
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeavier N-heterocyclic half-sandwich tetrylenes†
Inga-Alexandra
Bischoff
,
Bernd
Morgenstern
and
André
Schäfer
 *
*
Department of Chemistry, Faculty of Natural Sciences and Technology Saarland University Campus Saarbrücken, 66123 Saarbrücken, Germany. E-mail: andre.schaefer@uni-saarland.de
First published on 19th July 2022
Abstract
ansa-Half-sandwich complexes of the group 14 elements germanium, tin and lead are reported, which represent a new class of Lewis amphiphilic tetrylenes and bridge the gap between classical N-heterocyclic systems and group 14 metallocenes. These compounds can form complexes both with carbenes and transition metal fragments.
Divalent group 14 compounds with the tetrel element in the oxidation state +II, commonly referred to as tetrylenes, have attracted significant attention for several decades.1 Among other things, persistent examples of these species have been applied in bond activation processes and as ligands for various metal fragments.2 The discovery of the first stable singlet carbene dates back to the early work of Wanzlick et al. and was later complemented by the first structural authentications of such species by Bertrand et al. and Arduengo et al.3 The history of the discovery of the heavier diorgano congeners is a bit more convoluted, as reports of an N-heterocyclic silylene by Denk and West et al. in 1994, as well as Lappert et al.'s descriptions of bis(trimethylsilyl)methyl and bis(trimethylsilyl)-amido germylenes, stannylenes and plumbylenes in 1973/1974, and Veith's note of an N-heterocyclic stannylene in 1975 are often regarded as the first examples.4 However, it is worth noting that the description of heavier group 14 metallocenes (tetrelocenes) pre-date those reports in all cases (Cp*2Si: Jutzi et al., 1986;5 Cp2Ge: Scibelli et al., 1973;6 Cp2Sn: Fischer et al., 1956;7 Cp2Pb: Fischer et al., 19568), and there can be no doubt that tetrelocenes of the type Cp2E are also divalent diorgano tetrel(II) species, although their reactivities differ from that of “typical” tetrylenes in some cases.2b,9 Due to the frontier orbital configuration in singlet tetrylenes, with a lone pair and a vacant orbital of p symmetry at the low-valent tetrel atom, tetrylenes can exhibit Lewis amphiphilic character, although the Lewis acidity is sometimes quenched by donor ligands and/or substituents. Tetrelocenes on the other hand, do not exhibit significant σ donor character, but can act as electron acceptors, as highlighted by amine, bipyridine and NHC complexes of stannocenes and plumbocene.9,10 With the aim to merge the two fields of tetrelocenes and heavier bis(amido)tetrylenes, we investigated the possibility of applying ansa-half-sandwich ligands to Ge(II), Sn(II) and Pb(II). These ligands are already quite common in transition metal chemistry, for instance in group 4 complexes,11 but have not seen much application in main-group chemistry so far.12 Herein, we report the first heavier N-heterocyclic half-sandwich tetrylenes and reactivity studies of their donor and acceptor behaviour.
Reaction of the dilithiated ligands, bearing a (tetramethyl)-cyclopentadienide and a tert-butyl amido substituent, with GeCl2, SnCl2, and PbCl2 resulted in the formation of the corresponding ansa-half-sandwich tetrylenes 1a-d (Scheme 1). Whereas compounds 1b,c were obtained as orange to red oils, 1a was isolated in form of yellow crystals and 1d in form of dark red crystals. 1a-d were examined by multinuclear NMR spectroscopy, which in case of the tin and lead species, 1a,c,d, allows for comparison with related di-N-heterocyclic systems and tetrelocenes. In the 119Sn{1H} NMR spectrum a signal is observed at −296 ppm for 1a, and at −315 ppm for 1c. Quite interestingly, these shifts are positioned between the typical low-field shifted resonances usually observed for di-N-heterocyclic stannylenes, as for instance reported for the structurally closely related Veith stannylene, and related stannocenes, which typically exhibit very upfield-shifted resonances.
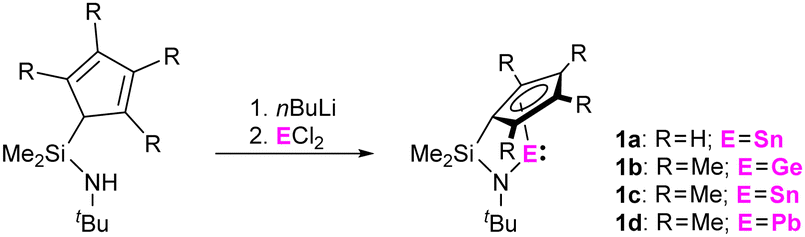 | ||
| Scheme 1 Synthesis of ansa-half-sandwich tetrylenes 1a-d. | ||
This clearly reflects the electronic influence of both substituent sides, as the deshielding of the tetrel atom in tetrylenes is usually associated with a large paramagnetic component to the NMR chemical shift, which relates to the energy gap between the lone pair and the vacant p orbital. This contribution is much smaller in tetrelocenes, as the lone pair is quite low in energy due to its high s character. The same phenomenon is observable for plumbylene 1d, for which a signal in the 207Pb{1H} NMR spectrum is observed at +1562 ppm, which is also located in-between the typical low-field shift range of diamido plumbylenes and high-field shifted resonances of plumbocenes (Fig. 1).
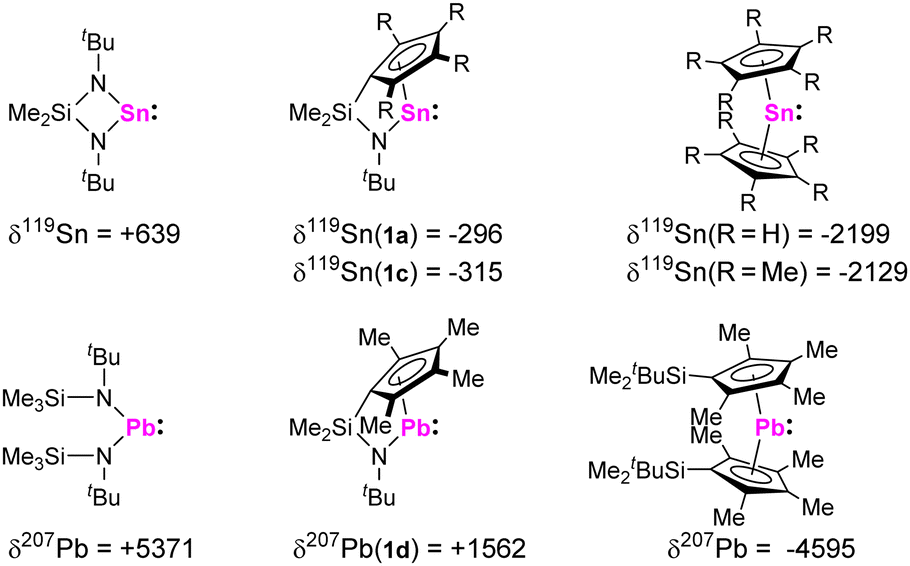 | ||
| Fig. 1 119Sn and 207Pb NMR chemical shifts of stannylenes 1a and 1c, plumbylene 1d, and related literature-known compounds.15 | ||
Structural characterization of 1a,d in the solid state by single crystal X-ray diffraction confirms the molecular structures of the compounds and reveals interesting intermolecular interactions (Fig. 2 and Fig. S51, S52, ESI†).13,14 In both structures, the tetrel atom is bound to the cyclopentadienide group in what might best be perceived as a strongly distorted η3 to η5 mode with unequal Sn/Pb–CCp bond distances (Sn–C: 249.54(16) to 338.86(18) pm; Pb–C: 249.37(23) to 339.50(21) pm) although the C–C distances within the cyclopentadienyl ring do not display isolated single and double bonds, but are rather relatively uniform, indicating a certain degree of aromaticity (1a: 139.71(20) to 143.22(21) pm; 1d: 139.60(33) to 146.82(28) pm). When interpreting these bonding situations, it must be taken into consideration that the Sn/Pb–Cp interaction is relatively ionic in nature10c and that there are significant intermolecular interactions in the solid state (Fig. S51–S52, ESI†). Thus, we believe drawing a circle within the Cp ring is justified. Furthermore, the shorter Sn/Pb–CCp contacts are in the same range as the bond lengths found in stannocene, Cp2Sn, (255.54(7) to 283.79(10) pm)9 and a related plumbocene, ((Me2tBuSi)Me4C5)2Pb, (270.71(44) to 277.30(48) pm).16 The Sn–N bond length in 1a is 210.64(12) pm, which is similar to Veith's stannylene (209.1(8) pm),17 and the Pb–N bond length in 1d is 221.47(19) to 222.74(23) pm, which is elongated compared to other N-heterocyclic plumbylenes, possessing Pb–N bond lengths between 208.7(5) and 213.6(5) pm.18 These elongations may result from the formation of polymeric chains in the solid state (Fig. S52, ESI†). Interestingly, such intermolecular interactions in the solid state are common for unsubstituted plumbocenes, but not for decamethylplumbocene or stannocenes.19
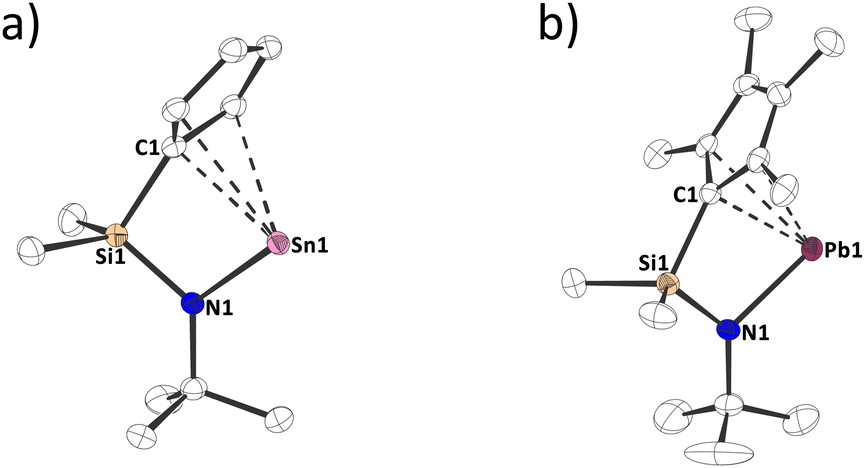 | ||
| Fig. 2 Molecular structure of (a) 1a and (b) 1d in the crystal (displacement ellipsoids at 50% probability level, hydrogen atoms omitted for clarity). | ||
The CCp–E–N angle around the tetrel, is 71.1° for 1a and 70.6° for 1d, which is similar to other four-membered N-heterocyclic stannylenes17 and plumbylenes.20
Following the NMR and structural characterizations, we inspected the molecular orbitals of 1a-d by DFT calculations14 (PBE0-D3/def2-TZVPP//PBE0-D3/def2-TZVP), which reveals that the orbital configuration is qualitatively identical in all four cases and that the LUMO corresponds to a vacant p orbital with a large coefficient at the central tetrel atom. This is typical for tetrelocenes as well as for “classical” tetrylenes. However, while in many tetrylenes the lone pair at the tetrel atom corresponds to the HOMO, it is energetically located significantly lower in tetrelocenes, due to higher s character. In 1a-d, the lone pair at the tetrel atom has a comparable shape to that in other N-heterocyclic tetrylenes and corresponds to the HOMO-2/3 (Fig. 3 and Table S1, ESI†).
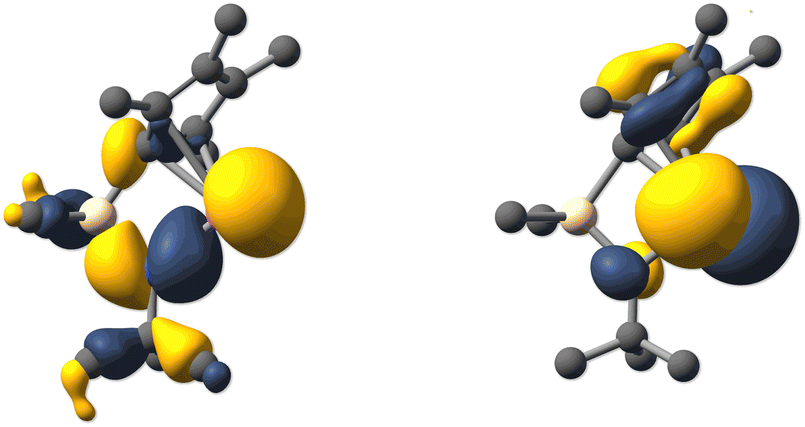 | ||
| Fig. 3 Kohn–Sham molecular orbital contours of the HOMO-3 (left) and LUMO (right) of germylene 1b (PBE0-D3/def2-TZVPP//PBE0-D3 def2-TZVP; isovalue 0.04). | ||
To study the reactivities of this new class of heavier N-heterocyclic tetrylenes and compare them with “classical” tetrylenes and tetrelocenes, three different reaction types were examined: oxidative additions, electron acceptor behavior (Lewis acidity) and electron donor behavior (Lewis basicity). To investigate oxidative addition reactions, 1a-d were treated with triethylsilane in toluene-D8, but no reactions were observed even after stirring for 4 h at 333 K. However, 1a,c were found to undergo rapid oxidative addition reactions with diphenyl diselenide, as indicated by 119Sn and 77Se NMR spectroscopy. When solutions of 1a,c in benzene-D6 were treated with equimolar amounts of diphenyl diselenide at ambient conditions, the 119Sn{1H} NMR spectra displayed new signals at −88.6 ppm21 (1a/2a) and at −70.5 ppm (1c/2b). Furthermore, in the 77Se{1H} NMR spectra, signals at 137.8 ppm21 (1a/2a) and 120.9 ppm (1c/2b) were observed, which displayed tin satellites corresponding to 1J77Se–Sn coupling constants of 1627 Hz (1a/2a) and 1631 Hz (1c/2b).
By comparison with related literature known compounds, we tentatively assign these signals to the corresponding bis(phenylseleniumyl)stannanes 2a,b (Scheme 2). These findings are in line with other N-heterocyclic stannylenes, which give the analogous oxidative addition products.22,23
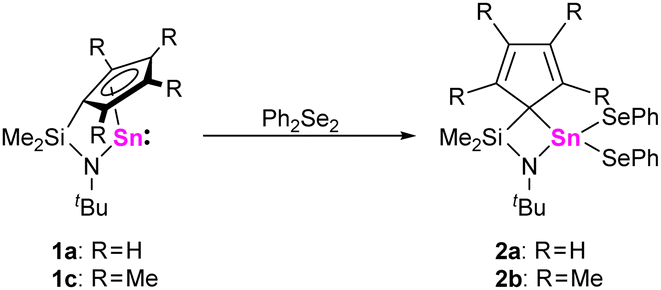 | ||
| Scheme 2 Reactions of 1a,c with diphenyl diselenide. | ||
To investigate the Lewis acidity of half-sandwich tetrylenes 1b-d, we reacted them with 4-(dimethylamino)-pyridine (DMAP) and 1,3-diisopropyl-4,5-dimethyl-imidazolin-2-ylidene (NHC). When mixtures of 1b-d and DMAP or NHC in benzene-D6 were inspected by multinuclear NMR spectroscopy, the formation of the corresponding adducts 4a-c and 5a-c was observed (Scheme 3). Most significantly, the carbene carbon atoms in 5a-c give rise to resonances at 179.4 ppm (5a), 182.9 ppm (5b) and 204.9 ppm (5c) in the 13C{1H} NMR spectra, clearly indicating a carbene metal coordination. Furthermore, crystals of 5a-c, suitable for single crystal X-ray diffraction could be obtained, allowing for solid-state structural characterization of these complexes (Fig. 4 and Fig. S54–S56, ESI†). Interestingly, the carbene coordination results in a change of bonding mode of the cyclopentadienyl substituent, as it adopts a σ bond to the central tetrel atom in all cases, 5a-c. Thus, a four-membered ring moiety consisting of the Si, N, CCp and Ge/Sn/Pb atoms is observed. This is remarkable, since such a change in bonding mode was not observed in stannocene and plumbocene NHC complexes, which we reported previously,9,10c and DFT calculations (PBE0-D3/def2-TZVP) suggest that the corresponding uncoordinated tetrylene with a σ bonded Cp ring is not a minimum on the potential energy surface.14
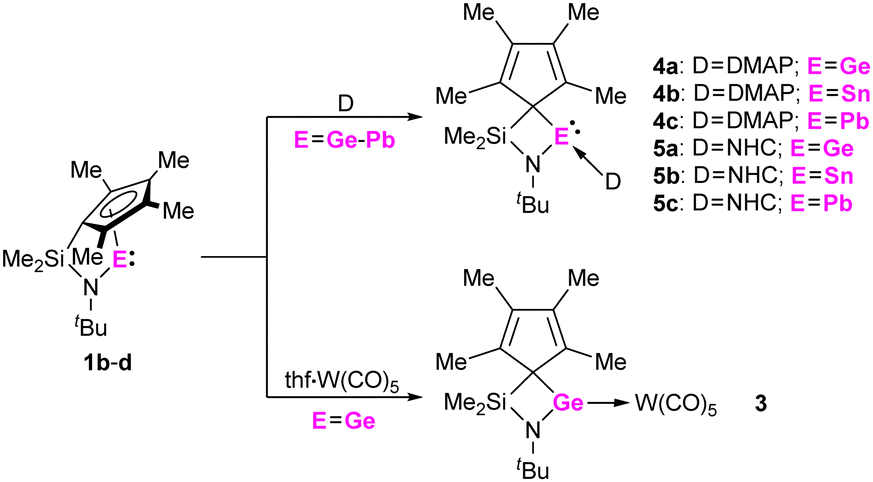 | ||
| Scheme 3 Reactions of 1b-d with 4-(dimethylamino)-pyridine (DMAP), 1,3-diisopropyl-4,5-dimethyl-imidazolin-2-ylidene (NHC) and thf·W(CO)5. | ||
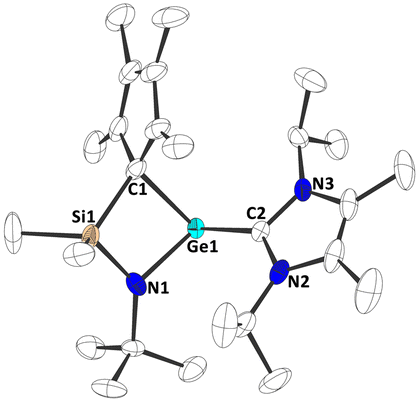 | ||
| Fig. 4 Molecular structure of 5a in the crystal (displacement ellipsoids at 50% probability level, hydrogen atoms omitted for clarity). | ||
The Ge–C1Cp bond length in 5a is 215.02(13) pm, the Sn–C1Cp bond length in 5b is 233.95(29) pm and the Pb–C1Cp bond length in 5c is 243.72(37) pm, which is slightly longer than typical Ge/Sn/Pb–C single bonds, although it must be mentioned that it is difficult to find structurally closely related examples,24–27 and the elongation might be related to steric reasons to some degree. The Ge/Sn/Pb–CNHC bond lengths are Ge–C2NHC (5a): 214.19(14) pm; Sn–C2NHC (5b): 237.23(28) pm and Pb–C2NHC (5c): 251.17(36) pm, which is comparable to diamino germylene NHC adducts (e.g. 214.9(3) to 219.2(3) pm),28 diamino stannylene NHC adducts (e.g. 239.9(4) pm),29 and diamino plumbylene NHC adducts (e.g. 258.6(7) pm).30 In contrast, these bonds are shorter than what was observed in related stannocene NHC complexes (245.06(10) ppm to 250.6 ppm) and a plumbocene NHC complex (272.5 pm), indicating a stronger binding of the NHC to the tetrel atom in 5a-c. This is also reflected by calculated (PBE0-D3/def2-TZVP) bond dissociation energies (BDEs), which are +110.4 kJ mol−1 (5a), +105.1 kJ mol−1 (5b) and +101.6 kJ mol−1 (5c).14 The increasingly stronger binding from 5c over 5b to 5a is presumably due to the elevated Lewis acidity. Noteworthy, comparable stannocene NHC complexes exhibit smaller BDEs (65.9 to 96.6 kJ mol−1),9 indicating a more pronounced Lewis acidity in 5a-c compared to their tetrelocene relatives, which may very well be related to the bonding flexibility of the ligand, with the Cp group being able to adopt different bonding modes, from η3–5 π complexation to σ bonding.
Finally, the Lewis basicity of germylene 1b and its concomitant ability to bind to a transition metal fragment was investigated. While complexation to a transition metal fragment is quite common for many di-N-heterocyclic tetrylenes,1 such complexes are unknown for tetrelocene, due to the almost non-existing σ donor abilities of the central atom in these systems.9 Two routes were found to yield the germylene–tungsten complex 3. Irradiation (365 nm) of a mixture of 1b and W(CO)6 in thf (Scheme 3), or treatment of Cl2Ge·W(CO)531 with the corresponding dilithiated ligand in thf gave 3. Orange crystals, suitable for single crystal X-ray diffraction, allowed for a structural characterization of this germylene tungsten complex (Fig. 5 and Fig. S53, ESI†). Interestingly, as in carbene complexes 5a-c, the Cp group adopts a σ bonding mode to the germanium atom. The Ge–W bond length is 252.92(5) pm, which is shorter than in a Cp*(Cl)Ge·W(CO)5 complex (257.1(1) pm),32 and in related NHGe·W(CO)5 complexes,33 which may indicate some π backbonding. This is in-line with the previously shown acceptor properties of 1b and with the discussion of π backbonding in a (RSe)2Ge·W(CO)5 complex with a similar Ge–W bond length (252.8(1) pm).34
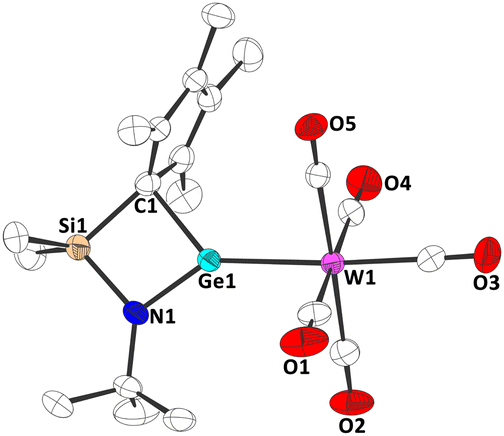 | ||
| Fig. 5 Molecular structure of 3 in the crystal (displacement ellipsoids at 50% probability level, hydrogen atoms omitted for clarity). | ||
In Summary, our work bridges the gap between tetrelocenes and “classical” N-heterocyclic tetrylenes, and shows that the applied ansa-half-sandwich ligands are also suitable ligand systems for stabilizing low valent tetrel(II) atoms. Additionally, we found that a key feature of these ligands in tetrylene chemistry is their ability to switch between different bonding types, as the Cp group can adopt both σ bonding and π complexation modes, depending on the electronic and steric situation at the tetrel atom. Furthermore, we could show that these N-heterocyclic half-sandwich tetrylenes possess Lewis amphiphilic character, as they can form complexes both with donor molecules such as DMAP or NHCs, and – in case of germylene 1b – with the transition metal fragment W(CO)5.
In the future, these compounds might be of interest as monomers in ring-opening polymerization or for applications in bond activation processes.
Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354 CrossRef CAS PubMed.
- (a) N. Sen and S. Khan, Chem. – Asian J., 2021, 16, 705 CrossRef CAS PubMed; (b) W. P. Neumann, Chem. Rev., 1991, 91, 311 CrossRef CAS.
- (a) H.-W. Wanzlick and E. Schikora, Angew. Chem., 1960, 72, 494 CrossRef CAS; (b) H.-W. Wanzlick and E. Schikora, Chem. Ber., 1961, 94, 2389 CrossRef CAS; (c) A. Igau, H. Grutzmacher, A. Baceiredo and G. Bertrand, J. Am. Chem. Soc., 1988, 110, 6463 CrossRef CAS; (d) A. J. Arduengo III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361 CrossRef.
- (a) M. Denk, R. Lennon, R. Hayashi, R. West, A. V. Belyakov, H. P. Verne, A. Haaland, M. Wagner and N. Metzler, J. Am. Chem. Soc., 1994, 116, 2691 CrossRef CAS; (b) P. J. Davidson and M. F. Lappert, J. Chem. Soc., Chem. Commun., 1973, 9, 317 RSC; (c) D. H. Harris and M. F. Lappert, J. Chem. Soc., Chem. Commun., 1974, 21, 895 RSC; (d) M. Veith, Angew. Chem., 1975, 87, 287 CrossRef CAS.
- P. Jutzi, D. Kanne and C. Krüger, Angew. Chem., Int. Ed. Engl., 1986, 25, 164 CrossRef.
- J. V. Scibelli and M. D. Curtis, J. Am. Chem. Soc., 1973, 95, 924 CrossRef CAS.
- E. O. Fischer and H. Grubert, Z. Naturforsch. B, 1956, 11, 423 Search PubMed.
- E. O. Fischer and H. Grubert, Z. Anorg. Allg. Chem., 1956, 286, 237 CrossRef CAS.
- C. Müller, A. Stahlich, L. Wirtz, C. Gretsch, V. Huch and A. Schäfer, Inorg. Chem., 2018, 57, 8050 CrossRef PubMed.
- (a) M. A. Beswick, N. L. Cromhout, C. N. Harmer, P. R. Raithby, C. A. Russell, J. S. B. Smith, A. Steiner and D. S. Wright, Chem. Commun., 1996, 1977 RSC; (b) D. R. Armstrong, M. A. Beswick, N. L. Cromhout, C. N. Harmer, D. Moncrieff, C. A. Russell, P. R. Raithby, S. Steiner, A. E. H. Wheatley and D. S. Wright, Organometallics, 1998, 17, 3176 CrossRef CAS; (c) S. Danés, C. Müller, L. Wirtz, V. Huch, T. Block, R. Pöttgen, A. Schäfer and D. M. Andrada, Organometallics, 2020, 39, 516 CrossRef.
- H. G. Alt, A. Reb, W. Milius and A. Weis, J. Organomet. Chem., 2001, 628, 169 CrossRef CAS.
- (a) J. M. Pietryga, J. D. Gorden, C. L. B. Macdonald, A. Voigt, R. J. Wiacek and A. H. Cowley, J. Am. Chem. Soc., 2001, 123, 7713 CrossRef CAS PubMed; (b) R. J. Wiacek, C. L. B. Macdonald, J. N. Jones, J. M. Pietryga and A. H. Cowley, Chem. Commun., 2003, 430 RSC; (c) M. Weger, P. Pahl, F. Schmidt, B. S. Soller, P. J. Altmann, A. Pöthig, G. Gemmecker, W. Eisenreich and B. Rieger, Macromolecules, 2019, 52, 7073 CrossRef CAS.
- DFT calculations at the PBE0-D3/def2-TZVP level of theory suggest 1b,c to have similar molecular structures.
- See ESI† for further details.
- (a) B. Wrackmeyer, A. Sebald and L. H. Merwin, Magn. Reson. Chem., 1991, 29, 260 CrossRef CAS; (b) C. Stader and B. Wrackmeyer, Z. Naturforsch. B, 1987, 42, 1515 CrossRef CAS; (c) C. Stader, B. Wrackmeyer and D. Schlosser, Z. Naturforsch. B, 1988, 43, 707 CrossRef CAS.
- S. P. Constantine, H. Cox, P. B. Hitchcock and G. A. Lawless, Organometallics, 2000, 19, 317 CrossRef CAS.
- M. Veith, Z. Naturforsch. B, 1978, 33, 7 CrossRef.
- J. P. H. Charmant, M. F. Haddow, F. E. Hahn, D. Heitmann, R. Fröhlich, S. M. Mansell, C. A. Russell and D. F. Wass, Dalton Trans., 2008, 6055 RSC.
- (a) J. S. Overby, T. P. Hanusa and V. G. Young, Inorg. Chem., 1998, 37, 1663 CrossRef PubMed; (b) J. L. Atwood, W. E. Hunter, A. H. Cowley, R. A. Jones and C. A. Stewart, J. Chem. Soc., Chem. Commun., 1981, 17, 925 RSC; (c) P. Jutzi, F. Kohl, P. Hofmann, C. Krüger and Y.-H. Tsay, Chem. Ber., 1980, 113, 757 CrossRef CAS.
- (a) D. Yang, J. Guo, H. Wu, Y. Ding and W. Zheng, Dalton Trans., 2012, 2187 RSC; (b) S.-J. Kim, Y.-J. Lee, S. H. Kim, J. Ko, S. Cho and S. O. Kang, Organometallics, 2002, 21, 5358 CrossRef CAS.
- Corresponds to the main products. Additional signals at δ77Se{1H} = 184.9; δ119Sn{1H} = −48.3, −133.7 were observed.
- R. Guthardt, D. Bachmann, C. Bruhn and U. Siemeling, Z. Anorg. Allg. Chem., 2020, 646, 761 CrossRef CAS.
- The reaction of Cp2Sn with Ph2Se2 in C6D6 at ambient conditions resulted in the formation of an analogous species as indicated by 77Se{1H} and 119Sn{1H} NMR spectroscopy, along other products: δ77Se{1H} = 85.7, 107.7, 140.7 (1J77Se–Sn = 1537 Hz) (major product), 185.1, 333.9; δ119Sn{1H} = −59.4 (1J119Sn–77Se = 1539 Hz; −27.7 (1J119Sn–77Se = 1496 Hz) (major product), −22.2, −133.7 (1J119Sn–77Se = 1585 Hz).
- (a) K. M. Baines and W. G. Stibbs, Coord. Chem. Ref., 1995, 145, 157 CrossRef CAS; (b) B. E. Eichler, D. R. Powell and R. West, Organometallics, 1998, 17, 2147 CrossRef CAS; (c) D. Ghereg, H. Gornitzka, H. Ranaivonjatovo and J. Escudié, Dalton Trans., 2010, 39, 2016 RSC; (d) M. Lazraq, J. Escudié, C. Couret, J. Satgé, M. Dräger and R. Dammel, Angew. Chem., Int. Ed. Engl., 1988, 27, 828 CrossRef.
- (a) A. Schäfer, M. Weidenbruch, W. Saak and S. Pohl, J. Chem. Soc., Chem. Commun., 1995, 11, 1157 RSC; (b) H. Grützmacher, S. Freitag, R. Herbst-Irmer and G. S. Sheldrick, Angew. Chem., Int. Ed. Engl., 1992, 31, 437 CrossRef; (c) R. K. Ingham, S. D. Rosenberg and H. Gilman, Chem. Rev., 1960, 60, 459 CrossRef CAS; (d) D. Ghazi, Z. Rasheed and E. Yousif, Arc. Org. Inorg. Chem. Sci., 2018, 3, 344 Search PubMed.
- F. Stabenow, W. Saak and M. Weidenbruch, Chem. Commun., 1999, 1131 RSC.
- P. Pyykkö, J. Phys. Chem. A, 2015, 119, 2326 CrossRef PubMed.
- S. Yao, Y. Xiong and M. Driess, Chem. Commun., 2009, 6466 RSC.
- F. E. Hahn, L. Wittenbecher, M. Kühn, T. Lügger and R. Fröhlich, J. Organomet. Chem., 2001, 617–618, 629 CrossRef CAS.
- B. Gehrhus, P. B. Hitchcock and M. F. Lappert, J. Chem. Soc., Dalton Trans., 2000, 18, 3094 RSC.
- P. Jutzi and W. Steiner, Chem. Ber., 1976, 109, 3473 CrossRef CAS.
- P. Jutzi, B. Hampel, K. Stroppel, S. Krüger, K. Angermund and P. Hofmann, Chem. Ber., 1985, 118, 2789 CrossRef CAS.
- (a) I. Saur, G. Rima, K. Miqueu, H. Gornitzka and J. Barrau, J. Organomet. Chem., 2003, 673, 77 CrossRef; (b) C. Jones, R. P. Rose and A. Stasch, Dalton Trans., 2008, 2871 RSC.
- W. W. Du Mont, L. Lange, S. Pohl and W. Saak, Organometallics, 1990, 9, 1395 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra, XRD data, computational details. CCDC 2174664, 2174665 and 2174672–2174675. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d2cc03107h |
| This journal is © The Royal Society of Chemistry 2022 |