
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePerspectives for polychlorinated trityl radicals
Imma
Ratera
 *a,
Jose
Vidal-Gancedo
a,
Daniel
Maspoch
bc,
Stefan T.
Bromley
cd,
Núria
Crivillers
*a and
Marta
Mas-Torrent
*a
*a,
Jose
Vidal-Gancedo
a,
Daniel
Maspoch
bc,
Stefan T.
Bromley
cd,
Núria
Crivillers
*a and
Marta
Mas-Torrent
*a
aInstitut de Ciència de Materials de Barcelona (ICMAB-CSIC) and CIBER-BBN, Campus de la UAB, 08193 Bellaterra, Spain. E-mail: iratera@icmab.es; ncrivillers@icmab.es; mmas@icmab.es
bCatalan Institute of Nanoscience and Nanotechnology (ICN2), CSIC and the Barcelona Institute of Science and Technology, Campus UAB, 08193 Bellaterra, Spain
cInstitució Catalana de Recerca i Estudis Avançats (ICREA), 08010 Barcelona, Spain
dDepartament de Ciència de Materials i Química Física & Institut de Química Teòrica i Computacional (IQTCUB), Universitat de Barcelona, 08028 Barcelona, Spain
First published on 14th July 2021
Abstract
An organic free radical is a molecule with one or more unpaired electrons. Although most free radicals are highly reactive, chemists have developed a few families of so-called persistent organic radicals with high kinetic stabilities. Polychlorinated trityl radicals, also known as polychlorotriphenylmethyl (PTM) radicals, are particularly chemically stable due to the high steric hindrance provided by the chlorine atoms in ortho positions which protect their single unpaired electron localised on the central carbon atom. In addition to their inherent magnetic spin due to the unpaired electron, PTMs exhibit other appealing properties such as a rich electrochemistry and characteristic optical properties (absorption and emission). Moreover, it has been shown that these properties can be tuned through the preparation of a large library of PTM-based derivatives. Here, we review recent developments employing PTM radicals, which include their implementation in molecular electronic junctions/switches, as building blocks for the preparation of magnetic networks and opto-electronic devices/materials and their exploitation in bio-applications.
1. Introduction
An organic free radical is a molecule with one or more unpaired electrons and is, generally, highly reactive. However, there are a few families of persistent organic radicals that have been developed over the last few decades that are stable at ambient conditions.1 These species have attracted great interest for a wide spectrum of applications including their use as building blocks for the preparation of magnetic and multifunctional materials, for obtaining structural, dynamical or chemical information from other systems (i.e. spin labelling, spin trapping), in catalysis and polymerisation processes, as antioxidants and as polarizing agents in Dynamic Nuclear Polarisation (DNP) to enhance Nuclear Magnetic Resonance (NMR) sensitivity.2One particularly stable and intensively investigated family of persistent organic free radicals is that of polychlorinated trityl radicals, also known as polychlorotriphenylmethyl (PTM) radicals. PTMs are composed of three totally or partially chlorinated phenyl rings connected to a central carbon atom with a sp2 hybridization where the spin is localised (Fig. 1a).3 These radicals are chemically and thermally stable due to the steric hindrance at the central carbon atom caused by the ortho chlorine atoms on the phenyl rings. The inter-chloro steric strain forces the molecule to adopt a propeller-like conformation. Thus, PTM radicals are inert to oxygen and many aggressive reagents and even stable under light in the solid state. As a consequence, PTM radicals can withstand standard synthetic procedures and, hence, a large variety of materials4 have been prepared exhibiting fascinating magnetic properties, such as polyradicals with high spin ground states.5,6 The magnetic properties of PTM derivatives in crystals and supra-molecular assemblies have also been a subject of intense research.7–9
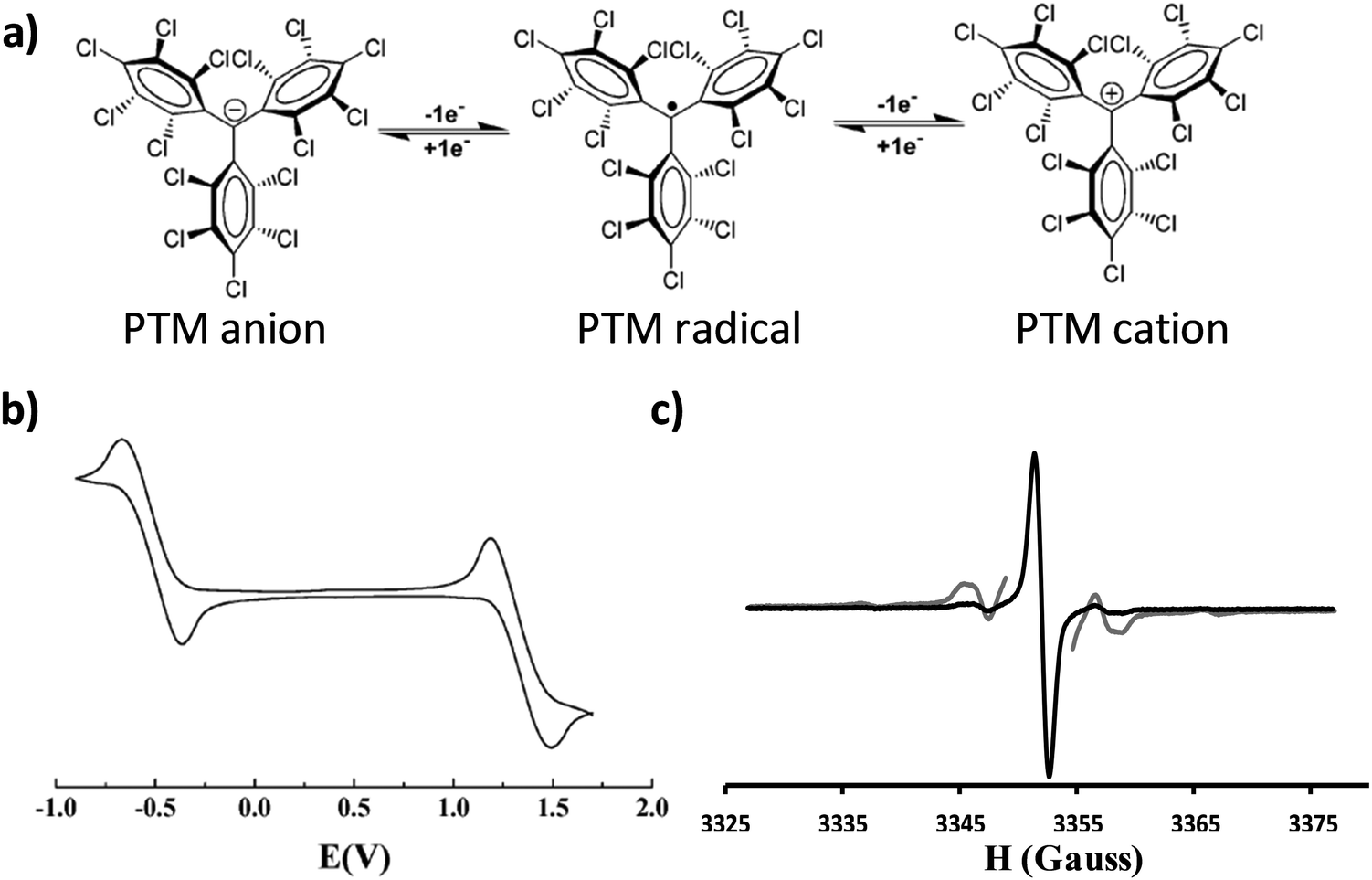 | ||
| Fig. 1 (a) Molecular structure of the PTM radical and its corresponding oxidation and reduction to the cation and anion, respectively. (b) Representative cyclic voltammogram of a PTM radical in CH2Cl2, with 0.1 M n-Bu4NPF6 (vs. Ag/AgCl). Adapted from ref. 4 Copyright 2012 Royal Society of Chemistry. (c) EPR spectrum of a PTM radical recorded at room temperature in deoxygenated CH2Cl2 (g = 2.0023). In grey the same spectrum recorded with a higher power to better appreciate the 13C hyperfine coupling. | ||
In addition to their inherent magnetic moment, PTM radicals possess a rich electrochemistry. PTM is a very good electron acceptor and can be easily chemically or electrochemically converted to its corresponding anion form in a reversible manner. Moreover, PTM radicals can also be oxidised to the cation species, although this process occurs at higher voltages (Fig. 1b). Accordingly, in solution PTM radicals have been extensively studied as molecular switches,10 as acceptor units in donor–acceptor dyads to investigate intramolecular charge transfer processes11–14 and non-linear optical properties,15 and as charge reservoirs in the preparation of mixed-valence systems to gain information about the transport properties of molecular wires.16,17 In most of these cases, the use of Electronic Paramagnetic Resonance (EPR) has been key to monitor the radical species and thus to follow the electronic processes taking place (Fig. 1c). More recently, PTMs have also been immobilized on solid supports in a step forward towards more advanced devices.18,19
PTM radicals offer a unique combination of magnetic, electrical and optical properties, which make these species hugely attractive for numerous ongoing investigations where they have the potential to renew or transform research fields. In this work, we give an overview of recent work employing PTM radicals systems including molecular electronic junctions, optical devices, active units in molecular switches, building blocks for magnetic networks and bio-applications.
2. Surface confined molecular switches based on PTM radicals
The functionalisation of surfaces with redox-active molecules allows for the modulation of the substrate properties upon the application of an electrical stimulus. Such hybrid systems have been focus of major interest for the development of switches and memories.20,21 In this sense, it has been shown that PTM radicals are appealing building blocks since they can be electrically interconverted between two easily accessible redox states (i.e. the PTM radical and PTM anion), which reveal distinct magnetic, optical and chemical properties. For this purpose, great efforts have been devoted over the last few years to synthesise PTM moieties bearing appropriate grafting groups to prepare chemically bonded surface self-assembled monolayers (SAMs) on Au, quartz, Si, and indium tin oxide (ITO) substrates.22–25 In particular, SAMs of a PTM radical bearing a silane group on ITO were reported by Rovira and co-workers to lead to robust electrochemical switches (Fig. 2a).24 The electrochemical characteristics of these PTM radical SAMs were investigated by cyclic voltammetry (CV). As expected, one reversible redox wave was observed with an oxidation peak at +53 mV and a reduction peak at −32 mV at a scan rate 100 mV s−1 (vs. Ag wire). The optical absorption, emission and magnetic responses were employed to read the state of this multi-channel switch. The absorption and emission bands characteristics from the two redox states (i.e., PTM radical: λabs = 365 nm, λem = 688 nm/PTM anion: λabs = 515 nm, no emission) changed simultaneously in opposite directions whilst applying write/erase cycles in a completely reversible way. In turn, the EPR signal observed in the PTM radical SAM disappeared upon reduction to the anionic form and appeared again after further oxidation, corroborating that the magnetic output was also a suitable read-out tool to determine the state of the switch. In analogous SAMs on gold, it was additionally proved that the modification of the redox state of the PTM SAM had also a strong impact on the surface wetting properties.26 The water contact angle of the highly hydrophobic PTM SAM was reported to be 102 ± 6°, but changed to 73 ± 3° after reducing the radical due to the formation of the more polar anionic state.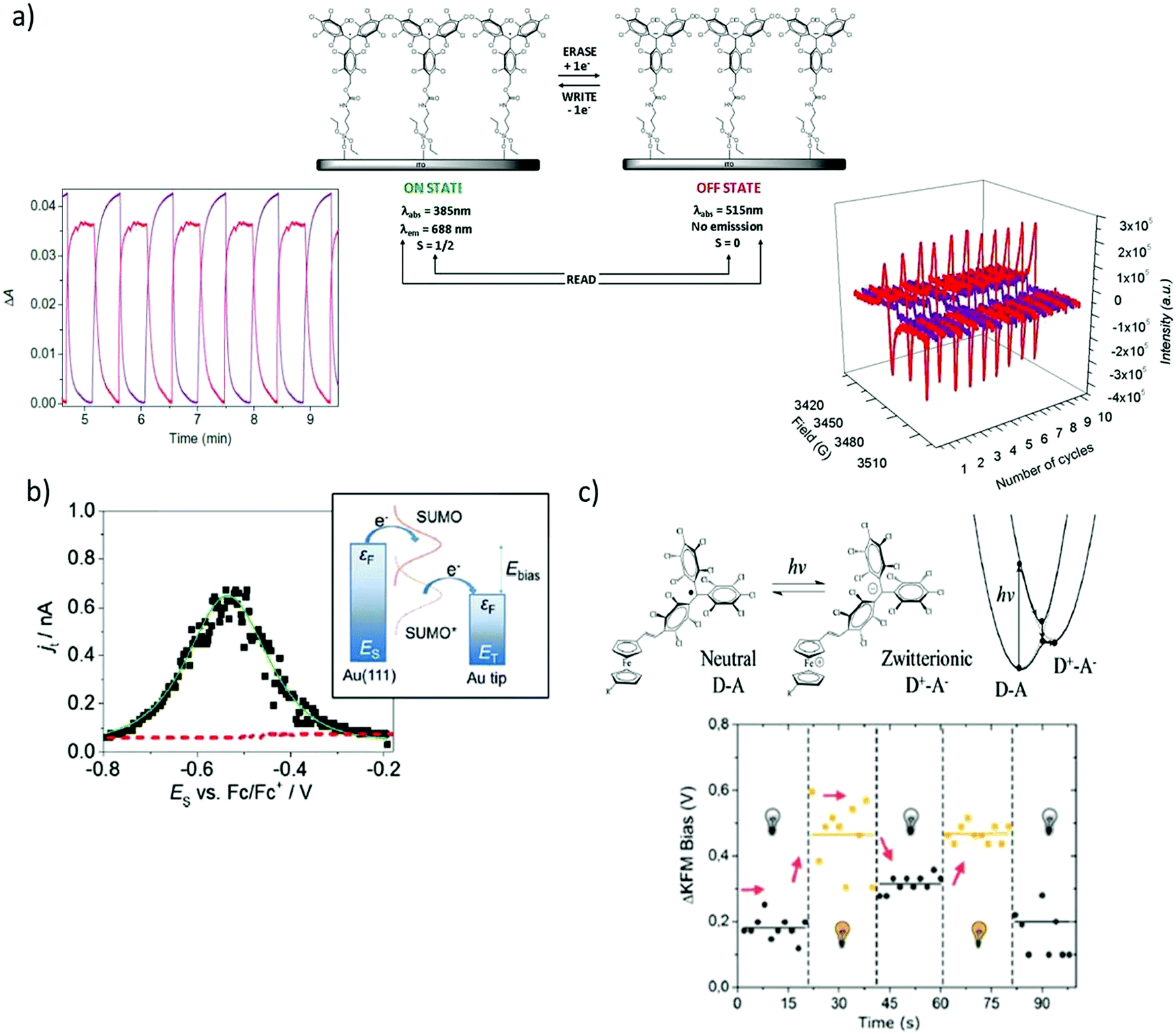 | ||
| Fig. 2 (a) Scheme of a self-assembled monolayer of a PTM radical on ITO and monitoring of the optical absorption (bottom left) and EPR signal (bottom right) outputs upon the application of write/erase voltage cycles to convert the PTM radical to the anion form. The optical absorption plot corresponds to the monitoring of the PTM radical absorption band at 365 nm (red) and the anion one at 515 nm (purple). In the EPR monitoring, red lines correspond to the radical SAM and the purple lines to the anion form. Reprinted with permission from ref. 24. Copyright 2011 Nature Group. (b) Current–voltage curve extracted by EC-STS from a PTM SAM on Au and schematic energy diagram of a two-step electron transfer process mediated by a redox active radical molecule. Reprinted with permission from ref. 30. Copyright 2016 Royal Society of Chemistry. (c) Top: Scheme of the neutral D–A and zwitterionic species and corresponding energy levels upon excitation. Bottom: Shift of the surface potential differences of PTM–Fc SAMs in the dark and under 950 nm illumination measured by KPFM. Reprinted with permission from ref. 35. Copyright 2019 Royal Society of Chemistry. | ||
Recently, SAMs of PTM have also been prepared on Si to fabricate a light-triggered capacitance switch under electrochemical conditions.25 The electronic properties of Si can be tuned by modifying the density and the nature of the charge carriers under illumination.27 In this way, the SAMs on Si in the dark showed negligible oxidation and reduction currents, whereas under illumination through a red filter, the radical/anion redox process could be observed in the CV voltammograms. To obtain further insights into the light dependence of the redox process in this PTM SAM, differential capacitance measurements were performed. Accordingly, in the dark, the measured capacitance values were small and did not exceed 1 mF cm−2, whereas under red light a much more intense capacitance peak (enhancement by a factor of 10) was found at the formal potential of the surface bound PTM.
The electrochemical switching ability of PTM radicals has also been implemented in other materials, such as in TiO2 and SiO2 porous thin films consisting of tilted nanocolumns supported on an ITO electrode.28 More recently, the modulation of the optical absorption and magnetic properties of three generations of PTM radical dendrimers has also been reported, in which up to 24 redox active units were incorporated.29 This work opens the door to the future possibility to gain control on the exact number of electrons transferred during the switching process, which could lead to a multistate switch.
The redox activity of the PTM SAMs have also been investigated at the nanoscale by electrochemical scanning tunnelling spectroscopy (EC-STS) in a ionic liquid medium.30,31 Electrochemical STS measurements in the constant bias mode were performed.32 In this mode, the potentials of the substrate ES and the tip ET were swept simultaneously while keeping the bias voltage Ebias = ET − ES constant. The STS results demonstrated a significant enhancement of the tunnelling current flowing between the substrate and the tip close to the equilibrium potential of the PTM radical/PTM anion redox pair, resembling a transistor like functionality (Fig. 2b).30 Such behaviour is typical of a redox mediated electron tunnelling (RMET) process,33,34 which is rationalised by a two-step electron transfer. In the first one, the electron transfers from the electrode with a higher Fermi level to the single-unoccupied molecular orbital (SUMO). Afterwards, the energy of the now occupied SUMO* decreases due to vibrational relaxation until the second electron transfer occurs towards the second electrode (Fig. 2b). Importantly, the high on/off current ratio of 13 (i.e., tunnelling increase ratio at the peak position) showed that, in comparison with other redox-active moieties, the PTM radical is among the most efficient redox mediators in RMET, demonstrating its applicability as active component in electronic devices of nanoscale size.
Finally, PTMs have also been exploited in light-triggered switches without the need of using electrochemical stimuli. In this case, the PTM was employed as the acceptor group in a donor–acceptor (D–A) dyad, where D was a ferrocene (Fc) moiety.35 The near-infrared (NIR) irradiation of PTM–Fc SAMs on gold in air led to the formation of a metastable zwitterionic state due to a light driven intramolecular charge transfer from the Fc to the PTM radical unit (Fig. 2c). This phenomenon had a significant impact on the metal work-function (WF). Hence, Kelvin Probe Force Microscopy (KPFM) was used to measure the contact potential difference changes (Fig. 2c). When switching cycles were applied, it was found that the WF increased when the sample was illuminated, while it decreased in the darkness in a reversible way.36 The value of the WF shift was reported to equal 250 meV, which was slightly higher than the largest shift reported for photoswitchable SAMs where structural isomerisations are place instead of electronic reorganisations.37
Considering all the above, clearly the high reversibility and stability of the PTM redox process that occurs at very low voltage, makes these materials very appealing for the development of a wide range of switches.
3. PTM radicals as active electronic components in molecular junctions
The incorporation of molecular species between two electrodes to evaluate their transport properties, primarily as molecular wires, has been pursued in the field of molecular electronics over the last few decades. Beyond the understanding of the charge transport mechanisms in non-functional molecules such as alkanethiols of different lengths38 and molecules with different degrees of conjugation,39 the study of more complex systems showing photo-,40 redox-41 and/or magnetic-activity42,43 has also attracted much attention. Such intrinsic molecular characteristics bring additional functionalities to the electrode/molecule/electrode structure since the conductivity can be modulated by external stimuli.In this regard, PTM radicals have shown to be fascinating candidates for preparing functional molecular junctions. Due to the open-shell nature of the PTM radicals, the most accessible molecular levels are the single-occupied molecular orbital (SOMO) and the SUMO. In the case of PTM radicals, the close energetic level alignment between the SUMO and the Fermi level of the gold contacts has been proven to significantly enhance the conductivity of the junction in comparison with the closed-shell hydrogenated PTM counterpart. This phenomenon was demonstrated for PTM radicals incorporated in mechanically controlled break junctions (MCBJ)44,45 as well as by top-contacting PTM-based SAMs on gold with an atomic force microscopy (AFM) tip (i.e., conducting-AFM)46,47 or with eutectic gallium–indium alloy (EGaIn).25,48,49 Importantly, the high chemical stability of the PTM radicals allowed the preparation of derivatives with different anchoring groups for gold (thiophene, thiol, disulfide and terminal alkyne) and with different linkers. In order to experimentally demonstrate the charge transport mechanism through PTM SAMs on Au, a family of PTM radical derivatives and their corresponding closed-shell form bearing long non-conjugated alkyl tether chains of different lengths were synthesized ensuring localization of the molecular frontier orbitals on the PTM moiety (Fig. 3a).49 The experimental results indicated that the charge transport mechanism across the junctions was SUMO-assisted coherent non-resonant tunnelling. This observation was further supported by a temperature independent transport behavior. Further, the transport through PTM-based D–A dyad SAMs having ferrocene48 or tetrathiafulvalene50 as donor units were also investigated, demonstrating the possibility to modulate the current rectification ratio between the radical and the non-radical (i.e., closed-shell) derivative.
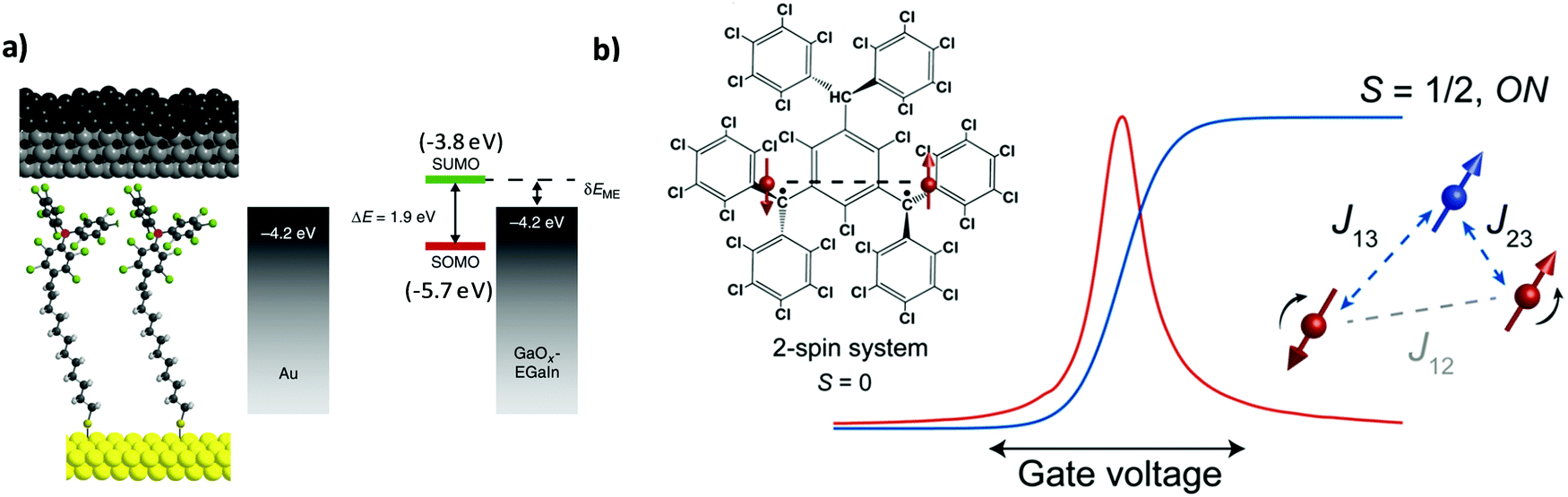 | ||
| Fig. 3 (a) Scheme of the Au/S-alkyl-PTM SAM/GaOx/EGaIn and the corresponding energy level diagram. Reprinted with permission from ref. 49. Copyright 2016 Nature Group. (b) Left: Structure and magnetism schematics of the neutral diradical investigated. The red dots and the dashed lines are the radical spins (2-spin system as synthesised, S = 0) and the exchange interactions, respectively. Right: Differential conductance (red) and corresponding redox center spin values (blue) as a function of gate voltage. The added electron is marked as the blue dot (3-spin system, S = 1/2). Reprinted with permission from ref. 52. Copyright 2017 ACS Publications. | ||
Beyond their relevance as molecular wires in the solid state, organic radicals are very appealing in the field of molecular spintronics because they show an intrinsic magnetic moment. In particular, compared to transition-metal-based magnetic compounds, they have low spin–orbit coupling and low hyperfine interactions suggesting relatively long spin lifetimes. The magnetic state of PTM radicals has been demonstrated to be stable in solid state single molecule junctions. Frisenda et al. reported the first work on the incorporation of a thiophene-terminated PTM radical in a single molecular junction.44 Here, the magnetic character of the PTM radical was undoubtedly identified by the presence of fully screened spin – ½ Kondo features, which were determined to be insensitive to the electrode displacement and background conductance. Furthermore, later on Bejarano et al. investigated the role of the binding motif in the electrical response of room temperature MCBJ.45 With a PTM radical having terminal alkynes, the formation of C–Au σ-bonds was demonstrated leading to more robust junctions with a better-defined geometry of the contact and a reduction in the variability of the conductance values. Remarkably, the tuning of the magnetic state of PTM-based molecular junctions could be achieved by employing neutral organic di- and triradical molecules embedded in a three-terminal device.51,52 The redox state of a PTM diradical could be reversibly modulated by charging the molecule with the application of an electric field. This induced the on and off switch of a parallel exchange path between the two radical spins. It was shown that the added electron led to the formation of a spin system with three antiferromagnetically coupled spins (Fig. 3b).52 In the case of the triradical system, a ferro-to-antiferromagnetic transition was observed and assigned to structural distortions. Additionally, the exchange coupling could also be tuned by electrical gating.51 In summary, all these findings constitute the bases of a promising future for integrating PTM radicals in electronic and spintronic nanodevices.
4. PTM radicals as molecular building blocks for reticular materials
Reticular materials are crystalline open-framework materials assembled from judiciously-designed molecular building blocks (MBBs) that are linked by strong bonds.53,54 Among the main families of reticular materials are metal–organic frameworks (MOFs) and covalent–organic frameworks (COFs):55,56 MOFs are 2D and 3D porous frameworks formed by joining organic MBBs to metal MBBs, whereas COFs exclusively comprise organic MBBs. All these materials share ultrahigh porosity (500 m2 g−1 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 m2 g−1), high thermal stability (at up to 200 °C to 500 °C), and good chemical stability in organic and aqueous media, including acids and bases.57 Based on these properties, reticular materials offer great potential for myriad applications, including storage of pollutants and/or hazardous gases (e.g. CO and CO2); fuel applications (e.g. with H2 or CH4); catalysis; biomedical applications; and gas/liquid separation (e.g. CO2/CH4, and xylene and alkane isomers).58–60 Beyond their inherent porosity, reticular materials offer rich chemical and structural versatility, allowing them to be conferred with additional functional properties such as magnetism and electrical or optical properties—typically, via fine-designing their frameworks.61–64 Such properties can in turn be modulated by using the intrinsic porosity of the frameworks to create devices such as sensors.64
000 m2 g−1), high thermal stability (at up to 200 °C to 500 °C), and good chemical stability in organic and aqueous media, including acids and bases.57 Based on these properties, reticular materials offer great potential for myriad applications, including storage of pollutants and/or hazardous gases (e.g. CO and CO2); fuel applications (e.g. with H2 or CH4); catalysis; biomedical applications; and gas/liquid separation (e.g. CO2/CH4, and xylene and alkane isomers).58–60 Beyond their inherent porosity, reticular materials offer rich chemical and structural versatility, allowing them to be conferred with additional functional properties such as magnetism and electrical or optical properties—typically, via fine-designing their frameworks.61–64 Such properties can in turn be modulated by using the intrinsic porosity of the frameworks to create devices such as sensors.64
The design of magnetic reticular materials, including those with cooperative or molecular properties, has attracted much attention over the past 20 years. The frameworks of reticular materials can be endowed with magnetism mainly by two strategies: by linking appropriate magnetically-active metal ions/clusters and/or MBBs; and by introducing magnetic guest species in the pores.63 To generate reticular materials with cooperative magnetic properties specifically, a particularly exotic strategy is to connect magnetically-active metallic nodes via magnetically-active organic-radical MBBs.62 MBBs based on PTM radicals are ideally suited to this application. The structure of the PTM molecule defines a rigid backbone that resembles those of many MBBs used to synthesize iconic MOFs and COFs.65,66 Another advantage of PTM-radical MBBs is that the meta and/or para positions (relative to the central carbon) of each phenyl ring can be functionalized with moieties that can coordinate to metal ions (e.g. COOH) or that can be linked via dynamic covalent bonds (e.g. NH2 or CHO). This versatility provides countless potential combinations to design MBBs with different connectivities, topologies and functionalities. For example, two or three carboxylic acid groups could be introduced at the para position of the phenyl rings to generate the (4,4′-dicarboxytridecachlorotriphenyl)methyl radical (PTMDC)67 or the (4,4′,4′′-tricarboxydodecachlorotriphenyl)methyl radical (PTMTC),68 respectively. Accordingly, the PTMDC radical can act as a two-connected (2-c) bent MBB, whereas PTMTC can act as a 3-c triangular MBB. Finally, PTM-radical MBBs are stable in the solid state up to relatively high temperatures.
Seeking to exploit the aforementioned advantages of PTM-radical MBBs, Veciana and co-workers synthesized the first-ever magnetic MOFs built from organic-radical MMBs. To this end, 3-c triangular PTMTC was reacted with either Cu(II) or Co(II) to obtain two 2-D MOFs exhibiting an underlying 3-c hcb topology (Fig. 4a).69,70 The first, a Cu(II)-based MOF (called MOROF-1; where MOROF = metal–organic radical open-framework), combines very large pores (diameter: 2.8 nm to 3.1 nm) with bulk magnetic ordering at low temperature (Fig. 4b).69 Remarkably, MOROF-1 was among the first reported breathing MOFs and sponge-like magnets: upon loss and reuptake of solvent molecules, it transforms between a crystalline and an amorphous phase, a process that leads to a reversible variation in the critical ordering temperature (Fig. 4b and c). Later on, 3-c PTMTC was also combined with two different 6-c Tb(III) clusters to obtain two 3-D MOFs: one with an underlying 3,6-c spn topology and one with a 3,6-c 3,3,3,5T33 topology, both showing antiferromagnetic interactions.71–73 The versatility of PTM-MBBs was further demonstrated by adding six carboxylic acid groups at the meta positions of the three phenyl rings to obtain a new MBB: a 3,3-c MBB.74 Reaction of this MBB with Cu(II) and 4,4′-bypiridine afforded a two-interpenetrated 3,3,3-c MOF. Finally, a new 3-c PTM-MBB built up with three pyridyl groups, tris(3,5-dichloro-4-pyridyl)methyl radical (trisPyM), was recently reacted with Zn(II) to synthesize a 2-D luminescent MOF also exhibiting an underlying 3-c hcb topology.75
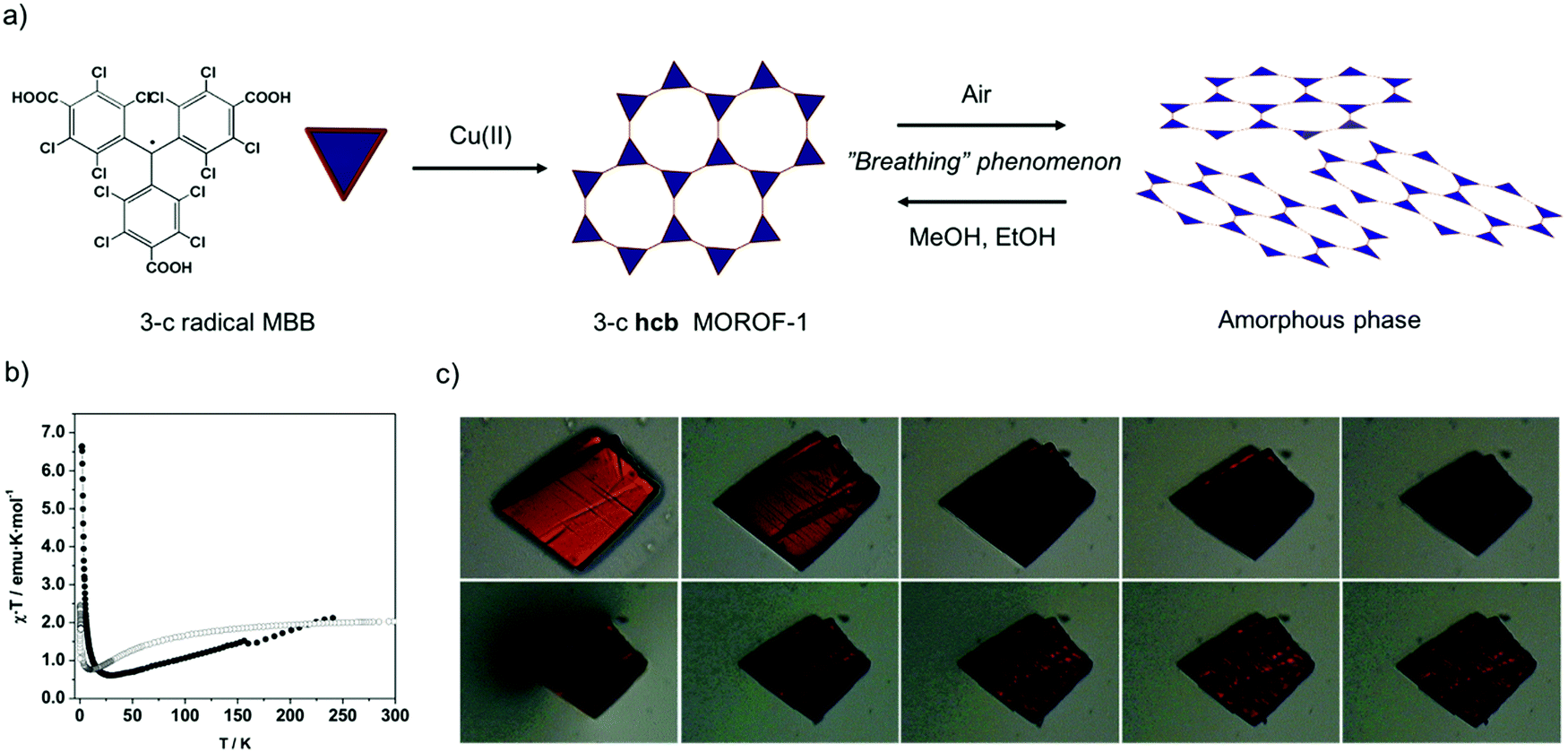 | ||
| Fig. 4 (a) Schematic representation of the formation of hcb MOROF-1 using the 3-c PTMTC and Cu(II), and its breathing behaviour. (b) Value of χT as a function of the temperature for MOROF-1 (filled circle, MOROF-1; open circle, evacuated and amorphous MOROF-1 phase). (c) Breathing phenomenon of a single crystal of MOROF-1 when exposed to the air (top) and re-exposed to ethanol liquid (bottom). (b and c) Adapted from ref. 69. Copyright 2003 Nature Group. | ||
Another option for making network materials of linked PTMs is to join them together using only covalent bonding and organic linkers. Joining PTMs (and/or similar triarylmethyls – TAMs) via organic linkers to form open-shell 2D COFs (or covalent organic radical frameworks – CORFs) was proposed theoretically in 2017.76,77 Hexagonally connected planar lattices of TAM radical MBBs are of particular interest for making 2D CORFs that could possess a range of energetically low-lying electronic states. Specifically, at low temperatures hexagonal 2D CORFs will tend to exhibit antiferromagnetic (AFM) semiconducting/insulating states, but, in principle, also have the potential to show diamagnetic quinoidal closed-shell, and graphene-like semimetallic behaviour.77 Soon after these predictive modelling studies, the first PTM-based 2D COF was synthesised (see Fig. 5a). This material (PTM-CORF) is based on layers of hexagonally ordered PTM MBBs linked together via acetylenic bridges and was found to be an AFM semiconductor.78,79 Interestingly, PTM-CORF was also found to be highly active electrochemically for the oxygen reduction reaction which was attributed to the energetically low lying unoccupied electronic levels of the regularly spaced and accessible radical centres which favours the acceptance of electrons from oxygen.78
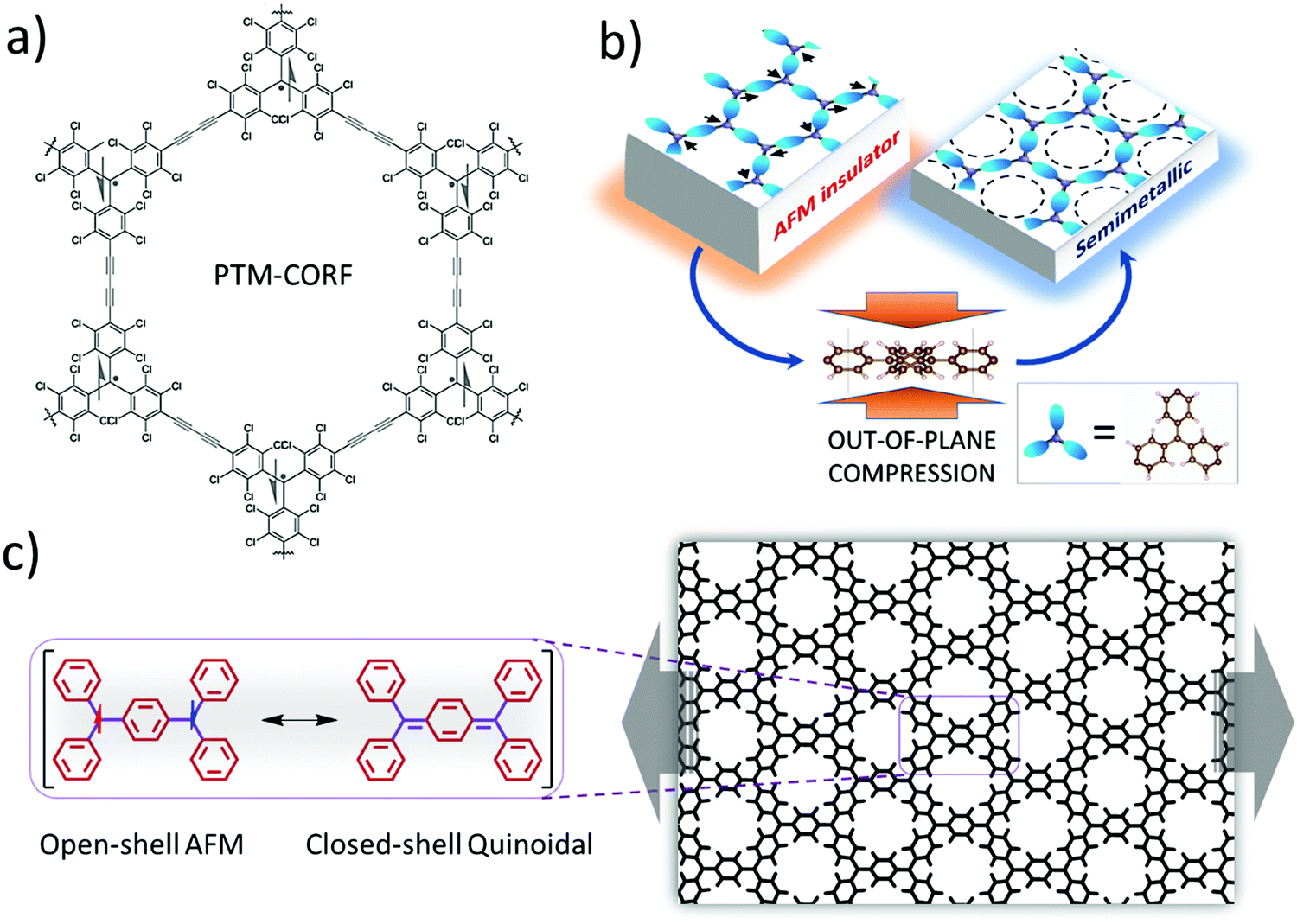 | ||
| Fig. 5 (a) Synthesised PTM-CORF. Adapted from ref. 78. Copyright 2018 Wiley-VCH Verlag GmbH & Co. (b) Mott-like transition in a ring-sharing hexagonal 2D CORF upon out-of-plane pressure. Reprinted from ref. 80. Copyright 2021 Wiley-VCH Verlag GmbH & Co. (c) Transition between open-shell AFM state and closed-shell quinoidal state in a ring-sharing 2D CORF induced by in-plane strain. Reprinted from ref. 84. Copyright 2021 Nature Group. | ||
Although COFs, due to their covalent bonding, tend to be structurally more rigid than MOFs, they are still more flexible and stretchable than most inorganic materials. To access different electronic states in hexagonal TAM-based 2D CORFs, it has been proposed that out-of-plane compression could help induce a crossover from the parent open-shell AFM insulating state towards a more delocalised graphene-like semimetallic state80 (see Fig. 5b). This Mott-like transition81 is favoured by the pressure-induced flattening (with respect to the plane of the CORF) of the aryl rings in each TAM centre, which leads to increased inter-radical conjugation. This behaviour is predicted to be enhanced in hexagonal 2D CORFs in which the TAMs share aryl rings. Although ring-sharing CORFs have not been yet synthesised, molecules with six ring-sharing TAMs have been reported,82 which potentially could be used as CORF MBBs. As the AFM insulating state is a correlated electronic state83 (i.e., due to strong inter-site electron–electron interactions) the ability to tune this state via external pressure implies that 2D CORFs could provide a new class of tuneable low dimensional correlated electronic material.81
An alternative means to externally induce changes in the electronic/chemical properties of TAM-based hexagonal 2D CORFs is to apply in-plane stress to strain the network. When uniaxially strained, the aryl rings in TAM-based 2D CORFs are affected in an anisotropic manner such that some rings flatten and some twist more out-of-plane.77,84,85 In hexagonal 2D CORFs this structural response can lead to pairing of electrons between neighbouring TAM centres and the transition from the open-shell multi-radical ground state to a diamagnetic closed-shell quinoidal state86 (see Fig. 5c). Low-lying open-shell radical and closed-shell quinoidal states are also found in di-radical molecules.87 Forming 2D hexagonal CORFs from TAM radical MBBs provides a means with which to reversibly control this fundamental chemical property.
Like MOFs, COFs are also porous materials, which also enhances their structural flexibility. Hexagonal 2D CORFs such as PTM-CORF possess regularly spaced arrays of radially symmetric micropores with diameters >1 nm. By joining PTMs together with triazine-based linkers, a 2D CORF (PTMR-CTF) was made with pores having both radially symmetric and elongated micropores with sizes of 1.1 nm and 1.2 nm diameter, respectively.86 Although the relatively large distance between PTM MBBs in PTMR-CTF leads to a paramagnetic electronic ground state, the ability to change micropore geometry via different chemical connectivities will be important for tailoring the future multifunctionality of 2D CORFs.
To date, only a few PTM-based MOROFs and two PTM-based 2D CORFs have been synthesised and thus there is huge potential for further exploration. For example, the use of PTM radicals functionalized with carboxylic acid groups at the para position of the phenyl rings as MBBs may be optimized by synthesizing them without chlorine atoms at the meta positions. This will reduce the steric effects allowing the carboxylate groups to participate in the formation of metallic clusters typically found in MOFs (e.g. Cu(II) paddle-wheel clusters, Fe(III) oxo clusters, etc.) and therefore, enhance the possibility to synthesize magnetic MOROFs with permanent porosity. In CORFs, their impressive electrocatalytic properties strongly suggest that a suitably optimised 2D CORF could have technological promise for fuel cell applications. Generally, the theoretically predicted electronic/structural responsiveness of TAM-based 2D CORFs to in-plane strain and out-of-plane compression shows that these materials could eventually find applications in the burgeoning fields of straintronics and biosensors. To progress towards these goals new TAM-based CORFs should aim to enhance the spin–spin coupling between radical centres through chemical design (e.g. TAM functionalisation, linker choice, aryl ring-sharing).
5. Optical applications for PTM radicals
PTMs have a number of promising optical properties associated with their doublet electronic configuration, like their emission at long wavelengths without an elongated π-system, large Stokes’ shift, and emission lifetimes of the order of tens of nanoseconds. However, PTMs also possess three principal drawbacks that have limited their use in optical applications for many years: (i) low values of luminescent quantum yield, ϕF = 0.02, especially in polar solvents, (ii) low photostability in solution giving the fluorenyl radical derivative88 and (iii) non-fluorescent characteristics in solid state despite being completely photostable. The absorption spectrum of the PTM radical presents a weak absorption in the visible region, due to a symmetry forbidden transition SOMO → SUMO (D0 → D1), and an intense absorption in the near ultraviolet due to the SOMO → LUMO+1 transition at 380 nm. PTM shows low room temperature fluorescence at 520 nm (SUMO → SOMO) but becomes more intense at low temperature (Fig. 6a and b).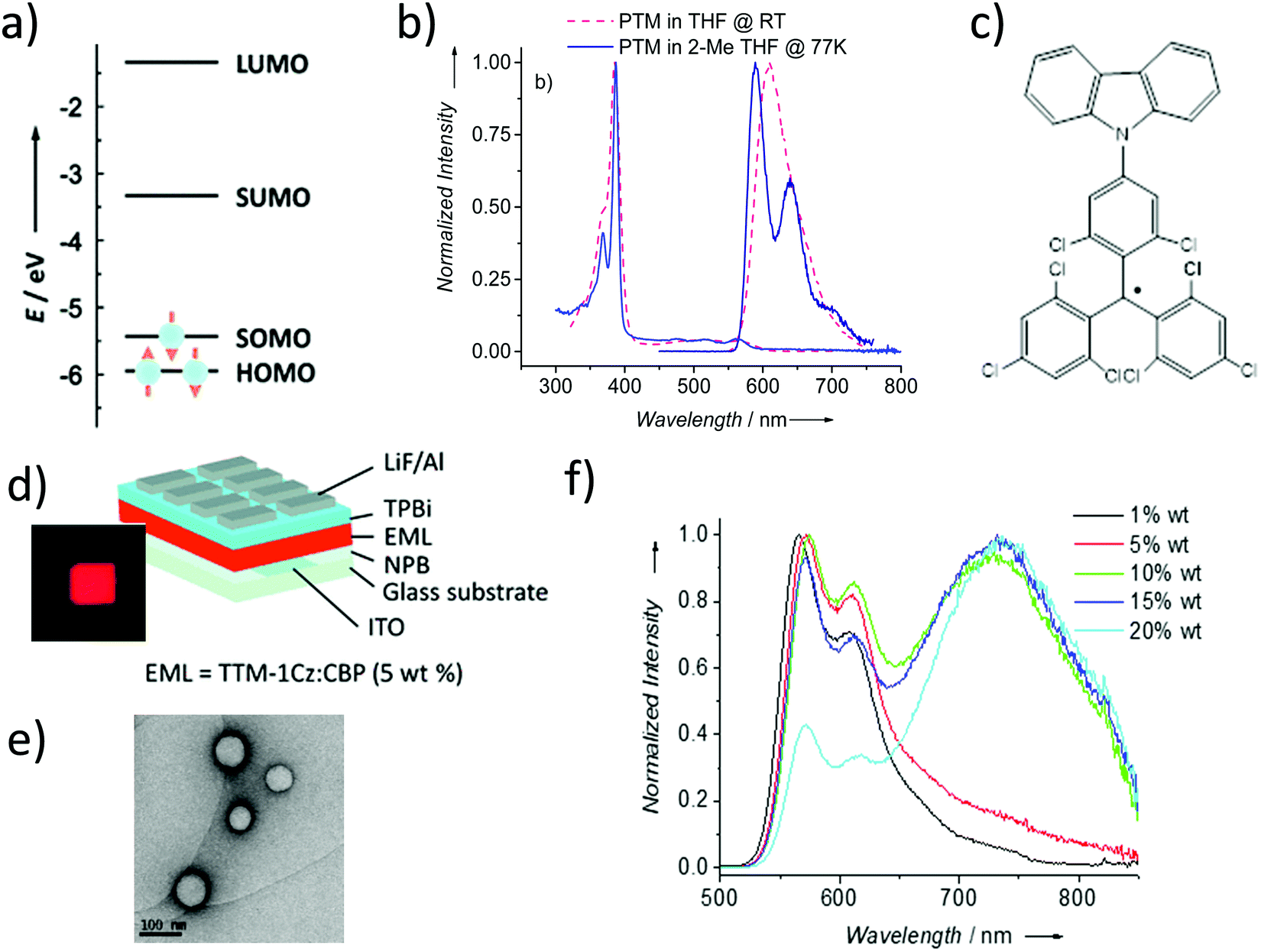 | ||
| Fig. 6 (a) Schematic representation of the frontier orbitals of PTM radicals. (b) Absorption and emission of PTM in THF at room temperature (ϕF = 0.02) and 2-methyl THF at 77 K (ϕF = 0.72). (c) Molecular structure of the TTM-1Cz. (d) Device architecture of the OLED based on TTM-1Cz. The inset shows a picture of the TTM-1Cz-based OLED operating at 7 V. Reprinted with permission from ref. 92. Copyright 2015 Wiley-VCH. (e) TEM image of PTMd-ONPs. (f) Normalized emission spectra of TTMd-ONPs. Reprint with permission from ref. 108. Copyright 2017 Royal Society of Chemistry. | ||
Recently, it has been shown that thanks to their doublet spin-configuration, organic radicals can avoid the formation of triplet excitons that limit the electroluminescence efficiency of non-radical emitters. Thus, organic radicals turned out to be a new solution to obtain new organic deep-red emitters able to exhibit internal quantum efficiencies of 100%. In this regard, in 2006, Julià and co-workers reported the PTM-carbazole derivative (TTM-1Cz) as possible molecular material for a deep-red organic light emitting diode (OLED) (Fig. 6c).89 TTM-1Cz presents an emission in cyclohexane at 628 nm with a ϕF = 0.64 due to an increase of the oscillator strength of the SOMO → SUMO transition.90,91 In 2015, Li et al. reported the development of the first doublet emitter OLED with deep red emission based on an organic radical using a blend containing TTM-1Cz (Fig. 6d).92 Recently, it has been showed that non-alternant systems with a conjugated hydrocarbon with an odd-membered ring., i.e. like TTM-1Cz, are necessary to lift the degeneracy of the lowest energy orbital excitations enhancing the oscillator strength of the emitter. In fact, the TTM radical's low emission is typical of alternant hydrocarbon systems in which conjugated atoms are divided into two groups and atoms of the same group are not directly connected. In these systems, the HOMO–SOMO and SOMO–LUMO gaps are identical. This is the reason why it is necessary to covalently attach electron-rich, non-alternant hydrocarbon groups to TTM or PTM units to form non-alternant donor–acceptor radicals to achieve stable and luminescent radicals. Radicals should be synthesized with a broken alternacy symmetry which, was not considered by the early theories on radical electronic structure. Thus, aza substitution of the Cz moiety boosts the radical emission to above 90%. OLEDs based on these molecules showed pure-red emission with more than 27% external quantum efficiency, the highest reported value for deep-red and infrared OLEDs. These insights have been beneficial for the rational design and discovery of new highly luminescent doublet emitters (Fig. 7).93–95 In fact, with the right choice of the hole-injection and hole-transporting layers, it is possible to generate the 100% of doublet excitons, achieving in this way an internal quantum efficiency of 100%.96 Finally, it is worth mentioning that the first discotic all-organic liquid crystal with the magnetic spin source placed in the core is also based on a para carbazolyl substituted dichlorotriphenyl methyl system.97
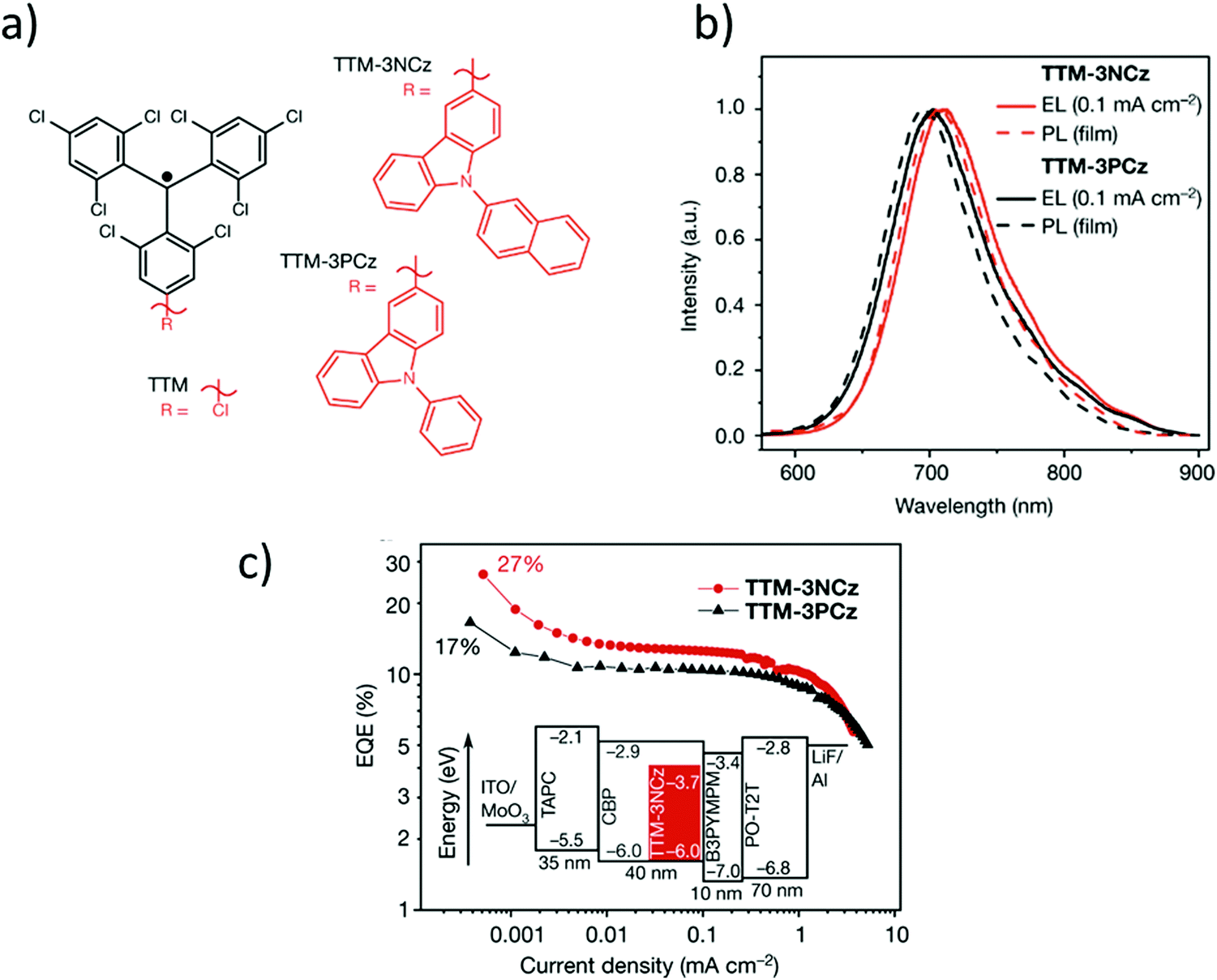 | ||
| Fig. 7 (a) Chemical structures of TTM, TTM-3NCz and TTM-3PCz; (b) electroluminescence (photoexcitation wavelength, 375 nm) spectra; (c) external quantum efficiency (EQE)–current density curves for TTM-3NCz (red) and TTM-3PCz (black) LEDs. The inset shows the TTM-3NCz device layout; the labels give the energy levels in electronvolts and the thickness of layers in nanometres. For the best TTM-3NCz LED architecture, SOMO = −3.7 eV and HOMO = −6.0 eV. Reprint with permission from ref. 95. Copyright 2018 Nature. | ||
In 2014, Nishihara et al. reported a PTM derivative in which one phenyl ring was substituted with a 3,5-dichloropyridyl one, which exhibited low emission at 585 nm in dichloromethane. The emission increased to 0.81 at 77 K and 0.26 when dispersed in a polymeric film.98 Its luminescence could also be increased from 0.02 to 0.2 by substituting the chlorine atoms in ortho by fluorine and making a pyridyl Au(I) complex.99,100 Also, cation-responsive turn-on fluorescence without heavy atoms101,102 and increased photostability has been reported for a radical containing two pyridyl groups.103
Electron-donating units quench the luminescence of PTM radicals due to the appearance of a charge transfer (CT)90 phenomena given by the HOMO → SUMO transition. This behaviour is observed in PTM-triphenylamine (PTM-TPA) derivatives, which exhibit high luminescence in the NIR region (760 nm) when dissolved in non-polar solvents (ϕF = 0.38 in cyclohexane) but when electron donating groups are added to the TPA moiety, the luminescence is quenched.104,105 In addition, it has been found that PTM radicals linked to electron-donating units (D-PTM) do not follow the Aufbau principle because the SOMO is found to lie below the HOMO. These D–A radicals have a strong emission yield (up to 54%) and high photostability. OLEDs based on them show deep-red emission with a maximal external quantum efficiency of 5.3%.106 Recently, Brédas and collaborators have reported on the impact of chemical substitution and electronic-state hybridization on the luminescence properties of D-PTM radicals.107 These results highlight that in order to understand the emissive properties of D–A radicals, the electronic hybridization of the CT states with both the ground and the low excitation states should be properly considered. This information will be very useful to design a new generation of efficient D–A radicals with high-energy emission.
Veciana and co-workers reported the luminescence properties of PTM-doped polymeric films and organic nanoparticles (PTMd-ONPs)108 (Fig. 6e). The confinement of radicals into organic rigid hosts decreases the non-radiative interactions, which affect the radicals in solid state enhancing their luminescence and photostability. ONPs exhibited uniform size distribution and high colloidal stability in water showing life times and ϕF much higher than PTMs in solution. When increasing the radical doping, a new excimeric band at the NIR region, which varied with temperature, appeared (Fig. 6f). Their water compatibility together with the emission in the first biological transparency window make these materials promising candidates for optoelectronics and also for bio-sensing and bio-imaging applications.108 The intensity of these monomer and excimeric organic radical emissions were modulated with a magnetic field (18 T at 4.2 K) for the first time.109 Further, the excited-state dynamics of such luminescent radicals doped in a host crystal has been measured using optically detected magnetic resonance (ODMR) and time-resolved emission spectroscopies corroborating the excimeric emission.110
Interestingly, PTMs also exhibit high values of hyperpolarizability opening the possibility to be explored in the field of non-linear optics.111–113 Moreover, in a recent pioneering study, PTMs have been described as the first organic radicals showing chiral emission with intrinsic Circularly Polarized Luminesce (CPL). Opposite signed CPL signs for each pair of conformers (the right and left-handed propeller-like PTM structures) have been obtained with acceptable luminescent dissymmetry factors.114,115 This study opens the foundations for the optimization of new magnetically active organic chiral emitters and the emergence of a new field in optoelectronics and spintronics, where chiral, magnetic and luminescent properties can be exploited in a single device.
The exploitation of the luminescent properties of PTM radicals in devices is still in its infancy. However, considering the previously mentioned promising results, progress in this direction will undoubtedly bring new insights in the field of organic optoelectronics.
6. PTM radicals for bio-applications
PTM radicals are appealing in bio-applications as polarizing agents in dynamic nuclear polarization (DNP), oxygen sensors or in spin labelling thanks to their high stability, chemical versatility and narrow EPR linewidth.DNP is used for the enhancement of nuclear magnetic resonance (NMR) signals both in liquids (dissolution DNP, dDNP) and solids (magic angle spinning DNP, MAS-DNP). DNP allows for the intrinsically large spin polarization of electrons to be transferred to nuclei, leading to a NMR signal enhancement of up to more than four orders of magnitude. DNP has permitted NMR studies of previously unreachable systems, such as material surfaces or low concentration biological samples.
The nature of the polarizing agent has a crucial role in the efficiency of DNP, as it determines the polarization-transfer mechanism. Supramolecular interactions take place between the radical, the glassing solvent, and the molecule undergoing polarization. In the case of dDNP, efforts have been focused on monoradicals, while biradicals have raised more interest as polarizing agents mainly for MAS-DNP experiments. In particular, the contribution of trityl and PTM radicals to DNP has been significant in solid state and in solution, using organic solvents116 and even water by synthesising suitably soluble derivatives. The most soluble trityl radicals in aqueous media are those based on the Finland trityl radical family like OX63, the benchmark radical in this field (Fig. 8a). Most of the current advances have been developed using these types of systems, especially those more soluble in aqueous media, which have been applied for the sensitivity improvement of a wide range of techniques (solid-state NMR, DNP, high field 13C DNP, EPR).117–122 Therefore, the current trend is to increasingly use DNP techniques in the study of complex biological systems both in vitro and in vivo. Especially promising is the use of DNP in nuclear magnetic resonance imaging (MRI), for example in metabolic imaging applications of 13C-labelled metabolites in vivo.123
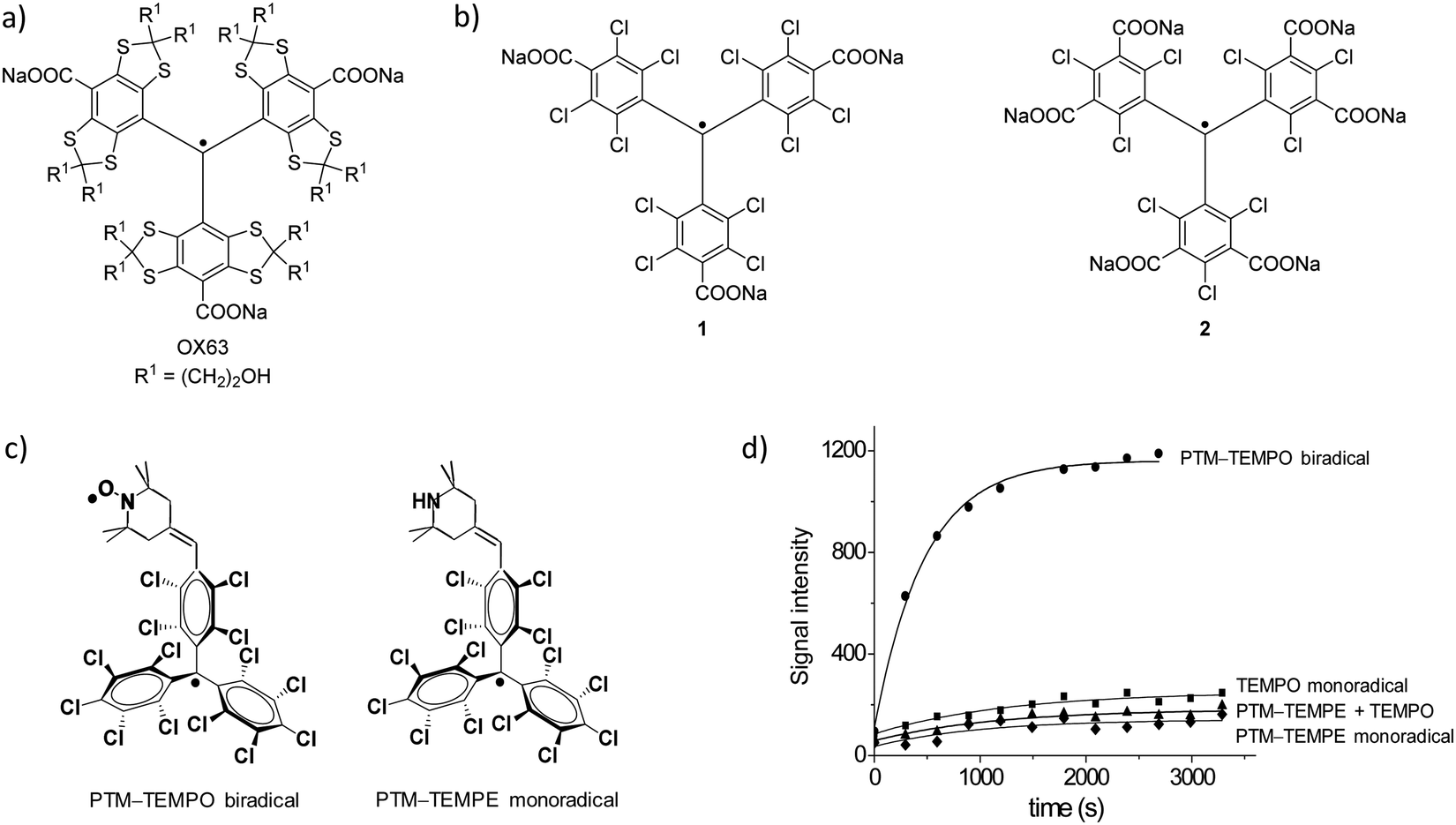 | ||
| Fig. 8 (a) OX63. (b) Two different PTM derivatives functionalized with three (1) and six (2) carboxylate groups.124 (c) Molecular structures of biradical PTM-TEMPO and monoradical PTM-TEMPE. (d) Polarization building curves (3.35 T, 1.4 K) of biradical PTM-TEMPO (90 mM) at 94.067 GHz, monoradical PTM-TEMPE (90 mM) at 94.08 GHz, 4-oxo-TEMPO (90 mM) at 94.120 GHz, and a mixture of monoradical PTM-TEMPE and 4-oxo-TEMPO both at 90 mM.116 | ||
Remarkably, PTM functionalized with three (1) or six carboxylic acids (2) and their corresponding sodium salt (Fig. 8b) have been show to be efficient DNP 13C polarizers.124–126 The polarization obtained was reported to be mediated by the Cl nuclei and it was dependent on the position of the chlorine substituents on the trityl skeleton. An interesting reported example was the use of the biradical PTM-TEMPO [i.e., (trans)-4-(2,2,6,6,-tetramethyl-1-piperidinyloxyl)-2,3,5,6-(tetrachlorophenyl)bis(pentachloro-phenyl)methyl biradical] (Fig. 8c).116 In this work, the DNP efficiency of this biradical was compared with two model systems representing the individual radical species: the monoradicals 4-oxo-TEMPO and (trans)-4-(2,2,6,6,-tetramethyl-1-piperidine)-2,3,5,6-(tetrachlorophenyl)bispentachlorophenyl)methyl radical PTM-TEMPE (Fig. 8c). A mixture of the two monoradicals at the same optimal concentration was also studied. Interestingly, it was observed that the polarization achieved with the biradical was much higher than with the individual monoradicals or their mixture, demonstrating the high efficiency of the biradical (Fig. 8d). With the PTM-TEMPO biradical almost 50% more polarization was achieved when compared to the golden standard OX63 under the same conditions.
Trityl radicals also have been found to be the most sensitive soluble EPR oximetric probes and with good stability in vivo. The EPR-based method, known as “EPR oximetry”, is becoming a fundamental tool in biology and medicine to monitor changes in the pO2. It uses oxygen-sensing spin probes whose EPR lines are broadened by molecular oxygen.127 It is even possible to perform in vivo oxygen mapping in animals,128,129 or to use two techniques at the same time such as dissolution dynamic nuclear polarization and Overhauser-enhanced magnetic resonance imaging (OMRI) for simultaneously visualizing tissue oxygen concentration and microvascular permeability.130
Trityl organic radicals can be used as spin labels to measure distances in bio-macromolecules. Using pulsed dipolar EPR spectroscopy (PDS) in conjunction with site-directed spin labelling (SDSL) techniques, these species have enabled distances measurements to be performed in DNA and proteins for structural biology studies. This approach has allowed structure and dynamics of these bio-molecules to be studied in order to better understand their biomolecular functions under native conditions.131–135
Finally, it has been shown that trityl radicals are also suitable for EPR imaging. Regarding this application, carboxy-substituted trityl radicals have proved to be highly appealing for in vivo probes. The narrow spectral widths they exhibit facilitate spectral–spatial rapid scan electron paramagnetic resonance imaging.136
7. Future innovative perspectives for PTM radicals
Even though PTM radicals have been known for a few decades, it is clear that they continue to be highly attractive species for a wide range of research fields. The unique electronic, magnetic and optical properties of PTMs, combined with their chemical and thermal stability and their amenability to functionalisation, have led to their innovative use in many systems of both fundamental and technological importance.As mentioned, the high reversibility of PTM redox processes that occur at low voltage has allowed the exploitation of these materials as active components in a variety of electrochemical switches. Particularly appealing in the future might be the synthesis of macromolecules containing a large number of PTM units to achieve high charge storage molecular systems and multistate switches. Furthermore, PTM radicals have been integrated in molecular junctions. The presence of the SOMO and SUMO energy levels has been shown to have a strong impact on the electronic transport properties. Remarkably, the interaction of the transported electrons with the unpaired spin offers great opportunities in the area of molecular spintronics.
PTMs have been employed as molecular building blocks for the fabrication of extended materials such as COFs and MOFs, which can show potential in a variety of applications in energy (i.e., fuel cells and electrocatalysis). Additionally, having 2-dimensional ordered radical networks might allow for simultaneously modifying their properties by external stimuli (e.g. moisture, pressure) with promising applications in sensing and switching devices.
Regarding the luminescent properties of PTM radicals, there is still plenty of room for their exploitation in devices bringing new insights in the field of organic optoelectronics. Of particular interest might be the unique combination of chirality, magnetism and luminescence in a single device.
Thanks to their narrow EPR linewidth, PTM and trityl radicals have also been shown to be appealing in bio-applications where EPR is employed as read-out signal. PTMs have been used as polarizing agents in dynamic nuclear polarization (DNP), as EPR oximetric probes and as spin labels. Further, trityl radicals have also proved to be highly promising as in vivo probes for EPR imaging.
Although not discussed in this review, PTM radicals are also likely to have an impact on the emerging field of quantum technologies.137,138 Organic radicals are potential candidates as molecular quantum bits,139,140 where PTMs derivatives have already shown promise.141 In the area of energy, PTM radicals can also play a key role. PTMs can act as redox mediators favouring the oxygen reduction reaction and thus, improving the capacity and potential of discharge of non-aqueous Li–O2 batteries.142 These findings open new ways to enhance the performance of batteries using organic radicals like PTM, with synthetically tuneable properties.
Considering all the above, we expect that PTM radical based systems will continue to be key in many new and established areas of pure and applied research for many years to come.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Prof. Jaume Veciana and Prof. Concepció Rovira for their continuous support along all these years. This work was funded by the Spanish Ministry of Science and Innovation with the projects GENESIS PID2019-111682RB-I00, Mol4Bio PID2019-105622RB-I00, RTI2018-095622-B-I00 and RTI2018-095460-B-100, and through the “Severo Ochoa” Programme for Centers of Excellence in R&D (FUNFUTURE CEX2019-000917-S and Grant No. SEV-2017-0706) and the Spanish Structures of Excellence María de Maeztu program (MDM-2017-0767). The authors also thank the Generalitat de Catalunya (2017-SGR-918, 2017SGR13 and 2017SGR238). It was also funded by the CERCA Program/Generalitat de Catalunya.References
- Stable Radicals. Fundamentals and Applied Aspects of Odd-Electron Compounds, ed. R. G. Hicks, John Wiley & Sons, Chichester, 2010 Search PubMed.
- R. G. Hicks, Org. Biomol. Chem., 2007, 5, 1321–1338 RSC.
- O. Armet, J. Veciana, C. Rovira, J. Riera, J. Castaner, E. Molins, J. Rius, C. Miravitlles, S. Olivella and J. Brichfeus, J. Phys. Chem., 1987, 91, 5608–5616 CrossRef CAS.
- I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303–349 RSC.
- J. Veciana, C. Rovira, N. Ventosa, M. I. Crespo and F. Palacio, J. Am. Chem. Soc., 1993, 115, 57–64 CrossRef CAS.
- D. Ruiz-Molina, J. Veciana, F. Palacio and C. Rovira, J. Org. Chem., 1997, 62(26), 9009–9017 CrossRef CAS.
- M. Souto, M. C. Gullo, H. Cui, N. Casati, F. Montisci, H. O. Jeschke, R. Valentí, I. Ratera, C. Rovira and J. Veciana, Chem. – Eur. J., 2018, 24, 5500–5505 CrossRef CAS PubMed.
- N. Crivillers, S. Furukawa, A. Minoia, A. Ver Heyen, M. Mas-Torrent, C. Sporer, M. Linares, A. Volodin, C. Van Haesendonck, M. Van Der Auweraer, R. Lazzaroni, S. De Feyter, J. Veciana and C. Rovira, J. Am. Chem. Soc., 2009, 131, 6246–6252 CrossRef CAS PubMed.
- F. Vera, M. Mas-Torrent, J. Esquena, C. Rovira, Y. Shen, T. Nakanishi and J. Veciana, Chem. Sci., 2012, 3, 1958–1962 RSC.
- C. Franco, M. Mas-Torrent, A. Caballero, A. Espinosa, P. Molina, J. Veciana and C. Rovira, Chem. – Eur. J., 2015, 21, 5504–5509 CrossRef CAS PubMed.
- A. Heckmann, C. Lambert, M. Goebel and R. Wortmann, Angew. Chem., Int. Ed., 2004, 43, 5851–5856 CrossRef CAS PubMed.
- M. Steeger, S. Griesbeck, A. Schmiedel, M. Holzapfel, I. Krummenacher, H. Braunschweig and C. Lambert, Phys. Chem. Chem. Phys., 2015, 17, 11848–11867 RSC.
- J. Guasch, L. Grisanti, V. Lloveras, J. Vidal-Gancedo, M. Souto, D. C. Morales, M. Vilaseca, C. Sissa, A. Painelli, I. Ratera, C. Rovira and J. Veciana, Angew. Chem., 2012, 124, 11186–11190 CrossRef.
- I. Ratera, C. Sporer, D. Ruiz-Molina, N. Ventosa, J. Baggerman, A. M. Brouwer, C. Rovira and J. Veciana, J. Am. Chem. Soc., 2007, 129, 6117–6129 CrossRef CAS PubMed.
- C. Sporer, I. Ratera, D. Ruiz-Molina, Y. Zhao, J. Vidal-Gancedo, K. Wurst, P. Jaitner, K. Clays, A. Persoons, C. Rovira and J. Veciana, Angew. Chem., Int. Ed., 2004, 43, 5266–5268 CrossRef CAS PubMed.
- V. Lloveras, J. Vidal-Gancedo, T. M. Figueira-Duarte, J. F. Nierengarten, J. J. Novoa, F. Mota, N. Ventosa, C. Rovira and J. Veciana, J. Am. Chem. Soc., 2011, 133, 5818–5833 CrossRef CAS PubMed.
- C. Franco, P. M. Burrezo, V. Lloveras, R. Caballero, I. Alcon, S. T. Bromley, M. Mas-Torrent, F. Langa, J. T. L. Navarrete, C. Rovira, T. J. Casado and J. Veciana, J. Am. Chem. Soc., 2017, 139, 686–692 CrossRef CAS PubMed.
- M. Mas-Torrent, N. Crivillers, V. Mugnaini, I. Ratera, C. Rovira and J. Veciana, J. Mater. Chem., 2009, 19, 1691–1695 RSC.
- M. Mas-Torrent, N. Crivillers, C. Rovira and J. Veciana, Chem. Rev., 2012, 112, 2506–2527 CrossRef CAS PubMed.
- Z. Liu, A. a. Yasseri, J. S. Lindsey and D. F. Bocian, Science, 2003, 302, 1543–1545 CrossRef CAS PubMed.
- M. Mas-Torrent, C. Rovira and J. Veciana, Adv. Mater., 2013, 25, 462–468 CrossRef CAS PubMed.
- N. Crivillers, M. Mas-Torrent, S. Perruchas, N. Roques, J. Vidal-Gancedo, J. Veciana, C. Rovira, L. Basabe-Desmonts, B. J. Ravoo, M. Crego-Calama and D. N. Reinhoudt, Angew. Chem., Int. Ed., 2007, 46, 2215–2219 CrossRef CAS PubMed.
- N. Crivillers, M. Mas-Torrent, J. Vidal-Gancedo, J. Veciana and C. Rovira, J. Am. Chem. Soc., 2008, 130, 5499–5506 CrossRef CAS PubMed.
- C. Simão, M. Mas-Torrent, N. Crivillers, V. Lloveras, J. M. Artés, P. Gorostiza, J. Veciana and C. Rovira, Nat. Chem., 2011, 3, 359–364 CrossRef PubMed.
- J. A. De Sousa, F. Bejarano, D. Gutiérrez, Y. R. Leroux, E. M. Nowik-Boltyk, T. Junghoefer, E. Giangrisostomi, R. Ovsyannikov, M. B. Casu, J. Veciana, M. Mas-Torrent, B. Fabre, C. Rovira and N. Crivillers, Chem. Sci., 2020, 11, 516–524 RSC.
- C. Simão, M. Mas-Torrent, J. Veciana and C. Rovira, Nano Lett., 2011, 11, 4382–4385 CrossRef PubMed.
- B. Fabre, Chem. Rev., 2016, 116, 4808–4849 CrossRef PubMed.
- M. Oliveros, L. González-García, V. Mugnaini, F. Yubero, N. Roques, J. Veciana, A. R. González-Elipe and C. Rovira, Langmuir, 2011, 27, 5098–5106 CrossRef CAS PubMed.
- V. Lloveras, F. Liko, J. L. Muñoz-Gómez, J. Veciana and J. Vidal-Gancedo, Chem. Mater., 2019, 31, 9400–9412 CrossRef CAS.
- A. V. Rudnev, C. Franco, N. Crivillers, G. Seber, A. Droghetti, I. Rungger, I. V. Pobelov, J. Veciana, M. Mas-Torrent and C. Rovira, Phys. Chem. Chem. Phys., 2016, 18, 27733–27737 RSC.
- G. Seber, A. V. Rudnev, A. Droghetti, I. Rungger, J. Veciana, M. Mas-Torrent, C. Rovira and N. Crivillers, Chem. – Eur. J., 2017, 23, 1415–1421 CrossRef CAS PubMed.
- I. V. Pobelov, Z. Li and T. Wandlowski, J. Am. Chem. Soc., 2008, 130, 16045–16054 CrossRef CAS PubMed.
- J. Zhang, A. M. Kuznetsov, I. G. Medvedev, Q. Chi, T. Albrecht, P. S. Jensen and J. Ulstrup, Chem. Rev., 2008, 108, 2737–2791 CrossRef CAS PubMed.
- T. Albrecht, K. Moth-Poulsen, J. B. Christensen, J. Hjelm, T. Bjørnholm and J. Ulstrup, J. Am. Chem. Soc., 2006, 128, 6574–6575 CrossRef CAS PubMed.
- V. Diez-Cabanes, A. Gómez, M. Souto, N. González-Pato, J. Cornil, J. Veciana and I. Ratera, J. Mater. Chem. C, 2019, 7, 7418–7426 RSC.
- V. Diez-Cabanes, D. C. Morales, M. Souto, M. Paradinas, F. Delchiaro, A. Painelli, C. Ocal, D. Cornil, J. Cornil, J. Veciana and I. Ratera, Adv. Mater. Technol., 2019, 4, 1–7 Search PubMed.
- A. M. Masillamani, S. Osella, A. Liscio, O. Fenwick, F. Reinders, M. Mayor, V. Palermo, J. Cornil and P. Samorì, Nanoscale, 2014, 6, 8969–8977 RSC.
- M. M. Thuo, W. F. Reus, C. A. Nijhuis, J. R. Barber, C. Kim, M. D. Schulz and G. M. Whitesides, J. Am. Chem. Soc., 2011, 133, 2962–2975 CrossRef CAS PubMed.
- R. Cohen, K. Stokbro, J. M. L. Martin and M. A. Ratner, J. Phys. Chem. C, 2007, 111, 14893–14902 CrossRef CAS.
- I. Hnid, D. Frath, F. Lafolet, X. Sun and J. C. Lacroix, J. Am. Chem. Soc., 2020, 142, 7732–7736 CrossRef CAS PubMed.
- Y. Han and C. A. Nijhuis, Chem. – Asian J., 2020, 15, 3752–3770 CrossRef CAS PubMed.
- G. Ke, C. Duan, F. Huang and X. Guo, InfoMat, 2020, 2, 92–112 CrossRef CAS.
- R. Hayakawa, M. A. Karimi, J. Wolf, T. Huhn, M. S. Zöllner, C. Herrmann and E. Scheer, Nano Lett., 2016, 16, 4960–4967 CrossRef CAS PubMed.
- R. Frisenda, R. Gaudenzi, C. Franco, M. Mas-Torrent, C. Rovira, J. Veciana, I. Alcon, S. T. Bromley, E. Burzurí and H. S. J. Van Der Zant, Nano Lett., 2015, 15, 3109–3114 CrossRef CAS PubMed.
- F. Bejarano, I. J. Olavarria-Contreras, A. Droghetti, I. Rungger, A. Rudnev, D. Gutiérrez, M. Mas-Torrent, J. Veciana, H. S. J. Van Der Zant, C. Rovira, E. Burzurĺ and N. Crivillers, J. Am. Chem. Soc., 2018, 140, 1691–1696 CrossRef CAS PubMed.
- N. Crivillers, M. Paradinas, M. M. Torrent, S. T. Bromley, C. Rovira, C. Ocal and J. Veciana, Chem. Commun., 2011, 47, 4664–4666 RSC.
- N. Crivillers, C. Munuera, M. Mas-Torrent, C. Simão, S. T. Bromley, C. Ocal, C. Rovira and J. Veciana, Adv. Mater., 2009, 21, 1177–1181 CrossRef CAS.
- M. Souto, L. Yuan, D. C. Morales, L. Jiang, I. Ratera, C. A. Nijhuis and J. Veciana, J. Am. Chem. Soc., 2017, 139, 4262–4265 CrossRef CAS PubMed.
- L. Yuan, C. Franco, N. Crivillers, M. Mas-Torrent, L. Cao, C. S. S. Sangeeth, C. Rovira, J. Veciana and C. A. Nijhuis, Nat. Commun., 2016, 7, 12066 CrossRef CAS PubMed.
- M. Souto, V. Díez-Cabanes, L. Yuan, A. R. Kyvik, I. Ratera, C. A. Nijhuis, J. Cornil and J. Veciana, Phys. Chem. Chem. Phys., 2018, 20, 25638–25647 RSC.
- R. Gaudenzi, E. Burzurí, D. Reta, I. D. P. R. Moreira, S. T. Bromley, C. Rovira, J. Veciana and H. S. J. Van Der Zant, Nano Lett., 2016, 16, 2066–2071 CrossRef CAS PubMed.
- R. Gaudenzi, J. De Bruijckere, D. Reta, I. D. P. R. Moreira, C. Rovira, J. Veciana, H. S. J. Van Der Zant and E. Burzurí, ACS Nano, 2017, 11, 5879–5883 CrossRef CAS PubMed.
- O. M. Yaghi, M. J. Kalmutzki and C. S. Diercks, Introduction to Reticular Chemistry, Wiley, 2019 Search PubMed.
- O. M. Yaghi, M. O’Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341 Search PubMed.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- O. M. Yaghi, J. Am. Chem. Soc., 2016, 138, 15507–15509 CrossRef CAS PubMed.
- L. Zhu, X. Q. Liu, H. L. Jiang and L. B. Sun, Chem. Rev., 2017, 117, 8129–8176 CrossRef CAS PubMed.
- S. Rojas, A. Arenas-Vivo and P. Horcajada, Coord. Chem. Rev., 2019, 388, 202–226 CrossRef CAS.
- L. S. Xie, G. Skorupskii and M. Dincă, Chem. Rev., 2020, 120, 8536–8580 CrossRef CAS PubMed.
- R. Medishetty, J. K. Zareba, D. Mayer, M. Samoć and R. A. Fischer, Chem. Soc. Rev., 2017, 46, 4976–5004 RSC.
- M. Souto, K. Strutyński, M. Melle-Franco and J. Rocha, Chem. – Eur. J., 2020, 26, 10912–10935 CrossRef CAS PubMed.
- G. Mínguez Espallargas and E. Coronado, Chem. Soc. Rev., 2018, 47, 533–557 RSC.
- E. Coronado and G. M. Espallargas, Chem. Soc. Rev., 2013, 42, 1525–1539 RSC.
- H. Ghasempour, K. Y. Wang, J. A. Powell, F. ZareKarizi, X. L. Lv, A. Morsali and H. C. Zhou, Coord. Chem. Rev., 2021, 426, 213542 CrossRef CAS.
- M. S. Lohse and T. Bein, Adv. Funct. Mater., 2018, 28, 1705553 CrossRef.
- D. Maspoch, N. Domingo, D. Ruiz-Molina, K. Wurst, J. Tejada, C. Rovira and J. Veciana, J. Am. Chem. Soc., 2004, 126, 730–731 CrossRef CAS PubMed.
- D. Maspoch, N. Domingo, D. Ruiz-Molina, K. Wurst, G. Vaughan, J. Tejada, C. Rovira and J. Veciana, Angew. Chem., Int. Ed., 2004, 43, 1828–1832 CrossRef CAS PubMed.
- D. Maspoch, D. Ruiz-Molina, K. Wurst, N. Domingo, M. Cavallini, F. Biscarini, J. Tejada, C. Rovira and J. Veciana, Nat. Mater., 2003, 2, 190–195 CrossRef CAS PubMed.
- D. Maspoch, N. Domingo, D. Ruiz-Molina, K. Wurst, J. M. Hernández, G. Vaughan, C. Rovira, F. Lloret, J. Tejada and J. Veciana, Chem. Commun., 2005, 5035–5037 RSC.
- N. Roques, D. Maspoch, I. Imaz, A. Datcu, J. P. Sutter, C. Rovira and J. Veciana, Chem. Commun., 2008, 3160–3162 RSC.
- A. Datcu, N. Roques, V. Jubera, I. Imaz, D. Maspoch, J. Sutter, C. Rovira and J. Veciana, Chem. – Eur. J., 2011, 17, 3644–3656 CrossRef CAS PubMed.
- A. Datcu, N. Roques, V. Jubera, D. Maspoch, X. Fontrodona, K. Wurst, I. Imaz, G. Mouchaham, J.-P. Sutter, C. Rovira and J. Veciana, Chem. – Eur. J., 2012, 18, 152–162 CrossRef CAS PubMed.
- N. Roques, D. Maspoch, F. Luis, A. Camón, K. Wurst, A. Datcu, C. Rovira, D. Ruiz-Molina and J. Veciana, J. Mater. Chem., 2008, 18, 98–108 RSC.
- S. Kimura, M. Uejima, W. Ota, T. Sato, S. Kusaka, R. Matsuda, H. Nishihara and T. Kusamoto, J. Am. Chem. Soc., 2021, 143, 4329–4338 CrossRef CAS PubMed.
- I. Alcón, D. Reta, I. de P. R. Moreira and S. T. Bromley, Chem. Sci., 2017, 8, 1027–1039 RSC.
- I. Alcón, F. Viñes, I. D. P. R. Moreira and S. T. Bromley, Nat. Commun., 2017, 8, 1957 CrossRef PubMed.
- S. Wu, M. Li, H. Phan, D. Wang, T. S. Herng, J. Ding, Z. Lu and J. Wu, Angew. Chem., Int. Ed., 2018, 57, 8007–8011 CrossRef CAS PubMed.
- Y. Yang, C. Liu, X. Xu, Z. Meng, W. Tong, Z. Ma, C. Zhou, Y. Sun and Z. Sheng, Polym. Chem., 2018, 9, 5499–5503 RSC.
- R. Santiago, I. Alcón, J. Ribas-Arino, M. Deumal, I. P. R. Moreira and S. T. Bromley, Adv. Funct. Mater., 2021, 31, 2004584 CrossRef CAS.
- N. F. Mott, Rev. Mod. Phys., 1968, 40, 677–683 CrossRef CAS.
- Z. Li, T. Y. Gopalakrishna, Y. Han, Y. Gu, L. Yuan, W. Zeng, D. Casanova and J. Wu, J. Am. Chem. Soc., 2019, 141, 16266–16270 CrossRef CAS PubMed.
- S. Thomas, H. Li and J. Bredas, Adv. Mater., 2019, 31, 1900355 CrossRef PubMed.
- I. Alcón, R. Santiago, J. Ribas-Arino, M. Deumal, I. de, P. R. Moreira and S. T. Bromley, Nat. Commun., 2021, 12, 1–9 CrossRef PubMed.
- I. Alcón and S. T. Bromley, Phys. Chem. Chem. Phys., 2018, 20, 5028–5035 RSC.
- Y. Jiang, I. Oh, S. H. Joo, O. Buyukcakir, X. Chen, S. H. Lee, M. Huang, W. K. Seong, J. H. Kim, J. U. Rohde, S. K. Kwak, J. W. Yoo and R. S. Ruoff, ACS Nano, 2019, 13, 5251–5258 CrossRef CAS PubMed.
- P. Ravat and M. Baumgarten, Phys. Chem. Chem. Phys., 2015, 17, 983–991 RSC.
- M. A. Fox, E. Gaillard and C. C. Chen, J. Am. Chem. Soc., 1987, 109, 7088–7094 CrossRef CAS.
- D. Velasco, S. Castellanos, M. López, F. López-Calahorra, E. Brillas and L. Juliá, J. Org. Chem., 2007, 72, 7523–7532 CrossRef CAS PubMed.
- L. Fajarí, R. Papoular, M. Reig, E. Brillas, J. L. Jorda, O. Vallcorba, J. Rius, D. Velasco and L. Juliá, J. Org. Chem., 2014, 79, 1771–1777 CrossRef PubMed.
- S. Castellanos, D. Velasco, F. López-Calahorra, E. Brillas and L. Julia, J. Org. Chem., 2008, 73, 3759–3767 CrossRef CAS PubMed.
- Q. Peng, A. Obolda, M. Zhang and F. Li, Angew. Chem., Int. Ed., 2015, 54, 7091–7095 CrossRef CAS PubMed.
- A. Abdurahman, T. J. H. Hele, Q. Gu, J. Zhang, Q. Peng, M. Zhang, R. H. Friend, F. Li and E. W. Evans, Nat. Mater., 2020, 19, 1224–1229 CrossRef CAS PubMed.
- X. Ai, Y. Chen, Y. Feng and F. Li, Angew. Chem., Int. Ed., 2018, 57, 2869–2873 CrossRef CAS PubMed.
- X. Ai, E. W. Evans, S. Dong, A. J. Gillett, H. Guo, Y. Chen, T. J. H. Hele, R. H. Friend and F. Li, Nature, 2018, 563, 536–540 CrossRef CAS PubMed.
- A. Obolda, X. Ai, M. Zhang and F. Li, ACS Appl. Mater. Interfaces, 2016, 8, 35472–35478 CrossRef CAS PubMed.
- S. Castellanos, F. López-Calahorra, E. Brillas, L. Julia and D. Velasco, Angew. Chem., Int. Ed., 2009, 48, 6516–6519 CrossRef CAS PubMed.
- Y. Hattori, T. Kusamoto and H. Nishihara, Angew. Chem., Int. Ed., 2014, 53, 11845–11848 CrossRef CAS PubMed.
- Y. Hattori, T. Kusamoto and H. Nishihara, Angew. Chem., Int. Ed., 2015, 54, 3731–3734 CrossRef CAS PubMed.
- Y. Hattori, T. Kusamoto, T. Sato and H. Nishihara, Chem. Commun., 2016, 52, 13393–13396 RSC.
- Y. Hattori, S. Kimura, T. Kusamoto, H. Maeda and H. Nishihara, Chem. Commun., 2018, 54, 615–618 RSC.
- T. Kusamoto, S. Kimura, Y. Ogino, C. Ohde and H. Nishihara, Chem. – Eur. J., 2016, 22, 17725–17733 CrossRef CAS PubMed.
- S. Kimura, A. Tanushi, T. Kusamoto, S. Kochi, T. Sato and H. Nishihara, Chem. Sci., 2018, 9, 1996–2007 RSC.
- A. Heckmann and C. Lambert, J. Am. Chem. Soc., 2007, 129, 5515–5527 CrossRef CAS PubMed.
- Y. Hattori, E. Michail, A. Schmiedel, M. Moos, M. Holzapfel, I. Krummenacher, H. Braunschweig, U. Müller, J. Pflaum and C. Lambert, Chem. – Eur. J., 2019, 25, 15463–15471 CrossRef CAS PubMed.
- H. Guo, Q. Peng, X. K. Chen, Q. Gu, S. Dong, E. W. Evans, A. J. Gillett, X. Ai, M. Zhang, D. Credgington, V. Coropceanu, R. H. Friend, J. L. Brédas and F. Li, Nat. Mater., 2019, 18, 977–984 CrossRef CAS PubMed.
- E. Cho, V. Coropceanu and J. L. Brédas, J. Am. Chem. Soc., 2020, 142, 17782–17786 CrossRef CAS PubMed.
- D. Blasi, D. M. Nikolaidou, F. Terenziani, I. Ratera and J. Veciana, Phys. Chem. Chem. Phys., 2017, 19, 9313–9319 RSC.
- S. Kimura, T. Kusamoto, S. Kimura, K. Kato, Y. Teki and H. Nishihara, Angew. Chem., Int. Ed., 2018, 57, 12711–12715 CrossRef CAS PubMed.
- K. Kato, S. Kimura, T. Kusamoto, H. Nishihara and Y. Teki, Angew. Chem., Int. Ed., 2019, 58, 2606–2611 CrossRef CAS PubMed.
- I. Ratera, S. Marcen, S. Montant, D. Ruiz–Molina, C. Rovira, J. Veciana, J.-F. Létard and E. Freysz, Chem. Phys. Lett., 2002, 363, 245–251 CrossRef CAS.
- I. Ratera, D. Ruiz-Molina, C. Sporer, S. Marcen, S. Montant, J.-F. Létard, E. Freysz, C. Rovira and J. Veciana, Polyhedron, 2003, 22, 1851–1856 CrossRef CAS.
- M. Souto, J. Calbo, I. Ratera, E. Ortí and J. Veciana, Chem. – Eur. J., 2017, 23, 11067–11075 CrossRef CAS PubMed.
- P. Mayorga-Burrezo, V. G. Jiménez, D. Blasi, T. Parella, I. Ratera, A. G. Campaña and J. Veciana, Chem. – Eur. J., 2020, 26, 3776–3781 CrossRef CAS PubMed.
- P. Mayorga Burrezo, V. G. Jiménez, D. Blasi, I. Ratera, A. G. Campaña and J. Veciana, Angew. Chem., Int. Ed., 2019, 58, 16282–16288 CrossRef CAS PubMed.
- J.-L. Muñoz-Gómez, I. Marín-Montesinos, V. Lloveras, M. Pons, J. Vidal-Gancedo and J. Veciana, Org. Lett., 2014, 16, 5402–5405 CrossRef PubMed.
- V. K. Michaelis, A. A. Smith, B. Corzilius, O. Haze, T. M. Swager and R. G. Griffin, J. Am. Chem. Soc., 2013, 135, 2935–2938 CrossRef CAS PubMed.
- L. Lumata, Z. Kovacs, A. D. Sherry, C. Malloy, S. Hill, J. Van Tol, L. Yu, L. Song and M. E. Merritt, Phys. Chem. Chem. Phys., 2013, 15, 9800–9807 RSC.
- S. Macholl, H. Jóhannesson and J. H. Ardenkjaer-Larsen, Phys. Chem. Chem. Phys., 2010, 12, 5804–5817 RSC.
- G. Mathies, M. A. Caporini, V. K. Michaelis, Y. Liu, K.-N. Hu, D. Mance, J. L. Zweier, M. Rosay, M. Baldus and R. G. Griffin, Angew. Chem., 2015, 127, 11936–11940 CrossRef.
- A. S. Lilly Thankamony, J. J. Wittmann, M. Kaushik and B. Corzilius, Prog. Nucl. Magn. Reson. Spectrosc., 2017, 102–103, 120–195 CrossRef CAS PubMed.
- P. Demay-Drouhard, H. Y. V. Ching, C. Decroos, R. Guillot, Y. Li, L. C. Tabares, C. Policar, H. C. Bertrand and S. Un, Phys. Chem. Chem. Phys., 2020, 22, 20792–20800 RSC.
- E. M. Serrao and K. M. Brindle, Porto Biomed. J., 2017, 2, 71–75 CrossRef PubMed.
- C. Gabellieri, V. Mugnaini, J. C. Paniagua, N. Roques, M. Oliveros, M. Feliz, J. Veciana and M. Pons, Angew. Chem., 2010, 122, 3432–3434 CrossRef.
- J. C. Paniagua, V. Mugnaini, C. Gabellieri, M. Feliz, N. Roques, J. Veciana and M. Pons, Phys. Chem. Chem. Phys., 2010, 12, 5824–5829 RSC.
- D. Banerjee, J. C. Paniagua, V. Mugnaini, J. Veciana, A. Feintuch, M. Pons and D. Goldfarb, Phys. Chem. Chem. Phys., 2011, 13, 18626–18637 RSC.
- A. Bratasz, A. C. Kulkarni and P. Kuppusamy, Biophys. J., 2007, 92, 2918–2925 CrossRef CAS.
- V. V. Khramtsov, A. A. Bobko, M. Tseytlin and B. Driesschaert, Anal. Chem., 2017, 89, 4758–4771 CrossRef CAS.
- U. Sanzhaeva, M. Poncelet, O. Tseytlin, M. Tseytlin, M. Gencheva, T. D. Eubank, V. V. Khramtsov and B. Driesschaert, J. Org. Chem., 2020, 85, 10388–10398 CrossRef CAS PubMed.
- S. Matsumoto, H. Yasui, S. Batra, Y. Kinoshita, M. Bernardo, J. P. Munasinghe, H. Utsumi, R. Choudhuri, N. Devasahayam, S. Subramanian, J. B. Mitchell and M. C. Krishna, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17898–17903 CrossRef CAS PubMed.
- J. J. Jassoy, A. Berndhäuser, F. Duthie, S. P. Kühn, G. Hagelueken and O. Schiemann, Angew. Chem., 2017, 129, 183–187 CrossRef.
- N. Fleck, C. A. Heubach, T. Hett, F. R. Haege, P. P. Bawol, H. Baltruschat and O. Schiemann, Angew. Chem., Int. Ed., 2020, 59, 9767–9772 CrossRef CAS PubMed.
- Y. Yang, B. Bin Pan, X. Tan, F. Yang, Y. Liu, X. C. Su and D. Goldfarb, J. Phys. Chem. Lett., 2020, 11, 1141–1147 CrossRef CAS PubMed.
- N. Wili, H. Hintz, A. Vanas, A. Godt and G. Jeschke, Magn. Reson., 2020, 1, 75–87 CrossRef.
- V. M. Tormyshev, A. S. Chubarov, O. A. Krumkacheva, D. V. Trukhin, O. Y. Rogozhnikova, A. S. Spitsyna, A. A. Kuzhelev, V. V. Koval, M. V. Fedin, T. S. Godovikova, M. K. Bowman and E. G. Bagryanskaya, Chem. – Eur. J., 2020, 26, 2705–2712 CrossRef CAS PubMed.
- H. B. Elajaili, J. R. Biller, M. Tseitlin, I. Dhimitruka, V. V. Khramtsov, S. S. Eaton and G. R. Eaton, Magn. Reson. Chem., 2014, 53, 280–284 CrossRef PubMed.
- A. Gaita-Ariño, F. Luis, S. Hill and E. Coronado, Nat. Chem., 2019, 11, 301–309 CrossRef PubMed.
- M. Atzori and R. Sessoli, J. Am. Chem. Soc., 2019, 141, 11339–11352 CrossRef CAS PubMed.
- F. Ciccullo, A. Calzolari, K. Bader, P. Neugebauer, N. M. Gallagher, A. Rajca, J. Van Slageren and M. B. Casu, ACS Appl. Mater. Interfaces, 2019, 11, 1571–1578 CrossRef CAS PubMed.
- Y. Wu, J. Zhou, J. N. Nelson, R. M. Young, M. D. Krzyaniak and M. R. Wasielewski, J. Am. Chem. Soc., 2018, 140, 13011–13021 CrossRef CAS PubMed.
- Y. Z. Dai, B. W. Dong, Y. Kao, Z. Y. Wang, H. I. Un, Z. Liu, Z. J. Lin, L. Li, F. B. Xie, Y. Lu, M. X. Xu, T. Lei, Y. J. Sun, J. Y. Wang, S. Gao, S. Da Jiang and J. Pei, ChemPhysChem, 2018, 19, 2972–2977 CrossRef CAS PubMed.
- A. Y. Tesio, D. Blasi, M. Olivares-Marín, I. Ratera, D. Tonti and J. Veciana, Chem. Commun., 2015, 51, 17623–17626 RSC.
| This journal is © The Royal Society of Chemistry 2021 |