
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSteric influence on solvate formation – a comparison of resorcylic acid and two brominated derivatives†
Katharina
Edkins
 *a,
Jane
Tweedy
b,
Stephanie
Fung
b and
Robert M.
Edkins
c
*a,
Jane
Tweedy
b,
Stephanie
Fung
b and
Robert M.
Edkins
c
aSchool of Health Sciences, The University of Manchester, Oxford Road, Manchester M13 9PT, UK. E-mail: katharina.edkins@manchester.ac.uk
bSchool of Medicine, Pharmacy and Health, Durham University Queen's Campus, University Boulevard, Stockton-on-Tees TS17 6BH, UK
cWestCHEM Department of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow G1 1XL, UK
First published on 19th January 2022
Abstract
The molecular structure of a candidate pharmaceutical drug compound is routinely modified during lead optimisation. While the difference in pharmacological effect may be subtle, the effect on the solid-state landscape can be unpredictable and dramatic. In the present study we investigate the effect of small changes in steric bulk of a substituent on solvate formation with the model compounds β-resorcylic acid, 5-bromo-β-resorcylic acid and 3,5-dibromo-β-resorcylic acid. Overall, 18 solvates were structurally characterised, half of those are observed for the monobrominated compound. Although none of the crystal structures show isostructurality, there are three major interaction motifs: centrosymmetric carboxylic acid homodimer formation with solvent interaction through the 4-hydroxyl group into finite tetramers, infinite chain formation of these tetramers, and no homodimer formation. Using the first interaction shell around the host (resorcylic acid) molecules, the effect of the steric hindrance by blocking of certain interaction sites can be visualised. The realisation of halogen bonds leads to a loosening of the molecular packing, which likely leads to increased solvent inclusion. It can be concluded that the introduction of steric bulk furthers solvent inclusion, a factor that must be considered during lead optimisation.
Introduction
The number and nature of crystal forms that a single molecule can adopt is of importance for a variety of applications. In the development of a pharmaceutical, the crystal form can either be a curse due to instability or low solubility, or a blessing in the form of a prolonged patent coverage.1,2 The crystal form can also majorly influence manufacturability.3 But crystal forms also impact other areas of applied chemistry. For example, the colour of a pigment strongly depends on its crystal form,4 while in opto-electronics, the molecular arrangement in the solid state and thin films has significant impact on the efficiency of a dye.5Pharmaceutically active organic molecules are prone to form a number of different crystal forms. These range from unsolvated polymorphs containing only the drug compound to inclusion compounds, such as solvates and hydrates, to multi-component co-crystals.1 Each of these crystal forms exhibits its own physicochemical characteristics with significant influence on the manufacturability, such as melting point, physical and chemical stability, solubility, and colour.6,7 The standard approach to investigate the crystal-form landscape of a given drug compound is an experimental crystallisation screening,8,9 either manually or with the help of robotics, to explore experimental space and thus find as many experimentally accessible crystal forms as possible. In fact, the US Food and Drug Administration (FDA) requests a completed crystallisation screening before any new drug entity can enter the market.10 However, there is no clear guideline as to how extensive such a screen should be, and to paraphrase the famous quote of Walter McCrone: ‘the more time and money we invest, the more crystal forms we will find’.11 The profundity of this statement has been shown for olanzapine, which forms over 60 polymorphs and solvates.12 Nowadays, an experimental screen is often supplemented by computational crystal-structure prediction, which can guide the experimental work.13,14 However, both approaches are work- and time intensive.
To complicate matters, the development of a novel drug entity includes the process of lead optimisation, where chemical derivatives of the same molecule are investigated to find the most appropriate compound for further development. Each of these derivatives shows small changes in the chemical structure, mainly introduced through bio-isosteric replacement.15,16 With this tool, one functional group can be exchanged for another without changing the overall pharmacological response. The bio-isosteres can improve solubility or metabolic stability and hence alter the pharmacokinetics of the new drug. But each replacement changes the molecular shape and interactions and, with that, the crystal-form landscape. It has been shown that the change of a single to double bond can change the solid-state landscape significantly,17 while bio-isosteric exchange within the halogen series has significant influence on solvent inclusion and the physical stability of the respective solvates.18 In addition, change in conformation can also favour solvent inclusion due to generation of pockets in the crystal packing.19 The impact of molecular shape on crystallisation has also been shown especially for steric hindrance close to hydrogen bonding site, which can cause a switch from catemer to dimer formation.20,21
As part of our project to systematically investigate the influence of molecular shape on crystallisation, crystal-form landscape and solvent inclusion,18,21 we present here the study of steric influence on solvent inclusion in a small organic molecule, 2,4-dihydroxy benzoic acid (β-resorcylic acid, RA). The unsubstituted RA has been reported previously to crystallise in at least nine different crystal forms, five of which are solvates.22 To introduce steric bulk, we introduced a single bromo-substitution in the 5-position (5-bromoresorcylic acid, 5-BRA, Fig. 1), or two bromo-substituents in the 3- and 5-positions of the aromatic ring (3,5-dibromoresorcylic acid, 3,5-BRA). Halogen-substitution is common in pharmaceuticals, and H/F and F/Cl are bio-isosteric pairs, and the influence of halogen substitution on water inclusion upon crystallisation has previously been demonstrated in phloroglucinols by Saha and Nangia.23 While bromination is rare in pharmaceuticals, we have chosen this particular substitution due to the chemically soft nature of bromine-substituents to avoid a significant change in the electronic structure of the molecules, especially around the hydrogen-bonding functionalities, as well as the relative ease of synthesis.
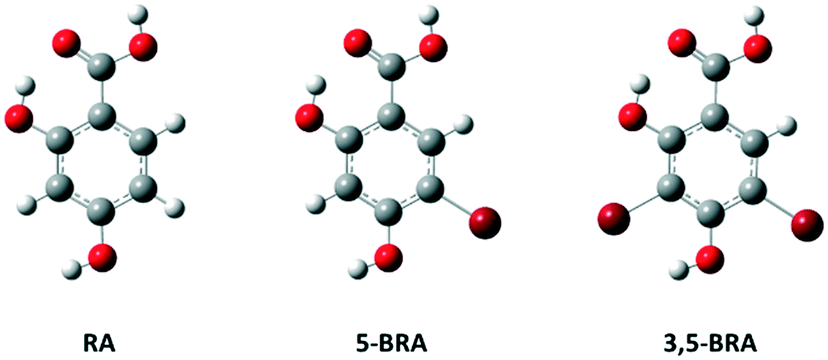 | ||
| Fig. 1 Molecular structure of RA, 5-BRA and 3,5-BRA. Colour code: carbon (grey), hydrogen (light grey), oxygen (red), bromine (brown). | ||
Experimental
Materials
β-Resorcylic acid (2,4-dihydroxy benzoic acid), elemental bromine and all organic solvents (analytical grade) were purchased from Sigma Aldrich (UK) and used without further purification.Synthesis
5-BRA and 3,5-BRA were synthesised according to the literature by brominating RA with bromine in acetic acid at room temperature.24Crystallisation methods
All three compounds were crystallised from the same set of 21 solvents (Table S9†) using the following four crystallisation methods to ensure complete comparability of the obtained solvates. For slow and fast cooling experiments, sufficient solvent was added to 15–20 mg compound to generate a saturated solution at boiling point. The solution was then either cooled in an insulating wooden block to room temperature (slow cooling) or quenched in an ice-water bath (fast cooling). For evaporation, sufficient solvent was added to 15–20 mg compound to generate an undersaturated solution at room temperature. The solution was filtered into a glass vial and allowed to evaporate to dryness. Precipitation was conducted on a hot saturated solution of 15–20 mg compound with at least twice the volume of a miscible non-solvent at room temperature. The crystals obtained from fast cooling and precipitation were immediately removed from the solution, dried on a filter paper and directly analysed. The solid resulting from slow cooling was allowed to stand for at least 24 hours to reach equilibrium before removal from the mother liquor.Thermogravimetry
Measurements were performed on a Q5000 thermogravimetric system (TA Instruments, New Castle, USA). Samples of approximately 2–6 mg were accurately weighed into 50 μL platinum pans and subjected to non-isothermal TGA at a heating rate of 10 °C min−1. Nitrogen was used as purge gas at a flow rate of 60 mL min−1.X-ray diffraction
Powder X-ray diffraction patterns were recorded on an Empyrean diffractometer (Panalytical, Almelo, Netherlands) in Bragg–Brentano geometry. The instrument used Cu-Kα radiation (λ = 1.5406 Å), a graphite monochromator, 0.2 mm fixed Soller slits and a PIXcel detector. The X-ray tube was operated at 40 kV and 40 mA. The dry powder samples were prepared on a zero-background silicon slide sample holder.Single crystals of the solvates were picked and coated in perfluoro polyether oil, then mounted on MiTeGen sample holders. For most solvates, these were placed directly into a precooled cryostream on a D8 Quest diffractometer (Bruker, Karlsruhe, Germany) using Mo-Kα radiation (λ = 0.71073 Å). Single-crystal diffraction of RA dioxane hemisolvate and RA DMF monosolvate was carried out on a Saturn 724+ diffractometer (Rigaku, Sevenoaks, UK) at station I19 of the Diamond Light Source synchrotron (undulator, λ = 0.71073 Å, ω-scan, 1.0° per frame). All data were reduced using Bruker Apex-III software, and the structure was solved using Olex.solve25 and refined using ShelXL.26 All non-hydrogen atoms were picked from the electron density map and refined anisotropically. Hydrogen atoms were added in geometrical positions and refined riding on the heavy atom.
Results & discussion
Crystal structures of the solvates
Slow cooling of a solution in acetic acid yields the acetic acid monosolvate (RA SAcOH), which was previously observed and its crystal structure reported here for the first time (see ESI† Table S1).22RA SAcOH crystallises in the monoclinic space group P21/n with one formula unit in the asymmetric unit. RA and acetic acid form heterodimers through the centrosymmetric carboxylic acid dimer, while two heterodimers are connected through a hydrogen bond from the RA 4-OH group to the carbonyl oxygen atom of the acetic acid. This results in infinite hydrogen-bonded chains of zigzag pattern and almost complete flatness. There are no directed interactions between the layers or stacks of these chains.
Fast and slow cooling of a hot saturated DMF solution of RA results in the new DMF monosolvate (RA SDMF). This needle-shaped crystal form crystallises in the monoclinic space group P21/c with two formula units in the asymmetric unit. The structure does not show the centrosymmetric carboxylic acid dimer. Instead, each RA molecule hydrogen bonds from the carboxylic OH group and 4-OH group to an independent DMF molecule. Each DMF molecule is thus accepting a HB from one 4-OH and one carboxylic acid OH group, resulting in infinite hydrogen bonded zigzag chains of almost flat conformation. There are no strong or directed interactions between the chains or layers of the crystal structure.
Finally, slow or fast cooling of a hot saturated dioxane solution of RA yields the dioxane hemisolvate (RA SDIO0.5). This solvate has been previously observed by Braun et al.22 and its crystal structure could here be determined with the help of synchrotron radiation. The needle-shaped crystal form crystallises in the monoclinic space group P21/c with one formula unit in the asymmetric unit. The RA molecules connect through the homomeric carboxylic acid dimer, while the 4-OH group hydrogen bonds to the dioxane molecule. This generates infinite chains of planar RA dimers with the dioxane molecules being twisted out of this plane by approximately 63°.
Crystallising 5-BRA from ethanol by fast cooling results in the monoclinic monosolvate 5-BRA SEtOH in the space group P21/c. The hydrogen bonding formed in the structure resembles that of 5-BRA SMeOH, with the carboxylic acid dimer hydrogen bonding to ethanol through the 4-OH group. However, the solvent does not form a hydrogen-bonded square in SEtOH, but connects 5-BRA molecules in an infinite chain, with each 5-BRA phenyl ring at 83° to the neighbouring molecule. This hydrogen-bonding motif generates a 3D network of directed interactions.
By precipitation with acetone from toluene solution, we only once obtained single crystals of the acetone monosolvate 5-BRA SAce. This solvate crystallises in the monoclinic space group P21/n with one formula unit in the asymmetric unit. The structure shows the 5-BRA homodimer through the centrosymmetric carboxylic acid dimer, while a further HB is donated by the 4-OH group to the solvent molecule.
Slow/fast cooling or precipitation with toluene of a DMF solution results in the monosolvate 5-BRA SDMF. The monoclinic crystal structure in P21/n contains two formula units in the asymmetric unit. Two 5-BRA molecules form the homomeric carboxylic acid dimer with the respective 4-OH group hydrogen bonding to the DMF carbonyl oxygen atom. These finite tetramers are almost completely planar and stack into a layered structure without further strong interactions between the tetramers or the stacks.
Crystallisation from DMSO by any method tried results in the monosolvate 5-BRA SDMSO crystallising in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) with one formula unit in the asymmetric unit. This crystal form shows infinite hydrogen-bonded chains of 5-BRA and DMSO, in which the DMSO oxygen atom accepts hydrogen bonds from both the 5-BRA carboxylic acid and the 4-OH group, resulting in the planar arrangement of the 5-BRA host molecules with the DMSO in between planes of the host.
with one formula unit in the asymmetric unit. This crystal form shows infinite hydrogen-bonded chains of 5-BRA and DMSO, in which the DMSO oxygen atom accepts hydrogen bonds from both the 5-BRA carboxylic acid and the 4-OH group, resulting in the planar arrangement of the 5-BRA host molecules with the DMSO in between planes of the host.
Fast cooling of a hot butanone (ethyl methyl ketone, EMK) solution resulted in the hemisolvate 5-BRA SEMK (Table S3†). The triclinic structure in the space group P contains two formula units in the asymmetric unit. Both symmetry-independent 5-BRA molecules are involved in the centrosymmetric acid homodimer, while one hydrogen bonds through the 4-OH group to the 2-OH group of the next 5-BRA, the other donates a hydrogen bond to the solvent molecule. This arrangement results in planar stacks of 5-BRA molecules that form steep herringbone stacking with the next stack with a twist angle of 87° between neighbouring 5-BRA molecules.
Crystallisation from dioxane results in two different solvates: slow and fast cooling of a hot solution results in the orthorhombic monosolvate 5-BRA SDIO1 in the space group Pnma. The asymmetric unit contains two formula half-units on special positions. Each of the symmetry-independent 5-BRA molecules hydrogen bonds to a dioxane molecule through the carboxylic acid, while the 4-OH group of one 5-BRA molecule connects to another 5-BRA, that of the other connects to a solvent molecule. This generates a planar network of host and guest molecules in the [0 1 0] plane, in which the host molecules are arranged completely planar and the dioxane molecules are twisted out of plane by 90°. No further strong interactions exist between the planes. By precipitation from dioxane with toluene, the hemisolvate 5-BRA SDIO0.5 can be obtained, which crystallises in the triclinic space group P. The asymmetric unit is a single formula unit, for which the dioxane lies on the inversion centre. The 5-BRA molecules interact through the centrosymmetric carboxylic acid dimer with each of the molecules donating a HB to dioxane through the 4-OH group. The resulting infinite chains arrange in a layered structure without further strong interactions between them.
Slow and fast cooling of a hot THF solution yields the monosolvate 5-BRA STHF, which crystallises in the monoclinic space group P21/c. The asymmetric unit contains one formula unit, of which the carbon backbone of the THF molecule is disordered over two positions. The 5-BRA molecules interact through the centrosymmetric acid homodimer while the 4-OH groups donate a HB to the solvent. The 5-BRA dimers are planar while the solvent is twisted out of plane by approximately 90°. These tetramers are stacked along [1 0 0] with adjacent stacks forming a herringbone pattern.
In addition to these crystal forms, a nitromethane solvate crystallises by fast cooling from nitromethane solution and an acetonitrile solvate could be obtained from slow/fast cooling and by precipitation with chloroform or toluene, but no crystal structures could be obtained.
Fast cooling of a hot THF solution results in the crystallisation of the monosolvate 3,5-BRA STHF in the monoclinic space group P21/n. The asymmetric unit contains one formula unit in which both molecules are disordered. The THF molecule shows disorder in the carbon backbone with a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 distribution over two sites, while the 3,5-BRA molecule shows a 180° flip with an approximately 9:1 distribution between the two orientations. The structure shows 3,5-BRA forming the centrosymmetric acid dimer with the respective 4-OH group hydrogen bonding to the THF molecules. These tetramers then form steep stacks along [1 0 0] with no further strong interaction between them or between stacks. The THF molecules of one stack are accommodated in the space closest to the acid groups of the next stack, allowing sufficient space for motion within the solvent backbone leading to the observed disorder.
:1 distribution over two sites, while the 3,5-BRA molecule shows a 180° flip with an approximately 9:1 distribution between the two orientations. The structure shows 3,5-BRA forming the centrosymmetric acid dimer with the respective 4-OH group hydrogen bonding to the THF molecules. These tetramers then form steep stacks along [1 0 0] with no further strong interaction between them or between stacks. The THF molecules of one stack are accommodated in the space closest to the acid groups of the next stack, allowing sufficient space for motion within the solvent backbone leading to the observed disorder.
Cooling of hot saturated solutions in DMF results in the monosolvate 3,5-BRA SDMF, which crystallises in the orthorhombic space group Pna21 with one formula unit in the asymmetric unit. The structure shows each 3,5-BRA hydrogen bonding to two DMF molecules through the acid moiety and the 4-OH group. Each DMF accepts hydrogen bonds from two different 3,5-BRA molecules, leading to infinite chains in the crystallographic b-plane. No further strong interaction exists between the chains.
Fast cooling of a 1-pentanol solution results in the monosolvate 3,5-BRA SPeOH which crystallises in the monoclinic space group P21/c with one formula unit in the asymmetric unit. The structure again is formed by infinite hydrogen-bonded chains but unlike any other solvate, these are formed purely from 3,5-BRA hydrogen bonding through the 4-OH group to the acid carbonyl. The acid OH-group hydrogen bonds to the solvent OH-group, which in turn donates to the adjacent 3,5-BRA 4-OH group to form an R33(8) motif.28 The flat and rigid infinite chains stack along [0 0 1] without further interaction between them, with the solvent located between individual stacks. This generates a certain degree of phase separation with the highly ordered and rigid host layers interspersed with more flexible solvent layers. This flexibility leads to disorder in the solvent molecules, which adopt at least two major positions with close to 1:1 probability.
From the reaction mixture, the monosolvate 3,5-BRA SAcOH crystallises in the monoclinic space group I2/a with one formula unit in the asymmetric unit. The structure shows both the host and the solvent individually forming centrosymmetric acid dimers which are connected through a HB donated by the 4-OH group of 3,5-BRA. These flat infinite chains pack on top of each other along [0 1 0] and stack in a shallow herringbone pattern with no further strong interactions between the chains or stacks.
A second acetic acid solvate, namely the hemisolvate 3,5-BRA SAcOH0.5, exists in this system, as identified by TGA and PXRD of the reaction mixture after storage for 24 hours (ESI† Fig. S10 and S25).
Packing considerations
Despite the wide range of solvates, none of the structures are isostructural with another, but there are three major interaction motifs that can be identified (Fig. 2). From the total of 18 crystal structures of all three resorcylic acid derivatives, 11 show the formation of the centrosymmetric acid homodimers (ESI† Table S5). Even though any statistics based on such a small dataset is inherently limited, the four RA structures show 50% dimer formation, monobromination increases this behaviour to 78% (7 out of 9), while dibromination reduces this behaviour to 40% (2 out of 5). This points to a preference of dimer formation for the resulting crystal packing in the case of one sterically hindering substituent and a resulting unbalanced, or ‘awkward’ molecular structure. This concept of awkwardness has previously been identified as one of the driving factors of molecules forming crystal structures with higher Z′,29 and it seems also to further homomeric acid dimer formation in the case of sterically hindered resorcylic acid derivatives. The reason for this is currently unclear and needs a larger dataset for further investigation.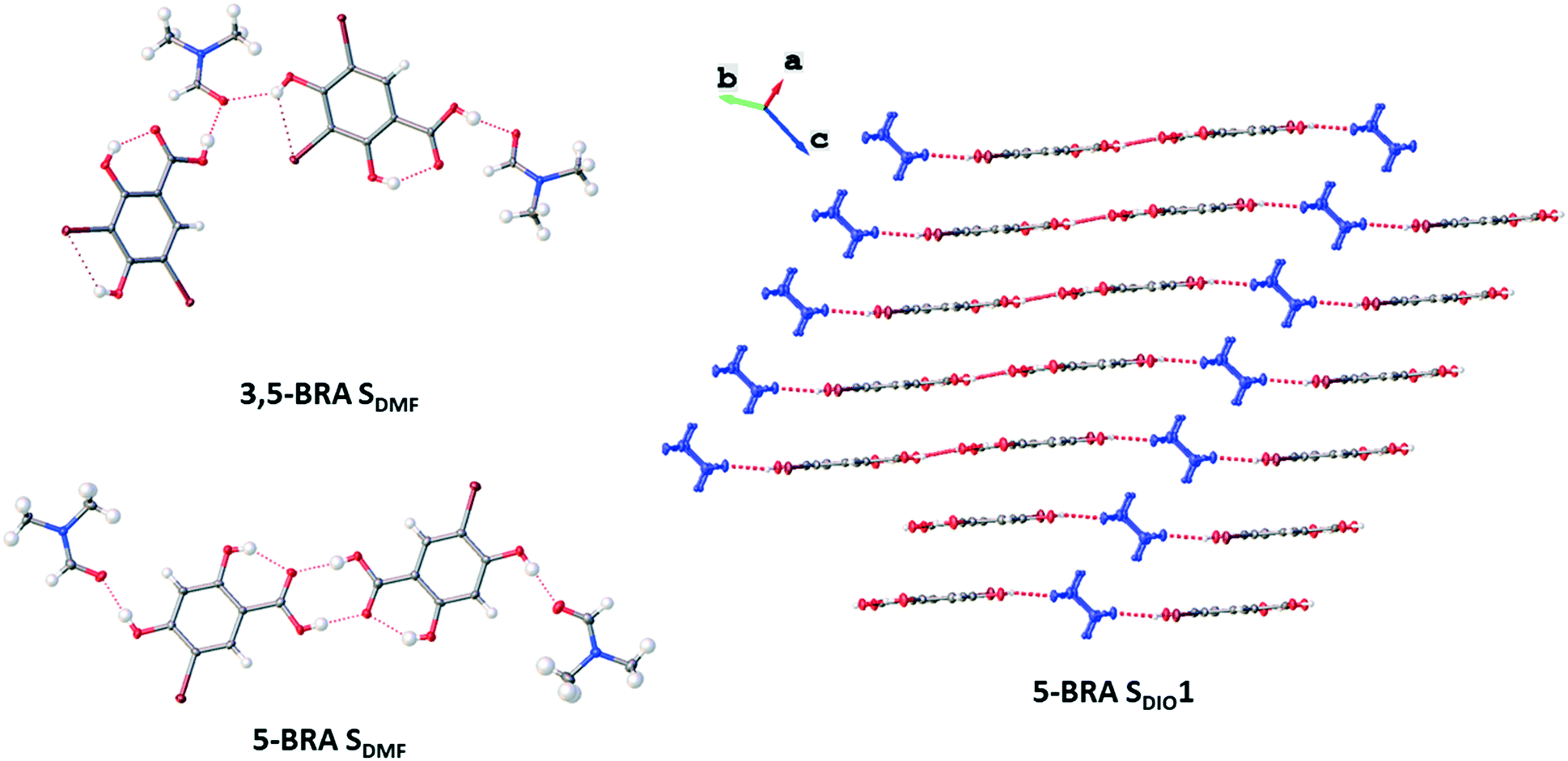 | ||
| Fig. 2 Molecular interaction motifs as observed in the different crystal forms: no dimer formation in 3,5-BRA SDMF, dimer formation into finite tetramers with solvate molecules in 5-BRA SDMF, and dimer formation into infinite chains in 5-BRA SDIO1. | ||
Of the seven solvated crystal structures not forming the acid dimer motif, five show infinite chains of the host connected through the solvent by hydrogen bonds donated both by the carboxylic acid as well as the 4-OH group (Fig. S1†). These chains can either be linear, such as in the case of 5-BRA SDMSO, or be zigzagged, such as in RA SAcOH. 5-BRA SDIO1 shows both the infinite chain along [1 0 0] as well as finite chains of 5-BRA and dioxane connected to the main chain through the 4-OH group of the host molecule. These finite chains terminate with the dioxane after one unit. The second motif is observed in 3,5-BRA SPeOH, where the solvent forms the mentioned cyclic motif with the carboxylic acid and the 4-OH group of two host molecules (vide supra). This motif is repeated infinitely, comparable to the other crystal forms in this group.
Of the 11 acid dimer structures, five form finite tetramers in which the solvent hydrogen-bonds to the 4-OH groups of the host dimers (Fig. S2†). This is due to the nature of the solvents, which only have one HB acceptor group, and thus terminate the motif. The packing of these finite tetramers can follow two options: they can either be stacked, forming phase separated layers of the host and the guest molecules, or tessellated where the guest molecules locate in pockets or channels surrounded by host dimers (Fig. S3†). However, the angle between adjacent layers or stacks varies between the five solvates. The remaining six solvates revealing tetramers show infinite chain formation where the solvent links the host dimers. The incorporated solvents of these crystal forms can either accept two hydrogen bonds, such as dioxane, or accept and donate one HB each, such as the lower alcohols. All but one of the five forms showing infinite chains also reveal a layered structure with no strong interactions between the chains or layers (Fig. S4a†). The exception is 5-BRA SEtOH, in which the ethanol molecules connect different layers through the 21-screw axis along [0 1 0]. This leads to highly hydrogen-bonded layers, but no further strong interaction between these sheets, comparable to the remaining four forms showing the infinite chain motif (Fig. S4b and c†).
While it is interesting that these three major motifs (no dimers, dimers in finite tetramers or dimers in infinite tetramer chains) exist, there is no obvious trend in motif formation based on the nature of the incorporated solvents. For example, dioxane is incorporated in three crystal forms, and while the hemi-solvates of RA and 5-BRA show infinite tetramer chain formation, the monosolvate of 5-BRA does not form dimers. In the case of 5-BRA, the alcohol solvates show infinite chain formation, but in 3,5-BRA, the inclusion of 1-pentanol leads to a completely different motif. Finally, DMSO incorporation can either lead to infinite chain or to no dimer formation, and the incorporation of DMF leads mostly to infinite chain but also to finite tetramers. There is also no correlation between the stoichiometry of the incorporated solvent and the motif, with hemisolvates showing the same spread of motifs as monosolvates. It is clear that in these small and rigid host molecules there are more important structure-driving forces than the hydrogen bonding of the guest molecules.
The packing similarity between all reported forms was investigated using the crystal packing similarity search option in Mercury.30 A cluster of 30 molecules of each structure was chosen with the tolerances on distance and angle given as 20%. The search ignored molecular difference and hydrogen count to allow for comparison between the different derivatives. Overall, similarities of up to 23 of 30 molecules could be found (RA SDMSO and RA SDIO0.5), but all of these are limited to finite rods of stacked host monomers or dimers and no further overarching cluster could be identified.
Hirshfeld surface analysis
To better understand the relationship within this set of diverse solvate structures, a Hirshfeld surface analysis has been undertaken and the fingerprint plots are presented in the ESI† (Tables S6–S8). All structures showing the centrosymmetric acid dimer also have two significant short-range interactions in the fingerprint plots. This is also true for the two acetic acid solvates, which form heteromeric acid dimers. For the structures not forming these dimers, only one short-range interaction shows in the fingerprint plot, representing the hydrogen bond donated through the acid OH group. In addition, the surfaces are moving further away from the central molecule upon substitution with bromine. In the parent compound, the Hirshfeld surfaces are rarely further away than 2.4 Å from the internal atom, while this lengthens to 2.6 Å for the brominated compounds. This is solely due to the bromine substituents as can be seen by the partial surfaces. It is also clear that with the introduction of bromine, this substituent is taking an increasing role in the intermolecular interactions. While in RA these are dominated by oxygen contacts making up between 28.5 and 29.9% of the surface, with the introduction of one bromine substituent this contribution is reduced to between 22.6 and 26.1%. Bromine contacts contribute between 18.4 and 20.0% of the overall Hirshfeld surface. This indicates that inclusion of a bromine substituent is by no means insignificant when considering potential crystal forms. Addition of two bromine substituents effects the dominance of bromine contacts on the Hirshfeld surface (31.9 to 33.8%) while oxygen contacts only make between 22.7 and 25.2% of the surface.Non-covalent interactions (radial distribution)
An alternative way to look at similarity in this set of very diverse crystal structures is to investigate the radial distribution of the molecular interactions. These distributions show the localisation of adjacent molecules around a central molecule in the structural overlay of all structures, and for clarity has been divided into groups of the same host molecule. Despite the lack of isostructurality in this set of crystal forms, the impact of steric bulk becomes apparent through this analysis, as the distribution of solvent around the central resorcylic acid derivative shows clear differences between the non-brominated, mono-brominated and dibrominated species (Fig. 3).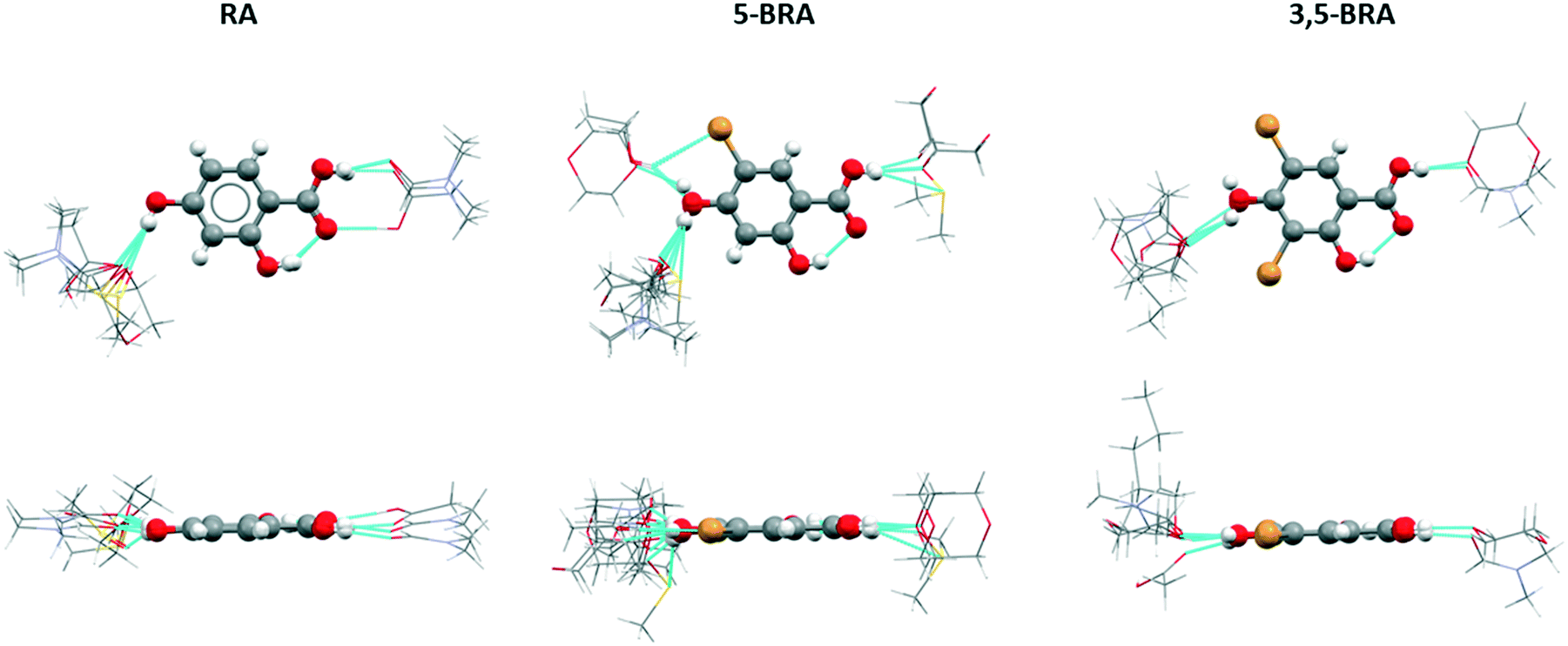 | ||
| Fig. 3 First interaction shell with solvent of RA, 5-BRA and 3,5-BRA for all known crystal structures. Only hydrogen bonded solvent molecules are shown, solvent molecules are presented as wireframe for clarity. | ||
RA structures show a directed interaction with solvent through the acid moiety only with acetic acid, forming the acid heterodimer, or with DMF in a closely related motif. All other solvent molecules, i.e. DMSO, dioxane, and a second DMF molecule, are located in the vicinity of the 4-OH group. Surprisingly, this latter group of solvents occupies a small space off the central axis of the molecule and on the side of the 2-OH group. In addition, all solvent molecules are in the same plane as the host molecule. The DMF and acetic acid molecules bound to the RA acid moiety show a small deviation of 12–16° between their molecular plane and that of the host RA, while the heavy atoms of the solvent around the 4-OH group occupy a compact space of only 1.2–1.4 Å above and below the plane of RA.
The introduction of unbalanced steric bulk in the case of 5-BRA softens the interaction shell. The binding sites to solvent remain the acid and the 4-OH groups, with most of the solvent molecules interacting with the latter. This is due to the increased occurrence of the centrosymmetric acid homodimer of the host in 5-BRA compared to RA. While in RA the orientation of the solvent around the host acid group is strongly confined, in 5-BRA this orientation is wider with none of the solvents adopting the acid dimer orientation. The width of the solvent space occupied is 5.6 Å (furthest apart heavy atoms) in 5-BRA. Around the 4-OH group, there are now two locations of solvent binding. One is the orientation towards the side of the 2-OH group, which is also observed in RA. In addition, some solvent molecules locate towards the bromo-substituent, indicating that there is now space available due to the steric bulk of the halogen. Interestingly, this new position contains both hydrogen-bond donating and accepting solvents (ethanol and dioxane). Compared to RA, the solvation shells extend 1.2–3.6 Å further above and below the host molecule. This is partly due to the increased size of the incorporated solvent and partly due to the orientation of the solvent molecules, especially in the case of the DMSO solvate which locates completely below the plane of the host and protrudes out by 2.6–3.6 Å with its furthest heavy atom.
The solvation shell of 3,5-BRA resembles more that of RA than that of 5-BRA, which could be explained by balancing the steric bulk provided by the dibromination. All solvent molecules either HB to the acid or the 4-OH group, and the orientation of the solvent around the acid resembles that found in 5-BRA. The 4-OH group connects to incorporated solvent only on the side of the 2-OH group, comparable to RA; however, the angles of these hydrogen bonds are with 133–141° shallower than those of the unsubstituted host (111–121°). This is due to the steric bulk of the second bromo substituent. Incorporated solvent also extends the furthest above and below the host molecule indicating that the face-to-face stacking of 3,5-BRA is less efficient than in the other derivatives. This enables the inclusion of larger solvents, such as 1-pentanol that orients orthogonal to the plane of the host molecules.
The increased steric hindrance of the bromo-substituents can also be observed in the location of neighbouring host molecules. In the case of RA, most neighbouring molecules in the different forms are connected through the acid group. However, there are weaker non-classical hydrogen bonds through both the C–H bonds in the 3- and 5-positions (Fig. S5†). On the side of the 2-OH group, these interactions are to co-planar molecules, while the interactions on the opposite side lead to a more angled orientation of the RA molecules. Introduction of one bromo-substituent leads to a significant change in the host–host interactions. Firstly, due to the wider first solvation shell around the 4-OH group, the 3-position of the ring is sterically obstructed and thus does not show any interactions with neighbouring molecules. These interactions are shifted to the 2-OH group, which accepts hydrogen bonds from neighbouring 5-BRA molecules oriented orthogonal to the central molecule (Fig. S6†). Secondly, the 5-position of the ring has been brominated and hence clearly cannot donate a C–H hydrogen bond. However, the bromine atom can interact through halogen bonds, which is realised in four of nine solvates of 5-BRA only, and the interaction are only between neighbouring host molecules but not to solvent molecules. In 5-BRA SDMF, STHF and SMeOH, the halogen bond is donated from the central molecule to accepting 2-OH groups exclusively. In 5-BRA SEtOH, the bromo-substituents form a halogen-bonded chain along [0 1 0]. Finally, the introduction of the larger bromine atom leads to a lengthening of the interaction of neighbouring host molecules, with the centroid distances changing from 5.8–6.9 Å in RA to 7.8–8.7 Å in 5-BRA. This loosening of the host network can be seen as one of the reasons for the increased solvent inclusion in 5-BRA.
The inclusion of a second bromo-substituent introduces steric bulk around the 2-OH group, which can be observed by a widened interaction shell of the 3,5-BRA solvates. While the hydrogen bonds around the 4-OH and the carboxyl groups show a similar planar orientation as observed in RA (Fig. S7†), the 2-OH group is involved in interactions covering a comparable space to the 5-BRA solvates. However, while 5-BRA only shows host–host interactions through the 2-OH group, 3,5-BRA shows interactions with solvent in the space close to the steric bulk of the bromo-substituent, and host–host interactions are further pushed towards the side of the carboxylic acid group (Fig. S8†). In addition, the presence of two bromine atoms in the molecule enables a stronger presence of halogen bonds around the central molecule (Fig. S8†), which now show a wide variation of orientations and one case of centrosymmetric dimer formation. The distance between host molecules in 3,5-BRA is with 8.7–9.1 Å even farther apart than in 5-BRA. The exception is 3,5-BRA SDIO, which shows two different halogen bonding interactions with shorter distances. Firstly, two 3,5-BRA molecules show a centrosymmetric dimer formation through halogen bonding with the carboxylic acid group leading to a centroid distance of only 7.3 Å. On the other side of the molecule, the halogen atoms form infinite chains by bonding to the carboxylic acid of neighbouring host molecules of orthogonal orientation leading to a centroid distance of 8.5 Å. This shows that there are closer packing arrangements possible, even in the presence of increased steric bulk, but this strongly depends on the incorporated solvent.
Physicochemical stability
The thermal stability of the solvates does not show any significant difference between the different packing motifs (Fig. S9 and S10†). The absence of carboxylic acid homodimer structures in 5-BRA SDMSO and SDIO1 leads to the highest and lowest thermal stability of all 5-BRA solvates. Even though it seems that the formation of finite dimers leads to a reduced thermal stability in 5-BRA SEMK, STHF and 3,5-BRA STHF, 5-BRA SDMF shows one of the highest thermal stabilities of the series. There is also no connection between the steric bulk of the host molecule and the thermal stability of the respective solvates. Both 5-BRA and 3,5-BRA solvate stabilities are spread over a thermal range from room temperature to over 100 °C. It is thus clear that the overall differences in crystal packing, as well as the electrostatic interactions between the solvent, plays a dominant role in determining the thermal stability, rather than the steric effect of the host molecules.A second consideration is to link the ease of nucleation/growth of the solvate to the packing motif. While no in-depth nucleation and growth studies have been conducted in this project, the appearance of the solvates during the crystallisation screen can be taken as preliminary data (Table S9†). Again, no clear trend can be observed with most of the solvates found crystallising by the kinetically controlled methods of fast cooling and precipitation. This fact points towards molecular interactions present already in solution that can be kinetically trapped when crystallisation happens far from equilibrium. A related fact has been observed for molecular solvates with a gradual desolvation in a fashion reminiscent of Ostwald's rule of stages.31,32 On the other hand, a large range of the solvates can be crystallised by methods close to the thermodynamic equilibrium, such as slow cooling and evaporation. Again, there is no correlation between structural motif and crystallisation method.
Conclusions
In this study we investigated the influence of steric bulk on the formation and structure of crystalline solvates in a related series of non-, mono- and dibrominated β-resorcylic acids. A total of 18 crystal structures of solvates of the three compounds were compared, of which 17 are newly reported here. The majority of the forms (9) are observed in the monobrominated derivative 5-BRA. None of the crystal structures show isostructurality, even though RA SDIO0.5 and RA SDMSO show a close structural relationship. Three different packing motifs can be distinguished: the host acid molecules can either show the centrosymmetric carboxylic acid dimer or not. When showing the dimer formation, this motif can either form finite tetramers with solvent at the terminal OH groups or infinite chains through bridging solvent molecules.While the impact of the steric bulk introduced by the bromo-substituents is not clearly recognisable through packing studies, it becomes more obvious from the analysis of the first interaction shell around a central host molecule. While both RA and 3,5-BRA as host molecules without or with more balanced steric bulk show a compact interaction shell with the solvent molecules through either the acid or 4-OH groups, the more ‘awkward’ 5-BRA has a widened interaction shell. The steric bulk additionally pushes host–host interactions to occur through the 2-OH group.
The introduction of bromo substituents leads to the formation of halogen bonds, either between two bromo-substituents, but more often between bromine and oxygen of neighbouring molecules. This stabilising but longer interaction leads to a lengthening of the centroid distance of neighbouring host molecules, and we hypothesise that this loosening of the molecular packing further enables the incorporation of solvent molecules.
Overall, it is clear that steric bulk furthers the formation of solvates in this system, and we surmise this may be more general. This insight is particularly important for pharmaceutical lead optimisation, during which a frequently used tool is bio-isosteric replacement. Bio-isosteric groups can vary significantly in bulk and thus can introduce steric imbalance into the lead compound. Thus, such changes may inadvertently induce a greater propensity to form solvates. The resulting solvent inclusion can lead to the carryover of potentially toxic solvents into different manufacturing stages, or even into the product where it can cause severe harm to the patient. The results of this study add to the overall understanding of the influence of molecular structure on crystallisation behaviour and thus can be used to generate a more informed lead-optimisation process.
Author contributions
J. T., S. F. and R. M. E. were responsible for investigation, K. E. and R. M. E. were responsible for writing and editing, K. E. further provided conceptualisation, methodology, investigation and resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Diamond Light Source for access to its single crystal beamline granted to the Durham–Newcastle BAG application.Notes and references
- R. Hilfiker, Polymorphism: In the Pharmaceutical Industry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2006 Search PubMed.
- H. G. Brittain, Polymorphism in pharmaceutical solids, CRC Press, 2018 Search PubMed.
- G. G. Z. Zhang, D. Law, E. A. Schmitt and Y. Qiu, Adv. Drug Delivery Rev., 2004, 56, 371–390 CrossRef CAS PubMed.
- K. Hunger, Rev. Prog. Color. Relat. Top., 1999, 29, 71–84 CAS.
- M. Li, A. H. Balawi, P. J. Leenaers, L. Ning, G. H. L. Heintges, T. Marszalek, W. Pisula, M. M. Wienk, S. C. J. Meskers, Y. Yi, F. Laquai and R. A. J. Janssen, Nat. Commun., 2019, 10, 2867 CrossRef PubMed.
- J. Haleblian and W. McCrone, J. Pharm. Sci., 1969, 58, 911–929 CrossRef CAS PubMed.
- D. Singhal and W. Curatolo, Adv. Drug Delivery Rev., 2004, 56, 335–347 CrossRef CAS PubMed.
- C.-H. Gu, V. Young and D. J. W. Grant, J. Pharm. Sci., 2001, 90, 1878–1890 CrossRef CAS PubMed.
- P. Láng, V. Kiss, R. Ambrus, G. Farkas, P. Szabó-Révész, Z. Aigner and E. Várkonyi, J. Pharm. Biomed. Anal., 2013, 84, 177–183 CrossRef PubMed.
- FDA Guidance for Industry: ANDAs: Pharmaceutical Solid Polymorphism, July 2007.
- W. C. McCrone, in Physics and Chemistry of the Organic Solid State, ed. D. Fox, M. M. Labes and A. Weissberger, Interscience, New York, 1965, vol. II, pp. 725–767 Search PubMed.
- S. M. Reutzel-Edens and R. M. Bhardwaj, IUCrJ, 2020, 7, 955–964 CrossRef CAS PubMed.
- D. E. Braun, P. G. Karamertzanis and S. L. Price, Chem. Commun., 2011, 47, 5443–5445 RSC.
- P. T. Galek, F. H. Allen, L. Fábián and N. Feeder, CrystEngComm, 2009, 11, 2634–2639 RSC.
- H. L. Friedman, Influence of isosteric replacements upon biological activity, NAS-NRS Publication, 1951, pp. 295–358 Search PubMed.
- P. H. Olesen, Curr. Opin. Drug Discovery Dev., 2001, 4, 471–478 CAS.
- A. B
![[e with combining overline]](https://www.rsc.org/images/entities/char_0065_0305.gif) rziņš and A. Actiņš, Cryst. Growth Des., 2016, 16, 1643–1653 CrossRef.
rziņš and A. Actiņš, Cryst. Growth Des., 2016, 16, 1643–1653 CrossRef. - T. Kitchen, C. Melvin, M. N. Mohd Najib, A. S. Batsanov and K. Edkins, Cryst. Growth Des., 2016, 16, 4531–4538 CrossRef CAS.
- G. T. Gál, N. V. May, L. Trif, J. Mihály and P. Bombicz, CrystEngComm, 2021, 23(42), 7425–7441 RSC.
- M. T. Kirchner, L. S. Reddy, G. R. Desiraju, R. K. R. Jetti and R. Boese, Cryst. Growth Des., 2004, 4, 701–709 CrossRef CAS.
- R. M. Edkins, E. Hayden, J. W. Steed and K. Edkins, Chem. Commun., 2015, 51, 5314–5317 RSC.
- D. E. Braun, P. G. Karamertzanis, J. B. Arlin, A. J. Florence, V. Kahlenberg, D. A. Tocher, U. J. Griesser and S. L. Price, Cryst. Growth Des., 2011, 11, 210–220 CrossRef CAS PubMed.
- B. K. Saha and A. Nangia, Chem. Commun., 2005, 3024–3026 RSC.
- E. J. Tisdale, B. G. Vong, H. Li, S. H. Kim, C. Chowdhury and E. A. Theodorakis, Tetrahedron, 2003, 59, 6873–6887 CrossRef CAS.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. Leiserowitz, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1976, 32, 775–802 CrossRef.
- M. C. Etter, J. C. MacDonald and J. Bernstein, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 256–262 CrossRef PubMed.
- K. M. Anderson, M. R. Probert, A. E. Goeta and J. W. Steed, CrystEngComm, 2011, 13, 83–87 RSC.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- K. Fucke, J. A. K. Howard and J. W. Steed, Chem. Commun., 2012, 48, 12065–12067 RSC.
- R. J. Davey, M. Brychczynska, G. Sadiq, G. Dent and R. G. Pritchard, CrystEngComm, 2013, 15, 856–859 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: For supplementary figures and tables. CCDC numbers 2120714–2120730. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ce01592c |
| This journal is © The Royal Society of Chemistry 2022 |