
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Thermo-chemical conversion of carbonaceous wastes for CNT and hydrogen production: a review
Ye Shui
Zhang
 ab,
Hua Lun
Zhu
c,
Dingding
Yao
d,
Paul T.
Williams
*e,
Chunfei
Wu
f,
Dan
Xu
ag,
Qiang
Hu
d,
George
Manos
a,
Lu
Yu
h,
Ming
Zhao
i,
Paul R.
Shearing
ab and
Dan J. L.
Brett
*ab
ab,
Hua Lun
Zhu
c,
Dingding
Yao
d,
Paul T.
Williams
*e,
Chunfei
Wu
f,
Dan
Xu
ag,
Qiang
Hu
d,
George
Manos
a,
Lu
Yu
h,
Ming
Zhao
i,
Paul R.
Shearing
ab and
Dan J. L.
Brett
*ab
aElectrochemical Innovation Lab (EIL), Department of Chemical Engineering, University College London, London, WC1E 7JE, UK. E-mail: d.brett@ucl.ac.uk
bThe Faraday Institution, Quad One, Harwell Science and Innovation Campus, Didcot, OX11 0RA, UK
cDepartment of Chemical Engineering, Imperial College London, London, SW7 2AZ, UK
dEnvironmental Research Institute, National University of Singapore, Singapore
eEnergy Research Institute, School of Chemical and Process Engineering, University of Leeds, Leeds, LS2 9JT, UK. E-mail: p.t.williams@leeds.ac.uk
fSchool of Chemistry and Chemical Engineering, Queen's University of Belfast, Belfast, BT9 5AG, UK
gSchool of Energy and Environment, Southeast University, Nanjing, P. R. China
hBureau of Ecology and Environment in Shashi District, Jingzhou, P. R. China
iSchool of Environment, Tsinghua University, Beijing 100084, China
First published on 21st June 2021
Abstract
Thermo-chemical conversion of carbonaceous wastes such as tyres, plastics, biomass and crude glycerol is a promising technology compared to traditional waste treatment options (e.g. incineration and landfill). The process promotes the sustainable management of carbonaceous wastes and realizes the potential value of these wastes. Carbon nanotubes (CNTs) are one of the most extensively investigated and high-value materials due to their featured electrical, mechanical and physical properties. Producing CNTs from waste materials could solve the issues of waste management and simultaneously bring down the cost of CNT production. This review focuses on the four most abundant waste carbonaceous materials (waste tyres, plastics, biomass and crude glycerol) which have great potential to be alternative feedstocks for CNT production. The review considers the background of these four major types of waste to highlight the incentives in using thermo-chemical conversion to deal with these waste materials. Catalyst development for thermo-chemical conversion is discussed to summarize the most common catalysts and provide guidance for future novel catalyst improvement. Current research studies regarding CNT and hydrogen production from waste materials have been reviewed which show that the topic is highly attractive for researchers. The applications of CNTs have also been grouped based on different properties, aiming to guide the future research to explore the potential applications of CNTs synthesized from wastes. This review provides an overview of the recent developments in this research area and stimulates research to promote the deployment of the technology.
Dr Zhang is a research fellow based at University College London UK. She has 20 publications since 2015 and her research covers a broad range of catalysis and pyrolysis, especially pyrolysis-catalysis of waste for obtaining CNTs. |
Dr Yao is a research fellow at National University of Singapore, Singapore, who has expertise in converting plastic wastes into CNTs. She has four related research articles published in Appl. Catal., B in the last 4 years. |
Prof. Williams is a Professor of Environmental Engineering at the University of Leeds, UK, and has a research background in both applied chemistry and process engineering. He is a Chartered Engineer and Fellow of the Institute of Energy. He has published more than 400 academic papers in the area of environmental engineering, including waste and biomass pyrolysis, waste incineration and the development of analytical methodologies for the detailed analysis of complex organic liquids, characterisation of the organic fraction of particulate emissions and the speciation of heavy metals in flue gases. He has also authored a second edition of a text book entitled ‘Waste Treatment and Disposal’ (John Wiley & Sons, 2005). He has an ‘h’ index of 50 and more than 10 |
Dr Wu is a Reader at Queen's University of Belfast, UK. His research interests include energy/chemical production from the catalytic thermo-chemical conversion of biomass and municipal solid wastes, and multifunctional catalytic sorbent development for carbon capture and utilisation. He has published >150 peer-reviewed journal papers (with an h-index of 45 in Google Scholar). |
Prof. Brett specialises in electrochemical materials science and technology development. He is the co-founder of the UCL Electrochemical Innovation Lab (www.ucl.ac.uk/eil) that provides a sandpit environment where basic science meets industrial development leading to exploitation of new technologies. He has published >600 peer-reviewed journal papers (with an h-index of 56 in Google Scholar) and was awarded the 2009 De Nora Prize in recognition of his ‘outstanding contribution to fuel cell and battery research’, along with the 2011 Baker Medal from the Institute of Civil Engineers for his published work on fuel cells. |
1 Introduction
An enormous amount of waste is produced globally every year which attracts people's concern in terms of waste management, cost and pollution. There are four abundant carbonaceous wastes (waste tyres, plastics, biomass and crude glycerol) from different sectors which will be introduced in this section regarding waste generation and management.1.1 Waste tyre production and management
A large number of used tyres are generated around the world each year. It has been reported that the annual global waste tyre production is approximately 17 million tonnes.1 The automotive retail economy is still in high demand throughout Europe accompanied by the growth of the transport sector; therefore, the increasing trend of end-of-life tyre waste generated in the EU is still expected to show an upward trend.2Waste tyres are a mixture of elastomers (e.g. natural rubber, butadiene and styrene–butadiene rubbers), carbon black filler/strengthener, metal reinforcements, zinc, sulphur and other additives.3 They are considered as one of the most difficult waste materials to degrade due to chemical and biological resistance to degradation, which results in extremely long periods in waste landfill sites. Moreover, the waste tyre dumps could be the habitats for mosquitos and rodents that induce many diseases.1 Therefore, most developed countries have banned the landfilling of waste tyres. In the EU, the Waste Landfill Directive has banned whole used tyres going to landfill since 2003 and shredded used tyres going to landfill since 2006.3 The uncontrolled combustion of waste tyres produces smoke, oil and other toxic substances that pollute the atmosphere, soil, surface water and groundwater. With the demand for tyres growing globally each year, in 2020, ETRMA reported that 324 million tonnes of tyres were sold, representing 20% of the world tyre market,4 and the environmental issues caused by waste tyre disposal is becoming more serious.3 There is a need to find alternative methods to retreat the waste tyres. The approaches to manage the waste tyre issue include energy recovery, recycling and reuse.1
1.2 Waste plastic production and management
The large quantity of plastic consumption around the world results in the production of enormous amounts of waste plastics. Global annual commercial plastic production reached 359 million tonnes (Mt) in 2018 (ref. 5) and is estimated to double within the next 20 years.6 It was reported that plastic converter demand in European countries was 51.2 million tonnes, and 29.1 million tonnes of waste plastics were generated in 2018.5 The amount of plastic waste will inevitably increase further in 2020 because of the outbreak of COVID-19, as more plastic materials are needed whether in medical areas (especially for personal protective equipment like masks) or in daily packing areas (for food hygiene and increasing online purchases).7 There are growing concerns that the current consumption and disposal of plastics bring substantial problems. A vast unmanaged plastic waste stream which leaks into the environment can be transformed into micro-/nano-plastics, posing a threat to the terrestrial and oceanic ecosystems.8,9 For those collected plastic post-consumer waste, there are three options that are commonly used for disposal, including landfill, energy recovery and recycling. In Europe, though the recycling rate has doubled since 2006, the amount of plastic waste sent to landfill still accounts for 25%.10 The landfill represents the cheapest and the least environmentally harmful waste tyre management method, while it requires hundreds of years for degradation and calls for much land area which is unrealistic in land-scarce areas such as Singapore. Energy recovery (also known as incineration) would significantly reduce the volume of plastic waste with simultaneous generation of heat, but hazardous substances may be released during incineration. Mechanical recycling provides a solution to recover some industrial plastic waste (especially PET and HDPE) into new low-grade plastic materials; however, the feasibility of such technology highly depends on the purity of waste and the availability of cost-efficient sorting equipment.10 Chemical recycling, especially pyrolysis is another option for plastic waste management, which would be discussed in detail in a later section. It should be noted that most currently available recycling options are not cost competitive, hampering the commercialization of plastic recycling. More efficient and sustainable recycling options, which ideally not only solve the environmental issues caused by plastic but also generate value-added materials or fuels are needed to ensure the circular economy economy of plastics.111.3 Biomass waste production and management
Biomass is one of the most abundant and renewable carbon sources around the world that originates from the photosynthesis process taking place in plants utilizing carbon dioxide, water and solar light.12 In 2017, electricity generated from biomass-based sources was the third largest renewable electricity source after hydropower and wind (596 TW h). In 2018, 35.4 million tonnes of wood pellets were produced, of which 55% of the production was from Europe and 31% from the Americas (mainly USA).13 Biomass waste as a low-value material has raised urgent environmental concern. The devastating environmental and health issues are caused by the disposal of very large quantities of biomass;14,15 it is estimated to be 100 billion metric tonnes every year, including forestry residues, agricultural wastes and waste generated from food processing.16,17 The food and green waste make up 44% of global waste.18 Nearly 36% of waste generated globally has been landfilled and 33% of waste is disposed in open dumpsites. A limited amount of waste (∼19%) can be recovered through recycling and composting, and ∼11% of global waste is sent to incinerators.19 In most developed countries, the recycling rate of the waste is higher than the global rate. In 2016, 47.4% of waste generated from 28 EU member countries has been sent to landfill or incinerators without energy recovery, and 52.6% of the waste was treated by recycling (36.7%), backfilling (10.1%) or energy recovery (5.8%).20 However, waste management in most developing countries such as South Asia is still facing serious disposal issues as the most widespread disposal method is to dump in open sites.21,221.4 Crude glycerol production and management
Recently, biodiesel is increasingly used as a transport fuel since the derived engine emissions are much lower than the use of fossil fuels; in particular, biodiesel utilisation has a low overall carbon dioxide (CO2) emission.23,24 Also, biodiesel is biodegradable and nontoxic. It can be made from domestic or renewable resources, such as plain oil, animal fat, used cooking oil and algae. Biodiesel can be blended with regular diesel and used in diesel engines.24 It can be produced by micro-emulsions, thermal cracking (pyrolysis) and trans-esterification, which is the most common method to produce biodiesel, yielding 10 wt% of glycerol as a by-product.25,26 Considering all the benefits from biodiesel, the large scale production of biodiesel dominates the renewable energy industry in the EU. The EU expects 8.6% of its overall transport consumption in 2020 to be from biodiesel and its capacity of biodiesel production needs to reach around 23 million tonnes.27,28 However, there is the expectation that a large amount of glycerol produced from the biodiesel production process will be usefully employed, making biodiesel more cost-competitive with the conventional diesel fuel.29As a by-product of biodiesel manufacturing, the large amount of glycerol can influence the development of the biodiesel industry.28 Volatility in the glycerol market can lead to a decrease in price, thereby bringing a series of problems including waste storage and treatment. The disposal or use of glycerol has become a challenge to make it a competitive commodity in the market. One issue is the presence of impurities in crude glycerol.29 The impurities include water, ash, methanol and certain fatty materials. The impurities could cause catalyst deactivation and impede the performance of catalysts used in the glycerol steam reforming process.30 There is a need to unlock the potential value of the crude glycerol as a by-product of the biodiesel industry.31
An enormous amount of waste is produced globally every year and is attracting people's concern in terms of waste management, cost and pollution. Four abundant carbonaceous wastes (waste tyres, plastics, biomass and crude glycerol) from different sectors are introduced in the review. Thermo-chemical conversion of waste is more desirable compared with traditional recycling methods, as it could ease the environmental issues caused by landfilling or incineration. Also, the waste hydrocarbons can be present as an alternative feedstock for CNT and hydrogen production due to the much lower costs of feedstock.
The review summarizes the research works published in the last 20 years on the use of thermo-chemical conversion to produce high value products from waste materials, especially for obtaining CNTs. The development of this research field is discussed critically. There is a lack of research covering the applications of the CNTs produced from waste materials due to the impurities and un-controllable quality of the CNT production. So, a broad range of applications of CNTs have been covered in this review that would guide the researchers for extending the investigation to further imply the CNT products. There is potential to achieve commercialization of this technique for converting waste to high-value products.
This review would guide the researchers for further developing the technique for scaling-up. A larger scale of research would further boost the commercialization of this technique to extract values from waste materials. Also, the technique could guide the government sector to promote sustainable techniques for waste management.
The products from thermo-chemical conversion process consist of liquid, solid and gaseous products. Recently, Nanda and Berruti32 reviewed the thermo-chemical conversion of plastic wastes to liquid fuels. Paul33 reviewed the topic of pyrolysis-catalysis of waste plastics for hydrogen and CNT production. However, the previous reviews did not cover the applications of the CNTs which could be the future direction of this research. Solis and Silveira34 critically reviewed different techniques based on thermo-chemical conversion and highlighted the economic feasibility of chemical recycling, which is still challenging to assess. The most important reasons are the uncontrollable quality of the products, purification methods of the products and potential applications. So, our review covers a more comprehensive summary regarding the most up-to-date research outcomes and critically discusses the research gaps to guide the researchers for next stage investigation. Although, thermo-chemical conversion of other waste materials has also been reviewed, such as biomass,35 tyres36 and glycerol,37 they all focus on one single feedstock with a limited insight of the overall field. The catalyst development has been mentioned as a small proportion in the published reviews; here, we have made a more comprehensive catalyst development table to cover as much catalysts as possible. The future outlook highlights the future directions of this research, which show the potential of this technique for future development to financially and environmental benefit mankind.
2 Thermo-chemical conversion of carbonaceous wastes
Pyrolysis/gasification of waste is a thermo-chemical process which can be used for energy recovery from waste tyres, plastics, biomass and crude glycerol to prevent the waste going to landfill or incinerators.38,392.1 Thermo-chemical conversion of waste tyres
A typical tyre has a high carbon content of ∼81.2 wt% and a hydrogen content of ∼7.2 wt%.40 The recovery of valuable products from tyres has been studied by many researchers, including the production of hydrogen, aromatic compounds, activated carbons, and others.40–43 Pyrolysis as a thermal degradation process for recovering more valuable products from waste tyres has been investigated as a process option for waste tyre recycling.41,43 The typical pyrolysis temperature for pyrolysis of tyres is around 500 °C, where the tyre is heated in an inert atmosphere for tyre degradation to produce gases, solid chars and oils. The gaseous products contain hydrogen and C1–C4 hydrocarbons (methane, ethene, ethane, propene, propane, butene, butane, and butadiene) and have a high calorific value of up to 65 MJ m−3 under atmospheric conditions, depending on the process conditions.41 The solid char contains carbon black fillers that could be used as solid fuels or upgraded to activated carbons, which have been widely used for purification and separation.44 Oils from pyrolysis of waste tyres are complex liquids with the texture of oil/wax, dark brown or black colours and with a sulphurous odour. The oil consists of over 100 identified compounds with chemical structures between C5–C60. The fractionation of these oils shows the presence of aliphatic hydrocarbons (including decane, undecane, dodecane, tridecane, tetradecane and others), aromatic hydrocarbons (including toluene, ethylbenzene, xylene, propylbenzene, ethylbenzene, naphthalene, phenanthrene, and pyrene), heteroatoms and polar fractions.45Different compositions of tyre pyrolysis oils have been reported by different researchers. Using a circulating fluidized-bed reactor at 500 °C, Dai et al.46 obtained a tyre pyrolysis oil which contained 26.77 wt% alkanes, 42.09 wt% aromatics, 26.64 wt% non-hydrocarbons and 4.05 wt% asphalt. Conesa et al.47 reported that a pyrolysis oil produced with a fluidized-bed reactor at 700 °C consisted of 39.5 wt% aliphatic fraction, 19.1 wt% aromatic fraction, 21.3 wt% hetero-atom fraction and 20.1 wt% polar fraction. Aylón et al.48 produced a tyre pyrolysis oil at 600 °C by a screw kiln reactor and contained 6.7 wt% alkane fraction, 65.5 wt% aromatic fraction and 27.8 wt% polar fraction. The difference in oil compositions is due to various tyre compositions and reaction temperatures.
Pyrolysis processes are normally carried out at relatively low temperatures (around 500 to 600 °C) with a higher yield of oils than gaseous products.41 The typical gasification process is usually carried out at temperatures between 800 and 1200 °C, yielding more gaseous products than pyrolysis.49 During gasification of waste tyres, hydrogen-enriched syngas is normally the target product. Syngas can be used for power generation using internal combustion gas engines or for producing chemicals through the Fischer–Tropsch process.50–53 Hydrogen is regarded as a clean energy carrier for a projected future hydrogen economy, as it can be produced from many sources, its combustion only generates water and it has broad applications e.g. fuel cells.54
Tyres are a type of polymeric material made of single or double-bonded carbon atoms, where the rubbers are characterized by carbon–carbon double bonds.55 The thermal decomposition of tyre rubber produces sub-units of the tyre rubber's molecular structure which are highly reactive free radicals.56 The mechanism of tyre decomposition can be explained by reactions (1)–(5):57
 | (1) |
| C + CO2 ↔ 2CO, ΔHr = 172.5 kJ mol−1 | (2) |
| C + H2O ↔ CO + H2, ΔHr = 131.3 kJ mol−1 | (3) |
| CH4 + H2O ↔ CO + 3H2, ΔHr = 205.8 kJ mol−1 | (4) |
| CO + H2O ↔ CO2 + H2, ΔHr = −50.8 kJ mol−1 | (5) |
2.2 Thermo-chemical conversion of waste plastics
Thermo-chemical recycling has been seen as a promising technology with a low carbon footprint for disposing of waste plastics. Quite a number of projects have been conducted on chemical recycling of waste plastics to produce liquid fuels. However, many challenges still exist in the product homogeneity from a mixed feedstock, optimization of liquid quality, and minimization of handling cost, before such technology is accepted and widely applied for plastic waste.58 Many researchers also investigated the gasification of plastic waste for producing hydrogen or syngas.59–63 It is often suggested that large quantities of waste plastics are available and can be used to produce significant amounts of hydrogen. Polyethylene, including HDPE and LDPE, is one of the main types of waste plastics.64 Many researchers have focused on the thermal decomposition of plastics, and their results are indicative of the viability of using plastics to produce hydrogen.59–61,65 Gasification of waste plastics to produce synthesis gases (H2, CH4 and CO) could effectively convert all of the carbon compounds in waste plastics to valuable products.66 The reactions can be explained by eqn (6)–(9).67 By using waste plastics to replace the conventional feedstock for hydrogen production, there is a promising potential to mitigate the high demand for fossil fuels.65| C + H2O ↔ CO + H2, ΔH = 131.3 kJ mol−1 | (6) |
 | (7) |
| H2O + CO ↔ H2 + CO2, ΔH = −41.2 kJ mol−1 | (8) |
| C + CO2 ↔ 2CO, ΔH = 162.4 kJ mol−1 | (9) |
In the thermo-chemical conversion of waste plastics to produce hydrogen, catalysts play a key role in maximizing hydrogen production. Also, two-stage pyrolysis-catalysis systems are more controllable than one-stage catalysis processes because they separate the pyrolysis residues containing contaminants from catalysts.64 From previous studies, nickel-based catalysts are the most common catalysts used for hydrogen production from plastics by thermal processing, mainly because of their high thermal stabilities and hydrogen selectivity.59,62,64 Many types of Ni-based catalysts have been investigated such as Ni/Al2O3,68 Ni–Mg–Al64 and Ni/MgO catalysts.69
2.3 Thermo-chemical conversion of waste biomass
Biomass waste contains a higher fraction of oxygen and a lower percentage of hydrogen and carbon compared with petroleum resources.70 Thermo-chemical conversion/pyrolysis-gasification/reforming of biomass has been extensively studied by researchers through supercritical water gasification,71 microwave-assisted gasification72 and chemical looping gasification.71 Pagliaro23 summarized the chemistry of energy from biomass and highlighted that it will play a crucial role in future bio-refineries. The energy from biomass includes biomass for glycerol and biodiesel production; the biodiesel can be used to produce energy, or the CO2 and H2O could be the source of biomass through photosynthesis, and glycerol can be catalytically converted to chemicals, fuels and drugs. A bio-refinery can be considered as an integral unit that can accept different biological feedstocks and convert them to a range of useful products including chemicals, energy and materials.23 The chemical reactions occurring during thermo-chemical conversion of biomass can be described through eqn (10)–(15):73| CxHyOz → (H2 + CO2 + CO + CH4 +C2…) + tar + char, ΔH298 K > 0 | (10) |
| C + H2O ↔ CO + H2, ΔH = 131.3 kJ mol−1 | (11) |
 | (12) |
| CH4 + H2O ↔ 3H2 + CO, ΔH = 206.3 kJ mol−1 | (13) |
| C + CO2 ↔ 2CO, ΔH = 162.4 kJ mol−1 | (14) |
| CO + H2O ↔ CO2 + H2, ΔH = −41.2 kJ mol−1 | (15) |
2.4 Thermo-chemical conversion of crude glycerol
Increasing the value of glycerol is an effective way to meet the challenges of the future development of biodiesel production.15 Considering the relatively high costs of producing biodiesel and the purification of crude glycerol, the development of converting glycerol to value-added chemicals is important.15,21,22 Many reactions occur during the pyrolysis of glycerol across the temperature range of 650 to 800 °C. Products from pyrolysis of glycerol include gases, liquids and chars. Gas components include H2, CO, CO2, CH4, and C2H2. The liquid components include methanol, ethanol, acetone, acetaldehyde, acrolein, acetic acid and water.74 The liquid, gas and char are produced by dehydration reactions when the temperature is relatively low. When the temperature is relatively high, H2, CO and char are produced as the main products by consecutive thermal cracking reactions.74 Valliyappan et al.74 surmised that the total char production from pyrolysis of glycerol is below 10 wt%, which means that the process is quite feasible.The chemical process for conversion of glycerol to CO and H2 is shown in eqn (16). CO and H2 are the main gaseous products from thermo-chemical conversion of glycerol. There are also small contents of other gases such as CO2, CH4, C2H4, C2H5 and C3H6. There are also simultaneous reactions happening during the decomposition of glycerol, as shown in eqn (17).74 Subsequent reactions, such as water–gas shift and methanation reactions, are shown as eqn (18) and (19).30,75
Decomposition of glycerol:
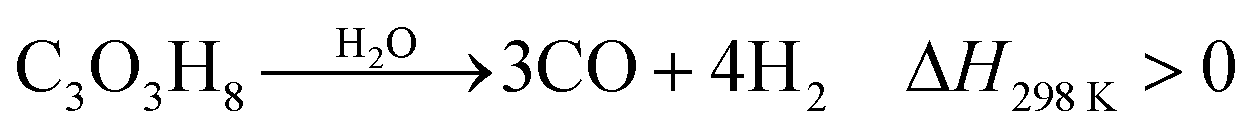 | (16) |
Simultaneous reactions:
| CmOnHk → CxHyOz + gas (H2, CO, CH4, CO2…) + coke, ΔH298 K > 0 | (17) |
Water–gas shift reaction:
| CO + H2O ⇔ CO2 + H2, ΔH = −41.2 kJ mol−1 | (18) |
Methanation reaction:
| CO + 3H2 → CH4 + H2O, ΔH = −206.1 kJ mol−1 | (19) |
2.5 Discussion of thermo-chemical conversion of carbonaceous waste
The production and disadvantages of conventional waste management for the four most abundant waste carbonaceous materials (waste tyres, waste plastics, biomass and crude glycerol from the biodiesel industry) have been discussed in Sections 1.1–1.4. There is a need to develop recovery methods to further improve the energy recovery efficiency from these waste materials. Thermo-chemical conversion/pyrolysis–gasification as a relatively mature and effective route to convert these four waste materials to profitable liquid, gaseous and solid products is explained in Sections 2.1–2.4, which are summarized in Fig. 1.76,77 Liquid products containing naphtha, tars and phenols can be applied straightaway or upgraded as fuels.78 Gas products containing syngas (H2 + CO), methane and other gases can also be used as fuels;79 and solid products are mainly chars that can be upgraded to activated carbons.80 As gasification is different from pyrolysis, it requires gasifying agents such as water, air or CO2 to enhance the gaseous products, and this consequently results in lower production of oils. The gaseous products are mainly H2, CO, CO2, CH4 and H2O.76 As there are many products produced through thermo-chemical conversion of carbonaceous waste, the development of the technique should focus on the most valuable products which have great potential to reach a higher recovery rate or financial values. So, hydrogen gas and CNTs (CNTs) will be further discussed as the targeting products.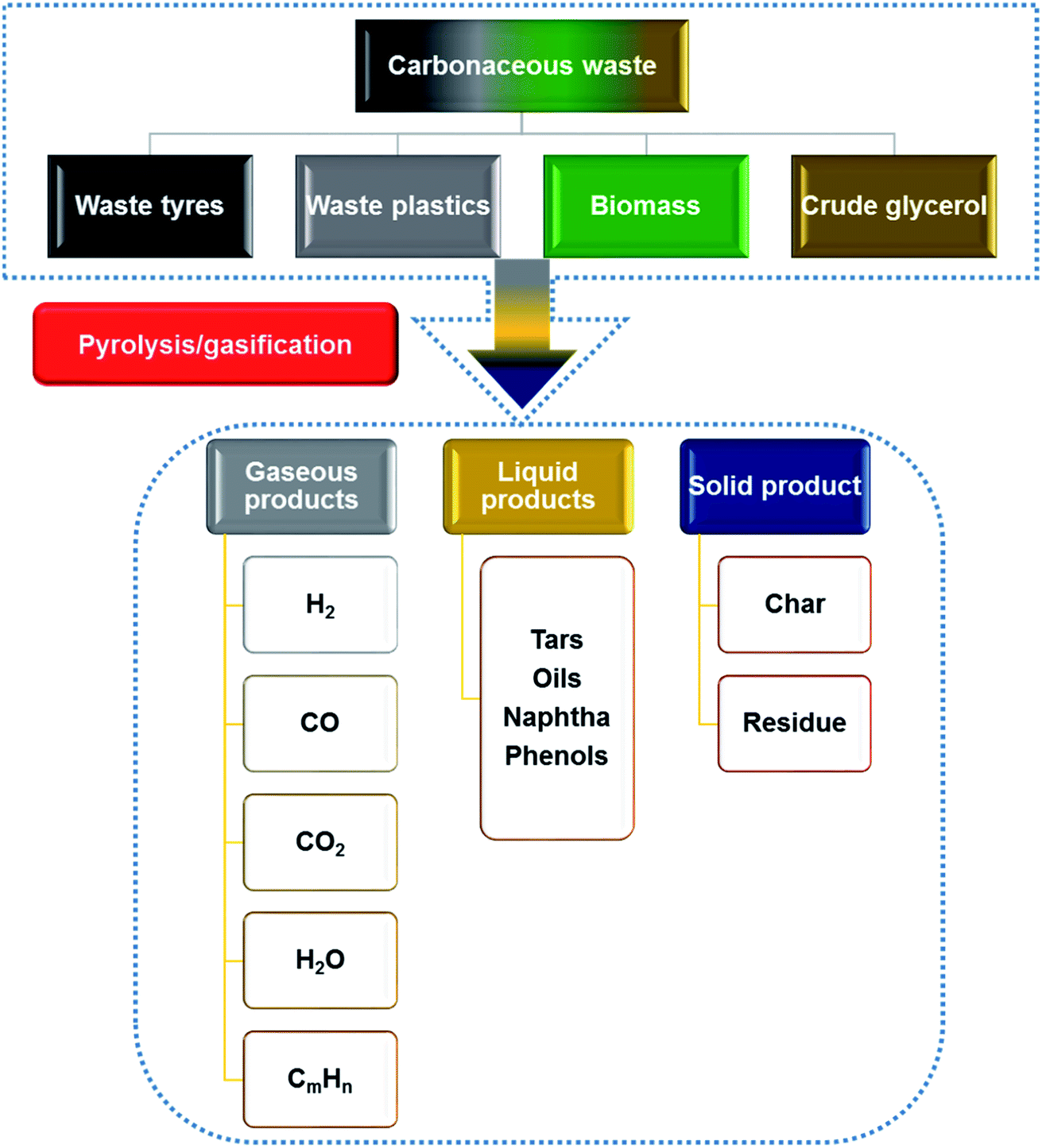 | ||
| Fig. 1 Products from the pyrolysis and gasification of carbonaceous materials. | ||
3 Hydrogen and CNT production
As discussed in Section 2.5, the products from thermo-chemical conversion of carbonaceous waste are generally grouped as gaseous, liquid and solid products, and the targeting products of hydrogen and CNTs are mainly covered in this review. Therefore, the production and background of hydrogen and CNTs are discussed in this section to explain the reasons for targeting these two products, especially CNTs from the thermo-chemical conversion of pure gas precursors and carbonaceous wastes.3.1 Hydrogen
There is around 5 × 1011 N m3 of hydrogen produced in the world, and around 96% of hydrogen is produced from fossil fuels. Hydrogen is mainly produced through methane reforming (48%), oil/naphtha reforming (30%), coal gasification (18%) and electrolysis (3.9%).81 Costs and alternative material sources are key challenges for the development of a sustainable hydrogen economy. Therefore, using alternative sources to generate hydrogen is imperative.Hydrogen is considered as a clean energy fuel which has the potential to reduce the world consumption of fossil fuels to meet sustainable development targets. Currently, the methods to produce hydrogen energy are not renewable. The costs and alternative sustainable energy sources are issues for hydrogen economy development.82 There is increasing research interest in investigating new feedstocks to produce hydrogen. Waste hydrocarbons are a significant potential source as it can help solve waste disposal issues and maximise the value of wastes by producing hydrogen and value-added products such as CNTs.
3.2 CNTs
Since the discovery of CNTs in the early 1990s, CNTs have been popularly applied in a wide range of science and engineering fields due to their unique physical and chemical properties.83,84 Single-walled carbon nanotubes (SWCNTs) are formed by a single graphite sheet wrapped around to form a cylinder with the diameter in the range of 0.8–1 nm. Multi-walled carbon nanotubes (MWCNTs) are formed by nested cylinders of graphene sheets in which the diameters are typically in the range of 5–20 nm but can also exceed 100 nm. The length of CNTs varies from 100 nanometers to centimeters.85 CNTs have several featured properties including relatively high tensile strength (around 100 GPa) which is 100 times greater than that of stainless steel, high modulus (around 1 TPa),86 a large aspect ratio of length to diameter, a cylindrical structure, low density (around 1100–1300 kg m−3) which equates to one-sixth of that of stainless steel, small size on the nano-scale, and good chemical and environmental stability. CNTs have relatively high thermal conductivities of about 3500 W m−1 K−1 at room temperature, similar to those of diamond and a relatively high electrical conductivity of 109 A cm−2 comparable to that of copper.87,88 The applications of CNTs are widely varied, making CNTs a high-value material.Bulk CNTs have been used for a wide range of applications including rechargeable batteries, automotive parts, sporting goods and boat hulls.86 The initial applications of CNTs on super-capacitors, actuators and lightweight electromagnetic shields have already achieved commercial impact. CNTs can be used as multifunctional coating materials. For example, MWCNTs can be added to paint which can discourage algae and barnacles attached on boat hulls and therefore reduce bio-fouling.89 MWCNTs have been widely used in lithium-ion batteries by blending MWCNTs with active materials and a polymer binder.90,91 MWCNTs can increase the electrical connectivity and mechanical integrity; therefore, the rate capacity and life cycle of batteries can be enhanced.92,93 Considering the low electron scattering and band gap of high quality SWCNTs, they have been used in transistors. CNTs have also been used in biosensors and medical devices because of their chemical and dimensional compatibility with biomolecules.94
However, the commercial applications of CNTs have still not reached their full potential, and there is still room for the development of CNT production from wastes as a complementary process for large-scale CNT production. The existing methods for CNT production are energy and resource intensive and include but are not limited to the electric arc-discharge method, laser ablation method, catalytic chemical vapour deposition (CVD), flame synthesis and a solar energy route.95 Among these methods, CVD is currently the most promising and preferred method for large-scale production. The typical reactor used for CVD is a fluidized bed reactor which improves the gas diffusion and heat transfer to catalyst nanoparticles.95 The low-cost feedstock, efficiency improvement, energy consumption reduction and waste reduction are the main factors that affect the scale-up of CNTs produced by CVD.96 There is a successful example of scaled-up camphor CVD for MWCNT production that has been commercialized in Japan (Meijo Nano Carbon Co. Ltd.).97 It has been reported that due to the relatively low price of camphor, the cost for producing CNTs has been reduced to around $100 kg−1, which is the lowest commercial price reported for purified CNTs.85
Nevertheless, researchers are still looking for more efficient and cost-effective ways for large-scale production of CNTs which has been defined as the production of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tonnes of CNTs per year. Hence, in recent years, there have been efforts regarding possible alternative routes for producing CNTs or investigating alternative feedstocks.85
000 tonnes of CNTs per year. Hence, in recent years, there have been efforts regarding possible alternative routes for producing CNTs or investigating alternative feedstocks.85
Dasgupta et al.101 noted that both the arc discharge and laser ablation methods have the issue of requiring a large amount of energy to induce the reorganization of carbon atoms into CNTs. The temperature for the process needs to reach 3000 °C or higher for effective crystallization and good graphite alignment of CNTs. Difficulties have emerged in meeting the basic required conditions for the large-scale production of CNTs, such as a vacuum environment and continuous graphite target replacement. CNTs produced by the arc-discharge method and laser ablation are normally in a carbonaceous soot form, which consists of amorphous carbon and metal particles from catalysts. This will significantly affect the purity and quality of the products. Transition metal carbide has been considered by many researchers to promote filamentous carbon deposition.102–104 Aligned SWCNTs can be synthesised by an attractive coagulation-based spinning of CNT suspensions, which has the potential to enlarge the scale and be extended to the production of MWCNTs.105
CVD is the most common and popular method to commercially synthesise CNTs. The CVD method is based on the thermal decomposition of hydrocarbon vapour, and the process can be promoted by adding metal-based catalysts. The general process includes the passing of a hydrocarbon vapour through a tubular reactor at temperatures of 600 to 1200 °C, in the presence of a catalyst. As the hydrocarbon decomposes in the reactor, CNTs grow on the surface of the catalyst. The growth ends with the system cooling down to room temperature. Precursors for CNT synthesis by the CVD method can be of various phases, such as liquid and solid hydrocarbons.97 The CVD method is more energy-saving compared with the arc discharge method and the laser ablation method. Also, the structure of CNTs can be properly controlled in the CVD process, such as the wall number, length, diameter and alignment. In consideration of the advantages of CVD, such as mild operations, low cost and a well-controlled process, CVD has great promise as a feasible method for large-scale production of CNTs.106 However, the disadvantage still exists that the CNTs produced are a mixture of MWCNTs and SWCNTs.99
Weidenkaff et al.117 produced MWCNTs with diameters in the range of 5 to 20 nm from carbon monoxide and gaseous hydrocarbons by CVD in the presence of a Fe-based catalyst in a fluidized bed reactor. Venegoni et al.118 produced MWCNTs from a mixture of hydrogen and ethylene in the presence of an Fe/SiO2 catalyst by a CVD method in a fluidized bed reactor. Homogeneous MWCNTs were produced with diameters in the range of 10 to 20 nm. Morançais et al.119 selectively synthesised MWCNTs from ethylene by the CVD method in a fluidized bed reactor, in the presence of a Fe/Al2O3 catalyst. The improved CVD process with efficient mixing of carbon precursors and catalysts resulted in a high selectivity of MWCNTs and a high purity. Philippe et al.120 also produced MWCNTs by a CVD method with a fluidized bed reactor in the presence of a Fe/Al2O3 catalyst. They also proposed a two-stage MWCNT growth mechanism based on their experiments and characterizations, which started with the MWCNT nucleation and grew by reconstruction as well as simultaneous carburization of the catalytic film. When the catalytic film was consumed, the catalyst particles inside of the mesoporous support were reduced and tangled CNTs were formed. Li et al.121 used Fe/Al2O3 catalysts prepared by an ion-beam assisted deposition method to produce well-aligned CNT arrays with lengths in a range of 500 μm to 1.5 mm by the CVD method. See et al.122 used Fe/Co/Al2O3 catalysts in their experiments to investigate process parameters for CNT synthesis in a fluidized bed. The results showed that the synthesis temperature affected the formation of CNTs greatly while the influence on the CNT diameter, quality, and yield was not clear. With the increase of synthesis temperature in all of their experiments, the yield of carbon increased rapidly and the increment of the CNT yield is small. It was indicated that the selectivity to CNTs decreased when the catalytic temperature increased. Nevertheless, See and co-researchers123 found that the quality of CNTs improved with the increase of reaction temperature in relation to the graphitization of CNTs. The results also showed that a higher fluidization ratio resulted in a pronounced increase of carbon yield compared with the increase of the deposition time. The type of catalyst and the interaction between the catalyst and the temperature were proved to have significant effects on carbon yields. The selectivity to CNTs significantly depended on the type of catalyst. In addition, their experimental results showed that the Fe–Co/Al2O3 catalyst had a relatively higher selectivity toward CNT formation than the Fe/Al2O3 catalyst.
Some researchers have used Co-based catalysts to enhance the growth of CNTs obtained from hydrocarbon reforming.124 Qian et al.124 used Co- and Ni-based catalysts in methane decomposition to enhance CNT production in a fluidized bed reactor. They compared the decomposition of methane with and without catalyst reduction. The yield of CNTs produced by the process without catalyst reduction was 3 to 4 times less than the CNT yield from the methane decomposition process with catalyst reduction. They explained that the in situ catalyst reduction provides energy for endothermic methane decomposition. Also, the in situ catalyst reduction consumed hydrogen and carbon to form water and carbon monoxide, which promoted hydrogen and CNT formation. Kong et al.110 synthesised CNTs by CVD from methane in the presence of different catalysts. They reported the effect of different metals (Fe-, Ni-, Co- and Fe/Co-based) and different supports (alumina and silica) on the formation of CNTs.
Wei et al.125 investigated the thickness of Fe- and Ni-based catalyst films on CNT formation by the CVD method. They found that there was no correlation between the thickness of the catalyst film and the formation of CNTs. However, the vertically oriented CNTs formed by using plasma-enhanced CVD with the nickel catalyst showed a strong correlation between the diameter of CNTs and the thickness of the catalyst film. Fang et al.126 reported that Ce–Ni mixed oxide can be one of the most effective and stable catalysts in steam reforming of ethanol to produce hydrogen and carbon nano-materials. The results from their research showed that the Ce–Ni catalyst is not only active for hydrogen production but also is the most effective catalyst to produce carbon nano-materials. They also found that co-precipitation is the most suitable method for catalyst preparation in terms of hydrogen and carbon nano-material production for ethanol steam reforming. The co-precipitation method can form small size NiO with a diameter of 15 nm and CeO2 with a diameter of 4 nm. These led to less formation of nano-fibrous carbon materials. In addition, the size of nano-fibrous carbon materials depended on the size of Ni nanoparticles. Ce–Ni catalysts prepared by the co-precipitation method can form smaller and more homogeneous carbon filaments compared to the catalysts prepared by other methods. The graphitic filaments are CNTs and carbon nanofibres (CNFs).
Ni-based catalysts have significant catalytic activity; their catalytic activity and stability are determined by the size of NiO and CeO2 and the interaction between nickel and cerium species, which are defined by the method of preparation. Considering the strong relationship between the catalytic stability and the type of carbon formation, catalytic stability can be analysed according to the size of graphitic filaments.127
Fe, Co and Ni are the most common metals used in CNT synthesis for two reasons: first, the carbon solubility can reach high levels at high temperature and second, carbon diffusion can attain high rates in these metals. Apart from the common transition metals used in CNT production, Cu, Au, Ag, Pt and Pd were also found to catalyse hydrocarbon decomposition to form CNTs.128 Kong et al.129 synthesised single-walled CNTs (SWCNTs) of high quality by CVD of methane with Fe-based catalysts at 1000 °C. Different catalyst supports were investigated, and the authors concluded that the catalysts supported by amorphous silica particles could produce SWCNT bundles. However, the catalysts supported by crystalline alumina nanoparticles produced individual SWCNTs and small bundles. Fan et al.111 produced self-oriented regular arrays of CNTs by CVD of ethylene with patterned porous silicon as the substrate. Satishkumar et al.112 produced bundles of aligned CNTs by pyrolysis of ferrocene and hydrocarbon mixtures. They found that the ferrocene–acetylene mixture is ideal for producing compact aligned nanotube bundles. The bundles of CNTs were associated with nanoparticles in a size range of 2–13 nm, and the alignment of catalyst nanoparticles was dominated by the ferromagnetism of transition metal nanoparticles. Li et al.113 enlarged the scale of aligned CNT production by CVD of xylene with iron nanoparticle embedded mesoporous silica catalyst. The growth direction of aligned CNTs was controlled by the pores of the mesoporous silica catalyst support. Sen et al.114 investigated the effects of metallocenes such as ferrocene, cobaltocene and nickelocene on CNT synthesis by pyrolysis of benzene. The wall thickness of nanotubes was associated with the metallocene content.
Organometallocenes have also been widely used as catalysts to produce CNTs because the metal particles can be liberated in situ and effectively promote hydrocarbon decomposition to form CNTs.97 Wei et al.115 synthesised multi-walled CNTs using a promoted method by exposing a silica substrate to a xylene and ferrocene mixture at 800 °C. The authors reported that the mixture of xylene and ferrocene vapour enhanced the selectivity to multi-walled CNTs. Nikolaev et al.116 synthesised SWCNTs by gas-phase catalytic reforming of carbon monoxide. The catalysts were obtained by in situ decomposition of iron pentacarbonyl in a hot carbon monoxide flow.
Cyclohexane130,131 and fullerene132,133 are also commonly used as carbon precursors to produce multi-walled CNTs. Liu et al.130 prepared CNTs by catalytic decomposition of cyclohexane. Li et al.131 synthesised three-dimensional hierarchical CNTs by electrochemical iron deposition of cyclohexane. The CNTs were shown to have high electrical conductivity. Nerushev et al.132 used fullerene and acetylene as carbon sources to investigate the dependence of catalytic particle size in a CVD process. They found that the diameter of CNTs increased when the catalytic particle size increased. Morjan et al.133 used fullerene as a carbon precursor to synthesize multi-walled CNT films by an iron-catalysed thermo-chemical vapour deposition process. The structural properties of CNTs produced from fullerene were different from the CNTs produced from acetylene.
Catalyst supports used in CNT production by the chemical vapour deposition process include graphite,134 quartz,135,136 silicon,137,138 silicon carbide,139,140 silica,141,142 alumina,143–146 alumina-silicate (zeolite),147,148 CaCO3,149 and magnesium oxide,150–152 among others. The interactions between catalytic particles and supports play an important role in CNT formation. The formation of chemical bonds between catalytic metal particles and supports could inhibit the catalytic ability of metal particles. Also, the morphology and textural properties of catalyst supports could affect the yield and quality of CNTs.97 For example, zeolite with nano-scale pores can significantly enhance CNT yields with a relatively small particle size.148
Alumina materials have been reported to be better catalyst supports than silica due to their strong metal–support interaction which could promote metal dispersion on catalysts.153 Stainless steel meshes have been applied by many researchers in CNT production from different sources.154–158 For example, the use of a stainless steel mesh as a catalyst support has been introduced in CNT and hydrogen production from the reforming of toluene, which can easily separate the CNT products from the catalysts.154 Alves et al.155 produced CNTs using a stainless steel type 304 alloy which consisted of 67% iron, 18–20% chromium and 11% nickel. They concluded that stainless steel can promote CNT growth which has also been reported by other researchers.156,157 Sano et al.156 produced aligned multi-walled CNTs on the surface of stainless steel by phenol decomposition. The stainless steel mesh was activated by intensive oxidation in air and then reduced in H2. Wal and Hall158 used an activated type 304 stainless steel mesh as a catalyst to produce CNTs from a mixture of C2H2/benzene or CO/benzene mixture by CVD.
Overall, common CNT synthesis methods like CVD always come with impurities from the production process, such as amorphous carbon and catalyst metal particles. Furthermore, the incorporation of dry and wet treatments can improve the purity of CNTs dramatically.
 | ||
| Fig. 2 The most accepted mechanism for CNT growth: (a) tip-growth model and (b) base-growth model.162 (This publication is distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike-3.0 License, which permits use, distribution and reproduction for non-commercial purposes, provided the original is properly cited and derivative works building on this content are distributed under the same license.) | ||
Fig. 2(a) shows the tip-growth model and describes the growth of CNTs on the catalyst where there is a weak metal–support interaction when the metal particles are at an acute contact angle with the support. The hydrocarbons decompose on the tip of the metal particle, then carbon atoms diffuse down through the metal particles and accumulate at the bottom of the metal particles. Because of the weak interaction between the metal particles and the support, the accumulated carbon on the bottom of the particle would push the metal particle away from the support. The accumulated carbon are hollow carbon CNTs which can keep growing until they reach the carbon solubility of the metal particle. Fig. 2(b) illustrates the base-growth model which describes the growth of CNTs on the catalyst where there is a strong metal–support interaction. The strong metal–support interaction indicates that the metal particle has an obtuse contact angle with the support. The beginning stage of hydrocarbon decomposition and carbon atom diffusion is similar to that of the tip-growth model. Due to the strong interaction between metal particles and support, the carbon precipitates out on top of the metal particles as a hemispherical dome. As more and more hydrocarbons decompose and diffuse to the lower peripheral surface of the metal particles, the carbon atoms accumulate as crystallized CNTs.97,162 The authors also broadly concluded that SWCNTs are formed when the catalyst particle sizes are of a few nanometers in size and the MWCNTs are formed when the catalyst particle sizes are of a few tens of nanometers. CNT growth is affected by a number of parameters, including the hydrocarbon precursors and the type of catalyst. Other factors such as the synthesis temperature, pressure, residence time, reactor type and flow rate of reactant can also influence the quality of CNT formation. Tessonnier and Su164 reached a similar conclusion that the diameter of the SWCNTs increases as the size of the catalyst particles increases. However, the nature of the CNTs would change at a certain point where double walled carbon nanotubes and MWCNTs are formed. They also highlighted that large metal particles at diameters above micro-meter size could dominate graphene or graphite formation. The shapes of catalyst particles also affect the features of filamentous carbon formation; round shape catalyst particles lead to hollow CNTs, while irregular metal particles with sharp edges lead to fishbone carbon nanofibers.165
In 2011, Tessonnier and Su164 proposed a vapour–solid–solid growth mechanism based on the vapour–liquid–solid mechanism proposed by Baker et al. in 1970s,134 as shown in Fig. 3(I)–(III). The vapour–liquid–solid mechanism includes three main steps. The first step (I) is the elementary carbon atom formation by the absorption and dissociation of a gaseous carbon precursor on the surface of the catalyst particle. In the second step (II), the carbon atoms dissolve in the bulk of nanoparticles, the liquid metastable carbide forms and carbon diffusion occurs within the particles. Finally, in the third step (III), the carbon atoms precipitate out at the side of the catalyst particles to accumulate as carbon filaments. Steps (II) and (III) could be alternatively described as (IV) and (V) that carbon species diffuse only on the surface of the catalyst particle. The vapour–liquid–solid mechanism has been supported by many other researchers as the calculated activation energy for carbon nanofiber formation agrees with the calculation of carbon dissolution in metals.134,166,167
 | ||
| Fig. 3 The vapour–liquid–solid mechanism of carbon nanotube growth, which is reproduced from ref. 164 with permission from John Wiley and Sons. | ||
There are still debates on the vapour–liquid–solid mechanism because the diffusion step is not clear, especially the nature of the driving force pushing the carbon atoms to diffuse on the catalyst particle surface has not been stated clearly. The vapour–liquid–solid mechanism could explain the CNT growth mechanism when the metal particles melt. The study of the CNT growth mechanism has been modified as the carbon diffusion occurs at the surface of the catalyst particle instead of through the bulk of catalyst particles, which has been supported by many researchers. Tessonnier and Su164 investigated the vapour–solid–solid mechanism including the carbon precursor dissociation, carbon atom diffusion on the surfaces of catalyst particles and carbon precipitation to form CNTs. They also proposed a hypothesis about the sub-diffusion of carbon atoms according to the calculation that each carbon atom could gain 0.3 eV energy by diffusing on the sub-surface.
In 2018, Yang et al.168 proposed a multi-route reaction in the gas-phase-assisted vertically aligned CNT growth mechanism in temperature gradient chemical vapour deposition of acetylene, whose growth kinetic characterization shifted from a single rate-limiting reaction to a multi-route reaction. C2H2 contributed to CNT growth through dissociation, followed by path one of bulk diffusion or path two of surface diffusion without gas-phase reactions. The secondary products (C4 and C6 species) were formed and approached the Fe-based catalyst substrate by the enhanced gas-phase reactions, which could be path three or others. The authors highlighted that the findings could help understand the vertically aligned CNT growth from thermally rearranged precursors, which would then guide the diameter-controlled and low-temperature growth.
In 2019, Li et al.169 reported the vaporizing phenomenon of the Na catalyst in low-temperature (∼400 °C) CNTs catalyzed by sodium-based ingredients coupled with an oxidative dehydrogenation of acetylene, which is feasible for the synthesis of metal-free CNTs. The authors proposed a plausible CNT growth mechanism as C2H2 reacts with metallic liquid Na to produce Na2C2 where sodium carbide could have several stoichiometries, then Na2C2 decomposes in the presence of H2 to produce metallic Na and a free C precursor. The graphitic structures of carbon will start assembling once free condensed C is present on the surface.
In 2020, Ashok et al.170 proposed the growth mechanism of bamboo-like N-doped CNTs from the polymerization of melamine. The C atoms keep accumulating at the base of the NiCo–graphene interface, and more atoms are nucleated on the edge and bottom of the deformed particle with the formation of bamboo knots over the outer layer. A restoring cohesive force developed through an increase in the surface tension force and the compressive stress from the growth walls of the CNTs. This could provoke the catalytic particle to be released from the newly formed graphene layer, and the growth process is repeated to form a complete bamboo-like structure with multiple knots. The authors also mentioned that the growth mechanism of CNTs is specifically different from bamboo-like CNTs whose most favourable route is a chain of adsorption–decomposition–diffusion–nucleation. The nucleated carbon atoms that are excreted through the surface of the catalyst and form a multiple graphene layer that is in line with the diameter of the catalytic nanoparticle form MWCNTs.
4 Influence of waste feedstock on CNT and hydrogen production
It is known that both amorphous and filamentous carbons are formed during the catalytic conversion of carbonaceous feedstocks.171 As the carbon deposition deactivates the catalyst and reduces the efficiency of thermo-chemical conversion of waste feedstock, enhancing the production of CNTs as well as reducing the production of amorphous carbon will add significant value to the process of hydrogen production from waste carbonaceous feedstocks.The catalyst deactivation resulting from carbon formation on the surface of catalyst would decrease the efficiency of the catalytic process.29,172 However, CNTs produced in the process of tyre gasification for hydrogen production can be regarded as a secondary product, instead of considering it as unwanted coke. It is therefore interesting to manipulate the gasification process e.g. using a catalyst to maximize the production of CNTs. Thus, the economic feasibility of hydrogen production would be effectively increased from waste hydrocarbons by thermo-chemical conversion.
Although the most common method to synthesise CNTs is via a CVD process from hydrocarbons like methane, benzene, xylene or other hydrocarbons,173,174 waste hydrocarbons such as waste tyres, plastics and crude glycerol, with their high content of hydrocarbons also represent a potential feedstock for the production of CNTs. A large amount of publications have been reviewed by Bazargan98 in relation to the synthesis of CNTs from waste plastics.
4.1 Waste tyres for CNT and hydrogen production
As tyres have high carbon and hydrogen contents,81 the recovery of valuable products from tyres has been studied by many researchers.6,29,31,81 Pyrolysis as a thermal degradation process to recover more valuable products from waste tyres has been investigated.29,31 Recent works50,175–178 have shown that under certain process conditions, carbon deposited on the surfaces of catalysts during pyrolysis catalysis of waste tyres is composed mainly of CNTs. Yang et al.179 successfully used waste tyres as an alternative carbon source to produce CNTs by a CVD method over a cobalt-based catalyst. This indicated that there is potential for waste tyres to be used as carbon precursors to synthesise CNTs. Murr et al.180 investigated a novel electric arc discharge method for the synthesis of MWCNTs from tyre powder. Poyraz et al.181 produced CNTs from devulcanized ground tyre rubber particles. The short-term microwave irradiation devulcanization of ground tyre rubber was first carried out in a microwave for less than 4 min. The microwave-treated ground tyre rubber particles were coated with polypyrrole as a substrate to grow CNTs by a secondary microwave treatment. The successful growth of CNTs was confirmed by TGA, FTIR, SEM and TEM techniques.4.2 Waste plastics for CNT and hydrogen production
Co-production of hydrogen and CNTs by pyrolysis-catalysis of plastics has become more attractive to researchers for simultaneously solving the plastic waste related issues and upcycling the waste to get value-added products.182 Among all kinds of plastics, PE (LDPE and HDPE), PP, PS and their mixtures have displayed their superior potential to be used as feedstocks for the synthesis of carbon nanomaterials. Kukovitskii et al.183 confirmed that crooked CNTs with 10–40 nm diameters were produced from polyethylene by pyrolysis at 420–450 °C and 4 atm in a quartz tube in the presence of a nickel plate catalyst in the early 90s. The CNT production rate could reach up to 200–300 mg h−1. Recently, quite a few researchers have worked on the production of CNTs within a flexible pyrolysis-catalysis system at atmospheric pressure. Liu et al.184 investigated the influence of temperature on MWCNTs and hydrogen-rich syngas production from polypropylene by a pyrolysis catalytic-reforming process. The two-stage reaction process involved polypropylene pyrolysis over a HZSM-5 zeolite catalyst in a crew kiln reactor and further catalysis of the pyrolysis vapour over a nickel-based catalyst in a moving-bed reactor. They found that 700 °C was the optimum temperature for MWCNT and hydrogen production. Recently, Jie et al.185 also used waste plastics as the feedstock to produce hydrogen and MWCNTs but using a different technique of microwave-initiated catalytic deconstruction of plastic waste under 1000 W microwave irradiation.4.3 Waste biomass (including bio-oil) for CNT and hydrogen production
Waste biomass is a biodegradable, eco-friendly resource, which is a low-cost, readily available, widely distributed, renewable carbon source with the ability to make renewable fuels and high-value carbon materials.12 Wang et al.12 recently reviewed the technology of the preparation of CNTs from biomass in the last few decades and summarized the CNT production from biomass, together with the application of biomass as a catalyst and a catalyst support, which have been extensively investigated. The authors also pointed out the potential of the biomass-based carbon nano-material market.He et al.186 utilized the Ni/α-Al2O3 catalyst for catalytic reforming of biomass-derived organics (toluene, 1-methylnaphthalene, phenol, ethanol, etc.). It was found that phenol and ethanol yielded a higher amount of hydrogen and CNTs than the other two organics, due to the enhancement of the oxygen functional group in phenol and ethanol. In addition, the lower molecular weight and the higher degree of saturation of organics stimulated CNT growth. Zhang et al.187 prepared high-quality three-dimensional graphene foams by utilizing waste biomass pyrolysis gases. The resulting 3DGFs exhibited excellent performance in environmental and energy-storage applications. Nevertheless, considering the congenital characteristic of biomass that contains sufficient oxygen and strong chemical bonding, as well as multiple ash components, it is difficult to realize direct conversion from biomass to CNMs.
4.4 Crude glycerol for CNT and hydrogen production
Glycerol can be a potential feedstock for producing H2, with a theoretical production ratio of glycerol/hydrogen of 1:4.188,189 Comparing all the processes of glycerol production, the recovery of hydrogen from the glycerol reforming process is the most effective way to improve the utilization of glycerol.190–192 Avasthi et al.29 summarized that hydrogen production from glycerol could be a very good option because the amount of hydrogen produced from glycerol reforming processes was stoichiometrically higher than the hydrogen production from conventional methods e.g. methane reforming. At high temperatures, gasification of glycerol can produce H2 and CO. The Fischer–Tropsch synthesis is a way to produce green biodiesel using syngas as the feedstock, at a H2/CO ratio of around 2:1.188,193 The gas produced from glycerol can be used to produce electricity because of the medium heating value.188,194 Valliyappan et al.195 produced syngas by pyrolysis of glycerol at atmospheric pressure and at different nitrogen flow rates and temperatures with different types or sizes of packing materials in the reactor. Simonetti et al.196 focused on syngas production from glycerol by gas-phase reforming. They used carbon-supported platinum (Pt) and platinum–rhenium (Pt–Re) catalysts in the experiments and measured the rates of glycerol conversion and H2/CO ratio by controlling the reaction conditions. The interaction of CO with the surface was decreased by the primary promotional effect of Re. Therefore, the coverage of CO decreased and the catalyst operated more effectively when the gaseous CO was presented. Peres et al.197 found that the optimized output of converting glycerol to gas in their pyrolysis of glycerol (they used commercial glycerol with a purity of 99% in experiments) reached up to 80 wt%. The highest hydrogen concentration was up to 40%, and the highest CO concentration was up to 48%.
Similar to carbon deposition in waste tyre and plastic pyrolysis catalysis/catalytic reforming processes, coke formation on the surface of catalysts during hydrogen production from glycerol reforming processes is also a challenge which cannot be avoided. Efficiencies of the reforming process could be reduced dramatically due to catalyst deactivation. Therefore, it would reduce the heat transfer from gas to catalyst.29,172 Ebshish et al.172,198 reported that coke formation during their glycerol steam reforming process was due to the acidic properties of the catalyst support, which affected the glycerol conversion process including hydrogen production. Chiodo et al.199 concluded that coke formation is mainly due to the large amounts of olefins that exist in the reaction streams. Investigations on how to convert coke to value-added products would increase the feasibility of glycerol reforming to such products.27 Wu et al.27 reported that 500 N m3 hydrogen fuel produced from 1 tonne of glycerol through gas-phase catalytic reforming along with ∼2.8 kg CNT production could have a considerable impact on the economics of glycerol utilization. Charisiou et al.200 also detected CNT formation through the glycerol steam reforming reaction. However, for future applications, this process needs further investigation because of the impurities contained in the crude glycerol which include spent and excess alkali metal catalysts, salts, excess methanol, fatty acids and esters.
5 Catalyst development for thermo-chemical conversion of carbonaceous wastes
5.1 Active metal types (monometallic type – Ni-, Fe-, and Co-based – and multi-metal based)
Ni-based catalysts are some of the most widely used catalysts in the catalytic reforming of tyres. Several Ni-based catalysts have been investigated by Elbaba et al.50,175,177,178 to improve the production of hydrogen from waste tyres. They used a two-stage pyrolysis coupled with a catalytic steam reforming reactor in the presence of nickel catalysts to produce a syngas with a high content of hydrogen (65 vol%) from waste tyres.178 The authors50,178 reported that hydrogen production could be increased with a higher nickel content in catalysts. Zhang et al.189,203–206 also used Ni-based catalysts to produce hydrogen and CNTs from waste tyres by catalytic pyrolysis/catalytic steam reforming. Ni-based catalysts are also the most common catalysts used in the glycerol reforming process.207 Different catalyst supports have been investigated in steam reforming/catalytic-gasification processes. Adhikari et al.208 considered that active catalysts for ethanol steam reforming could also be active in glycerol steam reforming. They used Ni-based catalysts in glycerol reforming experiments to produce hydrogen. The recent research they carried out on a crude glycerol conversion process showed that the maximum hydrogen production and purity of 68% could be achieved at 600 °C. Czernik et al.209 investigated the glycerol steam reforming process in a fluidized-bed reactor by using a commercial Ni-based catalyst. The hydrogen production efficiency reached around 80% of the theoretical yield. Adhikari et al.210 focused their research on the kinetics and reactor modelling of hydrogen production using Ni/MgO, Ni/TiO2 and Ni/CeO2 catalysts in a glycerol reforming process. They reported an activation energy of 103 kJ mol−1. The surface area of the Ni/CeO2 catalyst was the highest (67.0 m2 g−1) and therefore it gave the maximum hydrogen selectivity (74.7%) compared with the Ni/MgO and Ni/TiO2 catalysts under same experimental conditions: 600 °C, a water to glycerol molar ratio of 12:1 and a feed flow rate of 0.5 ml min−1.210,211
Alkali metals are effective for eliminating tar formation or upgrading the gaseous products formed in the thermo-chemical conversion of biomass, and the presence of alkali carbonates increased the carbon conversion to gases from biomass gasification processes.212 Hauserman213 investigated primary alkali catalysts for hydrogen production from wood gasification. The alkali catalyst from wood ash after gasification showed a high content of alkali metals (CaO, 44.3 wt%; MgO, 15 wt%; and K2O, 14.5 wt%).
Although the Ni-based catalyst is effective in the catalytic conversion process, there is still room for development of the catalysts. As the current research indicates that when using Ni-based catalysts in the reforming process, efficiency mainly depends on the temperature of the reaction which needs to be at a minimum of 550 °C. An impregnation method was used by Buffoni et al.214 to modify a catalyst by adding ZrO2 and CeO2 oxides onto a commercial α-Al2O3 support to boost the activity and stability of the Ni catalyst. The results proved that the modified Ni–Ce/α-Al2O3 catalyst could reduce the coke formation and was more stable in the reforming process. This is because the character of Ni/Ce restrains lateral dehydration, rearrangement and condensation reactions which result in coke formation with intermediate components.214 A precipitation method was used by Zhang et al.215 to prepare an M/CeO2 (M = 2% Ir, 15% Co and 15% Ni in weight) catalyst. The conversions of glycerol to hydrogen achieved up to 85% at a temperature as low as 400 °C using the Ir/CeO2 catalyst in the glycerol reforming process.215 To achieve the same conversion efficiency using Co/CeO2 and Ni/CeO2 catalysts required temperatures of 425 and 450 °C, respectively.172,215
Modifications of the Ni-based catalysts by adding promoters have been investigated by researchers to improve the catalytic activity. Wu et al.27 used Ni–Mg–Al catalysts for a glycerol pyrolysis/catalytic-gasification process. They found that the catalysts stayed effective after six hours of testing in terms of hydrogen production and the concentrations of gases. Iriondo et al.216 investigated hydrogen production from a glycerol reforming process by using an alumina supported nickel-based catalyst modified with Mg, Zr, Ce, and La. They found that the addition of promoters to catalysts could promote hydrogen selectivity in the glycerol reforming process. The results showed that when Mg was used as a promoter in the catalyst, the improved surface area of the catalyst resulted in high hydrogen selectivity. Also, when Zr was used as a promoter in the catalyst, the capacity of activating steam increased. Higher catalyst stability was achieved when Ce and La were used as promoters. The conversion of glycerol could stay at 100% over 50 h when using a Ni/Al2O3–ZrO2 catalyst.172 Iriondo et al.172,217 also investigated the possibility of improving the activity of an alumina supported Ni-based catalyst by adding intermediate amounts of La to the catalyst. Hao et al.218 synthesised CNTs by using an Fe/Mo/Al2O3 catalyst in the catalytic pyrolysis of polypropylene in a nano-agglomerate fluidized bed reactor. The researchers218 studied the formation of CNTs during this process including the initial fragmentation of the support of the catalyst, sub-agglomerate formation and the growth of CNTs which expanded the agglomerates. The CNT product yield was high when the agglomerates were fully developed. Ateyya A.219 used a Fe–Mo/MgO catalyst with different Fe/Mo ratios for the catalytic pyrolysis of polyethylene waste to produce carbon nanomaterials. MWCNTs, CNFs and graphene nanosheets were detected individually, or as a binary or ternary mixture depending on the Fe/Mo ratio. Higher Fe or Mo ratios were found to be optimal for enhancing the growth of CNTs and CNFs.
Fe- and Ni-based bimetallic catalysts have also been investigated in waste plastic pyrolysis-catalysis processes.203,220–224 He et al.221 recently studied the effect of the addition of Fe to the Ni catalyst on CNT growth. The interaction in the Ni–Fe alloy promoted the generation of metal carbide for enhancing CNT growth and H2 production. Yao et al.220 investigated the effects of Ni:Fe molar ratios at 1:3, 1:2, 1:1, 2:1 and 3:1 on the production of hydrogen and CNTs from waste plastics in a two-stage pyrolysis-catalysis steam reforming process at 800 °C. The results showed that the ratio of Ni:Fe at 1:3 supported by γ-Al2O3 gave the highest H2 yield of 84.72 mg g−1 plastic. Zhang et al.203 also studied the effect of different Fe/Ni weight ratios (00:20, 05:15, 10:10, 05:15, and 20:00) on hydrogen production from waste plastics in a two-stage fixed bed reactor. The authors pointed out that the presence of Fe and Ni together on the MCM-41 supported catalysts produced a synergistic enhancement of the total gas yield, especially for hydrogen production. The Fe:Ni weight ratio of 10:10 produced the highest gas yield of 95 wt% and the highest H2 production of 46.1 mmol H2 g−1 plastic. Yao et al.224 co-produced hydrogen and CNTs from the steam gasification of real-world waste plastics (a mixture of disposable drink cups, lunch boxes and plastic wraps) over Fe/Ni supported Al2O3 catalysts, yielding 287 mg gplastic−1 of CNTs and 31.8 mmol gplastic−1 of H2 for the bimetallic Ni–Fe/γ-Al2O3 catalyst at 800 °C in the absence of steam.
:1, it gave a better reaction stability, higher activity and selectivity of H2/CO at the desired temperature. The authors prepared the carbon supported Pt–Re catalysts by the incipient wetness method, impregnating carbon black with aqueous solutions of H2PtCl6·6H2O and HReO4. The carbon support needed to be dried up for 12 hours at 373 K before impregnation. For every gram of support, 1.7 g of solvent was needed. After impregnation and drying at 403 K for 12 hours, catalyst preparation was done.192,196 Slinn et al.226 found that when using a Pt/Al2O3 catalyst in the glycerol reforming process, optimum hydrogen production could be achieved at 860 °C with a flow rate of 0.12 mol glycerol per min per kg catalyst and a steam to carbon (S/C) ratio of 2.5. The optimized hydrogen selectivity was 70% and the glycerol conversion to gas was 100%.
As Ni-based catalysts are some of the most widely used and effective catalysts in the catalytic conversion process, various kinds of noble metal-based catalysts have been investigated to compare the catalytic activities with the Ni/Al2O3 catalyst. Adhikari et al.208 performed different experiments at 900 °C, with a water to glycerol molar ratio of 9:1 and a feed flow rate of 0.15 ml min−1 by using 14 different catalysts in a glycerol steam reforming process. These included Al2O3, Rh/Al2O3, Pt/Al2O3, Pd/Al2O3, Ir/Al2O3, Ru/Al2O3, Ni/Al2O3, Ce/Al2O3, Rh/Ce/Al2O3, Pt/Ce/Al2O3, Pd/Ce/Al2O3, Ir/Ce/Al2O3, Ru/Ce/Al2O3, and Ni/Ce/Al2O3. The catalysts were prepared on 92% alumina ceramic foam monoliths, which contained 8% silica, by the wetness technique using nitrate and chlorate precursors. Among the 14 different catalysts, Ni/Al2O3 and Rh/CeO2/Al2O3 resulted in the best H2 selectivity and glycerol conversion. The results showed that the highest hydrogen selectivity of 80% was achieved by using Ni/Al2O3, and the H2 selectivity realized using Rh/CeO2/Al2O3 was 71%. They also found that the increase in the water/feedstock ratio led to an increase of H2 selectivity and glycerol conversion. However, the efficiency of H2 production from the glycerol conversion process was reduced because of the increase of energy needed for water evaporation. Sanchez et al.225 found that the glycerol reforming efficiency could increase from 96.8% to 99.4% when the temperature was increased from 600 to 700 °C at 1 atm pressure and a 16:1 water/feedstock ratio using a Ni/Al2O3 catalyst (5.8 wt% Ni). The maximum glycerol conversion efficiency reached 99.7% at 650 °C and started to decrease at 600 °C over time. However, Chiodo et al.199 found that a Rh/Al2O3 catalyst showed higher activity and better stability compared to a Ni/Al2O3 catalyst in hydrogen production from their glycerol steam reforming process.
Hirai et al.227 found that H2 selectivity could reach up to 90% in the glycerol steam reforming process with complete conversion at 600 °C by using a Ru/Y2O3 catalyst. Also, a Ru/Y2O3 catalyst with 3 wt% Ru loading was considered as a more durable catalyst for limiting deactivation of catalysts caused by carbon deposition in the glycerol steam reforming process. They reported that this catalyst demonstrated a very high activity in a prolonged experiment. In their experiments, Groups 8–10 metals were used to prepare the catalysts over Y2O3, ZrO2, CeO2, La2O3, SiO2, MgO, and Al2O3 supports. The results showed that the order of catalyst activity was as follows: Ru ≈ Rh > Ni > Ir > Co > Pt > Pd > Fe. In addition, Kikuchi et al.228 found the order of the activity of the catalyst on silica in steam reforming methane was as follows: Ru ≈ Rh > Ni > Ir > Pt ≈ Pd > Co ≈ Fe. Hirai et al.227 summarized that active metals in steam reforming of methane also afforded high activity in the glycerol steam reforming process. Their results showed that Ru exhibited the highest H2 yield at a reaction temperature of 600 °C. Although Al2O3 could be a favorable support for the steam reforming of hydrocarbons, Ru on an Al2O3 support gave the lowest conversion in the glycerol steam reforming process. The greater the CH4 produced and the lower the CO2 produced, the less the amount of H2 was produced. So, the Ru/Y2O3 catalyst showed the best performance in the glycerol steam reforming process. The optimal Ru loading was attained at 500 °C. The results also showed that when the Ru loading increased, the H2 yield increased until the Ru loading was up to 3 wt%. The further increase of Ru loading up to 5 wt% had no significant effect on the H2 yield.227
Noble metals have also been added into the Ni-based catalysts as promoters to investigate the effects on the catalytic activity. Catalytic performances of Ni/CeO2–Al2O3 catalysts were promoted by the addition of noble metals (Pt, Pd, Ru, Ir). The results showed that the modified catalyst with noble metals led to higher H2 yields than the catalysts without noble metal promoters. The best result from their glycerol reforming process was achieved when the Ni/Pt/CeO2–Al2O3 catalyst was used at 700 °C.229 Profeti et al.229 found that a Ni/CeO2–Al2O3 catalyst can be promoted by noble metals (Pt, Ir, Pd, and Ru) since the dispersed CeO2 on alumina can prevent the formation of active nickel aluminate. The addition of noble metals could stabilize Ni sites in the reduced state in the reforming process, leading to a decrease in coke formation and an increase in glycerol conversion. In their experiments, a higher catalytic performance together with the highest H2 yield and a lower CO yield was achieved by using the Ni/Pt catalyst. Ni/CeO2–Al2O3 catalysts were prepared by a sequential impregnation method. The first step was to incorporate CeO2 on γ-Al2O3 using the incipient wetness method. An aqueous solution of Ce(NO3)2 prepared in a rotary evaporator at 60 °C was also required. For the removal of adsorbed contaminants, the γ-Al2O3 pellets needed to be sieved to 80–100 mesh particles and treated at 550 °C for three hours under the synthetic flow. The sample was calcined at 550 °C for 3 hours under a 20 cm3 min−1 airflow after drying at 80 °C for 10 hours. Then, Ni was incorporated on the CeO2–Al2O3 support by using the incipient wetness method with an aqueous solution of Ce(NO3)2·6H2O. Finally, the catalysts were obtained by calcination at 550 °C for three hours and drying at 80 °C for 10 hours. Cui et al.230 investigated the performance of a La1−xCexNiO3 catalyst in the glycerol steam reforming process by comparing its catalytic activity with that of a Pt metal catalyst. They found that Ni can be easily reduced in the La0.3Ce0.7NiO3 structure. The results were calculated by a non-stoichiometric method and compared with the thermodynamic equilibrium. The result showed that the catalyst had the highest activity in the glycerol steam reforming process. The glycerol conversion efficiency was close to the thermodynamic equilibrium when the temperature was in the range of 500 to 700 °C. The minimum carbon formation on the surface of the catalyst was achieved by using the La0.3Ce0.7NiO3 catalyst.172
5.2 Catalyst supports (artificial – Al-, Si, Si–Al, and activated carbon – and natural-based supports)
Alumina support has been applied widely in many reactions and considered to be the most effective support for catalysts. Mirodatos et al.145 found that Ni/Al2O3 catalysts demonstrated the highest amount of carbon deposition in the methanation reaction compared to Ni/SiO2 catalysts. Ni/Al2O3 catalysts presented the highest catalytic stability in reforming processes. A similar situation occurred in the reforming of methane using Ni/Al2O3 and Ru/Al2O3 catalysts carried out by Zhang et al.146 and Slagtern et al.231There are also some other common catalyst supports which have been used in hydrocarbon reforming processes to obtain hydrogen, such as γ/α-Al2O3, MgO, MgAl2O4, SiO2, ZrO2, CeO2 and TiO2.232 Adhikari et al.210 found that hydrogen production from glycerol reforming reached a maximum of 56.5% using a Ni/MgO catalyst and compared it with that from Ni/TiO2 and Ni/CeO2 catalysts at 650 °C and 1 atm.233 The catalysts were prepared using Ni(NO3)2·6H2O over three supports including MgO, TiO2 and CeO2, by the wet impregnation method. The same content of Ni loading (15 wt%) was used in all prepared catalysts. All catalysts were dried at 110 °C for 12 hours and then calcined at 500 °C in air for 6 hours. The final step was to sieve the catalyst using sieves with 16–35 mesh sizes. Dou et al.198 used a commercial Ni-based catalyst and dolomite sorbent in the glycerol conversion process. CO2 removal progressed simultaneously with hydrogen production from the glycerol conversion process. The results showed that the optimum temperature for the reactions was around 500 °C. Rossetti et al.234 prepared Ni-based catalysts supported on TiO2, ZrO2 and SiO2 by synthesizing supports in a liquid phase. These were followed by impregnation with the active phase and calcination at 800 °C. The metal–support interaction and surface acidity are the most important parameters for assessing catalysts. The metal–support interaction strongly depends on the catalyst preparation procedure. If the metal–support interaction is stronger, the activity and the stability of catalysts will be relatively higher. The surface acidity of catalysts can be modified using different catalyst supports.
Different catalyst supports also have an effect on the performance of a catalyst through the interaction of the active metal with the support, surface area and porosity of the support material, among others. Miyazawa et al.235 investigated the performance of nickel catalysts on various supports for steam reforming of tars derived from biomass pyrolysis. Ni/Al2O3, Ni/ZrO2, Ni/TiO2, Ni/CeO2 and Ni/MgO catalysts were studied. The Ni–Al2O3 catalyst was reported as the most active one and the Ni–MgO catalyst showed the lowest activity in relation to hydrogen production. It was suggested that the type of support influenced the nickel metal particle size, which was key to the catalyst activity. Inaba et al.236 investigated Ni/SiO2, Ni–ZrO2, Ni–CeO2 and a series of Ni-zeolites for hydrogen production from the gasification of cellulose. The production of hydrogen followed the order Ni/SiO2 > Ni/ZrO2 > Ni/CeO2. The production of hydrogen using the Ni-zeolites depended on the type of zeolite. Wu and Williams59 used a two-stage pyrolysis-catalytic steam reforming process to produce hydrogen from polypropylene using various substrate supports with nickel, including Ni/Al2O3, Ni/MgO, Ni/CeO2 and Ni/ZSM-5. The ZSM-5 zeolite performed as a relatively good support for the Ni-based catalyst with a high rate of hydrogen production; this could be due to the high surface area compared with other catalysts. He et al.237 synthesized CNTs through toluene steam reforming with Ni/γ-Al2O3 and Ni/α-Al2O3 catalysts. They reported that the strong interaction between Ni particles and γ-Al2O3 led to the base-growth mechanism of CNTs that the active sites of Ni-based catalyst will be covered. In contrast, the Ni/α-Al2O3 catalyst with a weaker interaction would lead to the tip-growth mechanism that would increase the Ni dispersion and promote the reforming reaction. Similar results were also reported by Yao et al.224 in the co-production of hydrogen and carbon nanotubes from real-world waste plastics in a fixed-bed reactor.
MCM-41 is a mesoporous material with a high surface area (up to 1000 m2 g−1), pore diameters of ∼2–10 nm and a flexible structure of amorphous silica walls.238 It has been used as a catalyst for hydrogen production, Wu et al.238 investigated Ni on a MCM-41 support for H2 production from biomass and Zhao et al.239 compared Ni–Al2O3 and Ni/MCM-41 for hydrogen production from cellulose. Zhao et al.239 reported that the highly ordered mesoporous structure of an MCM-41 support improved the dispersion of active nickel particles and subsequently increased the interaction between the nickel sites and gaseous products. However, few studies have investigated the production of H2 from waste plastics using Ni/MCM-41 or the role of the addition of metal promoters such as Fe- to Ni-catalysts. Yao et al.240 investigated a series of zeolite supported Ni-based catalysts (Ni/ZSM5-30, Ni/β-zeolite-25 and Ni/Y-zeolite-30) for the catalytic steam reforming of polyethylene process with a two-stage pyrolysis-catalytic steam reforming reactor. The authors found the ranking of syngas selectivity of each catalyst as Ni/ZSM5-30 > Ni/β-zeolite-25 > Ni/Y-zeolite-30 catalyst. In addition, the Ni/ZSM-5 catalyst showed excellent coke resistance and thermal stability.
5.3 Discussion of catalyst development for thermo-chemical conversion of carbonaceous waste
Catalysts play an important role in the waste hydrocarbon pyrolysis-catalysis/catalytic gasification process/thermo-chemical conversion processes. A comprehensive table and a summarized figure about different catalysts have been reviewed in the thermo-chemical conversion process and are shown in Table 1 and Fig. 4, respectively. The most common catalysts used in this process are transition metals-based catalysts like Ni-, Co-, Cu- and Fe-based catalysts. Noble metal-based catalysts, such as Rh, Pt, Pd, Ir, Ru and Ce also promote the catalysis or reforming process. However, the cost of noble metals is considerable, and as such, noble metal-based catalysts could not be commercialized for the large-scale production, for thermo-chemical conversion of waste feedstock.| Catalysts | Active content | Precursors | Catalyst synthesis method | Feedstock | Reaction conditions | Reactor |
|---|---|---|---|---|---|---|
| Ni-based | — | — | C11-NK, a commercial nickel-based naphtha reforming catalyst | Pine sawdust derived bio-oil, crude glycerine, trap grease | 750–850 °C | Bench-scale bubbling fluidized-bed209 |
| Ni–Mg–Al | Molar ratio 1:1:1 |
Ni(NO3)·6H2O, Al(NO3)3·9H2O, Mg(NO3)2·6H2O | Co-precipitation | Plastics (PP, PS and HDPE); waste tyres (natural rubber styrene–butadiene rubber and polybutadiene rubber); crude glycerol | 800–850 °C | Two-stage fixed-bed38,59,64,171,178 |
| Ni–Al | Molar ratio 1:2 |
Ni(NO3)2·6H2O, Al(NO3)3·9H2O | Precipitation | PP | 800 °C | Two-stage fixed-bed59 |
| Ni/CaO | 17 wt% | Ni(NO3)2·6H2O, CaO | Impregnation | Methane | 550–750 °C | Fixed-bed146 |
| Ni/CaO/C | Ni(NO3)2·6H2O, CaO, activated carbon | Impregnation, increasing pH and sol–gel | LDPE + pine sawdust | 800 °C | Two-stage fixed-bed63 | |
| Ni/MgO | 9.62–15 wt% | Ni(NO3)2·6H2O; MgO | Incipient wetness | PP, glycerol, wood-derived tar | 500–800 °C | Two-stage fixed-bed;59 tubular furnace;211,233 laboratory-scale continuous feeding dual-bed reactor235 |
| Ni/zeolites | 7.5–10 wt% | Ni(NO3)2·6H2O; H-β (27); H-mordenite (18.3); Na-mordenite (18.3); H-ZSM-5 (29); Na-ZSM-5 (29); USY (14); Na–Y (5.7); ZY-5.2; ZY-30; ZY-80 Ni/chabazite; ZSM5-30; ZSM5-50; ZSM5-80; β-zeolite-25; Y-zeolite-30 (the numbers in parentheses denote Si/Al2 ratios) | Impregnation | Cellulose, naphthalene, HDPE | 500–800 °C | Fixed-bed;236 two-stage fixed-bed;240 clear fused quartz compression tube248 |
| Ni/ZSM-5 | 10 wt% | Ni(NO3)2·6H2O; ZSM-5 | Incipient wetness | PP | 800 °C | Two-stage fixed-bed59 |
| Ni/MCM-41 | 5–20 wt% | Ni(NO3)2·6H2O; MCM-41 | Impregnation | Wood, cellulose | 200–800 °C | Two-stage fixed-bed;238 TGA-MS239 |
| Ni/CeO2 | 10–15 wt% | Ni(NO3)2·6H2O; CeO2 | Incipient wetness/impregnation; deposition–precipitation | PP, glycerol, ethanol | 250–800 °C | Two-stage fixed-bed,59 fixed-bed,210,225 tubular reactor;210,211,233 fixed-bed quartz micro-reactor;215 laboratory-scale continuous feeding dual-bed reactor235 |
| Ni/Al2O3 | 2–30 wt%; 18% NiO | Ni(NO3)2·6H2O; α/γ-Al2O3; Al2O3 ceramic foam monoliths (92% Al2O3 and 8% SiO2) | Incipient wetness/impregnation; commercial catalyst (Ni-3288 Engelhard) | PP, LDPE, toluene, cellulose, glycerol waste tyres, styrene–butadiene rubber, butadiene rubber and natural rubber, tyre oil, methane, wood derived tar | 200–900 °C; N2 | Two-stage fixed-bed;59,175,177,202,205 fixed-bed;68,146,198,214,225 conventional autoclave with a magnetic stirrer;69 tubular furnace;208,225 fixed-bed linear quartz micro-reactor;199 laboratory-scale continuous feeding dual-bed reactor;235 TGA-MS;239 packed bed reactor248 |
| Ni/CeO2–Al2O3 | Ni 2–20 wt%, NiO 16 wt%, CeO2 0–30 wt% | Ni(NO3)2·6H2O, Ce(NO3)3·6H2O, γ-Al2O3, Ce(NO3)2 | Co-impregnation; incipient wetness/impregnation; sequential impregnation | PP, waste tyres, glycerol | 450–900 °C | Two-stage fixed-bed;50,59 fixed-bed;214,216,225,229 tubular furnace208 |
| Ni–Pt/CeO2–Al2O3 | Ni 5 wt%, Pt 0.3 wt%, CeO2 10 wt% | Ni(NO3)2·6H2O, Ce(NO3)2; γ-Al2O3, H2PtCl6·2H2O | Sequential impregnation | Glycerol | 450–600 °C | Fixed-bed229 |
| Ni–Ir/CeO2–Al2O3 | Ni 5 wt%, Ir 0.3 wt%, CeO2 10 wt% | Ni(NO3)2·6H2O, Ce(NO3)2; γ-Al2O3, IrCl4·xH2O | Sequential impregnation | Glycerol | 450–600 °C | Fixed-bed229 |
| Ni–Pd/CeO2–Al2O3 | Ni 5 wt%, Pd 0.3 wt%, CeO2 10 wt% | Ni(NO3)2·6H2O, Ce(NO3)2; γ-Al2O3, PdCl2 | Sequential impregnation | Glycerol | 450–600 °C | Fixed-bed229 |
| Ni–Ru/CeO2–Al2O3 | Ni 5 wt%, Ru 0.3 wt%, CeO2 10 wt% | Ni(NO3)2·6H2O, Ce(NO3)2; γ-Al2O3, RuCl3·H2O | Sequential impregnation | Glycerol | 450–600 °C | Fixed-bed229 |
| Ni/natural olivine | 2.8 wt% | Ni(NO3)2·6H2O; (Mg0.92Fe0.08)2SiO4 | Impregnation | Methane | 600–850 °C; steam or CO2 | Fixed-bed249 |
| Ni/dolomite | 5–20 wt% | Ni(NO3)2·6H2O; (NH4)2CO3; dolomite | Impregnation | Toluene, waste tyres | 550–800 °C | Fixed-bed;68 two-stage fixed-bed176,177 |
| Ni/SiO2 | 8.9 wt% | Ni(NO3)2·6H2O; H4O4Si | Impregnation | Glycerol | 650–700 °C | Fixed bed234 |
| Ni/SiO2–Al2O3 | 5–20 wt%, Al2O3 to SiO2 mole ratios (3:5, 1:1, 3:2, 2:1) |
Ni(NO3)2·6H2O; SiO2–Al2O3, C9H21O3Al; SiO2 | Impregnation, sol–gel | Toluene, waste tyres | 550–800 °C | Fix-bed;68 two-stage fixed-bed250 |
| Ni/ZrO2 | 8.8–15 wt% | Ni(NO3)2·6H2O; ZrOCl2·8H2O; ZrO2 | Precipitation, impregnation | Glycerol, wood-derived tar | 500–700 °C | Fixed-bed;234,251 laboratory-scale continuous feeding dual-bed reactor235 |
| Ni/ZrO2/Al2O3 | Ni 2 wt%, NiO 17 wt%, ZrO2 7 wt% | Ni(NO3)2·6H2O; Zr(NO3)4·5H2O; γ-Al2O3 | Impregnation | Glycerol | 600–850 °C | Fixed-bed214,216 |
| Ni/La2O3 | 3–17 wt% | Ni(NO3)2·6H2O; La2O3 | Impregnation | Methane | 550–750 °C | Fixed-bed146 |
| Ni/La2O3/Al2O3 | Ni 16 wt%, NiO 17 wt%, La2O3 3–15 wt% | Ni(NO3)2·6H2O; La(NO3)3·6H2O; CeO2; γ-Al2O3 | Impregnation/co-impregnation | Glycerol | 500–600 °C | Fixed-bed216,217 |
| Ni/MgO/Al2O3 | NiO 16.2 wt%, MgO 2.5 wt% | Ni(NO3)2·6H2O; Mg(NO3)2·6H2O; γ-Al2O3 | Impregnation | Glycerol | 600 °C | Fixed-bed216 |
| Na2CO3 | — | Sodium carbonate | Commercial catalysts | Cellulose | 200–350 °C | Conventional autoclave with a magnetic stirrer69 |
| Ir/CeO2 | 2 wt% | H2Ir4Cl6·6H2O; CeO2 | Deposition–precipitation | Ethanol, glycerol | 250–600 °C | Fixed-bed quartz micro-reactor215 |
| Ir/Al2O3 | 2.5% | H2Cl6Ir; Al2O3 | Incipient wetness | Glycerine | 600–900 °C | Tubular furnace208 |
| Ir/Ce/Al2O3 | 2.5% | H2Cl6Ir; Ce(NO3)3·6H2O; Al2O3 | Incipient wetness | Glycerine | 600–900 °C | Tubular furnace208 |
| Ce/Al2O3 | 2.5% | Ce(NO3)3·6H2O; Al2O3 | Incipient wetness | Glycerine | 600–900 °C | Tubular furnace208 |
| Ce/zeolites | 5–30 wt% | Ce(NO3)3·6H2O | Impregnation | Cellulose | 500–600 °C | Fixed-bed236 |
| Co/CeO2 | 15 wt% | Co(OAc)2·4H2O; CeO2 | Deposition–precipitation | Ethanol, glycerol | 250–600 °C | Fixed-bed quartz micro-reactor215 |
| Rh/Al2O3 | 2.5–5% | Rh(NO3)3; Al2O3 | Incipient wetness | Glycerine | 600–900 °C | Tubular furnace;208 fixed-bed linear quartz micro-reactor199 |
| Rh/SiO2 | 30 wt% | HN4O10Ru; SiO2 | Impregnation | Cellulose | 500–600 °C | Fixed-bed236 |
| Rh/Ce/Al2O3 | 2.5% | Rh(NO3)3; Ce(NO3)3·6H2O; Al2O3 | Incipient wetness | Glycerine | 600–900 °C | Tubular furnace208 |
| Rh/Ce/SiO2 | Rh 2 wt%, Ce 30 wt% | C15H21O6Rh, SiO2; Ce(NO3)3·6H2O | Impregnation | Cellulose | 500–600 °C | Fixed-bed236 |
| Ru/Al2O3 | 2.5% | HN4O10Ru; Al2O3 | Incipient wetness | Glycerine | 600–900 °C | Tubular furnace208 |
| Ru/Y2O3 | 3 wt% | — | Impregnation | Glycerol | 600 °C | Fixed-bed flow type reactor227 |
| Ru/Ce/Al2O3 | 2.5% | HN4O10Ru; Ce(NO3)3·6H2O; Al2O3 | Incipient wetness | Glycerine | 600–900 °C | Tubular furnace208 |
| Pt/Al2O3 | 2.5% | H2PtCl6, Al2O3; [Pt(NH3)]4(NO3) | Incipient wetness | Glycerine | 600–900 °C; 225–300 °C | Tubular furnace;208 tubular stainless steel reactor192 |
| Pt/Ce/Al2O3 | 2.5% | H2PtCl6; Ce(NO3)3·6H2O; Al2O3 | Incipient wetness | Glycerine | 600–900 °C | Tubular furnace208 |
| Pt–Ni/La2O3/Al2O3 | Pt 2 wt%, NiO 2.5–12.6 wt%, 6 wt% La2O3 | Ni(NO3)2·6H2O; La(NO3)3·6H2O; CeO2; γ-Al2O3 | Consecutive wet impregnation | Glycerol | 500–600 °C | Fixed-bed217 |
| Pd/Al2O3 | 2.5% | Pd(NO3)2; Al2O3 | Incipient wetness | Glycerine | 600–900 °C | Tubular furnace208 |
| Pd/Ce/Al2O3 | 2.5% | Pd(NO3)2; Ce(NO3)3·6H2O; Al2O3 | Incipient wetness | Glycerine | 600–900 °C | Tubular furnace208 |
| Pt–Re/C | 5.6 wt% (Pt:Re 1:1, 10:1 and 1:2) |
H2PtCl6·6H2OH; ReO4; carbon black; HReO4 | Incipient wetness impregnation | Glycerol | 700 °C | Fixed-bed196 |
| Pt/C | 5 wt% | — | Commercial catalysts from E-TEK | Glycerol | 225–300 °C | Tubular stainless steel reactor192 |
| Pt–Ru/C | — | — | Commercial catalysts from E-TEK | Glycerol | 225–300 °C | Tubular stainless steel reactor192 |
| Pt/ZrO2 | — | H2PtCl6; ZrO(NO3)2·xH2O | Incipient wetness impregnation | Glycerol | 225–300 °C | Tubular stainless steel reactor192 |
| Pt/CeO2/ZrO2 | — | H2PtCl6; (NH4)2Ce(NO3)6; ZrO(NO3)2·xH2O | Incipient wetness impregnation; co-precipitation | Glycerol | 225–300 °C | Tubular stainless steel reactor192 |
| Pt/MgO/ZrO2 | — | [Pt(NH3)]4(NO3)2; Mg(NO3)2·6H2O; ZrO(NO3)2·xH2O | Incipient wetness impregnation; co-precipitation | Glycerol | 225–300 °C | Tubular stainless steel reactor192 |
| Fe-based | 99.99 wt% | — | Commercial iron powder from Johnson Matthey | Ethylene | 600 °C | Tube reactor103 |
| Fe/Al2O3 | 10 wt% | Fe(NO3)3·9H2O; γ-Al2O3 | Impregnation | Tyre, LDPE | 800 °C | Two-stage fixed-bed reactor189,202 |
| Fe/Ni/MCM-41 | Total 20 wt%, Fe/Ni ratios (00:20, 05:15, 10:10, 15:05, 20:00) |
Fe(NO3)3·9H2O; Ni(NO3)2·6H2O; MCM-41 | Impregnation | Simulated waste plastics | 800 °C | Two-stage fixed-bed reactor252 |
| Fe/Ni/Al2O3 | 10 wt%, molar ratio of Ni:Fe at (1:3, 1:2, 1:1, 2:1 and 3:1) |
Ni(NO3)3·6H2O; Fe(NO3)3·9H2O; α/γ-Al2O3 | Impregnation | Simulated waste plastics, real-world waste plastics | 800 °C | Two-stage fixed-bed reactor203,220–224 |
| Co/Al2O3 | 10 wt% | Co(NO3)2·6H2O; γ-Al2O3 | Impregnation | Tyre, LDPE | 800 °C | Two-stage fixed-bed reactor189,202 |
| Cu/Al2O3 | 10 wt% | Cu(NO3)2·3H2O; γ-Al2O3 | Impregnation | Tyre, LDPE | 800 °C | Two-stage fixed-bed reactor189,202 |
 | ||
| Fig. 4 A summary of how reviewed catalyst materials have been applied in catalytic conversion processes. | ||
Catalyst supports also affect the process and play an important role in terms of hydrogen selectivity and catalyst stability because of their basic characteristics and redox properties.241 The most commonly used catalyst supports are alumina, zeolite and silica. Catalyst synthesis methods are also very important which mainly include incipient wetness impregnation, precipitation and sol–gel. Although the impregnation is the most used method, the reported sol–gel method could significantly improve the catalyst porosity and nickel dispersion, which leads to the highest activity among the catalysts prepared by impregnation and co-precipitation.242
In addition, a fixed-bed reactor is the most typical reactor used in thermo-chemical conversion, which also has been modified and renamed as two-stage fixed-bed, tubular reactor, clear fused quartz compression tube, fixed-bed quartz micro-reactor, fixed-bed linear quartz micro-reactor, packed-bed reactor, fixed-bed quartz micro-reactor and tubular stainless steel reactor. There are also other types of reactors, such as bench-scale bubbling fluidized-bed, laboratory-scale continuous feeding dual-bed reactor, conventional autoclave with a magnetic stirrer and TGA-MS. Kikuchi et al.228 summarized a table about the types and methods of gasification and smelting processes for a semi-pilot scale test for hydrogen-rich fuel gas produced from different wastes. Muhammad et al.243 reviewed different pyrolysis techniques for synthesizing carbon nanomaterials.228 Williams33 recently reviewed the specific reactor designs for hydrogen production from waste plastics. So, the types of reactors of pyrolysis techniques are not included in this review.
Catalyst deactivation resulting from coke formation on the surface of the catalyst is one of the challenges in hydrogen production from tyre gasification.235,244–247 Catalyst deactivation is also affected by sulphur poisoning. Elbaba et al.50 found that the deactivation of a Ni/Al2O3 catalyst in gasification of a waste tyre for hydrogen production was due to sulphur poisoning and carbon deposition. It was noted that there were different forms of carbons generated in the process, including amorphous carbon and graphite carbons, for instance, CNTs. Giannakeas et al.248 found the evidence from X-ray diffraction results regarding the deposition of carbons on the surfaces of reacted catalysts, which caused catalyst deactivation in waste tyre reforming.
6 Evaluation of process parameters on CNT and hydrogen production from thermo-chemical conversion of carbonaceous waste
6.1 Process conditions for thermo-chemical conversion of carbonaceous wastes
Pyrolysis of waste tyres can be influenced by process conditions including the type of raw material, sample size, residence time, temperature and feeding rate that have been studied by many researchers.47,51,190,253–255 Dai et al.46 investigated the pyrolysis temperatures, residence times and particle sizes of samples by using a circulating fluidized-bed reactor in which pyrolysis and secondary reactions occurred. They reported that secondary reactions were favored by a longer residence time, and carbonization is favored at lower temperature and heating rates. Gas product yields increased with the increment of temperature from 400 to 800 °C. The yields of CH4, H2 and CO were also increased with the increment of temperature. However, the yields of CO2 and heavier hydrocarbon gases decreased. As the residence time increased, the production of syngas and light hydrocarbon increased, while CO2 production decreased. This is because the enhanced secondary reactions, such as further cracking of heavy pyrolysis oils, char reduction and shift reactions. The temperature and residence time played predominant roles in the production of gas and tar/oil. Furthermore, the particle size of feedstock showed only a slight influence on gaseous production.Aylón et al.48 carried out tyre pyrolysis using a moving-bed reactor at different temperatures (600, 700 and 800 °C). The yield of pyrolysis oil decreased dramatically from 41.5 to 27.5 wt% as the temperature increased from 600 to 800 °C. However, gaseous production significantly increased from 17.9 to 31.5 wt%. The reason for the promoted tar cracking reactions at higher temperatures was due to the enhanced primary cracking of heavy hydrocarbons. Cunliffe and Williams255 investigated the temperature and chemical class fractions of tyre pyrolysis oil using a fixed-bed reactor. They found a clear increase of the aromatic fraction from 36.7 to 45.6 wt% and a decrease of the aliphatic fraction from 51.3 to 36.1 wt% as the temperature increased from 450 to 600 °C. They also pointed out that the extended residence time of pyrolysis gases in the reactor could increase the fraction of aromatic compounds. Kyari et al.253 compared pyrolysis products from seven different types of tyres (countries of origin: Poland, Korea, Japan, South Africa, Italy and Great Britain) also with a fixed-bed reactor to investigate the influence of tyre origin on the yields of pyrolysis products. They found that the yields of gases, chars and oil products were not significantly affected by the type of tyres. However, the composition of the pyrolysis gas and pyrolysis oil varied between different tyres. Leung et al.51 studied the influence of operational parameters (equivalence ratio, tyre feed rate, temperatures and particle sizes) on hydrogen production from waste tyre gasification using a tubular reactor. They concluded that the yield of gaseous product was proportional to the equivalence ratio, tyre feed rate and tyre particle size. The yield of char decreased slightly when the equivalence ratio, feed rate or particle size increased. The yield of oil reduced considerably with the increase of the equivalence ratio and tyre feeding rate. The gaseous products were mainly H2, CO, CO2, H2S and other light hydrocarbons (C2–C4), with relatively high heating values ranging between 20 and 37 MJ m−3. Rodriguez et al.254 used an unstirred stainless steel 0.0035 m3 autoclave to run tyre pyrolysis experiments at temperatures in the range of 300 to 700 °C with 100 °C intervals. They found that the pyrolysis temperature significantly affected the gas production in the waste tyre pyrolysis process, and the highest temperature of 700 °C resulted in the highest gas yield. The calorific value of the pyrolysis gases was in the range of 68 to 84 MJ m−3, which is much higher than the values found in the literature (20–37 MJ m−3).256–258 Ucar et al.190 also investigated the pyrolysis of two types of tyres (passenger car tyre and truck tyre) using a fixed-bed reactor. They reported that there was no significant difference in the composition of gases. Hydrogen production from the waste car tyre was higher than that from the waste truck tyre. The composition of pyrolysis oil obtained from the two types of tyres was different. For example, the oil produced from the passenger car tyre contained more sulphur and aromatic compounds compared to the oil produced from the truck tyre. Solid char obtained from the truck tyre contained less ash, which is more suitable for upgrading to activated carbon.190
6.2 Effects of reaction conditions on CNT production
In addition to the effect of catalysts and precursors on CNT production, process conditions such as temperature and addition of oxidants also play dominant roles. Zhou et al.259 and Zhao et al.260 concluded that the orientation of graphene sheets of CNTs can be turned to 0–90° relative to the filament axis. The nature of the carbon precursor and synthesis temperature could lead to the re-construction of carbon particles.Hata et al.137 investigated water addition in SWCNT formation by a CVD method. The results showed that water could stimulate the catalyst activity and increase the catalysis lifetime by an etching effect, which oxidized the carbon encapsulated on the catalyst particles. The enhanced aligned SWCNTs with 2.5 mm height were formed within 10 minutes with 99.98% carbon purity by this water-assisted CVD of ethylene, also called “super growth”. However, the oxidant (normally less than 1000 ppm) could also etch CNTs. Magrez et al.261 mixed CO2 with ethylene to grow CNTs at a ratio of 1:1. Zhang et al.262 added ethanol (C2H5OH) to the CNT formation process to grow vertically aligned CNT forests. The modified process had insufficient H2O or CO for the etching effect in this process with ethanol addition. However, there was still a small amount of water formed by the ethanol decomposition. The results showed that the walls of the CNTs could be reduced by ethanol addition and the catalyst lifetime also increased by more than three times. They used online dewpoint and mass spectrometry measurements and found that the decomposition of ethanol forming active carbons could enhance the growth of CNTs, and the water was used to etch the amorphous carbons accumulated on the surface of catalysts, whereas the catalyst activity was subsequently improved and the lifetime was prolonged. Motta et al.263 investigated the effects of sulphur on SWCNT and MWCNT production at high temperatures between 1200 and 1300 °C in the presence of Fe-based catalysts. The results showed that sulphur could enhance the growth of CNTs by the diffusion of sulphur atoms into the first layers of iron atoms to form Fe–S. The surface energy could potentially be modified by the liquid Fe–S layer due to the lower melting point compared to iron. Also, the presence of sulphur prevented the diffusion of carbon inside bulk catalyst particles, promoting the production of SWCNTs instead of MWCNTs which need sub-surface diffusion of carbon atoms.
In 2017, Zhang et al.189,204,264,265 completed a more comprehensive study on MWCNTs produced from waste tyres, and a summary of the results obtained under different process conditions is presented in Fig. 5. The preliminary investigations concerned different metal catalysts (Ni/Al2O3, Co/Al2O3/Fe/Al2O3 and Cu/Al2O3), which were investigated to determine the effect on the production of CNTs and hydrogen by pyrolysis-catalysis of waste truck tyres. The results showed that the addition of catalysts in the pyrolysis-catalysis of waste tyre process can increase hydrogen production. The Ni/Al2O3 catalyst gave the highest hydrogen production at 18.14 mmol gtyre−1 along with the production of relatively high-quality CNTs which were homogeneous.189
 | ||
| Fig. 5 A summary of the findings obtained by Zhang et al.189,204,264 about CNT and hydrogen production from pyrolysis-catalysis of waste tyres. (a) Optimum conditions for CNT and hydrogen production under different process conditions (catalysts, temperatures, sample-to-catalyst ratios and water injection rates); (b)-1 DTG of carbon produced with different catalysts (Ni/Al2O3, Co/Al2O3/Fe/Al2O3 and Cu/Al2O3); (b)-2 and (b)-3 SEM and TEM micrographs of CNTs produced with Ni/Al2O3; (c)-2 and (c)-3 SEM and TEM micrographs of CNTs produced at 900 °C; (d)-2 and (d)-3 SEM and TEM micrographs of CNTs produced with the sample-to-catalyst ratio of 1:0.5; (e)-2 and (e)-3 SEM and TEM micrographs of CNTs without water. | ||
The influence of catalyst support was investigated with different SiO2:Al2O3 ratios (3:5, 1:1, 3:2, 2:1) with nickel. The results showed that the Ni-based SiO2:Al2O3 supported catalyst at a 1:1 ratio at 900 °C with the tyre sample to catalyst ratios at 1:2 gave the highest hydrogen production at 27.41 mmol g−1, and the 1:1 ratio gave the highest filamentous carbon production at 201.5 mg g−1.250 The influence of process parameters on hydrogen and CNT production was investigated with the Ni/Al2O3 catalyst. Hydrogen production reached the highest level of 27.41 mmol g−1 at 900 °C with a sample-to-catalyst ratio of 1:2. The highest filamentous carbon production was obtained with the sample to catalyst ratio of 1:1 at 900 °C. The influence of water injection rate was also investigated that water introduction inhibited filamentous carbon production but increased hydrogen production.204
Three different tyre rubbers were investigated to understand the mechanism of CNT formation from the pyrolysis-catalysis of waste tyres.205,264 The results showed that natural rubber, which is the main component of most tyres, dominated hydrogen production at 25 mmol g−1 and styrene–butadiene rubber gave the highest carbon formation at 40 wt%. The study was further extended to investigate waste plastics and different types of waste plastic feedstocks used in the pyrolysis catalysis/catalytic reforming process to produce hydrogen and CNTs.265 As the separation of CNTs from catalysts is a challenge, a nickel metal catalyst was loaded on a stainless steel mesh and applied in the high-density polyethylene pyrolysis-catalysis process. The benefit of this catalyst has shown that the formation of carbon could be easily separated by physical shaking from the stainless steel–nickel mesh catalyst.265 However, further investigation on waste plastics was concentrated on hydrogen production and where CNTs were the by-products of the process. Fe–Ni bimetallic catalysts supported by MCM-41 with different Fe:Ni ratios were investigated using simulated mixed waste plastics. A synergistic effect of iron and nickel was observed, particularly for the (10:10) Fe/Ni/MCM-41 catalyst, where the highest gas yield (95 wt%) and the highest H2 production (46.1 mmol gplastic−1) have been achieved, along with the lowest carbon deposition, which was 6 wt% with the formation of CNTs.252
6.3 Discussion of process parameters on CNT and hydrogen production from thermo-chemical conversion of carbonaceous waste
A summary of the influence of process parameters on the production of CNT and hydrogen is shown in Fig. 6. Relevant process conditions include the temperature, purging gas, flow rate, heating rate and type of reactors. There are various parameters involved in the catalysis process, such as the type of active metals and supports, active metal contents, and feedstock-to-catalyst ratios. The precursor also plays a dominant role in the formation of CNTs, and the production of hydrogen including four most abundant waste carbonaceous materials (biomass, tyres, plastics and crude glycerol) has been reviewed. | ||
| Fig. 6 Summary of the relevant process parameters that can affect CNT and hydrogen production. | ||
From the literature, reaction parameters such as the temperature and the addition of oxidants such as water and CO2 could improve the yield of CNTs by improving the catalyst activity and prolonging the catalyst life. The most common precursors for CNT synthesis are ethanol, methane, ethylene, acetylene, benzene, xylene and carbon monoxide, while ethanol has become the most popular precursor for SWCNTs synthesised at low temperatures.85,140 The molecular structure of the precursors affects the morphology of CNTs directly. Hydrocarbons with linear structures such as methane, ethylene, acetylene result in a dominance of linear structured CNTs, since the hydrocarbon could decompose into atomic carbons, linear dimers or trimers of carbon and form the straight hollow filamentous carbons. Likewise, cyclic hydrocarbons such as benzene, xylene and cyclohexane would lead to a dominance of curved CNT formation, with bridges inside of the tubes.132 SWCNT formation generally requires a higher temperature than for MWCNTs, which is 600–900 °C and 900–1200 °C, respectively.97
The most popular transition metals used for the production of CNTs and hydrogen from waste polymers are Fe, Ni, and Co due to their high solubility of carbon at high temperatures, high diffusion rates, relatively high melting points and low equilibrium-vapour pressures. It is also known that Fe, Ni and Co have stronger adhesion to CNTs. These transition metal-based catalysts are also suitable for CNTs formulated by arc-discharge and laser-vaporization methods in addition to the CVD method. Solid organometallocenes like ferrocene, cobaltocene and nickelocene are used as catalysts for CNT formation because they could liberate metal particles in situ and effectively improve the catalytic activity.97 It has been reported that the same metal-based catalyst supported on different supports could have different catalytic activities. The common catalyst supports are graphite, quartz, silicon, silicon carbide, silica, alumina, zeolite, calcium oxide, and magnesium oxide, among others. The quality and yield of CNTs are affected by the morphology and textures of supports. Aluminium supports are more suitable for CNT formation compared to silica supports, because a stronger metal interaction would promote a higher metal dispersion.153
7 Applications of CNTs
In recent years, CNTs have been one of the most frequently investigated materials due to their physical, chemical, mechanical and thermal properties, and their potential applications in different industries which have grown rapidly since they were first discovered by Iijima.83 The commercial interest in CNTs is reflected in their production of thousands of tonnes every year. The wide range of applications will be reviewed in this section and summarized into different groups based on their different properties.7.1 Applications based on electrical properties
To date, lithium-ion batteries (LIBs) have dominated the global battery market. They have a higher energy density and a longer lifespan compared to lead-acid, nickel–cadmium, or nickel–metal hydride batteries.266,267 However, some issues such as resource availability, limited energy density, etc. are being exposed. Lithium–sulfur batteries (LSBs) possess a much higher theoretical energy density of 2600 W h kg−1 than that of LIBs.268 However, the high solubility of polysulfide intermediates and the consumption of metallic lithium anode restrain the large-scale production.MWCNTs can be blended with active materials and polymer binders in LIBs for laptops and mobile phones, since they can substantially enhance electrical connectivity and mechanical integrity, consequently increasing the electrochemical performance of the cells due to three reasons.269,270 First, CNTs can act as electron- and ion-transport facilitators to construct a robust and interpenetrating conductive network when integrated with electroactive phases. Therefore, the diffusion of Li+ can be improved to increase the energy density and the specific capacity of LIBs.271 Cao et al.272 synthesized Fe2O3 hollow spheres anchored on CNT composites for the application in LIBs. The Fe2O3/CNT hybrid delivered a reversible specific capacity of 1176 mA h g−1, which is approximately a 40-fold enhancement compared to bare Fe2O3 (31.2 mA h g−1). Second, the structure characteristics of MWCNT also play an important role in electrochemical performance. Li et al.273 found that highly fluorinated MWCNTs with larger diameters of around 50 nm displayed better electrochemical performance in lithium/solid cathode systems because the larger diameter of MWCNTs could effectively avoid the diffusion path of lithium ions from being plugged by the swelled LiF crystals. The high strength and one-dimensional structure of CNTs can buffer the volume change of materials like sulphur, silicon, metal oxides, etc. in the process of battery cycling. The addition of CNTs can also maintain a complete conductive network and improve the cycling performance of these materials.274–278 Zhang et al.276 prepared nickel sulfide anchor on CNT nanocomposites for all-solid-state LIBs. The NiS-CNT nanocomposites exhibited a much higher reversible capacity of 259.6 mA h g−1 at 1 A g−1 than that of bulk-NiS owing to the presence of MWCNTs that increased the specific area of NiS-CNT composites, accommodated the volume changes and prevented the structural distortion. Third, when metallic lithium is used as the negative electrode, the formation of lithium dendrites results in durability limitations and safety issues. The introduction of CNTs with a high specific surface area and a superior electrical conductivity to those of lithium metal anodes can suppress the dendrite growth by means of changing the local current density, controlling the lithium nucleation and accommodating lithium deposition.279 In addition, due to its fascinating features, including lightweight, mechanical durability and chemical stability, CNTs can be easily assembled into freestanding films for use as current collectors. CNT-based current collectors can serve as a porous reservoir to accommodate polysulfides in Li–S batteries.280
Sodium-ion and potassium-ion batteries (SIBs and PIBs) are considered as low-cost alternatives.281 However, their poor transport kinetics and low power density of anode materials hinder the wide application of SIBs and PIBs. Incorporating CNTs into battery electrodes is a feasible strategy for alleviating the problem. Zhang et al.282 employed CNTs with heterostructure bimetallic sulfides to form a special three-dimensional hierarchical structure, which was used as the anode for LIBs, SIBs, and PIBs and PIBs. CNTs as conductive bridges interconnected the mixed Ni–Fe–S units and shortened the diffusion path of lithium/sodium/potassium ions, leading to an enhanced charge capacity of 1535 mA h g−1 after 100 cycles at 0.2 A g−1 for LIBs, 431 mA h g−1 after 100 cycles at 0.1 A g−1 for SIBs, and 181 mA h g−1 at 0.1 A g−1 after 50 cycles for PIBs. Additionally, other types of batteries such as lead-acid batteries (LABs) are still widely employed in vehicles and popular in developing countries. Carbon nanomaterials such as MWCNTs and SWCNTs can be embedded in negative plates as sulfation-suppressing additives for high-performance LABs.283
CNTs can be used as catalyst supports for fuel cell electrodes since they can reduce more than half of Pt usage compared with normal carbon black.284 Further research has also proven that the application of doped CNTs in fuel cells may not need Pt.285,286 Veksha et al.287 processed plastic packaging waste (11.8 wt% polyethylene terephthalate) through catalytic-pyrolysis to produce MWCNTs and prepared them as electrode materials for the hydrogen evolution and oxygen reduction reactions (HER and ORR), respectively. Although the catalytic activity of the obtained MWCNT materials is lower than that of Pt/C electrodes in the HER, MWCNTs exhibited a higher electro-catalytic performance for ORR, where the overpotentials of MWCNT electrodes reached −0.033 V, lower than those of reported graphene-based materials.
CNTs can also be used in supercapacitors and hybrid supercapacitors.288–290 SWCNTs have been studied for packaged cells which show a remarkable performance of using forest-grown SWCNT application. An energy density of 16 W h kg−1 and a power density of 10 kW kg−1 have been achieved for a 40 F supercapacitor, with a maximum voltage of 3.5 V. The lifetime of this supercapacitor has been forecasted to reach 16 years at 105 °C.85 Zhou et al.291 synthesized three-dimensional hierarchical hybrid CNT nanostructures and used them for high-performance supercapacitors. The electrical conductivity of electrode after CNT growth was elevated to 0.1 S cm−1, in comparison to the initial value of 9 × 10−5 S cm−1. The inter-tube structure of the open-tipped CNTs provided fast transfer channels for electrolyte ions, increasing the specific capacitance to 440 F g−1 at 1 A g−1, as well as the enhancement of cycling stability (98.4% retention of initial capacitance after 3000 cycles at 5 A g−1). Natalia et al.292 found that the addition of a small percentage of MWCNTs to a glucose-derived carbon material not only reduced the cell resistance but also promoted the formation of phenol/carbonyl surface functionalities. Correspondingly, a better capacitance of 206 F g−1 and a high rate cyclability (97% after 5000 cycles) were achieved. Wu et al.293 prepared core–shell Bi–Bi2O3/MWCNT composites by directly annealing Bi2O3/MWCNTs due to the high reducibility of CNTs. A three-dimensional structure of CNTs was used as cell bodies to support Bi–Bi2O3 nanospheres. The Bi–Bi2O3/CNT electrode delivered an excellent capacitance of 714 F g−1 at 30 A g−1. When it was used as the negative electrode for an asymmetric supercapacitor, a high energy density of 36.7 W h kg−1 and a maximum power density of 8000 W kg−1 were obtained. Wen294 used carbon black combined with the Ni2O3 catalyst for catalytic carbonization of plastics and obtained CNTs with high yield and quality. Electrodes made from the synthesized CNTs displayed a higher specific capacitance than those of commercial CNTs and carbon black electrodes.
CNT-based electrochemical sensors have also been investigated extensively in recent decades, owing to their remarkable electrical and mechanical performances.295,296 Zorica et al.297 incorporated SWCNTs into carbon paste electrodes (CPEs) for the voltammetric determination of histamine. The SWCNT-CPE showed 50 times higher sensitivity than that observed for the blank CPE to histamine, due to enhanced conductivity and a large specific surface area. Moreover, CNTs have been used as the support of metal and metal oxides. Najari et al.298 utilized gold nanoparticles–MWCNTs to modify a glassy carbon electrode (Au-MWCNTs/GCE) for the determination of an anti-cancer drug. The results showed a sharp decline in the charge-transfer resistance (RCT). As a result, Au-MWCNTs/GCE has good selectivity, sensitivity and reproducibility due to the large surface area and rapid electron-transfer rate.
7.2 Applications based on physical and mechanical properties
000 S m−1.299 MWCNTs have also been applied in the automotive industry. By mixing CNTs into plastics, it can prevent electrostatic charge in the electrostatic-assisted painting of mirror housings, fuel lines and filters. MWCNTs can also be added to products in the microelectronic industry, such as electromagnetic interference shielding packages and wafer carriers.85
Due to the different features of CNTs with different diameters, aspect ratios, alignment, dispersion and interfacial interaction with the matrix, CNTs mixed with polymers or precursor resins can meaningfully improve the stiffness, strength and toughness of composite materials. It is reported in the literature that with a 1 wt% addition of CNTs into epoxy resins, the stiffness was increased by 6% and the fracture toughness was increased by 23%.300,301 The resins mixed with CNTs have recently been used to manufacture lightweight and strong wind turbine blades and boat hulls.
MWCNTs can also be added to plastics as flame-retardants, which can potentially replace environmentally hazardous halogenated flame retardants. The reasons for use of CNTs as flame-retardant additives are due to the changes in rheology.302 CNTs can be added not only to polymers to form composite materials but also to metals to enhance the modulus and tensile strength. These metals with improved features can be applied in the aerospace and automotive industries.303 For example, commercial aluminium and MWCNT mixtures have strengths close to that of stainless steel which are in the range of 0.7 to 1 GPa. However, the density is only 2.6 g cm−3. The mixture of MWCNTs and aluminium provides higher strength with a lower cost than those of the Al–Li alloy.
Indium tin oxide is commonly used in displays, touch-screen devices and photovoltaics. The price of indium tin oxide continues to rise due to a scarcity of indium.304 As an alternative, CNT-based transparent conducting films could take the place of indium tin oxide to form more flexible transparent conductors for displays. Currently, SWCNTs films have been commercially produced. The surface resistivity is suitable for some applications such as CNT thin-film heaters, defrosting windows and sidewalks, but the price is considerably higher than those of the indium tin oxide coatings.303
7.3 Discussion of CNT applications
A comprehensive and most up to date list of CNT applications is summarized in Table 2 and Fig. 7. The applications of the most typical types of CNTs are summarized in Table 2 including advantages of using CNTs in the specific industry and the correlated properties with the applications. This section aims to guide the researchers to further investigate the potential applications of CNTs produced from waste materials. The initial applications of CNTs in super-capacitors, microelectronic industry and lightweight electromagnetic shields appear to have been successful. For example, using CNTs as a conductivity additive for LIB electrodes to boost the electrical connectivity269,270 results in improved specific capacity.269,270 CNTs can be used as multifunctional coating materials. For example, MWCNTs can be added to paints to discourage the attachment of algae and barnacles to the hulls of boats and therefore reduce bio-fouling.89,305 MWCNTs have been used widely in LIBs by blending MWCNTs with active materials and polymer binders.41,305,306 MWCNTs can increase the electrical connectivity and mechanical integrity with the result that the rate capacity and the life cycle of batteries have been enhanced.93,305,307 Considering the low electron scattering and band gap of high-quality SWCNTs, they have been used in making transistors. CNTs could also be used in biosensors and medical devices due to their chemical and dimensional compatibility with biomolecules.305,308| Type of CNT | Application | Applied industries | Advantages |
|---|---|---|---|
| MWCNTs | Electrical/physical | Conductive additive for LIB electrodes | Enhance the electrical connectivity and mechanical integrity;269,270 boost the specific capacity272 |
| MWCNTs | Physical | Lithium/carbon fluoride batteries, lithium-metal anodes, flexible lithium-organic batteries, solid-state lithium batteries, LIB anodes | Avoid the diffusion path of lithium ions from being plugged by the swelled LiF crystals.273 Buffer the volume change of materials (sulphur, silicon, metal oxides, etc.) during cycling, to maintain a complete conductive network and improve the cycling performance.274–278 Boost reversible capacity, accommodate volume changes and prevent structural distortion276 |
| MWCNTs | Electrical/physical | Additive to LIB anodes | Suppress the dendrite growth by means of changing the local current density, controlling the lithium nucleation and accommodating the lithium deposition279 |
| MWCNTs | Electrical/physical/mechanical | Assembled into current collectors for Li–S batteries | CNT-based current collectors can serve as a porous reservoir to accommodate polysulfides in Li–S batteries280 |
| MWCNTs | Electrical/physical/mechanical | Incorporating CNTs into sodium-ion and potassium-ion battery (SIBs and PIBs) anodes | Alleviate poor transport kinetics and low power density of anode materials281 |
| MWCNTs | Electrical | As conductive bridges of LIBs, SIBs, and PIBs | To interconnect the mixed Ni–Fe–S units and shorten the diffusion path of lithium/sodium/potassium ions, leading to enhanced charge capacity282 |
| MWCNTs/SWCNTs | Electrical/physical | Additive to lead-acid batteries (LABs) | Increase the electronic conductivity and the contact among the particles. SWCNTs mitigate pronouncedly the detrimental sulfation phenomena283 |
| MWCNTS | Physical | Catalyst support in fuel cells | Reduce Pt usage compared with normal carbon black.284 The doped CNTs may not need Pt285,286 |
| MWCNTS | Physical | Electrode materials for hydrogen evolution and oxygen reduction reactions (HER and ORR) | MWCNTs exhibit a high electrocatalytic performance for ORR287 |
| SWCNTs | Electrical | Supercapacitors | Enhance the energy density, power density and lifetime85 |
| Hierarchical hybrid nanostructure CNTs | Electrical/physical | Supercapacitors | Increase the electrical conductivity. The inter-tube structure of the open-tipped CNTs provide fast transfer channels for electrolyte ions291 |
| MWCNTs | Electrical | Supercapacitors, hybrid supercapacitors, anodes of asymmetric supercapacitors | Reduce the resistance, enhance the specific capacitance and high rate cyclability.288–290,292,294 Enhance the energy and power densities293 |
| SWCNTs | Electrical/mechanical | CNT-based electrochemical sensors | Remarkable electrical and mechanical performances295,296 |
| SWCNTs | Electrical/physical | Additive for carbon paste electrode (CPE) bulk for the voltammetric determination of histamine | Increased sensitivity than blank CPE297 |
| MWCNTs | Electrical/physical | Gold nanoparticles-MWCNTs to modify the glassy carbon electrode (Au-MWCNTs/GCE) for the determination of anti-cancer drugs | Improve the selectivity, sensitivity and reproducibility298 |
| MWCNTs | Electrical/physical | Electrically conductive fillers in plastics | To form a percolation network and enhance the conductivity of disordered polymers299 |
| MWCNTs | Physical/mechanical | Composition of plastics; microelectronic industry (electromagnetic interference shielding packages and wafer carriers) | To enable electrostatic-assisted painting of mirror housings, fuel lines and filters in order to prevent electrostatic charge85 |
| MWCNTs | Physical/mechanical | Resins mixed with CNTs, multifunctional coating materials (additive of paints and CNT-based transparent conducting films) | To manufacture lightweight and strong wind turbine blades and boat hulls;300,301 reduce biofouling of ship hulls which could possibly replace the conventional environmentally hazardous biocide-containing paints.89 A potential replacement of expensive and scarce indium tin oxide to form more flexible transparent conductors for displays304 |
| MWCNTs | Physical/mechanical | As flame-retardants in plastics | Potential to replace environmentally hazardous halogenated flame retardants302 |
| MWCNTs | Physical/mechanical | Additive to metals | To enhance the modulus and tensile strength303 |
| SWCNTs | Physical/mechanical | Thin film coating | The surface resistivity is suitable for some applications such as CNT thin-film heaters, defrosting windows and sidewalks, but are of considerably higher price than those of the indium tin oxide coatings309 |
 | ||
| Fig. 7 Grouped applications of CNTs according to electrical, physical and mechanical properties. | ||
As shown in Fig. 7, most of the CNT applications are correlated with the physical properties, or electrical and mechanical properties. The applications rely on both the electrical and physical properties, such as their use as a conductive additive for LIBs, LABs and CPEs in the microelectronic industry, the composition of plastics, the anode of supercapacitors, and glassy carbon electrodes. The applications that rely on both physical and mechanical properties include their use as transparent conducting films, additives to metals and paints, flame-retardants and resins. There are a few applications that rely on the combination of electrical, physical and mechanical properties, including SIB and PIB anodes, and Li–S battery composites. There are still many applications that relate to the physical properties only, which mainly focus on battery applications, such as LIB anodes, Li-metal anodes, flexible Li-organic batteries, catalyst supports for fuel cells, Li/C fluoride batteries and all-solid-state batteries. Applications that only rely on electrical properties include supercapacitors (also include hybrid supercapacitors and asymmetric supercapacitors) and LIB anodes. There are limited applications that solely rely on their mechanical and electrical properties, which is in electrochemical sensors.
8 Conclusions, challenges and future prospects
8.1 Conclusions
Thermo-chemical conversion of waste hydrocarbons including waste tyres, plastics, biomass and crude glycerol is more desirable, compared with traditional recycling methods, as it could ease the environmental issues caused by landfilling or incineration. Also, the waste hydrocarbons can be used as an alternative feedstock for CNT production due to the much lower cost of feedstocks. Therefore, this work targets reviewing CNT production from thermo-chemical conversion of carbonaceous waste streams including waste tyres, plastics, biomass and crude glycerol and guides the researchers to use alternative resources to manufacture high-quality CNTs for furthering a broad range of applications.The production and management of waste tyres, plastics, biomass and crude glycerol show the necessity of treatment to deal with these wastes. First, the influence of feedstocks on CNT and hydrogen production by a thermo-chemical conversion process is introduced. Then, the associated catalyst development is deliberated, including the influence of active metals, catalyst supports, synthesis methods, catalysis temperatures and reactors. The process parameters that are involved in the thermo-chemical conversion process and the effects on CNT and hydrogen production are also discussed in detail. Then, the existing applications of the CNTs were discussed according to their electrical, physical and mechanical properties, which could be the future direction of the research, aiming to further apply the CNTs synthesised from wastes in different sectors. Finally, the review ends up with a discussion of challenge and future outlook about thermo-chemical conversion of carbonaceous waste.
8.2 Challenges and future prospects
CNTs produced from waste tyres, plastics, biomass and crude glycerol have been extensively studied for many years. However, there are still challenges to develop a techno-economic feasible process to realize the value of CNTs produced from waste materials. The CNT growth mechanism is complex and it varies with different feedstocks and reactors. Investigations are mainly carried out with lab-scale reactors and lack of research covering the applications of the CNTs produced from waste materials. Therefore, this review discusses the key challenges and future prospects on CNTs produced from waste carbonaceous materials which include the mechanism study, CNT purification and applications, and quality control and larger scale of CNT production.More research studies could be conducted using pyrolysis of oil model compounds to simplify the mechanism studies. There might be differences in the behavior of different model compounds; therefore, more complicated structures of model compounds should be investigated for CNT production derived from waste carbonaceous materials. The investigation on the formation of CNTs from waste materials is worthy of further efforts, which could help to compare the particular CNT formation mechanism under different reaction regimes with the mechanism of CNT formation from the conventional CVD method. Applications of advanced metrology, such as in situ TEM, X-ray diffraction, X-ray photoelectron spectroscopy, Raman spectroscopy or other metrology could help to understand the growth mechanism of CNTs formed by pyrolysis-catalysis of wastes to monitor the carbon formation process on metal catalysts. There is also a need to explore a more advanced metrology to study the CNT growth mechanism.
Author contributions
Conceptualization: Y. S. Zhang, P. T. Williams and D. J. L. Brett; visualization: Y. S. Zhang and L. Yu; writing – original draft preparation: Y. S. Zhang, H. L. Zhu and D. Xu; funding acquisition: D. J. L. Brett and P. R. Shearing; writing – review & editing: all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Faraday Institution (grant number FIRG015). Special thanks are due to Miss Ruike Zhang from Nanyang Junhao Chemical Co. Ltd., P. R. China, for contributing to the graphical designs.References
- D. Czajczyńska, R. Krzyżyńska, H. Jouhara and N. Spencer, Energy, 2017, 134, 1121–1131 CrossRef.
- ETRMA, European Tyre & Rubber Industry Statistics 2019, European Tyre & Rubber Manufactures' Association, 2019 Search PubMed.
- T. A. Saleh and V. K. Gupta, Adv. Colloid Interface Sci., 2014, 211, 93–101 CrossRef CAS PubMed.
- ETRMA, The European Tyre Industry Facts and Figures 2020, European Tyre & Rubber Manufactures Association, 2019 Search PubMed.
- PlasticsEurope, Plastics-The Facts 2019, Plastics Europe, 2019 Search PubMed.
- L. Lebreton and A. Andrady, Palgrave Commun., 2019, 5, 6 CrossRef.
- K. R. Vanapalli, H. B. Sharma, V. P. Ranjan, B. Samal, J. Bhattacharya, B. K. Dubey and S. Goel, Sci. Total Environ., 2020, 750, 141514 CrossRef PubMed.
- M. C. Rillig and A. Lehmann, Science, 2020, 368, 1430–1431 CrossRef CAS PubMed.
- S. Sharma and S. Chatterjee, Environ. Sci. Pollut. Res., 2017, 24, 21530–21547 CrossRef PubMed.
- J. J. Klemeš, Y. V. Fan and P. Jiang, Energy Sources, Part A, 2020, 1–17 Search PubMed.
- B. M. Weckhuysen, Science, 2020, 370, 400–401 CrossRef PubMed.
- Z. Wang, D. Shen, C. Wu and S. Gu, Green Chem., 2018, 20, 5031–5057 RSC.
- WBA, Global Bioenergy Statistics 2019, World Bioenergy Association, 2019 Search PubMed.
- J. Lehmann and S. Joseph, Biochar for Environmental Management: Science and Technology, 2009, vol. 1, pp. 1–12 Search PubMed.
- A. A. Koutinas, A. Vlysidis, D. Pleissner, N. Kopsahelis, I. L. Garcia, I. K. Kookos, S. Papanikolaou, T. H. Kwan and C. S. K. Lin, Chem. Soc. Rev., 2014, 43, 2587–2627 RSC.
- S. Alatzas, K. Moustakas, D. Malamis and S. Vakalis, Energies, 2019, 12, 1095 CrossRef CAS.
- E. J. Cho, L. T. P. Trinh, Y. Song, Y. G. Lee and H.-J. Bae, Bioresour. Technol., 2020, 298, 122386 CrossRef CAS PubMed.
- S. Kaza, L. Yao, P. Bhada-Tata and F. Van Woerden, What a Waste 2.0: a Global Snapshot of Solid Waste Management to 2050, The World Bank, 2018 Search PubMed.
- H. Daniel and B.-T. Perinaz, What a Waste: A Global Review of Solid Waste Management, Urban development series, knowledge papers no. 15, World Bank, Washington, DC, 2012, © World Bank, License: CC BY 3.0 IGO, https://openknowledge.worldbank.org/handle/10986/17388 Search PubMed.
- Eurostat, Waste Statistics, https://ec.europa.eu/eurostat/statistics-explained/index.php/Waste_statistics#Waste_treatment Search PubMed.
- P. Agamuthu, Solid Waste: Principles and Management: with Malaysian Case Studies, University of Malaya Press, Kuala Lumpur, 2001 Search PubMed.
- N. Gupta, K. K. Yadav and V. Kumar, J. Environ. Sci., 2015, 37, 206–217 CrossRef PubMed.
- M. Pagliaro, R. Ciriminna, H. Kimura, M. Rossi and C. Della Pina, Angew. Chem., Int. Ed., 2007, 46, 4434–4440 CrossRef CAS PubMed.
- National Biodiesel Board, Biodiesel Myths, http://www.biodiesel.org/docs/default-source/ffs-basics/biodiesel-myths-vs-facts.pdf?sfvrsn=14, accessed 25/06/2014, 2014 Search PubMed.
- F. Ma and M. A. Hanna, Bioresour. Technol., 1999, 70, 1–15 CrossRef CAS.
- X. Lang, A. K. Dalai, N. N. Bakhshi, M. J. Reaney and P. B. Hertz, Bioresour. Technol., 2001, 80, 53–62 CrossRef CAS PubMed.
- C. Wu, Z. Wang, P. T. Williams and J. Huang, Sci. Rep., 2013, 3, 2742 CrossRef PubMed.
- European Bioenergy Board, Biodiesel Industry calls for ambitious post 2020 transportation renewable targets, 2014 Search PubMed.
- K. S. Avasthi, R. N. Reddy and S. Patel, Procedia Eng., 2013, 51, 423–429 CrossRef CAS.
- S. Adhikari, S. D. Fernando and A. Haryanto, Energy Convers. Manage., 2009, 50, 2600–2604 CrossRef CAS.
- O. Babajide, J. Energy, 2013, 7 Search PubMed.
- S. Nanda and F. Berruti, Environ. Chem. Lett., 2020, 1–26 Search PubMed.
- P. T. Williams, Waste Biomass Valorization, 2021, 12(1), 1–28 CrossRef CAS.
- M. Solis and S. Silveira, Waste Manage., 2020, 105, 128–138 CrossRef CAS PubMed.
- A. Ochoa, J. Bilbao, A. G. Gayubo and P. Castaño, Renewable Sustainable Energy Rev., 2020, 119, 109600 CrossRef CAS.
- M. Arabiourrutia, G. Lopez, M. Artetxe, J. Alvarez, J. Bilbao and M. Olazar, Renewable Sustainable Energy Rev., 2020, 129, 109932 CrossRef CAS.
- M. Checa, S. Nogales-Delgado, V. Montes and J. M. Encinar, Catalysts, 2020, 10, 1279 CrossRef CAS.
- C. Wu, Z. Wang, P. T. Williams and J. Huang, Sci. Rep., 2013, 3, 2742 CrossRef PubMed.
- W. M. Lewandowski, M. Ryms and W. Kosakowski, Processes, 2020, 8, 516 CrossRef CAS.
- Y. Zhang, C. Wu, M. A. Nahil and P. Williams, Proc. Inst. Civ. Eng.: Waste Resour. Manage., 2016, 169, 137–145 Search PubMed.
- P. T. Williams, Waste Manage., 2013, 33, 1714–1728 CrossRef CAS PubMed.
- P. Williams, A. Cunliffe and A. Brindle, J. Inst. Energy, 2001, 74, 100–112 CAS.
- M. Sienkiewicz, J. Kucinska-Lipka, H. Janik and A. Balas, Waste Manage., 2012, 32, 1742–1751 CrossRef CAS PubMed.
- M. Suzuki, Carbon, 1994, 32, 577–586 CrossRef CAS.
- P. T. Williams and A. J. Brindle, J. Anal. Appl. Pyrolysis, 2003, 67, 143–164 CrossRef CAS.
- X. Dai, X. Yin, C. Wu, W. Zhang and Y. Chen, Energy, 2001, 26, 385–399 CrossRef CAS.
- J. A. Conesa, R. Font and A. Marcilla, Energy Fuels, 1996, 10, 134–140 CrossRef CAS.
- E. Aylón, A. Fernández-Colino, R. Murillo, M. Navarro, T. García and A. Mastral, Waste Manage., 2010, 30, 1220–1224 CrossRef PubMed.
- C. J. Roos, Clean Heat and Power Using Biomass Gasification for Industrial and Agricultural Projects, Northwest CHP Application Center, 2010 Search PubMed.
- I. F. Elbaba, C. Wu and P. T. Williams, Int. J. Hydrogen Energy, 2011, 36, 6628–6637 CrossRef CAS.
- D. Y. C. Leung and C. L. Wang, Fuel Process. Technol., 2003, 84, 175–196 CrossRef CAS.
- G. Xiao, M.-J. Ni, Y. Chi and K.-F. Cen, Energy Convers. Manage., 2008, 49, 2078–2082 CrossRef CAS.
- S. Galvagno, G. Casciaro, S. Casu, M. Martino, C. Mingazzini, A. Russo and S. Portofino, Waste Manage., 2009, 29, 678–689 CrossRef CAS PubMed.
- R. M. Dell, P. T. Moseley and D. A. J. Rand, in Towards Sustainable Road Transport, ed. R. M. Dell, T. Moseley and A. J. Rand, Academic Press, Boston, 2014, pp. 260–295, DOI:10.1016/B978-0-12-404616-0.00008-6.
- A. Zadgaonkar, Feedstock Recycling and Pyrolysis of Waste Plastics: Converting Waste Plastics into Diesel and Other Fuels, 2006, pp. 709–728 Search PubMed.
- I. de Marco Rodriguez, M. Laresgoiti, M. Cabrero, A. Torres, M. Chomon and B. Caballero, Fuel Process. Technol., 2001, 72, 9–22 CrossRef.
- A. Donatelli, P. Iovane and A. Molino, Fuel, 2010, 89, 2721–2728 CrossRef CAS.
- M. S. Qureshi, A. Oasmaa, H. Pihkola, I. Deviatkin, A. Tenhunen, J. Mannila, H. Minkkinen, M. Pohjakallio and J. Laine-Ylijoki, J. Anal. Appl. Pyrolysis, 2020, 104804 CrossRef CAS.
- C. Wu and P. T. Williams, Appl. Catal., B, 2009, 87, 152–161 CrossRef CAS.
- S. Czernik and R. J. French, Energy Fuels, 2006, 20, 754–758 CrossRef CAS.
- P. T. Williams, Waste Treatment and Disposal, 2nd edn, 2005, pp. 245–323 Search PubMed.
- C. Wu and P. T. Williams, Appl. Catal., B, 2009, 90, 147–156 CrossRef CAS.
- Y. Chai, N. Gao, M. Wang and C. Wu, Chem. Eng. J., 2020, 382, 122947 CrossRef CAS.
- C. Wu and P. T. Williams, Fuel, 2010, 89, 3022–3032 CrossRef CAS.
- S. L. Lakhapatri and M. A. Abraham, Appl. Catal., A, 2011, 405, 149–159 CrossRef CAS.
- G. Sierra Gallego, J. Barrault, C. Batiot-Dupeyrat and F. Mondragón, Catal. Today, 2010, 149, 365–371 CrossRef CAS.
- M. He, B. Xiao, Z. Hu, S. Liu, X. Guo and S. Luo, Int. J. Hydrogen Energy, 2009, 34, 1342–1348 CrossRef CAS.
- J. Srinakruang, K. Sato, T. Vitidsant and K. Fujimoto, Catal. Commun., 2005, 6, 437–440 CrossRef CAS.
- T. Minowa, F. Zhen and T. Ogi, J. Supercrit. Fluids, 1998, 13, 253–259 CrossRef CAS.
- F. H. Isikgor and C. R. Becer, Polym. Chem., 2015, 6, 4497–4559 RSC.
- Y. Lin, H. Wang, Y. Wang, R. Huo, Z. Huang, M. Liu, G. Wei, Z. Zhao, H. Li and Y. Fang, Energy Fuels, 2020, 34, 7847–7862 CrossRef CAS.
- Y. Zhang, C. Ke, W. Fu, Y. Cui, M. A. Rehan and B. Li, Renewable Energy, 2020, 154, 488–496 CrossRef CAS.
- D. Yao, Q. Hu, D. Wang, H. Yang, C. Wu, X. Wang and H. Chen, Bioresour. Technol., 2016, 216, 159–164 CrossRef CAS PubMed.
- T. Valliyappan, N. N. Bakhshi and A. K. Dalai, Bioresour. Technol., 2008, 99, 4476–4483 CrossRef CAS PubMed.
- J. Gao, Y. Wang, Y. Ping, D. Hu, G. Xu, F. Gu and F. Su, RSC Adv., 2012, 2, 2358–2368 RSC.
- I. Ahmed and A. Gupta, Appl. Energy, 2009, 86, 1813–1821 CrossRef CAS.
- P. T. Williams, Waste Treatment and Disposal, John Wiley & Sons, 2005 Search PubMed.
- A. Demirbas, J. Anal. Appl. Pyrolysis, 2004, 72, 243–248 CrossRef CAS.
- Y. Kodera, Y. Ishihara and T. Kuroki, Energy Fuels, 2006, 20, 155–158 CrossRef CAS.
- A. Esfandiari, T. Kaghazchi and M. Soleimani, J. Taiwan Inst. Chem. Eng., 2012, 43, 631–637 CrossRef CAS.
- B. C. R. Ewan and R. W. K. Allen, Int. J. Hydrogen Energy, 2005, 30, 809–819 CrossRef CAS.
- S. Authayanun, A. Arpornwichanop, W. Paengjuntuek and S. Assabumrungrat, Int. J. Hydrogen Energy, 2010, 35, 6617–6623 CrossRef CAS.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- M. Monthioux and V. L. Kuznetsov, Carbon, 2006, 44, 1621–1623 CrossRef CAS.
- M. F. De Volder, S. H. Tawfick, R. H. Baughman and A. J. Hart, Science, 2013, 339, 535–539 CrossRef CAS PubMed.
- B. Peng, M. Locascio, P. Zapol, S. Li, S. L. Mielke, G. C. Schatz and H. D. Espinosa, Nat. Nanotechnol., 2008, 3, 626–631 CrossRef CAS PubMed.
- B. Wei, R. Vajtai and P. Ajayan, Appl. Phys. Lett., 2001, 79, 1172–1174 CrossRef CAS.
- E. Pop, D. Mann, Q. Wang, K. Goodson and H. Dai, Nano Lett., 2006, 6, 96–100 CrossRef CAS PubMed.
- A. Beigbeder, P. Degee, S. L. Conlan, R. J. Mutton, A. S. Clare, M. E. Pettitt, M. E. Callow, J. A. Callow and P. Dubois, Biofouling, 2008, 24, 291–302 CrossRef PubMed.
- L. Dai, D. W. Chang, J.-B. Baek and W. Lu, Small, 2012, 8, 1130–1166 CrossRef CAS PubMed.
- A. R. Köhler, C. Som, A. Helland and F. Gottschalk, J. Cleaner Prod., 2008, 16, 927–937 CrossRef.
- K. Evanoff, J. Khan, A. A. Balandin, A. Magasinski, W. J. Ready, T. F. Fuller and G. Yushin, Adv. Mater., 2012, 24, 533–537 CrossRef CAS PubMed.
- C. Sotowa, G. Origi, M. Takeuchi, Y. Nishimura, K. Takeuchi, I. Y. Jang, Y. J. Kim, T. Hayashi, Y. A. Kim, M. Endo and M. S. Dresselhaus, ChemSusChem, 2008, 1, 911–915 CrossRef CAS PubMed.
- D. A. Heller, S. Baik, T. E. Eurell and M. S. Strano, Adv. Mater., 2005, 17(23), 2793–2799 CrossRef CAS.
- M. Endo, T. Hayashi and Y.-A. Kim, Pure Appl. Chem., 2006, 78, 1703–1713 CAS.
- Q. Zhang, J. Q. Huang, M. Q. Zhao, W. Z. Qian and F. Wei, ChemSusChem, 2011, 4, 864–889 CrossRef CAS PubMed.
- M. Kumar and Y. Ando, J. Nanosci. Nanotechnol., 2010, 10, 3739–3758 CrossRef CAS PubMed.
- A. Bazargan and G. McKay, Chem. Eng. J., 2012, 195, 377–391 CrossRef.
- W.-W. Liu, S.-P. Chai, A. R. Mohamed and U. Hashim, J. Ind. Eng. Chem., 2014, 20, 1171–1185 CrossRef CAS.
- P. N. H. D. P. Petit, Science, 1996, 273, 483–487 CrossRef PubMed.
- K. Dasgupta, J. B. Joshi and S. Banerjee, Chem. Eng. J., 2011, 171, 841–869 CrossRef CAS.
- E. Boellaard, P. K. de Bokx, A. J. H. M. Kock and J. W. Geus, J. Catal., 1985, 96, 481–490 CrossRef CAS.
- N. M. Rodriguez, M. S. Kim and R. T. K. Baker, J. Catal., 1993, 144, 93–108 CrossRef CAS.
- H. Boehm, Carbon, 1973, 11, 583IN1587–1586IN5590 CrossRef.
- N. Behabtu, C. C. Young, D. E. Tsentalovich, O. Kleinerman, X. Wang, A. W. Ma, E. A. Bengio, R. F. ter Waarbeek, J. J. de Jong and R. E. Hoogerwerf, Science, 2013, 339, 182–186 CrossRef CAS PubMed.
- E. L. K. Mui, V. K. C. Lee, W. H. Cheung and G. McKay, Energy Fuels, 2008, 22, 1650–1657 CrossRef CAS.
- V. B. Mortola, S. Damyanova, D. Zanchet and J. M. C. Bueno, Appl. Catal., B, 2011, 107, 221–236 CrossRef CAS.
- J. Nishikawa, K. Nakamura, M. Asadullah, T. Miyazawa, K. Kunimori and K. Tomishige, Catal. Today, 2008, 131, 146–155 CrossRef CAS.
- K. Hernadi, A. Fonseca, J. Nagy, D. Bernaerts and A. Lucas, Carbon, 1996, 34, 1249–1257 CrossRef CAS.
- J. Kong, A. M. Cassell and H. Dai, Chem. Phys. Lett., 1998, 292, 567–574 CrossRef CAS.
- S. Fan, M. G. Chapline, N. R. Franklin, T. W. Tombler, A. M. Cassell and H. Dai, Science, 1999, 283, 512–514 CrossRef CAS PubMed.
- B. C. Satishkumar, A. Govindaraj and C. N. R. Rao, Chem. Phys. Lett., 1999, 307, 158–162 CrossRef CAS.
- W. Z. Li, S. S. Xie, L. X. Qian, B. H. Chang, B. S. Zou, W. Y. Zhou, R. A. Zhao and G. Wang, Science, 1996, 274, 1701–1703 CrossRef CAS PubMed.
- R. Sen, A. Govindaraj and C. N. R. Rao, Chem. Phys. Lett., 1997, 267, 276–280 CrossRef CAS.
- B. Q. Wei, R. Vajtai, Y. Jung, J. Ward, R. Zhang, G. Ramanath and P. M. Ajayan, Nature, 2002, 416, 495–496 CrossRef CAS PubMed.
- P. Nikolaev, M. J. Bronikowski, R. K. Bradley, F. Rohmund, D. T. Colbert, K. A. Smith and R. E. Smalley, Chem. Phys. Lett., 1999, 313, 91–97 CrossRef CAS.
- A. Weidenkaff, S. G. Ebbinghaus, P. Mauron, A. Reller, Y. Zhang and A. Züttel, Mater. Sci. Eng., C, 2002, 19, 119–123 CrossRef.
- D. Venegoni, P. Serp, R. Feurer, Y. Kihn, C. Vahlas and P. Kalck, Carbon, 2002, 40, 1799–1807 CrossRef CAS.
- A. Morançais, B. Caussat, Y. Kihn, P. Kalck, D. Plee, P. Gaillard, D. Bernard and P. Serp, Carbon, 2007, 45, 624–635 CrossRef.
- R. Philippe, B. Caussat, A. Falqui, Y. Kihn, P. Kalck, S. Bordère, D. Plee, P. Gaillard, D. Bernard and P. Serp, J. Catal., 2009, 263, 345–358 CrossRef CAS.
- Q. Li, X. Zhang, R. F. DePaula, L. Zheng, Y. Zhao, L. Stan, T. G. Holesinger, P. N. Arendt, D. E. Peterson and Y. T. Zhu, Adv. Mater., 2006, 18, 3160–3163 CrossRef CAS.
- C. H. See and A. T. Harris, Aust. J. Chem., 2007, 60, 541–546 CrossRef CAS.
- C. H. See, O. M. Dunens, K. J. MacKenzie and A. T. Harris, Ind. Eng. Chem. Res., 2008, 47, 7686–7692 CrossRef CAS.
- W. Qian, T. Liu, F. Wei, Z. Wang and Y. Li, Appl. Catal., A, 2004, 258, 121–124 CrossRef CAS.
- Y. Wei, G. Eres, V. Merkulov and D. Lowndes, Appl. Phys. Lett., 2001, 78, 1394–1396 CrossRef CAS.
- W. Fang, C. Pirez, M. Capron, S. Paul, T. Raja, P. L. Dhepe, F. Dumeignil and L. Jalowiecki-Duhamel, RSC Adv., 2012, 2, 9626–9634 RSC.
- W. Li, J. Wen and Z. Ren, Appl. Phys. A: Mater. Sci. Process., 2002, 74, 397–402 CrossRef CAS.
- A. Moisala, A. G. Nasibulin and E. I. Kauppinen, J. Phys.: Condens. Matter, 2003, 15, S3011 CrossRef CAS.
- J. Kong, A. M. Cassell and H. Dai, Chem. Phys. Lett., 1998, 292, 567–574 CrossRef CAS.
- Z.-J. Liu, R. Che, Z. Xu and L.-M. Peng, Synth. Met., 2002, 128, 191–195 CrossRef CAS.
- N. Li, X. Chen, L. Stoica, W. Xia, J. Qian, J. Aßmann, W. Schuhmann and M. Muhler, Adv. Mater., 2007, 19, 2957–2960 CrossRef CAS.
- O. A. Nerushev, S. Dittmar, R.-E. Morjan, F. Rohmund and E. E. B. Campbell, J. Appl. Phys., 2003, 93, 4185–4190 CrossRef CAS.
- R. E. Morjan, O. A. Nerushev, M. Sveningsson, F. Rohmund, L. K. L. Falk and E. E. B. Campbell, Appl. Phys. A: Mater. Sci. Process., 2004, 78, 253–261 CrossRef CAS.
- R. T. K. Baker, M. A. Barber, P. S. Harris, F. S. Feates and R. J. Waite, J. Catal., 1972, 26, 51–62 CrossRef CAS.
- R. Andrews, D. Jacques, A. M. Rao, F. Derbyshire, D. Qian, X. Fan, E. C. Dickey and J. Chen, Chem. Phys. Lett., 1999, 303, 467–474 CrossRef CAS.
- M. Kumar and Y. Ando, Chem. Phys. Lett., 2003, 374, 521–526 CrossRef CAS.
- K. Hata, D. N. Futaba, K. Mizuno, T. Namai, M. Yumura and S. Iijima, Science, 2004, 306, 1362–1364 CrossRef CAS PubMed.
- M. Kumar, K. Kakamu, T. Okazaki and Y. Ando, Chem. Phys. Lett., 2004, 385, 161–165 CrossRef CAS.
- D. Ding, J. Wang, Z. Cao and J. Dai, Carbon, 2003, 41, 579–582 CrossRef CAS.
- T. Murakami, T. Sako, H. Harima, K. Kisoda, K. Mitikami and T. Isshiki, Thin Solid Films, 2004, 464–465, 319–322 CrossRef CAS.
- B. Kitiyanan, W. E. Alvarez, J. H. Harwell and D. E. Resasco, Chem. Phys. Lett., 2000, 317, 497–503 CrossRef CAS.
- C. Mattevi, C. T. Wirth, S. Hofmann, R. Blume, M. Cantoro, C. Ducati, C. Cepek, A. Knop-Gericke, S. Milne, C. Castellarin-Cudia, S. Dolafi, A. Goldoni, R. Schloegl and J. Robertson, J. Phys. Chem. C, 2008, 112, 12207–12213 CrossRef CAS.
- C. L. Cheung, A. Kurtz, H. Park and C. M. Lieber, J. Phys. Chem. B, 2002, 106, 2429–2433 CrossRef CAS.
- H. Hongo, M. Yudasaka, T. Ichihashi, F. Nihey and S. Iijima, Chem. Phys. Lett., 2002, 361, 349–354 CrossRef CAS.
- C. Mirodatos, H. Praliaud and M. Primet, J. Catal., 1987, 107, 275–287 CrossRef CAS.
- Z. Zhang and X. E. Verykios, Appl. Catal., A, 1996, 138, 109–133 CrossRef CAS.
- I. Willems, Z. Kónya, J. F. Colomer, G. Van Tendeloo, N. Nagaraju, A. Fonseca and J. B. Nagy, Chem. Phys. Lett., 2000, 317, 71–76 CrossRef CAS.
- M. Kumar and Y. Ando, Carbon, 2005, 43, 533–540 CrossRef CAS.
- E. Couteau, K. Hernadi, J. W. Seo, L. Thiên-Nga, C. Mikó, R. Gaál and L. Forró, Chem. Phys. Lett., 2003, 378, 9–17 CrossRef CAS.
- J. F. Colomer, C. Stephan, S. Lefrant, G. Van Tendeloo, I. Willems, Z. Kónya, A. Fonseca, C. Laurent and J. B. Nagy, Chem. Phys. Lett., 2000, 317, 83–89 CrossRef CAS.
- J. W. Ward, B. Q. Wei and P. M. Ajayan, Chem. Phys. Lett., 2003, 376, 717–725 CrossRef CAS.
- H. Ago, K. Nakamura, S. Imamura and M. Tsuji, Chem. Phys. Lett., 2004, 391, 308–313 CrossRef CAS.
- N. Nagaraju, A. Fonseca, Z. Konya and J. B. Nagy, J. Mol. Catal. A: Chem., 2002, 181, 57–62 CrossRef CAS.
- G. Busca, U. Costantino, T. Montanari, G. Ramis, C. Resini and M. Sisani, Int. J. Hydrogen Energy, 2010, 35, 5356–5366 CrossRef CAS.
- J. O. Alves, Y. A. Levendis and J. A. S. Tenório, Carbon, 2012, 85, 82 Search PubMed.
- N. Sano, Y. Hori, S. Yamamoto and H. Tamon, Carbon, 2012, 50, 115–122 CrossRef CAS.
- J. Amadou, D. Begin, P. Nguyen, J. P. Tessonnier, T. Dintzer, E. Vanhaecke, M. J. Ledoux and C. Pham-Huu, Carbon, 2006, 44, 2587–2589 CrossRef CAS.
- R. L. Vander Wal and L. J. Hall, Carbon, 2003, 41, 659–672 CrossRef CAS.
- T. Ebbesen, P. Ajayan and K. Tanigaki, Nature, 1994, 367, 519 CrossRef.
- Y.-Q. Xu, H. Peng, R. H. Hauge and R. E. Smalley, Nano Lett., 2005, 5, 163–168 CrossRef CAS PubMed.
- J.-M. Moon, K. H. An, Y. H. Lee, Y. S. Park, D. J. Bae and G.-S. Park, J. Phys. Chem. B, 2001, 105, 5677–5681 CrossRef CAS.
- M. Kumar and Y. Ando, in Nanotechnology and Nanomaterials; Carbon Nanotubes-Synthesis, Characterisation, Application, ed. S. Yellampalli, INTECH Open Access Publisher, 2011, pp. 148–170 Search PubMed.
- L. Ma, X. Dong, M. Chen, L. Zhu, C. Wang, F. Yang and Y. Dong, Membranes, 2017, 7, 16 CrossRef PubMed.
- J. P. Tessonnier and D. S. Su, ChemSusChem, 2011, 4, 824–847 CrossRef CAS PubMed.
- A. Rinaldi, J. P. Tessonnier, M. E. Schuster, R. Blume, F. Girgsdies, Q. Zhang, T. Jacob, S. B. Abd Hamid, D. S. Su and R. Schlögl, Angew. Chem., Int. Ed., 2011, 50, 3313–3317 CrossRef CAS PubMed.
- R. Baker, P. Harris, R. Thomas and R. Waite, J. Catal., 1973, 30, 86–95 CrossRef CAS.
- S. Shukrullah, N. M. Mohamed, M. S. Shaharun and M. Y. Naz, Mater. Chem. Phys., 2016, 176, 32–43 CrossRef CAS.
- N. Yang, S. K. Youn, C. E. Frouzakis and H. G. Park, Carbon, 2018, 130, 607–613 CrossRef CAS.
- R. Li, E. F. Antunes, E. Kalfon-Cohen, A. Kudo, L. Acauan, W.-C. D. Yang, C. Wang, K. Cui, A. H. Liotta, A. G. Rajan, J. Gardener, D. C. Bell, M. S. Strano, J. A. Liddle, R. Sharma and B. L. Wardle, Angew. Chem., Int. Ed., 2019, 58, 9204–9209 CrossRef CAS PubMed.
- A. Ashok, A. Kumar, J. Ponraj and S. A. Mansour, Carbon, 2020, 170, 452–463 CrossRef CAS.
- C. Wu and P. T. Williams, Appl. Catal., B, 2010, 96, 198–207 CrossRef CAS.
- A. Ebshish, Z. Yaakob, Y. H. Taufiq-Yap, A. Bshish and S. M. Tasirin, Proc. Inst. Mech. Eng., Part A, 2012, 226, 1060–1075 CrossRef CAS.
- N. Das, A. Dalai, J. S. Soltan Mohammadzadeh and J. Adjaye, Carbon, 2006, 44, 2236–2245 CrossRef CAS.
- H. Ago, N. Uehara, N. Yoshihara, M. Tsuji, M. Yumura, N. Tomonaga and T. Setoguchi, Carbon, 2006, 44, 2912–2918 CrossRef CAS.
- I. F. Elbaba and P. T. Williams, Appl. Catal., B, 2012, 125, 136–143 CrossRef CAS.
- I. F. Elbaba and P. T. Williams, Fuel, 2013, 106, 528–536 CrossRef CAS.
- I. F. Elbaba and P. T. Williams, Energy Fuels, 2014, 28, 2104–2113 CrossRef CAS.
- I. F. Elbaba, C. Wu and P. T. Williams, Energy Fuels, 2010, 24, 3928–3935 CrossRef CAS.
- W. Yang, W. J. Sun, W. Chu, C. F. Jiang and J. Wen, Chin. Chem. Lett., 2012, 23, 363–366 CrossRef CAS.
- L. E. Murr, D. K. Brown, E. V. Esquivel, T. D. Ponda, F. Martinez and A. Virgen, Mater. Charact., 2005, 55, 371–377 CrossRef CAS.
- S. Poyraz, Z. Liu, Y. Liu and X. Zhang, Curr. Org. Chem., 2013, 17, 2243–2248 CrossRef CAS.
- C. Wu, Z. Wang, L. Wang, P. T. Williams and J. Huang, RSC Adv., 2012, 2, 4045–4047 RSC.
- E. F. Kukovitskii, L. A. Chernozatonskii, S. G. L'Vov and N. N. Mel'nik, Chem. Phys. Lett., 1997, 266, 323–328 CrossRef CAS.
- J. Liu, Z. Jiang, H. Yu and T. Tang, Polym. Degrad. Stab., 2011, 96, 1711–1719 CrossRef CAS.
- X. Jie, W. Li, D. Slocombe, Y. Gao, I. Banerjee, S. Gonzalez-Cortes, B. Yao, H. AlMegren, S. Alshihri and J. Dilworth, Nat. Catal., 2020, 1–11 Search PubMed.
- L. He, S. Hu, L. Jiang, G. Liao, L. Zhang, H. Han, X. Chen, Y. Wang, K. Xu, S. Su and J. Xiang, Fuel, 2017, 210, 307–314 CrossRef CAS.
- S. Zhang, S.-F. Jiang, B.-C. Huang, X.-C. Shen, W.-J. Chen, T.-P. Zhou, H.-Y. Cheng, B.-H. Cheng, C.-Z. Wu, W.-W. Li and H. Jiang, Nat. Sustain., 2020, 3(9), 753–760 CrossRef.
- A. Romero, M. Jobbágy, M. Laborde, G. Baronetti and N. Amadeo, Appl. Catal., A, 2014, 470, 398–404 CrossRef CAS.
- Y. Zhang, C. Wu, M. A. Nahil and P. Williams, Energy Fuels, 2015, 29, 3328–3334 CrossRef CAS.
- S. Ucar, S. Karagoz, A. R. Ozkan and J. Yanik, Fuel, 2005, 84, 1884–1892 CrossRef CAS.
- E. Kwon and M. J. Castaldi, Environ. Sci. Technol., 2008, 42, 2175–2180 CrossRef CAS PubMed.
- R. R. Soares, D. A. Simonetti and J. A. Dumesic, Angew. Chem., Int. Ed., 2006, 45, 3982–3985 CrossRef CAS PubMed.
- H. Brown and Y. Shah, in Clean Production, ed. K. B. Misra, Springer Berlin Heidelberg, 1996, ch. 28, pp. 647–683, DOI:10.1007/978-3-642-79940-2_28.
- S. Claude, Lipid/Fett, 1999, 101(3), 101–104 CrossRef CAS.
- T. Valliyappan, N. N. Bakhshi and A. K. Dalai, Bioresour. Technol., 2008, 99, 4476–4483 CrossRef CAS PubMed.
- D. Simonetti, E. Kunkes and J. Dumesic, J. Catal., 2007, 247, 298–306 CrossRef CAS.
- A. P. G. Peres, N. D. L. Da Silva and M. R. W. Maciel, Int. J. Energy Convers., 2013, 1(1), 2013 Search PubMed.
- B. Dou, V. Dupont, G. Rickett, N. Blakeman, P. T. Williams, H. Chen, Y. Ding and M. Ghadiri, Bioresour. Technol., 2009, 100, 3540–3547 CrossRef CAS PubMed.
- V. Chiodo, S. Freni, A. Galvagno, N. Mondello and F. Frusteri, Appl. Catal., A, 2010, 381, 1–7 CrossRef CAS.
- N. D. Charisiou, K. N. Papageridis, G. Siakavelas, V. Sebastian, S. J. Hinder, M. A. Baker, K. Polychronopoulou and M. A. Goula, Catal. Today, 2019, 319, 206–219 CrossRef CAS.
- Z. Abu El-Rub, E. A. Bramer and G. Brem, Ind. Eng. Chem. Res., 2004, 43, 6911–6919 CrossRef CAS.
- J. C. Acomb, C. Wu and P. T. Williams, Appl. Catal., B, 2016, 180, 497–510 CrossRef CAS.
- Y. Zhang, J. Huang and P. T. Williams, Energy Fuels, 2017, 31(8), 8497–8504 CrossRef CAS.
- Y. Zhang and P. T. Williams, J. Anal. Appl. Pyrolysis, 2016, 122, 490–501 CrossRef CAS.
- Y. Zhang, C. Wu, M. A. Nahil and P. Williams, Proc. Inst. Civ. Eng.: Waste Resour. Manage., 2016, 169, 137–145 Search PubMed.
- Y. Zhang, Y. Tao, J. Huang and P. Williams, Waste Manage. Res., 2017, 35(10), 1045–1054 CrossRef CAS PubMed.
- A. C. Freitas and R. Guirardello, Int. J. Hydrogen Energy, 2014, 39(31), 17969–17984 CrossRef CAS.
- S. Adhikari, S. Fernando and A. Haryanto, Catal. Today, 2007, 129, 355–364 CrossRef CAS.
- S. Czernik, R. French, C. Feik and E. Chornet, Ind. Eng. Chem. Res., 2002, 41, 4209–4215 CrossRef CAS.
- S. Adhikari, S. D. Fernando and A. Haryanto, Chem. Eng. Technol., 2009, 32, 541–547 CrossRef CAS.
- S. Adhikari, S. D. Fernando, S. F. To, R. M. Bricka, P. H. Steele and A. Haryanto, Energy Fuels, 2008, 22(2), 1220–1226 CrossRef CAS.
- D. Sutton, B. Kelleher and J. R. H. Ross, Fuel Process. Technol., 2001, 73, 155–173 CrossRef CAS.
- W. B. Hauserman, Int. J. Hydrogen Energy, 1994, 19, 413–419 CrossRef CAS.
- I. N. Buffoni, F. Pompeo, G. F. Santori and N. N. Nichio, Catal. Commun., 2009, 10, 1656–1660 CrossRef CAS.
- B. Zhang, X. Tang, Y. Li, Y. Xu and W. Shen, Int. J. Hydrogen Energy, 2007, 32, 2367–2373 CrossRef CAS.
- A. Iriondo, V. L. Barrio, J. F. Cambra, P. L. Arias, M. B. Güemez, R. M. Navarro, M. C. Sánchez-Sánchez and J. L. G. Fierro, Top. Catal., 2008, 49, 46–58 CrossRef CAS.
- A. Iriondo, V. L. Barrio, J. F. Cambra, P. L. Arias, M. B. Güemez, R. M. Navarro, M. C. Sanchez-Sanchez and J. L. G. Fierro, Catal. Commun., 2009, 10, 1275–1278 CrossRef CAS.
- Y. Hao, Z. Qunfeng, W. Fei, Q. Weizhong and L. Guohua, Carbon, 2003, 41, 2855–2863 CrossRef.
- A. A. Aboul-Enein and A. E. Awadallah, Chem. Eng. J., 2018, 354, 802–816 CrossRef CAS.
- D. Yao, C. Wu, H. Yang, Y. Zhang, M. A. Nahil, Y. Chen, P. T. Williams and H. Chen, Energy Convers. Manage., 2017, 148, 692–700 CrossRef CAS.
- L. He, S. Hu, X. Yin, J. Xu, H. Han, H. Li, Q. Ren, S. Su, Y. Wang and J. Xiang, Fuel, 2020, 276, 118116 CrossRef CAS.
- D. Yao, H. Li, Y. Dai and C.-H. Wang, Chem. Eng. J., 2020, 127268, DOI:10.1016/j.cej.2020.127268.
- N. Cai, H. Yang, X. Zhang, S. Xia, D. Yao, P. Bartocci, F. Fantozzi, Y. Chen, H. Chen and P. T. Williams, Waste Manage., 2020, 109, 119–126 CrossRef CAS PubMed.
- D. Yao, Y. Zhang, P. T. Williams, H. Yang and H. Chen, Appl. Catal., B, 2018, 221, 584–597 CrossRef CAS.
- A. Iriondo, V. L. Barrio, J. F. Cambra, P. L. Arias, M. B. Guemez, M. C. Sanchez-Sanchez, R. M. Navarro and J. L. G. Fierro, Int. J. Hydrogen Energy, 2010, 35, 11622–11633 CrossRef CAS.
- M. Slinn, K. Kendall, C. Mallon and J. Andrews, Bioresour. Technol., 2008, 99, 5851–5858 CrossRef CAS PubMed.
- T. Hirai, N.-o. Ikenaga, T. Miyake and T. Suzuki, Energy Fuels, 2005, 19, 1761–1762 CrossRef CAS.
- R. Kikuchi, H. Sato, Y. Matsukura and T. Yamamoto, Fuel Process. Technol., 2005, 86, 1279–1296 CrossRef CAS.
- L. P. R. Profeti, E. A. Ticianelli and E. M. Assaf, Int. J. Hydrogen Energy, 2009, 34, 5049–5060 CrossRef CAS.
- Y. Cui, V. Galvita, L. Rihko-Struckmann, H. Lorenz and K. Sundmacher, Appl. Catal., B, 2009, 90, 29–37 CrossRef CAS.
- A. Slagtern, Y. Schuurman, C. Leclercq, X. Verykios and C. Mirodatos, J. Catal., 1997, 172, 118–126 CrossRef CAS.
- A. Le Valant, N. Bion, F. Can, D. Duprez and F. Epron, Appl. Catal., B, 2010, 97(1–2), 72–81 CrossRef CAS.
- S. Adhikari, S. D. Fernando and A. Haryanto, Renewable Energy, 2008, 33(5), 1097–1100 CrossRef CAS.
- I. Rossetti, A. Gallo, V. Dal Santo, C. L. Bianchi, V. Nichele, M. Signoretto, E. Finocchio, G. Ramis and A. Di Michele, ChemCatChem, 2013, 5, 294 CrossRef CAS.
- T. Miyazawa, T. Kimura, J. Nishikawa, S. Kado, K. Kunimori and K. Tomishige, Catal. Today, 2006, 115, 254–262 CrossRef CAS.
- M. Inaba, K. Murata, M. Saito and I. Takahara, Energy Fuels, 2006, 20, 432–438 CrossRef CAS.
- L. He, S. Hu, L. Jiang, G. Liao, X. Chen, H. Han, L. Xiao, Q. Ren, Y. Wang, S. Su and J. Xiang, Fuel Process. Technol., 2018, 176, 7–14 CrossRef CAS.
- C. Wu, L. Dong, J. Onwudili, P. T. Williams and J. Huang, ACS Sustainable Chem. Eng., 2013, 1, 1083–1091 CrossRef CAS.
- M. Zhao, N. H. Florin and A. T. Harris, Appl. Catal., B, 2009, 92, 185–193 CrossRef CAS.
- D. Yao, H. Yang, H. Chen and P. T. Williams, Appl. Catal., B, 2018, 227, 477–487 CrossRef CAS.
- S. Bepari and D. Kuila, Int. J. Hydrogen Energy, 2020, 45, 18090–18113 CrossRef CAS.
- D. Yao, H. Yang, H. Chen and P. T. Williams, Appl. Catal., B, 2018, 239, 565–577 CrossRef CAS.
- M. U. Zahid, E. Pervaiz, A. Hussain, M. I. Shahzad and M. B. K. Niazi, Mater. Res. Express, 2018, 5, 052002 CrossRef.
- P. A. Simell, J. O. Hepola and A. O. I. Krause, Fuel, 1997, 76, 1117–1127 CrossRef CAS.
- A. M. Venezia, Catal. Today, 2003, 77, 359–370 CrossRef CAS.
- H. E. Lim, Y. Miyata, R. Kitaura, Y. Nishimura, Y. Nishimoto, S. Irle, J. H. Warner, H. Kataura and H. Shinohara, Nat. Commun., 2013, 4(1), 1–7 Search PubMed.
- L. Bokobza and J. Zhang, eXPRESS Polym. Lett., 2012, 6, 601–608 CrossRef CAS.
- N. Giannakeas, A. Lea-Langton, V. Dupont and M. V. Twigg, Appl. Catal., B, 2012, 126, 249–257 CrossRef CAS.
- C. Courson, E. Makaga, C. Petit and A. Kiennemann, Catal. Today, 2000, 63, 427–437 CrossRef CAS.
- Y. Zhang, Y. Tao, J. Huang and P. Williams, Waste Manage. Res., 2017, 35, 1045–1054 CrossRef CAS PubMed.
- X. Wang, M. Li, S. Li, H. Wang, S. Wang and X. Ma, Fuel Process. Technol., 2010, 91, 1812–1818 CrossRef CAS.
- Y. Zhang, J. Huang and P. T. Williams, Energy Fuels, 2017, 31, 8497–8504 CrossRef CAS.
- M. Kyari, A. Cunliffe and P. T. Williams, Energy Fuels, 2005, 19, 1165–1173 CrossRef CAS.
- I. de Marco Rodriguez, M. F. Laresgoiti, M. A. Cabrero, A. Torres, M. J. Chomón and B. Caballero, Fuel Process. Technol., 2001, 72, 9–22 CrossRef.
- A. M. Cunliffe and P. T. Williams, J. Anal. Appl. Pyrolysis, 1998, 44, 131–152 CrossRef CAS.
- D. Leung, X. Yin, Z. Zhao, B. Xu and Y. Chen, Fuel Process. Technol., 2002, 79, 141–155 CrossRef CAS.
- C. Roy, B. Labrecque and B. de Caumia, Resour., Conserv. Recycl., 1990, 4, 203–213 CrossRef.
- A. Napoli, Y. Soudais, D. Lecomte and S. Castillo, J. Anal. Appl. Pyrolysis, 1997, 40, 373–382 CrossRef.
- J.-H. Zhou, Z.-J. Sui, P. Li, D. Chen, Y.-C. Dai and W.-K. Yuan, Carbon, 2006, 44, 3255–3262 CrossRef CAS.
- T. Zhao, W. Sun, X. Gu, M. Rönning and D. Chen, Appl. Catal., A, 2007, 323, 135–146 CrossRef CAS.
- A. Magrez, J. W. Seo, R. Smajda, B. Korbely, J. C. Andresen, M. Mionic, S. Casimirius and L. s. Forró, ACS Nano, 2010, 4, 3702–3708 CrossRef CAS PubMed.
- Y. Zhang, J. M. Gregoire, R. Van Dover and A. J. Hart, J. Phys. Chem. C, 2010, 114, 6389–6395 CrossRef CAS.
- M. Motta, A. Moisala, I. Kinloch and A. Windle, J. Nanosci. Nanotechnol., 2008, 8, 2442–2449 CrossRef CAS PubMed.
- Y. Zhang, Hydrogen and carbon nano-materials from the pyrolysis-catalysis of wastes, University of Leeds, 2017 Search PubMed.
- Y. Zhang, M. A. Nahil, C. Wu and P. T. Williams, Environ. Technol., 2017, 38, 1–9 CrossRef PubMed.
- R. Schmuch, R. Wagner, G. Hörpel, T. Placke and M. Winter, Nat. Energy, 2018, 3, 267–278 CrossRef CAS.
- R. Fang, K. Chen, L. Yin, Z. Sun, F. Li and H. M. Cheng, Adv. Mater., 2019, 31, e1800863 CrossRef PubMed.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19–29 CrossRef CAS PubMed.
- L. Dai, D. W. Chang, J. B. Baek and W. Lu, Small, 2012, 8, 1122 CrossRef CAS.
- K. Evanoff, J. Khan, A. A. Balandin, A. Magasinski, W. J. Ready, T. F. Fuller and G. Yushin, Adv. Mater., 2012, 24, 533–537 CrossRef CAS PubMed.
- J. Wang, X. Yan, Z. Zhang, H. Ying, R. Guo, W. Yang and W. Q. Han, Adv. Funct. Mater., 2019, 29, 1904819 CrossRef.
- Y. Cao, A.-Q. Zhang, H. Zhang, G.-Q. Ding and L.-S. Zhang, Inorg. Chem. Commun., 2020, 111, 107633 CrossRef CAS.
- Y. Li, Y. Feng and W. Feng, Electrochim. Acta, 2013, 107, 343–349 CrossRef CAS.
- Y. Wu, J. Wang, K. Jiang and S. Fan, Front. Phys., 2014, 9, 351–369 CrossRef.
- M. Zhang, R. Lu, H. Yuan, K. Amin, L. Mao, W. Yan and Z. Wei, ACS Appl. Mater. Interfaces, 2019, 11, 20873–20880 CrossRef CAS PubMed.
- Q. Zhang, G. Peng, J. P. Mwizerwa, H. Wan, L. Cai, X. Xu and X. Yao, J. Mater. Chem. A, 2018, 6, 12098–12105 RSC.
- D. Gueon and J. H. Moon, Chem. Commun., 2019, 55, 361–364 RSC.
- W. Mao, G. Ai, Y. Dai, Y. Fu, Y. Ma, S. Shi, R. Soe, X. Zhang, D. Qu, Z. Tang and V. S. Battaglia, J. Power Sources, 2016, 310, 54–60 CrossRef CAS.
- Y. Li, X. Wu, C. Liu, S. Wang, P. Zhou, T. Zhou, Z. Miao, W. Xing, S. Zhuo and J. Zhou, J. Mater. Chem. A, 2019, 7, 7128–7137 RSC.
- L. Zhang, R. A. Senthil, J. Pan, A. Khan, X. Jin and Y. Sun, Ionics, 2019, 25, 4761–4773 CrossRef CAS.
- P. K. Nayak, L. Yang, W. Brehm and P. Adelhelm, Angew. Chem., 2018, 57, 102–120 CrossRef CAS PubMed.
- S. Zhang, G. Wang, B. Wang, J. Wang, J. Bai and H. Wang, Adv. Funct. Mater., 2020, 30, 2001592 CrossRef CAS.
- A. Banerjee, B. Ziv, E. Levi, Y. Shilina, S. Luski and D. Aurbach, J. Electrochem. Soc., 2016, 163, A1518–A1526 CrossRef CAS.
- T. Matsumoto, T. Komatsu, K. Arai, T. Yamazaki, M. Kijima, H. Shimizu, Y. Takasawa and J. Nakamura, Chem. Commun., 2004, 840–841 RSC.
- A. Le Goff, V. Artero, B. Jousselme, P. D. Tran, N. Guillet, R. Métayé, A. Fihri, S. Palacin and M. Fontecave, Science, 2009, 326, 1384–1387 CrossRef CAS PubMed.
- K. Gong, F. Du, Z. Xia, M. Durstock and L. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- A. Veksha, K. Yin, J. G. S. Moo, W. D. Oh, A. Ahamed, W. Q. Chen, P. Weerachanchai, A. Giannis and G. Lisak, J. Hazard. Mater., 2020, 387, 121256 CrossRef CAS PubMed.
- T. Kshetri, D. T. Tran, D. C. Nguyen, N. H. Kim, K.-t. Lau and J. H. Lee, Chem. Eng. J., 2020, 380, 122543 CrossRef CAS.
- A. Afzal, F. A. Abuilaiwi, A. Habib, M. Awais, S. B. Waje and M. A. Atieh, J. Power Sources, 2017, 352, 174–186 CrossRef CAS.
- Y. Liu, K. Shi and I. Zhitomirsky, Electrochim. Acta, 2017, 233, 142–150 CrossRef CAS.
- F. Zhou, Q. Liu, D. Kang, J. Gu, W. Zhang and D. Zhang, J. Mater. Chem. A, 2014, 2, 3505–3512 RSC.
- N. Rey-Raap, M. Enterria, J. I. Martins, M. F. R. Pereira and J. L. Figueiredo, ACS Appl. Mater. Interfaces, 2019, 11, 6066–6077 CrossRef CAS PubMed.
- H. Wu, J. Guo and D. a. Yang, J. Mater. Sci. Technol., 2020, 47, 169–176 CrossRef.
- X. Wen, X. Chen, N. Tian, J. Gong, J. Liu, M. H. Rummeli, P. K. Chu, E. Mijiwska and T. Tang, Environ. Sci. Technol., 2014, 48, 4048–4055 CrossRef CAS PubMed.
- N. X. Viet, S. Kishimoto and Y. Ohno, ACS Appl. Mater. Interfaces, 2019, 11, 6389–6395 CrossRef CAS PubMed.
- T. Rasheed, F. Nabeel, M. Adeel, K. Rizwan, M. Bilal and H. M. N. Iqbal, J. Mol. Liq., 2019, 292, 111425 CrossRef CAS.
- Z. S. Stojanović, E. Mehmeti, K. Kalcher, V. Guzsvány and D. M. Stanković, Food Anal. Methods, 2016, 9, 2701–2710 CrossRef.
- S. Najari, H. Bagheri, Z. Monsef-Khoshhesab, A. Hajian and A. Afkhami, Ionics, 2018, 24, 3209–3219 CrossRef CAS.
- W. Bauhofer and J. Z. Kovacs, Compos. Sci. Technol., 2009, 69, 1486–1498 CrossRef CAS.
- T.-W. Chou, L. Gao, E. T. Thostenson, Z. Zhang and J.-H. Byun, Compos. Sci. Technol., 2010, 70, 1–19 CrossRef CAS.
- F. Gojny, M. Wichmann, U. Köpke, B. Fiedler and K. Schulte, Compos. Sci. Technol., 2004, 64, 2363–2371 CrossRef CAS.
- T. Kashiwagi, F. Du, J. F. Douglas, K. I. Winey, R. H. Harris and J. R. Shields, Nat. Mater., 2005, 4, 928–933 CrossRef CAS PubMed.
- S. R. Bakshi and A. Agarwal, Carbon, 2011, 49, 533–544 CrossRef CAS.
- Z. Wu, Z. Chen, X. Du, J. M. Logan, J. Sippel, M. Nikolou, K. Kamaras, J. R. Reynolds, D. B. Tanner and A. F. Hebard, Science, 2004, 305, 1273–1276 CrossRef CAS PubMed.
- M. F. De Volder, S. H. Tawfick, R. H. Baughman and A. J. Hart, Science, 2013, 339(6119), 535–539 CrossRef CAS PubMed.
- P. T. Williams, S. Besler and D. T. Taylor, Proc. Inst. Mech. Eng., Part A, 1993, 207, 55–63 CrossRef.
- J. D. Martínez, N. Puy, R. Murillo, T. García, M. V. Navarro and A. M. Mastral, Renewable Sustainable Energy Rev., 2013, 23, 179–213 CrossRef.
- A. Evans and R. Evans, The Composition of a Tyre: Typical Components, the Waste & Resource Action Programme, Banbury, 2006 Search PubMed.
- S. De and J. N. Coleman, MRS Bull., 2011, 36, 774–781 CrossRef CAS.
- X. Liu, B. Shen, Z. Wu, C. M. Parlett, Z. Han, A. George, P. Yuan, D. Patel and C. Wu, Chem. Eng. Sci., 2018, 192, 882–891 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |