
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A review of functional linear carbon chains (oligoynes, polyynes, cumulenes) and their applications as molecular wires in molecular electronics and optoelectronics†
Martin R.
Bryce

Department of Chemistry, Durham University, Stockton Road, Durham DH1 3LE, UK. E-mail: m.r.bryce@durham.ac.uk
First published on 30th April 2021
Abstract
Linear, sp-hybridised, 1-dimensional (1D) all-carbon wires are conceptually the simplest π-conjugated organic molecules. Their fundamental experimental and theoretical properties and their materials chemistry applications are under-developed compared to many other π-conjugated systems. The emphasis of this review is on the emerging applications of functional oligoynes and polyynes in molecular electronics and optoelectronics. The molecules considered herein contain at least two contiguous acetylene units (that is, a conjugated diacetylene or butadiynyl fragment). Relevant examples of cumulenes will also be included. Oligoynes/polyynes/cumulenes represent unique carbon nanostructures, among the so-called “synthetic carbon allotropes”. The molecules are structurally versatile because the extent of conjugation and the electronic and photophysical properties can be tailored by judicious selection of end-groups, the number of alkyne units, and the option to incorporate metal atoms into the backbone. Their applications will be considered as molecular wires in electrode|(single)-molecule|electrode architectures, and as active components for non-linear optics, electrochromism, redox-activity, organic light emitting devices, photoinduced electron transfer systems, liquid crystals, bioimaging and biosensing, with emphasis on the role played by the special longitudinal properties of the carbon-wire units. Four to six contiguous triple bonds is generally the length limit of oligoynes for practical applications, due to the synthetic challenges and inherent instability of longer homologues under ambient conditions. The review will also outline appealing future directions for next-generation linear, sp-hybridised, all-carbon molecular wires with new and enhanced properties.
 Martin R. Bryce | Martin Bryce received a BSc from Wolverhampton Polytechnic and a DPhil from the University of York, UK. After postdoctoral work at the University of British Columbia, Vancouver, with Prof. Larry Weiler, and at Bristol University with Prof. Roger Alder, Martin moved to Durham University. Since 1995 he has been a full professor of chemistry at Durham. He has held visiting scientist positions at the University of California, Santa Barbara, the University of Copenhagen and the University of Florida, Gainesville. Martin's research covers molecular design, synthesis and properties of optoelectronic materials (small molecules and polymers), molecular electronics and nano-scale chemistry. |
1. Introduction
In the past decades, π-conjugated semiconducting organic materials have developed rapidly in both experimental and theoretical aspects. They have been widely exploited by the academic and industrial communities in fields such as light-emitting diodes, field-effect transistors, solar cells, photodetectors, bioimaging, sensors, logic circuits, memory devices, batteries, and thermoelectric devices. The materials can be small molecules, oligomers and (co)polymers, typically comprising aromatic or heteroaromatic units (phenyl, fluorene, pyrene, thiophene, carbazole, porphyrin, triazine and others) with varied extent of conjugation and tailored frontier molecular orbital energy distribution. Alkyne, alkene, alkane or other linkers are sometimes incorporated into the molecular backbones to spacially separate the (hetero)aromatic units and/or provide additional conformational flexibility.1–8Conceptually, the simplest π-conjugated organic molecules are linear, sp-hybridised, 1-dimensional (1D) all-carbon wires. A model all-carbon wire can be constructed with two distinct bonding patterns (Fig. 1). These are: (i) alternating single (longer) and triple (shorter) carbon–carbon bonds to give oligoynes, polyynes and ultimately carbyne (for an infinite chain length). (ii) Consecutive, equalised carbon–carbon double bonds to give cumulenes. Generally, experimental and theoretical data show that the oligo/polyynic structure is more stable than the corresponding cumulenic structure. This was initially predicted by Karpfen in 1979 for carbyne using ab initio Hartree–Fock (HF) theory.9 A Peierls distortion (dimerisation) favours the electronic structure of polyyne with bond length alternation, which leads to opening of a band gap and semiconducting behaviour. In contrast, an infinitely-long, symmetric cumulene would have a metallic band stucture.10 Because of the rather small energy difference between cumulene and polyyne it was suggested that both structures could coexist in appropriate experimental conditions.11 A recent theoretical study by Solomon et al. demonstrates that polyynes, odd-[n]cumulenes, and even-[n]cumulenes exhibit distinctively different transmission profiles under axial torsion.12
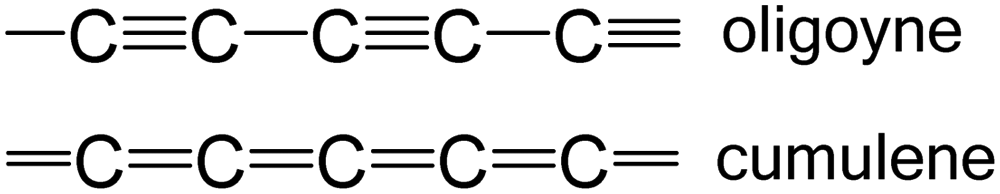 | ||
| Fig. 1 The two possible structures of an sp carbon-atom wire. | ||
The synthesis and fundamental materials chemistry applications of these 1D carbon chains are under-developed compared to many π-conjugated systems, including polyacetylene (sp2 hybridised)13,14 and polydiacetylene (sp/sp2 hybridised) counterparts.15–17 This discrepancy is primarily because of the considerable challenges encountered in the synthesis and purification of extended sp chains: the stability of these molecules rapidly decreases with chain elongation beyond only a few repeating units. Nonetheless, in recent years laboratory-scale synthetic routes to end-capped oligoynes and cumulenes have been developed and these advances have led to the increased use of sp carbon chains as building blocks in materials chemistry. Oligoynes/polyynes/cumulenes represent unique carbon nanostructures, among the so-called “synthetic carbon allotropes” which include sp2 hybridised fullerenes, carbon nanotubes, nanoonions, nanohorns and graphene.18–21
Before progressing further, it should be noted that the literature terminology ‘oligo’ or ‘poly’ for sp-hybridised carbon chains of various lengths has been rather arbitrary and inconsistent. The molecules considered in the present review contain at least two contiguous acetylene units (that is, a conjugated diacetylene or butadiynyl fragment). They will be referred to as oligoynes, except for a few cases where long polyynes have been observed experimentally (sometimes as ill-defined compounds with random lengths) or have been studied theoretically. Based on standard oligomer/polymer nomenclature for π-conjugated molecules2,3,22 the prefix ‘oligo’ is more appropriate than ‘poly’ for all the end-capped sp carbon chains that have been purified and well-characterised.23 Monoynes will be included in this review only as model compounds for oligoyne analogues.
Oligoynes are naturally-occurring compounds. They have been detected in interstellar space using Fourier-transform microwave spectroscopy.24–26 They have been isolated from bacteria, plants, fungi and marine organisms.27,28 However, these aspects of oligoyne chemistry are outside the scope of the present review. Also, the details of laboratory synthetic routes to oligo/polyynes will not be considered; key synthetic contributions are highlighted in Section 2. General reviews on linear carbon chains have been published.29–33 The emphasis of the present review is on the emerging applications of functional oligoynes and polyynes in molecular electronics and optoelectronics. Relevant examples of cumulenes will also be included. This emphasis gives the present work a different flavour compared to previous reviews on sp carbon chains. The author hopes that researchers will be stimulated to engage in the challenges and opportunities that these fascinating compounds offer in materials chemistry.
2. Highlights of oligoyne synthesis
Authoritative reviews on oligoyne synthesis are recommended.34–37 The following brief historical perspective is informative. In 1869 Glaser reported that the copper derivative of phenylacetylene dimerised in the presence of oxygen to give diphenylbutadiyne.38 This protocol of metal-mediated oxidative couplings of terminal acetylenes is still widely used today in oligoyne synthesis. In 1885 Baeyer published an article entitled “Ueber Polyacetylenverbindungen” [“About polyacetylene compounds”].39 The article emphasises “der Explosivität der Acetylenverbindungen” [“the explosiveness of the acetylene compounds”]. For example, “silver diacetylene is extraordinarily explosive” and “diiododiacetylene explodes with great violence producing a red flash.” Nonetheless, a C and I analysis consistent with the formula I–(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C)2–I was reported! Baeyer states that diacetylene monocarboxylic acid “could not be obtained analytically pure because of the high temperature and much light of the summer months.” However, a compound was obtained “which without doubt is tetraacetylene dicarboxylic acid,” i.e., HO2C–(CC)4–CO2H. Also “an acid crystallises into beautiful needles which are extraordinarily explosive.” With this inauguration it is not surprising that oligoynes received scant attention in the decades following Baeyer's seminal studies! CAUTION! There are more recent reports of the explosive properties of similar oligoyne compounds and their intermediates.40–44
C)2–I was reported! Baeyer states that diacetylene monocarboxylic acid “could not be obtained analytically pure because of the high temperature and much light of the summer months.” However, a compound was obtained “which without doubt is tetraacetylene dicarboxylic acid,” i.e., HO2C–(CC)4–CO2H. Also “an acid crystallises into beautiful needles which are extraordinarily explosive.” With this inauguration it is not surprising that oligoynes received scant attention in the decades following Baeyer's seminal studies! CAUTION! There are more recent reports of the explosive properties of similar oligoyne compounds and their intermediates.40–44
The synthesis of methyl end-capped oligoynes Me–(CC)n–Me (n = 2–6)45 and tBu analogues (n = 2–7)46 by oxidative coupling of acetylenic units were reported in 1951 and 1953, respectively. Incremental red-shifts were seen in the fine-structured UV-visible absorption bands with increasing chain length. A correct C, H elemental analysis was reported for the first isolated heptayne tBu–(CC)7–tBu.47 These pioneering studies gave evidence that bulky end-groups could stabilise longer oligoynes. In 1972 Walton and coworkers reported a series of bis(triethylsilyl) end-capped oligoynes [Et3Si–(CC)n–SiEt3] which were desilylated to give either the mono- or bis-deprotected oligoyne, culminating in the parent dodecayne [H–(CC)12–H] which was characterised by its UV-vis absorption spectrum in methanol.48 Cataldo reported that mixtures of oligoynes were obtained through vapourisation of graphite by an electric arc: separation was achieved by HPLC, and the hydrogen-terminated hexayne H–(CC)6–H was identified.49 Casari et al. recently reported the detection of hydrogen-terminated oligoynes as long as H–(CC)11–H via pulsed laser ablation of a graphite target in acetonitrile or alcohol solvents.50
For the controlled laboratory synthesis of longer oligoynes attention turned to attaching very bulky aryl end-groups to prevent intermolecular reactions. Hirsch et al. characterised a (CC)10 oligoyne with dendron-substituted aryl end-groups.51 Cox et al. reported an unstable (CC)12 oligoyne with 3,5-bis(trifluoromethyl)phenyl end-groups (UV-vis absorption and mass spectrometric evidence).52 Recently, Anderson and coworkers reported a (CC)8 oligoyne with tris(t-butylphenyl)methyl end-groups which was synthesised by photochemically unmasking alkyne equivalents in the backbone.53 The 4-(3,6-di-tert-butyl-N-carbazolyl)phenyl end-group was shown to stabilise terminal oligoynes, Ar–(CC)n–H (up to n = 4), in solution and in the solid state.54
The most spectacular achievements in the syntheses of isolable, monodisperse oligoynes have come from independent work in the Gladysz55–57 and Tykwinski groups.58–60 Gladysz et al. focussed on bulky organometallic end-groups, [highest homologue, (CC)14] 1 (Fig. 2).57 More recently, related longitudinal arrays based on –L2Pt–(CC)4– repeat units, up to the tetraplatinum homologue, have been synthesised.61 Tykwinski et al. employed tri(isopropyl)silyl (TIPS), adamantyl and “super-trityl” end-groups [highest homologue, (CC)24] 2.60 The Fritsch–Buttenberg–Wiechell rearrangement was used to avoid the need to synthesise long unstable terminal oligoyne intermediates.62 In all the above cases, the oligoyne chains are terminated by single C–C or C–metal bonds. An interesting structural variation has recently been reported by Hill and Manzano: namely oligoyne complexes with terminal C–tungsten triple bonds, e.g.3.63
 | ||
| Fig. 2 A selection of stable long oligoyne structures. | ||
The synthetic achievements highlighted above have given unprecedented opportunities to explore the effect of increasing oligoyne length on the molecules’ spectroscopic properties in solution, and on the bond length alternation and geometry in the solid state, allowing extrapolation to the properties of carbyne. UV-visible and Raman spectroscopy are commonly used techniques, although a complicating factor is that some end-capping units can participate in intramolecular charge transfer, with an obvious larger effect in shorter wires. A combined experimental and theoretical study of oligoynes terminated with various aromatic groups (phenyl, biphenyl, naphthalene, coronene, etc.) concluded that the π-conjugated, sp2-hybridised, end-groups communicate with the sp backbone and influence the bond length alternation towards a more cumulene-like structure.64 Experimental and theoretical studies have consistently shown that with increasing chain length there is a decrease in the optical energy band gap.65–67 It was observed that based on the convergence limit of the UV-vis absorption spectra the properties of Tykwinski's longer oligoynes 2 tend towards that of polyynes.60 A recent combined experimental and computational study has reassessed the estimates for both the optical and fundamental gap of carbyne to below 1.6 eV, which is significantly lower than previously suggested values.68 An interesting feature of single-crystal X-ray crystallographic studies of oligoynes with various end-groups is that the backbones often have a nonlinear curved geometry.69,70
It is noteworthy that the selection of oligoynes shown in Fig. 2 all have an even number of acetylene units and the same terminal group at both ends. Unsymmetrical derivatives, i.e. those with two different terminal groups, or an odd number of acetylene units, tend to be more challenging to synthesise, as they typically require cross-coupling reactions of two different alkyne intermediates as some stage on route to the final oligoyne.
Intramolecular or supramolecular encapsulation has proved to be a successful strategy for stabilising longer homologues by, in effect, insulating the oligoyne and preventing intermolecular interactions. Examples include the (CC)8 derivative 4 with a very long alkylene bridge which can wrap around the oligoyne,71 and the (CC)10 rotaxane 5.72 The environment around the oligoyne chain in 5 was further elaborated by metal complexation to the phenanthroline unit to give the intricate structure shown in Fig. 2. However, all synthetic studies on lengthening oligoyne chains invariably reach limits of stability. There is, however, convincing evidence from a combination of transmission electron microscopy (TEM), X-ray diffraction and (near-field) resonance Raman spectroscopy that long polyyne molecules can be synthesised inside double-walled carbon nanotubes (DWCNTs) by very high temperature and high vacuum annealing.73 These polyynes can be extracted together with the inner tubes from the hosting outer tube of the DWCNTs.74 The CNTs act as a cage to protect the polyyne chains.
This review will now focus on examples of oligoynes in molecular electronic devices, both at the single-molecule level and as multi-molecule arrays assembled between electrodes. This topic has progressed rapidly in recent years due to highly interdisciplinary collaborations involving synthesis, physical chemistry, device engineering and computational studies.
3. Oligoynes as molecular wires in electronic devices
3.1 General considerations
Understanding and controlling charge transport through molecules that are assembled between two electrodes is essential for the development of molecular electronic devices.75–77 Although practical applications are more likely to involve large-area addressable arrays of molecules,78,79 studies on single-molecule junctions have received great attention as a means to establish well-defined structure–property relationships in electrode|single-molecule|electrode architectures, without complications associated with cooperative effects between molecules.80–82 These studies are important for the fundamental development of topics such as sensors based on detection of individual binding events,83,84 logic operations and information storage,85 and thermoelectric devices in the quest for new energy-saving technologies.86The very bulky end-groups that are required to stabilise longer oligoynes (Section 2 and Fig. 2) are obviously not suitable for binding molecules to electrodes. Therefore, shorter oligoynes with less bulky, more polar end-groups have been synthesised for this purpose. Several experimental studies have focussed on transport properties of oligoynes in single-molecule junctions, using scanning tunnelling microscopy break junction (STM-BJ) and mechanically controlled break junction (MCBJ) techniques. The STM-BJ approach comprises the repeated formation and breaking of a vertical single-molecule junction between an atomically-sharp gold STM tip and a flat Au(111) substrate, and the simultaneous monitoring of the current (iT) or conductance G = iT/Vbias at constant bias voltage Vbias.87 The MCBJ technique relies on the formation and rupture of molecular junctions between horizontally-suspended gold wires with in situ electrical characterisation.88
At this point the significance of the conductance data that are discussed below should be briefly explained. Charge transport through a molecular junction is a complex process. Experimentally it is not possible to precisely control, or to know, the atomic and molecular features of the junction. Consequently, there is significant measurement-to-measurement variability between experiments. In general, this can arise as a result of fluctuations in the conformation of the molecular backbone, the anchor-electrode contact geometry and the nanoscale geometry of the electrode surface.89 To account for this variability, and to more accurately assign the conductance to a single molecule assembled in the junction, many hundreds or thousands of individual measurements are analysed by compiling them into a conductance histogram to show a distribution of all the measured conductance values. The quoted conductance values refer to the peak values obtained from these histograms, and they are reported in units of G0. G0 is the conductance quantum unit and is defined as G0 = 2e2/h = 77.5 μS, (e = charge; h = Planck's constant). G0 is the conductance of a single gold atom contact, and it corresponds to 100% transmission of electrons through that point contact from one electrode to the other. Therefore, the conductance of a molecule given in units of G0 defines its conductance in comparison with a metal atom. The ability of different backbones to transport charge can be evaluated by comparing how their conductance decays with increasing oligomer length. Backbones that are strongly conjugated and effective at transporting charge have a shallow conductance decay and, consequently, a low attenuation constant, termed the β value.
Experimental measurements of transport properties of molecular junctions are often carried out hand-in-hand with theoretical calculations. Authoritative reviews on the theoretical approaches used in single-molecule electronics are recommended.75,90,91 In particular, van Ruitenbeek and coworkers have recently critically compared theoretical and experimental data for several molecular systems.91 The authors noted that density functional theory (DFT) cannot reliably reproduce essential features such as the positions of the highest occupied and lowest unoccupied molecular orbitals (HOMOs and LUMOs, respectively) relative to the Fermi energy (EF) of the electrodes. This deficiency is exacerbated by the inherent variability in the different experimental parameters involved in the measurements, as noted above. Consequently, quantitative agreement between theory and experiment is often not achieved. Indeed, Lambert and coworkers have used oligoynes as a model system to illustrate the wide range of predictions of the HOMO–LUMO gap obtained from different theoretical approaches.67,90
3.2 Oligoynes in metal|molecule|metal junctions
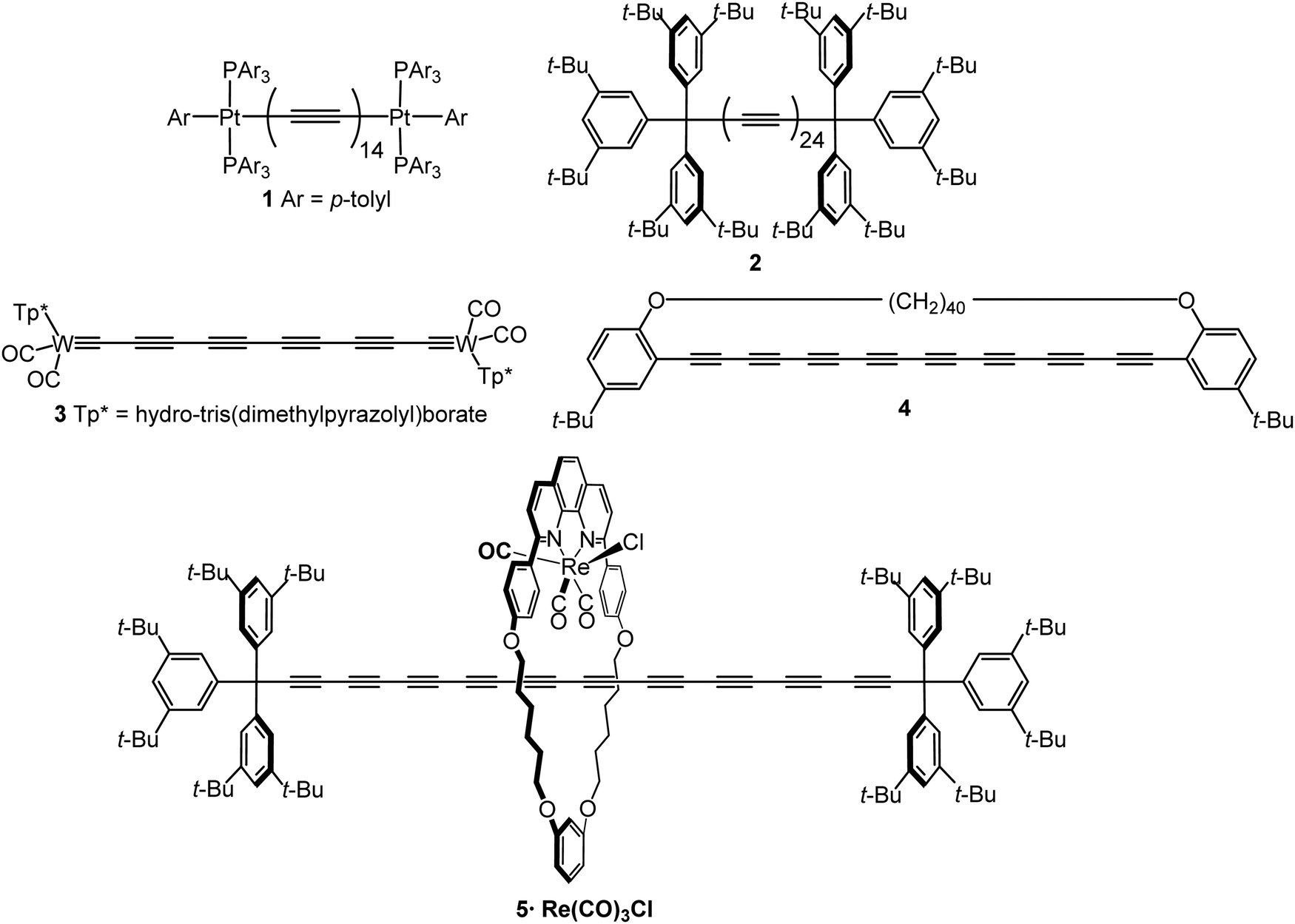
An sp-carbon wire can be regarded as the ultimate basic component of a molecular device. More than 20 years ago theoretical calculations by Lang and Avouris predicted remarkable conductance properties of carbon atomic wires (3–9 carbon atoms in length) directly linking a pair of metal electrodes.92,93 Oligoynes are well-suited as components for molecular junction studies for the following reasons. (i) The length of the molecules [ca. 1.0–3.0 nm (including anchor groups)] is a good match for the distance between the two electrodes in STM-BJ and MCBJ experiments. (ii) Oligoynes are rigid and length-persistent molecules; therefore, this key junction parameter is well-defined, and fluctuations due to conformational changes of the assembled molecule within the junction are minimised. (iii) The single-molecule techniques have the benefit of excluding any molecular aggregation and unwanted cross-linking between the oligoyne units in the junctions. The big challenge has been to design and synthesise stable oligoynes with end-groups that will anchor between the metal electrodes. The key findings of a selection of studies will now be presented.The first experimental report of oligoynes in single-molecule junctions between gold electrodes concerned the stable pyridyl end-capped series 6 (Fig. 3).94 These molecules and analogues 7–10 with different anchoring groups were subsequently studied in detail in Wandlowski's laboratory using both MCBJ and STM-BJ techniques.95,96 The different series 6–10 comprised mono-, di- and tetraynes. The single-molecule conductance data for the BT-end-capped series 9 are shown in Fig. 4A and B.95 The length dependence of the conductance values for 6–10 is shown in Fig. 4C.96 Data for analogues with –C6H4–SH and –C6H4–NH2 end-groups (n = 1 and 2) are included in Fig. 4C for comparison: the tetrayne homologues with these two end-groups could not be purified, so conductance measurements were not possible. Fig. 4C shows that series 9 (BT anchor) gave the highest conductance values and series 8 (NO2 anchor) was the least conductive. The experimental β values range between (1.75 ± 0.1) nm−1 (CN series 7) and (5.4 ± 0.3) nm−1 (NO2 series 8).
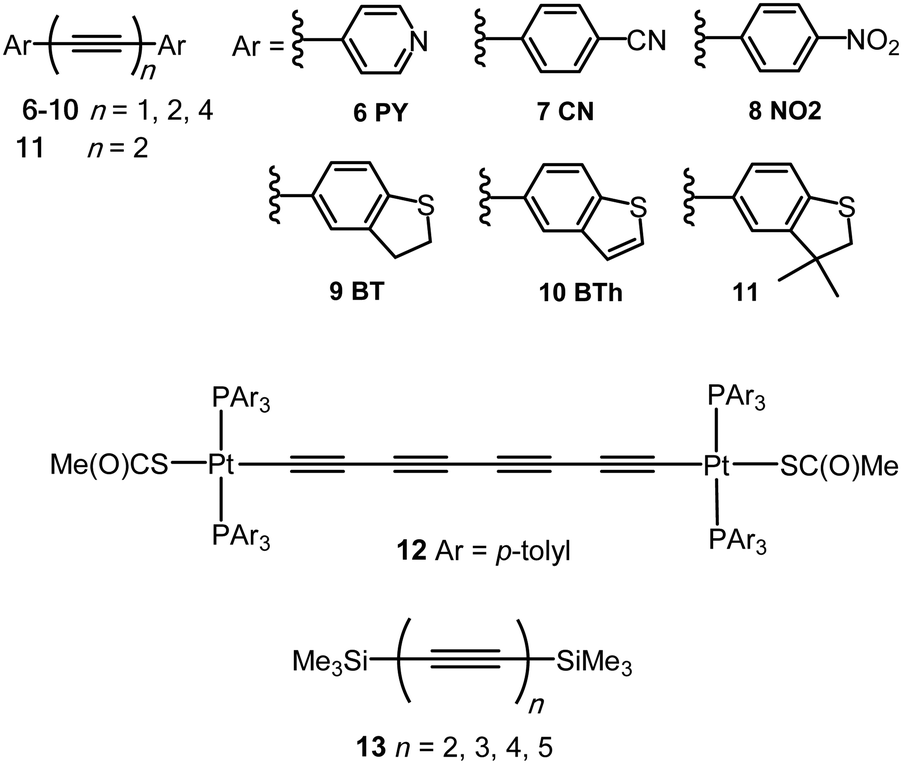 | ||
| Fig. 3 Structures of 6–13. | ||
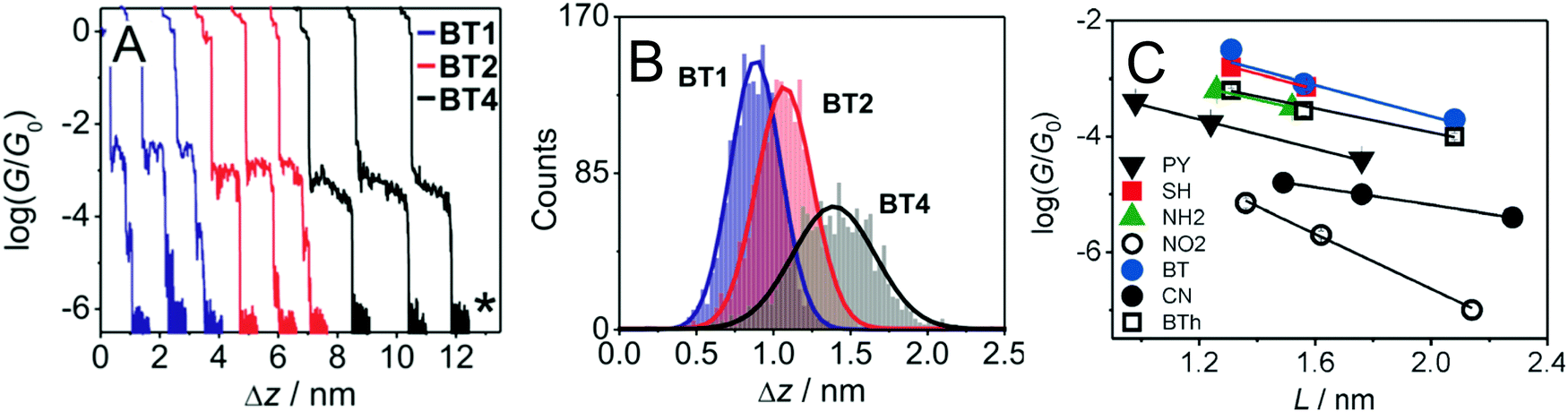 | ||
Fig. 4 (A) STM-BJ conductance measurements and (B) characteristic displacement histograms for the BT series 9 in the high conductance region; data in mesitylene/tetrahydrofuran (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) containing 0.1 mM of the target molecules. The asterisk in (A) indicates the noise level of the setup. Reproduced from ref. 95 with permission from the American Chemical Society. (C) Single-molecule conductance values (logG/G0) of oligoynes obtained from the analysis of the conductance histograms in dependence of the length of the molecule. Molecular length is defined as the distance between the centre of the anchor atom at one end of a fully extended isolated molecule to the centre of the anchor atom at the other end. Reproduced from ref. 96 with permission from John Wiley and Sons. :1 v/v) containing 0.1 mM of the target molecules. The asterisk in (A) indicates the noise level of the setup. Reproduced from ref. 95 with permission from the American Chemical Society. (C) Single-molecule conductance values (logG/G0) of oligoynes obtained from the analysis of the conductance histograms in dependence of the length of the molecule. Molecular length is defined as the distance between the centre of the anchor atom at one end of a fully extended isolated molecule to the centre of the anchor atom at the other end. Reproduced from ref. 96 with permission from John Wiley and Sons. | ||
The distinctly different β values for 6–10 demonstrated that the nature of the anchor group controls the strength of the electronic coupling to the gold leads, the position of the energy levels involved in the electron transport across the single-molecule junction, as well as their coupling into the oligoyne backbone. Theoretical calculations qualitatively supported the trends in experimental data, and suggested that sliding of the anchor groups on the electrode during stretching of the junction leads to oscillations in both the electrical conductance and the binding energies of the molecules.95 The β values of oligoynes are comparable with those of other π-conjugated molecular wires, such as oligo(phenyleneethynylene)s, oligo(phenyleneimine) and oligo(phenylenevinylene)s.81
A recent study reported the single-molecule transport properties of the butadiynyl analogue 11.97 Measurement of the thermopower (Seebeck coefficient, S) of the Au|11|Au junction gave a small, positive value (S = 3.7 μV K−1) consistent with transmission by tunneling through the tail of the HOMO resonance close to the middle of the HOMO–LUMO gap.
Electronic-vibrational coupling is a particularly intricate aspect of charge transport in molecular junctions. Weber and coworkers studied diyne and tetrayne 6 (n = 2, 4) in temperature-dependent MCBJ experiments (at 8–40 K) to probe the role of vibrations in charge transport.98,99 The absence of temperature dependence in the current–voltage (I–V) plots indicated that interference effects and vibrationally-induced decoherence mechanisms were strongly suppressed in these junctions. Oligoynes with their unique “string-like” conjugated backbone provide a counter-example to the majority of molecules previously investigated: inelastic vibrational sidepeaks were measured as satellites to the electronic transitions and were assigned to longitudinal excitations of the oligoyne backbone of 6 in the junction. In agreement with the DFT-based predictions, the reduction of the molecule's length from tetrayne to diyne resulted in an increase in the vibrational energies of the longitudinal modes. A similar assignment of vibronic features to longitudinal modes was made in an MCBJ study of the tetrayne 12 (related to structure 1) where the molecule was anchored in the junction via S–Au bonding after cleavage of the thioacetyl groups.100
The solvent dependence of the single-molecule conductance for a homologous series of trimethylsilyl-terminated oligoynes 13 has been studied using STM-based techniques.101 Trimethylsilylethynyl groups (CC–SiMe3) bind to gold through the silicon atom. The resulting junctions give well-defined conductance features, attributed to the steric bulk of the trimethylsilyl group, although conductances are lower than for more usual anchoring groups. Measurements were made in dry mesitylene, 1,2,4-trichlorobenzene and propylene carbonate, which were chosen for their differing polarities. The oligoyne series 13 represented ideal candidates for investigations of solvent effects. The molecules should be only minimally affected by any trace amounts of water present in thoroughly-dried solvents, as there are no functional groups in the backbone, or polar anchor groups, to coordinate any water molecules. In mesitylene, a lower conductance was obtained for each member of series 13, with a length decay value β = 0.94 nm−1, compared to the measurements in 1,2,4-trichlorobenzene and propylene carbonate which gave higher conductance values and lower β values of 0.13 and 0.54 nm−1, respectively. DFT calculations showed that electrostatic interactions between the solvent and the oligoyne backbone strongly impacted the computed transmission curves for the junctions. This behaviour was described as “solvent-induced gating of the molecular junction electrical properties”.101 Higher β values were found computationally using DFT when the contact Fermi energies were close to the middle of the HOMO–LUMO gap; β decreased as the Fermi energies approached resonance with either the occupied or unoccupied frontier orbitals. In the light of the results with 13 the different β values observed experimentally in the two previous studies of the series 694,95 could now be ascribed to solvent effects. This work contributed to the growing recognition that solvation can significantly affect the conductance of molecular junctions formed from various families of molecules.102
The assembly and measurement of the parent tetrayne obtained via the in situ fluoride-induced removal of trimethylsilyl end-groups from compound 13 (n = 4) has been reported.103 The sequential attachment of a tetrayne chain to a gold substrate and to gold nanoparticles (GNPs) gave the two-terminal Au|C8|GNP monolayer device shown in Fig. 5. Surface-enhanced Raman spectroscopy (SERS) confirmed the formation of the terminal carbon–metal bonds. I–V curves were recorded for the Au|C8|GNP structures using a conductive atomic force microscope (c-AFM) (Fig. 5a). Importantly, no low resistance traces were recorded which would be characteristic of metallic short circuits. Transition voltage spectroscopic (TVS) data confirmed that the electrical behaviour was consistent with that of a molecular junction rather than a metallic junction for which ohmic behaviour and much higher conductance would be expected. Fig. 5b shows a representative I–V trace of ca. 150 recorded curves and the conductance histogram built from the experimental data in the −0.5 to +0.5 V ohmic region for each of the 150 I–V curves obtained at the stated set-point force. These I–V curves exhibit a linear response only at relatively low bias voltages. Increasing curvature was observed at higher bias which is the usual behaviour for metal|molecule|metal junctions. This work demonstrated a novel and simple methodology for the fabrication of highly-conductive nanoscale junctions based on oligoynes. In this context it should be noted that other workers have shown that fluoride-induced desilylation of terminal trimethylsilylethynyl units in phenylene–ethynylene molecules during MCBJ measurements is not straightforward. The process can lead to oxidative dimerization of the terminal alkyne, presumably mediated by the gold surface. The product molecules containing butadiynyl units in the backbone subsequently anchored in the junction, which complicated the interpretation of the conductance behaviour.104
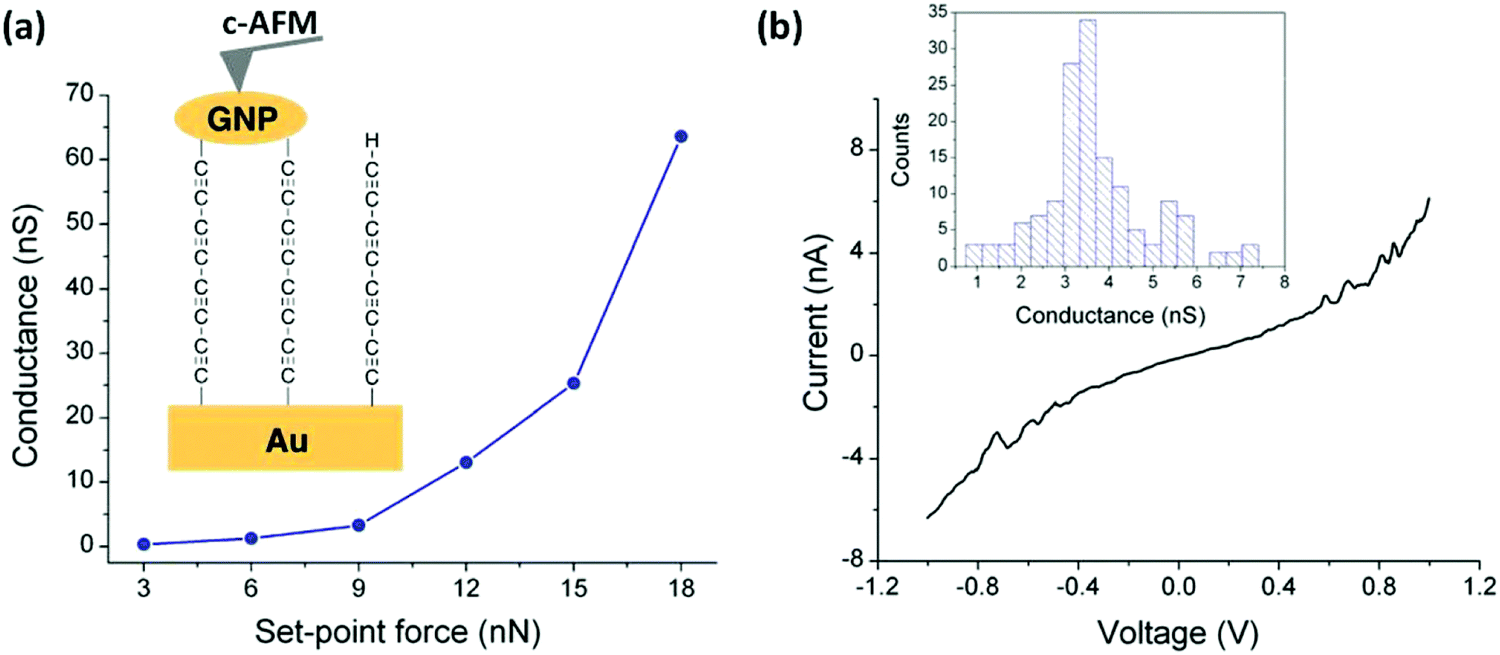 | ||
| Fig. 5 (a) Average conductance values measured by locating the tip of the c-AFM on top of GNPs at the indicated set-point forces. Inset: A scheme of the Au|C8|GNP structures contacted by the c-AFM tip. (b) Representative I–V curve experimentally obtained by positioning the c-AFM tip on top of a GNP when a set-point force of 9 nN was applied. Inset: Conductance histogram built by adding all the experimental data from −0.5 to 0.5 V for each I–V curve obtained (ca. 150 curves). Reproduced from ref. 103 with permission from the Royal Society of Chemistry. | ||
Steric shielding of the end-most ethynyl units of an oligoyne wire by installing two phenyl groups on the terminal pyridyl units led to kinetic stabilisation of the hexayne derivative 14 (Fig. 6). This molecule was also constructed within a macrocycle (M) to give the corresponding rotaxane 14·M in which the phenyl groups act as stoppers to prevent the macrocycle sliding off the oligoyne axle. These two molecules (14 and 14·M) were studied in single-molecule junctions by STM-BJ techniques and were found to have broadly similar conductance values of about 10−5G0.105
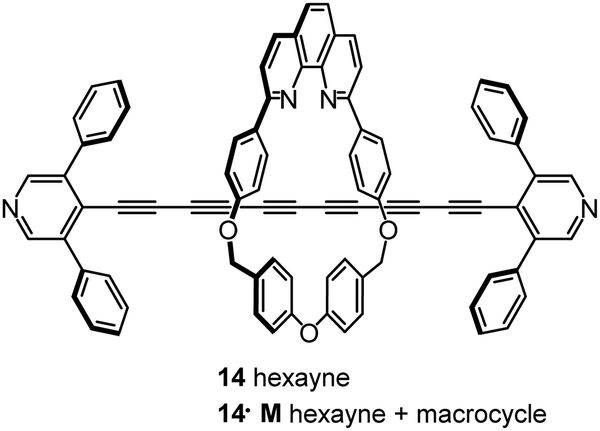 | ||
| Fig. 6 Structures of 14 and 14·M. | ||
Computational data showed that the macrocycle in 14·M had little effect on the energetics or densities of the charge transporting molecular orbitals of the oligoyne backbone. A drawback of the rotaxane strategy was revealed by a marked decrease in the propensity of 14·M to form molecular junctions, compared to 14. This difference was explained by steric effects of the bulky macrocycle which can slide along the oligoyne rod and impede contact between a pyridyl anchor and the electrode.
3.3 Organometallic groups in the oligoyne backbone
Several studies have incorporated organometallic groups into the backbones of oligoyne wires and it is well established that metal centres can enhance the single-molecule conductance compared to all-organic counterparts. Concepts and design strategies in this area have been reviewed by Low and coworkers,106,107 by Higgins and Nichols,108 and by Tanaka et al.109 Terminal arylalkynyl species (Ar–CC–H) are versatile ligands for transition metals, readily giving trans-Ar–CC–[MLx]–CC–Ar species (M = metal, L = ligand).110 A key feature of this generic structure is that the metal dxy and dyz orbitals have the correct π-type symmetry to interact with the alkynyl π-orbitals, thereby extending the π-conjugation of the backbone. Important molecular electronic properties, such as the energies of the frontier orbitals and the HOMO–LUMO gap, can be systematically varied by the choice of the metal, the choice of ligands attached to the metal, and by the terminal contacts. Oligoynes can be considered as the ultimate unsaturated bridging ligands as they cannot be twisted out of conjugation. The different accessible redox states of the metal centre can also be exploited for electrochemical gating of transport properties relevant to switching and memory applications.111
The single-molecule conductance of a series of platinum(II) trans-bis(alkynyl) complexes with terminal trimethylsilyl contacting groups has been reported, including the bis(butadiynyl) structures 15 and 16 (Fig. 7).112 The experimentally measured value for 15 (ca. 5 × 10−5G0) indicates that introduction of the trans-Pt(PEt3)2 fragment within the chain gives enhanced conductance compared with the analogous octatetrayne 13 (n = 4) (1.42 × 10−5G0) or decapentayne 13 (n = 5) (0.90 × 10−5G0) measured in the same solvent. It was shown that the use of PEt3 (15) rather than PPh3 (16) as ancillary ligands is beneficial, as orthogonal contacts of the STM tip occurred with the phenyl groups of 16, which complicated analysis of the conductance data.
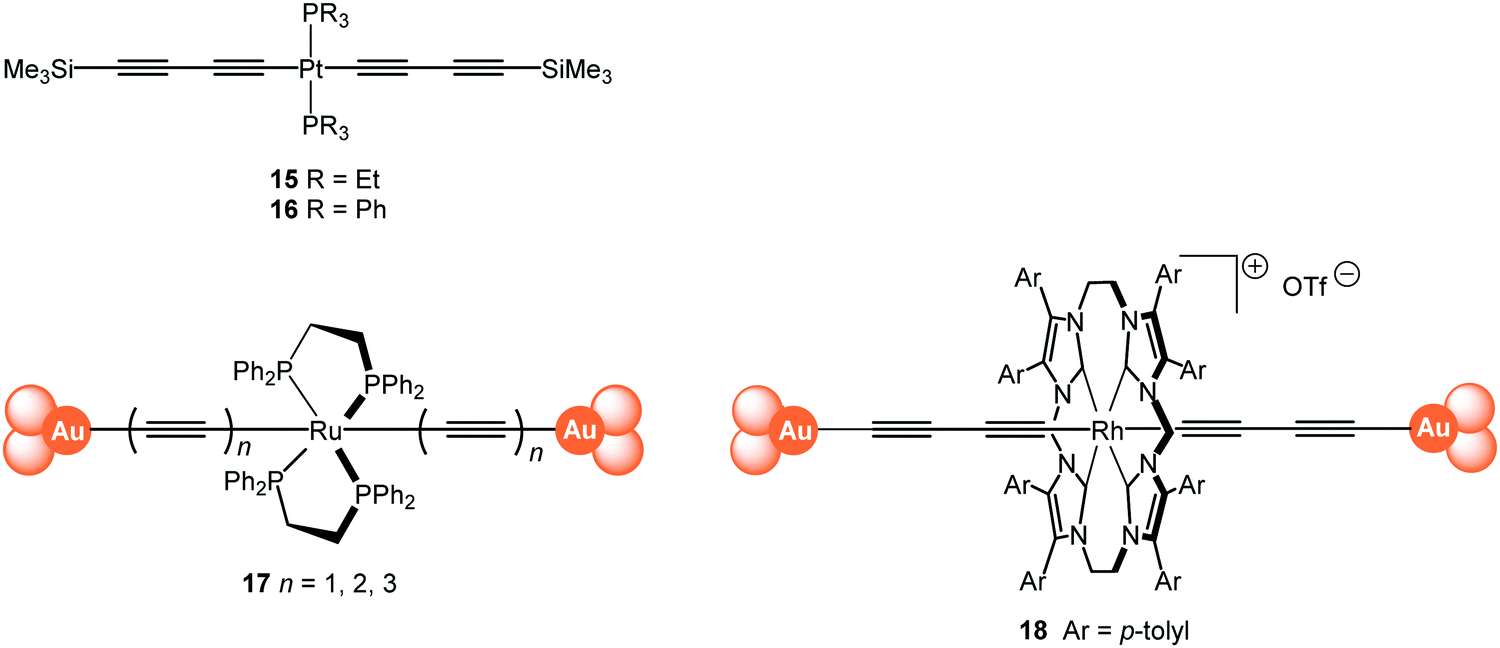 | ||
| Fig. 7 Structures of 15–18, after removal of the end-groups from 17 and 18 and their assembly in Au|molecule|Au junctions. | ||
Tanaka and coworkers studied a series of oligoynyl wires with Ru(dppe)2 cores and Au–ligand end-groups which were removed in situ to form covalent C–Au(electrode) bonds, as shown in structure 17.113 The conduction pathways of 17 are based on the HOMO orbitals, to which the metal centres contribute significantly. It was suggested that a unique “doping” effect of the electron-donating Ru(dppe)2 fragment accounts for the significantly higher conductance for this series, compared to the previously-reported oligoyne analogues of similar molecular lengths: 6,95995 and 13 (n = 5).101 The experimentally-determined conductance values for 17n = 1, 2, 3 were 2.1 × 10−2, 5.0 × 10−3 and 1.6 × 10−3G0, giving a β value of 2.5 nm−1. It is also notable that the conductance of 17 (n = 2) is significantly higher than that of other all-organic molecular wires of similar length with direct C–Au(electrode) bonding.114
Theoretical calculations have recently shown that single-molecule junctions formed by Ru(dppe)2-oligoyne molecules 17 are candidates to display negative differential resistance (NDR) behaviour under low bias.115 NDR is an interesting phenomenon where current decreases with an increase of the bias, with potential applications in data storage and logic operations. The NDR behaviour in 17 was assigned to suppression of the transmission peaks due to decay of the local density of states that originate from coupling of the Ru d and the carbon π orbitals of the oligoyne.
The cationic bis(butadiynyl)rhodium(III) wire 18 containing a cyclic tetracarbene ligand to stabilise the Rh(III) state has recently been studied using STM-BJ techniques in tetraglyme solution.116 The Rh(III) system is isoelectronic with the Ru(II) derivatives. Assembly in junctions was achieved from both the terminal (unprotected) butadiyne and a terminal Au-ligand precursor. The conductance of the single-molecule Rh+ wire junction [(6–7) × 10−3G0] was very similar from both precursors, suggesting the formation of the same C–Au(electrode) covalent bonds. Notably, the probability of molecular junction formation was much higher from the Au-ligand precursor molecule than from the free terminal alkyne. The conductance value for 18 was about one-third that of the Ru analogue 17 (n = 2) of the same molecular length. These studies demonstrated that both the transmission profiles and the accompanying conductance values can be tuned by the metal fragments installed in organometallic-oligoynyl wires.
Chen et al. demonstrated that Ru(II) promotes conduction and alleviates length attenuation in oligoyne chains with MeS–C6H4 end-groups (Fig. 8).117 Implanting a trans-Ru(dppe)2 moiety into the chains resulted in 2.6 to 5.8-fold enhancement of the conductance, even though the molecular length increased by ca. 0.25 nm as a result of Ru insertion. More importantly, the Ru moiety dramatically alleviated the length-dependent conductance attenuation, with β reduced to 2.6 nm−1 for series 20 compared to 4.6 nm−1 for the all-carbon oligoynyl compounds 19 (Fig. 8). Theoretical calculations suggested that the Ru(II) unit raised the HOMO level. The energy barrier between the HOMO and the Fermi energy, EF, was thus lowered so that the HOMO-dominated charge transport was facilitated (Fig. 8).
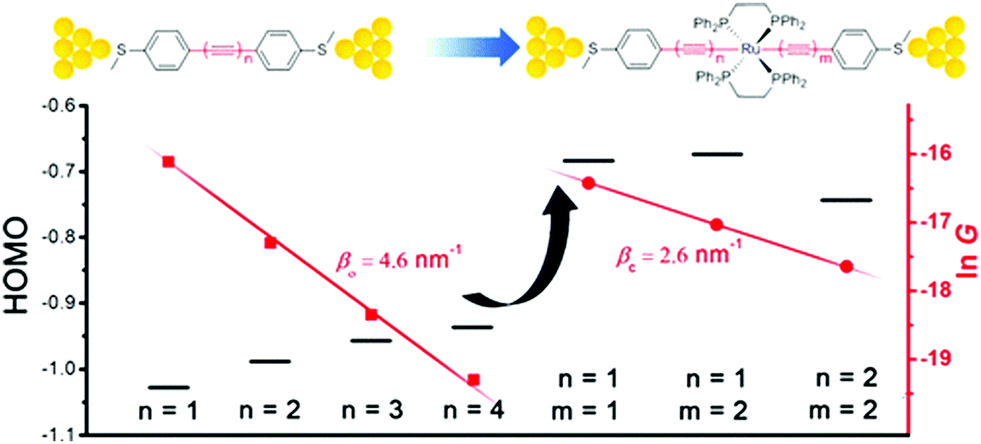 | ||
| Fig. 8 Single-molecule conductance data and HOMO levels of 19 and 20. Reproduced from ref. 117 with permission from the American Chemical Society. | ||
Multinuclear complexes in which the metal centres are linked with oligoynyl units have been studied. For example, STM-based experiments were reported on a homologous series of three oligo-Zn porphyrins separated by butadiynyl linkages and terminated with thioacetate groups (as protected thiol)118 or pyridyl anchor groups 21,119 with molecular lengths up to ca. 50 nm (Fig. 9). Remarkably, precisely the same β value of 0.040 ± 0.006 Å−1 (i.e., 0.40 ± 0.06 nm−1) was reported for both series, demonstrating that the attenuation of the bridges remained low, even with substantially different anchors at the metal leads. DFT calculations and an accompanying analytical model suggested that the observed transport data were consistent with phase-coherent tunnelling through the whole molecular junction, rather than an incoherent thermally-assisted hopping mechanism.119
 | ||
| Fig. 9 Structure of 21. | ||
Tian and coworkers studied a series of three butadiynyl-linked dinuclear dicationic Ru(II) complexes 22–24 of 2–3 nm length within single-molecule break junctions (Fig. 10).120 After deprotection of the thiolate groups, 22–24 displayed distinct conductance peaks at 3.0 × 10−4, 1.0 × 10−3 and 1.4 × 10−3G0, respectively. The conductance trend was ascribed to the different auxiliary terpyridyl ligands of the complexes. Predictably, the methylene group at each end of 22 significantly inhibited charge transport along the junction, hence the conductance was the lowest for this complex. The increased values of G0 for 23 and 24 are consistent with the enhanced coupling of both molecules to the electrodes, combined with the shorter length of 24. The calculated transmission spectra indicated that 24 has a three-fold higher zero bias conductance than the corresponding (hypothetical) tetrayne HS–H4C6–(CC)4–C6H4–SH, although both molecules have the same intamolecular S⋯S length (2.09 nm).
 | ||
| Fig. 10 Structures of 22–24. | ||
These results further established that incorporating organometallic centres into π-conjugated organic backbones is a viable way to construct long molecular wires with higher conductance and weaker length dependence than their non-organometallic counterparts of comparable length. In addition, electrochemical studies on SAMs of complexes 22 and 23 on Au(111) electrodes demonstrated two, one-electron redox waves, consistent with coupling between the two ruthenium centres. This data suggested the possibility of electrochemical gating of the molecular conductance within an experimentally-accessible electrochemical window.120
MCBJ experiments have been reported on metal complex wires 25–29 featuring two Fe(depe)2 moieties in the backbone (Fig. 11).121 Groups X served as the electrode contacting group for 25–27; for 28 and 29 elimination of the trimethyltin moiety resulted in direct CC–Au contacts. The cyano-contacted compound 25 gave unstable junctions with very low conductance (8.1 × 10−7G0). However, stable junctions were obtained with conductance values for 26 = 7.9 × 10−6G0 and 27 = 3.8 × 10−4G0 at 0.2 V bias. At higher bias (1 V) the complexes exhibited conductance values as high as 1.3 × 10−2G0 for 28 and 8.9 × 10−3G0 for the longer molecule 29. Narrower conductance histograms were observed for 28 and 29, compared to 26 and 27. Lower conductance peaks (ca. 10−5–10−6G0) with less prevalence, were also observed for 28 and 29 and were ascribed to different bonding scenarios of the CC–Au end groups, or to incomplete cleavage of the SnMe3 groups. Computational analysis suggested that junction transmission occurs through the tails of the resonances arising from the HOMO and HOMO−1, which are delocalised over the molecular backbone through π–d orbital overlaps.
 | ||
| Fig. 11 Structures of 25–29. | ||
3.4 “All-carbon” molecular junctions.
The fabrication of “all-carbon” molecular junctions comprised of atomic carbon chains embedded between carbon electrodes is a fascinating topic. In 2008 Bockrath et al. reported a nonvolatile memory element based on graphene break junctions.122 The model for device operation involved the formation and breaking of carbon atomic chains (ca. 1 nm long) that bridged the junctions under a strong electric field. The authors noted that the experimental ON/OFF ratios were relatively modest (ca. 50–100 while 105–106 can be achieved in CMOS devices). The switches were operated for many thousands of cycles without degradation, with the conductance state persisting for >24 h.In 2009 Peng and coworkers experimentally demonstrated the formation of 1D free-standing carbon atomic chains between two graphene edges by removing carbon atoms row-by-row from graphene with the electron beam of a transmission electron microscope (TEM).123 Chains up to approximately 2.1 nm in length (corresponding to about 16 carbon atoms) were observed in the TEM images. The bonding between the chain ends and the graphene edges was not stable, and the chain was observed to migrate along the edge. The type of carbon atomic chain (oligoyne or cumulene) could not be experimentally determined. No transport measurements were reported in this work.
In 2013 Prasongkit et al. proposed a theoretical mechano-switching device comprising a short carbon wire covalently-bonded between two zigzag carbon nanotubes. (CNTs).124 Upon stretching the junction (i.e. varying the gap width between the electrodes) and without breaking the wire, the computed I–V characteristics showed high- and low-conductance states (named as ON and OFF states) corresponding to cumulene and oligoyne structures, respectively. The calculations showed that cumulene wires do not always give high conductance: the electrode binding geometry is important. In the case of armchair CNTs the difference between the conductance of cumulene and oligoyne wires was insufficient to show switching characteristics useful for electronic applications.
By using an STM tip in a TEM stage both the in situ synthesis and the electrical characterisation of free-hanging atomic carbon chains were accomplished by Banhart and coworkers in 2013.125 The chains were synthesised by pulling out carbon atoms from few-layer graphene ribbons while an electrical current flowed through the ribbons and, subsequently, through the chain from the integrated sp2 carbon electrodes (Fig. 12). The lengths of the carbon chains could only be estimated due to projection effects in the TEM images: the chains appeared to be ca. 1–2 nm long. The chains were stable for a few seconds then ruptured and concomitantly the current dropped to zero. The measured large fluctuations of the current, and the low conductance values, were attributed to the changing characteristics of the contacts and to changes in strain within the carbon chain. Simulations suggested that a unique oligoyne or cumulene electronic configuration might not exist due to the variation of strain during the experiment. A subsequent experimental study established that the type of contact has an important influence on the I–V characteristics of the chains. A release of strain, due to a slight movement of one of the electrodes, led to an observable semiconductor-to-metal transition, corresponding to an oligoyne-to-cumulene structural transition in the chain.126 The experimental results were supported by theoretical transport calculations.
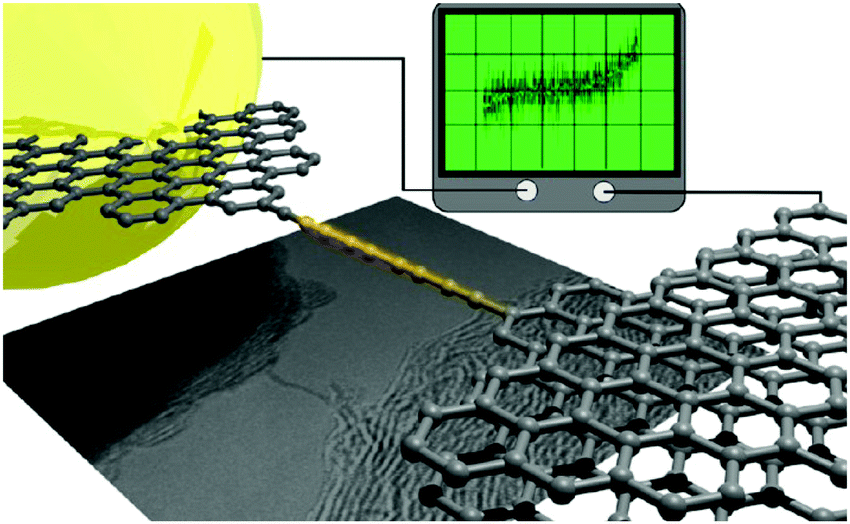 | ||
| Fig. 12 Schematic representation and TEM image of a conducting monoatomic carbon chain between graphene electrodes and an I–V trace of the device. Reproduced from ref. 125 with permission from the American Chemical Society. | ||
3.5 Cumulenes in metal|molecule|metal junctions
There are far fewer studies on cumulenes in molecular junctions, compared to oligoynes. It should be noted that there are significant structural and electronic differences between even-numbered and odd-numbered [n]cumulenes, ([n] = number of double bonds) due to their spatially degenerate and nondegenerate π-systems, respectively.12 [9]Cumulenes are the longest derivatives that have been experimentally characterised to date.127 Early theoretical studies showed that odd-numbered [n]cumulenes should have higher conductance than even-numbered homologues.92 More recently, Solomon and coworkers predicted that cumulenes could show increased electronic transmission with increasing length, i.e. a negative decay constant β.128Two recent experimental studies have demonstrated remarkable conductance properties for cumulenes in single-molecule junctions. STM-BJ measurements on the series of molecules 30–33 (Fig. 13) under solvent-free conditions showed that 32 and 33 had almost the same conductance, and they were both more conductive than the alkene 30 (Fig. 14A and B). The lower conductance of the allene analogue 31 was explained by its twisted geometry: theoretical calculations predicted that this relatively low conductance is a general feature of [n]cumulenes where n is an even number. The lack of length dependence in the conductance of 32 and 33 was attributed to the strong decrease in the HOMO–LUMO gap with increasing cumulene length.129
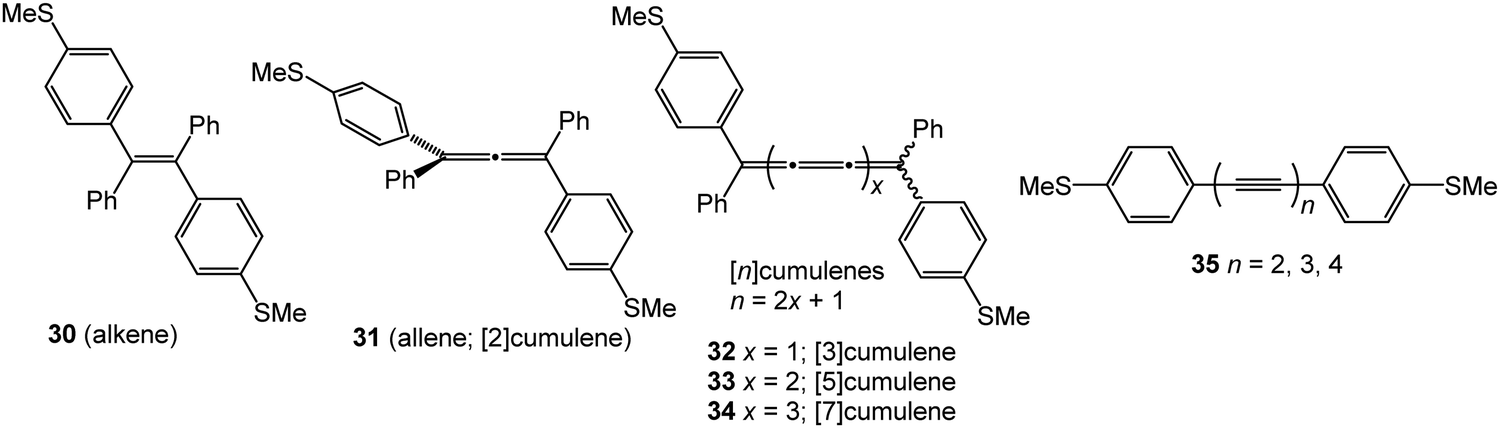 | ||
| Fig. 13 Structures of 30–35. | ||
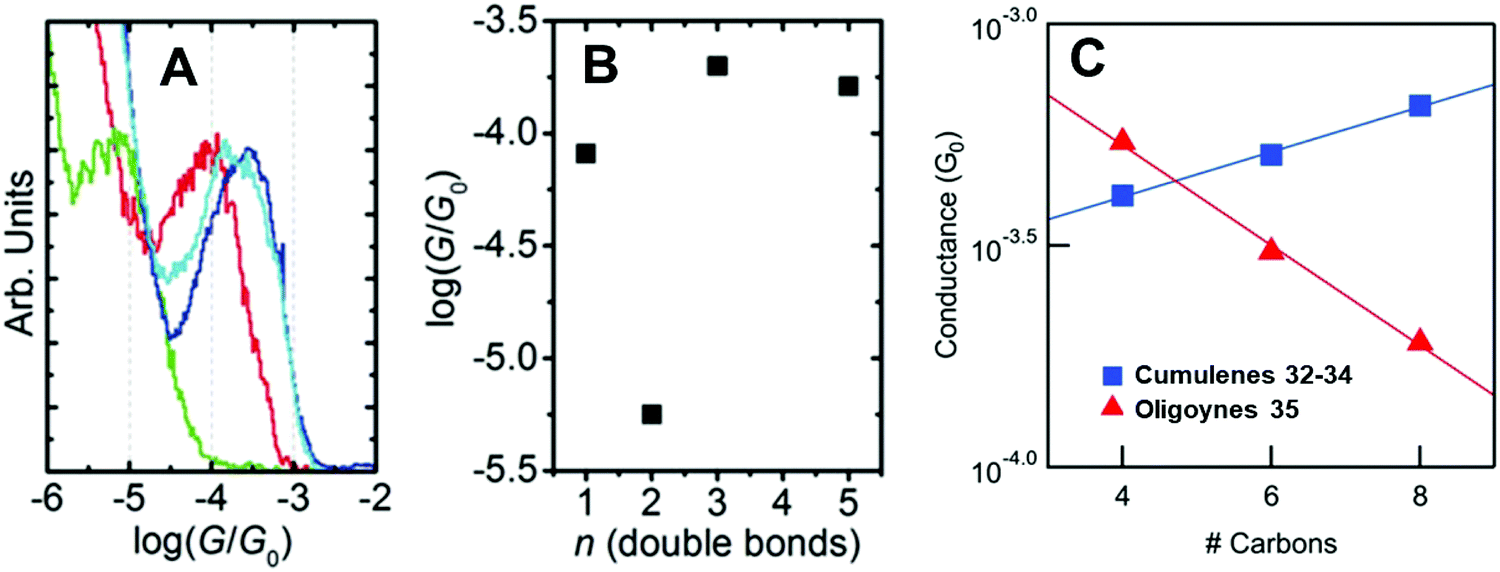 | ||
| Fig. 14 (A) 1D conductance histograms and (B) conductance values for single-molecule data of cumulenes 30–33. Reproduced with permission from ref. 129 with permission from John Wiley and Sons. (C) Single-molecule conductance values of cumulenes 32–34 (blue) and oligoynes 35 (red) as a function of the number of carbons in the chain. Reproduced from ref. 130 with permission from the American Chemical Society. | ||
An independent STM-BJ study included the [7]cumulene homologue 34. An incremental increase in conductance with increasing length, i.e. a rare negative β value, was observed for the [3], [5] and [7]cumulene series 32–34 in both tetradecane (non-polar) and propylene carbonate (polar) solvents (Fig. 14C).130 This length dependence of conductance of cumulenes is in marked contrast to comparable oligoynes 35 which have a positive β value (Fig. 14C),130 as was expected from previous studies on oligoynes 6–10.96 It is, therefore, firmly established theoretically and experimentally that cumulenes are excellent wires for molecular electronics applications.
4. Oligoynes as optoelectronic materials
4.1 General considerations
The unique combination of electronic and structural properties of oligoynes has led to many interesting optoelectronic applications. The molecules are structurally versatile because the extent of conjugation and photophysical properties can be modulated by judicious selection of end-groups, the number of alkyne units, and the option to incorporate metal atoms into the backbone. Moreover, rotation about single bonds in oligoynes does not affect orbital overlap and does not interrupt conjugation, in contrast with oligoenes. Leading examples of oligoynes will now be considered in the areas of nonlinear optics, redox activity and electrochromism, photoinduced intramolecular charge- and energy-transfer, organic light emitting devices, liquid crystals and bioimaging and biosensing. Each section begins with a very brief introduction, including review references, to place the oligoyne work in context.4.2 Nonlinear optical (NLO) properties
The optical response of a material usually scales linearly with the amplitude of the electric field. However, when high-powered light passes through a material, its electric field can result in a nonlinear response. For example, the original beam can be changed in phase, frequency, amplitude or polarisation. Typically, only laser light is sufficiently intense to modify the optical properties of a material in this way. Interest in NLO materials developed rapidly in the 1980s because the telecommunications industry needed high-bandwidth optical switching, and transmission and processing devices commensurate with the burgeoning computer age.131,132 The source of the large nonlinearity in organic molecules is the polarisability of the π-electrons in conjugated systems. A large displacement of charge in the presence of a small applied field results in easy modulation of the optical properties, for example, by inducing intramolecular charge-transfer transitions and a large change in dipole moment. A second-order NLO response requires asymmetric charge distribution in the molecule, which is often achieved by incorporation of donor and acceptor moieties, and the molecules must be packed in a non-centrosymmetric way. An extended delocalised π-electron backbone, as found for example in conjugated polymers, typically gives rise to third-order NLO properties.The third-order NLO properties of the symmetrically end-capped oligoyne series 36133,134 and 37135 (Fig. 15) were reported by Tykwinski and coworkers. Both series showed a substantial increase in molecular second hyperpolarizability (γ) as a function of increasing length. The magnitude of this power-law relationship for series 36 was described as “surprisingly high in comparison to that of other conjugated materials.”133 This observation was explained by considering that the major component to molecular hyperpolarisability stems from electron delocalisation along a conjugated backbone (longitudinal hyperpolarisability). Oligoynes undergo minimal conformational distortion in solution, therefore, orbital overlap along the molecular axis is well maintained which should enhance γ compared to other structures. The authors also noted that the longitudinal hyperpolarisability of an oligoyne will be less dependent on orientation with respect to the electric field of the incident light source – a factor which should also enhance γ values. Consistent with the linear absorption data, the NLO trend in the TIPS series 36 showed no sign of the onset of saturation up to n = 10. An advantage of oligoynes 36, compared to most other conjugated organic molecules, is that the high-energy region of the UV spectra (220–270 nm) is nearly transparent.
 | ||
| Fig. 15 Structures of 36–45. | ||
Although the power-law increase in γ values determined for the phenyl series 37 was slightly lower than that for the TIPS series 36, the values for 37 were comparable to, or higher than, those of other conjugated oligomers, including polytriacetylenes, oligo(p-phenyleneethynylene)s and polyenes. These data provided further evidence that oligoynes closely model a true 1-D conjugated system.134 A subsequent study included the adamantyl-series 38 and confirmed the large second hyperpolarizabilities and a large power-law increase in γ values versus length. The data showed that the measurement of absolute Raman intensity is a convenient method to characterise the nonlinear response of oligoynes in solution.136 Oligoynes achieve sizable γ values without a significant lowering of the electronic absorption energy found for polyenes of equal length. Studies with series 39 (Tr* = super-trityl, as drawn in structure 2) concluded that the origin of the NLO response was derived almost exclusively from the sp-carbon framework with a minimal contribution from the end-groups.137
Experiments by Stiegman et al. provided evidence that the NLO response in unsymmetrical push–pull oligoynes, including 40–42, saturates at short conjugation lengths. The second-order molecular hyperpolarizabilities (β) were dominated by the energy of the intramolecular charge transfer (ICT) band with strong donor–acceptor pairs producing large values of β primarily because they have low-energy absorption bands.138 Increasing the conjugation length from one to two alkyne linkers in 40–42 (and other analogues) did not significantly increase the value of β measured in N-methylpyrrolidone solution; however, β increased on going to the hexatriynyl homologue 43 (n = 3). This increase was attributed to the superposition of several nearly isoenergetic excited states. The alkene-bridged D–B–A counterparts had larger β values than the alkyne analogues. This was explained by the greater degree of conjugation inherent in the alkene-linked systems. However, an unavoidable consequence for enhancement of β in alkene compounds is the red-shift in their optical absorption into the visible part of the spectrum, thus limiting their practical application in this frequency range. The alkyne/oligoyne bridges significantly reduce the intramolecular charge-transfer character and thus give compounds with improved optical transparency. Weak second harmonic generation was observed from powders of 43 (n = 1–3) which was ascribed to the centrosymmetric space groups observed in their X-ray crystal structures.139
Diederich et al. studied the series of oligoynes 44.140 These molecules were designed as non-planar chromophores that could form amorphous thin films which could achieve high homogeneity and optical quality over large areas suitable for practical applications. Electrochemical and UV-Vis absorption studies in solution showed that the HOMO–LUMO gap decreased with increasing oligoyne length, although the effect levelled off in 44, and in a related series,141 at four alkyne units. High third-order nonlinearities were observed for 44 in dichloromethane solution, with the optimal conjugation length being n = 2 and 3.
Gauthier et al. reported a series of linear mono- and di-platinum complexes end-capped with diphenylpyranylidene (donor) and pentafluorophenyl (acceptor) moieties, including 45 which gave a low second-order NLO response in chloroform solution.142 The length of the oligoyne linkers, and the number of platinum centres, did not significantly affect the NLO responses of these complexes. Their structural configuration plays a significant role, as a related V-shaped complex exhibited an NLO response twice as high as that of the linear counterpart.
4.3 Redox-active and electrochromic oligoynes
Electrochromism is the ability of a material to change its absorbance or transmittance upon reduction or oxidation that occurs on the passage of an electrical current when an external voltage is applied. Electrochromic materials are utilised in applications including smart windows for energy-efficient buildings, information displays, electronic paper, and camouflage materials.143 π-Conjugated oligomers and polymers are widely-studied in this context due to low operational driving voltages for visual colour changes, high contrast and good reversibility of their redox states.144 Unlike many typical organic electrochromic materials, such as viologens or oligo/polythiophenes, oligoynes are not intrinsically redox-active. Therefore, to endow oligoynes with electrochromism, redox-active substituents need to be attached at the terminal positions. However, the limited stability of oxidised (p-doped) or reduced (n-doped) species – especially for longer oligoynes – may restrict their practical applications. Nonetheless, there are some interesting proof-of-concept reports on electrochromic oligoynes. Compounds 46–55 (Fig. 16) provide examples where spectroelectrochemical properties have been reported in some detail. | ||
| Fig. 16 Structures of 46–57. | ||
The two-stage oxidation behaviour of the tetrathiafulvalene (TTF) unit was exploited in electrochromic studies of the donor–bridge–acceptor (D–B–A) molecule 47.145 Spectroelectrochemical data in dichloromethane solution gave λmax values for neutral (462 nm), cation radical (468, 596, 669, 714 nm) and dication species (507 nm). The dark-green cation radical was a prominent feature of this redox cycle. The absorption spectrum of neutral 47 was red-shifted by 15 nm compared to the mono-alkyne analogue 46 due to extended conjugation through the butadiynyl system.
Zhao et al. described a series of diaryloligoynes with terminal electron-donating 1,3-dithiol-2-ylidene units.146 The absorption wavelength incrementally red-shifted in the series with 2, 4 and 6 triple bonds. DFT calculations showed that the electron accepting ability of the oligoyne increased with its increasing length. Both electrochemical and chemical oxidation of the monomers (n = 2 and 4) gave films of the corresponding polymerised species 48 and 49. Thin films of 48, prepared by electrodeposition on ITO glass, gradually changed colour from pale yellow to dark green upon oxidation with the appearance of a strong broad band in the 500–850 nm range. For polymer 49 this band was red-shifted further into the near-IR region, in the range 550–900 nm, peaking at 691 nm (Fig. 17). Fine vibrational structure was present in the high-energy region, which is the signature of a tetraynyl unit, indicating that, significantly, the tetraynyl units in polymer 49 remained intact upon oxidation. The reversibility of this redox process was not demonstrated.
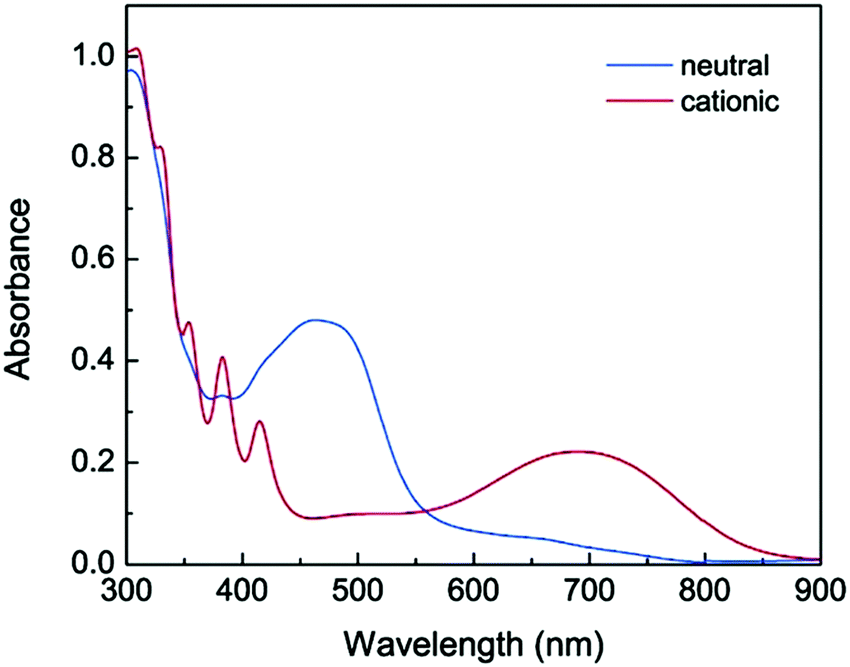 | ||
| Fig. 17 UV-vis absorption of polymer 49 on ITO glass in the neutral (blue spectrum) and cationic (red spectrum) states. Reproduced from ref. 146 with permission from the American Chemical Society. | ||
The highly delocalised electronic structures often associated with [{LnM}{μ-(CC)x}{MLn}] complexes has led to many studies of intramolecular electron transfer and mixed valence characteristics.147,148 Experimentally, even-numbered carbon chains with predominantly oligoynyl character have been most commonly studied, notably butadiynyl complexes, due to their relative ease of synthesis. For compounds which feature an odd number of carbon atoms in the chain, at least one of the metal centres must be involved in an M–C multiple bond. Relevant organometallic systems, where detailed redox and electrochromic properties have been reported include molecules 50–55. The series 50–52 with a Ru2-oligoyne-Ru2 backbone has been studied by Ren et al.149 Two, stepwise, one-electron reductions are localised on the Ru2 termini, indicating a significant electronic coupling between the termini. In particular, the rich spectroelectrochemistry of 51 with a Ru2–C6–Ru2 backbone showed good reversibility in THF, revealing intervalence charge-transfer (IVCT) transitions mediated by the hexatriynyl bridge.150 As shown in Fig. 18, reduction to the anion 51− resulted in a decrease of the absorbance bands at ca. 600 nm and 800 nm and the growth of a band centred at 1200 nm, with a new broad absorption at ca. 1900 nm. These low energy bands were lost on further reduction to 512−. Electronic delocalisation in cationic species was also observed.149
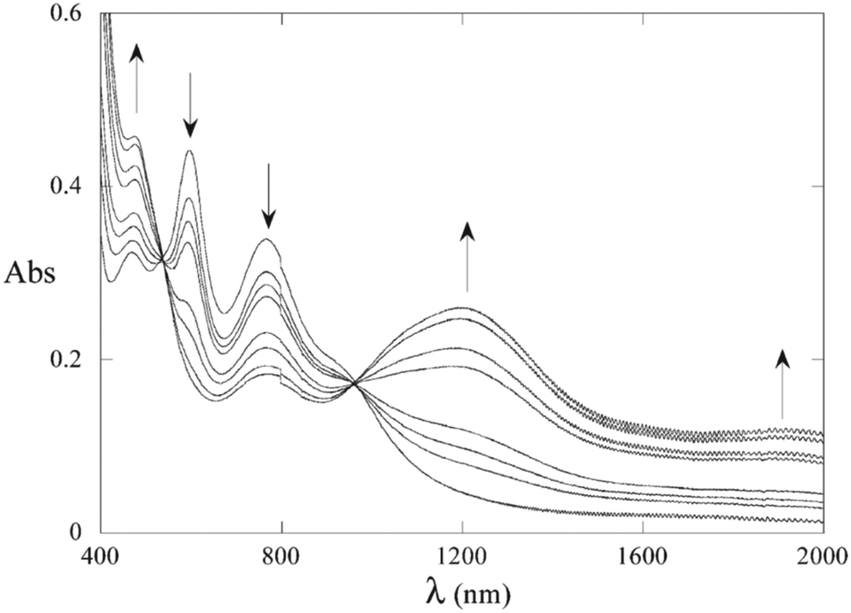 | ||
| Fig. 18 Spectroelectrochemical reduction (0/−1 states) of complex 51 (X = iBuO) in THF and 0.2 M TBAPF6. Reproduced from ref. 150 with permission from the American Chemical Society. | ||
In related trimeric [Ru2]3 complexes the butadiynyl and hexatriynyl linkers effectively mediated intermetallic electronic couplings between the [Ru2] units, although this coupling was weakened in the longer bridges. In comparison, para-phenylenediethynyl bridges did not mediate significant electronic coupling among three Ru2 units.151
Bruce, Halet and coworkers showed that the C4 bridged complex 53 displayed rich electrochemical and spectroelectrochemical behaviour throughout four oxidative processes. The experimental data, supported by quantum chemical calculations, demonstrated a progressive shift in the bonding in the carbon chain upon oxidation from neutral butadiynyl (–CC–CC–), through dicationic butatrienylidene/cumulene (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCCC) towards butynediyldiide (C–CC–C) in the tetracationic state.152 Subsequent studies by Kaupp, Low and coworkers153 identified an IVCT absorption band in the NIR region and concluded that the carbon chain plays a significant role in stabilising the unpaired electron in 53+. A comparison of 53 with the tethered analogue 55 established that the appearance of the IVCT band, and the underlying electronic structure, is dependent on the various rotameric forms, e.g. with differing P–Ru–Ru–P dihedral angle. These conclusions likely apply to many other mixed-valence complexes with low axial symmetry and relatively free rotational elements. The hexatriynyl analogue 54 underwent similar spectroelectrochemistry to 53, although the longer, more exposed, oligoyne chain of 54 led to an interesting intermolecular coupling reaction upon chemical oxidation.154
CCCC) towards butynediyldiide (C–CC–C) in the tetracationic state.152 Subsequent studies by Kaupp, Low and coworkers153 identified an IVCT absorption band in the NIR region and concluded that the carbon chain plays a significant role in stabilising the unpaired electron in 53+. A comparison of 53 with the tethered analogue 55 established that the appearance of the IVCT band, and the underlying electronic structure, is dependent on the various rotameric forms, e.g. with differing P–Ru–Ru–P dihedral angle. These conclusions likely apply to many other mixed-valence complexes with low axial symmetry and relatively free rotational elements. The hexatriynyl analogue 54 underwent similar spectroelectrochemistry to 53, although the longer, more exposed, oligoyne chain of 54 led to an interesting intermolecular coupling reaction upon chemical oxidation.154
A study of the dimeric Co(III)(cyclam) species 56 and 57 demonstrated how terminal substituents on the metal atoms can significantly influence the intramolecular interactions along an oligoynyl bridge.155 Voltammetric analysis of Cl-terminated 56 revealed a significant Co–Co interaction which was attenuated by the length of the oligoyne. In contrast, the butadiynyl derivative 57 with phenylacetylene termini lacked any observable Co–Co coupling. The different behaviour was rationalised through DFT analysis of the frontier orbitals of 56 and 57.
4.4 Photoinduced intramolecular charge and energy transfer in D–B–A triad systems
A comprehensive understanding of photoinduced electron-transfer and electron-transport processes is crucial for progress in many fields of science that depend on charge separation and recombination processes, such as artificial photosynthesis, photovoltaics, field effect transistors and molecular switches. Many studies have focussed on covalently-linked donor–bridge–acceptor (D–B–A) triad systems. Key questions are: (i) how fast can electrons or excitation energy be transferred between the donor and acceptor units, and (ii) what is the role played by the bridge (the molecular wire) in charge separation and recombination? The goal of charge management in these types of molecules is usually to facilitate thermodynamically-driven charge separation to yield a high-energy radical ion pair. Suppression of charge recombination is desirable for most applications; this process should ideally be exergonic (release energy) and be shifted into the Marcus “inverted region”.156,157 The standard techniques for in-depth analysis of charge-separation, charge-transfer, and charge-recombination events are transient absorption pump–probe measurements on the femto-, pico-, nano-, and micro-second time-scales, combined with fluorescence spectroscopy, and supported by computational calculations. Many conjugated oligomers have been used as the bridge between D and A units, such as oligo-phenylenes, -phenylenevinylenes, -phenyleneethynylenes, -fluorenes, -thiophenes and others.158,159 This section will now consider oligoynes in this context.Early indications of interesting photoinduced charge-transfer properties through oligoynyl bridges came from two independent reports in 1997. The first report from Khundkar et al.160 showed that 43n = 2 was more weakly emitting (lower photoluminescent quantum yield, PLQY) than 43n = 1. For both of these molecules photoexcitation induced rapid and efficient charge separation, as evidenced by the large solvatochromism observed in their emission spectra. The increase in the emission lifetime with increasing solvent polarity was attributed to dipolar stabilisation of the emitting state. The rate of nonradiative charge recombination was faster for 43n = 2 than for 43n = 1 and this was ascribed to stronger electronic coupling(s) in the butadiynyl derivative. Stiegman et al. subsequently reported comparable results for the extended series 43 (n = 1, 2, 3). The series showed strong emission from a charge-transfer state with “unusual effects in varying n”.161 In particular: (i) the emission maxima, half-width and Stokes shift showed little change in a given solvent within the series; (ii) the emission intensity decreased dramatically in going from n = 1 to n = 3; (iii) the rate constant for non-radiative decay increased with increasing oligoyne length.
Mishra et al. studied the photoinduced charge transfer in butadiynyl-bridged biaryl derivatives 58 and 59 (Fig. 19).162 Pyrene behaved as a donor unit in 58 with a cyano acceptor attached to the phenyl ring, whereas pyrene behaved as an acceptor in the dimethylamino analogue 59. In mixed water-acetonitrile solvents with 80–99% water, the derivatives showed emissions from aggregate states, centred in the range 510–560 nm, in addition to LE and ICT states. The aggregate state emissions were found to originate from excimers involving pyrene–pyrene stacking interactions. The solid-state fluorescence of 59 deposited on a quartz plate could be switched reversibly through at least five cycles between green and greenish-yellow (Δλmaxca. 15 nm) upon thermal and mechanical stimuli.163 Reversible fluorescence switching was not observed for the analogues with pyrenyl replaced by phenyl.
 | ||
| Fig. 19 Structures of 58–60. | ||
Studies in our laboratory on an extended series of compounds with triphenylamine donor and oxadiazole acceptor 60 confirmed: (i) strong solvatochromism of photoluminescence; (ii) a dramatic reduction in PLQY with increased oligoyne length, and (iii) that strong non-radiative pathways dominated the excited-state decay for the more conjugated systems. A sequential reduction in the HOMO–LUMO gap of 60 was observed along the series n = 1–4. The data showed that ICT was equally efficient for the shorter and longer molecules.164,165
The analogous N-arylcarbazolyl series 61 (Fig. 20) was probed recently using in-depth photophysical and time-dependent DFT (TD-DFT) techniques.166 The overall absorption characteristics gave rise to a strong length dependence of the bridge, and, as above, a dramatic reduction in PLQY accompanied increasing oligoyne length. Solvent-polarity-dependent emission measurements were reported. For all the conjugates in non-polar methylcyclohexane, the fluorescence spectra were dominated by a rather sharp peak at λmax 385 nm, together with vibrational fine structure and broad, low energy bands. The low energy bands red shift in more polar solvents, consistent with their ICT nature. Representative data for 61n = 3 are shown in Fig. 20. The noise level in the spectra increased along the series n = 1–4, in line with dramatically decreasing PLQY. In this work, we observed the unusual feature of continuing acceleration of the charge-recombination dynamics along the series 61n = 1–4. Significant delocalisation of the molecular orbitals along the bridge indicated that the oligoynyl states come into resonance with either the D or A units, thereby accelerating the charge transfer. One possible explanation was a better electronic coupling once planarisation had occurred in the excited state. Another notable result was the computed differences in the single and triple bond lengths between the ground and excited states as the bridge length increased. The ratio of the averaged C–C and CC bond distances (d) approached unity in both the singlet and triplet charge-separated states (1CSS and 3CSS), representing significant cumulenic character in the photoexcited state, compared to the oligoynyl ground state (S0). Specifically, compare 61n = 1: d(C–C)/d(CC) = 1.18, 1.11 and 1.05 for S0, 1CSS and 3CSS with 61n = 4 where these values are 1.14, 1.07, 1.07, respectively. A cumulenyl rather than an oligoynyl bridge is expected to accelerate electron transfer processes, and charge recombination in particular.
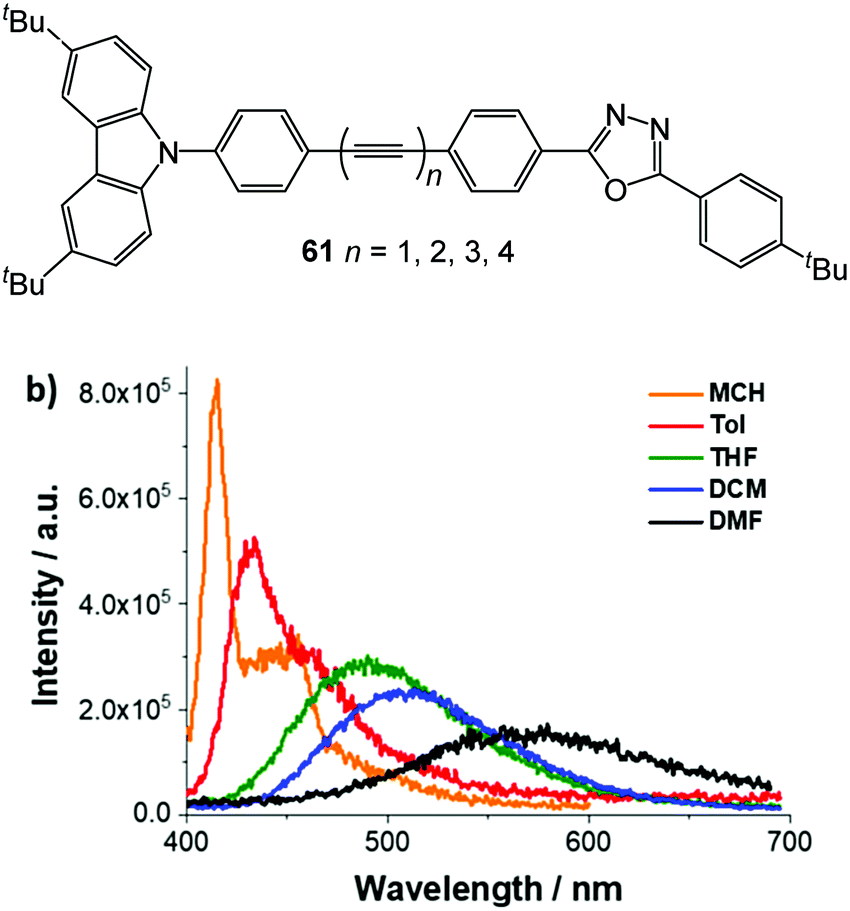 | ||
| Fig. 20 Structure of 61. Fluorescence spectra of 61n = 3 in solvents of different polarity (MCH, methylcyclohexane; Tol, toluene; THF, tetrahydrofuran; DCM, dichloromethane; DMF, N,N-dimethylformamide) with matching absorption of OD = 0.05 upon 350 nm excitation. Reproduced from ref. 166 with permission from the American Chemical Society. | ||
(Metallo)porphyrin derivatives have been the subject of many reports of photoinduced charge- and energy-transfer. Their synthetic versatility (of the central metal and peripheral substituents) combined with outstanding visible light-harvesting and electron-donating properties make them ideal building blocks for D–B–A conjugates.167 Two noteworthy studies incorporate oligoynyl bridges.
Schuster and coworkers studied the D–B–A series 62 with Zn–porphyrin (ZnP) as the D unit (Fig. 21).168 C60 is an excellent acceptor in D–A ensembles designed to mimic the photosynthetic reaction centre. The long-lived charge-separated (CS) states generally observed in fullerene ensembles can be attributed to the low reorganisation energy of the fullerene and the suppression of back electron transfer by forcing charge recombination into the Marcus inverted region.169 Monitoring the first reduction potentials by solution electrochemistry of 62 confirmed that the extent of electronic interaction between porphyrin and fullerene units decreased with increasing length of the bridge. These molecules exhibited very rapid photoinduced electron transfer in tetrahydrofuran and benzonitrile, which is consistent with normal Marcus behaviour. It was proposed that charge separation from photoexcited 1ZnP* to the fullerene in polar solvents directly gave the ZnP+˙ (cation radical)/C60−˙ (anion radical) pair, bypassing 1C60*. The rates for charge recombination indicated that this process was occurring in the Marcus inverted region. A plot of the back electron transfer rates as a function of D–A separation led to a distance dependence with a very small attenuation factor (β) of 0.06 ± 0.005 Å−1 demonstrating that oligoynes effectively mediate electronic interactions in this series. The efficient molecular wire behaviour was rationalised in terms of the extensive π-conjugation achieved between the phenyl ring of ZnP, the oligoynyl bridge, and the C60 moiety.
 | ||
| Fig. 21 Structures of 62 and 63. | ||
Anderson, Albinsson and coworkers investigated the series of butadiynyl-linked porphyrin oligomers 63.170 In 63, the ZnP unit(s) are an integral part of the backbone, not the terminal donor unit, as in 62. A fully charge-separated state (Fc+˙–wire–C60−˙) was formed after photo-excitation of the porphyrin oligomer and subsequent charge transfer steps (Fig. 22). The rates of long-range charge recombination through the bridging oligomers were remarkably fast and exhibited very weak distance dependence, giving a very small apparent attenuation factor β of 0.003 Å−1. It was concluded that this long-range charge recombination process occurred via coherent electron tunneling rather than via hopping or triplet recombination. The observation that the longest wire in the series mediated fast long-range charge transfer over 65 Å indicates that the inter-porphyrin electronic coupling was strong due to the significant π-interaction between the porphyrin macrocycles mediated by the butadiynyl bridges. The low β value for 63 is reminiscent of the low β value obtained from single-molecule conductance studies of the porphyrin series 21 in Au|molecule|Au junctions.119 However, the data are not directly comparable because different exchange interactions and tunneling energy barriers through the bridge are expected in the very different experimental conditions of the two studies.171
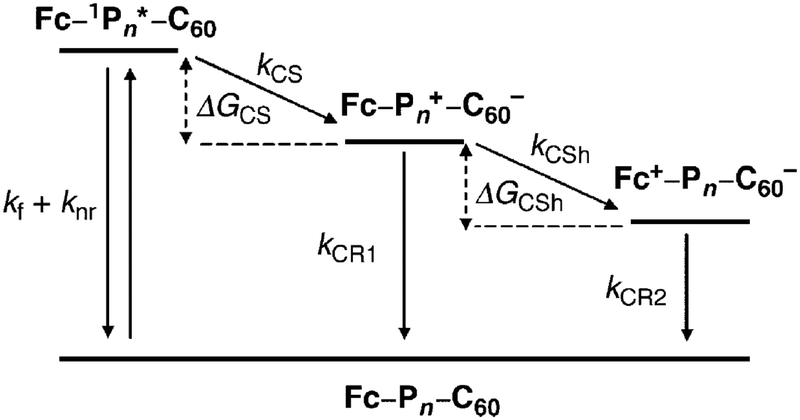 | ||
| Fig. 22 Schematic energy level diagram showing the charge separation (CS), charge shift (CSh) and charge recombination (CR1) and (CR2) reactions for molecules 63. Reproduced from ref. 170 with permission from the American Chemical Society. | ||
4.5 Oligoynes in OLEDs
During the last decade organic light-emitting devices (OLEDs) have grown into a competitive technology for full-colour displays and lighting panels.172 Oligoynes have been exploited only rarely for organic electroluminescence (EL), and due to the low PLQYs reported for longer oligoynes, they may not find widespread practical use for EL. Nonetheless, two interesting prototype examples will now be summarised.A fluorescent solution-processed OLED using butadiynyl derivative 61n = 2 was reported in 2014.173 Neat thin films of 61n = 2 gave blue photoluminescence (λmax 428 nm). However, the EL spectrum of 61n = 2 blended in poly(N-vinylcarbazole) host material unexpectedly showed two main peaks at λmaxca. 450 and 575 nm, corresponding to blue and orange-red emission, respectively. This resulted in a high colour rendering index (CRI) of 84–88 for white light emission [CIEx,y coordinates: (0.31, 0.33), close to the ideal for “pure” white (0.33, 0.33)] with a very small offset of (ΔCIEx,y 0.034, 0.033) over the driving bias range 9–15 V (Fig. 23). These data suggest that a balanced distribution of holes and electrons was achieved within the recombination zone in the emitting layer which could be facilitated by the donor–acceptor (bipolar) structure of the emitter. The strong orange-red emission band in the EL, which was not observed in the PL, was consistent with the formation of electric field-induced electromers during device operation. Although the maximum brightness was only 116 cd m−2, this study is an unusual example of a single-component emitter for white EL.174
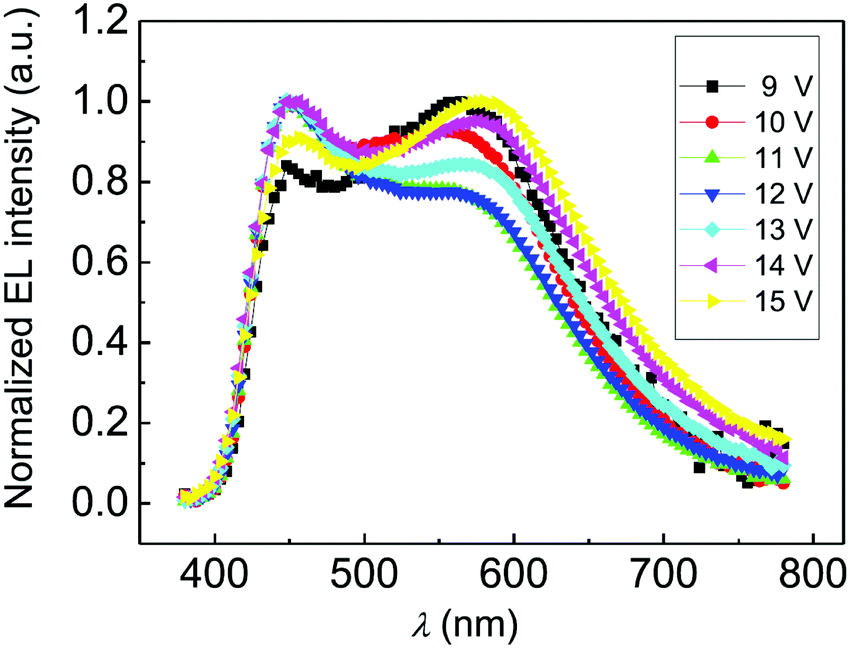 | ||
| Fig. 23 Normalised EL spectra of a white OLED of 61n = 2 under driving voltages of 9–15 V. Reproduced from ref. 173 with permission from Elsevier. | ||
Very recently, Schanze and co-workers reported solution-processed phosphorescent OLEDs (PhOLEDs) of the trans-Pt(II) complexes 64 and 65 (Fig. 24).175 The EL spectra closely matched the PL spectra in both THF solution and doped into poly(methylmethacrylate) films. The emission from 64 originated from butadiynyl ligand-centred 3(π → π*) transitions with a very minor contribution from 3MLCT character. Compared to phenylacetylene ligands, the phenylbutadiynyl units of 65 gave sharper emission due to the well-defined CC vibronic modes that dominate the phosphorescence spectrum. The PhOLEDs with 65 as the emitter doped into DPEPO host had blue-green EL (Fig. 24). The comparable PhOLEDs of 64 were unusually deep-blue [λmax 415 nm, with very narrow full-width at half-maximum (fwhm) = 12 nm] and CIEx,y of (0.172, 0.086), approaching the National Television System Committee (NTSC) coordinates for “pure” blue of (0.14, 0.08) (Fig. 24). The 64 PhOLEDs had a maximum external quantum efficiency (EQE) of 3.2% at very low brightness, and low device efficiency at higher, practically-useful brightness levels (0.5% EQE at 100 cd m−2) – a common effect in blue OLEDs known as efficiency roll-off.
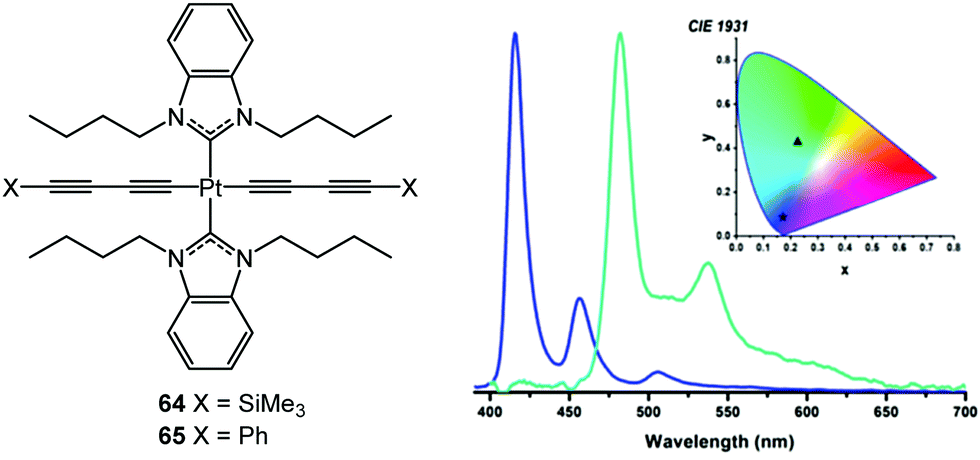 | ||
| Fig. 24 Structures of 64 and 65, and EL spectra of PhOLEDs of 64 (blue) and 65 (green). Reproduced with permission from ref. 175 with permission from the American Chemical Society. | ||
4.6 Liquid crystalline oligoynes
Liquid crystals (LCs) are an intermediate state of matter between that of a three-dimensionally ordered crystalline solid and a disordered isotropic liquid phase. A review of LCs by Steinstrasse and Pohl in 1973 concluded by stating: “Looking further into the future we can also anticipate [applications in] optical memory devices and flat large area color television screens.”176 More recent reviews177,178 highlight how LCs have indeed been of paramount importance for the last few decades in optoelectronic devices, including those technologies anticipated in 1973. Moreover, liquid crystals have new emerging applications in areas such as separation technology for environmental and healthcare benefits, chromism, sensing, electrooptical effects, actuation, and templating.176The preferred compounds for high birefringence (i.e., high anisotropy of the refractive index, Δn) are conjugated in the direction of the long molecular axis. Therefore, oligoynes are obvious candidates. Butadiynes in particular have been successfully exploited. In 1978 Grant reported the preliminary mesomorphic properties of two LC butadiynyl derivatives 66 and 67 (Fig. 25).179
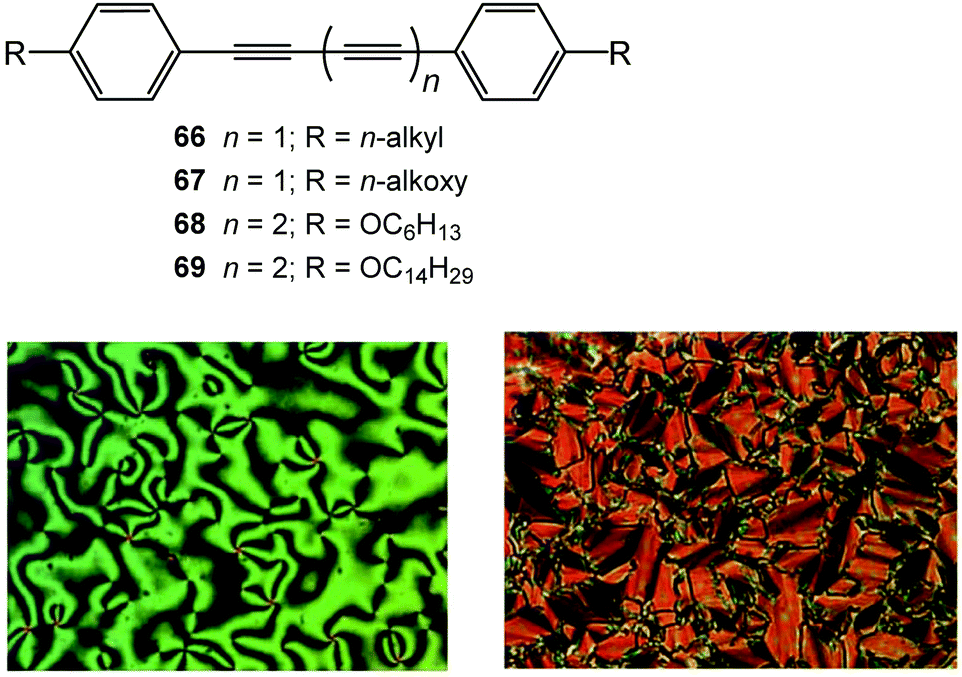 | ||
| Fig. 25 Structures of 66–69. Polarised-light optical microscopy images of 69 at 130 °C (left) and 117 °C (right). Reproduced from ref. 183 with permission from the Royal Society of Chemistry. | ||
Related dialkyl and unsymmetrical alkyl/alkoxy derivatives were studied in more detail by Wu et al. more than a decade later.180,181 These LCs had the benefits of higher Δn and lower rotational viscosity, compared to related tetraphenyl analogues. Applications of these LCs were anticipated in high-definition displays, modulating IR radiation, high-speed electrooptic modulators, long-wavelength applications where photostability is not a problem, and as host candidates for eutectic mixtures. The alkoxy series exhibited higher Δn than alkyl analogues.182 Hexatriynyl derivatives 68 and 69 gave further improved LC behaviour.183 An increasing number of alkyne units (n = 1–3) led to wider nematic phases and a linear increase in birefringence; the increment in Δn per alkyne unit was ca. 0.14. An odd–even oscillation, depending on alkyl chain length, was observed for transition temperature values of derivatives with relatively short alkoxy tails. Measurements of 68 yielded a notably high Δn of 0.45 (at 550 nm, 140 °C), and a refractive index (ne) of >2.0. Polarised-light optical microscopy (POM) images of 69 are shown in Fig. 25. At 130 °C a nematic phase was clearly seen, exhibiting a schlieren texture which is typical for a nematic phase. At 117 °C a typical smectic-C phase with a focalconic texture was observed.
There is much current interest in the topochemical polymerisation of LC molecules containing butadiynyl units to yield polydiacetylenes. This well-known 1,4-addition reaction occurs with only minor structural changes and yields a semiconducting polymer and a chromophore by virtue of the extended alternating alkyne–alkene conjugation in the backbone.15,16 Three examples illustrate the varied structures that have been studied recently. Dumbbell-shaped LC monomers 70 were photopolymerised (using 254 nm irradiation) in a thin solid film state (Fig. 26).184 Hydrogen bonding between the amide units assisted the long-range ordering that correctly aligned the butadiynyl units for polymerisation. The monomer 70 had no absorption band in the 350–750 nm region; upon irradiation a peak at λmax = 530 nm gradually appeared, arising from electronic transitions of the π-orbitals of the polymer chain. The gelation properties of 70 in toluene solution and subsequent photopolymerisation in the gel state to give a thermochromic polymer were also reported.183
 | ||
| Fig. 26 Molecular packing of self-assembled butadiynyl derivative 70 before and after photopolymerisation. The red circles represent dendron units. Reproduced from ref. 184 with permission from the Royal Society of Chemistry. | ||
Tetraazaporphyrin derivative 71 (Fig. 27) self-assembled in a columnar nematic mesophase state; photopolymerisation (254 nm irradiation) gave a multi-functional, soluble nematic polydiacetylene derivative.185 The polymer was sufficiently soluble for solution processing into thin films. This concept was developed further by Tao et al. in 2020 with hexa-peri-hexabenzocoronene derivative 72. Both film and solution states of the columnar LCs of 72 were polymerised thermally or photochemically to afford a polydiacetylene derivative locked into a columnar structure.186 Further work can be expected on exploiting monomeric oligoynes that possess LC properties to achieve both local and long-range charge-transport in polydiacetylene derivatives.
 | ||
| Fig. 27 Structures of 71 and 72. | ||
4.7 Biomedical applications of oligoynes
Recent reports of the use of oligoyne derivatives in photodynamic therapy, endothelial cell staining and imaging of single organelles in live cells all suggest this is a growing area of oligoyne chemistry with promising future biomedical applications. In all cases the oligoyne unit plays a beneficial role in extending the π-system of the chromophore. Photodynamic therapy (PDT) utilises photosensitizers (PSs) under light irradiation to generate reactive oxygen species (ROS) that are cytotoxic against solid tumours. PSs with long-wavelength excitation enhance therapeutic efficacy, as they have lower autofluorescence, deeper tissue penetration and cause less photodamage than higher energy emitters.187In 2017 Zhang, Tang and coworkers reported fluorophore 73 (Fig. 28) containing an octatetraynyl bridge: the terminal tetraphenylethylene units prevent intramolecular π–π stacking and impart aggregation induced emission (AIE) properties due to restriction of intramolecular motion.188 Solvatochromism studies and DFT calculations revealed that the oligoyne unit extends the π-conjugation and acts as an electron acceptor to promote intramolecular charge transfer: these effects combine to red shift the emission of 73 to λmax 630 nm in the solid state. AIE dots of 73 were fabricated and conjugated with the monoclonal antibody cetuximab affording dots with high photostability and excellent specificity for imaging lung cancer cells in vitro through receptor-mediated endocytosis. AIE dots incorporating compound 73 have subsequently been used for in vivo tumour metabolic labelling and targeted imaging in mice.189 Almost no side toxicity was detected in the major organs of the mice.
 | ||
| Fig. 28 Structures of 73–75. | ||
The dianthrylbutadiynyl derivative 74 functionalized with water-solubilising polymer chains is a red emitter (λmax 654 nm in water) and an agent for two-photon microscopy with good selectivity for high-contrast endothelial staining of cerebral blood vessels.190 The dye 74 showed sufficient PLQY and high photostability to allow pathological phenomena to be monitored over several hours. After intravenous injection the brain tissue of a mouse was imaged to a depth of 620 μm, which is significantly deeper than many other red/NIR-emitting two-photon fluorophores. 74 was identified as a versatile compound for a broad range of applications related to microscopic vascular imaging, both in vivo and ex vivo.
The intensity of the Raman bands of oligoynes is much enhanced compared to monoynes. Consequently, butadiynyl derivatives have been used as covalently-bound tags for Raman imaging and semi-quantitative estimation of mobile small molecules, such as coenzyme Q analogues, in HeLa cells.191 Marder and coworkers have recently extended this concept by combining Raman imaging and fluorescence spectroscopy to study the tetracationic bis-triarylborane derivative 75 for intracellular selective sensing of several types of DNA, RNA and proteins.192 A comparison with model analogues showed that the rod-like dumbbell structure of 75 was crucial for strong interactions with ss-RNA. Circular dichroism results suggested that the ss-RNA chain wrapped around 75, which is an unusual mode of binding of small molecules to RNA. Preliminary screening showed that 75 very efficiently entered human HeLa cells without affecting cell viability up to 10−4 M concentrations.
Optical multiplexing bioassays are important for in vitro and in vivo bioimaging and biosensing. Fluorescence and surface enhanced Raman scattering (SERS) are the key readout signals.193 In 2018 Min et al. reported a library of oligoynes (from butadiynyl to dodecahexaynyl derivatives) as optical barcodes for super-multiplexing based on the molecules’ distinct Raman frequencies. By engineering the conjugation length, bond-selective 13C isotope doping, and various end-capping groups, twenty distinct barcodes were obtained – termed a “Carbon rainbow” or “Carbow”.194 In particular, the frequency of the single, intense, narrow Raman peak decreased almost linearly on progressing from C4 to C12 derivatives making these oligoynes an ideal scaffold for optical multiplexing in the cell-silent Raman spectral window. The oligoynes were converted into live-cell organelle-specific imaging probes by functionalisation of the terminal hydroxy groups to give e.g. structure 76 for mitochondria and 77 for endoplasmic reticulum (Fig. 29). Five organelle-targeted probes and five fluorescent reporters were combined to achieve tandem ten-colour optical imaging of subcellular structures in live cells, without any mixing or colour compensation. The desirable combination of good specificity, sensitivity, photostability and live-cell compatibility was demonstrated.
 | ||
| Fig. 29 Structures of 76 and 77. | ||
5. Summary and outlook
In the last twenty years oligoynes have developed from being curious and unpredictable structures of specialised interest into a versatile mainstream component of molecular electronic and optoelectronic materials. Ingenious synthetic chemistry has made oligoynes widely available for experimental studies in fundamental and applied sciences. The judicious choice of end-groups and encapsulation techniques have successfully overcome the inherent instability of longer oligoynes. Nonetheless, 4–6 contiguous triple bonds is generally the length limit for practical applications. Long polyynes and the elusive carbyne remain alluring targets for dedicated synthetic chemists and theoreticians. The unique longitudinal properties of sp-hybridised carbon wires will continue to offer new prospects for device applications of carbon nanostructures.In the next few years, oligoynes are likely to continue to gain momentum in the fields of molecular electronics and optoelectronics discussed above. New applications are eagerly anticipated in topics such as organic field effect transistors,195 solar cells,196 thermoelectrics,86 spintronics197 and nanomechanical systems,198 where they are essentially unexplored. Remarkable properties have been theoretically predicted, but not yet realised, for sp-hybridised carbon wires, including negative differential resistance (NDR),115 rectifying behaviour,199 nanoelectronic/spintronic devices,200,201 hydrogen storage202 and thermoelectric power generation.203 Magnetic interactions in sp carbon wires have been only rarely reported, although there are some promising results for butadiynyl systems with terminal spin labels.204,205
This review will conclude by summarising two inspiring theoretical studies which give a glimpse into a possible future of polyynes. Calculations suggested that carbyne is one of the strongest materials ever considered, with a very high value of the normal Young's modulus (up to 32.7 TPa), extreme tensile stiffness, and a specific stiffness which is a 2–3 fold improvement over the stiffest known materials, including CNTs, graphene, and diamond.206 Thermal conductivity has been estimated to reach extremely high values (200–80 kW m−1 K−1 for cumulenes and polyynes at room temperature) that are well above the maximum values of graphene (5 kW m−1 K−1) and nanotubes (3.5 kW m−1 K−1). Such super-high thermal conductivity was attributed to high phonon frequencies and a long phonon mean free path allowing ballistic thermal transport up to the micron-scale.207 Perhaps true 1D systems like oligoynes and polyynes will soon enter the vast playground occupied by 2D graphene and its molecular counterparts on equal terms!
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by EPSRC grant EP/P027520/1 and EC H2020 FET Open projects: grant agreement numbers 767187 ‘QuIET’ and 766853 ‘EFINED’.References
- H. Bronstein, C. B. Nielsen, B. C. Schroeder and I. McCulloch, Nat. Rev. Chem., 2020, 4, 66–77 Search PubMed.
- Electronic Materials: The Oligomer Approach, ed. K. Mullen and G. Wegner, Wiley, Weinheim, 1998 Search PubMed.
- Handbook of Conducting Polymers, ed. J. R. Reynolds, B. C. Thompson and T. A. Skotheim, CRC Press, 4th edn, 2019 Search PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 Search PubMed.
- M. B. Avinash and T. Govindaraju, Acc. Chem. Res., 2018, 51, 414–426 Search PubMed.
- J. Li and K. Pu, Chem. Soc. Rev., 2019, 8, 38–71 Search PubMed.
- J. Roncali, Chem. Rev., 1997, 97, 173–205 Search PubMed.
- Organic Synthesis and Molecular Engineering, ed. M. B. Nielsen, Wiley, 2013 Search PubMed.
- A. Karpfen, J. Phys. C: Solid State Phys., 1979, 12, 3227–3237 Search PubMed.
- F. Banhart, Beilstein J. Nanotechnol., 2015, 6, 559–569 Search PubMed.
- S. Cahangirov, M. Topsakal and S. Ciraci, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 195444 Search PubMed.
- M. H. Garner, W. Bro-Jørgensen and G. C. Solomon, J. Phys. Chem. C, 2020, 124, 18968–18982 Search PubMed.
- H. A. Pohl, J. Polym. Sci., Part C: Polym. Symp., 1967, 17, 13–40 Search PubMed.
- D. J. Berets and D. S. Smith, Trans. Faraday Soc., 1968, 64, 823–828 Search PubMed.
- G. Wegner, Z. Naturforsch. B, 1969, 24, 824–832 Search PubMed.
- R. H. Baughman, J. Appl. Phys., 1972, 43, 4362–4370 Search PubMed.
- S. Lee, J.-Y. Kim, X. Chen and J. Yoon, Chem. Commun., 2016, 52, 9178–9196 Search PubMed.
- J. L. Delgado, M. A. Herranz and N. Martín, J. Mater. Chem., 2008, 18, 1417–1426 Search PubMed.
- A. Hirsch, Nat. Mater., 2010, 9, 868–871 Search PubMed.
- Y. Segawa, H. Ito and K. Itami, Nat. Rev. Mater., 2016, 1, 15002 Search PubMed.
- C. S. Casari and A. Milani, MRS Commun., 2018, 8, 207–219 Search PubMed.
- R. E. Martin and F. Diederich, Angew. Chem., Int. Ed., 1999, 38, 1350–1377 Search PubMed.
- In early literature the name “polyacetylene” was used for compounds with contiguous acetylene units. This is, however, no longer appropriate because polyacetylene has become the accepted nomenclature for polymerised acetylene, formula (–HCCH)n.
- W. W. Duley and A. Hu, Astrophys. J., 2009, 698, 808–811 Search PubMed.
- J. August, H. W. Kroto and N. Trinajstic, Astrophys. Space Sci., 1986, 128, 411–419 Search PubMed.
- P. Thaddeus, M. C. McCarthy, M. J. Travers, C. A. Gottlieb and W. Chen, Faraday Discuss., 1998, 109, 121–135 Search PubMed.
- A. L. K. Shi Shun and R. R. Tykwinski, Angew. Chem., Int. Ed., 2006, 45, 1034–1057 Search PubMed.
- B. W. Gung, C. R. Chim., 2009, 12, 489–505 Search PubMed.
- A. M. Sladkov and Y. P. Kudryavtsev, Russ. Chem. Rev., 1963, 32, 229–243 Search PubMed.
- C. S. Casari, M. Tommasini, R. R. Tykwinski and A. Milani, Nanoscale, 2016, 8, 4414–4435 Search PubMed.
- J. A. Januszewski and R. R. Tykwinski, Chem. Soc. Rev., 2014, 43, 3184–3203 Search PubMed.
- K. Zhang, Y. Zhang and L. Shi, Chin. Chem. Lett., 2020, 31, 1746–1756 Search PubMed.
- Polyynes: Synthesis, Properties, and Applications, ed. F. Cataldo, CRC, Boca Raton, FL, 2005 Search PubMed.
- W. A. Chalifoux and R. R. Tykwinski, C. R. Chim., 2009, 12, 341–358 Search PubMed.
- S. Frankenberger, J. A. Januszewski and R. R. Tykwinski, in Fullerenes and other Carbon-Rich Nanostructures, Series: Structure and Bonding, ed. J.-F. Nierengarten, Springer, 2014, vol. 159, pp. 219–256 Search PubMed.
- P. Siemsen, R. C. Livingston and F. Diederich, Angew. Chem., Int. Ed., 2000, 39, 2633–2657 Search PubMed.
- M. Jevric and M. B. Nielsen, Asian J. Org. Chem., 2015, 4, 286–295 Search PubMed.
- C. Glaser, Ber. Dtsch. Chem. Ges., 1869, 2, 422–424 Search PubMed.
- A. Baeyer, Ber. Dtsch. Chem. Ges., 1885, 18, 2269–2281 Search PubMed.
- J. B. Armitage, N. Entwistle, E. R. H. Jones and M. C. Whiting, J. Chem. Soc., 1954, 147–154 Search PubMed.
- D. F. Perepichka and S. Jeeva, Chem. Eng. News, 2010, 88(3), 2 Search PubMed . http://cen.acs.org/articles/88/i3/Chemical-Safety-Trimethylsilylacetylene-Explosion.html.
- K. Gao and N. S. Goroff, J. Am. Chem. Soc., 2000, 122, 9320–9321 Search PubMed.
- R. H. Baughman, Science, 2006, 312, 1009–1010 Search PubMed.
- A safe and simple synthesis of the key building block 1,4-bis(trimethylsilyl)buta-1,3-diyne has been reported recently: S. Bock and P. J. Low, Aust. J. Chem., 2018, 71, 307–310 Search PubMed.
- E. R. H. Jones, M. C. Whiting, J. B. Armitage, C. L. Cook and N. Entwhistle, Nature, 1951, 168, 900–903 Search PubMed.
- F. Bohlmann, Angew. Chem., Int. Ed. Engl., 1953, 65, 385–408 Search PubMed.
- F. Bohlmann, Chem. Ber./Recl., 1953, 86, 657–667 Search PubMed.
- R. Eastmond, T. R. Johnson and D. R. M. Walton, Tetrahedron, 1972, 28, 4601–4616 Search PubMed.
- F. Cataldo, Polyhedron, 2004, 23, 1889–1896 Search PubMed.
- S. Peggiani, P. Marabotti, R. A. Lotti, A. Facibeni, P. Serafini, A. Milani, V. Russo, A. L. Bassi and C. S. Casari, Phys. Chem. Chem. Phys., 2020, 22, 26312–26321 Search PubMed.
- T. Gibtner, F. Hampel, J.-P. Gisselbrecht and A. Hirsch, Chem. – Eur. J., 2002, 8, 408–432 Search PubMed.
- S. M. E. Simpkins, M. D. Weller and L. R. Cox, Chem. Commun., 2007, 4035–4037 Search PubMed.
- S. L. Woltering, P. Gawel, K. E. Christensen, A. L. Thompson and H. L. Anderson, J. Am. Chem. Soc., 2020, 142, 13523–13532 Search PubMed.
- C. Wang, A. S. Batsanov, K. West and M. R. Bryce, Org. Lett., 2008, 10, 3069–3072 Search PubMed.
- T. Bartik, B. Bartik, M. Brady, R. Dembinski and J. A. Gladysz, Angew. Chem., Int. Ed. Engl., 1996, 35, 414–417 Search PubMed.
- W. Mohr, J. Stahl, F. Hampel and J. A. Gladysz, Chem. – Eur. J., 2003, 9, 3324–3340 Search PubMed.
- Q. Zheng, J. C. Bohling, T. B. Peters, A. C. Frisch, F. Hampel and J. A. Gladysz, Chem. – Eur. J., 2006, 12, 6486–6505 Search PubMed.
- S. Eisler, N. Chahal, R. McDonald and R. R. Tykwinski, Chem. – Eur. J., 2003, 9, 2542–2550 Search PubMed.
- W. A. Chalifoux and R. R. Tykwinski, Chem. Rec., 2006, 6, 169–182 Search PubMed.
- W. A. Chalifoux and R. R. Tykwinski, Nat. Chem., 2010, 2, 967–971 Search PubMed.
- Q. Zheng, J. F. Schneider, H. Amini, F. Hampel and J. A. Gladysz, Dalton Trans., 2019, 48, 5800–5816 Search PubMed.
- P. Bichler, W. A. Chalifoux, S. Eisler, A. L. K. Shi Shun, E. T. Chernick and R. R. Tykwinski, Org. Lett., 2009, 11, 519–522 Search PubMed.
- A. F. Hill and R. A. Manzano, Angew. Chem., Int. Ed., 2019, 58, 15354–15357 Search PubMed.
- A. Milani, M. Tommasini, V. Barbieri, A. Lucotti, V. Russo, F. Cataldo and C. S. Casari, J. Phys. Chem. C, 2017, 121, 10562–10570 Search PubMed.
- S. Yang and M. Kertesz, J. Phys. Chem. A, 2006, 110, 9771–9774 Search PubMed.
- E. Mostaani, B. Monserrat, N. D. Drummond and C. J. Lambert, Phys. Chem. Chem. Phys., 2016, 18, 14810–14821 Search PubMed.
- A. Al-Backri, V. Zólyomi and C. J. Lambert, J. Chem. Phys., 2014, 140, 104306 Search PubMed.
- J. Zirzlmeier, S. Schrettl, J. C. Brauer, E. Contal, L. Vannay, É. Brémond, E. Jahnke, D. M. Guldi, C. Corminboeuf, R. R. Tykwinski and H. Frauenrath, Nat. Commun., 2020, 11, 4797 Search PubMed.
- A. Lucotti, M. Tommasini, D. Fazzi, M. Del Zoppo, W. A. Chalifoux, M. J. Ferguson, G. Zerbi and R. R. Tykwinski, J. Am. Chem. Soc., 2009, 131, 4239–4244 Search PubMed.
- S. Szafert and J. A. Gladysz, Chem. Rev., 2003, 103, 4175–4205 Search PubMed.
- C. Klinger, O. Vostrowsky and A. Hirsch, Eur. J. Org. Chem., 2006, 1508–1524 Search PubMed.
- L. D. Movsisyan, D. V. Kondratuk, M. Franz, A. L. Thompson, R. R. Tykwinski and H. L. Anderson, Org. Lett., 2012, 14, 3424–3426 Search PubMed.
- L. Shi, P. Rohringer, K. Suenaga, Y. Niimi, J. Kotakoski, J. C. Meyer, H. Peterlik, M. Wanko, S. Cahangirov, A. Rubio, Z. J. Lapin, L. Novotny, P. Ayala and T. Pichler, Nat. Mater., 2016, 15, 634–640 Search PubMed.
- L. Shi, K. Yanagi, K. Cao, U. Kaiser, P. Ayala and T. Pichler, ACS Nano, 2018, 12, 8477–8484 Search PubMed.
- E. Scheer and J. C. Cuevas, Molecular Electronics: An Introduction to Theory and Experiment, World Scientific, Singapore, 2nd edn, 2017 Search PubMed.
- M. Ratner, Nat. Nanotechnol., 2013, 8, 378–381 Search PubMed.
- S. Marquez-Gonzalez and P. J. Low, Aust. J. Chem., 2016, 69, 244–253 Search PubMed.
- L. Herrer, S. Martin and P. Cea, Appl. Sci., 2020, 10, 6064 Search PubMed.
- X. Wang, T. L. R. Bennett, A. Ismael, L. A. Wilkinson, J. Hamill, A. J. P. White, I. M. Grace, O. V. Kolosov, T. Albrecht, B. J. Robinson, N. J. Long, L. F. Cohen and C. J. Lambert, J. Am. Chem. Soc., 2020, 142, 8555–8560 Search PubMed.
- E. Leary, A. La Rosa, M. T. González, G. Rubio-Bollinger, N. Agraït and N. Martín, Chem. Soc. Rev., 2015, 44, 920–942 Search PubMed.
- T. A. Su, M. Neupane, M. L. Steigerwald, L. Venkataraman and C. Nuckolls, Nat. Rev. Mater., 2016, 1, 16002 Search PubMed.
- L. Sun, Y. A. Diaz-Fernandez, T. A. Gschneidtner, F. Westerlund, S. Lara-Avilab and K. Moth-Poulsen, Chem. Soc. Rev., 2014, 43, 7378–7411 Search PubMed.
- J. Liu, X. Zhao, Q. Al-Galiby, X. Huang, J. Zheng, R. Li, C. Huang, Y. Yang, J. Shi, D. Z. Manrique, C. J. Lambert, M. R. Bryce and W. Hong, Angew. Chem., Int. Ed., 2017, 56, 13061–13065 Search PubMed.
- M. Baghernejad, C. Van Dyck, J. Bergfield, D. R. Levine, A. Gubicza, J. D. Tovar, M. Calame, P. Broekmann and W. Hong, Chem. – Eur. J., 2019, 25, 15141–15146 Search PubMed.
- P. Li, C. Jia and X. Guo, Chem. Rec., 2020 DOI:10.1002/tcr.202000114.
- N. Mosso, H. Sadeghi, A. Gemma, S. Sangtarash, U. Drechsler, C. Lambert and B. Gotsmann, Nano Lett., 2019, 19, 7614–7622 Search PubMed.
- B. Q. Xu and N. J. Tao, Science, 2003, 301, 1221–1223 Search PubMed.
- W. Hong, H. Valkenier, G. Meszaros, D. Z. Manrique, A. Mishchenko, A. Putz, P. M. García, C. J. Lambert, J. C. Hummelen and T. Wandlowski, Beilstein J. Nanotechnol., 2011, 2, 699–713 Search PubMed.
- F. Chen, J. Hihath, Z. Huang, X. Li and N. J. Tao, Annu. Rev. Phys. Chem., 2007, 58, 535–564 Search PubMed.
- C. J. Lambert, Chem. Soc. Rev., 2015, 44, 875–888 Search PubMed.
- F. Evers, R. Korytar, S. Tewari and J. M. van Ruitenbeek, Rev. Mod. Phys., 2020, 92, 035001 Search PubMed.
- N. D. Lang and P. Avouris, Phys. Rev. Lett., 1998, 81, 3515–3518 Search PubMed.
- N. D. Lang and P. Avouris, Phys. Rev. Lett., 2000, 84, 358–361 Search PubMed.
- C. Wang, A. S. Batsanov, M. R. Bryce, S. Martin, R. J. Nichols, S. J. Higgins, V. M. García-Suárez and C. Lambert, J. Am. Chem. Soc., 2009, 131, 15647–15654 Search PubMed.
- P. Moreno-Garcia, M. Gulcur, D. Z. Manrique, T. Pope, W. Hong, V. Kaliginedi, C. Huang, A. S. Batsanov, M. R. Bryce, C. Lambert and T. Wandlowski, J. Am. Chem. Soc., 2013, 135, 12228–12240 Search PubMed.
- M. Gulcur, P. Moreno-Garcia, X. Zhao, M. Baghernejad, A. S. Batsanov, W. Hong, M. R. Bryce and T. Wandlowski, Chem. – Eur. J., 2014, 20, 4653–4660 Search PubMed.
- M. Naher, D. C. Milan, O. A. Al-Owaedi, I. J. Planje, S. Bock, J. Hurtado-Gallego, P. Bastante, Z. M. A. Dawood, L. Rincón-García, G. Rubio-Bollinger, S. J. Higgins, N. Agraït, C. J. Lambert, R. J. Nichols and P. J. Low, J. Am. Chem. Soc., 2021, 143, 3817–3829 Search PubMed.
- S. Ballmann, R. Härtle, P. B. Coto, M. Elbing, M. Mayor, M. R. Bryce, M. Thoss and H. B. Weber, Phys. Rev. Lett., 2012, 109, 056801 Search PubMed.
- S. Ballmann, W. Hieringer, R. Härtle, P. B. Coto, M. R. Bryce, A. Görling, M. Thoss and H. B. Weber, Phys. Status Solidi B, 2014, 250, 2452–2457 Search PubMed.
- S. Ballmann, W. Hieringer, D. Secker, Q. Zheng, J. A. Gladysz, A. Gorling and H. B. Weber, ChemPhysChem, 2010, 11, 2256–2260 Search PubMed.
- D. C. Milan, O. A. Al-Owaedi, M.-C. Oerthel, S. Marques-Gonzalez, R. J. Brooke, M. R. Bryce, P. Cea, J. Ferrer, S. J. Higgins, C. J. Lambert, P. J. Low, D. Z. Manrique, S. Martin, R. J. Nichols, W. Schwarzacher and V. M. Garcia-Suarez, J. Phys. Chem. C, 2016, 120, 15666–15674 Search PubMed.
- Z. Tang, S. Hou, Q. Wu, Z. Tan, J. Zheng, R. Li, J. Liu, Y. Yang, H. Sadeghi, J. Shi, I. Grace, C. J. Lambert and W. Hong, Sci. Bull., 2020, 65, 944–950 Search PubMed.
- A. Moneo, A. González-Orive, S. Bock, M. Fenero, L. Herrer, D. C. Milan, M. Lorenzoni, R. J. Nichols, P. Cea, F. Perez-Murano, P. J. Low and S. Martin, Nanoscale, 2018, 10, 14128–14138 Search PubMed.
- W. Hong, H. Li, S.-X. Liu, Y. Fu, J. Li, V. Kaliginedi, S. Decurtins and T. Wandlowski, J. Am. Chem. Soc., 2012, 134, 19425–19431 Search PubMed.
- D. C. Milan, M. Krempe, A. K. Ismael, L. D. Movsisyan, M. Franz, I. Grace, R. J. Brooke, W. Schwarzacher, S. J. Higgins, H. L. Anderson, C. J. Lambert, R. R. Tykwinski and R. J. Nichols, Nanoscale, 2017, 9, 355–361 Search PubMed.
- P. J. Low, Dalton Trans., 2005, 2821–2824 Search PubMed.
- D. C. Milan, A. Vezzoli, I. J. Planje and P. J. Low, Dalton Trans., 2018, 47, 14125–14138 Search PubMed.
- S. J. Higgins and R. J. Nichols, Polyhedron, 2018, 140, 25–34 Search PubMed.
- Y. Tanaka, M. Kiguchi and M. Akita, Chem. – Eur. J., 2017, 23, 4741–4749 Search PubMed.
- A. Haque, R. A. Al-Balushi, I. J. Al-Busaidi, M. S. Khan and P. R. Raithby, Chem. Rev., 2018, 118, 8474–8597 Search PubMed.
- F. Schwarz, G. Kastlunger, F. Lissel, C. Egler-Lucas, S. N. Semenov, K. Venkatesan, H. Berke, R. Stadler and E. Lörtscher, Nat. Nanotechnol., 2016, 11, 170–176 Search PubMed.
- O. A. Al-Owaedi, S. Bock, D. C. Milan, M.-C. Oerthel, M. S. Inkpen, D. S. Yufit, A. N. Sobolev, N. J. Long, T. Albrecht, S. J. Higgins, M. R. Bryce, R. J. Nichols, C. J. Lambert and P. J. Low, Nanoscale, 2017, 9, 9902–9912 Search PubMed.
- Y. Tanaka, Y. Kato, T. Tada, S. Fujii, M. Kiguchi and M. Akita, J. Am. Chem. Soc., 2018, 140, 10080–10084 Search PubMed.
- I. J. Olavarria-Contreras, M. L. Perrin, Z. Chen, S. Klyatskaya, M. Ruben and H. S. J. van der Zant, J. Am. Chem. Soc., 2016, 138, 8465–8469 Search PubMed.
- Y.-D. Guo, J.-J. Wang, H.-L. Zeng, Y.-S. Ran, Z.-C. Liang and X.-H. Yan, J. Appl. Phys., 2020, 128, 184301 Search PubMed.
- Y. Tanaka, K. Ohmura, S. Fujii, T. Tada, M. Kiguchi and M. Akita, Inorg. Chem., 2020, 59, 13254–13261 Search PubMed.
- L.-Y. Zhang, P. Duan, J.-Y. Wang, Q.-C. Zhang and Z.-N. Chen, J. Phys. Chem. C, 2019, 123, 5282–5288 Search PubMed.
- G. Sedghi, K. Sawada, L. J. Esdaile, M. Hoffmann, H. L. Anderson, D. Bethell, W. Haiss, S. J. Higgins and R. J. Nichols, J. Am. Chem. Soc., 2008, 130, 8582–8583 Search PubMed.
- G. Sedghi, V. M. Garcia-Suarez, L. J. Esdaile, H. L. Anderson, C. J. Lambert, S. Martin, D. Bethell, S. J. Higgins, M. Elliott, N. Bennett, J. E. Macdonald and R. J. Nichols, Nat. Nanotechnol., 2011, 6, 517–523 Search PubMed.
- H.-M. Wen, Y. Yang, X.-S. Zhou, J.-Y. Liu, D.-B. Zhang, Z.-B. Chen, J.-Y. Wang, Z.-N. Chen and Z.-Q. Tian, Chem. Sci., 2013, 4, 2471–2477 Search PubMed.
- F. Schwarz, G. Kastlunger, F. Lissel, H. Riel, K. Venkatesan, H. Berke, R. Stadler and E. Lortscher, Nano Lett., 2014, 14, 5932–5940 Search PubMed.
- B. Standley, W. Bao, H. Zhang, J. Bruck, C. N. Lau and M. Bockrath, Nano Lett., 2008, 8, 3345–3349 Search PubMed.
- C. Jin, H. Lan, L. Peng, K. Suenaga and S. Iijima, Phys. Rev. Lett., 2009, 102, 205501 Search PubMed.
- J. Prasongkit, A. Grigoriev and R. Ahuja, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 155434 Search PubMed.
- O. Cretu, A. R. Botello-Mendez, I. Janowska, C. Pham-Huu, J.-C. Charlier and F. Banhart, Nano Lett., 2013, 13, 3487–3493 Search PubMed.
- A. La Torre, A. Botello-Mendez, W. Baaziz, J.-C. Charlier and F. Banhart, Nat. Commun., 2015, 6, 6636 Search PubMed.
- D. Wendinger and R. R. Tykwinski, Acc. Chem. Res., 2017, 50, 1468–1479 Search PubMed.
- M. H. Garner, W. Bro-Jørgensen, P. D. Pedersen and G. C. Solomon, J. Phys. Chem. C, 2018, 122, 26777–26789 Search PubMed.
- W. Xu, E. Leary, S. Hou, S. Sangtarash, M. T. Gonzalez, G. Rubio-Bollinger, Q. Wu, H. Sadeghi, L. Tejerina, K. E. Christensen, N. Agrait, S. J. Higgins, C. J. Lambert, R. J. Nichols and H. L. Anderson, Angew. Chem., Int. Ed., 2019, 58, 8378–8382 Search PubMed.
- Y. Zang, T. Fu, Q. Zou, F. Ng, H. Li, M. L. Steigerwald, C. Nuckolls and L. Venkataraman, Nano Lett., 2020, 20, 8415–8419 Search PubMed.
- D. J. Williams, Angew. Chem., Int. Ed. Engl., 1984, 23, 690–703 Search PubMed.
- N. J. Long, Angew. Chem., Int. Ed. Engl., 1995, 34, 21–38 Search PubMed.
- A. D. Slepkov, F. A. Hegmann, S. Eisler, E. Elliott and R. R. Tykwinski, J. Chem. Phys., 2004, 120, 6807–6810 Search PubMed.
- S. Eisler, A. D. Slepkov, E. Elliott, T. Luu, R. McDonald, F. A. Hegmann and R. R. Tykwinski, J. Am. Chem. Soc., 2005, 127, 2666–2676 Search PubMed.
- T. Luu, E. Elliott, A. D. Slepkov, S. Eisler, R. McDonald, F. A. Hegmann and R. R. Tykwinski, Org. Lett., 2005, 7, 51–54 Search PubMed.
- A. Lucotti, M. Tommasini, D. Fazzi, M. Del Zoppo, W. A. Chalifoux, R. R. Tykwinski and G. Zerbi, J. Raman Spectrosc., 2012, 43, 1293–1298 Search PubMed.
- N. R. Agarwal, A. Lucotti, M. Tommasini, W. A. Chalifoux and R. R. Tykwinski, J. Phys. Chem. C, 2016, 120, 11131–11139 Search PubMed.
- A. E. Stiegman, E. Graham, K. J. Perry, L. R. Khundkar, L.-T. Cheng and J. W. Perry, J. Am. Chem. Soc., 1991, 113, 7658–7666 Search PubMed.
- E. M. Graham, V. M. Miskowski, J. W. Perry, D. R. Coulter, A. E. Stiegman, W. P. Schaefer and R. E. Marsh, J. Am. Chem. Soc., 1989, 111, 8771–8779 Search PubMed.
- M. Stefko, M. D. Tzirakis, B. Breiten, M.-O. Ebert, O. Dumele, W. B. Schweizer, J.-P. Gisselbrecht, C. Boudon, M. T. Beels, I. Biaggio and F. Diederich, Chem. – Eur. J., 2013, 19, 12693–12704 Search PubMed.
- B. B. Frank, P. R. Laporta, B. Breiten, M. C. Kuzyk, P. D. Jarowski, W. B. Schweizer, P. Seiler, I. Biaggio, C. Boudon, J.-P. Gisselbrecht and F. Diederich, Eur. J. Org. Chem., 2011, 4307–4317 Search PubMed.
- S. Gauthier, A. Porter, S. Achelle, T. Roisnel, V. Dorcet, A. Barsella, N. Le Poul, P. G. Level, D. Jacquemin and F. Robin-Le Guen, Organometallics, 2018, 37, 2232–2244 Search PubMed.
- H. Wang, C.-J. Yao, H.-J. Nie, L. Yang, S. Meia and Q. Zhang, J. Mater. Chem. C, 2020, 8, 15507–15525 Search PubMed.
- R. J. Mortimer, A. L. Dyer and J. R. Reynolds, Displays, 2006, 27, 2–18 Search PubMed.
- C. Wang, A. S. Batsanov and M. R. Bryce, Chem. Commun., 2004, 578–579 Search PubMed.
- G. Chen, I. Mahmud, L. N. Dawe, L. M. Daniels and Y. Zhao, J. Org. Chem., 2011, 76, 2701–2715 Search PubMed.
- F. Paul and C. Lapinte, Coord. Chem. Rev., 1998, 178–180, 431–509 Search PubMed.
- M. I. Bruce and P. J. Low, Adv. Organomet. Chem., 2004, 50, 179–444 Search PubMed.
- G.-L. Xu, G. Zou, Y.-H. Ni, M. C. DeRosa, R. J. Crutchley and T. Ren, J. Am. Chem. Soc., 2003, 125, 10057–10065 Search PubMed.
- B. Xi, I. P.-C. Liu, G.-L. Xu, M. M. R. Choudhuri, M. C. DeRosa, R. J. Crutchley and T. Ren, J. Am. Chem. Soc., 2011, 133, 15094–15104 Search PubMed.
- J.-W. Ying, Z. Cao, C. Campana, Y. Song, J.-L. Zuo, S. F. Tyler and T. Ren, Polyhedron, 2015, 86, 76–80 Search PubMed.
- M. I. Bruce, P. J. Low, K. Costuas, J.-F. Halet, S. P. Best and G. A. Heath, J. Am. Chem. Soc., 2000, 122, 1949–1962 Search PubMed.
- M. Parthey, J. B. G. Gluyas, P. A. Schauer, D. S. Yufit, J. A. K. Howard, M. Kaupp and P. J. Low, Chem. – Eur. J., 2013, 19, 9780–9784 Search PubMed.
- M.-C. Oerthel, D. S. Yufit, M. A. Fox, M. R. Bryce and P. J. Low, Organometallics, 2015, 34, 2395–2405 Search PubMed.
- T. D. Cook, S. N. Natoli, P. E. Fanwick and T. Ren, Organometallics, 2016, 35, 1329–1338 Search PubMed.
- J. B. Derr, J. Tamayo, J. A. Clark, M. Morales, M. F. Mayther, E. M. Espinoza, K. Rybicka-Jasinska and V. I. Vullev, Phys. Chem. Chem. Phys., 2020, 22, 21583–21629 Search PubMed.
- H. Imahori, K. Tamaki, D. M. Guldi, C. Luo, M. Fujitsuka, O. Ito, Y. Sakata and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 2607–2617 Search PubMed.
- B. Albinsson, M. P. Eng, K. Pettersson and M. U. Winters, Phys. Chem. Chem. Phys., 2007, 9, 5847–5864 Search PubMed.
- C. Schubert, J. T. Margraf, T. Clark and D. M. Guldi, Chem. Soc. Rev., 2015, 44, 988–998 Search PubMed.
- M. Biswas, P. Nguyen, T. B. Marder and L. R. Khundkar, J. Phys. Chem. A, 1997, 101, 1689–1695 Search PubMed.
- A. E. Stiegman, V. M. Miskowski, J. W. Perry and D. R. Coulter, J. Am. Chem. Soc., 1987, 109, 5884–5886 Search PubMed.
- A. K. Pati, S. J. Gharpure and A. K. Mishra, Faraday Discuss., 2015, 177, 213–235 Search PubMed.
- A. K. Pati, S. J. Gharpure and A. K. Mishra, J. Phys. Chem. A, 2015, 119, 10481–10493 Search PubMed.
- C. Wang, L.-O. Pålsson, A. S. Batsanov and M. R. Bryce, J. Am. Chem. Soc., 2006, 128, 3789–3799 Search PubMed.
- L.-O. Pålsson, C. Wang, A. S. Batsanov, S. M. King, A. Beeby, A. P. Monkman and M. R. Bryce, Chem. – Eur. J., 2010, 16, 1470–1479 Search PubMed.
- A. Zieleniewska, X. Zhao, S. Bauroth, C. Wang, A. S. Batsanov, C. K. Calderon, A. Kahnt, T. Clark, M. R. Bryce and D. M. Guldi, J. Am. Chem. Soc., 2020, 142, 18769–18781 Search PubMed.
- G. Bottari, G. de la Torre, D. M. Guldi and T. Torres, Coord. Chem. Rev., 2021, 428, 213605 Search PubMed.
- S. A. Vail, P. J. Krawczuk, D. M. Guldi, A. Palkar, L. Echegoyen, J. P. C. Tomé, M. A. Fazio and D. I. Schuster, Chem. – Eur. J., 2005, 11, 3375–3388 Search PubMed.
- A. Zieleniewska, F. Lodermeyer, A. Roth and D. M. Guldi, Chem. Soc. Rev., 2018, 47, 702–714 Search PubMed.
- M. U. Winters, E. Dahlstedt, H. E. Blades, C. J. Wilson, M. J. Frampton, H. L. Anderson and B. Albinsson, J. Am. Chem. Soc., 2007, 129, 4291–4297 Search PubMed.
- R. Venkatramani, E. Wierzbinski, D. H. Waldeck and D. N. Beratan, Faraday Discuss., 2014, 174, 57–78 Search PubMed.
- S.-J. Zou, Y. Shen, F.-M. Xie, J.-D. Chen, Y.-Q. Li and J.-X. Tang, Mater. Chem. Front., 2020, 4, 788–820 Search PubMed.
- X. Wu, L. Wang, Y. Hua, C. Wang, A. S. Batsanov and M. R. Bryce, Tetrahedron, 2014, 70, 2015–2019 Search PubMed.
- K. T. Kamtekar, A. P. Monkman and M. R. Bryce, Adv. Mater., 2010, 22, 572–582 Search PubMed.
- R. He, Z. Xu, S. Valandro, H. D. Arman, J. Xue and K. S. Schanze, ACS Appl. Mater. Interfaces, 2021, 13, 5327–5337 Search PubMed.
- R. Steinstrasser and L. Pohl, Angew. Chem., Int. Ed. Engl., 1973, 12, 617–630 Search PubMed.
- G. W. Gray and S. M. Kelly, J. Mater. Chem., 1999, 9, 2037–2050 Search PubMed.
- T. Kato, J. Uchida, T. Ichikawa and T. Sakamoto, Angew. Chem., Int. Ed., 2018, 57, 4355–4371 Search PubMed.
- B. Grant, Mol. Cryst. Liq. Cryst., 1978, 48, 175–182 Search PubMed.
- S.-T. Wu, H.-H. B. Meng and L. R. Dalton, J. Appl. Phys., 1991, 70, 3013–3017 Search PubMed.
- S.-T. Wu, J. D. Margerum, B. H. Meng, L. R. Dalton, C.-S. Hsu and S.-H. Lung, Appl. Phys. Lett., 1992, 61, 630–632 Search PubMed.
- Y. Arakawa, S. Nakajima, R. Ishige, M. Uchimura, S. Kang, G. Konishi and J. Watanabe, J. Mater. Chem., 2012, 22, 8394–8398 Search PubMed.
- Y. Arakawa, S. Kang, S. Nakajima, K. Sakajiri, Y. Cho, S. Kawauchi, J. Watanabe and G. Konishi, J. Mater. Chem. C, 2013, 1, 8094–8102 Search PubMed.
- D.-Y. Kim, S.-A. Lee, D. Jung, J. Koo, J. S. Kim, Y.-T. Yu, C.-R. Lee and K.-U. Jeong, Soft Matter, 2017, 13, 5759–5766 Search PubMed.
- M. N. Tahir, E. Abdulhamied, A. Nyayachavadi, M. Selivanova, S. H. Eichhorn and S. Rondeau-Gagné, Langmuir, 2019, 35, 15158–15167 Search PubMed.
- Y.-L. Kuo, C.-Y. Tseng, C.-W. Tseng, K.-T. Chu, Y.-C. Liu, M.-H. Chiang, A. Saeki, Y.-T. Tao and H.-H. Chen, ACS Appl. Polym. Mater., 2020, 2, 248–255 Search PubMed.
- S. S. Lucky, K. C. Soo and Y. Zhang, Chem. Rev., 2015, 115, 1990–2042 Search PubMed.
- Z. Zhao, H. Su, P. Zhang, Y. Cai, R. T. K. Kwok, Y. Chen, Z. He, X. Gu, X. He, H. H. Y. Sung, I. D. Willimas, J. W. Y. Lam, Z. Zhang and B. Z. Tang, J. Mater. Chem. B, 2017, 5, 1650–1657 Search PubMed.
- P. Zhang, T. Jiang, Y. Li, Z. Zhao, P. Gong, L. Cai, R. T. K. Kwok, J. W. Y. Lam, X. Gu and B. Z. Tang, Chem. – Asian J., 2019, 14, 770–774 Search PubMed.
- B. Mettra, F. Appaix, J. Olesiak-Banska, T. Le Bahers, A. Leung, K. Matczyszyn, M. Samoc, B. van der Sanden, C. Monnereau and C. Andraud, ACS Appl. Mater. Interfaces, 2016, 8, 17047–17059 Search PubMed.
- H. Yamakoshi, K. Dodo, A. Palonpon, J. Ando, K. Fujita, S. Kawata and M. Sodeoka, J. Am. Chem. Soc., 2012, 134, 20681–20689 Search PubMed.
- H. Amini, Z. Ban, M. Ferger, S. Lorenzen, F. Rauch, A. Friedrich, I. Crnolatac, A. Kendel, S. Miljanic, I. Piantanida and T. B. Marder, Chem. – Eur. J., 2020, 26, 6017–6028 Search PubMed.
- Y. Fan, S. Wang and F. Zhang, Angew. Chem., Int. Ed., 2019, 58, 13208–13219 Search PubMed.
- F. Hu, C. Zeng, R. Long, Y. Miao, L. Wei, Q. Xu and W. Min, Nat. Methods, 2018, 15, 194–200 Search PubMed.
- A. D. Scaccabarozzi, A. Milani, S. Peggiani, S. Pecorario, B. Sun, R. R. Tykwinski, M. Caironi and C. S. Casari, J. Phys. Chem. Lett., 2020, 11, 1970–1974 Search PubMed.
- B. J. Eckstein, F. S. Melkonyan, N. Zhou, E. F. Manley, J. Smith, A. Timalsina, R. P. H. Chang, L. X. Chen, A. Facchetti and T. J. Marks, Macromolecules, 2017, 50, 1430–1441 Search PubMed.
- L. Guo, X. Gu, X. Zhu and X. Sun, Adv. Mater., 2019, 31, 1805355 Search PubMed.
- Y. Han, J. Zhou, H. Wang, L. Gao, S. Feng, K. Cao, Z. Xu and Y. Lu, Appl. Nanosci., 2021 DOI:10.1007/s13204-021-01702-0.
- M. Qiu, Z. H. Zhang, X. Q. Deng and J. B. Pan, Appl. Phys. Lett., 2010, 97, 242109 Search PubMed.
- Z. Zanolli, G. Onida and J.-C. Charlier, ACS Nano, 2010, 4, 5174–5180 Search PubMed.
- M. G. Zeng, L. Shen, Y. Q. Cai, Z. D. Sha and Y. P. Feng, Appl. Phys. Lett., 2010, 96, 042104 Search PubMed.
- P. B. Sorokin, H. Lee, L. Y. Antipina, A. K. Singh and B. I. Yakobson, Nano Lett., 2011, 11, 2660–2665 Search PubMed.
- H. Sadeghi, S. Sangtarash and C. J. Lambert, Nano Lett., 2015, 15, 7467–7472 Search PubMed.
- E. Hernandez-Gasio, M. Mas, E. Molins, C. Rovira, J. Veciana, J. J. Borras-Almenar and E. Coronado, Chem. Mater., 1994, 6, 2398–2411 Search PubMed.
- E. V. Tretyakov, S. E. Tolstikov, G. V. Romanenko, A. S. Bogomyakov, D. V. Stass, M. K. Kadirov, K. V. Holin, O. G. Sinyashin and V. I. Ovcharenko, Polyhedron, 2011, 30, 3232–3237 Search PubMed.
- M. Liu, V. I. Artyukhov, H. Lee, F. Xu and B. I. Yakobson, ACS Nano, 2013, 7, 10075–10082 Search PubMed.
- M. Wang and S. Lin, Sci. Rep., 2015, 5, 18122 Search PubMed.
Footnote |
| † This article honours the pioneering contributions of Professor Jaume Veciana and Professor Concepció Rovira in materials chemistry. |
| This journal is © The Royal Society of Chemistry 2021 |