
The role of polymers in lithium solid-state batteries with inorganic solid electrolytes
Sudeshna
Sen
,
Enrico
Trevisanello
 ,
Elard
Niemöller
,
Bing-Xuan
Shi
,
Fabian J.
Simon†
and
Felix H.
Richter
*
,
Elard
Niemöller
,
Bing-Xuan
Shi
,
Fabian J.
Simon†
and
Felix H.
Richter
*
Center for Materials Research, Institute of Physical Chemistry, Justus-Liebig-Universität Gießen, Heinrich-Buff-Ring 17, 35392 Gießen, Germany. E-mail: felix.h.richter@pc.jlug.de
First published on 20th July 2021
Abstract
Solid-state batteries have gained increasing attention with the discovery of new inorganic solid electrolytes, some of which rival the ionic conductivity of liquid electrolytes. With the additional benefit of being single-ion conductors, several inorganic solid electrolytes achieve the lithium ion conduction required for solid-state batteries to become the next generation of energy storage device in combination with lithium metal. However, the challenges faced when preparing thin layers and stable interfaces of solely inorganic and brittle materials limit the performance of lithium solid-state batteries that are made purely of inorganic materials. Therefore, the best-performing solid-state batteries also introduce polymers to the system to improve the interfaces, cohesion, manufacture and mechanical properties of the cell as a whole. This article highlights recent developments made with the combination of polymer and inorganic materials in the form of composite electrolytes, interlayers, protective coatings and binders. The role of polymers regarding interface chemistry, interface resistance and lithium transfer is discussed and the importance of polymers for the processing of solid-state batteries is described. Taken as a whole, the article surveys the relevance of polymers at each cell component and discerns how polymers may provide the key to access the full potential of solid-state batteries with inorganic solid electrolytes.
 Sudeshna Sen | Dr Sudeshna Sen obtained her PhD degree from Indian Institute of Science in Bangalore, India. She pursued her postdoctoral studies at GSK's Carbon Neutral Laboratory, University of Nottingham and University College London, United Kingdom. She has been awarded Royal Society-SERB Newton fellowship from University of Glasgow. Currently, she is working in the junior research group of Dr Felix H. Richter and the research group of Professor Jürgen Janek at the Justus-Liebig-University Gießen. Her current research involves the development of polymer protective layers for solid-state batteries and the degradation analysis of solid-state battery interfaces. |
 Enrico Trevisanello | Enrico Trevisanello studied Materials Science at the University of Padua, where he received his BSc and MSc working in the field of electrochemical synthesis of polymers. His doctoral research, supervised by Dr Felix H. Richter and Professor Jürgen Janek at the Justus-Liebig-University Gießen, focuses on the study of hybrid electrolytes for lithium batteries, investigating the electro-chemo-mechanical interplay between polymers, ceramic electrolyte and cathode active materials. |
 Elard Niemöller | Elard Niemöller received his MSc at the Friedrich-Schiller University Jena in Materials Science. He completed his master thesis at the Center for Energy and Environmental Chemistry, investigating the protection of the sodium metal anode in room-temperature sodium-sulfur batteries. His doctoral research in the junior research group of Dr Felix H. Richter and the research group of Professor Jürgen Janek at the Justus-Liebig-University Gießen focuses on the development of polymeric materials as protective interlayer between the lithium metal anode and the solid electrolyte in lithium solid-state batteries. |
 Bing-Xuan Shi | Bing-Xuan Shi received his Master of Science in Chemical Engineering from the National Cheng Kung University, Taiwan in 2018. After graduation, he worked at the Battery Research Center of Green Energy in Taiwan and conducted the research of solid-state lithium-ion batteries and Zn/LiFePO4 aqueous rechargeable batteries including making them into pouch cells. He is now a PhD candidate in Physical Chemistry in the junior research group of Dr Felix H. Richter and the research group of Professor Jürgen Janek at the Justus-Liebig-University Gießen. He investigates polymer coatings on cathode active materials in solid-state batteries. |
 Fabian J. Simon | Dr Fabian J. Simon received his PhD degree in chemistry from Justus-Liebig-University Gießen in 2020. His research focused on the (electro-)chemical analysis of interfaces between solid polymer electrolytes and thiophosphate-based solid electrolytes. He then joined Robert Bosch GmbH as a development engineer for MEMS technologies. |
 Felix H. Richter | Dr Felix H. Richter is a junior research group leader in Physical Chemistry, Materials Science and Characterization at the Center for Materials Research of the Justus-Liebig-University Gießen. He is particularly interested in concepts that bridge the properties of inorganic materials and polymers. Currently, the Richter workgroup specializes in analyzing the fundamental transport mechanisms of ions across the interfaces of different battery components and the development of hybrid approaches for solid-state batteries. In March 2020, he received the NanoMatFutur funding by the Federal Ministry for Education and Research on the topic of Solid-State Batteries with Lithium Metal and Polymer Protective Coatings. |
Introduction
Electrochemical cells are ideal for electrical energy storage because they have excellent energy efficiency and can be prepared in diverse shapes and sizes at a reasonable cost. Thus, they are widely used to power mobile electric appliances from hearing aids to electric vehicles and can even be made in sufficient quantities to balance large power grids. Amongst the different types of cell chemistries, lithium-ion batteries achieve exceptional performance by weight and volume.1 Even 30 years after their first commercialization, they are still improved continuously and form a moving target to be overcome by developers of other cell technologies.The challenge remains the desire to create a cell with still higher energy and power density by packing materials with exceptional reductive and oxidative potential in an increasingly compact cell format while maintaining safe device operation.2 One of the most prominent emerging alternatives is the lithium solid-state battery (LiSSB), which uses solid electrolytes and a lithium metal anode instead of liquid electrolytes and graphite anode that are typically used in lithium-ion batteries.3 The incentive for their development is based on the expectation that solid electrolytes allow the reversible cycling of a lithium metal anode, which increases the attainable energy density.4
Therefore, the development of a suitable interface of lithium metal and solid electrolyte is one of the central topics in this research field. Most LiSSBs use a lithium foil or film that is added during cell construction, which provides a reservoir of excess lithium that helps with the formation of a stable interface and stable cell cycling.5 Ideally, this would not be required as the lithium to be cycled in the cell is already present in the cathode active material (CAM). If the lithium metal anode can be formed as a uniform layer on the pristine solid electrolyte during charging of the cell this would maximize energy density. The uniform and reversible plating and stripping of lithium metal is at the heart of enabling the lithium metal anode. As already demonstrated with liquid electrolyte,6,7 polymer coatings at the interface between the solid electrolyte and the current collector may prove promising here.
In a LiSSB, the cathode layer determines the attainable cell capacity and potential. This sets a clear target for cathode development, which is to maximize the specific capacity, cathode layer thickness, content of active material and cell potential. These requirements are the same as for lithium-ion batteries, which is why LiSSBs mostly rely on the same types of CAM as lithium-ion batteries: the intercalation-type and conversion-type CAMs. Intercalation-type CAMs are lithium transition metal oxides that can store lithium ions by intercalation in the interstitial layers of the crystal structure.8 They typically have high electrochemical potential, but low specific capacity, which makes them more suited to create LiSSBs with high energy density (by volume).9 The conversion-type CAMs can be transition metal oxides, sulfides, fluorides, phosphides, and nitrides, which react during lithiation to form entirely new products. They typically have intermediate electrochemical potential, but high specific capacity, which makes them more suited to create LiSSBs with high specific energy (by mass).10,11
While it is straightforward to increase the cathode layer thickness and CAM content, theoretical models show that more than about 5 mS cm−1 of lithium ionic conductivity is required for the solid electrolyte in the composite cathode to obtain a competitive LiSSB.12 To date, only few inorganic solid electrolytes (ISEs) exceed this conductivity. Unless, new solid polymer electrolytes (SPEs) with much improved ionic conductivity are discovered, it appears that LiSSBs must rely on ISEs to provide the ion transport in the composite cathode if the cells are to have competitive performance at ambient temperature.
Of course, LiSSBs that use a polymer electrolyte have already been brought to market.13 This demonstrates the key advantage of polymers: processing as solution or melt opens a range of preparative tools not always applicable to inorganic solids. The preparation of thin films of polymer electrolyte is more straightforward than that of inorganic solid electrolytes and can often be carried out at lower temperature. Hence, polymers are expected to facilitate the manufacture of LiSSBs with inorganic solid electrolytes and may provide solutions to the remaining interface challenges.14Fig. 1 depicts a schematic of a LiSSB and highlights how polymers can be employed to improve the performance of LiSSBs with ISEs by acting as protective coatings, interlayers or binders to stabilize the cell.
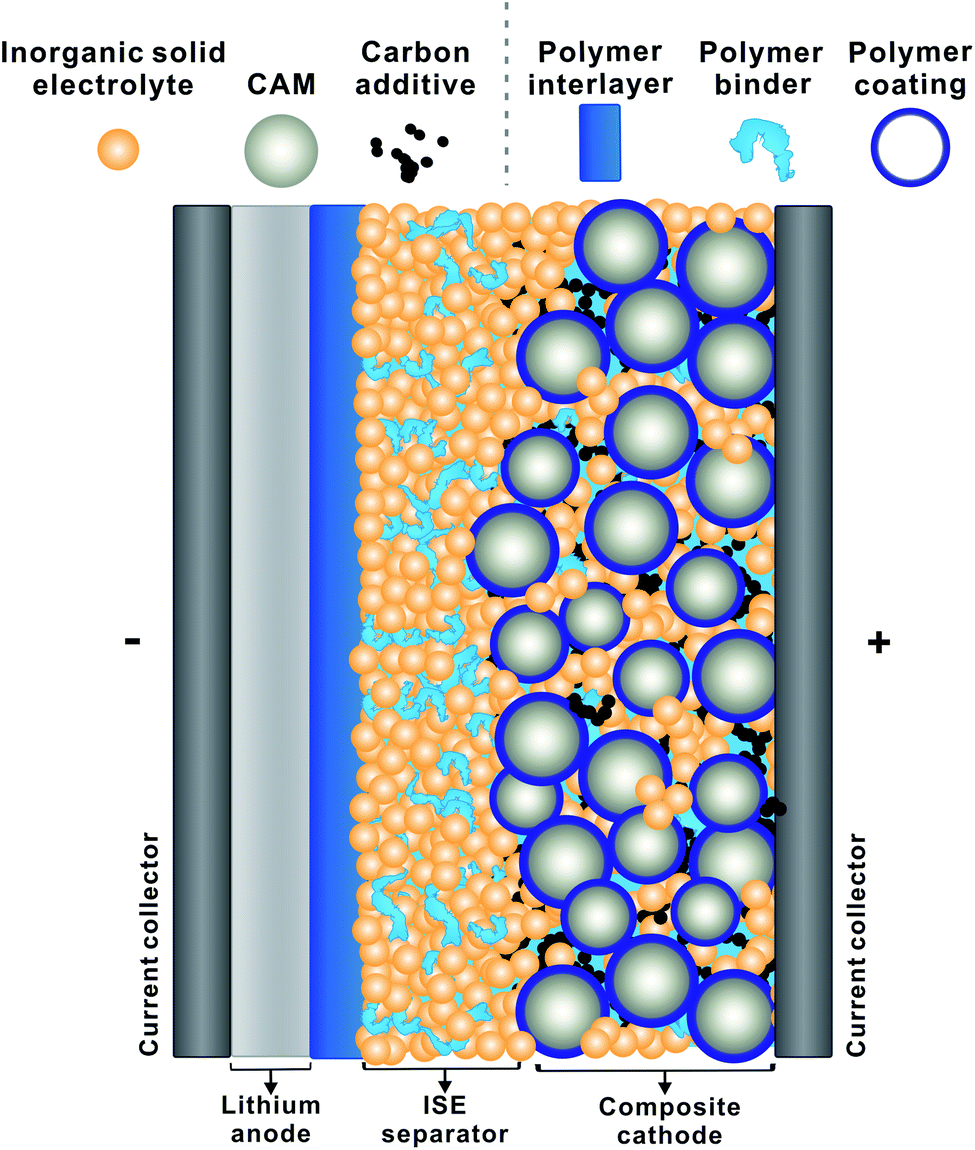 | ||
| Fig. 1 Schematic of a solid-state battery with polymer coating, polymer interlayer and binder (CAM: cathode active material, ISE: inorganic solid electrolyte). | ||
For example, the preparation of thin layers of inorganic solid electrolyte and SSB electrodes over a large area relies on the use of polymer binders.15,16 The SSB with the probably best overall performance published in the academic literature to date (Lee et al.) uses three different binders: polyvinylidene fluoride, polytetrafluoroethylene and a non-aqueous acrylate-type binder in the anode, cathode and solid electrolyte layers, respectively.17 For the successful implementation of polymer functionality, knowledge of the interface and interactions of polymers with inorganic materials is essential. In the following, the types of solid electrolytes and development challenges of LiSSBs with inorganic solid electrolytes are summarized first, after which the role of polymer–inorganic interfaces, composite electrolytes (CEs), interlayers, protective coatings and binders are highlighted.
Comparison of inorganic solid electrolytes with solid polymer electrolytes
While a high ionic conductivity is one of the key properties for solid electrolytes (SEs), also a low electronic conductivity, thermal and chemical stability, easy device integration and facile processing are important. Several material classes have been investigated in recent years, each with their own strengths and weaknesses. SEs are primarily divided into inorganic and polymer SEs. Fig. 2 gives an overview of the predominantly investigated classes of SEs. The group of ISEs encompasses oxide-, phosphate- and thiophosphate-based compounds,18 halides19 and hydrides/borohydrides.19,20 SPEs are classified either as salt-in-polymer electrolytes composed of a lithium salt dissolved in a polymer in which both cations and anions are mobile or as single-ion-conducting polymers, in which the anion is immobilized as it is covalently bound to the polymer backbone, allowing only the transfer of lithium ions.21,22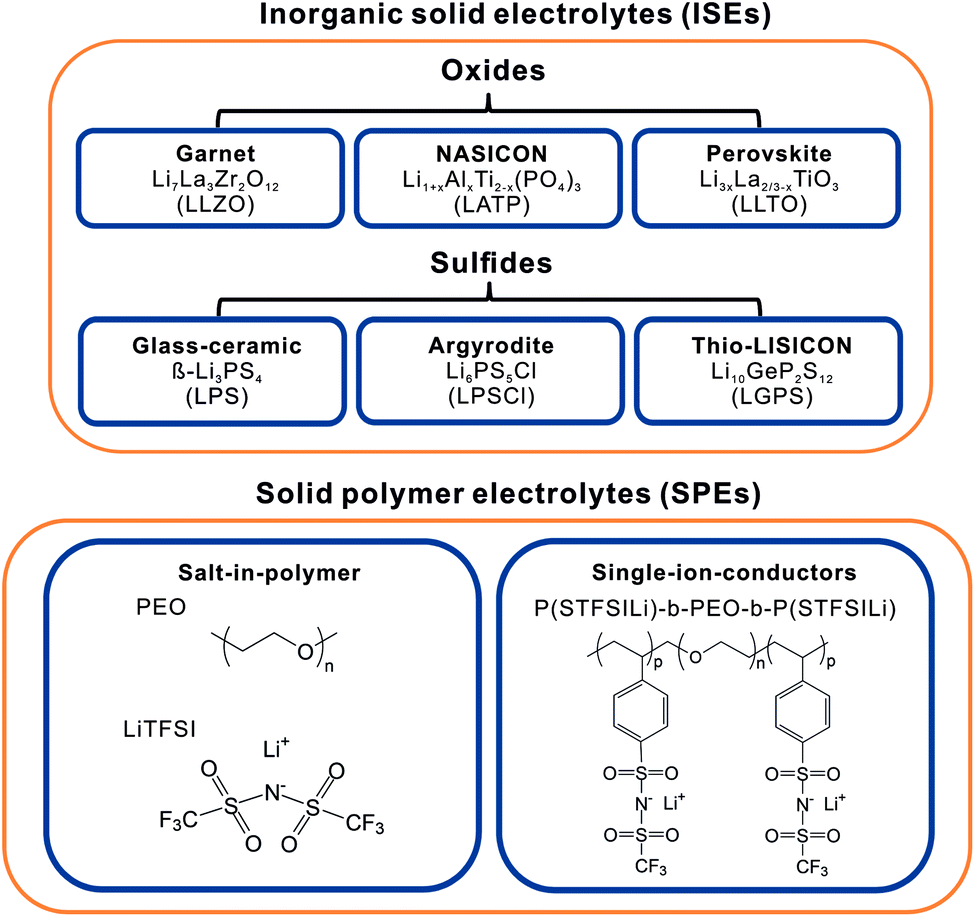 | ||
| Fig. 2 Overview of the primarily investigated classes of solid electrolytes and specific examples. | ||
The best ionic conductivity and mechanical properties are achieved with thiophosphate ISEs.24 Their malleability at ambient temperature facilitates the preparation of model cells by pelletization and creates good contact with the anode and cathode active materials. However, thiophosphate ISEs have only a limited intrinsic electrochemical window of stability and react with most CAMs and lithium metal, resulting in the formation of an interphase layer of decomposition products.25,26 Oxide and phosphate-based ISEs of perovskite, garnet and NASICON structure types have moderate lithium ionic conductivity, high mechanical strength27 and a larger intrinsic electrochemical window of stability.28 However, the need to sinter oxide ISEs at high temperatures to create good ion conduction and contact with the active materials makes cell preparation and cycling very challenging.29 For further reading about ISEs, we refer to a recent review that summarizes fundamentals, challenges and perspectives of SEs.30
In contrast, SPEs comprise good stability with lithium metal while being flexible and cost-efficient to produce.31 Polyethylene oxide (PEO) with lithium bis(trifluoro-methane-sulfonyl)-imide salt (LiTFSI) is arguably the most extensively studied polymer electrolyte due to its low cost, good electrochemical stability and excellent compatibility with lithium salts.21 Their largest impediment is their low room temperature conductivity (<10−4 S cm−1) that necessitates operation temperatures of about 60 °C in order to achieve sufficient ionic conductivity.
Fig. 3 exemplarily summarizes key properties for the most studied solid electrolytes: Li1+xAlxTi2−x(PO4)3 (LATP), Li7La3Zr2O12 (LLZO), Li6PS5Cl (LPSCl) and PEO-LiTFSI solid electrolytes. The ISEs exhibit higher ionic conductivity and elastic modulus than PEO-LiTFSI, but the latter can be prepared easiest as films at low temperature.18,32 Unsurprisingly, Lee et al. use Li6PS5Cl solid electrolyte as it scores reasonably to excellent for all properties: it has high ionic conductivity, cells can be prepared at a temperature of <100 °C and the limited electrochemical stability with anode and cathode can be enhanced with suitable protective coatings (5 nm thick Li2O–ZrO2-coated CAM) and innovative anode concepts (Ag–C nanocomposite).17
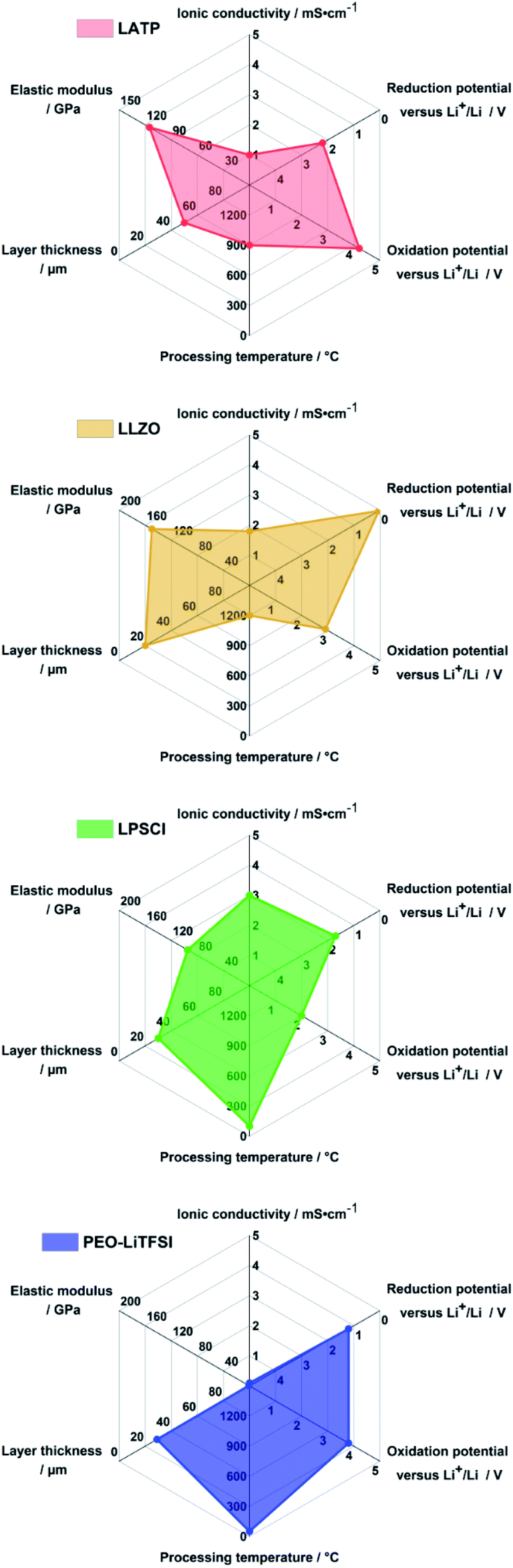 | ||
| Fig. 3 Radar charts of the properties of Li1+xAlxTi2−x(PO4)3 (LATP), Li7La3Zr2O12 (LLZO), Li6PS5Cl (LPSCl) and PEO-LiTFSI solid electrolytes. Data plotted is obtained from the literature.18,23 | ||
Development challenges for solid-state batteries with inorganic solid electrolytes
Overview of challenges in lithium solid-state batteries with inorganic solid electrolytes
Fig. 4 schematically depicts the three major challenges arising in LiSSBs with inorganic solid electrolytes. Due to the limited electrochemical stability of ISEs, decomposition reactions occur at the anode and cathode interfaces, forming interphases of decomposition products.33 Additionally, inhomogeneous lithium stripping/plating for lithium metal anodes and the volume change of CAMs during cycling result in cracking and contact loss, thereby limiting the accessible capacity.34,35 Finally, a mechanically thin ISE separator layer that prevents the formation of uncontrolled lithium growth also needs to be developed.31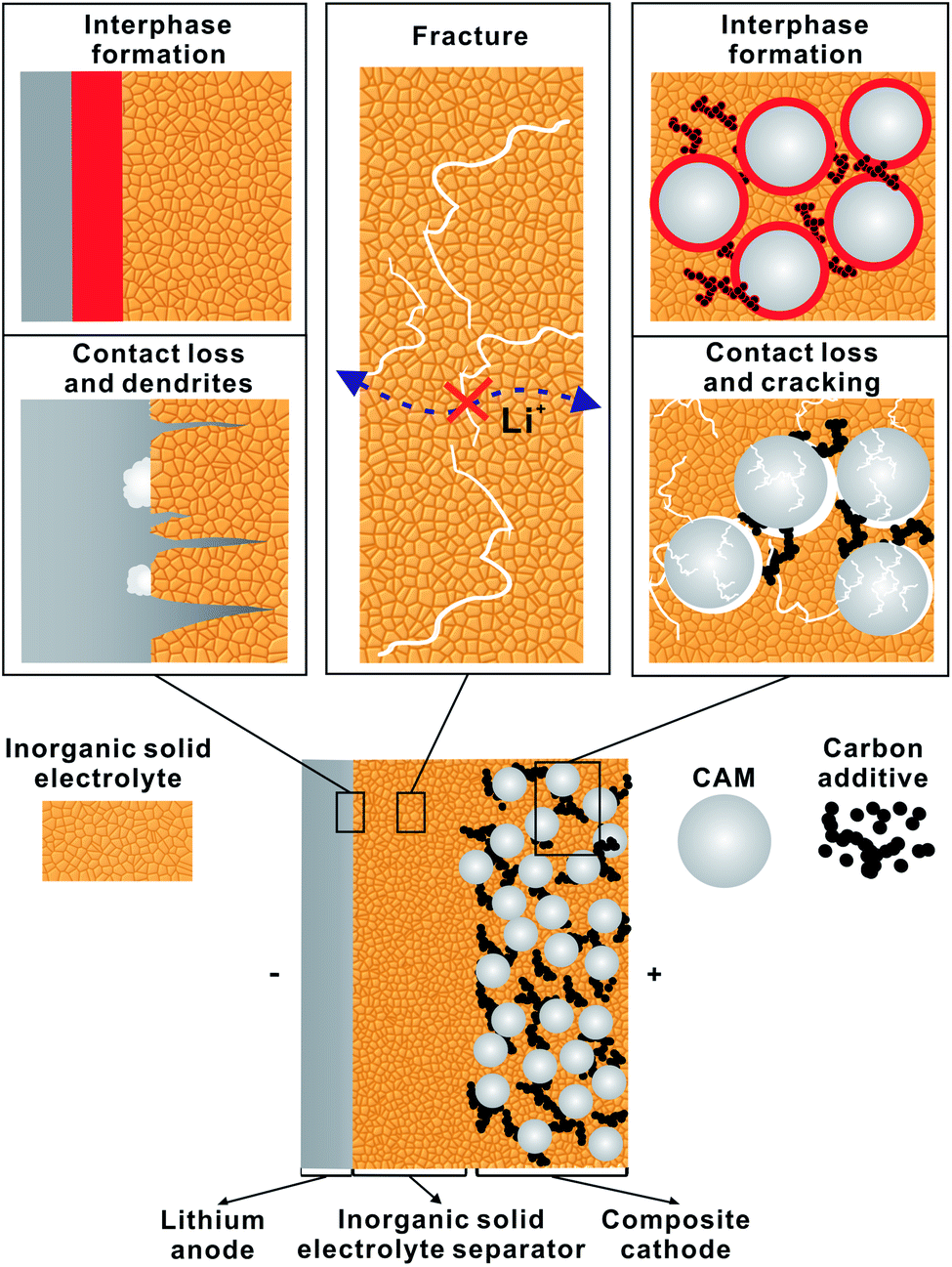 | ||
| Fig. 4 Schematic of the major development challenges in LiSSBs with ISEs: (left) preventing chemical degradation and contact loss of the lithium metal interface, (right) preventing chemical degradation and contact loss of the cathode active material interface, and (center) the preparation of a thin inorganic solid electrolyte separator layer without fracture. | ||
Electrochemical stability and interface degradation of inorganic solid electrolytes
Most solid electrolytes are electrochemically reduced or oxidized when in contact with anode or cathode active materials, respectively, forming an interphase of decomposition products with low ionic conductivity, which increases the lithium transfer resistance during cell operation.33 First-principles calculations obtained the thermodynamic stability window for a number of ISEs.28 For example, the argyrodite-type LPSCl has reduction and oxidation potentials of 1.71 V and 2.01 V versus Li+/Li, respectively. Also, NASICON-type ISEs are reduced by lithium metal, forming a mixed conducting interlayer, which is detrimental to cell performance. Of these ISEs, only LLZO is stable against lithium metal. This makes LLZO a uniquely suitable ISE to study the lithium metal interface. In addition, both LLZO and NASICON-type ISEs are more stable towards oxidation than lithium thiophosphates.Auvergniot et al. experimentally confirmed the oxidation of LPSCl when contacted with intercalation-type cathode active materials.26 This results in the formation of an interphase predominantly consisting of sulfur, lithium polysulfides, phosphor polysulfides, phosphates, and lithium chloride.36 Walther et al. have located the distribution of decomposition products at the interface of CAM and ISE via 3D secondary-ion mass spectrometry (SIMS).37 In addition, the ISE decomposes at the interfaces with conducting carbon additives and current collectors during cell cycling.38 In order to prevent the decomposition of the ISE, a number of inorganic coatings with superior oxidation stabilities have been developed for CAMs and carbon additives.39,40 For example, Lee et al. use 5 nm thick Li2O–ZrO2 coated high-Ni Li(NixMnyCoz)O2 (NMC) as CAM.17
Contact loss, particle cracking and short-circuit formation
Besides interphase formation, volume changes in the anode and cathode during cell cycling create mechanical stress, leading to cracking, pore formation and contact loss, which limit the performance and lifetime during SSB operation.41 Indirectly, these changes can be monitored during cycling using a pressure sensor attached to the SSB.42 From these results, it appears that the overall volume change of LiSSBs is dominated by the changes at the lithium metal anode.35 However, the volume changes sustained by the CAM may be smaller overall, but they locally impact lithium transfer and diffusion paths significantly.43 Hence, both mechanisms in anode and cathode are critical.In the anode, negligible lithium transfer resistance between lithium metal and garnet-type solid electrolyte demonstrates that fast lithium transfer is in principle possible across the interface between two solids.34 However, this relies on an intimate and pristine contact between the two materials, which can be obtained by applying high isostatic pressure. Three caveats of cycling lithium metal with ISEs are non-uniform stripping and plating of lithium, contact loss at the interface, and the formation of short-circuits at a practically relevant current density due to lithium growth through the solid electrolyte.44,45 The application of external pressure on the cell maintains contact at the interface and improves cycling performance.45 However, the requirement for high external pressures poses challenges for the practical application of LiSSBs.
In contrast, CAMs undergo volume change during cycling because the CAM particle volume depends on the lithium content (state of charge). This plays an important role at the interface with the ISE and contact loss can degrade cell performance.46 In addition, polycrystalline CAM particles often sustain cracks within the particle already during the first charge.47 This is less of a concern for lithium-ion batteries as liquid electrolyte can infiltrate these cracks, but for cells with solid electrolytes, infiltration of cracks is impossible, which results in a large resistance contribution from lithium diffusion in the CAM.48 This demonstrates that CAMs and suitable coatings or interlayers need to be tailored to the application in SSBs. Possibly, hybrid interlayers can be developed to circumvent this problem. For further information on degradation mechanisms at LiSSB interfaces, we refer to recent reviews.49–52
Preparation of thin and mechanically stable layers of inorganic solid electrolyte
In order to be competitive with state-of-the-art lithium-ion batteries regarding energy and power density, thin ISE layers (<30 μm layer thickness) with low resistance for lithium ion transport between anode and cathode are required. The powder pressing method is widely used for the convenient preparation of model cells with thick ISE separators (>100 μm layer thickness) for fundamental interface and cycling studies.5Tape casting of slurries is well-known from the manufacturing of lithium-ion batteries and provides electrode sheets with suitable layer thicknesses. It can also be applied to ISEs, however, the as-prepared thin films are fragile and prone to fracture due to the brittleness of the ISEs. Therefore, ISE particles are combined with polymers in the form of composite electrolytes (CEs) resulting in robust and bendable thin films.53 Both insulating polymer binders and ion-conducting polymer electrolytes have been used to help with the preparation of cells. In the case of polymer binders that are not able to conduct lithium ions, larger ISE contents are required to ensure effective ion conduction through the ISE particle network. In contrast, when using polymer electrolytes, lithium ion conduction is possible, independent of the ISE content.
The introduction of composites, polymer coatings and binders creates additional polymer–ISE interfaces. To assess possible applications of polymer-based interlayers and binders, knowledge about the polymer–ISE interface chemistry, mechanics, lithium transfer resistance and possible decomposition reactions is crucial.
Combining polymers and inorganic materials
Interfaces of polymers and inorganic materials
In SSBs with polymers and inorganic components, there inevitably exist interfaces between the two material types. The broad range of possible interactions between a polymeric matrix and inorganic components can be described as interface phenomena regulating contact and adhesion between the two materials. Independent of the crystalline or amorphous nature of the materials involved, adhesion usually describes the set of mechanisms and interactions that couple two surfaces.54 If the materials are immiscible, possible interactions between the atoms and molecules of the two phases include:54,55 (1) chemical interactions, where electrons from both materials are shared between the two phases in the form of covalent bonds, hydrogen bonds and Lewis acid–base interactions. A typical example is the grafting of polymer chains on ceramic particles;56,57 (2) electrostatic interactions, where localized charged terminal groups in the ceramic and/or polymer interact with each other. The electrostatic forces are especially relevant with ceramic nanoparticles given their high surface to volume ratio and specific localized charge;58 (3) dispersive interactions, where van der Waals forces between the atoms and molecules of the two materials dominate the interaction – the geckos' ability to adhere to almost any surface has been shown to be linked to this type;59 and (4) mechanical interactions, where the microstructure at the micro- and nanoscale mechanically interlocks the two phases, this being the main mechanism of common polymer-based coatings, such as polytetrafluoroethylene coatings on non-stick cookware.The strength of the interactions depends significantly on the chemical composition, morphology and preparation method of the polymer and inorganic materials. In the context of the polymer–ISE interfaces for electrochemical applications, the nature of such interactions is rarely investigated.56 However, such a mechanistic understanding of the phenomena governing the atomic and molecular dynamics at the interface is key for the design of better performing composites. In practical electrochemical devices, close contact between the inorganic and polymer phases is necessary, as fast transfer of the charge carriers across the boundary between the two materials is required to achieve good battery performance.
Distinctions need to be made between electron conduction and ion conduction mechanisms due to the different nature of the charge carries, i.e. electrons/holes or ions, respectively. In the early models proposed by Mott for electron conducting polymers, delocalized and doped π-orbital systems in the macromolecule facilitate the transport of the electrons/holes along the polymer backbone.60 Occasionally, electrons can transfer (tunnel) from one polymer chain to the next, giving rise to a percolating network for electronic conduction.60 Long-range transport however can be hindered by the presence of amorphous domains in the polymer.61 Therefore, recent efforts focused on understanding and controlling the microstructure of these materials or developing new polymers where semi-crystallinity is not required for fast electronic transport.62
In contrast, the predominant mechanism of ion transport in typical salt-in-polymer electrolytes such as PEO functions through ion solvation by the polymer in a predominantly amorphous matrix.63,64 In this type of electrolytes, ions are transported from site to site following the energy landscape created by the local α-relaxation of the polymer backbone (Fig. 5).65–68 This process is generally referred to as segmental motion and its magnitude, and the conductivity of the electrolyte, depend on the molecular weight of the polymer.69,70 Research efforts to lower the timescale of the backbone relaxation or completely decouple ion dynamics from it have been met with moderate success to increase ionic conductivity.63,71 Furthermore, the low transference number of PEO-based electrolytes and the associated concentration polarization during cycling limit the maximum current density in practical devices.72
 | ||
| Fig. 5 (a) Schematic of the transport mechanisms and energy landscape for PEO-based polymer electrolyte (adapted from ref. 60 with permission from The Royal Society of Chemistry). (b) Schematic representation of pristine (top) and degraded (bottom) interfaces between SPE and ISE forming a solid-polymer electrolyte interphase (SPEI), adapted from ref. 78. (c) Schematic of the transport mechanisms and energy landscape for LPSCl argyrodite ISE (adapted with permission from ref. 75, Copyright 2018 American Chemical Society). | ||
In recent years, the field of single-ion conducting polymer electrolytes with a transference number close to unity has received particular interest. These polymers, often referred to as ionomers, are characterized by electrically charged moieties immobilized by covalent bonds to the polymer backbone.63,73,74 The high molecular weight of the macromolecule reduces the mobility of the ions connected to it. Therefore, long range transport in this system is dominated by the mobile, non-covalently bound ion, allowing for transference number close to unity.63,73 While this avoids concentration polarization in the electrolyte when currents are applied to the electrochemical devices, long rage transport is still limited by the segmental motion of the solvating host polymer, so that temperatures above 40 °C are still necessary to reach practical conductivity.
In ISEs, the high concentration of ionic defects together with their high diffusivity allows for fast ion transport. As a prerequisite for an electrolyte, ion mobility needs to be decoupled from the electronic charge carrier concentration and mobility. In lithium conducting ISEs, the main mechanism for transport involves ion hopping from site to site in the crystal framework (Fig. 5).75 To give rise to long-range transport, ion hops need to take place not only locally within the anion crystal cages (intracage jumps) but also from cage to cage (intercage jumps).75 While ion diffusivity is largely determined by the energy landscape, the high concentration of ionic charge carriers is mostly a feature of the material's stoichiometry. For example, in the highly conducting LPSCl the concentration of Li+ is about 37 mol dm−3. In comparison, typical concentrations of salt in liquid and polymer (PEO with EO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Li = 10:1) electrolytes are 1 mol dm−3 and 2.5 mol dm−3, respectively.
:Li = 10:1) electrolytes are 1 mol dm−3 and 2.5 mol dm−3, respectively.
The ion transport mechanism further complicates at the interface between polymer and inorganic electrolyte. For ion transfer between the two electrolytes, desolvation/solvation of the ion from/by the polymeric matrix and its transfer between the two electrolyte phases take place. Given the higher mass of ions compared to electrons, charge transfer can happen only through available coordination/defect sites in the polymer and inorganic phases with ion-tunneling not playing a significant role in the dynamics of the interface.76 However, the charge-transfer can still be modelled as an activated process between the energy landscapes of the mobile ions in the two phases.77 The resulting equations describing the transport kinetics are similar to Butler–Volmer kinetics at an electrode–electrolyte-interface.77,78 In this scenario, space-charge layers likely do not play a significant role as the concentration of ‘unbalanced’ localized charge is expected to be negligible in an ISE.78,79
However, lithium transfer can be significantly hindered by the formation of an insulating decomposition layer between the two electrolytes (see Fig. 5). In this scenario, ionic transport between the two electrolytes can be slowed down by the lower ionic conductivity of the interphase layer of decomposition products.78,80 The formation of such interphases was also observed in several studies of the ISE interface with liquid electrolytes, and the so-called solid–liquid electrolyte interphase contributes significantly to the total impedance of the cell.77,81–83
At the electrodes of an electrochemical device, metallic or semiconducting inorganic active materials are in contact with the electrolyte. The mechanism governing the charge transfer depends greatly on the chemical environment of the participating ions (and electrons). At a metal electrode in contact with an electrolyte, electrons tunneling from the metal into the electrolyte are the main charge transfer mechanism.84 However, most alkali metals form a solid electrolyte interphase (SEI) that prevents/slows down further reduction of the electrolyte.84,85 In this case, charge transfer occurs directly at the interface between the metal and the ISE in the SEI.
Intercalation-type CAMs are of particular relevance for battery applications. At the interface of the active materials and the electrolyte, a coupled ion-electron transfer takes place through a concerted process involving a redox reaction and the charge-balancing uptake or release of a mobile ion species.76 This interfacial process is generally limited by the contact area between electrolyte and active material. To achieve good contact between ISE and the active material, high pressures and/or high temperatures are usually required. In contrast, the malleability of SPEs allows for uniform coating of the active materials, homogenizing ion flux at the surface of the active material. However, electrolyte decomposition and interphase formation can slow ion and electron transfer at the interface.86
Characterization of the interface
Experimental characterization of interphases and interface dynamics is often a difficult task, as interfacial phenomena are usually limited to sub-micron range around the phase boundary. Still, several techniques are used to study the interface/interphase between inorganic and polymer electron or ion conductors. With a special focus on the transport at the interface, the most common method employed for the characterization of these systems is electrochemical impedance spectroscopy (EIS). For example, this technique in combination with purpose-built cells allows the measurement of transport resistance at the interface of SPE and ISE. The employed cells usually consist of two or more layers of the conductors stacked on each other, thereby controlling the system geometry and contact area.87,88With the setup shown in Fig. 6a, the Li+ transfer resistance between PEO-LiTFSI and LPSCl was only 0.3 Ω cm2 at 80 °C.80 Measurement of the interface resistance was possible by introducing reference electrodes in the polymer layers, thereby excluding the contribution from the electrodes to the EIS spectra. Without employing reference electrodes, many authors were able to determine the interfacial resistance between PEO-type SPE and LLZO to be in the range between 100 Ω cm2 to 10 kΩ cm2 at 80 °C, which is strongly dependent on the salt concentration of the SPE and annealing treatment of the ISE.89–91 For LATP, the interfacial resistance determined by Chen et al. with PEO-based electrolyte was reported to be 30 Ω cm2 at 80 °C.92
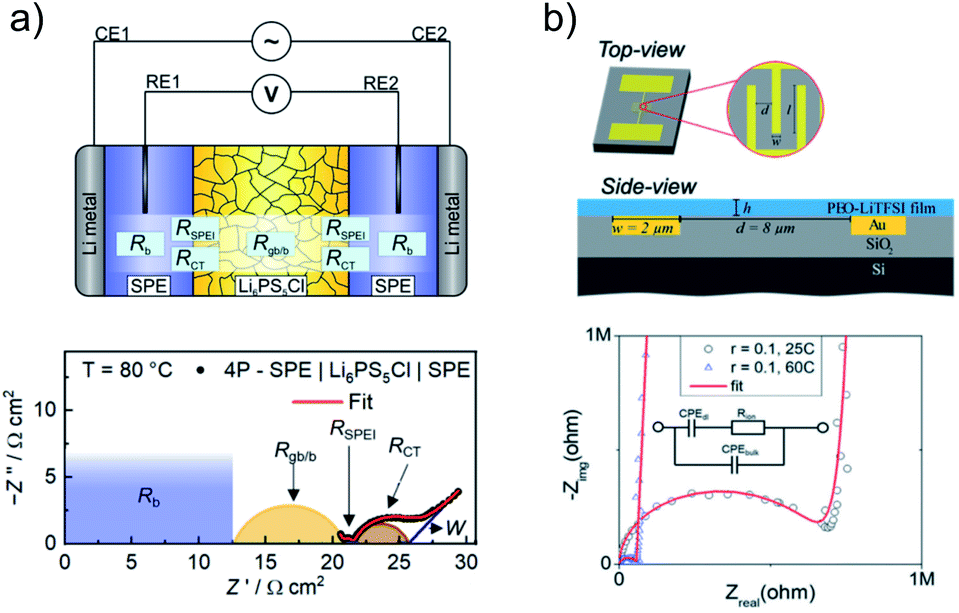 | ||
| Fig. 6 Cell types and electrochemical techniques employed for the characterization of the transport dynamics at the interface between SPE and ISE. (a) Schematic of a cell setup employing reference electrodes (top) on which EIS was used to determine the interface resistance between PEO-LiTFSI SPE and LPSCl (bottom). Key properties such as bulk resistance (RB) and grain-boundary resistance (RGB), the solid-polymer electrolyte interphase resistance (RSPEI), the charge transfer resistance (RCT) and Warburg diffusion (W) are highlighted in the Nyquist plot. Adapted with permission from ref. 80, copyright 2019 American Chemical Society. (b) Schematic of a micro-device developed for the determination of the conduction properties along the interface between SPE and ISE (top) and exemplary Nyquist plot of the EIS data recorded with such interdigitated electrodes (bottom). Reproduced from ref. 93 with permission from The Royal Society of Chemistry. | ||
Another setup to study ion transport along the interface between inorganic materials and polymer electrolytes of sub-micron thickness is shown in Fig. 6b.93 With this setup, Dong et al. were able to determine the effects that a SiO2 substrate has on the ionic conductivity of a few nanometers thick layer of PEO-based electrolyte. They concluded that grafting of the polymer to the inorganic material reduced its segmental motion and its ionic conductivity. At the same time, they could show that transport along the interface is influenced strongly by the salt concentration and PEO end-chain functionality.
Another technique to study the movement of ions in their chemical environments is nuclear magnetic resonance spectroscopy (NMR). Zagórski et al. successfully measured the diffusion of lithium from PEO into the garnet LLZO solid electrolyte.94 With this method, they also showed that the timescale of the charge transfer across the interface is around 0.1 s, which is in good agreement with EIS results.90 To probe the relaxation dynamics of the polymer and the effect of an inorganic filler on such dynamics, a composite electrolyte was analyzed with quasi-elastic neutron scattering (QENS) by Chen et al.95 They showed that segmental motion of the polymer chain is progressively reduced when PEO is mixed with LiTFSI and when the PEO-LiTFSI is further mixed with NASICON-type Ohara glass. Interestingly, they noticed that the polymer relaxation did not change when only Ohara glass was added to the polymer, suggesting an interplay between the lithium salt and the surface of the ISE in slowing down polymer segmental dynamics.
In addition to spectroscopic methods, atomic force microscopy (AFM) was used by Dixit et al. to measure the local mechanical properties of a PEO and LLZO composite electrolyte.96 They were able to correlate battery performance to the properties of the extrinsic surface (i.e. the surface of the whole CE). The degree of ISE dispersion and size under the surface and the polymer molecular weight are significant metrics predicting the delivered capacity and capacity retention in assembled batteries.96
Composite electrolytes
Attempting to combine the favorable properties of ISEs and SPEs in CEs is an extensively researched topic aimed at increasing ionic conductivity and easing the processing of the ISEs.97–99 Generally, CEs can be grouped into inorganic-rich (>50 vol% ISE content) and polymer-rich (<50 vol% ISE content) CEs.99,100 The ISE content and morphology determine the path of lithium ion transport, as spherical particles or fibrous morphologies can be organized in different arrangements.101,102 A schematic overview of the different ISE morphologies, configurations and pathways for lithium ion conduction is given in Fig. 7. One of the major characteristics of a CE is its conductivity. Therefore, an accurate description of the ion dynamics in the CE is necessary. Four main ion transport processes can take place in a composite: transport (1) only in the polymeric phase, (2) only in the inorganic phase, (3) along the interface/interphase and (4) across both phases.99 Based on these four main transport types, CEs and electrochemical cells can be optimized.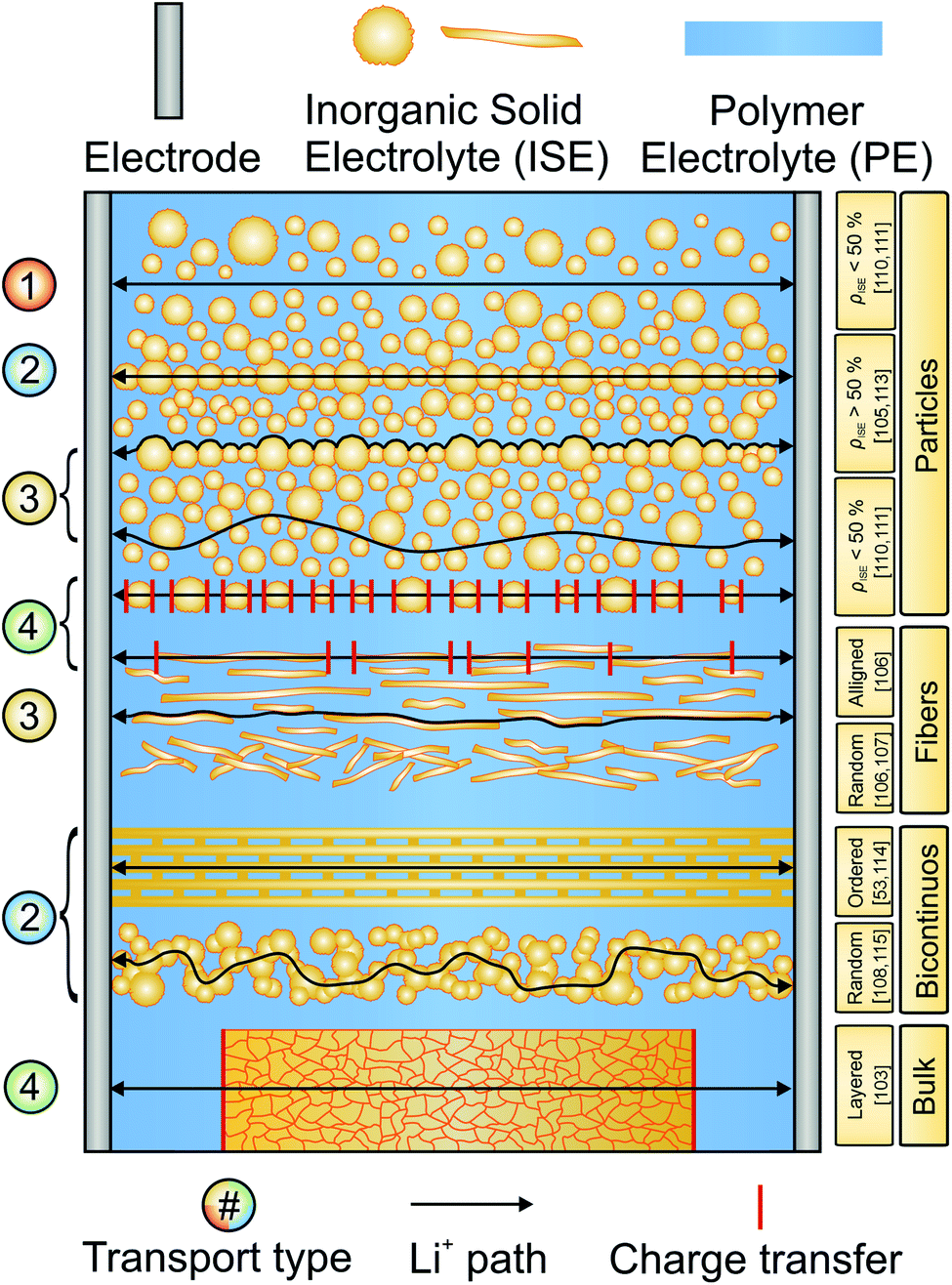 | ||
| Fig. 7 Schematic representation of the surveyed composite electrolyte architectures. The possible pathways for lithium transport are represented by black lines. In some cases, only one possible pathway is shown for clarity. | ||
In polymer-rich CEs, transport can proceed with mechanism 1, 3 and 4 and the preferred pathway greatly depends on the SPE and ISE employed. In bicontinuous CEs, as well as in inorganic-rich CEs, the existence of a 3D interconnected bicontinuous percolation network of the two phases allows that all four mechanisms can contribute to the total conductivity. If the lithium transfer between ISE grains is rapid, the polymer mostly serves as binder and does not contribute significantly to ionic conductivity.
CEs made of fibrous ISEs in a SPE matrix are of particular interest, in which the aspect ratio of the ISE component can increase the mean free path for ion transport in the more conductive ISE. Both in randomly oriented architectures and in aligned microstructures, the number of ion-transfer steps between the two phases is reduced compared to polymer-rich CEs with spherical ISE particles. Careful consideration of the main transport dynamics can improve the conductivity of the composites and it is crucial to consider the interface resistance when designing the CE structure.
In layered cell design (Fig. 6a), all the current applied between two electrodes needs to pass through both the ISE and the SPE layers and across the interfaces. This arrangement has been shown to be particularly effective in decreasing the concentration polarization resistance in SPEs due to the accumulation of anions at the electrodes, thanks to the almost unity transference number of the ISE.103
Now, the properties of several examples of oxide-based CEs are presented. An overview of the obtained overall conductivity for different compositions of particle- or fiber-in-polymer composite electrolytes is given in Fig. 8, where only polymers are considered that are themselves ionic conductors (i.e. salt-in-polymer electrolytes, single-ion conductors). Multiple reports of polymer-rich CEs can be found in literature for LLZO, with overall conductivities often reported in the range of 0.1 mS cm−1 or below at room temperature in the case of PEO-based composites.104,105 Also, ISE-rich composites were prepared, however in this case, the conductivity did not exceed the conductivity of the pure PEO-based electrolyte.94 The ineffectiveness of LLZO, even91 at high volume fractions suggest that a percolating network cannot be formed by simple ISE particle contact. Furthermore, the high interfacial resistance reported between PEO and LLZO means that transport across the two phases is unlikely even when the particles are tightly packed and separated by a thin layer of SPE. The low ionic conductivity of these composites at room temperature reflects the sluggish transport in the polymer phase, with the ISE mainly acting as plasticizer.100 To overcome these limitations, both aligned and random distribution of LLZO fibers were investigated by several research groups.106–109 The reported conductivity values match well with each other, with a 3D network of nanofibers reaching a conductivity of 0.025 mS cm−1 at 30 °C.
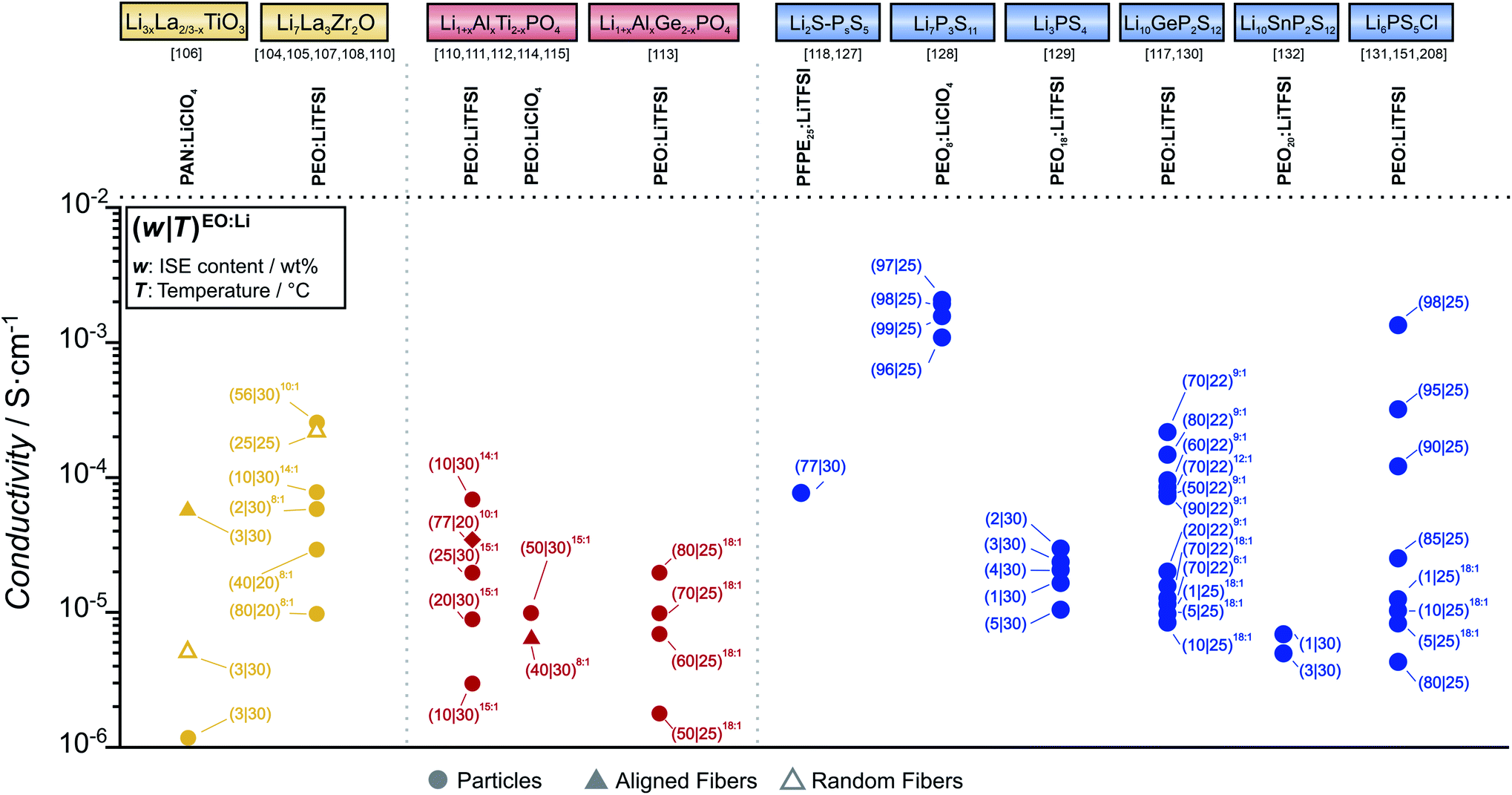 | ||
| Fig. 8 Ionic conductivity survey of oxide- (yellow), phosphate- (red) and thiophosphate-based (blue) particle-in-polymer or fiber-in-polymer composite electrolytes. Data for different polymers, conducting salt and ISE morphology/microstructure are shown. Measurement temperature and composition of the composite electrolyte are highlighted for each point. | ||
Composites of SPE and NASICON-type ISE have also been extensively studied. Examples for each architecture described in the previous section are reported in Fig. 8. Polymer-rich composites showed limited conductivity improvements, with conductivities in the range of 0.1 mS cm−1.110–112 ISE-rich CEs with PEO-based electrolyte showed moderate improvements over the bare SPE, with conductivities at room temperature in the order of 0.1 mS cm−1, as demonstrated by Jung et al.113 Recently, increasing attention to the ISE architecture in the polymer has lead scientists to the preparation of hybrid electrolytes where the microstructure of the ISE is controlled. A range of LAGP-based microarchitectures was studied and developed by Zekoll et al., with the best performing one showing a conductivity of 0.14 mS cm−1.53 One interesting approach is the one employed by Zhai et al. where ice-templated ISE powders allowed the preparation of a porous, interconnected network of ISE that could be infiltrated by a SPE.114 To achieve a percolation network in a 3D structure of LATP, Palmer et al. used a spray-coating technique.115
Compared to the phosphate and oxide classes of ISEs, thiophosphate-based ISEs are characterized by a much higher chemical reactivity towards solvent and polymers used in CE preparation. In the case of polyether based molecules, the reactivity was associated to interactions between the oxygen of the ether group and the oxophilic phosphorus in the thiophosphate.116 Conveniently, a possible mitigation pathway involves the addition of lithium salts, as the lithium ions are complexed by the ether groups, thus reducing the nucleophilic properties of the ether oxygen.116 Still, the formation of an interphase complicates the description of the conduction mechanism in the CE, as mobile species such as polysulfides are formed and can contribute to the total ionic conductivity.
This was experimentally shown in a study on CEs based on PEO-LiTFSI as SPE and LGPS as ISE by Hu et al.117 Varying the EO:Li salt concentration between 6:1 and 18:1, the authors showed a maximum lithium ionic conductivity for the 9:1 ratio.117 A similar positive influence on the conductivity was observed for 78Li2S–22P2S5 based CEs when adding a lithium salt. In their work, Zhang et al. showed that replacing PEO with PEO8:LiTFSI resulted in an increase in ionic conductivity from 0.2 mS cm−1 to 0.5 mS cm−1 at room temperature.118
Regarding the CE composition and its influence on ion transport, ISE-rich CEs generally have a much higher conductivity compared to polymer-rich CEs. Generally, the ionic conductivity of the polymer-rich CE is independent from the ISE used and in the order of 0.1 mS cm−1, suggesting that transport through the PE matrix is still predominant.119–124 However, conductivities in the order of 1 mS cm−1 were reported for ISE-rich composites, implying fast transport in the percolating thiophosphate ISE. At the same time, the low fractions of SPE electrolyte included in recent works used the polymer matrix simply as a binder for the ISE particles, rather than contributing significantly to ion transport.
Future perspective and comments
Research results in the field often attribute the better performance of CEs compared to the single materials to interfacial effects between the organic and inorganic materials. However, those claims are often based on improved bulk properties, such as conductivity or electrochemical stability, which do not necessarily represent a true improvement at the interface level. Thorough interfacial characterization with some of the techniques presented in the previous section is still missing for many ISE and SPE combinations.One of the major challenges in combining ISEs and SPEs is the transfer resistance between the two materials. While at elevated temperatures (>60 °C) it can be reduced to few Ω cm2 for ISEs in contact with PEO-LiTFSI, the ambient temperature values are often in the order of kΩ cm2. Such a high resistance practically isolates the embedded ISE particles from participating in the total ionic conductivity of particles-based CEs. In most polymer-rich composites presented in the literature, the small improvement of the ionic conductivity can be ascribed to the formation of a plasticized polymer zone in proximity to the ISE particles.125 Such plasticization dynamics are dependent on the thermal history of the CE below the melting point of the SPE.126 However, the thermal history of CEs is often underreported, leaving the exact causality unclear.
The chemical reactivity between ISEs and SPEs also plays an important role in the dynamics taking place at the contact between the two materials. In particular, chemical interaction and or reaction between the components can significantly influence ion transport. This was also shown to be detrimental for ISEs in contact with liquid electrolytes, and similar reactivity was observed for thiophosphates in contact with SPEs. While minimizing side-reactions between the components of the electrolyte is necessary for long term stability, the slow kinetics at the interface is still the strongest limitation for CE applicability in devices working at ambient temperature.
Overall, a trend towards architecture-engineered ISEs can be observed for both phosphate- and oxide-based CEs, focusing on the transport of ions in the ISE. This can be achieved by employing ISE fibers in the CE or by structuring the ISE to allow for polymer (and active materials) infiltration into an ISE-based porous structure. At the same time, a porous framework can increase the interface area available for charge transfer from the SPE to the ISE compared to traditional layered concepts, overcoming the limitation of the charge transfer between the two electrolytes. However, high-temperature processing seems to be unavoidable for these ISEs and the scalability of these concepts is likely limited to low volume applications. On the other hand, thiophosphate-based CEs show promising results when combined with SPEs. The main open challenge is reducing their chemical reactivity towards polymers and (polar) solvents. The trend in terms of composition goes towards maximizing the ISE content in the CE, with the SPE serving mostly as binder.
Binders
The role of polymers as binders
Binders are used to maintain cohesion between inorganic materials that would otherwise not remain in contact. For example, in a cathode for lithium-ion batteries, binders are added in small quantity to ensure the cohesion of the composite cathode and its adhesion to the current collector foil. Extensive knowledge about binder chemistry is already available from studies focused on the development of cathodes and anodes for lithium-ion batteries, as recently reviewed.127,128 The most commonly used binder in lithium-ion batteries with liquid electrolyte is polyvinylidene fluoride (PVDF) due to its chemical, thermal and electrochemical stability.129–131 The inert fluorinated backbone, non-toxicity and ability to form homogeneous cathode slurry with polar solvent N-methyl-2-pyrrolidone (NMP) make it viable for LIB application. However, it offers only weak interactions between polymer and CAM particles, which can result in contact loss during cycling.132 Alternatively, carboxymethyl cellulose (CMC), sodium carboxymethyl chitosan (CCTS), sodium alginate (SA), styrene–butadiene rubber (SBR), polytetrafluoroethylene (PTFE), or conducting polymers are also used.133–135 Among them, hydroxyl and carboxylic acid terminated binders form polar interactions and hydrogen bonds with the surface of electrode active materials.132 The good dispersion in polar solvents make them suitable for slurry-based electrode casting.132Chemical, electrostatic, dispersive and/or mechanical interactions between binder and the CAM surface are responsible for good binding ability.136 Mechanically, the binder needs to be strong, yet allow for accommodation of volume change without fracture during cycling. Tuning the type and strength of the interactions by varying the binder chemistry and functionality is required to obtain the optimum binding properties for each cell chemistry. According to Nguyen and Kuss, the “ideal electrode matrix should be able to (1) form strong interactions with active materials to maintain adhesion over cycling; (2) offer strong adhesion towards current collectors to prevent electrode delamination; (3) provide a continuous conductive network within the electrode; (4) exhibit sufficiently high failure strain to accommodate volume changes during charge/discharge cycling without breaking; (5) be electrochemically and chemically stable in the harsh battery environment and with high voltage cathodes (6) be applicable with the slurry casting method to be compatible with current electrode fabrication facilities; (7) be accessible at low cost to commercialize at wide scale”.132
For LiSSBs, binders are integrated with battery components as ISE-binder composite in the ISE separator and in the composite cathode (CAM, binder, ISE, carbon additive). Fig. 9 sketches the role of binders in the ISE separator (Fig. 9a) and composite cathode (Fig. 9b) and highlights the respective interfaces. Binder polymers may, but usually do not contain lithium salts, as ion conduction is not their primary role. Instead, they maintain the percolation network between the ISE, CAM, carbon additives and current collector to ensure efficient ion and electron transport through the inorganic cell components. Bielefeld et al. correlated binder content and ion percolation in a SSB cathode composite of NMC and LPS with PVDF and acrylonitrile butadiene rubber (NBR) binders.12 The authors point out that the presence of non-conducting binders blocks ion percolation pathways and reduces the total effective ionic conductivity of the ISE. Therefore, it is important that either low binder content or ion conducting binder is used for SSBs. Also, binder distribution in the composite is very important.
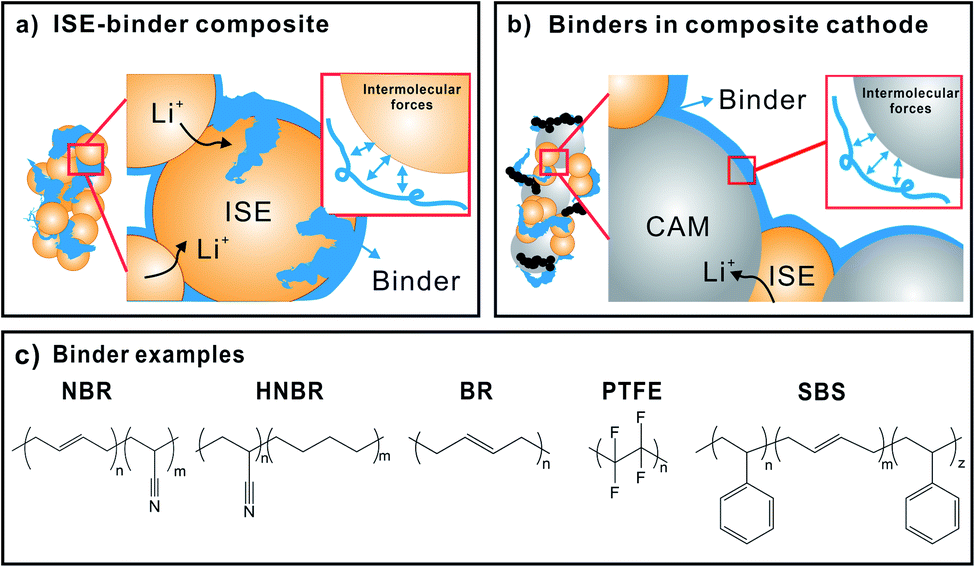 | ||
| Fig. 9 (a) Schematic illustration of the role of binders in ISE–binder composites and (b) composite cathode (CAM–binder–ISE). The presence of intermolecular forces at the binder–inorganic interface, which governs the binding ability of polymers is also depicted. (c) Structures of common binders utilized in SSBs with ISE as separator. | ||
There are three important differences to be considered when using binders in cells with solid instead of liquid electrolyte: (1) solid electrolyte cannot flow around or infiltrate the binder or cracks; (2) the prepared ISE separator layer needs to be made as thin as possible (replacing liquid electrolyte infiltrated separators used in lithium-ion batteries); and (3) anode, ISE separator and composite cathode need to be consolidated in one cell with low internal resistance. In addition to improving cohesion of composite cathodes, the binder also influences the ionic and electronic percolation networks. These points pose a significant challenge for cell preparation. Commonly, slurry-based film casting is used to make ISE films with binder and appropriate solvents, but dry processing methods have also been developed. In the case of SSBs, the most used binders for intercalation-type cathodes are PEO, NBR, polyvinylpyrrolidone, PTFE and PVDF, and the most used for ISE–binder separators are NBR, polyacrylonitrile butadiene (HNBR), PTFE, polystyrene-block-polybutadiene-block-polystyrene (SBS) (Fig. 9c).137
Binder–solvent–ISE compatibility in ISE–binder composite
To prepare a suitable slurry for the preparation of ISE sheets, the chemical composition, polymer structures, functional groups, mechanical properties and surface interactions are to be considered. Especially, the compatibility of binder, solvent, ISE, carbon additive and CAM is a prerequisite to create separator and cathode layers with good ionic conductivity and cycling performance. Generally, cathode slurries for LIBs are prepared in polar solvents such as NMP. However, ISEs are often unstable in polar solvents and care needs to be taken in finding appropriate solvents to solubilize the binder and suspend the ISE.137–139Herein, the choice of solvent is crucial.137,138 For example, polar solvents with high dielectric constants such as acetonitrile or dimethyl carbonate interact with Li7P3S11, resulting in the formation of PS43− and P2S64− units and in a reduced conductivity.140 A similar decomposition is obtained when ethyl acetate is used with 78Li2S–22P2S5 and PEO. As a result, the ionic conductivity was reduced by almost 75% compared with the pure ISE.118 Degradation of LPS and LGPS with ethers, for example triethylene glycol dimethyl ether, was also demonstrated, but could be prevented by addition of LiTFSI, which coordinates to the ether oxygen, thus reducing its reactivity towards nucleophilic attack on the ISE.116
No significant degradation was observed with the less polar solvents toluene and xylene. This was demonstrated for CEs comprised of 75Li2S–25P2S5 and NBR or polybutadiene processed with either xylene or toluene, which showed only minor change in conductivity compared to the pure ISE.137 Very recently, a blend of the less polar dibromomethane solvent and the more polar hexyl butyrate solvents was shown to work synergistically to dissolve NBR and LiTFSI, while preserving the argyrodite-type ISE, forming a SSB with the Li+-conducting NBR-LiTFSI binder.141
In addition to solvents, the functionality of the polymer also influences the properties of the ISE–binder composite. Riphaus et al. compared the results of several binders poly(styrene-co-butadiene) (SBR), polyisobutene (PIB), polymethyl methacrylate (PMMA), HNBR, polyethylene vinyl acetate (PEVA) with Li10SnP2S12 (LSPS) ISE.142 The LSPS–HNBR composite showed the highest conductivity (3.2 mS cm−1) and PEVA the lowest one (1.5 mS cm−1). This is in line with previous reports, stating that incorporation of a binder leads to a reduced effective ionic conductivity compared to the pure ISE (here: LSPS with 3.6 mS cm−1).
Fig. 10 summarizes the ionic conductivity of selected thiophosphate-ISE-binder combinations prepared with different solvents by film casting. The influence of binder content on ionic conductivity is discussed. We highlight only thiophosphate-based ISE-binder composites as oxide-based ISE-binder composites have too low effective conductivity due to the high transfer resistance from particle to particle without sintering at high temperature. Due to the superior ionic conductivity and malleability of sulfide ISEs at ambient temperature, ion transport from particle to particle can be ensured more easily. Where available, the conductivity is also compared dependent on the binder content.
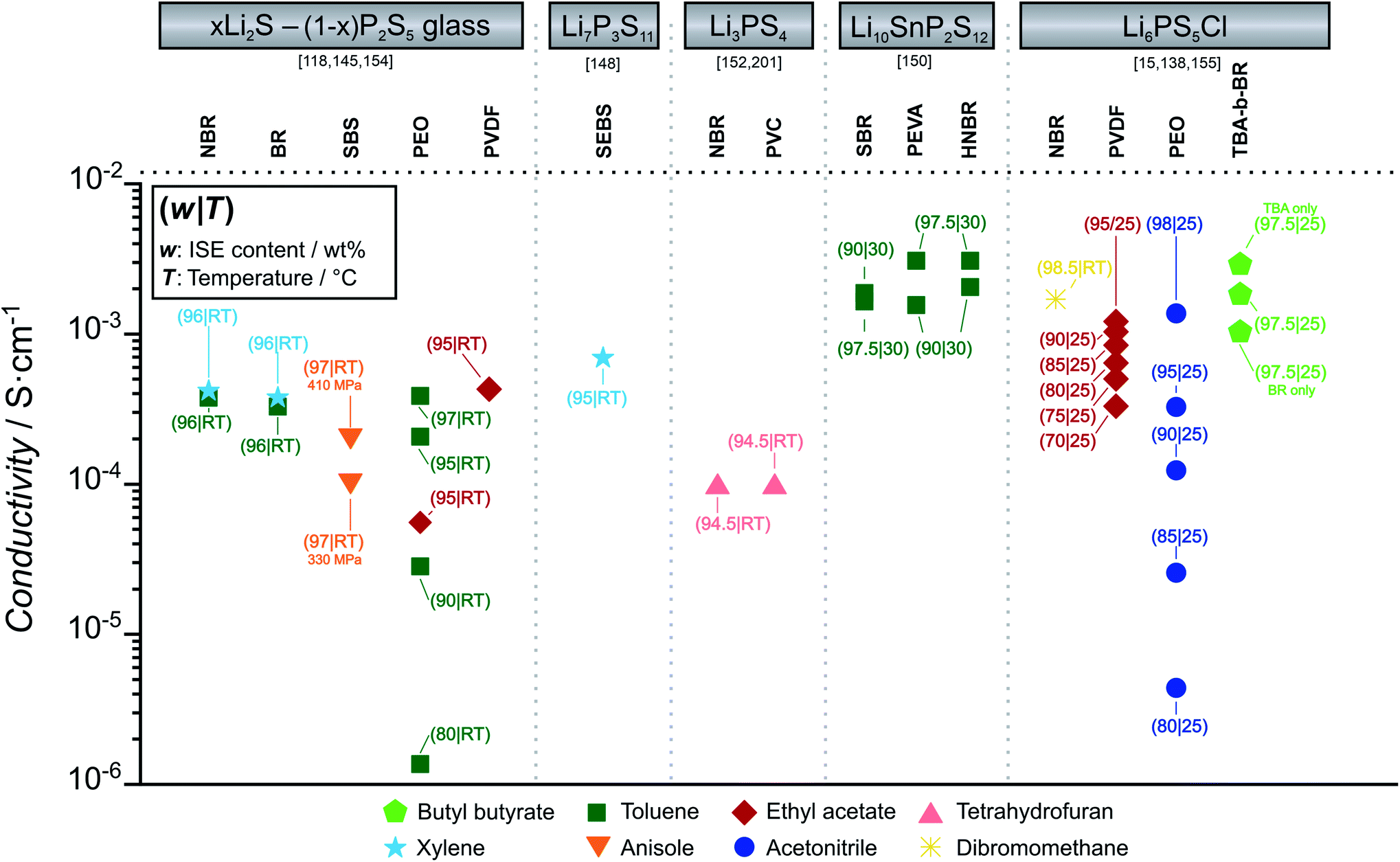 | ||
| Fig. 10 The figure summarizes ionic conductivity of thiophosphate ISE (xLi2S – (1 − x)P2S5 glass ceramics, Li7P3S11, Li3PS4, Li12SnP2S12, Li6PS5Cl) composites with binders poly(ethylene oxide) (PEO), poly(acrylonitrile-co-butadiene) (NBR), polystyrene-block-polybutadiene (SBS), poly-styrene-ethylene-butylene-styrene (SEBS), 1,4-butadiene rubber (BR), poly(tert-butyl acrylate)-b-poly(1,4-butadiene) (TBA-b-BR), poly(tert-butyl acrylate) (TBA). The solvents used for composite preparation (toluene, ethyl acetate, tetrahydrofuran, butyl butyrate, xylene, anisole, acetonitrile, dibromomethane) are highlighted by different colors and symbols. Measurement temperature and composition of the composite electrolyte are highlighted for each point. | ||
For all the ISE-binder composites, a common trend is observed. As evident in Fig. 10, the presence of binders generally decreases the overall ionic conductivity as compared with the pure ISE. For the compositions with the highest ISE content, the respective ionic conductivities are similar for all types of binders. However, with increasing binder content, the drop in ionic conductivity of composites is different for polar and non-polar binders. In the case of LPSCl-PEO, almost an order of magnitude lower conductivity is observed, if we compare LPSCl-PEO with 98 wt% and with 90 wt% LPSCl.143 A similar sharp drop in conductivity is observed for 78Li2S–22P2S5–PEO composites, when increasing PEO content from 3 wt% to 20 wt%.118
For non-polar binders, when we compare LPSCl-PVDF with 90 wt% and with 80 wt% LPSCl, the drop in conductivity is not as significant.133 In the case of two different non-polar binders with same ISE, (75Li2S–25P2S5–NBR versus 75Li2S–25P2S5–BR and Li3PS4–NBR versus Li3PS4–PVC) conductivity is similar, even though composites are made in two different non polar solvents (xylene and toluene).137,144 However, if we compare the same binders with different ISEs (NBR-LPSCl, NBR-75Li2S-25P2S5 and NBR-Li3PS4 (ref. 137)), ionic conductivity is determined by the ISEs.15,137,144 For the same ISEs, polar binders show slightly lower conductivity than non-polar binders (e.g. 78Li2S–22P2S5 – 95% PVDF versus 78Li2S–22P2S5 – 95% PEO).118 Polar binders accelerate the localized ISE-binder agglomeration due to the strong electrophilic interaction between electron rich functional groups of binders (O, N, F) with aliovalent atoms such as P or transition metals.139 The favored binders with thiophosphate ISEs are those containing nonpolar groups, such as poly(acrylonitrile-co-butadiene) (NBR), polystyrene-block-polybutadiene (SBS) or poly(styrene-ethylene-butylene-styrene) (SEBS).140 In addition to composition, compactness and density of the ISE films are also important. In case of 75Li2S–25P2S5 in styrene-butadiene-styrene copolymer (SBS), the application of 410 MPa instead of 330 MPa to compress the CE sheet resulted in a doubling of the conductivity.144
Binders in composite cathodes
In addition to maintaining ionic conductivity in the ISE separator layer, the binder in the composite cathode needs to fulfill additional requirements: adhesion to CAM, carbon additive and current collector and high oxidation stability. The adhesion of binders can be varied by structural modification of binders to modify the strength of interactions between CAM and binder. Lee et al. demonstrated a new binder (C10) to improve interfacial resistance and increase adhesion between electrode components (Fig. 11a).145 The styrene-butadiene-block-copolymers (SBS) are functionalized with an optimal level of carboxylic acid groups by employing thiol–ene click reaction. The polystyrene-block-polybutadiene-block-polystyrene (SBS) rubber was reacted with 3-mercaptocarboxylic acid (3-MPA) via a photolysis reaction. This reaction generates alkyl sulfide from the reaction between thiol and alkene.145 | ||
| Fig. 11 (a) Surface interaction of click binders (C10) with NMC cathode and SEM image of cathode composite layer. Solubility of binders (BR, NBR, C4, C10, C40, PAN, PVDF) with different solvents (heptane, isopropyl ether, p-xylene, DCM, THF, NMP) of varying polarity. Reproduced with permission from ref. 145, copyright @ 2018 American Chemical Society. (b) Schematic diagram illustrating microstructures of NMC-Li6PS5Cl-NBR without and with addition of LiG3 to NBR binder. The diagram depicts transition from non-conducting to conducting composite by addition of LiG3 to NBR binder. The Li+ ionic pathways are indicated by blue arrows. (c) The charge–discharge voltage profiles for NMC711 at first cycle without and with LiG3 (1.6 to 2.5 wt%) addition to composite cathode. Reproduced with permission from ref. 15, copyright JohnWiley and Sons and CCC, 2019. | ||
The molar ratio between the 3-MPA to butadiene of SBS was varied as 4:100, 10:100 and 40:100. The corresponding SBS-COOH click binders are denoted as C4, C10, and C40, respectively. The finely tuned polarity of C10 is compatible to LPSCl ISE and p-xylene solvent. Fig. 11a presents the solubility of binders (BR, NBR, C4, C10, C40, PAN, PVDF) in several solvents of varying polarity and the ISE compatibility. The ISE is stable in polarity up to p-xylene (blue region). The C10 is soluble in p-xylene, where ISE is stable. More polar binders (C40, PAN, PVDF) are only soluble in more polar NMP, where ISE is not stable. Following the favorable solubility and ISE compatibility, the C10 binder is used in a SSB with NMC and LPSCl.
The C10 click binder offers the H-bond between the surface of NMC cathode and the carboxylic acid groups of the binder (Fig. 11a).145 With increasing content of carboxylic acid groups, adhesion to the CAM increases, even in the presence of ISE. The favorable adhesion and stable interface between ISE-binder-CAM is beneficial to address volume changes and contact loss of cathode particles during cycling. As a result, the C10 exhibits superior capacity retention (78.8% capacity retention) as compared to conventional NBR binder (57.8% capacity retention).
To enhance ion transport, Oh et al. prepared a binder based on solvated ionic liquid incorporated in NBR polymer, applicable for an SSB with LPSCl (Fig. 11b).15 This offers additional functionality in SSB cathodes, such as ionic conductivity, buffering volume changes and stabilization of the cathode–electrolyte interface. The solvated ionic liquid (Li(G3)4) employs a saturated salt solution of glyme-based solvent (triglyme: G3).15 A sheet-type slurry-fabricated cathode composite, NMC–LPSCl–NBR–LiG3, where NBR–LiG3 is used as ion conducting gel-type binder (0.17 mS cm−1) is prepared.15 The presence of LiG3 with NBR results in the increase in ionic conductivity of the composite cathode. The solvated ionic liquid is stable in LPSCl and exhibits negligible volatility. Such improvement in conductivity is reflected in the improvement of rate capability and capacity retention in the SSB with LPSCl. With the presence of LiG3 in NBR–LiG3 (1.6 or 2.5 wt% LiG3) in NMC–NBR composite, the first cycle discharge capacity at 0.025C was increased from 131 mA h g−1 (0 wt% LiG3) to 172 mA h g−1 (1.6 wt%) or 164 mA h g−1 (2.5 wt%), respectively. The LiG3-NBR binder plays a multifunctional role to improve ion conduction and maintain stable contact between ISE and NMC particles.15
Processing of SSBs with binders
To make SSBs viable on large scale, high energy density, high power density, long life time, safety, and low cost are required.29 This calls for high content of CAM, thick electrodes and thin ISE separator layers. However, SSB research has so far mostly focused on binder-free pellet-type model cells with comparatively thick ISE separator layers and thin electrode layers for ease of fabrication, handling and investigation. However, to achieve practical energy densities, ISE separators need to be thinner than 50 μm, which is where the binder becomes relevant due to the brittleness of ISEs.139 Inspired by the slurry coating method used for lithium-ion battery cathodes, the development of processes to prepare sheet-type ISE separators and composite cathodes is aspired in SSB research. Photographs of the dry mixing of cathode components, preparation of the cathode slurry and the resulting sheet-type electrode are exemplarily shown in Fig. 12. Because of the reactivity of most ISEs, it is challenging to find the right combination of compounds for the slurry mixture containing CAM, carbon additive, ISE, binder and solvent. | ||
| Fig. 12 Photographs of (a) cathode composite (active material, ISE, binder, super C65 carbon additive) prepared by dry mixing, (b) cathode composite slurry with xylene solvent and (c) sheet type NMC electrode film as prepared by wet slurry process. Reproduced with permission from ref. 16. Copyright © 2017 Elsevier B.V. license @ CCC, Elsevier. | ||
For example, Sakuda et al. prepared a sheet-type SSB with 75Li2S-25P2S5, graphite composite anode and NMC composite cathode.146 This preparation is similar to that of lithium-ion battery electrodes. For the cathode slurries, acetylene black (AB) as additive and a styrene-butadiene-based binders (SEBS or SBS), are added and mixed with ISE, C and NMC. The cathode slurries are made with a suitable choice of solvents, based on the binder selection: heptane for SEBS and anisole for SBS. For the SE and anode sheets, SBS is used as binder and anisole as solvent to make the slurries. The electrode slurries are coated on current collectors (copper foil for the anode and aluminum foil for the cathode), followed by drying. All the sheets were stacked and pressed at 330 MPa to construct a full sheet-type SSB.146
Lee et al. prepared a new class of polymer binder-ISE-CAM composite by slurry-based method by mixing LPSCl electrolyte and binders in butyl butyrate solvent.147 The structure of the binder was based on polytert-butyl acrylate (TBA) and poly(tert-butyl acrylate)-b-poly(1,4-butadiene) (TBA-b-BR) and their deprotected forms (exposing t-butyl protected acid groups). Both the ISE-TBA and ISE-TBA-b-BR composites show similar conductivity with LPSCl as ISE (about 1 mS cm−1) (Fig. 10).147 The cathode composites with NMC with deprotected binders are operated in a full cell. Due to the presence of more carboxylic acid groups, binders improve adhesion between CAM and ISE particles, preventing further contact loss during battery operation.
Due to the incompatibility of sulfide-based electrolytes with the majority of polar solvents, a versatile alternative preparation route is required. The alternative route should be scalable and applicable to many cell chemistries, operable under continuous conditions, and solvent-free to avoid ISE decomposition in organic polar solvent.16 For both the cathode and ISE separator sheets, the dry approach is superior as compared to conventional wet chemical method. According to Nam et al., a sheet-type electrode fabricated using a wet-slurry process showed poorer rate capability and capacity due to the poor ionic contact in cathode components when compared to that of dry mixing process.16 However, the non-scalable dry method is as yet not perfect since the added binder partially blocks ion conduction pathways.
In this regard, Hippauf et al. introduced a scalable dry film approach to prepare cathode sheets for successfully operable practical cell size (9 cm2).148 This method employs dry premixing of NMC, LPSCl, carbon additive and fibrous PTFE, followed by applying shear force to prepare films. The dry film approach can effectively reduce the surface blocking of active materials with fibrous binders. In comparison to the slurry method, the dry film approach results in superior rate performance, even with lowest binder content. Lee et al. adopted the dry film approach to prepare a sheet-type NMC and LPSCl composite cathode and prototype SSB with exceptional performance (0.6 A h and > 900 W h l−1).
Future perspective and comments
Revisiting the seven points made by Nguyen and Kuss about the binder properties with a particular perspective on SSBs, we draw the following conclusions.132 Points (1) and (2) to form strong interactions with the CAM and current collector hold true in SSBs as well, yet, this poses a particular challenge in combination with the ISE. Stronger interactions tend to be obtained with polar binders, which require polar solvents for the slurry process, but these also show higher degradation with the ISE. Solving this will be a key challenge for binder development and processing. In line with these considerations, Lee et al. rely on PTFE and dehydrated xylene to prepare the cathode composite, followed by a dry film process to prepare the cathode sheet.17 Thus, avoiding solvents as much as possible may be the way forward to minimize degradation of the ISE.Regarding point (3) to maintain the electron conducting network in the electrode also holds true for SSBs, but this requirement needs to be extended by the prerequisite to also ensure the ion conducting network across the entire cell including the ISE separator. Dual conducting binders with ionic and electronic conductivity may help with maintaining both percolation pathways in the composite cathode. Interestingly, Lee et al. use a non-aqueous acrylate-type binder and anhydrous xylene and anhydrous isobutyl isobutyrate solvents for the preparation of the ISE separator.17 In order to minimize loss of ionic conductivity due to the binder, use of ion conducting binders, in particular single-ion-conductors, may prove fruitful.
Point (4), the accommodation of volume change during cycling is especially relevant for SSBs as any cracking would immediately and irrevocably reduce cell performance. Using binder alternatives like self-healing polymers, hyper branched or crosslinked polymers, polymer gels or solvated ionic liquids are possibilities to mitigate this problem. As for point (5), electrochemical and chemical stability in the harsh battery environment is equally relevant. Degradation of the ISE, CAM and binder must be avoided. This requires careful investigation of the electrochemical stability of especially the ISE and binder in a strongly oxidizing environment. In particular, further theoretical investigations into the stability of these components will be very helpful, especially if accompanied by matching experiments.
Regarding point (6), slurry casting capability looks to be complemented by recent progress with the dry film process for the preparation of SSBs. It is imperative that the cathode components including CAM, carbon additive, binder, ISE and solvents are carefully matched regarding the (electro)chemical stability of all components. The slurry casting process is at least equally challenging for SSBs as for lithium-ion batteries, if not more so, as most ISEs are exceptionally air and moisture sensitive, as well as instable with many solvents and common battery materials. And point (7), cost and scaling is difficult to estimate at present, as production processes have not yet been finalized and the potential for cost reduction during scaling of SSB construction is still unclear.
Transferring innovations and concepts from LIB research has the potential to accelerate SSB development. Recently, Niu et al. reported a polyelectrolyte binder, synthesized by the polymerization of bis(2-hydroxyethyldisulfide) (HEDS), hexamethylene diisocyanate (HMDI) and poly(tetramethylene ether glycol) (PTMEG).149 It contains hard and soft phases, which offer excellent mechanical strength and flexibility to accommodate volume expansion in sulfur-based cathodes with liquid electrolytes.149 Mechanical flexibility and a wide electrochemical window are also beneficial for high voltage cathodes to prevent cell performance loss during SSB operation. In reference to binders for batteries with liquid electrolyte, the functional binders with self-healing property,149 hyper branched/crosslinked polymers,150 or solvated ionic liquids151 may also create new opportunities in SSBs. The full scope of the versatile properties of polymers regarding solubility, cross-linking, gel formation, and a range of mechanical properties (soft, hard, elastic, or swellable) and interactions (hydrophobic to hydrophilic) will be beneficial to SSB research. Particular focus needs to be placed on the development of (electro)chemically stable sheet type cathodes and thin ISE sheets for which the selection and development of binder will be crucial.
Protective coatings and interlayers in cathode
Role of polymer interlayers in cathode
The cathode composite layer for LiSSBs with ISEs is composed of CAM, ISE, carbon additives and binders. Due to the reactivity of many ISEs at high potential versus Li+/Li, and crack formation and volume changes of the CAMs during cell cycling, the interfaces degrade causing the cell performance to deteriorate. Therefore, amongst many inorganic coatings,39,152 several polymer coatings have also been applied to protect the interface between electrolyte, CAMs and carbon additives. Fig. 13 schematically shows the three interfaces present in the cathode composite with polymer interlayers: CAM-polymer coating, carbon-polymer coating and ISE-polymer coating. The first two are discussed in this chapter, whereas the third was indirectly already discussed in the previous sections.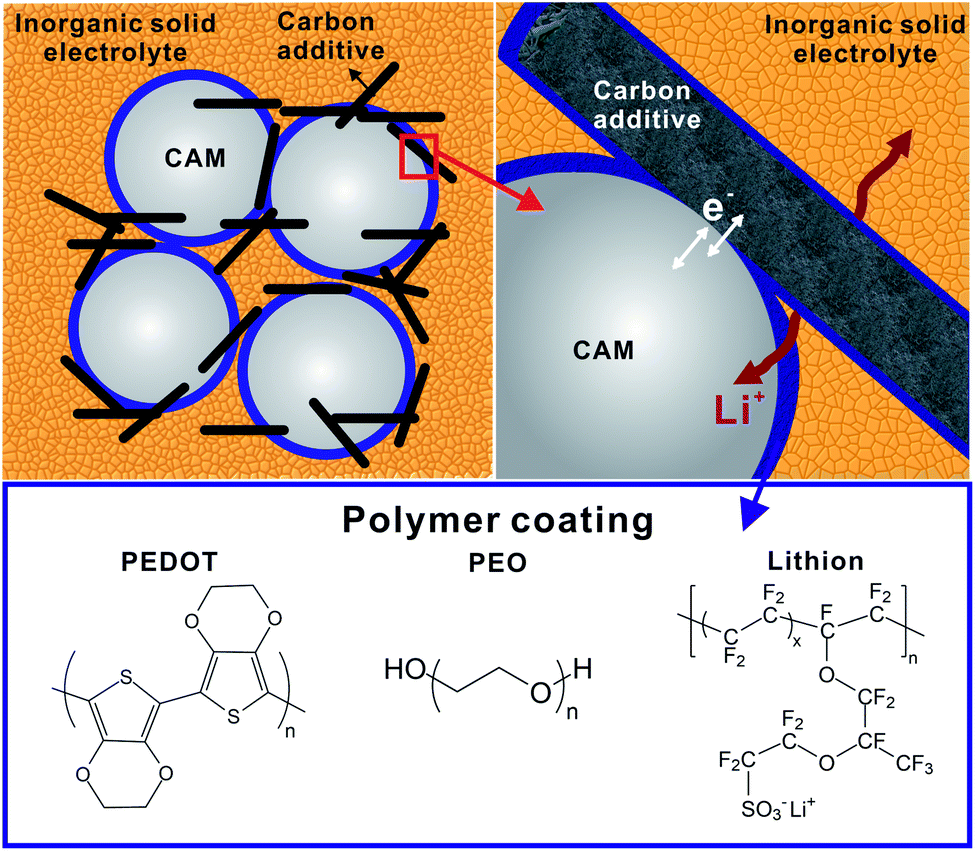 | ||
| Fig. 13 Schematic representation of ternary polymer interlayer in SSB composite cathode (ISE-CAM-polymer). Insights into the respective interfaces between cathode active material (CAM)-polymer, polymer–carbon additives and inorganic solid electrolyte (ISE)-polymer interlayer are highlighted. Structure of potential polymers (PEDOT, PEO, lithion) as CAM coating interlayer in SSB. | ||
The coating on CAM should primarily serve three functions: exhibit wide electrochemical window to offer protection for the ISE against high voltage cathodes, decrease the charge transfer resistance and maintain contact with the ISE. Especially during cycling, crack formation and contact loss is observed due to the volume changes of the CAM. Therefore, rheological properties of the polymers can play an important role to compensate contact loss and maintain the ion percolation network.
Different types of dry and wet coating procedures153,154 have been reported to prepare coatings on CAMs: solvent casting,153 electrostatic spray deposition,155in situ crosslinking of polymer with CAM,156 chemical vapor deposition for conformal coating of polymers on cathodes.157 Examples of polymers utilized as coating in the SSB cathode are shown in Fig. 13. The classes of polymers utilized are polymers with polar functional groups,23 poly(ionic liquid),23 poly(electrolytes) (e.g. lithion),158 electron conducting polymers (e.g. poly(3,4-ethylenedioxythiophene). PEDOT)159 and dual polymers combining conducting polymer and insulating polymers (e.g. polyaniline/PEO,84 PEDOT/PEG159), most of which have been tested in cells with polymer or liquid electrolytes. Therefore, we build our discussion on the extensive knowledge base already available on batteries with solid polymer and liquid electrolytes as this gives important insight into the CAM polymer interactions and may provide a directive on how polymer coatings can also benefit SSBs with ISEs.13,14,160
Electrochemical stability window of polymer interlayers
For application in composite cathodes, polymer coatings should be stable at high potential versus Li+/Li to avoid electrochemical oxidation within the potential range of intercalation-type CAMs. In general, to prevent oxidation (and reduction) of the polymer coating in a battery, the intrinsic electrochemical stability window (ESW) of the polymer should extend beyond the electrochemical potential of the electrodes.23,84 The ESW is defined as the gap between the oxidation and reduction potentials of the polymer. The wider the gap between HOMO and LUMO orbitals of the polymer or polymer salt complex, the wider the ESW. As shown in Fig. 14, if the electrochemical potential of the anode is above the polymer reduction potential (μa > E (LUMO) of polymer), the polymers could be reduced by the anode. A cathode could oxidize the polymer if its electrochemical potential is below the oxidation potential of the polymer (μc < E (HOMO) of polymer), unless a passivation layer prevents electron transfer.23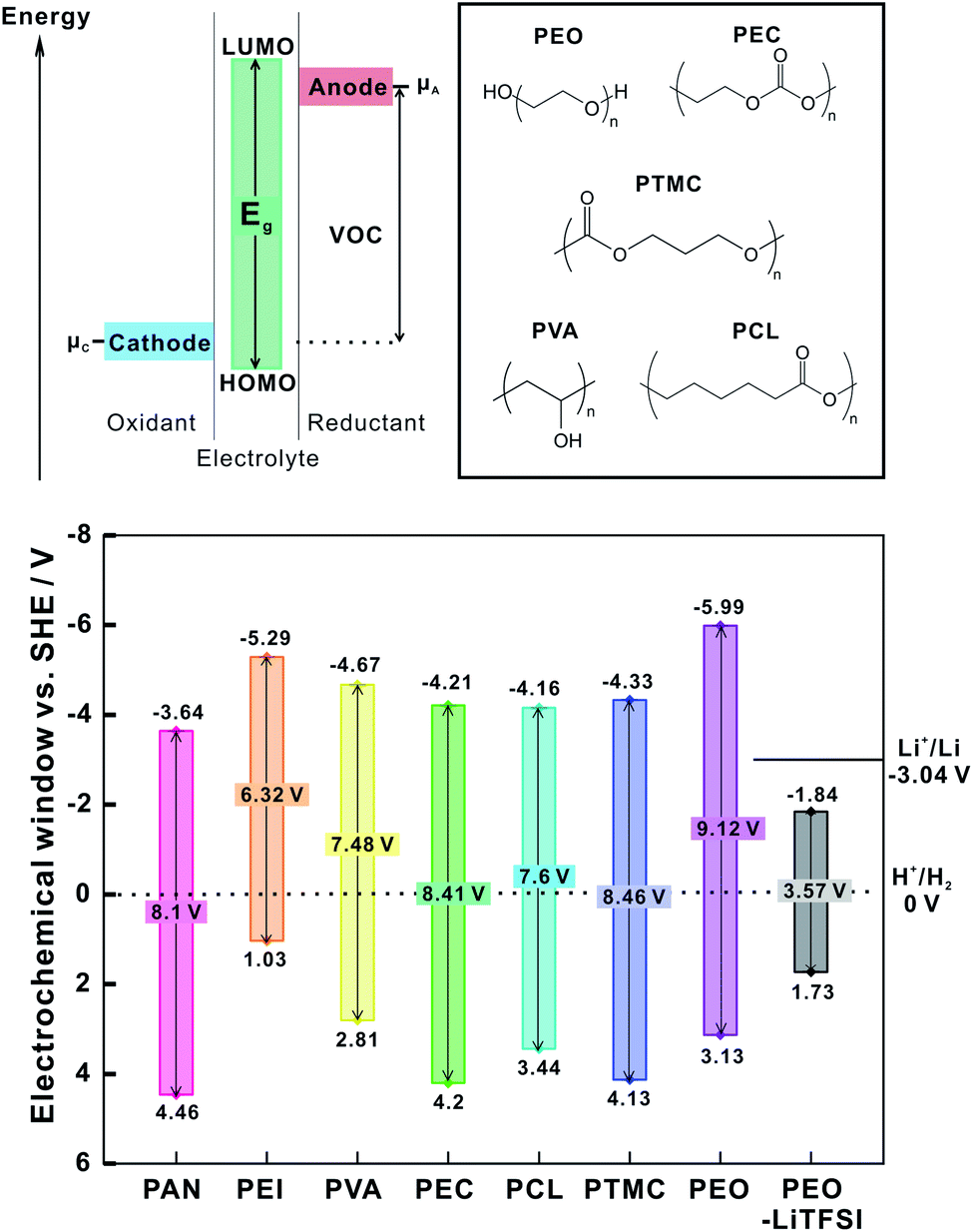 | ||
| Fig. 14 General representation of energy level diagram denoting HOMO and LUMO energy level and electrochemical window of polymers. The oxidation and reduction potentials as referenced to the SHE potential and the electrochemical window of pristine polymers and PEO-LiTFSI complex (as theoretically calculated in ref. 156). The electrochemical potential of cathode: μc, electrochemical potential of anode: μa. The structure of the polymers and the values of electrochemical stability are presented as reported in ref. 23. | ||
Marchiori et al. calculated the oxidation and reduction potentials (ESW = Eox − Ered) of several polymers with different polarity and structure: polyacrylonitrile (PAN), polyethylene imine (PEI), poly(vinyl alcohol) (PVA), polyethylene oxide (PEO), polycaprolactone (PCL), polyethylene carbonate (PEC), poly(trimethylene carbonate) (PTMC) (Fig. 14).23 The redox potentials are obtained through calculation of Gibb's free energy of the redox reaction and by using the Nernst equation. The corresponding redox pair is N/Red (Ox/N) where, N, Red and Ox are neutral, reduced and oxidized state of polymers, respectively.
The reduction (Ered) and oxidation (Eox) potentials of pristine polymers referenced to standard hydrogen electrode (SHE) potential are shown in Fig. 14. Of the selected polymers, PAN exhibits the highest oxidation potential (4.46 V versus SHE), and reduction potential (−3.65 V versus SHE) close to the Li+/Li potential. The PEI showed lower reduction potential than lithium with narrow ESW of about 6.32 V, restricting their application as interlayer. PTMC has a very large ESW (8.46 V), which means that negligible oxidation of PTMC is expected when used as coating for high voltage cathodes. However, Marchiori et al. also point out that the reactivity observed in experiments may be significantly different than the thermodynamic calculations suggest.23 For example, it is known that PAN reacts with lithium metal even though the reduction potential of PAN (−3.65 versus SHE) is quite close to the electrochemical potential of the lithium metal anode.161
Additionally, the presence of LiTFSI salt in PEO-LiTFSI decreases the electrochemical stability window (Fig. 14) as compared to pristine PEO.162 The higher diffusivity of the anion towards positive electrodes prompts additional oxidation of the salt anion. Such effects of salt decomposition result in capacity fade of LiFePO4 (LFP)-PEO-LiTFSI cathodes.162 Nie et al. also showed that LiCoO2 (LCO) serves as catalytic sites for PEO-LiTFSI decomposition in the cathode with SPE as separator.163
Still, the most used polymer coating is PEO-LiTFSI, as employed with LCO, LFP and NMC cathodes.84,160,164,165 The ESW of PEO-LiTFSI (3.8 V versus Li+/Li with carbon electrode) limits their application for high voltage cathodes.166 With LCO and LiNi0.5Mn1.5O4 (LNMO) cathodes, PEO-LiTFSI electrolyte exhibits a noisy voltage profile, which is interpreted as possible decomposition of PEO-LiTFSI in contact with a high voltage cathode. However, Homann et al. recently presented evidence that PEO-LiTFSI exhibits an oxidation potential of 4.6 V versus Li+/Li under galvanostatic cycling and potentio-dynamic conditions with LCO and NMC CAMs.167 Thus, the oxidation of PEO-LiTFSI was not found to be the cause for the noisy galvanostatic voltage profile of high voltage cathodes, potentially reopening the discussion on the electrochemical stability of PEO. Still, the possibility of polymer decomposition at high voltage cathodes with high surface area under repetitive cycling cannot be ruled out.
Irrespective of this, a conformal polymer coating has already been applied as protection layer on CAM and carbon additives in a SSB. It reduces the decomposition at the interfaces with the ISE. Deng et al. employed molecular layer deposition (MLD) to form an efficient conformal coating of 5 nm to 10 nm thickness with conducting polymer PEDOT on NMC and carbon nanotubes, and prepared a composite cathode with LGPS (Fig. 15a).157 The PEDOT served as protective layer on NMC to prevent LGPS degradation at NMC. The first charge–discharge voltage profile of composite cathode containing LGPS, PEDOT@NMC-811 and PEDOT@CNT in comparison to bare NMC and CNT is shown in Fig. 15b. At the first discharge, the polymer coated cathode (5 nm thickness) exhibited almost 2 times increment in capacity as compared to bare NMC at 0.05C rate (Fig. 15b). The performance enhancement is more distinct at higher C rate. The 5 nm PEDOT coated cathode delivers a capacity of over 100 mA h g−1 at 1C, which is 10 times higher than that of the bare cathode with LGPS. The capacity retention of 51% in PEDOT modified cathode composite is higher than that of the uncoated analogue (13.6%). The LGPS was electrochemically stable with PEDOT@CNT inside composite cathode as depicted from cyclic voltammograms (Fig. 15c).
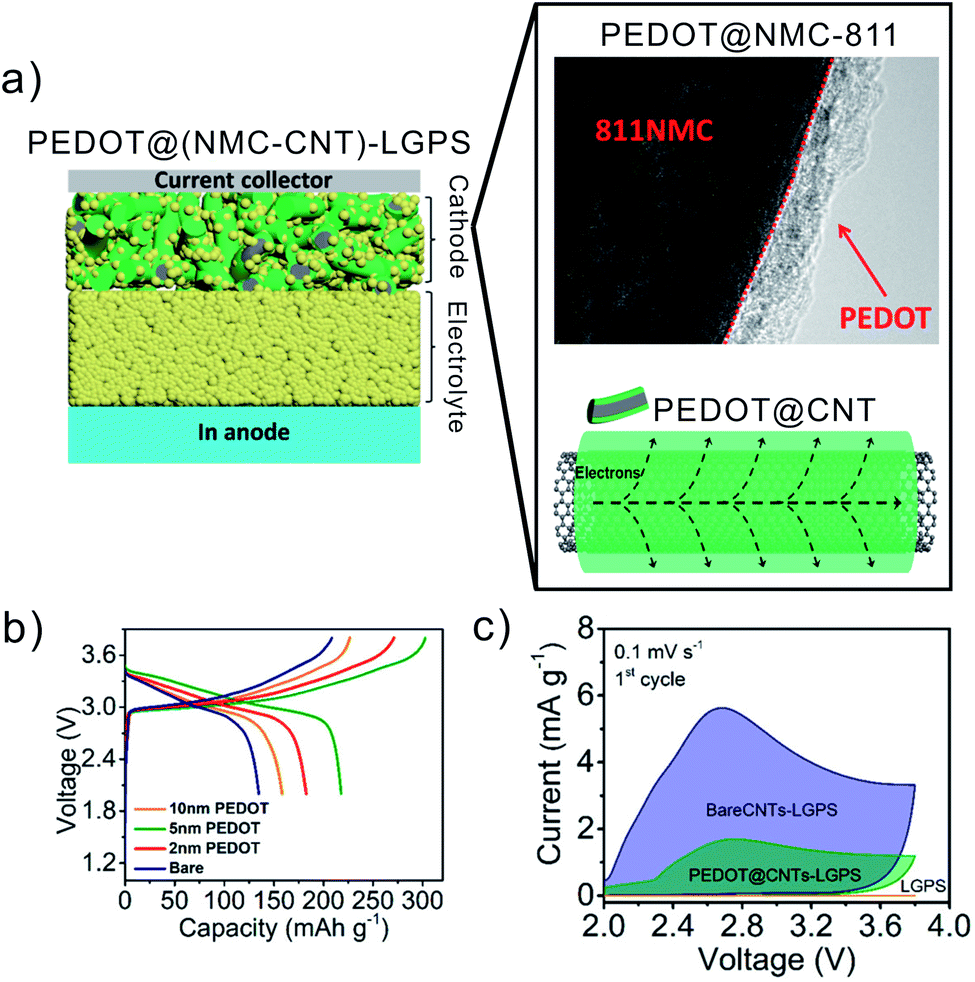 | ||
| Fig. 15 (a) Schematic illustration of the designed solid-state battery (Li/LGPS/PEDOT@NMC-CNT) with PEDOT interlayer on CAM. The composite cathode is denoted as PEDOT@(NMC-CNT)-LGPS. The conformal polymer coating onto cathode surface (PEDOT@Ni0.8Mn0.1Co0.1O2) (NMC-811) and onto CNT additive (PEDOT@CNT) is presented. (b) Charge–discharge profile of PEDOT@(NMC-CNT) (with a coating thickness 2 nm to 10 nm) along with bare cathode active material at the first cycle at 0.05C rate. (c) Cyclic voltammograms of In/LGPS/(LGPS-CNT) cell with and without PEDOT coating on CNT, recorded at 0.1 mV s−1 at the first cycle. Reproduced with permission from ref. 157. CCC, copyright @ 2020, American Chemical Society. | ||
The strategy of polymer coating on CAM has so far been more used in liquid and polymer electrolytes, where high voltage CAMs degrade liquid or polymer electrolytes. The effect of polymer coating on electrochemical stabilization of the cathode electrolyte interface is prominent with high voltage cathodes. For example, the anionic polyelectrolyte lithion (lithiated nafion) coating on NMC prevents degradation of ether-based electrolytes at the cathode interface.158 As a second example, polyethyl-α-cyanoacrylate coating on LCO exhibits better capacity retention with PEO-based electrolyte due to enhanced interface stability.168 The –CN group improves salt dissolution, ion conduction, and prevents transition metal ion dissolution from the cathode.168 In the same context, polymers with higher stability are desirable candidates for which some examples are reported for use as CAM coatings in batteries with liquid electrolyte. The dual core shell coating with an inner core coating with lithium polyacrylate (LiPAA) and an outer shell coating with cross-linked polymer based on silane-coupling agent (KH550) and poly(acrylic acid) (PAA), or single ion conducting block-co-poly(ionic liquid) coating are prominent examples.169,170Table 1 summarizes the cell cycling data of several composite cathodes with a wide range of polymer coatings, employed with liquid, polymer and inorganic solid electrolyte.
| CAMreference | Polymer coating | Electrolyte | Capacity | Capacity retention |
|---|---|---|---|---|
| a PEDOT: poly(3,4-ethylenedioxythiophene). LGPS: Li10GeP2S12 : NMC: nickel manganese cobalt oxide. Diglyme: bis(2-methoxyethyl) ether. LiNO3: lithium nitrate. HFiP: tris(hexafluoro-iso-propyl)phosphate. Lithion: lithiated Nafion. PEG: polyethylene glycol. EC: ethylene carbonate, DMC: dimethyl carbonate, EMC: ethyl methyl carbonate. PECA: polyethyl α-cyanoacrylate. LiDFOB: lithium difluoro(oxalato)borate. LiPAA: lithiated polyacrylic acid. KH550: 3-aminopropyltriethoxy silane. LiPF6: lithium hexafluorophosphate. PANI: polyaniline. The specific capacity of all the cathode active materials are reported with lithium metal as anode material except for ref. 157. In ref. 157, Li-In is used as anode material for LGPS solid electrolyte and PEDOT coated composite cathode. | ||||
| LiNi0.8Co0.1Mn0.1O2 (ref. 157) | PEDOT | LGPS | 100 mA h g−1 at 20th cycle at 1C | 51% after 23 cycles at 0.1C |
| NMC158 | Lithion | Diglyme–LiNO3–HFiP | 1.6 mA h cm−2 at 1st cycle at 0.1C | 80% after 200 cycles at 0.2C |
| LiNi0.6Co0.2Mn0.2O2 (ref. 159) | PEDOT-co-PEG | LiPF6 in EC:DMC (1:1) |
184.3 mA h g−1 at 1st cycle at 0.1C | 94% after 100 cycles at 0.5C |
| LiCoO2 (ref. 168) | PECA | PEO-LiDFOB | 172.8 mA h g−1 at 1st cycle at 0.1C | 39% after 75 cycles at 0.1C |
| LiNi0.8Co0.15Al0.05O2 (ref. 169) | LiPAA/KH550 | LiPF6 in DC:EMC:DMC (1:1:1) |
201.3 mA h g−1 at 1st cycle at 1C | 82.5% after 100 cycles at 1C |
| LiNi0.8Co0.1Mn0.1O2 (ref. 171) | PANI-PEG | LiPF6 in EMC:DMC:EC (1:1:1) |
201.6 mA h g−1 at 1st cycle at 0.2C | 93.4% after 100 cycles, 0.2C |
Maintaining percolation networks in cathode
For the successful operation of LiSSBs, the primary requirement in the cathode is good contact between the CAM and ISEs to maintain easy ionic and electronic conduction. The concepts of polymer interlayers employed in batteries with polymer and liquid electrolytes and their underlying chemistries are the obvious choice to be transferred to SSB cathodes. For example, mixed ionic and electronic conductivity through the respective percolation networks in the composite cathode is crucial. Both conduction mechanisms may be addressed by dual conducting polymers, such as PEDOT/PEG blends or polyaniline and PEO.159,171 One polymer serves as electron conducting pathway and the other gives mechanical strength and offers sites for ion conduction. In this way, the impedance of the cathode composite in SSBs can be reduced.Alternatively, ion-conducting polymer interlayers that have suitable rheological properties may serve as a soft adaptive buffer to decrease the interface resistance between CAM particles, mitigate crack formation and improve ion transfer between CAM and ISE. For example, coating with ionogel electrolytes,172 self-healing polymers,173,174 or by in situ polymerization,156 may prove to be very beneficial. Even though these have already been developed for batteries with liquid and polymer electrolyte, their application in SSBs with ISEs has yet to be tested. However, a potential caveat to the use of these more complex polymer chemistries is the reactivity of many ISEs with polar compounds, which we have discussed in the binder section. More research is required for a better understanding of the complex interplay of materials including polymers in the composite cathode.
Future perspective and comments
Although polymer coatings on CAMs have been investigated to improve the cycling stability using cells with liquid electrolytes and SPEs, to the best of our knowledge, their use in SSBs with ISEs appears to be limited to only one example at present. This surprising gap will hopefully incentivize research in this direction, especially considering the promising examples, which have already been demonstrated for batteries with liquid or polymer electrolytes. More research with ISE-based SSBs is needed to explore the interplay of polymers and inorganic materials in the composite cathode. It is as yet unclear, which are the most suitable mechanical properties for interlayers and coatings to mitigate cracks in the cathode. But, considering the full scope of polymer mechanical properties covering viscous melts, rigid, brittle, or rubbery solids as well as cross-linked and swollen gels, there is clearly a large parameter space for further investigation.Evidently, chemical compatibility of all materials in the composite cathode needs to be ensured, which will not be simple to achieve considering the reactivity of the compounds involved. Nonetheless, progress has been made on increasing the electrochemical stability of polymers. However, the plethora of chemical, rheological and mechanical properties of polymers offers a large area for innovation. In particular, research on charge transport and mechanical properties at the polymer/ISE, polymer/CAM and polymer/carbon additive interfaces is required. The key aspects include improving electrochemical stability, understanding the mechanics of the interfaces, and optimizing the electronic and ionic transport in the composite cathode. This challenge offers scientists the opportunity to pursue a truly interdisciplinary approach, develop new hybrid materials and create multifunctional cell designs.
Protective coatings and interlayers in anode
Solid electrolyte interface and role of polymer interlayer
In the previous chapters, we discussed the roles of polymer interlayers in the separator and cathode. Now, we will conclude our journey through the different cell components of LiSSBs with an overview on polymer materials when used as a protective coating on the lithium metal anode. A crucial prerequisite to achieve a sufficient electrochemical performance of lithium metal batteries is the formation of a stable interface between lithium metal and the electrolyte. Three principal problems can be identified: formation of an unstable SEI, dendrite growth, and pore formation at the interface with the electrolyte. Accordingly, the incorporation of a protective interlayer is suggested to address these problems and may facilitate the employment of lithium metal as anode material. Polymer interlayers are utilized to decrease the interface resistance by facilitating uniform ion diffusion, reducing the overpotential and maintaining the contact between lithium metal and the ISE. As there are yet only few examples of polymer interlayers used with ISEs, we also refer to polymer coatings for lithium tested in cells with liquid electrolyte to give a more complete overview of materials, concepts and opportunities of polymer coatings on lithium metal.Several preparation methods have been used to create polymer coatings on lithium metal foil. For example, solution based coating techniques such as spin coating,175,176 solution casting177 and tape casting178,179 are applied to achieve thin coatings without expensive equipment. The choice of process parameters and materials affects the uniformity and thickness of the coating layers,180 which generally range from submicron to a few micrometers.181 In addition to the usual procedure to introduce a coating on lithium metal, as is required for use with liquid electrolytes, polymer interlayer deposition on ISE was also reported to enable straightforward coating by dipping the ISE into the polymer solution177,182 and to prevent the reaction of acetonitrile in contact with lithium metal.177 In addition, molecular layer deposition (MLD) allows to prepare uniform coatings with a thickness of a few nanometers and provide the possibility to combine inorganic and organic precursors at low growth temperatures.183,184 However, the MLD process suffers from moisture sensitivity of the coatings, a limited number of available organic precursors with sufficient vapor pressure and the permeability of MLD coatings, resulting in a precursor gas absorption and subsequent layer growth.185
For the analysis of the chemical composition and microstructure of the interface between lithium metal and liquid electrolytes, XPS and SIMS measurements were used186 and more recently also cryo-TEM.187 The formation of a SEI passivation layer is almost inevitable when using lithium metal as anode material. The absence of a stable SEI would lead to a continuous reaction of lithium metal with almost all liquid and solid electrolytes.33,188 Furthermore, short-circuits caused by dendrite growth during cell cycling can be prevented by an ideal SEI.189,190 However, the potentially infinite relative volume change during lithium stripping and plating still challenges the mechanical stability of the thin native SEI layer.191 Thus, the performance of a protective layer largely depends on its capability to accommodate the stress resulting from the lithium anode volume change. Elastic polymers can address the existing interfacial challenges and facilitate the formation of a low resistive and stable SEI.192,193
Initially, protective polymer coatings were introduced to protect lithium in liquid electrolytes. Due to its lithium affinity, ether-based polymers appeared to be suitable coating materials. Gao et al. coated a lithium anode with the cyclic ether containing poly((N-2,2-dimethyl-1,3-dioxolane-4-methyl)-5-norbornene-exe-2,3-dicarbox-imide) and cells with a NMC cathode with liquid electrolyte provided a capacity of 1.2 mA h cm−2 with high capacity retention of 90% after 400 cycles.194Fig. 16a exemplarily depicts the structure of the polymer coating, a schematic illustration of the optimized SEI and protected lithium in combination with liquid electrolyte. As a second example, Dong et al. deposited a flexible, 5.1 μm thick polymer film composed of 18-crown-6 and PVDF via spin coating on lithium metal and achieved an enhanced cycling stability in Li–Cu half-cells with liquid electrolyte.175 The assumption that the Li+ affinity of oxygen atoms of 18-crown-6 improves the homogeneity and kinetics of the Li+ transport is confirmed by a reduced resistance compared to a pure PVDF layer. XPS measurements of the coated lithium anode revealed the presence of LiF and the authors attributed it to a stabilization of the SEI and prevention of dendrite growth.195–197 Other polymers including aluminium alkoxide (alucone),183 poly(diallyldimethylammonium chloride) (PDADMA),196 poly(dimethylsiloxane) (PDMS),198 poly(vinylidene fluoride-co-hexafluoropropylene) (PVDF-HFP),199 or cross-linked polymeric ionic liquid178 have also been used as protective coatings in lithium metal batteries with liquid electrolyte.
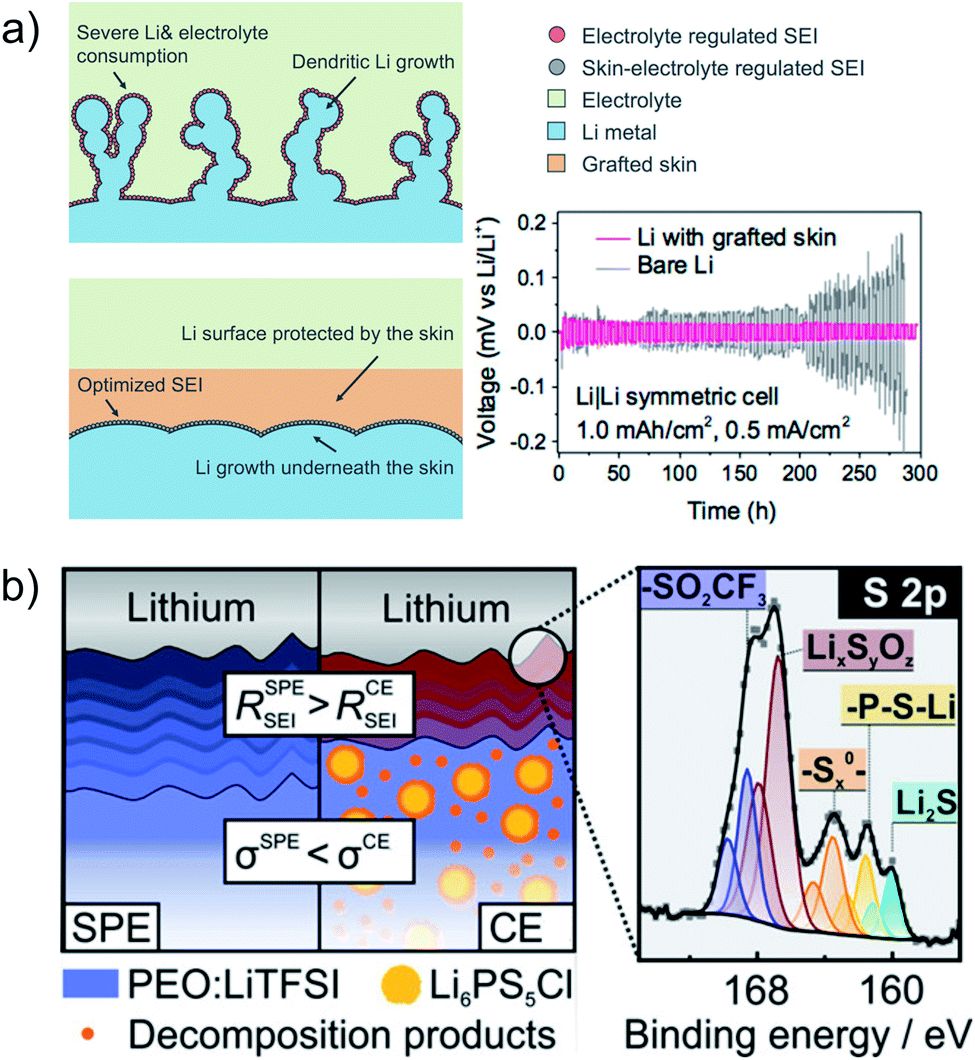 | ||
| Fig. 16 (a) Schematic illustration of the formed protective surface layer and the voltage profile of a symmetric Li metal cell. Adapted with permission from ref. 194 CCC, copyright @ 2017, American Chemical Society. (b) Schematic representation of the contribution of composite electrolyte decomposition products on SEI formation and corresponding X-ray photoelectron spectroscopy measurement of decomposition products observed in the S 2p signal. Adapted with permission from ref. 200 CCC, copyright @ 2020, American Chemical Society. | ||
The most extensively investigated polymer coatings on lithium are PEO-based SPEs.201 Since the Li+ ion transport in PEO is facilitated by segmental motion and relaxation of the polymer backbone in the amorphous regions, a higher amorphous fraction will be advantageous for ion conduction, but the mechanical stability decreases. Early studies reported that lithium metal in contact with PEO-based electrolytes can suffer from a gradually increasing interfacial impedance.192,202 In addition, a study on the interface between lithium metal and pure PEO without lithium salt revealed that the dissolution of lithium into PEO is associated with good adhesion of lithium and PEO.203 Additionally, the authors reported an increased conductivity in Li/PEO/Li cells by a factor of 16 after annealing at 120 °C for 7 days and suggested that a lithium cation and a free electron is dissolved in PEO.
Fig. 16b depicts the XPS analysis of decomposition products at the interface of lithium with LPSCl and PEO-LiTFSI CE by Simon et al.200 The authors found that the LPSCl particles increased the lithium ionic conductivity from 0.84 mS cm−1 (PEO-LiTFSI) to 3.6 mS cm−1 (40 wt% LPSCL in PEO-LiTFSI – CE 40) at 80 °C. The formed decomposition products, predominantly LiF and Li2S, were incorporated in the SEI, resulting in a reduced interface resistance from 5.8 Ω cm2 (PEO-LiTFSI) to 3.3 Ω cm2 (CE 40) and a reduced growth rate of SEI resistance from 1.2 Ω cm2 h−0.5 (PEO-LiTFSI) to 0.57 Ω cm2 h−0.5 (CE 40) at 80 °C. In a second example, a cross-linked poly(ethylene glycol) methyl ether acrylate SPE layer was used to protect LATP ISE, which would be reduced in direct contact with lithium metal.103 A PEO-LiTFSI interlayer has also been used to improve the cycling stability of Li/PEO-LiTFSI/LPS/NMC with β-Li3PS4, which is known to chemically decompose in direct contact with lithium.193 Additionally, Tang et al. reported the use of 15 vol% of a liquid carbonate based electrolyte to protect the chemically instable LATP ISE and the interfacial resistance was decreased from 1400 Ω (pristine LATP) to 90 Ω.204
The incorporation of fillers can improve the chemical stability and mechanical strength of the SPEs.201,205 Additionally, a ceramic filler (e.g. Al2O3, TiO2) increases the low-temperature ionic conductivity of PEO-based SPEs by hampering recrystallization via Lewis acid–base interactions.206,207 Chen et al. deployed 50 wt% LATP ISE filler in a cross-linked PEO-based polymer gel electrolyte plasticized with tetraethylene glycol dimethyl ether (TEGDME).208 It was found that the cycling performance at room temperature was increased significantly with the introduction of the LATP filler, resulting in decreased overpotentials (0.04 V compared to 0.13 V without filler), reduced interfacial impedance (360 Ω cm2 compared to 750 Ω cm2 without filler) and doubled Li+ transference number. Huo et al. showed that the incorporation of 20 vol% of LLZTO filler in a PEO matrix results in a CE with an ionic conductivity of 0.16 mS cm−1 at 30 °C.209 Recently, Xu et al. reported a CE of PEO-LiClO4 containing nanosized SiO2 with an ionic conductivity of 0.11 mS cm−1 at 30 °C and a high capacity retention of 70% after 100 cycles at 0.3C and 90 °C.210
Despite its comparatively low ionic conductivity of 10−6 S cm−1, which restricts its application to thin film batteries, glassy lithium phosphorous oxynitride (LiPON) offers stable and fast cycling with lithium metal anodes. Therefore, it is used as solid electrolyte in commercial thin film batteries. LiPON exhibits a wide voltage window (0–5.5 V) in contact with lithium metal and Li/LiPON/LiCoO2 cells achieve more than 50% of the maximum capacity at a high discharge rate of 5 mA cm−2. For a lithium free Cu/LiPON/LiCoO2 cell the capacity decreased only by 20% over 1000 cycles at a discharge current of 1 mA cm−2.211 It was also reported that LiPON is capable to be cycled at remarkable current densities up to 10 mA cm−2.212 These results have inspired the development of LiPON-like polymers with decent ionic conductivities (around 10−5 S cm−1) synthesized from molecular precursors like hexachlorophosphazene (Cl2PN)3.213 Additionally, Abels et al. synthesized soluble polyphosphazenes ((H2PO2N)n) with a chemical composition similar to glassy LiPON by a two-step one pot synthesis.214 Such LiPON-inspired polymers may facilitate a stable SEI formation on lithium by decomposing into Li2O, Li3P and Li3N, which are decomposition products of glassy LiPON.215
Role of polymer interlayer for preventing dendrite and pore formation
Owing to the favorable mechanical properties of polymers, their incorporation as interlayer between lithium anode and ISE can enable enhanced wetting and prevent contact loss at the interfaces. For example, to compensate the roughness of a garnet ISE surface and decrease the interfacial impedance, Fu et al. applied a 2 μm thick conformal PEO-based polymeric gel layer between lithium metal and LLZO.216 Symmetric Li/PEO/LLZO/PEO/Li cells exhibit a low and smooth voltage hysteresis of ±0.3 V at a current density of 0.3 mA cm−2, which confirms a decreased interfacial resistance and a homogenous lithium ion flux. Fig. 17a illustrates schematically the incorporation of a uniform PEO-LiTFSI interlayer to improve the poor interfacial contact of lithium metal and Ta-doped LLZO (LLZTO) by Chi et al.177 To achieve a better wetting, the Li/PEO-LiTFSI/LLTZO/PEO-LiTFSI/Li cells were pre-heated to 90 °C for 5 h and a subsequent cycling at 0.2 mA cm−2 delivered a flat and stable voltage plateau and reduced interfacial resistance from 2000 Ω (without PEO-LiTFSI layer) to 750 Ω (with PEO-LiTFI layer) after 100 h cycling at 90 °C. The 6 μm thick PEO-LiTFSI coating was stable up to 4.6 V vs. Li+/Li. In situ SEM studies of a 20 μm thick PEO-LiTFSI SPE in a Li/PEO-LiTFSI/Li1.2V3O8 cell assembled with a pressure of 35 psi showed a contraction of about 8 μm when the temperature was set at 80 °C during the first cycle.13 The authors concluded that the SPE has filled the remaining pores when the temperature was raised. The working principle appears to be similar to that of an ionic liquid interlayer, which has also been used to mitigate pore formation at the lithium ISE interface.217,218 | ||
| Fig. 17 Schematic illustration of the benefit of polymer interlayers between lithium anode and solid electrolyte. (a) A polymer interlayer is employed to fill the voids at the lithium metal LLZTO interface and thereby improve the interfacial contact. Adapted from ref. 177, Copyright © 2019 Elsevier B.V. license @ CCC, Elsevier. (b) Inhomogeneous ion flux will result in dendrite growth (left), whereas a polymer coating, such as PEO, PVDF-HFP or PVDF, on the lithium anode can facilitate a homogeneous ion flux (right). | ||
Huo et al. reported the use of an electron-blocking interfacial shield comprising a PAA interlayer between lithium anode and LLZO (Fig. 18a), which decreased the interfacial resistance from 1104 Ω cm2 without PAA interlayer to 55 Ω cm2 with PAA interlayer.219 Symmetric Li/PAA/LLZTO/PAA/Li cells showed stable cycling for 400 h at a current density of 0.5 mA cm−2 whereas a Li/LZZTO/Li cell could not be cycled (Fig. 18b).
 | ||
| Fig. 18 Schematic illustration of the electron-blocking interlayer shield between lithium anode and LLZTO ISE and the electrochemical performance of a Li symmetrical cell. (a) A PAA interlayer is employed to hamper the dendrite growth at the lithium metal LLZTO interface. (b) Voltage profile of lithium symmetric cells with and without PAA interlayer at a current density of 0.5 mA cm−2. Adapted from ref. 219. | ||
Since the mechanical properties of thermoplastic polymers depend on temperature and pressure, an applied pressure at elevated temperatures should improve the interfacial contact. Gupta et al. investigated the influence of applied pressure and temperature on the interface stability between lithium metal and SPE (PEO-LiTFSI).220 They found that the conductivity increased with temperature from 10−4 S cm−1 at 60 °C to 10−3 S cm−1 at 100 °C and that the interfacial impedance decreased exponentially with increasing stack pressure up to an asymptote at 400 kPa at 60 °C and 200 kPa at 80 °C. However, the critical current density (CCD) for initiation of dendritic growth (0.5 mA cm−2) was invariant with temperature. This is contradictory to the trend of an increased CCD with increasing temperature that was observed for LLZO in Li/LLZO/Li symmetrical cells.221 An improved wetting and interfacial contact between lithium metal and LLZO with elevated temperature is suggested to account for a decreased interfacial charge-transfer resistance and accordingly a more uniform current density.222 In contrast, PEO-LiTFSI may not follow this trend since it exhibits a too low shear modulus to hamper the dendrite growth at elevated temperature.220 Regarding the hampering of dendrite growth, theoretical models for LiSSBs with the lithium polymer interface proposed a suppression of lithium dendrites can be obtained by a polymer electrolyte with a sufficiently high shear modulus (Gelectrolyte > 2 × Glithium),223–225 which leads to a required shear modulus of 6 GPa that is challenging to achieve for PEO above the glass transition temperature.167
A comprehensive overview of the interface of lithium metal to ISEs by Krauskopf et al. showed that the mechanisms of lithium growth through ISEs cannot be explained exclusively by this criterion.44 Additionally, Porz et al. reported a study of the lithium ISE interface.226 They concluded that interfacial defects favor dendrite propagation in high modulus, brittle inorganic solid electrolytes. Therefore, uniform Li+ flux is required for dendrite-free plating of lithium. SPE interlayers between the lithium metal anode and the ISE are considered a promising approach to prevent electrolyte decomposition and dendrite growth, as illustrated in Fig. 17b. Zhou et al. reported a stable and dendrite free cycling of LATP enclosed between two cross-linked poly(ethylene glycol) methyl ether acrylate SPE layers in a Li/SPE/LATP/SPE/LiFePO4 cell.103 The conductivity of the SPE was around 0.1 mS cm−1 at 65 °C and cycling of the cell obtained a CE of 99.9% at 0.2C with a slightly increased cell polarization (0.22 V) compared to a Li/SPE/LiFePO4 cell (0.15 V). The polymer layer provided a uniform Li+ flux by an efficient wetting of the lithium anode and the anion blocking ceramic reduced the anion depletion at the Li/PEO interface, which contributed to dendrite suppression and an enhanced Coulomb efficiency by preventing ISE degradation.103
‘Lithium free’ approach
The ‘lithium free’ approach describes a LiSSB based on an in situ formed lithium anode that is created during the first charge, instead of placing a lithium foil as anode. Thereby, the energy density is increased and the handling of highly reactive lithium metal during cell assembly is no longer necessary.227 This cell concept is particularly challenging to realize as it requires careful control of the nucleation process during plating, which needs to result in a uniform layer of deposited lithium. In recent years, several concepts of ‘lithium free’ cells have been reported that stabilize lithium stripping and plating, and accommodate the high volume changes during cycling. Nanda et al. summarized the strategies to improve ‘lithium free’ cell performance as the modification of electrolyte and current collector substrate and the optimization of formation and cycling parameters.228 The authors reported the use of organic polymer coatings, a secondary inorganic solid electrolyte, lithiophilic materials (Si, Ge, Au, Ag) and polymer–ceramic composites as buffer layer to maintain the pristine contact between lithium metal and ISE in ‘lithium free’ SSBs.With liquid electrolytes, Sahalie et al. employed a composite coating containing Al2O3 particles in a polyacrylonitrile (PAN) matrix on a copper current collector.229 The composite layer showed a more homogeneous lithium ion flux, which was explained by the beneficial effect of aluminum- and fluoride-based decomposition compounds of the LiPF6 salt in the SEI, and exhibited a sufficient strength to prevent dendrite formation. Cycling experiments with a NMC cathode and liquid electrolyte at 0.2 mA cm−2 achieved an initial discharge capacity of 160 mA h g−1 and a capacity retention of 30% after 82 cycles. As a second example with liquid electrolyte, solvent processed polyelectrolyte (poly(N,N-dially-N,N-dimethylammonium bis-(trifluoromethylsulfonylimide)) (PDADMA)) was coated on copper and employed as host for lithium deposition in a Li/Cu cell with liquid electrolyte.230 The PDADMA coating provided dendrite-free cycling at a current density of 1 mA cm−2 and high Coulomb efficiency for 80 cycles.
Lee et al. recently reported the successful operation of a ‘lithium free’ SSB by electroplating lithium metal at the interface of a stainless-steel current collector (SUS) and a Ag–C nanocomposite in a SUS/Ag–C/LPSCl/NMC cell.17 The lithiophilic silver nanoparticles and carbon black formed a composite with PVDF that facilitated dendrite free and uniform lithium plating. The authors demonstrated more than 1000 cycles of the SUS/Ag-C/LPSCl/NMC cell at 60 °C while achieving a remarkably high energy density (900 W h l−1) and Coulomb efficiency (99.8%). Notably, this cell concept relies on the use of a PVDF–Ag–C nanocomposite coated on the current collector, which allows the reversible plating and stripping of lithium. As a second example, the use of a garnet PEO-LiTFSI composite layer both on the copper current collector and the cathode was recently reported by Zegeye et al.231 The garnet-PEO CE exhibited an electrochemical window of 4.75 V, an ambient temperature conductivity of 0.48 mS cm−1 and cycling with a NMC cathode delivered a CE of 98.8% and a capacity retention of 41.2% after 65 cycles at 0.2 mA cm−2 at 55 °C. Table 2 summarizes the cell cycling data of polymer coated anodes using liquid, polymer and inorganic solid electrolyte.
| AnodeReference | Polymer coating | Electrolyte | CAM | Capacity | Capacity retention |
|---|---|---|---|---|---|
| a Polycyclic polymer: poly((N-2,2-dimethyl-1,3-dioxolane-4-methyl)-5-norboronene-exo-2,3-dicarboximide). PVDF: polyvinylidene fluoride. 18-crown-6: 1,4,7,10,13,16-hexaoxacyclooctadecane. CPMEA: cross-linked poly(ethylen glycol) methyl ether acrylate LiTFSI: lithium bis(trifluoromethanesulfonyl)imide. PEO: polyethylene oxide. PAA: polyacrylic acid. PAN: polyacrylonitrile. Al2O3: aluminium oxide. LiNO3: lithium nitrate. KaNO3: potassium nitrate. Li3PO4: lithium phosphate. LiFePO4: lithium iron phosphate. EC: ethylene carbonate. DMC: dimethyl carbonate. EMC: ethyl methyl carbonate. FEC: fluoroethylene carbonate. DOL: dioxolane. LiPF6: Lithium hexafluorophosphate. VGCF: vapor grown carbon fibers. CNT: carbon nanotube. | |||||
| Lithium194 | Polycyclic polymer | LiPF6 in EC:EMC:FEC (3:7:1) |
LiNi0.5Co0.2Mn0.3O2 | 1.0 mA h cm−2 at 1st cycle at 0.3 mA cm−2 | 90% after 400 cycles at 0.3 mA cm−2 |
| Lithium175 | 18-crown-6, PVDF | LiTFSI inDOL:DME (1:1), LiNO3 |
S | 530 mA h g−1 at 1st cycle at 0.2C | 78% after 200 cycles at 0.2C |
| Lithium103 | CPMEA | Li1.3Al0.3Ti1.7(PO4)3 | LiFePO4 | 120 mA h g−1 at 1st cycle at 0.6C | 85% after 640 cycles at 0.6C |
| Lithium193 | LiTFSI-PEO | β-Li3PS4 | LiNi0.6Mn0.2Co0.2O2, VGCF | 140 mA h g−1 at 1st cycle at 0.05C | 90% after 50 cycles at 0.05C |
| Lithium216 | PEO | Li7La2.75Ca0.25Zr1.75Nb0.25O12, LiTFSI in DOL:DME (1:1) |
S (7.5 mg cm−2), CactNT | 645 mA h g−1 at 1st cycle at 0.2 mA cm−2 | 75% after 32 cycles at 0.2 mA cm−2 |
| Lithium177 | LiTFSI-PEO | Li6.4La3Zr1.4Ta0.6O12 | LiFePO4 | 149 mA h g−1 at 1st cycle at 0.2C | 90% after 200 cycles at 0.2C |
| Lithium219 | PAA | Li6.4La3Zr1.4Ta0.6O12 | LiFePO4 | 120 mA h g−1 at 1st cycle at 0.5C | 83% after 200 cycles at 0.5C |
| Anode free229 | PAN–Al2O3 | LiPF6 in EC:DEC (1:1), KNO3 |
LiNi0.3Mn0.3Co0.3O2 | 160 mA h g−1 at 1st cycle at 0.2 mA cm−2 | 30% after 82 cycles at 0.2 mA cm−2 |
| Anode free17 | Ag–C–PVDF | Li6PS5Cl | LiNi0.9Co0.05Mn0.05O2 | 146 mA h g−1 at 1st cycle at 0.5C | 89% after 1000 cycles at 0.5C |
| Anode free231 | LiTFSI-PEO-Li6.75La3Zr1.75Ta0.25O12 | LiTFSI-PEO-Li6.75La3Zr1.75Ta0.25O12 | LiNi0.3Co0.3Mn0.3O2 | 2.20 mA h cm−2 at 1st cycle at 0.2 mA cm−2 | 41.2% after 65 cycles at 0.2 mA cm−2 |
Future perspective and comments
The interfacial instability of lithium metal anodes in contact with SEs remains a challenge to be overcome on the pathway to develop competitive LiSSBs. Due to their versatile and tunable properties, there is no doubt that polymers can contribute to stabilize the lithium metal interface in energy storage devices. To date, polymers have been used as protective coating on lithium metal with a variety of solid and liquid based electrolytes. For both, it has been found that decomposition compounds such as LiF and LiS2 can effectively improve interfacial stability, reduce interfacial resistance and provide uniform Li+ ion flux.197,200 In the case of LiSSBs, SPEs have also been applied to enhance the often poor contact between two inorganic solids and to facilitate the protection of chemically unstable ISEs such as LATP and β-Li3PS4. Despite their use as protective interlayers, it must be noted that the electrochemical stability against lithium metal often remains unreported for the applied SPEs.Composite electrolytes exhibit enhanced mechanical properties that are required for sufficient suppression of dendrite growth and obtain the possibility of an improved room temperature ionic conductivity compared to SPEs. Nevertheless, the incorporation of filler particles in the polymer matrix can result in a trade-off in properties since the modification of the polymer structure can have an opposing effect on ionic conductivity and mechanical strength. Recently, Lee et al. demonstrated a ‘lithium free’ cell with a remarkably high overall performance and a Coulomb efficiency of 99.8% that used a PVDF–Ag–C nanocomposite as coating on the current collector.17 The trend to ‘lithium free’ cell concepts has become dynamic in recent years and the efforts to a fundamental understanding will certainly provide new challenges for polymer interlayers. Particularly, the effect of a polymer coating on the nucleation of lithium needs to be intensively studied and a better understanding will help to achieve the required homogenous and stable lithium plating. While most of the reported literature focuses on SPEs based on PEO, other polymers are still underrepresented. Obviously, polymers combine convenient solution or melt processing with the possibility to tune the mechanical properties. Structures can range from an elastic to a rigid cross-linked network. In addition to the properties of the polymer layer, the decomposition products also affect the stability of the lithium polymer interface.
Conclusions
The roles of polymers in solid-state batteries with inorganic solid electrolytes are as numerous as the polymer chemistry and properties are varied. Following this idea, we give a general overview of polymer–inorganic composites and highlight recent literature to demonstrate the manifold use of polymers in composite electrolytes, as binders in separators and cathodes, as interlayers in composite cathodes, and as coatings on lithium metal. The surveyed literature demonstrates the increasing importance given to polymer-modified LiSSBs with inorganic solid electrolytes to help with cell preparation and with stabilizing the electrode interfaces with the inorganic solid electrolyte.Over time, it seems to become increasingly evident that no single solid electrolyte combines all required properties in one material and that it takes the right combination and handling of all types of materials to create a SSB with competitive energy density, rate capability and cycling stability. However, our review also shows that the number of investigations is not evenly distributed among the SSB components. In particular, there seems to be a striking lack of investigations on the use of polymers as coatings in the composite cathode. This may be because of the long-standing teaching that particularly PEO-based solid polymer electrolytes are not stable at high potential versus Li+/Li, but recent studies appear to open up new avenues of research regarding the development and application of new and more stable polymers.
Much may already be learnt also from lithium-ion battery research, particularly regarding the use of binders for cell preparation and efforts to make the ‘lithium-free’ anode a scalable reality using polymer coatings. For SSB preparation, reactivity between polymer, cathode active material, carbon additive, inorganic solid electrolyte and solvent plays a crucial role, incentivizing research on dry processing methods, avoiding the use of polar solvents in particular. Another important topic is the electrochemical stability of polymers at cathode and anode interfaces. Only those with wider electrochemical stability window are expected to be suitable as SSB interlayer, as chemical reaction and surface degradation between battery materials usually slows ion transport. However, it is important to keep an open mind about this, as interphases of degradation products sometimes turn out to be suitable protective layers that form in situ. Therefore, the degradation products that are formed at the interfaces also need to be studied carefully, to finely correlate chemical functionality, mechanical properties and SSB performance.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are very grateful to Professor Jürgen Janek and his research group at the Institute of Physical Chemistry of the Justus-Liebig-University Gießen for valuable discussions. We would also like to thank the Federal Ministry of Education and Research (BMBF) for funding of the projects FLiPS (03XP0261) and Adambatt (03XP0305C).References
- B. D. McCloskey, Expanding the Ragone Plot: Pushing the Limits of Energy Storage, J. Phys. Chem. Lett., 2015, 6, 3592–3593 CrossRef CAS PubMed.
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X.-Q. Yang and J.-G. Zhang, Pathways for practical high-energy long-cycling lithium metal batteries, Nat. Energy, 2019, 4, 180–186 CrossRef CAS.
- M. Pasta, D. Armstrong, Z. L. Brown, J. Bu, M. R. Castell, P. Chen, A. Cocks, S. A. Corr, E. J. Cussen, E. Darnbrough, V. Deshpande, C. Doerrer, M. S. Dyer, H. El-Shinawi, N. Fleck, P. Grant, G. L. Gregory, C. Grovenor, L. J. Hardwick, J. T. S. Irvine, H. J. Lee, G. Li, E. Liberti, I. McClelland, C. Monroe, P. D. Nellist, P. R. Shearing, E. Shoko, W. Song, D. S. Jolly, C. I. Thomas, S. J. Turrell, M. Vestli, C. K. Williams, Y. Zhou and P. G. Bruce, 2020 roadmap on solid-state batteries, J. Phys. Energy, 2020, 2, 32008 CrossRef CAS.
- J. Janek and W. G. Zeier, A solid future for battery development, Nat. Energy, 2016, 1(9), 1–4 CrossRef.
- S. Randau, D. A. Weber, O. Kötz, R. Koerver, P. Braun, A. Weber, E. Ivers-Tiffée, T. Adermann, J. Kulisch, W. G. Zeier, F. H. Richter and J. Janek, Benchmarking the performance of all-solid-state lithium batteries, Nat. Energy, 2020, 5, 259–270 CrossRef CAS.
- J. Li, Y. Cai, H. Wu, Z. Yu, X. Yan, Q. Zhang, T. Z. Gao, K. Liu, X. Jia and Z. Bao, Polymers in Lithium-Ion and Lithium Metal Batteries, Adv. Energy Mater., 2021, 11, 2003239 CrossRef CAS.
- S. Li, F. Lorandi, J. F. Whitacre and K. Matyjaszewski, Polymer Chemistry for Improving Lithium Metal Anodes, Macromol. Chem. Phys., 2020, 221, 1900379 CrossRef CAS.
- W. Li, B. Song and A. Manthiram, High-voltage positive electrode materials for lithium-ion batteries, Chem. Soc. Rev., 2017, 46, 3006–3059 RSC.
- W. Li, E. M. Erickson and A. Manthiram, High-nickel layered oxide cathodes for lithium-based automotive batteries, Nat. Energy, 2020, 5, 26–34 CrossRef CAS.
- F. Wu and G. Yushin, Conversion cathodes for rechargeable lithium and lithium-ion batteries, Energy Environ. Sci., 2017, 10, 435–459 RSC.
- S.-H. Yu, X. Feng, N. Zhang, J. Seok and H. D. Abruña, Understanding Conversion-Type Electrodes for Lithium Rechargeable Batteries, Acc. Chem. Res., 2018, 51, 273–281 CrossRef CAS PubMed.
- A. Bielefeld, D. A. Weber and J. Janek, Modeling Effective Ionic Conductivity and Binder Influence in Composite Cathodes for All-Solid-State Batteries, ACS Appl. Mater. Interfaces, 2020, 12, 12821–12833 CrossRef.
- P. Hovington, M. Lagacé, A. Guerfi, P. Bouchard, A. Mauger, C. M. Julien, M. Armand and K. Zaghib, New lithium metal polymer solid state battery for an ultrahigh energy: nano C-LiFePO4 versus nano Li1.2V3O8, Nano Lett., 2015, 15, 2671–2678 CrossRef CAS.
- J. Lopez, D. G. Mackanic, Y. Cui and Z. Bao, Designing polymers for advanced battery chemistries, Nat. Rev. Mater., 2019, 4, 312–330 CrossRef CAS.
- D. Y. Oh, Y. J. Nam, K. H. Park, S. H. Jung, K. T. Kim, A. R. Ha and Y. S. Jung, Slurry-Fabricable Li + -Conductive Polymeric Binders for Practical All-Solid-State Lithium-Ion Batteries Enabled by Solvate Ionic Liquids, Adv. Energy Mater., 2019, 9, 1802927 CrossRef.
- Y. J. Nam, D. Y. Oh, S. H. Jung and Y. S. Jung, Toward practical all-solid-state lithium-ion batteries with high energy density and safety: Comparative study for electrodes fabricated by dry- and slurry-mixing processes, J. Power Sources, 2018, 375, 93–101 CrossRef CAS.
- Y.-G. Lee, S. Fujiki, C. Jung, N. Suzuki, N. Yashiro, R. Omoda, D.-S. Ko, T. Shiratsuchi, T. Sugimoto, S. Ryu, J. H. Ku, T. Watanabe, Y. Park, Y. Aihara, D. Im and I. T. Han, High-energy long-cycling all-solid-state lithium metal batteries enabled by silver–carbon composite anodes, Nat. Energy, 2020, 5, 299–308 CrossRef CAS.
- K. J. Kim, M. Balaish, M. Wadaguchi, L. Kong and J. L. M. Rupp, Solid-State Li–Metal Batteries: Challenges and Horizons of Oxide and Sulfide Solid Electrolytes and Their Interfaces, Adv. Energy Mater., 2021, 11, 2002689 CrossRef CAS.
- X. Li, J. Liang, X. Yang, K. R. Adair, C. Wang, F. Zhao and X. Sun, Progress and perspectives on halide lithium conductors for all-solid-state lithium batteries, Energy Environ. Sci., 2020, 13, 1429–1461 RSC.
- H. Liu, Z. Ren, X. Zhang, J. Hu, M. Gao, H. Pan and Y. Liu, Incorporation of Ammonia Borane Groups in the Lithium Borohydride Structure Enables Ultrafast Lithium Ion Conductivity at Room Temperature for Solid-State Batteries, Chem. Mater., 2020, 32, 671–678 CrossRef CAS.
- Z. Xue, D. He and X. Xie, Poly(ethylene oxide)-based electrolytes for lithium-ion batteries, J. Mater. Chem. A, 2015, 3, 19218–19253 RSC.
- H. Zhang, C. Li, M. Piszcz, E. Coya, T. Rojo, L. M. Rodriguez-Martinez, M. Armand and Z. Zhou, Single lithium-ion conducting solid polymer electrolytes: advances and perspectives, Chem. Soc. Rev., 2017, 46, 797–815 RSC.
- C. F. N. Marchiori, R. P. Carvalho, M. Ebadi, D. Brandell and C. M. Araujo, Understanding the Electrochemical Stability Window of Polymer Electrolytes in Solid-State Batteries from Atomic-Scale Modeling: The Role of Li-Ion Salts, Chem. Mater., 2020, 32, 7237–7246 CrossRef CAS.
- A. Hayashi, A. Sakuda and M. Tatsumisago, Development of Sulfide Solid Electrolytes and Interface Formation Processes for Bulk-Type All-Solid-State Li and Na Batteries, Front. Energy Res., 2016, 4, 25 Search PubMed.
- S. Wenzel, S. J. Sedlmaier, C. Dietrich, W. G. Zeier and J. Janek, Interfacial reactivity and interphase growth of argyrodite solid electrolytes at lithium metal electrodes, Solid State Ionics, 2018, 318, 102–112 CrossRef CAS.
- J. Auvergniot, A. Cassel, J.-B. Ledeuil, V. Viallet, V. Seznec and R. Dedryvère, Interface Stability of Argyrodite Li6PS5Cl toward LiCoO2 LiNi1/3Co1/3Mn1/3O 2 and LiMn2O4 in Bulk All-Solid-State Batteries, Chem. Mater., 2017, 29, 3883–3890 CrossRef CAS.
- J. Wolfenstine, J. L. Allen, J. Sakamoto, D. J. Siegel and H. Choe, Mechanical behavior of Li-ion-conducting crystalline oxide-based solid electrolytes: a brief review, Ionics, 2018, 24, 1271–1276 CrossRef CAS.
- Y. Zhu, X. He and Y. Mo, Origin of Outstanding Stability in the Lithium Solid Electrolyte Materials: Insights from Thermodynamic Analyses Based on First-Principles Calculations, ACS Appl. Mater. Interfaces, 2015, 7, 23685–23693 CrossRef CAS.
- J. Schnell, F. Tietz, C. Singer, A. Hofer, N. Billot and G. Reinhart, Prospects of production technologies and manufacturing costs of oxide-based all-solid-state lithium batteries, Energy Environ. Sci., 2019, 12, 1818–1833 RSC.
- Q. Zhao, S. Stalin, C.-Z. Zhao and L. A. Archer, Designing solid-state electrolytes for safe, energy-dense batteries, Nat. Rev. Mater., 2020, 5, 229–252 CrossRef CAS.
- J. Schnell, T. Günther, T. Knoche, C. Vieider, L. Köhler, A. Just, M. Keller, S. Passerini and G. Reinhart, All-solid-state lithium-ion and lithium metal batteries – paving the way to large-scale production, J. Power Sources, 2018, 382, 160–175 CrossRef CAS.
- A. Manthiram, X. Yu and S. Wang, Lithium battery chemistries enabled by solid-state electrolytes, Nat. Rev. Mater., 2017, 2(4), 1–16 Search PubMed.
- Y. Zhu, X. He and Y. Mo, First principles study on electrochemical and chemical stability of solid electrolyte–electrode interfaces in all-solid-state Li-ion batteries, J. Mater. Chem. A, 2016, 4, 3253–3266 RSC.
- T. Krauskopf, H. Hartmann, W. G. Zeier and J. Janek, Toward a Fundamental Understanding of the Lithium Metal Anode in Solid-State Batteries-An Electrochemo-Mechanical Study on the Garnet-Type Solid Electrolyte Li6.25Al0.25La3Zr2O12, ACS Appl. Mater. Interfaces, 2019, 11, 14463–14477 CrossRef CAS.
- R. Koerver, W. Zhang, L. de Biasi, S. Schweidler, A. O. Kondrakov, S. Kolling, T. Brezesinski, P. Hartmann, W. G. Zeier and J. Janek, Chemo-mechanical expansion of lithium electrode materials – on the route to mechanically optimized all-solid-state batteries, Energy Environ. Sci., 2018, 11, 2142–2158 RSC.
- J. Auvergniot, A. Cassel, D. Foix, V. Viallet, V. Seznec and R. Dedryvère, Redox activity of argyrodite Li6PS5Cl electrolyte in all-solid-state Li-ion battery: An XPS study, Solid State Ionics, 2017, 300, 78–85 CrossRef CAS.
- F. Walther, R. Koerver, T. Fuchs, S. Ohno, J. Sann, M. Rohnke, W. G. Zeier and J. Janek, Visualization of the Interfacial Decomposition of Composite Cathodes in Argyrodite-Based All-Solid-State Batteries Using Time-of-Flight Secondary-Ion Mass Spectrometry, Chem. Mater., 2019, 31, 3745–3755 CrossRef CAS.
- F. Walther, S. Randau, Y. Schneider, J. Sann, M. Rohnke, F. H. Richter, W. G. Zeier and J. Janek, Influence of Carbon Additives on the Decomposition Pathways in Cathodes of Lithium Thiophosphate-Based All-Solid-State Batteries, Chem. Mater., 2020, 32, 6123–6136 CrossRef CAS.
- S. P. Culver, R. Koerver, W. G. Zeier and J. Janek, On the Functionality of Coatings for Cathode Active Materials in Thiophosphate-Based All-Solid-State Batteries, Adv. Energy Mater., 2019, 9, 1900626 CrossRef.
- S. Randau, F. Walther, A. Neumann, Y. Schneider, R. S. Negi, B. Mogwitz, J. Sann, K. Becker-Steinberger, T. Danner, S. Hein, A. Latz, F. H. Richter and J. Janek, On the Additive Microstructure in Composite Cathodes and Alumina-Coated Carbon Microwires for Improved All-Solid-State Batteries, Chem. Mater., 2021, 33, 1380–1393 CrossRef CAS.
- A. O. Kondrakov, A. Schmidt, J. Xu, H. Geßwein, R. Mönig, P. Hartmann, H. Sommer, T. Brezesinski and J. Janek, Anisotropic Lattice Strain and Mechanical Degradation of High- and Low-Nickel NCM Cathode Materials for Li-Ion Batteries, J. Phys. Chem. C, 2017, 121, 3286–3294 CrossRef CAS.
- W. Zhang, D. Schröder, T. Arlt, I. Manke, R. Koerver, R. Pinedo, D. A. Weber, J. Sann, W. G. Zeier and J. Janek, (Electro)chemical expansion during cycling: monitoring the pressure changes in operating solid-state lithium batteries, J. Mater. Chem. A, 2017, 5, 9929–9936 RSC.
- T. Shi, Y.-Q. Zhang, Q. Tu, Y. Wang, M. C. Scott and G. Ceder, Characterization of mechanical degradation in an all-solid-state battery cathode, J. Mater. Chem. A, 2020, 8, 17399–17404 RSC.
- T. Krauskopf, F. H. Richter, W. G. Zeier and J. Janek, Physicochemical Concepts of the Lithium Metal Anode in Solid-State Batteries, Chem. Rev., 2020, 120, 7745–7794 CrossRef CAS.
- J. Kasemchainan, S. Zekoll, D. Spencer Jolly, Z. Ning, G. O. Hartley, J. Marrow and P. G. Bruce, Critical stripping current leads to dendrite formation on plating in lithium anode solid electrolyte cells, Nat. Mater., 2019, 18, 1105–1111 CrossRef CAS.
- R. Koerver, I. Aygün, T. Leichtweiß, C. Dietrich, W. Zhang, J. O. Binder, P. Hartmann, W. G. Zeier and J. Janek, Capacity Fade in Solid-State Batteries: Interphase Formation and Chemomechanical Processes in Nickel-Rich Layered Oxide Cathodes and Lithium Thiophosphate Solid Electrolytes, Chem. Mater., 2017, 29, 5574–5582 CrossRef CAS.
- R. Ruess, S. Schweidler, H. Hemmelmann, G. Conforto, A. Bielefeld, D. A. Weber, J. Sann, M. T. Elm and J. Janek, Influence of NCM Particle Cracking on Kinetics of Lithium-Ion Batteries with Liquid or Solid Electrolyte, J. Electrochem. Soc., 2020, 167, 100532 CrossRef CAS.
- E. Trevisanello, R. Ruess, G. Conforto, F. H. Richter and J. Janek, Polycrystalline and Single Crystalline NCM Cathode Materials—Quantifying Particle Cracking, Active Surface Area, and Lithium Diffusion, Adv. Energy Mater., 2021, 2003400 CrossRef CAS.
- Z. Ding, J. Li, J. Li and C. An, Review—Interfaces: Key Issue to Be Solved for All Solid-State Lithium Battery Technologies, J. Electrochem. Soc., 2020, 167, 70541 CrossRef.
- R. Chen, Q. Li, X. Yu, L. Chen and H. Li, Approaching Practically Accessible Solid-State Batteries: Stability Issues Related to Solid Electrolytes and Interfaces, Chem. Rev., 2020, 120, 6820–6877 CrossRef CAS.
- S. Wang, R. Fang, Y. Li, Y. Liu, C. Xin, F. H. Richter and C.-W. Nan, Interfacial challenges for all-solid-state batteries based on sulfide solid electrolytes, J. Materiomics, 2021, 7, 209–218 CrossRef.
- Y. Xiao, Y. Wang, S.-H. Bo, J. C. Kim, L. J. Miara and G. Ceder, Understanding interface stability in solid-state batteries, Nat. Rev. Mater., 2020, 5, 105–126 CrossRef CAS.
- S. Zekoll, C. Marriner-Edwards, A. K. O. Hekselman, J. Kasemchainan, C. Kuss, D. E. J. Armstrong, D. Cai, R. J. Wallace, F. H. Richter, J. H. J. Thijssen and P. G. Bruce, Hybrid electrolytes with 3D bicontinuous ordered ceramic and polymer microchannels for all-solid-state batteries, Energy Environ. Sci., 2018, 11, 185–201 RSC.
- F. Awaja, M. Gilbert, G. Kelly, B. Fox and P. J. Pigram, Adhesion of polymers, Prog. Polym. Sci., 2009, 34, 948–968 CrossRef CAS.
- A. Pizzi and K. L Mittal, Handbook of Adhesive Technology, CRC Press, 3rd edn, 2017 Search PubMed.
- E. Kuhnert, L. Ladenstein, A. Jodlbauer, C. Slugovc, G. Trimmel, H. M. R. Wilkening and D. Rettenwander, Lowering the Interfacial Resistance in Li6.4La3Zr1.4Ta0.6O12|Poly(Ethylene Oxide) Composite Electrolytes, Cell Rep. Phys. Sci., 2020, 1, 100214 CrossRef CAS.
- F. Colombo, S. Bonizzoni, C. Ferrara, R. Simonutti, M. Mauri, M. Falco, C. Gerbaldi, P. Mustarelli and R. Ruffo, Polymer-in-Ceramic Nanocomposite Solid Electrolyte for Lithium Metal Batteries Encompassing PEO-Grafted TiO 2 Nanocrystals, J. Electrochem. Soc., 2020, 167, 70535 CrossRef.
- R. A. Gage, E. P. K. Currie and M. A. Cohen Stuart, Adsorption of Nanocolloidal SiO 2 Particles on PEO Brushes, Macromolecules, 2001, 34, 5078–5080 CrossRef CAS.
- A. P. Russell, A. Y. Stark and T. E. Higham, The Integrative Biology of Gecko Adhesion: Historical Review, Current Understanding, and Grand Challenges, Integr. Comp. Biol., 2019, 59, 101–116 CrossRef CAS.
- N. F. Mott and E. A. Davis, Electronic processes in non-crystalline materials, Clarendon Press, Oxford, 2nd edn, 2012 Search PubMed.
- S. Fratini, M. Nikolka, A. Salleo, G. Schweicher and H. Sirringhaus, Charge transport in high-mobility conjugated polymers and molecular semiconductors, Nat. Mater., 2020, 19, 491–502 CrossRef CAS PubMed.
- T. M. Swager, 50th Anniversary Perspective : Conducting/Semiconducting Conjugated Polymers. A Personal Perspective on the Past and the Future, Macromolecules, 2017, 50, 4867–4886 CrossRef CAS.
- D. Bresser, S. Lyonnard, C. Iojoiu, L. Picard and S. Passerini, Decoupling segmental relaxation and ionic conductivity for lithium-ion polymer electrolytes, Mol. Syst. Des. Eng., 2019, 4, 779–792 RSC.
- K. I. S. Mongcopa, M. Tyagi, J. P. Mailoa, G. Samsonidze, B. Kozinsky, S. A. Mullin, D. A. Gribble, H. Watanabe and N. P. Balsara, Relationship between Segmental Dynamics Measured by Quasi-Elastic Neutron Scattering and Conductivity in Polymer Electrolytes, ACS Macro Lett., 2018, 7, 504–508 CrossRef CAS.
- M. Armand, Polymers with Ionic Conductivity, Adv. Mater., 1990, 2, 278–286 CrossRef CAS.
- M. A. Ratner, P. Johansson and D. F. Shriver, Polymer Electrolytes: Ionic Transport Mechanisms and Relaxation Coupling, MRS Bull., 2000, 25, 31–37 CrossRef CAS.
- S. Dongmin Kang and G. Jeffrey Snyder, Charge-transport model for conducting polymers, Nat. Mater., 2017, 16, 252–257 CrossRef CAS PubMed.
- M. A. Webb, Y. Jung, D. M. Pesko, B. M. Savoie, U. Yamamoto, G. W. Coates, N. P. Balsara, Z.-G. Wang and T. F. Miller, Systematic Computational and Experimental Investigation of Lithium-Ion Transport Mechanisms in Polyester-Based Polymer Electrolytes, ACS Cent. Sci., 2015, 1, 198–205 CrossRef CAS.
- A. A. Teran, M. H. Tang, S. A. Mullin and N. P. Balsara, Effect of molecular weight on conductivity of polymer electrolytes, Solid State Ionics, 2011, 203, 18–21 CrossRef CAS.
- D. Devaux, R. Bouchet, D. Glé and R. Denoyel, Mechanism of ion transport in PEO/LiTFSI complexes: Effect of temperature, molecular weight and end groups, Solid State Ionics, 2012, 227, 119–127 CrossRef CAS.
- Q. Zheng, D. M. Pesko, B. M. Savoie, K. Timachova, A. L. Hasan, M. C. Smith, T. F. Miller, G. W. Coates and N. P. Balsara, Optimizing Ion Transport in Polyether-Based Electrolytes for Lithium Batteries, Macromolecules, 2018, 51, 2847–2858 CrossRef CAS.
- K. Pożyczka, M. Marzantowicz, J. R. Dygas and F. Krok, Ionic conductivity and lithium transference number of poly(ethylene oxide):litfsi system, Electrochim. Acta, 2017, 227, 127–135 CrossRef.
- V. Bocharova and A. P. Sokolov, Perspectives for Polymer Electrolytes: A View from Fundamentals of Ionic Conductivity, Macromolecules, 2020, 53, 4141–4157 CrossRef CAS.
- R. Bouchet, S. Maria, R. Meziane, A. Aboulaich, L. Lienafa, J.-P. Bonnet, T. N. T. Phan, D. Bertin, D. Gigmes, D. Devaux, R. Denoyel and M. Armand, Single-ion BAB triblock copolymers as highly efficient electrolytes for lithium-metal batteries, Nat. Mater., 2013, 12, 452–457 CrossRef CAS.
- T. Bernges, S. P. Culver, N. Minafra, R. Koerver and W. G. Zeier, Competing Structural Influences in the Li Superionic Conducting Argyrodites Li6PS5- xSe xBr (0 ≤ x ≤ 1) upon Se Substitution, Inorg. Chem, 2018, 57, 13920–13928 CrossRef CAS PubMed.
- D. Fraggedakis, M. McEldrew, R. B. Smith, Y. Krishnan, Y. Zhang, P. Bai, W. C. Chueh, Y. Shao-Horn and M. Z. Bazant, Theory of coupled ion-electron transfer kinetics, Electrochim. Acta, 2021, 367, 137432 CrossRef CAS.
- M. Schleutker, J. Bahner, C.-L. Tsai, D. Stolten and C. Korte, On the interfacial charge transfer between solid and liquid Li+ electrolytes, Phys. Chem. Chem. Phys., 2017, 19, 26596–26605 RSC.
- M. Weiss, F. J. Simon, M. R. Busche, T. Nakamura, D. Schröder, F. H. Richter and J. Janek, From Liquid- to Solid-State Batteries: Ion Transfer Kinetics of Heteroionic Interfaces, Electrochem. Energ. Rev., 2020, 3, 221–238 CrossRef.
- N. J. J. de Klerk and M. Wagemaker, Space-Charge Layers in All-Solid-State Batteries; Important or Negligible?, ACS Appl. Energy Mater., 2018, 1, 5609–5618 CAS.
- F. J. Simon, M. Hanauer, A. Henss, F. H. Richter and J. Janek, Properties of the Interphase Formed between Argyrodite-Type Li6PS5Cl and Polymer-Based PEO10:LiTFSI, ACS Appl. Mater. Interfaces, 2019, 11, 42186–42196 CrossRef CAS PubMed.
- M. R. Busche, T. Drossel, T. Leichtweiss, D. A. Weber, M. Falk, M. Schneider, M.-L. Reich, H. Sommer, P. Adelhelm and J. Janek, Dynamic formation of a solid-liquid electrolyte interphase and its consequences for hybrid-battery concepts, Nat. Chem., 2016, 8, 426–434 CrossRef CAS PubMed.
- M. R. Busche, M. Weiss, T. Leichtweiss, C. Fiedler, T. Drossel, M. Geiss, A. Kronenberger, D. A. Weber and J. Janek, The Formation of the Solid/Liquid Electrolyte Interphase (SLEI) on NASICON-Type Glass Ceramics and LiPON, Adv. Mater. Interfaces, 2020, 7, 2000380 CrossRef CAS.
- J. Liu, X. Gao, G. O. Hartley, G. J. Rees, C. Gong, F. H. Richter, J. Janek, Y. Xia, A. W. Robertson, L. R. Johnson and P. G. Bruce, The Interface between Li6.5La3Zr1.5Ta0.5O12 and Liquid Electrolyte, Joule, 2020, 4, 101–108 CrossRef CAS.
- A. Wang, S. Kadam, H. Li, S. Shi and Y. Qi, Review on modeling of the anode solid electrolyte interphase (SEI) for lithium-ion batteries, npj Comput. Mater., 2018, 4(15), 1–26 CAS.
- E. Peled and S. Menkin, Review—SEI: Past, Present and Future, J. Electrochem. Soc., 2017, 164, A1703–A1719 CrossRef CAS.
- M. Gauthier, T. J. Carney, A. Grimaud, L. Giordano, N. Pour, H.-H. Chang, D. P. Fenning, S. F. Lux, O. Paschos, C. Bauer, F. Maglia, S. Lupart, P. Lamp and Y. Shao-Horn, Electrode-electrolyte interface in Li-ion batteries: current understanding and new insights, J. Phys. Chem. Lett., 2015, 6, 4653–4672 CrossRef CAS PubMed.
- T. Abe, M. Ohtsuka, F. Sagane, Y. Iriyama and Z. Ogumi, Lithium Ion Transfer at the Interface between Lithium-Ion-Conductive Solid Crystalline Electrolyte and Polymer Electrolyte, J. Electrochem. Soc., 2004, 151, A1950 CrossRef CAS.
- W. E. Tenhaeff, K. A. Perry and N. J. Dudney, Impedance Characterization of Li Ion Transport at the Interface between Laminated Ceramic and Polymeric Electrolytes, J. Electrochem. Soc., 2012, 159, A2118–A2123 CrossRef CAS.
- D. Brogioli, F. Langer, R. Kun and F. La Mantia, Space-Charge Effects at the Li7La3Zr2O12/Poly(ethylene oxide) Interface, ACS Appl. Mater. Interfaces, 2019, 11, 11999–12007 CrossRef CAS.
- F. Langer, M. S. Palagonia, I. Bardenhagen, J. Glenneberg, F. La Mantia and R. Kun, Impedance Spectroscopy Analysis of the Lithium Ion Transport through the Li 7 La 3 Zr 2 O 12/P(EO) 20 Li Interface, J. Electrochem. Soc., 2017, 164, A2298–A2303 CrossRef CAS.
- A. Gupta and J. Sakamoto, Controlling Ionic Transport through the PEO-LiTFSI/LLZTO Interface, Electrochem. Soc. Interface, 2019, 28, 63–69 CrossRef CAS.
- X. C. Chen, X. Liu, A. Samuthira Pandian, K. Lou, F. M. Delnick and N. J. Dudney, Determining and Minimizing Resistance for Ion Transport at the Polymer/Ceramic Electrolyte Interface, ACS Energy Lett., 2019, 4, 1080–1085 CrossRef CAS.
- B. X. Dong, P. Bennington, Y. Kambe, D. Sharon, M. Dolejsi, J. Strzalka, V. F. Burnett, P. F. Nealey and S. N. Patel, Nanothin film conductivity measurements reveal interfacial influence on ion transport in polymer electrolytes, Mol. Syst. Des. Eng., 2019, 4, 597–608 RSC.
- J. Zagórski, J. M. Del López Amo, M. J. Cordill, F. Aguesse, L. Buannic and A. Llordés, Garnet–Polymer Composite Electrolytes: New Insights on Local Li-Ion Dynamics and Electrodeposition Stability with Li Metal Anodes, ACS Appl. Energy Mater., 2019, 2, 1734–1746 CrossRef.
- X. C. Chen, R. L. Sacci, N. C. Osti, M. Tyagi, Y. Wang, M. J. Palmer and N. J. Dudney, Study of segmental dynamics and ion transport in polymer–ceramic composite electrolytes by quasi-elastic neutron scattering, Mol. Syst. Des. Eng., 2019, 4, 379–385 RSC.
- M. B. Dixit, W. Zaman, N. Hortance, S. Vujic, B. Harkey, F. Shen, W.-Y. Tsai, V. de Andrade, X. C. Chen, N. Balke and K. B. Hatzell, Nanoscale Mapping of Extrinsic Interfaces in Hybrid Solid Electrolytes, Joule, 2020, 4, 207–221 CrossRef CAS.
- G. Yang, Y. Song, Q. Wang, L. Zhang and L. Deng, Review of ionic liquids containing, polymer/inorganic hybrid electrolytes for lithium metal batteries, Mater. Des., 2020, 190, 108563 CrossRef CAS.
- N. Boaretto, L. Meabe, M. Martinez-Ibañez, M. Armand and H. Zhang, Review—Polymer Electrolytes for Rechargeable Batteries: From Nanocomposite to Nanohybrid, J. Electrochem. Soc., 2020, 167, 70524 CrossRef CAS.
- M. Keller, A. Varzi and S. Passerini, Hybrid electrolytes for lithium metal batteries, J. Power Sources, 2018, 392, 206–225 CrossRef CAS.
- W. Zaman, N. Hortance, M. B. Dixit, V. de Andrade and K. B. Hatzell, Visualizing percolation and ion transport in hybrid solid electrolytes for Li–metal batteries, J. Mater. Chem. A, 2019, 7, 23914–23921 RSC.
- L. Han, M. L. Lehmann, J. Zhu, T. Liu, Z. Zhou, X. Tang, C.-T. Heish, A. P. Sokolov, P. Cao, X. C. Chen and T. Saito, Recent Developments and Challenges in Hybrid Solid Electrolytes for Lithium-Ion Batteries, Front. Energy Res., 2020, 8, 202 CrossRef.
- S.-J. Tan, X.-X. Zeng, Q. Ma, X.-W. Wu and Y.-G. Guo, Recent Advancements in Polymer-Based Composite Electrolytes for Rechargeable Lithium Batteries, Electrochem. Energ. Rev., 2018, 1, 113–138 CrossRef CAS.
- W. Zhou, S. Wang, Y. Li, S. Xin, A. Manthiram and J. B. Goodenough, Plating a Dendrite-Free Lithium Anode with a Polymer/Ceramic/Polymer Sandwich Electrolyte, J. Am. Chem. Soc., 2016, 138, 9385–9388 CrossRef CAS.
- J. Zhang, N. Zhao, M. Zhang, Y. Li, P. K. Chu, X. Guo, Z. Di, X. Wang and H. Li, Flexible and ion-conducting membrane electrolytes for solid-state lithium batteries: Dispersion of garnet nanoparticles in insulating polyethylene oxide, Nano Energy, 2016, 28, 447–454 CrossRef CAS.
- L. Chen, Y. Li, S.-P. Li, L.-Z. Fan, C.-W. Nan and J. B. Goodenough, PEO/garnet composite electrolytes for solid-state lithium batteries: From “ceramic-in-polymer” to “polymer-in-ceramic”, Nano Energy, 2018, 46, 176–184 CrossRef CAS.
- W. Liu, S. W. Lee, D. Lin, F. Shi, S. Wang, A. D. Sendek and Y. Cui, Enhancing ionic conductivity in composite polymer electrolytes with well-aligned ceramic nanowires, Nat. Energy, 2017, 2(5), 1–7 Search PubMed.
- K. K. Fu, Y. Gong, J. Dai, A. Gong, X. Han, Y. Yao, C. Wang, Y. Wang, Y. Chen, C. Yan, Y. Li, E. D. Wachsman and L. Hu, Flexible, solid-state, ion-conducting membrane with 3D garnet nanofiber networks for lithium batteries, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7094–7099 CrossRef CAS.
- D. Cai, D. Wang, Y. Chen, S. Zhang, X. Wang, X. Xia and J. Tu, A highly ion-conductive three-dimensional LLZAO-PEO/LiTFSI solid electrolyte for high-performance solid-state batteries, Chem. Eng. J., 2020, 394, 124993 CrossRef CAS.
- J. Wan, J. Xie, X. Kong, Z. Liu, K. Liu, F. Shi, A. Pei, H. Chen, W. Chen, J. Chen, X. Zhang, L. Zong, J. Wang, L.-Q. Chen, J. Qin and Y. Cui, Ultrathin, flexible, solid polymer composite electrolyte enabled with aligned nanoporous host for lithium batteries, Nat. Nanotechnol., 2019, 14, 705–711 CrossRef CAS.
- R. F. Samsinger, S. O. Schopf, J. Schuhmacher, P. Treis, M. Schneider, A. Roters and A. Kwade, Influence of the Processing on the Ionic Conductivity of Solid-State Hybrid Electrolytes Based on Glass-Ceramic Particles Dispersed in PEO with LiTFSI, J. Electrochem. Soc., 2020, 167, 120538 CrossRef CAS.
- X. Yu and A. Manthiram, A Long Cycle Life, All-Solid-State Lithium Battery with a Ceramic–Polymer Composite Electrolyte, ACS Appl. Energy Mater., 2020, 3, 2916–2924 CrossRef CAS.
- X. Ban, W. Zhang, N. Chen and C. Sun, A High-Performance and Durable Poly(ethylene oxide)-Based Composite Solid Electrolyte for All Solid-State Lithium Battery, J. Phys. Chem. C, 2018, 122, 9852–9858 CrossRef CAS.
- Y.-C. Jung, S.-M. Lee, J.-H. Choi, S. S. Jang and D.-W. Kim, All Solid-State Lithium Batteries Assembled with Hybrid Solid Electrolytes, J. Electrochem. Soc., 2015, 162, A704–A710 CrossRef CAS.
- H. Zhai, P. Xu, M. Ning, Q. Cheng, J. Mandal and Y. Yang, A Flexible Solid Composite Electrolyte with Vertically Aligned and Connected Ion-Conducting Nanoparticles for Lithium Batteries, Nano Lett., 2017, 17, 3182–3187 CrossRef CAS.
- M. J. Palmer, S. Kalnaus, M. B. Dixit, A. S. Westover, K. B. Hatzell, N. J. Dudney and X. C. Chen, A three-dimensional interconnected polymer/ceramic composite as a thin film solid electrolyte, Energy Storage Materials, 2020, 26, 242–249 CrossRef.
- D. Y. Oh, Y. J. Nam, K. H. Park, S. H. Jung, S.-J. Cho, Y. K. Kim, Y.-G. Lee, S.-Y. Lee and Y. S. Jung, Excellent Compatibility of Solvate Ionic Liquids with Sulfide Solid Electrolytes: Toward Favorable Ionic Contacts in Bulk-Type All-Solid-State Lithium-Ion Batteries, Adv. Energy Mater., 2015, 5, 1500865 CrossRef.
- J. Zheng, P. Wang, H. Liu and Y.-Y. Hu, Interface-Enabled Ion Conduction in Li 10 GeP 2 S 12 –Poly(ethylene Oxide) Hybrid Electrolytes, ACS Appl. Energy Mater., 2019, 2, 1452–1459 CrossRef CAS.
- Y. Zhang, R. Chen, S. Wang, T. Liu, B. Xu, X. Zhang, X. Wang, Y. Shen, Y.-H. Lin, M. Li, L.-Z. Fan, L. Li and C.-W. Nan, Free-standing sulfide/polymer composite solid electrolyte membranes with high conductance for all-solid-state lithium batteries, Energy Storage Materials, 2020, 25, 145–153 CrossRef.
- I. Villaluenga, K. H. Wujcik, W. Tong, D. Devaux, D. H. C. Wong, J. M. DeSimone and N. P. Balsara, Compliant glass-polymer hybrid single ion-conducting electrolytes for lithium batteries, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 52–57 CrossRef CAS.
- X. Xu, G. Hou, X. Nie, Q. Ai, Y. Liu, J. Feng, L. Zhang, P. Si, S. Guo and L. Ci, Li7P3S11/poly(ethylene oxide) hybrid solid electrolytes with excellent interfacial compatibility for all-solid-state batteries, J. Power Sources, 2018, 400, 212–217 CrossRef CAS.
- S. Chen, J. Wang, Z. Zhang, L. Wu, L. Yao, Z. Wei, Y. Deng, D. Xie, X. Yao and X. Xu, In-situ preparation of poly(ethylene oxide)/Li3PS4 hybrid polymer electrolyte with good nanofiller distribution for rechargeable solid-state lithium batteries, J. Power Sources, 2018, 387, 72–80 CrossRef CAS.
- Y. Zhao, C. Wu, G. Peng, X. Chen, X. Yao, Y. Bai, F. Wu, S. Chen and X. Xu, A new solid polymer electrolyte incorporating Li10GeP2S12 into a polyethylene oxide matrix for all-solid-state lithium batteries, J. Power Sources, 2016, 301, 47–53 CrossRef CAS.
- J. Zhang, C. Zheng, J. Lou, Y. Xia, C. Liang, H. Huang, Y. Gan, X. Tao and W. Zhang, Poly(ethylene oxide) reinforced Li6PS5Cl composite solid electrolyte for all-solid-state lithium battery: Enhanced electrochemical performance, mechanical property and interfacial stability, J. Power Sources, 2019, 412, 78–85 CrossRef CAS.
- X. Li, D. Wang, H. Wang, H. Yan, Z. Gong and Y. Yang, Poly(ethylene oxide)-Li10SnP2S12 Composite Polymer Electrolyte Enables High-Performance All-Solid-State Lithium Sulfur Battery, ACS Appl. Mater. Interfaces, 2019, 11, 22745–22753 CrossRef CAS.
- X. Mei, Y. Wu, Y. Gao, Y. Zhu, S.-H. Bo and Y. Guo, A quantitative correlation between macromolecular crystallinity and ionic conductivity in polymer-ceramic composite solid electrolytes, Mater. Today Commun., 2020, 24, 101004 CrossRef CAS.
- B. Kumar, Polymer–ceramic composite electrolytes: conductivity and thermal history effects, Solid State Ionics, 1999, 124, 239–254 CrossRef CAS.
- F. Zou and A. Manthiram, A Review of the Design of Advanced Binders for High-Performance Batteries, Adv. Energy Mater., 2020, 10, 2002508 CrossRef CAS.
- S. Li, Y.-M. Liu, Y.-C. Zhang, Y. Song, G.-K. Wang, Y.-X. Liu, Z.-G. Wu, B.-H. Zhong, Y.-J. Zhong and X.-D. Guo, A review of rational design and investigation of binders applied in silicon-based anodes for lithium-ion batteries, J. Power Sources, 2021, 485, 229331 CrossRef CAS.
- R. Wang, L. Feng, W. Yang, Y. Zhang, Y. Zhang, W. Bai, B. Liu, W. Zhang, Y. Chuan, Z. Zheng and H. Guan, Effect of Different Binders on the Electrochemical Performance of Metal Oxide Anode for Lithium-Ion Batteries, Nanoscale Res. Lett., 2017, 12, 575 CrossRef.
- S. Wang, X. Zhang, S. Liu, C. Xin, C. Xue, F. Richter, L. Li, L. Fan, Y. Lin, Y. Shen, J. Janek and C.-W. Nan, High-conductivity free-standing Li6PS5Cl/poly(vinylidene difluoride) composite solid electrolyte membranes for lithium-ion batteries, J. Materiomics, 2020, 6, 70–76 CrossRef.
- X. Zhang, T. Liu, S. Zhang, X. Huang, B. Xu, Y. Lin, B. Xu, L. Li, C.-W. Nan and Y. Shen, Synergistic Coupling between Li6.75La3Zr1.75Ta0.25O12 and Poly(vinylidene fluoride) Induces High Ionic Conductivity, Mechanical Strength, and Thermal Stability of Solid Composite Electrolytes, J. Am. Chem. Soc., 2017, 139, 13779–13785 CrossRef CAS PubMed.
- A. van Nguyen and C. Kuss, Review—Conducting Polymer-Based Binders for Lithium-Ion Batteries and Beyond, J. Electrochem. Soc., 2020, 167, 65501 CrossRef.
- Y. Shi, X. Zhou and G. Yu, Material and Structural Design of Novel Binder Systems for High-Energy, High-Power Lithium-Ion Batteries, Acc. Chem. Res., 2017, 50, 2642–2652 CrossRef CAS PubMed.
- J. Liang, D. Chen, K. Adair, Q. Sun, N. G. Holmes, Y. Zhao, Y. Sun, J. Luo, R. Li, L. Zhang, S. Zhao, S. Lu, H. Huang, X. Zhang, C. V. Singh and X. Sun, Insight into Prolonged Cycling Life of 4 V All-Solid-State Polymer Batteries by a High-Voltage Stable Binder, Adv. Energy Mater., 2021, 11, 2002455 CrossRef CAS.
- D. Bresser, D. Buchholz, A. Moretti, A. Varzi and S. Passerini, Alternative binders for sustainable electrochemical energy storage – the transition to aqueous electrode processing and bio-derived polymers, Energy Environ. Sci., 2018, 11, 3096–3127 RSC.
- H. Chen, M. Ling, L. Hencz, H. Y. Ling, G. Li, Z. Lin, G. Liu and S. Zhang, Exploring Chemical, Mechanical, and Electrical Functionalities of Binders for Advanced Energy-Storage Devices, Chem. Rev., 2018, 118, 8936–8982 CrossRef CAS PubMed.
- K. Lee, S. Kim, J. Park, S. H. Park, A. Coskun, D. S. Jung, W. Cho and J. W. Choi, Selection of Binder and Solvent for Solution-Processed All-Solid-State Battery, J. Electrochem. Soc., 2017, 164, A2075–A2081 CrossRef CAS.
- J. Ruhl, L. M. Riegger, M. Ghidiu and W. G. Zeier, Impact of Solvent Treatment of the Superionic Argyrodite Li 6 PS 5 Cl on Solid-State Battery Performance, Adv. Energy Mater, 2021, 2, 2000077 Search PubMed.
- D. H. S. Tan, A. Banerjee, Z. Chen and Y. S. Meng, From nanoscale interface characterization to sustainable energy storage using all-solid-state batteries, Nat. Nanotechnol., 2020, 15, 170–180 CrossRef CAS PubMed.
- D. H. S. Tan, A. Banerjee, Z. Deng, E. A. Wu, H. Nguyen, J.-M. Doux, X. Wang, J. Cheng, S. P. Ong, Y. S. Meng and Z. Chen, Enabling Thin and Flexible Solid-State Composite Electrolytes by the Scalable Solution Process, ACS Appl. Energy Mater., 2019, 2, 6542–6550 CrossRef CAS.
- K. T. Kim, D. Y. Oh, S. Jun, Y. B. Song, T. Y. Kwon, Y. Han and Y. S. Jung, Tailoring Slurries Using Cosolvents and Li Salt Targeting Practical All-Solid-State Batteries Employing Sulfide Solid Electrolytes, Adv. Energy Mater., 2021, 2003766 CrossRef CAS.
- N. Riphaus, P. Strobl, B. Stiaszny, T. Zinkevich, M. Yavuz, J. Schnell, S. Indris, H. A. Gasteiger and S. J. Sedlmaier, Slurry-Based Processing of Solid Electrolytes: A Comparative Binder Study, J. Electrochem. Soc., 2018, 165, A3993–A3999 CrossRef CAS.
- D. Li, L. Cao, C. Liu, G. Cao, J. Hu, J. Chen and G. Shao, A designer fast Li-ion conductor Li6.25PS5.25Cl0.75 and its contribution to the polyethylene oxide based electrolyte, Appl. Surf. Sci., 2019, 493, 1326–1333 CrossRef CAS.
- D. Y. Oh, D. H. Kim, S. H. Jung, J.-G. Han, N.-S. Choi and Y. S. Jung, Single-step wet-chemical fabrication of sheet-type electrodes from solid-electrolyte precursors for all-solid-state lithium-ion batteries, J. Mater. Chem. A, 2017, 5, 20771–20779 RSC.
- K. Lee, J. Lee, S. Choi, K. Char and J. W. Choi, Thiol–Ene Click Reaction for Fine Polarity Tuning of Polymeric Binders in Solution-Processed All-Solid-State Batteries, ACS Energy Lett., 2019, 4, 94–101 CrossRef CAS.
- A. Sakuda, K. Kuratani, M. Yamamoto, M. Takahashi, T. Takeuchi and H. Kobayashi, All-Solid-State Battery Electrode Sheets Prepared by a Slurry Coating Process, J. Electrochem. Soc., 2017, 164, A2474–A2478 CrossRef CAS.
- J. Lee, K. Lee, T. Lee, H. Kim, K. Kim, W. Cho, A. Coskun, K. Char and J. W. Choi, In Situ Deprotection of Polymeric Binders for Solution-Processible Sulfide-Based All-Solid-State Batteries, Adv. Mater., 2020, 32, e2001702 CrossRef.
- F. Hippauf, B. Schumm, S. Doerfler, H. Althues, S. Fujiki, T. Shiratsuchi, T. Tsujimura, Y. Aihara and S. Kaskel, Overcoming binder limitations of sheet-type solid-state cathodes using a solvent-free dry-film approach, Energy Storage Materials, 2019, 21, 390–398 CrossRef.
- C. Niu, J. Liu, T. Qian, X. Shen, J. Zhou and C. Yan, Single lithium-ion channel polymer binder for stabilizing sulfur cathodes, Natl. Sci. Rev., 2020, 7, 315–323 CrossRef CAS.
- C. Chen, F. Chen, L. Liu, J. Zhao and F. Wang, Cross-linked hyperbranched polyethylenimine as an efficient multidimensional binder for silicon anodes in lithium-ion batteries, Electrochim. Acta, 2019, 326, 134964 CrossRef CAS.
- K. Ueno, J. Murai, K. Ikeda, S. Tsuzuki, M. Tsuchiya, R. Tatara, T. Mandai, Y. Umebayashi, K. Dokko and M. Watanabe, Li + Solvation and Ionic Transport in Lithium Solvate Ionic Liquids Diluted by Molecular Solvents, J. Phys. Chem. C, 2016, 120, 15792–15802 CrossRef CAS.
- J. Liang, Y. Sun, Y. Zhao, Q. Sun, J. Luo, F. Zhao, X. Lin, X. Li, R. Li, L. Zhang, S. Lu, H. Huang and X. Sun, Engineering the conductive carbon/PEO interface to stabilize solid polymer electrolytes for all-solid-state high voltage LiCoO 2 batteries, J. Mater. Chem. A, 2020, 8, 2769–2776 RSC.
- B. Ludwig, Z. Zheng, W. Shou, Y. Wang and H. Pan, Solvent-Free Manufacturing of Electrodes for Lithium-ion Batteries, Sci. Rep., 2016, 6, 23150 CrossRef CAS.
- N. Verdier, G. Foran, D. Lepage, A. Prébé, D. Aymé-Perrot and M. Dollé, Challenges in Solvent-Free Methods for Manufacturing Electrodes and Electrolytes for Lithium-Based Batteries, Polymers, 2021, 13(3), 323 CrossRef CAS PubMed.
- C. Zhu, Y. Fu and Y. Yu, Designed Nanoarchitectures by Electrostatic Spray Deposition for Energy Storage, Adv. Mater., 2019, 31, e1803408 CrossRef PubMed.
- A. Vizintin, R. Guterman, J. Schmidt, M. Antonietti and R. Dominko, Linear and Cross-Linked Ionic Liquid Polymers as Binders in Lithium–Sulfur Batteries, Chem. Mater., 2018, 30, 5444–5450 CrossRef CAS.
- S. Deng, Y. Sun, X. Li, Z. Ren, J. Liang, K. Doyle-Davis, J. Liang, W. Li, M. Norouzi Banis, Q. Sun, R. Li, Y. Hu, H. Huang, L. Zhang, S. Lu, J. Luo and X. Sun, Eliminating the Detrimental Effects of Conductive Agents in Sulfide-Based Solid-State Batteries, ACS Energy Lett., 2020, 5, 1243–1251 CrossRef CAS.
- S. Choudhury, Z. Tu, A. Nijamudheen, M. J. Zachman, S. Stalin, Y. Deng, Q. Zhao, D. Vu, L. F. Kourkoutis, J. L. Mendoza-Cortes and L. A. Archer, Stabilizing polymer electrolytes in high-voltage lithium batteries, Nat. Commun., 2019, 10, 3091 CrossRef PubMed.
- S. H. Ju, I.-S. Kang, Y.-S. Lee, W.-K. Shin, S. Kim, K. Shin and D.-W. Kim, Improvement of the cycling performance of LiNi(0.6)Co(0.2)Mn(0.2)O(2) cathode active materials by a dual-conductive polymer coating, ACS Appl. Mater. Interfaces, 2014, 6, 2546–2552 CrossRef CAS.
- S. N. Patel, 100th Anniversary of Macromolecular Science Viewpoint: Solid Polymer Electrolytes in Cathode Electrodes for Lithium Batteries. Current Challenges and Future Opportunities, ACS Macro Lett., 2021, 10, 141–153 CrossRef CAS.
- M. Ebadi, C. Marchiori, J. Mindemark, D. Brandell and C. M. Araujo, Assessing structure and stability of polymer/lithium-metal interfaces from first-principles calculations, J. Mater. Chem. A, 2019, 7, 8394–8404 RSC.
- M. Nakayama, S. Wada, S. Kuroki and M. Nogami, Factors affecting cyclic durability of all-solid-state lithium polymer batteries using poly(ethylene oxide)-based solid polymer electrolytes, Energy Environ. Sci., 2010, 3, 1995 RSC.
- K. Nie, X. Wang, J. Qiu, Y. Wang, Q. Yang, J. Xu, X. Yu, H. Li, X. Huang and L. Chen, Increasing Poly(ethylene oxide) Stability to 4.5 V by Surface Coating of the Cathode, ACS Energy Lett., 2020, 5, 826–832 CrossRef CAS.
- K. Hanai, M. Ueno, N. Imanishi, A. Hirano, O. Yamamoto and Y. Takeda, Interfacial resistance of the LiFePO4-C/PEO-LiTFSI composite electrode for dry-polymer lithium-ion batteries, J. Power Sources, 2011, 196, 6756–6761 CrossRef CAS.
- W. Zhou, Z. Wang, Y. Pu, Y. Li, S. Xin, X. Li, J. Chen and J. B. Goodenough, Double-Layer Polymer Electrolyte for High-Voltage All-Solid-State Rechargeable Batteries, Adv. Mater., 2019, 31, e1805574 CrossRef.
- M. Wetjen, G.-T. Kim, M. Joost, G. B. Appetecchi, M. Winter and S. Passerini, Thermal and electrochemical properties of PEO-LiTFSI-Pyr14TFSI-based composite cathodes, incorporating 4 V-class cathode active materials, J. Power Sources, 2014, 246, 846–857 CrossRef CAS.
- G. Homann, L. Stolz, J. Nair, I. C. Laskovic, M. Winter and J. Kasnatscheew, Poly(Ethylene Oxide)-based Electrolyte for Solid-State-Lithium-Batteries with High Voltage Positive Electrodes: Evaluating the Role of Electrolyte Oxidation in Rapid Cell Failure, Sci. Rep., 2020, 10, 4390 CrossRef PubMed.
- J. Ma, Z. Liu, B. Chen, L. Wang, L. Yue, H. Liu, J. Zhang, Z. Liu and G. Cui, A Strategy to Make High Voltage LiCoO 2 Compatible with Polyethylene Oxide Electrolyte in All-Solid-State Lithium Ion Batteries, J. Electrochem. Soc., 2017, 164, A3454–A3461 CrossRef CAS.
- G. Hu, J. Fan, Y. Lu, Y. Zhang, K. Du, Z. Peng, L. Li, B. Zhang, Y. Shi and Y. Cao, Surface Architecture Design of LiNi0.8 Co0.15 Al0.05 O2 Cathode with Synergistic Organics Encapsulation to Enhance Electrochemical Stability, ChemSusChem, 2020, 13, 5699–5710 CrossRef CAS.
- L. Porcarelli, A. S. Shaplov, M. Salsamendi, J. R. Nair, Y. S. Vygodskii, D. Mecerreyes and C. Gerbaldi, Single-Ion Block Copoly(ionic liquid)s as Electrolytes for All-Solid State Lithium Batteries, ACS Appl. Mater. Interfaces, 2016, 8, 10350–10359 CrossRef CAS PubMed.
- Y. Cao, X. Qi, K. Hu, Y. Wang, Z. Gan, Y. Li, G. Hu, Z. Peng and K. Du, Conductive Polymers Encapsulation To Enhance Electrochemical Performance of Ni-Rich Cathode Materials for Li-Ion Batteries, ACS Appl. Mater. Interfaces, 2018, 10, 18270–18280 CrossRef CAS.
- N. Chen, H. Zhang, L. Li, R. Chen and S. Guo, Ionogel Electrolytes for High-Performance Lithium Batteries: A Review, Adv. Energy Mater., 2018, 8, 1702675 CrossRef.
- Y. H. Jo, S. Li, C. Zuo, Y. Zhang, H. Gan, S. Li, L. Yu, D. He, X. Xie and Z. Xue, Self-Healing Solid Polymer Electrolyte Facilitated by a Dynamic Cross-Linked Polymer Matrix for Lithium-Ion Batteries, Macromolecules, 2020, 53, 1024–1032 CrossRef CAS.
- B. B. Jing and C. M. Evans, Catalyst-Free Dynamic Networks for Recyclable, Self-Healing Solid Polymer Electrolytes, J. Am. Chem. Soc., 2019, 141, 18932–18937 CrossRef CAS PubMed.
- H. Dong, X. Xiao, C. Jin, X. Wang, P. Tang, C. Wang, Y. Yin, D. Wang, S. Yang and C. Wu, High lithium-ion conductivity polymer film to suppress dendrites in Li metal batteries, J. Power Sources, 2019, 423, 72–79 CrossRef CAS.
- J. Lopez, A. Pei, J. Y. Oh, G.-J. N. Wang, Y. Cui and Z. Bao, Effects of Polymer Coatings on Electrodeposited Lithium Metal, J. Am. Chem. Soc., 2018, 140, 11735–11744 CrossRef CAS.
- S.-S. Chi, Y. Liu, N. Zhao, X. Guo, C.-W. Nan and L.-Z. Fan, Solid polymer electrolyte soft interface layer with 3D lithium anode for all-solid-state lithium batteries, Energy Storage Materials, 2019, 17, 309–316 CrossRef.
- C. A. Calderón, A. Vizintin, J. Bobnar, D. E. Barraco, E. P. Leiva, A. Visintin, S. Fantini, F. Fischer and R. Dominko, Lithium Metal Protection by a Cross-Linked Polymer Ionic Liquid and Its Application in Lithium Battery, ACS Appl. Energy Mater., 2020, 3, 2020–2027 CrossRef.
- C. Yang, B. Liu, F. Jiang, Y. Zhang, H. Xie, E. Hitz and L. Hu, Garnet/polymer hybrid ion-conducting protective layer for stable lithium metal anode, Nano Res., 2017, 10, 4256–4265 CrossRef CAS.
- X.-B. Cheng, R. Zhang, C.-Z. Zhao and Q. Zhang, Toward Safe Lithium Metal Anode in Rechargeable Batteries: A Review, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS PubMed.
- H. Zhou, S. Yu, H. Liu and P. Liu, Protective coatings for lithium metal anodes: Recent progress and future perspectives, J. Power Sources, 2020, 450, 227632 CrossRef CAS.
- Y. Yin, C.-S. Jiang, H. Guthrey, C. Xiao, N. Seitzman, C. Ban and M. Al-Jassim, Improved Stability and Cyclability of Ceramic Solid Electrolyte by Coating Polymer, J. Electrochem. Soc., 2020, 167, 20519 CrossRef CAS.
- L. Chen, Z. Huang, R. Shahbazian-Yassar, J. A. Libera, K. C. Klavetter, K. R. Zavadil and J. W. Elam, Directly Formed Alucone on Lithium Metal for High-Performance Li Batteries and Li-S Batteries with High Sulfur Mass Loading, ACS Appl. Mater. Interfaces, 2018, 10, 7043–7051 CrossRef CAS PubMed.
- Y. Zhao, L. V. Goncharova, Q. Sun, X. Li, A. Lushington, B. Wang, R. Li, F. Dai, M. Cai and X. Sun, Robust Metallic Lithium Anode Protection by the Molecular-Layer-Deposition Technique, Small Methods, 2018, 2, 1700417 CrossRef.
- Y. Zhao and X. Sun, Molecular Layer Deposition for Energy Conversion and Storage, ACS Energy Lett., 2018, 3, 899–914 CrossRef CAS.
- C. Fiedler, B. Luerssen, M. Rohnke, J. Sann and J. Janek, XPS and SIMS Analysis of Solid Electrolyte Interphases on Lithium Formed by Ether-Based Electrolytes, J. Electrochem. Soc., 2017, 164, A3742–A3749 CrossRef CAS.
- D. T. Boyle, W. Huang, H. Wang, Y. Li, H. Chen, Z. Yu, W. Zhang, Z. Bao and Y. Cui, Corrosion of lithium metal anodes during calendar ageing and its microscopic origins, Nat. Energy, 2021, 6(5), 487–494 CrossRef CAS.
- K. Xu, Nonaqueous Liquid Electrolytes for Lithium-Based Rechargeable Batteries, Chem. Rev., 2004, 104, 4303–4417 CrossRef CAS PubMed.
- P. Zhai, L. Liu, X. Gu, T. Wang and Y. Gong, Interface Engineering for Lithium Metal Anodes in Liquid Electrolyte, Adv. Energy Mater., 2020, 10, 2001257 CrossRef CAS.
- C. Yang, K. Fu, Y. Zhang, E. Hitz and L. Hu, Protected Lithium-Metal Anodes in Batteries: From Liquid to Solid, Adv. Mater., 2017, 29(36), 1701169 CrossRef.
- D. Lin, Y. Liu and Y. Cui, Reviving the lithium metal anode for high-energy batteries, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- R. Bouchet, S. Lascaud and M. Rosso, An EIS Study of the Anode Li/PEO-LiTFSI of a Li Polymer Battery, J. Electrochem. Soc., 2003, 150, A1385–A1389 CrossRef CAS.
- T. Ates, M. Keller, J. Kulisch, T. Adermann and S. Passerini, Development of an all-solid-state lithium battery by slurry-coating procedures using a sulfidic electrolyte, Energy Storage Materials, 2019, 17, 204–210 CrossRef.
- Y. Gao, Y. Zhao, Y. C. Li, Q. Huang, T. E. Mallouk and D. Wang, Interfacial Chemistry Regulation via a Skin-Grafting Strategy Enables High-Performance Lithium-Metal Batteries, J. Am. Chem. Soc., 2017, 139, 15288–15291 CrossRef CAS PubMed.
- S. Choudhury and L. A. Archer, Lithium Fluoride Additives for Stable Cycling of Lithium Batteries at High Current Densities, Adv. Electron. Mater., 2016, 2, 1500246 CrossRef.
- S.-Y. Lee, J. Shangguan, J. Alvarado, S. Betzler, S. J. Harris, M. M. Doeff and H. Zheng, Unveiling the mechanisms of lithium dendrite suppression by cationic polymer film induced solid–electrolyte interphase modification, Energy Environ. Sci., 2020, 13, 1832–1842 RSC.
- X. Fan, L. Chen, X. Ji, T. Deng, S. Hou, J. Chen, J. Zheng, F. Wang, J. Jiang, K. Xu and C. Wang, Highly Fluorinated Interphases Enable High-Voltage Li-Metal Batteries, Chem, 2018, 4, 174–185 CAS.
- B. Zhu, Y. Jin, X. Hu, Q. Zheng, S. Zhang, Q. Wang and J. Zhu, Poly(dimethylsiloxane) Thin Film as a Stable Interfacial Layer for High-Performance Lithium-Metal Battery Anodes, Adv. Mater., 2017, 29(2), 1603755 CrossRef.
- S. Guo, L. Wang, Y. Jin, N. Piao, Z. Chen, G. Tian, J. Li, C. Zhao and X. He, A polymeric composite protective layer for stable Li metal anodes, Nano Convergence, 2020, 7(1), 1–10 CrossRef.
- F. J. Simon, M. Hanauer, F. H. Richter and J. Janek, Interphase Formation of PEO20:LiTFSI-Li6PS5Cl Composite Electrolytes with Lithium Metal, ACS Appl. Mater. Interfaces, 2020, 12, 11713–11723 CrossRef CAS PubMed.
- S. Nematdoust, R. Najjar, D. Bresser and S. Passerini, Understanding the Role of Nanoparticles in PEO-Based Hybrid Polymer Electrolytes for Solid-State Lithium–Polymer Batteries, J. Phys. Chem. C, 2020, 124, 27907–27915 CrossRef CAS.
- P. Bruce and F. Krok, Characterisation of the electrode/electrolyte interfaces in cells of the type Li/PEO LiCF3SO3/V6O13 by ac impedance methods, Solid State Ionics, 1989, 36, 171–174 CrossRef CAS.
- M. D. Galluzzo, D. M. Halat, W. S. Loo, S. A. Mullin, J. A. Reimer and N. P. Balsara, Dissolution of Lithium Metal in Poly(ethylene oxide), ACS Energy Lett., 2019, 4, 903–907 CrossRef CAS.
- J. Tang, L. Wang, L. You, X. Chen, T. Huang, L. Zhou, Z. Geng and A. Yu, Effect of Organic Electrolyte on the Performance of Solid Electrolyte for Solid-Liquid Hybrid Lithium Batteries, ACS Appl. Mater. Interfaces, 2021, 13, 2685–2693 CrossRef CAS.
- Y. Hu, Y. Zhong, L. Qi and H. Wang, Inorganic/polymer hybrid layer stabilizing anode/electrolyte interfaces in solid-state Li metal batteries, Nano Res., 2020, 13, 3230–3234 CrossRef CAS.
- F. Croce, G. Appetecchi, L. Persi and B. Scrosati, Nanocomposite polymer electrolytes for lithium batteries, Nature, 1998, 394, 456–458 CrossRef CAS.
- F. Croce, L. Persi, B. Scrosati, F. Serraino-Fiory, E. Plichta and M. Hendrickson, Role of the ceramic fillers in enhancing the transport properties of composite polymer electrolytes, Electrochim. Acta, 2001, 46, 2457–2461 CrossRef CAS.
- X. C. Chen, Y. Zhang, L. C. Merrill, C. Soulen, M. L. Lehmann, J. L. Schaefer, Z. Du, T. Saito and N. J. Dudney, Gel composite electrolyte – an effective way to utilize ceramic fillers in lithium batteries, J. Mater. Chem. A, 2021, 9, 6555–6566 RSC.
- H. Huo, Y. Chen, J. Luo, X. Yang, X. Guo and X. Sun, Rational Design of Hierarchical “Ceramic-in-Polymer” and “Polymer-in-Ceramic” Electrolytes for Dendrite-Free Solid-State Batteries, Adv. Energy Mater., 2019, 9, 1804004 CrossRef.
- Z. Xu, T. Yang, X. Chu, H. Su, Z. Wang, N. Chen, B. Gu, H. Zhang, W. Deng, H. Zhang and W. Yang, Strong Lewis Acid-Base and Weak Hydrogen Bond Synergistically Enhancing Ionic Conductivity of Poly(ethylene oxide)@SiO2 Electrolytes for a High Rate Capability Li-Metal Battery, ACS Appl. Mater. Interfaces, 2020, 12, 10341–10349 CrossRef.
- J. B. Bates, N. J. Dudney, B. Neudecker, A. Ueda and C. D. Evans, Thin-film lithium and lithium-ion batteries, Solid State Ionics, 2000, 135, 33–45 CrossRef CAS.
- J. Li, C. Ma, M. Chi, C. Liang and N. J. Dudney, Solid Electrolyte: the Key for High-Voltage Lithium Batteries, Adv. Energy Mater., 2015, 5, 1401408 CrossRef.
- X. Zhang, E. Temeche and R. M. Laine, Design, Synthesis, and Characterization of Polymer Precursors to LixPON and LixSiPON Glasses: Materials That Enable All-Solid-State Batteries (ASBs), Macromolecules, 2020, 53, 2702–2712 CrossRef CAS.
- G. Abels, I. Bardenhagen and J. Schwenzel, One-pot synthesis of polymeric LiPON, Polymer, 2020, 192, 122300 CrossRef CAS.
- A. Schwöbel, R. Hausbrand and W. Jaegermann, Interface reactions between LiPON and lithium studied by in situ X-ray photoemission, Solid State Ionics, 2015, 273, 51–54 CrossRef.
- K. Fu, Y. Gong, G. T. Hitz, D. W. McOwen, Y. Li, S. Xu, Y. Wen, L. Zhang, C. Wang, G. Pastel, J. Dai, B. Liu, H. Xie, Y. Yao, E. D. Wachsman and L. Hu, Three-dimensional bilayer garnet solid electrolyte based high energy density lithium metal–sulfur batteries, Energy Environ. Sci., 2017, 10, 1568–1575 RSC.
- S. A. Pervez, G. Kim, B. P. Vinayan, M. A. Cambaz, M. Kuenzel, M. Hekmatfar, M. Fichtner and S. Passerini, Overcoming the Interfacial Limitations Imposed by the Solid-Solid Interface in Solid-State Batteries Using Ionic Liquid-Based Interlayers, Small, 2020, 16, e2000279 CrossRef PubMed.
- T. Fuchs, B. Mogwitz, S.-K. Otto, S. Passerini, F. H. Richter and J. Janek, Working Principle of an Ionic Liquid Interlayer during Pressureless Lithium Stripping on Li6.25Al0.25La3Zr2O12 LLZO Garnet-Type Solid Electrolyte, Batteries Supercaps, 2021, 4(7), 1145–1155 CrossRef CAS.
- H. Huo, J. Gao, N. Zhao, D. Zhang, N. G. Holmes, X. Li, Y. Sun, J. Fu, R. Li, X. Guo and X. Sun, A flexible electron-blocking interfacial shield for dendrite-free solid lithium metal batteries, Nat. Commun., 2021, 12, 176 CrossRef CAS.
- A. Gupta, E. Kazyak, N. Craig, J. Christensen, N. P. Dasgupta and J. Sakamoto, Evaluating the Effects of Temperature and Pressure on Li/PEO-LiTFSI Interfacial Stability and Kinetics, J. Electrochem. Soc., 2018, 165, A2801–A2806 CrossRef CAS.
- N. J. Taylor, S. Stangeland-Molo, C. G. Haslam, A. Sharafi, T. Thompson, M. Wang, R. Garcia-Mendez and J. Sakamoto, Demonstration of high current densities and extended cycling in the garnet Li7La3Zr2O12 solid electrolyte, J. Power Sources, 2018, 396, 314–318 CrossRef CAS.
- A. Sharafi, H. M. Meyer, J. Nanda, J. Wolfenstine and J. Sakamoto, Characterizing the Li–Li7La3Zr2O12 interface stability and kinetics as a function of temperature and current density, J. Power Sources, 2016, 302, 135–139 CrossRef CAS.
- C. Monroe and J. Newman, The Effect of Interfacial Deformation on Electrodeposition Kinetics, J. Electrochem. Soc., 2004, 151, A880–A886 CrossRef CAS.
- C. Monroe and J. Newman, The Impact of Elastic Deformation on Deposition Kinetics at Lithium/Polymer Interfaces, J. Electrochem. Soc., 2005, 152, A396–A404 CrossRef CAS.
- P. Barai, K. Higa and V. Srinivasan, Lithium dendrite growth mechanisms in polymer electrolytes and prevention strategies, Phys. Chem. Chem. Phys., 2017, 19, 20493–20505 RSC.
- L. Porz, T. Swamy, B. W. Sheldon, D. Rettenwander, T. Frömling, H. L. Thaman, S. Berendts, R. Uecker, W. C. Carter and Y.-M. Chiang, Mechanism of Lithium Metal Penetration through Inorganic Solid Electrolytes, Adv. Energy Mater., 2017, 7, 1701003 CrossRef.
- T. Krauskopf, R. Dippel, H. Hartmann, K. Peppler, B. Mogwitz, F. H. Richter, W. G. Zeier and J. Janek, Lithium-Metal Growth Kinetics on LLZO Garnet-Type Solid Electrolytes, Joule, 2019, 3, 2030–2049 CrossRef CAS.
- S. Nanda, A. Gupta and A. Manthiram, Anode-Free Full Cells: A Pathway to High-Energy Density Lithium-Metal Batteries, Adv. Energy Mater., 2021, 11, 2000804 CrossRef CAS.
- N. A. Sahalie, Z. T. Wondimkun, W.-N. Su, M. A. Weret, F. W. Fenta, G. B. Berhe, C.-J. Huang, Y.-C. Hsu and B. J. Hwang, Multifunctional Properties of Al 2 O 3/Polyacrylonitrile Composite Coating on Cu to Suppress Dendritic Growth in Anode-Free Li-Metal Battery, ACS Appl. Energy Mater., 2020, 3, 7666–7679 CrossRef CAS.
- L. Li, L. Ma and B. A. Helms, Architected Macroporous Polyelectrolytes That Suppress Dendrite Formation during High-Rate Lithium Metal Electrodeposition, Macromolecules, 2018, 51, 7666–7671 CrossRef CAS.
- T. A. Zegeye, W.-N. Su, F. W. Fenta, T. S. Zeleke, S.-K. Jiang and B. J. Hwang, Ultrathin Li6.75La3 Zr1.75 Ta0.25O12 -Based Composite Solid Electrolytes Laminated on Anode and Cathode Surfaces for Anode-free Lithium Metal Batteries, ACS Appl. Energy Mater., 2020, 3, 11713–11723 CrossRef CAS.
Footnote |
| † Current address: Robert Bosch GmbH, Tübingerstr. 123, 72762, Reutlingen, Germany. |
| This journal is © The Royal Society of Chemistry 2021 |