
Effect of dendron structure on the luminescent and charge transporting properties of solution processed dendrimer-based OLEDs†
Mile
Gao
 a,
Van T. N.
Mai
a,
Junhyuk
Jang
a,
Chandana Sampath Kumara
Ranasinghe
a,
Ronan
Chu
a,
Paul L.
Burn
*a,
Ian R.
Gentle
a,
Almantas
Pivrikas
b and
Paul E.
Shaw
a
a,
Van T. N.
Mai
a,
Junhyuk
Jang
a,
Chandana Sampath Kumara
Ranasinghe
a,
Ronan
Chu
a,
Paul L.
Burn
*a,
Ian R.
Gentle
a,
Almantas
Pivrikas
b and
Paul E.
Shaw
a
aCentre for Organic Photonics & Electronics, The School of Chemistry and Molecular Biosciences, The University of Queensland, Queensland 4072, Australia. E-mail: p.burn2@uq.edu.au
bPhysics Department, Murdoch University, Perth, Western Australia 6150, Australia
First published on 10th November 2021
Abstract
The photophysical and charge transport properties of neat and blend films of fac-tris[2-phenylpyridinato-C2,N]iridium(III) [Ir(ppy)3]-cored light-emitting dendrimers and tris(4-carbazoyl-9-ylphenyl)amine (TCTA)-based hosts, with each combination having the same first generation dendrons were measured and compared. The first generation dendrons were composed of bis(fluorenyl)carbazolyl with n-propyl surface groups, or biphenyl with 2-ethylhexyloxy or t-butyl surface groups. Dendronisation of the emitter and host was found to decrease the energy transfer efficiency from the host to the guest relative to an evaporated Ir(ppy)3:TCTA blend film, with the blend composed of materials with biphenyl dendrons and t-butyl surface groups having the largest decrease. The hole mobilities of the solution processed neat and blend layers were found to be 1–2 orders of magnitude lower than those of the equivalent evaporated films. The blend film containing the host and dendrimer with first generation biphenyl dendrons and 2-ethylhexyloxy surface groups had the highest photoluminescence quantum yield (PLQY), comparable to evaporated Ir(ppy)3:TCTA-based films but the lowest hole mobility (≈10−8 cm2 V−1 s−1). In contrast, the blend with the dendrimers composed of the bis(fluorenyl)carbazolyl dendrons with n-propyl surface groups had a low PLQY but higher hole mobility. It was found for the combinations of these solution processable materials that the hole mobility of the blend film was the limiting factor in OLED performance. Devices containing an emissive layer of the materials with the bis(fluorenyl)carbazolyl dendrons with n-propyl surface groups (11 mole percent of the emitter) had a PLQY of 40.8% but the highest external quantum efficiency of 10.0 ± 0.4%, reaching a maximum luminance of almost 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cd m−2.
000 cd m−2.
Introduction
The growing demand for lighting and displays has led to the development of highly efficient phosphorescent-based organic light-emitting diodes (OLEDs). A common feature of these OLEDs is that the emitter is blended at a low concentration into a host material to prevent intermolecular interactions that can lead to the quenching of the emitter luminescence. Efficient phosphorescent emitters are complexes with a heavy metal atom at the centre, which facilitates the intersystem crossing between singlet and triplet states and enables harvesting of both singlet and triplet excitons. Examples include iridium(III) complexes such as fac-tris[2-phenylpyridinato-C2,N]iridium(III) [Ir(ppy)3],1–3 and other heavy metal complexes such as those containing platinum(II).4OLED fabrication by sequential vapour deposition of each organic layer has enabled OLED displays to enter the commercial market. However, for applications that require large-area fabrication, solution coating techniques may have an advantage. The heavy metal complexes used for manufacturing OLEDs by evaporation cannot be easily transferred to solution coating due to their poor solubility. This poor solubility in processing solvents means that films with the desired quality and thickness cannot be formed. Consequently, there have been efforts in developing phosphorescent materials that have increased solubility and can be coated from solution to form good quality thin films. One strategy to increase the solubility of phosphorescent organometallic emitters is to attach solubilising dendrons.5–9 In common with light-emitting layers used in OLEDs formed by vacuum evaporation, the emissive films formed using solution-processing found in the most efficient solution processed OLEDs are also composed of a blend of emissive and host materials.10–12 However, in common with the emissive materials, most host materials are also designed to be evaporated and generally do not have the solubility required to provide good quality films of the required thickness for use in OLEDs without a solubilising guest. For example, tris(4-carbazoyl-9-ylphenyl)amine (TCTA) is a common host material for phosphorescent OLEDs fabricated by vacuum deposition but has low room temperature solubility in solvents commonly used for OLEDs fabricated using solution processing techniques.13,14 One strategy to improve the solubility of the host materials is to follow the approach used for the light-emitting materials, namely dendronisation.7 However, at this stage there is not a clear understanding of the effects of dendronisation on the optoelectronic properties of blends composed of light-emitting dendrimers and dendritic hosts.
In this manuscript we compare the charge transporting and photophysical properties of solution processed first generation dendrimeric TCTA hosts and their blends with first generation light-emitting dendrimers with the same “dendron” structure. The dendrimeric materials had first-generation bis(fluorenyl)carbazolyl dendrons with n-propyl surface groups (1 and 4), or biphenyl dendrons with 2-ethylhexyloxy (2 and 5) or t-butyl (3 and 6) surface groups. The light-emitting dendrimers all had an Ir(ppy)3-based core with the dendrons attached at the same point on the ligands.15,16 That is, each light-emitting dendrimer and host pair had the same dendrons and solubilising groups (see Fig. 1).
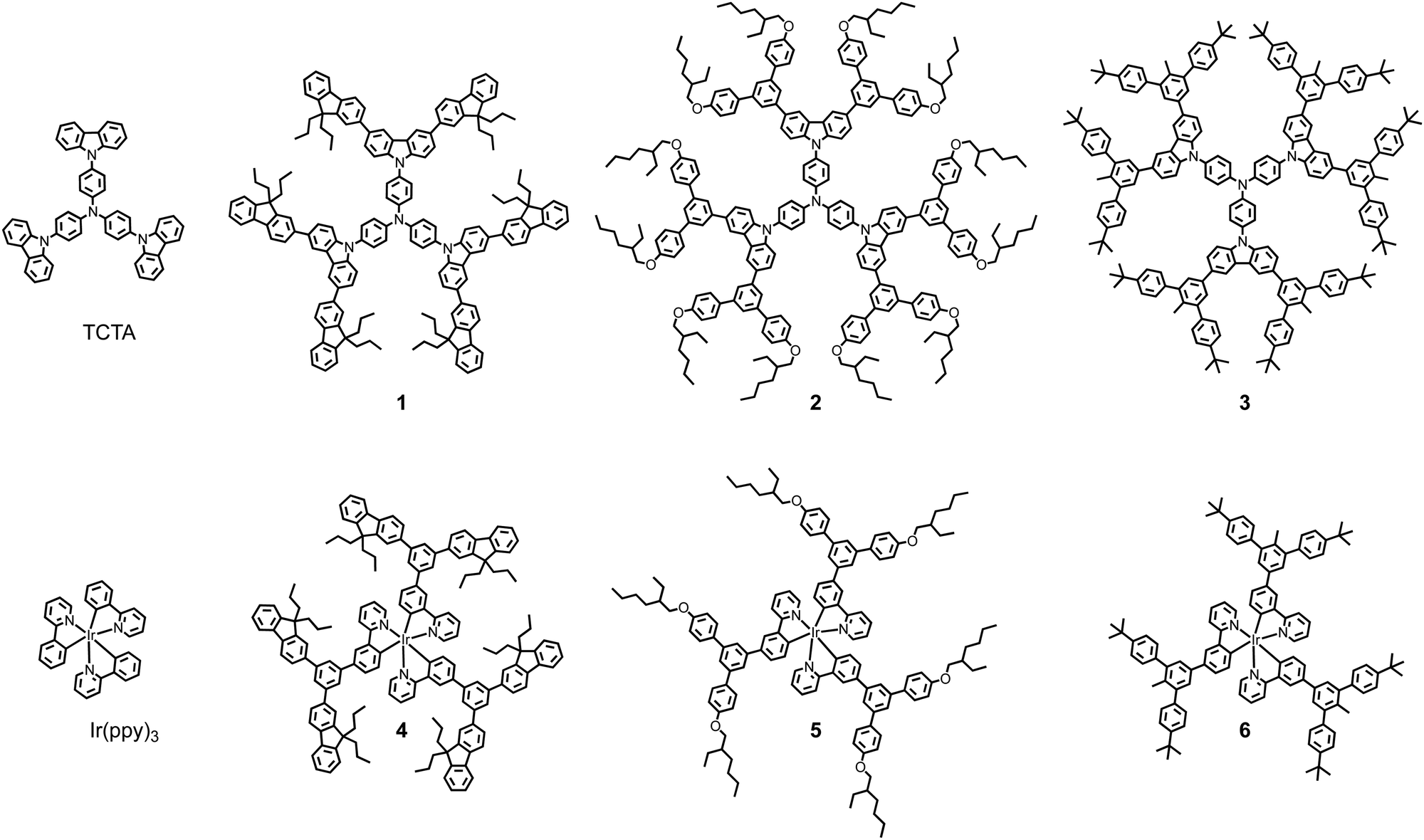 | ||
| Fig. 1 Structures of the host materials [TCTA, and first generation TCTA-based hosts 1, 2, and 3] and green phosphorescent light-emitting materials [Ir(ppy)3, and first-generation dendrimers 4, 5, and 6]. | ||
Results and discussion
Materials synthesis and thermal properties
The first-generation host 1 and phosphorescent dendrimers 4, 5, and 6 were synthesised according to previously reported methods15–18 while the syntheses of the new TCTA-based hosts 2 and 3 are shown in Scheme 1. The first step in the sequence were Suzuki cross-coupling reactions between commercial 3,6-dibromo-9H-carbazole and boronic esters 7 and 8.15,16 Under these conditions, the carbazolyl derivatives 9 and 10 were isolated in yields of 25% and 76%, respectively. Subsequently, Buchwald-type aminations between 9 and 10 and tris(4-iodophenyl)amine19 produced the target dendrimers 2 and 3 in isolated yields of 46%, and 68%, respectively. All the dendrimeric materials had good solubility in standard processing solvents and could be spin-coated from solution at room temperature to form good quality thin films.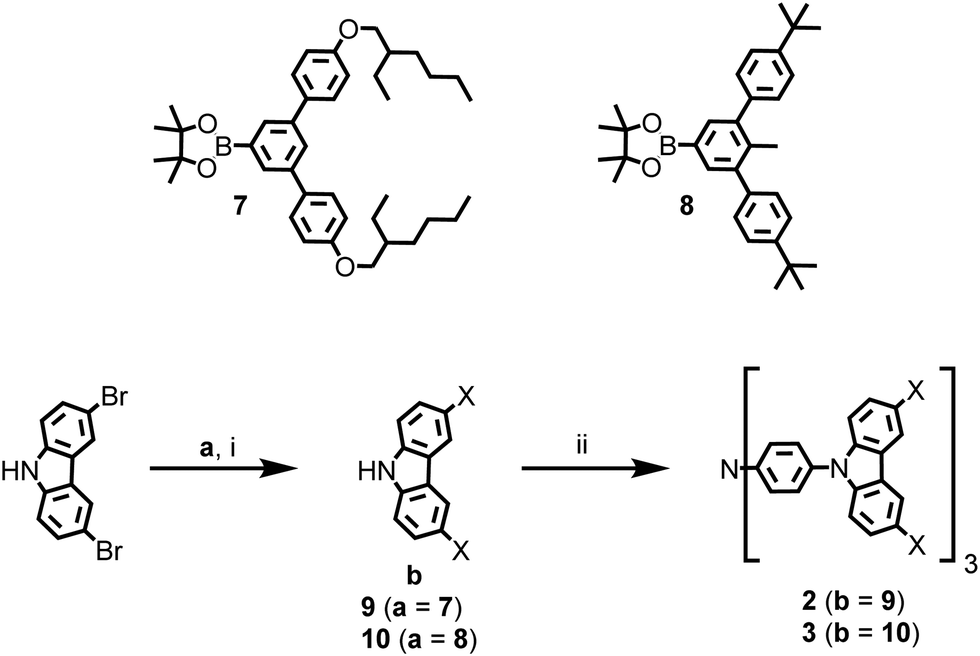 | ||
| Scheme 1 Synthetic routes to the dendritic hosts 2 and 3. Reagents and conditions: (i) K2CO3, Pd(PPh3)4, toluene/ethanol/water, Ar(g), Δ; (ii) tBuONa, Pd2(dba)3, [(tBu)3PH]·BF4, toluene, Ar(g), Δ. X = the dendron from the corresponding boronate ester, 7 or 8. | ||
Thermogravimetric analysis (TGA) (Fig. S1, ESI†) and differential scanning calorimetry (DSC) (Fig. S2 and S3, ESI†) were used to determine the thermal properties of the dendrimeric hosts 2 and 3. Both 2 and 3 were found to have excellent thermal stability with their decomposition temperatures (Td), corresponding to a 5% weight loss, at 413 °C, and 500 °C, respectively. These decomposition temperatures were comparable to that of 1 (443 °C). While TCTA and 1 have been reported to have glass transition temperatures (Tg) of 152 °C and 220 °C,17,20 no thermal transition between −30 and 240 °C was observed for dendrimers 2 and 3.
Solution optical and electronic properties
The redox properties of hosts 2 and 3 were investigated using solution cyclic voltammetry. While TCTA is generally chemically unstable to oxidation in solution,21 by capping the 3- and 6-positions of the carbazole moieties we were able to measure chemically reversible oxidations for both 2 and 3 within the solvent window. The E1/2(ox)s for 2 and 3 were 0.4 V, 0.6 V and 0.9 V, and 0.4 V, 0.6 V, 0.8 V and 1.0 V versus the ferrocene/ferrocenium couple, respectively (Fig. S4 and S5, ESI†). Based on the first oxidation potential and the reported ionisation potential (IP) for ferrocene we estimated that the IPs for 2 and 3 were 5.2 eV, which are similar within experimental uncertainty to that of TCTA (5.3 eV) and 1 (5.2 eV).17 No reduction potentials were measured under the same conditions, which is consistent with TCTA having negligible electron transport.22Film photoluminescence and charge mobility properties
The photoluminescence (PL) of the thin films was measured with the excitation at 330 nm, where both the light-emitting guests and hosts absorb (see Fig. S6, ESI†). Fig. 2 shows the PL spectra of the neat and blend films at various doping concentrations. The dendrimeric host and light-emitting dendrimer films were solution processed, while the Ir(ppy)3-based films (including the neat TCTA film) were thermally vacuum deposited. All the films were optically clear without observable crystallisation. The neat host films all had a PL maximum at around 400 nm arising from the “TCTA core”. In addition, weak luminescence from excimer or aggregate emission was observed at wavelengths longer than 450 nm. The intensity of the long wavelength emission relative to the peak at 400 nm differed, with TCTA having the smallest intensity and 3 having the largest. A greater intensity is indicative of more aggregation in the dendritic host films, particularly for 3, which is somewhat surprising as normally dendronisation reduces intermolecular interactions of electroactive cores. The neat films of the light-emitting dendrimers all had a PL peak at around 510 nm arising from the Ir(ppy)3 core. The similarity of the PL emission of the light-emitting dendrimers is consistent with the attachment of the dendrons to the Ir(ppy)3 core having minimal effect on the excited state energy, which arises from the fact that the dendrons on the ligand phenyl group were attached meta to the pyridine ring and do not increase the ligand conjugation length.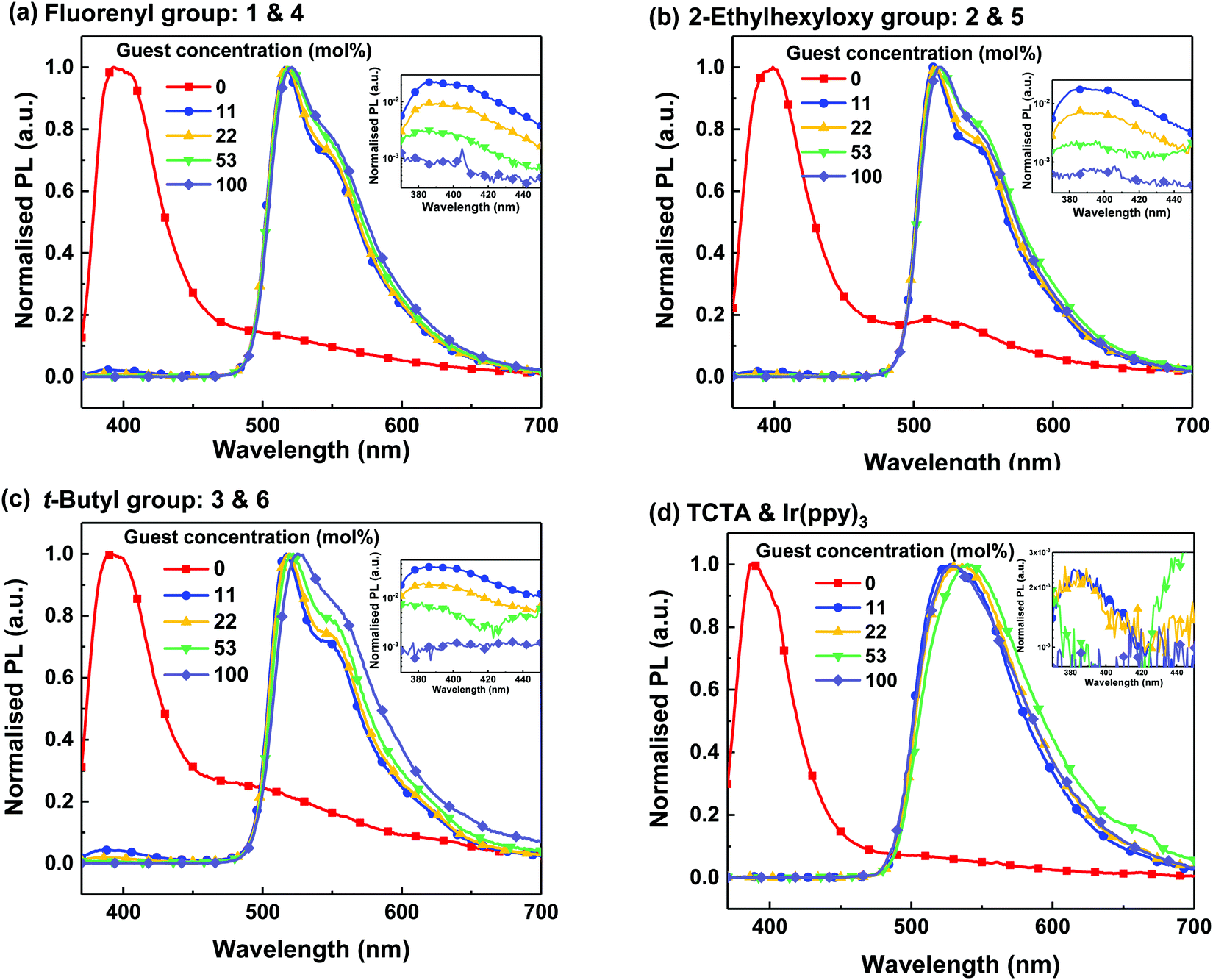 | ||
| Fig. 2 PL spectra of neat and blend films of 1 and 4 (a), 2 and 5 (b), 3 and 6 (c) and TCTA and Ir(ppy)3 (d) at different concentrations excited at 330 nm. The PL data of neat Ir(ppy)3 was taken from the literature.23 Inset figures show the PL spectra at shorter wavelengths with the PL intensity on a logarithmic scale. | ||
When the PL spectra of the blend films were plotted on a logarithmic scale (Fig. 2, insets) residual emission at around 400 nm from the host could be observed. Residual host emission was clearly visible for all concentrations of the films containing the dendrimeric hosts. In comparison, only the 11 and 22 mol% Ir(ppy)3:TCTA blend films showed residual emission from the host within the sensitivity of the measurement. The amount of residual host emission relative to the guest emission is an indication of the efficiency of energy transfer from the host to the guest, which is directly related to the distribution of the guest in the host. The fact that they are different for each of the blends suggests that the transfer efficiency is dependent on dendron type.
To quantitatively determine the effect of dendron structure on the energy transfer from host to guest, the energy transfer efficiency was calculated (see Experimental section), and the results are shown in Fig. 3. It is important to note that the molecular structures and volumes of the molecules used were different and hence the guest number density of the blend films, measured using X-ray reflectometry (XRR, Fig. S7, ESI†), rather than the weight or mole percent was used for the comparison. From Fig. 3 it can be seen that the energy transfer efficiency for the evaporated Ir(ppy)3:TCTA blend films was the highest, even at the lowest guest number density of 11 × 1019 cm−3 (corresponding to 11 mol%) having an energy transfer efficiency of nearly 100%. The solution processed dendrimeric host-based blend films had comparable energy transfer efficiency to the evaporated Ir(ppy)3:TCTA films at similar number densities. The slightly reduced transfer is likely related to differences in the closest separation of the Ir(ppy)3-core of the light-emitting dendrimers and the emissive chromophore of the hosts. The three dendritic host materials have similar emissive chromophores composed of a TCTA-like core, as indicated by the similarity of their PL spectra. Therefore, if the light-emitting dendrimer guests were evenly distributed throughout the film, the decrease in energy transfer efficiency would be directly related to the steric bulk of the dendrons both on the host and guest, which increases the distance between the guest and host emissive chromophores. The energy transfer efficiencies for the dendrimer combinations containing the bis(fluorenyl)carbazolyl dendrons with n-propyl surface groups, or biphenyl dendrons with 2-ethylhexyloxy surface groups were the highest for the solution processed blend films, with the latter consistently the highest. The blend films containing the biphenyl dendrons with t-butyl surface groups had consistently lower efficiencies. For example, comparing similar number densities (3.1 × 1019 cm−3, 1.8 × 1019 cm−3, and 2.3 × 1019 cm−3), the energy transfer efficiency was 97.5 ± 1.3%, 98.0 ± 1.1% and 94.4 ± 1.5% for 1 and 4, 2 and 5, and 3 and 6, respectively. However, the key point of these measurements is that despite the steric bulk of the dendrimeric emissive guest and host structures there was still relatively efficient transfer from the host to guest such that in a working device, any excitons formed on the host should readily transfer to the guest.
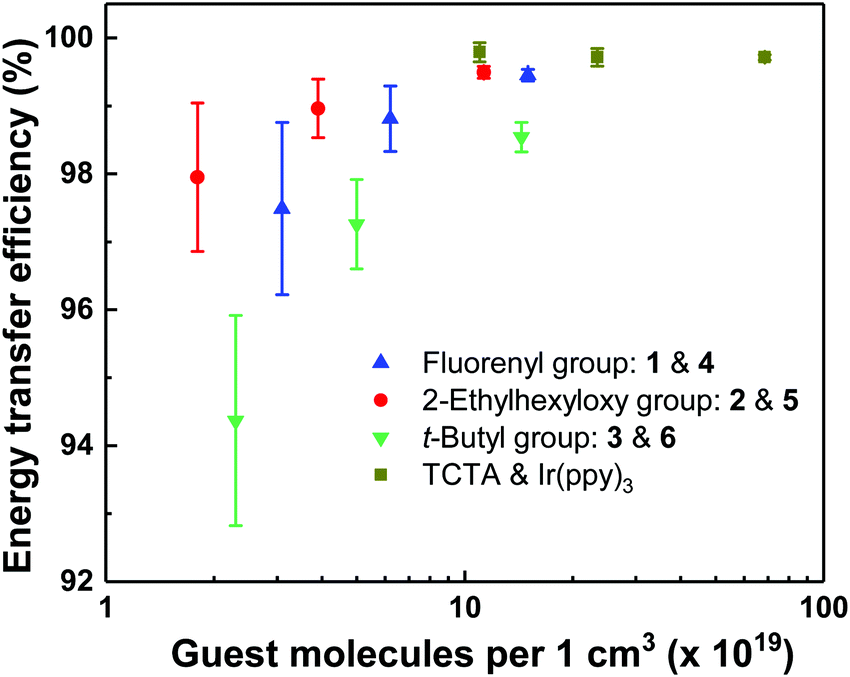 | ||
| Fig. 3 Energy transfer efficiency from the dendritic hosts to the emissive dendrimers with the same dendrons in blend films versus guest number density. | ||
We next measured the photoluminescence quantum yield (PLQY) of each film, with the results shown in Fig. 4. The excitation wavelength for the PLQY measurements of the blend films was 325 nm and hence the host was also excited. To minimise the effect of the host emission a 400 nm cut-off filter was used in the measurement, and hence the PLQYs measured are effectively those of the light-emitting dendrimer guests. The PLQYs of the blend films had their maxima at the lower concentrations studied (11–22 mol%). Increasing the guest concentration resulted in a reduction of the PLQYs due to the intermolecular interactions of the emissive chromophores of adjacent light-emitting dendrimers. The maximum PLQY values were 40.8 ± 3.1% (1 and 4), 82.5 ± 7.5% (2 and 5), 43.3 ± 3.7% (3 and 6) and 77.9 ± 4.6% [TCTA and Ir(ppy)3], respectively. In comparison, the solution PLQYs of 4, 5, 6 and Ir(ppy)3 were 69%, 78 ± 8%, 79 ± 8% and ∼100%, respectively.16,24,25 Of the solution processed films, only the blend containing 2 and 11 mol% of 5 (materials composed of biphenyl dendrons with 2-ethylhexyloxy surface groups) had a PLQY close to that of the light-emitting dendrimer (5) in solution. Even at the lowest concentration measured, the blend films containing dendrimers composed of the bis(fluorenyl)carbazolyl dendrons with n-propyl surface groups or biphenyl dendrons with t-butyl surface groups had PLQYs lower than the light-emitting dendrimer in solution. This is indicative of the light-emitting dendrimers not being evenly distributed throughout the host, which is consistent with the poorer energy transfer from host to guest for these latter materials. Interestingly, despite each of the combinations of 1 and 4, and 3 and 6 having similar dendrons, the guest was not evenly distributed through the emissive film. That is, not all the light-emitting dendrimers can be viewed as isolated emitters in the blends.
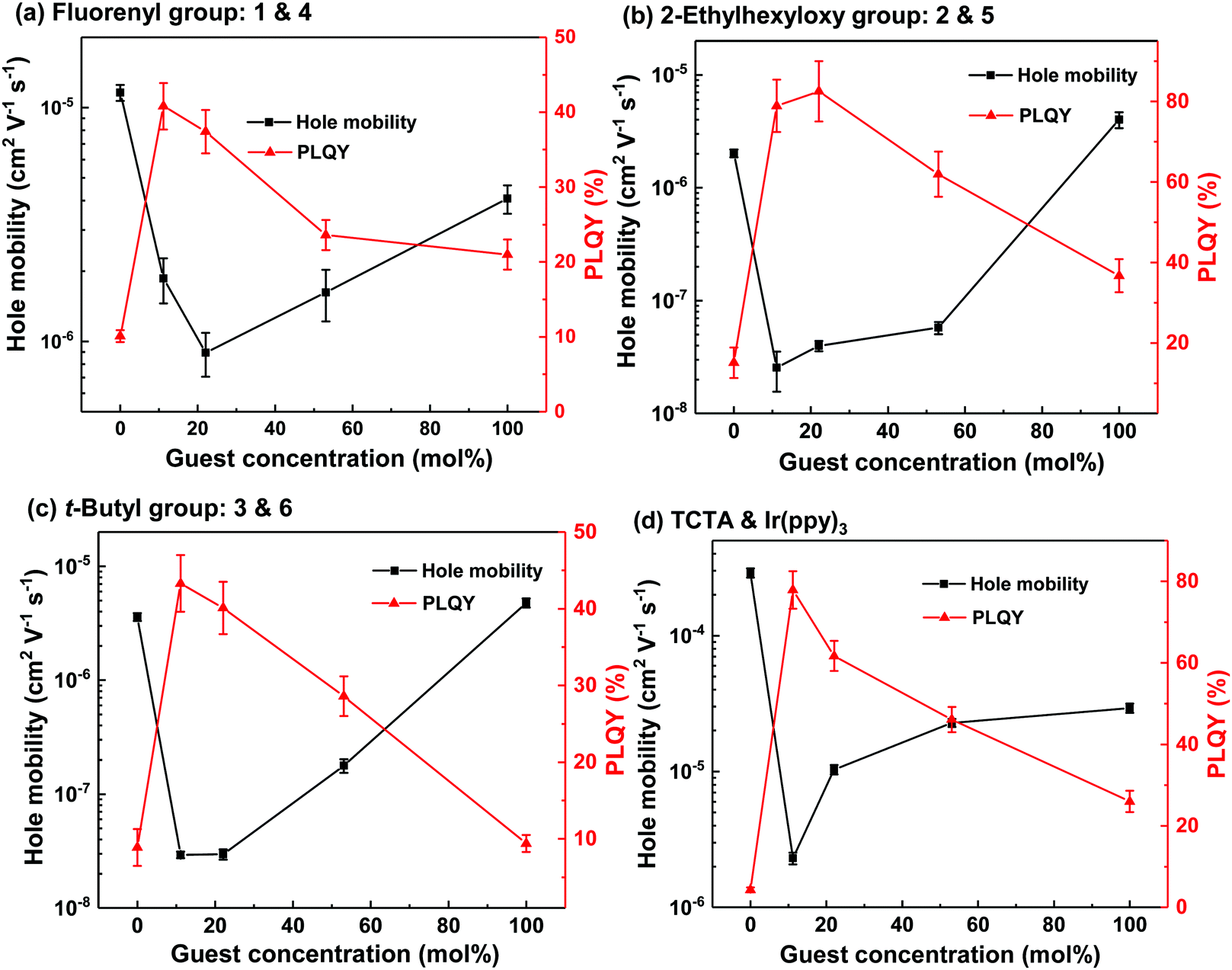 | ||
| Fig. 4 Mobility and PLQY values of neat and blend films of 1 and 4 (a), 2 and 5 (b), 3 and 6 (c) and TCTA and Ir(ppy)3 (d) at different concentrations. Fig. (d) was reproduced with permission.26,27 | ||
The results of the PLQY measurements indicated that the interactions between the light-emitting dendrimers were different for the blends based on the dendrons. We were therefore interested to see if charge transport within the blend films was similarly affected. Metal–Insulator–Semiconductor Charge Extraction by Linear Increasing Voltage (MIS-CELIV) was used to measure the hole mobility of the neat and blend films at different doping ratios with the results summarised in Fig. 4. As mentioned previously, the Ir(ppy)3:TCTA films were vacuum evaporated while the others were solution processed. For the neat host and light-emitting dendrimer films, it can be clearly seen that attachment of dendrons to the TCTA core decreases the hole mobility significantly, with the value for evaporated TCTA being (2.9 ± 0.1) × 10−4 cm2 V−1 s−1, while that of 1, 2 and 3 were one to two orders of magnitude lower at (1.2 ± 0.1) × 10−5, (2.0 ± 0.2) × 10−6 and (3.6 ± 0.3) × 10−6 cm2 V−1 s−1, respectively. The hole mobility of Ir(ppy)3 was (2.9 ± 0.2) × 10−5 cm2 V−1 s−1, while that of 4, 5, and 6 were (4.0 ± 0.7) × 10−6, (4.8 ± 0.4) × 10−6, and (4.1 ± 0.6) × 10−6 cm2 V−1 s−1, respectively. Interestingly, the hole mobility of solution processed TCTA has been reported to be (9.2 ± 0.8) × 10−6 cm2 V−1 s−1, which is also an order of magnitude lower than that of the evaporated TCTA film.17 The lower mobility of the solution processed TCTA was ascribed to the film having greater disorder leading to an increase in trap sites and more dispersive transport. Therefore, the lower hole mobility of the neat dendrimeric host and light-emitting dendrimer guest films is at least in part due to greater disorder as well as the fact that the electroactive cores of the materials are on average held further apart by the dendrons. Thus, charge hopping between adjacent cores is decreased, leading to lower mobilities. All the blend films showed a similar trend in mobility versus light-emitting guest concentration. The addition of 11 mol% of the dendrimeric guests caused a significant decrease in mobility (approximately two orders of magnitude in each case) relative to the neat host film. The hole mobility of films composed of 2 and 5, and 3 and 6 blend films decreased to ≈10−8 cm2 V−1 s−1 at 11 mol% while that of 1 and 4 decreased to around 10−6 cm2 V−1 s−1 at a guest concentration of 22 mol%. The decrease in mobility upon addition of the light-emitting dendrimer guest is due to them acting as charge traps, which is consistent with the “Ir(ppy)3” having a smaller IP than “TCTA”. The fact that the dendrimeric guest:host combinations with the largest and most sterically hindering dendrons have the lowest hole mobilities indicates that the Ir(ppy)3 cores responsible for charge transport and trapping are on average held further apart. Increasing the concentration of the guest in the host led to an increase in the hole mobility, which is consistent with the light-emitting dendrimers forming larger interconnected pathways through the blend films.26
OLED fabrication and characterisation
The final aspect of the study was the OLED performance of devices composed of light-emitting films with the maximum PLQY and/or minimum hole mobility (11 or 22 mol% light-emitting dendrimer guest concentration). The light-emitting layers used in the OLEDs were studied using atomic force microscopy (AFM, see Fig. S8, ESI†) to ensure that differences in performance were not related to surface topology. It was found that all the neat and blend films had smooth surfaces (root mean square roughness of between 0.6 and 0.7 nm) with no significant surface phase separation in the plane of the substrate. Furthermore, grazing incidence X-ray diffraction spectra (GIXRD, Fig. S9, ESI†) showed that the blend films used as the light-emitting layers in the OLEDs were amorphous. The device architecture was glass/ITO/poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS)/emissive layer/3,3′,5,5′-tetra[(m-pyridyl)-phen-3-yl]biphenyl (BP4mPy)/lithium fluoride (LiF)/aluminum (Al) and all the layers were of similar thicknesses for the different devices. The external quantum efficiency (EQE), current efficiency (CE), power efficiency (PE) and J–V–L characteristics of the OLEDs are shown in Fig. 5 with the key parameters summarised in Table 1. For both blend concentrations of the bis(fluorenyl)carbazolyl-containing 1 and 4 based devices, the EQE increases and then rolls off with increasing luminance (maximum luminance >104 cd m−2). The initial low efficiency at low bias is due to the imbalanced charge injection and transport, while at higher luminance triplet–triplet annihilation (TTA) and/or exciton-polaron quenching occurs, leading to the roll off in performance. Comparison of the 11 mol% and 22 mol% devices shows that the 22 mol% had slightly better performance with a maximum EQE of 10.0 ± 0.4%. It is interesting to note that based on the PLQY of the 22 mol% blend film of 1 and 4 (40.8 ± 3.1%) and a light outcoupling efficiency of 20% from the bottom emitting device, the maximum EQE should be ≈8%. Thus, the results suggest that for the blend of 1 and 4, there was increased outcoupling of the light generated in the device. For OLEDs based on blends of 2 and 5, and 3 and 6, the efficiencies were highest at low luminance (<10 cd m−2). However, for these blends the EQE decreased with increasing luminance. At 100 cd m−2, the EQE of all the devices dropped to around, or less than, 2%. The OLEDs composed of 2 and 5 or 3 and 6 had maximum luminance of <1000 cd m−2. Such severe efficiency roll-off at low luminance is ascribed to the low hole mobility of these amorphous blend films (≈10−8 cm2 V−1 s−1) as evidenced by the low current density shown in Fig. 5(b). These results suggest that while a high film PLQY is important for device efficiency (note that the blend containing 2 and 5 had the highest PLQY) the low hole mobility is likely the limiting factor for solution processed OLEDs, particularly at higher luminance. To set these device results in context, a more complex multilayer OLED composed of an Ir(ppy)3:TCTA emissive layer has previously been reported to have an EQE of 15.8%.28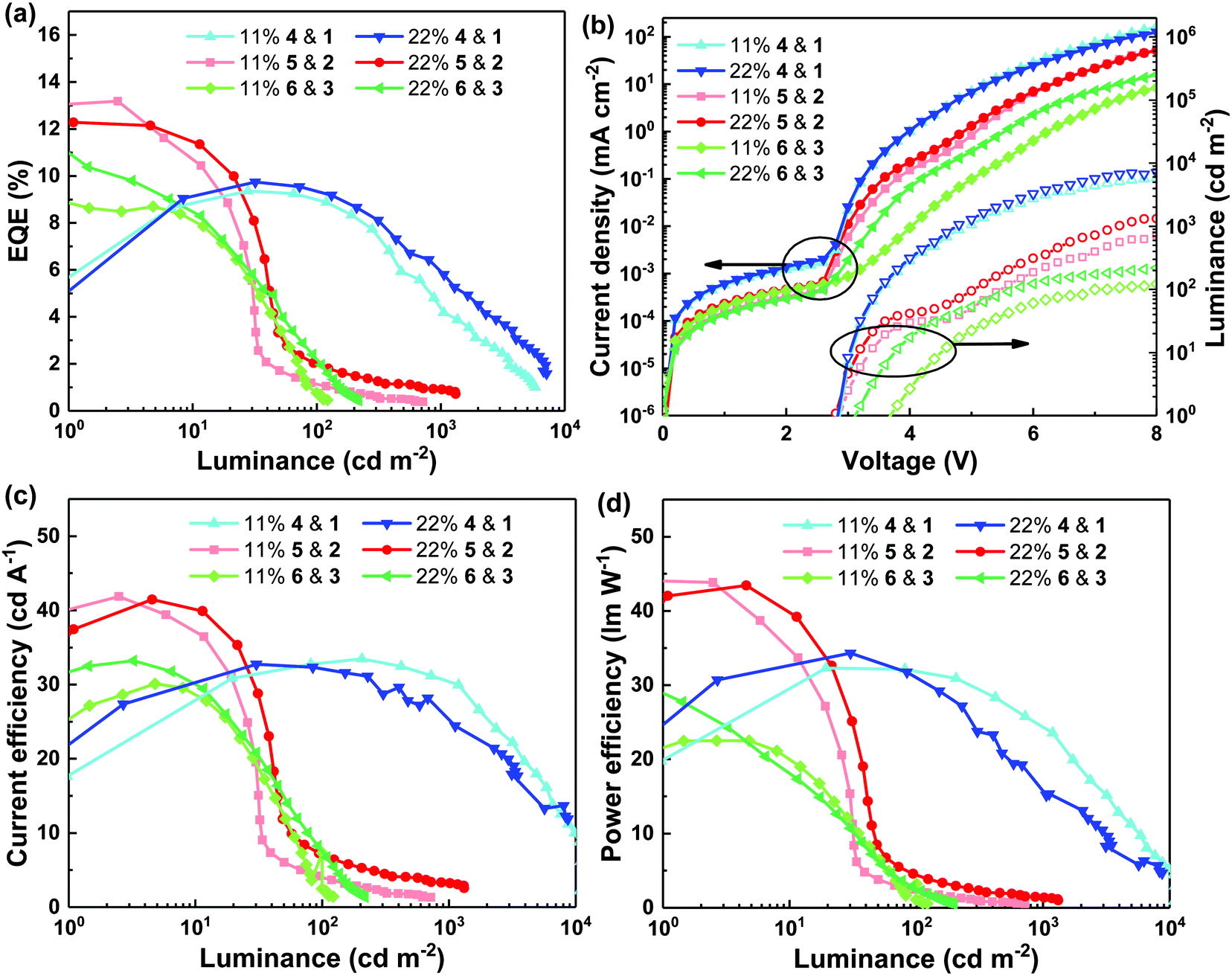 | ||
| Fig. 5 EQE (a), CE (c) and PE (d) versus luminance and J–V–L characteristics (b) of OLEDs composed of different material combinations at different ratios. | ||
| Materials | Guest ratio (mol%) | V on (V) | EQE (%) | CE (cd A−1) | PE (lm W−1) |
|---|---|---|---|---|---|
| Max/100/1000c | Max/100/1000c | Max/100/1000c | |||
| a The data comes from between 3–5 pixels from 2 device chips and the error is the standard deviation. b Turn on voltage at 1 cd m−2. c EQE, CE and PE at the maximum value/at 100 cd m−2/at 1000 cd m−2. | |||||
| 1 & 4 | 11 | 2.8 | 9.3 ± 0.0/9.0 ± 0.1/4.4 ± 0.1 | 33.0 ± 0.5/32.5 ± 0.5/30.0 ± 0.5 | 31.9 ± 0.5/31.6 ± 0.5/24.0 ± 0.5 |
| 22 | 2.8 | 10.0 ± 0.4/9.3 ± 0.1/5.4 ± 0.4 | 32.3 ± 0.5/31.5 ± 0.8/25.9 ± 1.9 | 32.8 ± 1.3/30.4 ± 1.1/16.9 ± 1.0 | |
| 2 & 5 | 11 | 3.0 | 12.5 ± 0.6/1.1 ± 0.1/N/A | 39.5 ± 1.8/3.8 ± 0.3/N/A | 41.4 ± 1.9/2.1 ± 0.2/N/A |
| 22 | 2.8 | 12.5 ± 0.3/2.0 ± 0.1/0.9 ± 0.0 | 41.2 ± 0.2/7.0 ± 0.2/3.2 ± 0.1 | 43.2 ± 0.2/4.3 ± 0.2/1.3 ± 0.1 | |
| 3 & 6 | 11 | 3.8 | 9.2 ± 0.2/1.1 ± 0.4/N/A | 30.3 ± 0.3/3.5 ± 1.4/N/A | 25.0 ± 3.5/1.6 ± 0.8/N/A |
| 22 | 3.2 | 10.6 ± 1.3/2.1 ± 0.1/N/A | 31.9 ± 1.1/7.3 ± 0.5/N/A | 31.2 ± 1.7/4.0 ± 0.3/N/A | |
Conclusions
The charge transport and photophysical properties of neat Ir(ppy)3 complex-cored light-emitting dendrimers and TCTA-based dendrimeric hosts and blends where the emitter and host have the same first generation dendrons and surface groups were measured and compared. It was found that the film PL spectra of the blends were generally the same but with different levels of residual host emission. It was also found that the dendron type affected the efficiency of energy transfer to different extents, with those composed of the first-generation biphenyl dendrons with t-butyl surface groups having the poorest host to guest energy transfer. The dendrimeric hosts and light-emitting dendrimers all had lower hole mobilities than evaporated TCTA or Ir(ppy)3, which was due to a combination of increased disorder from the solution processing and the fact that the electroactive components were partially shielded by the dendrons, increasing the average hopping distance between the cores. At the low guest concentrations typically used in light-emitting layers to maximise the PLQYs, the hole mobility of blends composed of hosts and light-emitting guests with biphenyl dendrons and 2-ethylhexyloxy or t-butyl surface groups decreased to 10−8 cm2 V−1 s−1. The decrease in hole mobility could be ascribed to the more disordered structure in the solution processed films and the encapsulation of the Ir(ppy)3 and TCTA cores by the dendrons. Finally, OLEDs containing an emissive layer composed of 1 and 4 [bis(fluorenyl)carbazolyl dendrons] had the highest hole mobility and EQE, although the devices had imbalanced charge injection at low luminescence and showed roll-off in the efficiency at higher luminescence. Critically, the results show that the hole mobility can be the limiting factor affecting device performance even for a film with a high PLQY, with a mobility of ≈10−6 cm2 V−1 s−1 being the apparent lower limit for efficient devices with reduced roll-off at higher luminance. Thus, when considering the design of solution processed guest–host blends, it is critical that they have charge mobilities as high as possible.Experimental
All reagents were purchased from commercial sources and were used as received unless otherwise stated. Dichloromethane was distilled prior to use. Anhydrous xylene was stirred over sodium before distillation prior to use. Toluene was dried using a LC Technology SPBT-1 solvent purification system. Medium pressure liquid chromatography (MPLC) was performed on a Grace Reveleris X2 system using silica cartridges. Thin layer chromatography (TLC) was performed using aluminium backed silica gel 60 F254 plates from Merck. Size exclusion chromatography was performed with a BioRad Laboratories Bio-Beads S-X1 Support, using toluene as eluent. NMR spectra were recorded using an Avance 300 MHz, Avance 500 MHz, or Ascend 500 MHz spectrometer with chemical shifts (δ) reported in parts per million (ppm) and referenced to the residual solvent peak. Deuterated chloroform was referenced to 7.26 ppm and 77.0 ppm for the proton and carbon NMR spectra, respectively. Coupling constants (J) are given in Hertz (Hz) and are quoted to the nearest 0.5 Hz. SPhH = dendron surface phenyl H, BPhH = dendron branch phenyl H, CzH = carbazolyl H, and NPhH = H of phenyl ring attached to central nitrogen atom. Melting points (m.p.) were measured in a glass capillary on a Büchi B-545 melting point apparatus and are uncorrected. Elemental microanalyses were carried out on a Thermo Scientific FlashSmart CHNS/O Elemental Analyzer. Mass spectra (ESI, both LRMS and HRMS) were performed using a Bruker micrOTOF-Q (quadrupole – Time of Flight) instrument with a Bruker ESI source (ESI-micrOTOF-Q). Accurate mass measurements were carried out with direct infusion of 10 nM to 10 μM solutions of the sample in dichloromethane and with external calibration using an Agilent Tune Mix as the reference. Solution (dichloromethane distilled from calcium hydride) UV-vis absorption measurements were recorded with a Cary Varian 5000 UV-vis-NIR spectrophotometer. Differential scanning calorimetry (DSC) was performed on a PerkinElmer Pyris Diamond DSC. Thermogravimetric analysis (TGA) was carried out on a PerkinElmer STA 6000 Simultaneous Thermal Analyser. The decomposition temperature (Td) is reported for a 5% decrease in sample weight corrected for the crucible. Electrochemistry was performed using a BAS Epsilon electrochemistry station, a standard three-electrode system, at room temperature using a solution comprising approximately 1 mM of the sample and 0.1 M tetra-n-butylammonium perchlorate (TBAP, Alfa Aesar, electrochemical grade) as the electrolyte in distilled dichloromethane (from calcium hydride) or distilled tetrahydrofuran (from lithium aluminium hydride). Glassy carbon working, platinum wire counter, and a silver/silver nitrate solution reference electrodes were used. Scan rates between 50 and 200 mV s−1 were used to obtain the optimal voltammograms. The glassy carbon electrode was polished between measurements with a polishing pad and rinsed with the solvent used for the measurement. The solutions were sparged with argon and measured under an argon atmosphere. Values were corrected against ferrocene as an external standard and expressed against the ferrocene/ferrocenium (Fc/Fc+) couple.3,6-Bis[4,4′′-bis({2-ethylhexyl}oxy)-(1,1′:3′,1′′-terphenyl)-5′-yl]-9H-carbazole 9
A mixture of 3,6-dibromo-9H-carbazole (290 mg, 0.892 mmol), 7 (1.21 g, 1.97 mmol),15 tetrakis(triphenylphosphine)palladium(0) (81 mg, 0.07 mmol), and potassium carbonate (952 mg, 6.90 mmol) was deoxygenated by placing under vacuum and backfilling with argon three times. Toluene (8.0 mL), ethanol (4.0 mL), and water (4.0 mL) were separately sparged with nitrogen for 30 min and then added to the mixture. The resulting solution was carefully deoxygenated by placing under vacuum before being backfilled with argon. The reaction mixture was then stirred in an oil bath held at 90 °C for 54 h under argon. The reaction was allowed to cool to room temperature and then ethyl acetate (50 mL) and water (40 mL) were added. The two layers were separated and then the aqueous layer was extracted with ethyl acetate (2 × 30 mL). The organic layers were combined, washed with brine (2 × 50 mL), dried over anhydrous sodium sulfate and filtered. The filtrate was collected, and solvent removed under reduced pressure. The crude was purified using MPLC over silica with dichloromethane:n-hexane mixtures (0:1–1:3–1:1) as eluent to give 9 as a colourless solid (256 mg, 25%); m.p.: 65.8–67.3 °C; λmax (CH2Cl2)/nm: 268 (logε/dm3 mol−1 cm−1 5.10), 285sh (5.05), 306sh (4.81); 1H NMR (500 MHz, CDCl3) δ 0.93–1.00 (24H, m, CH3), 1.33–1.62 (32H, m, CH2), 1.76–1.81 (4H, m, CH), 3.90–3.96 (8H, m, OCH2), 7.05 and 7.69 (16H, AA′BB′, SPhH), 7.53 (2H, d, J = 8.5, CzH), 7.72 (2H, brs, BPhH), 7.80 (2H, dd, J = 8.5 & 1.5, CzH), 7.85 (4H, d, J = 1.0, BPhH), 8.13 (1H, brs, NH), 8.47 (2H, s, CzH); 13C NMR (125 MHz, CDCl3) δ 11.1, 14.1, 23.1, 23.9, 29.1, 30.5, 39.4, 70.6, 111.0, 114.9, 119.1, 123.7, 124.0, 124.3, 125.8, 128.3, 133.3, 133.7, 139.5, 142.0, 142.9, 159.1; m/z (ESI): calculated for C80H97NO4 [M]: 1135.7418 (100%), 1136.7451 (87%), 1137.7485 (37%), 1138.7518 (10%); found: 1135.7417 (65%), 1136.7476 (100%), 1137.7518 (67%), 1138.7551 (25%); C80H97NO4 requires C, 84.5; H, 8.6; N, 1.2; found: C, 84.5; H, 8.6; N, 1.2%.
Tris[4-(3,6-bis{4,4′′-bis[(2-ethylhexyl)oxy]-[1,1′:3′,1′′-terphenyl]-5′-yl}-9H-carbazol-9-yl)phenyl]amine 2
A mixture of tris(4-iodophenyl)amine (33.7 mg, 54.1 μmol), 9 (256 mg, 0.225 mmol), tris(dibenzylideneacetone)dipalladium(0) (5.50 mg, 6.01 μmol), tri-tert-butylphosphonium tetrafluoroborate (8.90 mg, 30.7 μmol), and sodium tert-butoxide (35.7 mg, 0.371 mmol) was deoxygenated by placing under vacuum and backfilling with argon three times. Anhydrous toluene (7.0 mL) was added and the resultant mixture was subjected to three cycles of freeze–pump–thaw, followed by backfilling with argon. The reaction mixture was stirred in an oil bath held at 110 °C for 39 h. The mixture was allowed to cool to room temperature and the solvent was removed. Dichloromethane (40 mL) and water (40 mL) were added to the mixture and the two layers were separated. The aqueous layer was extracted with dichloromethane (2 × 30 mL). The organic layers were combined, washed with water (2 × 60 mL), brine (1 × 60 mL), dried over anhydrous sodium sulfate and filtered. The filtrate was collected, and solvent removed under reduced pressure. The crude was first purified using MPLC over silica with dichloromethane:n-hexane mixtures (0:1–1:2) as eluent, then size-exclusion chromatography using toluene as eluent to give 2 as a colourless solid (91.0 mg, 46%); m.p.: 145.8–147.3 °C; Td(5%) = 413 °C; λmax (CH2Cl2)/nm: 269 (logε/dm3 mol−1 cm−1 5.57), 293sh (5.44), 307sh (5.32), 330sh (5.09); 1H NMR (500 MHz, CDCl3) δ 0.90–0.97 (72H, m, CH3), 1.30–1.57 (96H, m, CH2), 1.73–1.80 (12H, m, CH), 3.88–3.94 (24H, m, OCH2), 7.03 (24H, 1/2AA′BB′, SPhH), 7.64–7.73 (48H, m, CzH, SPhH, BPhH and NPhH), 7.82–7.86 (18H, m, CzH and BPhH), 8.54 (6H, d, J = 1.5, CzH); m/z (MALDI-ToF): calculated for C258H300N4O12 [M]: 3646.3 (26%), 3647.3 (72%), 3648.3 (100%), 3649.3 (92%), 3650.3 (64%), 3651.3 (35%); found: 3646.2 (29%), 3647.3 (70%), 3648.2 (93%), 3649.2 (100%), 3650.3 (78%), 3651.2 (40%); C258H300N4O12 requires C, 84.9; H, 8.3; N, 1.5; found: C, 84.85; H, 8.1; N, 1.5%.
3,6-Bis[4,4′′-di-tert-butyl-2′-methyl-(1,1′:3′,1′′-terphenyl)-5′-yl]-9H-carbazole 10
A mixture of 3,6-dibromo-9H-carbazole (285 mg, 0.877 mmol), 8 (941 mg, 1.95 mmol),16 tetrakis(triphenylphosphine)palladium(0) (80.3 mg, 0.07 mmol), and potassium carbonate (1.00 g, 7.25 mmol) was deoxygenated by placing under vacuum and backfilling with argon three times. Toluene (8.0 mL), ethanol (4.0 mL), and water (4.0 mL) were separately sparged with nitrogen for 30 min and then added to the mixture. The resulting mixture was carefully placed under vacuum and then backfilled with argon. The reaction mixture was then stirred in an oil bath held at 90 °C for 51 h before being allowed to cool to room temperature. The solvent was removed and dichloromethane (50 mL) and water (50 mL) were added. The layers were separated and the aqueous layer was extracted with dichloromethane (2 × 50 mL). The organic layers were combined, washed with brine (2 × 60 mL), dried over anhydrous sodium sulfate and filtered. The filtrate was collected and solvent removed under reduced pressure. The crude residue was purified using MPLC over silica with dichloromethane:n-hexane mixtures (0:1–1:9–1:3) as eluent to give 10 as a white solid (584 mg, 76%); m.p.: 267.3–268.3 °C; λmax (CH2Cl2)/nm: 254 (logε/dm3 mol−1 cm−1 5.00), 262sh (4.97), 297 (4.77), 345sh (3.57); 1H NMR (500 MHz, CDCl3) δ 1.40 (36H, s, t-butylH), 2.23 (6H, brs, CH3), 7.42–7.50 (18H, m, SPhH and CzH), 7.64 (4H, brs, BPhH), 7.75 (2H, brd, J = 8.0, CzH), 8.03 (1H, brs, NH), 8.38 (2H, brs, CzH); 13C NMR (125 MHz, CDCl3) δ 18.5, 31.4, 34.5, 110.8, 118.7, 124.1, 125.0, 125.4, 127.6, 129.1, 131.4, 132.4, 138.6, 139.3, 139.7, 143.2, 149.6; m/z (ESI): calculated for C66H69N [M]: 875.5430 (100%), 876.5464 (71%), 877.5497 (25%); found: 875.5430 (100%), 876.5463 (89%), 877.5498 (35%); C66H69N requires C, 90.5; H, 7.9; N, 1.6; found: C, 89.8; H, 8.0; N, 1.6.
Tris[4-(3,6-bis{4,4′′-di-tert-butyl-2′-methyl-[1,1′:3′,1′′-terphenyl]-5′-yl}-9H-carbazol-9-yl)phenyl]amine 3
A mixture of tris(4-iodophenyl)amine (88.3 mg, 0.142 mmol), 10 (448 mg, 0.511 mmol), tris(dibenzylideneacetone)dipalladium(0) (7.80 mg, 8.52 μmol), tri-tert-butylphosphonium tetrafluoroborate (9.30 mg, 32.1 μmol), and sodium tert-butoxide (104 mg, 1.08 mmol) was deoxygenated by placing under vacuum and backfilling with argon three times. Anhydrous xylene (12.0 mL) was added and the resulting mixture was subjected to three cycles of freeze–pump–thaw, and then backfilled with argon. The reaction mixture was then stirred in an oil bath held at 135 °C for 16 h. The mixture was allowed to cool to room temperature and the solvent was removed. Dichloromethane (50 mL) and water (50 mL) were added and the two layers were separated. The aqueous layer was extracted with dichloromethane (2 × 40 mL). The organic layers were combined, washed with brine (2 × 60 mL), dried over anhydrous sodium sulfate and filtered. The filtrate was collected and solvent removed under reduced pressure. The crude was first purified using MPLC over silica with dichloromethane:n-hexane mixtures (3:17–1:3) as eluent, then size-exclusion chromatography using toluene as eluent, then MPLC over silica using dichloromethane:n-hexane mixtures (3:17–1:4) as eluent to give 3 as a colourless solid (276 mg, 68%); m.p. >300 °C; Td(5%) = 500 °C; λmax(CH2Cl2)/nm: 246sh (logε/dm3 mol−1 cm−1 5.42), 254 (5.44), 264sh (5.40), 305 (5.23), 332sh (5.03); 1H NMR (300 MHz, CDCl3) δ 1.42 (108H, s, t-ButylH), 2.24 (18H, s, CH3), 7.44 and 7.50 (48H, AA′BB′, SPhH), 7.55–7.66 (18H, m, NPhH and CzH), 7.68 (12H, s, BPhH), 7.78 (6H, dd, J = 8.5 & 1.5, CzH), 8.47 (6H, d, J = 1.5, CzH); m/z (ESI): calculated for C216H216N4 [M]: 2865.7 (37%), 2866.7 (86%), 2867.7 (100%), 2868.7 (58%), 2869.7 (30%); found: 2865.7 (9%), 2866.7 (36%), 2867.7 (100%), 2868.7 (87%), 2869.7 (35%); C216H216N4 requires C, 90.5; H, 7.6; N, 1.95; found: C, 90.1; H, 7.7; N, 1.9%.
MIS-CELIV device fabrication and characterisation
Prepatterned ITO substrates were sequentially ultrasonically cleaned in acetone and 2-propanol for 10 min. A 30 nm insulating layer was prepared by spin-coating Cyclotene 3022-35 (BCB, Dow Chemical Company) at 5000 rpm for 60 s and baked at 300 °C for 10 min. After cooling to room temperature, the substrates were transferred into an evaporation chamber to deposit a 70 nm protective magnesium fluoride (MgF2) layer when the vacuum was less than 1.3 × 10−6 mbar to prevent the insulating BCB layer from being damaged by the spin-coating process. The substrates were then transferred to a nitrogen-filled glovebox and a series of 20 mg mL−1 light-emitting dendrimer, host, and guest:host solutions in toluene were spin-coated onto the substrates at 1000 rpm for 60 s. Finally, the substrates were transferred into an evaporation chamber and a 7.5 nm molybdenum oxide (MoO3) layer and a 100 nm Ag layer were sequentially deposited to complete the device under vacuum (less than 1.3 × 10−6 mbar). A Tektronix 3052C function generator, a WaveRunner 6200A oscilloscope (2 GHz) and a Falco Systems WMA-320 voltage amplifier were used for the MIS-CELIV measurements, which were carried out at room temperature with the sample held under vacuum.OLED fabrication and characterisation
The ITO substrates were cleaned using the same procedures as used for MIS-CELIV substrates. The substrates were further cleaned with an UV-Ozone cleaner for 30 min. Then PEDOT:PSS (Clevious P Al4083) was spin-coated at 4000 rpm for 40 s in a class 1000 cleanroom before being annealed at 200 °C for 10 min. The light-emitting dendrimers and TCTA-based dendrimeric hosts were dissolved in toluene at a concentration of 7.5 mg mL−1 in a nitrogen-filled glovebox (O2 < 0.1 ppm and H2O < 0.1 ppm), and then mixed at the ratios which gave the desired blend mole percent (mol%). Blend solutions were spin-coated immediately after preparation at 3000 rpm for 40 s onto the PEDOT:PSS layer, forming 30–40 nm thick uniform films. After spin-coating, all the edge regions of the film were wiped with a cotton swab containing toluene to expose the ITO. The substrates were then transferred into an evaporation chamber and BP4mPy (65 nm) (from Luminescence Technology Corporation), LiF (1 nm) and Al (100 nm) layers were deposited sequentially by thermal vacuum evaporation using a Kurt. J. Lesker SPECTROS evaporation system at high vacuum (5 × 10−7 mbar). The J–V–L and efficiency measurements of the OLEDs were carried out using a Keithley 2400 source meter and an absolute EQE measurement system with a calibrated integrating sphere (Hamamatsu Photonics C9920-12).Photophysical measurements
Fused silica substrates were cleaned, and the films deposited using the same procedures as used for MIS-CELIV devices. The film PLQY measurements were carried using an integrating sphere purged with nitrogen and a 325 nm HeCd laser with beam power of 0.2 mW was used to excite the samples.29 A Newport 818-UV photodetector and a KEITHLEY 4210 source meter were used to quantify the photoluminescence. The PL spectra were measured using an FS5 spectrofluorometer from Edinburgh Instruments. The film absorbance spectra were measured using a Cary 5000 UV-vis-NIR from Agilent Technologies.Structural characterisation [GIXRD, XRR, and AFM] and number density calculation
Silicon wafers (50 mm diameter) were cleaned with piranha solution, ultrasonicated in acetone and 2-propanal, and dried under a flow of nitrogen. The compounds were dissolved in toluene (10 mg mL−1) and spin-coated at 2000 rpm for 60 s. GIXRD and XRR measurements were conducted on a Rigaku Smartlab X-ray diffractometer with a 9 kW rotating anode (Cu) source. Cu Kα (λ = 1.54 Å) radiation was focused into a parallel beam onto the samples, measuring in a θ/2θ configuration using a Hypix3000 detector. The GIXRD measurements were conducted with a fixed incidence angle of 0.3°, measuring over a range of scattering angles (2θ) from 2°–35° in a continuous scan mode every 0.02° at a scan rate of 1.5° min−1. XRR experiments were measured over a range of 2θ from 0.05–4.0° at a scan rate of 0.6° min−1. The data were then converted to momentum transfer, Q [Q = (4π/λ)sinθ], and fitted using the Refnx reflectometry analysis program.30 The XRR profiles were modelled with a silicon substrate, native silicon oxide layer, bulk organic layer, and in some cases a substrate interfacial layer (Fig. S7, ESI†). The number of guest molecules per 1 cm3 (number density) was derived from the molecular volume, Vm, which was calculated by the following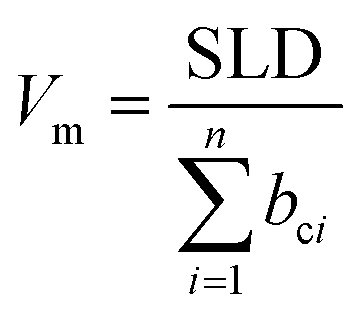 | (1) |
The films for the AFM measurements were prepared using the same procedures as those used for the OLEDs. The AFM images were acquired using an Asylum Research Cypher S in tapping mode with the scan rate of 1.0 Hz and with a measurement area of 8 μm × 8 μm.
Energy transfer efficiency calculation
As the first step in calculating the energy transfer efficiency, the expected PL contribution (PLexp) was calculated considering a situation where there is no energy transfer from the TCTA-based dendrimeric host to light emissive dendrimer guest. The percentage of light absorbed by the host (Ahost) and guest (Aguest) were calculated from the absorption coefficient (Fig. S6, ESI†) at the corresponding excitation wavelength. With the PLQY values of the host (ηhost) and guest (ηguest) materials, PLexp was calculated using the following equation| PLexp = Ahost × ηhost/(Ahost × ηhost + Aguest × ηguest) | (2) |
Finally, the energy transfer efficiency was calculated using the following equation
| Energy transfer efficiency = (PLexp − PLact)/PLexp | (3) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was carried at the Centre for Organic Photonics & Electronics at The University of Queensland. P. L. B. is an Australian Research Council Laureate Fellow (FL160100067), and the work was supported by the Fellowship. We acknowledge the financial support from the China Scholarship Council program (CSC, No. 201707090077) for M. G. This work was performed in part at the Queensland node of the Australian National Fabrication Facility (ANFF-Q), a company established under the National Collaborative Research Infrastructure Strategy to provide nano and micro fabrication facilities for Australia's researchers. The authors acknowledge the facilities, and the scientific and technical assistance, of the Centre for Microscopy and Microanalysis, The University of Queensland, the Queensland node of Microscopy Australia.References
- T. Zhang, M. Zhu, J. Li, Y. Zhang and X. Wang, Dyes Pigm., 2021, 192, 109426 CrossRef CAS.
- T.-Y. Li, J. Wu, Z.-G. Wu, Y.-X. Zheng, J.-L. Zuo and Y. Pan, Coord. Chem. Rev., 2018, 374, 55–92 CrossRef CAS.
- J.-H. Zhao, Y.-X. Hu, H.-Y. Lu, Y.-L. Lü and X. Li, Org. Electron., 2017, 41, 56–72 CrossRef CAS.
- C. Cebrián and M. Mauro, Beilstein J. Org. Chem., 2018, 14, 1459–1481 CrossRef PubMed.
- S. M. Russell, A. M. Brewer, D. M. Stoltzfus, J. Saghaei and P. L. Burn, J. Mater. Chem. C, 2019, 7, 4681–4691 RSC.
- A. S. Abd-El-Aziz, A. A. Abdelghani, B. D. Wagner and R. Bissessur, Macromol. Rapid Commun., 2019, 40, 1800711 CrossRef PubMed.
- Z. Ma, W. Dong, J. Hou, Q. Duan, S. Shao and L. Wang, J. Mater. Chem. C, 2019, 7, 11845–11850 RSC.
- X. Liu, Z. Yu, M. Yu, X. Zhang, Y. Xu, P. Lv, S. Chu, C. Liu, W. Lai and W. Huang, ACS Appl. Mater. Interfaces, 2019, 11, 26174–26184 CrossRef CAS PubMed.
- W.-Y. Lai, M. N. Balfour, J. W. Levell, A. K. Bansal, P. L. Burn, S.-C. Lo and I. D. W. Samuel, Macromolecules, 2012, 45, 2963–2971 CrossRef CAS.
- Y. H. Kim, T. H. Han, C. Lee, Y. H. Kim, Y. Yang and T. W. Lee, Adv. Funct. Mater., 2020, 30, 2005292 CrossRef CAS.
- S. V. Dayneko, M. Rahmati, M. Pahlevani and G. C. Welch, J. Mater. Chem. C, 2020, 8, 2314–2319 RSC.
- C. You, D. Liu, J. Yu, H. Tan, M. Zhu, B. Zhang, Y. Liu, Y. Wang and W. Zhu, Adv. Opt. Mater., 2020, 8, 2000154 CrossRef CAS.
- Y. W. Park, Y. M. Kim, J. H. Choi, T. H. Park, J.-W. Jeong, M. J. Cho, D. H. Choi and B. K. Ju, J. Nanosci. Nanotechnol., 2010, 10, 3250–3253 CrossRef CAS PubMed.
- Y. Feng, T. Lu, D. Liu, W. Jiang and Y. Sun, Org. Electron., 2019, 67, 136–140 CrossRef CAS.
- S.-C. Lo, E. B. Namdas, P. L. Burn and I. D. W. Samuel, Macromolecules, 2003, 36, 9721–9730 CrossRef CAS.
- D. M. Stoltzfus, W. Jiang, A. M. Brewer and P. L. Burn, J. Mater. Chem. C, 2018, 6, 10315–10326 RSC.
- M. Gao, J. Jang, T. Leitner, V. T. N. Mai, C. S. K. Ranasinghe, R. Chu, P. L. Burn, A. Pivrikas and P. E. Shaw, Adv. Mater. Interfaces, 2021, 2100820 CrossRef CAS.
- I. D. W. Samuel, P. L. Burn and S.-C. Lo, WO 2004029134, 2004.
- R. Singh, J. S. Meena, C. S. Wu and F.-H. Ko, Phys. Chem. Chem. Phys., 2015, 17, 5227–5235 RSC.
- B. Pan, H. Huang, X. Yang, J. Jin, S. Zhuang, G. Mu and L. Wang, J. Mater. Chem. C, 2014, 2, 7428–7435 RSC.
- K. Karon and M. Lapkowski, J. Solid State Electrochem., 2015, 19, 2601–2610 CrossRef CAS.
- M. Rahmati, S. Dayneko, M. Pahlevani and Y. Shi, Adv. Funct. Mater., 2019, 29, 1906742 CrossRef CAS.
- N. Ide, N. Matsusue, T. Kobayashi and H. Naito, Thin Solid Films, 2006, 509, 164–167 CrossRef CAS.
- S.-C. Lo, T. D. Anthopoulos, E. B. Namdas, P. L. Burn and I. D. W. Samuel, Adv. Mater., 2005, 17, 1945–1948 CrossRef CAS.
- X. Yang, H. Guo, X. Xu, Y. Sun, G. Zhou, W. Ma and Z. Wu, Adv. Sci., 2019, 6, 1801930 CrossRef PubMed.
- M. Gao, T. Lee, P. L. Burn, A. E. Mark, A. Pivrikas and P. E. Shaw, Adv. Funct. Mater., 2020, 30, 1907942 CrossRef CAS.
- T. Kobayashi, N. Ide, N. Matsusue and H. Naito, Jpn. J. Appl. Phys., 2005, 44, 1966 CrossRef CAS.
- S. Reineke, G. Schwartz, K. Walzer and K. Leo, Appl. Phys. Lett., 2007, 91, 123508 CrossRef.
- N. C. Greenham, I. D. W. Samuel, G. R. Hayes, R. T. Phillips, Y. A. R. R. Kessener, S. C. Moratti, A. B. Holmes and R. H. Friend, Chem. Phys. Lett., 1995, 241, 89–96 CrossRef CAS.
- A. R. Nelson and S. W. Prescott, J. Appl. Crystallogr., 2019, 52, 193–200 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc03949k |
| This journal is © The Royal Society of Chemistry 2021 |