. Chem. Soc., 2018, 140, 176–184 CrossRef CAS PubMed.
- Z. Luo, V. Nachammai, B. Zhang, N. Yan, D. T. Leong, D.-E. Jiang and J. Xie, J. Am. Chem. Soc., 2014, 136, 10577–10580 CrossRef CAS PubMed.
- T. Chen, V. Fung, Q. Yao, Z. Luo, D.-E. Jiang and J. Xie, J. Am. Chem. Soc., 2018, 140, 11370–11377 CrossRef CAS PubMed.
- K. R. Krishnadas, A. Baksi, A. Ghosh, G. Natarajan and T. Pradeep, Nat. Commun., 2016, 7, 13447 CrossRef CAS PubMed.
- K. R. Krishnadas, A. Baksi, A. Ghosh, G. Natarajan, A. Som and T. Pradeep, Acc. Chem. Res., 2017, 50, 1988–1996 CrossRef CAS PubMed.
- A. Baksi, E. K. Schneider, P. Weis, K. R. Krishnadas, D. Ghosh, H. Hahn, T. Pradeep and M. M. Kappes, J. Phys. Chem. C, 2019, 123, 28477–28485 CrossRef CAS.
- B. Zhang, G. Salassa and T. Bürgi, Chem. Commun., 2016, 52, 9205–9207 RSC.
- B. Huang and Y. Pei, J. Mater. Chem. A, 2020, 8, 10242–10251 RSC.
- X. Kang and M. Zhu, Chem. Mater., 2019, 31, 9939–9969 CrossRef CAS.
- J. E. McMurry, Organic Chemistry 9th Edition, Cengage Learning, Boston, 2015 Search PubMed.
- J. R. Gispert, Coordination Chemistry, Wiley-VCH, Weinheim, 2008 Search PubMed.
- S. Takano and T. Tsukuda, J. Am. Chem. Soc., 2021, 143, 1683–1698 CrossRef CAS PubMed.
- D. E. Bergeron, P. J. Roach, A. W. Castleman, Jr., N. O. Jones and S. N. Khanna, Science, 2005, 307, 231–235 CrossRef CAS PubMed.
- T. Omoda, S. Takano and T. Tsukuda, Small, 2021, 17, 2001439 CrossRef CAS PubMed.
- K. Yamamoto, T. Imaoka, M. Tanabe and T. Kambe, Chem. Rev., 2020, 120, 1397–1437 CrossRef CAS PubMed.
- T. Kawawaki, M. Kawachi, D. Yazaki, Y. Akinaga, D. Hirayama and Y. Negishi, Nanomaterials, 2022, 12, 344 CrossRef.
| This journal is © the Owner Societies 2022 |